
COCIR/EUROM VI contribution on the Revision of the ...
36
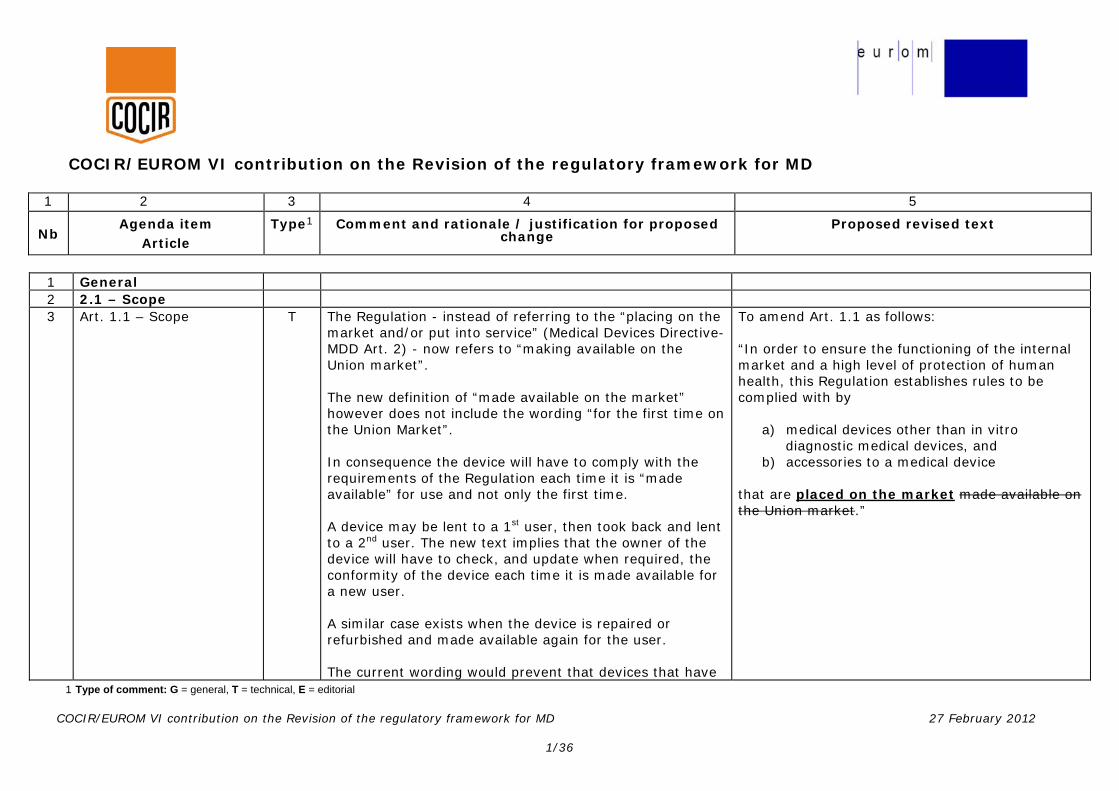
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 1 2 3 4 5 Nb Agenda item Article Type 1 Comment and rationale / justification for proposed change Proposed revised text 1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012 1/36 1 General 2 2.1 – Scope 3 Art. 1.1 – Scope T The Regulation - instead of referring to the “placing on the market and/or put into service” (Medical Devices Directive- MDD Art. 2) - now refers to “making available on the Union market”. The new definition of “made available on the market” however does not include the wording “for the first time on the Union Market”. In consequence the device will have to comply with the requirements of the Regulation each time it is “made available” for use and not only the first time. A device may be lent to a 1 st user, then took back and lent to a 2 nd user. The new text implies that the owner of the device will have to check, and update when required, the conformity of the device each time it is made available for a new user. A similar case exists when the device is repaired or refurbished and made available again for the user. The current wording would prevent that devices that have To amend Art. 1.1 as follows: “In order to ensure the functioning of the internal market and a high level of protection of human health, this Regulation establishes rules to be complied with by a) medical devices other than in vitro diagnostic medical devices, and b) accessories to a medical device that are placed on the market made available on the Union market .”
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of COCIR/EUROM VI contribution on the Revision of the ...
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
1/36
1 General 2 2.1 – Scope 3 Art. 1.1 – Scope
T The Regulation - instead of referring to the “placing on the
market and/or put into service” (Medical Devices Directive-MDD Art. 2) - now refers to “making available on the Union market”. The new definition of “made available on the market” however does not include the wording “for the first time on the Union Market”. In consequence the device will have to comply with the requirements of the Regulation each time it is “made available” for use and not only the first time. A device may be lent to a 1st user, then took back and lent to a 2nd user. The new text implies that the owner of the device will have to check, and update when required, the conformity of the device each time it is made available for a new user. A similar case exists when the device is repaired or refurbished and made available again for the user. The current wording would prevent that devices that have
To amend Art. 1.1 as follows: “In order to ensure the functioning of the internal market and a high level of protection of human health, this Regulation establishes rules to be complied with by
a) medical devices other than in vitro diagnostic medical devices, and
b) accessories to a medical device that are placed on the market made available on the Union market.”
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
2/36
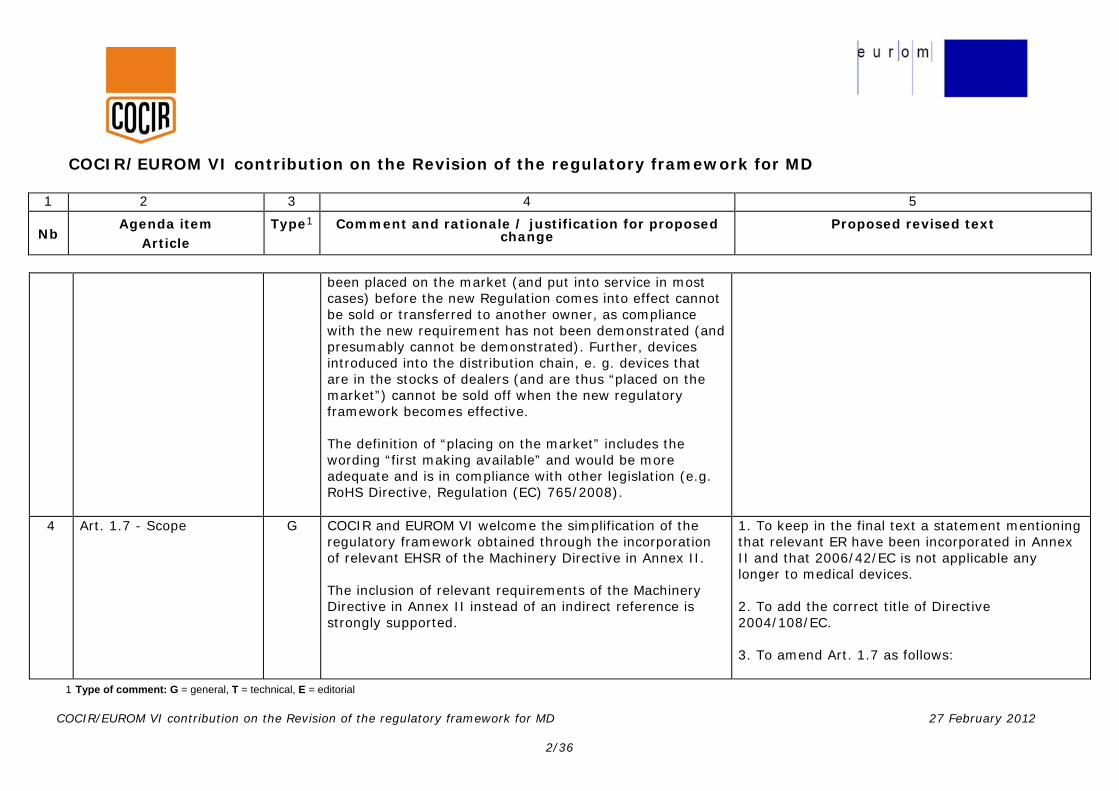
been placed on the market (and put into service in most cases) before the new Regulation comes into effect cannot be sold or transferred to another owner, as compliance with the new requirement has not been demonstrated (and presumably cannot be demonstrated). Further, devices introduced into the distribution chain, e. g. devices that are in the stocks of dealers (and are thus “placed on the market”) cannot be sold off when the new regulatory framework becomes effective. The definition of “placing on the market” includes the wording “first making available” and would be more adequate and is in compliance with other legislation (e.g. RoHS Directive, Regulation (EC) 765/2008).
4 Art. 1.7 - Scope G COCIR and EUROM VI welcome the simplification of the regulatory framework obtained through the incorporation of relevant EHSR of the Machinery Directive in Annex II. The inclusion of relevant requirements of the Machinery Directive in Annex II instead of an indirect reference is strongly supported.
1. To keep in the final text a statement mentioning that relevant ER have been incorporated in Annex II and that 2006/42/EC is not applicable any longer to medical devices. 2. To add the correct title of Directive 2004/108/EC. 3. To amend Art. 1.7 as follows:

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
3/36
“This Regulation is a specific Union legislation within the meaning of Article 1(4) of Directive 2004/108/EC of the European Parliament and of the Council on the approximation of the laws of the Member States relating to personal protective equipment electromagnetic compatibility (EMC) and within the meaning of Article 3 of Directive 2006/42/EC of the European Parliament and of the Council of 17 May 2006 on machinery. Relevant specific ER of the Machinery Directive and of EMC Directive are set out in Annex II.”
5 Art. 2 – Definitions (b) "accessory"
T The proposed definition leaves the room for interpretation related to major components, which are necessary for using a Medical Devices in accordance with their primary intended use. It should be made clear that an accessory is not necessary to qualify a device as a medical device while it depends on the product design defined by the manufacturer; in such case it would be in most of the cases an integral part of the Medical Device and should be considered as a component/part of this device.
To amend as follows: “(b) accessory to a ‘parent’ medical device” means an article which, whilst not being a medical device, is intended specifically by its manufacturer to be used together with a particular device to enable in conjunction with one or several medical devices to enable or assist the device to be used in accordance with its intended use;”
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
4/36
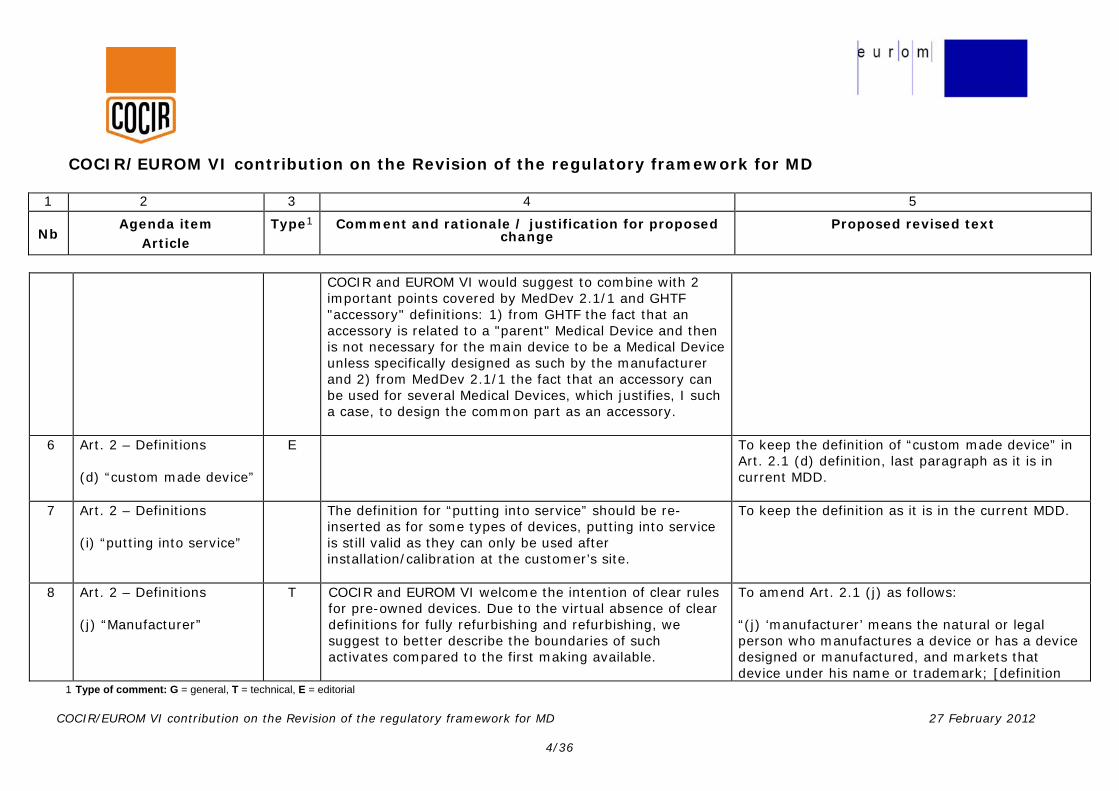
COCIR and EUROM VI would suggest to combine with 2 important points covered by MedDev 2.1/1 and GHTF "accessory" definitions: 1) from GHTF the fact that an accessory is related to a "parent" Medical Device and then is not necessary for the main device to be a Medical Device unless specifically designed as such by the manufacturer and 2) from MedDev 2.1/1 the fact that an accessory can be used for several Medical Devices, which justifies, I such a case, to design the common part as an accessory.
6 Art. 2 – Definitions (d) “custom made device”
E To keep the definition of “custom made device” in Art. 2.1 (d) definition, last paragraph as it is in current MDD.
7 Art. 2 – Definitions (i) “putting into service”
The definition for “putting into service” should be re-inserted as for some types of devices, putting into service is still valid as they can only be used after installation/calibration at the customer’s site.
To keep the definition as it is in the current MDD.
8 Art. 2 – Definitions (j) “Manufacturer”
T COCIR and EUROM VI welcome the intention of clear rules for pre-owned devices. Due to the virtual absence of clear definitions for fully refurbishing and refurbishing, we suggest to better describe the boundaries of such activates compared to the first making available.
To amend Art. 2.1 (j) as follows: “(j) ‘manufacturer’ means the natural or legal person who manufactures a device or has a device designed or manufactured, and markets that device under his name or trademark; [definition
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
5/36
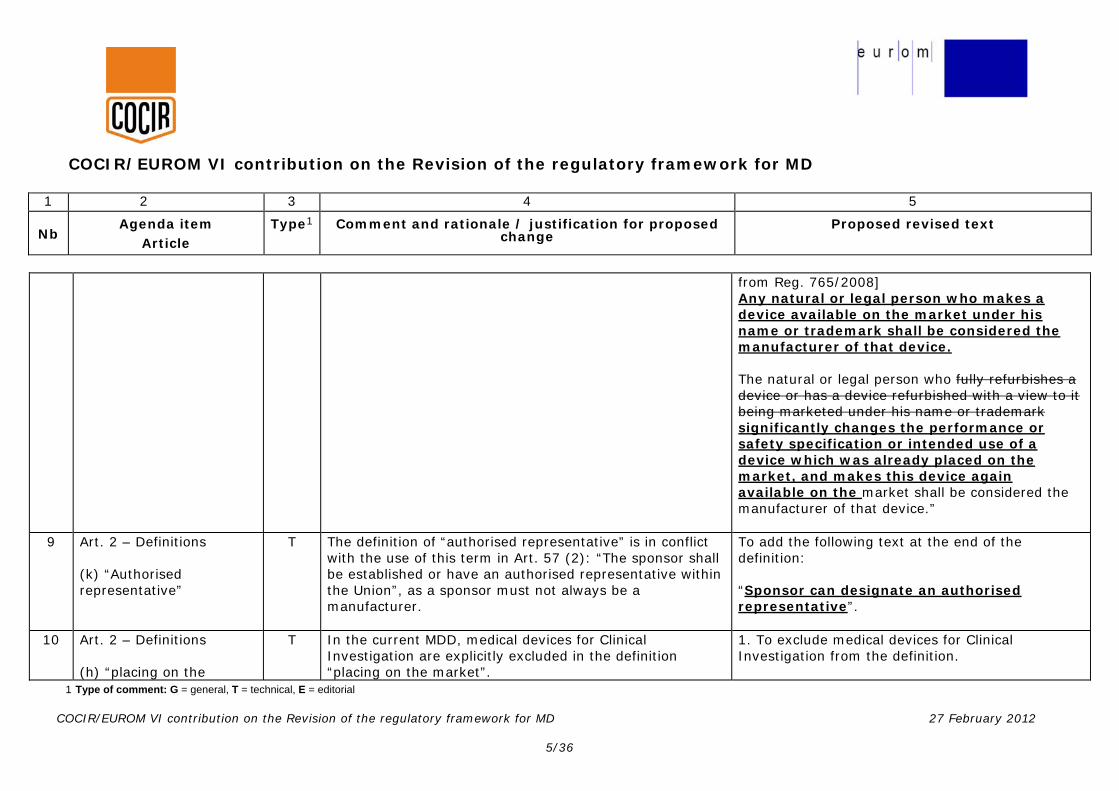
from Reg. 765/2008] Any natural or legal person who makes a device available on the market under his name or trademark shall be considered the manufacturer of that device. The natural or legal person who fully refurbishes a device or has a device refurbished with a view to it being marketed under his name or trademark significantly changes the performance or safety specification or intended use of a device which was already placed on the market, and makes this device again available on the market shall be considered the manufacturer of that device.”
9 Art. 2 – Definitions (k) “Authorised representative”
T The definition of “authorised representative” is in conflict with the use of this term in Art. 57 (2): “The sponsor shall be established or have an authorised representative within the Union”, as a sponsor must not always be a manufacturer.
To add the following text at the end of the definition: “Sponsor can designate an authorised representative”.
10 Art. 2 – Definitions (h) “placing on the
T In the current MDD, medical devices for Clinical Investigation are explicitly excluded in the definition “placing on the market”.
1. To exclude medical devices for Clinical Investigation from the definition.
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
6/36
market” 2. To add the following text at the end of the definition: “other than a device intended for clinical investigation".
11 Art. 2 – Definitions (s) “recall”
The definition of recall does not cover the correction of a device on site. “Recall” refers only to device returned back but not to corrections made on site.
To add the following definition of “field safety corrective action” as used in Art. 44 (1) b) or as used in current MEDDEV 2.12-1 rev 6/7: “A field safety corrective action is an action taken by a manufacturer to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market.”
12 Art. 2 - Definitions Deletion of (p) “devices subcategory” and (q) “generic device group”
T Definitions “devices subcategory” and “generic device group” have been deleted and are intended to be incorporated by a separate implementing act. In the current MDD these definitions constitute the basis for Notified Bodies sampling procedure.
To clarify why the definitions have been taken out and how is planned proceed with this issue.
13 Art. 18 - Systems and procedure packs
T Article 18.1 second indent of the proposed text enhances the scope of Article 12 in the current MDD to other products which are not in the scope of this Regulation (not
To either clarifying the scope in Art. 18.1 of the proposed text, or deleting the following text in Art 18.2: “where the system or procedure pack

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
7/36
necessarily bearing a CE mark, but in conformity with the applicable legislation). The term ‘other products’ needs further clarification. The enhancement to other products not bearing a CE mark may be in conflict with Art. 18.2 with regard to “as in cases where the system or procedure pack incorporate devices which do not bear a CE marking”.
incorporate medical devices which do not bear a CE marking or”.
14 Art. 18 - Systems and procedure packs
G The new Art. 18 does not cover the possibility for the assembler of a system or procedure pack to sterilize this pack in accordance with the original manufacturer’s instructions.
If not covered elsewhere in the completed proposed text, to re-integrate the text from Art. 12.3 of the current MDD.
15 Art. 18a – Parts and components
T Definitions for part and components (P&C) are missing. They are different from accessories to a medical device and thus should not be considered per se as medical devices in their own right even if from a 3rd party supplier, also because: - There is no classification rule adapted for these P&C, nor are there conformity assessment schemes or (harmonized) standards applicable to them.
1. To add “on the market” to be inline with the definition of “making available on the market” in Article 1 (g). 2. To replace “article” by “item” as this might be confused with definitions in other directives, e.g., REACH, where it has a different meaning. 3. To amend as follows:

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
8/36
- Enforcement of the original text is almost impossible (and the MDD does not impose obligations on the user of the device, i.e., the MDD does not forbid replacing parts by fake or unsafe replacements or to forsake maintenance schedules that are part of risk reduction schemes, etc.) If such an article is considered a device in its own right a (fragmentary) conformity assessment procedure for the part or component should be performed. It is unclear how the resulting modification after integration of the part/component into the device shall be made obvious.
“1. Any natural or legal person who makes available on the market an article item intended to specifically replace an identical or similar integral part or component of a device that is defective or worn in order to maintain or re-establish the functionality of the device without significantly changing its characteristics or performances, shall ensure that the article item does not negatively affect the safety and performance of the device. If the item is made available on the market outside the responsibility of the manufacturer of the device, a declaration of equivalence shall be made up by the person making this item available on the market, stating that the item does not negatively affect the safety and performance of the device. Substantiating evidence shall be available to the competent authorities. 2. In the following cases, an article shall be considered a device in its own right:
a) where an article referred to in paragraph 1 is manufactured and/or placed on the

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
9/36
market outside the responsibility of the manufacturer of the device, or
b) b) where an article, intended to specifically replace a part of a device, significantly changes the characteristics or performances of the device.
2. Where an item, intended to specifically replace a part or component of a device, significantly changes the characteristics or performances of the device, it shall be considered a device in its own right.”
16 Annex I - List of products covered by Article 2.1(a)
T Point 5. Surgical laser equipment: the term “surgical” already indicates a medical purpose
To amend point 5 as follows: “Surgical Laser equipment for invasive procedure”.
17 2.2. - Clinical investigations, clinical evaluation, and post-market surveillance plan, incl. post-market clinical follow-up
18 Art. 4 - Making available T Only Member State can ensure that the requirements laid 1. To amend title of Art. 4 as follows:

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
10/36
on the market (related to Art. 1 point 1, Art. 2 (g), (h)
down in this Regulation are ultimately being respected. This is in line with the current Art. 2.
See rationale in Art. 1.1 – Scope (line 3).
“Making available Placing on the market.” 2. To amend Art. 4.1. as follows: “1. Member States shall take all necessary steps to ensure that a device may only be placed made available on the market when it complies with the requirements laid down in this Regulation when duly supplied and properly installed, maintained and used in accordance with its intended purpose”. 3. To amend Art. 4.2. as follows: “2. In particular, a device placed made available on the market must meet the essential requirements set out in Annex II which apply to it, taking into account the intended purpose of the device concerned.”
19 Art. 57 - General requirements regarding clinical investigations
T Section 1 of art. 57 states that Clinical investigations (CI) that do not aim at generating clinical data for CE marking are not covered by the Regulation. This would leave the door open to national requirements
To draft a new MEDDEV addressing other CI which are not CE relevant (pre- or post-market), in analogy to the MEDDEV 2.12/2 rev 2 on PMCF that was just released.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
11/36
for the other types of investigation, like Post market Clinical Follow-up (PCMF) studies, investigations on CE marked devices, etc. The Regulation should cover all the investigations and define the different levels of controls for the different types of clinical trials, in order to ensure harmonization and avoid fragmentation among Member States.
20 Art. 57 - General requirements regarding clinical investigations Art. 57.1 (b): “To verify that devices achieve the intended benefits to the patient as specified by the manufacturer”.
T This requirement refers directly to claims and could lead to an increased number of Clinical Investigations.
To clarify the definition of “intended benefits to the patient”.
21 Art. 58 - Application for clinical investigations
T Annex XV, Part B (1) last indent: “This statement may be supported by an attestation issued by a notified body.” In combination with Annex XV, Part B point 3, “The sponsor must ensure that the assessment, or audit where
In Art. 58.3: 1. to replace "and" by "or": “In the case of devices falling within class III or implantable or long-term invasive devices falling within class IIa or IIb, the competent authorities

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
12/36
necessary, of the effectiveness of these measures are authorised.” In this case, i.e. attestation of statement by NB and e.g. the certification of the manufacturers QMS by a NB, the competent authority should replace or at least reduce the assessment referred in Art. 58.3., as it significantly reduces the workload of the CA. As good practice this would avoid double investigations/assessments. Art. 58.3. states that “In the case of devices falling within class III and implantable and long-term invasive devices falling within class IIa or IIb, the competent authorities shall, within the period mentioned in paragraph 2, carry out an assessment of the application, and in particular as regards the sponsor's statement referred to in Annex XV that the device in question conforms to the essential requirements…”
shall, […]”. 2. To replace “may” by “shall”: “… the competent authorities of the Member States may shall, where appropriate, enter into agreements of work-sharing and administrative assistance between each others.” 3. To introduce the following text after the sentence ending by “…every precaution has been taken to protect the health and safety of the patient.”: “In case the sponsor fulfils Annex XV, Part B point 1. last sentence and Annex XV, Part B point 3, the competent authorities shall consider this in their assessment of the application.”
22 Art. 61 - Reporting of events occurring during clinical investigations
We welcome the statement “When setting up the database mentioned in the previous sentence, registers for clinical investigations established at international level shall be taken into account with a view to avoid duplication of registration of clinical investigations” as this reduces workload to sponsors. However, it is unclear how clinical
1. To amend Art. 61.1 (a) as follows: “a) serious unanticipated adverse event, or” 2. The definitions provided in Art. 61.2 should be listed in Art. 2.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
13/36
investigations shall be registered if performed in one or more Member States and outside the EU. Art. 61 (a): The words “of any serious adverse event” are not clear. Better is the term “of any serious unanticipated adverse event” (see 21 Code of Federal Regulation (CFR) 812). Reports based on “any serious adverse event” will result in a tremendous number of reports, especially in combination with the requirement of immediate reporting.
3. The paragraph following Art. 61.2 (c) “In respect to subjects to a clinical investigation …” is not easy to understand.
23 Annex XV Part A : Clinical investigations
To align point 2.5 with “single notification” procedure of Art. 60.
To amend Annex XV, point 2.5 as follows: “All serious adverse events must be fully recorded and immediately notified to the competent authority of the Member States in which the incident occurred in which the Clinical investigation is being performed. Where a clinical investigation is being conducted in more than one Member State, the Commission shall, in collaboration with the Member States, ensure that a report of an adverse event received by electronic means, is transmitted electronically to the competent authorities of
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
14/36
the other Member States concerned.”
24 Annex XV Part B : Statement concerning devices intended for clinical investigation
E See comments in line 21.
1. To insert numbering at the beginning of the section: “Part B: statement concerning devices intended for clinical investigation 1.” 2. To insert numbering at the beginning of the section: “- the results of the design calculations, and of the inspections and technical tests carried out, etc. 3. The sponsor must ensure that all necessary….”
25 2.3 – Essential requirements
26 Art. 4 - Making available on the market
See comments on Art. 1 (line 3) and on clinical investigations (line 21).
27 Annex II - Essential safety and performance requirements
T The definition of “state of the art” is missing.
To add the definition from EN ISO 14971: “State of the art” means what is currently and

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
15/36
generally accepted as good practice. Various methods can be used to determine "state of the art" for a particular medical device. Examples are: • standards used for the same or similar
devices; • best practices as used in other devices of
the same or similar type; • results of accepted scientific research. State of the art does not necessarily mean the most technologically advanced solution."
28 Annex II - Essential safety and performance requirements ER2
G 1. The wording changed from “as far as possible” to “as far as reasonably practicable” is a very useful clarification of what can be expected in a practical risk reduction process, and is consistent with international definitions. 2. The step indicated under (a) is a necessary step in risk management and is not a step in a priority scheme for implementation of risk reduction.
1. To maintain the wording “as far as reasonably practicable” instead of “as far as possible” and amend other ERs, where necessary for consistency (as proposed in comments below) 2. To consider blending (a) in body of text of ER 2.
29 ER2 d) G A reference to training as one possible risk reduction step should be included.
To amend as follows: "d) inform users of any residual risks including,

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
16/36
where appropriate, the need for specific training".
30 ER6 G The wording is not consistent with the risk reduction. To insert the following text: “shall be minimised as far as reasonably practicable and be acceptable”.
31 ER7.2 G The wording is not consistent with the risk reduction. To insert the following text: “…such a way as to minimize as far as reasonably practicable the risk posed by...”
32 ER7.3 G Incorrect wording. To amend as follows: “with the materials and substances, including gases, with”
33 ER7.4 G To move the second and third paragraphs to the respective labeling ERs.
34 ER7.6 G Consistency with ER7.5 To amend the text as follows: “such a way as to reduce as far as reasonably
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
17/36
practicable the risks linked”.
35 ER10.2 (a) T Not all animals can meet the requirement stipulated in this paragraph, e.g. sharks.
To amend as follows: “Where appropriate, tissues and cells of animal origin shall originate from animals that have been subjected …”
36 ER11.1 G No definition of “equipment”. The wording is not consistent with the risk reduction.
To amend the text as follows: “such a way as to minimize as far as reasonably practicable all possible risks from incorrect connection.”
37 ER 11.2 c) E The listing can lead to different interpretations (see brackets): …(radiation associated with diagnostic or therapeutic procedures, pressure, humidity, temperature, variations in pressure) and acceleration) or radio signal interferences)
To restructure the text as follows: “c) risks connected with reasonably foreseeable external influences or environmental conditions, such as - magnetic fields, - external electrical and electromagnetic effects, - electrostatic discharge, - radiation associated with diagnostic or therapeutic procedures, - pressure,
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
18/36
- humidity, - temperature, variations in pressure and acceleration, - or radio signal interferences;”
38 ER11.3 G The wording is not consistent with the risk reduction. To amend as follows: “…such a way as to minimise as far as reasonably practicable the risks of fire or explosion...”
39 ER11.3 G The phrase “single fault condition” is introduced here, as well as in a few other instances. It is not clear if this has the same meaning as in EN 60601-1.
To be clarified.
40 ER11.6 G No ER on interoperability is necessary as it is already addressed by the compatibility statements made in the Instructions for Use (IfU)'s of the device. Otherwise, all compatibility types should be addressed individually with the risk to forget some not yet known.
To remove 11.6.
41 ER13.2 (a) G The applicability of this ER is unclear. It mentions “for a specific medical purpose” that can be interpreted as devices of Annex I being excluded from the scope of this
To be clarified.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
19/36
ER.
42 ER14.1 G “Single fault condition” for standalone software is unclear.
To be clarified.
43 ER17.2 G The wording is not consistent with the risk reduction To amend as follows: “…to prevent, as far as reasonably practicable, the accidental...”
44 ER19.1 (f) E To change “offset out” to “set out”. “… under the conditions offset out in Commission Regulation (EU) …”
45 ER 19.2 (b) T “...identify the device and its use.” It is unclear what additional requirement “its use” means: i.e. should we indicate for an Magnetic Resonance (MR) equipment that it is used for image acquisition? Since the name or trade name of the device is required per 19.2.a), it is to clarify what particulars are covered now by 19.2 b). In case the “intended use” is meant by this, that is already
In 19.2 (b) to delete: “… and its use.”
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
20/36
stated in the IFU. It would create an unnecessary burden to translate the “use” into all EU languages on the label.
46 ER19.2 (d) T The necessity to add local label related to importer should not be needed. The authorised representative should remain sufficient. Furthermore, in case of parallel imports the authorised representative will usually not know the importer. From an importer’s point of view - who is not the authorised representative - placing on the market occurs with release for free circulation by customs (Interpretative Document of the Commission’s Services on Placing on the Market of Medical Devices, 2010-11-16). To fulfill the requirements of ER19.2d) the importer would have to place his label on the devices as long as they are in customs quarantine. This article would establish rules which are a disadvantage for manufacturers based outside the EU. Note: Requirement from Decision 768/2008/EC (Art. R14)
To delete the requirement for a label of the importer.
47 ER19.2 (i)
T The requirement for providing the year of manufacture was limited in the previous MDD to active devices. Very similar requirement is as well already defined in EN
To keep the original text of actual MDD: “(l) year of manufacture for active devices other

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
21/36
1041 (not mandatory). Mandatory information on manufacturing year will not lead to more safe devices or useful information for users.
than those covered by (19.2 h). This indication may be included in the batch or serial number;”
48 ER19.3 (a) E The reference to “17.1 a) seems incorrect.
To change “17.1 a)…” to “19.1 a)…”
49 19.3 (s) E The wording could be improved to make it clearer. To amend as follows: “Date of issue or the latest revision status of the instructions for use. If appropriate, a document identification number shall be added.”
50 2.4 – Classification rules
51 Art. 35 - Classification of medical devices
G This ER needs more details on the process of escalation, such as who, and how, as well as timing aspects, whether the national CA can decide by itself or not, etc. While such details might be considered for a guidance document, industry believes it IS appropriate to have this directly in the legal text for maximum transparency and coherence across the EU.
To amend Art. 35.2 as follows: “In the event of a dispute between the manufacturer or his authorised representative and the notified body concerned, resulting from the application of the classification rules, the matter shall be referred by both parties jointly for decision to the national authority responsible for the notified body. Within 60 calendar days

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
22/36
upon receipt of the matter, the competent authority shall submit its envisaged decision to the MDEG and inform the involved manufacturer and notified body. If the envisaged decision is not challenged by MDEG within 30 calendar days, it will become final. Prior to any decision, the competent authority shall submit its envisaged decision to the MDEG.”
52 Annex IX - Classification rules II. Implementing rules for the classification rules 3. Stand alone software
“3. Stand alone software, which drives a device or influences the use of a device, is classified according to the intended use of the combination.” The new intended use of the combination is already incorporated in the intended use of the stand alone software. Thus, any new risks that might occur in the combination are foreseeable during design of the stand alone software. The stand alone software risk management process ensures that foreseeable risks – in the context of its foreseeable use - are addressed. Further, the MEDDEV 2.1/6 of January 2012 also suggests differently. Finally, the case where a software is included in a device
To amend as follows: “2.3. Stand alone Software, which drives a device or influences the use of a device, falls automatically in the same class. If stand alone software is independent of any other device, it is classified in its own right.”
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
23/36
and drives another device is covered by the current MDD and is not covered anymore in the new proposal.
53 Annex IX Classification rules III. Classification rules 2.4. Rule 8, 5th dash
Only those accessories that are necessary for the proper functioning of the active implantable device should be classified as class III.
To amend as follows:
“- intended to be active implantable medical devices, or an accessory to an active implantable medical device necessary for the proper functioning of the active implantable device, in which case they are in class III,”
54 3.2. Rule 10 T Definitions for “Direct Diagnosis” and “Direct Monitoring” are needed. Please refer to Joint medical industry interpretation of the term “direct diagnosis” (Doc. Feb.2012/SANCO/BC/06) An active device is defined in MDD Annex IX, point 1.6: “Any active medical device, whether used alone or in combination with other medical devices, to supply information for detecting, diagnosing, monitoring or treating physiological conditions, states of health, illnesses or congenital deformities.”
This is to be interpreted as to supply information so a
To add the following definition: “A device is considered to allow direct diagnosis when it provides the diagnosis of the disease or condition by itself or when it provides decisive information for the diagnosis.”

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
24/36
medical professional can use the information for the purposes mentioned (detecting, etc.). “Diagnosis” is generally and commonly interpreted as “the process of attempting to determine and/or identify a possible disease or condition and determine treatment. It includes follow-up of the progression of the disease, condition and treatment.”
The diagnostic process begins by observing the patient for specific signs and symptoms and by taking a specific history, e.g. how did these signs and symptoms come about, etc. Specific signs, symptoms and historical clues allow the physician to perform a specific physical examination and order specific diagnostic investigations. The physician usually formulates a "short list" of likely diagnoses and requests further testing to confirm or rule-out competing diagnoses before providing treatment.
“Direct” is to be interpreted with regard to completeness, i.e., without the necessity to acquire or take into account additional information. Then diagnosis can be made by a medical professional or by the medical device itself. Note that the information available may not be absolutely

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
25/36
complete (i.e. other parameters may be measured, the anamnesis may not be complete), but that the information is sufficient to imply a specific diagnosis. A device may provide information with varying medical relevance: • Indicative information: the information can be used in
a decision tree along with several other clinical and technical patient data for a healthcare professional to arrive at a diagnosis.
• Decisive information: the information is one of the critical elements in determining the diagnosis.
A device is considered to “allow direct diagnosis” when it provides the diagnosis of the disease or condition by itself or when it provides decisive information for the diagnosis. Indicative information is not sufficient to imply a “direct diagnosis”.
55 Annex IX - Classification rules III. Classification rules 2. Invasive devices
T Rule 5 last paragraph: • “biological effect” needs either to be defined or
deleted. • If the intention is to create a special rule for ingested
devices, it should be considered to extend this rule to administration routes which potentially have a similar impact, such as inhalation and rectal administration.
To amend as follows: “All invasive devices with respect to body orifices, other than surgically invasive devices, that are intended to have a biological effect or that are intended to be orally ingested, inhaled or administered rectally and to be wholly or mainly

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
26/36
2.1 Rule 5 • It should be avoided to unintentionally upclassify products such as artificial tears or comfort drops.
• The text could be clarified by mentioning the administration route instead of the body orifice.
absorbed, are in Class IIb if they are for transient use, and in class III if they are for short-term or long-term use.”
56 Annex IX - Classification rules III. Classification rules 4. Special rules 4.4. Rule 16
T The use of X-ray photographic material is less extensive than it used to be, but still substantial throughout the EU.
It is justified to maintain rule 16.
57 2.5 – Conformity assessment procedures
58 Art. 36 – Conformity assessment
E
10. Design dossier for class lla and llb devices. At the end of Art. 36 referring to Art. 12.3, sterilization of procedure packs under this article is a current practice. Deleting it would impact products currently on the market. We welcome the fact that for class I devices which are placed on the market in sterile condition or have a measuring function, a notified body can be involved
1. To replace “design dossier” by “technical documentation”. 2. See comment for Art. 18 (line 14).
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
27/36
according to Annex XII or Annex X (full quality management system), as the latter is formally not the case in the current MDD.
59 Art. 38 (a) – Rotation of Notified bodies involvement
G The concept of rotation of Notified Bodies should be abandoned, as the Notify Body number is printed with the CE mark on the device and its labeling. Changing Notify Body would require changing of the labeling, an activity that requires usually a long time and lots of resources with no additional benefit for the patient. Further, such requirement would void out any planning of Notified Bodies staff. It would make more sense to require that Notified Bodies should have minimum personnel to allow for rotation within their staff.
To delete this article.
60 Art. 39 - Mechanism for the scrutiny of certain conformity assessments
T/G 1. Art. 39 introduces the possibility for MDEG or Commission to comment on the preliminary assessment of class III devices conducted by the Notified Bodies. This will in practice establish a pre-market approval procedure that will substantially delay the placing on the market. Art. 39. 1, 2, 3 apply to applications for new class III devices: Any comments and additional requirements or requests received in this phase of the development of a
1. To delete Art. 39 or alternatively to include in Article 70 - Scientific Advice - an early advice procedure. 2. To move Art. 39.4 under the section on market Surveillance.
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
28/36
medical device are too late in the process. The consultation should happen earlier in the process, and cannot be part of the formal conformity assessment procedure. 2. Point 4: It is difficult to understand how this measure will affect conformity assessment, as the trigger for such a decision would be devices already on the market i.e. for which the conformity assessment has already been completed. The additional review is based on evidence or data coming from market surveillance and cannot be part of the conformity assessment procedure.
61 Art. 43 – Certificate of free sale
Art. 43 (2) Certificates of free sale issued by notified bodies are not expected to be accepted by most of the relevant countries.
To delete the delegation to notified bodies.
62 Annex V - EC Declaration of conformity
Having the unique identification number of a product on the Declaration of Conformity (including serial no.) will not be practicable for mass produced devices. Product or trade name, or all trade names are not a requirement in Decision 768/2008/EC Annex III (recommended to delete this requirement).
To be clarified.
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
29/36
The generic requirements taken from Decision 768/2008/EC Annex III should be adapted to the medical devices in a pragmatic manner. If the corresponding legislation identifies the manufacturer as the only responsible for his products it is not necessary to have “under our sole responsibility” on the Declaration of Conformity. The conformity assessment procedure shall demonstrate compliance with the Essential Safety and Performance Requirements, and only this should be stated on the Declaration of Conformity. If harmonized standards have been used to demonstrate compliance with the ERs, this may be stated on e.g. a separate document, but should not be mandatory part of the Declaration of Conformity.
63 Annex V EC Declaration of conformity
T Standards are listed in the Technical Documentation (see also Blue Guide (2000) point 5.2), which are assessed by the NB (see Annex II, point 7.2). The manufacturer is further obliged to conduct market surveillance activities incl. monitoring of applicable standards assuring that the products fulfils the Essential Requirements, hence the state of the art.
To delete requirement 7.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
30/36
Thus, there is no additional safety benefit by repeating on the DoC the relevant harmonised standards. It must be also considered, that the requirement forces to update the DoC whenever a harmonized standard is revised. This too does not increase the safety of a device. For highly innovative products, where no relevant harmonised standards exists, it could be a disadvantage for a manufacturer who cannot reference to a harmonized standard e.g. when the product is more state of the art than the standard.
64 Annex X - Conformity assessment based on full quality assurance plus design examination
4.4: Unannounced visits by the Notified Body should not be a routine practice, but the exception.
To keep the wording as in the current MDD (Annex II, 5.4): “The notified body may pay unannounced visits to the manufacturer.”
65 Annex X - Conformity assessment based on full quality assurance plus design examination Chapter II 6.1 d) and 6.2 b)
The time intervals of 90 days or 210 days for the medicinal products competent authority and/or EMA to issue an opinion is too long.
To shorten the time intervals.
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
31/36
66 Annex XIV - Minimum
content of certificates issued by a notified body
T The data listed in point 6. may be required for product related certificates according to Annexes X Chapter II, XI or XII Part B, but not all these data (e.g. intended purpose of the devices, GMDN codes) make sense in case of certificates related to the quality assurance system of manufacturers (e.g. certificates according to Annex X Chapter I, Annex XII Part A).
To differentiate between certificates related to the manufacturer’s quality assurance system and certificates related to devices, according to NBOG BPG 2010-3.
67 3.1 – Roles and obligations of economic operators
68 Art. 7.9 - Obligations of the manufacturer
T The requirement in Art. 7.9: “Manufacturers shall, further to a reasoned request from a competent authority, provide it with information and documentation … in an official Union language which can be easily understood by that authority” may represent an unnecessary burden for manufacturers. In general, English language is accepted.
To add the following text: “Generally, information provided in English shall be accepted.”
69 Art. 7.10 - Obligations of the manufacturer
A manufacturer (own brand labeler) cannot legally be required to provide data or information related to devices not in his responsibility, as these data or information is owned by the Original Equipment manufacturer and may
To delete Art. 7.10.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
32/36
be confidential.
70 Art. 8 - Authorised representative
T Art. 8.3: Manufacturers should not be able to delegate to the authorised representative the actions listed in paragraphs 5 and 6 as providing proper labeling falls under Art. 7.1 (“… manufactured in accordance with the requirements of this Regulation”) which cannot be delegated.
Delegation of such activities would require that they must be performed under the manufacturer’s quality management system (“extended work bench”).
To amend Art. 8.3, second sentence as follows: “…the obligations of the manufacturer laid down in Article 7.1, 2, 4, 5 and 6 shall not form part of the authorised representative’s mandate.”
71 Art. 9 - Obligations of importers
T 1. Art. 9.3: There is no need that the importer appears on the label; this would only foster parallel trade without manufacturer and authorized representative being involved; to show the manufacturer and the authorized representative on the label provides all information that is required by users, patients and authorities; further, it appears not practicable in many cases to have three different addresses on the label of small medical devices). 2. Art. 9.5: The tasks listed in Art. 9.5 are in the responsibility of the manufacturer and can usually only be
1. To delete Art. 9.3. 2. To delete Art. 9.5.

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
33/36
performed by the manufacturer because of special knowledge and equipment needed. 3. Art. 9.6: Importers should not act without having a clear mandate from the responsible manufacturer (or from the competent authority). Importers shall by no means be allowed to perform any changes on the devices or perform any corrective measures without having a clear order from the responsible manufacturer. 4. Art. 9.7: As keeping the declaration of conformity as well as the technical documentation is clearly assigned to the authorized representative, this should not be assigned to the importer in parallel. 5. Art. 9.8: The responsibilities addressed in Art 9.8 are already assigned to the authorised representative and would lead to double responsibilities.
3. To delete “take the necessary measures to bring that device into conformity. New text: “… and, if appropriate, withdraw or recall it if requested by the manufacturer or the competent authority.” 4. To delete Art. 9.7. 5. To delete Art. 9.8.
72 Art. 10.1- Obligations of distributors
T The requirements in Art. 10.2 (a) and (c) cannot be applied to devices introduced into the distribution chain or legally placed on the Union market before the new Regulation comes into effect. Therefore, Art. 10.2 should be restricted to devices placed
To limit Art. 10.1 to device falling under this Regulation and amend as follows: “1. When making a device available on the market falling under the requirements of this regulation, distributors shall act with due care in
COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
34/36
on the market after the new Regulation has become effective.
relation to the requirements applicable.”
73 Art. 10.2 (c) - Obligations of distributors
G Importers should not be labeled on devices.
To delete “importer” and reference to Art. 9.
74 Art. 10.4 - Obligations of distributors
Art. 10.4 overlaps with the obligations of the authorised representative.
To delete “importer”.
75 Art. 10.5 - Obligations of distributors
Art. 10.5 overlaps with the obligations of the authorised representative who resumes manufacturer’s obligations with regard to authorities.
To delete Art 10.5 or alternatively to amend as follows: “They shall cooperate with that authorities, at its their request, on any action taken to eliminate the risks posed by devices which they have made available on the market.”
76 3.2 – Traceability /UDI 77 Art. 13 - Traceability
To ensure that requirements related to UDI are
consistent with the work done by GHTF/IMDRF.
78 3.3 – Vigilance procedure and market surveillance

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
35/36
79 Art. 44.1 - Reporting of incidents and corrective actions
T COCIR and EUROM VI welcome the creation of the central database on vigilance which will avoid duplication of reporting. Article 44.1 states that he manufacturer shall immediately report to the database, without specifying the delay. MEDDEV 2.12-1 rev 7 requires: - Serious public health threat: IMMEDIATELY (without any
delay that could not be justified) but not later than 2 calendar days after awareness by the MANUFACTURER of this threat.
- Death or UNANTICIPATED serious deterioration in state of health: IMMEDIATELY (without any delay that could not be justified) after the MANUFACTURER established a link between the device and the event but not later than 10 elapsed calendar days following the date of awareness of the event.
-Others: IMMEDIATELY (without any delay that could not be justified) after the MANUFACTURER established a link between the device and the event but not later than 30 elapsed calendar days following the date of awareness of the event.
1. To clarify the reporting timing, to incorporate the requirements already included in the MEDDEV 2.12-1 rev 6/7 on Vigilance. 2. To add a clarification in 44.1: “…shall immediately (without any delay that could not be justified) report electronically to the database...” 3. To add at the end of 44.1: “Timing for reporting: - Serious public health threat: IMMEDIATELY
(without any delay that could not be justified) but not later than 2 calendar days after awareness by the MANUFACTURER of this threat.
- Death or UNANTICIPATED serious deterioration in state of health: IMMEDIATELY (without any delay that could not be justified) after the MANUFACTURER established a link between the device and the event but not later than 10 elapsed calendar days following the date of awareness of the event.
-Others: IMMEDIATELY (without any delay

COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD
1 2 3 4 5
Nb Agenda item
Article Type1 Comment and rationale / justification for proposed
change Proposed revised text
1 Type of comment: G = general, T = technical, E = editorial COCIR/EUROM VI contribution on the Revision of the regulatory framework for MD 27 February 2012
36/36
that could not be justified) after the MANUFACTURER established a link between the device and the event but not later than 30 elapsed calendar days following the date of awareness of the event.”
80 Art. 44.2 - Reporting of incidents and corrective actions
T Extending the possibility to report electronically any incident according to point a) of Art. 44 (1) to patients and nonprofessional users might lead to a lot of unqualified reports, resulting in unnecessary administrative workload for competent authorities and manufacturers.
To delete reference to patients and non-professional users.
81 Art. 45.3 - Database on vigilance
T Art. 45.3 is redundant if there is a centralised database. Only one communication should be sufficient and the notification of the relevant CA automatic…
To clarify that the manufacturer has only to report once in the database on Vigilance, and that the information will be transmitted to the relevant competent authorities by the organization managing the database.




















