
CL22 – a novel cationic peptide for efficient transfection of mammalian cells
12
Gene Therapy (2001) 8, 99–110 2001 Nature Publishing Group All rights reserved 0969-7128/01 $15.00 www.nature.com/gt RESEARCH ARTICLE CL22 – a novel cationic peptide for efficient transfection of mammalian cells AMR Haines 1 , AS Irvine 1 , A Mountain 1 , J Charlesworth 1 , NA Farrow 1 , RD Husain 1 , H Hyde 1 , H Ketteringham 1 , RH McDermott 2 , AF Mulcahy 1 , TL Mustoe 1 , SCH Reid 1 , M Rouquette 1 , JC Shaw 1 , DR Thatcher 1 , JH Welsh 1 , DE Williams 1 , W Zauner 3 and RO Phillips 1 1 Cobra Therapeutics Ltd, Keele, UK Condensing peptide–DNA complexes have great potential as nonviral agents for gene delivery. To date, however, such complexes have given transfection activities greatly inferior to adenovirus and somewhat inferior to cationic lipid–DNA complexes, even for cell lines and primary cells in vitro. We report here the identification of a novel condensing peptide, CL22, which forms DNA complexes that efficiently transfect many cell lines, as well as primary dendritic and endothelial cells. We report studies with sequence and structure vari- Keywords: gene delivery; non-viral; condensing peptide; transfection Introduction Vectors giving efficient delivery of DNA from an extra- cellular location to the nucleus of target cells are required for gene therapy, for other modern approaches to drug discovery (such as functional genomics) and for funda- mental research into the function and control of mam- malian genes. Although several viral vectors can give efficient gene delivery, their use for research applications is usually slow and labour-intensive, and their utility for gene therapy limited by DNA carrying capacity, diffi- culties in reliable and cost-effective manufacture, and by immunogenicity and other safety concerns. 1–3 At the present time nonviral vectors typically give lower trans- fection efficiencies than adenovirus (the most efficient viral vector), and also give large variability in transfec- tion efficiencies between cell types. They have the advan- tages, however, of increased DNA carrying capacity, lower immunogenicity, greater flexibility and greater simplicity of preparation, purification and storage. Most nonviral gene delivery strategies use synthetic molecules to condense DNA to reduce its size and alter its charge, thus facilitating nuclear access across cell membrane barriers. Nonviral transfection agents reported in the literature include polydisperse cationic polymers such as polylysine, 4,5 defined length oligolys- ines, 6,7 polyarginine, 8 polyethyleneimine, 9–12 Starburst dendrimers 13,14 or cationic lipids. 15–17 Although many of Correspondence: A Haines, Cobra Therapeutics Ltd, Stephenson Building, Keele University Science Park, Keele, Staffordshire, ST5 5SP, UK 2 Current addresses: Manchester Innovation Ltd, Manchester, UK; and 3 Intercell GmbH, Wien, Austria Received 15 March 2000; accepted 24 July 2000 ants that define some properties of the peptide that contrib- ute to efficient transfection. We demonstrate that the superior transfection activity of CL22 compared with other DNA condensing peptides is conferred at a step after uptake of the complexes into cells. We show that CL22–DNA com- plexes have transfection activity that is at least equivalent to the best available nonviral agents. Gene Therapy (2001) 8, 99–110. these are reported to transfect various cell lines in vitro with useful efficiency, few give reproducible transfection of primary cells ex vivo. Only a few cationic lipids, poly- ethyleneimine and combinations of cationic polymer and lipid give significant transfection in vivo, and even for these agents transfection in vivo is substantially poorer than in vitro. Although used less commonly, cationic peptides of defined length have much potential as transfection agents. They offer greater reproducibility of synthesis, lower heterogeneity and hence greater reliability of trans- fection than most nonviral alternatives, as well as biodeg- radability and low toxicity. A large number of commer- cially available natural and non-natural amino acid building blocks enable the preparation and testing of peptide–DNA complexes differing substantially in physicochemical or biological character. Modern peptide synthesis and bioconjugation strategies, which can utilise the different reactive groups available on peptides, allow for facile, site-specific attachment of additional peptidic or nonpeptidic entities to the cationic condensing pep- tide. 18 Such entities may be attached to reduce unwanted, nonspecific interactions between the complex and plasma components or extracellular matrix in vivo, and confer specific uptake into target cells or improved intra- cellular trafficking. In this paper, we describe a 35 amino acid cationic peptide, CL22, which forms complexes with DNA that give efficient transfection of mammalian cells in vitro and ex vivo. The sequence of CL22, NH 2 -KKKKKK GGFLGFWRGENGRKTRSAYERMCNILKGKCOOH, was designed to incorporate three domains. It has a lysine- rich N-terminus to facilitate DNA condensation and a C- terminal sequence derived largely from influenza nucleo- protein, joined by a sequence GGFLGF designed for
Transcript of CL22 – a novel cationic peptide for efficient transfection of mammalian cells

Gene Therapy (2001) 8, 99–110 2001 Nature Publishing Group All rights reserved 0969-7128/01 $15.00
www.nature.com/gt
RESEARCH ARTICLE
CL22 – a novel cationic peptide for efficienttransfection of mammalian cells
AMR Haines1, AS Irvine1, A Mountain1, J Charlesworth1, NA Farrow1, RD Husain1, H Hyde1,H Ketteringham1, RH McDermott2, AF Mulcahy1, TL Mustoe1, SCH Reid1, M Rouquette1,JC Shaw1, DR Thatcher1, JH Welsh1, DE Williams1, W Zauner3 and RO Phillips1
1Cobra Therapeutics Ltd, Keele, UK
Condensing peptide–DNA complexes have great potentialas nonviral agents for gene delivery. To date, however, suchcomplexes have given transfection activities greatly inferiorto adenovirus and somewhat inferior to cationic lipid–DNAcomplexes, even for cell lines and primary cells in vitro. Wereport here the identification of a novel condensing peptide,CL22, which forms DNA complexes that efficiently transfectmany cell lines, as well as primary dendritic and endothelialcells. We report studies with sequence and structure vari-
Keywords: gene delivery; non-viral; condensing peptide; transfection
IntroductionVectors giving efficient delivery of DNA from an extra-cellular location to the nucleus of target cells are requiredfor gene therapy, for other modern approaches to drugdiscovery (such as functional genomics) and for funda-mental research into the function and control of mam-malian genes. Although several viral vectors can giveefficient gene delivery, their use for research applicationsis usually slow and labour-intensive, and their utility forgene therapy limited by DNA carrying capacity, diffi-culties in reliable and cost-effective manufacture, and byimmunogenicity and other safety concerns.1–3 At thepresent time nonviral vectors typically give lower trans-fection efficiencies than adenovirus (the most efficientviral vector), and also give large variability in transfec-tion efficiencies between cell types. They have the advan-tages, however, of increased DNA carrying capacity,lower immunogenicity, greater flexibility and greatersimplicity of preparation, purification and storage.
Most nonviral gene delivery strategies use syntheticmolecules to condense DNA to reduce its size and alterits charge, thus facilitating nuclear access across cellmembrane barriers. Nonviral transfection agentsreported in the literature include polydisperse cationicpolymers such as polylysine,4,5 defined length oligolys-ines,6,7 polyarginine,8 polyethyleneimine,9–12 Starburstdendrimers13,14 or cationic lipids.15–17 Although many of
Correspondence: A Haines, Cobra Therapeutics Ltd, Stephenson Building,Keele University Science Park, Keele, Staffordshire, ST5 5SP, UK2Current addresses: Manchester Innovation Ltd, Manchester, UK; and3Intercell GmbH, Wien, AustriaReceived 15 March 2000; accepted 24 July 2000
ants that define some properties of the peptide that contrib-ute to efficient transfection. We demonstrate that thesuperior transfection activity of CL22 compared with otherDNA condensing peptides is conferred at a step after uptakeof the complexes into cells. We show that CL22–DNA com-plexes have transfection activity that is at least equivalent tothe best available nonviral agents. Gene Therapy (2001) 8,99–110.
these are reported to transfect various cell lines in vitrowith useful efficiency, few give reproducible transfectionof primary cells ex vivo. Only a few cationic lipids, poly-ethyleneimine and combinations of cationic polymer andlipid give significant transfection in vivo, and even forthese agents transfection in vivo is substantially poorerthan in vitro.
Although used less commonly, cationic peptides ofdefined length have much potential as transfectionagents. They offer greater reproducibility of synthesis,lower heterogeneity and hence greater reliability of trans-fection than most nonviral alternatives, as well as biodeg-radability and low toxicity. A large number of commer-cially available natural and non-natural amino acidbuilding blocks enable the preparation and testing ofpeptide–DNA complexes differing substantially inphysicochemical or biological character. Modern peptidesynthesis and bioconjugation strategies, which can utilisethe different reactive groups available on peptides, allowfor facile, site-specific attachment of additional peptidicor nonpeptidic entities to the cationic condensing pep-tide.18 Such entities may be attached to reduce unwanted,nonspecific interactions between the complex and plasmacomponents or extracellular matrix in vivo, and conferspecific uptake into target cells or improved intra-cellular trafficking.
In this paper, we describe a 35 amino acid cationicpeptide, CL22, which forms complexes with DNA thatgive efficient transfection of mammalian cells in vitro andex vivo. The sequence of CL22, NH2-KKKKKKGGFLGFWRGENGRKTRSAYERMCNILKGKCOOH, wasdesigned to incorporate three domains. It has a lysine-rich N-terminus to facilitate DNA condensation and a C-terminal sequence derived largely from influenza nucleo-protein, joined by a sequence GGFLGF designed for
CL22 transfection peptideAMR Haines et al
100
Gene Therapy
cleavage by the abundant endosomal endoproteasecathepsin D.19,20 The C-terminal domain corresponds toresidues 206–225 of influenza nucleoprotein, a sequencebelieved to comprise an epitope presented on class IImolecules of the human leukocyte antigen (HLA) com-plex.21 CL22 was originally designed to explore the con-cept of using peptide–DNA complexes to deliver bothpeptide epitopes and plasmid encoding intact nucleo-protein to professional antigen presenting cells in orderto achieve presentation of epitopes on HLA class I andII molecules for vaccine purposes. CL22 complexes givemaximal transfection when the peptide is in the dimericform, [CL22]2. Spontaneous dimerisation occurs throughformation of a disulphide bond between cysteine resi-dues close to the C-terminus. We report here that[CL22]2–DNA complexes unexpectedly give transfectionefficiencies for immortalised cell lines in vitro and pri-mary human cells ex vivo that are greatly superior tothose of defined polylysine complexes. We present datarelating transfection efficiency to biophysical propertiesof the [CL22]2–DNA complexes, and which show thatthese complexes compare favourably for transfectionefficiency with the best commercially available nonpep-tide transfection agents.
Results
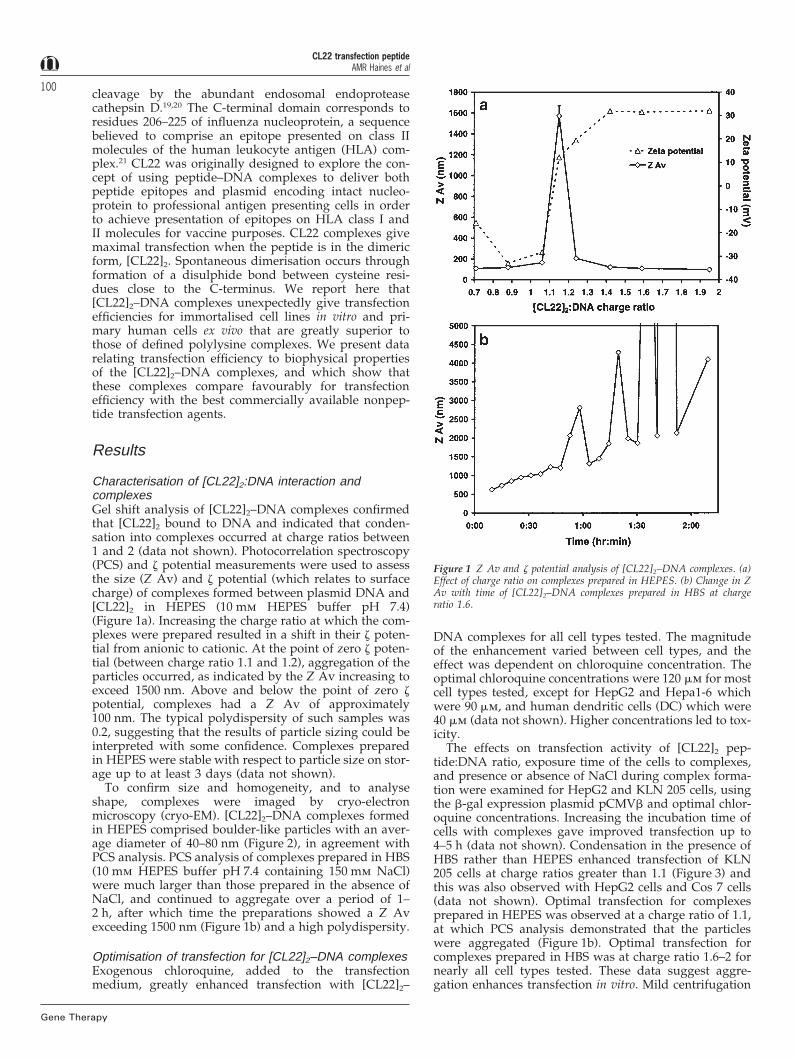
Characterisation of [CL22]2:DNA interaction andcomplexesGel shift analysis of [CL22]2–DNA complexes confirmedthat [CL22]2 bound to DNA and indicated that conden-sation into complexes occurred at charge ratios between1 and 2 (data not shown). Photocorrelation spectroscopy(PCS) and z potential measurements were used to assessthe size (Z Av) and z potential (which relates to surfacecharge) of complexes formed between plasmid DNA and[CL22]2 in HEPES (10 mm HEPES buffer pH 7.4)(Figure 1a). Increasing the charge ratio at which the com-plexes were prepared resulted in a shift in their z poten-tial from anionic to cationic. At the point of zero z poten-tial (between charge ratio 1.1 and 1.2), aggregation of theparticles occurred, as indicated by the Z Av increasing toexceed 1500 nm. Above and below the point of zero zpotential, complexes had a Z Av of approximately100 nm. The typical polydispersity of such samples was0.2, suggesting that the results of particle sizing could beinterpreted with some confidence. Complexes preparedin HEPES were stable with respect to particle size on stor-age up to at least 3 days (data not shown).
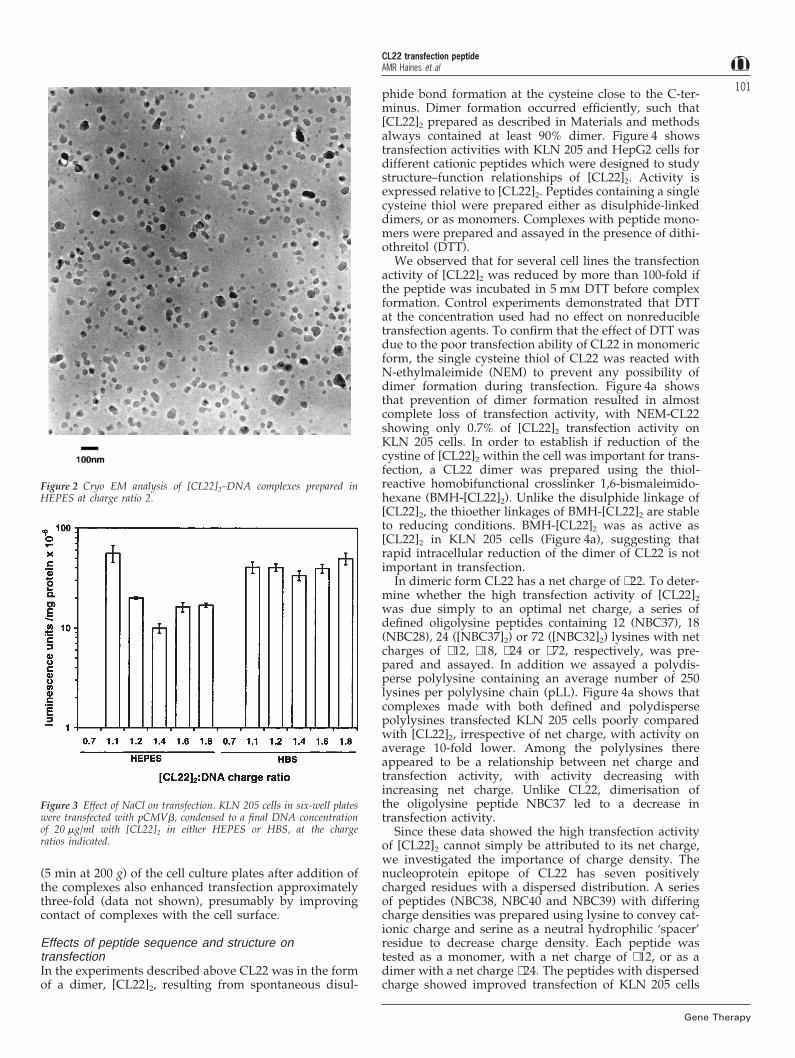
To confirm size and homogeneity, and to analyseshape, complexes were imaged by cryo-electronmicroscopy (cryo-EM). [CL22]2–DNA complexes formedin HEPES comprised boulder-like particles with an aver-age diameter of 40–80 nm (Figure 2), in agreement withPCS analysis. PCS analysis of complexes prepared in HBS(10 mm HEPES buffer pH 7.4 containing 150 mm NaCl)were much larger than those prepared in the absence ofNaCl, and continued to aggregate over a period of 1–2 h, after which time the preparations showed a Z Avexceeding 1500 nm (Figure 1b) and a high polydispersity.
Optimisation of transfection for [CL22]2–DNA complexesExogenous chloroquine, added to the transfectionmedium, greatly enhanced transfection with [CL22]2–
Figure 1 Z Av and z potential analysis of [CL22]2–DNA complexes. (a)Effect of charge ratio on complexes prepared in HEPES. (b) Change in ZAv with time of [CL22]2–DNA complexes prepared in HBS at chargeratio 1.6.
DNA complexes for all cell types tested. The magnitudeof the enhancement varied between cell types, and theeffect was dependent on chloroquine concentration. Theoptimal chloroquine concentrations were 120 mm for mostcell types tested, except for HepG2 and Hepa1-6 whichwere 90 mm, and human dendritic cells (DC) which were40 mm (data not shown). Higher concentrations led to tox-icity.
The effects on transfection activity of [CL22]2 pep-tide:DNA ratio, exposure time of the cells to complexes,and presence or absence of NaCl during complex forma-tion were examined for HepG2 and KLN 205 cells, usingthe b-gal expression plasmid pCMVb and optimal chlor-oquine concentrations. Increasing the incubation time ofcells with complexes gave improved transfection up to4–5 h (data not shown). Condensation in the presence ofHBS rather than HEPES enhanced transfection of KLN205 cells at charge ratios greater than 1.1 (Figure 3) andthis was also observed with HepG2 cells and Cos 7 cells(data not shown). Optimal transfection for complexesprepared in HEPES was observed at a charge ratio of 1.1,at which PCS analysis demonstrated that the particleswere aggregated (Figure 1b). Optimal transfection forcomplexes prepared in HBS was at charge ratio 1.6–2 fornearly all cell types tested. These data suggest aggre-gation enhances transfection in vitro. Mild centrifugation
CL22 transfection peptideAMR Haines et al
101
Figure 2 Cryo EM analysis of [CL22]2–DNA complexes prepared inHEPES at charge ratio 2.
Figure 3 Effect of NaCl on transfection. KLN 205 cells in six-well plateswere transfected with pCMVb, condensed to a final DNA concentrationof 20 mg/ml with [CL22]2 in either HEPES or HBS, at the chargeratios indicated.
(5 min at 200 g) of the cell culture plates after addition ofthe complexes also enhanced transfection approximatelythree-fold (data not shown), presumably by improvingcontact of complexes with the cell surface.
Effects of peptide sequence and structure ontransfectionIn the experiments described above CL22 was in the formof a dimer, [CL22]2, resulting from spontaneous disul-
Gene Therapy
phide bond formation at the cysteine close to the C-ter-minus. Dimer formation occurred efficiently, such that[CL22]2 prepared as described in Materials and methodsalways contained at least 90% dimer. Figure 4 showstransfection activities with KLN 205 and HepG2 cells fordifferent cationic peptides which were designed to studystructure–function relationships of [CL22]2. Activity isexpressed relative to [CL22]2. Peptides containing a singlecysteine thiol were prepared either as disulphide-linkeddimers, or as monomers. Complexes with peptide mono-mers were prepared and assayed in the presence of dithi-othreitol (DTT).
We observed that for several cell lines the transfectionactivity of [CL22]2 was reduced by more than 100-fold ifthe peptide was incubated in 5 mm DTT before complexformation. Control experiments demonstrated that DTTat the concentration used had no effect on nonreducibletransfection agents. To confirm that the effect of DTT wasdue to the poor transfection ability of CL22 in monomericform, the single cysteine thiol of CL22 was reacted withN-ethylmaleimide (NEM) to prevent any possibility ofdimer formation during transfection. Figure 4a showsthat prevention of dimer formation resulted in almostcomplete loss of transfection activity, with NEM-CL22showing only 0.7% of [CL22]2 transfection activity onKLN 205 cells. In order to establish if reduction of thecystine of [CL22]2 within the cell was important for trans-fection, a CL22 dimer was prepared using the thiol-reactive homobifunctional crosslinker 1,6-bismaleimido-hexane (BMH-[CL22]2). Unlike the disulphide linkage of[CL22]2, the thioether linkages of BMH-[CL22]2 are stableto reducing conditions. BMH-[CL22]2 was as active as[CL22]2 in KLN 205 cells (Figure 4a), suggesting thatrapid intracellular reduction of the dimer of CL22 is notimportant in transfection.
In dimeric form CL22 has a net charge of +22. To deter-mine whether the high transfection activity of [CL22]2
was due simply to an optimal net charge, a series ofdefined oligolysine peptides containing 12 (NBC37), 18(NBC28), 24 ([NBC37]2) or 72 ([NBC32]2) lysines with netcharges of +12, +18, +24 or +72, respectively, was pre-pared and assayed. In addition we assayed a polydis-perse polylysine containing an average number of 250lysines per polylysine chain (pLL). Figure 4a shows thatcomplexes made with both defined and polydispersepolylysines transfected KLN 205 cells poorly comparedwith [CL22]2, irrespective of net charge, with activity onaverage 10-fold lower. Among the polylysines thereappeared to be a relationship between net charge andtransfection activity, with activity decreasing withincreasing net charge. Unlike CL22, dimerisation ofthe oligolysine peptide NBC37 led to a decrease intransfection activity.
Since these data showed the high transfection activityof [CL22]2 cannot simply be attributed to its net charge,we investigated the importance of charge density. Thenucleoprotein epitope of CL22 has seven positivelycharged residues with a dispersed distribution. A seriesof peptides (NBC38, NBC40 and NBC39) with differingcharge densities was prepared using lysine to convey cat-ionic charge and serine as a neutral hydrophilic ‘spacer’residue to decrease charge density. Each peptide wastested as a monomer, with a net charge of +12, or as adimer with a net charge +24. The peptides with dispersedcharge showed improved transfection of KLN 205 cells

CL22 transfection peptideAMR Haines et al
102
Gene Therapy
Figure 4 Comparison of [CL22]2 with other cationic peptides for transfection activity. The results are from separate assays and are the average oftriplicates at the optimal charge ratio for each peptide (between 1.2 and 2). Error bars are standard deviation of the mean of the triplicates. (a) Transfectionof KLN 205 cells with pCMVb plasmid in six-well plates, in the presence of 120 mm chloroquine and absence of FCS. Complexes were prepared inHEPES and transfection expressed as a percentage of bgal luminescence compared with that obtained with [CL22]2–pCMVb complexes prepared in thesame manner in parallel with each assay. (b) Transfection of HepG2 cells with pCMVluc plasmid in 96-well plates in the presence of 90 mm chloroquineand 10% FCS. Complexes were prepared in HBS and the results are expressed as a percentage of the luciferase luminescence compared with that obtainedwith [CL22]2–pCMVLuc complexes prepared in the same manner in parallel with each assay.
over corresponding high charge density oligolysine pep-tides with the same net charge (Figure 4a). Dimerisationof NBC38 and NBC40, which effects an increase in netcharge of each peptide, again resulted in a decrease intransfection activity. Dimerisation of NBC39, however,was not detrimental to transfection activity. These datasuggest the dispersed charge distribution in the nucleop-rotein epitope of CL22 may contribute to its high trans-fection activity. The possibility that the exact amino acidsequence of the nucleoprotein epitope is important wasexamined using a peptide, CL28, in which the order wasrandomised. The transfection activity of [CL28]2 on KLN205 cells was similar to that of [CL22]2, as indicated inFigure 4a. Transfection assays with HepG2 cells gaveresults similar to those with KLN 205. As shown inFigure 4b, the high charge density oligolysines [NBC37]2
(24 lysines) and [NBC28]2 (36 lysines) gave substantiallylower transfection activity than [CL22]2, and reducingcharge density with serine spacers using the peptides[NBC38]2, [NBC40]2 and [NBC39]2 improved activitycompared with [NBC37]2.
We next investigated the requirement for dense cat-ionic charge in a terminal position of CL22, using HepG2cells. A series of peptides was prepared in which the sixlysine N-terminal stretch of [CL22]2, was replaced with1, 2, 3, 5 or 12 lysines, respectively, in [CL35]2, [CL36]2,[CL37]2, [CL39]2 and [CL26]2. Figure 4b shows that aminimum N-terminal stretch of four or five lysines isrequired for optimal transfection activity. Even though
the net charge of [CL36]2 is +14, transfection activity wasnegligible compared with that of [CL22]2. Increasing thenumber of N-terminal lysines to 12, in the peptide CL26,did not significantly improve transfection activity.
In order to investigate the possible contribution of theprotease cleavage sequence ‘GGFLGF’ to transfectionactivity, we made and tested a variant (CL25) in whichthis motif was replaced by FGLGGF. The transfectionactivity of [CL25]2 was 40% of that displayed by [CL22]2
and substantially greater than the oligolysines(Figure 4b), suggesting that efficient release of the nucleo-protein epitope by protease cleavage in the endosome isnot an absolute requirement for transfection activity. Weobserved that a variant [CL41]2 in which the entire CL22sequence was reversed, gave transfection activity verysimilar to that of [CL22]2. The result with this peptidesuggested first that the precise order of residues in thenucleoprotein epitope is not crucial, and second that hightransfection activity does not require the dense cationiccharge to be N-terminal; a C-terminal location is alsocompatible with good transfection.
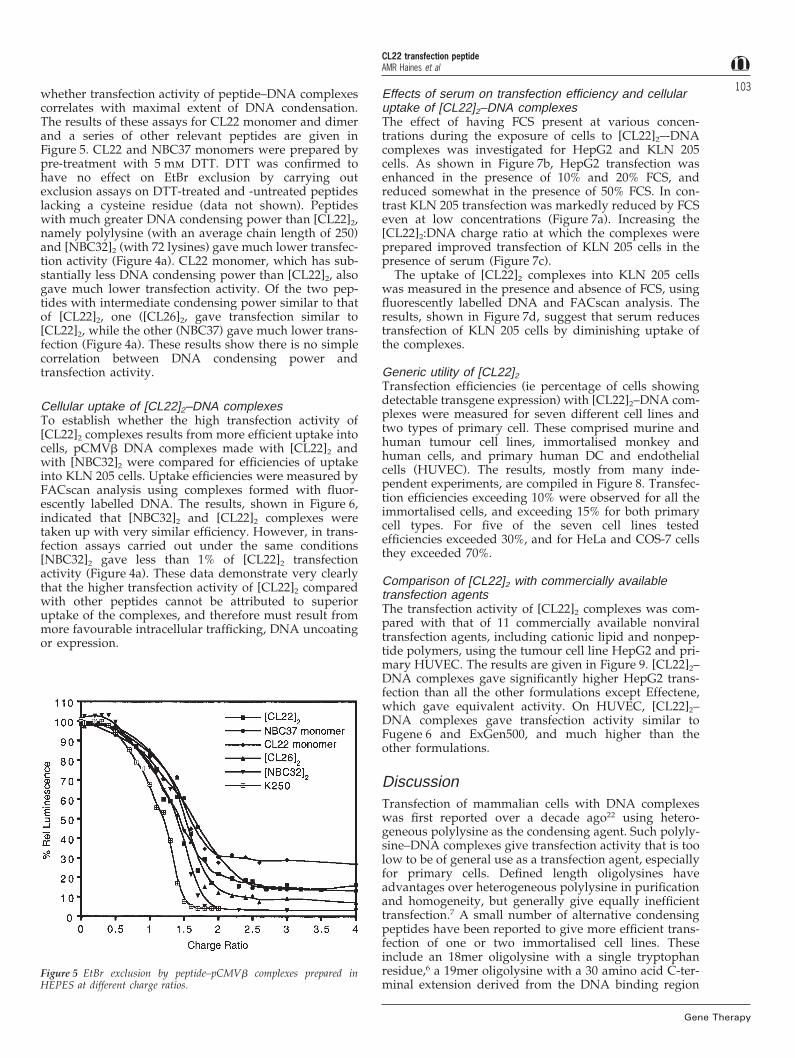
Finally, we observed that the variant CL33, in whichthe charged residues of the nucleoprotein epitope werereplaced with neutral serine residues, gave very lowtransfection activity (Figure 4b). These results raised thepossibility that the overall DNA condensing power ofCL22 is the key factor conferring its high transfectionactivity. To investigate this possibility ethidium bromide(EtBr) exclusion assays were conducted to investigate
CL22 transfection peptideAMR Haines et al
103whether transfection activity of peptide–DNA complexescorrelates with maximal extent of DNA condensation.The results of these assays for CL22 monomer and dimerand a series of other relevant peptides are given inFigure 5. CL22 and NBC37 monomers were prepared bypre-treatment with 5 mm DTT. DTT was confirmed tohave no effect on EtBr exclusion by carrying outexclusion assays on DTT-treated and -untreated peptideslacking a cysteine residue (data not shown). Peptideswith much greater DNA condensing power than [CL22]2,namely polylysine (with an average chain length of 250)and [NBC32]2 (with 72 lysines) gave much lower transfec-tion activity (Figure 4a). CL22 monomer, which has sub-stantially less DNA condensing power than [CL22]2, alsogave much lower transfection activity. Of the two pep-tides with intermediate condensing power similar to thatof [CL22]2, one ([CL26]2, gave transfection similar to[CL22]2, while the other (NBC37) gave much lower trans-fection (Figure 4a). These results show there is no simplecorrelation between DNA condensing power andtransfection activity.
Cellular uptake of [CL22]2–DNA complexesTo establish whether the high transfection activity of[CL22]2 complexes results from more efficient uptake intocells, pCMVb DNA complexes made with [CL22]2 andwith [NBC32]2 were compared for efficiencies of uptakeinto KLN 205 cells. Uptake efficiencies were measured byFACscan analysis using complexes formed with fluor-escently labelled DNA. The results, shown in Figure 6,indicated that [NBC32]2 and [CL22]2 complexes weretaken up with very similar efficiency. However, in trans-fection assays carried out under the same conditions[NBC32]2 gave less than 1% of [CL22]2 transfectionactivity (Figure 4a). These data demonstrate very clearlythat the higher transfection activity of [CL22]2 comparedwith other peptides cannot be attributed to superioruptake of the complexes, and therefore must result frommore favourable intracellular trafficking, DNA uncoatingor expression.
Figure 5 EtBr exclusion by peptide–pCMVb complexes prepared inHEPES at different charge ratios.
Gene Therapy
Effects of serum on transfection efficiency and cellularuptake of [CL22]2–DNA complexesThe effect of having FCS present at various concen-trations during the exposure of cells to [CL22]2–-DNAcomplexes was investigated for HepG2 and KLN 205cells. As shown in Figure 7b, HepG2 transfection wasenhanced in the presence of 10% and 20% FCS, andreduced somewhat in the presence of 50% FCS. In con-trast KLN 205 transfection was markedly reduced by FCSeven at low concentrations (Figure 7a). Increasing the[CL22]2:DNA charge ratio at which the complexes wereprepared improved transfection of KLN 205 cells in thepresence of serum (Figure 7c).
The uptake of [CL22]2 complexes into KLN 205 cellswas measured in the presence and absence of FCS, usingfluorescently labelled DNA and FACscan analysis. Theresults, shown in Figure 7d, suggest that serum reducestransfection of KLN 205 cells by diminishing uptake ofthe complexes.
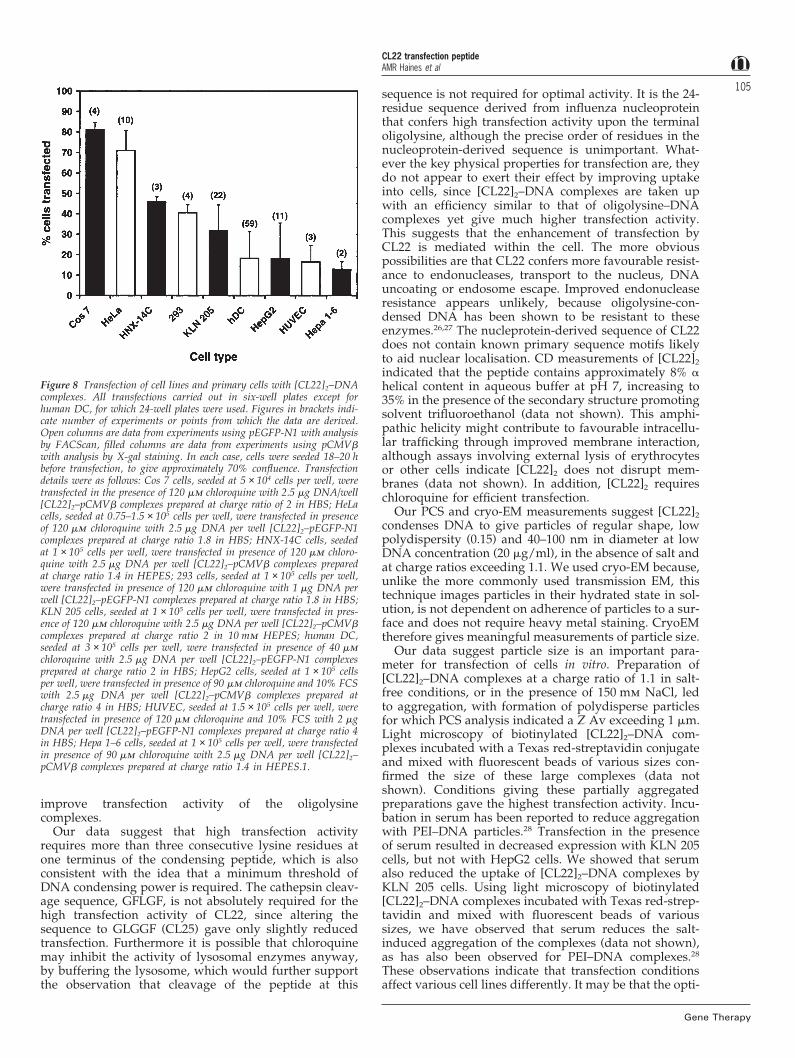
Generic utility of [CL22]2Transfection efficiencies (ie percentage of cells showingdetectable transgene expression) with [CL22]2–DNA com-plexes were measured for seven different cell lines andtwo types of primary cell. These comprised murine andhuman tumour cell lines, immortalised monkey andhuman cells, and primary human DC and endothelialcells (HUVEC). The results, mostly from many inde-pendent experiments, are compiled in Figure 8. Transfec-tion efficiencies exceeding 10% were observed for all theimmortalised cells, and exceeding 15% for both primarycell types. For five of the seven cell lines testedefficiencies exceeded 30%, and for HeLa and COS-7 cellsthey exceeded 70%.
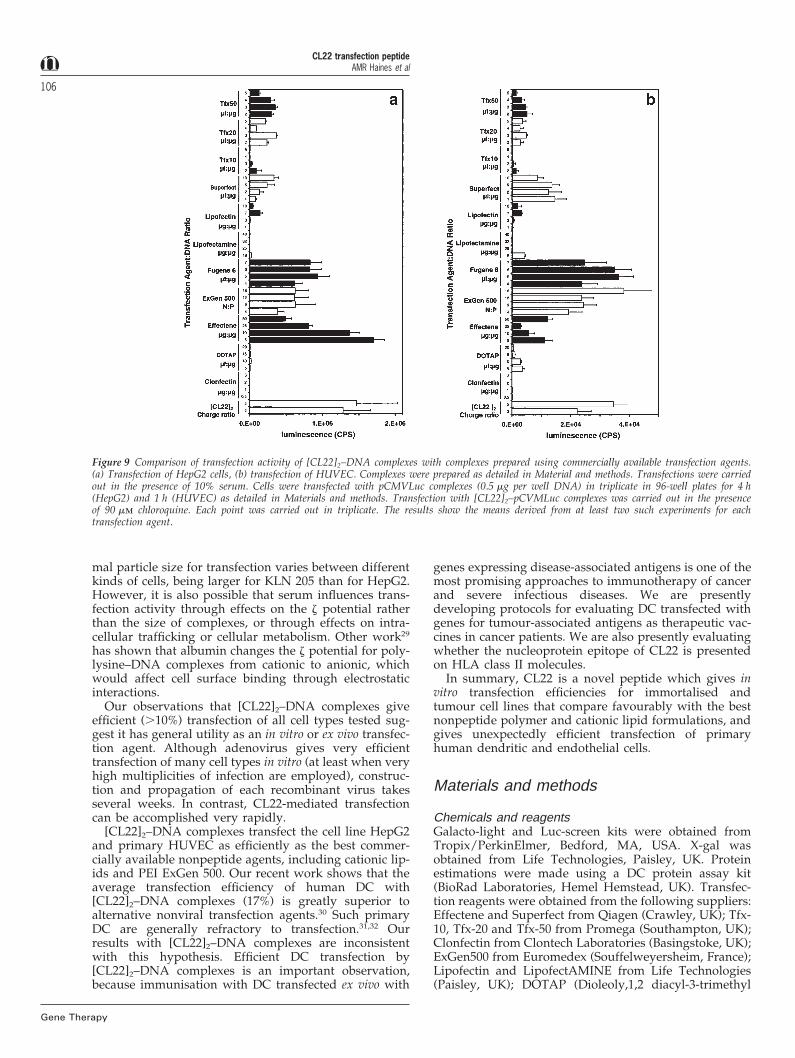
Comparison of [CL22]2 with commercially availabletransfection agentsThe transfection activity of [CL22]2 complexes was com-pared with that of 11 commercially available nonviraltransfection agents, including cationic lipid and nonpep-tide polymers, using the tumour cell line HepG2 and pri-mary HUVEC. The results are given in Figure 9. [CL22]2–DNA complexes gave significantly higher HepG2 trans-fection than all the other formulations except Effectene,which gave equivalent activity. On HUVEC, [CL22]2–DNA complexes gave transfection activity similar toFugene 6 and ExGen500, and much higher than theother formulations.
DiscussionTransfection of mammalian cells with DNA complexeswas first reported over a decade ago22 using hetero-geneous polylysine as the condensing agent. Such polyly-sine–DNA complexes give transfection activity that is toolow to be of general use as a transfection agent, especiallyfor primary cells. Defined length oligolysines haveadvantages over heterogeneous polylysine in purificationand homogeneity, but generally give equally inefficienttransfection.7 A small number of alternative condensingpeptides have been reported to give more efficient trans-fection of one or two immortalised cell lines. Theseinclude an 18mer oligolysine with a single tryptophanresidue,6 a 19mer oligolysine with a 30 amino acid C-ter-minal extension derived from the DNA binding region

CL22 transfection peptideAMR Haines et al
104
Gene Therapy
Figure 6 Binding and uptake of peptide–DNA complexes in KLN 205 cells. (a) [CL22]2–YOYO-1 labelled DNA complexes prepared in HEPES atcharge ratio 1.6. (b) [NBC32]2–YOYO-1 labelled DNA complexes prepared in HEPES at charge ratio 2. Cellular autofluorescence control from untreatedcells (dotted line); binding from 4°C control (thin line); binding and uptake at 37°C (thick line).
Figure 7 Effects of serum on transfection and uptake with [CL22]2–DNA complexes. Transfection of KLN 205 cells (a) and HepG2 cells (b) in six-wellplates in the presence of the indicated amounts of FCS. [CL22]2–pCMVb complexes were prepared in HEPES at charge ratio of 1.6. (c) Transfectionof KLN 205 cells in six-well plates in the presence of 10% FCS. [CL22]2–pCMVb complexes were prepared in HEPES at the indicated charge ratios.(d) Uptake of [CL22]2–YOYO-1 labelled DNA complexes into KLN 205 cells in presence (red) and absence (black) of 10% FCS. Complexes were preparedin HEPES at a charge ratio of 1.6. Cellular autofluorescence control from untreated cells (dotted lines); binding from 4°C control (thin lines); bindingand uptake at 37°C (thick lines).
of an anti-DNA antibody,23 a 19mer oligolysine with a20 amino acid extension derived from adenovirus fibreprotein,24 and a peptide designed from the natural DNAbinding protein, human histone H1.25 The basis of theimproved transfection activity for these peptides has notbeen elucidated, but in the cases of the extended oligolys-ines it was speculated to involve enhanced intracellulartrafficking.
The condensing peptide [CL22]2 unexpectedly givesefficient transfection of all immortalised cell lines andboth primary human cell types we have tested. Ourefforts to define precisely the physical properties of[CL22]2 which endow it with good transfection activityare still ongoing, but the studies reported in this paper
provide some insight. It is striking that dimerisation isrequired for high transfection activity. Since our studiesshow there is no simple relationship between DNA con-densing activity and transfection activity of condensingpeptides, the results imply that a certain minimum thres-hold of condensing power is required for efficient trans-fection. CL22 monomer is below the threshold and CL22dimer above it. The possibility that intracellularreduction of the disulphide linking the two monomers of[CL22]2 plays a part in releasing DNA within the cell isunlikely, because the thioether-linked dimer, BMH-[CL22]2, which should be much more resistant to intra-cellular reduction, gave transfection activity equivalent to[CL22]2. In addition, disulphide dimerisation did not
CL22 transfection peptideAMR Haines et al
105
Figure 8 Transfection of cell lines and primary cells with [CL22]2–DNAcomplexes. All transfections carried out in six-well plates except forhuman DC, for which 24-well plates were used. Figures in brackets indi-cate number of experiments or points from which the data are derived.Open columns are data from experiments using pEGFP-N1 with analysisby FACScan, filled columns are data from experiments using pCMVbwith analysis by X-gal staining. In each case, cells were seeded 18–20 hbefore transfection, to give approximately 70% confluence. Transfectiondetails were as follows: Cos 7 cells, seeded at 5 × 104 cells per well, weretransfected in the presence of 120 mm chloroquine with 2.5 mg DNA/well[CL22]2–pCMVb complexes prepared at charge ratio of 2 in HBS; HeLacells, seeded at 0.75–1.5 × 105 cells per well, were transfected in presenceof 120 mm chloroquine with 2.5 mg DNA per well [CL22]2–pEGFP-N1complexes prepared at charge ratio 1.8 in HBS; HNX-14C cells, seededat 1 × 105 cells per well, were transfected in presence of 120 mm chloro-quine with 2.5 mg DNA per well [CL22]2–pCMVb complexes preparedat charge ratio 1.4 in HEPES; 293 cells, seeded at 1 × 105 cells per well,were transfected in presence of 120 mm chloroquine with 1 mg DNA perwell [CL22]2–pEGFP-N1 complexes prepared at charge ratio 1.8 in HBS;KLN 205 cells, seeded at 1 × 105 cells per well, were transfected in pres-ence of 120 mm chloroquine with 2.5 mg DNA per well [CL22]2–pCMVbcomplexes prepared at charge ratio 2 in 10 mm HEPES; human DC,seeded at 3 × 105 cells per well, were transfected in presence of 40 mmchloroquine with 2.5 mg DNA per well [CL22]2–pEGFP-N1 complexesprepared at charge ratio 2 in HBS; HepG2 cells, seeded at 1 × 105 cellsper well, were transfected in presence of 90 mm chloroquine and 10% FCSwith 2.5 mg DNA per well [CL22]2–pCMVb complexes prepared atcharge ratio 4 in HBS; HUVEC, seeded at 1.5 × 105 cells per well, weretransfected in presence of 120 mm chloroquine and 10% FCS with 2 mgDNA per well [CL22]2–pEGFP-N1 complexes prepared at charge ratio 4in HBS; Hepa 1–6 cells, seeded at 1 × 105 cells per well, were transfectedin presence of 90 mm chloroquine with 2.5 mg DNA per well [CL22]2–pCMVb complexes prepared at charge ratio 1.4 in HEPES.1.
improve transfection activity of the oligolysinecomplexes.
Our data suggest that high transfection activityrequires more than three consecutive lysine residues atone terminus of the condensing peptide, which is alsoconsistent with the idea that a minimum threshold ofDNA condensing power is required. The cathepsin cleav-age sequence, GFLGF, is not absolutely required for thehigh transfection activity of CL22, since altering thesequence to GLGGF (CL25) gave only slightly reducedtransfection. Furthermore it is possible that chloroquinemay inhibit the activity of lysosomal enzymes anyway,by buffering the lysosome, which would further supportthe observation that cleavage of the peptide at this
Gene Therapy
sequence is not required for optimal activity. It is the 24-residue sequence derived from influenza nucleoproteinthat confers high transfection activity upon the terminaloligolysine, although the precise order of residues in thenucleoprotein-derived sequence is unimportant. What-ever the key physical properties for transfection are, theydo not appear to exert their effect by improving uptakeinto cells, since [CL22]2–DNA complexes are taken upwith an efficiency similar to that of oligolysine–DNAcomplexes yet give much higher transfection activity.This suggests that the enhancement of transfection byCL22 is mediated within the cell. The more obviouspossibilities are that CL22 confers more favourable resist-ance to endonucleases, transport to the nucleus, DNAuncoating or endosome escape. Improved endonucleaseresistance appears unlikely, because oligolysine-con-densed DNA has been shown to be resistant to theseenzymes.26,27 The nucleprotein-derived sequence of CL22does not contain known primary sequence motifs likelyto aid nuclear localisation. CD measurements of [CL22]2
indicated that the peptide contains approximately 8% ahelical content in aqueous buffer at pH 7, increasing to35% in the presence of the secondary structure promotingsolvent trifluoroethanol (data not shown). This amphi-pathic helicity might contribute to favourable intracellu-lar trafficking through improved membrane interaction,although assays involving external lysis of erythrocytesor other cells indicate [CL22]2 does not disrupt mem-branes (data not shown). In addition, [CL22]2 requireschloroquine for efficient transfection.
Our PCS and cryo-EM measurements suggest [CL22]2
condenses DNA to give particles of regular shape, lowpolydispersity (0.15) and 40–100 nm in diameter at lowDNA concentration (20 mg/ml), in the absence of salt andat charge ratios exceeding 1.1. We used cryo-EM because,unlike the more commonly used transmission EM, thistechnique images particles in their hydrated state in sol-ution, is not dependent on adherence of particles to a sur-face and does not require heavy metal staining. CryoEMtherefore gives meaningful measurements of particle size.
Our data suggest particle size is an important para-meter for transfection of cells in vitro. Preparation of[CL22]2–DNA complexes at a charge ratio of 1.1 in salt-free conditions, or in the presence of 150 mm NaCl, ledto aggregation, with formation of polydisperse particlesfor which PCS analysis indicated a Z Av exceeding 1 mm.Light microscopy of biotinylated [CL22]2–DNA com-plexes incubated with a Texas red-streptavidin conjugateand mixed with fluorescent beads of various sizes con-firmed the size of these large complexes (data notshown). Conditions giving these partially aggregatedpreparations gave the highest transfection activity. Incu-bation in serum has been reported to reduce aggregationwith PEI–DNA particles.28 Transfection in the presenceof serum resulted in decreased expression with KLN 205cells, but not with HepG2 cells. We showed that serumalso reduced the uptake of [CL22]2–DNA complexes byKLN 205 cells. Using light microscopy of biotinylated[CL22]2–DNA complexes incubated with Texas red-strep-tavidin and mixed with fluorescent beads of varioussizes, we have observed that serum reduces the salt-induced aggregation of the complexes (data not shown),as has also been observed for PEI–DNA complexes.28
These observations indicate that transfection conditionsaffect various cell lines differently. It may be that the opti-
CL22 transfection peptideAMR Haines et al
106
Gene Therapy
Figure 9 Comparison of transfection activity of [CL22]2–DNA complexes with complexes prepared using commercially available transfection agents.(a) Transfection of HepG2 cells, (b) transfection of HUVEC. Complexes were prepared as detailed in Material and methods. Transfections were carriedout in the presence of 10% serum. Cells were transfected with pCMVLuc complexes (0.5 mg per well DNA) in triplicate in 96-well plates for 4 h(HepG2) and 1 h (HUVEC) as detailed in Materials and methods. Transfection with [CL22]2–pCVMLuc complexes was carried out in the presenceof 90 mm chloroquine. Each point was carried out in triplicate. The results show the means derived from at least two such experiments for eachtransfection agent.
mal particle size for transfection varies between differentkinds of cells, being larger for KLN 205 than for HepG2.However, it is also possible that serum influences trans-fection activity through effects on the z potential ratherthan the size of complexes, or through effects on intra-cellular trafficking or cellular metabolism. Other work29
has shown that albumin changes the z potential for poly-lysine–DNA complexes from cationic to anionic, whichwould affect cell surface binding through electrostaticinteractions.
Our observations that [CL22]2–DNA complexes giveefficient (.10%) transfection of all cell types tested sug-gest it has general utility as an in vitro or ex vivo transfec-tion agent. Although adenovirus gives very efficienttransfection of many cell types in vitro (at least when veryhigh multiplicities of infection are employed), construc-tion and propagation of each recombinant virus takesseveral weeks. In contrast, CL22-mediated transfectioncan be accomplished very rapidly.
[CL22]2–DNA complexes transfect the cell line HepG2and primary HUVEC as efficiently as the best commer-cially available nonpeptide agents, including cationic lip-ids and PEI ExGen 500. Our recent work shows that theaverage transfection efficiency of human DC with[CL22]2–DNA complexes (17%) is greatly superior toalternative nonviral transfection agents.30 Such primaryDC are generally refractory to transfection.31,32 Ourresults with [CL22]2–DNA complexes are inconsistentwith this hypothesis. Efficient DC transfection by[CL22]2–DNA complexes is an important observation,because immunisation with DC transfected ex vivo with
genes expressing disease-associated antigens is one of themost promising approaches to immunotherapy of cancerand severe infectious diseases. We are presentlydeveloping protocols for evaluating DC transfected withgenes for tumour-associated antigens as therapeutic vac-cines in cancer patients. We are also presently evaluatingwhether the nucleoprotein epitope of CL22 is presentedon HLA class II molecules.
In summary, CL22 is a novel peptide which gives invitro transfection efficiencies for immortalised andtumour cell lines that compare favourably with the bestnonpeptide polymer and cationic lipid formulations, andgives unexpectedly efficient transfection of primaryhuman dendritic and endothelial cells.
Materials and methods
Chemicals and reagentsGalacto-light and Luc-screen kits were obtained fromTropix/PerkinElmer, Bedford, MA, USA. X-gal wasobtained from Life Technologies, Paisley, UK. Proteinestimations were made using a DC protein assay kit(BioRad Laboratories, Hemel Hemstead, UK). Transfec-tion reagents were obtained from the following suppliers:Effectene and Superfect from Qiagen (Crawley, UK); Tfx-10, Tfx-20 and Tfx-50 from Promega (Southampton, UK);Clonfectin from Clontech Laboratories (Basingstoke, UK);ExGen500 from Euromedex (Souffelweyersheim, France);Lipofectin and LipofectAMINE from Life Technologies(Paisley, UK); DOTAP (Dioleoly,1,2 diacyl-3-trimethyl

CL22 transfection peptideAMR Haines et al
107ammonium propane); FuGENE 6 from Boehringer,Mannheim, Germany and poly-l-lysine (pLL) 250merHCl from Sigma. Peptide synthesis reagents were fromPE Biosystems (Warrington, UK), and all other chemicalswere from Aldrich (Poole, UK) unless otherwise stated.
Peptide synthesis, purification and analysisPeptides were prepared by solid phase peptide synthesisusing standard Fmoc chemistry. HPLC analysis indicatedall were at least 90% pure. The synthesis wasaccomplished using a Biosearch 9050 plus Pepsynthesizerin extended synthesis cycle mode. Lysine and tryptophanside chains were Boc protected; arginine side chains werePbf protected; serine, threonine and tyrosine side chainswere tBu protected; asparagine and cysteine side chainswere Trt protected and glutamate side chains were tBuprotected. The amino acid derivatives were coupled in afour-fold molar excess using 0.6 m O-(1H-benzotriazo-1-yl)tetramethyluronium tetrafluoroborate (TBTU)/0.9 mN-ethyldiisopropylamine in dimethylformamide as activ-ating agents. TBTU was obtained from Alexis Biochemi-cals (Nottingham, UK). Deprotection of the N-terminalFmoc group before each coupling was achieved using asolution of 20% piperidine in dimethylformamide (1 minat high flow rate followed by 10 min at 3 ml/min). Thecoupling time for each residue was 1.5 h. On completion,the resin-conjugated peptide was washed with dichloro-methane and dried. The peptides were cleaved from theresin using trifluoroacetic acid/triisopropylsilane/thioanisole/1,2-ethanedithiol (92.5: 2.5: 2.5: 2.5) for 1.5 hat room temperature, which simultaneously deprotectedthe amino acid side chains. The resin was then removedby filtration and washed with trifluoroacetic acid. Thecombined filtrate and washings were concentrated byevaporation then precipitated using diethyl ether fol-lowed by centrifugation to give the crude peptides. Thesewere dissolved in a minimum volume of 20 mmammonium acetate; pH 4.6 and purified using aSephadex G25 (Superfine, Amersham Pharmacia Biotech,Little Chalfont, UK) gel filtration column (100 × 2.6 cm)run in the same buffer. The fractions containing peptide,as determined by analytical RP-HPLC, were pooled andlyophilised. Further purification was achieved by pre-parative HPLC using a C18 RP-HPLC column (Dynamax83-221-C) (Rainin Instruments, Woburn, MA, USA) anda gradient of 20–50% acetonitrile (0.1% trifluoroaceticacid) in water (0.1% trifluoroacetic acid) over 30 min. Thefractions corresponding to the major peak on the chroma-tograph were pooled and lyophilised. Finally, the pep-tides were desalted using a Sephadex G15 (medium) gelfiltration column (70 × 2.6 cm) run in 20 mm ammoniumacetate, pH 4.6. The peptide fractions, which weredetected by analysis at 226 nm, were pooled andlyophilised. The pure peptides were stored at −20°C. Thepeptides were characterised using MALDI-TOF (matrixassisted laser desorption/ionisation time of flight) massspectrometry. 0.1–0.5 mg of peptides was dissolved in1 ml of 0.1% trifluoroacetic acid, and 0.5 ml applied to thetarget and analysed using a Kratos Kompact MALDI II-tDE spectrometer (Kratos Analytical, Manchester, UK). Ineach case the observed mass was within one mass unitof that expected.
Preparation of NEM-CL22Six mg (1.5 mmol) CL22 was dissolved in 0.9 ml PBS andthe cysteine reduced by adding 3 mg (20 mmol) DTT in
Gene Therapy
0.1 ml water. After 2 h at 22°C the DTT was removed bygel filtration using a 1.6 × 30 cm column packed withSephadex G25 Fine, equilibrated in 20 mm ammoniumacetate, pH 5.0. Fractions containing peptide were col-lected, pooled and lyophilised. The molar ratio of freethiol to peptide, as determined by an Ellman’s assay, was0.88. To 0.9 ml of freshly reduced CL22 resuspended to4 mg/ml in PBS, 100 ml water containing 0.8 mg(6.4 mmol) N-ethylmaleimide (NEM) was added. After3 h incubation at 22°C an Ellman’s assay failed to detectthe presence of free thiols. The excess NEM was removedby gel filtration, as described above, before lyophilisationto give 2.1 mg thiol-blocked CL22. Analysis by MALDI-TOF mass spectrometry gave an observed molecularweight of 4227.8 (expected molecular weight 4227.0).
Preparation of peptide dimersTo form disulphide linked peptide dimers, typically 0.5–1.0 ml of 5 mg/ml of the peptide in water was added to1.0 ml 0.1 m sodium borate pH 8.0. The cysteine thiolswere left to oxidise in air at 22°C. The progress ofdimerisation was followed by observing the change inretention time by capillary electrophoresis. After 16 h thepeptide was judged to have dimerised completely.Addition of 10 mm DTT to a peptide sub-sample reversedthe observed shift in retention time. Dimer formation wasalso confirmed by gel filtration analysis using a Superdex(HR10/30) column (Amersham-Pharmacia). Finally, thesodium borate salt was removed by gel filtration inammonium acetate followed by lyophilisation. The finaldimer products were confirmed by gel filtration on aSuperdex HR10/30 column to be .90% in dimeric form,and by MALDI-TOF mass spectroscopy to have molecu-lar weights within one mass unit of that predicted.
Preparation of BMH-[CL22]2To prepare cross-linked [CL22]2, 20 mg (4.9 mmol) ofCL22 was made up to 1.0 ml with 25 mm HEPES, pH 7.2.The homobifunctional cross-linker, 1,6 bis-(maleimido)hexane (0.7 mg; 2.5 mmol, from Molecular Biosciences,Colorado, USA), was added as a solid to the stirred pep-tide solution, and left for 1 h at 22°C. The reaction mix-ture was purified by gel filtration on Sephadex G-25 toremove any unreacted linker. No further purification wasnecessary. The peptide was characterised using capillaryelectrophoresis, HPLC and MALDI-TOF mass spec-trometry. Mass spectrometry gave an observed molecularweight of 8480.4 (expected molecular weight = 8480.0).Capillary electrophoresis analysis was used to confirmthat the BMH-[CL22]2 was free from monomeric anddisulphide-linked peptide.
DNApCMVb and pEGFP-N1 plasmids were obtained fromCambridge Biosciences, UK. pCMVluc was obtained byligating the luciferase gene from pGL3 (Promega) intopCMV.33 Plasmids were grown in DH5a cells in 5 litrefermentations in T broth (Gibco) containing ampicillin at100 mg/ml and were purified using Ultrapure 100 kit andcolumn (Qiagen).
Ethidium bromide exclusion assayspCMVb DNA at 14 mg/ml in 10 mm HEPES was labelledwith ethidium bromide (EtBr) by mixing with an equalvolume of 4 mm EtBr in 10 mm HEPES pH 7.4. EtBr-lab-

CL22 transfection peptideAMR Haines et al
108
Gene Therapy
elled DNA was then condensed by adding an equal vol-ume of condensing agent, (typically 1 ml + 1 ml), to givethe desired charge ratio. Fluorescence was read 30 minafter addition on a Jenway 6200 fluorimeter (Jenway,Dunmow, UK), lex = 492 nm, lem = 520 nm, using narrowband pass filters, and then re-checked 1 h later. In nocases did the fluorescence change significantly from theinitial reading. Monomer peptides were prepared by pre-incubation overnight with 5 mm DTT before conden-sation. DTT did not affect ethidium bromide exclusion ofnon-dimerized peptides. The fluorimeter was calibratedto 1 unit with 1 mm ethidium bromide, and to 1000 unitswith 3.5 mg/ml DNA and 1 mm EtBr.
Cryo-electron microscopyPlasmid DNA was condensed as described for transfec-tion assays. A drop (approximately 4 ml) of sample wasloaded on to both sides of either bare or holey carbon-coated copper 400-mesh grids. The drop was briefly blot-ted and grids immediately plunged into freezing ethanecooled by liquid nitrogen. Sample grids were stored inliquid nitrogen and analysed on a Philips CM10 electronmicroscope (FEI Company, Eindhoven, The Netherlands)under low-dose mode.
Photocorrelation spectroscopy and z potentialmeasurementsAll buffers, DNA and peptides were filtered through0.22 mm filters (Millipore) before DNA condensation. Theconcentrations of DNA and peptide stocks wererechecked after filtering, by measuring absorbance at260 nm and 280 nm, respectively. Plasmid DNA was con-densed as described for transfection assays. For PCS,200 ml of sample was analysed in a small volume cuvettein an Autosizer 4700 (Malvern Instrument) at 25°C, at anangle of 90°, and 100 mm aperture. For z potentialmeasurements, 3 ml of sample was analysed using a ZetaSizer 3000 (Malvern Instruments) at 25°C. A minimum ofthree measurements consisting of 10 sub-runs each wastaken for both PCS and z analysis.
Cells and cell cultureThe cell lines HeLa (human cervical adenocarcinoma),HepG2 (human hepatoma), KLN 205 (mouse lungsquamous carcinoma), COS-7 (monkey kidney), 293(human kidney), and Hepa1-6 (mouse hepatoma) wereobtained from the American Type Culture Collection.HNX-14C (human oral cavity squamous carcinoma) wasa gift from the Cancer Research Unit, University ofNewcastle upon Tyne, UK. Human umbilical vein endo-thelial cells (HUVEC) were purchased from Promocell,Heidelburg, Germany. Human DC were preparedaccording to the procedure of Romani et al.34 HepG2,COS-7 and Hepa1-6 were grown in DMEM (Sigma) sup-plemented with 10% fetal calf serum (FCS). HeLa, KLN205 and 293 cells were grown in EMEM (Sigma) sup-plemented with 1% non-essential amino acids (NEAA)(Sigma) and 10% FCS. HNX-14C were grown in RPMI-1640 (Sigma) supplemented with 10% FCS. HUVEC weregrown in endothelial growth medium (Promocell), andall flasks or wells used for HUVEC culture were pre-coated with 0.1% porcine gelatin. Human DC weregrown in RMPI-1640 supplemented with 50 mm b-mer-captoethanol, 1% NEAA (Sigma), 800 U/ml GM-CSF(LeucoMax) (Sandoz/Schering-Plough, Horsham, UK)and 500 U/ml IL-4 (PeproTech, London, UK).
Preparation of DNA complexes for transfectionCharge ratios were calculated as previously described.35
The average molecular weight of a DNA base was takenas 330, the mass of CL22 was taken as 4101.9 with a netcharge of +11.
Peptide–DNA complexes: Peptides were diluted to anappropriate concentration for final charge ratio in eitherHEPES (10 mm HEPES buffer pH 7.4) or in some casesHBS (10 mm HEPES buffer, 150 mm NaCl pH 7.4). Thiswas rapidly mixed with an equal volume of plasmidDNA at 40 mg/ml in the same buffer and incubated for1 h at room temperature before transfection. To preparecomplexes with monomers, peptides at 200 mg/ml weretreated overnight with 5 mm DTT.
Polylysine–DNA complexes: These were prepared in anidentical manner as for peptide–DNA complexes.
Clonfectin: Two hundred mg Clonfectin was resuspendedin 180 ml 10 mm HEPES-NaCl (Clontech), pre-warmed to45–55°C. pCMVLuc, at 6.65 mg/ml in HEPES-bufferedRPMI-1640 in polystyrene tubes was mixed with an equalvolume of Clonfectin solution diluted appropriately inHEPES-buffered RPMI (Clonfectin added to medium) togive ratios of 0.5:1, 1:1, 2:1, and 3:1 (mg Clonfectin:mgDNA). Complexes were incubated for 10–30 min beforeaddition to cells. Cells were transfected in the absenceof antibiotics.
DOTAP: Forty mg/ml pCMVLuc was mixed with anequal volume of DOTAP diluted appropriately inHEPES-buffered RPMI-1640 (DOTAP added to buffer) togive ratios of 5:1, 10:1, 15:1, 20:1 (mg DOTAP:mg DNA).Complexes were incubated for 15 min at room tempera-ture before addition to cells.
Effectene: pCMVLuc at 0.32 mg/ml in HEPES wasdiluted with EC buffer (Qiagen) to 7.2 mg/ml and treatedwith 8 ml enhancer (Qiagen) per mg DNA to a final con-centration of 6.7 mg/ml and incubated for 2–5 min. 320 mlEffectene, appropriately diluted in EC buffer, was addedto 480 ml pCMVLuc/enhancer to give ratios of 5:1, 10:1,25:1 and 50:1 (mg Effectene: mg DNA) and a final DNAconcentration of 4 mg/ml. This was incubated for 5–10min at room temperature before addition to cells.
ExGen 500: pCMVLuc at 40 mg/ml in 150 mm NaCl wasmixed with an equal volume of ExGen 500 diluted in150 mm NaCl to appropriate concentrations to give ratiosof 3:1, 9:1, 12:1 and 15:1 (N:P). Complexes were incubatedfor 10 min at room temperature before addition to cells.
FuGENE 6: FuGENE 6 was diluted appropriately inHEPES-buffered RPMI-1640, with care taken to ensurethat it did not come into contact with plastic walls of ves-sel, such that the dilutions would give final ratios of 4:1,5:1, 6:1 and 7:1 (ml FuGENE 6:mg DNA). These were leftfor 5 min at room temperature. Each FuGENE 6 dilutionwas added drop wise to pCMVLuc at 400 mg/ml inHEPES-buffered RPMI-1640 to give a final concentrationof 20 mg/ml DNA. Complexes were incubated for 15 minat room temperature before addition to cells.
Lipofectin and LipofectAMINE: pCMVLuc at 20 mg/ml inHEPES-buffered RMPI-1640 was mixed with an equal

CL22 transfection peptideAMR Haines et al
109volume of Lipofectin or LipofectAMINE diluted appro-priately in HEPES-buffered RPMI-1640, to give finalratios of 18:1, 25:1, 32:1 and 40:1 (mg LipofectAMINE:mgDNA) or 1:1, 3:1, 7:1 and 10:1 (mg Lipofectin:mg DNA)Complexes were incubated for 45 min at room tempera-ture before addition to cells. Cells were transfected in theabsence of antibiotics.
Superfect: pCMVLuc at 16.6 mg/ml in HEPES-bufferedRMPI-1640 was mixed with Superfect diluted appropri-ately in HEPES-buffered RPMI-1640 to give ratios of 1:1,2:1, 5:1 and 10:1 (ml Superfect:mg DNA), and a final DNAconcentration of 10 mg/ml. Complexes were incubatedfor 10 min then diluted with transfection media (withoutFCS) to 3.3 mg/ml before addition to cells.
Tfx reagents: 3.1 mg, 1.6 mg and 0.7 mg of Tfx-10, -20and -50 respectively, were dissolved in nuclease-freewater to 1 mm. Samples were vortexed, incubated at 65°Cfor 1 min, vortexed again and frozen overnight at −20°Cfor 30 min, thawed out and re-vortexed. Volumes of eachTfx reagent were added to 300 ml pCMVLuc DNA at10 mg/ml in HEPES-buffered RPMI-1640 to give ratios of3:1, 4.5:1 6:1 and 7.5:1 (ml Tfx reagent:mg DNA), equiva-lent to charge ratios of 2, 3, 4 and 5. HEPES-bufferedRPMI-1640 was added to maintain final DNA concen-tration at 9.3 mg/ml. Complexes were allowed to incu-bate for 10–15 min at room temperature before additionto cells.
TransfectionsAll bgal transfections were carried out in six-well plates.HepG2 and KLN 205 were seeded at 1 × 105 cells per well,18–20 h before transfection. Immediately before transfec-tion, cells were washed once with 1–2 ml of RPMI-1640and 875 ml of RA transfection medium (RPMI-1640 con-taining 0.1% human serum albumin), containing whereappropriate 90 mm chloroquine for HepG2 and 120 mmchloroquine KLN 205, was added to each well followedby 125 ml of DNA complexes at 20 mg DNA/ml(prepared as described above). Medium in wells wasmixed up and down with a pipette tip to ensure thoroughdispersion of the complexes. All points in transfectionassays were carried out in triplicate. Transfectionmedium was used as a negative control. Plates were thenincubated at 37°C for 5 h (unless otherwise specified) andmedia replaced with 3 ml fresh growth medium andincubated for a further 18–24 h before bgal reporterassay.
For all cell types except human DC, GFP transfectionswere carried out in six-well plates, seeded 18–20 h beforetransfection at the densities indicated in the Figures.Immediately before transfection, cells were washed oncewith 1–2 ml of PBS and typically 875 ml of RA transfec-tion medium (or M199 containing 10% FCS for HUVEC)was added to each well followed by 125 ml of DNA com-plexes at 20 mg/ml (prepared as described above). Tovary DNA dose the volume of DNA complexes wasvaried, maintaining the final volume on cells at 1 ml.Medium in wells was agitated as above to ensure thor-ough dispersion of the complexes. Plates were incubatedat 37°C for 5 h (or 2 h for HUVEC) and media replacedwith 3 ml fresh growth medium appropriate for the celltype and incubated for a further 18–24 h before FACScananalysis. For GFP transfection of human DC, cells were
Gene Therapy
washed with PBS, resuspended in RA transfectionmedium containing nonessential amino acids, 50 ng/mltransferrin (Sigma) and 50 mm b-mercaptoethanol. DNAcomplexes were then added followed by chloroquine to40 mm and the cells plated 1 ml/well into 24-well platesat 3 × 105 cells per well, 2.5 mg DNA per well, followedby centrifugation at 1000 r.p.m. for 5 min to sedimentcells and complexes. Plates were incubated at 37°C for1.5 h and media replaced with 1 ml fresh human DCgrowth medium, then incubated for a further 18–24 hbefore FACScan analysis.
Luciferase transfections were carried out in 96-wellplates. HepG2 cells and HUVEC were seeded at 5 × 104
and 1 × 104 cells per well, respectively, 18–20 h beforetransfection. Immediately before transfection, cells werewashed once with 100 ml of PBS and then placed in trans-fection media (HEPES-buffered RPMI-1640 for HepG2and M199 for HUVEC, both containing antibiotics: peni-cillin and streptomycin, unless otherwise stated). Trans-fection complexes were added to the cells in triplicatewells, to give a final dose of 0.2 mg per well DNA, anda final volume of 100 ml. FCS was included in the trans-fection media such that after addition of complexes thefinal FCS concentration was 10%. After addition of com-plexes, plates were centrifuged for 5 min at 200 g to pelletthe complexes on to the surface of cells. HepG2 cells weretransfected for 4 h at 37°C under atmospheric CO2, andHUVEC for 1 h at 37°C under 5% CO2. After the transfec-tion period, cells were washed with 100 ml per well PBS,and incubated overnight at 37°C in cell growth mediabefore luciferase reporter assay.
Reporter assaysTotal bgal enzyme activity was measure using Galacto-Light reagents (PE-Biosystems). Cells (in six-well plates)were washed once with PBS and lysed with 250 ml perwell lysis buffer. Cell debris was removed by centrifug-ation at 12 000 g and 10 ml of the resulting supernatantadded to 100 ml Galacto-Light reaction buffer, incubatedfor 1 h, then assayed on Lumat LB9501 luminometer(Berthold, Pforzheim, Germany), with light emissionmeasured over 10 s. Samples giving off-scale readingswere diluted appropriately and re-read. The percentageof cells transfected with bgal was determined by X-galstaining. Cells (in six-well plates) were washed once withPBS, fixed with 0.05% glutaraldehyde for 10 min, washedagain, then stained with X-gal solution (LifeTechnologies) and incubated overnight at 37°C. Posi-tively-stained cells were counted and expressed as per-centage of total cells in a field of view.
For analysis of GFP expression (in six-well plates), cellswere removed with trypsin-EDTA, washed with PBS,and the percentage of expressing cells determined usinga Becton Dickinson FACscan, with mock-transfected cellsas autofluorescence controls. A marker region was set ona FL-1H histogram plot that contained none or very fewof the mock-transfected cells, and this marker was thenused to measure the number of GFP-expressing cells inthe transfected populations. For human DC (in 24-wellplates) the procedure was similar except that cells weredislodged by pipette and washed once with PBS beforeFACScan analysis.
Total luciferase enzyme activity was determined usingLuc Screen reagents (PE-Biosystems). Two hundredml/well of Luc Screen reagent was added directly to the

CL22 transfection peptideAMR Haines et al
110
Gene Therapy
100 ml tissue culture media on cells in 96-well plates andthe plates shaken gently for 10 min at room temperature.250 ml was removed and transferred to correspondingwells of black 96-well plates (Packard Bioscience, Pang-bourne, UK) (to prevent well to well cross-talk) and lumi-nescence read using a TopCount scintillation counter(Packard Bioscience) in SPC mode and a count time of0.02 min. Results expressed as c.p.s. per well.
Cellular uptake of complexesUptake experiments were carried out with KLN 205 cellsseeded 18–24 h previously at 1 × 105 cells per well in six-well plates, using the method for transfections except thatplasmid DNA was pre-labelled with YOYO-1 (MolecularProbes, Leiden, The Netherlands) 1 molecule per 100 basepairs, and then condensed with peptides. YOYO-1 lab-elled complexes were incubated with cells for 4 h at 4°Cand 37°C in RPMI-1640 containing 120 mm chloroquine,after which cells were removed with trypsin-EDTA,washed twice with PBS, and fluorescence determined byFACScan analysis.
AcknowledgementsWe thank Dr Tim Booth (currently at CFIA, NationalCentre for Foreign Animal Disease,Winnipeg, Canada)and Dr Svetla Stoilova (Leeds University, UK) for Cryo-Electron microscopy. We would also like to thank Pro-fessor Nick Price and Dr Sharon Kelly at BBSRC ScottishCircular Dichroism Facility for their generous help andadvice.
References1 Treco DA, Selden RF. Non-viral gene therapy. Mol Med Today
1995; 1: 314–321.2 Cristiano RJ. Targeted, non-viral gene delivery for cancer gene
therapy. Front Biosci 1998; 3: D1161–D1170.3 Mahato RI, Takakura Y, Hashida M. Non-viral vectors for in
vivo gene delivery: physicochemical and pharmacokinetic con-siderations. Crit Rev Ther Drug Carrier Syst 1997; 14: 133–172.
4 Wu CH, Wilson JM, Wu GY. Targeting genes: delivery and per-sistent expression of a foreign gene driven by mammalian regu-latory elements in vivo. J Biol Chem 1989; 264: 16985–16987.
5 Perales JC, Ferkol T, Molas M, Hanson RW. An evaluation ofreceptor-mediated gene transfer using synthetic DNA–ligandcomplexes. Eur J Biochem 1994; 226: 255–266.
6 Wadhwa MS et al. Peptide-mediated gene delivery: influence ofpeptide structure on gene expression. Bioconjug Chem 1997; 8:81–88.
7 Gottschalk S et al. A novel DNA–peptide complex for efficientgene transfer and expression in mammalian cells. Gene Therapy1996; 3: 48–57.
8 Emi N, Kidoaki S, Yoshikawa K, Saito H. Gene transfermediated by polyarginine requires a formation of big carrier-complex of DNA aggregate. Biochem Biophys Res Commun 1997;231: 421–424.
9 Boussif O et al. A versatile vector for gene and oligonucleotidetransfer into cells in culture and in vivo: polyethylenimine. ProcNatl Acad Sci USA 1995; 92: 7297–7301.
10 Chemin I et al. Liver-directed gene transfer: a linear polyethleni-mine derivative mediates highly efficient DNA delivery to pri-mary hepatocytes in vitro and in vivo. J Viral Hepat 1998; 5:369–375.
11 Goula D et al. Polyethylenimine-based intravenous delivery oftransgenes to mouse lung. Gene Therapy 1998; 5: 1291–1295.
12 Ferrari S et al. ExGen 500 is an efficient vector for gene delivery
to lung epithelial cells in vitro and in vivo. Gene Therapy 1997; 4:1100–1106.
13 Haensler J, Szoka FC Jr. Polyamidoamine cascade polymersmediate efficient transfection of cells in culture. Bioconjug Chem1993; 4: 372–379.
14 Tang MX, Redemann CT, Szoka FC Jr. In vitro gene delivery bydegraded polyamidoamine dendrimers. Bioconjug Chem 1996; 7:703–714.
15 Felgner PL et al. Lipofection: a highly efficient, lipid-mediatedDNA-transfection procedure. Proc Natl Acad Sci USA 1987; 84:7413–7417.
16 Behr JP, Demeneix B, Loeffler JP, Perez-Mutul J. Efficient genetransfer into mammalian primary endocrine cells with lipopoly-amine-coated DNA. Proc Natl Acad Sci USA 1989; 86: 6982–6986.
17 Lasic DD, Templeton NS. Liposomes in gene therapy. Adv DrugDel Rev 1996; 20: 221–226.
18 Lemieux GA, Bertozzi CR. Chemoselective ligation reactionswith proteins, oligosaccharides and cells. Trends Biotechnol 1998;16: 506–513.
19 Keilova H. On the specificity and inhibition of cathepsins D andB. In: Barrett AJ, Dingle JT (eds). Tissue Proteinases. North Hol-land Publishing: Amsterdam, 1971, pp 45–68.
20 Kirschke H, Barrett AJ. Chemistry of lysosomal proteases. In:Glaumann H, Ballard FJ (eds). Lysosomes: Their Role in ProteinBreakdown. Academic Press: London, 1987, pp 193–238.
21 Brett SJ et al. Human T cell recognition of influenza A nucleo-protein. Specificity and genetic restriction of immunodominantT helper cell epitopes. J Immunol 1991; 147: 984–991.
22 Wu GY, Wu CH. Receptor-mediated in vitro gene transform-ation by a soluble DNA carrier system (published erratumappears in J Biol Chem 1988; 263: 588). J Biol Chem 1987; 262:4429–4432.
23 Avrameas A et al. Polyreactive anti-DNA monoclonal antibodiesand a derived peptide as vectors for the intracytoplasmic andintranuclear translocation of macromolecules. Proc Natl Acad SciUSA 1998; 95: 5601–5606.
24 Zhang F et al. A transfecting peptide derived from adenovirusfiber protein. Gene Therapy 1999; 6: 171–181.
25 Thatcher DR, Haines A, Phillips R. Peptide-mediated gene deliv-ery. In: Kabanov AV, Felgner PL, Seymour LW (eds). Self-Assembling Complexes for Gene Delivery: From Laboratory to ClinicalTrial. John Wiley: Chichester, 1998, pp 331–350.
26 Adami RC et al. Stability of peptide-condensed plasmid DNAformulations. J Pharm Sci 1998; 87: 678–683.
27 Dash PR, Toncheva V, Schacht E, Seymour LW. Synthetic poly-mers for vectorial delivery of DNA: characteristics of polymerDNA complexes by photon correlation spectroscopy and stab-ility to nuclease degradation and disruption by polyanions invitro. J Control Rel 1997; 48: 269–276.
28 Ogris M et al. The size of DNA/transferrin-PEI complexes is animportant factor for gene expression in cultured cells. Gene Ther-apy 1998; 5: 1425–1433.
29 Dash PR et al. Factors affecting blood clearance and in vivo distri-bution of polyelectrolyte complexes for gene delivery. GeneTherapy 1999; 6: 643–650.
30 Irvine AS et al. Efficient nonviral transfection of dendritic cellsand their use for in vivo immunization. Nat Biotechnol 2000; 18:1273–1278.
31 Arthur JF et al. A comparison of gene transfer methods inhuman dendritic cells. Cancer Gene Ther 1997; 4: 17–25.
32 Philip R et al. Transgene expression in dendritic cells to induceantigen-specific cytotoxic T cells in healthy donors. Cancer GeneTher 1998; 5: 236–246.
33 Hanke T et al. DNA multi-CTL epitope vaccines for HIV andPlasmodium falciparum: immunogenicity in mice. Vaccine 1998;16: 426–435.
34 Romani N et al. Proliferating dendritic cell progenitors in humanblood. J Exp Med 1994; 180: 83–93.
35 Felgner PL et al. Nomenclature for synthetic gene delivery sys-tems (editorial). Hum Gene Ther 1997; 8: 511–512.




















