
CLIC2-RyR1 Interaction and Structural Characterization by Cryo-electron Microscopy
16
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of CLIC2-RyR1 Interaction and Structural Characterization by Cryo-electron Microscopy

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
CLIC2-RyR1 Interaction and Structural Characterizationby Cryo-electron Microscopy
Xing Meng1†, Guoliang Wang2†, Cedric Viero3†, Qiongling Wang2,Wei Mi4, Xiao-Dong Su4, Terence Wagenknecht1, Alan J. Williams3⁎,Zheng Liu1⁎ and Chang-Cheng Yin2⁎1Wadsworth Center, New YorkState Department of Health,Albany, NY 12201, USA2Department of Biophysics,Peking University Health ScienceCenter, Peking University,Beijing 100191, China3Department of Cardiology,Wales Heart Research Institute,Cardiff University School ofMedicine, Heath Park, CardiffCF14 4XN, UK4National Laboratory of ProteinEngineering and Plant GeneticEngineering, College of LifeSciences, Peking University,Beijing 100871, China
Received 24 August 2008;received in revised form18 January 2009;accepted 27 January 2009Available online4 February 2009
Chloride intracellular channel 2 (CLIC2), a newly discovered small proteindistantly related to the glutathione transferase (GST) structural family, ishighly expressed in cardiac and skeletal muscle, although its physiologicalfunction in these tissues has not been established. In the present study, [3H]ryanodine binding, Ca2+ efflux from skeletal sarcoplasmic reticulum (SR)vesicles, single channel recording, and cryo-electron microscopy wereemployed to investigate whether CLIC2 can interact with skeletalryanodine receptor (RyR1) and modulate its channel activity. We foundthat: (1) CLIC2 facilitated [3H]ryanodine binding to skeletal SR and purifiedRyR1, by increasing the binding affinity of ryanodine for its receptorwithout significantly changing the apparent maximal binding capacity; (2)CLIC2 reduced the maximal Ca2+ efflux rate from skeletal SR vesicles; (3)CLIC2 decreased the open probability of RyR1 channel, through increasingthe mean closed time of the channel; (4) CLIC2 bound to a region betweendomains 5 and 6 in the clamp-shaped region of RyR1; (5) and in the sameclamp region, domains 9 and 10 became separated after CLIC2 binding,indicating CLIC2 induced a conformational change of RyR1. These datasuggest that CLIC2 can interact with RyR1 and modulate its channelactivity. We propose that CLIC2 functions as an intrinsic stabilizer of theclosed state of RyR channels.
© 2009 Elsevier Ltd. All rights reserved.
Edited by W. BaumeisterKeywords: Ca2+-release channel; Ca2+ signaling; chloride intracellularchannel 2; cryo-electron microscopy; ryanodine receptor
Introduction
The chloride ion is the most abundant anion in thetissues of animals and plants, and anion channels inthe cells are often referred to as chloride channels.Several classes of chloride channels have beenfound, of which three are well-characterized: the
ligand-gated receptors, the cystic fibrosis transmem-brane conductance regulators (CFTR), and thechloride ion channels (CLC).1 Chloride ion channelsare involved in regulation of absorption andsecretion of Na+, setting the cell membrane poten-tial, acidification of cytoplasmic organelles, andregulation of cell volume.2As a new class of chloride channels, chloride
intracellular channel (CLIC) proteins, differ from theother classes of chloride ion channels in primarystructure and in the transmembrane regions of thetertiary structure.3 Since the first member of CLIC,p64 (CLIC5), was discovered in bovine kidney,several members of the CLIC family have beenfound in other tissues from many species, includingNCC27 (CLIC1), CLIC2, CLIC3, mtCLIC (CLIC4),and parchorin (CLIC6).4–11 With the exception of p64
*Corresponding authors. E-mail addresses:[email protected]; [email protected];[email protected]; [email protected].† X.M., G.W. and C.V. contributed equally to this work.Abbreviations used: cryo-EM, cryo-electron
microscopy; CLIC, chloride intracellular channel; RyR,ryanodine receptor; SR, sarcoplasmic reticulum; Po, openprobability; To, mean open time; Tc, mean closed time.
doi:10.1016/j.jmb.2009.01.059 J. Mol. Biol. (2009) 387, 320–334
Available online at www.sciencedirect.com
0022-2836/$ - see front matter © 2009 Elsevier Ltd. All rights reserved.

Author's personal copy
and parchorin, these proteins are composed of∼240 amino acid residues. The CLIC proteinsshow sequence homology with members of theglutathione-S-transferase (GST) superfamily.12
Another feature of CLIC proteins distinguishablefrom other ion channels is that they exist in twodifferent forms: either as soluble globular proteins, or
as an integral membrane protein that is incorporatedinto lipid bilayers and forms ion channels.7,10,13–19Human CLIC2 protein is composed of 247 amino
acid residues and is found in many organs, includingthe spleen, lung, liver, and in both skeletal and cardiacmuscles.6,20 Consistent with their high degree ofprimary structure homology, CLIC2 is similar toCLIC1 andCLIC4 in terms of tertiary structure.17,21–23
Like other members of the CLIC family, CLIC2 canexist as a soluble globular protein, or incorporate intoa lipid bilayer to form a Cl– channel.17
While the membrane-incorporated CLIC2 proteinsfunction as Cl– channels, the physiological functionof the soluble formCLIC proteins is less well defined.Recently, Dulhunty et al showed that CLIC2 caninteract with the cardiac ryanodine receptor (RyR2)and modulate its calcium release channel activity,implying that CLIC proteins may play a role in theregulation of Ca2+ signaling.20,24 Ryanodine recep-tors are the major Ca2+ release channels in bothcardiac and skeletal muscle, and they play a crucialrole in the Ca2+ signaling pathway that governs themuscle excitation–contraction coupling.25 The gatingof RyRs is regulated with a various intracellularmessengers, including calmodulin, FK-binding pro-tein, ATP, Ca2+, Mg2+, and protein kinase A.26 Theinvestigations reported by Dulhunty et al raiseseveral issues: (1) can CLIC2 act as a modulator ofthe skeletal ryanodine receptor channel (RyR1) and,if so, is CLIC2 a common regulator of both RyR1 andRyR2? (2) Does CLIC2 modulate RyR1 differentlyfrom RyR2? (3) How does CLIC2 interact with RyRand regulate its channel activity?In this study, we attempted to answer the above
questions by employing several biochemical andelectrophysiological approaches, including [3H]
Fig. 1. Effects of CLIC2 on [3H]ryanodine binding toskeletal heavy SR and purified RyR1. (a) CLIC2 increases[3H]ryanodine binding to skeletal heavy SR. Equilibriumbinding experiments were performed in the absence or inthe presence of CLIC2. The binding buffer contained 2 nM[3H]ryanodine and 10 μM Ca2+. CLIC2 increased [3H]ryanodine binding to skeletal heavy SR from 1.31±0.1pmol/mg (buffer, n=6) to 1.57±0.03 pmol/mg (15 μMCLIC2, n=6), and to 1.7±0.01 pmol/mg (30 μM CLIC2,n=6). (b) CLIC2 increases [3H]ryanodine binding topurified RyR1. Equilibrium [3H]ryanodine binding experi-ments were carried out in binding buffer containing 2 nM[3H]ryanodine and various concentrations of Ca2+, in theabsence or in the presence of 15 μM CLIC2. [Ca2+] wasmaintained, in the range 0.1 μM – 10mM, by a combinationof EGTA and CaCl2. Free Ca2+ concentrations werecalculated as described.55 Data points shown are themean±S.E.M. from three separate experiments. (c and d)Equilibrium saturation assay of [3H]ryanodine binding topurified RyR1. Experiments were carried out in bindingbuffer containing 10 μMCa2+, and various concentrations of[3H]ryanodine (1–24 nM), in the absence or in the presenceof 15 μMCLIC2, as described in Materials andMethods. (c)The saturation curves for [3H]ryanodine binding to purifiedRyR1. Inset are the best-fit values of Bmax and Kd. (d) TheScatchard analysis of the data in (c). Data points shown arethe mean±S.E.M., from three separate experiments.
321CLIC2-RyR1 Interaction
Author's personal copy
ryanodine binding, Ca2+ efflux from skeletal sarco-plasmic reticulum (SR) vesicles, and single channelrecording; and ultimately by using the structuralapproach of 3D cryo-electron microscopy (cryo-EM)and single-particle image processing. Takentogether, our results provide for the first time adirect evidence for a physical interaction of CLIC2with RyR1, and they suggest that the interactionbetween CLIC2 and RyR1 stabilizes the closed stateof the Ca2+ release channel.
Results
CLIC2 facilitated [3H]ryanodine binding toskeletal heavy SR and purified RyR1
Ryanodine is a plant alkaloid that binds to the openstate RyR/Ca2+ release channel with high affinity.27
To test whether CLIC2 can interact with RyR1 andmodify its function, we first carried out [3H]ryano-dine binding experiments. As shown in Fig. 1a, at a[Ca2+] of 10 μM, CLIC2 increased [3H]ryanodinebinding to skeletal heavy SR; binding rose from 1.31±0.1 pmol/mg (buffer, n=6) to 1.57±0.03 pmol/mg(15 μM CLIC2, n=6) and then to 1.7±0.01 pmol/mg(30 μM CLIC2, n=6).To assess whether this effect is due to a direct
interaction of CLIC2 with RyR1, we next undertook[3H]ryanodine binding to purified RyR1. As shownin Fig. 1b, CLIC2 increased [3H]ryanodine bindingto purified RyR1, the binding rose from 7.42±0.48(n=6) to 8.28±1.39 (n=6) pmol/mg at 10 μM [Ca2+],and from 5.01±0.62 (n=6) to 6.90±1.31 (n=6)pmol/mg at 100 μMCa2+. These data demonstratedthat CLIC2 can indeed interact with RyR1 and affect[3H]ryanodine binding to skeletal heavy SR andpurified RyR1.To characterize quantitatively how CLIC2 inter-
acts with RyR1, we did equilibrium saturationexperiments and Scatchard analysis. As shown inFigs. 1c and d, the interaction of CLIC2 withpurified RyR1 resulted in an increase in the bindingaffinity, without changing the apparent maximalbinding capacity significantly. In the absence ofCLIC2, the dissociation constant Kd for purifiedRyR1 was 21.92±6.91 nM (n=6). In the presence of15 μM CLIC2, however, the dissociation constantKd for purified RyR1 was reduced to 15.00±2.07 nM(n=6).Although the dissociation constantKd changedsignificantly, the apparent maximal binding capacity,Bmax, remained virtually unchanged (62.89±11.96(control, n=6) versus 62.83±4.68 (15 μM CLIC2,n=6) pmol/mg). These data provided evidence thatCLIC2 facilitated [3H]ryanodine binding to skeletalheavy SR and purified RyR1.Time-dependent association and dissociation
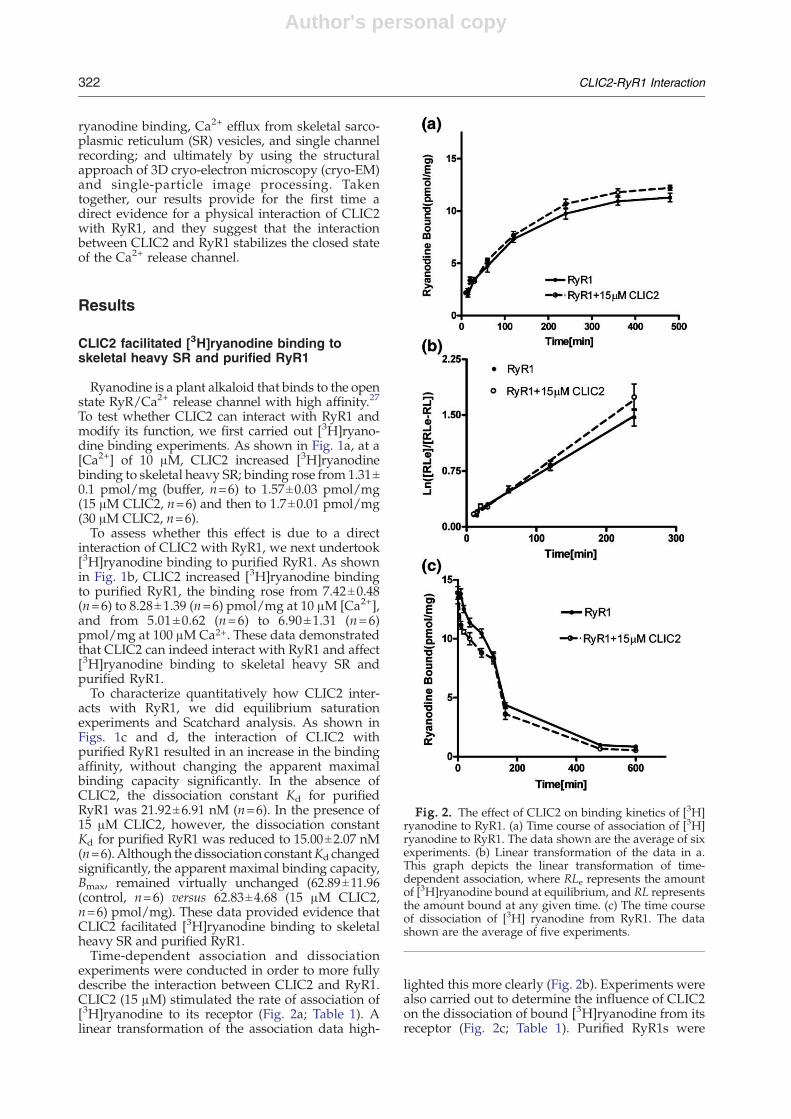
experiments were conducted in order to more fullydescribe the interaction between CLIC2 and RyR1.CLIC2 (15 μM) stimulated the rate of association of[3H]ryanodine to its receptor (Fig. 2a; Table 1). Alinear transformation of the association data high-
lighted this more clearly (Fig. 2b). Experiments werealso carried out to determine the influence of CLIC2on the dissociation of bound [3H]ryanodine from itsreceptor (Fig. 2c; Table 1). Purified RyR1s were
Fig. 2. The effect of CLIC2 on binding kinetics of [3H]ryanodine to RyR1. (a) Time course of association of [3H]ryanodine to RyR1. The data shown are the average of sixexperiments. (b) Linear transformation of the data in a.This graph depicts the linear transformation of time-dependent association, where RLe represents the amountof [3H]ryanodine bound at equilibrium, and RL representsthe amount bound at any given time. (c) The time courseof dissociation of [3H] ryanodine from RyR1. The datashown are the average of five experiments.
322 CLIC2-RyR1 Interaction

Author's personal copy
labeled with [3H]ryanodine in the absence of CLIC2,or with 15 μM CLIC2, and allowed to equilibrate for12 h at 24 °C. Aliquots were subsequently diluted100-fold into a buffer without ryanodine. CLIC2 hadno effect on the dissociation of [3H]ryanodine fromits receptor (Fig. 2c; Table 1). These data coupledwith the CLIC2-dependent increase in the rate ofassociation suggested that the increase in [3H]ryanodine binding affinity caused by CLIC2 wasdue solely to an enhancement of the ryanodineassociation kinetics.
CLIC2 diminished Ca2+ efflux from skeletalheavy SR vesicles
The effect of CLIC2 on [3H]ryanodine binding toskeletal heavy SR vesicles and purified RyR1 isindicative that CLIC2 can interact with RyR1. Wethen performed experiments of Ca2+ efflux fromskeletal SR vesicles, to determine whether theinteraction between CLIC2 and RyR1 affects thechannel activity of RyR1 (Fig. 3). After skeletal heavySR vesicles were partially loaded by the addition offour consecutive aliquots of CaCl2, thapsigargin wasadded to block the SR Ca2+-ATPase/pump, and thenCa2+ efflux was monitored. After the blockade of the
Ca2+-ATPase/pump, the medium [Ca2+] increaseddue to Ca2+ efflux from heavy SR vesicles, asmonitored by the Ca2+ indicator antipyrylazo III.The rise in extravesicular [Ca2+] was stopped byruthenium red, an RyR channel blocker, indicatingthat Ca2+ efflux occurred through the RyR1 channel.The presence of CLIC2 diminished Ca2+ efflux, asindicated by the reduced rate of rise of the curve. Themaximal Ca2+ efflux rate decreased from 57.08±7.77 nmol min–1 mg–1 (control, n=11) to 42.58±5.37 nmol min–1 mg–1 (30 μM CLIC2, n=6). Thesedata showed that CLIC2 is an inhibitor of Ca2+ effluxfrom skeletal heavy SR vesicles.
Single-channel recordings revealed that CLIC2stabilized the closed state of the RyR1 channel
To confirm that the inhibitory effect of CLIC2 onCa2+ efflux from heavy SR vesicles is indeed due to adirect interaction of CLIC2 with RyR1, and tocharacterize the effect of CLIC2 on the RyR1 channelactivity, we assessed the effect of CLIC2 on thegating properties of single RyR1 channels incorpo-rated into planar lipid bilayers. To minimizevariability in open probability (Po), single-channelrecordings were performed in the presence of EMD41000, a derivative of caffeine that acted as an RyRchannel stimulator.28 As shown in Fig. 4, EMD 41000raised Po to 0.55±0.13 (n=8 channels), 0.55±0.13(n=6 channels), and 0.51±0.10 (n=11 channels),when the holding potential was +20 mV, +30 mVand +40 mV, respectively (Fig. 4a–c, upper traces).CLIC2 (7 μM) drastically reduced Po by 70%, 70%,and 72% at +20 mV, +30 mV and +40 mV,respectively (Fig. 4a–c 4, lower traces). Statisticalanalysis indicated that CLIC2 decreased Po byprolonging the mean closed time Tc (increased by afactor of 9, 15, and 30 at +20 mV, +30 mV, and+40 mV, respectively), without affecting the meanopen time To significantly (Fig. 5). It is noteworthythat CLIC2 did not change the conductance of theRyR1 channel, since the current–voltage relationship
Table 1. The observed kinetics data for untreated RyR1and RyR1 treated with 15 μM CLIC2
Treatmentkobs
(min-1)k–1
(min-1)k+1
(min-1nM-1)Kd(k–1/k+1)
(nM)
Control 0.005642 0.004959 0.0003415 14.52±0.00068 ±0.00033(n=6) (n=5)
15 μM CLIC2 0.006791 0.004934 0.0009285 5.314±0.00082 ±0.00065(n=6) (n=5)
kobs was determined from linear transformation of the data shownin Fig. 2a, k–1 was calculated according to k–1= ln 2/t1/2 from thedata shown in Fig. 2c, where t1/2 is the half dissociation time. k+1=(kobs − k–1)/[L], where [L] is the total ligand concentration.
Fig. 3. Effects of CLIC2 on Ca2+ efflux from skeletal SR vesicles. (a) Representative traces of extravesicular variations of[Ca2+] measured with Antipyrylazo III as a Ca2+ indicator. These traces depict the process of Ca2+ uptake by Ca2+-ATPase/pump after four applications of 7.5 μM CaCl2, Ca2+ efflux through RyR1 in the presence of Thapsigargin, andtotal Ca2+ release from skeletal heavy SR vesicles at the end evoked by A23187. Experiments were divided into twogroups, buffer (black) and 30 μM CLIC2 (gray), respectively. Arrows indicate the time points at which each reagent wasadded. (b) Histogram of the maximal Ca2+ efflux rate from skeletal heavy SR vesicles. The asterisk denotes that the valuesare significantly different between the two groups, as assessed by Student’s t-test (pb0.05).
323CLIC2-RyR1 Interaction

Author's personal copy
Fig. 4. Single-channel activity ofpurified RyR1 in the presence ofCLIC2. Representative single chan-nel recordings (current flow versustime) under EMD 41000 stimulation,with or without the addition of 7 μMCLIC2 at three different holdingpotentials: (a) +20 mV; (b) +30 mV;(c) +40 mV. Both substances wereapplied to the cytosolic (cis) side ofthe channels. The closing and open-ing levels are indicated by an arrowand the letter C or O, respectively.The traces were taken from datafiltered at 1 kHz.
Fig. 5. CLIC2 modifies gating parameters of the skeletal RyR1 channel. The analysis of three parameters issummarized in the histograms: mean open probability (Po), mean open time (To), and mean closed time (Tc). Comparisonsof data that showed a significant difference from the value with EMD alone (filled bars) have been marked with asterisksas follows: ⁎ p≤0.05, ⁎⁎ p≤0.02, ⁎⁎⁎ p≤0.01. The open bars represent parameters following the addition of CLIC2. Thedata were taken from: (a) 6–8 single-channel experiments for +20 mV; (b) 5 or 6 single-channel experiments for+30 mV; (c) 6–11 single-channel experiments for +40 mV.
324 CLIC2-RyR1 Interaction

Author's personal copy
was not modified (data not shown). Furthermore,we observed neither sub-conductance openings norcoupled gating between RyR1 channels in thepresence of CLIC2, whereas CLIC2 had beenshown to induce sub-state activity of the RyR2channel, and coupled gating between RyR2channels.24 These data indicated that CLIC2 is aninhibitor of RyR1 channel opening, and that thereduced Ca2+ efflux from skeletal heavy SR vesiclesobserved in the presence of CLIC2 is likely to be adirect consequence of this inhibition; the inhibitoryeffect of CLIC2 on the RyR channel may be isoform-dependent, given that it seems to have somewhatdifferent effects on RyR1 and RyR2.
Cryo-EM and 3D reconstructions of theRyR1–CLIC2 complex
Our [3H]ryanodine binding, Ca2+ efflux, andsingle-channel recording experiments showed thatCLIC2 could indeed interact with RyR1 and modifyits channel activity. To better assess how CLIC2causes these effects, we studied the 3D structure ofthe RyR1–CLIC2 complex by cryo-EM and imagereconstruction. Figure 6 shows a typical electronmicrograph of frozen-hydrated RyR1–CLIC2 com-plexes. The particle images displayed characteristicRyR1 appearances, with multiple orientations simi-lar to those observed for the uncomplexed RyR1.29-31
Figure 7a shows 2D averages of the RyR1–CLIC2complexes, and Fig. 7b shows 2D averages of theRyR1 molecules, computed using images of selectedparticles, which were lying with their 4-fold sym-metry axes oriented perpendicular to the carbonsupport film. These images were aligned by cross-correlation methods. By visual inspection, theaveraged images of RyR1 and RyR1–CLIC2 complexappear very similar to each other, and to the 2D
averages determined for RyR1.29 Subtraction of the2D average of RyR1 from that of the RyR1–CLIC2complex provides a difference map (Fig. 7c) thatshould resolve the position of bound CLIC2, and thatperhaps should reveal conformational differencesbetween the two averaged projection structures. InFig. 7c, the brightest white areas, corresponding tothe most significant positive densities, representingprotein mass present in the RyR1–CLIC2 complexbut absent from RyR1. These areas (one of which ishighlighted by a circle in Fig. 7a–c) are located in aregion between domains 5 and 6 (see the 3Dreconstruction in Fig. 7d), part of an assemblage ofdomains that form the corners of the square-shapedcytoplasmic assembly and that have been termed theclamp.32 A statistical analysis of the differencebetween the two averaged images indicated thatthese areas show significant differences at a con-fidence level greater than 99.9%.33 These differencesalmost certainly correspond to the additional masscontributed by bound CLIC2 in the RyR1–CLIC2complex. This interpretation was confirmed, and thesite of difference was mapped more precisely in the3D reconstructions.Before computing the 3D reconstruction of the
RyR1–CLIC2 complex, we had to address a problemof heterogeneity of the dataset, which is caused bypartial occupancy of the four CLIC2 binding sites onone RyR1 homo-tetramer. Since the binding affinity
Fig. 6. Cryo-EM of RyR1–CLIC2 complexes. A portionof a cryo-EMmicrograph of RyR1–CLIC2 complexes, withthe protein particles embedded in a thin layer of vitreousice. The tetrameric structure of RyR1 is well preserved, asindicated by the characteristic square appearance, whichrepresents the images of the particles lying with their 4-fold symmetry axes oriented perpendicular to the carbonsupport film. Several individual particles are marked withwhite circles. The scale bar represents 500 Å.
Fig. 7. Two-dimensional averages of RyR1 and theRyR1–CLIC2 complex. (a) The 2D average of the RyR1–CLIC2 complex (n=297 particle images) in top view; (b)top view of the 2D average of RyR1 control (n=317 particleimages); (c) the difference map obtained by subtracting thedata in b from those in a. The top view represents theprojection of the channel as seen from the cytoplasmicside, as shown in the cartoon in d. The largest differencesshown in c correspond to the four additional massescontributed by binding of CLIC2, are seen as bright whiteareas, one of which is circled. The corresponding locationof the major difference in c is highlighted with green dotsin (a, b, and d). The scale bar represents 100 Å.
325CLIC2-RyR1 Interaction

Author's personal copy
between CLIC2 and purified RyR1 is in the micro-molar range, we estimate that, under the conditionsused for cryo-EM, ∼90% of CLIC2 binding sites onRyR1 should be occupied. However, this estimate isfor optimal binding conditions, and the actualbinding on the EM grids is likely to be lower. ForRyR1, the case is even more complicated, becauseeach RyR1 tetramer has four CLIC2 binding sites.Besides the RyR1 molecules showing full occupancywhen bound with four CLIC2 molecules, there arefive other categories of RyR1 molecules that showless than the full complement of four bound CLIC2molecules (i.e., with zero, one, two, or three CLIC2
molecules). We used a 2D classification analysis toestimate the actual percentages of RyR1 with thevarious complements of bound CLIC2 molecules.Top images of RyR1–CLIC2 complexes (n=789)were subjected to a multi-reference supervisedclassification,34 and the result showed that ∼70%of the particles had four CLIC2 molecules bound.The classification allowed us to eliminate most ofthe incompletely occupied RyR1 particles in thesubsequent image processing. We used only theparticles with CLIC2 binding sites fully occupiedto compute the final 3D reconstruction of theRyR1–CLIC2 complex.
Fig. 8. Three-dimensional surface representations of RyR1 and the RyR1–CLIC2 complex, and the 3D difference map.The 3D reconstruction of RyR1 is shown in yellow (a) and the RyR1–CLIC2 complex (b) is shown in orange. (c) Thedifference map (RyR1–CLIC2 minus RyR1) shown in orange is superimposed on the 3D reconstruction of RyR1 alone(yellow), another minor negative difference (RyR1 minus RyR1–CLIC2) shown in blue-violet, is also superimposed. The3D reconstructions are shown in three views: left, top view from the cytoplasmic surface, which in situ would face thetransverse tubule; middle, side view; right, bottom view showing the surface that would face the SR lumen. The numerals1–11 on the cytoplasmic assembly indicate the distinguishable domains, according to our earlier nomenclature.29 Thescale bar represents 100 Å.
326 CLIC2-RyR1 Interaction

Author's personal copy
In Fig. 8a and b, the 3D reconstructions of RyR1and the RyR1–CLIC2 complex are displayed insurface representation, in three orientations. Bothreconstructed structures consist of two majorcomponents: a large cytoplasmic assembly com-posed of at least 10 distinct domains (labeled bynumerals 1–10)29 and a smaller transmembraneassembly. For the reconstruction of the RyR1–CLIC2 complex, the final resolution was estimatedto be 25 Å (see Materials and Methods). Thedifference map was masked by simply increasingthe threshold of the 3D volume. The densitythreshold for the 3D volume of control RyR1 is4.2×10-4, in which the volume of the 3D reconstruc-tion matches the molecular mass of the tetramerRyR, 2.26 MDa. The density threshold for the 3Dvolume of RyR1–CLIC2 complex is 4.1×10-4, inwhich the 3D volume matches the molecular mass,2.37 MDa, of the tetramer RyR1 plus four CLIC2molecules. Overall, the 3D reconstruction of RyR1–CLIC2 is very similar to that of RyR1, but closeexamination reveals some subtle differences. Themost noticeable difference was found betweendomains 5 and 6, within the clamp-shaped struc-tures that form each of the corners of the square-shaped cytoplasmic assembly of RyR1.29 Specifi-cally, the mass between domains 5 and 6 of theRyR1–CLIC2 complex appears to be larger than themass in the corresponding region of the controlRyR1 structure. These differences could resultdirectly from the CLIC2 binding, and/or fromconformational changes of RyR1 caused by CLIC2binding. To determine the differences more pre-cisely, we generated a 3D difference map, bysubtracting the 3D volume of RyR1 from that ofthe RyR1–CLIC2 complex. The difference regionsare displayed in orange and superimposed on the3D reconstruction of RyR1 in Fig. 8c. The densitythreshold for the 3D difference map is also 4.1×10-4,identical with the threshold for the RyR1–CLIC2complex. At a lower density threshold, other minordifference showed up; however, it also makes theCLIC2 volume oversized. We simply masked thedifference map by increasing the threshold thatmatches to the RyR1–CLIC2 complex, at this thresh-old, no other difference was presented. The differencemap clearly showed four significant differencefeatures, one associated with each of the copies inthe region between domains 5 and 6 in thecytoplasmic assembly. RyR1 is a homotetramercomposed of four identical monomers, and oneCLIC2 molecule binds to each RyR1 monomer, thedifference would thus be expected to repeat fourtimes in the 3D difference map. We are confident thatthe four significant difference features are attributabledirectly to the excess mass contributed by the CLIC2binding to RyR1, because they are the most sig-nificant differences that appear when the 3D differ-ence map is displayed at a density threshold valuealmost the same as the level that was used to imagethe RyR1–CLIC2 complex and RyR1 structures.Apart from this area of extra mass between
domains 5 and 6, the other notable difference is
that the linkage between domains 9 and 10 in RyR1has broken in the RyR1–CLIC2 complex (Fig. 8b).Domains 9 and 10 are also located in the clamp-shaped region of RyR1. The separation betweendomains 9 and 10 is interpreted as a conformationalchange in RyR1 itself caused by the binding ofCLIC2. A similar feature has been observed in theopen state RyR1.35,36 To further clarify this minordifference, we computed a reverse difference mapby subtracting the 3D volume of the RyR1–CLIC2complex from that of the RyR1 control, anddisplayed the volume in another color (blue violetin Fig. 8c).Other minor differences between the two struc-
tures can be regarded as insignificant, both in massand density, and are unlikely to relate specifically tothe binding of CLIC2. Reassuringly, the locations ofthe four major binding sites of CLIC2 on the 3Dreconstruction are consistent with the major differ-ences seen in the 2D analysis (Fig. 7).
Docking the crystal structure of CLIC2 into thecryo-EM density map
The crystal structure of CLIC2 was solvedrecently by two research groups.17,23 We haveused one of these atomic structures of CLIC2 toperform an interactive docking by fitting theatomic coordinates (PDB code 2PER) into thecryo-EM surface envelope of the difference mapbetween RyR1–CLIC2 and RyR1 control. Figure 9illustrates the fitting of CLIC2, in a view in whichthe cytoplasmic side of RyR1–CLIC2 complex istilted from the 4-fold symmetry axis. We foundthat this orientation of the crystal structure ofCLIC2 fitted quite well into the cryo-EM densitythat was assigned to the CLIC2 molecule, asassessed by the cross-correlation coefficient values(0.67) between the difference map and the fittedatomic coordinates. Other docking orientations, forexample, a 180° rotation around the vertical axis(switch the N-terminal domain and foot loop) or180° rotation around the horizontal axis (switch theN-terminal and C-terminal domains), gave lowervalues of the cross-correlation coefficient.37 Ourdocking result illustrates clearly that CLIC2 likelyinteracts directly with domain 5 and domain 6 ofRyR1, with the foot loop interacting with domain5, and the N-terminal domain and the joint loopregion interacting with domain 6.
Discussion
CLIC proteins are found in both skeletal andcardiac muscle of humans and other vertebrates.6,20
A feature of CLIC proteins that is distinctive fromother chloride channels is that they exist in twodifferent forms: soluble and membrane-bound.While the membrane-bound CLIC proteins functionas Cl– channels, the function of soluble CLICproteins is not known. Dulhunty et al. showedrecently that CLIC2 could interact with RyR2 and
327CLIC2-RyR1 Interaction

Author's personal copy
modulate its channel activity, demonstrating thatCLIC2 is a RyR2 channel regulator, and implyingthat CLIC proteins may play a role in the regulationof Ca2+ signaling.20,24 Since CLIC2 exists in bothcardiac and skeletal muscle, and RyR2 shares a highdegree of homology with RyR1, we speculate thatCLIC2 may also interact with and modulate thechannel activity of RyR1.In this study, we investigated the interaction of
CLIC2 with the skeletal RyR/Ca2+ release channel.We have shown the following: (1) CLIC2 increases[3H]ryanodine binding to skeletal heavy SR andpurified RyR1; (2) CLIC2, in equilibrium saturation[3H]ryanodine binding experiments, increases thebinding affinity of ryanodine for its receptor (i.e., itdecreased Kd) without changing the maximal bind-ing capacity (Bmax) significantly; (3) CLIC2 reducesthe maximal Ca2+ efflux rate from skeletal heavy SRvesicles; (4) CLIC2 decreases the open probability ofRyR1, through increasing the mean closed time ofthe channel; (5) 3D cryo-EM of RyR1–CLIC2 com-plex shows that CLIC2 binds to a region betweendomains 5 and 6 in the clamp-shaped, cytoplasmic
regions of RyR1; (6) and the interaction betweendomains 9 and 10 in the clamp-shaped regions ofRyR1 appears to be absent or reduced when CLIC2binds to RyR1 under conditions that should favorthe closed state of RyR1, indicating that CLIC2induces conformational changes of RyR1 that aredistal to its binding site. Our data indicate thatCLIC2 can interact with RyR1 and modulate itschannel activity.As CLIC2 has been shown to interact with RyR2,
this protein may therefore be a common modulatorof Ca2+ release channels in both cardiac and skeletalmuscle.
Effect of CLIC2 on the structure of RyR1
By using cryo-EM and image reconstruction, wedetermined the binding site of CLIC2 on RyR1 asbeing located in an area between domains 5 and 6 inthe clamp-shaped region of RyR1. Previous studieshave shown that the region around domains 5 and 6contains “hotspots” of RyRs, which play importantroles in the regulation of these channels. Manycritical residues, such as divergent region 2,38phosphorylation site S2808,39 the central disease-causing mutation region,40 and one proposedcoupling site with DHPR,41 are located in thisregion. It is not surprising that the binding ofCLIC2 to this region could affect the function ofRyR1. Intriguingly, domains 5 and 6 in RyR1, withwhich CLIC2 interacts directly, contain the twomutation hotspots that we have previously mapped,the N-terminal disease-causing mutation region andthe central disease-causing mutation region.40,42
More than 100 mutations in RyR1 have beenidentified in families with malignant hyperthermiaand central core disease, and these mutations arelargely clustered into three regions of the ∼5000amino acid RyR1 sequence: the amino-terminalregion (residues 35–614), the central region (1728–2728), and the carboxyl-terminal region (3348–4973).43 The two mutation hotspots that lie in thecytoplasmic region, the N-terminal region and thecentral region, are well separated in the primarysequence (N1100 amino acids). According to thehypothesis proposed by Ikemoto and colleagues,44
the two mutation hotspots occur in structuraldomains that interact physically in the 3D structure,and changes in the strength of their interaction affectchannel gating. In that hypothesis, the N-terminaldomain and central domain interact in a way thatserves as a regulatory switch for channel gatingactivity; a tight “zipping” of the interacting domainsstabilizes the channel in the closed state. A mutationin either domain weakens the domain–domaininteraction, thus increasing the tendency toward“unzipping”; such an unzipping causes activationand leakiness of the Ca2+ release channel.Our structural information, together with the
domain switch hypothesis, leads us to propose amolecular mechanism for inhibition of the RyR1 Ca2+
release channel by CLIC2. As illustrated in Fig. 9b,the N-terminal domain and the joint loop of CLIC2
Fig. 9. Docking of the X-ray crystal structure of CLIC2into the cryo-EM density map of the RyR1–CLIC2 complex.The atomic coordinates of the CLIC2 (PDB code 2PER)werefit manually into our cryo-EM density map of RyR1–CLIC2complex, using the programO. (a) A tilted view of the RyR1+ CLIC2 complex, with one CLIC2 molecule docked in thebinding site of one subunit in the RyR1 homotetramer. (b) Amagnified view of the clamp region from a, showing thatCLIC2binds to domains 5 and 6 ofRyR1.Domain renderingof the CLIC2 molecule: N-terminal domain (amino acidresidues 11–94), red; joint loop (residues 95–106), yellow;C-terminal domain (residues 107–152 and 179–246),green; and foot loop, (residues 153–178), blue.
328 CLIC2-RyR1 Interaction

Author's personal copy
interact with domain 6, where the central region islocated, and the foot loop of CLIC2 interacts withdomain 5, where theN-terminal region is located. It islikely that binding of CLIC2 to domains 5 and 6 ofRyR1 concurrently strengthens the interactionbetween these two domains, and thereby minimizesdomain switch unzipping and stabilizes the closedstate of the RyR1 channel. Previously, we showedthat natrin, a toxin protein from the venom of thesnake Naja naja atra, also binds to domains 5 and 6 ofRyR1 concurrently, and inhibits the channel activityof RyR1.45 Our structural information suggests thatnatrin and CLIC2 interact with and inhibit RyR1channel in a similar manner, which conforms to thedomain switch mechanism.Domains 9 and 10 in the clamp-shaped region of
RyR1 are thought to be involved in the gating of theRyR1 channel.35,36 It was reported that domains 9and 10 were linked in the closed state of RyR1 andthe central channel in the transmembrane domainappeared to be closed, whereas they were separatedin an open state of RyR1 and the central channelappeared open.36 We observed that domains 9 and10 were separated upon CLIC2 binding to the closedstate RyR1, but the central channel appears to beclosed (see Fig. 8a and b).
Effect of CLIC2 on the function of RyR1
As a Ca2+ release channel in the skeletal SR, theactivity of RyR1 is modulated by many endogenousand exogenous regulators. In general, channelactivators such as cytosolic Ca2+ (in the micromolarrange), ATP and caffeine increase the Po of the RyR1channel and enhance [3H]ryanodine binding; chan-nel inhibitors such as ruthenium red, and Mg2+ andCa2+ (in the millimolar range) lower RyR1 Po andreduce [3H]ryanodine binding to the channel. As aconsequence, it has become largely accepted that theryanodine binding site on the RyR channel isaccessible only when the channel is in an openconformation. The results presented here demon-strate that CLIC2 is a RyR channel ligand that doesnot conform to this general pattern. CLIC2 reducessingle channel Po and RyR1-mediated Ca2+ effluxfrom heavy SR but unexpectedly increases theaffinity of the receptor for [3H]ryanodine. Similar,seemingly contradictory, effects were observedwhen CLIC2 was added to RyR2.20,24
The novel cryo-EM data presented here provideinformation that may contribute towards an under-standing of this phenomenon. Previous investiga-tions have defined specific structural rearrangementsassociated with the transition of the RyR1 channelfrom a closed to an open conformation; these includeseparation of domains 9 and 10 in the cytoplasmicclamp region of the channel, and the appearance of acentral opening in the transmembrane region.36 Theinteraction of CLIC2 with the closed RyR1 channelinduces a separation of domains 9 and 10 of themolecule but no equivalent change in the transmem-brane region. The cryo-EM results on RyR1-CLIC2,albeit at a resolution that is too low to provide a
precise molecular mechanism of the effect of CLIC2on RyR1, nevertheless show structural features thathave been attributed to the open and to the closedforms of the channel,35,36 which might be related tothe biochemical/functional assays that showedcharacteristics of both open (in the clamp regions)and closed (in the central region of the transmem-brane domain) channel states in the RyR1–CLIC2complex. We suggest that the binding of CLIC2 tothe closed RyR1 induces a conformational changethat resembles the open configuration in the clamp-shaped region of RyR1, which may facilitate theassociation of ryanodine with the binding site, andconsequently increase its [3H]ryanodine bindingaffinity. However, the binding of CLIC2 to domains5 and 6 of RyR1 concurrently strengthens theinteraction between these two domains, therebyminimizes domain switch unzipping, stabilizes theclosed state of the RyR1 channel, and consequentlyreduces the rate of Ca2+ efflux from skeletal heavy SR.Finally, CLIC2 resembles the effect of FKBP12 on
RyR1.46,47 In the latter work, FKBP12 was shown todecrease Po after caffeine activation of skeletal RyRchannels; in the absence of FKBP12, RyR1 channelsexhibit increased gating frequency, suggesting thatFKBP12 qstabilizesq the channel in the open andclosed states. Our results suggest that CLIC2, in amanner quite similar to that of FKBP12, may be anintrinsic stabilizer of the RyR1 channel.FKBP12 interacts, as CLIC2 does, with the RyR1
clamp regions, but at a distinct site (involvingdomains 9, 10, and 3) from thatwhere CLIC2 interacts(involving domains 5 and 6).48 FKBP12 binds to theperiphery between domains 3 and 9, whereas theCLIC2 we mapped is located on top of domains 5and 6. A sequence between residues 216 and 572 inRyR1 has sequence homology to an IP3-binding coreregion, and a homology model was docking insidedomain 5.49 In another report, the N-terminalresidues 41–420 of RyR1 was found has a high levelof sequence homology to phosphorylated isocitratedehydrogenase, a homologymodel was also dockinginside domains 5 and 9.50 Both of these dockings arein close proximity to the mapped CLIC2 binding site,but at distinct sites. These structural studies lendfurther support to the hypothesis that the clampregions of RyR are of critical importance to regulatingthe channel activity of RyR1, even though they arespatially far remote from the actual ion channel.Understanding how these rather remote regulatorysites can affect the conformations in the transmem-brane domains bringing the channel into more openor closed state, however, require further high-resolu-tion structural work.
Materials and Methods
Chemicals
All chemicals were of analytical grade or above andwere purchased from Sigma-Aldrich (St. Louis, Missouri,USA) unless specified otherwise.
329CLIC2-RyR1 Interaction

Author's personal copy
CLIC2 expression and purification
CLIC2 was expressed and purified as described.23
Briefly, CLIC2 was expressed with an N-terminal His6tag and purified by Ni2+-chelating column chromatogra-phy and gel-filtration chromatography. The final buffer inwhich purified CLIC2 was dissolved was 20 mM Tris–HCl(pH 7.5), 200 mM NaCl.
Preparation of skeletal heavy SR vesicles
Skeletal heavy SR (heavy SR) vesicles were preparedfromNew Zealand white rabbits essentially as described51
but with some modifications. Briefly, skeletal muscle washomogenized in five volumes (v/w) of buffer A (0.3 Msucrose, 10 mM Hepes, pH 7.0, 0.5 mM EDTA, 2 mMPMSF and 1:1000 diluted Protease Inhibitory Cocktail(Sigma P8340). Two steps of differential centrifugationwere carried out in sequence, 15 min at 11,000 g followedby 1 h at 110,000 g. The pellet from the secondcentrifugation was collected and suspended in buffer Awith 0.65 M KCl, with a ratio of 1:1 (w/v: weight ofskeletal muscle used initially /volume of KCl extractionbuffer), and kept on ice for 1 h. The suspension was thenre-pelleted and re-homogenized, and aliquots werelayered onto a sucrose step gradient. The gradient stepsfrom bottom to top are 45%, 38%, 32%, and 27% (w/v)sucrose. After centrifugation for 16 h in a Beckman SW28rotor at 20,000 rpm (70,000 g), the membrane fraction(heavy SR) at the interface between 38% and 45% sucrosewas collected, diluted approximately twofold with bufferA and centrifuged again for 1 h at 110,000 g. The pelletswere suspended in buffer A without EDTA, quick-frozenin liquid nitrogen, and stored at –80 oC. Proteinconcentration was measured with the BCA methodaccording to the manufacturer’s instruction.
RyR1 purification
RyR1 was purified from Chaps-solubilized skeletalheavy SR essentially as described52-54 but with somemodifications. Briefly, skeletal heavy SR vesicles (50 mg)were suspended in 20 ml of buffer B (1 M NaCl, 20 mMHepes, pH 7.0, 2 mM DTT, 2 mM PMSF, 1:1000 dilutedProtease Inhibitory Cocktail) and appropriate Chaps-soy-bean phospholipids mixture (10% Chaps and 5% soybeanphospholipids, all from Calbiochem, La Jolla, CA). TheChaps/protein ratio was 13.3 (w/w). After incubation onice for 30 min with shaking, the sample was centrifugedfor 1 h at 110,000 g. The supernatant was loaded onto ahydroxyapatite ceramic (Bio-Rad, Hercules, California,USA) column (5 ml) equilibrated with buffer C (10 mMK2HPO4, pH 7.0, 0.5% Chaps, 0.25% soybean lecithin,2 mM DTT). The column was washed sequentially with15 ml of buffer C and buffer D (10 mM K2HPO4, 200 mMNaCl, pH 7.0, 0.5% Chaps, 0.25% soybean lecithin, 2 mMDTT) and buffer E (50 mM K2HPO4, 200 mM NaCl, pH7.0, 0.5% Chaps, 0.25% soybean lecithin, 2 mM DTT). The15 ml elute was collected and concentrated by centrifuga-tion at 1000 g in a Centricon concentrator (100 kDa cut-off;Millipore, Billerica, MA.) for further purification on a 5% –20% (w/v) linear sucrose gradient buffered with buffer Band 0.5% Chaps, 0.25% soybean lecithin. After centrifuga-tion for 16 h in a Beckman SW28 rotor at 26,000 rpm(110,000 g), the gradient was fractionated into 2 mlportions. After checking by SDS-PAGE, the RyR1-contain-ing fractions were pooled and concentrated as described,53
followed by division into 50 μl aliquots, quick-frozen inliquid nitrogen, and stored at −80 °C.
[3H]ryanodine binding assay
The [3H]ryanodine binding experiments were done asdescribed.24 Skeletal heavy SR vesicles (0.25 mg/ml) orpurified RyR1 (25 μg/ml) was incubated with 0.2 M KCl,20 mMHepes, pH 7.0, with various concentrations of Ca2+
and [3H]ryanodine (Perkin-Elmer, Waltham, MA), andwith different concentrations of CLIC2 (15 μM or 30 μM)or buffer only without CLIC2, various concentrations ofCa2+ and [3H]ryanodine were present during the incuba-tion period. The binding reaction was stopped by rapidfiltration through Whatman GF/B glass-fiber filters pre-soaked with 1% (w/v) polyethyleneimine, which werethen rinsed twice with 10 ml of ice-cold buffer containing0.2 MKCl, 20 mMHepes, pH 7.0. The filters were air-driedand placed into 20 ml scintillation vials with 5 ml ofscintillation solution (2.5 g of PPO and 150 mg of POPOPin 500 ml of dimethyl benzene), incubated overnight, andthe radioactivity was counted the following day. Free Ca2+
concentrations were calculated using the computer pro-gram described by Fabiato & Fabiato.55 Non-specificbinding was measured in the presence of a 1000-foldexcess of unlabeled ryanodine (Calbiochem, La Jolla, CA).The experiments were repeated at least twice on twodifferent skeletal heavy SR preparations and on purifiedRyR1 preparations. For details of individual experiments,refer to the figure legends.
Measurement of association/dissociation kinetics
The [3H]ryanodine association/dissociation kineticsexperiments were done as described.56 The association rateof [3H]ryanodine (2 nM) was measured by quenching thebinding reaction by filtration at times ranging from 1min to500min after the addition of purifiedRyR1 (25 μg/ml) to the[3H]ryanodine reactionmedium (1mMCaCl2, 1 mMEGTA,0.2 M KCl, 20 mM Hepes, 0.5% Chaps, 0.25% soybeanlecithin, pH 7.0). Dissociation of [3H]ryanodine from theequilibrium complex followed equilibration of [3H]ryano-dine with purified RyR1 in the presence of 0, or 15 μMCLIC2, and allowed to equilibrate for 12 h at 24 °C. Aliquotswere subsequently diluted 100-fold into a buffer withoutryanodine. Determinations of residual specific bindingweremade at times ranging from 5 min to 600 min.
Ca2+ efflux from skeletal heavy SR vesicles
Experiments were done as described.57 Briefly, skeletalheavy SR vesicles (100 μg/ml) were added to a 2 mlsolution containing 100 mM KH2PO4 (pH 7); 4 mMMgCl2; 1 mM Na2ATP, and 0.5 mM antipyrylazo III. Thetemperature was maintained at 25 °C. Extra-vesicular[Ca2+] was monitored at 710 nm using a HITACHI U2010spectrophotometer. Vesicles were loaded with Ca2+ byaddition of four aliquots of CaCl2, each initially increas-ing the extra-vesicular [Ca2+] by 7.5 μM. Sufficient time(2–5 min) was allowed between one addition of Ca2+ andthe next, so that the uptake reduced the Ca2+ concentrationto baseline level. Thapsigargin (TG, 200 nM,) was added toblock the Ca2+-ATPase/pump, and Ca2+ efflux was thenmeasured for 10 min. Ruthenium red (5 μM) was added toconfirm that Ca2+ efflux was through RyR1. Finally, theCa2+ ionophore A23187 (3 μg/ml) was added, to release
330 CLIC2-RyR1 Interaction

Author's personal copy
all the Ca2+ remaining in the vesicles. Experiments wereperformed with buffer containing no CLIC2, or with30 μM CLIC2, added together with skeletal heavy SR. Acalibration curve was established at the start of eachexperiment by measuring changes in antipyrylazo IIIabsorption in response to four sequential additions of7.5 μM CaCl2.
Single-channel recordings
Single-channel recordings were done as described.58
Planar phospholipid bilayers were formed from suspen-sions of phosphatidylethanolamine (Avanti Polar Lipids,Alabaster, AL) in n-decane (35 mg/ml) across a 200 μmdiameter hole in a polystyrene copolymer partition thatseparated two chambers referred to as cis (0.5 ml) andtrans (1.0 ml). The trans chamber was held at virtualground, whereas the cis chamber could be clamped atholding potentials relative to ground. Current flow acrossthe bilayer was monitored by an operational amplifier thatserved as a current–voltage converter.59 Bilayers wereformed with solutions containing 600 mM KCl, 20 mMHepes, titrated to pH 7.2 with KOH, resulting in a solutioncontaining 610 mM K+ in both chambers. An osmoticgradient was created by the addition of two aliquots(100 μl each) of 3 M KCl to the cis chamber. Purified RyR1proteins were added to the cis chamber and stirred. Underthese conditions, channels usually incorporated into thebilayer within 5 min. If channels did not incorporate, athird aliquot of 3 M KCl was added to the cis chamber.After channel incorporation, further fusion was preventedby perfusion of the cis chamber with 610 mM K+. Channelproteins incorporate into the bilayer in a fixed orientation,so that the cytosolic face of the channel is exposed to thesolution in the cis chamber, and the luminal face of thechannel is exposed to the solution in the trans chamber.Single-channel open probability Po was increased by theaddition to the cis chamber of 20 – 100 μM EMD 41000 inall experiments, to minimize Po variability.
28 Experimentswere done at room temperature (22 °C). CLIC2 was addedto the cis chamber 5 min before recording.Single-channel current fluctuations were filtered at
1 kHz with a low-pass filter, digitized and displayed inthe program Acquire 5.0 (Bruxton Corporation, Seattle,WA). For analysis, filtered data representing 30 – 120 s ofchannel activity were digitized and replayed. Single-channel current amplitudes and lifetimes were measuredfrom digitized data with the software TAC 3.0 (BruxtonCorporation, Seattle, WA).
Cryo-electron microscopy and image processing
For preparation of the RyR1–CLIC2 complex, RyR1(50 μg/ml) and CLIC2 (9.28 μM)were incubated in 20 mMNa-Mops, pH 7.4, 200 mM NaCl, 2.0 mM EGTA, 0.5%Chaps, 2 mM DTT, 2.0 μg/ml leupeptin for 30 min at 4°C.EM grids were prepared for cryo-EM by an FEI Vitrobotcomputer-controlled freeze-plunging instrument (FEICompany, Hillsboro, OR). Micrographs were recordedwith low-dose protocols on an FEI Tecnai F20 fieldemission gun transmission electron microscope operatedat 200 kV, equipped with an Oxford CT3500 cryo-transferholder (Gatan, Inc., Warrendale, PA). The temperature ofthe grids was maintained at around –170 °C. The defocusof the micrographs ranged between –1.5 and –4.5 μm, at amagnification of 50,760× (±2%). The RyRs have apreferred orientation on the carbon support film of the
EM grids, and so to obtain an adequate sampling oforientation, we have collected additional EM data with thespecimen grids tilted up to 50°. Tilting provides theadditional orientational views of the RyR molecule thatare required to compute accurate 3D reconstructions (seeFig. S1 in the Supplementary Data). Each exposurecorresponded to an electron dose of ∼10 e–/Å2. Micro-graphs were checked for drift, astigmatism, and thepresence of Thon rings by optical diffraction. Selectedelectron micrographs were digitized on a Zeiss/Imagingscanner (Z/I Imaging Corporation, Huntsville, Alabama,USA) with a step size of 14 μm.Images were processed using the SPIDER/WEB soft-
ware package.60 The contrast transfer function effect wascorrected for each individual particle using the phaseflipping method.61 For tilted images, eachmicrograph wasdivided into small pieces (about 1500 pixel×800 pixel), thedefocus value was estimated for each piece using theSPIDER program, and the defocus value for a particularpiece was assigned to all particles lying within the piece.The error of defocus values between particles at differentlocations within one piece of titled micrograph is about0.1 μm, similar to the error range of the defocus values.The 3D reconstructions were obtained from images ofparticles lying in all available orientations, through aprojection matching procedure.62 The final 3D reconstruc-tions of RyR1 control (un-complexed) and of the RyR1–CLIC2 complex were computed from 16,519 and 15,889particle images, respectively; and 4-fold symmetry wasenforced in all 3D reconstructions. The final resolutionswere estimated by the Fourier shell correlation with a cut-off of 0.5,63 for RyR1 control structure, the resolution is21 Å; and 25 Å for the RyR1–CLIC2 complex structure. AnX-ray structure of CLIC2 (PDB code 2PER) was dockedinto the cryo-EM density of the 3D difference mapmanually, using the molecular graphics programs O andChimera. Various possible fitting orientations were eval-uated by the correlation coefficient using the SPIDERprogram, and best fitting orientation was chosen by thehighest correlation coefficient.37
Statistics
Results were analyzed using the Mann–Whitney ranksum test (SigmaStat software, Systat Software Inc., SanJose, CA). Furthermore, if a normality test was successful,Student’s t-test was undertaken. Diffrenceswere regarded assignificant when pb0.05. The results are expressed as meanvalues±S.E.M.
Acknowledgements
This work was supported by research grants fromthe Natural Science Foundation of China, the Trans-Century Talent Awarding Program, Ministry ofEducation, and the National Basic Research Program(973 Program), Ministry of Science and Technology,P. R. China to C.C.Y., research grants from the BritishHeart Foundation to A.J.W., by American HeartAssociation grant 0430076N to Z.L., and by NationalInstitutes of Health grant AR40615 to T.W. We thankBin Geng (Department of Physiology, The HealthScience Centre, Peking University) for excellenttechnical assistance, and the Wadsworth Center’s
331CLIC2-RyR1 Interaction

Author's personal copy
Electron Microscopy Core Facility and the Resourcefor Visualization of Biological Complexity (NIHBiotechnological Resource grant RR01219).
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2009.01.059
References
1. Jentsch, T. J. (2008). CLC chloride channels andtransporters: from genes to protein structure, pathol-ogy and physiology. Crit. Rev. Biochem. Mol. Biol. 43,3–36.
2. Strange, K., Emma, F. & Jackson, P. S. (1996). Cellularand molecular physiology of volume-sensitive anionchannels. Am. J. Physiol. 270, C711–C730.
3. Ashley, R. H. (2003). Challenging accepted ionchannel biology: p64 and the CLIC family of putativeintracellular anion channel proteins. Mol. Membr. Biol.20, 1–11.
4. Landry, D., Sullivan, S., Nicolaides, M., Redhead,C., Edelman, A., Field, M. et al. (1993). Molecularcloning and characterization of p64, a chloride channelprotein from kidney microsomes. J. Biol. Chem. 268,14948–14955.
5. Valenzuela, S. M., Martin, D. K., Por, S. B., Robbins,J. M., Warton, K., Bootcov, M. R. et al. (1997).Molecular cloning and expression of a chloride ionchannel of cell nuclei. J. Biol. Chem. 272, 12575–12582.
6. Heiss, N. S. & Poustka, A. (1997). Genomic structure ofa novel chloride channel gene, CLIC2, in Xq28.Genomics, 45, 224–228.
7. Qian, Z., Okuhara, D., Abe, M. K. & Rosner, M. R.(1999). Molecular cloning and characterization of amitogen-activated protein kinase-associated intracel-lular chloride channel. J. Biol. Chem. 274, 1621–1627.
8. Duncan, R. R., Westwood, P. K., Boyd, A. &Ashley, R. H. (1997). Rat brain p64H1, expressionof a new member of the p64 chloride channel proteinfamily in endoplasmic reticulum. J. Biol. Chem. 272,23880–23886.
9. Berryman, M. & Bretscher, A. (2000). Identification ofa novel member of the chloride intracellular channelgene family (CLIC5) that associates with the actincytoskeleton of placental microvilli. Mol. Biol. Cell, 11,1509–1521.
10. Nishizawa, T., Nagao, T., Iwatsubo, T., Forte, J. G. &Urushidani, T. (2000). Molecular cloning and character-ization of a novel chloride intracellular channel-relatedprotein, parchorin, expressed in water-secreting cells.J. Biol. Chem. 275, 11164–11173.
11. Berry, K. L., Bulow, H. E., Hall, D. H. & Hobert, O.(2003). A C. elegans CLIC-like protein required forintracellular tube formation and maintenance. Science,302, 2134–2137.
12. Dulhunty, A., Gage, P., Curtis, S., Chelvanayagam, G.& Board, P. (2001). The glutathione transferasestructural family includes a nuclear chloride channeland a ryanodine receptor calcium release channelmodulator. J. Biol. Chem. 276, 3319–3323.
13. Edwards, J. C. (1999). A novel p64-related Cl− channel:subcellular distribution and nephron segment-specificexpression. Am. J. Physiol. 276, F398–F408.
14. Tulk, B. M., Schlesinger, P. H., Kapadia, S. A. &Edwards, J. C. (2000). CLIC-1 functions as a chloridechannel when expressed and purified from bacteria.J. Biol. Chem. 275, 26986–26993.
15. Tulk, B. M., Kapadia, S. & Edwards, J. C. (2002).CLIC1 inserts from the aqueous phase into phospho-lipid membranes, where it functions as an anionchannel. Am. J. Physiol. Cell Physiol. 282, C1103–C1112.
16. Littler, D. R., Harrop, S. J., Fairlie, W. D., Brown, L. J.,Pankhurst, G. J., Pankhurst, S. et al. (2004). Theintracellular chloride ion channel protein CLIC1undergoes a redox-controlled structural transition.J. Biol. Chem. 279, 9298–9305.
17. Cromer, B. A., Gorman, M. A., Hansen, G., Adams, J.J., Coggan, M., Littler, D. R. et al. (2007). Structure ofthe Janus protein human CLIC2. J. Mol. Biol. 374,719–731.
18. Proutski, I., Karoulias, N. & Ashley, R. H. (2002).Overexpressed chloride intracellular channel proteinCLIC4 (p64H1) is an essential component of novelplasma membrane anion channels. Biochem. Biophys.Res. Commun. 297, 317–322.
19. Berryman, M., Bruno, J., Price, J. & Edwards, J. C.(2004). CLIC-5A functions as a chloride channel invitro and associates with the cortical actin cytoske-leton in vitro and in vivo. J. Biol. Chem. 279,34794–34801.
20. Board, P. G., Coggan, M., Watson, S., Gage, P. W. &Dulhunty, A. F. (2004). CLIC-2 modulates cardiacryanodine receptor Ca2+ release channels. Int. J.Biochem. Cell Biol. 36, 1599–1612.
21. Harrop, S. J., DeMaere, M. Z., Fairlie, W. D., Reztsova,T., Valenzuela, S. M., Mazzanti, M. et al. (2001). Crystalstructure of a soluble form of the intracellular chlorideion channel CLIC1 (NCC27) at 1.4-Å resolution. J. Biol.Chem. 276, 44993–45000.
22. Littler, D. R., Assaad, N. N., Harrop, S. J., Brown, L. J.,Pankhurst, G. J., Luciani, P. et al. (2005). Crystalstructure of the soluble form of the redox-regulatedchloride ion channel protein CLIC4. FEBS J. 272,4996–5007.
23. Mi, W., Liang, Y. -H., Li, L. & Su, X. -D. (2008). Thecrystal structure of human chloride intracellularchannel protein 2: a disulfide bond with functionalimplications. Proteins:Struct. Funct. Genet. 71, 509–513.
24. Dulhunty, A. F., Pouliquin, P., Coggan, M., Gage, P. W.& Board, P. G. (2005). A recently identified member ofthe glutathione transferase structural family modifiescardiac RyR2 substate activity, coupled gating andactivation by Ca2+ and ATP. Biochem. J. 390, 333–343.
25. Fill, M. & Copello, J. A. (2002). Ryanodine receptorcalcium release channels. Physiol. Rev. 82, 893–922.
26. Meissner, G. (2004). Molecular regulation of cardiacryanodine receptor ion channel. Cell Calcium, 35,621–628.
27. Meissner, G. & el-Hashem, A. (1992). Ryanodine as afunctional probe of the skeletal muscle sarcoplasmicreticulumCa2+ release channel.Mol. Cell Biochem. 114,119–123.
28. McGarry, S. J. & Williams, A. J. (1994). Activation ofthe sheep cardiac sarcoplasmic reticulum Ca2+-releasechannel by analogues of sulmazole. Br. J. Pharmacol.111, 1212–1220.
29. Radermacher, M., Rao, V., Grassucci, R., Frank, J.,Timerman, A. P., Fleischer, S. &Wagenknecht, T. (1994).Cryo-electron microscopy and three-dimensionalreconstruction of the calcium release channel/ryano-dine receptor from skeletal muscle. J. Cell Biol. 127,411–423.
332 CLIC2-RyR1 Interaction

Author's personal copy
30. Sharma, M. R., Penczek, P., Grassucci, R., Xin, H.B., Fleischer, S. & Wagenknecht, T. (1998). Cryoe-lectron microscopy and image analysis of thecardiac ryanodine receptor. J. Biol. Chem. 273,18429–18434.
31. Sharma, M. R., Jeyakumar, L. H., Fleischer, S. &Wagenknecht, T. (2000). Three-dimensional structureof ryanodine receptor isoform three in two conforma-tional states as visualized by cryo-electron micro-scopy. J. Biol. Chem. 275, 9485–9491.
32. Serysheva, I. I., Orlova, E. V., Chiu, W., Sherman,M. B.,Hamilton, S. L. & van Heel, M. (1995). Electroncryomicroscopy and angular reconstitution used tovisualize the skeletal muscle calcium release channel.Nat. Struct. Biol. 2, 18–24.
33. Wagenknecht, T., Frank, J., Boublik, M., Nurse, K. &Ofengand, J. (1988). Direct localization of the tRNA–anticodon interaction site on the Escherichia coli 30S ribosomal subunit by electron microscopy andcomputerized image averaging. J. Mol. Biol. 203,753–760.
34. Frank, J. (2006). Multivariate data analysis andclassification of images. In Three-dimensional ElectronMicroscopy of Macromolecular Assemblies (Frank, J., ed.),pp. 145–192, Oxford University Press, New York.
35. Orlova, E. V., Serysheva, II, van Heel, M., Hamilton,S. L. & Chiu, W. (1996). Two structural configurationsof the skeletal muscle calcium release channel. Nat.Struct. Biol. 3, 547–552.
36. Serysheva, I. I., Schatz, M., van Heel, M., Chiu, W. &Hamilton, S. L. (1999). Structure of the skeletal musclecalcium release channel activated with Ca2+ andAMP-PCP. Biophys. J. 77, 1936–1944.
37. Agrawal, R. K., Penczek, P., Grassucci, R. A. &Frank, J. (1998). Visualization of elongation factor Gon the Escherichia coli 70S ribosome: the mechan-ism of translocation. Proc. Natl Acad. Sci. USA, 95,6134–6138.
38. Liu, Z., Zhang, J., Wang, R. W., Chen, S. R. W. &Wagenknecht, T. (2004). Location of divergent region 2on the three-dimensional structure of cardiac muscleryanodine receptor/calcium release channel. J. Mol.Biol. 338, 533–545.
39. Meng, X., Xiao, B., Cai, S., Huang, X., Li, F., Bolstad, J.et al. (2007). Three-dimensional localization of serine2808, a phosphorylation site in cardiac ryanodinereceptor. J. Biol. Chem. 282, 25929–25939.
40. Liu, Z., Wang, R. W., Zhang, J., Chen, S. R. W. &Wagenknecht, T. (2005). Localization of a disease-associated mutation site in the three-dimensionalstructure of the cardiac muscle ryanodine receptor.J. Biol. Chem. 280, 37941–37947.
41. Perez, C. F., Mukherjee, S. & Allen, P. D. (2003). Aminoacids 1-1,680 of ryanodine receptor type 1 hold criticaldeterminants of skeletal type for excitation-contrac-tion coupling. Role of divergence domain D2. J. Biol.Chem. 278, 39644–39652.
42. Wang, R., Chen, W., Cai, S., Zhang, J., Bolstad, J.,Wagenknecht, T. et al. (2007). Localization of an NH2-terminal disease-causing mutation hotspot to the“clamp” region in the three-dimensional structure ofthe cardiac ryanodine receptor. J. Biol. Chem. 282,17785–17793.
43. McCarty, T. V., Quane, K. A. & Lynch, P. J. (2000).Ryanodine receptormutations inmalignant hyperther-mia and central core disease. Hum. Mutat. 15, 410–417.
44. Ikemoto, N. & Yamamoto, T. (2002). Regulation ofcalcium release by interdomain interaction withinryanodine receptors. Front. Biosci. 7, d671–d683.
45. Zhou, Q., Wang, Q. L., Meng, X., Shu, Y., Jiang, T.,Wagenknecht, T. et al. (2008). Structural and functionalcharacterization of ryanodine receptor-natrin toxininteraction. Biophys J. 95, 4289–4299.
46. Brillantes, A. B., Ondrias, K., Scott, A., Kobrinsky, E.,Ondriasova, E., Moschella, M. C. et al. (1994).Stabilization of calcium release channel (ryanodinereceptor) function by FK506-binding protein. Cell, 77,513–523.
47. Gaburjakova, M., Gaburjakova, J., Reiken, S., Huang,F., Marx, S. O., Rosemblit, N. & Marks, A. R. (2001).FKBP12 binding modulates ryanodine receptor chan-nel gating. J. Biol. Chem. 276, 16931–16935.
48. Samsó, M., Shen, X. & Allen, P. D. (2006). Structuralcharacterization of the RyR1-FKBP12 interaction. J. Mol.Biol. 356, 917–927.
49. Serysheva, I. I., Hamilton, S. L., Chiu, W. & Ludtke,S. J. (2005). Structure of Ca2+ release channel at 14 Åresolution. J. Mol. Biol. 345, 427–431.
50. Baker, M. L., Serysheva, I. I., Sencer, S., Wu, Y.,Ludtke, S. J. et al. (2002). The skeletal muscle Ca2+
release channel has an oxidoreductase-like domain.Proc. Natl Acad. Sci. USA, 99, 12155–12160.
51. Chu, A., Dixon, M. C., Saito, A., Seiler, S. &Fleischer, S. (1988). Isolation of sarcoplasmic reticu-lum fractions referable to longitudinal tubules andjunctional terminal cisternae from rabbit skeletalmuscle. Methods Enzymol. 157, 36–46.
52. Inui, M., Saito, A. & Fleischer, S. (1987). Purification ofthe ryanodine receptor and identity with feet struc-tures of junctional terminal cisternae of sarcoplasmicreticulum from fast skeletal muscle. J. Biol. Chem. 262,1740–1747.
53. Lai, F. A., Erickson, H. P., Rousseau, E., Liu, Q. Y. &Meissner, G. (1988). Purification and reconstitution ofthe calcium release channel from skeletal muscle.Nature, 331, 315–319.
54. Yin, C. C. & Lai, F. A. (2000). Intrinsic lattice formationby the ryanodine receptor calcium-release channel.Nat. Cell Biol. 2, 669–671.
55. Fabiato, A. & Fabiato, F. (1979). Calculator programsfor computing the composition of the solutionscontaining multiple metals and ligands used forexperiments in skinned muscle cells. J. Physiol.(Paris), 75, 463–505.
56. Favero, T. G., Zable, A. C. & Abramson, J. J. (1995).Hydrogen peroxide stimulates the Ca2+ releasechannel from skeletal muscle sarcoplasmic reticulum.J. Biol. Chem. 270, 25557–25563.
57. Dulhunty, A. F., Laver, D. R., Gallant, E. M., Casarotto,M. G., Pace, S. M. & Curtis, S. (1999). Activation andinhibition of skeletal RyR channels by a part of theskeletal DHPR II-III loop: effects of DHPR Ser687 andFKBP12. Biophys. J. 77, 189–203.
58. Tanna, B., Welch, W., Ruest, L., Sutko, J. L. &Williams,A. J. (2006). The interaction of an impermeant cationwith the sheep cardiac RyR channel alters ryanoidassociation. Mol. Pharmacol. 69, 1990–1997.
59. Miller, C. (1982). Open-state substructure of singlechloride channels from Torpedo electroplax. Phil.Trans. Roy. Soc. ser. B, 299, 401–411.
60. Frank, J., Radermacher, M., Penczek, P., Zhu, J., Li, Y. H.,Ladjadj, M. & Leith, A. (1996). SPIDER and WEB:processing and visualization of images in 3D electronmicroscopyand related fields. J. Struct. Biol.116, 190–199.
61. Frank, J. (2006). Computational correction of thecontrast transfer function. In Three-dimensional ElectronMicroscopy of Macromolecular Assemblies (Frank, J., ed.),pp. 58–62, Oxford University Press, New York.
333CLIC2-RyR1 Interaction

Author's personal copy
62. Liu, Z., Zhang, J., Sharma, M. R., Li, P., Chen, S. R. W.& Wagenknecht, T. (2001). Three-dimensional recon-struction of the recombinant type 3 ryanodinereceptor and localization of its amino terminus. Proc.Natl Acad. Sci. USA, 98, 6104–6109.
63. Malhotra, A., Penczek, P., Agrawal, R. K., Gabashvili,I. S., Grassucci, R. A., Jünemann, R. et al. (1998).Escherichia coli 70 S ribosome at 15 Å resolution bycryo-electron microscopy: localization of fMet-tRNAf-Met and fitting of L1 protein. J. Mol. Biol. 280, 103–116.
334 CLIC2-RyR1 Interaction




















