
Co-Treatment with Single and Ternary Mixture Gas of ... - MDPI

Biochem. J. (2009) 424, 357–366 (Printed in Great Britain) doi:10.1042/BJ20091020 357
Binary and ternary crystal structure analyses of a novel inhibitor with17β-HSD type 1: a lead compound for breast cancer therapyMausumi MAZUMDAR, Diane FOURNIER, Dao-Wei ZHU, Christine CADOT, Donald POIRIER and Sheng-Xiang LIN1
Laboratory of Molecular Endocrinology and Oncology, CHUL Research Center (Centre Hospitalier de Universite Laval, CHUQ) and Laval University, Quebec, Canada, G1V 4G2
Oestradiol is a well-characterized sex hormone that stimulatesbreast cancer and other oestrogen-related diseases. 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) catalyses thelast step in the synthesis of oestradiol and androstenediol inbreast tumour tissue. The enzyme’s high expression and acti-vity after simultaneous blockade of oestrogen receptors andinhibition of aromatase in the tumour shows the necessity forits inhibition as a requirement for breast cancer therapy. In thepresent paper, we report structures of the binary and ternarycomplexes of 17β-HSD1 with a new inhibitor E2B {3-[3′,17′β-dihydroxyestra-1′,3′,5′(10′)-trien-16′β-methyl]benzamide}, andthe enzyme inhibition by the later. The IC50 value for E2B wasdetermined to be 42 nM in T47D cells. Multiple interactionsbetween E2B and the enzyme include hydrogen bonds andhydrophobic interactions, as well as π–π interactions. A kinetic
study demonstrated that E2B inhibits the enzyme’s reductionforming oestradiol from oestrone, with a K i of 0.9 +− 0.15 nM.Such strong inhibition is in agreement with its extensiveinteraction with the enzyme, suggesting its potential as a leadcompound for breast cancer therapy. In fact, this possibility isenhanced by its capacity for cell penetration similar to naturalsteroids. Such inhibitors that block oestrogen synthesis to suppressthe sulfatase pathway producing oestradiol can be used in adjuvanttherapies with oestrogen receptor blockade, opening a neworientation of breast cancer treatment.
Key words: breast cancer, 3-[3′,17′β-dihydroxyoestra-1′,3′,5′(10′)-trien-16′β-methyl]benzamide (E2B), 17β-hydroxysteroiddehydrogenase, kinetic inhibition, oestrogen, π–π interaction,protein–ligand interaction.
INTRODUCTION
The potent oestrogen, E2 (oestradiol), has been a well-knownfactor responsible for the stimulation of breast cancer and is alsoimplicated in other oestrogen-related diseases. It is also involvedin the genesis and development of diseases in certain genetic andphenotypic cases. The American Cancer Society estimated thatwithin 2008, 182460 cases of breast cancer would be reported,leading to 40480 deaths. In 2008, an estimated 22400 womenwere diagnosed with breast cancer in Canada alone, resulting in5300 deaths, of which most were E2-dependent. Breast cancercontinues to lead in incidence among North American women,with more than twice as many new cases as lung cancer.
The biological activity of steroid hormones is regulated at thepre-receptor level by several enzymes, including 17β-HSDs (17β-hydroxysteroid dehydrogenases). 17β-HSD1 is the first memberknown in this family to catalyse the last step in the synthesis of E2
[1–4]. To date, it also possesses the highest activity for oestrogensynthesis in the 17β-HSD family towards the reduction reaction[5]. The first detailed enzyme activity measurements applied tothe homogeneous and fully active enzyme preparation appearedin 1992 [6], which laid down the basis of crystallization and theenzyme homodimeric structure solution.
Most 17β-HSDs belong to the SDR (short-chain dehydro-genase–reductase) family, with the exception of type 5, whichis an aldo–keto reductase. 17β-HSDs of the SDR family shareseveral common amino acid sequence motifs [7–11]. Even thoughmany 17β-HSD members are ubiquitously present in differenttissues, they demonstrate distinct preferences for either the
activation or inactivation of different steroid hormones [12].For the enzymes using triphosphate cofactors, they are usuallyreductive in cells, where [NADPH]>[NADP] [13]. On the otherhand, those using diphosphate cofactors are generally oxidative incells as [NAD]>[NADH] ([13] and references therein). Besidesthe general gonadal supply of sex steroids, there is the otherand important intracrinological provision by peripheral steroidsynthesis (e.g. adrenal, skin or brain) and steroid activation–inactivation (epithelial cells of breast, uterus, prostate or kidney).In addition adrenal steroid precursor is delivered in the formof DHEA (dihydroepiandrosterone) sulfate and E1S (oestronesulfate) to the peripheral tissue where they are converted intooestrogens and androgens.
Two principal pathways are involved in the last steps ofE2 synthesis in breast cancer: the ‘aromatase pathway’ whichtransforms androgens into oestrogens and the ‘sulfatase pathway’which converts E1S into E1 (oestrone) and then, by the actionof 17β-HSD1, into E2. Previous studies have reported that, inhuman breast tumours, the sulfatase pathway predominates overthe aromatase pathway [14,15]. Despite low levels of circulatingoestrogens, the concentration of E2 in cancer tissue is severaltimes higher than found in plasma and is also higher in post-menopausal than in pre-menopausal patients, suggesting a specifictumoral biosynthesis and hormone accumulation [16,17]. Inaddition to the synthesis of E2, which stimulates breast cancer[18–20], higher mRNA expression levels for 17β-HSD1, butnot for aromatase or sulfatase, is found in post-menopausalbreast cancers [18]. The latter indicates the important role playedby 17β-HSD1. Oestrogens are reported to be essential for the
Abbreviations used: DHEA, dihydroepiandrosterone; E1, oestrone; E1S, oestrone sulfate; E2, oestradiol; E2B, 3-[3′,17′β-dihydroxyoestra-1′,3′,5′(10′)-trien-16′β-methyl]benzamide; ER, oestrogen receptor; HEK, human embryonic kidney; 17β-HSD, 17β-hydroxysteroid dehydrogenase; IGF, insulin-likegrowth factor; RMSD, root mean square deviation; SDR, short-chain dehydrogenase–reductase.
1 To whom correspondence should be addressed (email [email protected]).The atomic co-ordinates for 17β-HSD1–E2B–NADP+ and 17β-HSD1–E2B have been deposited in the PDB under codes 3HB5 and 3HB4 respectively.
c© The Authors Journal compilation c© 2009 Biochemical Society
www.biochemj.org
Bio
chem
ical
Jo
urn
al

358 M. Mazumdar and others
growth and differentiation of the mammary gland, and tumourformation may originate from excessive hormonal stimulationof breast epithelium [18]. Moreover 17β-HSD1 mRNA is aprognostic marker for breast cancer progression regardless of theER (oestrogen receptor) status [19]. A high level of 17β-HSD1correlates with an increased risk of developing a late relapseof breast cancer in ER-positive breast cancer patients [21,22].Purohit et al. [23] have shown that, in breast tumour tissues, thereductive direction of oestrogen metabolism prevails and thereis the possibility that tumour-derived factors might modulatethe activity of 17β-HSD1. Growth factors [e.g. IGF (insulin-like growth factor)-I and IGF-II] and cytokines [IL (interleukin)-6 and TNFα (tumour necrosis factor α)] have been shown tostimulate the activity of 17β-HSD1 in breast tumour tissue [23].Sasano et al. [24,25] have analysed clinical data and shownthat no correlation was detected for intratumoral aromatase and17β-HSD1, demonstrating that intratumoral E2 was producedby a pathway other than aromatase in certain breast cancerpatients [25]. For patients in whom overexpression of intratumoral17β-HSD1, but not aromatase, is detected, inhibition of 17β-HSD1 could be of much more importance than inhibition ofaromatase.
In gonadal and peripheral tissues, 17β-HSD1 is the mostimportant oestrogen-activating enzyme [3,26]. 17β-HSD1 canconvert less active oestrogens such as E1 into more potentforms, first from E1 into E2 and secondly from DHEA intoandrostenediol [3], whereas 17β-HSD7 possesses a significantlylower activity for conversion of E1 into E2. Both 17β-HSD1 and17β-HSD2 are known to be expressed in normal breast tissue, buthigher expression of 17β-HSD1 mRNA has been demonstrated inalmost 50% of breast cancer tissues [20]. This suggests that theaccumulation of E2 in breast cancer cells as 17β-HSD2 is knownto deactivate E2 by its conversion into E1, which is less potent andhas lower oestrogenic activity. In addition, 17β-HSD1 has beenreported to be responsible for intracellular accumulation of E2 inmalignant breast epithelial cells. This suggests that the presence of17β-HSD1 in cancerous tissue leads to increased proliferationof oestrogen-dependent cancer cells and breast cancer devel-opment [3,20,23].
Knowledge of the function of 17β-HSD1 was quite limitedbefore its three-dimensional structure was determined. The firstdiffraction-quality crystals were obtained in the early 1990s [27],and the enzyme structure was determined to a resolution of 2.2 Å(1 Å = 0.1 nm), becoming the first three-dimensional structure ofany human steroid-converting enzyme [9,28]. A dozen differentcomplex structures have been solved to date [9,28–37]. Althoughthe enzyme can bind both NADH and NADPH, NADPH is thecognate cofactor rich in cells and drives the substrate reductionin vivo [13], as NADPH is the major form of the triphosphatecofactor.
The enzyme’s structure–activity relationship has been welldocumented, but to date progress in the design of an inhibitorthat is effective for clinical trials has not been achieved [38–42].However, inhibitors of 17β-HSDs constitute a growing interest inbiomedical research, and new compounds have been developedin recent years [38–42]. One of these inhibitors, the E2-adenosinehybrid compound EM-1745, revealed key interactions with twodifferent enzyme-binding sites, namely the substrate- and thecofactor-binding sites [36]. For the phenylmethyl derivatives(such as E2B {3-[3′,17′β-dihydroxyoestra-1′,3′,5′(10′)-trien-16′β-methyl]benzamide}), our results suggest that the presence of ashort flexible 16β-methylene group allows improved positioningof the phenyl ring to facilitate interaction with loop residues inthe presence of the cofactor along with high affinity and drug-likequality.
In the present study, we have solved crystal structures of 17β-HSD1 in binary and ternary complexes with the new inhibitorE2B, and the cofactor NADP+ along with a detailed kinetic study.The crystal structures of both binary and ternary structures of anyinhibitor complex with 17β-HSD1 reported in the present studydemonstrated the high affinity of the inhibitor for the enzymeand also provides an atomic-level insight into the structure-basedefficiency of the inhibitor, detailing its interaction and scope forfurther development.
MATERIALS AND METHODS
Protein purification
Oestrogenic 17β-HSD1 was purified from human placenta by aprocedure consisting of three FPLC steps using Q-Sepharose,Blue-Sepharose and phenyl-Sepharose affinity columns asdescribed in the protocol used by Lin and colleagues [6]. β-OG(β-octylglucoside) was added to the protein thus obtained, whichwas then concentrated to 16 mg/ml. The protein concentration wasdetermined by spectrophotometry and its activity was measuredby the oxidation of E2 to E1.
Inhibitor E2B
The inhibitor E2B (Figure 1A) was easily synthesized from E1 bythree chemical reactions consisting of an aldol condensation with3-amidobenzaldehyde, a stereoselective carbonyl reduction and astereoselective double-bond hydrogenation, which was publishedpreviously for closely related compounds [40]. The inhibitorycurves of E2B and E1 show the transformation of [4-14C]E1
(60 nM) into [4-14C]E2 in T47D intact cells. This experimentwas performed as described previously [40]. The IC50 value (theinhibition concentration at which 50% of the enzyme activityis inhibited) was calculated using an unweighted iterative least-squares method for four-parameter logistic curve fitting (DE50program; CHUL Research Center).
Inhibition study
A radioactive assay was used to determine the enzyme steady-state kinetics first to calculate the Km for E1 reduction into E2 by17β-HSD1 and then the same kinetics in the presence of E2B atdifferent concentrations for the K i determination, similar to Qiuet al. [43]. The reactions were carried out in the presence of anexcess of NADPH (50 μM) at pH 7.4 at 37 +− 0.5 ◦C. The Km forE1 reduction into E2 in the absence of E2B and different apparentKm values in the presence of 0.01–5 nM E2B were determined. Atconcentrations above 2 nM of E2B, total inhibition of the enzymeactivity was obtained (results not shown).
The stock enzyme at 1.06 mg/ml was diluted with a buffercontaining 50 mM Tris/HCl (pH 7.5), 20% glycerol, 1 mMEDTA and 0.2 mM DTT (dithiothreitol) to a final concentrationof 1.06 × 10−5 mg/ml for kinetic studies. Reactions were initiatedby the addition of the diluted enzyme sample to the reactionmixture resulting in the conversion of E1 into E2. Aliquots weretaken and the reaction was stopped at different time intervals(0, 3, 8 and 20 min) followed by steroid extractions with 2.5 ×volume of diethyl ether. The steroids were separated by TLCmigration in 80% toluene and 20% acetone, and analysed byphosphoimaging. Initial velocities were measured with less than5% substrate consumption. The experiments were repeated withdifferent concentrations of E1 ranging from 0.01 to 0.2 μM. Toobtain an optimal signal, larger reaction mixture volumes wereused for low substrate concentrations (i.e. 1 ml reaction mixturesfor 0.01, 0.02 and 0.05 μM E1, and 0.5 ml reaction mixture for
c© The Authors Journal compilation c© 2009 Biochemical Society
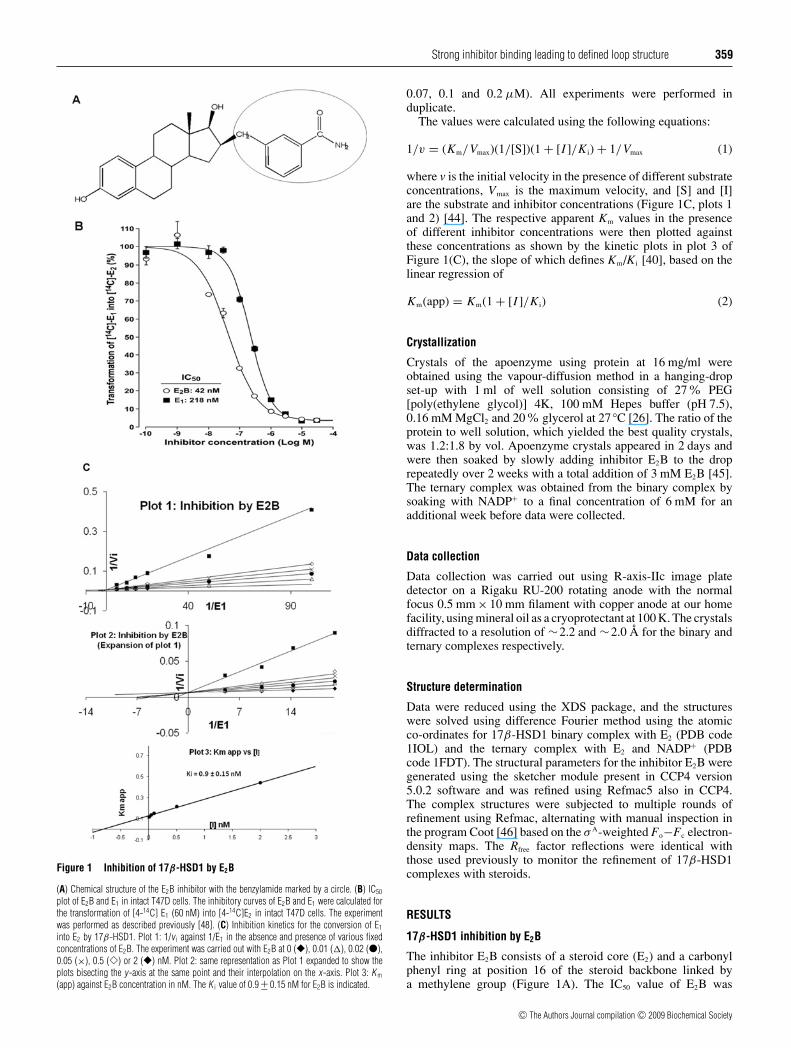
Strong inhibitor binding leading to defined loop structure 359
Figure 1 Inhibition of 17β-HSD1 by E2B
(A) Chemical structure of the E2B inhibitor with the benzylamide marked by a circle. (B) IC50
plot of E2B and E1 in intact T47D cells. The inhibitory curves of E2B and E1 were calculated forthe transformation of [4-14C] E1 (60 nM) into [4-14C]E2 in intact T47D cells. The experimentwas performed as described previously [48]. (C) Inhibition kinetics for the conversion of E1
into E2 by 17β-HSD1. Plot 1: 1/v i against 1/E1 in the absence and presence of various fixedconcentrations of E2B. The experiment was carried out with E2B at 0 (�), 0.01 (�), 0.02 (�),0.05 (×), 0.5 (�) or 2 (�) nM. Plot 2: same representation as Plot 1 expanded to show theplots bisecting the y-axis at the same point and their interpolation on the x-axis. Plot 3: K m
(app) against E2B concentration in nM. The K i value of 0.9 +− 0.15 nM for E2B is indicated.
0.07, 0.1 and 0.2 μM). All experiments were performed induplicate.
The values were calculated using the following equations:
1/v = (Km/Vmax)(1/[S])(1 + [I ]/K i) + 1/Vmax (1)
where v is the initial velocity in the presence of different substrateconcentrations, Vmax is the maximum velocity, and [S] and [I]are the substrate and inhibitor concentrations (Figure 1C, plots 1and 2) [44]. The respective apparent Km values in the presenceof different inhibitor concentrations were then plotted againstthese concentrations as shown by the kinetic plots in plot 3 ofFigure 1(C), the slope of which defines Km/K i [40], based on thelinear regression of
Km(app) = Km(1 + [I ]/K i) (2)
Crystallization
Crystals of the apoenzyme using protein at 16 mg/ml wereobtained using the vapour-diffusion method in a hanging-dropset-up with 1 ml of well solution consisting of 27% PEG[poly(ethylene glycol)] 4K, 100 mM Hepes buffer (pH 7.5),0.16 mM MgCl2 and 20% glycerol at 27 ◦C [26]. The ratio of theprotein to well solution, which yielded the best quality crystals,was 1.2:1.8 by vol. Apoenzyme crystals appeared in 2 days andwere then soaked by slowly adding inhibitor E2B to the droprepeatedly over 2 weeks with a total addition of 3 mM E2B [45].The ternary complex was obtained from the binary complex bysoaking with NADP+ to a final concentration of 6 mM for anadditional week before data were collected.
Data collection
Data collection was carried out using R-axis-IIc image platedetector on a Rigaku RU-200 rotating anode with the normalfocus 0.5 mm × 10 mm filament with copper anode at our homefacility, using mineral oil as a cryoprotectant at 100 K. The crystalsdiffracted to a resolution of ∼2.2 and ∼2.0 Å for the binary andternary complexes respectively.
Structure determination
Data were reduced using the XDS package, and the structureswere solved using difference Fourier method using the atomicco-ordinates for 17β-HSD1 binary complex with E2 (PDB code1IOL) and the ternary complex with E2 and NADP+ (PDBcode 1FDT). The structural parameters for the inhibitor E2B weregenerated using the sketcher module present in CCP4 version5.0.2 software and was refined using Refmac5 also in CCP4.The complex structures were subjected to multiple rounds ofrefinement using Refmac, alternating with manual inspection inthe program Coot [46] based on the σ A-weighted Fo−Fc electron-density maps. The Rfree factor reflections were identical withthose used previously to monitor the refinement of 17β-HSD1complexes with steroids.
RESULTS
17β-HSD1 inhibition by E2B
The inhibitor E2B consists of a steroid core (E2) and a carbonylphenyl ring at position 16 of the steroid backbone linked bya methylene group (Figure 1A). The IC50 value of E2B was
c© The Authors Journal compilation c© 2009 Biochemical Society

360 M. Mazumdar and others
Table 1 Data collection and refinement statistics of 17β-HSD1–E2B–NADP+ and 17β-HSD1–E2B crystal structures
Rcryst;free = �hkl ||F calc(hkl)|−|F obs(hkl)||/�hkl |F obs|, where the crystallographic and free R-factors are calculated, including and excluding refined reflections respectively. The Rfree reflections setconstituted 5 % of the total number of reflections.
Value
Parameter 17β-HSD1–E2B–NADP+ 117β-HSD1–E2B
Space group C21 C21
Unit cell a = 123.200, b = 44.000, c = 60.80, α = γ = 90.00◦, β = 100.30◦ a = 123.080, b = 43.700, c = 60.510, α = γ = 90.00◦, β = 100.74◦
Resolution range (A) 19.94–2 18.98–2.21Highest shell range (A) 2.052–2.000 2.263–2.207Total reflections 95369 15658Unique reflections 21662 15658Completeness (%) 96.2 97.3Multiplicity 3.2 (2.3) 3.2 (2.1)Rmerge 0.037 0.045Mean (I)/σ (I) 27.02 22.03RMSD of bond length (A) and bond angle (◦) 0.022 and 2.135 0.024 and 2.235Rcryst (%) 20.3 19.1Rfree (%) 25 26Average B-value (A2) 30.978 29.8Water molecules 94 45Ramachandran plot (%) 98 97.8
determined with intact cells expressing 17β-HSD1 as mentionedby Laplante et al. [40]. Although E2B inhibited the transformationof E1 into E2 in transfected HEK (human embryonic kidney)-293cells, T47D and MCF-7 breast cancer cells, its best potency wasobtained with intact T47D cells. This is in correlation with thestudy which details the significantly higher expression of 17β-HSD1 in T47D cells than in MCF-7 cells, and also, in transfectedHEK-293 cells, the presence of the cofactor can drive the reactionto either direction [21]. In the latter cells, the IC50 value wasdetermined to be 42 nM (Figure 1B). To assess the unwantedoestrogenic activity as E2B is a steroid derivative, a 10-daytreatment with E2B at a concentration of 0.5 μM, which is morethan 10-fold higher than its IC50 with T47D cells, induced someproliferation (38%) of T47D oestrogen-receptor-positive breastcancer cells. Interestingly, when E2B (0.5 μM) was given with E1
(0.1 nM) in a 10-day treatment, it blocked 62 % of the T47D cellproliferation induced by E1 after its reduction to E2 by 17β-HSD1[40]. To verify whether E2B was acting as an anti-oestrogen, aninhibition study in the presence of E2 along with anti-oestrogenEM-139 was carried out. E2B did not reverse the proliferativeeffect on oestrogen-receptor-positive cells by E2 (0.1 nM) as doesthe pure anti-oestrogen EM-139, thereby confirming that E2Bdoes not act as an anti-oestrogenic compound, but as an inhibitorof 17β-HSD1 [40].
As shown in Figure 1(C), plots 1 and 2, the Lineweaver–Burkplots for E1 reduction to E2 in the absence and the presence ofdifferent E2B concentrations intersect at the same point on the 1/vaxis. For the reciprocal plots corresponding to different inhibitorconcentrations, the curves in the presence of increasing inhi-bitor concentration result in a higher slope, pivoting aroundthe intersection point on the control curve. This demonstratesa typical reversible and competitive inhibition by E2B againstE1, in agreement with equation [1]. E2B, as expected from theformation of the ternary complex, showed non-competitivenesswith NADPH (results not shown). The results also agree with theinhibitor-binding position in the substrate site shown by thecrystallographic structure. A K i value of 0.9 +− 0.15 nM wascalculated from the plot (Figure 1C, plot 3) by the intersectionof the above line on the left of the [I] axis, which demonstratesa higher affinity than any other available inhibitor for 17β-HSD1in the literature. E2B shows a strong enzyme inhibition and a
significantly higher apparent affinity for the enzyme than doesE1. The Km for E1 reduction by 17β-HSD1 was measured to beapprox. 0.03 μM, a result similar to that found by Jin and Lin[5]. Such an inhibition by E2B is in agreement with its extensiveinteraction with the 17β-HSD1-binding pocket as observed in theenzyme–E2B complex structures.
Structure determination of the enzyme–inhibitor complexes
Both the binary and ternary complexes crystallized in space groupC21 with one molecule in the asymmetric unit. The R-factor valuesfor 17β-HSD1–E2B and 17β-HSD1–E2B–NADP+ are shown inTable 1. The resulting model showed very clear electron densityfor almost all residues except for Ala191, Phe192, Met193, Leu197
and Gly198 in the binary complex at 1.8 σ . In the ternary complexat 1.8 σ level, all residues showed clear electron density exceptfor Met193. These residues which did not show clear electrondensity in other structures constitute the active-site loop regionfrom residues 189 to 200 of the enzyme. Electron density wasclearly seen in the 2Fo−Fc map for the region constituting theloop at 1σ level (Figures 2A and 2B). After the initial refinement,the Fo−Fc electron-density map is unambiguous for inhibitorE2B at the substrate-binding site in both the binary and ternarycomplexes. In the ternary complex, the electron density of theadenosine ring, the nicotinamide ring and the phosphate groupsof NADP+ was obvious, whereas that for ribose was unclear evenafter the final refinement. In the binary complex, the density ofthe benzylamide group of the inhibitor did suggest the presenceof two conformations (Figure 2C).
E2B interactions with 17β-HSD1 in the binary complex
In the binary structure of the inhibitor with the enzyme, a stronghydrogen bond is present between Oγ of Ser142 and O-17 of thesteroidal backbone of E2B (2.58 Å) (Figure 3). Water moleculeHOH938 makes two bifurcated hydrogen bonds, one with O-17 ofE2B (2.86 Å) and other with O of Gly186 (3.10 Å) (water-mediatedinteractions are not shown in the Figure). In the binary complex,Asn152 forms water-mediated intramolecular hydrogen bonds withTyr218.
c© The Authors Journal compilation c© 2009 Biochemical Society
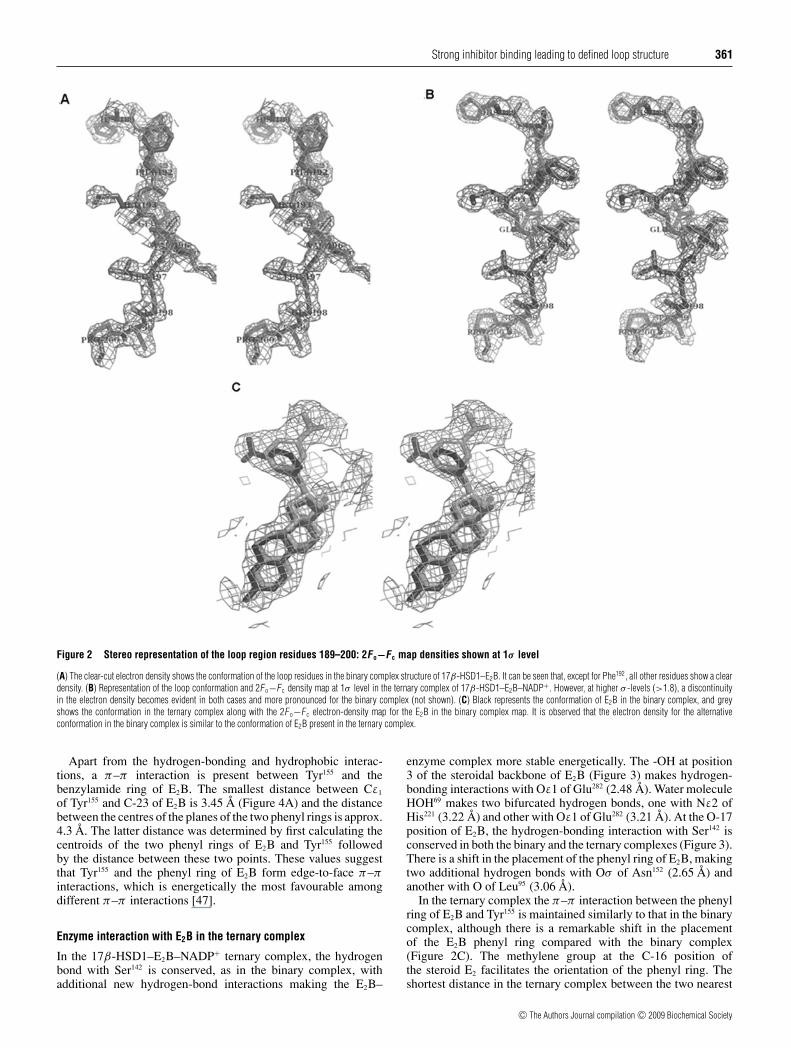
Strong inhibitor binding leading to defined loop structure 361
Figure 2 Stereo representation of the loop region residues 189–200: 2Fo−Fc map densities shown at 1σ level
(A) The clear-cut electron density shows the conformation of the loop residues in the binary complex structure of 17β-HSD1–E2B. It can be seen that, except for Phe192, all other residues show a cleardensity. (B) Representation of the loop conformation and 2F o−F c density map at 1σ level in the ternary complex of 17β-HSD1–E2B–NADP+. However, at higher σ -levels (>1.8), a discontinuityin the electron density becomes evident in both cases and more pronounced for the binary complex (not shown). (C) Black represents the conformation of E2B in the binary complex, and greyshows the conformation in the ternary complex along with the 2F o−F c electron-density map for the E2B in the binary complex map. It is observed that the electron density for the alternativeconformation in the binary complex is similar to the conformation of E2B present in the ternary complex.
Apart from the hydrogen-bonding and hydrophobic interac-tions, a π–π interaction is present between Tyr155 and thebenzylamide ring of E2B. The smallest distance between Cε1
of Tyr155 and C-23 of E2B is 3.45 Å (Figure 4A) and the distancebetween the centres of the planes of the two phenyl rings is approx.4.3 Å. The latter distance was determined by first calculating thecentroids of the two phenyl rings of E2B and Tyr155 followedby the distance between these two points. These values suggestthat Tyr155 and the phenyl ring of E2B form edge-to-face π–πinteractions, which is energetically the most favourable amongdifferent π–π interactions [47].
Enzyme interaction with E2B in the ternary complex
In the 17β-HSD1–E2B–NADP+ ternary complex, the hydrogenbond with Ser142 is conserved, as in the binary complex, withadditional new hydrogen-bond interactions making the E2B–
enzyme complex more stable energetically. The -OH at position3 of the steroidal backbone of E2B (Figure 3) makes hydrogen-bonding interactions with Oε1 of Glu282 (2.48 Å). Water moleculeHOH69 makes two bifurcated hydrogen bonds, one with Nε2 ofHis221 (3.22 Å) and other with Oε1 of Glu282 (3.21 Å). At the O-17position of E2B, the hydrogen-bonding interaction with Ser142 isconserved in both the binary and the ternary complexes (Figure 3).There is a shift in the placement of the phenyl ring of E2B, makingtwo additional hydrogen bonds with Oσ of Asn152 (2.65 Å) andanother with O of Leu95 (3.06 Å).
In the ternary complex the π–π interaction between the phenylring of E2B and Tyr155 is maintained similarly to that in the binarycomplex, although there is a remarkable shift in the placementof the E2B phenyl ring compared with the binary complex(Figure 2C). The methylene group at the C-16 position ofthe steroid E2 facilitates the orientation of the phenyl ring. Theshortest distance in the ternary complex between the two nearest
c© The Authors Journal compilation c© 2009 Biochemical Society

362 M. Mazumdar and others
Figure 3 Ligplots showing the interactions of E2B with 17β-HSD1 in binary (right) and ternary (left) complexes
The residue number ‘700’ and chain-id ‘A & X’ of E2B in the PDB file is labelled.
phenyl rings is 3.80 Å, compared with 4.3 Å in the binarycomplex. In both complexes, they form an edge-to-face π–πinteraction, the most favoured π–π interaction reported by Hunteret al. [47].
Enzyme interaction with NADP+ in ternary complex
The major hydrogen-bond interactions for the cofactor are notedat the nicotinamide end of NADP+. Two strong hydrogen bondsare observed with NO7 of the nicotinamide of NADP+ with theN and O of Val188, with distances of 3.06 and 2.94 Å respectively.The other two possible hydrogen bonds are observed at NO2*of nicotinamide with -OH of Tyr155 and Nσ of Lys159. The NO7also forms a hydrogen bond with the water molecule HOH76.The other important interaction at this end of NADP+ is thatof NN7 with Oγ 1 of Thr190. Other than these hydrogen-bondinteractions at the nicotinamide end of NADP+, the residues whichinteract with the cofactor are Thr41, Arg37, Asp38, Val66, Gly92,Ser11, Ser12, Gly13, Ile14, Gly141, Thr140, Cys185 and Lys159 alongwith additional water-mediated hydrogen-bonding interactions.In the former structure–function study [13], it was found thatthe major interaction between the cofactor and enzyme happensat the ADP part. This is why the enzyme binding has a similaraffinity to ADP or NADP+ [13], suggesting that the nicotinamideis relatively flexible in the complex. In the E2B ternary complex,it was observed that the benzylamide ring of the inhibitor in turnstabilizes this flexible nicotinamide part of the cofactor, anotherstrong interaction leading to the compact fit in the active site.
DISCUSSION
Comparison of the binary and ternary complex interactions
The size of E2B is greater than E2 due to the additionalbenzylamide group at the C-16 position. However, the volume of
the active-site cavity, calculated using Pocket-finder, did not differsignificantly between the E2 ternary, E2B binary and E2B ternarystructures. This suggests the volume occupancy at the active siteof the enzyme by E2B is higher when compared with E2, as thebenzylamide ring of E2B occupies a space in the active site whichis not occupied by the native substrates of the enzyme. This isalso confirmed by comparison of the E2 ternary and E2B ternarystructures (Figure 4B). The extra fitting of E2B does not interferewith the NADP+-binding site as confirmed by the structures aswell as kinetic studies.
In the binary complex, the steroidal scaffold of inhibitor E2Bshows one strong hydrogen bond directly with the -OH of Ser142
(2.58 Å) which is also found in the ternary complex. In theternary complex, E2B interacts with the enzyme by forming threeadditional hydrogen bonds, of which two result from the -OH of itsbenzylamide ring (Figure 3). The electron-density map stronglysuggests that, in the binary complex, the benzylamide ring canmanifest two conformations (Figure 2C). In the ternary complex,the presence of the nicotinamide ring of the cofactor stabili-zes the positioning of the inhibitor, ensuring a more compact fit.The B-factor average of the E2B inhibitor is higher in the binarythan in the ternary complex, with the highest difference comingfrom the benzylamide part of E2B.
The phenyl ring of Phe192 in the binary and ternary complexshows a displacement (Figure 4C). In the absence of the cofactor(NADP+), there is a strong possibility of forming a hydrogen bondbetween the amide group of E2B and Phe192. This interaction isabsent from the ternary complex where the E2B displacementcompared with its position in binary makes possible the hydrogenbond between the NO* of NADP+ and -OH of Tyr155 (3.32 Å).The aromatic rings of the E2B inhibitor with Phe192 and the Tyr155
demonstrate a π-stacking interaction, similar to that defined byHunter et al. [47]. From the ternary complex, it is clear that E2Bdoes not compete with the cofactor.
c© The Authors Journal compilation c© 2009 Biochemical Society

Strong inhibitor binding leading to defined loop structure 363
Figure 4 Structure and intraction of E2B with 17 β-HSD1
(A) π–π interaction between E2B and Tyr155 in the binary complex. (B) Superposition of the E2B binary and ternary complexes along with 17β-HSD1–E2–NADP+ (PDB code 1FDT). E2B-T andE2B-B show the conformation of E2B in the ternary and binary complex respectively. Blue represents the NADP+ conformation in 17β-HSD1–E2B–NADP+, and pink represents the conformation in1FDT. (C) Superposition of the binary (in light blue) and ternary complex (in cyan). The shift in the conformation of benzylamide of E2B for both complexes results in a slight orientation difference,while still maintaining the π–π interaction. Also the shift in the Phe192 is seen. (D) Superposition of the ternary complex of 17β-HSD1 with E2 and NADP+ in blue, the ternary complex with E2B inred and the binary complex with E2B in green showing the loop conformation.
Comparison of 17β-HSD1 structures in complex with E2B and thosewith other ligands
EM-1745 is the only inhibitor of 17β-HSD1 with which anenzyme–inhibitor complex structure has been reported [35].Although the K i for EM-1745 and E2B are similar, i.e. 3.0 +− 0.8and 0.9 +− 0.15 nM respectively, the IC50 values of EM-1745 forcell homogenate is 52 nM, but for intact cells, it is 8 μM; theinhibitory curves were determined at 0.1 μM E1 [48], comparedwith an IC50 of 42 nM for E2B in intact cells. The activity of E2Bin intact cells as well as cell homogenate was almost the same(M. Mazumdar, D. Fournier, D.-W. Zhu, C. Cadot, D. Poirier andS.X. Lin, unpublished work), suggesting that its capacity for cellpenetration is similar to that of natural steroids. This suggests thatE2B is more accessible for the cells compared with EM-1745 inthe present form, making E2B more drug-like.
As seen from the superposition of the substrates and thecofactors from the E2 ternary with the E2B in binary and ternarycomplexes, the E2 backbone in E2B and the NADP+ of both
ternary structures occupy almost the same position except thatthe benzylamide ring of E2B occupies a new position. Themaximum variation in the active-site residues are observedin the loop region consisting of residues 189–201, whichis the only flexible area surrounding the active site of theenzyme (Figure 4D). This additional interaction enhances theaffinity of E2B with 17β-HSD1. EM-1745 with an adenineconnected to the steroidal backbone, occupies partially theNADP+-binding site, whereas E2B does not interfere with the co-factor binding.
The aromatic–aromatic interactions between phenylalanineresidues in proteins are known to play an important role in proteinconformation. Although the energy of the π–π interaction isless than the electrostatic energy, being comparable with thatof van der Waals interactions [47], the formation of these π–π interactions between the phenyl rings plays an importantrole towards stabilizing and increasing the affinity of E2B forthe enzyme. Hydrophobic interactions in the substrate-bindingsite of 17β-HSD1 contribute to the major binding energy. In
c© The Authors Journal compilation c© 2009 Biochemical Society

364 M. Mazumdar and others
this case, the placement of the three phenyl rings is stabilizedby the π–π interactions [46]. This may be an importantfactor resulting in the high affinity and potency of this newinhibitor.
In both the binary and ternary structures, the phenyl ring ofTyr155 and the benzylamide of E2B show π–π interactions, butwhen the distance between the two centroids is considered, theinteraction is stronger in the ternary complex. The flexibility atthe methylene group (C-21 position) of E2B allows the shift of thebenzylamide ring in the presence of NADP+ without perturbingthe π–π interactions with Tyr155 (Figure 4C). The side chain ofPhe192 is displaced to accommodate the nicotinamide ring, butthe π–π stacking interaction between Tyr155, E2B and Phe192
is maintained. NADP+ also shows a modest shift in the E2Bternary complex when compared with its placement in the ternarycomplex of 17β-HSD1 and E2, its RMSD (root mean squaredeviation) being 0.528 Å.
When the position of the E2 core in E2B is compared withthe other available crystal structures in the PDB, it can be seenthat there is a shift in the structures along the line of the steroidbackbone (Figure 4B). A shift is also observed between the binaryand ternary complexes of E2B with 17β-HSD1 (in the absence andpresence of the cofactor). In the ternary complex, the movement ismore towards the active-site core with respect to Tyr155, resultingin new interactions at the O3 position mentioned above. The E2
backbone of E2B shows a stronger interaction between its O-17and the Oγ of Ser142 in the E2B complex with the enzyme thanthe same backbone in either the E2 or the EM-1745 complexesconsidering the distance [36].
In both binary and ternary complexes of 17β-HSD1 with E2B,the presence of a hydrogen-bond interaction with Oγ of Ser142
renders the selectivity criterion for the present inhibitor. Theinteractions with His221 are absent from the E2B complexes, and itwas reported previously in the variation studies that His221 is notan important residue for substrate recognition or binding [37].
When the other complex structures of 17β-HSD1 with E2
(1IOL), E2 and NADP+ (1FDT), and EM-1745 (1I5R) aresuperimposed, a shift is seen at the active site for the E2 corealong the horizontal axis as mentioned. In the EM-1745 complex,the O-3 of the steroid scaffold is seen to be displaced away fromTyr155 in order to adjust for the other interactions at the adenosineend of the inhibitor. The O-17 interaction with Ser142 is lacking inthe case of the EM-1745 complex [35], which has been identifiedas an important interaction for substrate selectivity by theenzyme.
Loop region of 17β-HSD1
The loop region (residues 189–200) shows a shift in the ternarycomplex when compared with the binary complex. The maximumshift is observed for residues Phe192 to Leu195. Comparison of theRMSD values from the various regions is presented in Table 2.In the 2Fo − Fc map of both binary and ternary complexes, allresidues in the loop have a clear electron density at the 1σ level,except for Met193 in the ternary complex. The RMSD of the entiremolecule and that of the loop region shows a significant difference(Table 2). The loop seen in the ternary complex is adjusted so asto accommodate more space for the cofactor in the active sitewhen compared with the binary complex. This loop region showsdifferent conformation in various complexes solved to date [27–37] and is the only flexible region of the enzyme. In both complexstructures of E2B the loop region is in, as seen in Figure 4(D), awider and more extended conformation, compared with the openconformation in the E2 ternary complex. This additional space
Table 2 RMSD of the given residues of the binary and ternary complexesof E2B with 17β-HSD1 compared with various other complex structures
RMSD with E2B RMSD with E2BPDB code Residue number binary complex (A) ternary complex (A)
1IOL 1–284 0.416 0.419188–201 3.184 3.069192 0.624 0.592195 1.342 1.088
1FDT 1–284 0.393 0.384188–201 2.604 2.098192 4.016 4.302195 1.224 1.077
1DHT 1–284 0.486 0.495188–201 3.355 3.303192 0.929 0.730195 0.918 1.211
in the active site by the placement of the loop residues provides,first, enough space for the accommodation of the benzylamidering of E2B and secondly, a compact fit of the substrate (E2B inthis case) in the active site. In the complexes with E2B, it cannot bepredicted whether the loop is in the closed or open conformationas reported by Sawicki et al. [32], but the observed shift definitelyresults in a more spacious volume of the active site than that inthe E2 ternary complex.
Conclusion
Both hydrogen-bonding as well as van der Waals interactions arepresent in the interaction of the steroidal part of E2B, similar tothose reported previously in other binary and ternary complexes ofnatural steroids, in 17β-HSD1 complexes. In the ternary complex,we observe that the presence of the cofactor results in a twist at theC-21 (methylene group linker), with the adjustment leading to anadditional hydrogen-bonding interaction. The hydrogen-bondingsuggests that the steroidal core of E2B interacts with the substrate-binding site and the phenylamide group interacts with a secondsubsite. The latter is neither the site of the natural substrate nor thatof the cofactor, but an additional space in the active site to interactwith the shifted residues in the loop region. This produces a morecompact occupation, thereby leaving very little space unoccupiedat the active site. 17β-HSD1 is known to obey a Bi Bi mechanism,by which it binds to the cofactor first followed by the substrate,hence a desirable candidate inhibitor should be non-competitivewith respect to the cofactor [49]. A hybrid inhibitor having boththe substrate and cofactor parts, such as EM-1745, can competewith the substrate as well as the cofactor cores which is not thecase with E2B.
From the ternary and binary structures, the following propertiesof E2B can be seen: (i) lower B-factor of E2B in the ternarycomplex structure than in the binary; (ii) unique electron densityfor the single conformation at the benzylamide of E2B in theternary complex compared with the binary complex which showstwo possible conformations; (iii) additional hydrogen bond at thesteroidal core and the benzylamide ring with the enzyme in theternary complex; and (iv) the non-competitive nature of E2B withthe cofactor, as seen from the ternary structure, renders E2B apotential candidate as an inhibitor of 17β-HSD1 in breast cancercells and maybe also in animal models. This compound needs tobe explored further for the development of additional 17β-HSD1inhibitors in order to advance adjuvant therapy for breast cancer
c© The Authors Journal compilation c© 2009 Biochemical Society

Strong inhibitor binding leading to defined loop structure 365
along with SERMs (selective ER modulators), by suppressing theE2 accumulation.
AUTHOR CONTRIBUTION
Mausumi Mazumdar carried out the experiments including crystallization, data collectionand solution, kinetic studies and wrote the paper, Diane Fournier took part in the kineticstudies, and Dao-Wei Zhu helped in protein purification. The inhibitor was designed andsynthesized by Christine Cadot and Donald Poirier. Sheng-Xiang Lin designed/supervisedthe project and wrote/revised the paper.
ACKNOWLEDGEMENTS
We thank Dr Muriel Kelly for the excellent pre-submission editing service.
FUNDING
We acknowledge the support of Canadian Institutes of Health Research to S.-X.L. [grantnumber MOP57892].
REFERENCES
1 Strickler, R. C. and Tobias, B. (1980) Estradiol 17β-dehydrogenase and20α-hydroxysteroid dehydrogenase from human placental cytosol: one enzyme with twoactivities? Steroids 36, 243–253
2 Engel, L. L. and Groman, E. V. (1974) Human placental 17β-estradiol dehydrogenase:characterization and structural studies. Recent Prog. Horm. Res. 30, 139–169
3 Dumont, M., Luu-The, V., de Launoit, Y. and Labrie, F. (1992) Expression of human17β-hydroxysteroid dehydrogenase in mammalian cells. J. Steroid Biochem. Mol. Biol.41, 605–608
4 Moeller, G. and Adamski, J. (2009) Integrated view on 17β-hydroxysteroiddehydrogenases. Mol. Cell. Endocrinol. 301, 7–19
5 Jin, J. Z. and Lin, S. X. (1999) Human estrogenic 17β-hydroxysteroid dehydrogenase:predominance of estrone reduction and its induction by NADPH. Biochem. Biophys. Res.Commun. 259, 489–493
6 Yang, F., Zhu, D. W., Wang, J. Y. and Lin, S. X. (1992) Rapid purification yielding highlyactive 17β-hydroxysteroid dehydrogenase: application of hydrophobic interaction andaffinity fast protein liquid chromatography. J. Chromatogr. 582, 71–76
7 Oppermann, U., Salim, S., Hult, M., Eissner, G. and Jornvall, H. (1999) Regulatory factorsand motifs in SDR enzymes. Adv. Exp. Med. Biol. 463, 365–371
8 Oppermann, U., Filling, C., Hult, M., Shafqat, N., Wu, X., Lindh, M., Shafqat, J., Nordling,E., Kallberg, Y., Persson, B. and Jornvall, H. (2003) Short-chaindehydrogenases/reductases (SDR): the 2002 update. Chem. Biol. Interact. 143–144,247–253
9 Persson, B., Kallberg, Y., Oppermann, U. and Jornvall, H. (2003) Coenzyme-basedfunctional assignments of short-chain dehydrogenases/reductases (SDRs). Chem. Biol.Interact. 143–144, 271–278
10 Baker, M. E. (2001) Evolution of 17β-hydroxysteroid dehydrogenases and their role inandrogen, estrogen and retinoid action. Mol. Cell. Endocrinol. 171, 211–215
11 Ghosh, D., Pletnev, V. Z., Zhu, D. W., Wawrzak, Z., Duax, W. L., Pangborn, W., Labrie, F.and Lin, S. X. (1995) Structure of human estrogenic 17β-hydroxysteroid dehydrogenaseat 2.20 A resolution. Structure 3, 503–513
12 Luu-The, V. (2001) Analysis and characteristics of multiple types of human17β-hydroxysteroid dehydrogenase. J. Steroid Biochem. Mol. Biol. 76, 143–51
13 Huang, Y.-W., Pineau, I., Chang, H.-J., Azzi, A., Bellemare, V., Laberge, S. and Lin, S.-X.(2001) Critical residues for specificity toward NADH or NADPH in human estrogenic17β-hydroxysteroid dehydrogenase: site-directed mutagenesis designed from thethree-dimensional structure of the enzyme. Mol. Endocrinol. 15, 2010–20
14 Gunnarsson, C., Olsson, B. M. and Stal, O. (2001) Abnormal expression of17β-hydroxysteroid dehydrogenases in breast cancer predicts late recurrence.Cancer Res. 61, 8448–8451
15 Vihko, P., Herrala, A., Harkonen, P., Isomaa, V., Kaija, H., Kurkela, R. and Pulkka, A.(2006) Control of cell proliferation by steroids: the role of 17HSDs. Mol. Cell. Endocrinol.248, 141–148
16 Suzuki, T., Moriya, T., Ariga, N., Kaneko, C., Kanazawa, M. and Sasano, H. (2000)17β-hydroxysteroid dehydrogenase type 1 and type 2 in human breast carcinoma:a correlation to clinicopathological parameters. Br. J. Cancer 82, 518–523
17 Miyoshi, Y., Ando, A., Shiba, E., Taguchi, T., Tamaki, Y. and Nogushi, S. (2001)Involvement of up-regulation of 17β-hydroxysteroid dehydrogenase type 1 inmaintenance of intratumoral high estradiol levels in postmenopausal breast cancers.Int. J. Cancer 94, 685–689
18 Pasqualini, J. R., Cortes-Prieto, J., Chetrite, G., Talbi, M. and Ruiz, A. (1997)Concentrations of estrone, estradiol and their sulfates, and evaluation of sulfatase andaromatase activities in patients with breast fibroadenoma. Int. J. Cancer 70, 639–643
19 Oduwole, O. O., Li, Y., Isomaa, V. V., Mantyniemi, A., Pulkka, A. E., Soini, Y. and Vihko,P. T. (2004) 17β-Hydroxysteroid dehydrogenase type 1 is an independent prognosticmarker in breast cancer. Cancer Res. 64, 7604–7609
20 Vihko, P., Harkonen, P., Soronen, P., Torn, S., Herrala, A., Kurkela, R., Pulkka, A.,Oduwole, O. and Isomaa, V. (2004) 17β-Hydroxysteroid dehydrogenases: their role inpathophysiology. Mol. Cell. Endocrinol. 215, 83–88
21 Day, J. M., Tutill, H. J., Newman, S. P., Purohit, A., Lawrence, H. R., Vicker, N., Potter,B. V. and Reed, M. J. (2006) 17β-Hydroxysteroid dehydrogenase Type 1 and Type 2:association between mRNA expression and activity in cell lines. Mol. Cell. Endocrinol.248, 246–249
22 Vihko, R. and Apter, D. (1989) Endogenous steroids in the pathophysiology of breastcancer. Crit. Rev. Oncol. Hematol. 9, 1–16
23 Purohit, A., Tutill, H. J., Day, J. M., Chander, S. K., Lawrence, H. R., Allan, G. M., Fischer,D. S., Vicker, N., Newman, S. P., Potter, B. V. and Reed, M. J. (2006) The regulation andinhibition of 17β-Hydroxysteroid dehydrogenase in breast cancer. Mol. Cell. Endocrinol.248, 199–203
24 Sasano, H., Suzuki, T., Nakata, T. and Moriya, T. (2006) New development inIntracrinology of breast carcinoma. Breast Cancer 13, 129–136
25 Sasano, H., Suzuki, T., Miki, Y. and Moriya, T. (2008) Intracrinology of estrogens andandrogens in breast carcinoma. J. Steroid Biochem. Mol. Biol. 108, 181–185
26 Langer, L. J. and Engel, L. L. (1958) Human placental estradiol-17β dehydrogenase.I. Concentration, characterization and assay. J. Biol. Chem. 233, 583–588
27 Zhu, D. W., Lee, X., Labrie, F. and Lin, S. X. (1994) Crystal growth of human estrogenic17β-hydroxysteroid dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 50,550–555
28 Azzi, A., Rehse, P. H., Zhu, D. W., Campbell, R. L., Labrie, F. and Lin, S. X. (1996) Crystalstructure of human estrogenic 17β-hydroxysteroid dehydrogenase complexed with17β-estradiol. Nat. Struct. Biol. 3, 665–668
29 Puranen, T., Poutanen, M., Ghosh, D., Vihko, R. and Vihko, P. (1997) Origin of substratespecificity of human and rat 17β-hydroxysteroid dehydrogenase type 1, using chimericenzymes and site-directed substitutions. Endocrinology 138, 3532–3539
30 Breton, R., Housset, D., Mazza, C. and Fontecilla-Camps, J. C. (1996) The structure of acomplex of human 17β-hydroxysteroid dehydrogenase with estradiol and NADP+
identifies two principal targets for the design of inhibitors. Structure 4, 905–91531 Lin, S. X., Zhu, D. W., Azzi, A., Campbell, R. L., Breton, R., Labrie, F., Ghosh, D., Pletnev,
V., Duax, W. L. and Pangborn, W. (1996) Studies on the three-dimensional structure ofestrogenic 17β-hydroxysteroid dehydrogenase. J. Endocrinol. 150 (Suppl.),S13–S20
32 Sawicki, M. W., Erman, M., Puranen, T., Vihko, P. and Ghosh, D. (1999) Structure of theternary complex of human 17β-hydroxysteroid dehydrogenase type 1 with3-hydroxyestra-1,3,5,7-tetraen-17-one (equilin) and NADP+. Proc. Natl. Acad. Sci.U.S.A. 96, 840–845
33 Lin, S. X., Han, Q., Azzi, A., Zhu, D., Gangloff, A. and Campbell, R. L. (2000) 3D-structureof human estrogenic 17β-HSD1: binding with various steroids. J. Steroid Biochem. Mol.Biol. 73, 183
34 Gangloff, A., Shi, R., Nahoum, V. and Lin, S. X. (2003) Pseudo-symmetry of C19 steroids,alternative binding orientations, and multispecificity in human estrogenic17β-hydroxysteroid dehydrogenase. FASEB J. 17, 274–276
35 Shi, R. and Lin, S. X. (2004) Cofactor hydrogen bonding onto the protein main chain isconserved in the short chain dehydrogenase/reductase family and contributes tonicotinamide orientation. J. Biol. Chem. 279, 16778–16785
36 Qiu, W., Campbell, R. L., Gangloff, A., Dupuis, P., Boivin, R. P., Tremblay, M. R., Poirier,D. and Lin, S. X. (2002) A concerted, rational design of type 1 17β-hydroxysteroiddehydrogenase inhibitors: estradiol–adenosine hybrids with high affinity. FASEB J. 16,1829–1831
37 Mazza, C., Breton, R., Housset, D. and Fontecilla-Camps, J. C. (1998) Unusual chargestabilization of NADP+ in 17β-hydroxysteroid dehydrogenase. J. Biol. Chem. 273,8145–8152
38 Poirier, D. (2003) Inhibitors of 17β-hydroxysteroid dehydrogenases. Curr. Med. Chem.10, 453–477
39 Poirier, D., Boivin, R. P., Tremblay, M. R., Berube, M., Qiu, W. and Lin, S. X. (2005)Estradiol–adenosine hybrid compounds designed to inhibit type 1 17β-hydroxysteroiddehydrogenase. J. Med. Chem. 48, 8134–8147
c© The Authors Journal compilation c© 2009 Biochemical Society

366 M. Mazumdar and others
40 Laplante, Y., Cadot, C., Fournier, M. A. and Poirier, D. (2007) Estradiol and estrone C-16derivatives as inhibitors of type 1 17β-hydroxysteroid dehydrogenase: blocking of ER+
breast cancer cell proliferation induced by estrone. Bioorg. Med. Chem. 16,1849–1860
41 Brozic, P., Lanisnik Risner, T. and Gobec, S. (2008) Inhibitors of 17β-hydroxysteroiddehydrogenase type 1. Curr. Med. Chem. 15, 137–150
42 Day, J. M., Foster, P. A., Tutill, H. J., Parsons, M. F., Newman, S. P., Chander, S. K., Allan,G. M., Lawrence, H. R., Vicker, N., Potter, B. V. et al. (2008) 17β-Hydroxysteroiddehydrogenase Type 1, and not Type 12, is a target for endocrine therapy ofhormone-dependent breast cancer. Int. J. Cancer 122, 1931–1940
43 Qiu, W., Zhou, M., Mazumdar, M., Azzi, A., Ghanmi, D., Luu-The, V., Labrie, F. and Lin,S. X. (2007) Structure-based inhibitor design for an enzyme that binds different steroids:a potent inhibitor for human type 5 17β-hydroxysteroid dehydrogenase. J. Biol. Chem.282, 8368–8379
44 Segel, L. A. (1976) Incorporation of receptor kinetics into a model for bacterialchemotaxis. J. Theor. Biol. 57, 23–42
45 Mazumdar, M., Zhou, M., Zhu, D., Azzi, A. and Lin, S.-X. (2007) Crystallogenesis ofsteroid-converting enzymes and their complexes: enzyme-ligand interaction studiesand inhibitor design facilitated by complex structures. Cryst. Growth Des. 7,2206–2212
46 Emsley, P. and Cowtan, K. (2004) Coot: model-building tools for molecular graphics. ActaCrystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132
47 Hunter, C. A., Singh, J. and Thornton, J. M. (1991) π–π interactions: the geometry andenergetics of phenylalanine–phenylalanine interactions in proteins. J. Mol. Biol. 218,837–846
48 Fournier, D., Poirier, D., Mazumdar, M. and Lin, S.-X. (2008) Design and synthesis ofbisubstrate inhibitors of type 1 17β-hydroxysteroid dehydrogenase: overview andperspectives. Eur. J. Med. Chem. 43, 2298–2306
49 Hara, A., Nakayama, T., Nakagawa, M., Inoue, Y., Tanabe, H. and Sawada, H.(1987) Kinetic and stereochemical studies on reaction mechanism of mouseliver 17β-hydroxysteroid dehydrogenases. J. Biochem. (Tokyo) 102,1585–1592
Received 7 July 2009/28 August 2009; accepted 14 September 2009Published on the Internet 7 December 2009, doi:10.1042/BJ20091020
c© The Authors Journal compilation c© 2009 Biochemical Society
Copyright © 2022 FDOKUMEN




















