
Let's Talk about Endings A document to help parents, children ...
Upload
assumptionunivCategory
view
0download
0
ORIGINAL PAPER
Characterization of Phenytoin, Carbamazepine, Vinpocetineand Clorgyline Simultaneous Effects on Sodium Channelsand Catecholamine Metabolism in Rat Striatal Nerve Endings
Marıa Sitges Æ Blanca I. Aldana Æ Luz M. Chiu ÆVladimir Nekrassov
Accepted: 7 July 2008 / Published online: 19 August 2008
� Springer Science+Business Media, LLC 2008
Abstract The effects of two classic antiepileptic drugs
(carbamazepine and phenytoin), a potential antiepileptic
(vinpocetine) and a monoamine-oxidase inhibitor (clorgy-
line) on the simultaneous changes (detected by HPLC) on
Glu, Asp, dopamine and DOPAC inside and outside striatal
isolated nerve endings were investigated. Under resting
conditions phenytoin, carbamazepine and clorgyline
increased dopamine release. Phenytoin and clorgyline
increased internal dopamine and decreased DOPAC for-
mation. Carbamazepine decreased internal dopamine and
practically did not change DOPAC formation. Glu and Asp
release was unchanged. Neurotransmitter release induced
by the Na+ channel opener veratridine was reduced by all
the antiepileptic drugs tested, except phenytoin which, like
clorgyline, facilitated veratridine-induced dopamine
release. We conclude that besides the antagonism exerted
by carbamazepine, phenytoin and vinpocetine on excitatory
neurotransmitters release triggered by Na+ channel acti-
vation, that might importantly contribute to their
anticonvulsant action, they exert different actions on stri-
atal dopamine distribution, that might explain their
different side effect profiles.
Keywords Antiepileptic drugs � Monoamino-oxidase �Dopamine � DOPAC � Glutamate � Aspartate � Veratridine
Introduction
Several of the most effective antiepileptic drugs are
believed to stop the paroxysmal neuronal activity acting as
sodium channel blockers. For instance, the involvement of
voltage-gated sodium channels in the mechanism of action
of the anticonvulsants, carbamazepine and phenytoin, was
indicated by the inhibition of the sustained repetitive firing
in spinal cord neurons and neurons in culture from the
seminal work of Macdonald and McLean [1].
Veratridine is a toxin of natural origin that binds to Na+
channel site 2 (voltage sensor) impeding its inactivation
and by this mean increases the rate of Na+ entry. The
increase in the internal Na+ concentration induced by
veratridine in cerebral isolated nerve endings (synapto-
somes) preloaded with the Na+ selective indicator dye,
SBFI is inhibited by the Na+ channel blocker tetrodotoxin
and independent of external Ca2+ [2]. Also neurotrans-
mitter release induced by veratridine in synaptosomes is
sensitive to tetrodotoxin, is independent of external Ca2+
and depends on the presence of external Na+ [3–5].
Therefore sensitivity of veratridine-induced responses to a
drug usually indicates Na+ channel antagonism.
In hippocampal synaptosomes preloaded with radioac-
tive Glu ([3H]Glu) several of the most amply used
antiepileptic drugs and the new potential antiepileptic
vinpocetine (ethyl apovincamine-22-oate) inhibited
[3H]Glu release induced by veratridine in the absence of
external Ca2+; vinpocetine being the most potent [6, 7].
Also in the guinea pig in vivo phenytoin and carbamaze-
pine were less potent than vinpocetine to inhibit the
epileptic cortical activity induced by the convulsing agent,
pentylenetetrazole [8].
Vinpocetine is well tolerated in humans [9] and to our
knowledge no reports of motor disturbances caused by
M. Sitges (&) � B. I. Aldana � L. M. Chiu
Depto. de Biologıa Celular y Fisiologıa, Instituto de
Investigaciones Biomedicas, Universidad Nacional Autonoma de
Mexico, Apartado Postal 70228, Ciudad Universitaria,
04510 Mexico, D.F., Mexico
e-mail: [email protected]
V. Nekrassov
Division de Investigacion Basica y Aplicada, Instituto Nacional
de Rehabilitacion, SSA, Mexico, D.F., Mexico
123
Neurochem Res (2009) 34:470–479
DOI 10.1007/s11064-008-9805-7

vinpocetine exist. In contrast, several reports indicate that
phenytoin can induce dyskinesia [10–16] and some reports
have implicated carbamazepine in the production of
abnormal involuntary movements [17, 18].
Under conditions in which the energy supply surpasses
the energy demand, such as in models of experimental
epilepsy or ischemia, a massive release of striatal dopa-
mine accompanied by a decrease of its metabolites,
DOPAC and HVA takes place [19–24]. Interestingly
vinpocetine protects against neuronal damage induced by
cerebral ischemia [25–29], and increases the external
concentration of the DOPAC under baseline as well as
under depolarized conditions without changing dopamine
baseline release [30].
In an attempt to explore the possibility of a dopamine
involvement in the eventual motor disturbances caused by
the classic antiepileptic drugs, besides testing phenytoin
and carbamazepine effects under resting and veratridine-
depolarized conditions on dopamine release simultaneously
with their effects on Glu and Asp release, their effects on
the concentrations of dopamine and its main metabolite
(DOPAC) inside and outside striatal isolated nerve endings
were measured, and compared with the effects of vinp-
ocetine and of the MAO-I inhibitor, clorgyline.
Materials and Methods
Source of Materials
Drugs used in the experiments were acquired from the
following companies: vinpocetine (eburnamenine-14-car-
boxylic acid ethyl ester) was obtained either from Sigma
Chemical Co. (St. Louis, MO) or from Psicofarma S.A. de
C.V. (Mexico). Carbamazepine was obtained either from ICN
Biomedicals Inc. (Ohio) or from Psicofarma S.A. de C.V.
(Mexico). Phenytoin (5,5-diphenylhydantoin sodium salt)
was from Psicofarma S.A. de C.V. (Mexico). Veratridine,
n-methyl-n-propargyl-3-(2,4-dichloro-phenoxy)propylamine
(clorgyline) and 1-octanesulfonic acid were from Sigma
Chemical Co. (St. Louis, MO). The o-phthalaldehyde reagent
solution was from Pierce (Rockford, ILL). All other reagents
were of analytical grade.
Preparation of Synaptosomes
Dissected striata of 4 male Wistar rats (250–300 g) were
immediately placed in cold isotonic sucrose (1:10, w/v) and
homogenized (6 strokes at 2000 rpm, 0.15 mm pestle-
vessel clearance). The resulting suspensions were centri-
fuged at 1100g for 10 min, and supernatants obtained
from this centrifugation were centrifuged for another 20
min at 8200g. The resulting pellets containing striatal
synaptosomes were resuspended in standard Kreb’s Ringer
HEPES (KRH). The composition of the KRH is, in mM:
127 NaCl, 1.2 KH2PO4, 3.37 KCl, 1 CaCl2, 1.18 MgSO4,
20 HEPES and 5.6 mM dextrose, pH 7.4, bubbled with a
O2/CO2 mixture.
Release Experiments
Aliquots of striatal synaptosomes (450 ± 43 lg) suspended
in 500 ll of KRH were pre-incubated at 37�C for 5 min
before exposure to the experimental conditions to be
studied. Namely, carbamazepine (500 and 1500 lM),
phenytoin (range from 70 to 1500 lM), vinpocetine (15
lM), or clorgyline (0.1–10 lM), in the absence or in the
presence of 5 lM veratridine, and then incubated at 37�C
for 10 min. Incubation was stopped by centrifugation. The
supernatants resulting from this centrifugation containing
the released neurotransmitters were transferred to clean
vials and treated with an aliquot of a 0.1 M perchloric acid
(PCA)/0.1 mM EDTA solution, and stored at -40�C for
later analysis.
The resulting pellets were resuspended in 1 ml of the
PCA/EDTA solution pH 1.4 and vigorously vortex mixed.
These drastic conditions guarantee a complete discharge of
the synaptosomal content. The vortex mixed suspension
containing the disrupted synaptosomes was centrifuged.
The supernatants resulting from this centrifugation (con-
taining the neurotransmitters that were inside of each
synaptosomal sample) were also stored at -40�C for later
analysis. In order to standardize neurotransmitter release
per mg of synaptosomal protein, the resulting pellets were
suspended in 2 ml of a NaOH 5 mM solution and used for
protein determination. The samples containing the neuro-
transmitters released were injected into the HPLC system
within the 2 weeks after the experiment. Results are
expressed as the concentration of Glu (in nmol), Asp (in
nmol), dopamine (in pmol) or DOPAC (in pmol) released
or retained inside striatal synaptosomes per mg of synap-
tosomal protein.
Determination of the Concentrations of Dopamine and
DOPAC
Twenty microliters of sample suspended in 0.1 M PCA/0.1
mM EDTA were injected directly into a Waters HPLC
system for analysis. The HPLC system consists of a
delivery pump (model 600), a Rheodyne injector, an ana-
lytical column (resolve, C18, 150 9 3.9 mm internal
diameter, particle size 5 lm) controlled at 30�C, and an
electrochemical detector (model DECADE) with glassy
carbon used at a voltage of +0.8 V vs. a KCl (3 M) ref-
erence electrode (range 1 nA). A mobile phase composed
of 50 mM ortho-phosphoric acid/50 mM citric acid buffer,
Neurochem Res (2009) 34:470–479 471
123
pH 3.1 adjusted with KOH, containing 5% (v/v) methanol,
100 mg/l octanesulfonic acid and 20 mg/l EDTA, at a flow
rate of 1 ml/min, was applied for catecholamine elution.
Dopamine and DOPAC concentrations in the experimental
samples were calculated with calibration curves obtained
from the injection of increasing concentrations of the
external standard mixture (containing dopamine, DOPAC,
Glu and Asp) into the HPLC system.
Determination of the Concentrations of Glu and Asp
Ten microliters of sample suspended in 0.1 M PCA/0.1
mM EDTA was mixed with 20 ll of o-phthalaldehyde
reagent. After 120 s (strict time), a 10 ll aliquot of that
mixture was injected into the HPLC system. An analytical
column (Nova-pak C-18, 75 9 3.9 mm internal diameter,
particle size 10 lm) set at 25�C and a fluorescence detec-
tor set at 360 nm excitation wavelength and 450 nm
emission wave length were used. A lineal gradient elution
program performed over 30 min was applied for amino
acid elution: eluent A (30 mM sodium acetate buffer,
pH 6.8) from 100 to 50%, and eluent B (methanol) from
0 to 50%, at a flow rate of 1 ml/min. The concentrations of
Glu and Asp in the experimental samples were calculated
with calibration curves obtained from the injection of
increasing concentrations of the external standard mixture
after o-phthalaldehyde reagent derivatization into the
HPLC system.
Statistical Analysis
Paired Student’s t test was used for statistical evaluations.
From P \ 0.05 differences between data were considered
statistically significant. However most of the significant
differences between paired data obtained in parallel (i.e.
control and a specific condition) were below this value, as
indicated in some of the figures.
Results
Changes Induced by Carbamazepine, Phenytoin and
Vinpocetine on Glu, Asp and Dopamine Release in
Striatum Isolated Nerve Endings Incubated in the
Absence and Presence of Veratridine
When control neurotransmitter release values under resting
conditions (first line on Table 1) are compared with neu-
rotransmitter release values in the presence of the
antiepileptic drugs under resting conditions, a selective
increase exerted by carbamazepine and phenytoin on
dopamine baseline release becomes evident. Glu and Asp
baseline release remain unchanged by the presence of the
drugs tested.
The concentrations of glutamate, aspartate and dopa-
mine released for 10 min under resting conditions from
striatal synaptosomes (first line on Table 1) were markedly
increased by 5 lM veratridine (second line on Table 1). In
the presence of carbamazepine (1500 lM), phenytoin (200
lM) and vinpocetine (15 lM) the veratridine-induced
release of the excitatory amino acids was significantly
reduced (first two columns on Table 1). The veratridine-
induced release of dopamine also was reduced in the
presence of carbamazepine and vinpocetine at the above
concentrations, whereas phenytoin was paradoxically
unable to prevent the veratridine-induced increase in
dopamine release (third column on Table 1).
Table 1 Effect of AEDs on veratridine-evoked release of excitatory amino-acid neurotransmitters and dopamine in striatal synaptosomes
Glutamate Aspartate Dopamine
Control 16.0 ± 2.2 7.6 ± 1.3 9.1 ± 0.9
Veratridine 34.1 ± 2.8a 12.9 ± 1.0a 43.7 ± 7.6a
Carbamazepine 1500 lM 14.9 ± 2.3 6.5 ± 0.9 20.8 ± 4.5b
Carbamazepine 1500 lM + veratridine 15.4 ± 2.2c 5.9 ± 0.9c 30.2 ± 4.9a,c
Phenytoin 200 lM 14.6 ± 1.7 8.6 ± 1.6 19.6 ± 3.4b
Phenytoin 200 lM + veratridine 24.9 ± 2.0a,c 9.8 ± 1.1c 52.3 ± 5.5a,c
Vinpocetine 15 lM 13.7 ± 2.1 5.9 ± 0.7 8.1 ± 0.7
Vinpocetine 15 lM + veratridine 21.1 ± 2.1a,c 7.9 ± 0.6a,c 16.6 ± 0.9a,c
Amino acids are in nmol/mg and dopamine in pmol/mg synaptosomal protein. Results are the mean ± SEM values of 6 independent experiments
in parallela P \ 0.03 between neurotransmitter release in the absence and presence of veratridine under the indicated conditionb P \ 0.03 between the baseline release in the absence (control) and presence of the indicated AEDc P \ 0.03 between veratridine-induced release in the absence and presence of the indicated AED
472 Neurochem Res (2009) 34:470–479
123

Phenytoin, Clorgyline and Carbamazepine Effects
on Dopamine Distribution Inside and Outside
Synaptosomes Under Resting Conditions
In an attempt to get a better understanding of the selective
increase on dopamine baseline release exerted by phenyt-
oin and carbamazepine, their effects, at increasing
concentrations, on dopamine release and on the internal
concentrations of dopamine and its metabolite DOPAC
were measured, and compared with the effect of increasing
concentrations of clorgyline.
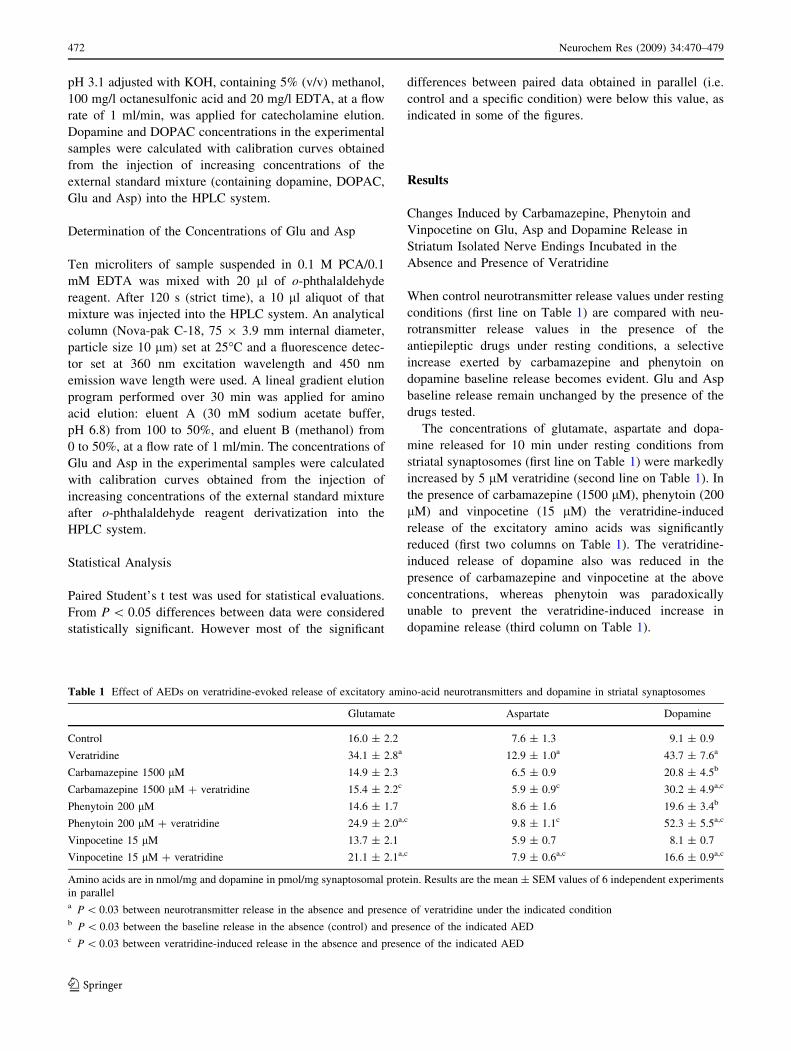
The progressive increase induced by phenytoin in the
range from 70 to 1500 lM on dopamine baseline release
and on the internal dopamine concentration is shown in
Fig. 1a, b, respectively. Similarly to phenytoin, increasing
concentrations of clorgyline (from 0.1 to 10 lM) progres-
sively increased both the external (Fig. 1c) as well as the
internal dopamine concentration (Fig. 1d). Carbamazepine
at high doses (500 and 1500 lM) also raised dopamine
baseline release dose dependently (Fig. 1e). However, in
contrast to phenytoin and clorgyline, the internal dopamine
concentration was lowered by carbamazepine (Fig. 1f).
Effect of Phenytoin, Clorgyline and Carbamazepine on
the Concentration of DOPAC Inside Synaptosomes
Under Resting Conditions
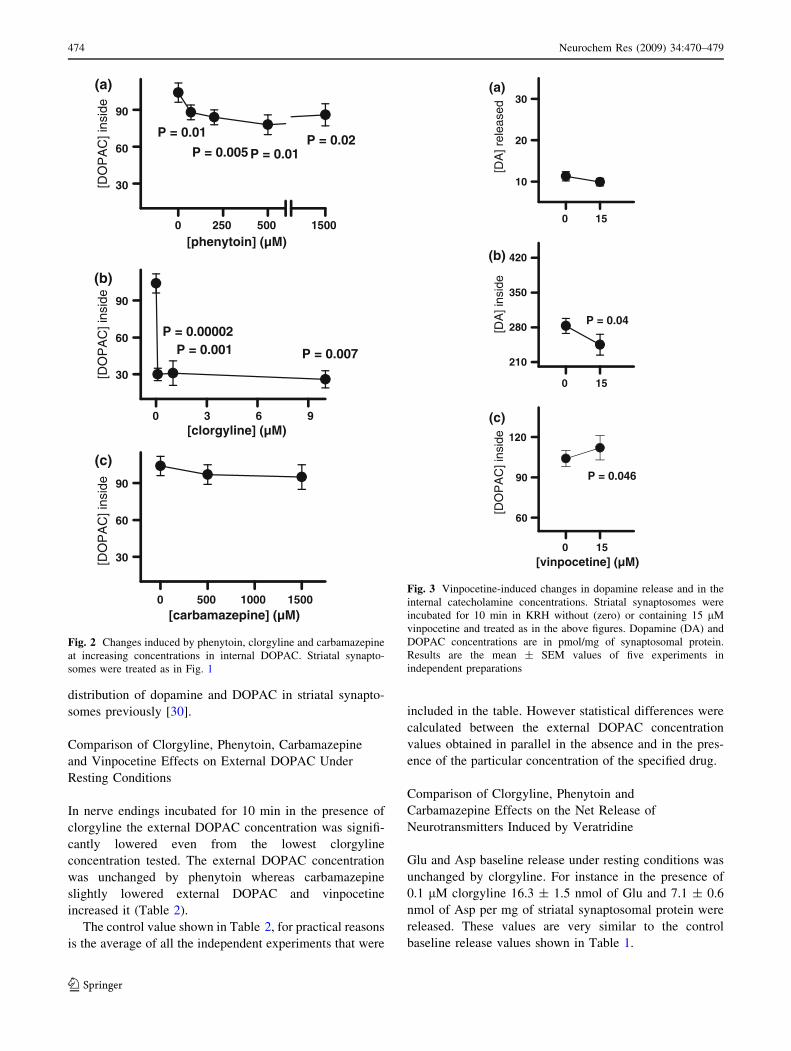
Oppositely to the raise in the internal dopamine concen-
tration, the increasing concentrations of phenytoin and
clorgyline progressively lowered the internal DOPAC
concentration under resting conditions (Fig. 2a, b). Car-
bamazepine in contrast failed in modifying internal
DOPAC significantly (Fig. 2c).
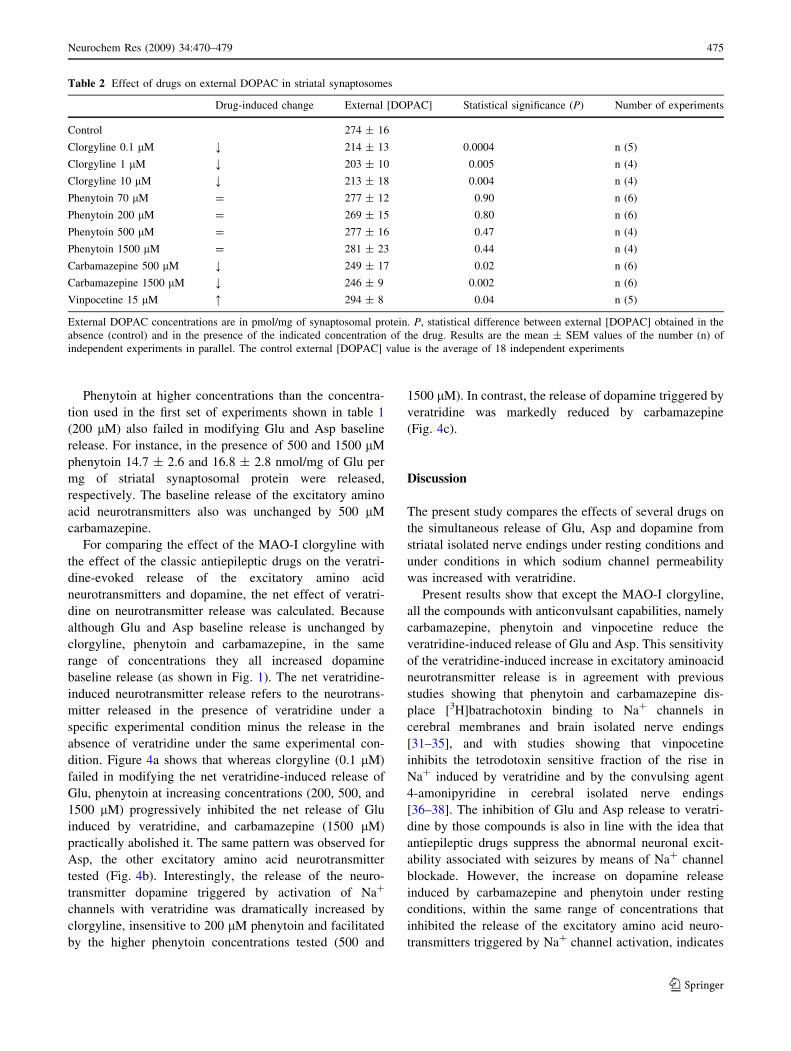
Figure 3 shows that the baseline release of dopamine
was unchanged by vinpocetine (15 lM), and that oppo-
sitely to phenytoin and clorgyline, vinpocetine decreased
internal dopamine and increased internal DOPAC.
We just tested vinpocetine at one concentration here
because we have already characterized the dose response
effects of vinpocetine on the internal and external
[clorgyline] (µM)
[DA
] re
leas
ed
10
20
30P = 0.002
P = 0.0002
P = 0.02
[DA
] in
sid
e
210
280
350
420
[clorgyline] (µM)
P = 0.00001
P = 0.004P = 0.0001
0 500 1000 1500
[DA
] in
sid
e
210
280
350
420
[carbamazepine] (µM)
P = 0.049P = 0.02
[carbamazepine] (µM) 0 500 1000 1500
[DA
] re
leas
ed
10
20
30P = 0.03
P = 0.001
0 250 500 1500
[DA
] in
sid
e
210
280
350
420
[phenytoin] (µM)
P = 0.03P = 0.001
P = 0.0001
P = 0.03
[phenytoin] (µM)0 250 500 1500
[DA
] re
leas
ed
10
20
30 P = 0.01P = 0.01
P = 0.0003
P = 0.01(a) (b)
(d)(c)
(e) (f)
0 3 6 9 0 3 6 9
Fig. 1 Changes induced by
phenytoin, clorgyline and
carbamazepine at increasing
concentrations in dopamine
distribution. Striatal
synaptosomes were incubated
(37�C) for 10 min in: KRH
without (zero) or containing
increasing concentrations of:
phenytoin (upper graphs),
clorgyline (middle graphs) or
carbamazepine (bottom graphs).
Incubation was stopped by
centrifugation. Samples
containing dopamine released
and retained were prepared for
HPLC analysis (see section
‘‘Release Experiments’’ for
details) and the pellets used for
protein determination.
Dopamine (DA) released (left
columns) or retained (right
columns) is in pmol/mg of
synaptosomal protein. Results
are the mean ± SEM values of
at least 4 experiments in
independent preparations for
each point. The statistical
significance (P) between a
particular drug concentration
and its respective control is
indicated
Neurochem Res (2009) 34:470–479 473
123
distribution of dopamine and DOPAC in striatal synapto-
somes previously [30].
Comparison of Clorgyline, Phenytoin, Carbamazepine
and Vinpocetine Effects on External DOPAC Under
Resting Conditions
In nerve endings incubated for 10 min in the presence of
clorgyline the external DOPAC concentration was signifi-
cantly lowered even from the lowest clorgyline
concentration tested. The external DOPAC concentration
was unchanged by phenytoin whereas carbamazepine
slightly lowered external DOPAC and vinpocetine
increased it (Table 2).
The control value shown in Table 2, for practical reasons
is the average of all the independent experiments that were
included in the table. However statistical differences were
calculated between the external DOPAC concentration
values obtained in parallel in the absence and in the pres-
ence of the particular concentration of the specified drug.
Comparison of Clorgyline, Phenytoin and
Carbamazepine Effects on the Net Release of
Neurotransmitters Induced by Veratridine
Glu and Asp baseline release under resting conditions was
unchanged by clorgyline. For instance in the presence of
0.1 lM clorgyline 16.3 ± 1.5 nmol of Glu and 7.1 ± 0.6
nmol of Asp per mg of striatal synaptosomal protein were
released. These values are very similar to the control
baseline release values shown in Table 1.
[DO
PA
C] i
nsid
e
30
60
90
[clorgyline] (µM)
P = 0.001 P = 0.007
0 500 1000 1500
[DO
PA
C] i
nsid
e
30
60
90
[carbamazepine] (µM)
0 250 500 1500
[DO
PA
C] i
nsid
e
30
60
90
[phenytoin] (µM)
P = 0.02P = 0.01
P = 0.01
P = 0.005
(a)
(b)
(c)
P = 0.00002
0 3 6 9
Fig. 2 Changes induced by phenytoin, clorgyline and carbamazepine
at increasing concentrations in internal DOPAC. Striatal synapto-
somes were treated as in Fig. 1
15
[DA
] rel
ease
d
10
20
30
[DA
] ins
ide
210
280
350
420
[DO
PA
C] i
nsid
e
60
90
120
P = 0.046
[vinpocetine] (µM)
(a)
(b)
(c)
P = 0.04
0
150
150
Fig. 3 Vinpocetine-induced changes in dopamine release and in the
internal catecholamine concentrations. Striatal synaptosomes were
incubated for 10 min in KRH without (zero) or containing 15 lM
vinpocetine and treated as in the above figures. Dopamine (DA) and
DOPAC concentrations are in pmol/mg of synaptosomal protein.
Results are the mean ± SEM values of five experiments in
independent preparations
474 Neurochem Res (2009) 34:470–479
123
Phenytoin at higher concentrations than the concentra-
tion used in the first set of experiments shown in table 1
(200 lM) also failed in modifying Glu and Asp baseline
release. For instance, in the presence of 500 and 1500 lM
phenytoin 14.7 ± 2.6 and 16.8 ± 2.8 nmol/mg of Glu per
mg of striatal synaptosomal protein were released,
respectively. The baseline release of the excitatory amino
acid neurotransmitters also was unchanged by 500 lM
carbamazepine.
For comparing the effect of the MAO-I clorgyline with
the effect of the classic antiepileptic drugs on the veratri-
dine-evoked release of the excitatory amino acid
neurotransmitters and dopamine, the net effect of veratri-
dine on neurotransmitter release was calculated. Because
although Glu and Asp baseline release is unchanged by
clorgyline, phenytoin and carbamazepine, in the same
range of concentrations they all increased dopamine
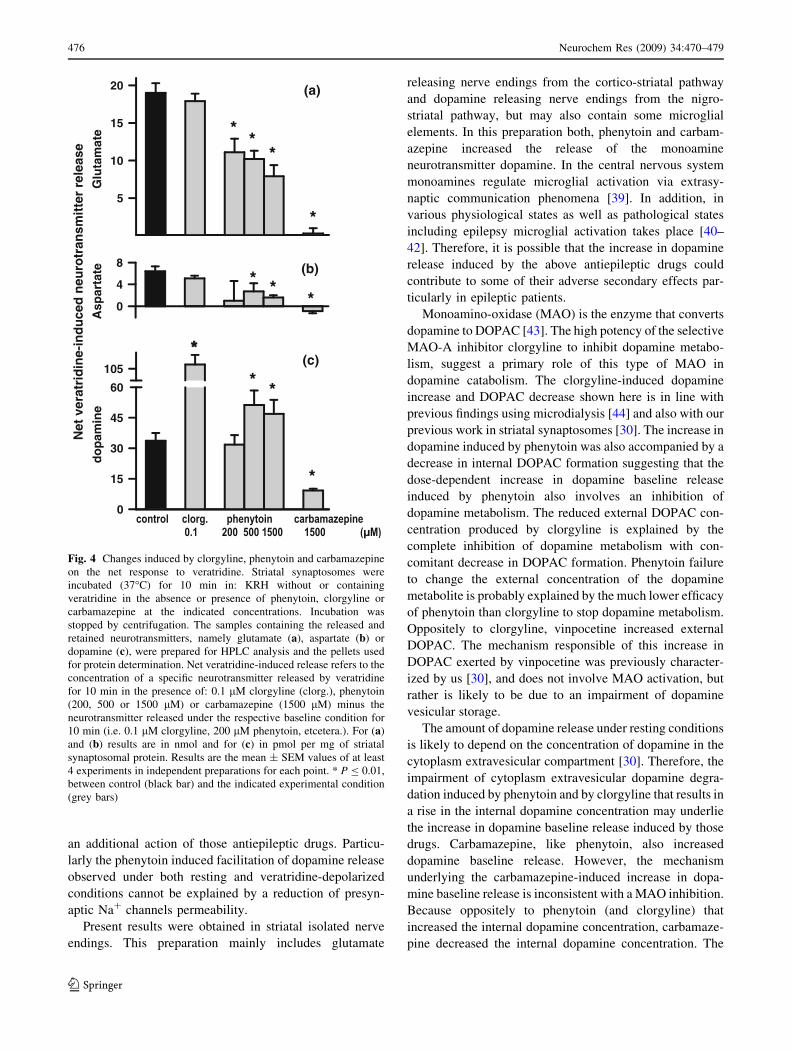
baseline release (as shown in Fig. 1). The net veratridine-
induced neurotransmitter release refers to the neurotrans-
mitter released in the presence of veratridine under a
specific experimental condition minus the release in the
absence of veratridine under the same experimental con-
dition. Figure 4a shows that whereas clorgyline (0.1 lM)
failed in modifying the net veratridine-induced release of
Glu, phenytoin at increasing concentrations (200, 500, and
1500 lM) progressively inhibited the net release of Glu
induced by veratridine, and carbamazepine (1500 lM)
practically abolished it. The same pattern was observed for
Asp, the other excitatory amino acid neurotransmitter
tested (Fig. 4b). Interestingly, the release of the neuro-
transmitter dopamine triggered by activation of Na+
channels with veratridine was dramatically increased by
clorgyline, insensitive to 200 lM phenytoin and facilitated
by the higher phenytoin concentrations tested (500 and
1500 lM). In contrast, the release of dopamine triggered by
veratridine was markedly reduced by carbamazepine
(Fig. 4c).
Discussion
The present study compares the effects of several drugs on
the simultaneous release of Glu, Asp and dopamine from
striatal isolated nerve endings under resting conditions and
under conditions in which sodium channel permeability
was increased with veratridine.
Present results show that except the MAO-I clorgyline,
all the compounds with anticonvulsant capabilities, namely
carbamazepine, phenytoin and vinpocetine reduce the
veratridine-induced release of Glu and Asp. This sensitivity
of the veratridine-induced increase in excitatory aminoacid
neurotransmitter release is in agreement with previous
studies showing that phenytoin and carbamazepine dis-
place [3H]batrachotoxin binding to Na+ channels in
cerebral membranes and brain isolated nerve endings
[31–35], and with studies showing that vinpocetine
inhibits the tetrodotoxin sensitive fraction of the rise in
Na+ induced by veratridine and by the convulsing agent
4-amonipyridine in cerebral isolated nerve endings
[36–38]. The inhibition of Glu and Asp release to veratri-
dine by those compounds is also in line with the idea that
antiepileptic drugs suppress the abnormal neuronal excit-
ability associated with seizures by means of Na+ channel
blockade. However, the increase on dopamine release
induced by carbamazepine and phenytoin under resting
conditions, within the same range of concentrations that
inhibited the release of the excitatory amino acid neuro-
transmitters triggered by Na+ channel activation, indicates
Table 2 Effect of drugs on external DOPAC in striatal synaptosomes
Drug-induced change External [DOPAC] Statistical significance (P) Number of experiments
Control 274 ± 16
Clorgyline 0.1 lM ; 214 ± 13 0.0004 n (5)
Clorgyline 1 lM ; 203 ± 10 0.005 n (4)
Clorgyline 10 lM ; 213 ± 18 0.004 n (4)
Phenytoin 70 lM = 277 ± 12 0.90 n (6)
Phenytoin 200 lM = 269 ± 15 0.80 n (6)
Phenytoin 500 lM = 277 ± 16 0.47 n (4)
Phenytoin 1500 lM = 281 ± 23 0.44 n (4)
Carbamazepine 500 lM ; 249 ± 17 0.02 n (6)
Carbamazepine 1500 lM ; 246 ± 9 0.002 n (6)
Vinpocetine 15 lM : 294 ± 8 0.04 n (5)
External DOPAC concentrations are in pmol/mg of synaptosomal protein. P, statistical difference between external [DOPAC] obtained in the
absence (control) and in the presence of the indicated concentration of the drug. Results are the mean ± SEM values of the number (n) of
independent experiments in parallel. The control external [DOPAC] value is the average of 18 independent experiments
Neurochem Res (2009) 34:470–479 475
123
an additional action of those antiepileptic drugs. Particu-
larly the phenytoin induced facilitation of dopamine release
observed under both resting and veratridine-depolarized
conditions cannot be explained by a reduction of presyn-
aptic Na+ channels permeability.
Present results were obtained in striatal isolated nerve
endings. This preparation mainly includes glutamate
releasing nerve endings from the cortico-striatal pathway
and dopamine releasing nerve endings from the nigro-
striatal pathway, but may also contain some microglial
elements. In this preparation both, phenytoin and carbam-
azepine increased the release of the monoamine
neurotransmitter dopamine. In the central nervous system
monoamines regulate microglial activation via extrasy-
naptic communication phenomena [39]. In addition, in
various physiological states as well as pathological states
including epilepsy microglial activation takes place [40–
42]. Therefore, it is possible that the increase in dopamine
release induced by the above antiepileptic drugs could
contribute to some of their adverse secondary effects par-
ticularly in epileptic patients.
Monoamino-oxidase (MAO) is the enzyme that converts
dopamine to DOPAC [43]. The high potency of the selective
MAO-A inhibitor clorgyline to inhibit dopamine metabo-
lism, suggest a primary role of this type of MAO in
dopamine catabolism. The clorgyline-induced dopamine
increase and DOPAC decrease shown here is in line with
previous findings using microdialysis [44] and also with our
previous work in striatal synaptosomes [30]. The increase in
dopamine induced by phenytoin was also accompanied by a
decrease in internal DOPAC formation suggesting that the
dose-dependent increase in dopamine baseline release
induced by phenytoin also involves an inhibition of
dopamine metabolism. The reduced external DOPAC con-
centration produced by clorgyline is explained by the
complete inhibition of dopamine metabolism with con-
comitant decrease in DOPAC formation. Phenytoin failure
to change the external concentration of the dopamine
metabolite is probably explained by the much lower efficacy
of phenytoin than clorgyline to stop dopamine metabolism.
Oppositely to clorgyline, vinpocetine increased external
DOPAC. The mechanism responsible of this increase in
DOPAC exerted by vinpocetine was previously character-
ized by us [30], and does not involve MAO activation, but
rather is likely to be due to an impairment of dopamine
vesicular storage.
The amount of dopamine release under resting conditions
is likely to depend on the concentration of dopamine in the
cytoplasm extravesicular compartment [30]. Therefore, the
impairment of cytoplasm extravesicular dopamine degra-
dation induced by phenytoin and by clorgyline that results in
a rise in the internal dopamine concentration may underlie
the increase in dopamine baseline release induced by those
drugs. Carbamazepine, like phenytoin, also increased
dopamine baseline release. However, the mechanism
underlying the carbamazepine-induced increase in dopa-
mine baseline release is inconsistent with a MAO inhibition.
Because oppositely to phenytoin (and clorgyline) that
increased the internal dopamine concentration, carbamaze-
pine decreased the internal dopamine concentration. The
Net
ver
atri
din
e-in
du
ced
neu
rotr
ansm
itte
r re
leas
ed
op
amin
e
0
15
30
45
60
105
**
**
Glu
tam
ate
5
10
15
20
**
*
Asp
arta
te
0
4
8
**
*
*
*
(a)
(b)
(c)
Fig. 4 Changes induced by clorgyline, phenytoin and carbamazepine
on the net response to veratridine. Striatal synaptosomes were
incubated (37�C) for 10 min in: KRH without or containing
veratridine in the absence or presence of phenytoin, clorgyline or
carbamazepine at the indicated concentrations. Incubation was
stopped by centrifugation. The samples containing the released and
retained neurotransmitters, namely glutamate (a), aspartate (b) or
dopamine (c), were prepared for HPLC analysis and the pellets used
for protein determination. Net veratridine-induced release refers to the
concentration of a specific neurotransmitter released by veratridine
for 10 min in the presence of: 0.1 lM clorgyline (clorg.), phenytoin
(200, 500 or 1500 lM) or carbamazepine (1500 lM) minus the
neurotransmitter released under the respective baseline condition for
10 min (i.e. 0.1 lM clorgyline, 200 lM phenytoin, etcetera.). For (a)
and (b) results are in nmol and for (c) in pmol per mg of striatal
synaptosomal protein. Results are the mean ± SEM values of at least
4 experiments in independent preparations for each point. * P B 0.01,
between control (black bar) and the indicated experimental condition
(grey bars)
476 Neurochem Res (2009) 34:470–479
123

reduction produced by carbamazepine in the internal dopa-
mine concentration, i.e. in the MAO substrate, also may
explain the slight decrease in the amount of external DOPAC
observed in carbamazepine exposed nerve endings. Car-
bamazepine-induced increase in dopamine baseline release
at expense of internal dopamine may involve mechanisms
such as inhibition of the dopamine transporter or carbam-
azepine hetero-exchange with internal dopamine, but not
inhibition of dopamine metabolism. It is important to realize
that high K+ depolarization increases dopamine baseline
release about threefold [45], and the drugs tested here in
about the same extent. Moreover, on the basis of evidence
indicating the presence of presynaptic autoreceptors that
enable catecholamines, including dopamine, to reduce their
own release via feedback modulation [46, 47], it is possible
that the rise on external dopamine induced by depolarization
or by some of the drugs tested here under the present
experimental conditions is underestimated.
Another difference between the two classic antiepileptic
drugs is that phenytoin failed to inhibit and at higher
concentrations even facilitated the veratridine-induced
release of dopamine. Although, the possibility of a cross-
talk between glutamatergic and dopaminergic nerve
endings via presynaptic hetero-receptors underlying this
particular effect of phenytoin cannot be discarded, it is
unlikely. Because carbamazepine, that like phenytoin also
increases dopamine baseline release and inhibits Glu
release to veratridine, does not facilitate the veratridine
induced release of dopamine, and rather decreased it like
vinpocetine. The phenytoin facilitation of veratridine-
induced dopamine release found here also was observed in
microdialysis experiments of the hippocampus in vivo [48].
Our findings that carbamazepine inhibits the release of all
the neurotransmitters tested including dopamine in striatal
isolated nerve endings also are in line with the sensitivity to
carbamazepine of veratrine-induced release of [3H]dopa-
mine in rat cortical slices preloaded with the labeled
neurotransmitter [49], but contrast with the failure of car-
bamazepine to inhibit dopamine release to veratridine in
the hippocampus [48]. One possible explanation of this
failure could be the high veratridine concentration used,
that in neurons of the hippocampal formation in culture
was shown to induce leakage of the cytoplasmic protein
lactate dehydrogenase [50].
Glu followed by Asp are by far the most abundant neu-
rotransmitters even in cerebral nerve endings arriving to the
striatum [45]. Thus, the inhibition of the veratridine induced
release of those excitatory amino acid neurotransmitters
produced by the anticonvulsants carbamazepine, phenytoin
and vinpocetine (and not by clorgyline) might be of par-
ticular importance to stop the paroxysmal neuronal activity.
Lamotrigine is another antiepileptic drug that antagonizes
Glu release induced by activation of cerebral Na+ channels
with a similar potency to carbamazepine and phenytoin [7].
Interestingly, in the presence of a monoamine agonist,
lamotrigine increased its potency to antagonize spinal Na+
channels [51]. Since microglial activation modifies the
accumulation and diffusion parameters of brain neuroactive
substances [40], it is possible that the monoamine-induced
microglial activation produced in the central nervous sys-
tem by cross talk non-synaptic mechanisms [52, 53], further
increases the potency of antiepileptic drugs on Na+ chan-
nels in the epileptic tissue, where inflammatory pathways
are also chronically activated [41].
Our findings that phenytoin failed to inhibit the release
of dopamine triggered by veratridine at the same concen-
trations and under the same experimental conditions that
reduced the veratridine induced release of Glu and Asp
lead, however, to speculate whether different populations
of Na+ channels control the release of the excitatory amino
acid neurotransmitters and dopamine. Nonetheless, in
addition to this antagonism on presynaptic Na+ channels
controlling Glu and Asp release, the classic antiepileptic
drugs, carbamazepine and phenytoin produced an addi-
tional effect on dopamine release and/or metabolism that
may possibly contribute to some of their adverse secondary
effects, such as the dyskinesias and paradoxical seizures
observed in some phenytoin treated patients or the abnor-
mal involuntary movements less frequently caused by
carbamazepine [10–18, 54, 55]. Supporting this idea the
NMDA receptor antagonist MK-801, that has anticonvul-
sant effects [56, 57] and causes disturbances in motor
coordination [58], inhibited the veratridine-evoked release
of Glu and Asp and selectively increased dopamine base-
line release in striatal synaptosomes [45].
Like carbamazepine, phenytoin and MK-801, vinpoce-
tine inhibited veratridine-evoked Glu and Asp release, and
also like carbamazepine and MK-801, vinpocetine inhib-
ited the veratridine-evoked release of dopamine,
confirming its antagonistic action on presynaptic Na+
channels. But oppositely to carbamazepine, phenytoin and
MK-801, vinpocetine did not increase dopamine baseline
release. This failure of vinpocetine to increase dopamine
baseline levels along with its higher potency and efficacy
than several commonly used antiepileptic drugs to prevent
Glu release induced by activation of voltage sensitive
presynaptic channels in cerebral isolated nerve endings
[59] and to prevent seizures in animal models of epilepsy
[8, 60, 61] further supports the vinpocetine potential as an
alternative antiepileptic drug.
Acknowledgments The authors thank Araceli Guarneros for her
excellent technical assistance. This work was partially supported by
grants 225008 from PAPIIT and D-48695 from SEP-CONACYT.
Blanca I. Aldana Garcıa scholarship was also supported by grant
225008 from PAPIIT.
Neurochem Res (2009) 34:470–479 477
123

References
1. Macdonald RL, McLean MJ (1986) Anticonvulsant drugs:
mechanisms of action. Adv Neurol 44:713–736
2. Sitges M, Pena F, Chiu LM et al (1998) Study on the possible
involvement of protein kinases in the modulation of brain presyn-
aptic sodium channels; comparison with calcium channels.
Neurochem Int 32:177–190. doi:10.1016/S0197-0186(97)00065-X
3. Sitges M (1989) Effect of organic and inorganic calcium channel
blockers on s-amino-n-butyric acid release induced by monensin
and veratrine in the absence of external calcium. J Neurochem
53:436–441. doi:10.1111/j.1471-4159.1989.tb07353.x
4. Sitges M, Chiu LM (1995) w-Aga IVA selectively inhibits the
calcium dependent fraction of the evoked release of [3H]GABA
from synaptosomes. Neurochem Res 20:1065–1071. doi:
10.1007/BF00995561
5. Sitges M, Galindo C (2005) Omega-agatoxin-TK is a useful tool
to study P-type Ca2+ channel-mediated changes in internal
Ca2+ and glutamate release in depolarised brain nerve terminals.
Neurochem Int 46:53–60. doi:10.1016/j.neuint.2004.07.004
6. Sitges M, Chiu LM, Nekrassov V (2006) Single and combined
effects of carbamazepine and vinpocetine on depolarization-
induced changes in Na(+), Ca(2+) and glutamate release in
hippocampal isolated nerve endings. Neurochem Int 49:55–61.
doi:10.1016/j.neuint.2005.12.019
7. Sitges M, Chiu LM, Nekrassov V (2007) Effects of carbamaze-
pine, phenytoin, lamotrigine, oxcarbazepine, topiramate and
vinpocetine on Na+ channel-mediated release of [3H]glutamate
in hippocampal nerve endings. Neuropharmacology 52:598–605.
doi:10.1016/j.neuropharm.2006.09.002
8. Nekrassov V, Sitges M (2006) Additive effects of antiepileptic
drugs and pentylenetetrazole on hearing. Neurosci Lett 406:276–
280. doi:10.1016/j.neulet.2006.07.042
9. Hindmarch I, Fuchs HH, Erzigkeit H (1991) Efficacy and toler-
ance of vinpocetine in ambulant patients suffering from mild to
moderate organic psychosyndromes. Int Clin Psychopharmacol
6:31–34. doi:10.1097/00004850-199100610-00005
10. Caksen H, Odabas D, Anlar O (2003) Use of biperiden hydro-
chloride in a child with severe dyskinesia induced by phenytoin. J
Child Neurol 18:494–496. doi:10.1177/08830738030180070101
11. Girija A (2002) Paroxysmal dyskinesia in phenytoin toxicity. J
Assoc Physicians India 50:1449–1450
12. Montenegro M, Scotoni A, Cendes F (1999) Dyskinesia induced
by phenytoin. Arq Neuropsiquiatr 57:356–360
13. Chaudhary N, Ravat S, Shah P (1998) Phenytoin induced dys-
kinesia. Indian Pediatr 35:274–276
14. Sethi K, Hitri A, Diamond B (1990) Phenytoin potentiation of
neuroleptic-induced dyskinesias. Mov Disord 5:325–327. doi:
10.1002/mds.870050413
15. Dravet C, Dalla Bernardina B, Mesdjian E et al (1980) Parox-
ysmal dyskinesia during treatment with diphenylhydantoin. Rev
Neurol (Paris) 136:1–14
16. Nausieda P, Koller W, Weiner W et al (1979) Clinical and
experimental studies of phenytoin-induced hyperkinesias. J
Neural Transm 45:291–305. doi:10.1007/BF01247146
17. Jacome D (1979) Carbamazepine-induced dystonia. JAMA
241:2263. doi:10.1001/jama.241.21.2263b
18. Schwartzman M, Leppik I (1990) Carbamazepine-induced dys-
kinesia and ophthalmoplegia. Cleve Clin J Med 57:367–372
19. Yao H, Sadoshima S, Ishitsuka T et al (1988) Massive striatal
dopamine release in acute cerebral ischemia in rats. Experientia
44:506–508. doi:10.1007/BF01958929
20. Kawano T, Tsutsumi K, Miyake H et al (1988) Striatal dopamine
in acute cerebral ischemia of stroke-resistant rats. Stroke
19:1540–1543
21. Slivka A, Brannan T, Weinberger J et al (1988) Increase in
extracellular dopamine in the striatum during cerebral ischemia: a
study utilizing cerebral microdialysis. J Neurochem 50:1714–
1718. doi:10.1111/j.1471-4159.1988.tb02468.x
22. Hillered L, Hallstrom A, Segersvard S et al (1989) Dynamics of
extracellular metabolites in the striatum after middle cerebral
artery occlusion in the rat monitored by intracerebral microdi-
alysis. J Cereb Blood Flow Metab 9:607–616
23. Toner CC, Stamford JA (1999) Effects of metabolic alterations on
dopamine release in an in vitro model of neostriatal ischaemia.
Brain Res Bull 48:395–399. doi:10.1016/S0361-9230(99)00016-7
24. Freitas R, Oliveira Ade A, Vasconcelos SM et al (2006)
Expression of muscarinic and dopaminergic receptors and
monoamine levels frontal cortex of epileptic rats. Pharmacol
Biochem Behav 83:302–306. doi:10.1016/j.pbb.2006.02.011
25. Balestreri R, Fontana L, Astengo F (1987) A double-blind pla-
cebo controlled evaluation of the safety and efficacy of
vinpocetine in the treatment of patients with chronic vascular
senile cerebral dysfunction. J Am Geriatr Soc 35:425–430
26. King GA (1987) Protective effects of vinpocetine and structurally
related drugs on the lethal consequences of hypoxia in mice. Arch
Int Pharmacodyn Ther 286:299–307
27. Sauer D, Rischke R, Beck T et al (1988) Vinpocetine prevents
ischemic cell damage in rat hippocampus. Life Sci 43:1733–
1739. doi:10.1016/0024-3205(88)90485-7
28. Rischke R, Krieglstein J (1990) Effects of vinpocetine on local
cerebral blood flow and glucose utilization seven days after
forebrain ischemia in the rat. Pharmacology 41:153–160. doi:
10.1159/000138712
29. Araki T, Kogure K, Nishioka K (1990) Comparative neuropro-
tective effects of pentobarbital, vinpocetine, flunarizine and
ifenprodil on ischemic neuronal damage in the gerbil hippo-
campus. Res Exp Med (Berl) 190:19–23
30. Trejo F, Nekrassov V, Sitges M (2001) Characterization of vinp-ocetine effects on DA and DOPAC release in striatal isolated nerve
endings. Brain Res 909:59–67. doi:10.1016/S0006-8993(01)
02621-X
31. Willow M, Kuenzel EA, Catterall WA (1984) Inhibition of
voltage-sensitive sodium channels in neuroblastoma cells and
synaptosomes by the anticonvulsant drugs diphenylhydantoin and
carbamazepine. Mol Pharmacol 25:228–234
32. Deffois A, Fage D, Carter C (1996) Inhibition of synaptosomal
veratridine-induced sodium influx by antidepressants and neuro-
leptics used in chronic pain. Neurosci Lett 220:117–120. doi:
10.1016/S0304-3940(96)13227-4
33. Bonifacio MJ, Sheridan RD, Parada A et al (2001) Interaction of
the novel anticonvulsant, BIA 2–093, with voltage-gated sodium
channels: comparison with carbamazepine. Epilepsia 42:600–
608. doi:10.1046/j.1528-1157.2001.43600.x
34. Santangeli S, Sills GJ, Thompson GG et al (2002) Na+ channel
effects of remacemide and desglycinyl-remacemide in rat cortical
synaptosomes. Eur J Pharmacol 438:63–68. doi:10.1016/S0014-
2999(02)01297-9
35. Lingamaneni R, Hemmings HCJ (2003) Differential interaction
of anaesthetics and antiepileptic drugs with neuronal Na+
channels, Ca2+ channels, and GABA(A) receptors. Br J Anaesth
90:199–211. doi:10.1093/bja/aeg040
36. Tretter L, Adam-Vizi V (1998) The neuroprotective drug vinpoce-
tine prevents veratridine-induced [Na+]i and [Ca2+]i rise in
synaptosomes. NeuroReport 9:1849–1853. doi:10.1097/00001756-
199806010-00034
37. Sitges M, Nekrassov V (1999) Vinpocetine selectively inhibits
neurotransmitter release triggered by sodium channel activa-
tion. Neurochem Res 24:1585–1591. doi:10.1023/A:102116
4418478
478 Neurochem Res (2009) 34:470–479
123

38. Sitges M, Galvan E, Nekrassov V (2005) Vinpocetine blockade
of sodium channels inhibits the rise in sodium and calcium
induced by 4-aminopyridine in synaptosomes. Neurochem Int
46:533–540. doi:10.1016/j.neuint.2005.02.001
39. Selmeczy Z, Vizi E, Csoka B et al (2008) Role of nonsynaptic
communication in regulating the immune response. Neurochem
Int 52:52–59. doi:10.1016/j.neuint.2007.06.001
40. Sykova E, Vargova L (2008) Extrasynaptic transmission and the
diffusion parameters of the extracellular space. Neurochem Int
52:5–13. doi:10.1016/j.neuint.2007.04.007
41. Ravizza T, Gagliardi B, Noe F et al (2008) Innate and adaptive
immunity during epileptogenesis and spontaneous seizures: evi-
dence from experimental models and human temporal lobe epilepsy.
Neurobiol Dis 29:142–160. doi:10.1016/j.nbd.2007.08.012
42. Vezzani A, Balosso S, Ravizza T (2008) The role of cytokines in
the pathophysiology of epilepsy. Brain Behav Immun 22:797–
803
43. Shih J, Chen K, Ridd M (1999) Monoamine oxidase: from genes
to behavior. Annu Rev Neurosci 22:197–217
44. Adachi Y, Watanabe K, Higuchi H et al (2001) Oxygen inhalation
enhances striatal dopamine metabolism and monoamineoxidase
enzyme inhibition prevents it: a microdialysis study. Eur J Phar-
macol 422:61–68. doi:10.1016/S0014-2999(01)01074-3
45. Sitges M, Nekrassov V, Guarneros A (2000) Simultaneous action
of MK-801 (dizclopine) on dopamine, glutamate, aspartate and
GABA release from striatum isolated nerve endings. Brain Res
854:48–56. doi:10.1016/S0006-8993(99)02282-9
46. Langer S (2008) Presynaptic autoreceptors regulating transmitter
release. Neurochem Int 52:26–30
47. Lajtha A (2008) Interrelated mechanisms in reward and learning.
Neurochem Int 52:73–79. doi:10.1016/j.neuint.2007.08.019
48. Ahmad S, Fowler L, Whitton P (2005) Lamotrigine, carbamaz-
epine and phenytoin differentially alter extracellular levels of
5-hydroxytryptamine, dopamine and amino acids. Epilepsy Res
63:141–149. doi:10.1016/j.eplepsyres.2005.02.002
49. Waldmeier P, Baumann P, Wicki P et al (1995) Similar potency
of carbamazepine, oxcarbazepine, and lamotrigine in inhibiting
the release of glutamate and other neurotransmitters. Neurology
45:1907–1913
50. Pauwels P, Van Assouw L, Peeters L et al (1990) Neurotoxic
action of veratridine in rat brain neuronal cultures: mechanism of
neuroprotection by Ca++ antagonists nonselective for slow
Ca++ channels. J Pharmacol Exp Ther 255:1117–1122
51. Than M, Kocsis P, Tihany K et al (2007) Concerted action of
antiepileptic and antidepressant agents to depress spinal
neurotransmission: possible use in the therapy of spasticity and
chronic pain. Neurochem Int 50:642–652. doi:10.1016/j.neuint.
2006.12.008
52. Orio L, O’Shea E, Pradillo J et al (2004) 3,4-Methylenedioxy-
methamphetamine increases interleukin-1beta levels and
activates microglia in rat brain: studies on the relationship with
acute hyperthermia and 5-HT depletion. J Neurochem 89:1445–
1453. doi:10.1111/j.1471-4159.2004.02443.x
53. Zhang L, Shirayama Y, Shimizu E et al (2006) Protective effects
of minocycline on 3,4-methylenedioxymethamphetamine-
induced neurotoxicity in serotonergic and dopaminergic neurons
of mouse brain. Eur J Pharmacol 544:1–9. doi:10.1016/j.ejphar.
2006.05.047
54. Lazarus A (1994) Tardive dyskinesia-like syndrome associated
with lithium and carbamazepine. J Clin Psychopharmacol
14:146–147. doi:10.1097/00004714-199404000-00012
55. Chua H, Venketasubramanian N, Tan C et al (1999) Paradoxical
seizures in phenytoin toxicity. Singapore Med J 40:276–277
56. Clifford D, Olney J, Benz A et al (1994) Ketamine, phencycli-
dine, and MK-801 protect against kainic acid-induced seizure-
related brain damage. Epilepsia 31:382–390. doi:10.1111/
j.1528-1157.1990.tb05492.x
57. Peterson S (1995) Infusion of NMDA antagonists into the nucleus
reticularis pontis oralis inhibits the maximal electroshock seizure
response. Brain Res 702:101–109. doi:10.1016/0006-8993(95)
01026-2
58. Carter A (1994) Many agents that antagonize the NMDA
receptor-channel complex in vivo also cause disturbances of
motor coordination. J Pharmacol Exp Ther 269:573–580
59. Sitges M, Guarneros A, Nekrassov V (2007) Effects of carbam-
azepine, phenytoin, valproic acid, oxcarbazepine, lamotrigine,
topiramate and vinpocetine on the presynaptic Ca2+ channel-
mediated release of [3H]glutamate: comparison with the Na+
channel-mediated release. Neuropharmacology 53:854–862. doi:
10.1016/j.neuropharm.2007.08.016
60. Nekrassov V, Sitges M (2004) Vinpocetine inhibits the epileptic
cortical activity and auditory alterations induced by pentylen-
etetrazole in the guinea pig in vivo. Epilepsy Res 60:63–71. doi:
10.1016/j.eplepsyres.2004.05.005
61. Sitges M, Nekrassov V (2004) Vinpocetine prevents 4-amino-
pyridine-induced changes in the EEG, the auditory brainstem
responses and hearing. Clin Neurophysiol 115:2711–2717. doi:
10.1016/j.clinph.2004.06.019
Neurochem Res (2009) 34:470–479 479
123
Copyright © 2022 FDOKUMEN




















