
Cloning, expression, and characterization of a xylanase 10 from Aspergillus terreus (BCC129) in...
7
Protein Expression and PuriWcation 46 (2006) 143–149 www.elsevier.com/locate/yprep 1046-5928/$ - see front matter 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.pep.2005.09.013 Cloning, expression, and characterization of a xylanase 10 from Aspergillus terreus (BCC129) in Pichia pastoris Duriya Chantasingh a , Kusol Pootanakit b , Verawat Champreda a , Pattanop Kanokratana a , Lily Eurwilaichitr a,¤ a BIOTEC Central Research Unit, 113 Paholyothin Road, Klong 1, Klong Luang, Pathumthani, 12120, Thailand b Institute of Molecular Biology and Genetics, Mahidol University, Salaya Campus, Nakhon Pathom 73170, Thailand Received 10 June 2005, and in revised form 11 August 2005 Available online 11 October 2005 Abstract A full-length xylanase gene, encoding 326 amino acids belonging to the fungal glycosyl hydrolase family 10, from Aspergillus terreus BCC129 was cloned and sequenced. Sequence analysis suggested that the Wrst 25 amino acids of this enzyme is the signal peptide. There- fore, only the mature xylanase gene of 906 bp was cloned into a yeast expression vector, pPICZA, for heterologous expression in Pichia pastoris. A band of approximately, 33 kDa was observed on the SDS–PAGE gel after one day of methanol induction. The expressed enzyme was puriWed by gel Wltration chromatography. The puriWed recombinant xylanase demonstrated optimal activity at 60 °C, pH 5.0 and a K m of 4.8 § 0.07 mg/ml and a V max of 757 § 14.54 mol/min mg, using birchwood xylan as a substrate. Additionally, the puriWed enzyme demonstrated broad pH stability from 4 to 10 when incubated at 40 °C for 4 h. It also showed a moderate thermal stability since it retained 90% of its activity when incubated at 50 °C, 30 min, making this enzyme a potential use in the animal feed and paper and pulp industries. 2005 Elsevier Inc. All rights reserved. Keywords: Aspergillus terreus; Xylanase; Glycosyl hydrolase 10; Pichia pastoris expression; Xylan Xylanases (endo--1,4-xylanase; EC 3.2.1.8) randomly hydrolyse the -(1,4) glycosidic linkages of xylan, the heter- ogeneous polysaccharide in plant cell wall, into short oligo- saccharide. The complete hydrolysis of xylan requires a combination of enzymes, dependent on substrate [1,2]. In hardwoods, xylan exists as O-acetyl-4-O-methylglucuron- oxylan. Arabino-4-O-methylglucuronoxylan is found in softwood, while arabinoxylan is typically present in grasses and annual plants. The variety of xylans resulted in a diver- sity of xylanases, which are classiWed into families 10 and 11 of the glycosyl hydrolases, based on catalytic domain, pri- mary structure and amino acid sequence similarity [3–6]. The majority of family 10 members are endo--1,4-xylan- ases, which have a high molecular weight (>30 kDa) and a low pI. Family 11 consists mainly of xylanases with a high pI and a low molecular weight (<30 kDa) [1,4,5]. Xylanases are found in plants, algae, insects, protozoans, and microorganisms [7]. Among microbial sources, the Wla- mentous fungi are well known as secretor of high level of xylanase enzymes into the culture medium. This property makes fungi economically eVective producers of xylanases, which are widely used in various industrial applications. For instance, in the pulp and paper industry, xylanases are employed in the prebleaching process to reduce the use of the toxic chlorine chemicals [8]. In the animal feed industry, xylanases are used to increase the body weigh gains of the animals [9,10]. In the bread and bakery industry, xylanases are used to decrease the dough viscosity while increasing the bread volume and shelf life [11]. As Thailand lies near the equator, she experiences hot and humid climes, resulting in high fungal diversity within her border. Here at BIOTEC, we have accumulated thousands of * Corresponding author. Fax: +66 2 5646707. E-mail address: [email protected] (L. Eurwilaichitr).
Transcript of Cloning, expression, and characterization of a xylanase 10 from Aspergillus terreus (BCC129) in...

Protein Expression and PuriWcation 46 (2006) 143–149
www.elsevier.com/locate/yprep
Cloning, expression, and characterization of a xylanase 10 from Aspergillus terreus (BCC129) in Pichia pastoris
Duriya Chantasingh a, Kusol Pootanakit b, Verawat Champreda a, Pattanop Kanokratana a, Lily Eurwilaichitr a,¤
a BIOTEC Central Research Unit, 113 Paholyothin Road, Klong 1, Klong Luang, Pathumthani, 12120, Thailandb Institute of Molecular Biology and Genetics, Mahidol University, Salaya Campus, Nakhon Pathom 73170, Thailand
Received 10 June 2005, and in revised form 11 August 2005Available online 11 October 2005
Abstract
A full-length xylanase gene, encoding 326 amino acids belonging to the fungal glycosyl hydrolase family 10, from Aspergillus terreusBCC129 was cloned and sequenced. Sequence analysis suggested that the Wrst 25 amino acids of this enzyme is the signal peptide. There-fore, only the mature xylanase gene of 906 bp was cloned into a yeast expression vector, pPICZ�A, for heterologous expression in Pichiapastoris. A band of approximately, 33 kDa was observed on the SDS–PAGE gel after one day of methanol induction. The expressedenzyme was puriWed by gel Wltration chromatography. The puriWed recombinant xylanase demonstrated optimal activity at 60 °C, pH 5.0and a Km of 4.8 § 0.07 mg/ml and a Vmax of 757 § 14.54 �mol/min mg, using birchwood xylan as a substrate. Additionally, the puriWedenzyme demonstrated broad pH stability from 4 to 10 when incubated at 40 °C for 4 h. It also showed a moderate thermal stability sinceit retained 90% of its activity when incubated at 50 °C, 30 min, making this enzyme a potential use in the animal feed and paper and pulpindustries. 2005 Elsevier Inc. All rights reserved.
Keywords: Aspergillus terreus; Xylanase; Glycosyl hydrolase 10; Pichia pastoris expression; Xylan
Xylanases (endo-�-1,4-xylanase; EC 3.2.1.8) randomlyhydrolyse the �-(1,4) glycosidic linkages of xylan, the heter-ogeneous polysaccharide in plant cell wall, into short oligo-saccharide. The complete hydrolysis of xylan requires acombination of enzymes, dependent on substrate [1,2]. Inhardwoods, xylan exists as O-acetyl-4-O-methylglucuron-oxylan. Arabino-4-O-methylglucuronoxylan is found insoftwood, while arabinoxylan is typically present in grassesand annual plants. The variety of xylans resulted in a diver-sity of xylanases, which are classiWed into families 10 and 11of the glycosyl hydrolases, based on catalytic domain, pri-mary structure and amino acid sequence similarity [3–6].The majority of family 10 members are endo-�-1,4-xylan-ases, which have a high molecular weight (>30 kDa) and a
* Corresponding author. Fax: +66 2 5646707.E-mail address: [email protected] (L. Eurwilaichitr).
1046-5928/$ - see front matter 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.pep.2005.09.013
low pI. Family 11 consists mainly of xylanases with a highpI and a low molecular weight (<30 kDa) [1,4,5].
Xylanases are found in plants, algae, insects, protozoans,and microorganisms [7]. Among microbial sources, the Wla-mentous fungi are well known as secretor of high level ofxylanase enzymes into the culture medium. This propertymakes fungi economically eVective producers of xylanases,which are widely used in various industrial applications.For instance, in the pulp and paper industry, xylanases areemployed in the prebleaching process to reduce the use ofthe toxic chlorine chemicals [8]. In the animal feed industry,xylanases are used to increase the body weigh gains of theanimals [9,10]. In the bread and bakery industry, xylanasesare used to decrease the dough viscosity while increasingthe bread volume and shelf life [11].
As Thailand lies near the equator, she experiences hot andhumid climes, resulting in high fungal diversity within herborder. Here at BIOTEC, we have accumulated thousands of

144 D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149
locally found fungi within our collection (http://mycol-ogy.biotec.or.th/current_research/diversity/maindiversity.htm). Preliminary screening of approximately 766 fungishowed that one of these, Aspergillus terreus (BCC129), pos-sessed high xylanolytic activity at a wide pH range (pH 3–10).This strain may therefore be a potential source of industrialxylanase. However, A. terreus was reported to produce myco-toxins, which are harmful to animals and humans [12,13].Thus, in this paper, heterologous expression of BCC129xylanase was performed in Pichia pastoris to eliminate theunwanted eVects of the mycotoxins. SpeciWcally, the geneencoding this broad range pH xylanase was isolated andexpressed using P. pastoris expression system. The secretedheterologous protein is produced as a major component in theculture supernatant, facilitating downstream enzyme puriWca-tion. Moreover, biochemical characterization of the puriWedrecombinant BCC129 xylanase was also performed. Further-more, of the numerous fungal xylanases found in the data-base, this is the Wrst report from the A. terreus species.
Materials and methods
Strains, plasmids, culturing conditions, and primers
Aspergillus terreus (BCC129) was obtained from BIO-TEC Culture Collection (BCC), Thailand. It was isolatedfrom soil in the central region of Thailand at Pa Sak, LopBuri province. It was aerobically cultured in 100 ml, 5%wheat bran at 30 °C with 200 rpm shaking. After 2 days ofcultivation, the culture was harvested by Wltration througha 0.5 mm diameter mesh. Escherichia coli, DH5�, was usedas host for plasmid propagation. It was cultured in low saltLB medium. P. pastoris KM71 (Invitrogen, USA), used as ahost for expression of xylanase, was grown in YEPD [2%(w/v) peptone, 2% (w/v) glucose (Sigma, USA), and 1% (w/v) yeast extract]. The P. pastoris transformants were cul-tured under a selective condition in YEPD containing100�g/ml Zeocin. pGEM-T Easy vector (Promega, USA)was used in cloning of PCR fragments. pPICZ�A vector(Invitrogen) was used in P. pastoris expression. All syn-thetic oligonucleotides used in this study were obtainedfrom the Bio Service Unit (BSU), BIOTEC, Thailand(Table 1).
Total RNA isolation, cDNA synthesis, and semi-nested PCR
BCC129 mycelia were frozen in liquid nitrogen beforegrounded up in an ice-cold mortar until powdery consis-tency was achieved. Tri-Reagent (Molecular Research Cen-ter, USA) was used for total RNA extraction frompowdered BCC129 mycelia, following the manufacturer’sinstruction. The quality and integrity of RNA was deter-mined by gel electrophoresis in 1% agarose containing 3.5%formaldehyde as described by Sambrook et al. [14].
First-strand cDNA from BCC129 was synthesised usingRevertAid H Minus First Strand cDNA Synthesis Kit (Fer-mentas, Lithuania) according to the supplier. First-strand
cDNAs were then used as template in the semi-nested PCR.The Wrst round of PCR was performed with 1 U of DyNA-zyme EXT DNA polymerase (Finnzyme, Finland) in a totalvolume of 50�l containing 20�M of Xyl2-F1 and Xyl2-B1degenerate primers, 1£ DyNAzyme buVer, 2.5 mM each ofdNTPs, 2 mM MgCl2, and 4�l cDNA. The PCR conditionwas: one cycle of 4 min at 94 °C, 35 cycles of 30 s at 94 °C,30 s at 40 °C, and 30 s at 72 °C, and Wnally 10 min at 72 °C.The second PCR condition was the same as in the Wrstround ampliWcation except here it contained 4�l of the pre-vious PCR products as template and Xyl2-F1 and Xyl2-B2as primers. The PCR products were analyzed by agarose gelelectrophoresis. The amplicon of the expected size was gel-puriWed using QIAquick gel extraction kit (Qiagen, Ger-many) and cloned into pGEM-T Easy vector (Promega,USA). Sequencing was performed by Macrogen (Korea).
Isolation of the full-length xylanase gene by 3� RACE and 5� RACE
For the 3� RACE, Wrst-strand cDNAs were generatedfrom 4 �g of total RNA from BCC129 mycelia using oligo-(dT) adaptor (PM1 primer) and MMLV-H minus reversetranscriptase, under the conditions suggested by the manu-facturer (Fermentas, Lithuania). PCR was performed asdescribed above, using 3� RazXyl-Ast1 and PM2 primerswith an annealing temperature of 55 °C.
For the 5� RACE, Wrst-strand cDNAs were synthesized asdescribed above, except here, random hexamer instead ofgene speciWc primer, as was commonly employed at this step,was used in the reaction. The RNA template was eliminatedby adding NaOH to the Wnal concentration of 0.5 M andincubated at 55 °C for 30 min. The reaction was then neutral-ized with 72�l of 1% (v/v) acetic acid and gel-puriWed. Next,poly(A) tail was added to the 3� end of the obtained Wrst-strand cDNAs using terminal deoxynucleotidyl transferase(Promega, USA) according to the supplier’s instruction. Toamplify the 5� end of the cDNA, nested-PCR was performedusing 5� RazXyn-Ast1 and PM1, and 5� RaceXyn-Ast2 andPM2 primers consecutively. The PCR conditions for both
Table 1Oligonucleotides used in the studies
For degenerate primer, the following abbreviations are used (N D A, T, C,G; H D A, T, G; Y D C, T; M D A, C; S D C, G; R D A, G).
Primer names Sequence 5� ! 3�
PM1 CCGGAATTCAAGCTTCTAGAGGATCCTTTTTTTTTTTTTTTT
PM2 CCGGAATTCAAGCTTCTAGAGGATCCXyl2-F1 ACNCCNGARAAYTCNATGAARXyl2-B1 HATYTCRTTNACNACRTCCCAXyl2-B2 RTCRTTHATRTANAGYTTNGC3� RazXyl-Ast1 TCGGTCTTGAAGAACCACATC5� RazXyl-Ast1 TCAGCACCACCAAAGCTGAAC5� RazXyl-Ast2 GATTCGGCTCAGTCGCATCAstXyl-F1 CCGCTCGAGAAAAGACAGGCGGCTTCGAGCAstXyl-R1 CTAGTCTAGATTACAAGGCGGAGATAATTG

D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149 145
round of PCR were the same as described above for 3�RACE PCR. The obtained PCR product was puriWed,cloned and sequenced.
The DNA sequence obtained was translated intoamino acid sequence by using the Bioedit Sequence Align-ment Editor program. The nucleotide and the deducedamino acid sequences were searched against GenBankdatabase and compared to other related sequences usingClustal X program. The nucleotide sequence of the full-length BCC129 xylanase cDNA was submitted to Gen-Bank (Accession No. DQ087436).
Cloning and expression of BCC129 xylanase in P. pastoris
The mature xylanase gene was ampliWed by DyNA-zymeEXT DNA polymerase using gene-speciWc primers, Ast-Xyl-F1 and AstXyl-R1. The PCR condition was: one cycle at94°C for 1min, 35 cycles at 94°C for 30s, 55°C for 30s and72°C for 45s, and followed by the Wnal extension for 10minat 72°C. The PCR product was gel-puriWed and digested withXhoI and XbaI (Promega, USA) before subcloning intopPICZ�A. Transformation was then performed as describedin the Invitrogen’s instruction manual. The integration of thexylanase gene into the genome of P. pastoris was determinedby PCR using 5� AOX1 and 3� AOX1 primers.
To induce xylanase production in P. pastoris, the yeastcells were grown at 30 °C in 5 ml of fresh buVered minimalglycerol complex medium (BMGY)1 until the culturereached an OD600 of 5–6. Then, the cell pellet was harvestedand resuspended in buVered minimal methanol medium(BMMY) using 1/5th volume of the original culture (1 ml).The cell suspension was placed in a 50 ml tube. Absolutemethanol was added every 24 h to a Wnal concentration of3% (v/v) to maintain induction. The culture supernatantwas collected at 0, 24, 72, and 120 h. Secreted proteins wereanalyzed by using SDS–PAGE [15].
Agar plate diVusion assay
Agar plate, containing 0.2% AZCL-xylan (Megazyme,Ireland) in screening buVer of pH 3, 7, or 10 (a 100ml buVercontained 0.92ml of 85% H3PO4, 0.92ml of glacial acetic acidand 0.99g of H3BO3, the pH was adjusted to 3, 7 or 10 with 4NNaOH) was used to identify xylanase expressing clones. SpeciW-
cally, the cultured supernatants were loaded onto the wells ofthe AZCL-xylan agar plate and incubated for 6h at 30°C. Theblue zone around the well indicated xylanase activity.
PuriWcation and characterization of BCC129 xylanase from P. pastoris
The cell-free medium was loaded in a gel Wltration col-umn (Superose 12 10/300 GL, Amersham Bioscience,
1 Abbreviations used: BMGY, buVered minimal glycerol complex medi-um; BMMY, buVered minimal methanol medium; DNS, 3,5-dinitrosali-cylic acid.
USA), equilibrated with 100 mM potassium phosphatebuVer pH 6.5 and 50 mM NaCl, and run on an AKTAExplorer (Pharmacia Biotech, USA) at a Xow rate of 0.5 ml/min. The protein fractions were collected and assayed forxylanase activity using 3,5-dinitrosalicylic acid (DNS) [16].The xylanase activity was determined from the amount ofreducing sugar liberated from birchwood xylan (Sigma,USA). Xylose was used as a reference for preparing thestandard curve. One unit of enzyme activity was deWned asthe quantity of enzyme that liberated reducing sugar at therate of 1�mol/min. To determine the optimal pH and tem-perature proWles, the enzymatic reaction was carried out atdiVerent pHs and temperatures as indicated in the Wgurelegends. The Km and Vmax of the enzyme were analyzed byusing KaleidaGraph version 3.51 data analysis software(Synergy Software, USA).
Results
Isolation of the full-length cDNA of xylanase gene from BCC129
Based on the conserved sequence of xylanases from vari-ous Sordariomycetes, degenerate primers (Xyl2-F1, Xyl2-B1 and Xyl2-B2) were designed and used in semi-nestedPCR ampliWcation of xylanase gene from BCC129.Sequence analysis of the 260 bp ampliWed fragment showed87% amino acid identity to xylanase F3 of A. oryzae (datanot shown). This obtained partial sequence was used todesign gene-speciWc primer for 3� and 5� RACE to obtainthe sequence of the full-length gene.
The 3� and 5� RACE yielded a 771 bp and a 327 bp DNAfragments, respectively (data not shown). Sequence analysisindicated that there was a start codon (ATG) in the 5�-frag-ment and a stop codon (TAA) in the 3�-fragment. Aftersequence assembling, the putative full-length xylanasecDNA of 981 bp was obtained. Comparison analysis usingblastx revealed that its deduced amino acid sequence (326amino acids) contained conserved domains of glycosylhydrolase family 10, and showed 82% identity to xylanaseF3 of A. oryzae [17], 81% identity to endo-1,4�-D-xylanaseof A. sojae (GenBank Accession No. AB040414), 73% iden-tity to endo-1,4�-D-xylanase A of Penicillium purpurogenum[18] and 73% identity to endo-1,4�-D-xylanase of P. canes-cens [19]. Furthermore, identiWcation of signal peptidecleavage site with SignalP program [20] suggested that theWrst 25 amino acids are the signal sequence (Fig. 1). Conse-quently, the predicted mature xylanase consisted of 301amino acids with a molecular weight of approximately33 kDa.
Cloning and expression of BCC129 xylanase gene in P. pastoris
The DNA coding for mature xylanase, fused in-frameto the �-factor secretion signal was obtained by perform-ing RT-PCR on the puriWed BCC129 total RNA by using,

146 D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149
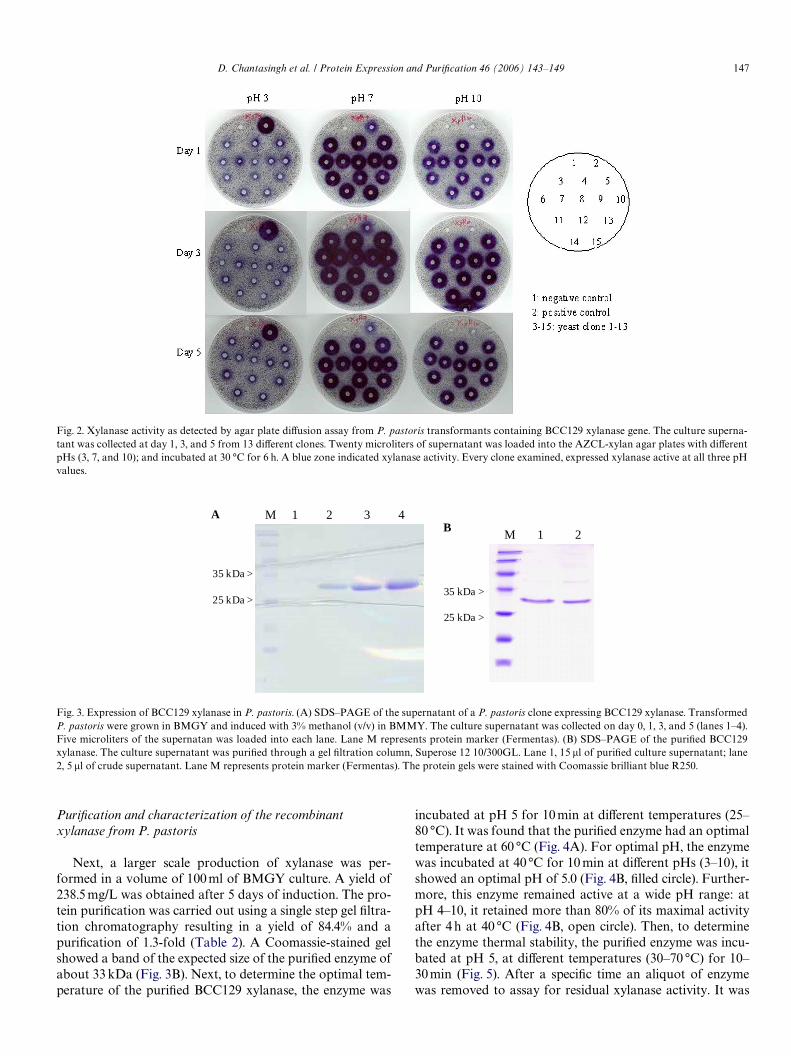
AstXyl-F1 and AstXyl-R1 primers. A fragment of 920 bpwas obtained and cloned into the yeast expression vector,pPICZ�A. Four positive clones were randomly chosen forsequence analysis to obtain the consensus sequence(Fig. 1). One of the two clones that showed 100% sequenceidentity to the consensus sequence was introduced intoP. pastoris KM71. The transformants were screened onYEPD plate containing 0.1 mg/ml Zeocin and further con-Wrmed by PCR using 5�- and 3�-AOX primers. Thirteenintegrants were randomly chosen for small-scale induc-tion. After 1, 3 and 5 days of induction, xylanase activitywas monitored by agar plate diVusion assay at pH 3, 7,and 10. Hydrolysis areas, appearing as blue zones, were
measured semi-quantitatively to identify the expressedclones. The results showed that all transformants pro-duced about the same level of xylanase as soon as day 1 ofinduction as measured by the radius of the zone (Fig. 2).Next, SDS–PAGE was performed to determine the size ofthe recombinant xylanase from the culture supernatant ofclone number 1 at various days of induction (0, 1, 3, and 5days) (Fig. 3A). The results showed a major band ofapproximately 33 kDa, which was the predicted size fromthe deduced amino acid sequence. The optimal inductiontime was also determined and the result showed that thisclone gave the highest level of the recombinant proteinafter 5 days of induction.
Fig. 1. Sequence alignment of the deduced amino acid of BCC129 xylanase with other mature fungal xylanases: A. oryzae xylanase F3 [17], A. sojae endo-1,4�-D-xylanase (AB040414), P. purpurogenum endo-1,4�-D-xylanase A [18] and P. canescens endo-1,4�-D-xylanase [19]. The arrows indicate the positionsof the oligonuleotide primers used in this study. Arrowhead indicates the putative start position of BCC129 mature xylanase. The conserved domains ofglycosyl hydrolase family 10 are boxed.
Xyl2-F1 5’RazXyl-Ast2 5’RazXyl-Ast1
Xyl2-R2
Xyl2-R1
3’RazXyl-Ast1
AstXyl-F1
AstXyl2-R1
D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149 147
PuriWcation and characterization of the recombinant incubated at pH 5 for 10 min at diVerent temperatures (25–
xylanase from P. pastorisNext, a larger scale production of xylanase was per-formed in a volume of 100 ml of BMGY culture. A yield of238.5 mg/L was obtained after 5 days of induction. The pro-tein puriWcation was carried out using a single step gel Wltra-tion chromatography resulting in a yield of 84.4% and apuriWcation of 1.3-fold (Table 2). A Coomassie-stained gelshowed a band of the expected size of the puriWed enzyme ofabout 33 kDa (Fig. 3B). Next, to determine the optimal tem-perature of the puriWed BCC129 xylanase, the enzyme was
80 °C). It was found that the puriWed enzyme had an optimaltemperature at 60 °C (Fig. 4A). For optimal pH, the enzymewas incubated at 40 °C for 10 min at diVerent pHs (3–10), itshowed an optimal pH of 5.0 (Fig. 4B, Wlled circle). Further-more, this enzyme remained active at a wide pH range: atpH 4–10, it retained more than 80% of its maximal activityafter 4 h at 40 °C (Fig. 4B, open circle). Then, to determinethe enzyme thermal stability, the puriWed enzyme was incu-bated at pH 5, at diVerent temperatures (30–70 °C) for 10–30 min (Fig. 5). After a speciWc time an aliquot of enzymewas removed to assay for residual xylanase activity. It was
Fig. 2. Xylanase activity as detected by agar plate diVusion assay from P. pastoris transformants containing BCC129 xylanase gene. The culture superna-tant was collected at day 1, 3, and 5 from 13 diVerent clones. Twenty microliters of supernatant was loaded into the AZCL-xylan agar plates with diVerentpHs (3, 7, and 10); and incubated at 30 °C for 6 h. A blue zone indicated xylanase activity. Every clone examined, expressed xylanase active at all three pHvalues.
Fig. 3. Expression of BCC129 xylanase in P. pastoris. (A) SDS–PAGE of the supernatant of a P. pastoris clone expressing BCC129 xylanase. TransformedP. pastoris were grown in BMGY and induced with 3% methanol (v/v) in BMMY. The culture supernatant was collected on day 0, 1, 3, and 5 (lanes 1–4).Five microliters of the supernatan was loaded into each lane. Lane M represents protein marker (Fermentas). (B) SDS–PAGE of the puriWed BCC129xylanase. The culture supernatant was puriWed through a gel Wltration column, Superose 12 10/300GL. Lane 1, 15 �l of puriWed culture supernatant; lane2, 5 �l of crude supernatant. Lane M represents protein marker (Fermentas). The protein gels were stained with Coomassie brilliant blue R250.
M 1 2 3 4
35 kDa >
25 kDa >
M 1 2
35 kDa >
25 kDa >
AB
148 D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149
Table 2PuriWcation of BCC129 xylanase from P. pastoris
Protein concentration (�g/ml)
Total volume (ml)
Total protein (�g)
SpeciWc activity (U/mg)
Total activity (U)
Yield (%)
PuriWcationfold
Crude protein 238.5 4 954 420 400.7 100 1Superose 12 10/300 GL 52.9 12 634.8 533 338.4 84.4 1.3
120A 120
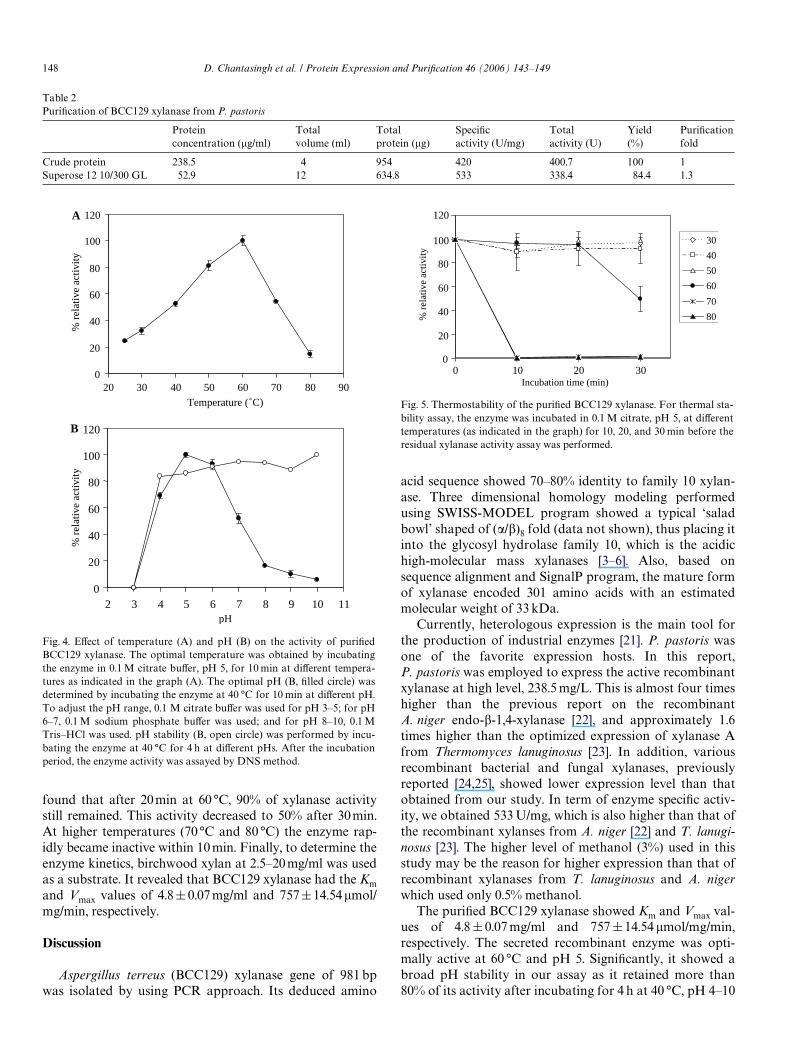
found that after 20 min at 60 °C, 90% of xylanase activitystill remained. This activity decreased to 50% after 30 min.At higher temperatures (70 °C and 80 °C) the enzyme rap-idly became inactive within 10 min. Finally, to determine theenzyme kinetics, birchwood xylan at 2.5–20 mg/ml was usedas a substrate. It revealed that BCC129 xylanase had the Kmand Vmax values of 4.8 § 0.07 mg/ml and 757§ 14.54�mol/mg/min, respectively.
Discussion
Aspergillus terreus (BCC129) xylanase gene of 981 bpwas isolated by using PCR approach. Its deduced amino
Fig. 4. EVect of temperature (A) and pH (B) on the activity of puriWedBCC129 xylanase. The optimal temperature was obtained by incubatingthe enzyme in 0.1 M citrate buVer, pH 5, for 10 min at diVerent tempera-tures as indicated in the graph (A). The optimal pH (B, Wlled circle) wasdetermined by incubating the enzyme at 40 °C for 10 min at diVerent pH.To adjust the pH range, 0.1 M citrate buVer was used for pH 3–5; for pH6–7, 0.1 M sodium phosphate buVer was used; and for pH 8–10, 0.1 MTris–HCl was used. pH stability (B, open circle) was performed by incu-bating the enzyme at 40 °C for 4 h at diVerent pHs. After the incubationperiod, the enzyme activity was assayed by DNS method.
0
20
40
60
80
100
20 30 40 50 60 70 80 90Temperature (˚C)
% r
elat
ive
activ
ity
0
20
40
60
80
100
120
2 3 4 5 6 7 8 9 10 11pH
% r
elat
ive
activ
ity
B
acid sequence showed 70–80% identity to family 10 xylan-ase. Three dimensional homology modeling performedusing SWISS-MODEL program showed a typical ‘saladbowl’ shaped of (�/�)8 fold (data not shown), thus placing itinto the glycosyl hydrolase family 10, which is the acidichigh-molecular mass xylanases [3–6]. Also, based onsequence alignment and SignalP program, the mature formof xylanase encoded 301 amino acids with an estimatedmolecular weight of 33 kDa.
Currently, heterologous expression is the main tool forthe production of industrial enzymes [21]. P. pastoris wasone of the favorite expression hosts. In this report,P. pastoris was employed to express the active recombinantxylanase at high level, 238.5 mg/L. This is almost four timeshigher than the previous report on the recombinantA. niger endo-�-1,4-xylanase [22], and approximately 1.6times higher than the optimized expression of xylanase Afrom Thermomyces lanuginosus [23]. In addition, variousrecombinant bacterial and fungal xylanases, previouslyreported [24,25], showed lower expression level than thatobtained from our study. In term of enzyme speciWc activ-ity, we obtained 533 U/mg, which is also higher than that ofthe recombinant xylanses from A. niger [22] and T. lanugi-nosus [23]. The higher level of methanol (3%) used in thisstudy may be the reason for higher expression than that ofrecombinant xylanases from T. lanuginosus and A. nigerwhich used only 0.5% methanol.
The puriWed BCC129 xylanase showed Km and Vmax val-ues of 4.8 § 0.07 mg/ml and 757 § 14.54 �mol/mg/min,respectively. The secreted recombinant enzyme was opti-mally active at 60 °C and pH 5. SigniWcantly, it showed abroad pH stability in our assay as it retained more than80% of its activity after incubating for 4 h at 40 °C, pH 4–10
Fig. 5. Thermostability of the puriWed BCC129 xylanase. For thermal sta-bility assay, the enzyme was incubated in 0.1 M citrate, pH 5, at diVerenttemperatures (as indicated in the graph) for 10, 20, and 30 min before theresidual xylanase activity assay was performed.
0
20
40
60
80
100
0 10 20 30Incubation time (min)
30
40
50
60
70
80 % r
elat
ive
activ
ity

D. Chantasingh et al. / Protein Expression and PuriWcation 46 (2006) 143–149 149
(Fig. 4B). Also, this enzyme demonstrated a moderate ther-mal stability since it still retained 90% of its activity whenincubated at 50 °C for 30 min or 60 °C for 20 min, suggest-ing that BCC129 xylanase is a mesophilic enzyme. Theseproperties should make it a good candidate in variousindustrial applications. For instance, with its broad pH sta-bility and moderate thermal stability properties, it is mostsuitable for used in the animal feed industry since the pHand temperature of the livestock digestive tract are approx-imately 4.8 and 40 °C, respectively [2]. Another potentialuse of the enzyme may be in the pulp and paper industriesin which alkaline pH and moderate to high temperatures ofaround 55–70 °C are employed [26,27].
In conclusion, we have successfully cloned and expresseda xylanase gene from A. terreus (BCC129). Even thoughvarious Aspergilli xylanases have been cloned andsequenced [22,28], this is the Wrst report from A. terreus.BCC129 xylanase was successfully expressed and secretedin the active form; its enzymatic properties were investi-gated. In the future, structure-function analysis of theenzyme will be performed to gain a better understanding ofthis enzyme that will lead to the modiWcation of the enzymeproperties by genetic engineering approach.
Acknowledgments
This study was supported by the National Center forGenetic Engineering and Biotechnology (BIOTEC),National Science and Technology Development Agency,Thailand, NSTDA (E25FJ0016).
References
[1] K.K.Y. Wong, L.U.L. Tan, J.N. Saddler, Multiplicity of �-1,4-xylan-ase in microorganisms: functions and applications, Microbiol. Rev. 52(1988) 305–317.
[2] T. Collins, C. Gerday, G. Feller, Xylanases, xylanase families andextremophilic xylanases, FEMS Microbiol. Rev. 29 (2005) 3–23.
[3] C. Gaboriaud, V. Bissey, T. Benchetrit, J.P. Mornon, Hydrophobiccluster analysis: an eYcient new way to compare and analyze aminoacid sequence, FEBS Lett. 224 (1987) 149–155.
[4] B. Henrissat, A. Bairoch, Updating the sequence-based classiWcationof glycosyl hydrolase, Biochem. J. 316 (1996) 695–696.
[5] B. Henrissat, G. Davies, Structural and sequence-based classiWcationof glycosyl hydrolases, Curr. Opin. Struct. Biol. 7 (1997) 637–644.
[6] P. Biely, M. Vrsanska, M. Tenkanen, D. Kluepfel, Endo-�-1,4 xylanase fam-ilies: diVerences in catalytic properties, J. Biotechnol. 57 (1997) 151–166.
[7] A. Sunna, G. Antranikian, Xylanolytic enzymes from fungi and bacte-rial, Crit. Rev. Biotechnol. 17 (1997) 39–67.
[8] P. Bajpai, Application of enzymes in the pulp and paper industry, Bio-technol. Prog. 15 (1999) 147–157.
[9] F.G. Silversides, M.R. Bedford, EVect of pelleting temperature on therecovery and eYcacy of a xylanase enzyme in wheat-based diets,Poult. Sci. 78 (1999) 1184–1190.
[10] L. Kung Jr., R.J. Treacher, G.A. Nauman, A.M. Smagala, K.M.Endres, M.A. Cohen, The eVect of treating forages with Wbrolyticenzymes on its nutritive value and lactation performance of dairycows, J. Dairy Sci. 83 (2000) 115–122.
[11] M.C. Figueroa-Espinoza, C. Poulsen, J. Borch Soe, M.R. Zargahi, X.Rouau, Enzymatic solubilization of arabinoxylans from native,extruded, and high-shear-treated rye bran by diVerent endo-xylanasesand other hydrolyzing enzymes, J. Agric. Food Chem. 52 (2004) 4240–4249.
[12] K.H. Ling, C.K. Yang, F.T. Peng, Territrems, tremorgenic mycotoxinsof Aspergillus terreus, Appl. Environ. Microbiol. 37 (1979) 355–357.
[13] F.C. Peng, K.H. Ling, Y. Wang, G.H. Lee, Isolation, chemicalstructure, acute toxicity, and some physicochemical properties of ter-ritrem B from Aspergillus terreus, Appl. Environ. Mcrobiol. 49 (1985)721–723.
[14] J. Sambrook, E.F. Fritsch, T. Maniatis, Molecular Cloning: A Labo-ratory Manual, Cold Spring Harbour Press, Cold Spring Harbour,New York, 1989.
[15] U.K. Laemmli, Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4, Nature 227 (1970) 680–685.
[16] M.J. Bailey, P. Biely, K. Poutanen, Interlaboratory testing ofmethods for assay of xylanase activity, J. Biotechnol. 23 (1992)257–270.
[17] T. Kimura, H. Suzuki, H. Furuhashi, T. Aburatani, K. Morimoto, K.Sakka, K. Ohmiya, Molecular cloning, characterization, and expres-sion analysis of the xynF3 gene from Aspergillus oryzae, Biosci. Bio-technol. Biochem. 66 (2002) 285–292.
[18] R. Chavez, C. Almarza, K. Schachter, A. Peirano, P. Bull, J. Eyzagu-irre, Structure analysis of the endoxylanase A gene from Penicilliumpurpurogenum, Biol. Res. 34 (2001) 217–226.
[19] V.A. Serebryanyi, E.A. Vavilova, A.M. Chulkin, Y.P. Vinetskii, Clon-ing of Penicillium canescens endo-1,4-beta-xylanase gene and con-struction of multicopy strains, Appl. Biochem. Microbiol. 38 (2002)420–426.
[20] J.D. Bendtsen, H. Nielsen, G.V. Heijne, S. Brunak, Improved predic-tion of signal peptides-SignalP 3.0, J. Mol. Biol. 340 (2004) 783–795.
[21] O. Kirk, T.V. Borchert, C.C. Fuglsang, Industrial enzyme applica-tions, Curr. Opin. Biotechnol. 13 (2002) 345–351.
[22] J-G. Berrin, G. Williamson, A. Puigserver, J-C. Chaix, W.R. McL-auchlan, N. Juge, High-level production of recombinant fungal endo-�-1,4-xylanase in the methylotrophic yeast Pichia pastoris, ProteinExpress. Purif. 19 (2000) 179–187.
[23] M.C.T. Damaso, M.S. Almeida, E. Kurtenbach, O.B. Martins, N.Pereira Jr., C.M.M.A. Andrade, R.M. Albano, Optimized expressionof a thermostable xylanase from Thermomyces lanuginosus in Pichiapastoris, Appl. Microbiol. Biotechnol. 69 (2003) 6064–6072.
[24] D.J. Walsh, P.L. Bergquist, Expression and secretion of a thermosta-ble bacterial xylanase in Kluyveromyces lactis, Appl. Environ. Micro-biol. 63 (1997) 3297–3300.
[25] X.-L. Li, L.G. Ljungdahl, Expression of Aureobasidium pullulansXynA in, and secretion of the xylanase from Saccharomyces cerevi-siae, Appl. Environ. Microbiol. 62 (1996) 209–213.
[26] L. Viikari, Xylanases in bleaching: from an idea to the industry,FEMS Microbiol. Rev. 13 (1994) 335–350.
[27] Q.K. Beg, M. Kapoor, L. Mahajan, G.S. Hoondal, Microbial xylan-ases and their industrial applications: a review, Appl. Microbiol. Bio-technol. 56 (2001) 326–338.
[28] K. Asano, R. Sriprang, J. Gobsuk, L. Eurwilaichitr, S. Tanapongpipat,K. Kirtikara, Endo-1,4-�-xylanase B from Aspergillus cf. nigerBCC14405 isolated in Thailand: puriWcation, characterization andgene isolation, J. Biochem. Mol. Biol. 38 (2005) 17–23.




















