
Characterization of spliced leader genes of Trypanosoma ( Megatrypanum) theileri: phylogeographical...
12
Characterization of spliced leader genes of Trypanosoma (Megatrypanum) theileri : phylogeographical analysis of Brazilian isolates from cattle supports spatial clustering of genotypes and parity with ribosomal markers A. C. RODRIGUES 1 , H. A. GARCIA 1 , J. S. BATISTA 2 , A. H. H. MINERVINO 3 , G. GO ´ ES-CAVALCANTE 4 , F. MAIA DA SILVA 1 , R. C. FERREIRA 1 , M. CAMPANER 1 , F. PAIVA 5 and M. M. G. TEIXEIRA 1 * 1 Departamento de Parasitologia, Universidade de Sa ˜o Paulo, Sa ˜o Paulo, SP, 05508-900, Brasil 2 Departamento de Cie ˆncias Animais, Laborato ´rio de Patologia Veterina ´ria, Universidade Federal Rural do Semi-A ´ rido, RN, 59625-900, Brasil 3 Faculdade de Medicina Veterina ´ria e Zootecnia, Universidade de Sa ˜o Paulo, SP, 05508-900, Brasil 4 Central de Diagno ´stico Veterina ´rio, Universidade Federal do Para ´ , PA, 66080-000, Brasil 5 Departamento de Parasitologia Veterina ´ria, Universidade Federal do Mato Grosso do Sul, MS, 79070-900, Brasil (Received 20 May 2009; revised 30 June 2009; accepted 1 July 2009; first published online 21 September 2009) SUMMARY Trypanosoma (Megatrypanum) theileri from cattle and trypanosomes of other artiodactyls form a clade of closely related species in analyses using ribosomal sequences. Analysis of polymorphic sequences of a larger number of trypanosomes from broader geographical origins is required to evaluate the clustering of isolates as suggested by previous studies. Here, we determined the sequences of the spliced leader (SL) genes of 21 isolates from cattle and 2 from water buffalo from distant regions of Brazil. Analysis of SL gene repeats revealed that the 5S rRNA gene is inserted within the intergenic region. Phylogeographical patterns inferred using SL sequences showed at least 5 major genotypes of T. theileri distributed in 2 strongly divergent lineages. Lineage TthI comprises genotypes IA and IB from buffalo and cattle, respectively, from the Southeast and Central regions, whereas genotype IC is restricted to cattle from the Southern region. Lineage TthII includes cattle genotypes IIA, which is restricted to the North and Northeast, and IIB, found in the Centre, West, North and Northeast. PCR-RFLP of SL genes revealed valuable markers for genotyping T. theileri. The results of this study emphasize the genetic complexity and corroborate the geographical structuring of T. theileri genotypes found in cattle. Key words: Trypanosoma theileri, spliced leader gene, ITS rDNA, phylogeny, evolution, phylogeography, bovids, populational structure. INTRODUCTION Trypanosoma (Megatrypanum) theileri is a cosmo- politan parasite of cattle with a high prevalence on every continent except Antarctica. Tabanidae (Diptera) are thought to be the most important vectors of T. theileri; the parasites develop in their digestive tract and are then transmitted by a con- taminative route (Hoare, 1972 ; Bose and Heister, 1993). This species is not considered pathogenic, despite the fact that it induces chronic infections and is a potential factor in illness when associated with concurrent diseases (Schafler, 1979 ; Doherty et al. 1993 ; Seifi, 1995). There are still many unclear aspects of host and parasite interactions, including immune response and evasion strategies, and the multiplication of T. theileri in mammalian hosts. Morphology is the traditional taxonomic criterion for the classification of T. theileri and allied species, which comprise trypanosomes that share large blood trypomastigotes, restricted mammalian hosts, world- wide distribution, lack of pathogenicity and con- taminative transmission by tabanid or hippoboscid flies (Hoare, 1972 ; Wells, 1976). We demonstrated through phylogenetic analysis that T. theileri of cattle (Bos taurus) from distinct regions of America (Brazil and USA), Europe (Scotland and Germany) and Asia (Japan) were highly related phylogenetically and tightly clustered into the T. theileri clade, a homogeneous and strongly supported monophyletic assemblage exclusive to artiodactyls whose position is well resolved in the genus Trypanosoma (Rodrigues et al. 2006). T. theileri from cattle grouped together with T. theileri-like trypanosomes from water buffalo * Corresponding author : Departamento de Parasitologia, Instituto de Cie ˆncias Biome ´dicas, Universidade de Sa ˜o Paulo, Sa ˜o Paulo, SP, 05508-900, Brasil. Tel : +55 11 3091 7268. Fax: +55 11 3091 7417. E-mail : mmgteix@icb. usp.br 111 Parasitology (2010), 137, 111–122. f Cambridge University Press 2009 doi:10.1017/S0031182009991053 Printed in the United Kingdom
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Characterization of spliced leader genes of Trypanosoma ( Megatrypanum) theileri: phylogeographical...

Characterization of spliced leader genes of Trypanosoma(Megatrypanum) theileri : phylogeographical analysis ofBrazilian isolates from cattle supports spatial clustering
of genotypes and parity with ribosomal markers
A. C. RODRIGUES1, H. A. GARCIA1, J. S. BATISTA2, A. H. H. MINERVINO3,
G. GOES-CAVALCANTE4, F. MAIA DA SILVA1, R. C. FERREIRA1, M. CAMPANER1,
F. PAIVA5 and M. M. G. TEIXEIRA1*
1Departamento de Parasitologia, Universidade de Sao Paulo, Sao Paulo, SP, 05508-900, Brasil2Departamento de Ciencias Animais, Laboratorio de Patologia Veterinaria, Universidade Federal Rural do Semi-Arido,RN, 59625-900, Brasil3Faculdade de Medicina Veterinaria e Zootecnia, Universidade de Sao Paulo, SP, 05508-900, Brasil4Central de Diagnostico Veterinario, Universidade Federal do Para, PA, 66080-000, Brasil5Departamento de Parasitologia Veterinaria, Universidade Federal do Mato Grosso do Sul, MS, 79070-900, Brasil
(Received 20 May 2009; revised 30 June 2009; accepted 1 July 2009; first published online 21 September 2009)
SUMMARY
Trypanosoma (Megatrypanum) theileri from cattle and trypanosomes of other artiodactyls form a clade of closely related
species in analyses using ribosomal sequences. Analysis of polymorphic sequences of a larger number of trypanosomes from
broader geographical origins is required to evaluate the clustering of isolates as suggested by previous studies. Here, we
determined the sequences of the spliced leader (SL) genes of 21 isolates from cattle and 2 from water buffalo from distant
regions of Brazil. Analysis of SL gene repeats revealed that the 5S rRNA gene is inserted within the intergenic region.
Phylogeographical patterns inferred using SL sequences showed at least 5 major genotypes of T. theileri distributed in
2 strongly divergent lineages. Lineage TthI comprises genotypes IA and IB from buffalo and cattle, respectively, from the
Southeast and Central regions, whereas genotype IC is restricted to cattle from the Southern region. Lineage TthII
includes cattle genotypes IIA, which is restricted to the North and Northeast, and IIB, found in the Centre, West, North
and Northeast. PCR-RFLP of SL genes revealed valuable markers for genotyping T. theileri. The results of this study
emphasize the genetic complexity and corroborate the geographical structuring of T. theileri genotypes found in cattle.
Key words: Trypanosoma theileri, spliced leader gene, ITS rDNA, phylogeny, evolution, phylogeography, bovids,
populational structure.
INTRODUCTION
Trypanosoma (Megatrypanum) theileri is a cosmo-
politan parasite of cattle with a high prevalence
on every continent except Antarctica. Tabanidae
(Diptera) are thought to be the most important
vectors of T. theileri; the parasites develop in their
digestive tract and are then transmitted by a con-
taminative route (Hoare, 1972; Bose and Heister,
1993). This species is not considered pathogenic,
despite the fact that it induces chronic infections and
is a potential factor in illness when associated with
concurrent diseases (Schafler, 1979; Doherty et al.
1993; Seifi, 1995). There are still many unclear
aspects of host and parasite interactions, including
immune response and evasion strategies, and the
multiplication of T. theileri in mammalian hosts.
Morphology is the traditional taxonomic criterion
for the classification of T. theileri and allied species,
which comprise trypanosomes that share large blood
trypomastigotes, restricted mammalian hosts, world-
wide distribution, lack of pathogenicity and con-
taminative transmission by tabanid or hippoboscid
flies (Hoare, 1972; Wells, 1976). We demonstrated
through phylogenetic analysis thatT. theileri of cattle
(Bos taurus) from distinct regions of America (Brazil
and USA), Europe (Scotland and Germany) and
Asia (Japan) were highly related phylogenetically
and tightly clustered into the T. theileri clade, a
homogeneous and strongly supported monophyletic
assemblage exclusive to artiodactyls whose position
is well resolved in the genus Trypanosoma (Rodrigues
et al. 2006). T. theileri from cattle grouped together
withT. theileri-like trypanosomes fromwater buffalo
* Corresponding author: Departamento de Parasitologia,Instituto de Ciencias Biomedicas, Universidade de SaoPaulo, Sao Paulo, SP, 05508-900, Brasil. Tel:+55 11 30917268. Fax: +55 11 3091 7417. E-mail : [email protected]
111
Parasitology (2010), 137, 111–122. f Cambridge University Press 2009
doi:10.1017/S0031182009991053 Printed in the United Kingdom

(Brazil), deer (Europe and Asia) and antelopes
(Africa), forming the T. theileri clade in phylogenies
inferred using SSU rRNA and gGAPDH genes. In
all analyses, this clade was positioned well apart from
species infecting orders other than Artiodactyla,
which were morphologically classified as T. (Mega-
trypanum) (Stevens et al. 1999; Rodrigues et al. 2006;
Hatama et al. 2007; Hamilton et al. 2007, 2009).
Discovery of a strongly supported monophyletic
assemblage formed by very closely phylogenetically
related and morphologically similar trypanosomes,
all from ruminant artiodactyls but from distinct
species and with widespread geographical distri-
bution (Rodrigues et al. 2006), allowed validation of
the subgenus T. (Megatrypanum) with T. theileri
as its type species, as established by Hoare (1972).
Experimental infections and positioning of tabanid-
infecting trypanosomes together with T. theileri in
phylogenetic analysis corroborated these flies as
vectors of T. theileri (Bose et al. 1987; Wells, 1976;
Rodrigues et al. 2006).
Phylogenies based on SSU and ITS1 rDNA se-
quences revealed a complex branching pattern within
the clade T. theileri, with separation of the isolates
into 5 lineages: (A) represented by genotypes found
in Brazilian water buffalo; (B) and (C) consisting
of 2 divergent genotypes found in Brazilian cattle
isolates; and (D) and (E), genotypes assigned to
European cattle and deer isolates, respectively
(Rodrigues et al. 2006). Recently, African T. theileri
trypanosomes of ‘antelopes’ were compared using
V7-V8 rDNA, and 1 isolate from sitatunga was
assigned to a new lineage F, whereas isolates from
duikers were assigned to lineages (C) or (E) despite
also showing unique genotypes (Hamilton et al.
2009). Description of species allied to T. theileri was
based on host restriction, which was experimentally
demonstrated for trypanosomes infecting cattle, sheep
and goats (Hoare, 1972; Wells, 1976). However, the
existence of separate species and/or lineages restric-
ted to host species and/or geographical origin has
not been corroborated by SSU rRNA phylogenies,
which support the polyphyly of cattle isolates and
close relationships among isolates from distinct host
species from distinct continents (Rodrigues et al.
2003, 2006; Hamilton et al. 2007, 2009).
Analysis of genes that are more polymorphic than
SSU rRNA of a larger number of trypanosomes of
the T. theileri clade, from broader host species and
geographical origins, can help to evaluate genotype
diversity and lineage segregation associated with host
and/or geographical origin. Previous analysis using
the more variable ITS rDNA sequences already
distinguished cryptic genotypes that had remained
undiscovered using conserved SSU rDNA genes in
T. theileri (Rodrigues et al. 2006),T. rangeli (Maia da
Silva et al. 2007) and T. vivax (Cortez et al. 2006).
Although the full phylogenetic range is unknown,
trans-splicing in Euglenozoa has been described as a
mechanism for the generation of mature messenger
RNAs (mRNAs): the 5k ends of precursor mRNAs
are replaced by a short spliced leader (SL) exon from
a small SL RNA. SL RNA repeats are organized as
large multicopy tandem arrays and have been used as
taxonomic markers for trypanosomatids of several
genera (Fernandes et al. 1997; Serrano et al. 1999a, b ;
Podlipaev et al. 2004; Westenberger et al. 2004;
Thomas et al. 2005; Maslov et al. 2007). Moreover,
SL sequences have been valuable for genotyping and
analysis of genetic relatedness and populational
structure of T. cruzi (Souto et al. 1996; Fernandes
et al. 2001; Brisse et al. 2001; Westenberger et al.
2006; Herrera et al. 2007; Mejıa-Jaramillo et al.
2009; Falla et al. 2009), T. rangeli (Urrea et al. 2005;
Maia da Silva et al. 2007, 2009) and T. vivax
(Ventura et al. 2001) isolates. The potential for using
the SL gene as a marker for phylogenetic analysis of
the genus Trypanosoma was assessed in a previous
study in which comparison of 27 trypanosomes re-
vealed the SL intergenic region to be far too variable
for informative comparison of any but the most
closely related trypanosomes (Gibson et al. 2000).
At present, available sequences (GenBank) of
whole repeats of the SL genes of trypanosomes from
mammals are restricted to T. cruzi, T. rangeli,
T. brucei, T. congolense, T. vivax, T. conorhini and
T. lewisi, while data from other species are limited to
the small transcript regions, which are only available
for European T. theileri from cattle and deer, and
consist of y200 bp from a whole SL repeat of
y900 bp (Gibson et al. 2000). Analysis of entire SL
repeats of T. theileri and related species are essential
to elucidate the organization of the SL repeats, and
intergenic regions of the SL gene are valuable for the
assessment of the genetic variability of trypanosomes
from different hosts and geographical regions. Here,
we characterized whole SL repeats of Brazilian iso-
lates of T. theileri, including 21 isolates from cattle
and 2 from water buffalo, from a wide range of
geographical origins, with the aim of investigating:
(a) genotype diversity; (b) phylogeographical pat-
terns; and (c) the congruence of phylogenetic analy-
ses and parity of taxonomic markers based on SL and
ribosomal genes.
MATERIALS AND METHODS
Origin and growth of T. theileri isolates
In this study, we characterized SL genes from a total
of 23 cultures of Brazilian T. theileri isolates.
Thirteen new isolates in this study were obtained
from cattle : 8 were from cross-bred dairy cattle from
the Northeast states of Paraıba (PB) and Rio Grande
do Norte (RN), 3 were from zebuine beef cattle, and
2 were from cross-bred dairy cattle from the state of
Para (PA). New isolates were obtained by haemo-
culture as previously described (Rodrigues et al.
A. C. Rodrigues and others 112
2003). We also included in this study 8 T. theileri
isolates from a previous study (Rodrigues et al.
2003, 2006), which were obtained from taurine beef
cattle from the Southern (states of Rio Grande do
Sul – RS, and Parana – PR) and Southeastern (Sao
Paulo – SP) regions, cross-bred beef cattle from the
Northwest (Rondonia – RO) and zebuine beef cattle
from the Central (Mato Grosso do Sul – MS) region.
We also characterized SL genes from 2 isolates from
water buffalo (Bubalus bubalis) from MS (Pantanal)
and SP (Vale do Ribeira), also from our previous
studies (Rodrigues et al. 2003, 2006) (Fig. 1;
Table 1). Additional cattle and buffalo isolates were
tested by genotyping methods based on SL and ITS
rDNA sequences (Table 1).
PCR amplification of SL and ribosomal sequences
Trypanosome DNA used as template for PCR am-
plification was obtained from cultured trypanosomes
by the classical method of phenol-chloroform ex-
traction. The location of the oligonucleotides em-
ployed as primers for PCR amplification of the SL
gene is depicted in Fig. 2C. Amplification of whole
SL gene repeats (y900 bp) was performed using
LSL1 (TTCTGTACTTCATGGTATG) and
LSL2 (CCAATGAAGTACAGAAACTG) primers
with 2.0 mM of each primer in 50 ml reaction mixtures
containing 50 ng of DNA, 200 mM of each dNTP
and 2.5 units of Taq DNA polymerase. PCR
amplification of partial SL sequences (y400 bp),
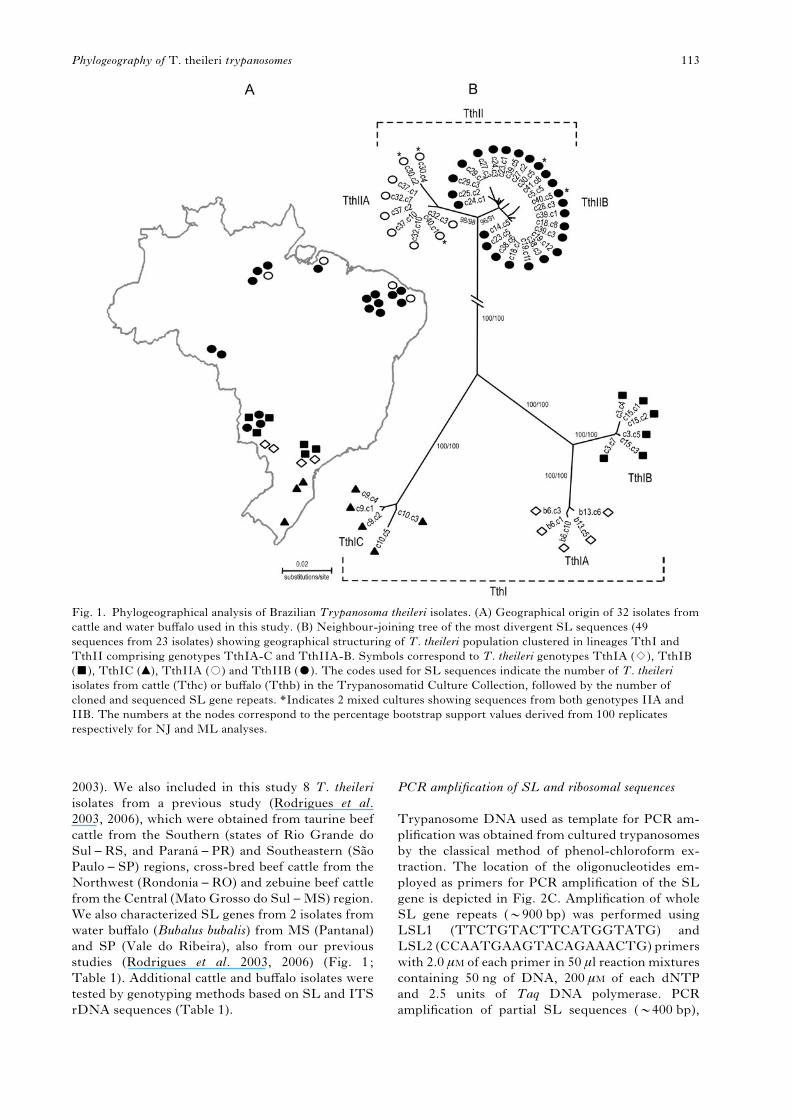
Fig. 1. Phylogeographical analysis of Brazilian Trypanosoma theileri isolates. (A) Geographical origin of 32 isolates from
cattle and water buffalo used in this study. (B) Neighbour-joining tree of the most divergent SL sequences (49
sequences from 23 isolates) showing geographical structuring of T. theileri population clustered in lineages TthI and
TthII comprising genotypes TthIA-C and TthIIA-B. Symbols correspond to T. theileri genotypes TthIA (1), TthIB
(&), TthIC (m), TthIIA (#) and TthIIB ($). The codes used for SL sequences indicate the number of T. theileri
isolates from cattle (Tthc) or buffalo (Tthb) in the Trypanosomatid Culture Collection, followed by the number of
cloned and sequenced SL gene repeats. *Indicates 2 mixed cultures showing sequences from both genotypes IIA and
IIB. The numbers at the nodes correspond to the percentage bootstrap support values derived from 100 replicates
respectively for NJ and ML analyses.
Phylogeography of T. theileri trypanosomes 113

Table 1. Trypanosoma theileri trypanosomes used in this study and genotypes (TthIA-C and TthIIA-B) defined based on SL and ribosomal (SSU rDNA and
ITS rDNA) sequences
T. theileriisolatesa TryCC
Hostorigin Geographical origin
Genotypingb based onribosomal and SL genes
V7V8rDNA SL
PCR-RFLP GenBank Accession number
ITSrDNA SL SL ITS SSU
Tthb4 162 buffalo Southeast (SP, Registro) A — IA IA Nd AY773701 AY773675Tthb6 165 buffalo Southeast (SP, Jacupiranga) A IA IA IA GQ162117-GQ162119* AY773699 AY773676Tthb13 — buffalo Central (MS, Dourados) A IA IA IA GQ162120-GQ162121* AY773703 AY773678Tthb12 — buffalo Central (MS, Dourados) A — IA IA Nd AY773702 AY773677Tthc22 — cattle Southeast (SP, Aracatuba) B2 — IB IB Nd Nd AY773688Tthc2 171 cattle Southeast (SP, Jacupiranga) B2 — IB IB Nd AY773707 AY773679Tthc3 161 cattle Southeast (SP, Eldorado) B2 IB IB IB GQ162125-GQ162127* AY773698 AY773681Tthc15 301 cattle Central (MS, Miranda) B2 IB IB IB GQ162122-GQ162124* Nd AY773686Tthc16 302 cattle Central (MS, Miranda) B2 — IB IB Nd AY773708 AY773687Tthc17 296 cattle Central (MS, Miranda) B2 — IB IB Nd Nd AY773693Tthc8 — cattle Southern (RS, Porto Alegre) B1 — IC IC Nd AY773704 AY773682Tthc9 — cattle Southern (PR, Londrina) B1 IC IC IC GQ162130-GQ162132* AY773705 AY773683Tthc10 — cattle Southern (PR, Londrina) B1 IC IC IC GQ162128-GQ162129* AY773706 AY773684
Tthc30 1460 cattle Northeast (RN, Mossoro) C IIA IIA IIA GQ162133-GQ162134* GQ176146* GQ176157*Tthc32 1462 cattle Northeast (RN, Mossoro) C IIA IIA IIA GQ162135-GQ162137* GQ176147* GQ176158*Tthc37 1787 cattle North (PA, Castanhal) C IIA IIA IIA GQ162138-GQ162140* GQ176148* GQ176159*Tthc5 — cattle Central (MS, Dourados) C IIB IIB IIB GQ162143* AY773711 AY773689Tthc12 298 cattle Central (MS, Miranda) C — IIB IIB Nd AY773712 AY773690Tthc14 299 cattle Central (MS, Miranda) C IIB IIB IIB GQ162144* AY773709 AY773692Tthc18 359 cattle West (RO, Monte Negro) C IIB IIB IIB GQ162145-GQ162146* AY773713 AY773694Tthc19 360 cattle West (RO, Monte Negro) C IIB IIB IIB GQ162147-GQ162148* AY773710 AY773695Tthc23 796 cattle Northeast (PB, Sao Mamede) — IIB IIB IIB GQ162149-GQ162151* Nd NdTthc24 797 cattle Northeast (PB, Sao Mamede) C IIB IIB IIB GQ162152-GQ162153* Nd GQ176152*Tthc25 798 cattle Northeast (PB, Patos) C IIB IIB IIB GQ162154* Nd GQ176154*Tthc26 799 cattle Northeast (PB, Catole-Rocha) C — IIB IIB Nd Nd GQ176153*Tthc27 1091 cattle Northeast (PB, Catole-Rocha) — IIB IIB IIB GQ162158-GQ162159* Nd NdTthc28 1458 cattle Northeast (RN, Mossoro) C IIB IIB IIB GQ162155* Nd GQ176155*Tthc29 1459 cattle Northeast (RN, Mossoro) C IIB IIB IIB GQ162156-GQ162157* Nd GQ176156*Tthc38 1788 cattle North (PA, Castanhal) C IIB IIB IIB GQ162141-GQ162142* GQ176149* GQ176160*Tthc39 — cattle North (PA, Santarem) — IIB IIB IIB GQ162160-GQ162162* GQ176150* NdTthc40 — cattle North (PA, Santarem) — IIB IIB IIB GQ162163-GQ162164* Nd NdTthc41 — cattle North (PA, Santarem) — IIB IIB IIB GQ162165-GQ162166* GQ176151* Nd
TryCC, code number at Trypanosomatid Culture Collection from the Department of Parasitology, University of Sao Paulo, ICBII, Brazil.a, Tthc, isolates from cattle; Tthb, isolates from water buffalo.b, Genotypes of T. theileri lineages TthI (IA-C) and TthII (IIA-B).Nd, sequences not determined, isolates were genotyped using one or more sequences from the different molecular markers used in this study: SL or V7V8 or ITSrDNA genes.* Sequences determined in the present study.
A.C.Rodrigu
esandoth
ers114

corresponding to the transcript region of SL plus
y270 bp of a contiguous intergenic region between
the intron and the 5S rDNA sequences, was carried
out using LSL1 and TthSLR (GGCAAAA(A/G)
T(G/C)ACCAA(A/C)AC) primers. PCR conditions
were the same for both reactions and consisted of 30
cycles as follows: 1 min at 94 xC, 2 min at 50 xC and
2 min at 72 xC (with an initial cycle of 3 min at 94 xC
and a final cycle of 10 min at 72 xC). PCR products
were eletrophoresed in a 2% agarose gel and stained
with ethidium bromide. PCR-amplification of V7-
V8 SSU rDNA and ITS rDNA of the newT. theileri
isolates were performed as previously described
(Rodrigues et al. 2006).
Sequencing and data analyses
PCR-amplified fragments of whole SL repeats from
cattle and buffalo isolates were purified (Spin-X,
Costar) from agarose gels and cloned, and 3–7 clones
from each isolate were sequenced using the internal
primersTthSLR (GGCAAAA(A/G)T(G/C)ACCAA
(A/C)AC) and TthSLF (GGGT(G/T)TTGGT
(G/C)AC(G/T)TTTTGCC). The sequences ob-
tained were aligned with sequences from GenBank
using ClustalX (Thompson et al. 1997). The result-
ing alignments were manually refined. Poly(A) tails
of SL transcripts were removed from final SL gene
alignments. Sequences of V7-V8 SSU rDNA and
Fig. 2. (A) Alignment of SL transcript sequences from 5 main genotypes Trypanosoma theileri trypanosomes. The 39 nt
corresponding to the exon sequences are in bold type within a grey box. Within the intron sequences, the conserved
CUUCC motif is boxed, and the GG motifs demarcating stem–loop II and the Sm binding sites are underlined. Dots
indicate identical nucleotides. (B) SL RNA structure of isolate Tthb6 from water buffalo with the splice donor site
indicated by an arrow. (C) Schematic diagram of the SL gene showing the exon, intron and intergenic regions, and
annealing sequences for oligonucleotides used as primers for PCRs. (D) Alignment of 5S rDNA sequences inserted
within the intergenic regions of SL repeats of T. theileri trypanosomes.
Phylogeography of T. theileri trypanosomes 115

ITS rDNAwere determined as previously described
(Rodrigues et al. 2006). Six alignments were created:
(1) whole SL (wSL) sequences; (2) exon and intron
(SL transcript) sequences; (3) SL transcript plus
intergenic sequence between intron and 5S rRNA;
(4) ITS1 rDNA sequences; (5) V7V8 SSU rDNA
sequences from 30 T. theileri trypanosomes from
distinct host species; and (6) concatenated alignment
generated by SL transcripts plus 5S and ITS1 rDNA
data sets. Accession numbers of the GenBank se-
quences, including those determined in this study,
are listed in Table 1.
Phylogenetic inferences were carried out by
the parsimony (P) and maximum likelihood (ML)
methods, and cluster analysis was performed using
neighbour-joining (NJ) distances. TheNJ trees were
estimated for alignments 1 and 3 using the Kimura 2
Parameter algorithm in Mega 4 software, and nodal
support was estimated with 500 bootstrap replicates.
Parsimony and bootstrap analyses were employed for
all alignments and carried out using PAUP* 4.0b10
(Swofford, 2002) with 100 replicates of a random
addition sequence followed by branch swapping
(RAS-TBR), as previously described (Ferreira et al.
2007). ML analyses were performed for alignments
1, 4, 5 and 6 using RAxML v.7.0.4 (Stamatakis,
2006), as previously shown (Ferreira et al. 2007).
Tree searches used the general time reversible (GTR)
model of nucleotide substitution with proportion
of invariable sites and gamma distribution, with 500
maximum parsimony starting trees. Model par-
ameters were estimated in RAxML over the duration
of the tree search and nodal support was estimated
with 300 bootstrap replicates. RNA structures were
predicted using the RNAdraw program (Matzura
and Wennborg, 1996).
Restriction analysis of PCR-amplified SL repeat and
ITS rDNA (PCR-RFLP)
Polymorphisms on the aligned whole SL gene se-
quences of T. theileri trypanosomes were investi-
gated using the software Webcutter 2.0 (Heiman,
1997) to detect restriction enzyme sites that gener-
ated DNA fragments of different length according
to clades/lineages. For restriction analysis, amplified
DNA corresponding to whole repeats of SL and
entire ITS rDNA genes were both digested with
BshI 1236 enzyme (Fermentas) ; the resulting DNA
fragments were analysed on 2.5% agarose gels stained
with ethidium bromide, as previously described
(Rodrigues et al. 2006).
RESULTS
Structural and sequence characterization of SL
repeats from T. theileri trypanosomes
Full-length SL unit repeats were PCR-amplified
from DNA of Brazilian T. theileri trypanosomes
from cattle (21 isolates) and water buffalo (2 isolates),
including isolates representative of lineages A, B
and C previously defined by ribosomal markers
(Rodrigues et al. 2006). SL repeats (y900 bp) of
similar lengths were observed for all T. theileri
trypanosomes in agarose gels, as was previously
shown for European T. theileri from cattle and re-
latedT. theileri-like trypanosomes fromdeer (Gibson
et al. 2000). However, sequences showed significant
length variability among previously defined ribo-
somal lineages (Rodrigues et al. 2006), enabling the
identification of the 5 genotypes defined in the
present study according to polymorphism on the
SL gene. The SL repeats of the various genotypes
ranged from 855 to 940 bp. SL repeats of y860 bp
were found for the genotypes TthIA/B, which cor-
respond to previously defined ribosomal lineages A
and B2; 927 bp for TthIC, formerly lineage B1;
855 bp for TthIIB, comprising isolates assigned to
ribosomal lineage C; and 940 bp for TthIIA isolates,
not included in our previous study using ribosomal
genes (Rodrigues et al. 2006) but also assigned to
ribosomal lineage C in the present study (Table 1).
We determined a total of 98 sequences of whole SL
genes, 3–5 from each of the 23 isolates examined, and
76 sequences were found to be unique. Comparison
of SL repeats revealed an identical exon (39 nucleo-
tides) shared by T. theileri from cattle and allied
species fromwater buffalo and fallow deer. However,
the intron sequences varied from 94 bp (TthI) to
98 bp (TthII). The intron and exon sequences
together account for 133 to 137 bp of the whole SL
transcripts of T. theileri lineages TthI and TthII,
respectively. T. theileri-like trypanosomes from deer
(Cervus dama) also had an SL intron of 94 bp
(Gibson et al. 2000). Sequence divergence in the
intergenic regions among different copies of SL re-
peats from the same isolate was due mostly to long
segments of variable numbers ofA andTnucleotides.
Simple sequence repeats such as AC3-8, GT3-8, CA3-5,
TG3-4 and three repeats each of TCG, TTC, CCA
and CAC were found in sequences from all isolates,
whereas long repeats, such as AAG>5, ACACCC>3,
CCCCT4 and GGGAG3 were exclusive to the TthII
lineage. The existence of various microsatellites in
the intergenic regions is also responsible for length
and sequence variability among the SL genes of other
trypanosomes (Gibson et al. 2000; Ventura et al.
2001; Maia da Silva et al. 2007, 2009).
Despite differences in length and sequence among
SL transcripts of T. theileri trypanosomes, isolates
from cattle, water buffalo and deer shared identical
secondary structures for the SL genes, with 3
stem–loop folded structures as reported forT. theileri
K127 and other trypanosomes (Gibson et al. 2000).
T. theileri from cattle and buffalo presented a second
stem–loop varying from 50 to 54 nucleotides and the
conserved motif GG(C/T)AAAUUUUGG of the
Sm binding site with C or T in the third position for
A. C. Rodrigues and others 116

TthI or TthII, respectively. The third stem–loop
exhibited a conserved UUCG motif and 21 nucleo-
tides for all T. theileri isolates. The secondary
structure of the SL transcript from T. theileri from
buffalo is represented in Fig. 2B.
Determination of whole sequences of SL repeats of
T. theileri trypanosomes revealed a copy of the 5S
ribosomal RNA (5S rRNA) gene inserted in the 3kend of the intergenic region of all isolates, in the same
orientation as the SL gene (Fig. 2D). Comparison
of 5S rDNA sequences of isolates from different
T. theileri genotypes revealed variable length of 121
(IA/B), 116 (IC) or 114 (IIA/B) bp. There was large
divergence in the 5S rDNA sequences between
lineages ThI and TthII. Identical 5S rDNA se-
quenceswereobservedforgenotypeswithintheTthII
contrasting to more divergent sequences from TthI
genotypes (Fig. 2D).
Phylogenetic analysis of SL sequences from
T. theileri isolates
Phylogenetic analysis of wSL (exon, intron and
intergenic region) sequences from trypanosomes
found in cultures from 21 isolates from cattle and 2
from buffalo corroborated data on length and se-
quence polymorphism of SL repeats, providing
evidence for 2 major lineages and 5 main genotypes
among Brazilian T. theileri trypanosomes. Although
different copies of the SL gene from a single isolate
might diverge in their intron and intergenic se-
quences, the sequenceswere similar enough to cluster
together individual repeats from the same isolate in
the same genotype, separated from the repeats from
isolates of other genotypes. According to the den-
drograms derived from wSL sequences, sequences
from all genotypes were nested in 2 major clades,
lineages TthI and TthII, separated by y33% di-
vergence (Fig. 1B). Dendrograms showing the same
topologies were constructed using the whole set of
76 unique wSL sequences from 23 isolates (data not
shown) or restricted to 49 sequences representing the
most divergent wSL sequences (Fig. 1B).
Lineage TthI includes 3 subclades: IA, exclus-
ively found in buffalo, and IB and IC, which
comprise genotypes restricted to cattle. Genotype IA
formed a very homogeneous clade (y0.4% diver-
gence) closest (y6.0%) to the genotype IB (y0.5%).
Genotypes IA and IB were found respectively in
buffalo and cattle from neighbouring regions in
the Central and Southeastern states of MS and SP.
Genotype IC harbours more heterogeneous se-
quences (y2.0% divergence) than cattle isolates from
the Southern region (states of RS and PR). Geno-
types IB and IC were separated by y26% diver-
gence, whereas IA diverged by y25% from IC.
The second assemblage within the cladeT. theileri,
lineage TthII, comprises 2 main genotypes found in
cattle. Lineage TthII showed lower overall internal
divergence (y5.0%) compared to TthI (y14%) but
more intra-genotype polymorphisms. One clade of
TthII contains sequences assigned to genotype IIA
(y4.0%), found in cattle from RN (Northeast) and
PA (North). Interestingly, sequences from primary
cultures (cattle isolates Tthc30 and Tthc40 from RN
and PA, respectively) were assigned to both IIA and
IIB, indicating the existence of mixed genotypes
(Fig. 1B). T. theileri of genotype IIB (y3.0%) were
from cattle from the Centre (MS), Northwest (RO),
North (PA) and Northeast (RN and PB). High intra-
genotype divergence in IIA and IIB suggests that
these genotypes may split into new subclades upon
analysis of more isolates from their regions of origin,
which are separated by large geographical distances.
Phylogenetic inferences using combined data set and
parity between taxonomic markers for T. theileri
lineages/genotypes based on SL and ITS1 rDNA
sequences
Phylogenetic analysis of wSL sequences confirmed 2
major lineages and resolved at least 5 main genotypes
(Fig. 1B). Parity analyses disclosed the same patterns
using wSL and ITS1 rDNA sequences (Fig. 3A,B).
Analyses using sequences more conserved than SL
repeats (5S rDNA and V7-V8 SSU rDNA) con-
firmed the 2 major lineages, but they were unable
to resolve the 5 main genotypes (data not shown).
Analysis restricted to conserved exon and intron
sequences (alignment 2) was unable to distinguish
TthIIA fromTthIIB (data not shown), but inclusion
of the whole (alignment 1, Fig. 1B) or partial inter-
genic sequence between the SL intron and 5S rRNA
(alignment 3, data not shown) resulted in dendro-
grams congruent with those from wSL sequences.
This finding dispensed the need of whole SL repeats
sequences demonstrating that partial sequences from
SL gene repeats, which are easily amplified and
sequenced, is enough for genotyping and phylogeo-
graphical analysis of T. theileri.
A combined data set of SL transcript plus 5S
and ITS1 rDNA sequences was employed to infer
phylogenetic trees aiming to corroborate the re-
lationships among T. theileri genotypes and to
improve support for all lineages/genotypes. All in-
ferences (P, ML and NJ) generated congruent
phylogenetic trees supporting both the 2 major
lineages and the 5 main genotypes. The phylogeo-
graphical pattern generated by the combined data set
was totally congruent with genotype relationships
and divergences based on independent sequences,
supporting the geographical structuring ofT. theileri
populations (Fig. 3C).
Parity of genotyping using PCR-RFLP of wSL and
ITS rDNA genes
Restriction patterns of PCR-amplified (PCR-RFLP)
wSL were evaluated for isolates representing all 5
Phylogeography of T. theileri trypanosomes 117

genotypes, with the aim of developing an easy tool for
genotyping T. theileri that dispenses with the need
for both established cultures and DNA sequencing.
Restriction patterns generated with BshI 1236 en-
zyme displayed 5 restriction profiles, each one
corresponding to one previously defined genotype.
The developed genotyping method separated even
the most closely related genotypes (Fig. 4A). Parity
analysis demonstrated that restriction analysis of
PCR-amplified ITS rDNA also displayed banding
patterns supporting the same 5 main genotypes de-
fined by the SL gene. However, genotypes within
TthII lineage were clearly distinguishable by SL
profiles, whereas they showed very similar patterns
of ITS rDNA PCR-RFLP (Fig. 4B). Both methods
proved to be useful for rapid genotyping ofT. theileri
and suitable for use with crude DNA templates from
easily obtained primary cultures (1–15 days), and
they are thus useful for field epidemiological surveys
of T. theileri populations.
DISCUSSION
Despite a broad geographical distribution around the
world and their ability to infect virtually all species
of domestic bovids (cattle, buffalo, goats, and sheep)
and wild species of cervids and antelopes, knowledge
about T. theileri and allied species is not sufficient
to make robust conclusions about the genetic varia-
bility and evolutionary relationships of these closely
Fig. 3. Phylogenetic trees inferred by maximum
parsimony showing total congruency among phylogenetic
relationships of Trypanosoma theileri isolates with parity
of all genotypes (TthIA-C and TthIIA-B) based on:
(A) whole SL sequences; (B) ITS1 rDNA sequences;
and (C) the combined data set of SL transcript, 5S
rDNA and ITS1 rDNA sequences. The numbers at the
nodes correspond to the percentage bootstrap support
values derived from 100 replicates for MP and ML
analyses.
Fig. 4. Agarose gels (2.5%) stained with ethidium
bromide (EtBr) showing DNA fragment profiles
resulting from restriction analyses of PCR-amplified
(PCR-RFLP) genes from Trypanosoma theileri isolates
selected to illustrate the 5 genotypes, TthIA-C and
TthIIA-B, described in this study. (A) PCR-RFLP of
whole SL gene repeats. (B) PCR-RFLP of ITS rDNA.
A. C. Rodrigues and others 118

related trypanosomes. Low genetic variability among
these trypanosomes, as revealed by conserved SSU
rRNA and gGAPDH genes in previous studies, is
insufficient to hypothesize about their distribution in
phylogenetic clades (lineages) (Rodrigues et al. 2006;
Hamilton et al. 2009). Data from these studies have
suggested that clades might not be strictly related
to either host species or geographical origin. How-
ever, data from zymodemes (Bose et al. 1993; Dirie
et al. 1990), RAPD patterns (Rodrigues et al. 2003),
and ribosomal sequences (Rodrigues et al. 2006;
Hamilton et al. 2007, 2009) have distinguished be-
tween trypanosomes from cattle, buffalo, cervids and
antelopes.
Understanding population structure and lineage
segregation within the clade T. theileri requires
genetic analysis of polymorphic genes of a larger
number of isolates from artiodactyls and vectors
from a broad geographical origin. Here, to further
evaluate genetic diversity, host association and geo-
graphical patterns within the clade T. theileri as
suggested in our previous studies (Rodrigues et al.
2006), and aiming for better-resolved intra-clade
phylogenetic relationships, we determined sequen-
ces of whole SL gene repeats. We characterized, for
the first time, whole SL repeats of T. theileri from
cattle (Bos taurus) and related trypanosomes from
water buffalo (Bubalus bubalis). Results showed that
the 5S rRNA gene is inserted within the intergenic
region of all isolates examined from cattle and
buffalo. This type of structural arrangement of 5S
rRNA in trypanosomes has thus far been reported
in SL repeats of T. rangeli, T. conorhini and T. vivax
(Aksoy et al. 1992; Roditi, 1992; Stevens et al. 1999;
Gibson et al. 2000; Ventura et al. 2001;Maia da Silva
et al. 2007). Contrasting to data from SSU rRNA
genes, divergence between 5S rDNA sequences was
found to be higher within the clade T. theileri com-
pared to lineages/genotypes of T. rangeli (Maia da
Silva et al. 2007) and T. cruzi (Westenberger et al.
2006). Although we also found the same organization
of 5S rRNA within the intergenic SL regions of
T. cyclops (data not shown), which is the species
closest to T. theileri in phylogenetic trees (Hamilton
et al. 2005, 2007), trypanosomes showing this
peculiar organization of 5S rRNA are not mono-
phyletic and were positioned in distant clades in the
phylogenetic tree of Trypanosoma. The nuclear 5S
rRNA genes are known to have moved in and out of
tandemly repeated eukaryotic gene families (rDNA,
SL and histones) during the evolution of fungus,
protist, nematode and arthropod species. To date,
these genes have been reported to be linked only to
the tandem repeats of SL genes in trypanosomatids
(Drouin and de Sa, 1995; Westenberger et al. 2004;
Thomas et al. 2005).
To assess the consistency of T. theileri lineages/
genotypes by comparing sequences showing differ-
ent evolutionary rates, we compared phylogenetic
analyses with SL and rDNA (SSU and ITS1) se-
quences. These genetic loci were analysed separately
as well as with a combined approach. The results
showed parity between phylogeographical patterns
based on SL and ITS1 rDNA sequences. Only
analyses including highly polymorphic sequences
from the intergenic region of SL and ITS1 rDNA
showed cryptic genotypes that were not found using
conserved SSU rDNA sequences (Rodrigues et al.
2006). T. theileri isolates showed relevant polymor-
phisms in the intergenic regions of SL gene repeats.
Phylogeographical analysis of 21T. theileri isolates
from Bos taurus from the North, South, West,
Southeast and Central regions of Brazil, plus 2 iso-
lates from Bubalus bubalis from the Southeast and
Central regions, was performed using phylogenetic
analysis of the SL gene. The phylogenies inferred
using either wSL (y900 bp) or partial SL repeat
(y400 bp) sequences were totally congruent, with a
total of 76 different sequences assigned to lineages
TthI and TthII, which comprise respectively 3 and
2 genotypes strongly supported in all analyses. TthI
comprises genotypes IA and IB from buffalo and
cattle, respectively, from the nearby Southeast and
Central regions, and IC, which is restricted to cattle
from the Southern region. TthII comprises geno-
types IIA, restricted to cattle from the North and
Northeast, and IIB, found in cattle from the Central,
West, North and Northeast regions. Besides the 5
main genotypes characterized in this study, analysis
of primary cultures from the North and Northeast
regions allowed the discovery of at least 3 sequences
that did not cluster with any of these genotypes,
indicating the existence of many more genotypes
(data not shown). Intra-array variation could affect
taxonomic interpretations made using a randomly
isolated repeats of the SL gene (Thomas et al. 2005;
Maslov et al. 2007). For this reason, we examined 3–7
cloned sequences from each culture. The polymor-
phisms found in different cloned SL sequences
from each established culture did not prevent their
clustering together. However, cattle harbouring
more than 1 genotype could be detected directly by
PCR of DNA from primary cultures, but not from
established lineages, thus demonstrating selection by
successive passages in culture. Parity of independent
molecular markers defining the same genotypes
indicated clonal propagation of T. theileri isolates.
Phylogeographical analysis reveals geographical
structuring of T. theileri genotypes found in cattle,
apparently, related to geographical distances and
livestock transit. Accordingly, genotypes restricted
to the Southern (IC) and Northern (IIA) regions,
which are too far from one another for normal live-
stock transportation, were separated by large genetic
distances and do not share the same area.
The states of MS and PA, together with adjacent
states in the Central and Northern (Amazonia) re-
gions, are the largest cattle production areas in
Phylogeography of T. theileri trypanosomes 119

Brazil, from which herds of cattle are sent to neigh-
bouring regions. The state of MS, in central Brazil,
was the only region showing cattle infected with both
TthI (IB) and ThII (IIB) lineages. The states of SP
and MS had both genotypes IB and IA, an expected
result since there is an intense transit of livestock
between these nearby regions. The states of PA, RN
and PB showed only TthII genotypes. The coexist-
ence of IIA and IIB genotypes in the Northeast and
North may be due to sporadic contact between cattle
from these regions. The constant high abundance of
tabanids in the Central (Pantanal), Southeasthern
(Vale do Ribeira) and Northern (Amazonia) regions
could play a role in the dispersion and heterogeneity
of T. theileri populations. The hypothesis that
hybridization events should occur in tabanids finds
support in the strong variability among sequences
of ribosomal genes amplified directly from isolates
parasitizing the gut of the same tabanid specimens
(Rodrigues et al. 2006). The species diversity and
amount of tabanids largely varied according to
geographical regions, probably determined by the
ecotopes and food availability. Therefore, different
genotypes could be at least partially related to dis-
tinct vector species. Further studies of T. theileri in
tabanid flies and other vectors are required to clarify
all these questions.
Buffalo isolates from SP and MS share one SL
genotype, so far exclusive to this host species.
Isolates from water buffalo clustered together in this
and in previous studies using ribosomal and RAPD
markers (Rodrigues et al. 2003, 2006). Clustering of
all buffalo genotypes in a homogeneous clade closest
to cattle genotype TthIB from the same regions
suggested both geographical and host species as-
sociation. However, elucidation of this question re-
quires further studies withmore buffalo isolates from
a broad range of geographical origins. Phylogenetic
studies using several genes showed that cattle isolates
of T. theileri are not monophyletic, and could
be closest to buffalo, cervid and antelope isolates.
However, isolates from distinct host species can be
differentiated by genetic polymorphisms detected
even in the conserved rRNA gene (Rodrigues et al.
2006; Hamilton et al. 2009). Findings from the
present study suggested that data from these pre-
vious studies using SSU rRNA and gGAPDH se-
quences might not reflect the real heterogeneity of
T. theileri populations from cattle and other artio-
dactyls around the world.
Altogether, data from the present study demon-
strate that the cladeT. theileri is composed of isolates
that can be genotyped into highly consistent phylo-
genetic lineages, defined by combinations of SL and
ribosomal markers. Overall, data demonstrated a
complex geographical clonal population structuring
of T. theileri found in cattle from distant geo-
graphical regions, independent of breed but appar-
ently shaped by geographical distance and livestock
transportation by man. SL sequences proved to be
excellent targets for understanding the populational
structure of T. theileri trypanosomes. The geno-
typing method that we have developed for T. theileri
lineages through PCR-RFLP of SL sequences,
which were totally congruent with the previously
described ITS rDNA-derived genotyping method
(Rodrigues et al. 2006), proved to be a valuable tool
for large studies of population structure of T. theileri
trypanosomes.
ACKNOWLEDGMENTS
We are indebted to several students and collaborators fromUSP, UNIFERSA, UFMS, and UFPA for their inesti-mable help in the fieldwork. We specially thank KedsonA. L.Neves, a recipient of scholarships fromFMVZ-USP,for his invaluable support in fieldwork in Santarem (PA).This work was supported by grants from the Brazilianagency CNPq within the UNIVERSAL program. A. C.Rodrigues and R. C. Ferreira are postdoctoral fellows ofPROTAX-CNPq and Maia da Silva F. from PNPD-CAPES. H. A. Garcia is a recipient of scholarships fromCDCH-UCV in Venezuela.
REFERENCES
Aksoy, S., Shay, G. L., Villanueva, M. S., Beard, C. B.
and Richards, F. F. (1992). Spliced leader RNA
sequences of Trypanosoma rangeli are organized within
the 5S rRNA-encoding genes. Gene 113, 239–243.
Bose, R. and Heister, N. C. (1993). Development of
Trypanosoma (Megatrypanum) theileri in tabanids.
The Journal of Eukaryotic Microbiology 40, 788–792.
doi:10.1111/j.1550-7408.1993.tb04475.x.
Bose, R., Petersen, K., Pospichal, H., Buchanan, N.
and Tait, A. (1993). Characterization of
Megatrypanum trypanosomes from European Cervidae.
Parasitology 107, 55–61. doi:10.1017/
S0031182000079403.
Bose, R., Friedhoff, K. T., Olbrich, S., Buscher, G. and
Domeyer, I. (1987). Transmission of Trypanosoma
theileri to cattle by Tabanidae. Parasitology Research 73,
421–424.
Brisse, S., Verhoef, J. and Tibayrenc, M. (2001).
Characterisation of large and small subunit rRNA and
mini-exon genes further supports the distinction of six
Trypanosoma cruzi lineages. International Journal
for Parasitology 31, 1218–1226. doi:10.1016/S0020-
7519(01)00238-7.
Cortez, A. P., Ventura, R. M., Rodrigues, A. C.,
Batista, J. S., Paiva, F., Anez, N., Machado, R. Z.,
Gibson, W. C. and Teixeira, M. M. G. (2006). The
taxonomic and phylogenetic relationships of
Trypanosoma vivax from South America and Africa.
Parasitology 133, 159–169. doi:10.1017/
S0031182006000254.
Dirie, M. F., Bornstein, S., Wallbanks, K. R., Stiles,
J. K. and Molyneux, D. H. (1990). Zymogram and
life-history studies on trypanosomes of the subgenus
Megatrypanum. Parasitology Research 76, 669–674.
Doherty, M. L., Windle, H., Voorheis, H. P., Larkin,
H., Casey, M., Clery, D. and Murray, M. (1993).
Clinical disease associated with Trypanosoma theileri
A. C. Rodrigues and others 120

infections in a calf in Ireland. Veterinary Record 26,
653–656.
Drouin, G. and de Sa, M. M. (1995). The concerted
evolution of 5S ribosomal genes linked to the repeat units
of other multigene families. Molecular Biology and
Evolution 12, 481–493.
Falla, A., Herrera, C., Fajardo, A., Montilla, M.,
Vallejo, G. A. and Guhl, F. (2009). Haplotype
identification within Trypanosoma cruzi I in Colombian
isolates from several reservoirs, vectors and humans.
Acta Tropica 110, 15–21. doi:10.1016/
j.actatropica.2008.12.003.
Fernandes, O., Santos, S. S., Cupolillo, E., Mendonca,
M. B. A., Junqueira, A., Santos, L. C., Derre, R.,
Sturm, N. R., Naiff, R. D., Barrett, T. V., Campbell,
D. A. and Coura, J. R. (2001). A mini-exon multiplex
polymerase chain reaction to distinguish the major
groups of Trypanosoma cruzi and T. rangeli in the
Brazilian Amazon. Transactions of the Royal Society of
Hygiene and Tropical Medicine 95, 97–99.
Fernandes, O., Teixeira, M. M. G., Sturm, N. R.,
Sousa, M. A., Camargo, E. P., Degrave, W. M. and
Campbell, D. A. (1997). Mini-exon gene sequence
define six groups within the genusCrithidia.The Journal
of Eukaryotic Microbiology 44, 535–539.
Ferreira, R. C., de Souza, A. A., Freitas, R. A.,
Campaner, M., Takata, C. S. A., Barrett, T. V.,
Shaw, J. J. and Teixeira, M. M. G. (2007). A
Phylogenetic lineage of closely related trypanosomes
(Trypanosomatidae, Kinetoplastida) of anurans and
sand flies (Psychodidae, Diptera) sharing the same
ecotopes in Brazilian Amazonia. The Journal
of Eukaryotic Microbiology 55, 427–435. doi:10.1111/
j.1550-7408.2008.00342.x.
Gibson, W., Bingle, L., Blendeman, W., Brown, J.,
Wood, J. and Stevens, J. (2000). Structure and
sequence variation of the trypanosome spliced leader
transcript. Molecular and Biochemical Parasitology 107,
269–277. doi:10.1016/S0166-6851(00)00193-6.
Hamilton, P. B., Adams, E. R., Njiokou, F., Gibson,
W. C., Cuny, G. and Herder, S. (2009). Phylogenetic
analysis reveals the presence of the Trypanosoma cruzi
clade in African terrestrial mammals. Infection, Genetics
and Evolution 9, 81–86. doi:10.1016/
j.meegid.2008.10.011.
Hamilton, P. B., Gibson, W. C. and Stevens, J. R.
(2007). Patterns of co-evolution between trypanosomes
and their hosts deduced from ribosomal RNA and
protein-coding gene phylogenies. Molecular
Phylogenetics and Evolution 44, 15–25. doi:10.1016/
j.ympev.2007.03.023.
Hamilton, P. B., Stevens, J. R., Gidley, J., Holz, P. and
Gibson, W. C. (2005). A new lineage of trypanosomes
from Australian vertebrates and terrestrial bloodsucking
leeches (Haemadipsidae). International Journal for
Parasitology 35, 431–443. doi:10.1016/
j.ijpara.2004.12.005.
Hatama, S., Shibahara, T., Suzuki, M., Kadota, K.,
Uchida, I. and Kanno, T. (2007). Isolation of a
Megatrypanum trypanosome from sika deer (Cervus
nippon yesoensis) in Japan. Veterinary Parasitology
149, 56–64. doi:10.1016/j.vetpar.2007.07.019.
Heiman, M. (1997). Webcutter 2.0. (http://
rna.lundberg.gu.se/cutter2/).
Herrera, C., Bargues, M. D., Fajardo, A., Montilla,
M., Triana, O., Vallejo, G. A. and Guhl, F. (2007).
Identifying four Trypanosoma cruzi I isolate haplotypes
from different geographic regions in Colombia. Infection
Genetic and Evolution 7, 535–539. doi:10.1016/
j.meegid.2006.12.003.
Hoare, C. A. (1972). Subgenus Megatrypanum. In The
Trypanosomes of Mammals: A Zoological Monograph
(ed. Hoare, C. A.), pp. 123–141. Blackwell Scientific
Publications, Oxford, UK.
Maia Da Silva, F., Junqueira, A. C., Campaner, M.,
Rodrigues, A. C., Crisante, G., Ramirez, L. E.,
Caballero, Z. C., Monteiro, F. A., Coura, J. R., Anez,
N. and Teixeira, M. M. G. (2007). Comparative
phylogeography of Trypanosoma rangeli and Rhodnius
(Hemiptera: Reduviidae) supports a long coexistence of
parasite lineages and their sympatric vectors. Molecular
Ecology 16, 3361–3373. doi:10.1111/j.1365-
294X.2007.03371.x.
Maia da Silva, F., Marcili, A., Lima, L., Cavazzana,
M Jr., Ortiz, P. A., Campaner, M., Takeda, G. F.,
Paiva, F., Nunes, V. L., Camargo, E. P. and
Teixeira, M. M. G. (2009). Trypanosoma rangeli
isolates of bats from Central Brazil : genotyping and
phylogenetic analysis enable description of a new lineage
using spliced-leader gene sequences. Acta Tropica 109,
199–207. doi:10.1016/j.actatropica.2008.11.005.
Maslov, D. A., Westenberger, S. J., Xu, X., Campbell,
D. A. and Sturm, N. R. (2007). Discovery and
barcoding by analysis of splice leader RNA gene
sequences of new isolates of Trypanosomatidae from
Heteroptera in Costa Rica and Ecuador. The Journal
of Eukaryotic Microbiology 54, 57–65. doi:10.1111/
j.1550-7408.2006.00150.x.
Matzura, O. and Wennborg, A. (1996). RNAdraw:
an Integrated Program for RNA Secondary Structure
Calculation and Analysis under 32-bit Microsoft
Windows.
Mejıa-Jaramillo, A. M., Arboleda-Sanchez, S.,
Rodrıguez, I. B., Cura, C., Salazar, A., del Mazo, J.,
Triana-Chavez, O. and Schijman, A. G. (2009).
Geographical clustering of Trypanosoma cruzi I groups
from Colombia revealed by low-stringency single
specific primer-PCR of the intergenic regions of
spliced-leader genes. Parasitology Research 104,
399–410. doi 10.1007/s00436-008-1212-0.
O’Connor, O., Bosseno, M-F., Barnabe, C., Douzery,
E. J. P. and Breniere, S. F. (2007). Genetic clustering
of Trypanosoma cruzi I lineage evidenced by intergenic
miniexon gene sequencing. Infection, Genetics and
Evolution 7, 587–593. doi:10.1016/
j.meegid.2007.05.003.
Podlipaev, S. A., Sturm, N. R., Fiala, I., Fernandes,
O., Westenberger, S. J., Dollet, M., Campbell, D. A.
and Lukes, J. (2004). Diversity of insect
trypanosomatids assessed from the spliced leader RNA
and 5S rRNA genes and intergenic regions. The Journal
of Eukaryotic Microbiology 51, 283–290. doi:10.1111/
j.1550-7408.2004.tb00568.x.
Roditi, I. (1992).Trypanosoma vivax : linkage ofmini-exon
(spliced leader) and 5S ribosomal RNA genes. Nucleic
Acids Research 20, 1995.
Rodrigues, A. C., Paiva, F., Campaner, M., Stevens,
J. R., Noyes, H. A. and Teixeira, M. M. G. (2006).
Phylogeography of T. theileri trypanosomes 121

Phylogeny of Trypanosoma (Megatrypanum) theileri and
related trypanosomes reveals lineages of isolates
associated with artiodactyl hosts diverging on SSU and
ITS ribosomal sequences. Parasitology 132, 215–224.
doi:10.1017/S0031182005008929.
Rodrigues, A., Campaner, M., Takata, C. S., Dell’
Porto, A., Milder, R. V., Takeda, G. F. and Teixeira,
M. M. G. (2003). Brazilian isolates of Trypanosoma
(Megatrypanum) theileri : diagnosis and differentiation of
isolates from cattle and water buffalo based on biological
characteristics and randomly amplified DNA sequences.
Veterinary Parasitology 116, 185–207. doi:10.1016/
S0304-4017(03)00236-X.
Schafler, D. H. (1979). Trypanosoma theileri : a literature
review and report of incidence in New York cattle.
Cornell Veterinarian 69, 411–425.
Seifi, H. A. (1995). Clinical trypanosomosis due to
Trypanosoma theileri in a cow in Iran. Tropical Animal
Health and Production 27, 93–94.
Serrano, M. G., Nunes, L. R., Campaner, M., Buck,
G. A., Camargo, E. P. and Teixeira, M. M. G.
(1999a). Trypanosomatidae: Phytomonas detection in
plants and phytophagous insects by PCR amplification
of a genus-specific sequence of the spliced leader gene.
Experimental Parasitology 91, 268–279. doi:10.1006/
expr.1998.4379.
Serrano, M. G., Campaner, M., Buck, G. A., Teixeira,
M. M. G. and Camargo, E. P. (1999b). PCR
amplification of the spliced leader gene for the diagnosis
of Trypanosomatid parasites of plants and insects in
methanol-fixed smears.FEMSMicrobiology Letters 176,
241–246.
Souto, R. P., Fernandes, O., Macedo, A. M.,
Campbell, D. and Zingales, B. (1996). DNA marker
define two major phylogenetic lineages of Trypanosoma
cruzi. Molecular and Biochemical Parasitology 83,
141–152. doi:10.1016/S0166-6851(96)02755-7.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum
likelihood-based phylogenetic analyses with thousands
of taxa and mixed models.Bioinformatics 22, 2688–2690.
doi:10.1093/bioinformatics/btl446.
Stevens, J. R., Teixeira, M. M. G., Bingle, L. E. and
Gibson, W. C. (1999). The taxonomic position and
evolutionary relationships of Trypanosoma rangeli.
International Journal for Parasitology 29, 749–757.
doi:10.1016/S0020-7519(99)00016-8.
Swofford, D. L. (2002). PAUP* Phylogenetic Analysis
using Parsimony (*and other Methods) Version 4.0b10.
Sinauer Associates, Sunderland, MA, USA.
Thomas, S., Westenberger, S. J., Campbell, D. A. and
Sturm, N. R. (2005). Intragenomic spliced leader RNA
array analysis of kinetoplastids reveals unexpected
transcribed region diversity in Trypanosoma cruzi. Gene
352, 100–108. doi:10.1016/j.gene.2005.04.002.
Thompson, J. D., Gibson, T. J., Plewniak, F.,
Jeanmougin, F. and Higgins, D. G. (1997). The
CLUSTAL X windows interface: flexible strategies for
multiple sequence alignment aided by quality analysis
tools. Nucleic Acids Research 25, 4876–4882.
Urrea, D. A., Carranza, J. C., Cuba, C. A.,
Gurgel-Goncalves, R., Guhl, F., Schofield, C. J.,
Triana, O. and Vallejo, G. A. (2005). Molecular
characterisation of Trypanosoma rangeli strains isolated
from Rhodnius ecuadoriensis in Peru, R. colombiensis in
Colombia and R. pallescens in Panama, supports a
co-evolutionary association between parasites and
vectors. Infection Genetic and Evolution 5, 123–129.
doi:10.1016/j.meegid.2004.07.005.
Ventura, R. M., Paiva, F., Silva, R. M. S., Takeda,
G. F., Buck, G. A. and Teixeira, M. M. G. (2001).
Trypanosoma vivax : characterization of the spliced-
leader gene of a Brazilian stock and species-specific
detection by PCR amplification of an intergenic spacer
sequence. Experimental Parasitology 99, 37–48.
doi:10.1006/expr.2001.4641.
Wells, E. A. (1976). Subgenus Megatrypanum. In
Biology of the Kinetoplastida (ed. Lumsden, W. H. R.
and Evans, D. A.), pp. 257–275. Academic Press,
London, UK.
Westenberger, S. J., Sturm,N. R. andCampbell, D. A.
(2006). Trypanosoma cruzi 5S rRNA arrays define five
groups and indicate the geographic origins of an ancestor
of the heterozygous hybrids. International Journal for
Parasitology 36, 337–346. doi:10.1016/
j.ijpara.2005.11.002.
Westenberger, S. J., Sturm, N. R., Yanega, D.,
Podlipaev, S. A., Zeledon, R., Campbell, D. A.
and Maslov, D. A. (2004). Trypanosomatid
biodiversity in Costa Rica: genotyping of parasites
from Heteroptera using the spliced leader RNA gene.
Parasitology 129, 537–547. doi: 10.1017/
S003118200400592X.
A. C. Rodrigues and others 122




















