
Blends of AB/BC Diblock Copolymers with a Large Interaction Parameter χ
Upload
independentCategory
view
0download
0
Reactive & Functional Polymers 69 (2009) 470–479
Contents lists available at ScienceDirect
Reactive & Functional Polymers
journal homepage: www.elsevier .com/ locate / react
Block copolymers containing pyridine moieties: Precise synthesis and applications
Nam-Goo Kang, Beom-Goo Kang, Haeng-Deog Koh, Mohammad Changez, Jae-Suk Lee *
Department of Material Science and Engineering and Department of Nanobio Materials and Engineering, Gwangju Institute of Science and Technology (GIST),261 Cheomdan-gwagiro (Oryong-dong), Buk-gu, Gwangju 500-712, Republic of Korea
a r t i c l e i n f o
Article history:Available online 23 April 2009
Keywords:Anionic polymerizationPoly(2-vinylpyridine)Amphiphilic block copolymerRod–coil block copolymerCoil–coil block copolymerSelf-assemblyMetal–pyridine complexMicelleNanoparticles
1381-5148/$ - see front matter � 2009 Elsevier Ltd. Adoi:10.1016/j.reactfunctpolym.2009.04.011
* Corresponding author.E-mail address: [email protected] (J.-S. Lee).
a b s t r a c t
This review highlights the precise synthesis and application of various well-defined rod–coil and coil–coilblock copolymers composed of poly(2-(or 4-)vinylpyridine) (P2VP or P4VP) block(s) with pyridine moi-eties. These polymers were synthesized by means of living anionic polymerization. Poly(hexyl isocya-nate) (PHIC) was used as the rod-like segment, because hexyl isocyanate undergoes living anionicpolymerization under carefully selected conditions. This review describes the syntheses of the blockcopolymers, polystyrene-b-P2VP, polystyrene-b-P4VP, polyisoprene-b-P2VP, P2VP-b-poly(methyl meth-acrylate), P2VP-b-poly(ethylene oxide), P2VP-b-poly(2-(N-carbazolyl)ethyl methacrylate), P2VP-b-PHIC,P2VP-b-PHIC-b-P2VP, and PHIC-b-P2VP-b-PHIC. The formation of self-organized nanostructured materi-als and molecular assemblies by such block copolymers and their possible applications are also described.These assemblies include monolayers, microphase-separated periodic-ordered nanostructures, micelles,polymer–metal complexes, nanoparticles, inorganic and metal layers, and nanoporous films withnanoparticles.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction
The synthesis and application of block copolymers have beenwidely studied in the last few decades. The well-defined, three-dimensional supramolecular architectures formed by block copoly-mers include coils (flexible), rods (rigid), and combinations thereof.These architectures offer appealing material properties, especiallyfor nano-material applications [1–3].
Differences between block segments in compatibility and ther-modynamic properties lead to the formation of a variety of self-assembled nanostructures by phase separation at the molecular le-vel. This self-assembly is followed by self-organization, which canproduce spherical, lamellar, cylindrical, and gyroid structures withperiodic nano-domains. The resulting morphology depends on thecomposition, interaction, and molecular weight of the blockcopolymers [4–7]. These block copolymer structures have beenused in electrochemical, electrical, and optical applications.
Block copolymers composed of poly(2-vinylpyridine) (P2VP)and/or poly(4-vinylpyridine) (P4VP) segment(s), which containpyridine moieties as side chains, have been used in many applica-tions. For example, these block copolymers have been used as tem-plates for metal complexes to prepare nanoparticles (NPs). TheseNP/copolymer assemblies improve the functionality of electronic,
ll rights reserved.
photonic, and chemical devices due to the presence of an unsharedelectron pair on the nitrogen atom of the pyridine ring and theresulting pH sensitivity [8–14]. Although the anionic polymeriza-tion of 2-vinylpyridine (2VP) and 4-vinylpyridine (4VP) werepreviously reported to be accompanied by side reactions, severalresearch groups have successfully achieved living anionic polymer-ization of both 2VP and 4VP. This polymerization wasaccomplished using sterically bulky, and relatively less reactive,anionic initiators such as 1,1-diphenylhexyllithium and diph-enylmethylpotassium in THF at low temperatures (�78 �C, forexample). Therefore, not only homopolymers but also blockcopolymers, with a well-controlled molecular weight and compo-sition, have been successfully prepared [15–21].
In this article, we focus on poly(alkyl isocyanate)s (PIC)s pos-sessing a rigid rod-like structure with a dynamic helical conforma-tion. Unfortunately, the anionic polymerization of alkyl isocyanate(IC) monomers was not controlled due to a serious side reaction.This side reaction, called ‘‘back-biting,” produces the trimmer dur-ing the course of the polymerization. For >10 years, Lee andcoworkers have successfully controlled the anionic polymerizationof n-hexyl isocyanate (HIC) and other similar monomers by addingeither NaBPh4 or 15-crown-5 to the anionic initiators or by devel-oping novel initiator systems to suppress back-biting. Through theuse of such systems, they were able to demonstrate the first prep-aration of homopolymers, as well as rod–coil block copolymers,with well-defined structures. Even beyond the living anionicpolymerization, PICs themselves are a fascinating and especially

C
O
N C
O
NHm
C
O
NC
O
HNm
CH
N
CH2 CH CH
N
CH2CH2 CH2 CH
N N
H2C CH2HC
N
CH
Nn n
N
CHCH2CHCH2 mn
N
CHCH2CHCH2 mn
N
C
N
CHCH2 n
O
Nm
CH2CHCn
O
NCm
C
N
CHCH2 n
O
N Cm
O
CH2
O
(a) (b) (c)
(e)
(d)
N
Fig. 1. The structures of block copolymers based on vinylpyridine; (a) polystyrene-b-poly(2-vinylpyridne) (PS-b-P2VP), (b) poly(2-(4-vinylphenyl)pyridine)-b-poly(2-vinylpyridne) (PVPPy-b-P2VP), (c) poly(2-vinylpyridine)-b-poly(n-hexyl isocyanate), (d) poly(n-hexyl isocyanate)-b-poly(2-vinylpyridine)-b-poly(n-hexyl isocyanate), and (e)poly(2-vinylpyridine)-b-poly(n-hexyl isocyanate)-b-poly(2-vinylpyridine).
N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479 471
functional class of polymers. PICs have many potential applica-tions, such as chiral recognition, optical switches, liquid crystals,and degradable materials. Furthermore, the block copolymerscomposed of rigid and helical PIC segments are of particular inter-est for fundamental studies of polymer behavior in solution and inbulk.
In this review, we highlight the synthesis of well-defined coil–coil, coil–rod, coil–rod–coil, and rod–coil–rod block copolymersbased on P2VP and PIC blocks by means of living anionic polymer-ization (Fig. 1). We also introduce possibly the applications of self-organized nanostructured materials and molecular assembliesformed from such block copolymers.
2. Synthesis of well-defined block copolymers containingpyridine moieties
2.1. Coil–coil block copolymers
As mentioned in the introduction, block copolymers containingpyridine moieties are interesting and versatile because of potentialapplications in nano-science and technology [14,22–29]. For thisreason, a variety of block copolymers composed of P2VP and/orP4VP blocks have been prepared by living anionic polymerization.Some examples are polystyrene-block-poly(2-vinylpyridne) (PS-b-P2VP), polystyrene-block-poly(4-vinylpyridne) (PS-b-P4VP),poly(1,3-butadiene)-block-poly(2-vinylpyridine) (PB-b-P2VP),poly(isoprene)-block-poly(2-vinylpyridne) (PI-b-P2VP), poly(2-vinylpyridine)-block-poly(methyl methacrylate) (P2VP-b-PMMA),poly(2-vinylpyridine)-block-poly(dimethylsiloxane) (P2VP-b-PDMS),poly(2-vinylpyridine)-block-poly(ethylene oxide) (P2VP-b-PEO),and poly(2-vinylpyridine)-block-poly(e-caprolactam) [17,20,30–37]. These block copolymers are usually synthesized withsequential living anionic polymerization, where the correspondingmonomers are sequentially added. In anionic polymerization, how-ever, the propagating chain-end anion can vary in reactivity.Choosing the proper order of monomer addition is therefore criti-cal to prepare the desired block copolymers. For example, living PS
polymerizes 2VP, while living P2VP cannot initiate the polymeriza-tion of styrene under the usual conditions. Accordingly, the addi-tion order must be styrene first and then 2VP, but not vice-versa,in order to prepare PS-b-P2VP [11,38–40]. Living PEO cannot ani-onically polymerize the 2VP monomer. Therefore, in order to pre-pare the corresponding block copolymer of P2VP-b-PEO, theaddition order must be 2VP, followed by ethylene oxide [41]. Theaddition order is also important for the preparation of ABA andABC triblock copolymers. The preparation of PS-b-P2VP-b-PS bysequential polymerization is difficult, because the living P2VP pro-duced at the 2nd-stage of polymerization cannot polymerize sty-rene, as mentioned above. The ABC triblock copolymer PS-b-P2VP-b-PtBMA is readily and quantitatively prepared by thesequential addition of styrene, then 2VP, followed by tert-butylmethacrylate. In contrast, the ABC triblock copolymer with a differ-ent block sequence, P2VP-b-PtBMA-b-PS, cannot be prepared bysequential addition in this order, since the styrene monomer doesnot react with the living PtBMA produced at the 2nd-stage of poly-merization [22].
The most utilized block copolymer at the moment is PS-b-P2VP(or P4VP in some cases). This copolymer has an amphiphilic char-acter and forms self-organized assemblies such as microphase-sep-arated nanostructures and micelles, which have widely beenstudied [20,30,41–45]. Similar to the case of PS-b-P2VP, bothwell-defined PI-b-P2VP and PB-b-P2VP could be prepared by thesequential polymerization of isoprene (or 1,3-butadiene), followedby the addition of 2VP [17,34]. Interestingly, an ABC triblockcopolymer, PS-b-P2VP-b-PB, was prepared by combining living an-ionic polymerization with end-group coupling. To accomplish this,living PS-b-P2VP was terminated with a large excess of p-xylenedichloride, which resulted in end-chlorinated PS-b-P2VP-Cl diblockprecursors. The resulting precursor was reacted with living PB,end-capped with 1,1-diphenylethylene, resulting in the formationof the desired PS-b-P2VP-b-PB triblock polymers [34].
In order to prepare P2VP-b-PDMS and P4VP-b-PDMS, it is neces-sary to first polymerize 2(or 4)VP. The cyclic dimethylsiloxanemonomer is then added at the 2nd-stage of polymerization. Thetriblock copolymer P2VP-b-PDMS-b-P2VP was successfully

CH2C CCH3
C OOCH3
N
H2CCH
CC
CH3
K H2CH
K
N
CH2CH
K CH2CH
K
HH K
Nucleophilicity
CH2C CCH3
C OOCH3
N
H2CCH
CC
CH3
K H2CH
K
N
CH2CH
K CH2CH
K
HH K
Nucleophilicity
Fig. 2. The nucleophilicity order of 2-(4-vinylphenyl)pyridine. (Reproduced with permission from Macromolecules (2007) American Chemical Society [14].)
472 N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479
prepared by such sequential polymerization, followed by an end-group coupling between the AB diblock copolymers thus prepared.Interestingly, P2VP-b-PDMS becomes water soluble following theoxidation of the P2VP block with peroxyacetic acid [36]. BothP2VP-b-PMMA and P2VP-b-PtBMA were prepared by sequentialanionic polymerization in a similar manner, i.e., 2VP first, followedby either of the methacrylate monomers. The PtBMA block ofP2VP-b-PtBMA is readily hydrolyzed under acidic conditions toproduce an amphiphilic, well-defined P2VP-block-poly(metha-crylic acid) with both basic and acidic segments [20]. An ABC tri-block copolymer, PS-b-P2VP-b-PtBMA, was successfully preparedby sequential anionic polymerization, where styrene, 2VP, andtert-butyl methacrylate were sequentially added. A variety ofmicrophase-separated periodic-ordered nanostructures, quite dif-ferent from those produced with AB diblock copolymers, were ob-served by varying the P(t-BMA) block weight fraction [22]. Thisphenomenon will be described in the proceeding section. Awater-soluble P2VP-b-PEO was also prepared by the anionic poly-merization of 2VP, followed by ethylene oxide. Because of itsamphiphilic character, the resulting polymer self-assembled toform onion-type micelles in aqueous solution [41].
A new monomer, 2-(4-vinylphenyl)pyridine (VPPy), a styrenemonomer p-substituted with a pyridine moiety, was synthesizedand anionically polymerized by Lee and coworkers [14]. Similarto styrene and 2VP, VPPy was found to undergo living anionic poly-merization with either sec-BuLi in the presence of LiCl or diphe-nylmethylpotasium in THF at �78 �C. Interestingly andimportantly, the resulting living PVPPy was more similar to livingP2VP than to living PS in reactivity of the chain-end anion, asshown in Fig. 2.
The preparation of a BAB triblock copolymer of 2VP withpoly(2-(N-carbazolyl)ethyl methacrylate) (PCzMA) by sequentialpolymerization has recently been reported by Lee and coworkers[12]. As in the case of P2VP-b-PMMA, 2VP was first polymerizedwith the 1,1-diphenylethlene (DPE)-derived bifunctional anionicinitiator prepared from DPE and potassium naphthalenide in THFat �78 �C. CzMA was subsequently added to polymerize underthe same conditions. In this polymerization, the addition of Et2Znwas essential at the 2nd-stage of polymerization to narrow themolecular weight distribution of the PCzMA block. Since the pyri-dine moiety of the resulting block copolymer forms a complex withruthenium, energy transfer from the CzMA side chain to the ruthe-nium complex can be observed. This phenomenon will be furtherdiscussed in the next section.
2.2. Rod and coil block copolymers
Block copolymers composed of rigid rod-like and coil segmentshave recently received considerable attention because of theirunique phase behavior at the molecular level and their physicalbehavior and properties in solution and in bulk. Although many
such block copolymers have been reported, we focus hereinmainly on block copolymers composed of P2VP and rod-likepoly(hexyl isocyanate) (PHIC) segment(s) prepared by livinganionic polymerization. The reason we have chosen PHIC is that,because the hexyl isocyanate (HIC) monomer is capable of livinganionic polymerization, it is possible to synthesize well-definedblock copolymers.
As mentioned in the introduction, Lee and coworkers haveachieved living anionic polymerization of HIC with sodium napht-halenide (Na-Naph) in THF at �98 �C using 15-crown-5 or NaBPh4
as an additive [46–52]. Both additives form a complex with thepropagating chain-end anion that may prevent back-biting due tosteric bulkiness. PHIC possessing well-controlled, high molecularweights up to 105 g/mol and narrow molecular weight distribu-tions (Mw/Mn < 1.1) has been obtained for the first time. More re-cently, they have demonstrated that sodium benzanilide is aneffective initiator of the living anionic polymerization of HIC. Inthis polymerization, it has been shown by several analytical resultsthat the sodium benzanilide clusters can protect the propagatingchain-end anion to prevent the back-biting and chain transferreactions. With the use of these effective initiator systems in theanionic sequential polymerization, various well-defined ABA rod–coil–rod triblock copolymers of isoprene or styrene with HIC wereprepared [53].
Rod–coil block copolymers composed of PHIC and P2VP are ofspecial interest as functional specialty polymers, since the PHICblock is a rigid rod-like polymer chain and can possibly be utilizedfor chiral recognition, optical switches, optical data storage, liquidcrystals, and degradable materials. Furthermore, the P2VP blockhas a basic, hydrophilic character and can coordinate many metalcations [54–60]. A rod–coil diblock copolymer, P2VP-b-PHIC, hasbeen recently prepared by sequential living anionic polymerization.2VP was first polymerized with sterically bulky diphenylmethylpo-tassium and HIC was subsequently added to polymerize in the pres-ence of NaBPh4. Finally, the living polymer was quenched with aCH3COOH/methanol mixture to end-cap the chain with a proton[13].
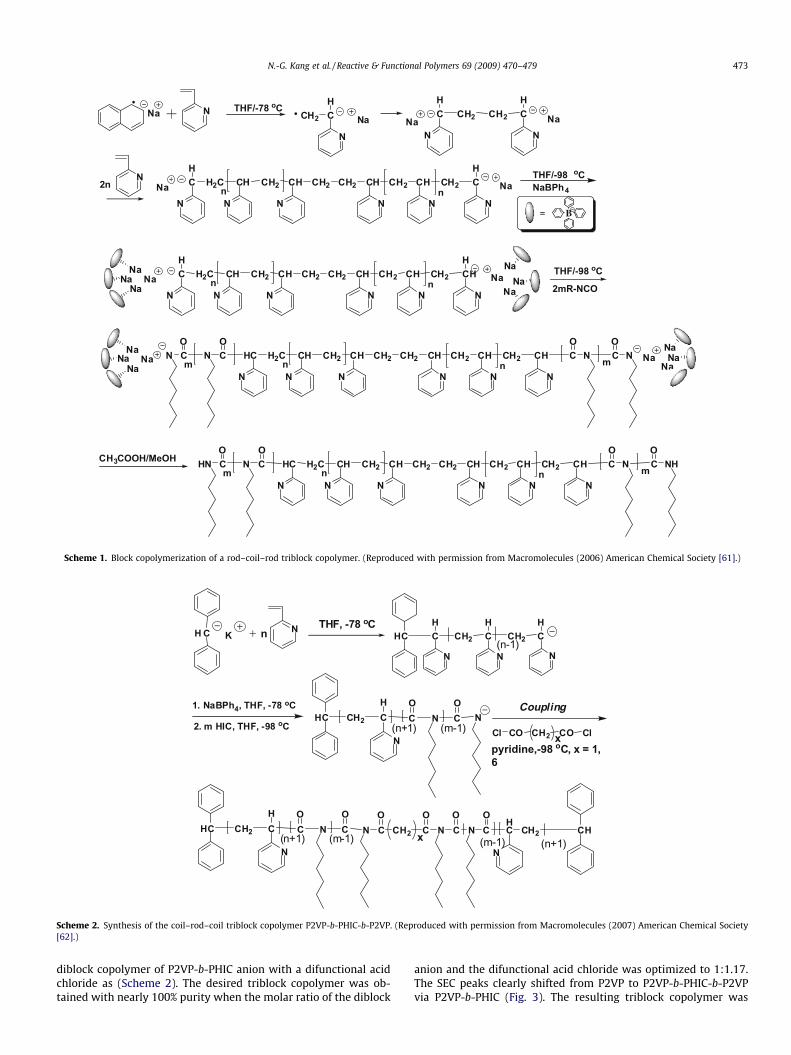
The rod–coil–rod triblock copolymer PHIC-b-P2VP-b-PHIC wasalso prepared by anionic polymerization of 2-VP with sodiumnaphthalenide at �78 �C in THF, followed by addition of HIC topolymerize in THF in the presence of NaBPh4 at �98 �C (Scheme 1)[61].
The coil–rod–coil triblock copolymer P2VP-b-PHIC-b-P2VP,with a reverse block order, cannot be prepared in a similar mannerby sequential anionic polymerization. Living PHIC is too weak innucleophilicity to initiate the polymerization of 2-VP at the 2nd-stage of the polymerization. Recently, Lee and coworkers havefound that living PHIC reacts quantitatively with various aliphaticacid chlorides to introduce useful functional groups at the PHICchain-ends. This efficient reaction has been extended to the prep-aration of a coil–rod–coil triblock copolymer by the reaction of a
Na N C
N
C C
NN
N
Na Na Na
B
Na
NaNaNa
Na
Na
Na
NaNaNaNa
Na
NaNa
2n
2mR-NCO
THF/-78 oC
=
THF/-98 oCNaBPh4
THF/-98 oC
CH3COOH/MeOH
CH2
H HCH2 CH2
H
CH
NNa CH2 CH CH
N
NaCH2CH2 CH2 CH
N N
Na Na
CO
N CO
NmCO
NCO
Nm
H2C CH2C
N
C
N
H H
n n
CH
N
CH2 CH CH
N
CH2CH2 CH2 CH
N N
H2C CH2C
N
CH
N
H H
n n
CH
N
CH2 CH CH
N
CH2CH2 CH2 CH
N N
H2C CH2HC
N
CH
Nn n
CO
N CO
NHmCO
NCO
HNm
CH
N
CH2 CH CH
N
CH2CH2 CH2 CH
N N
H2C CH2HC
N
CH
Nn n
Scheme 1. Block copolymerization of a rod–coil–rod triblock copolymer. (Reproduced with permission from Macromolecules (2006) American Chemical Society [61].)
C NKHTHF, -78 oC
CH
CH2
N
CH
N
CH2 CH
N(n-1)
2. m HIC, THF, -98 oCHC CH2 C C
N
HN
OCO
N(n+1) (m-1)
pyridine,-98 oC, x = 1,6
Coupling
Cl CO CH2 CO Clx
HC CH2 C C
N
HN
OCO
N(n+1) (m-1)
CO
CH2 CO
N Cx
ON C
OHC
N
CH2 CH(n+1)(m-1)
n
1. NaBPh4, THF, -78 oC
HC
Scheme 2. Synthesis of the coil–rod–coil triblock copolymer P2VP-b-PHIC-b-P2VP. (Reproduced with permission from Macromolecules (2007) American Chemical Society[62].)
N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479 473
diblock copolymer of P2VP-b-PHIC anion with a difunctional acidchloride as (Scheme 2). The desired triblock copolymer was ob-tained with nearly 100% purity when the molar ratio of the diblock
anion and the difunctional acid chloride was optimized to 1:1.17.The SEC peaks clearly shifted from P2VP to P2VP-b-PHIC-b-P2VPvia P2VP-b-PHIC (Fig. 3). The resulting triblock copolymer was

20 22 24 26 28 30Elution time (min)
abc
20 22 24 26 28 30
abc
Fig. 3. The shift of the SEC-LS profile of (a) P2VP, (b) P2VP-b-PHIC, and (c) P2VP-b-PHIC-b-P2VP. (Reproduced with permission from Macromolecules (2007) AmericanChemical Society [62].)
474 N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479
observed to have a predictable molecular weight and a narrowmolecular weight distribution [62].
Thus, Lee and coworkers have, under certain controlled condi-tions, successfully prepared a variety of well-defined rod–coildiblock and triblock copolymers based on PHIC and P2VP, inaddition to PHIC-b-PS-b-PHIC and PHIC-b-PI-b-PHIC, by means ofliving anionic polymerization [51,52]. These polymers are new,interesting, and versatile block copolymers that have characteristicrigid rod-like segments, multiphase architectures, and variousfunctionalities. Nano-scale self-assemblies form from such blockcopolymers, and their possible applications will be introduced inthe next section.
3. Self-assembly of block copolymers
3.1. Monolayer of block copolymers composed of PHIC and P2VPsegments
Recently, Park, Lee, and coworkers reported the liquid crystal-line behavior of diblock and triblock copolymers of PHIC andP2VP on a reactive substrate and the influence of annealing inthe presence of certain solvents. As shown in Fig. 4, a highly ori-ented, anisotropic self-assembled monolayer forms from the blockcopolymers [63]. In the SAMs, the PHIC block was tethered to thesurface through the adsorption of P2VP onto mica. The thicknessof the film was equal to the diameter of a single chain. Interest-ingly, the PHIC chains exhibited planar nematic ordering whenthe monolayer was exposed to the vapor of certain solvents. Theseresults offer a method for understanding the complex morpholog-
Fig. 4. Schematic structure of the rod-like PHIC chains tethered to the surface by adsoAmerican Chemical Society [63].)
ical evolution of amphiphilic rod–coil block copolymers on a sub-strate surface [64].
3.2. Microphase-separated structures of block copolymers
In certain diblock copolymers, the two blocks strongly phase-separate at the molecular level, followed by self-organization. Itis well known that an empirical relation exists between the volumefraction of the two blocks and the resulting morphology (micro-phase-separated structure) This morphology often takes the formof periodic, ordered supra-structures of nanometer size. In general,spherical domains, cylindrical domains, and then lamellar phasescan be observed with an increasing volume fraction of one block.In some cases, gyroid domains appear between the cylindricaland spherical domains [4–7]. Similar morphological behavior hasbeen observed in the diblock copolymers PS-b-P2VP and PVPPy-b-PMMA. It was reported by Bates and coworkers that four orderedmorphologies were observed in a series of block copolymers com-posed of PS and P2VP blocks by varying the PS composition. Thesemorphologies were lamellar, hexagonally perforated layers, bicon-tinuous cubic/gyroid, and hexagonally packed cylinders [30].
The morphologies of ABC triblock copolymers are much morecomplex because of the geometric restriction, in addition to thepresence of three components. Abetz and coworkers have recentlyreported the relationship between the generated morphologiesand volume fractions of the three blocks in the triblock copolymerPS-b-P2VP-b-P(tBMA). A variety of three-dimensional, periodic, or-dered microphase-separated supra-structures have been observedby increasing the PtBMA fraction while maintaining the block ratioof the PS/P2VP [20].
Many research groups have reported unique morphologicalbehavior in the rod–coil block copolymers. These structures arequite different from those formed by the usual coil–coil blockcopolymers. For example, rod–coil diblock copolymers of PS-b-PHIC (fPHIC = 0.90) showed a zigzag morphology with a high degreeof smectic-like long range order [2,6], while diblock copolymerswith fPHIC = 0.96 and 0.98 exhibited an arrowhead morphology[65]. On the other hand, a rod–coil triblock copolymer of oligosty-rene-b-oligoisoprene-b-biphenyl ester segments connected by es-ter linkages (fstyrene and fisoprene = 0.47) produced mushroom-shaped supramolecular structures [66].
In the rod–coil diblock copolymer P2VP-b-PHIC, Lee andcoworkers have observed various periodic microdomains with aPHIC volume fraction of 0.81 in chloroform [13]. Several interestingmorphologies have also been observed from the rod–coil–rod tri-block copolymers PHIC-b-P2VP-b-PHIC, as well as the coil–rod–coiltriblock copolymer P2VP-b-PHIC-b-P2VP [61,62]. As shown inFig. 5a and b, when using the latter block copolymer with PHIC vol-ume fractions (fPHIC) of 0.3 and 0.7, typical morphologies are ob-served in TEM micrographs. In Fig. 5, the P2VP domains arestained with I2 and show up as dark regions, while the PHIC do-mains are gray [62].
rption of P2VP coils. (Reproduced with permission from J. Am. Chem. Soc (2007)

Fig. 5. TEM micrographs of a P2VP-b-PHIC-b-P2VP film. The white regions are the PHIC domains, and the dark regions are the P2VP domains stained with I2 vapor. (a)fPHIC = 0.3 and (b) fPHIC = 0.7. (Reproduced with permission from Macromolecules (2007) American Chemical Society [62].)
Fig. 6. Schematic representation of the formation of solid micelles and vesicles [61].
Fig. 7. UV/Vis spectra of [Ru(tpy)(dmbpy)Cl] (- - -), PCzMA-b-P2VP-b-PCzMA (���),and RuIIL2bpolym (—). (Reproduced with permission from Macromol. RapidCommun. (2001) Wiley–VCH [12].)
N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479 475
3.3. Micellization of the block copolymers
The micellization behavior of amphiphilic PS-b-P2VP polymershas been widely studied for both basic and applied purposes. In-verse micelles of PS-b-P2VP have been recently used as switchabletemplates for nanofabrication with various topography and surfacechemistry [33,45]. Using a porous PS-b-P2VP monolayer micellarfilm, highly porous polyaniline nanofiber films were fabricatedfor potential applications in electrocatalysis, microreactor fabrica-tion, and sensing [67]. The blending of symmetric PS-b-P2VP di-block copolymers with surfactant nanoparticles of PS-Au formedbicontinuous morphologies that can potentially be used for photo-voltaic, fuel cell, and battery applications [68]. Lee and coworkersrecently reported the facile formation of clear core–shell micellesby quaternization of the P2VP block of PS-b-P2VP with methyl io-dide to increase the incompatibility between the blocks [11,38,69].
In certain solvents, the self-assembly of the rod–coil–rod tri-block copolymer PHIC-b-P2VP-b-PHIC led to the formation of mi-celles or vesicles (Fig. 6). With the use of the triblock copolymerin an equimolar ratio (fPHIC = 0.5) of the rod (PHIC) and coil(P2VP) blocks, spherical micelles with an average size of �20 nmwere produced in chloroform, since this solvent is selective forthe rod block. In contrast, vesicles of PHIC-b-P2VP-b-PHIC were ob-served in methanol, which is selective for the P2VP block and unfa-vorable for the PHIC block [61]. The PHIC-b-P2VP diblockcopolymer formed core–shell micelles, depending on the solventcomposition. For example, PHIC-b-P2VP (fPHIC = 0.26, Mw = 26 k)formed solid micelles with a PHIC core and a P2VP shell in metha-nol/THF (8/2, v/v), while the same block copolymer formed mi-celles with a PHIC shell and a P2VP core in THF/water (9/1, v/v).
Similarly, the triblock copolymer P2VP-b-PHIC-b-P2VP formed so-lid micelles in chloroform and vesicles in THF/methanol (2/8, v/v)[70].
Very recently, Lee and coworkers have successfully synthesizedunique branched polymers composed of PHIC and polyethyleneglycol (PEG), showing A2B and A2BA2 architecture, where A is ahydrophobic rod-like PHIC block, and B is a hydrophilic PEG block.These polymers were observed to form solid micelles in chloroformand vesicles in a THF/ethanol (1/9, v/v) mixture. Using the A2B,

Fig. 10. SEM image of an open-pored mesoscopic titania layer prepared with atemplate of PS-b-P2VP�MeI polymeric NPs. (Reproduced with permission from Adv.Mater. (2004) Wiley–VCH [11].)
Fig. 8. Functionalized copolymer containing a carbazole and an iridium complex asside groups, poly(Ir(ppy)2(2-(4-vinylphenyl)pyridine)-co-poly(vinylcarbazole).(Reproduced with permission from Opt. Mater. (2002) Elsevier [23].)
Fig. 9. PL spectra of an Ir complex copolymer in solution. (Reproduced withpermission from Opt. Mater. (2002) Elsevier [23].)
476 N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479
(PHIC)2–PEG (fPHIC = 0.73), a core–shell type micelle consisting ofPEG (core) and PHIC (shell) formed in CHCl3, which is a solventselective for the PHIC rod block. In CHCl3, the flexible PEG blockaggregated and the PHIC blocks remained fully extended, which
Fig. 11. Schematic of the formation of water-in-oil (W/O) emulsion-induced micelles (Langmuir (2007) American Chemical Society [39].)
led to the formation of well-defined spherical solid micelles.(PHIC)2–PEG (fPHIC = 0.73) formed a bilayer vesicle in THF/ethanol(2/8, v/v). The polymer adopted this structure, because ethanol isnot selective for PHIC and is favorable for PEG. On the other hand,in THF/ethanol (9/1–8/2, v/v), PHIC formed the interior of the ves-icle, whereas PEG was exposed to the solvent [71]. Thus, uniquemorphologies and molecular assembles (micelles) have beenformed by the incorporation of rigid rod-like PHIC segments intoblock copolymers and branched polymers.
4. Metal complexes with block copolymers
Lee and coworkers have prepared the block copolymer (PCzMA-b-P2VP-b-PCzMA)–metal complex via coordination of the pyridinemoiety to Ru. They then used this system to study the energy-transfer between PCzMA and the P2VP-Ru complex for use as aluminophore or quencher. The absorption spectra of the P2VP-Rucomplex, shown in Fig. 7, strongly indicate an inter- or
EIMs) and the synthesis of Au NPs using EIMs. (Reproduced with permission from

N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479 477
intra-molecular energy transfer. The maximum emission peakchanged from 352 and 370 to 466 nm, which confirmed that en-ergy transfer occurred from the PCzMA block to the Ru-coordi-nated P2VP block [12].
Lee and coworkers have also reported the synthesis of a randomcopolymer–metal complex, poly(Ir(ppy)2(2-(4-vinylphenyl)pyri-dine)-r-poly(vinyl carbazole), containing both the carbazole andIr(III) complex as side groups (Fig. 8). This Ir-coordinated copoly-mer was used to study the energy transfer from the carbazole tothe Ir-complex. The PL spectra clearly confirmed the energy trans-fer from the carbazole to the Ir complex (Fig. 9). Thus, even the ran-dom copolymer containing an Ir-complex has advanced optical andelectrical properties [23]. A study of the same subject using blockcopolymers is in progress.
5. Nanocomposites from block copolymers
5.1. Mono- and multi-layers of polymeric nanoparticles
Many research groups have attempted to prepare well-definedinorganic materials, such as TiO2 and SiO2, with ordered porosities.Such materials would have possible applications in catalysis and
Fig. 12. TEM image of an Au/nanoporous block copolymer film; the inset is the SEMimage at a 45� tilt. (Reproduced with permission from Langmuir (2007) AmericanChemical Society [39].)
Fig. 13. (a) TEM micrographs of arrays of iron oxide nanoparticles surrounded by gold naand a cross, respectively. (Reproduced with permission from J. Am. Chem. Soc. (2003) A
electronics, but the fabrication of such 2D porous inorganic mate-rials has not yet developed sufficiently to meet the demands ofthese applications. Moreover, aggregation among the particles atthe interface is often observed during the fabrication of 2D mono-layers with polystyrene and SiO2 particles [24–26].
Lee and coworkers have recently demonstrated that monodis-perse nano-sized TiO2 particles can be successfully prepared with-out aggregation using self-organized nanoparticles of the blockcopolymers, PS-b-P2VP and PS-b-poly(2-vinylpyridinum methyliodide). Nanoparticle monolayers of PS-b-P2VP�MeI were formedby the following procedure. First, a dispersion of nanoparticles(NPs) in toluene was spread on water to form a 2D monolayer atthe air–water interface. This monolayer was then transferred tothe substrate. The ordered array of polymeric NPs was infused withtitanium (IV) isopropoxide solution. Finally, the porous void-freeTiO2 layer was prepared through sintering. As shown in Fig. 10, a2D, open-pored, void-free, and mesoscopic TiO2 layer resultedwhen the latter block copolymer nanoparticles were used as tem-plates. The pore size and the wall thickness between the poreswere around 55 nm and 18 nm, respectively. Using this procedure,pore sizes in the 2D layer can be controlled down to dimensions ofless than 50 nm. This control is achievable in TiO2 layers as well asother porous inorganic materials [11]. A nanoporous structure ofAu-coated TiO2 has also been prepared, using 3D arrays of Au-coated PS-b-P2VP nanoparticles (80 ± 5 nm) as a template. Quat-ernized PS-b-P2VP powder was dissolved in toluene with stirringand sonication for 3 days to obtain core–shell-type nanoparticles.Au-coated PS-b-P2VP nanoparticles, prepared using 3-nm thiol-capped Au nanoparticles, were three-dimensionally well-orderedby the vertical immersion method on ITO glass at 40 �C for 5 days.Finally, the nanoporous Au-coated TiO2 layer was fabricated byremoving the PS-b-P2VP block copolymer nanoparticles with oxy-gen plasma etching [69].
The difference in surface energy between PS and P4PV blocks issubstantially enhanced by quaternization of the pyridine rings ofPS-b-P4VP. Using a quaternized block copolymer (Mw = 58 k, Mw/Mn = 1.1, and fPS = 0.95) as a template, Cho, Lee, and coworkershave demonstrated a new method to control the nucleation andgrowth of SiO2 to form nanopatterns in an oil phase. The SiO2
nucleates onto only the P4PV block and grows selectively on theprotonated P4VP block up to a length of 60 nm with a height of8 nm [72].
noparticles and (b) EDX spectra with gold and iron nanoparticles marked by circlesmerican Chemical Society [75].)

Fig. 14. (a) TEM micrographs of a single large core-embedded Au NP surrounded by smaller CdS NPs and (b) TEM micrographs of a larger core-embedded CdS NP surroundedby smaller Au NPs. (Reproduced with permission from Langmuir (2007) American Chemical Society [40].)
478 N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479
5.2. Nano-porous thin film with Au nanoparticles
In order to prepare nanoporous thin film with a precise controlof the pore diameter, Lee and coworkers have proposed a new pro-cedure in which H2O nanodroplets are stabilized by an amphiphilicblock copolymer in an organic solvent. These droplets are used toprepare a water-in-oil (W/O) inverse emulsion to obtain narrowsize distributions (Fig. 11) [39]. It is thus possible to prepare un-ique pyridine-functional nanoporous films. Furthermore, metalnanoparticles (NPs) could be selectively introduced into the nano-pore interiors by interaction between the pyridine moieties andthe metals. In fact, Au NPs have been introduced into the walls ofa nanoporous film thus prepared (Fig. 12).
Recently, self-assembly involving two different NPs has re-ceived significant attention because of the potential for advancedapplications, such as solar cells, electrochromic devices, light-emit-ting diodes, and sensors [29,73–75]. Sohn and coworkers have re-ported the preparation of a monolayer film with two metal NPs byintroducing two NPs into a monolayer film from PS-b-P2VP. The AuNPs were first physically introduced into the ordered hexagonalmicelles of the PS monolayer film. Subsequently, iron oxide NPswere chemically formed in the P2VP core, resulting in iron oxideNPs surrounded by Au NPs (Fig. 13) [76,77].
Lee and coworkers have proposed an in-situ synthetic method-ology to construct nanocomposites, embedding two NPs into thecore of PS-b-P2VP micellar templates. The location of the Au/CdSNPs is shown in Fig. 14a and b. One larger P2VP core is embeddedwith dozens of small CdS NPs, surrounding an Au NP. The structurecan also be assembled such that several small Au NPs surround alarger CdS NP. Thus, Au/CdS nanocomposites with selectively con-trolled sizes, numbers, and locations of Au and CdS NPs in the mi-celle cores have been stabilized. These structures are suitable forfuture optical, electronic, and catalytic applications [40]. Further-more, C60 molecules could be successfully incorporated into theP2VP cores of PS-b-P2VP/CdS micelles, resulting in well-organizedCdS and C60 structures in the micelle cores [78].
6. Conclusions
We have described a variety of well-defined block copolymerscomposed of P2VP and helical, rod-like segments. These helicalsegments were composed of either of coil block(s) or PHIC block(s).In both cases, the polymers were synthesized by means of livinganionic polymerization. The resulting block copolymers possessprecisely controlled molecular weight, composition, and a narrowmolecular weight distribution, as well as a well-defined blockarchitecture. Since most previously reported rod–coil block copoly-
mers have had an ill-defined structure and architecture, the rod–coil block copolymers composed of PHIC block(s) noted in thiswork are quite new and interesting. We have also mentioned thethree-dimensional periodic and ordered nano-size supra-struc-tures and molecular assemblies formed by these polymers. Thisassembly begins with phase-separation at the molecular level, fol-lowed by self-organization of the block copolymers. We have alsodiscussed potential applications of these structures in nano scienceand technology, though most of these ideas are preliminary at themoment. Many ordered nano-sized hybrid materials have beenprepared from pyridine-functionalized block copolymers and me-tal complexes. These materials will become increasingly impor-tant, as nano science and technology continue to develop, alongwith electronics, photonics, and display technologies.
Acknowledgements
This work was partially supported by the Program for Inte-grated Molecular Systems (PIMS)/(GIST), World Class University(WCU) Program, and the project for the synthesis of SIS block copo-lymers by anionic polymerization from the Kolong Company(2007-M-CC-24-P-04-3-050-2007).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.reactfunctpolym.2009.04.011.
References
[1] C. Park, J. Yoon, E.L. Thomas, Polymer 44 (2003) 6725.[2] J.T. Chen, E.L. Thomas, C.K. Ober, G.-P. Mao, Science 273 (1996) 343.[3] A.M. Urbas, E.L. Thomas, Adv. Mater. 14 (2002) 1850.[4] M.-S. Lee, B.-K. Cho, W.-C. Zin, Chem. Rev. 101 (2001) 3869.[5] S. Forster, M. Antonietti, Adv. Mater. 10 (1998) 195.[6] E.L. Thomas, J.T. Chen, M.J.E. O’Rourke, Macromol. Symp. 117 (1997) 241.[7] K. Ishizu, Prog. Polym. Sci. 23 (1998) 1383.[8] T. Hashimoto, M. Harada, N. Sakamoto, Macromolecules 32 (1999) 6867.[9] H. Zhao, E.P. Douglas, B.S. Harrison, K.S. Schanze, Langmuir 17 (2001) 8428.
[10] S.T. Selvan, T. Hayakawa, M. Nogami, M. Möller, J. Phys. Chem. B 103 (1999) 7441.[11] Y.-H. Cho, G. Cho, J.-S. Lee, Adv. Mater. 16 (2004) 1814.[12] Y.-S. Cho, C.-S. Ihn, H.-K. Lee, J.-S. Lee, Macromol. Rapid Commun. 22 (2001) 1249.[13] Y.-D. Shin, S.-H. Han, S. Samal, J.-S. Lee, J. Polym. Sci. Part A: Polym. Chem. 43
(2005) 607.[14] N.-G. Kang, M. Changez, J.-S. Lee, Macromolecules 40 (2007) 8553.[15] M. Tardi, P. Sigwalt, Eur. Polym. J. 8 (1972) 137.[16] C.J. Chang, T.E. Hogen Esch, J. Am. Chem. Soc. 97 (1975) 2805.[17] R.P. Quirk, S. Galvan-Corona, Macromolecules 34 (2001) 1192.[18] H. Watanabe, T. Amemiya, T. Shimura, T. Kotaka, Macromolecules 27 (1994)
2336.[19] M. Möller, R.W. Lenz, Makromol. Chem. 190 (1989) 1153.

N.-G. Kang et al. / Reactive & Functional Polymers 69 (2009) 470–479 479
[20] S. Ludwigs, A. Böker, V. Abetz, A.H.E. Müller, G. Krausch, Polymer 44 (2003)6815.
[21] E. Giebeler, R. Stadler, Macromol. Chem. Phys. 198 (1997) 3815.[22] R. Saito, A. Fujita, A. Ichimura, K. Ishizu, J. Polym. Sci. Part A: Polym. Chem. 38
(2000) 2091.[23] C.-L. Lee, N.-G. Kang, Y.-S. Cho, J.-S. Lee, J.-J. Kim, Opt. Mater. 21 (2002) 119.[24] A. Hagfeldt, M. Grätzel, Chem. Rev. 95 (1995) 49.[25] R. Aveyard, J.H. Clint, D. Nees, N. Quirke, Langmuir 16 (2000) 8820.[26] L.M. Goldenberg, J. Wagner, J. Stumpe, B. Paulke, E. Görnitz, Langmuir 18
(2002) 5627.[27] J.-S. Lee, A. Hirao, S. Nakahama, Macromolecules 21 (1988) 274.[28] J.-S. Lee, A. Hirao, S. Nakahama, Macromolecules 22 (1989) 2602.[29] N. Tessler, V. Medvedev, M. Kazes, S.H. Kan, U. Banin, Science 295 (2002) 1506.[30] M.F. Schulz, A.K. Khandpur, F.S. Bates, K. Almdal, K. Mortensen, D.A. Hajduk,
S.M. Gruner, Macromolecules 29 (1996) 2857.[31] J.C. Meiners, A. Quintel-Ritzi, J. Mlynek, H. Elbs, G. Krausch, Macromolecules 30
(1997) 4945.[32] K. Tsutsumi, Y. Funaki, Y. Hirokawa, T. Hashimoto, Langmuir 15 (1999) 5200.[33] C. Tsitsilianis, V. Sfika, Macromol. Rapid Commun. 22 (2001) 647.[34] H. Watanabe, T. Shimura, T. Kotaka, M. Tirrell, Macromolecules 26 (1993)
6338.[35] M. Kamachi, M. Kurihara, J.K. Stille, Macromolecules 5 (1972) 161.[36] J. Lee, T.E. Hogen-Esch, Macromolecules 34 (2001) 2805.[37] F.L. Duivenvoorde, J.J. Steven, S. van Es, C.F. van Nostrum, R. van der Linde,
Macromol. Chem. Phys. 201 (2000) 656.[38] Y.-H. Cho, J.-E. Yang, J.-S. Lee, Mater. Sci. Eng. C 24 (2004) 293.[39] H.-D. Koh, N.-G. Kang, J.-S. Lee, Langmuir 23 (2007) 12817.[40] H.-D. Koh, N.-G. Kang, J.-S. Lee, Langmuir 23 (2007) 11425.[41] M.R. Talingting, P. Munk, S.E. Webber, Z. Tuzar, Macromolecules 32 (1999)
1593.[42] J.J. Chiu, B.J. Kim, E.J. Kramer, D.J. Pine, J. Am. Chem. Soc. 127 (2005) 5036.[43] B.J. Kim, J. Bang, C.J. Hawker, E.J. Kramer, Macromolecules 39 (2006) 4108.[44] J. Peng, W. Knoll, C. Park, D.H. Kim, Chem. Mater. 20 (2008) 1200.[45] S. Krishnamoorthy, R. Pugin, J. Brugger, H. Heinzelmann, A.C. Hoogerwerf, C.
Hinderling, Langmuir 22 (2006) 3450.[46] J.-S. Lee, S.-W. Ryu, Macromolecules 32 (1999) 2085.[47] J.-S. Lee, S. Han, Y.-D. Shin, S.-Y. Kim, Polym. Prepr. 40 (1999) 1057.[48] Y.-D. Shin, S.-Y. Kim, J.-H. Ahn, J.-S. Lee, Macromolecules 34 (2001) 2408.
[49] Y.-D. Shin, J.-H. Ahn, J.-S. Lee, Macromol. Rapid Commun. 22 (2001) 1041.[50] Y.-D. Shin, J.-H. Ahn, J.-S. Lee, Polymer 42 (2001) 7979.[51] J.-H. Ahn, J.-S. Lee, Macromol. Rapid Commun. 24 (2003) 571.[52] J.-H. Ahn, Y.-D. Shin, S.-Y. Kim, J.-S. Lee, Polymer 44 (2003) 3847.[53] J.-H. Ahn, Y.-D. Shin, Y.G. Nath, S.-Y. Park, M.S. Rahman, S. Samal, J.-S. Lee, J.
Am. Chem. Soc. 127 (2005) 4132.[54] O. Vogl, G.D. Jaycox, Polymer 28 (1987) 2179.[55] G. Wulff, Angew. Chem., Int. Ed. Engl. 28 (1989) 21.[56] S. Mayer, R. Zentel, Prog. Polym. Sci. 26 (2001) 1973.[57] S.-H. Han, J.-W. Wu, J.-W. Kang, Y.-D. Shin, J.-S. Lee, J. Opt. Soc. Am. B 18 (2001) 298.[58] E.-M. Seo, M.-J. Kim, Y.-D. Shin, J.-S. Lee, D.-Y. Kim, Mol. Cryst. Liq. Cryst. 370
(2001) 143.[59] Y. Okamoto, T. Nakano, Chem. Rev. 94 (1994) 349.[60] Y. Okamoto, Macromol. Symp. 101 (1996) 343.[61] M.S. Rahman, S. Samal, J.-S. Lee, Macromolecules 39 (2006) 5009.[62] M.S. Rahman, S. Samal, J.-S. Lee, Macromolecules 40 (2007) 9279.[63] J.-H. Kim, M.S. Rahman, J.-S. Lee, J.-W. Park, J. Am. Chem. Soc. 129 (2007) 7756.[64] J.-H. Kim, M.S. Rahman, J.-S. Lee, J.-W. Park, Macromolecules 41 (2008) 3181.[65] J.T. Chen, E.L. Thomas, C.K. Ober, S.S. Hwang, Macromolecules 28 (1995) 1688.[66] S.I. Stupp, V. Lebonheur, K. Walker, L.S. Li, K.E. Huggins, M. Keser, A. Amstutz,
Science 276 (1997) 384.[67] X. Li, S. Tian, Y. Ping, D.H. Kim, W. Knoll, Langmuir 21 (2005) 9393.[68] B.J. Kim, G.H. Fredrickson, C.J. Hawker, E.J. Kramer, Langmuir 23 (2007) 7804.[69] W.-J. Shin, F. Basarir, T.-H. Yoon, J.-S. Lee, Langmuir 25 (2009) 3344.[70] Unpublished data.[71] M.S. Rahman, M. Changez, J.-W. Yoo, C.H. Lee, S. Samal, J.-S. Lee,
Macromolecules 41 (2008) 7029.[72] G. Cho, J. Jang, S. Jung, I.-S. Moon, J.-S. Lee, Y.-S. Cho, B.M. Fung, W.L. Yuan, E.A.
ORear, Langmuir 18 (2002) 3430.[73] D.L. Klein, R. Roth, A.K.L. Lim, A.P. Alivisatos, Nature 389 (1997) 699.[74] C. Bechinger, S. Ferrer, A. Zaban, J. Sprague, B.A. Gregg, Nature 383 (1996) 608.[75] M. Bruchez, M. Moronne, P. Giu, S. Weiss, A.P. Alivisatos, Science 281 (1998)
2013.[76] B.-H. Sohn, J.-M. Choi, S.-I. Yoo, S.-H. Yun, W.-C. Zin, J.-C. Jin, M. Kanehara, T.
Hirata, T. Teranish, J. Am. Chem. Soc. 125 (2003) 6368.[77] S.I. Yoo, J.-H. Kwon, B.-H. Sohn, J. Mater. Chem. 17 (2007) 2969.[78] H.-D. Koh, J.-P. Lee, J.-S. Lee, Macromol. Rapid Commun, in press, doi:10.1002/
marc.200900036.
Copyright © 2022 FDOKUMEN




















