
Cholinergic stimulation enhances bayesian belief updating in the deployment of spatial attention
Upload
independentCategory
view
0download
0
A R T I C L E S
1216 VOLUME 10 | NUMBER 11 | NOVEMBER 2004 NATURE MEDICINE
Sepsis, the third leading cause of death in developed societies, affects750,000 patients and accounts for 9% of the overall deaths in theUnited States annually1–3. Despite the recent advances in intensive caretreatment and the discovery of antibiotics, sepsis remains associatedwith a high mortality rate2–4. Although commonly initiated by aninfection, the pathogenesis of sepsis is characterized by an overwhelm-ing systemic inflammatory response that can lead to lethal multipleorgan failure. Endotoxin and other pathogen components stimulatemacrophages to produce proinflammatory cytokines such as TNF-αand interleukin-1β (IL-1β). Excessive production of these cytokinescan cause a lethal systemic inflammation that produces the most dra-matic pathological sequelae of sepsis, including systemic capillaryleakage syndrome, tissue injury and fatal organ failure5–10.Neutralization of TNF-α or IL-1β can prevent the development ofseptic shock in animal models5,6, but these strategies produced modestclinical effects in critically ill patients11–13. Several studies suggest thatother ‘late’ downstream proinflammatory cytokines may contribute tothe progression of sepsis. In agreement with this hypothesis, highdoses of endotoxin are lethal to TNF-α-deficient mice, and death fromendotoxemia and sepsis often occurs days after the ‘early’ TNF-α pro-duction, and long after serum TNF-α has returned to basal levels5,14,15.The identification of late downstream mediators represents a clinicaladvantage for the treatment of sepsis, because these mediators couldexpand the therapeutic time window5,15.
We recently identified high mobility group box 1 (HMGB1) proteinas a late mediator of lethal systemic inflammation in sepsis15.Originally described as an intracellular DNA-binding protein,
HMGB1 is released into the extracellular milieu by activatedmacrophages. Extracellular HMGB1 acts as a proinflammatorycytokine; it induces tissue-type plasminogen activator, stimulates theproduction of proinflammatory cytokines (TNF-α, IL-1β and IL-8)and promotes epithelial cell permeability16–21. Several observationsindicate that HMGB1 is a sufficient and necessary mediator of sepsis.First, systemic HMGB1 is found in animal models of sepsis, and in theserum of patients with bacteremia and sepsis-induced organ dysfunc-tion15. Second, administration of recombinant HMGB1 to mice reca-pitulates the most characteristic clinical signs of sepsis, including fever,malaise, derangement of the intestinal barrier function, acute lunginjury and lethal multiple organ failure18–21. Third, antibodies toHMGB1 prevent lethal endotoxemia and reverse the pathologicalcourse of established sepsis19. HMGB1 was defined as a late mediatorof sepsis because macrophages secrete HMGB1 ∼ 20 h after endotoxinstimulation. This delayed release is also observed in vivo, whereHMGB1 is released into the serum ∼ 20 h after the induction of endo-toxemia, bacteremia or peritonitis15–20. As a consequence, anti-HMGB1 therapeutics are successful in experimental models of sepsiseven if administered after the onset of the disease16–19. But becausemost antibody-blocking strategies do not inhibit the production of thecytokine and have shown limited efficacy in sepsis, we sought strategies to stop HMGB1 secretion from macrophages.
We recently discovered that the nervous system, through the vagusnerve, can modulate circulating TNF-α levels induced by endo-toxin22,23. This physiological mechanism is based on the release ofacetylcholine, the principal neurotransmitter of the vagus nerve22–25.
1The Center for Immunology and Inflammation, North Shore-LIJ Research Institute, 2Center for Patient-Oriented Research, 3Department of Surgery and 4Departmentof Emergency Medicine, North Shore University Hospital, 350 Community Drive, Manhasset, New York 11030, USA. 5These authors have contributed equally to thiswork. Correspondence should be addressed to L.U. ([email protected]).
Published online 24 October 2004; doi:10.1038/nm1124
Cholinergic agonists inhibit HMGB1 release and improvesurvival in experimental sepsisHong Wang1,5, Hong Liao1,5, Mahendar Ochani2, Marilou Justiniani1, Xinchun Lin3, Lihong Yang2,Yousef Al-Abed1, Haichao Wang4, Christine Metz1, Edmund J Miller3, Kevin J Tracey2 & Luis Ulloa1
Physiological anti-inflammatory mechanisms can potentially be exploited for the treatment of inflammatory disorders. Here we report that the neurotransmitter acetylcholine inhibits HMGB1 release from human macrophages by signaling through anicotinic acetylcholine receptor. Nicotine, a selective cholinergic agonist, is more efficient than acetylcholine and inhibitsHMGB1 release induced by either endotoxin or tumor necrosis factor-alpha (TNF-α). Nicotinic stimulation prevents activation ofthe NF-κB pathway and inhibits HMGB1 secretion through a specific ‘nicotinic anti-inflammatory pathway’ that requires the α7nicotinic acetylcholine receptor (α7nAChR). In vivo, treatment with nicotine attenuates serum HMGB1 levels and improvessurvival in experimental models of sepsis, even when treatment is started after the onset of the disease. These results revealacetylcholine as the first known physiological inhibitor of HMGB1 release from human macrophages and suggest that selectivenicotinic agonists for the α7nAChR might have therapeutic potential for the treatment of sepsis.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 11 | NOVEMBER 2004 1217
The therapeutic potential of this mechanism to improve survival insepsis is unknown. Here we report that acetylcholine suppressesHMGB1 release from macrophages signaling through a nicotinicacetylcholine receptor (nAChR). Nicotine, a more selective cholinergicagonist, acts on the α7nAChR, inhibits the NF-κB pathway, and sup-presses HMGB1 release from human macrophages. In vivo, treatmentwith nicotine attenuates serum HMGB1 levels and improves survivalin ‘established’ sepsis, even when treatment is initiated after the onsetof the disease. These results support acetylcholine as the first knownphysiological inhibitor of HMGB1 release from human macrophages,and establish a nicotinic anti-inflammatory pathway as a potentialpharmacological strategy for the treatment of sepsis.
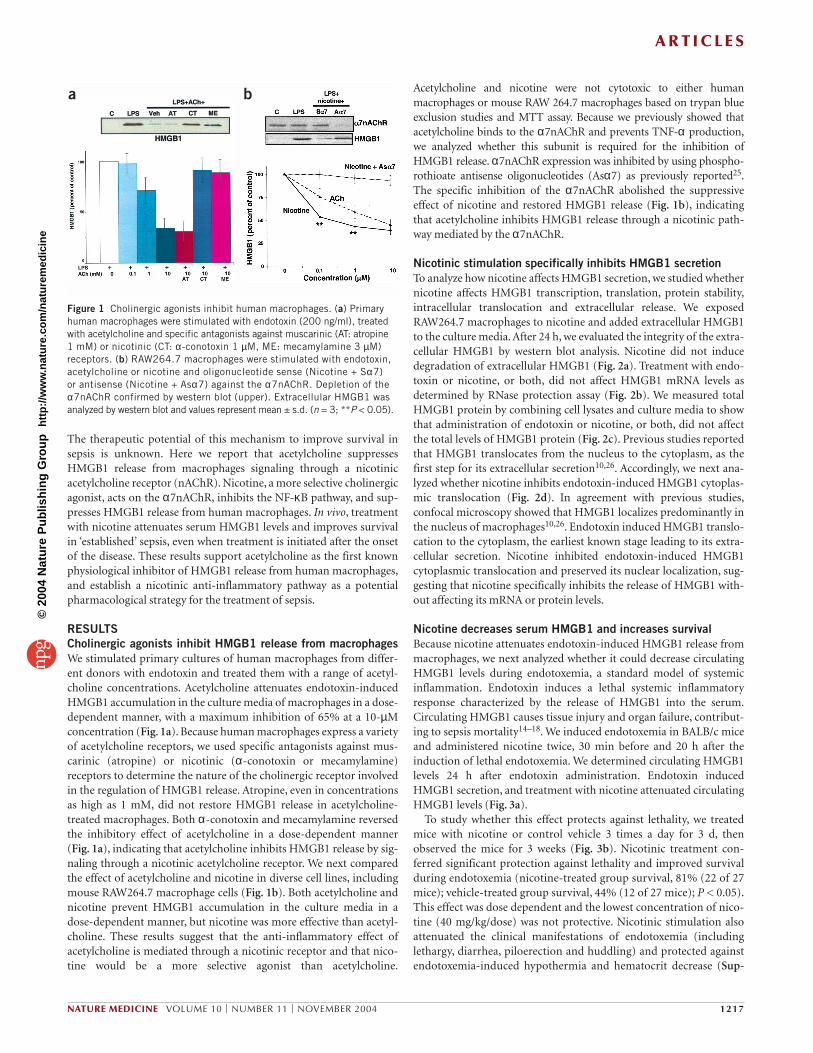
RESULTSCholinergic agonists inhibit HMGB1 release from macrophagesWe stimulated primary cultures of human macrophages from differ-ent donors with endotoxin and treated them with a range of acetyl-choline concentrations. Acetylcholine attenuates endotoxin-inducedHMGB1 accumulation in the culture media of macrophages in a dose-dependent manner, with a maximum inhibition of 65% at a 10-µMconcentration (Fig. 1a). Because human macrophages express a varietyof acetylcholine receptors, we used specific antagonists against mus-carinic (atropine) or nicotinic (α-conotoxin or mecamylamine)receptors to determine the nature of the cholinergic receptor involvedin the regulation of HMGB1 release. Atropine, even in concentrationsas high as 1 mM, did not restore HMGB1 release in acetylcholine-treated macrophages. Both α-conotoxin and mecamylamine reversedthe inhibitory effect of acetylcholine in a dose-dependent manner(Fig. 1a), indicating that acetylcholine inhibits HMGB1 release by sig-naling through a nicotinic acetylcholine receptor. We next comparedthe effect of acetylcholine and nicotine in diverse cell lines, includingmouse RAW264.7 macrophage cells (Fig. 1b). Both acetylcholine andnicotine prevent HMGB1 accumulation in the culture media in adose-dependent manner, but nicotine was more effective than acetyl-choline. These results suggest that the anti-inflammatory effect ofacetylcholine is mediated through a nicotinic receptor and that nico-tine would be a more selective agonist than acetylcholine.
Acetylcholine and nicotine were not cytotoxic to either humanmacrophages or mouse RAW 264.7 macrophages based on trypan blueexclusion studies and MTT assay. Because we previously showed thatacetylcholine binds to the α7nAChR and prevents TNF-α production,we analyzed whether this subunit is required for the inhibition ofHMGB1 release. α7nAChR expression was inhibited by using phospho-rothioate antisense oligonucleotides (Asα7) as previously reported25.The specific inhibition of the α7nAChR abolished the suppressiveeffect of nicotine and restored HMGB1 release (Fig. 1b), indicatingthat acetylcholine inhibits HMGB1 release through a nicotinic path-way mediated by the α7nAChR.
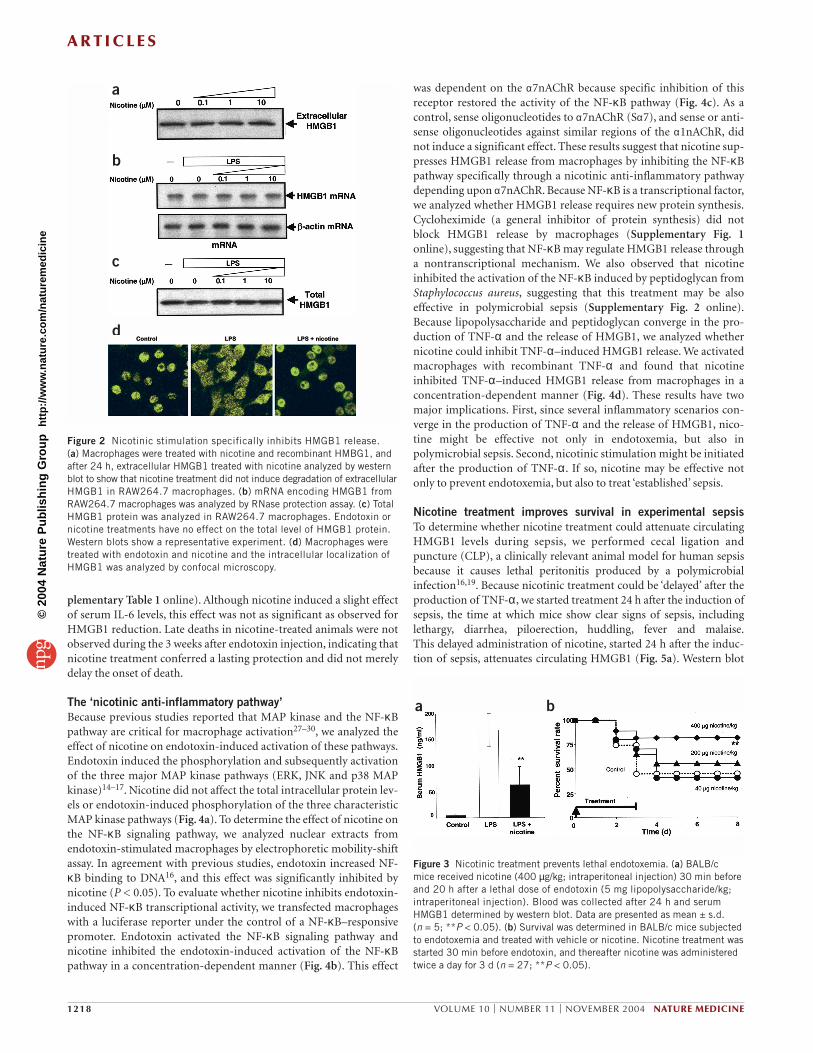
Nicotinic stimulation specifically inhibits HMGB1 secretionTo analyze how nicotine affects HMGB1 secretion, we studied whethernicotine affects HMGB1 transcription, translation, protein stability,intracellular translocation and extracellular release. We exposedRAW264.7 macrophages to nicotine and added extracellular HMGB1to the culture media. After 24 h, we evaluated the integrity of the extra-cellular HMGB1 by western blot analysis. Nicotine did not inducedegradation of extracellular HMGB1 (Fig. 2a). Treatment with endo-toxin or nicotine, or both, did not affect HMGB1 mRNA levels asdetermined by RNase protection assay (Fig. 2b). We measured totalHMGB1 protein by combining cell lysates and culture media to showthat administration of endotoxin or nicotine, or both, did not affectthe total levels of HMGB1 protein (Fig. 2c). Previous studies reportedthat HMGB1 translocates from the nucleus to the cytoplasm, as thefirst step for its extracellular secretion10,26. Accordingly, we next ana-lyzed whether nicotine inhibits endotoxin-induced HMGB1 cytoplas-mic translocation (Fig. 2d). In agreement with previous studies,confocal microscopy showed that HMGB1 localizes predominantly inthe nucleus of macrophages10,26. Endotoxin induced HMGB1 translo-cation to the cytoplasm, the earliest known stage leading to its extra-cellular secretion. Nicotine inhibited endotoxin-induced HMGB1cytoplasmic translocation and preserved its nuclear localization, sug-gesting that nicotine specifically inhibits the release of HMGB1 with-out affecting its mRNA or protein levels.
Nicotine decreases serum HMGB1 and increases survivalBecause nicotine attenuates endotoxin-induced HMGB1 release frommacrophages, we next analyzed whether it could decrease circulatingHMGB1 levels during endotoxemia, a standard model of systemicinflammation. Endotoxin induces a lethal systemic inflammatoryresponse characterized by the release of HMGB1 into the serum.Circulating HMGB1 causes tissue injury and organ failure, contribut-ing to sepsis mortality14–18. We induced endotoxemia in BALB/c miceand administered nicotine twice, 30 min before and 20 h after theinduction of lethal endotoxemia. We determined circulating HMGB1levels 24 h after endotoxin administration. Endotoxin inducedHMGB1 secretion, and treatment with nicotine attenuated circulatingHMGB1 levels (Fig. 3a).
To study whether this effect protects against lethality, we treatedmice with nicotine or control vehicle 3 times a day for 3 d, thenobserved the mice for 3 weeks (Fig. 3b). Nicotinic treatment con-ferred significant protection against lethality and improved survivalduring endotoxemia (nicotine-treated group survival, 81% (22 of 27mice); vehicle-treated group survival, 44% (12 of 27 mice); P < 0.05).This effect was dose dependent and the lowest concentration of nico-tine (40 mg/kg/dose) was not protective. Nicotinic stimulation alsoattenuated the clinical manifestations of endotoxemia (includinglethargy, diarrhea, piloerection and huddling) and protected against endotoxemia-induced hypothermia and hematocrit decrease (Sup-
a b
Figure 1 Cholinergic agonists inhibit human macrophages. (a) Primaryhuman macrophages were stimulated with endotoxin (200 ng/ml), treatedwith acetylcholine and specific antagonists against muscarinic (AT: atropine 1 mM) or nicotinic (CT: α-conotoxin 1 µM, ME: mecamylamine 3 µM)receptors. (b) RAW264.7 macrophages were stimulated with endotoxin,acetylcholine or nicotine and oligonucleotide sense (Nicotine + Sα7) or antisense (Nicotine + Asα7) against the α7nAChR. Depletion of theα7nAChR confirmed by western blot (upper). Extracellular HMGB1 wasanalyzed by western blot and values represent mean ± s.d. (n = 3; **P < 0.05).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
1218 VOLUME 10 | NUMBER 11 | NOVEMBER 2004 NATURE MEDICINE
plementary Table 1 online). Although nicotine induced a slight effectof serum IL-6 levels, this effect was not as significant as observed forHMGB1 reduction. Late deaths in nicotine-treated animals were notobserved during the 3 weeks after endotoxin injection, indicating thatnicotine treatment conferred a lasting protection and did not merelydelay the onset of death.
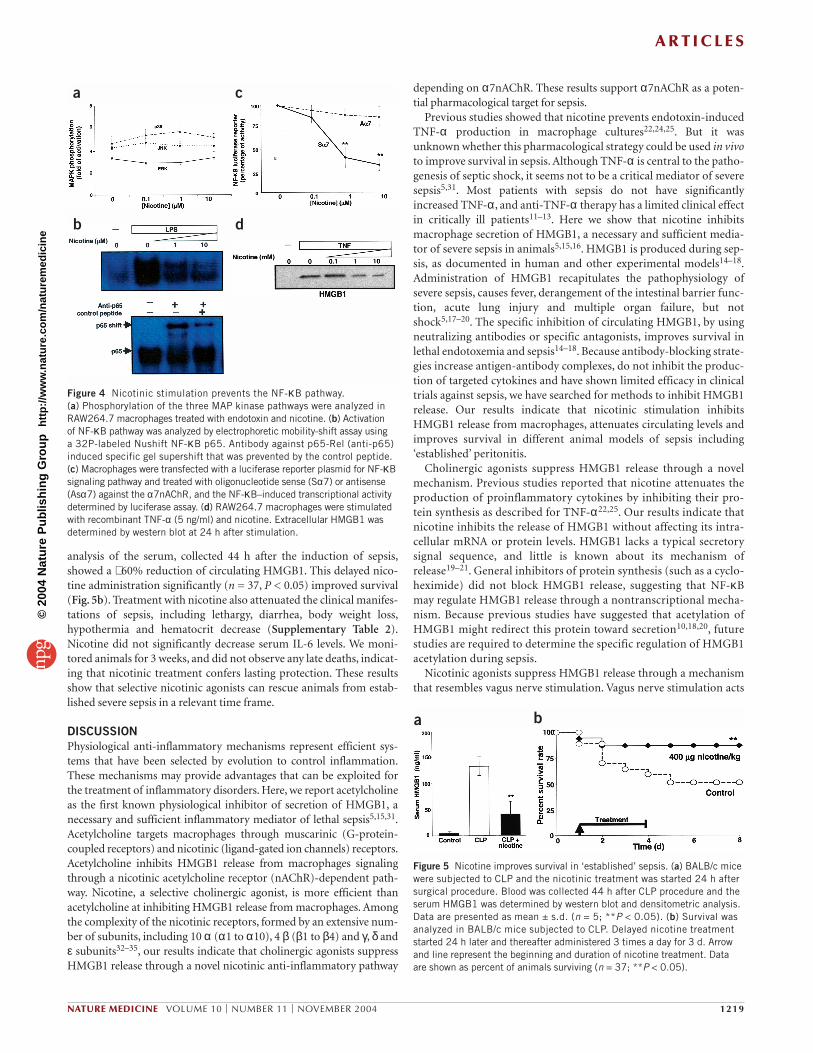
The ‘nicotinic anti-inflammatory pathway’ Because previous studies reported that MAP kinase and the NF-κBpathway are critical for macrophage activation27–30, we analyzed theeffect of nicotine on endotoxin-induced activation of these pathways.Endotoxin induced the phosphorylation and subsequently activationof the three major MAP kinase pathways (ERK, JNK and p38 MAPkinase)14–17. Nicotine did not affect the total intracellular protein lev-els or endotoxin-induced phosphorylation of the three characteristicMAP kinase pathways (Fig. 4a). To determine the effect of nicotine onthe NF-κB signaling pathway, we analyzed nuclear extracts fromendotoxin-stimulated macrophages by electrophoretic mobility-shiftassay. In agreement with previous studies, endotoxin increased NF-κB binding to DNA16, and this effect was significantly inhibited bynicotine (P < 0.05). To evaluate whether nicotine inhibits endotoxin-induced NF-κB transcriptional activity, we transfected macrophageswith a luciferase reporter under the control of a NF-κB–responsivepromoter. Endotoxin activated the NF-κB signaling pathway andnicotine inhibited the endotoxin-induced activation of the NF-κBpathway in a concentration-dependent manner (Fig. 4b). This effect
was dependent on the α7nAChR because specific inhibition of thisreceptor restored the activity of the NF-κB pathway (Fig. 4c). As acontrol, sense oligonucleotides to α7nAChR (Sα7), and sense or anti-sense oligonucleotides against similar regions of the α1nAChR, didnot induce a significant effect. These results suggest that nicotine sup-presses HMGB1 release from macrophages by inhibiting the NF-κBpathway specifically through a nicotinic anti-inflammatory pathwaydepending upon α7nAChR. Because NF-κB is a transcriptional factor,we analyzed whether HMGB1 release requires new protein synthesis.Cycloheximide (a general inhibitor of protein synthesis) did notblock HMGB1 release by macrophages (Supplementary Fig. 1online), suggesting that NF-κB may regulate HMGB1 release througha nontranscriptional mechanism. We also observed that nicotineinhibited the activation of the NF-κB induced by peptidoglycan fromStaphylococcus aureus, suggesting that this treatment may be alsoeffective in polymicrobial sepsis (Supplementary Fig. 2 online).Because lipopolysaccharide and peptidoglycan converge in the pro-duction of TNF-α and the release of HMGB1, we analyzed whethernicotine could inhibit TNF-α–induced HMGB1 release. We activatedmacrophages with recombinant TNF-α and found that nicotineinhibited TNF-α–induced HMGB1 release from macrophages in aconcentration-dependent manner (Fig. 4d). These results have twomajor implications. First, since several inflammatory scenarios con-verge in the production of TNF-α and the release of HMGB1, nico-tine might be effective not only in endotoxemia, but also inpolymicrobial sepsis. Second, nicotinic stimulation might be initiatedafter the production of TNF-α. If so, nicotine may be effective notonly to prevent endotoxemia, but also to treat ‘established’ sepsis.
Nicotine treatment improves survival in experimental sepsisTo determine whether nicotine treatment could attenuate circulatingHMGB1 levels during sepsis, we performed cecal ligation and puncture (CLP), a clinically relevant animal model for human sepsisbecause it causes lethal peritonitis produced by a polymicrobial infection16,19. Because nicotinic treatment could be ‘delayed’ after the production of TNF-α, we started treatment 24 h after the induction ofsepsis, the time at which mice show clear signs of sepsis, includinglethargy, diarrhea, piloerection, huddling, fever and malaise.This delayed administration of nicotine, started 24 h after the induc-tion of sepsis, attenuates circulating HMGB1 (Fig. 5a). Western blot
a
b
c
d
Figure 2 Nicotinic stimulation specifically inhibits HMGB1 release. (a) Macrophages were treated with nicotine and recombinant HMBG1, andafter 24 h, extracellular HMGB1 treated with nicotine analyzed by westernblot to show that nicotine treatment did not induce degradation of extracellularHMGB1 in RAW264.7 macrophages. (b) mRNA encoding HMGB1 fromRAW264.7 macrophages was analyzed by RNase protection assay. (c) TotalHMGB1 protein was analyzed in RAW264.7 macrophages. Endotoxin ornicotine treatments have no effect on the total level of HMGB1 protein.Western blots show a representative experiment. (d) Macrophages weretreated with endotoxin and nicotine and the intracellular localization ofHMGB1 was analyzed by confocal microscopy.
a b
Figure 3 Nicotinic treatment prevents lethal endotoxemia. (a) BALB/c mice received nicotine (400 µg/kg; intraperitoneal injection) 30 min beforeand 20 h after a lethal dose of endotoxin (5 mg lipopolysaccharide/kg;intraperitoneal injection). Blood was collected after 24 h and serumHMGB1 determined by western blot. Data are presented as mean ± s.d. (n = 5; **P < 0.05). (b) Survival was determined in BALB/c mice subjectedto endotoxemia and treated with vehicle or nicotine. Nicotine treatment wasstarted 30 min before endotoxin, and thereafter nicotine was administeredtwice a day for 3 d (n = 27; **P < 0.05).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 11 | NOVEMBER 2004 1219
analysis of the serum, collected 44 h after the induction of sepsis,showed a ∼ 60% reduction of circulating HMGB1. This delayed nico-tine administration significantly (n = 37, P < 0.05) improved survival(Fig. 5b). Treatment with nicotine also attenuated the clinical manifes-tations of sepsis, including lethargy, diarrhea, body weight loss,hypothermia and hematocrit decrease (Supplementary Table 2).Nicotine did not significantly decrease serum IL-6 levels. We moni-tored animals for 3 weeks, and did not observe any late deaths, indicat-ing that nicotinic treatment confers lasting protection. These resultsshow that selective nicotinic agonists can rescue animals from estab-lished severe sepsis in a relevant time frame.
DISCUSSIONPhysiological anti-inflammatory mechanisms represent efficient sys-tems that have been selected by evolution to control inflammation.These mechanisms may provide advantages that can be exploited forthe treatment of inflammatory disorders. Here, we report acetylcholineas the first known physiological inhibitor of secretion of HMGB1, anecessary and sufficient inflammatory mediator of lethal sepsis5,15,31.Acetylcholine targets macrophages through muscarinic (G-protein-coupled receptors) and nicotinic (ligand-gated ion channels) receptors.Acetylcholine inhibits HMGB1 release from macrophages signalingthrough a nicotinic acetylcholine receptor (nAChR)-dependent path-way. Nicotine, a selective cholinergic agonist, is more efficient thanacetylcholine at inhibiting HMGB1 release from macrophages. Amongthe complexity of the nicotinic receptors, formed by an extensive num-ber of subunits, including 10 α (α1 to α10), 4 β (β1 to β4) and γ, δ andε subunits32–35, our results indicate that cholinergic agonists suppressHMGB1 release through a novel nicotinic anti-inflammatory pathway
depending on α7nAChR. These results support α7nAChR as a poten-tial pharmacological target for sepsis.
Previous studies showed that nicotine prevents endotoxin-inducedTNF-α production in macrophage cultures22,24,25. But it wasunknown whether this pharmacological strategy could be used in vivoto improve survival in sepsis. Although TNF-α is central to the patho-genesis of septic shock, it seems not to be a critical mediator of severesepsis5,31. Most patients with sepsis do not have significantlyincreased TNF-α, and anti-TNF-α therapy has a limited clinical effectin critically ill patients11–13. Here we show that nicotine inhibitsmacrophage secretion of HMGB1, a necessary and sufficient media-tor of severe sepsis in animals5,15,16. HMGB1 is produced during sep-sis, as documented in human and other experimental models14–18.Administration of HMGB1 recapitulates the pathophysiology ofsevere sepsis, causes fever, derangement of the intestinal barrier func-tion, acute lung injury and multiple organ failure, but notshock5,17–20. The specific inhibition of circulating HMGB1, by usingneutralizing antibodies or specific antagonists, improves survival inlethal endotoxemia and sepsis14–18. Because antibody-blocking strate-gies increase antigen-antibody complexes, do not inhibit the produc-tion of targeted cytokines and have shown limited efficacy in clinicaltrials against sepsis, we have searched for methods to inhibit HMGB1release. Our results indicate that nicotinic stimulation inhibitsHMGB1 release from macrophages, attenuates circulating levels andimproves survival in different animal models of sepsis including‘established’ peritonitis.
Cholinergic agonists suppress HMGB1 release through a novelmechanism. Previous studies reported that nicotine attenuates theproduction of proinflammatory cytokines by inhibiting their pro-tein synthesis as described for TNF-α22,25. Our results indicate thatnicotine inhibits the release of HMGB1 without affecting its intra-cellular mRNA or protein levels. HMGB1 lacks a typical secretorysignal sequence, and little is known about its mechanism ofrelease19–21. General inhibitors of protein synthesis (such as a cyclo-heximide) did not block HMGB1 release, suggesting that NF-κBmay regulate HMGB1 release through a nontranscriptional mecha-nism. Because previous studies have suggested that acetylation ofHMGB1 might redirect this protein toward secretion10,18,20, futurestudies are required to determine the specific regulation of HMGB1acetylation during sepsis.
Nicotinic agonists suppress HMGB1 release through a mechanismthat resembles vagus nerve stimulation. Vagus nerve stimulation acts
a
b
c
d
Figure 4 Nicotinic stimulation prevents the NF-κB pathway. (a) Phosphorylation of the three MAP kinase pathways were analyzed inRAW264.7 macrophages treated with endotoxin and nicotine. (b) Activation of NF-κB pathway was analyzed by electrophoretic mobility-shift assay using a 32P-labeled Nushift NF-κB p65. Antibody against p65-Rel (anti-p65)induced specific gel supershift that was prevented by the control peptide. (c) Macrophages were transfected with a luciferase reporter plasmid for NF-κBsignaling pathway and treated with oligonucleotide sense (Sα7) or antisense(Asα7) against the α7nAChR, and the NF-κB–induced transcriptional activitydetermined by luciferase assay. (d) RAW264.7 macrophages were stimulatedwith recombinant TNF-α (5 ng/ml) and nicotine. Extracellular HMGB1 wasdetermined by western blot at 24 h after stimulation.
a b
Figure 5 Nicotine improves survival in ‘established’ sepsis. (a) BALB/c micewere subjected to CLP and the nicotinic treatment was started 24 h aftersurgical procedure. Blood was collected 44 h after CLP procedure and theserum HMGB1 was determined by western blot and densitometric analysis.Data are presented as mean ± s.d. (n = 5; **P < 0.05). (b) Survival wasanalyzed in BALB/c mice subjected to CLP. Delayed nicotine treatmentstarted 24 h later and thereafter administered 3 times a day for 3 d. Arrowand line represent the beginning and duration of nicotine treatment. Dataare shown as percent of animals surviving (n = 37; **P < 0.05).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
1220 VOLUME 10 | NUMBER 11 | NOVEMBER 2004 NATURE MEDICINE
on the α7nAChR to inhibit NF-κB signaling and prevents TNF-αproduction during endotoxemia36,37. In a similar mechanism, nico-tinic stimulation requires the α7nAChR to inhibit the NF-κB pathwayand suppresses HMGB1 release during sepsis (Fig. 6). These resultsalso imply that the α7nAChR can be used to control the NF-κB path-way, a critical step in the control of the immune response and a phar-macological target for several inflammatory disorders. Indeed, mostinflammatory scenarios and a broad range of toxins and cytokinesconverge at the activation of the NF-κB pathway. Accordingly, nico-tine can inhibit HMGB1 release induced not only by endotoxin, butalso by inflammatory cytokines such as TNF-α. Thus, nicotine treat-ment of animal models of sepsis can be delayed even after the produc-tion of TNF-α, and cholinergic agonists may halt HMGB1 release inother related inflammatory disorders such as rheumatoid arthri-tis38,39. Inhibition of the NF-κB pathway may also be the underlyingmechanism for the anti-inflammatory effect of nicotine in otherinflammatory scenarios. In fact, nicotine has been previously used inhuman clinical trials for the treatment of ulcerative colitis, an inflam-matory bowel disorder40,41. Several randomized, placebo-controlledtrials showed that nicotine treatment was associated with substantialremissions in ulcerative colitis severity42,43, suggesting that this nico-tinic anti-inflammatory pathway maybe a therapeutic target for sepsisand other inflammatory disorders.
Nicotinic stimulation attenuates circulating HMGB1 levels duringexperimental models of severe sepsis and nicotine can rescue animalsfrom established lethal peritonitis, even when the treatment wasstarted at 24 h after the induction of peritonitis by cecal puncture. Bycomparison with other targets, administration of antibodies to TNF-α can increase mortality when administered after cecal perfora-tion5,14,15. Antibodies to macrophage migration inhibitory factor areineffective if administered more than 8 h after the induction of peri-tonitis42,43. The therapeutic window for nicotine in animals is alsosubstantially wider than for lysophosphatidylcholine, which is onlyeffective when treatment occurs within 10 h after cecal puncture44.Our results suggest that nicotinic agonists can inhibit HMGB1 releasefrom human macrophages by signaling through a α7nAChR-depend-
ent nicotinic anti-inflammatory pathway. The α7nAChR represents apossible pharmacological target to control systemic inflammation,and further investigation is warranted to determine its potential clinical translation.
METHODSAnimal experiments. We performed all animal experiments in accordance withthe National Institutes of Health guidelines under protocols approved by theInstitutional Animal Care and Use Committee of the North Shore-LIJ ResearchInstitute. We randomly grouped 6–8-week-old male BALB/c mice and assignedthem to a specific experiment. Investigators who treated animals knew thetreatment groups and collected samples which were then analyzed by otherinvestigators blinded to the specific treatment. At the indicated time, we evalu-ated animals by analyzing body weight and temperature. Blood was collected bycardiac puncture. Neutrophils were evaluated using a Neubauer hemocytome-ter. We performed differentiate cell counts by counting at least 400 cells on acytospin preparation stained with Wright-Giemsa (Fisher Scientific), followingthe manufacturer’s instructions.
Endotoxemia. Mice received a dose of lipopolysaccharide (5 mg/kg, intraperi-toneal injection). We subjected mice to vehicle or nicotine (Sigma-Aldrich)treatment (40, 200 or 400 µg/kg, intraperitoneal injection), administered 30 min before lipopolysaccharide injection, given 3 times a day for 3 d.
Cecal ligation and puncture. We performed CLP as previously described16.Briefly, we anesthetized mice with ketamine (75 mg/kg, intramuscular injec-tion, Fort Dodge) and xylazine (20 mg/kg, intramuscular injection; BoehringerIngelheim). Then a midline incision was performed in the abdomen, and thececum was isolated. We placed a 6-0 prolene ligature 5.0 mm from the cecal tip,away from the ileocecal valve and the ligated cecal stump then was puncturedonce with a 22-gauge needle, and stool was extruded (1 mm). We then placedthe cecum back into its normal intra-abdominal position and closed theabdomen with a running suture of 6-0 prolene. All animals received subcuta-neous antibiotic treatment (Primaxin; 0.5 mg/kg, Merck), and resuscitativenormal saline (20 ml/kg body weight) 24 h after surgery. We subjected mice tovehicle or nicotine, started 24 h after surgery, and thereafter administered 3 doses a day for 3 d. We evaluated animals 44 h after surgery, collected bloodand analyzed it as indicated below. Serum HMGB1 levels were determined bywestern blot as we previously described16. HMGB1 concentrations were calcu-lated against standard curves of purified recombinant HMGB1, which waspurified as previously described21. HMGB1 immunofluorescence studies wereperformed as we described21,46 and analyzed by confocal microscope.
Macrophage culture, cell transfection and oligonucleotide treatment. Culturesof human macrophages and oligonucleotide treatment were performed as wepreviously described22. Briefly, we isolated primary blood mononuclear cells bydensity-gradient centrifugation. We collected and cultured cells in RPMI 1640medium with 10% heat-inactivated human serum (Gemini Bio-Products).After 2 h incubation at 37 °C, we detached adherent cells with 10mM EDTA,resuspended them (106 cells/ml) in medium supplemented with humanmacrophage colony-stimulating factor and allowed them to differentiate for 7 d. Specific antisense and proper control oligonucleotides were designed aspreviously described25,45 . Phosphorothioate oligonucleotides were synthesizedby Genosys. The sequences for the nucleotides were: Asα7nAChR, 5′-GCAGCGCATGTTGAGTCCCG-3′ ; Senα7nAChR, 5′-CGTCGCGTA-CAACTCAGGGC-3′; Asα1nAChR, 5′-GGGCTCCATGGGCTACCGGA-3′;Senα1nAChR, 5′-CCCGAGGTACCCGATGGCCT-3′. We pretreated macrophageswith 2 µM oligonucleotide for 24 h before treatment. All experiments usingacetylcholine included the acetylcholine esterase inhibitor pyridostigmine bro-mide (1mM, Sigma). For experiments using atropine, α-conotoxin or mecamy-lamine (Sigma), we added cholinergic antagonists to macrophage culture 30min before endotoxin stimulation. We collected conditioned culture media 24h after induction to measure extracellular HMGB1. The NF-κB pathway wasanalyzed by using the luciferase reporter pNF-κB-Luc (Clontech) following themanufacturer instructions. Briefly, we transfected cells 24 h before treatmentwith FuGENE 6 transfection reagent (Roche). After 3 h of treatment, we ana-lyzed luciferase activity using a standard Luciferase Assay System (Promega).
Figure 6 The ‘nicotinic anti-inflammatory pathway.’ During endotoxemia orsepsis, endotoxin, TNF-α and other stimuli converge in the activation of theNF-κB and induce HMGB1 translocation to cytoplasm and subsequentlyextracellular release. Extracellular HMGB1 functions as a proinflammatorycytokine that leads to lethal systemic inflammation. Acetylcholine andselective nicotinic agonists inhibit the NF-κB pathway through a specificnicotinic anti-inflammatory pathway mediated by the α7nAChR. Thispathway suppresses HMGB1 release induced by different inflammatoryscenarios including endotoxin- or TNF-α–induced stimulation ofmacrophages. Specific pharmacological stimulation of the α7nAChR inmacrophages might be a promising clinical approach to treat establishedsepsis and other HMGB1-related inflammatory disorders.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

A R T I C L E S
NATURE MEDICINE VOLUME 10 | NUMBER 11 | NOVEMBER 2004 1221
Electrophoretic mobility-shift assays were performed using Nushift NF-κB p65kit according to manufacturer’s instructions (Activemotif). We performedsupershift assays by incubating macrophage nuclear protein with anti-p65 NF-κB rabbit polyclonal antibody for 30 min before the incubation with the radio-labeled probe.
RNase protection assay. RNase protection assay was performed as previouslypublished43. Briefly, we extracted total RNA from cultured cells using RNAzol Baccording to manufacturer’s instructions (Tel-test). We verified the integrity ofthe RNA by electrophoresis on 1.2% agarose/17% formaldehyde gels. The levelsof HMGB1 were measured by using an RNase protection assay kit fromPharmingen following the manufacturer’s instructions. We labeled the anti-sense probe with [α-32P]-UTP (800 Ci/mmol; Amersham Pharmacia; 1 Ci = 37GBq) by using T7 RNA polymerase. Mouse HMGB1 probe was transcribed invitro from HMGB1 DNA fragment made by PCR using mouse HMGB1primers (sense: 5′-CCGAATTCGCTTCTGTCAACTTCTCAGAGTTTTCC-3′,antisense: 5′-GCGTAATACGACTCACTATAGGGCGAGGATCC-3′ containinga T7 promoter). This probe gave a 201-nt protection band after RNase diges-tion. We performed the RNase protection assay using an Ambion RNase protec-tion assay kit, according to manufacturer’s instructions.
Note: Supplementary information is available on the Nature Medicine website.
ACKNOWLEDGMENTSThe authors are grateful for the suggestions of J. Li, R. Wagner, P. Wang, L. Mantell,J. Peña and H. Yang. This research was supported by the Faculty Awards Program ofthe North Shore Research Institute, the North Shore-LIJ GCRC, NIGMS and DARPA.
COMPETING INTERESTS STATEMENTThe authors declare competing financial interests (see the Nature Medicine webwsitefor details).
Received 21 June; accepted 28 September 2004Published online at http://www.nature.com/naturemedicine/
1. Sands, K.E. et al. Epidemiology of sepsis syndrome in 8 academic medical centers.JAMA 278, 234–240 (1997).
2. Angus, D.C. et al. Quality-adjusted survival in the first year after the acute respiratorydistress syndrome. Am. J. Respir. Crit. Care Med. 163, 1389–1394 (2001).
3. Marshall, J.C. Inflammation, coagulopathy, and the pathogenesis of multiple organdysfunction syndrome. Crit. Care Med. 29, S99–S106 (2001).
4. Friedman, G., Silva, E. & Vincent, J.L. Has the mortality of septic shock changed withtime. Crit. Care Med. 26, 2078–2086 (1998).
5. Czura, C.J. et al. HMGB1 in the immunology of sepsis (not septic shock) and arthritis.Adv. Immunol. 84, 181–200 (2004).
6. Tracey, K.J. et al. Anti-cachectin/tnf monoclonal antibodies prevent septic shock dur-ing lethal bacteraemia. Nature 330, 662–664 (1987).
7. Dinarello, CA. The interleukin-1 family: 10 years of discovery. FASEB J. 8, 1314–1325(1994).
8. Dinarello, C.A. Therapeutic strategies to reduce IL-1 activity in treating local and sys-temic inflammation. Curr. Opin. Pharmacol. 4, 378–385 (2004).
9. Riedemann, N.C., Guo, R.F. & Ward, P.A. Novel strategies for the treatment of sepsis.Nat. Med. 9,517–524 (2003).
10. Scaffidi, P., Misteli, T. & Bianchi, M.E. Release of chromatin protein hmgb1 bynecrotic cells triggers inflammation. Nature 418, 191–195 (2002).
11. Abraham, E. et al. Double-blind randomised controlled trial of monoclonal antibody tohuman tumour necrosis factor in treatment of septic shock. Norasept ii study group.Lancet 351, 929–933 (1998).
12. Abraham, E., et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) insevere sepsis and early septic shock, A randomized, double-blind, placebo-controlled,multicenter phase iii trial with 1,342 patients. Crit. Care Med. 29, 503–510 (2001).
13. Fisher, C.J. et al. Recombinant human interleukin 1 receptor antagonist in the treat-ment of patients with sepsis syndrome. Results from a randomized, double-blind,
placebo-controlled trial. Phase iii rhil-1ra sepsis syndrome study group. JAMA 271,1836–1843 (1994).
14. Eskandari, M.K. et al. Anti-tumor necrosis factor antibody therapy fails to preventlethality after cecal ligation and puncture or endotoxemia. J. Immunol. 148,2724–2730 (1992).
15. Wang, H. et al. Hmg-1 as a late mediator of endotoxin lethality in mice. Science 285,248–251 (1999).
16. Andersson, U., et al. High mobility group 1 protein (hmg-1) stimulates proinflamma-tory cytokine synthesis in human monocytes. J. Exp. Med. 192, 565–570 (2000).
17. Ulloa, L. et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsisand systemic inflammation. Proc. Natl. Acad. Sci. USA 99, 12351–12356 (2002).
18. Bustin, M. At the crossroads of necrosis and apoptosis, signaling to multiple cellulartargets by HMGB1.Sci STKE 151 (2002).
19. Yang, H. et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. USA 101, 296–301 (2004).
20. Bonaldi, T. et al. (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 toredirect it towards secretion. EMBO J. 22, 5551–5560.
21. Li, J. et al. Structural basis for the proinflammatory cytokine activity of high mobilitygroup box 1. Mol. Med. 9, 37–45 (2003).
22. Borovikova, L.V. et al. Vagus nerve stimulation attenuates the systemic inflammatoryresponse to endotoxin. Nature 405, 458–462 (2000).
23. Bernik, T.R. et al. Pharmacological stimulation of the cholinergic antiinflammatorypathway. J. Exp. Med. 195, 781–788 (2002).
24. Tracey, K.J. The inflammatory reflex. Nature 420, 853–859 (2002).25. Wang, H. et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of
inflammation. Nature 421, 384–388 (2003).26. Rendon-Mitchell, B. et al. IFN-gamma induces high mobility group box 1 protein
release partly through a TNF-dependent mechanism. J. Immunol. 170, 3890–3897(2003).
27. Lee, J.C. et al. A protein kinase involved in the regulation of inflammatory cytokinebiosynthesis. Nature 372, 739–746 (1994).
28. Derijard, B. et al. Independent human map-kinase signal transduction pathwaysdefined by mek and mkk isoforms. Science 267, 682–685 (1995).
29. Baeuerle, P.A. & Henkel, T. Function and activation of NF-κB in the immune system.Annu. Rev. Immunol. 12, 141–179 (1994).
30. Li, Q. & Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2,725–734 (2002).
31. Wang, H., Czura, C. & Tracey, K.J. Lipid unites disparate syndromes of sepsis. Nat.Med. 10, 124–125 (2004).
32. Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev.Neurosci. 3, 102–114 (2002).
33. Wess, J. Muscarinic acetylcholine receptor knockout mice, novel phenotypes and clin-ical implications. Annu. Rev. Pharmacol. Toxicol. 44, 423–450 (2004).
34. Unwin, N. Acetylcholine receptor channel imaged in the open state. Nature 373,37–43 (1995).
35. Hogg, R.C., Raggenbass, M. & Bertrand, D. Nicotinic acetylcholine receptors, fromstructure to brain function. Rev. Physiol. Biochem. Pharmacol. 147, 1–46 (2003).
36. Ando, Y. Transdermal nicotine for ulcerative colitis. Ann. Intern. Med. 127, 491–492(1997).
37. Guarini, S. et al. Efferent vagal fibre stimulation blunts nuclear factor-κB activationand protects against hypovolemic hemorrhagic shock. Circulation 107, 1189–1194(2003).
38. Ulloa, L. Batliwalla, F.M., Andersson, U., Gregersen, P.K. & Tracey, K.J. High mobilitygroup box chromosomal protein 1 as a nuclear protein, cytokine, and potential thera-peutic target in arthritis. Arthritis Rheum. 48, 876–881 (2003).
39. Andersson, U. & Erlandsson-Harris, H. Hmgb1 is a potent trigger of arthritis. J. Intern. Med. 255, 344–350 (2004).
40. Jick, H. & Walker, A.M. Cigarette smoking and ulcerative colitis. N. Engl. J. Med. 308,261–263 (1983).
41. Rampton, D.S. Smoking and ulcerative colitis. Lancet 1,168 (1984).42. Martin, T.R. MIF mediation of sepsis. Nat. Med. 6, 140–141 (2000).43. Calandra, T. et al. Protection from septic shock by neutralization of macrophage migra-
tion inhibitory factor. Nat. Med. 6, 164–170 (2000).44. Yan, J. et al. Therapeutic effects of lysophosphatidylcholine in experimental sepsis.
Nat. Med. 10, 161–167 (2004).45. Ulloa, L., Diaz-Nido, J. & Avila, J. Depletion of casein kinase II by antisense oligonu-
cleotide prevents neuritogenesis in neuroblastoma cells. EMBO J. 12,1633–1640(1993).
46. Ulloa, L. et al. Inhibition of transforming growth factor-beta/SMAD signaling by theinterferon-gamma/STAT pathway. Nature 397, 710–713 (1999).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
Copyright © 2022 FDOKUMEN




















