
A biorefinery for integral valorisation of avocado peel and ...
Upload
independentCategory
view
1download
0
Carbon Dioxide in Biomass Processing: Contributions to the GreenBiorefinery ConceptAna R. C. Morais, Andre M. da Costa Lopes, and Rafał Bogel-Łukasik*
Unidade de Bioenergia, Laboratorio Nacional de Energia e Geologia, I.P., Estrada do Paco do Lumiar 22, 1649-038 Lisboa, Portugal
CONTENTS
1. Introduction A1.1. Biomass as Feedstock for Biorefinery A1.2. Supercritical Fluid Technology C
1.2.1. scCO2 and Its Place in Green Chemistry C1.2.2. High-Pressure CO2/H2O Technology D
2. Scope of the Review E3. Supercritical CO2 Pretreatment E
3.1. Process Condition Effects I3.1.1. Water Influence I3.1.2. Biomass Sources and Recalcitrance J3.1.3. CO2:Biomass Ratio J
3.2. Effect of High-Pressure CO2 on the Morphol-ogy of Lignocellulosic Biomass K
4. Enzymatic Hydrolysis in Supercritical CO2 K5. High-Pressure CO2/H2O Conversion Technologies M
5.1. Hydrolysis of Biomass-Derived Carbohy-drates M
5.1.1. Hemicellulose M5.1.2. Cellulose N5.1.3. Starch O
6. Hexose-Derived Sugar Conversion in High-Pressure CO2 and CO2/H2O Mixtures O
7. Conversion of Biomass-Derived Compounds inHigh-Pressure CO2 and CO2/H2O Mixtures Q7.1. Hydrogenation Q7.2. Conversion of Proteins R7.3. Delignification and Lignin Extraction under
scCO2 R7.4. Lignin Depolymerization S
8. Overview of CO2 Applications within the Bio-refinery Concept T
9. Economic Aspects of CO2 Processing of Biomass T10. Perspectives T11. Conclusions UAuthor Information U
Corresponding Author UNotes UBiographies U
Acknowledgments VReferences V
1. INTRODUCTION
The 21st century is witnessing a huge demand of fossil reservescoupled with a rapid reduction in readily and economicallyreachable oil feedstocks.1,2 The present energy demand is notfulfilled from fossil fuel sources, making the world exposed togeopolitical risk. Furthermore, concerns regarding the securityof the supply chain and the environmental impacts haveresulted in an ever-increasing shift of global energy policies toseek alternative technologies and sustainable sources of energy,materials, chemicals, and value-added products.1 Recently, theneed for development of an economy based on renewableresources has been recognized by society, and diverse R&Dactivities have started to be funded to accomplish this aim.3
However, generation of bioproducts based on sustainablesupply chains poses vast challenges for an eco-based economy.The simplest way to provide a supportable supply chain isthrough the employment of renewable biomass feedstocks,which is the only sustainable option to substitute for fossil fuelresources, as sources of organic compounds over a relativelyshort time scale and with limitless supply. All these factors havereinforced the need for research on production of biomass-derived commodities produced in a sustainable manner.4 Thebiorefinery concept considers the use of biomass as a low-costfeedstock for the chemical and biological industries. The mostwidely used description of biorefinery is a definition adopted byInternational Energy Agency Bioenergy Task 42. It states thatbiorefining is the sustainable processing of biomass into aspectrum of marketable products and energy.5 In other words,the biorefinery is a term used to define industrial facilities thatcover an extensive range of combined technologies in whichbiomass is transformed and converted, in a sustainable manner,into a wide range of value-added products, leading to directsimilarities to today’s petrorefineries. Following this idea, theaim of future biorefineries is the extraction of high-valuechemicals present in biomass, such as flavoring agents,fragrances, and nutraceuticals and, in the next step, processingof biomass-derived polysaccharides, lignin, and proteins towardbioderived materials, fuels, and other commodities.6
Received: June 23, 2014
Review
pubs.acs.org/CR
© XXXX American Chemical Society A dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXX

1.1. Biomass as Feedstock for Biorefinery
The worldwide production of lignocellulosic biomass isestimated as 1010 million tons.7 The lignocellulosic materials(LCMs) include hardwoods, softwoods, and residues fromagricultural and forest activity as well as energy crops. LCMsdemonstrate several advantages, namely, their lower price incomparison to that of traditional agricultural feedstocks, notrequiring arable land and fertilizers to grow, and not competingwith food and feed sectors.8,9 The primary metabolites ofLCMs are polysaccharides, for example, cellulose and hemi-cellulose, plus lignin as depicted in Figure 1. Secondary
compounds such as waxes, terpenoids, proteins, and phenoliccompounds such as lignans account for up to 10% of the dryweight of LCMs.10 All these fractions could be extracted andlater converted into valuable products, making the valorizationof biomass more sustainable. In addition, the starchylignocellulosic biomasses, such as waste from starch processingfactories, e.g., potato processing, are important sources ofanother carbohydrate for valorization.11
Cellulose is one of the main fractions present in biomass andis the world’s most abundant biopolymer. It is a homopoly-saccharide composed of glucose units linked by β-1,4-glycosidicbonds in which hydroxyl groups are oriented to arrange strongintra- and intermolecular hydrogen bonds. This internalorganization results in ordered micro- and macrofibrils, whichare present in crystalline and amorphous structures.14 Thepresence of intramolecular bonds inside glucose monomers andintermolecular bonds between various chains contributes to acomplex supramolecular structure. The cellulose linearity,crystallinity, and fibrous structure are responsible for themechanical strength of the plant cell.Hemicellulose in turn can be described as a heteropolymer
located in the primary and secondary plant cell walls. It iscomposed of various monosaccharides, including D-glucose, D-xylose, and D-mannose, and displays both branched andamorphous structures. The rate of hydration depends on thesugar type and decreases in the following order: xylose,mannose, and glucose. Thus, hemicellulose is more susceptibleto hydrolysis than cellulose, and dehydration of hexosesproduces 5-hydroxymethylfurfural (5-HMF), while furfural isproduced from the pentose fraction.15
Lignin is a complex network of three different phenylpropaneunits, namely, p-hydroxyphenyl, guaiacyl, and syringyl. Addi-tionally, lignin is attached to hemicellulose by covalent linkages,helping to establish the rigid matrix of LCMs.Starch is an α-liked polysaccharide composed of two
components with various molecular masses: 20−30% linearamylose and 70−80% 1,6-α-linked branched amylopectin. Theefficiency of starch hydrolysis depends on the potential for acidsto break down a starch granule and the physical distributionand structure of each starch component. As with cellulose,starch hydrolysis gives D-glucose and its degradation products,such as 5-HMF, levulinic acid, and formic acid.16 When starchis partially hydrolyzed, it produces maltodextrins, nutritivesaccharide polymers, and nonsweet products used astexturizers, fat substitutes, freezing aids, and filming controlagents among other applications.17
The diverse composition of biomass and the interactionsbetween fractions make its structure very complex and resistantto deconstruction. The molecular interactions define a complexand recalcitrant structure,18 which is difficult to hydrolyze andthus leads to harsh and expensive fractionation methods.19
Thus, processing of this feedstock into reactive intermediates,which can be used as building blocks for fuels and chemicals, isvery difficult.20 The production of biofuels or other value-addedproducts from these kinds of biomasses comprises several steps,such as hydrolysis of immobilized polysaccharides (mostlyhemicellulose) and their further conversion, separation of ligninfrom the remaining residue, and purification of the finalproducts.1 Despite dozens of years of research, the hydrolysis ofpolysaccharides into fermentable sugars is still the majorobstacle, since it depends on many structural, compositional,and physicochemical factors. These factors have a significanteffect on the entire biomass valorization process.1 The currentvalorization technologies are still mostly characterized by lowyields and high costs, which burden the production ofchemicals and fuels at competitive costs. Thus, the choice ofthe technology employed as a biomass processing methodshould be based on the characteristics of each biomass and theimpact of the process on the further valorization pathway by,for instance, production of toxic compounds for metabolism bymicroorganisms, energy demand, and waste production.21,22
Because of this, pretreatment is one of the most challengingsteps in biorefinery since it is among the most expensive andhas the most influence on subsequent stages such as hydrolysis,fermentation, and downstream processing.23−25
Several pretreatment technologies have been under ex-haustive research for many years. Among them are conven-tional (e.g., dilute acid hydrolysis, alkaline,20 hydrothermal(steam explosion,26 and autohydrolysis27)) and novel (ionicliquids28−30 and sub/supercritical fluids31−34) processes. Theconventional pretreatments lead generally to low sugar yields,are unselective, and normally involve more severe conditions.Thus, they favor the formation of degradation products,resulting in a decrease of the efficiency of the process, higherinvestment needs, and elevated processing costs.35,36 Addition-ally, there is a continuous need for newer and cleaner methodsof biomass processing with less energy demand and lower wastegeneration in which the product of interest can be formed,isolated, and separated from the biomass at competitive costs.Zhu and Pan published a review on conventional pretreatmentprocesses (supercritical CO2 (scCO2) is not included) in whichthe aspects related to energy demands of each technologydiscussed are presented. They showed serious concerns
Figure 1. Schematic presentation of the lignocellulosic biomassstructure. Adapted with permission from refs 12 and 13. Copyright1995 Elsevier and 2013 InTech, respectively. Chapter 16 of ref 13 wasdistributed under the terms of the Creative Commons AttributionLicense.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXB
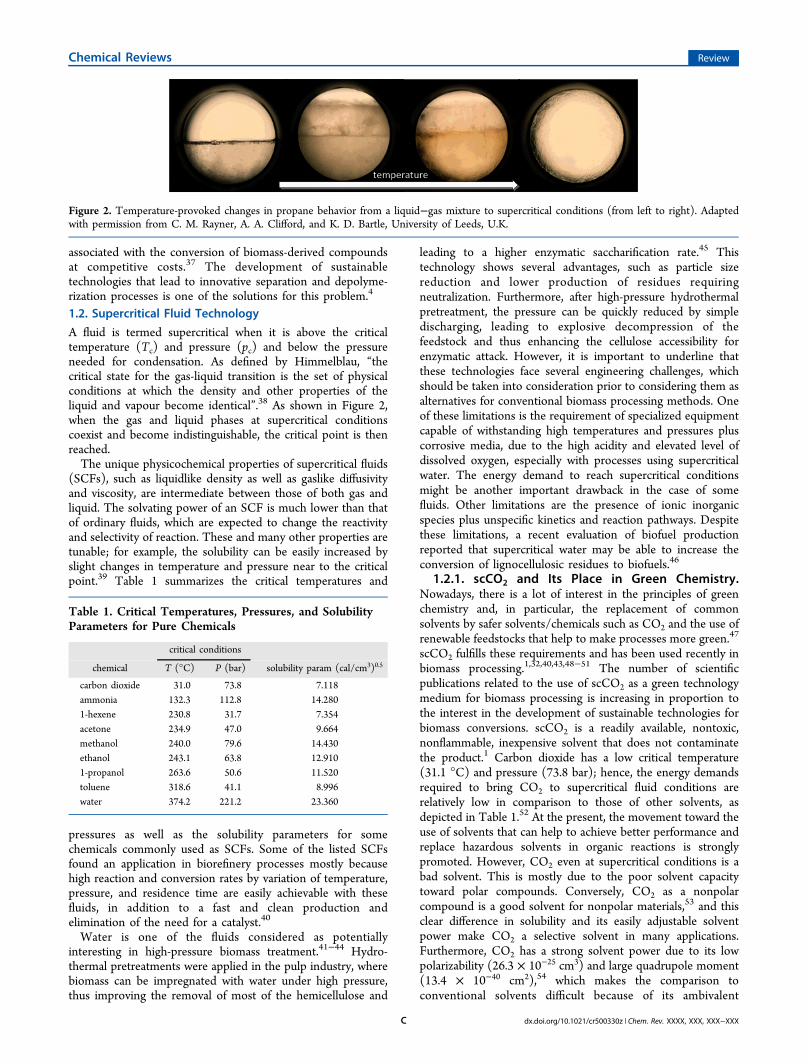
associated with the conversion of biomass-derived compoundsat competitive costs.37 The development of sustainabletechnologies that lead to innovative separation and depolyme-rization processes is one of the solutions for this problem.4
1.2. Supercritical Fluid Technology
A fluid is termed supercritical when it is above the criticaltemperature (Tc) and pressure (pc) and below the pressureneeded for condensation. As defined by Himmelblau, “thecritical state for the gas-liquid transition is the set of physicalconditions at which the density and other properties of theliquid and vapour become identical”.38 As shown in Figure 2,when the gas and liquid phases at supercritical conditionscoexist and become indistinguishable, the critical point is thenreached.The unique physicochemical properties of supercritical fluids
(SCFs), such as liquidlike density as well as gaslike diffusivityand viscosity, are intermediate between those of both gas andliquid. The solvating power of an SCF is much lower than thatof ordinary fluids, which are expected to change the reactivityand selectivity of reaction. These and many other properties aretunable; for example, the solubility can be easily increased byslight changes in temperature and pressure near to the criticalpoint.39 Table 1 summarizes the critical temperatures and
pressures as well as the solubility parameters for somechemicals commonly used as SCFs. Some of the listed SCFsfound an application in biorefinery processes mostly becausehigh reaction and conversion rates by variation of temperature,pressure, and residence time are easily achievable with thesefluids, in addition to a fast and clean production andelimination of the need for a catalyst.40
Water is one of the fluids considered as potentiallyinteresting in high-pressure biomass treatment.41−44 Hydro-thermal pretreatments were applied in the pulp industry, wherebiomass can be impregnated with water under high pressure,thus improving the removal of most of the hemicellulose and
leading to a higher enzymatic saccharification rate.45 Thistechnology shows several advantages, such as particle sizereduction and lower production of residues requiringneutralization. Furthermore, after high-pressure hydrothermalpretreatment, the pressure can be quickly reduced by simpledischarging, leading to explosive decompression of thefeedstock and thus enhancing the cellulose accessibility forenzymatic attack. However, it is important to underline thatthese technologies face several engineering challenges, whichshould be taken into consideration prior to considering them asalternatives for conventional biomass processing methods. Oneof these limitations is the requirement of specialized equipmentcapable of withstanding high temperatures and pressures pluscorrosive media, due to the high acidity and elevated level ofdissolved oxygen, especially with processes using supercriticalwater. The energy demand to reach supercritical conditionsmight be another important drawback in the case of somefluids. Other limitations are the presence of ionic inorganicspecies plus unspecific kinetics and reaction pathways. Despitethese limitations, a recent evaluation of biofuel productionreported that supercritical water may be able to increase theconversion of lignocellulosic residues to biofuels.46
1.2.1. scCO2 and Its Place in Green Chemistry.Nowadays, there is a lot of interest in the principles of greenchemistry and, in particular, the replacement of commonsolvents by safer solvents/chemicals such as CO2 and the use ofrenewable feedstocks that help to make processes more green.47
scCO2 fulfills these requirements and has been used recently inbiomass processing.1,32,40,43,48−51 The number of scientificpublications related to the use of scCO2 as a green technologymedium for biomass processing is increasing in proportion tothe interest in the development of sustainable technologies forbiomass conversions. scCO2 is a readily available, nontoxic,nonflammable, inexpensive solvent that does not contaminatethe product.1 Carbon dioxide has a low critical temperature(31.1 °C) and pressure (73.8 bar); hence, the energy demandsrequired to bring CO2 to supercritical fluid conditions arerelatively low in comparison to those of other solvents, asdepicted in Table 1.52 At the present, the movement toward theuse of solvents that can help to achieve better performance andreplace hazardous solvents in organic reactions is stronglypromoted. However, CO2 even at supercritical conditions is abad solvent. This is mostly due to the poor solvent capacitytoward polar compounds. Conversely, CO2 as a nonpolarcompound is a good solvent for nonpolar materials,53 and thisclear difference in solubility and its easily adjustable solventpower make CO2 a selective solvent in many applications.Furthermore, CO2 has a strong solvent power due to its lowpolarizability (26.3 × 10−25 cm3) and large quadrupole moment(13.4 × 10−40 cm2),54 which makes the comparison toconventional solvents difficult because of its ambivalent
Figure 2. Temperature-provoked changes in propane behavior from a liquid−gas mixture to supercritical conditions (from left to right). Adaptedwith permission from C. M. Rayner, A. A. Clifford, and K. D. Bartle, University of Leeds, U.K.
Table 1. Critical Temperatures, Pressures, and SolubilityParameters for Pure Chemicals
critical conditions
chemical T (°C) P (bar) solubility param (cal/cm3)0.5
carbon dioxide 31.0 73.8 7.118ammonia 132.3 112.8 14.2801-hexene 230.8 31.7 7.354acetone 234.9 47.0 9.664methanol 240.0 79.6 14.430ethanol 243.1 63.8 12.9101-propanol 263.6 50.6 11.520toluene 318.6 41.1 8.996water 374.2 221.2 23.360
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXC

properties. These characteristics make CO2 a special solvent formonomers but a poor solvent for polymers, leading to easyseparation as well as faster impregnation in the complexstructure of LCMs, in contrast to other solvents. Although theuse of scCO2 brings the environmental benefits depicted above,the economic viability should be demonstrated, to make thistechnology appropriate for use in industrial processes withinthe biorefinery concept.1.2.2. High-Pressure CO2/H2O Technology. High-
pressure CO2/H2O mixtures are broadly used in acid-catalyzedreactions.55,56 This is consistent with Sheldon’s statement in hisrecently published critical review, “...the use of water andsupercritical CO2 as reaction media is also consistent with thecurrent trend towards the use of renewable, biomass-based rawmaterials...”.57 His statement clearly shows that water is animportant medium and together with CO2 will play animportant role in novel methodologies of biomass processing.Thus, knowledge concerning the phase behavior (Figure 3) of
the mixture of CO2 and H2O is crucial for high efficiency of thehigh-pressure CO2/H2O processes. This knowledge could beuseful to tune the reaction conditions to the processrequirements for maximal efficiency.The updated research on the importance of water in biomass
processing reveals that the presence of H2O is an advantage,since it allows for high sugar yields, increasing the effectivenessof the process.59 At pressures ≥200 bar and temperatures
between 160 and 250 °C, the CO2-rich phase has a density verysimilar to that of a liquid containing at least 30 mol % water,while a water-rich phase contains up to 2 mol % CO2.
59
Another important aspect of this binary system is that CO2/H2O leads to in situ formation of carbonic acid, whichdissociates in two stages according to the following equation:
+ ↔ +
+ ↔ +
− +
− − +
CO 2H O HCO H O ;
HCO H O CO H O2 2 3 3
3 2 32
3 (1)
Thus, CO2 dissolved in water promotes acid-catalyzeddissolution of the biomass by formation of carbonic acid.When formation of carbonic acid occurs, an increase inhydronium ion concentration is observed due to thedissociation of the unstable acid. This leads to a decrease inthe pH value (slightly above 3), sufficient to promotedissolution and hydrolysis of hemicellulose32 into its corre-sponding sugars and leading, at the same time, to high celluloseenzymatic digestibility.60 Furthermore, the acidity of themedium does not constitute an environmental problem becausethe depressurization step removes CO2, thus increasing the pHof the solution. Apart from the chemical hydrolysis based onthe in situ acidic environment, the physical effect of CO2 is alsonoticeable. This physical phenomenon is based on the fact thatpolysaccharides react with liquid hot water and the CO2 acts asa catalyst, which under supercritical conditions leads to highdiffusivities and promotes the swelling effect of biomass.61,62
At the depressurization step, CO2 can also act as adetoxification medium, and due to the solubility of somecompounds in high-pressure CO2, it can help to removedegradation products such as furans from the hydrolysate.32,63
Furthermore, another complementary advantage of SCFs,including scCO2, is that the product streams are completelysterilized in regard to pathogens, viruses, and biotoxins.64−66
Summarizing, scCO2 pretreatment with H2O offers the samebenefits as acid hydrolysis without the typical drawbacksobserved for sulfuric acid application.
1.2.2.1. Reaction Severity. To evaluate the effect of CO2 onthe reaction severity, where the pH of the system isindispensable, a combined severity (CS) factor was proposed:60
= −RCS log( ) pH0 (2)
where R0 is the severity factor. Both the solubility of CO2 inwater and the dissociation of H2CO3 determine the hydroniumion concentration in the system. The solubility of CO2 in watercan be estimated using Henry’s constant and depends on thetemperature.60 Furthermore, the dissociation of H2CO3 inwater also depends on the temperature, and its constant can beexpressed by the following equation:60
= − +KT
Tp2382.3
8.513 0.02194a (3)
where the pKa is the negative decimal logarithm value of theacid dissociation constant (Ka) and T is the temperatureexpressed in degrees Celsius. Taking into account the effect ofthe temperature, partial pressure of CO2, solubility of CO2 inwater, and solubility of H2CO3 in water, van Walsum et al.60
proposed an equation which estimates the value of the pH inthe binary CO2/water system:
= × + −
+
− T T ppH (8.00 10 ) 0.00209 0.216 ln( )
3.92
6 2CO2
(4)
Figure 3. Temperature−pressure−composition phase diagram of thebinary system CO2/H2O. Reprinted with permission from ref 58.Copyright 1964 American Journal of Science.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXD

where the temperature was in the range of 100−250 °C and thepartial pressure of CO2 was up to 151.9 bar. To assess thecombined severity factor of the reaction at a specifictemperature, reaction time, and partial pressure of CO2, thecombined severity factor is depicted by the following equation:
= − × −
+ −
−R T T
p
CS log( ) (8.00 10 0.00209
0.216 ln( ) 3.92
p 06 2
CO
CO2
2 (5)
where CSpCO2 indicates the CS for reaction with CO2, T is the
temperature in degrees Celsius, and pCO2is the partial pressure
of CO2 expressed in atmospheres.60 The proposed combinedseverity factor reflects all the most important parametersaffecting the sugar production yields in the high-pressure CO2/H2O process. On the other hand, it is important to highlightthat the use of a severity factor should facilitate the comparisonof results between process conditions. However, very often alack of information about the pH value, process parameterssuch as the heating profile, and differences between substratesmake the comparison of literature data a difficult task.1.2.2.2. Corrosion in High-Pressure CO2/H2O Systems.
Considering the high compressibility of high-density fluids,special attention should be given to the possible corrosionissues caused by the reaction environment. Corrosion has asignificant influence on the safety of the high-pressureinstallation as well as on the economics of the process. Thereis no universal material which could be applied and whichwould withstand the reaction conditions across the existingdiverse technologies.67 The stainless steels are the mostcommonly used materials for reactor manufacturing due tothe relative resistance against reaction corrosive conditions andbecause of their much lower price than that of other alloys suchas Hastelloy, zirconium, or titanium. Considering the reactionscarried out in high-pressure CO2/H2O systems, it is worthemphasizing that the rate of material corrosion is predom-inantly affected by the solubility of the protective oxides.However, the oxide solubility is highly influenced by thedissociation of acids and bases and also by the pH of thesolutions thus formed. In high-pressure CO2/H2O systems oneof the main issues is an in situ formation of carbonic acidaccording to eq 1.68 The formation of carbonic acid depends onthe CO2 partial pressure and temperature, and consequently,the pH of the solution changes, affecting the corrosion rate.69 Itmust not be forgotten that, during biomass processing,corrosion induced by addition of CO2 is not a sole functionof the CO2 partial pressure and temperature but also of otherchemicals formed during reaction, e.g., organic acids such asacetic or formic acids. Both acids are known as strong organicacids and are recognized as strong corrosive agents with acorrosion power superior to that of carbonic acid.70 At certainconditions the corrosion force of acetic and formic acids can becompared even to the corrosive environment of sulfuric acidused often in acid hydrolysis.70
2. SCOPE OF THE REVIEWThe use of carbon dioxide under sub- or supercriticalconditions in biomass processing is a field of research still inits infancy but under rapid development in recent years. Thegrowing interest in the use of sub- or supercritical CO2 as aversatile tool in biomass processing for the hydrolysis ofbiomass fractions, such as cellulose, starch, hemicellulose,lignin, and proteins, to produce various bio-based materials and
chemicals is reflected in the number of publications available.The unique properties of CO2 as a supercritical fluid open awide variety of opportunities in the field of biorefinery, and it isexpected that the use of scCO2 is going to grow even more inthe near future.This review presents the current state of the art of biomass
processing using carbon dioxide and its mixture with water for awide range of raw materials. This work focuses on the role ofsupercritical CO2 as a tool for pretreatment to enhance theenzymatic digestibility to convert lignocellulosic/starch biomassinto biofuels and biochemicals. Especially, in the field ofenzymatic hydrolysis, the saccharification of biomass in high-pressure CO2 to improve bioconversion to ethanol is addressed.It also addresses the effect of high-pressure CO2 in theconversion of polysaccharides and proteins for the productionof various biochemicals. Additionally, this review depicts therole of CO2 in both the delignification process and conversionof lignin. Finally, critical issues and prospectives in the field ofCO2-assisted hydrothermal technologies are also enhanced.The present review summarizes the current achievements in theaforementioned areas published in scientific periodicals up tothe end of September 2014.
3. SUPERCRITICAL CO2 PRETREATMENTSupercritical carbon dioxide is mostly used as an extractionmedium but is considered for other nonextractive applications,such as pretreatment of biomass.40,71,72 This is mostly due tothe size of CO2 molecules being similar to that of both H2Oand ammonia. Also, at high pressures CO2 easily penetratesinto the small pores of recalcitrant LCMs, resulting in structuralchanges and enhancing the glucan and xylan accessibility forenzymatic hydrolysis. In the case of CO2 explosion, the quickrelease of CO2 facilitates the disruption of cellulose structure,decreasing the degree of crystallinity, enhancing the perme-ability of cellulose, and increasing the accessibility of enzymesto an extended surface area.73 Liu et al. performed a study onthe capacity of high-pressure CO2 to pretreat variouslignocellulosic residues such as corncob, cornstalk, and ricestraw.74 It was shown that, to reach the maximal reducing sugaryield, the most important factors were the temperature andmoisture content. However, they also highlighted theimportance of CO2 presence in biomass pretreatment for theaforementioned reasons.The interest in using enzymes to hydrolyze polysaccharides
arises because the overall cost of enzymatic hydrolysis is lowerthan that of acid hydrolysis. This is mostly because enzymes areusually used at milder conditions (pH 4.8 and temperatures of45−50 °C), thus causing less corrosion problems.75 Further-more, enzymes are considered as green catalysts since theyshow exceptional advantages, such as high chemo- andregioselectivity, minimizing the formation of coproducts, andcan be reused without loss of activity. Therefore, in the pastseveral years, a lot of works have been dedicated todemonstrating the ability of carbohydrate-degrading enzymes.However, the quick and efficient enzymatic hydrolysis ofbiomass using a low enzyme loading has proven to be one ofthe major technical and economical drawbacks in the overallbioconversion process for biofuel production and continues tobe a challenge for biofuel commercialization.76
Although the cellulose crystallinity and degree of polymer-ization (DP) are considered as the most important factorswhich determine the final rates of enzymatic hydrolysis, severalliterature reports showed that these factors alone do not explain
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXE

the low rates of enzymatic hydrolysis.77 For instance, for LCMs,a decrease of crystallinity accompanied by changes in thebiomass morphology, such as particle size reduction and anincrease of the available surface area, was observed. In addition,the removal of acetyl groups from hemicellulose reduces thesteric hindrance of hydrolytic enzymes and greatly enhancespolysaccharide digestibility.78 The use of enzymes to catalyzecellulose conversion is considered as a greener alternative incomparison to acid hydrolysis, which mostly requires two-stephydrolysis.79 Because of these benefits, the use of enzymes toconvert cellulose into glucose is the subject of numerousreviews.22,80 The high-pressure CO2/H2O process coupled withenzymatic hydrolysis is a very attractive approach for thedevelopment of green chemistry into a biorefinery concept.51
Moreover, high-pressure CO2 in the presence of water providesenvironmentally friendly pretreatment conditions for depoly-merization of hemicellulose and enables the enzymatic processwithout the necessity for pH adjustment of the medium for theenzyme-catalyzed hydrolysis.81 Less severe pretreatment ismore economically favorable, though the conditions must beoptimized since the mildness of the process leads to lower sugarrelease, and consequently, elevated amounts of enzymes arerequired to achieve high yields and thus increase the costs.Because of these benefits, a number of research groups havebeen actively studying green solvents, including ionic liquidsand high-pressure CO2, as innovative tools for lignocellulosicbiomass processing.36,82,83 For example, Zheng et al. exploredthe effect of high-pressure CO2 on the rate and degree ofcellulose hydrolysis in cellulose-rich materials such as Avicel (acommercial form of pure cellulose).73 They concluded that thepretreatment with high-pressure CO2 followed by quick releaseof CO2 leads to disruption of cellulosic structures, thusincreasing the accessible area of cellulose. Their results showedthat high-pressure CO2 is an effective pretreatment method forcellulose during which a pressure increase facilitates thepenetration of CO2 molecules into the crystalline celluloseand increases the rate of enzymatic hydrolysis, resulting in aglucose yield of approximately 50%. However, if pretreatment isconducted under subcritical conditions at 207 bar, then thereare no significant changes observed in the digestibility ofcellulose. The same Avicel sample treated with high-pressureCO2 at 35 °C showed a significant increase of glucose yieldwith increased pressure (76, 138, 207, and 276 bar). It isimportant to highlight that Avicel is pure cellulose; thus, it ismuch more susceptible to hydrolysis than recalcitrantlignocellulosic materials. To face this challenge, Zheng andco-workers also investigated the effect of high-pressure CO2 oncellulosic-rich materials, such as recycled paper mix andsugarcane bagasse. They found maximal glucan and glucoseyields of 75% and 22%.73 Furthermore, an increase of glucoseyield was found by changing the reaction conditions from 35°C at 69 bar to 80 °C at 207 bar. However, in this case thebiomass was subject to dilute-acid hydrolysis after high-pressureCO2, enhancing the cellulose susceptibility to enzyme attack:therefore, the effect is not straightforward.73 Dale and Moreirareported a great effect of CO2 explosion at low temperature (25°C) and 56.2 bar on enzymatic hydrolysis of alfalfa (75% sugaryield).84 Although a high glucose yield was obtained, thistechnology did not show a similar efficiency when compared tosteam explosion or ammonia fiber expansion (AFEX) pretreat-ment.84 However, in this case, the reduced effectiveness ofenzymatic hydrolysis can be counterbalanced by the fact that ina process with carbonated water the need for an enzyme is 33%
lower than when using dilute sulfuric acid pretreatment.85
Additionally, it was found that high-pressure CO2 can becompared to ionic liquid pretreatments,28−30,36 being moreeffective than ammonia explosion without the formation ofinhibitory compounds, which can be found after a steamexplosion.84 They proved that these fluids lead to an increasedefficiency of the enzymatic hydrolysis, though still less thanwith CO2. Hohlberg et al. reported an increase of digestibilityof pine sawdust after CO2 pretreatment, obtaining 32.5 g ofreducing sugars/100 g of substrate in comparison to 26 g·100g−1 for steam explosion after the substrate impregnation bySO2.
86 However, the opposite effect was found for hemi-cellulose hydrolysis in which the pretreatment with SO2 as theimpregnating agent was more efficient than with CO2. Betterresults were obtained with SO2, although severer conditionswere needed.87 At these conditions, 68.3% hemicelluloseremoval with SO2 was attained, in comparison to 40.5%attained from CO2 pretreatment. However, it is important tostress that the pretreatment with CO2 led to the production ofa xylose-rich fraction, mainly in oligomer form, in contrast topretreatment with SO2, which acted more as an impregnatingagent. Zhang and Wu demonstrated that biomass pretreatmentwith CO2 at subcritical conditions has a positive influence onsubsequent enzymatic hydrolysis of both eucalyptus chips andsugarcane bagasse.88,89 They reported maximum glucanconversions of 92.2% and 93% (at 180 °C and 50 bar for 80min), respectively, which are higher than for native eucalyptusfeedstock and sugarcane bagasse (46.8% and less than 10%,respectively). Matsushita et al. also found that the addition ofCO2 to hydrothermal technologies improved the saccharifica-tion rate of eucalyptus.90 Kim and Hong examined the effect ofCO2 on aspen (hardwood) and yellow pine (softwood) atdifferent temperatures (112−165 °C) and reaction pressures(214 and 276 bar) for 10−60 min processes with differentmoisture contents (0−73%, w/w).91 They showed that, fortemperatures below 120 °C and pressure up to 214 bar for 60min, no significant effect on the sugar yield was observed. Evena pressure increase to 276 bar did not affect the yields of thesugars released. However, the same study demonstrated thattemperature seems to have a more pronounced effect, becauseat 165 °C bar a higher glucose yield was achieved.91 It was alsodetermined that this treatment is much more effective for aspenthan for yellow pine, possibly because of structural differencesbetween both raw materials91 discussed in section 3.1.2. Aliniaet al. also investigated the effect of the residence pressure onwheat straw at constant temperature (190 °C) after 30 min.31
They concluded that the changes in pressure from 80 to 120bar caused an increase of the yield of reducing sugars. However,a pressure above 120 bar did not change the final reducingsugar concentration. Also Gurgel et al. examined the effect ofhot water coupled with high-pressure CO2 pretreatment onsugarcane bagasse.92 They used a flow-through supercriticalextraction stainless steel reactor pressurized with CO2 (68 barand initial temperature of 25 °C, liquid:solid ratio of 12:1).After high-pressure CO2/H2O pretreatment at 115 °C for 60min, a maximum cellulose conversion of 41.2% was achieved.It is important to underpin that all the studies presented
above were performed in externally heated reactors without anystirring system. Thus, hot spots, especially near the reactorwalls, and overheating may lead to local biomass pyrolysis. Thisincorrect processing may lead to the formation of degradationproducts from biomass subjected to high temperatures while atthe same time not being pretreated homogeneously, affecting
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXF
the process results. Additionally, the usefulness of theaforementioned works is reduced because the particle size ofbiomass used in these experiments, which was in the range of0.5 and 2 mm, would require an additional mechanicaltreatment step. Recently, Morais et al. demonstrated thatpressure changes from 0 (autohydrolysis) to 54 bar of initialCO2 (using a liquid:solid ratio of 10:1) at constant temperatureand nonisothermal conditions in wheat straw pretreatmentpromoted an increase of sugar yields.51 The glucose yield roseto 82.21% at the aforementioned conditions, which corre-sponds to an increase of 26% versus the yield from theautohydrolysis reaction at the same conditions.Some experiments related to biomass pretreatment in a two-
step temperature system were published. Luterbacher et al.used a biphasic CO2/H2O system in which two temperaturestages were used to pretreat hardwood and switchgrass.93 Inthis process, a short high-temperature stage at 210 °C for 16and 1 min for hardwood and switchgrass, respectively, followedby a long low-temperature stage at 160 °C for 60 min wasexamined.93 At a high solids loading (40 wt %), maximalglucose yields of 83% and 80% were achieved for hardwood andswitchgrass and overall sugar yields were as high as 65% and55%, respectively, for these materials. Compared with thepretreatment performed with only one temperature stage at170 °C for 60 min for which the maximal glucose yield was73% for hardwood,59 it can be concluded that, for a givenmaterial, either in an acidic or a neutral environment, the use ofa two-temperature pretreatment (which prevents formation ofunwanted products from hemicellulose, such as furfural) can beadvantageous.93 The hemicellulose depolymerization toproduce oligosaccharides occurs quickly during the pretreat-ment. These oligosaccharides are hydrolyzed to monomers,which can degrade to undesirable products such as furfural.94
With a single temperature stage it is difficult to simultaneouslyobtain both high glucan and high hemicellulose sugar yields.For hardwood, the hemicellulose sugar yields decreased from180 to 190 °C, with a contrary trend for the glucose yield. Thistendency was also observed with a two-temperature stage, butthe effect was less pronounced.95 Thus, in the biorefineryconcept, it may be advantageous to use high temperatures for ashort period of time during the initial lag in formation ofdegradation products and then continue with lower temper-
atures for a longer time, which prevents monomer degrada-tion.93 This procedure was applied in other types ofpretreatments, such as dilute-acid hydrolysis (190 °C for 150s with 1.1% acid and 210 °C for 115 s with 2.5% acid) for bark-rich biomass and aspen wood pretreated by autohydrolysis at175 °C.96−98 The results confirmed that a two-stage processgives better results than a typical single-stage pretreatment.Another interesting application is the use of high-pressure
CO2 with water/ethanol as a cosolvent system in enzymatichydrolysis of corn stover.99 The use of water/ethanol as acosolvent plays an important role in the yields of enzymatichydrolysis for several reasons: (i) high-pressure CO2 promotesa physical effect, which contributes to improvement of lignincleavage; (ii) carbonic acid formation helps hemicellulosehydrolysis; (iii) water as a polar solvent can break down thebonds between hemicellulose and lignin structures, increasingthe cellulose digestibility; (iv) CO2 enhances the capacity ofethanol to dissolve lignin. All these factors produce anadvantageous pretreatment in which potential inhibitors ofcellulose hydrolysis are eliminated. For instance, an improve-ment of 14% in glucose yield was achieved when 300 mL ofethanol was added to the CO2/water system, and after apretreatment at 150 bar and 180 °C for 60 min, a maximalglucose yield of 77.8% after enzymatic hydrolysis wasobtained.99
Ferreira-Leitao et al. studied the steam pretreatment ofsugarcane bagasse using CO2 as an impregnating agent in termsof glucose and xylose yields after enzymatic hydrolysis andformation of degradation products.87 The highest glucose yieldsof 86.6% and 97.2% (theoretical) were obtained for sugarcanebagasse and cane leaves, respectively.In some cases, the pretreatment of LCMs, such as southern
yellow pine, even at very severe conditions shows glucose yieldsvery similar to that for untreated biomass.91 Thus, it can bestated that the glucose yield following enzymatic hydrolysis,apart from the aforementioned factors, is also dependent on thecomposition and structure of biomass as presented in section3.1.2. It is important to remember that other parameters,discussed in sections 3.1.1−3.1.3, such as the moisture of theraw material, biomass loading and particle size, and CO2/biomass loading, have a great impact on glucose yields.
Table 2. scCO2 Coupled with Conventional Pretreatment Technologies
reaction conditions glucose yield/reducing sugar yield (%)
technology raw materialT
(°C) P (bar)t
(min)
conditions ofconventionaltechnology
moisturecontent(%)
CO2technology
conventionaltechnology
bothtechnologies
untreatedbiomass ref
Two-Step Pretreatment (scCO2 Followed by Conventional Technologies)ultrasound corncob 170 200 30 20 kHz, 600 W,
80 °C, 6 h50 31.0/62.0 42.0/87.0 10.0/12.5 62
cornstalk 170 200 30 20 kHz, 600 W,80 °C, 8 h
50 14.0a/25.5 16.0/30.0 13.5/16.6 62
sugarcanebagasse
80 65 120 40 kHz, 154 W,30 °C, 8 h
65 −/380 ± 9b −/350b −/300b −/127 ± 16b 100
sugarcanebagasse
180 206 60 35 °C, 4 h 80 61.3/− 20.2/− 97.8/− 13.4/− 101
steamc wheat straw 190 120 60 200 °C, 15 min 23 −/60.1 31alkaline(H2O2/NaOH)
sugarcanebagasse
180 206 60 0.6% H2O2,60 °C, 9 h
80 61.3/− 22.9/− 65.8/− 13.4/− 101
CO2-Assisted Conventional TechnologiesAFEX rice straw 175 7.5 30 15% NH4 46.75d/− 96.00e/− 99.04/− 102aCO2 pretreatment at 170 °C and 200 bar of CO2 pressure for 1 h.
bUnits of g·kg of dry biomass−1. cCO2 explosion was performed after the steamexplosion. dConditions: 160 °C, 50 min, 15 bar. eConditions: 160 °C, 50 min, 10% ammonia concentration.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXG

Table
3.Examples
oftheCO
2Effecton
BiomassPresented
asGlucanCon
versionYieldsafterEnzym
atic
Hydrolysis
reactio
nconditions
rawmaterial
T(°C)
P(bar)
t(m
in)
moisture
content
(%)
CO
2:biom
ass
ratio
(w/w
)
solid
loading
(w/w
)particlesize
(mm)
glucan
conversion
yield(%
)
glucan
conversion
yield(%
)from
untreatedbiom
ass
enzymatichydrolysisconditionsa
ref
ricestraw
110
300
305:1
1:1
<232.4±
0.5b
27.7±
0.5b
cellulase,3
0FP
U·g
−1(D
S);β-glucosidase,15
CBU·g
−1(D
S);50
°C;
150rpm;48
h104
eucalyptus
chips
160
5080
1:10
<192.2
cellulase,2
0FP
U·g
−1(D
S);50
°C;150rpm;60
h88
wheat
straw
225
540c
10:1
1:10
<1.5
82.2
34.3
cellulase,6
0FP
U·g
−1 ;β-glucosidase,64
p-NPG
U·g
−1 ;50
°C;200rpm;
96h
51
guayule
bagasse
175
262
3060
24:1
0.1−
0.3
56cellulase,0.92FP
U·mL−
1 ;β-glucosidase,0.76
U·mL−
1 ;d50
°C;200rpm;
72h
105
switchgrass
160
200
6060
8:1
40wt%
0.038−
181
±1
10.4±
0.4
cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59210/160
200
1/16
11:1
40wt%
0.038−
180
cellulase,15FP
U·g
−1(D
S);β-glucosidase,30
CBU·g
1(D
S);50
°C;7
2h
95150
220
6075
14.1:1
<1.18
14e
12e
cellulase,5
0U·100
mg−
1(D
S);β-glucosidase,20
U·100
mg−
1(D
S);
47°C
;24
h34
bigbluestem
grass
170
200
6060
8:1
40wt%
0.038−
166
±2
17±
1cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59
corn
stover
150
240
6075
14.2:1
<1.18
30e
12e
cellulase,50U·mg−
1(D
S);β-glucosidase,20
U·mg−
1(D
S);47
°C;24
h34
160
200
6060
8:1
0.038−
185
±2
36±
1cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59sugarcane
bagasse
180
206
6080
>0.9
61.3
13.4
cellulase,1
5FP
U·g
−1(D
S);50
°C;72
h101
136.2
6860
2.4:1
1:12
<1.19
42.8
9.54
cellulase,1
0FP
U·g
−1(D
S);β-glucosidase,20
U·g
1(D
S);50
°C;
150rpm;72
h92
205
f15
32.9
2−10
86.6
cellulase,6
5FP
U·g
−1(D
S);β-glucosidase,376U·g
−1(D
S);40
°C;
180rpm;96
h87
180
50100
1:15
<193.0
<10
cellulase,2
0FP
U·g
−1(D
S);50
°C;150rpm;72
h89
sugarcane
(leaves)
220
f5
112−
1097.2
cellulase,1
5FP
U·g
−1(D
S);β-glucosidase,18
U·g
−1(D
S);40
°C;
180rpm;96
h87
sugarcane
bagasse
(pulp)
60140
600.25−0.42
72cellulase,70FP
U·L
−1(C
C);β-glucosidase,250CBU·mL−
1 (CC);50
°C;
130rpm;8h
106
sugarcane
bagasse
(skin)
60140
50.25−0.42
76.1
cellulase,70FP
U·L
−1(C
C);β-glucosidase,250CBU·mL−
1 (CC);50
°C;
130rpm;8h
106
mixed
hardwood
170
145
606:1
20wt%
0.038−
177
±4
5.1±
0.3
cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59
170
145
608:1
40wt%
0.038−
173
±5
cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59210/160
200
16/60
11:1
40wt%
0.038−
183.3
cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
95mixed
perennial
grasses
170
145
608:1
40wt%
0.038−
168
±1
12.2±
0.4
cellulase,15FP
U·g
−1(D
S);β
-glucosidase,30CBU·g
1(D
S);50°C
;72h
59
aAbbreviations:FP
U,filterpaperunit;
CBU,cellobiaseunit;
p-NPG
U,p-nitrophenylglucosideunit;
U,internationalunit(μmol·(mg·s)
−1 );DS,
drysubstrate;
CC,cellulose
content.bGlucose
yield.
c Nonisotherm
alconditions.dNodataon
theenzymeloading.e G
lucose
yield(g
ofglucose/100gof
drybiom
ass).fCO
2addedto
thepretreatmentcorrespondsto
3%by
weighton
thebasisof
thewater
contentof
thematerial(30%
ofthedrymatter).
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXH
Combined pretreatments exhibited a synergistic effect oncarbohydrate recovery and enzymatic conversion of sugars.This could be considered as an option in the field of biomasspretreatments, as shown in Table 2. Additional research intothe influence of CO2, coupled with other technologies aimingto improve the biomass pretreatment efficiency, was performedby Benazzi et al.100 The Plackett−Burmann design wasemployed to describe the influence of each process variablein CO2 pretreatment on enzymatic hydrolysis. The resultsshowed that, when high-pressure CO2 is used, the amount offermentable sugars obtained was increased by 280%, whichcorresponds to a hydrolysis efficiency as high as 74.2%compared to that of nontreated bagasse. The addition ofhigh-pressure CO2 to ultrasound technology enhanced byabout 16% the amount of fermentable sugars compared to theultrasound process alone.100 Another example of CO2combined with ultrasound, for corncob and cornstalk pretreat-ment, was proposed by Yin et al.62 This group reported that theapplication of high-pressure CO2 enhanced the enzymatichydrolysis of corncob and cornstalk by 50% and 29.8%compared to that of the untreated materials, respectively. Atthe same time, high-pressure CO2 and ultrasound pretreatmentincreased the enzymatic hydrolysis yield by 75% and 13.4%,respectively. Recently, Phan and Tan compared the effect of asequential combination of CO2 explosion and alkaline hydro-gen peroxide (H2O2) with that of CO2 explosion followed byultrasound to pretreat sugarcane bagasse.101 They demon-strated that CO2 explosion (187 °C, 156 bar, and 40 minretention time) followed by 1% H2O2 pretreatment resulted inan enhancement of the maximal glucose yield by almost 50%(from 65.8% to 97.8%) compared to high-pressure CO2followed by ultrasound (the same conditions referred toabove plus 4 h of ultrasound). High-pressure CO2 promoted anincrease of the accessible biomass surface that was susceptibleto H2O2 contact. At alkaline conditions there was formation ofhydroxyl radicals and superoxide anion radicals from H2O2,which could oxidize and degrade the lignin. This enhanced thehydrolysis of hemicellulose and lignin dissolution, increasingthe digestibility of the remaining cellulose. When eachpretreatment was individually applied, high-pressure CO2 was
demonstrated to be the best method in which a maximalglucose yield of 61.3% was obtained, contrary to the lowglucose yield obtained individually by H2O2 (22.9%) andultrasound (20.2%).101 Cha et al. investigated the effect of CO2-added ammonia explosion on the glucan conversion from ricestraw in comparison to that of AFEX.102 Both methods whenused separately demonstrated an increase in the sugarconversion yield. Ammonia promoted both swelling oflignocellulosic biomass and removal of lignin, while high-pressure CO2 promoted pore opening in the lignocellulosiccomplex. The addition of high-pressure CO2 to ammoniapretreatment demonstrated an increase of the maximal glucoseyield to 99.04%; however, this increase was insignificant mostlydue to the very low CO2 pressure used (7.5 bar). Alinia et al.combined steam explosion and CO2 explosion. The bestconditions (200 °C and 15 min for the first stage, steaminjection and supercritical CO2 explosion at 120 bar and 190 °Cfor a 60 min retention time in the second stage) gave areducing sugar yield as high as 60%. Another approach wasexamined by Moscon et al., who integrated enzymatichydrolysis under high-pressure CO2. They examined ricehulls subjected to this treatment in comparison to thosesubjected to integrated enzymatic hydrolysis in an ultrasoundbath.103 They found that the addition of high-pressure CO2 as acosolvent does not have a positive effect since the maximumyield of fermentable sugars obtained from traditional enzymatichydrolysis was about 16 g·kg−1 versus 4.2 g·kg−1 obtained whenhigh-pressure CO2 was added to the saccharification process.Thus, observing data presented in Tables 3 and 4, it can bestated that the promising results obtained for CO2-mediatedenzymatic conversions presented in the literature shouldencourage research in this field, mostly due to lowertemperature requirements and lack of hazardous solvents.
3.1. Process Condition Effects
3.1.1. Water Influence. One of the most importantrequirements for the efficient utilization of lignocellulosicmaterials is removal of hemicellulose to provide a feasiblecellulose-rich cake for further enzymatic hydrolysis andfermentation to get, for example, ethanol.20,107 To obtain
Table 4. Overview of the High-Pressure CO2 Pretreatment Effect on Either Enzymatic Hydrolysis or Dilute-Acid Hydrolysis
reaction conditions
raw materialT
(°C)P
(bar)t
(min)
moisturecontent(%)
CO2:biomassratio (w/w)
solidloading(w/w)
particle size(mm)
reducingsugar yield
(%)
reducing sugaryield (%) from
untreated biomassenzymatic hydrolysis
conditionsa ref
High-Pressure CO2 Pretreatment Followed by Enzymatic Hydrolysis
aspen 165 214 30 73 <1 84.7 ± 2.6 14.5 ± 2.3 cellulase, 1000 U·g−1 (DS);50 °C; 100 rpm; 72 h
91
southernyellowpine
165 214 30 73 <1 27.3 ± 3.8 12.8 ± 2.7 cellulase, 1000 U·g−1 (DS);50 °C; 100 rpm; 72 h
91
sugarcanebagasse
80 250 120 65 74.2 19.6 b; 50 °C; 100 rpm; 8 h 100
cornstalk 170 200 150 50 0.39−0.83 25.5 16.6 cellulase, 4000 U·g−1 (DS);50 °C; 100 rpm; 72 h
62
corncob 170 200 60 50 0.39−0.83 62 12 cellulase, 4000 U·g−1 (DS);50 °C; 100 rpm; 72 h
62
wheat straw 185 120 30 23 11.5:1 3.3 0.42−0.83 53.4 cellulase, 15 FPU·g−1 (DS);50 °C; 240 rpm; 72 h
31
High-Pressure CO2 Pretreatment Followed by Dilute-Acid Hydrolysis
corncob 100 150 30 50 6:1 <0.417 39.6 26.2 1 wt % H2SO4; 160 °C; 40 min 74
cornstalk 100 150 30 50 6:1 <0.417 27.4 22.5 1 wt % H2SO4; 160 °C; 40 min 74
rice straw 100 150 30 50 6:1 <0.417 36.6 30.9 1 wt % H2SO4; 160 °C; 40 min 74aAbbreviations: U, international unit (μmol·(mg·s)−1); DS, dry substrate; FPU, filter paper unit. bNo data on enzyme activity and enzyme loading.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXI

high glucose yields from enzymatic hydrolysis, a sufficienthumidity of LCMs is required. Indeed, in the presence of water,high-pressure CO2 facilitates a swelling effect on biomass.Stamenic et al. showed that the structure of biomass andoperating conditions such as the pressure, temperature, andtime influence the degree of biomass swelling.61 Narayanasw-amy et al. found that for a wet corn stover (75%, w/w) at 240bar and 120 °C for 60 min, the glucose yield was doubled incomparison to that obtained for dry biomass.34 These resultsare consistent with the findings for high-pressure CO2pretreatment for dry Avicel.73 The intramolecular hydrogenbonds present in biomass decreased either the solubility inwater or the reactivity of the functional groups of the solvents.As a result, the development of any effective and environ-mentally benign recycling process for cellulose did not appearto be possible as long as it could be swollen or dissolved only inthose solvents. On the other hand, as previously described, thepresence of CO2 acidifies the aqueous environment, making theprocess similar to dilute-acid hydrolysis. In this context, Kim etal. reported no differences in sugar yields between drypretreated biomass and an untreated (control) aspen sample.91
The increase of the moisture content in aspen resulted in ageneral increase of the sugar yield obtained in enzymatichydrolysis.91 Alinia et al. investigated the effect of CO2pretreatment of dry and wet wheat straw by varying thetemperature (160−200 °C), pressure (80, 100, 120, 150, 170,and 200 bar), and residence time (10, 30, 60, and 70 min).31
They found that the high-pressure CO2 pretreatment withwater led to the best overall yield for sugar (208.4 g·kg of wheatstraw−1), which is more than 50 g·kg−1 higher than for drywheat straw (149.1 g·kg−1).The high solid loadings of substrates are desirable since the
sugar-rich hydrolysate is essential for the profitability of theprocess for a wide variety of products, including bioethanol.Nevertheless, due to the high viscosity of most lignocellulosicmaterials, it is difficult to pretreat with a solid content higherthan 10%.108 The lignocellulosic substrates have a high viscositybecause of the presence of considerable amounts of insolublecompounds and the water binding capacity of hemicellulose inthe material. Luterbacher et al. tested a biphasic CO2/H2Omixture in several biomass sources with a high solids content(40 wt %) over a range of temperatures between 150 and 250°C and residence times from 20 s to 60 min at 200 bar ofCO2.
59 Hardwood with a solids content of 20% (by weight) at170 °C, 60 min, and 200 bar of CO2 resulted in a 73% glucoseyield compared to 77% achieved for the same biomass with40% solids under equivalent conditions. This negligible increaseof the glucose yield shows that there is no clear evidence for abiomass loading effect on the sugar release. On the other hand,they found that a high solids content is less sensitive totemperature changes, which could be explained by a higherinertia of biomass as well as heat and mass transfer limitationscaused by the high solids content. Additionally, for a higherbiomass loading, a significant decrease of the hemicellulosesugar yield in the hydrolysate was accompanied by increasedhemicellulose degradation product yields.3.1.2. Biomass Sources and Recalcitrance. The
composition and concentration of the fractions vary, dependingon the biomass source, e.g., grass, hardwood, or softwood.Furthermore, these factors are dependent on externalparameters. The most important ones are climactericconditions, origin, age, harvesting method, drying, and storageof biomass. In addition, the recalcitrant structure of biomass is
influenced by inherent properties, such as the lignin content,the cellulose crystallinity and DP, cellulose accessibility toenzymes, sheathing of cellulose by hemicellulose and lignin, andthe fiber strength. All these factors influence the enzymaticdigestibility of a given biomass feedstock. It cannot be forgottenthat a major aim of biomass pretreatment with high-pressureCO2 is not to achieve a maximal glucose yield but rather toobtain satisfactory glucose yields at energy inputs as low aspossible. Thus, all these aforementioned parameters must beassessed before selection of the operation pretreatmentconditions. In addition, the physical properties of biomass areimportant; for example, softwoods are generally characterizedby a lower density than that of hardwoods, which facilitateshigh-pressure CO2 pretreatment. Different untreated biomassspecies, when subjected to enzymatic hydrolysis under the sameconditions, give substantially different saccharification yields.For instance, yields on enzymatic digestion of untreatedswitchgrass and corn stover were 10.4% and 36%, respec-tively.59 On the other hand, some biomasses are moresusceptible to processing and, independently of the processconditions, give higher sugar yields. As an example, the glucoseyields of switchgrass at 160 or 170 °C and 200 bar after 60 minin binary CO2/H2O pretreatment were always higher than forcorn stover, big bluestem, and mixed perennial grasses.59 Kimet al. compared the enzymatic digestibility of two differentbiomasses (aspen wood and southern yellow pine) pretreatedat 165 °C and 214 bar for 30 min with a moisture content of75%.91 The highest glucose yields were 84.7% and 27.3% foraspen and southern yellow pine, respectively. The more rigidand denser structure of yellow pine leads to more difficultaccess of CO2, and additionally, the large moisture contentnegatively influences the pretreatment, which results in muchlower enzymatic digestibility of this biomass. Thus, biomassspecies characterized by a low density are desirable to achievegood sugar yields while avoiding pretreatments at hightemperature and pressure. Santos et al. studied the high-pressure CO2 explosion of two different samples of sugarcane(bagasse pulp and skin), which were compared to Avicel (purecellulose).106 With these sugarcane samples, different glucoseyields were obtained; for instance, at 60 °C and 140 bar for 5min the best glucose yields were 76.10% and 28.39% forsugarcane skin and bagasse, respectively. However, the bestglucose yield (72.0%) for sugarcane bagasse was obtained at 60°C and 140 bar for 60 min. This result shows that the residencetime of pretreatment is an important variable.106 Puri et al.studied the effect of steam and CO2 under supercriticalconditions on cellulose hydrolysis before release of CO2.
109
The operating conditions used were 200 °C and between 34.5and 138 bar of CO2 pressure. The highest glucose yields were81%, 78%, and 75% for wheat straw, bagasse, and Eucalyptusregnans woodchips, respectively. Due to the structural andchemical composition differences between feedstocks, the timesrequired to obtain the maximal digestibility rate were 5 min forwheat straw and bagasse and 15 min for E. regnans.109
3.1.3. CO2:Biomass Ratio. One of the most importantfactors influencing the effectiveness of the pretreatment withhigh-pressure CO2 is the number of moles of CO2 and mass ofbiomass present in the reactor. The quantity of CO2 for aknown volume of reactor containing biomass is greatlydependent on the thermodynamics at the set point plus thepretreatment pressure and temperature. Unfortunately, fewresearch reports provide sufficient data for comparison.Narayanaswamy et al. used a CO2:dry biomass ratio close to
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXJ

14:1 (w/w) for high-pressure CO2 pretreatment and obtained ahigh glucose yield of 85% for corn stover.34 Also Walker andco-workers obtained the same glucose yield under the sameconditions but at lower CO2:dry biomass ratio of 8:1 (w/w).59
The ratio of the treatment medium (in this case CO2) tobiomass is lower than those for other pretreatment methodssuch as dilute-acid hydrolysis, autohydrolysis, or aqueousammonia. This shows that, even with a relatively low quantityof CO2, a satisfactory glucose yield can be achieved due to theimpregnation of the biomass with CO2 at supercriticalconditions.3.2. Effect of High-Pressure CO2 on the Morphology ofLignocellulosic Biomass
The morphological changes in biomass after pretreatment withhigh-pressure CO2 can be assessed by scanning electronmicroscopy (SEM).34,51,104 Figure 4 shows the structure of
untreated rice straw biomass (a) and morphological changesobserved after pretreatment (b) at 110 °C and 300 bar for 30min at a liquid:solid ratio of 1 g·g−1, plus samples of corn stoverwith 75% moisture before (c) and after (d) pretreatment at 150°C and 241.3 bar for 60 min.The untreated samples exhibited a rigid, tight, and
contiguous surface, while pretreated samples had fibrouscharacteristics with anomalous porosity and the lamellarstructures becoming fleecy. The fibers seem to be separatedfrom the initial structure and totally exposed, making thesurface more susceptible to cellulase enzyme attack.96 Somepretreatments facilitated hydrolysis, but at the same time, anincrease in cellulose crystallinity was observed. This may beexplained by the reduction or even total removal of amorphouscellulose after pretreatments.110 Unlike the results withcrystalline cellulose, the decrease of polymerization in pre-
treated solids suggests that hemicellulose removal has a greaterimpact than lignin removal.
4. ENZYMATIC HYDROLYSIS IN SUPERCRITICAL CO2
Supercritical carbon dioxide is a promising green solvent formany enzyme-catalyzed chemical reactions. The finding ofenhanced enzyme stability and activity in alternative solvents,e.g., with lipases,111 suggested a wide range of high-pressureCO2 applications as selective biocatalysts for synthesis and/orhydrolysis reactions. Recently, the number of studies involvingenzymatic catalysis under CO2 at supercritical conditions as asolvent have increased, since it has been demonstrated thatenzymes are stable and active in high-pressure CO2 and thatCO2 favors transport properties, accelerating mass transfer inenzymatic reactions.112 At the same time, high-pressure CO2was also successfully used as a medium for the recovery ofproducts and reactants.113
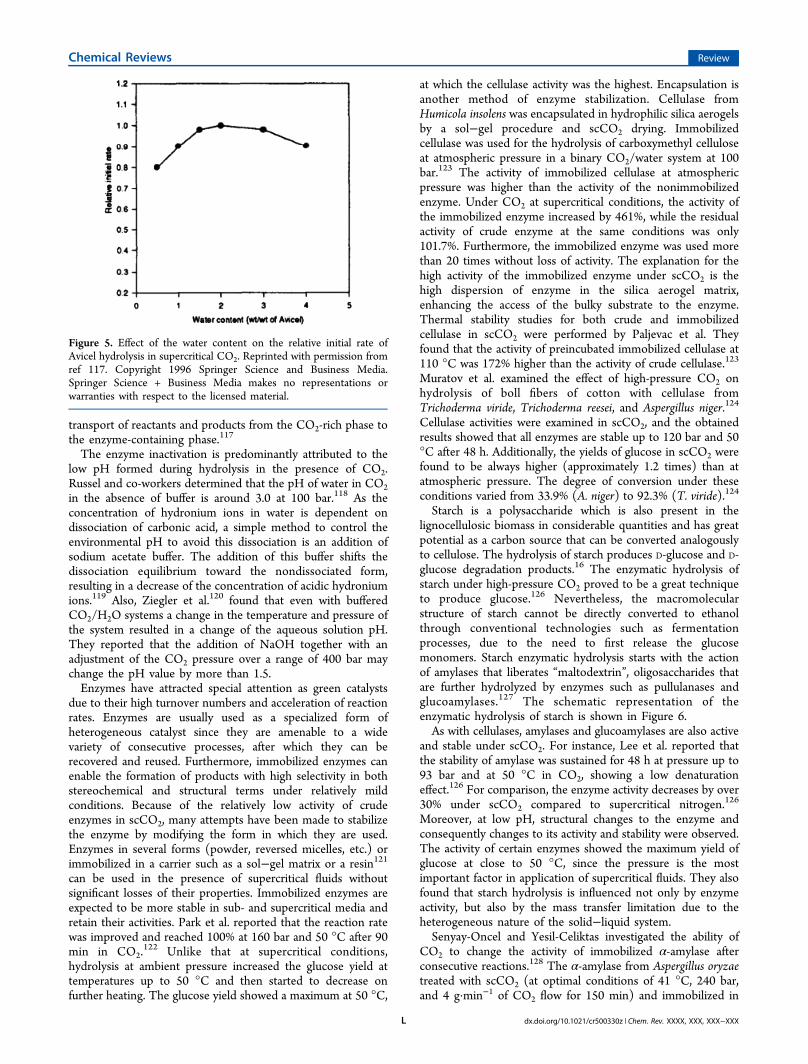
The most important factor in achieving high enzymatichydrolysis rates is the stability of the enzymes. This in turndepends on the pressure and temperature exerted, the pH, thenumber of depressurization steps, the water content, carbonicacid formation, the solid content, the enzyme species, and thesubstrates and types of end products due to the inhibition ofenzymatic processes by blocking the active sites of theenzyme.114 However, the use of CO2 at supercritical conditionsis not challenging from the enzyme point of view, becauseeither the temperature or pressure is within the operationalrange of most of the enzymes. However, enzyme activity ismore difficult to maintain than for other typical solvents, mostlycaused by lowering of the pH at higher CO2 pressures. TheCO2 pressure itself, even in the range of 200 bar, does not havean effect on enzyme denaturation, but the pH generated by thisexerted pressure may inactivate the enzyme. Additionally, thehigh substrate and product concentrations contribute to lowerenzyme effectiveness, which can be solved by a continuousmode of substrate feeding and removal of products by CO2extraction. In this way, the inhibitory effect can be minimized oreven eliminated. Xiao et al. investigated the effect of sugarinhibition of glucose on cellulase and β-glucosidase hydrolysisof both Avicel and acid-pretreated softwood.115 They foundthat the increase of the glucose concentration in the hydrolysateresulted in a dramatic decrease of both cellulase and glucosidaseactivities.115 Under the severe pretreatment conditionsexamined, the three-dimensional structure of the enzymesmay be irreversibly changed by denaturation, resulting in loss ofenzyme activity. With less severe conditions, the proteins mayrecover their structures and only minor changes are observed,which may lead to altered enzyme activity and stability. Thewater content in the system is also an important factor thatinfluences the activity of the enzyme. To maintain the enzymeactivity, a specific water activity is required, affecting diffusionand thus influencing both the reaction equilibrium and enzymestructure by noncovalent binding or disruption of hydrogenbonds.116 However, in a CO2/H2O system, the enzymes canchange their activity and stability with formation of carbonicacid due to a decrease of the pH in the environmental media.Zheng et al. reported that the presence of small amounts ofwater improves the reaction rate.117 However, the addition ofwater in quantities higher than necessary to form a micro-aqueous environment led to a decrease in rate (Figure 5).Additionally, a high quantity of water results in the formation ofan aqueous layer close to the active site, which inhibits the
Figure 4. Scanning electron microscopy pictures of rice straw samplesuntreated (a) and treated at 110 °C and 300 bar for 30 min with aliquid to solid ratio of 1 g·g−1 (b). Reprinted with permission from ref104. Copyright 2010 Elsevier. SEM images of corn stover samples with75% moisture before (c) and after (d) pretreatment at 150 °C and241.3 bar for 60 min. Reprinted with permission from ref 34.Copyright 2011 Elsevier.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXK
transport of reactants and products from the CO2-rich phase tothe enzyme-containing phase.117
The enzyme inactivation is predominantly attributed to thelow pH formed during hydrolysis in the presence of CO2.Russel and co-workers determined that the pH of water in CO2in the absence of buffer is around 3.0 at 100 bar.118 As theconcentration of hydronium ions in water is dependent ondissociation of carbonic acid, a simple method to control theenvironmental pH to avoid this dissociation is an addition ofsodium acetate buffer. The addition of this buffer shifts thedissociation equilibrium toward the nondissociated form,resulting in a decrease of the concentration of acidic hydroniumions.119 Also, Ziegler et al.120 found that even with bufferedCO2/H2O systems a change in the temperature and pressure ofthe system resulted in a change of the aqueous solution pH.They reported that the addition of NaOH together with anadjustment of the CO2 pressure over a range of 400 bar maychange the pH value by more than 1.5.Enzymes have attracted special attention as green catalysts
due to their high turnover numbers and acceleration of reactionrates. Enzymes are usually used as a specialized form ofheterogeneous catalyst since they are amenable to a widevariety of consecutive processes, after which they can berecovered and reused. Furthermore, immobilized enzymes canenable the formation of products with high selectivity in bothstereochemical and structural terms under relatively mildconditions. Because of the relatively low activity of crudeenzymes in scCO2, many attempts have been made to stabilizethe enzyme by modifying the form in which they are used.Enzymes in several forms (powder, reversed micelles, etc.) orimmobilized in a carrier such as a sol−gel matrix or a resin121
can be used in the presence of supercritical fluids withoutsignificant losses of their properties. Immobilized enzymes areexpected to be more stable in sub- and supercritical media andretain their activities. Park et al. reported that the reaction ratewas improved and reached 100% at 160 bar and 50 °C after 90min in CO2.
122 Unlike that at supercritical conditions,hydrolysis at ambient pressure increased the glucose yield attemperatures up to 50 °C and then started to decrease onfurther heating. The glucose yield showed a maximum at 50 °C,
at which the cellulase activity was the highest. Encapsulation isanother method of enzyme stabilization. Cellulase fromHumicola insolens was encapsulated in hydrophilic silica aerogelsby a sol−gel procedure and scCO2 drying. Immobilizedcellulase was used for the hydrolysis of carboxymethyl celluloseat atmospheric pressure in a binary CO2/water system at 100bar.123 The activity of immobilized cellulase at atmosphericpressure was higher than the activity of the nonimmobilizedenzyme. Under CO2 at supercritical conditions, the activity ofthe immobilized enzyme increased by 461%, while the residualactivity of crude enzyme at the same conditions was only101.7%. Furthermore, the immobilized enzyme was used morethan 20 times without loss of activity. The explanation for thehigh activity of the immobilized enzyme under scCO2 is thehigh dispersion of enzyme in the silica aerogel matrix,enhancing the access of the bulky substrate to the enzyme.Thermal stability studies for both crude and immobilizedcellulase in scCO2 were performed by Paljevac et al. Theyfound that the activity of preincubated immobilized cellulase at110 °C was 172% higher than the activity of crude cellulase.123
Muratov et al. examined the effect of high-pressure CO2 onhydrolysis of boll fibers of cotton with cellulase fromTrichoderma viride, Trichoderma reesei, and Aspergillus niger.124
Cellulase activities were examined in scCO2, and the obtainedresults showed that all enzymes are stable up to 120 bar and 50°C after 48 h. Additionally, the yields of glucose in scCO2 werefound to be always higher (approximately 1.2 times) than atatmospheric pressure. The degree of conversion under theseconditions varied from 33.9% (A. niger) to 92.3% (T. viride).124
Starch is a polysaccharide which is also present in thelignocellulosic biomass in considerable quantities and has greatpotential as a carbon source that can be converted analogouslyto cellulose. The hydrolysis of starch produces D-glucose and D-glucose degradation products.16 The enzymatic hydrolysis ofstarch under high-pressure CO2 proved to be a great techniqueto produce glucose.126 Nevertheless, the macromolecularstructure of starch cannot be directly converted to ethanolthrough conventional technologies such as fermentationprocesses, due to the need to first release the glucosemonomers. Starch enzymatic hydrolysis starts with the actionof amylases that liberates “maltodextrin”, oligosaccharides thatare further hydrolyzed by enzymes such as pullulanases andglucoamylases.127 The schematic representation of theenzymatic hydrolysis of starch is shown in Figure 6.As with cellulases, amylases and glucoamylases are also active
and stable under scCO2. For instance, Lee et al. reported thatthe stability of amylase was sustained for 48 h at pressure up to93 bar and at 50 °C in CO2, showing a low denaturationeffect.126 For comparison, the enzyme activity decreases by over30% under scCO2 compared to supercritical nitrogen.126
Moreover, at low pH, structural changes to the enzyme andconsequently changes to its activity and stability were observed.The activity of certain enzymes showed the maximum yield ofglucose at close to 50 °C, since the pressure is the mostimportant factor in application of supercritical fluids. They alsofound that starch hydrolysis is influenced not only by enzymeactivity, but also by the mass transfer limitation due to theheterogeneous nature of the solid−liquid system.Senyay-Oncel and Yesil-Celiktas investigated the ability of
CO2 to change the activity of immobilized α-amylase afterconsecutive reactions.128 The α-amylase from Aspergillus oryzaetreated with scCO2 (at optimal conditions of 41 °C, 240 bar,and 4 g·min−1 of CO2 flow for 150 min) and immobilized in
Figure 5. Effect of the water content on the relative initial rate ofAvicel hydrolysis in supercritical CO2. Reprinted with permission fromref 117. Copyright 1996 Springer Science and Business Media.Springer Science + Business Media makes no representations orwarranties with respect to the licensed material.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXL

calcium alginate beads and NaY zeolite yielded 67.7% higheractivity (29.7 mmol·mL−1·min−1) than the untreated enzyme(maximum of 17.7 mmol·mL−1·min−1).129 The immobilizedenzyme was reused in starch hydrolysis by consecutiveactivation by scCO2. This procedure was repeated until theactivity of the engaged enzyme was no longer re-established.The best results were achieved when α-amylase wasimmobilized in NaY zeolite and the enzyme was used in 17reactions and reactivated 4 times.128
5. HIGH-PRESSURE CO2/H2O CONVERSIONTECHNOLOGIES
5.1. Hydrolysis of Biomass-Derived Carbohydrates
Hydrothermal treatments were studied for the production ofhemicellulose-derived sugars in either oligomeric or monomericform. The monosaccharides thus produced can be convertedinto a wide variety of products such as biofuels (either cellulosicethanol or other furanic fuels),20,107 chemicals, and biomate-rials. Degradation products are also produced during hydrolysis,and they may inhibit the fermentative processes. pH loweringof the medium to slightly above 3 but with enough CO2 tofacilitate biomass hydrolysis was investigated as an alternativetechnology for hydrolysis of biomass.119,130 The solubility ofCO2 in H2O determines the pH of the medium, and unlikeacid-hydrolysis pretreatment, the acidity of the mediumproduced by the high-pressure CO2 process does not representan environmental problem, as after depressurization the pHbecomes solely dependent on the other compounds in thesolution. Therefore, the pH of the solution mixture can becontrolled by the degree of dissolution of CO2 in water, which,on the other hand, is dependent on the temperature andpressure. The CO2/H2O approach is a methodology that offers
the benefits of acid catalysis without the typical drawbacks ofsulfuric acid such as corrosion problems and/or the need foremployment of separation methods.
5.1.1. Hemicellulose. Due to a lack of repeating β-1,4-glycoside bonds, hemicellulose does not present the crystallinenor the resistant structure of cellulose.131 Thus, hemicellulose isa polymer most susceptible to hydrothermal treatments. Liu etal. reported a maximal hemicellulose yield of 99% asmonomeric sugars at 220 °C after a 15 min process.132 Theaddition of CO2 to the hydrothermal reaction led to the use oflower temperatures and shorter reaction times. van Walsum etal. observed that at elevated temperatures (above 200 °C)carbonic acid has a catalytic effect on pure xylan hydrolysis,allowing an increase of pentose release and a decrease of theDP of xylan oligomers when compared to those withautohydrolysis pretreatment.60 However, McWilliams et al.compared the hydrolysis of aspen wood under high-pressureCO2 at 180−220 °C to a water-only reaction and found thataddition of CO2 and consequent formation of carbonic acidimproved neither the xylose yield nor the additional formationof furan compounds.133
These results are not surprising because aspen wood containshighly acetylated hemicellulose; therefore, for water-onlytreatments at temperatures ≥170 °C hydrolysis of thesebonds occurs by hydronium ion formation originating fromwater autoionization, leading to a sufficiently strong catalyticeffect without the need for CO2-generated carbonic acid.135
McWilliams et al. also found that biomass processing with CO2produces a hydrolysate with a higher pH in comparison to theautohydrolysis experiments.133 In fact, these data can beelucidated by either a reduced CO2 pressure (55 bar) used inthe experiments or the low solubility of CO2 in the aqueousphase at the examined conditions. The literature studies reportthat, up to 100 °C, the CO2 solubility in water decreases withtemperature and increases with pressure.136 Therefore, experi-ments should be done at as a low temperature as possible andat relatively high CO2 pressures to obtain a high solubility ofCO2 in water and, consequently, a more pronounced effect ofCO2 on xylan hydrolysis. Chuang and Johanssen reported thatcarbonic acid has a great effect on the pH of aqueoussolutions.134 Figure 7 presents the effect of both thetemperature and pressure of CO2 on the pH of carbonatedwater reported by Chuang et al.On the other hand, the data reported by McWilliam et al.133
can also be explained by the fact that pH measurement wasdone after the experiments were finished. The depressurizationof the reaction mixture removes the greater part of CO2;however, the produced mixture is under nonequilibriumconditions, and a prolonged time is needed to attainequilibrium, which is achieved when the concentration ofCO2 in the liquor is equal to the partial pressure in thesurrounding atmosphere.32 Other studies showed the hydro-lytic effect of CO2 on a wide range of polysaccharides underhydrothermal conditions.137 Miyazawa and Funazukuri foundthat high-pressure CO2 hydrolyzed the polysaccharides into lowmolecular weight sugars. In the absence of CO2, the finalmonosaccharide yields from xylan were less than 5%, while, inthe presence of CO2, a significant yield improvement wasreached. Additionally, they reported that the CO2-assistedhydrothermal reaction did not produce degradation products insuch amounts as acid-catalyzed hydrolysis. Recently, Gurgel etal. reported the benefits of liquid hot water associated withhigh-pressure CO2 instead of a water-only reaction.92 They
Figure 6. Schematic presentation of starch hydrolysis. Reprinted withpermission from ref 125. Copyright 1997 Biochemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXM
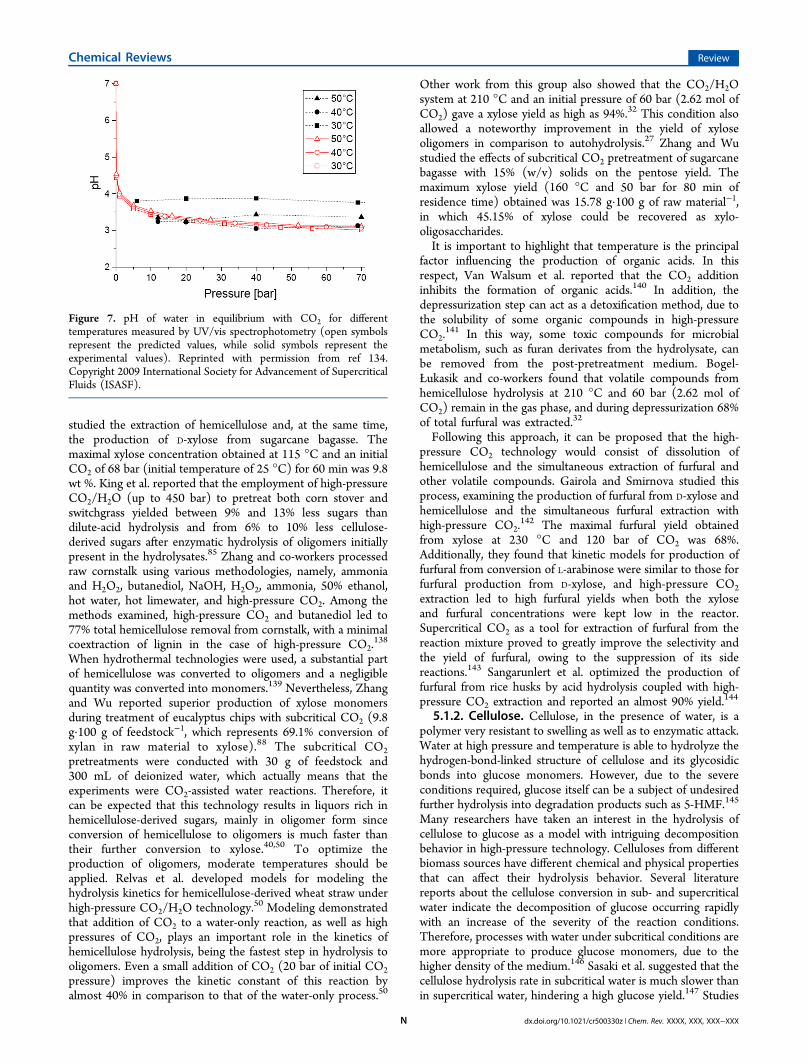
studied the extraction of hemicellulose and, at the same time,the production of D-xylose from sugarcane bagasse. Themaximal xylose concentration obtained at 115 °C and an initialCO2 of 68 bar (initial temperature of 25 °C) for 60 min was 9.8wt %. King et al. reported that the employment of high-pressureCO2/H2O (up to 450 bar) to pretreat both corn stover andswitchgrass yielded between 9% and 13% less sugars thandilute-acid hydrolysis and from 6% to 10% less cellulose-derived sugars after enzymatic hydrolysis of oligomers initiallypresent in the hydrolysates.85 Zhang and co-workers processedraw cornstalk using various methodologies, namely, ammoniaand H2O2, butanediol, NaOH, H2O2, ammonia, 50% ethanol,hot water, hot limewater, and high-pressure CO2. Among themethods examined, high-pressure CO2 and butanediol led to77% total hemicellulose removal from cornstalk, with a minimalcoextraction of lignin in the case of high-pressure CO2.
138
When hydrothermal technologies were used, a substantial partof hemicellulose was converted to oligomers and a negligiblequantity was converted into monomers.139 Nevertheless, Zhangand Wu reported superior production of xylose monomersduring treatment of eucalyptus chips with subcritical CO2 (9.8g·100 g of feedstock−1, which represents 69.1% conversion ofxylan in raw material to xylose).88 The subcritical CO2pretreatments were conducted with 30 g of feedstock and300 mL of deionized water, which actually means that theexperiments were CO2-assisted water reactions. Therefore, itcan be expected that this technology results in liquors rich inhemicellulose-derived sugars, mainly in oligomer form sinceconversion of hemicellulose to oligomers is much faster thantheir further conversion to xylose.40,50 To optimize theproduction of oligomers, moderate temperatures should beapplied. Relvas et al. developed models for modeling thehydrolysis kinetics for hemicellulose-derived wheat straw underhigh-pressure CO2/H2O technology.50 Modeling demonstratedthat addition of CO2 to a water-only reaction, as well as highpressures of CO2, plays an important role in the kinetics ofhemicellulose hydrolysis, being the fastest step in hydrolysis tooligomers. Even a small addition of CO2 (20 bar of initial CO2pressure) improves the kinetic constant of this reaction byalmost 40% in comparison to that of the water-only process.50
Other work from this group also showed that the CO2/H2Osystem at 210 °C and an initial pressure of 60 bar (2.62 mol ofCO2) gave a xylose yield as high as 94%.32 This condition alsoallowed a noteworthy improvement in the yield of xyloseoligomers in comparison to autohydrolysis.27 Zhang and Wustudied the effects of subcritical CO2 pretreatment of sugarcanebagasse with 15% (w/v) solids on the pentose yield. Themaximum xylose yield (160 °C and 50 bar for 80 min ofresidence time) obtained was 15.78 g·100 g of raw material−1,in which 45.15% of xylose could be recovered as xylo-oligosaccharides.It is important to highlight that temperature is the principal
factor influencing the production of organic acids. In thisrespect, Van Walsum et al. reported that the CO2 additioninhibits the formation of organic acids.140 In addition, thedepressurization step can act as a detoxification method, due tothe solubility of some organic compounds in high-pressureCO2.
141 In this way, some toxic compounds for microbialmetabolism, such as furan derivates from the hydrolysate, canbe removed from the post-pretreatment medium. Bogel-Łukasik and co-workers found that volatile compounds fromhemicellulose hydrolysis at 210 °C and 60 bar (2.62 mol ofCO2) remain in the gas phase, and during depressurization 68%of total furfural was extracted.32
Following this approach, it can be proposed that the high-pressure CO2 technology would consist of dissolution ofhemicellulose and the simultaneous extraction of furfural andother volatile compounds. Gairola and Smirnova studied thisprocess, examining the production of furfural from D-xylose andhemicellulose and the simultaneous furfural extraction withhigh-pressure CO2.
142 The maximal furfural yield obtainedfrom xylose at 230 °C and 120 bar of CO2 was 68%.Additionally, they found that kinetic models for production offurfural from conversion of L-arabinose were similar to those forfurfural production from D-xylose, and high-pressure CO2extraction led to high furfural yields when both the xyloseand furfural concentrations were kept low in the reactor.Supercritical CO2 as a tool for extraction of furfural from thereaction mixture proved to greatly improve the selectivity andthe yield of furfural, owing to the suppression of its sidereactions.143 Sangarunlert et al. optimized the production offurfural from rice husks by acid hydrolysis coupled with high-pressure CO2 extraction and reported an almost 90% yield.144
5.1.2. Cellulose. Cellulose, in the presence of water, is apolymer very resistant to swelling as well as to enzymatic attack.Water at high pressure and temperature is able to hydrolyze thehydrogen-bond-linked structure of cellulose and its glycosidicbonds into glucose monomers. However, due to the severeconditions required, glucose itself can be a subject of undesiredfurther hydrolysis into degradation products such as 5-HMF.145
Many researchers have taken an interest in the hydrolysis ofcellulose to glucose as a model with intriguing decompositionbehavior in high-pressure technology. Celluloses from differentbiomass sources have different chemical and physical propertiesthat can affect their hydrolysis behavior. Several literaturereports about the cellulose conversion in sub- and supercriticalwater indicate the decomposition of glucose occurring rapidlywith an increase of the severity of the reaction conditions.Therefore, processes with water under subcritical conditions aremore appropriate to produce glucose monomers, due to thehigher density of the medium.146 Sasaki et al. suggested that thecellulose hydrolysis rate in subcritical water is much slower thanin supercritical water, hindering a high glucose yield.147 Studies
Figure 7. pH of water in equilibrium with CO2 for differenttemperatures measured by UV/vis spectrophotometry (open symbolsrepresent the predicted values, while solid symbols represent theexperimental values). Reprinted with permission from ref 134.Copyright 2009 International Society for Advancement of SupercriticalFluids (ISASF).
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXN

of glucose hydrolysis over a wide range of temperatures,including for the use of supercritical water, depict the formationof various products according to the temperature used. Inaddition, the subcritical water technology is not able to separatelignin from the cellulose, which prevents a high productionyield of fermentable sugars. This problem can be resolved bythe introduction of CO2 into water-only processes, as this leadsto higher acidity of the reaction medium, creating a greaterconcentration of glucose monomers at lower temperatures. Tounderstand the behavior of glucose production in CO2-assistedhydrothermal technology, kinetic studies were performed forthe decomposition of cellulose into oligomers and monomers.For example, Schacht et al. demonstrated the enhancement ofcellulose dissolution in CO2-saturated water in comparison topure water over a temperature range of 240−260 °C.40 Fortemperatures higher than 260 °C, no further enhancement ofthe hydrolysis rate was obtained due to the pH drop in themedium, which leads to a less pronounced hydrolysis.Furthermore, the same authors stated that the CO2 solubilityat set conditions is more important than the amount added tothe water reaction, since undissolved CO2 does not have anyeffect on hydrolysis. Thus, CO2 has a great contribution to theacidity of the medium, but for a given CO2 pressure, the pHdepends mostly on the conditions applied. It is important toadd that the presence of CO2 in a hydrothermal process has notonly a chemical but also a physical effect. For instance, Moraiset al. found that, besides the CO2 pressure influencing thesolubility of CO2 in water, the pressure of CO2 exerts a force onthe CO2/water mixture, helping CO2 molecules to penetratethe porous biomass structure, and at the same time, due toCO2’s presence in the headspace of the reaction, it reduces thepartial pressure of water and keeps the hydronium ions, formedduring autohydrolysis of water, in the liquid phase.51 This alsoleads to higher glucose formation within shorter residencetimes, due to faster cleavage of glycosidic bonds than in the caseof a pure water reaction.41 Contrary to findings obtained byMcWilliam et al.133 and van Walsum et al.,60 a relationshipbetween the amount of CO2 loaded and the pH of the CO2-enriched water was attained.51 Therefore, various reactionswithin the pathways are sensitive to the pH, especially ifcompared to different pretreatment methods.5.1.3. Starch. Each type of polysaccharide needs different
reaction conditions. Many biomass sources contain starch asthe main polysaccharide, and the hydrolysis conditions requireoptimization to obtain a high yield of glucose, maltose, andfructose in a sustainable manner. Actually, the research onhydrolysis of starch started at the end of the 1990s.40 Unlikecellulose, starch is easily hydrolyzed under hydrothermalconditions without the addition of organic acids or enzymes.However, the starch to glucose yield is lower than that achievedby enzymatic technologies. It is supposed that starch is muchmore susceptible to hydrolysis, leading to a higher concen-tration of degradation products from glucose.148 Thangavelu etal. found that microwave hydrothermal hydrolysis of sago pith
waste (starchy lignocellulosic biomass) was improved whenCO2 (dry ice) was added. A maximum glucose yield of 43.8%was obtained when CO2 was applied at 900 W and 2 min ofirradiation.11 Nagamori and Funazukuri studied the hydrolysisof starch in water as a function of the glucose, fructose, maltose,and 5-HMF yields at 180−240 °C for various reaction times.They reported that high amounts of glucose were obtainedearly in the reaction, and with continuation of the reaction,glucose was degraded, mostly to 5-HMF.149 Miyazawa andFunazukuri reported significantly lower yields of glucose fromstarch (3.7%) at 200 °C after 15 min at unknown pressures.However, they found that the production of glucose increasessignificantly (14-fold) when CO2 is added (CO2:starch ratio of9, w/w) under similar conditions.137 They found that high CO2pressure promotes hydrolysis of polysaccharides into lowmolecular weight sugars, as mentioned earlier for xyloseproduction. The same tendency was found for glucose obtainedfrom starch, where the yield was increased from 3.7% to 53.0%by increasing the CO2:biomass loading from 0 to 10.7 g·g−1
when the reaction was conducted at 200 °C for 15 min.Moreschi et al. found that a CO2/H2O binary system undersubcritical conditions (200 °C and 150 bar of CO2) had a greateffect on the hydrolysis of starch-rich materials into a mixture ofgluco-oligosaccharides and glucose (above 97.1% after 15 minof residence time).150 Also, Liu et al. reported a higher yield ofstarch-derived sugars when CO2 was used as a soluble catalyst,which easily dissociates under the reaction conditions.151 Theyobserved an increase of the glucose yield by a factor of 50 quiteindependently of the initial starch concentration. Elevatedglucose yields were obtained when CO2 was used as a catalyst.An increased glucose yield of around 50% was obtained whenstarch was hydrolyzed at 230 °C and 240 bar (96% CO2saturation) in comparison to that of a water-only reactionunder the same conditions.40 In addition, they found that, at ahigher initial starch concentration (10%), hydrolysis with aCO2-saturated aqueous mixture resulted in a similar glucoseyield and selectivity toward glucose formation compared to thatof a lower starch loading (0.2%).40 Orozco et al. reported amaximum glucose yield of 548 g·kg of starch−1 from hydrolysisin hot water (200 °C) with 30 bar of CO2, accompanied bygeneration of 5-HMF.152
6. HEXOSE-DERIVED SUGAR CONVERSION INHIGH-PRESSURE CO2 AND CO2/H2O MIXTURES
The dehydration of biomass-derived cellulose and starch wasstudied in many systems using a large variety of catalysts,including organic acids153−155 or solid acids.156,157 Manysolvents, including supercritical water,158,159 acetone, andmethanol plus their mixtures, were employed in dehydrationof glucose and fructose.155,160 The decomposition of hexoseswas widely studied under both sub- and supercritical waterconditions for various reaction times.161 In high-pressure watersystems, the addition of CO2 has a large role in thedecomposition of hexoses due to the high concentration of
Scheme 1. Formation of 5-HMF in an Acid-Catalyzed Reaction
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXO

hydronium and hydroxide ions from autoionization of waterand from interaction between CO2 and water, which promotesthe dehydration reaction of glucose to a furan derivative(Scheme 1). Furthermore, in acid-catalyzed synthesis of 5-HMF from D-fructose, two different reactions can occurdepending on the pH of the medium. The first one, at lowpH (1.5), is 5-HMF rehydration into levulinic and formic acids,and the second is a polymerization reaction between 5-HMFand fructose taking place at pH > 2.158
According to a report of the U.S. Department of Energy, 5-HMF is considered one of the most versatile platformchemicals derived from biomass.162 The presence of twofunctional groups, combined with a furan ring, makes it aversatile precursor for a wide variety of chemicals relevant tothe fuel, polymer, and pharmaceutical industries as depicted inScheme 2.83
For this reason, diverse raw materials were used to produce5-HMF. For instance, lignocellulosic biomass, polysaccharideswith a high molecular weight (cellulose,163 starch,164 andinulin165), disaccharides (sucrose, maltose, and cello-biose166,167), and monosaccharides (glucose and fruc-
tose158,161) were transformed to 5-HMF, and the latter wasconverted to a wide variety of value-added products usingsupercritical fluids. Water under supercritical conditions is themost often used SCF to produce 5-HMF. However, high-pressure water technologies require temperatures in the rangeof 100−400 °C so that, on an industrial scale, a potentialreduction of the dehydration temperature means energysavings. The way to achieve this is the use of a CO2/watermixture. However, there is still little research in this field, butthe outcomes indicate promising results. For example, somegroups demonstrated that the production of 5-HMF fromhexoses can be achieved at lower temperatures in the presenceof CO2. Wu et al. reported the conversion of inulin, anoligosaccharide consisting of glucose−fructose or fructosepolymers, to 5-HMF using a CO2/water mixture.165 Such asystem led to a higher conversion of inulin to 5-HMF with ayield of 53%, an increase of 15% compared to that of reactionswithout CO2. Therefore, it can be assumed that the yieldenhancement is a consequence of the in situ carbonic acidformation. The same authors described that conversion ofinulin to 5-HMF is independent of the temperature used, since
Scheme 2. Chemistry and Applications of 5-HMF and Its Derivativesa
aA solid arrow indicates direct transformation and a broken arrow a multistep reaction. Abbreviations: 5-HMF, 5-(hydroxymethyl)furfuran; LEVA,levulinic acid; LEVE, levulinic ester; FA, formic acid; HFCA, 5-(hydroxymethyl)furoic acid; FDC, 2,5-furandicarbaldehyde; FDCA, 2,5-furandicarboxylic acid; DHMF, 2,5-bis(hydroxymethyl)furan; DHM-THF, 2,5-bis(hydroxymethyl)tetrahydrofuran; HMTHFA, 5-(hydroxymethyl)-tetrahydrofuran-2-carbaldehyde. Compound numbers: (1) 2-(hydroxy(5-(hydroxymethyl)tetrahydrofuran-2-yl)methyl)-5-(hydroxymethyl)-tetrahydrofuran-2-carbaldehyde; (2) (E)-4-(5-(hydroxymethyl)furan-2-yl)but-3-en-2-one; (3) (1E,4E)-1,5-bis(5-(hydroxymethyl)furan-2-yl)penta-1,4-dien-3-one; (4) tetrahydrofurfuryl alcohol; (5) 2,5-dimethyltetrahydrofuran; (6) furan; (7) 2-(hydroxymethyl)-5-vinylfuran; (8) furfuryl alcohol;(9) 2,5-bis(aminomethyl)furan; (10) 2-methyltetrahydrofuran; (11) 2,5-dimethylfuran; (12) 2-methylfuran. Reprinted from ref 83. Copyright 2011American Chemical Society.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXP

the CO2 pressure is optimized to obtain a maximum 5-HMFyield. The maximum yield of 5-HMF was obtained for 60 bar ofCO2 at 160, 180, and 200 °C. The main reason for this is thatthere is an optimal pH that can be achieved and can be adjustedby the CO2 pressure. Similar results were obtained in anaqueous solution using iron vanadyl phosphate as the catalyst(around 40% yield)168 and with ionic liquids/oxalic acid (56%yield).169 Recently, the influence of scCO2, in an ionic liquidsystem, on the formation of 5-HMF from hexoses was reportedby Machida et al.170 In a series of reactions with an ionic liquid(1-butyl-3-methylimidazolium chloride) as a solvent plusscCO2 as a cosolvent and D-fructose as the starting material,a strong effect of scCO2 on the reaction results was shown.scCO2 demonstrated the ability to decrease the viscosity ofionic liquids, which is favorable for a high-yielding conversionof D-fructose to 5-HMF (above 90%). In this case, CO2 acted asa viscosity-reducing agent for the ionic liquid reactions.170 Afterdehydration of hexoses to 5-HMF, the CO2 employed in thereaction can be used as a medium for extraction and recovery of5-HMF, analogous to the previously discussed furfuralrecovery.171
7. CONVERSION OF BIOMASS-DERIVEDCOMPOUNDS IN HIGH-PRESSURE CO2 ANDCO2/H2O MIXTURES
7.1. Hydrogenation
Most of the processes where gases react with liquids arecharacterized by low reaction rates. This is mostly caused by themass transfer limitations at the active sites of the catalysts. Thesolubility of some gases (for example, H2) in water or othertypical organic solvents is very low. scCO2 is an alternativesolvent that is feasible for carrying out these reactions witheither homogeneous or heterogeneous catalysts. CO2 under
supercritical conditions is still a liquid and is completelymiscible with H2. At the same time, scCO2 having a liquidlikedensity is a good solvent for nonpolar organics. Thus, takingthese dual properties of CO2 under supercritical conditions, itacts as a carrier for gas to the liquid phase, minimizing the masstransfer limitations.172−175 An additional benefit is thereduction of both flammability and catalyst fouling risks.Thus, CO2 can enhance the sustainable and economic viabilityof a hydrogenation process.176 In this section we aim todemonstrate hydrogenations in scCO2 with chemicals obtaineddirectly from biomass in the frame of the biorefinery concept.An example of application of scCO2 is a conversion of levulinicacid to γ-valerolactone (GVL) from glucose-derived biomass inwhich water was used as a cosolvent (Scheme 3).177 Poliakoff’sgroup developed an approach where a high GVL yield wasachieved (>99%) even at a lower pressure (around 100 bar)with a high concentration of levulinic acid.177 Furthermore, asGVL is soluble in water, the product accumulated in theaqueous phase can be easily extracted by scCO2 after thereaction.Levulinic acid is one of the most important platform
molecules produced from biomass, and the conversion toGVL represents an important step in its further conversion to aliquid transport fuel. However, this is very challenging becauseof the relatively low solubility of levulinic acid in CO2(xlevulinic acid = 0.009 at 182.5 bar at 39.85 °C), so a cosolvent(e.g., ethanol) is needed to improve this, giving a 3-foldincrease of the levulinic solubility at the same conditions(xethanol = 0.051).178
Outstanding results for the hydrogenation of 5-HMF under ahigh-pressure CO2/H2O mixture were presented by Chatterjeeet al., who designed a high-pressure CO2/water approach forthe conversion of 5-HMF into 2,5-dimethylfuran (DMF) with atotal yield of 100%.179 Various products can be obtained,
Scheme 3. γ-Valerolactone Production Pathwaysa
aA double-crossed arrow means several reaction steps.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXQ

depending on the CO2 pressure. At 80 °C for 2 h withpressures below 100 bar of CO2, tetrahydro-5-methyl-2-furanmethanol was produced, while pressures close to orabove 100 bar led to the production of DMF and 2,5-dimethyltetrahydrofuran, with high selectivity. Another inter-esting note is that, in the absence of CO2 or water, theselectivity toward DMF is negligible.179 The same approach wastested in the hydrogenation of furfural, which was quantitativelyconverted into 2-methylfuran at a very short reaction time (10min).179
7.2. Conversion of Proteins
Amino acids, the building blocks for proteins, have a highercommercial value than other biomass constituents. Amino acidsare interesting products for diverse industrial sectors such as thefood industry, the agroindustry, and pharmaceutical manufac-turing. Therefore, protein conversion into amino acids has beenextensively studied using hydrothermal technologies.180,181
Surprisingly, the hydrolysis of proteins with CO2 has rarelybeen studied. Rogalinski et al. demonstrated that hydrolysis ofproteins had a behavior similar to the hydrolysis ofpolysaccharides.182 Scheme 4 illustrates the reaction mecha-nism of acid-catalyzed hydrolysis of peptides.The addition of CO2 accelerates the hydrolysis reaction
because proton addition to the peptide is a determining step forthe reaction.182 However, the amino acid yields are fairly loweven under optimized conditions since the peptide bond in theprotein molecule is much more stable than β-1,4- and β-1,6-glycosidic linkages in polysaccharides.182
7.3. Delignification and Lignin Extraction under scCO2
Sustainable treatments of lignocellulosic materials becomeparticularly attractive when employed into a complete cycleof all components. Biomass is capable of being entirelyvalorized, enabling its components to be converted intovalue-added products, followed by “green” treatment of anywastes generated with renewed buildup of biomass.40 Thedevelopment of cost-effective pretreatment technologies toseparate polymers from biomass is needed.183 Most of thetechnologies allow for a high sugar yield, but the full recovery ofall components for maximal exploitation, including nonsugarfractions, should be one of the most important goals of bio-based processes from biomass. In this section, special attentionis given to the use of high-density fluids to remove lignin fromthe biomass matrix as well as prevent the degradation of woodpolysaccharides. The valorization of lignin plays an important
key role in the development of biorefinery processes for theproduction of biofuels, biomaterials, and biochemicals.The use of CO2 as a solvent for wood pulping demonstrated
several benefits, such as the generation of anomalous porosityand a lamellar structure of the biomass when subjected to thepretreatment. These effects make the organic matter moreaccessible to the solvents and/or to the pulping reagents,improving the pulping efficiency.104,184 The results of scCO2applications as a solvent for delignification and lignin extractionare compiled in Table 5.Schrems et al. investigated the effect of CO2 presence on an
organic solvent pulping process (ethanol/water) using modellignin substances.187 Carbon dioxide was found to increase theefficiency of the delignification process by a decrease of theactivation energy (42.0 and 53−68.9 kJ·mol−1 for processeswith and without scCO2, respectively) and by accelerating thebreakdown. While CO2 led to accelerated consumption of themodel compounds, the formation of stable end products wasslowed. scCO2 changed the chemical pathways and activationparameters and therefore the kinetics of the delignificationprocess. A high CO2 pressure was essential to promote thereaction between solvent and lignin. Pasquini et al. studied thepulp produced in an scCO2 process using the pulp of Pinustaeda wood chips and sugarcane bagasse with an ethanol/watermixture as a cosolvent.184 In this work, different ethanol/watermixtures (50−100% ethanol for sugarcane bagasse and 30−100% ethanol for P. taeda wood chips), temperatures (142−198°C), pressures (145−230 bar), and residence times (30−120and 30−150 min for sugarcane bagasse and P. taeda woodchips, respectively) were evaluated. They found that higherpressures led to an elevated residual lignin content, whilepulping yields were found to be similar. The process with 160bar of pressure at 190 °C gave residual Klason lignin contentsof 4.9% and 8.7% for P. taeda wood chips and sugarcanebagasse, respectively. The pulping yields were 43.7% and 32.7%for these materials, and delignification was in the range of 93%and 88%, respectively. They also observed that higher pressuresled to the same yields but lower delignification. Virtuallycomplete delignification occurred after 30 min of cooking time,mostly due to the high pressure and high acidity resulting fromthe in situ carbonic acid generation.184 Curvelo and co-workersstudied the delignification of sugarcane bagasse with sub- andsupercritical CO2 using a 1-butanol/water mixture as acosolvent.185 The highest delignification content (94.5%) wasobtained at 190 °C and 70 bar for 105 min using a mixture of 1-butanol/water (60%:40%, v/v). However, it is important to
Scheme 4. Schematic Acid-Hydrolysis Reaction of a Peptide Bond
Table 5. scCO2-Mediated Delignification from Lignocellulosic Materials
reaction conditions
lignin/model compd solvent T (°C) P (bar) t (min) pulp yield (%) delignification yield (%) ref
Pinus taeda wood CO2/C2H6O/H2O, 1:1, v/v 190 160 60 43.7 93.1 184sugarcane bagasse CO2/C2H6O/H2O, 1:1, v/v 190 160 60 32.7 88.4 184
1-butanol/H2O, 60:40, v/v 190 70 105 8.7 94.5 185aspen chips CO2/SO2, 98:2, mol/mol 140 138 240 84.0 186
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXR
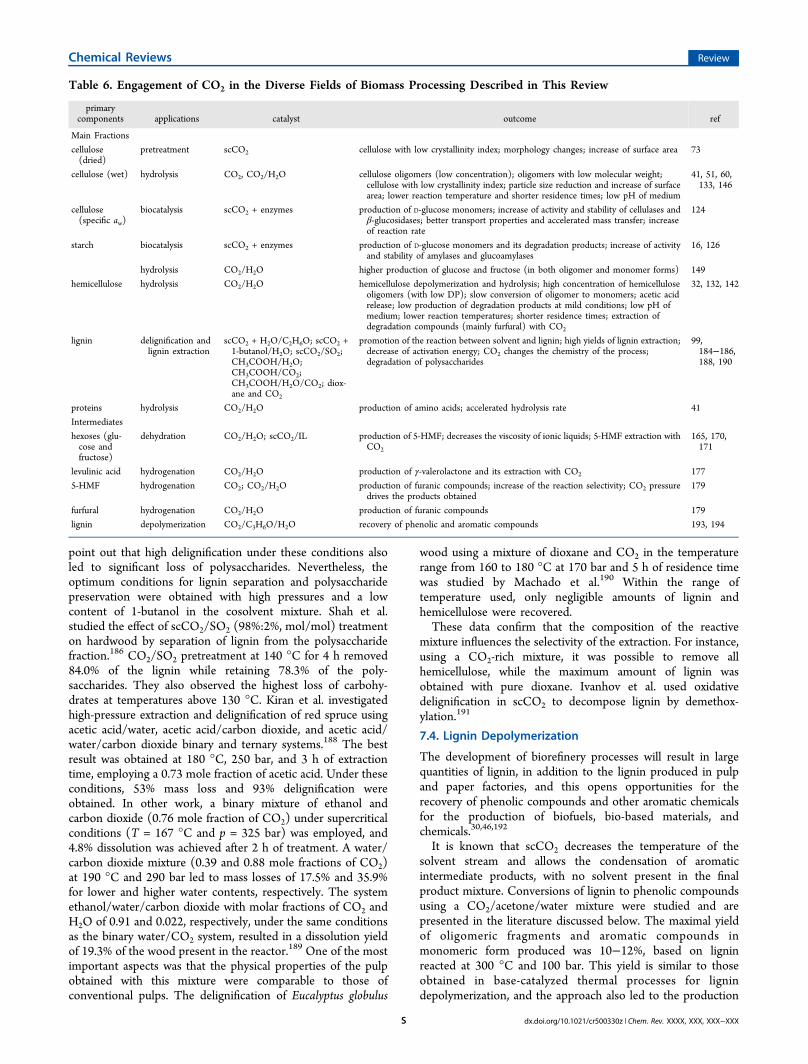
point out that high delignification under these conditions alsoled to significant loss of polysaccharides. Nevertheless, theoptimum conditions for lignin separation and polysaccharidepreservation were obtained with high pressures and a lowcontent of 1-butanol in the cosolvent mixture. Shah et al.studied the effect of scCO2/SO2 (98%:2%, mol/mol) treatmenton hardwood by separation of lignin from the polysaccharidefraction.186 CO2/SO2 pretreatment at 140 °C for 4 h removed84.0% of the lignin while retaining 78.3% of the poly-saccharides. They also observed the highest loss of carbohy-drates at temperatures above 130 °C. Kiran et al. investigatedhigh-pressure extraction and delignification of red spruce usingacetic acid/water, acetic acid/carbon dioxide, and acetic acid/water/carbon dioxide binary and ternary systems.188 The bestresult was obtained at 180 °C, 250 bar, and 3 h of extractiontime, employing a 0.73 mole fraction of acetic acid. Under theseconditions, 53% mass loss and 93% delignification wereobtained. In other work, a binary mixture of ethanol andcarbon dioxide (0.76 mole fraction of CO2) under supercriticalconditions (T = 167 °C and p = 325 bar) was employed, and4.8% dissolution was achieved after 2 h of treatment. A water/carbon dioxide mixture (0.39 and 0.88 mole fractions of CO2)at 190 °C and 290 bar led to mass losses of 17.5% and 35.9%for lower and higher water contents, respectively. The systemethanol/water/carbon dioxide with molar fractions of CO2 andH2O of 0.91 and 0.022, respectively, under the same conditionsas the binary water/CO2 system, resulted in a dissolution yieldof 19.3% of the wood present in the reactor.189 One of the mostimportant aspects was that the physical properties of the pulpobtained with this mixture were comparable to those ofconventional pulps. The delignification of Eucalyptus globulus
wood using a mixture of dioxane and CO2 in the temperaturerange from 160 to 180 °C at 170 bar and 5 h of residence timewas studied by Machado et al.190 Within the range oftemperature used, only negligible amounts of lignin andhemicellulose were recovered.These data confirm that the composition of the reactive
mixture influences the selectivity of the extraction. For instance,using a CO2-rich mixture, it was possible to remove allhemicellulose, while the maximum amount of lignin wasobtained with pure dioxane. Ivanhov et al. used oxidativedelignification in scCO2 to decompose lignin by demethox-ylation.191
7.4. Lignin Depolymerization
The development of biorefinery processes will result in largequantities of lignin, in addition to the lignin produced in pulpand paper factories, and this opens opportunities for therecovery of phenolic compounds and other aromatic chemicalsfor the production of biofuels, bio-based materials, andchemicals.30,46,192
It is known that scCO2 decreases the temperature of thesolvent stream and allows the condensation of aromaticintermediate products, with no solvent present in the finalproduct mixture. Conversions of lignin to phenolic compoundsusing a CO2/acetone/water mixture were studied and arepresented in the literature discussed below. The maximal yieldof oligomeric fragments and aromatic compounds inmonomeric form produced was 10−12%, based on ligninreacted at 300 °C and 100 bar. This yield is similar to thoseobtained in base-catalyzed thermal processes for lignindepolymerization, and the approach also led to the production
Table 6. Engagement of CO2 in the Diverse Fields of Biomass Processing Described in This Review
primarycomponents applications catalyst outcome ref
Main Fractions
cellulose(dried)
pretreatment scCO2 cellulose with low crystallinity index; morphology changes; increase of surface area 73
cellulose (wet) hydrolysis CO2, CO2/H2O cellulose oligomers (low concentration); oligomers with low molecular weight;cellulose with low crystallinity index; particle size reduction and increase of surfacearea; lower reaction temperature and shorter residence times; low pH of medium
41, 51, 60,133, 146
cellulose(specific aw)
biocatalysis scCO2 + enzymes production of D-glucose monomers; increase of activity and stability of cellulases andβ-glucosidases; better transport properties and accelerated mass transfer; increaseof reaction rate
124
starch biocatalysis scCO2 + enzymes production of D-glucose monomers and its degradation products; increase of activityand stability of amylases and glucoamylases
16, 126
hydrolysis CO2/H2O higher production of glucose and fructose (in both oligomer and monomer forms) 149
hemicellulose hydrolysis CO2/H2O hemicellulose depolymerization and hydrolysis; high concentration of hemicelluloseoligomers (with low DP); slow conversion of oligomer to monomers; acetic acidrelease; low production of degradation products at mild conditions; low pH ofmedium; lower reaction temperatures; shorter residence times; extraction ofdegradation compounds (mainly furfural) with CO2
32, 132, 142
lignin delignification andlignin extraction
scCO2 + H2O/C2H6O; scCO2 +1-butanol/H2O; scCO2/SO2;CH3COOH/H2O;CH3COOH/CO2;CH3COOH/H2O/CO2; diox-ane and CO2
promotion of the reaction between solvent and lignin; high yields of lignin extraction;decrease of activation energy; CO2 changes the chemistry of the process;degradation of polysaccharides
99,184−186,188, 190
proteins hydrolysis CO2/H2O production of amino acids; accelerated hydrolysis rate 41
Intermediates
hexoses (glu-cose andfructose)
dehydration CO2/H2O; scCO2/IL production of 5-HMF; decreases the viscosity of ionic liquids; 5-HMF extraction withCO2
165, 170,171
levulinic acid hydrogenation CO2/H2O production of γ-valerolactone and its extraction with CO2 177
5-HMF hydrogenation CO2; CO2/H2O production of furanic compounds; increase of the reaction selectivity; CO2 pressuredrives the products obtained
179
furfural hydrogenation CO2/H2O production of furanic compounds 179
lignin depolymerization CO2/C3H6O/H2O recovery of phenolic and aromatic compounds 193, 194
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXS

of a wide variety of monomeric compounds, giving maximumyields of 2% and 3.6% for syringic acid and syringol,respectively.193 Eckert et al. also demonstrated that CO2 is agas expander liquid to extract phenolic compounds such asvanillin, syringaldehyde, and syringol.194
8. OVERVIEW OF CO2 APPLICATIONS WITHIN THEBIOREFINERY CONCEPT
This review has shown that high-pressure CO2 is an interestingsolvent, which can be used in a great variety of reactions withinthe biorefinery concept starting from its use in direct biomassprocessing to its use as a catalyst for dehydration of biomass-derived carbohydrates and its use in specific applications forbiomass intermediary compounds. The application of high-pressure CO2 technology in such a wide variety of processes isstill in its infancy. However, due to the extraordinary propertiesof CO2, either acting individually or as a cosolvent in mixtures,CO2 (under both sub- and supercritical conditions) maycontribute to tackling the aforementioned challenges. Table 6shows the most representative examples of CO2 application inbiomass processing and the effects of sub/supercritical CO2 andmixtures of CO2 with other solvents (water, ethanol, etc.) oneach biomass fraction (either principal compounds orintermediary products).
9. ECONOMIC ASPECTS OF CO2 PROCESSING OFBIOMASS
A number of reports of catalytic reactions in high-pressure CO2confirm the commercial potential of these processes. In spite ofthe requirement for sustainable reactions, CO2 can be used as ahelpful chemical in green technologies. Furthermore, CO2-basedtechnologies can be designed to deliver optimal performancewith maximal energy savings. Furthermore, in terms of biomassprocessing, CO2-assisted technologies have benefits overconventional processes, as presented above.Considering the economic aspects of CO2 use in biomass
processing, it is important to mention that the biorefineryconcept assumes the integrated valorization of biomass towardenergy, materials, and chemicals. The production of bulky butlow-value energy sources such as ethanol can be counter-balanced by coproduction of small quantities of highly pricedchemicals, for instance, polyphenols and oligosaccharides. Thisjoint on-site valorization gives economic benefits, which arealready observed in classical petrorefineries. The production ofbulky low-price fuels is subsidized by production of lowamounts of high-value polymers. It is also very important topoint out that the price of feedstock is a large proportion of thefinal product price. In the case of fossil feedstock, the prices aresometimes far above $100 per barrel,195 while biomassfeedstock can be obtained at low or no cost. Furthermore,the great variety and versatility of existing biomass make futurebiorefineries less dependent on geopolitical turmoil.One of the important aspects that should be taken into
consideration when comparing classical petrochemical refin-eries and new biorefineries is the maturity of both technologies.The classical petrochemical refineries were developed andoptimized over more than a century,196 while biorefineries arestill in their infancy. Even nowadays, existing biorefineries donot valorize all production streams, or valorize them in a waythat is not the most economically feasible. Thus, because ofthese aspects, both classical petrochemical refineries andbiorefineries should not be compared straightaway.
The use of CO2 as a potential reactant in biorefineries mightbe especially beneficial, as CO2 is often a waste product frommany industrial processes (cement factories or power plantfactories) and, as such, can be obtained with little cost. At thesame time, it is considered as a green solvent, which definitivelyimproves the environmental aspects of the future biorefinery.Independently of the manner that CO2 pretreatment isemployed, current efforts to develop new methods of biomassvalorization with CO2 do not yet guarantee its economic andtechnical viability. These technologies require large investmentcosts, mostly related to the high-pressure equipment. On theother hand, these technologies demonstrated some advantages,e.g., in downstream processing for various applications,especially in comparison to other classical processes such asacid hydrolysis. Additionally, in the case of high-pressuretechnologies, higher yields and easier control of processselectivity can be obtained normally, which are beneficial forthe process economics.Therefore, to find the most adequate application of CO2 in
biorefineries, a large investment in R&D and later in pilot anddemonstration projects is needed.3 In addition, each processmust be analyzed separately to answer the question related tothe economic feasibility of the processes with CO2, consideringthe state of the art of the technology applied.
10. PERSPECTIVESThe present review shows the most important aspects of CO2use as a green solvent in biomass valorization. As mentionedbefore, the scope of this review does not include the extractionpart, as this was tackled by dozens of investigations during theprevious decades. The conclusion for CO2 use in biomassprocessing is that, in general, CO2 is still rarely used and that itsspecial benefits as a solvent under supercritical conditionswould justify its broader application. Thus, there is still placefor improvements, and the use of CO2 should be exploredfurther in several areas. Some of these are (i) direct hydrolysisof untreated biomass in the presence of CO2, (ii) use of otherpotentially beneficial cosolvents in mixtures with CO2 insteadof the CO2/H2O system, (iii) development of CO2-formedvalue-added products from all biomass fractions, (iv) processanalysis from a sustainability point of view, and (v) integratedvalorization of each biomass fraction.(i) A direct hydrolysis of untreated biomass in the presence
of CO2 would allow the integration and intensification of thebiomass valorization. In this way, biomass would not require acost-demanding pretreatment step, allowing the use of biomassdirectly in glucose production, which later could be convertedto energy sources such as biofuels. This challenging task hasbarely been tackled up to now, and only pure cellulose samples(cotton124 or Avicel117) have been examined. It is important tostress that this is one of the most important aspects in theeconomic and technological development of biomass process-ing, which also would require advances in enzyme engineeringto develop more resistant and efficient biological catalysts.127
(ii) Water is one of the best-known solvents; however, mostorganic compounds are insoluble in water. In addition, themutual solubility of water and CO2 is limited; thus, to takeadvantage of sub- or supercritical conditions, high pressures andtemperatures are required. Therefore, looking for alternativesfor water used in mixtures with CO2 is an interesting field ofresearch, which has not yet been properly explored. The latentsolution might be the use of carbonate-type solvents. This newclass of solvents is an alternative for many existing problems.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXT

For example, the synthesis of 1,2-glycerolcarbonate197,198 usesthe overproduced glycerol. In addition, the synthesis of thisclass of compounds also involves CO2 insertion, making thesynthesis greener and more sustainable. It cannot be forgottenthat other solvents can be used, although they have to fulfill therequirements of green chemistry and green chemical engineer-ing.47,199−202
(iii) Lignin was often considered as a less valuable fraction ofbiomass. Nowadays, however, it serves as a source ofpolyphenols.10,30,46,192,193 These references show the greatpotential in the CO2-assisted production of phenoliccompounds directly from biomass. It should also be mentionedthat this approach is different from the already well-known andexamined extraction of value-added phenolic-like compoundsfrom specific plants for pharmaceutical or food purposes.203,204
Hemicellulose is also very often considered as a fraction withlower value. The reason for this is its diverse composition,which is strongly dependent on the biomass type and otherexternal factors (e.g., climacteric and storage conditions). Thisdiversity makes the hemicellulose fraction valorization challeng-ing and less versatile, but it gives room for numerous productsthat can be obtained, depending on the technique applied.20,107
Among them are oligosaccharides,32,205 xylitol,206 and manyothers, including simple sugars,107 which can be produced withthe assistance of high-pressure CO2. Cellulose is mostly usedfor ethanol production, but there are many other chemicals thatcan be produced from this feedstock. Some examples (5-HMF,levulinic acid, formic acid, GVL) presented in this work showthe unrevealed potential of CO2 adjunctive application incellulose processing.16
(iv) SWOT (strengths, weaknesses, opportunities, andthreats) analysis of any process is crucial to find the pros andcons of each technology. This can be done in many waysconsidering various aspects. One of the easiest methods to doso is via the use of green metric techniques.207,208 Although theproposed manner of process analysis is much simpler than, forexample, life cycle analysis, it is important to add that it can beused successfully for very complex processes. It considersimportant metrics such as economic feasibility amd energy andmaterial efficiencies,209 depending on the particular needs ofthe examined process. In the case of processes with CO2 in thebiorefinery, it would be especially beneficial because thecomplexity of the processes and lack of many data would becompensated by the simplicity that the use of green metricsbrings.210
(v) As was already mentioned, LCMs are constituted bythree main fractions: cellulose, hemicellulose, and lignin. Up tonow the integrated valorization of these three fractions at theindustrial scale has not existed. The need for more research inthis direction is crucial to provide a sufficient knowledge in thisarea and to gain sufficient maturity to advance toward pilot ordemonstration scales of LCM biorefineries. The major problemis that, even at the bench-scale level, these kinds of processesare rarely presented in the literature; thus, academia needs tomake more effort to perform research that tackles the variousaspects of biomass processing.
11. CONCLUSIONSAwareness of today’s energy crisis and the climatic changes,which are threatening the world economy and sustainability,have increased the drive to substitute the current hazardoustechnologies by more sustainable, greener, and environmentallyfriendly processes. Moreover, the wide-world production, high
abundance, and great variety of LCMs makes them part of thesolution for the dependence on fossil fuel processes. Super-critical carbon dioxide as well as other nowadays broadlyexamined solvents and processes fits into this trend, recognizingCO2 as a nontoxic, green, and more sustainable solventfacilitating biomass processing. When in contact with water,which might be the moisture in biomass, this technology getsadditional benefits from the acidity introduced by the formationof carbonic acid. High-pressure CO2 has been demonstrated tobe very effective in fractionation and hydrolysis of lignocellu-losic fractions, showing its utility to achieve a range of goals inbiomass processing. Furthermore, after pretreatment, high-pressure CO2 can act as an extraction medium, removingproducts of interest or inhibitors of further processing, e.g.,fermentation. Thus, supercritical CO2 has proved to be anattractive alternative medium for integrated biomass processingwithin the biorefinery concept.
AUTHOR INFORMATION
Corresponding Author
*E-mail: [email protected]. Fax: +351217163636. Phone:+351210924600, ext 4224.
Notes
The authors declare no competing financial interest.
Biographies
Ana R. C. Morais received her M.Sc. and B.Sc. degrees in foodengineering and in biosciencesmicrobiology from the School ofAgronomy of the Technical University of Lisbon and PortugueseCatholic University. Currently, she is a Ph.D. researcher in sustainablechemistry in the Bioenergy Unit at the National Laboratory of Energyand Geology (LNEG), Portugal. She is experienced in high-pressurefluids, namely, CO2, working under the supervision of Dr. Rafał Bogel-Łukasik and Prof. Manuel Nunes da Ponte. Her research is focused onthe application of supercritical CO2 as a green solvent in processingbiomass in the frame of the biorefinery concept. She has given a seriesof lectures on her work to international congresses. Recently, she wasawarded the Innovation for Sustainability Award (2014) in thecategory processes for the RefinOleaIntegrated Valorization ofResidues and Sub-Products of Olive Oil Extraction Project. In 2012she was awarded the Poster Award at the third UBIOCHEM COSTAction Conference for the work entitled “Biological Pathways ofIsoprene Production”.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXU

Andre M. da Costa Lopes received both B.Sc. and M.Sc. degrees inbiotechnology from the University of Aveiro, Portugal. He gainedexperience in using alternative solvents for the valorization ofagricultural and industrial wastes, particularly pretreatment oflignocellulosic biomass using ionic liquids and hydrogenation ofanimal fat residues in supercritical CO2. Currently, he is a Ph.D.student in sustainable chemistry working under the supervision of Dr.Rafał Bogel-Łukasik and Prof. Manuel Nunes da Ponte. His research isfocused on valorization of lignocellulosic biomass using alternativesolvents in the frame of green and sustainable chemistry towardintegration in biorefineries.
Rafał Bogel-Łukasik graduated from the Warsaw University ofTechnology, Poland, with M.Sc. and B.Sc. degrees (2002). Hereceived his Ph.D. (2007) in chemical engineering from the NewUniversity of Lisbon, Portugal. Since 2009 he has held the position ofResearcher in the Bioenergy Unit at the National Laboratory of Energyand Geology (LNEG), Portugal. He was Marie Curie Fellow at TheQueen’s University Ionic Liquid Laboratories Research Centre, UnitedKingdom, at the Institute of Experimental Biology and Technology,Portugal, and at the Technical University of Hamburg, Germany. In2008 he was awarded The International Association of ChemicalThermodynamics Junior Award for Excellence in Thermodynamics.Recently, he was awarded the Innovation for Sustainability Award inthe category processes and the Green Project Award for theRefinOleaIntegrated Valorization of Residues and Sub-Products ofOlive Oil Extraction Project. Overall, he is the coauthor of more than60 peer-reviewed papers and a few book chapters and an invited editorof a book of the Royal Society of Chemistry Series, entitled IonicLiquids in the Bioref inery Concept. He is also a guest editor of the WhiteBiotechnologySustainability in Industry thematic issue of SustainableChemical Processes. His work is focused on biomass processing usingthe integrated approach of green chemistry in the biorefinery concept.
ACKNOWLEDGMENTS
This work was supported by the Fundacao para a Ciencia e aTecnologia (FCT, Portugal) through Bilateral CooperationProject FCT/CAPES 2014/2015 (Grant FCT/1909/27/2/2014/S) and Grants SFRH/BD/94297/2013 (A.R.C.M.),SFRH/BD/90282/2012 (A.M.d.C.L.), and IF/00424/2013(R.B.-Ł.) and by CAPES (Brazil) through the projectPesquisador Visitante Especial 155/2012. Special thanks aregiven to Mr. R. Andrew Wilbey for proof-reading this work.
REFERENCES(1) Clark, J. H.; Deswarte, F. E. I.; Farmer, T. J. Biofuels, Bioprod.Biorefin. 2009, 3, 72.(2) FitzPatrick, M.; Champagne, P.; Cunningham, M. F.; Whitney, R.A. Bioresour. Technol. 2010, 101, 8915.(3) HORIZON2020Proposal for a Council Decision Establishing theSpecific Programme Implementing Horizon 2020The FrameworkProgramme for Research & Innovation (2014−2020); EuropeanCommission: Brussels, Belgium, 2013.(4) Morais, A. R. C.; Bogel-Lukasik, R. Sustainable Chem. Processes2013, 1, 18.(5) Van Ree, R. IEA Bioenergy Task 42 Biorefining; InternationalEnergy AgencyIEA Bioenergy: Paris, 2014.(6) Elvnert, J.; Kleinschmit von Lengefeld, A.; Ringman-Beck, J.;Kerckow, B.; Sternberg, K.; Carrez, D.; Laurmaa, J.; Fehrmann, J.;Metzlaff, K.; Travella, S.; Perimenis, A.; Majer, S.; Meisel, K.; Muller-Langer, F.; Makinen, T.; Clark, J. H.; Kazmi, A.; Thomas, D.; van Ree,R. Joint European Biorefinery Vision for 2030, Star-COLIBRI, StrategicTargets for 2020Collaboration Initiative on Biorefineries; EuropeanUnion: Brussels, Belgium, 2011.(7) Roper, H. Starch/Staerke 2002, 54, 89.(8) Budarin, V. L.; Shuttleworth, P. S.; Dodson, J. R.; Hunt, A. J.;Lanigan, B.; Marriott, R.; Milkowski, K. J.; Wilson, A. J.; Breeden, S.W.; Fan, J. J.; Sin, E. H. K.; Clark, J. H. Energy Environ. Sci. 2011, 4,471.(9) Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev.2000, 100, 39.(10) Buranov, A. U.; Mazza, G. Ind. Crops Prod. 2008, 28, 237.(11) Thangavelu, S. K.; Ahmed, A. S.; Ani, F. N. Appl. Energy 2014,128, 277.(12) Tomme, P.; Warren, R. A. J.; Gilkes, N. R. Adv. Microb. Physiol.1995, 37, 1.(13) Ratanakhanokchai, K.; Waeonukul, R.; Pason, P.;Tachaapaikoon, C.; Kyu, K. L.; Sakka, K.; Kosugi, A.; Mori, Y. InBiomass NowCultivation and Utilization; Matovic, M. D., Ed.;InTech: Rijeka, Croatia, 2013.(14) Hsu, T. C.; Guo, G. L.; Chen, W. H.; Hwang, W. S. Bioresour.Technol. 2010, 101, 4907.(15) Saeman, J. F. Ind. Eng. Chem. 1945, 37, 43.(16) Moreschi, S. R. M.; Petenate, A. J.; Meireles, M. A. A. J. Agric.Food Chem. 2004, 52, 1753.(17) Theander, O.; Nelson, D. A. Adv. Carbohydr. Chem. Biochem.1988, 46, 273.(18) Garrote, G.; Dominguez, H.; Parajo, J. C. Holz Roh- Werkst.1999, 57, 191.(19) Lynd, L. R.; Weime, P. J.; Van Zyl, W. H.; Pretorius, I. S.Microbiol. Mol. Biol. Rev. 2002, 66, 506.(20) Girio, F. M.; Fonseca, C.; Carvalheiro, F.; Duarte, L. C.;Marques, S.; Bogel-Lukasik, R. Bioresour. Technol. 2010, 101, 4775.(21) Galbe, M.; Zacchi, G. Biofuels 2007, 108, 41.(22) Sun, Y.; Cheng, J. Y. Bioresour. Technol. 2002, 83, 1.(23) Wyman, C. E.; Dale, B. E.; Elander, R. T.; Holtzapple, M.;Ladisch, M. R.; Lee, Y. Bioresour. Technol. 2005, 96, 1959.(24) Wyman, C. E.; Dale, B. E.; Elander, R. T.; Holtzapple, M.;Ladisch, M. R.; Lee, Y. Y. Bioresour. Technol. 2005, 96, 2026.(25) Wooley, R.; Ruth, M.; Glassner, D.; Sheehan, J. Biotechnol. Prog.1999, 15, 794.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXV

(26) Ruiz, E.; Cara, C.; Manzanares, P.; Ballesteros, M.; Castro, E.Enzyme Microb. Technol. 2008, 42, 160.(27) Carvalheiro, F.; Silva-Fernandes, T.; Duarte, L. C.; Gírio, F. M.Appl. Biochem. Biotechnol. 2009, 153, 84.(28) da Costa Lopes, A. M.; Joao, K. G.; Rubik, D.; Bogel-Lukasik, E.;Duarte, L. C.; Andreaus, J.; Bogel-Lukasik, R. Bioresour. Technol. 2013,142, 198.(29) da Costa Lopes, A. M.; Joao, K. G.; Bogel-Lukasik, E.; Roseiro,L. B.; Bogel-Lukasik, R. J. Agric. Food Chem. 2013, 61, 7874.(30) Magalhaes da Silva, S. P.; da Costa Lopes, A. M.; Roseiro, L. B.;Bogel-Lukasik, R. RSC Adv. 2013, 3, 16040.(31) Alinia, R.; Zabihi, S.; Esmaeilzadeh, F.; Kalajahi, J. F. Biosyst. Eng.2010, 107, 61.(32) Magalhaes da Silva, S. P.; Morais, A. R. C.; Bogel-Lukasik, R.Green Chem. 2014, 16, 238.(33) Hendriks, A. T. W. M.; Zeeman, G. Bioresour. Technol. 2009,100, 10.(34) Narayanaswamy, N.; Faik, A.; Goetz, D. J.; Gu, T. Y. Bioresour.Technol. 2011, 102, 6995.(35) Eggeman, T.; Elander, R. T. Bioresour. Technol. 2005, 96, 2019.(36) da Costa Lopes, A. M.; Joao, K. G.; Morais, A. R. C.; Bogel-Lukasik, E.; Bogel-Lukasik, R. Sustainable Chem. Processes 2013, 1, 3.(37) Zhu, J. Y.; Pan, X. J. Bioresour. Technol. 2010, 101, 4992.(38) Johnson, H. F. AIChE J. 1967, 13, 1042.(39) King, J. W.; Srinivas, K. J. Supercrit. Fluids 2009, 47, 598.(40) Schacht, C.; Zetzl, C.; Brunner, G. J. Supercrit. Fluids 2008, 46,299.(41) Rogalinski, T.; Ingram, T.; Brunner, G. J. Supercrit. Fluids 2008,47, 54.(42) Sasaki, M.; Adschiri, T.; Arai, K. Bioresour. Technol. 2003, 86,301.(43) Zetzl, C.; Gairola, K.; Kirsch, C.; Perez-Cantu, L.; Smirnova, I.Chem. Ing. Tech. 2011, 83, 1016.(44) Zhao, Y.; Lu, W. J.; Wang, H. T.; Yang, J. L. Bioresour. Technol.2009, 100, 5884.(45) Zeng, M. J.; Mosier, N. S.; Huang, C. P.; Sherman, D. M.;Ladisch, M. R. Biotechnol. Bioeng. 2007, 97, 265.(46) Ragauskas, A. J.; Williams, C. K.; Davison, B. H.; Britovsek, G.;Cairney, J.; Eckert, C. A.; Frederick, W. J.; Hallett, J. P.; Leak, D. J.;Liotta, C. L.; Mielenz, J. R.; Murphy, R.; Templer, R.; Tschaplinski, T.Science 2006, 311, 484.(47) Anastas, P. T.; Warner, J. C. Green Chemistry: Theory andPractice; Oxford University Press: New York, 1998.(48) Deswarte, F. E. I.; Clark, J. H.; Hardy, J. J. E.; Rose, P. M. GreenChem. 2006, 8, 39.(49) Clark, J. H.; Budarin, V.; Deswarte, F. E. I.; Hardy, J. J. E.;Kerton, F. M.; Hunt, A. J.; Luque, R.; Macquarrie, D. J.; Milkowski, K.;Rodriguez, A.; Samuel, O.; Tavener, S. J.; White, R. J.; Wilson, A. J.Green Chem. 2006, 8, 853.(50) Morais, A. R. C.; da Costa Lopes, A. M.; Bogel-Lukasik, R.Green Technologies in the Valorization of Agrofood Wastes in theFrame of the Biorefinery Concept, Plenary lecture, II InternationalCongress Food Technology, Quality and Safety, Novi Sad, Serbia, 28-30.X.2014(51) Morais, A. R. C.; Mata, A. C.; Bogel-Lukasik, R. Green Chem.2014, 16, 4312.(52) Sheldon, R. A. Green Chem. 2005, 7, 267.(53) Morais, A. R. C.; da Costa Lopes, A. M.; Bogel-Lukasik, R.Monatsh. Chem. 2014, 145, 1555.(54) Chowdhury, P.; Bikkina, C.; Gumma, S. J. Phys. Chem. C 2009,113, 6616.(55) Hunter, S. E.; Savage, P. E. Ind. Eng. Chem. Res. 2003, 42, 290.(56) Hunter, S. E.; Savage, P. E. AIChE J. 2008, 54, 516.(57) Sheldon, R. A. Green Chem. 2014, 16, 950.(58) Takenouchi, S.; Kennedy, G. C. Am. J. Sci. 1964, 262, 1055.(59) Luterbacher, J. S.; Tester, J. W.; Walker, L. P. Biotechnol. Bioeng.2010, 107, 451.(60) van Walsum, G. P. Appl. Biochem. Biotechnol. 2001, 91−3, 317.
(61) Stamenic, M.; Zizovic, I.; Eggers, R.; Jaeger, P.; Heinrich, H.;Roj, E.; Ivanovic, J.; Skala, D. J. Supercrit. Fluids 2010, 52, 125.(62) Yin, J. Z.; Hao, L. D.; Yu, W.; Wang, E. J.; Zhao, M. J.; Xu, Q.Q.; Liu, Y. F. Chin. J. Catal. 2014, 35, 763.(63) Persson, P.; Larsson, S.; Jonsson, L. J.; Nilvebrant, N. O.; Sivik,B.; Munteanu, F.; Thorneby, L.; Gorton, L. Biotechnol. Bioeng. 2002,79, 694.(64) Zhang, J.; Davis, T. A.; Matthews, M. A.; Drews, M. J.; LaBerge,M.; An, Y. H. H. J. Supercrit. Fluids 2006, 38, 354.(65) Hong, S. I.; Pyun, Y. R. J. Food Sci. 1999, 64, 728.(66) Garcia-Gonzalez, L.; Geeraerd, A. H.; Spilimbergo, S.; Elst, K.;Van Ginneken, L.; Debevere, J.; Van Impe, J. F.; Devlieghere, F. Int. J.Food Microbiol. 2007, 117, 1.(67) Kritzer, P.; Dinjus, E. Chem. Eng. J. 2001, 83, 207.(68) Lopez, D. A.; Perez, T.; Simison, S. N.Mater. Des. 2003, 24, 561.(69) Videm, K.; Dugstad, A. Mater. Perform. 1989, 28, 63.(70) International Nickel Co., Inc. (INCO). Corrosion Resistance ofthe Austenitic Chromium-Nickel Stainless Steels in Chemical Environ-ments; INCO: New York, 1963.(71) Emodi, A. Food Technol. 1978, 32, 28.(72) Nigam, P.; Singh, D. Process Biochem. 1995, 30, 117.(73) Zheng, Y. Z.; Lin, H. M.; Tsao, G. T. Biotechnol. Prog. 1998, 14,890.(74) Liu, Y. F.; Luo, P.; Xu, Q. Q.; Wang, E. J.; Yin, J. Z. Cellul. Chem.Technol. 2014, 48, 89.(75) Duff, S. J. B.; Murray, W. D. Bioresour. Technol. 1996, 55, 1.(76) Arantes, V.; Saddler, J. N. Biotechnol. Biofuels 2010, 4, 3.(77) Puri, V. P. Biotechnol. Bioeng. 1984, 26, 1219.(78) Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P. I.;Geron, C. Atmos. Chem. Phys. 2006, 6, 3181.(79) Kim, K. H.; Tucker, M. P.; Nguyen, Q. A. Biotechnol. Prog. 2002,18, 489.(80) Brownell, H. H.; Saddler, J. N. Biotechnol. Bioeng. 1987, 29, 228.(81) Vonsivers, M.; Zacchi, G.; Olsson, L.; Hahnhagerdal, B.Biotechnol. Prog. 1994, 10, 555.(82) Gu, T. Y.; Held, M. A.; Faik, A. Environ. Technol. 2013, 34, 1735.(83) Zakrzewska, M. E.; Bogel-Lukasik, E.; Bogel-Lukasik, R. Chem.Rev. 2011, 111, 397.(84) Dale, B. E.; Moreira, M. J. Biotechnol. Bioeng. 1982, 31.(85) King, J. W.; Srinivas, K.; Guevara, O.; Lu, Y. W.; Zhang, D. F.;Wang, Y. J. J. Supercrit. Fluids 2012, 66, 221.(86) Hohlberg, A. I.; Aguilera, J. M.; Agosin, E.; Sanmartin, R.Biomass 1989, 18, 81.(87) Ferreira-Leitao, V.; Perrone, C. C.; Rodrigues, J.; Franke, A. P.M.; Macrelli, S.; Zacchi, G. Biotechnol. Biofuels 2010, 3, 7.(88) Zhang, H.; Wu, S. J. Chem. Technol. Biotechnol. 2014, DOI:10.1002/jctb.4470.(89) Zhang, H. D.; Wu, S. B. Bioresour. Technol. 2014, 158, 161.(90) Matsushita, Y.; Yamauchi, K.; Takabe, K.; Awano, T.; Yoshinaga,A.; Kato, M.; Kobayashi, T.; Asada, T.; Furujyo, A.; Fukushima, K.Bioresour. Technol. 2010, 101, 4936.(91) Kim, K. H.; Hong, J. Bioresour. Technol. 2001, 77, 139.(92) Gurgel, L. V. A.; Pimenta, M. T. B.; da Silva Curvelo, A. A. Ind.Crops Prod. 2014, 57, 141.(93) Luterbacher, J. S.; Chew, Q.; Li, Y.; Tester, J. W.; Walker, L. P.Energy Environ. Sci. 2012, 5, 6990.(94) Esteghlalian, A.; Hashimoto, A. G.; Fenske, J. J.; Penner, M. H.Bioresour. Technol. 1997, 59, 129.(95) Luterbacher, J. S.; Tester, J. W.; Walker, L. P. Biotechnol. Bioeng.2012, 109, 1499.(96) Kim, K. H.; Tucker, M.; Nguyen, Q. Bioresour. Technol. 2005,96, 1249.(97) Nguyen, Q. A.; Tucker, M. P.; Keller, F. A.; Eddy, F. P. Appl.Biochem. Biotechnol. 2000, 84−6, 561.(98) Mittal, A.; Chatterjee, S. G.; Scott, G. M.; Amidon, T. E. Chem.Eng. Sci. 2009, 64, 3031.(99) Lu, H. S.; Ren, M. M.; Zhang, M. H.; Chen, Y. Chin. J. Chem.Eng. 2013, 21, 551.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXW

(100) Benazzi, T.; Calgaroto, S.; Astolfi, V.; Dalla Rosa, C.; Oliveira,J. V.; Mazutti, M. A. Enzyme Microb. Technol. 2013, 52, 247.(101) Phan, D. T.; Tan, C.-S. Bioresour. Technol. 2014, 167, 192.(102) Cha, Y.-L.; Yang, J.; Ahn, J.-W.; Moon, Y.-H.; Yoon, Y.-M.; Yu,G.-D.; An, G. H.; Choi, I.-H. Bioprocess Biosyst. Eng. 2013, 36, 1913.(103) Moscon, J. M.; Priamo, W. L.; Bilibio, D.; Silva, J. R. F.; Souza,M.; Lunelli, F.; Foletto, E. F.; Kuhn, R. C.; Cancelier, A.; Mazutti, M.A. Biocatal. Agric. Biotechnol. 2014, DOI: 10.1016/j.bcab.2014.07.008.(104) Gao, M. A.; Xu, F.; Li, S. R.; Ji, X. C.; Chen, S. F.; Zhang, D. Q.Biosyst. Eng. 2010, 106, 470.(105) Srinivasan, N.; Ju, L. K. Biomass Bioenerg. 2012, 47, 451.(106) Santos, A. L. F.; Kawase, K. Y. F.; Coelho, G. L. V. J. Supercrit.Fluids 2011, 56, 277.(107) Girio, F. M.; Carvalheiro, F.; Duarte, L. C.; Bogel-Lukasik, R.In D-Xylitol Fermentative Production, Application and Commercializa-tion; Silverio da Silva, S., Chandel, A. K., Eds.; Springer-Verlag: Berlin,2012.(108) Varga, E.; Klinke, H. B.; Reczey, K.; Thomsen, A. B. Biotechnol.Bioeng. 2004, 88, 567.(109) Puri, V. P.; Mamers, H. Biotechnol. Bioeng. 1983, 25, 3149.(110) Mansfield, S. D.; Mooney, C.; Saddler, J. N. Biotechnol. Prog.1999, 15, 804.(111) Bogel-Lukasik, R.; Lourenco, N. M. T.; Vidinha, P.; da Silva,M. D. R. G.; Afonso, C. A. M.; Nunes da Ponte, M.; Barreiros, S. GreenChem. 2008, 10, 243.(112) Oliveira, J. V.; Oliveira, D. Ind. Eng. Chem. Res. 2000, 39, 4450.(113) Nakamura, K. Trends Biotechnol. 1990, 8, 288.(114) Habulin, M.; Primozic, M.; Knez, Z. Acta Chim. Slov. 2007, 54,667.(115) Xiao, Z. Z.; Zhang, X.; Gregg, D. J.; Saddler, J. N. Appl.Biochem. Biotechnol. 2004, 113, 1115.(116) Randolph, T. W.; Blanch, H. W.; Prausnitz, J. M. AIChE J.1988, 34, 1354.(117) Zheng, Y. Z.; Tsao, G. T. Biotechnol. Lett. 1996, 18, 451.(118) Kamat, S. V.; Beckman, E. J.; Russell, A. J. Crit. Rev. Biotechnol.1995, 15, 41.(119) Toews, K. L.; Shroll, R. M.; Wai, C. M.; Smart, N. G. Anal.Chem. 1995, 67, 4040.(120) Ziegler, K. J.; Hanrahan, J. P.; Glennon, J. D.; Holmes, J. D. J.Supercrit. Fluids 2003, 27, 109.(121) Novak, Z.; Habulin, M.; Krmelj, V.; Knez, Z. J. Supercrit. Fluids2003, 27, 169.(122) Park, C. Y.; Ryu, Y. W.; Kim, C. Korean J. Chem. Eng. 2001, 18,475.(123) Paljevac, M.; Primozic, M.; Habulin, M.; Novak, Z.; Knez, Z. J.Supercrit. Fluids 2007, 43, 74.(124) Muratov, G.; Seo, K. W.; Kim, C. J. Ind. Eng. Chem. 2005, 11,42.(125) Wimmer, B.; Lottspeich, F.; Ritter, J.; Bronnenmeier, K.Biochem. J. 1997, 328, 581.(126) Lee, H. S.; Lee, W. G.; Park, S. W.; Lee, H.; Chang, H. N.Biotechnol. Technol. 1993, 7, 267.(127) Krishna, P. N. Enzyme Technology: Pacemaker of Biotechnology;PHI Learning Private Ltd.: New Delhi, India, 2011.(128) Senyay-Oncel, D.; Yesil-Celiktas, O. J. Mol. Catal. B 2013, 91,72.(129) Senyay-Oncel, D.; Yesil-Celiktas, O. J. Biosci. Bioeng. 2011, 112,435.(130) Meyssami, B.; Balaban, M. O.; Teixeira, A. A. Biotechnol. Prog.1992, 8, 149.(131) Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure,Reactions; de Gruyter: New York, 1983.(132) Liu, C. G.; Wyman, C. E. Ind. Eng. Chem. Res. 2003, 42, 5409.(133) McWilliams, R. C.; van Walsum, G. P. Appl. Biochem.Biotechnol. 2002, 98, 109.(134) Chuang, M.-H.; Johannsen, M. 9th International Symposium onSupercritical Fluids (ISSF 2009), Arcachon, France, May 18−20, 2009;International Society for Advancement of Supercritical Fluids(ISASF): Nancy, France, 2009; Poster P004.
(135) Guenther, A. Atmos. Chem. Phys. 2007, 7, 4327.(136) Singh, R.; Shukla, A.; Tiwari, S.; Srivastava, M. RenewableSustainable Energy Rev. 2014, 32, 713.(137) Miyazawa, T.; Funazukuri, T. Biotechnol. Prog. 2005, 21, 1782.(138) Pang, J. F.; Zheng, M. Y.; Wang, A. Q.; Zhang, T. Ind. Eng.Chem. Res. 2011, 50, 6601.(139) Allen, S. G.; Schulman, D.; Lichwa, J.; Antal, M. J.; Laser, M.;Lynd, L. R. Ind. Eng. Chem. Res. 2001, 40, 2934.(140) van Walsum, G. P.; Shi, H. Bioresour. Technol. 2004, 93, 217.(141) Gamse, T.; Marr, R.; Froschl, F.; Siebenhofer, M. Sep. Sci.Technol. 1997, 32, 355.(142) Gairola, K.; Smirnova, I. Bioresour. Technol. 2012, 123, 592.(143) Sako, T.; Sugeta, T.; Nakazawa, N.; Okubo, T.; Sato, M.;Taguchi, T.; Hiaki, T. J. Chem. Eng. Jpn. 1992, 25, 372.(144) Sangarunlert, W.; Piumsomboon, P.; Ngamprasertsith, S.Korean J. Chem. Eng. 2007, 24, 936.(145) Mok, W. S. L.; Antal, M. J.; Varhegyi, G. Ind. Eng. Chem. Res.1992, 31, 94.(146) Ehara, K.; Saka, S. Cellulose 2002, 9, 301.(147) Sasaki, M.; Fang, Z.; Fukushima, Y.; Adschiri, T.; Arai, K. Ind.Eng. Chem. Res. 2000, 39, 2883.(148) Peterson, A. A.; Vogel, F.; Lachance, R. P.; Froling, M.; Antal,M. J.; Tester, J. W. Energy Environ. Sci. 2008, 1, 32.(149) Nagamori, M.; Funazukuri, T. J. Chem. Technol. Biotechnol.2004, 79, 229.(150) Moreschi, S. R. M.; Leal, J. C.; Braga, M. E. M.; Meireles, M. A.A. Braz. J. Chem. Eng. 2006, 23, 235.(151) Liu, K. Y.; Brunner, G. High Pressure Chem. Eng. 1999, 6271,123.(152) Orozco, R. L.; Redwood, M. D.; Leeke, G. A.; Bahari, A.;Santos, R. C. D.; Macaskie, L. E. Int. J. Hydrogen Energy 2012, 37,6545.(153) Kuster, B. F. M. Starch/Staerke 1990, 42, 314.(154) Roman-Leshkov, Y.; Barrett, C. J.; Liu, Z. Y.; Dumesic, J. A.Nature 2007, 447, 982.(155) Bicker, M.; Hirth, J.; Vogel, H. Green Chem. 2003, 5, 280.(156) Qi, X. H.; Watanabe, M.; Aida, T. M.; Smith, R. L. Green Chem.2008, 10, 799.(157) Nakamura, Y.; Morikawa, S. Bull. Chem. Soc. Jpn. 1980, 53,3705.(158) Asghari, F. S.; Yoshida, H. Ind. Eng. Chem. Res. 2006, 45, 2163.(159) Bicker, M.; Endres, S.; Ott, L.; Vogel, H. J. Mol. Catal. A 2005,239, 151.(160) Bicker, M.; Kaiser, D.; Ott, L.; Vogel, H. J. Supercrit. Fluids2005, 36, 118.(161) Kabyemela, B. M.; Adschiri, T.; Malaluan, R. M.; Arai, K. Ind.Eng. Chem. Res. 1999, 38, 2888.(162) Holladay, J. E.; Bozell, J. J.; White, J. F.; Johnson, D. DOE2203 Report PNNL-16983; 2007. http://www.pnl.gov/main/publications/ 2204 external/technical_reports/PNNL-16983.pdf. Ac-cessed Nov. 15, 2014.(163) Sasaki, M.; Adschiri, T.; Arai, K. AIChE J. 2004, 50, 192.(164) Hu, S. Q.; Zhang, Z. F.; Song, J. L.; Zhou, Y. X.; Han, B. X.Green Chem. 2009, 11, 1746.(165) Wu, S. X.; Fan, H. L.; Xie, Y.; Cheng, Y.; Wang, Q. A.; Zhang,Z. F.; Han, B. X. Green Chem. 2010, 12, 1215.(166) Binder, J. B.; Raines, R. T. J. Am. Chem. Soc. 2009, 131, 1979.(167) Yu, Y.; Shafie, Z. M.; Wu, H. W. Ind. Eng. Chem. Res. 2013, 52,17006.(168) Carlini, C.; Patrono, P.; Galletti, A. M. R.; Sbrana, G. Appl.Catal., A 2004, 275, 111.(169) Hu, S. Q.; Zhang, Z. F.; Zhou, Y. X.; Song, J. L.; Fan, H. L.;Han, B. X. Green Chem. 2009, 11, 873.(170) Machida, H.; Takesue, M.; Smith, R. L. J. Supercrit. Fluids 2011,60, 2.(171) Palma, M.; Taylor, L. T. J. Agric. Food Chem. 2001, 49, 628.(172) Bogel-Lukasik, E.; Bogel-Lukasik, R.; Kriaa, K.; Fonseca, I.;Tarasenko, Y.; Nunes da Ponte, M. J. Supercrit. Fluids 2008, 45, 225.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXX

(173) Bogel-Lukasik, E.; Wind, J.; Bogel-Lukasik, R.; Nunes daPonte, M. J. Supercrit. Fluids 2010, 54, 46.(174) Bogel-Lukasik, E.; Bogel-Lukasik, R.; Nunes da Ponte, M. Ind.Eng. Chem. Res. 2009, 48, 7060.(175) Bogel-Lukasik, E.; Bogel-Lukasik, R.; Nunes da Ponte, M.Monatsh. Chem. 2009, 140, 1361.(176) Morais, A. R. C.; da Costa Lopes, A. M.; Costa, P.; Fonseca, I.;Nogueira, I. N.; Oliveira, A. C.; Bogel-Lukasik, R. RSC Adv. 2014, 4,32081.(177) Bourne, R. A.; Stevens, J. G.; Ke, J.; Poliakoff, M. Chem.Commun. 2007, 4632.(178) Fan, J. L.; Hou, Y. C.; Wu, W. Z.; Zhang, J. W.; Ren, S. H.;Chen, X. T. J. Chem. Eng. Data 2010, 55, 2316.(179) Chatterjee, M.; Ishizaka, T.; Kawanami, H. Green Chem. 2014,16, 1543.(180) Rogalinski, T.; Herrmann, S.; Brunner, G. J. Supercrit. Fluids2005, 36, 49.(181) Yoshida, H.; Terashima, M.; Takahashi, Y. Biotechnol. Prog.1999, 15, 1090.(182) Rogalinski, T.; Liu, K.; Albrecht, T.; Brunner, G. J. Supercrit.Fluids 2008, 46, 335.(183) Mosier, N.; Wyman, C.; Dale, B.; Elander, R.; Lee, Y.;Holtzapple, M.; Ladisch, M. Bioresour. Technol. 2005, 96, 673.(184) Pasquini, D.; Pimenta, M. T. B.; Ferreira, L. H.; Curvelo, A. A.S. J. Supercrit. Fluids 2005, 36, 31.(185) Pasquini, D.; Pimenta, M. T. B.; Ferreira, L. H.; Curvelo, A. A.S. J. Supercrit. Fluids 2005, 34, 125.(186) Shah, M. M.; Song, S. K.; Lee, Y. Y.; Torget, R. Appl. Biochem.Biotechnol. 1991, 28−9, 99.(187) Schrems, M.; Liebner, F.; Betz, M.; Zeilinger, M.; Bohmdorfer,S.; Rosenau, T.; Potthast, A. J. Wood Chem. Technol. 2012, 32, 225.(188) Kiran, E.; Balkan, H. J. Supercrit. Fluids 1994, 7, 75.(189) Li, X.; Kiran, E. Ind. Eng. Chem. Res. 1988, 27, 1301.(190) Machado, A. S. R.; Sardinha, R. M. A.; de Azevedo, E. G.; daPonte, M. N. J. Supercrit. Fluids 1994, 7, 87.(191) Ivahnov, A. D.; Bogolitsyn, K. G.; Skrebets, T. E. Russ. J. Phys.Chem. B 2010, 4, 1077.(192) van Dam, J. E. G.; de Klerk-Engels, B.; Struik, P. C.; Rabbinge,R. Ind. Crops Prod. 2005, 21, 129.(193) Gosselink, R. J. A.; Teunissen, W.; van Dam, J. E. G.; de Jong,E.; Gellerstedt, G.; Scott, E. L.; Sanders, J. P. M. Bioresour. Technol.2012, 106, 173.(194) Eckert, C.; Liotta, C.; Ragauskas, A.; Hallett, J.; Kitchens, C.;Hill, E.; Draucker, L. Green Chem. 2007, 9, 545.(195) Oil-price.net: The No. 1 Oil Price Source. www.oil-price.net.Accessed Aug. 1, 2014.(196) Thomas Eakins: Scenes from Modern Life. http://www.pbs.org/eakins/we_1844.htm. Accessed Sept. 1, 2014.(197) Carrera, G. V. S. M.; Visak, Z.; Bogel-Lukasik, R.; Nunes daPonte, M. Fluid Phase Equilib. 2011, 303, 180.(198) George, J.; Patel, Y.; Pillai, S. M.; Munshi, P. J. Mol. Catal. A2009, 304, 1.(199) Anastas, P. T.; Zimmerman, J. B. Environ. Sci. Technol. 2003,37, 94a.(200) McDonough, W.; Braungart, M.; Anastas, P. T.; Zimmerman, J.B. Environ. Sci. Technol. 2003, 37, 434a.(201) Poliakoff, M. Green Chem. 2012, 14, 2089.(202) Tang, S. L. Y.; Smith, R. L.; Poliakoff, M. Green Chem. 2005, 7,761.(203) Manpong, P.; Douglas, S.; Douglas, P. L.; Pongamphai, S.;Teppaitoon, W. J. Food Process Eng. 2011, 34, 1408.(204) Murga, R.; Ruiz, R.; Beltran, S.; Cabezas, J. L. J. Agric. FoodChem. 2000, 48, 3408.(205) Carvalheiro, F.; Esteves, M. P.; Parajo, J. C.; Pereira, H.; Girio,F. M. Bioresour. Technol. 2004, 91, 93.(206) Carvalheiro, F.; Duarte, L. C.; Medeiros, R.; Girio, F. M.Biotechnol. Lett. 2007, 29, 1887.(207) Lapkin, A.; Constable, D. Green Chemistry Metrics: Measuringand Monitoring Sustainable Processes; Wiley-Blackwell: Ames, IA, 2008.
(208) Sheldon, R. A. Catal. Today 2011, 167, 3.(209) Matos, C. T.; Gouveia, L.; Morais, A. R. C.; Reis, A.; Bogel-Lukasik, R. Green Chem. 2013, 15, 2854.(210) Sheldon, R. A.; Sanders, J. P. M. Catal. Today 2014, 239, 3−6.
Chemical Reviews Review
dx.doi.org/10.1021/cr500330z | Chem. Rev. XXXX, XXX, XXX−XXXY
Copyright © 2022 FDOKUMEN




















