
Analysis and simulation of frontal affinity chromatography of proteins
Upload
khangminh22Category
view
0download
0
Bicyclic RGD peptides: Novel high-affinity ligands forselective integrin-binding and integrin-mediated cell
adhesion
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTHAachen University zur Erlangung des akademischen Grades eines Doktors der
Naturwissenschaften genehmigte Dissertation
vorgelegt von
Dominik Bernhagen M.Sc.aus
Lippstadt
Berichter: Univ.-Prof. Dr. rer. nat. Martin MöllerProf. Dr. Peter Timmerman
Tag der mündlichen Prüfung: 18. Oktober 2019
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek verfügbar.


Your future hasn’t been written yet – noone’s has. Your future is whatever you make it, so make it a good one.
Dr. Emmett Lathrop (“Doc”) Brown


Für meine Familie.


Summary
In this work, bicyclic Arg-Gly-Asp (RGD) peptides are established as novel se-lective, high-affinity integrin ligands, which can be applied for membrane-integrinvisualization and integrin-mediated cell adhesion in 2D and 3D polymer matrices.For the affinity-based screening and selection of hundreds of potential RGD-bicyclesfor their binding to integrins αvβ3, αvβ5 and α5β1, a fast and cost-efficient ELISAmethod was developed applying a high-affinity, biotinylated cysteine-knot RGD pep-tide (knottin-RGD) as a benchmark ligand.Screening and optimization of integrin αvβ3-, αvβ5- and α5β1-affinity and -selecti-vity of RGD-bicycle libraries, comprising the RGD motif in one loop and a randomXXX motif in the second, yielded three high-affinity and high-selectivity αvβ3-binders (CT3HPQcT3RGDcT3,CT3HPQCT3RGDcT3,CT3HSQCT3RGDcT3; IC50:30-42 nM), one medium-affinity and non-selective αvβ5-binding peptide (CT3RGD-cT3NWaCT3; IC50: 650 nM) and three high-affinity and medium-selectivity α5β1-binding peptides (CT3RGDcT3AY(D-Leu)CT3, CT3RGDcT3AWGCT3, CT3RGD-cT3AYaCT3; IC50: 90-173 nM). Selected αvβ3- and α5β1-binders were further char-acterized via surface plasmon resonance-enhanced fluorescence spectroscopy (SPFS)and 2D NMR spectroscopy.Membrane integrin staining with fluorescently labeled RGD-bicycles, analyzed viaconfocal microscopy, revealed high staining levels for adipose-derived stem cellswhen applying the α5β1-selective bicycle CT3RGDcT3AWGCT3, while most ef-ficient HT29 cell membrane staining was observed for the αvβ3-selective bicycleCT3HPQcT3RGDcT3.Moreover, αvβ3- and α5β1-selective bicycles were covalently conjugated to elastin-like recombinamer (ELR) polymers and polyisocyanopeptide hydrogels (PIC) in or-der to investigate the 2D and 3D cell adhesion and proliferation properties of thesematerials. Both αvβ3- and α5β1-selective bicycles promoted HUVEC adhesion andgrowth on 2D ELR surfaces with much higher efficiencies than linear benchmarkGRGDS, and with at least equal or even higher efficiencies as compared to mono-
I

cyclic (cyclo-[KRGDf]) and knottin-RGD benchmark peptides. In 3D PIC hydrogels,the α5β1-selective bicycle CT3RGDcT3AWGCT3 promoted superior cell adhesionand the formation of numerous protrusions already after one day as compared tohydrogels functionalized with linear, monocyclic and knottin-RGD benchmarks.The overall results of this work reveal that bicyclic RGD peptides represent an en-tirely novel and valuable platform with high potential for the development of newcell integrin biomarkers as well as cell adhesion-promoting compounds.
II

Zusammenfassung
In dieser Arbeit werden bizyklische Arg-Gly-Asp (RGD)-Peptide als neuartige se-lektive und hochaffine Integrin-Liganden beschrieben, die für die Visualisierung vonMembran-Integrinen und Integrin-vermittelte Zelladhäsion in 2D und 3D Polymer-matrices verwendet werden können. Für das affinitätsbasierte Screening und dieAuswahl hunderter potenzieller RGD-Bizyklen gemäß ihrer Bindung an Integrinαvβ3, αvβ5 und α5β1 wurde eine schnelle und kosteneffiziente ELISA-Methodeentwickelt, die ein hochaffines, biotinyliertes und gefaltetes Peptid (‘knottin-RGD’)als Benchmark-Liganden verwendet.Nach Screening und Optimierung der αvβ3-, αvβ5- und α5β1-Affinität und -Selekti-vität der bizyklischen RGD-Peptid-Bibliotheken, die das RGD-Motif in einem Ringund ein willkürliches XXX-Motif in dem zweiten Ring aufweisen, wurden drei hoch-affine und hochselektive αvβ3-Liganden (CT3HPQcT3RGDcT3, CT3HPQCT3RG-DcT3, CT3HSQCT3RGDcT3; IC50: 30-42 nM), ein semi-affines und nicht selektivesαvβ5-bindendes Peptid (CT3RGDcT3NWaCT3; IC50: 650 nM) und drei hochaffineund semi-selektive α5β1-Liganden (CT3RGDcT3AY(D-Leu)CT3, CT3RGDcT3AW-GCT3, CT3RGDcT3AYaCT3; IC50: 90-173 nM) ermittelt. Weiterhin wurden aus-gewählte αvβ3- und α5β1-bindende Peptide durch Oberflächenplasmonenresonanz-verbesserte Fluoreszenzspektroskopie (SPFS) und 2D NMR Spektroskopie charak-terisiert.Die Integrin-Färbung auf Zellmembranen mithilfe fluoreszenz-modifizierter RGD-Bizyklen, analysiert durch konfokale Mikroskopie, offenbarte hohe Färbungslevelsfür “Adipose-derived” Stammzellen bei Anwendung des α5β1-selektiven bizyklischenPeptids CT3RGDcT3AWGCT3, wohingegen die effizienteste Färbung von HT29-Zellmembranen bei Anwendung des αvβ3-selektiven Bizyklus CT3HPQcT3RGDcT3
beobachtet wurde.Ebenso wurden αvβ3- und α5β1-selektive Bizyklen kovalent an Elastin-ähnlicheRekombinamere (ELR) und Polyisocyanopeptid (PIC)-Hydrogele gebunden, umdie 2D- und 3D-Zelladhäsion und -proliferation dieser Materialien zu untersuchen.
III

Sowohl αvβ3- als auch α5β1-selektive Bizyklen förderten Adhäsion und Proliferationvon Endothelzellen (HUVEC) auf 2D-ELR-Oberflächen mit deutlich höherer Effek-tivität als die lineare Benchmark GRGDS und mit mindestens gleichwertiger odersogar höherer Effektivität verglichen mit den monozyklischen bzw. ‘knottin-RGD’Benchmarks. In 3D-PIC-Hydrogelen förderte das α5β1-selektive bizyklische PeptidCT3RGDcT3AWGCT3 schon nach einem Tag die Zelladhäsion und die Formationzahlreicher Auswüchse in überlegener Art und Weise verglichen mit den Hydroge-len, die mit linearen, monozyklischen ([RGDfK]) oder ‘knottin-RGD’-Benchmarksfunktionalisiert wurden.Insgesamt deuten die Ergebnisse dieser Arbeit darauf hin, dass bizyklische RGD-Peptide eine gänzlich neuartige und nützliche Plattform darstellen, die ein hohesPotenzial sowohl für die Entwicklung neuer Biomarker für Zellintegrin-Rezeptorenals auch für Zelladhäsion-fördernde Komponenten aufweist.
IV

Samenvatting
In dit werk worden bicyclische Arg-Gly-Asp (RGD) peptiden geïntroduceerd alsnieuwe selectieve en hoog-affiene liganden voor integrines, die kunnen worden toege-past voor visualisatie van integrine-gemedieerde hechting aan celmembranen in 2D-and 3D-polymeermatrices. Om de binding van honderden potentiële RGD-bicyclesaan de integrines αvβ3-, αvβ5- en α5β1 te bepalen, werd een snelle en kostenbe-sparende ELISA-methode ontwikkeld door toepassing van een hoog-affiene, gebi-otinyleerde en gevouwen RGD-peptide (‘knottin-RGD’) als referentie ligand.De screening en optimalisatie van de affiniteit en selectiviteit van honderden RGD-bicycles, bestaande uit het RGD-motief in één lus en een random XXX-motiefin de tweede, voor de integrines αvβ3-, αvβ5- en α5β1 leverde drie hoog-affieneen zeer selectieve αvβ3-binders op (CT3HPQcT3RGDcT3, CT3HPQCT3RGDcT3,CT3HSQCT3RGDcT3; IC50: 30-42 nM), een medium-affiene en niet selectieve αvβ5-bindende peptide (CT3RGD-cT3NWaCT3; IC50: 650 nM) en drie hoog-affiene enmedium-selectieve α5β1-binders (CT3RGDcT3AY(D-Leu)CT3, CT3RGDcT3AWG-CT3, CT3RGDcT3AYaCT3; IC50: 90-173 nM). De geselecteerde αvβ3- en α5β1-binders werden verder gekarakteriseerd door middel van Surface Plasmon Resonantie-versterkte Fluorescentie Spectroskopie (SPFS) en 2D NMR spectroskopie.De kleuring van integrine-receptoren in celmembranen werd gemeten met behulpvan fluorescent-gelabelde RGD-bicycles en geanalyseerd met behulp van confocalemicroskopie. Dit onthulde hoge kleurniveaus voor Adipose-derived stamcellen bijtoepassing van de α5β1-selectieve bicycle CT3RGDcT3AWGCT3, terwijl de meestefficiënte kleuring van HT29 cellen werd waargenomen voor de αvβ3-selectieve bi-cycle CT3HPQcT3RGDcT3.Bovendien werden αvβ3- en α5β1-selectieve bicycles covalent geconjugeerd aanelastine-like recombinamers (ELR) en polyisocyanopeptide-hydrogelen (PIC) om de2D en 3D cel adhesie en proliferatie van deze materialen te onderzoeken. Zowelαvβ3- en α5β1-selectieve bicycles bevorderden de adhesie en groei van endotheelcellen (HUVEC) op 2D ELR oppervlakken met veel hogere effectiviteiten dan het
V

lineaire (GRGDS) referentie peptide en in vergelijkbare mate als de monocyclis-che ([RGDfK]) en de ‘knottin-RGD’ referentie peptiden. In de 3D PIC hydrogelenbevorderde de α5β1-selectieve bicycle CT3R-GDcT3AWGCT3 snelle stamceladhesieen superieure vorming van uitstulpingen al naar één dag in vergelijking met lineaire,monocyclische en ‘knottin-RGD’ peptiden. De algehele resultaten van dit werk on-thullen dat bicyclische RGD-peptiden een nieuw en waardevol platform vormen meteen enorme potentie voor de ontwikkeling van nieuwe celintegrine biomarkers evenalsceladhesiebevorderende verbindingen.
VI

Contents1. General Introduction 1
2. An Overview of Integrin-Targeting Molecules and their Application inBiomaterials 72.1. Peptides and peptidomimetics as high-affinity protein binders . . . . 8
2.1.1. Advantages and limitations of natural peptides . . . . . . . . . 82.1.2. Peptide antagonists vs. peptide agonists . . . . . . . . . . . . 102.1.3. How peptidomimetics overcome the therapeutic limitations of
natural peptides . . . . . . . . . . . . . . . . . . . . . . . . . . 102.1.4. Multicyclic peptides and the CLIPS platform . . . . . . . . . 14
2.2. Integrins and the role of Arg-Gly-Asp (RGD) as an integrin-bindingECM protein mimic . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.2.1. Integrin structure, activation and the formation of focal adhe-
sions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.2.2. The four groups of integrins . . . . . . . . . . . . . . . . . . . 192.2.3. Expression and role of RGD-binding integrins . . . . . . . . . 192.2.4. RGD peptides: ECM protein mimics with broad integrin se-
lectivities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 222.2.5. Methods for measuring RGD peptide-integrin interactions . . 24
2.3. Applications of RGD peptides in biomaterials . . . . . . . . . . . . . 252.3.1. Nature-inspired polymers and hydrogels . . . . . . . . . . . . 262.3.2. Synthetic polymers and hydrogels . . . . . . . . . . . . . . . . 292.3.3. Inorganic materials . . . . . . . . . . . . . . . . . . . . . . . . 32
2.4. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3. Development of an ELISA Setup for the Detection of RGD-binding toVarious Integrins 493.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 503.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 503.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
3.3.1. Preliminary experiments . . . . . . . . . . . . . . . . . . . . . 523.3.2. Binding of knottin-RGD peptide to integrin αvβ3 . . . . . . . 523.3.3. Role of bivalent cations on integrin binding . . . . . . . . . . . 533.3.4. Role of spacer length between Biotin and RGD moiety . . . . 553.3.5. Binding of knottin-RGD peptide to integrins αvβ5, and α5β1 573.3.6. Role of detection tag . . . . . . . . . . . . . . . . . . . . . . . 573.3.7. Binding of cyclo-[RGD] peptides to integrins αvβ3, αvβ5, and
α5β1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 583.3.8. Determination of IC50 values via competition ELISA . . . . . 60
VII

Contents
3.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 623.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 633.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 673.7. Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
4. Screening for High Affinity and Selectivity Bicyclic RGD-Binders toIntegrin αvβ3 734.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 744.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 744.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
4.3.1. General procedure for library screening . . . . . . . . . . . . . 764.3.2. Design & synthesis of RGD peptide libraries . . . . . . . . . . 774.3.3. Screening for αvβ3-binding peptides . . . . . . . . . . . . . . 804.3.4. Amino acid replacement analysis for cysteines . . . . . . . . . 834.3.5. Screening of control single-loop peptides . . . . . . . . . . . . 844.3.6. Testing streptavidin-binding of ‘HPQ’-containing bicycles . . . 854.3.7. Determination of affinity binding constants (Kd) . . . . . . . . 864.3.8. Selectivity experiments (ELISA) . . . . . . . . . . . . . . . . . 884.3.9. Conformational analysis of αvβ3-binding bicycles . . . . . . . 894.3.10. Binding ELISA studies on biotinylated αvβ3-binding RGD
bicycles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 924.3.11. Trimerization of RGD peptides and its effect on αvβ3-affinity 93
4.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 954.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 964.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1014.7. Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
5. Screening for High Affinity and Selectivity Bicyclic RGD-Binders toIntegrins α5β1 and αvβ5 1115.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1125.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1125.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
5.3.1. Design & synthesis of randomized RGD-peptide libraries . . . 1145.3.2. General procedure for library screening . . . . . . . . . . . . . 1155.3.3. Screening for α5β1-binding peptides . . . . . . . . . . . . . . 1155.3.4. α5β1-binding of single-loop variants . . . . . . . . . . . . . . . 1195.3.5. Ala-replacement study for selected α5β1-binders . . . . . . . . 1195.3.6. Replacement of the non-RGD loop of 2nd generation α5β1-
binders with non-canonical amino acids . . . . . . . . . . . . . 1215.3.7. Determination of affinity binding constants (Kd) . . . . . . . . 1245.3.8. Selectivity experiments (ELISA) . . . . . . . . . . . . . . . . . 1245.3.9. Conformational analysis of α5β1-binding bicycles . . . . . . . 1265.3.10. Binding ELISA studies with biotinylated α5β1-binding RGD
bicycles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1295.3.11. Screening for αvβ5-binding peptides . . . . . . . . . . . . . . 1305.3.12. αvβ5-binding of single-loop variants . . . . . . . . . . . . . . 132
VIII

Contents
5.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 1335.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 1345.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1355.7. Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
6. Visualization of Integrin-Binding on Cells using Fluorescently LabeledRGD Bicyclic Peptides 1416.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1426.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1436.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 145
6.3.1. Selection of a fluorescent label . . . . . . . . . . . . . . . . . . 1456.3.2. Synthesis of Cy5-labeled peptides . . . . . . . . . . . . . . . . 1466.3.3. Experimental setup of membrane-labeling experiments . . . . 1466.3.4. HT29 cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1476.3.5. HeLa cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1526.3.6. Adipose-derived stem cells . . . . . . . . . . . . . . . . . . . . 1566.3.7. Inhibition properties of linker-modified and Cy5-labeled bi-
cyclic peptides . . . . . . . . . . . . . . . . . . . . . . . . . . . 1606.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 1636.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 1646.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
7. Synthesis and Biological Evaluation of ELR Surfaces Modified withIntegrin αvβ3- and α5β1-Selective RGD-Bicycles 1697.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1707.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1717.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
7.3.1. Selection of RGD peptides and cyclooctyne conjugation . . . . 1737.3.2. Synthesis of RGD peptide-functionalized ELRs . . . . . . . . . 1737.3.3. Time-dependent proliferation studies . . . . . . . . . . . . . . 1767.3.4. Morphology studies . . . . . . . . . . . . . . . . . . . . . . . . 183
7.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 1857.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 1867.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1897.7. Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 190
8. In Vitro Evaluation of Mechanical Properties of PolyisocyanopeptideHydrogels Modified with Integrin αvβ3- and α5β1-Selective RGD-Bicycles 1918.1. Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1928.2. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1928.3. Results & discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 194
8.3.1. Rheological characterization . . . . . . . . . . . . . . . . . . . 1948.3.2. Imaging of hydrogel-cell scaffolds . . . . . . . . . . . . . . . . 1978.3.3. Proliferation (WST-1) assay . . . . . . . . . . . . . . . . . . . 200
8.4. Conclusion & outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
IX

Contents
8.5. Materials & methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 2028.6. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 205
A. Supplementary Information 207A.1. Absorbance data (OD405 values) for all library screenings for αvβ3-
binders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 207A.1.1. First generation screening of 672 random-diversity peptides . . 207A.1.2. Second generation screening of 260 peptides . . . . . . . . . . 215A.1.3. Third generation screening of 199 peptides . . . . . . . . . . . 218
A.2. Absorbance data (OD405 values) for all library screenings for α5β1-binders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221A.2.1. First generation screening of 672 random-diversity peptides . . 221A.2.2. Second generation screening of 197 peptides . . . . . . . . . . 228
A.3. Absorbance data (OD405 values) for all library screenings for αvβ5-binders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 231A.3.1. First generation screening of 672 random-diversity peptides . . 231A.3.2. Second generation screening of 198 peptides . . . . . . . . . . 238
B. List of abbreviations 241
C. Acknowledgements 247
X

1
1. General Introduction

1 The development of peptides that bind with high affinity and selectivity to aparticular protein target, or interfere with a particular protein-protein interactionrepresents a novel area of therapeutic research. Protein-protein interactions (PPIs),the total number of which amounts to approximately 650,000 in humans,[1] are vitalprocesses to living systems. In the late 20th century, the increasing knowledge aboutPPIs has led to the development of therapeutic peptides, such as desmopressin, acyclic nonapeptidic derivative of endogenous hormone vasopressin that is used forthe treatment of nocturnal enuresis,[2] or atosiban, a cyclic nonapeptidic inhibitorof oxytocin that is used to delay birth.[3] Very recently, the discovery of the PD-1–PD-L1 interaction, which plays a vital role in T cell regulation and immunity wasone of the seminal discoveries of cancer therapies by inhibition of negative immuneregulation, for which James P. Allison and Tasuku Honjo were awarded the NobelPrize in Physiology or Medicine in 2018.[4, 5]
The integrin–extracellular matrix protein (ECM protein) interactions, which arecrucial for cell adhesion, migration and proliferation within the extracellular ma-trix, provides another interesting example of PPIs, which is the central topic of thisthesis.[6, 7] Integrins are a group of 24 αβ heterodimeric transmembrane proteinsthat mediate the cell attachment to the extracellular matrix. In 1984, Erkki Ru-oslahti, a Finnish-born cell biologist, discovered that the minimum binding motif, bywhich the ECM protein vitronectin binds to cellular integrins is the tripeptide Arg-Gly-Asp (RGD).[8, 9] Interestingly, it was found that various other ECM proteinscomprise RGD as their cellular recognition site, for example, vitronectin, fibrino-gen, collagens, and von Willebrand factor (vWF).[10] Remarkably, ECM proteinsbind to 8 out of the 24 different integrins via the RGD sequence. Well-known ex-amples of ECM protein-integrin interactions are the vitronectin-integrin αvβ3-,[11]
the fibronectin-α5β1-,[12] and the fibrinogen-αvβ3 interaction.[13]
In addition to applications in the therapeutics field, the importance of PPIs has alsobeen acknowledged in the field of tissue engineering, more precisely, in the mimicryof extracellular matrix proteins for tissue regeneration.[14] When encapsulated inECM-mimicking materials, for example, soft hydrogels, ECM proteins can improvecellular adhesion and proliferation, and the tissue regeneration properties of therespective material.[14] However, the application of complete proteins has variousdrawbacks, for example, the risks of denaturation and immune reactivity,[15] butalso the synthesis costs, and relatively few options for synthetic modifications. Thisprompted researchers to investigate if small peptides with molecular weights up to
2

15 kDa could replace proteins of up to 100 kDa in a PPI.The idea is to identify a relatively small peptide sequence in a protein that is primar-ily responsible for its interaction with the other protein. This peptide sequence canthen be optimized in terms of structure and flexibility, for example, using chemicalconstraining techniques, in order to further increase its binding affinity. A famousexample is the abovementioned Arg-Gly-Asp (RGD) peptide, the minimum integrin-binding motif from ECM proteins fibronectin and vitronectin.[10] Since its discovery,the integrin affinity and selectivity of RGD has continuously been optimized, forexample, by chemical cyclization[16] or peptide engineering.[17, 18] Until today, RGDis applied in numerous variants of biomaterials to improve their cellular adhesionand proliferation properties.[19, 20]
Cyclic peptides are accessible, for example, via backbone-cyclization of the C- andN-termini of the linear precursor peptide. The incorporation of two or more cyclicmotifs, however, is more challenging and can be performed, for example, via selectivebridging of multiple intraloop lysine and glutamine residues,[21] or the formation ofmultiple intramolecular disulfide bonds via cysteine residues.[17, 18] In 2005, Timmer-man and coworkers reported a very efficient approach, applying the trivalent scaffold1,3,5-tris(bromomethyl)benzene to convert cysteine-containing peptides into bicyclicpeptides under very mild conditions and with very high yields of up to 100%.[22]
Using this technology, brought to the market as CLIPS (Chemical Linkage of Pep-tides onto Scaffolds), various high-affinity and high-selectivity protein binders wereidentified. For example, Heinis and Winter combined the CLIPS platform with thephage-display technology in order to identify bicyclic, nanomolar inhibitors of humanplasma kallikrein[23] and human urokinase-type plasminogen activator (uPA).[24]
The purpose of the work described in this thesis was to bring the development ofpotent RGD-based ECM-mimics to the next level by introducing bicyclic CLIPS [22]
RGD peptides as a novel family of integrin-specific peptides, and establish thesemolecules as selective in vitro integrin-markers and promotors of integrin-mediatedcell adhesion and proliferation in biomaterials.In the theoretical introduction (Chapter 2) three main topics will be approached.In the first section, peptides and peptidomimetics as high-affinity protein binderswill be discussed. In the second section, integrins and the role of the RGD motifas an integrin-binding extracellular matrix (ECM) mimic will be elucidated. In thefinal section, an overview of the applications of RGD in nature-inspired, syntheticand inorganic biomaterials will be given.
3

1 In Chapter 3, the development of a competitive ELISA setup based on a high-affinity,biotinylated knottin-RGD peptide is described, which can be applied to screen hun-dreds of peptides for affinity to three different integrins that play an important rolein the mediation of cell adhesion in the ECM, i.e. integrins αvβ3, αvβ5 and α5β1.In Chapter 4, the high-throughput screening of partly-randomized bicyclic RGDpeptide libraries (672 compounds) and the identification of three high-affinity andhigh-selectivity integrin αvβ3-binders is described, the affinities of which are com-parable to that of the high-affinity benchmark knottin-RGD.The high binding affinities and selectivities for integrin αvβ3 as observed for theRGD-bicycles initiated the idea for affinity-screening of the peptide libraries alsoto integrins α5β1 and αvβ5. In Chapter 5, the identification of a single medium-affinity αvβ5-binding bicycle, and three high-affinity α5β1-binders is described, oneof which showing an even higher α5β1-affinity than the knottin-RGD benchmark.In Chapter 6, the in vitro integrin staining properties of selected fluorescently la-beled, integrin-selective RGD-bicycles towards three cell lines comprising differentintegrin expression patterns (HT29 carcinoma cells, HeLa cells, adipose-derived stemcells) are discussed. This work identified one of the αvβ3-selective bicycles as a veryefficient HT29 membrane integrin binder.The final two chapters approach the question, whether integrin-selective RGD-bicycles have potential as efficient mediators of cell adhesion and proliferation in2D and 3D polymer networks. In Chapter 7, investigations on endothelial celladhesion and proliferation on elastin-like recombinamer (ELR) surfaces, function-alized with αvβ3- and α5β1-selective RGD-bicycles, are described, suggesting theαvβ3-selective bicycles to promote HUVEC adhesion and proliferation even moreefficiently than monocyclic and linear RGD benchmarks.In Chapter 8, the evaluation of mechanical properties (rheology) and 3D adipose-derived stem cell adhesion properties of soft polyisocyanopeptide (PIC) hydrogels,also functionalized with the exact same selection of RGD-bicycles, is described in-dicating that α5β1-selective bicycles are able to significantly improve the stem celladhesion and growth properties in soft hydrogels.Overall, this thesis should convince the reader that bicyclic RGD peptides representa novel family of selective integrin binders and cell adhesion mediators with highpotential for applications in integrin-mediated therapeutics as well as in materialsfor tissue engineering.
4

1References[1] M. P. H. Stumpf, T. Thorne, E. D. Silva, et al., Proc. Natl. Acad. Sci. USA
2008, 105, 6959–6964.[2] A. Rembratt, C. Graugaard-Jensen, T. Senderovitz, et al., Eur. J. Clin. Phar-
macol. 2004, 60, 397–402.[3] P. Vlieghe, V. Lisowski, J. Martinez, et al., Drug Discov. Today 2010, 15,
40–56.[4] L. M. Francisco, P. T. Sage, A. H. Sharpe, Immnunol. Rev. 2010, 236, 219–
242.[5] C. K. Wang, D. J. Craik, Biopolymers 2016, 106, 901–909.[6] S. K. Akiyama, K. Olden, M. Yamada, Cancer Metast. Rev. 1995, 14, 173–
189.[7] Y. Takada, X. Ye, S. Simon, Genome Biol. 2007, 8, 215.[8] M. D. Pierschbacher, E. Ruoslahti, Proc. Natl. Acad. Sci. USA 1984, 81,
5985–5988.[9] M. D. Pierschbacher, E. Ruoslahti, Nature 1984, 309, 30–33.[10] E. Ruoslahti, M. D. Pierschbacher, Science 1987, 238, 491–497.[11] R. Pytela, M. D. Pierschbacher, E. Ruoslahti, Proc. Natl. Acad. Sci. USA
1985, 82, 5766–5770.[12] R. Pytela, M. D. Pierschbacher, E. Ruoslahti, Cell 1985, 40, 191–198.[13] J. Gailit, C. Clarke, D. Newman, et al., Exp. Cell. Res. 1997, 126, 118–126.[14] J. J. Rice, M. M. Martino, L. De Laporte, et al., Adv. Healthc. Mater. 2013,
2, 57–71.[15] S. L. Bellis, Biomaterials 2011, 32, 4205–4210.[16] M. A. Dechantsreiter, E. Planker, B. Matha, et al., J. Med. Chem. 1999, 42,
3033–3040.[17] R. H. Kimura, A. M. Levin, F. V. Cochran, et al., Proteins 2009, 77, 359–
369.[18] J. W. Kim, F. V. Cochran, J. R. Cochran, J. Am. Chem. Soc. 2015, 137,
6–9.[19] U. Hersel, C. Dahmen, H. Kessler, Biomaterials 2003, 24, 4385–4415.[20] M. Ø. Dalheim, J. Vanacker, M. A. Najmi, et al., Biomaterials 2016, 80,
146–156.[21] M. Bartoloni, X. Jin, M. J. Marcaida, et al., Chem. Sci. 2015, 6, 5473–5490.[22] P. Timmerman, J. Beld, W. C. Puijk, et al., ChemBioChem 2005, 6, 821–
824.
5

1 [23] C. Heinis, T. Rutherford, S. Freund, et al., Nat. Chem. Biol. 2009, 5, 502–507.
[24] A. Angelini, L. Cendron, S. Chen, et al., ACS Chem. Biol. 2012, 7, 817–821.
6
222. An Overview of Integrin-Targeting
Molecules and their Application inBiomaterials

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
The first section of this chapter discusses the use, advantages and limitationsof peptides as protein binders in general. Following this, an introduction to (RGD-binding) integrin receptors, integrin structure and activation, and the wide spectrumof RGD peptides will be presented. The final section of this chapter focuses on theapplications of RGD peptides in nature-inspired, synthetic and inorganic bioma-terials. It should be noted that this chapter provides selected examples from theabovementioned fields of research and does not cover the entire published body ofliterature that is available on these topics. The single-letter amino acid codes are be-ing used (unless stated otherwise), whereby the L-amino acids are written in capitalletters, and D-amino acids in small letters.
2.1. Peptides and peptidomimetics as high-affinityprotein binders
Peptides have found application in various fields in science, such as therapeutics,diagnostics and biomaterials. The great success of peptides is largely owed to thedevelopment of a technically-feasible and cost-efficient method for their productionvia solid-phase synthesis as described by Robert B. Merrifield in 1963,[1] for whichhe was awarded the Nobel Prize in Chemistry in 1984,[2] and which nowadays isthe preferred method for synthesis of oligo- and polypeptides. Numerous naturaland non-natural amino acids, either alone or in combination with various protectivegroups, can be incorporated to design peptides with a broad range of different prop-erties.
2.1.1. Advantages and limitations of natural peptides
The majority of therapeutic drugs belongs to either the group of small moleculedrugs (<500 Da), or to that of the recombinant proteins (>5000 Da), also referredto as ‘biologicals’. Naturally occurring peptides have the potential to combine thestability and bioavailability of small molecule drugs with that of the high bindingaffinities and specificities observed for protein drugs, thereby representing a novelpowerful platform for therapeutic intervention.[3] In order to assess the therapeuticpotential of nature-derived (small) peptides, it is important to evaluate the benefitsand limitations as compared to small molecules and biologicals.
8

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
Advantages/limitations as compared to small molecules
(Small) peptides have several benefits over small molecules when applied therapeuti-cally. Due to their natural occurrence, their application is safer,[4, 5] and they usuallyexhibit much higher binding selectivities to their targets, which generally results inmore efficient in vivo target binding[3] and in turn translates into considerably im-proved safety and tolerability.[6] Moreover, the degradation products of peptides –proteinogenic amino acids – are non-toxic.[4]
One of the major limitations of (small) peptides is their short half-lives due to fastliver and renal clearance, and, more importantly, proteolytic degradation in thedigestive system and the blood plasma.[5, 7]∗ This is the reason for their low oralbioavailability, precluding their application as orally available drugs.[8] The shorthalf-lives also represent one of the reasons why peptides are more costly than smallmolecule drugs.[4] Another disadvantage is their low membrane permeability, whichespecially presents a hurdle for peptides inducing their effect within the centralnervous system, or in the brain.[9] Moreover, some peptides have a tendency toaggregate, which can lead to poor water solubility.[10]
Advantages/limitations as compared to biologicals
In general, peptides are considerably less immunogenic than biologicals, such as re-combinant proteins or monoclonal antibodies.[11] And because of their smaller size,they are able to penetrate into solid tissues (e.g. tumor tissue) with much higherefficiency.[4] Moreover, they comprise much better long-term stabilities as comparedto e.g. antibodies.[4] Finally, their chemical synthesis is generally considered to bemuch cheaper as compared to the recombinant production of proteins.[3, 7]
One limitation of (small) peptides is that they cannot mimic the complex 3D con-formation of biologicals, such as therapeutic monoclonal antibodies, which usuallyhave unprecedented target affinities and specificities.[12] Moreover, the conforma-tional flexibility of (small) peptides can result in a lack of selectivity and thereforeto side effects.[4]
The abovementioned limitations towards small molecules and biologicals often pre-clude the use of peptides as therapeutics. However, there are a number of modifi-cations that can improve the performance of peptides as therapeutics.
∗A list of human proteolytic enzymes involved in peptide degradation is depicted in reference[7].
9

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
2.1.2. Peptide antagonists vs. peptide agonists
Natural and synthetic peptides can be applied as antagonists (also referred to as in-hibitors) or as agonists (also referred to as activators). Antagonists bind to a proteinto block the receptor for binding of a (natural) ligand, while agonists bind to andactivate a protein. Carriero et al. reported the N -acetylated tetrapeptide RERFto be an active inhibitor of urokinase-type plasminogen activator receptor (uPAR),a cell membrane receptor that promotes tumor metastasis.[13] Another protein tar-get involved in cancer development, i.e. glucose-regulated protein 78 (GRP78),[14, 15]
was targeted with a chimeric peptide combining the GRP78-selective sequence WIF-PWIQL with the pro-apoptotic klaklakklaklak sequence, suppressing tumor growthin mouse prostate and breast cancer.[16] More recently, it was shown in preclin-ical metastasis models that this chimeric peptide inhibits primary tumor growthand reduces outgrowth of lung and bone micrometastases, prolonging their overallsurvival.[17]
Peptides as activators find application in e.g. biosensors, i.e. molecules comprisingboth a target-binding sequence, and a transducer unit that transmits analytical in-formation via e.g. a fluorescent tag.[18, 19] It is a different form of activation, i.e. thepeptides’ transducer unit (and not a particular protein) is activated upon bindingof the target protein to the peptides’ target-binding sequence. In this way, Saps-ford et al. developed a sensor for HIV-1 specific monoclonal antibodies, comprising(from N- to C-terminus) a Cy3-labeled antibody recognition motif C(Cy3)EKIRLR,an α-helical spacer SGLG{Aib}AAAWGG (Aib: α-aminoisobutyric acid), and aHis tag HHHHHH that allows for affinity purification.[20] More recently, the peptideQHIMHLPHINTL was found to bind with submicromolar affinity to the 6H-P2 pro-teins of the norovirus, and might therefore serve as a lead for developing biosensorsthat detect gastroenteritis.[21]
2.1.3. How peptidomimetics overcome the therapeuticlimitations of natural peptides
The term “peptidomimetics” covers a range of different approaches to create struc-tural and/or functional derivatives of peptides that overcome the limitations de-scribed in section 2.1.1, and therefore make them more suitable for the use as ther-apeutics. Pelay-Gimeno et al. recently suggested to subdivide peptidomimetics intofour classes.[22] Class A represents peptides basically containing α-amino acids with
10

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
slight backbone or side chain modifications. Class B covers peptides comprising sev-eral side chain or backbone modifications, as well as foldamers and peptoids (videinfra). In contrast to this, classes C and D involve small molecules, representingstructural mimetics with similar 3D orientation as the original peptide (class C), ormimetics that mimic the mode of action without a link to the original peptide 3Dorientation (class D).[22] Since the mimetics in classes C and D are less relevant forthis overview, the discussions in the next subsections will focus on class A and Bpeptidomimetics.
Peptoids and N-methylated peptides
Peptoids, which are N -functionalized oligomers of glycine,[23] are proteolytically sta-ble peptide derivatives that have been widely studied as novel therapeutics[24] as wellas structural mimics of antimicrobial peptides.[25]
N -methylated peptides can be applied in order to improve the bioavailability of apeptide, for example in cyclosporine A (Figure 2.1), an inhibitor of calcineurin usedto treat several diseases and to prevent rejections during organ transplants.[26–28]
Selective N -methylation can result in higher target affinities, for example, as for thehigh-affinity integrin αvβ3/αvβ5-binder cilengitide (cyclo-[V(N -Me)RGDf], Fig-ure 2.1), which was tested but failed in phase III clinical studies for the treatmentof glioblastomas.[29, 30] Moreover, N -methylation potentially improves the oral avail-ability of cyclic peptide drugs.[31] Furthermore, it is an effective tool to preventpeptide aggregation by disrupting inter- and intra-molecular hydrogen bonding.[32]
For a comprehensive summary on the synthesis and perspectives of N -methylatedpeptides, the reader is kindly referred to a review by Chatterjee et al.[32]
Use of D- and non-natural amino acids
Peptides containing D-amino acids represent another platform for future therapeu-tics. For example, Welch et al. developed D-amino acid peptides that bind withhigh affinity (IC50: 250 pM) to the gp41 N-trimer pocket region of HIV-1, thereforerepresenting potentially promising leads for HIV/AIDS therapy.[37] The applicationof D-amino acid substitutions can also prevent the proteolysis of peptides as shown,for example, in the abovementioned gp41-specific peptide,[37] as well as for the hu-man granulysin-derived antimicrobial peptide RVCRTGRSRWR.[38] Furthermore,the application of D-amino acids can improve the stability of peptides as well as the
11

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
Figure 2.1.: Chemical structures of cyclic, N -methylated peptides cyclosporineA,[26] cilengitide,[29] and teixobactin,[33], cyclic desmopressin[34, 35] andlipidated peptide human ghrelin.[36]
affinity towards protein targets. For example, the replacement of glycine with D-serine in a bicyclic peptide inhibitor of cancer-related protease urokinase-type plas-minogen activator (uPA) led to a four-fold improvement in stability and a 1.75-foldimprovement in activity.[39] Moreover, the abovementioned high-affinity integrin-binder cilengitide exhibits a D-phenylalanine (f) residue that is not only importantfor the binding conformation of the RGD sequence, but is also essential for integrinbinding since it interacts with a tyrosine residue on the β-subunit.[32, 40] Anotherexample of improved target affinities is the N -acetylated, tumor-suppressing pep-tide FM123E4L (1: α-aminoisobutyric acid, 2: 4-phosphonomethylphenylalanine,3: 6-chloro-L,D-tryptophan, 4: 1-amino-cyclopropanecarboxylic acid, IC50: 5 nM)reported by García-Echeverría et al., binding with an approximate 60-fold higheraffinity to its target protein Hdm2 as compared to the non-optimized precursor
12

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
MPRFMRYWEGLN (IC50: 313 nM).[41]
Lipidation
Lipidated peptides represent a potentially powerful platform for drug leads.[42, 43]
In particular, lipidation can be applied to i) increase membrane-permeability forefficient cargo-transport into cells,[44] and ii) increase the metabolic stability ofpeptides.[43] For example, the endogeneous, appetite-regulating peptide hormoneghrelin (28 aa, Figure 2.1) exhibits an O-n-octanoylated serine residue that is es-sential for activity,[36] and allows the peptide to cross the blood-brain barrier.[45]
An example of the increased stability of lipidated peptides is the palmitoylatedanalogue of stromal cell-derived factor-1 α (SDF1 α, 70 aa), which showed an al-most seven-fold increase in life-time in porcine liver as compared to the naturalvariant.[46] Furthermore, lipidated peptides showed improved plasma stabilities overthe non-lipidated variants. For example, the caprylated WMRF amide (derivedfrom gastrin) showed an approx. 300-fold increased half-life as compared to thenon-caprylated variant.[47] In another degradation study, the lipidated variant of an-tiproliferative somatostatin derivative fCoxYwKVCoxW (Cox indicate cysteines aspart of a disulfide bond) was still detectable after 24 hours, while the non-lipidatedvariant was entirely degraded within 2 hours.[48] Moreover, Amon et al. showed thatT-cell antigen receptor-targeting peptides (core peptide: GLRILLLKV) exhibiteddramatically improved in vitro and in vivo function when functionalized with atleast two palmitoyl chains, while exhibiting cytotoxicity when functionalized withonly one palmitoyl chain.[49] These findings exemplify that for potential drug deliv-ery applications, it needs to be carefully verified (and guaranteed) that lipidationdoes not increase the cytotoxicity under any circumstances.
Cyclization
Cyclic peptides represent an attractive format for therapeutics, since they usuallyprovide high target binding affinities and selectivities, together with improved pro-teolytic stabilities and low toxicities.[50] They are accessible via head-to-tail-, head-to-side chain-, side chain-to-side chain-, and side chain-to-tail-cyclization of lin-ear precursor peptides.[51] One of the first cyclic peptide inhibitors was the 10-merGACoxRGDCoxLGA that, when applied in sub-µM concentrations, inhibited bind-ing of fibronectin to α5β1 integrin, a transmembrane protein involved in tumor
13

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
development (see Section 2.2.3, p.19).[52] Recently, Ling et al. reported the very ef-fective antibiotic teixobactin (Figure 2.1, p.12), a cyclized and N -methylated 11-merdepsipeptide discovered during screening of uncultured bacteria, and comprising ac-tivity towards a broad range of pathogenic micro-organisms (Figure 2.1).[33] Rinket al. synthesized a thioether-bridged analog of decapeptide luteinizing hormonerelease hormone (LHRH) that showed much higher proteolytic stability than thenatural hormone LHRH.[53]
Cyclic peptides also provide a potentially powerful platform for orally bioavailabledrugs, the development of which focused for a long time on molecules that fulfill Lip-inski’s “rule-of-five” (RO5, among others MW <500 Da, H-bond donors ≤5, H-bondacceptors ≤10, log P ≤5).[54, 55] Nowadays, there are numerous examples of cyclicpeptides that are orally absorbed by mammals,[56] covering a range from 4-mer pep-tides (for example, potential anti-atherosclerotic beauveriolides[57]) up to multicyclic>25-mer peptides, such as the kalata B1 derivative ckb-kal for inflammatory paintreatment.[58] Another example of a cyclic peptide drug is desmopressin (Figure 2.1,p.12), a derivative of vasopressin that is used to treat nocturnal enuresis.[34, 35]
2.1.4. Multicyclic peptides and the CLIPS platform
Multicyclic peptides
Multicyclic peptides have recently raised considerable interest as novel peptidetherapeutics.[59, 60] Established multicyclic peptide-based drugs are, for example,the nature-derived multicyclic peptides romidepsin (bicyclic) and vancomycin (tri-cyclic), as well as semisynthetic multicycles such as dalbavancin.[3, 61, 62]
There are various routes to generate multicyclic peptides. The formation of multiple,intramolecular disulfide bonds via cysteine oxidation is straightforward, however,the resulting products are prone to reduction, as can be initiated e.g. by variousenzymes.[63, 64] Another route to peptide multicycles is to crosslink the side chainof natural amino acids in backbone-cyclized peptides. For example, Bartoloni et al.recently reported the conversion of monocyclic into bicyclic peptides via bridgingof an intraloop lysine and glutamine residue with alanine.[65] These norbornapep-tides† showed high proteolytic stability in serum, one of which bound messenger pro-tein calmodulin with high affinity.[65] Another variant is the conversion of disulfide
†The expression “norbornapeptide” refers to the structural topology of the bicyclic alkane‘norbonane’.
14

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
bridges into more stable methylene thioacetals using methylene iodide as performedfor the neurotoxic bicyclic peptide α-conotoxin.[66] Multicylic peptides containingolefin-, ether-, biaryl-, imidazole- and triazole crosslinks (click chemistry) as well asthioether linkages (CLIPS chemistry) have also been reported.[67]
The CLIPS platform
In 1981, Kemp and McNamara reported a tandem ligation-cyclization of the cysteine-containing peptide cyclo-[GCGGCGGCG] applying 1,3,5-tris(bromomethyl)benzene,the yields of which, however, were very poor and the product entirely insoluble.[68]
More than 20 years later, Timmerman et al. significantly optimized the experi-mental conditions by converting linear Cys-containing peptides to bicyclic peptidesunder very mild conditions (room temperature, <1 hour, pH 8) and with very highconversions of up to 100% (for the formation of monocyclic and bicyclic CLIPS pep-tides, see Figure 2.2A).[69] Using this technology – brought to market under the name‘CLIPS ’ (Chemical Linkage of Peptides onto Scaffolds) – it was not only possible tosynthesize a novel group of bicyclic peptides (via trivalent (bromomethyl)benzenescaffolds), but also monocyclic and tricyclic peptides using bivalent and tetravalent(bromomethyl)benzene scaffolds, respectively.[70–73] Another benefit of this technol-ogy involves its compatibility with phage library screening[71] and ribosomal peptidesynthesis.[74]
The development of novel, versatile CLIPS scaffolds for the formation of multicyclicpeptides is still ongoing. Very recently, the synthesis of isomerically pure tricyclicpeptides was reported via application of scaffolds that combine the CLIPS tech-nology with Cu-assisted azide-alkyne cycloaddition (CuAAC), one of the so-called‘click’ reactions.[75] An overview of available CLIPS scaffolds is shown in Figure 2.2B.CLIPS scaffolds significantly control the conformation and activity of a peptide. Forexample, bivalent bromomethyl scaffolds have been applied to stabilize α-helical in-hibitors of the cysteine protease calpain.[72] In another study, it was shown that theactivity of plasma kallikrein inhibitor ACT3SDRFRNCT3PADEALCT3G‡ (IC50:2.82 nM) dropped by a factor of >1,000 when the linear peptides were constrainedwith 1,3,5-triacryloyl-1,3,5-triazinane (TATA, IC50: 3.61 µM) or N,N’,N”-(benzene-1,3,5-triyl)tris(2-bromoacetamide) (TBAB, IC50: 4.92 µM) instead.[76]
Applications of CLIPS peptides also cover cell penetrating peptides (CPPs),[73] pro-‡CT3: Cysteine residues that are constrained via trivalent scaffold 1,3,5-
tris(bromomethyl)benzene (Figure 2.2B).
15

22
2.1. PEPTIDES AND PEPTIDOMIMETICS AS HIGH-AFFINITY PROTEIN BINDERS
tein inhibitors,[71] and protein mimics of the iron-regulating hormone hepcidin.[77]
Figure 2.2.: (A) Formation of a monocyclic CLIPS peptide (example by Timmer-man et al.[69]), and a bicylic CLIPS peptide (example by Heinis etal.[71]) via bi- and trivalent bromomethyl benzene scaffolds, respec-tively; (B) Chemical structures of selected CLIPS scaffolds to createmono-, bi- or tricyclic CLIPS peptides. The CLIPS -CuAAC scaffold,reported by Richelle et al., can be used to synthesize isomerically-puretricycles when the linear precursor peptide comprises two cysteine andtwo azidohomoalanine residues.[75]
16

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
2.2. Integrins and the role of Arg-Gly-Asp (RGD) asan integrin-binding ECM protein mimic
2.2.1. Integrin structure, activation and the formation of focaladhesions
Structure of integrins
Integrins represent a large group of 24 αβ heterodimeric transmembrane proteinsthat mediate intercellular interactions and cell attachment to the extracellular ma-trix (ECM). They comprise large extracellular α (~1000 amino acids) and β (~750amino acids) domains, and short C-terminal cytoplasmic tails (~ 30-40 amino acids,except β4, ~1000).[78, 79] The α subunit determines the specificity of the integrinreceptor.[80] Starting at the N-terminus, it consists of a seven-bladed β-propellerdomain, followed by a thigh, a calf-1 and calf-2 domain, a transmembrane domainand a cytoplasmic domain (Figure 2.3A).[81] The three (or four) C-terminal bladesof the β-propeller domain exhibit a binding site for Ca2+, by which binding ofother ligands is allosterically affected.[80] Nine of the 18 integrin α subunits (α1,α10, α11, α2, αL, αM, αX, αD, αE) comprise an additional inserted domain (αIdomain, ~200 amino acids), which is the exclusive, or at least major binding sitein integrins containing this domain.[81] Notably, binding of ligands is realized viacoordination of a Mg2+ ion in the metal-ion-dependent adhesion site (MIDAS). Incontrast, the β subunit connects the integrin to the cytoskeleton and affects vari-ous signaling pathways.[80] Starting at the N-terminus, the β subunit is composedof a plexin-semaphorin-integrin (PSI) domain, a hybrid domain, a β1 domain, andfour epidermal growth factor (EGF) repeats, followed by a β tail, a transmembranedomain and a cytoplasmic domain (Figure 2.3A).[81] The β1 domain is involved inligand binding for integrins that do not exhibit an αI domain.[80] As for the αI do-main, the β1 domain also comprises a MIDAS, and additionally an adjacent region(ADMIDAS) that binds an inhibitory Ca2+ ion. Via binding of an Mn2+ ion to theADMIDAS, a conformational change is induced that leads to the activated formof that particular integrin.[80] For more detailed information on the structure andmechanics of integrins, the reader is kindly referred to excellent reviews by Arnaoutet al.[82] and Campbell et al.[83]
17

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
Integrin activation
Integrin activation, a bidirectional process characterized by outside-in and inside-out signaling cascades, involves several proteins and enzymes such as talin, paxilin,vinculin, FAK and p130CAS.[84, 85] During this process, the integrin and its ligandsadopt different conformations. In the inactivated state, the integrin exists in a bentconformation, while the extracellular ligand fibronectin is present in a globular shape(Figure 2.3B). Integrin activation leads to a more extended conformation, and is ei-ther triggered by binding of talin to the intracellular β-tail (inside-out signaling)or by binding of extracellular fibronectin to the integrin head piece (outside-in sig-naling). Subsequently, applied force results in extension of fibronectin and lateralpolymerization into fibers, while mechanical stress applied on talin leads to bridg-ing of the ECM and the actin-myosin cytoskeleton, which opens vinculin bindingsites.[86, 87] Thereby, the integrin changes conformation to a tensioned state.
Figure 2.3.: (A) Schematic representation of the general structure of integrins,reprint from [80]; (B) Illustration of integrin activation as a functionof intracellular (talin) or extracellular (ECM-proteins) signals. Theillustration was created on the basis of other illustrations that werereported in references [87–89].
Focal adhesions
Focal adhesions are protein complexes mediating connection between a cell and itssubstrate (e.g. ECM or biomaterial) via anchoring of the actin filaments (stressfibers).[90] They are formed upon extracellular ligand binding to integrins leadingto integrin clustering and subsequent migration of adhesome proteins towards thecell adhesion site.[91] Subsequent to talin activation, a weak talin-integrin interaction
18

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
leads to the formation of focal complexes followed by mechanically-controlled open-ing of talin rod domain binding sites for other proteins, e.g. vinculin.[87] It has tobe noted that the talin-integrin interaction represents only one out of approx. 700known interactions during integrin-mediated cell adhesion, involving in total approx.160 different activating, binding and inhibiting components, which are summarizedas the “integrin adhesome”.[85, 90]
2.2.2. The four groups of integrins
The integrins can be subdivided into four groups, i.e. RGD-binding, collagen-binding, laminin-binding and leucocyte-specific integrins (Figure 2.4A).§ The RGD-binding and leucocyte-specific integrins each cover 8 heterodimers, whereas thelaminin-binding and collagen-binding integrins each cover 4 protein complexes. 12out of the 24 integrins comprise a β1 group, and each 4 integrins a β2 and anαv chain. RGD-binding integrins appear to be the most investigated integrins interms of numbers of publications, followed by the laminin-binding integrins (Fig-ure 2.4B). Research focusing on RGD-binding heterodimers αvβ3, α5β1, collagen-binding α2β1 and leucocyte-specific α4β1 cover already > 50% of the publicationssince 1975 (considering the research parameters as defined in Figure 2.4B).The huge interest in RGD-binding integrins can probably not only be ascribed totheir critical role in cell-cell and cell-ECM signaling, but also to their impact on ap-plications such as drug targeting, therapeutics, and biomaterials. In the next section,the expression and role of RGD-binding human integrins will briefly be discussed,followed by an overview of the RGD-integrin binding interactions. Further informa-tion on collagen-binding integrins,[93, 94] laminin-binding[95–97] and leucocyte-specificintegrins[98–100] can be found elsewhere.
2.2.3. Expression and role of RGD-binding integrins
Integrin αIIbβ3
This integrin is overexpressed on circulating platelets where it can switch from an in-activated (OFF) state to an activated (ON) state, resulting in binding to fibrinogen,von Willebrand factor (vWF) and fibronectin, and subsequently in platelet aggrega-tion through crosslinking with fibrinogen.[79, 101, 102] Furthermore, it binds membrane
§Alternatively, Humphries et al. classified the integrins by RGD-binding, LDV-binding, A-domain β1, and non-αA-domain-containing laminin-binding integrins.[92]
19

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
Figure 2.4.: (A) Schematic representation of all 24 αβ integrin receptors, sub-divided into RGD-binding, collagen-binding, laminin-binding andleucocyte-specific integrins; (B) Number of Web of Science search re-sults for each integrin (search term example: “integrin alphaVbeta3”,research period: 1975 – April 28th, 2018).
glycoprotein ICAM-4 (LW blood group glycoprotein), which is considered importantfor cell adhesion and cell interaction events.[103]
Integrin αvβ1
Despite the fact that almost every cell expresses both αv and β1 monomers, onlyfew cell lines express heterodimer αvβ1, e.g. tissue fibroblasts.[104] Typical αvβ1-binding ligands are TGF-β-latency-associated peptide (LAP-TGF-β), fibronectinand osteopontin.[105] It was suggested that this integrin might be an auspicioustarget for treating diseases accompanied by excessive tissue fibrosis.[104]
Integrin αvβ3
Integrin αvβ3 is probably the most investigated heterodimer, since it is expressedin a variety of different tissues and exhibits important functions. For example, itis strongly expressed in glioblastoma,[106, 107] breast cancer cells,[108] blood vesselsin human wound granulation,[109] while minimally expressed on osteoclasts[110] andintestinal, vascular and uterine smooth muscle cells.[111] It plays an essential role intumor-induced angiogenesis (growth of new blood vessels)[109] and promotes breastcancer migration.[112]
20

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
Integrin αvβ3 binds various ligands, such as fibronectin, vitronectin, fibrinogen,vWF and tenascin.[105] Due to the overexpression in cancer tissues, inhibition of thisparticular integrin is considered an important strategy to the development of novelcancer therapeutics. [113, 114]
Integrin αvβ5
This integrin is overexpressed on scleroderma fibroblasts[115] and is also moderatelyexpressed on adipose-derived and bone marrow-derived stem cells.[116] Furthermore,it is expressed in melanoma and CNS (central nervous system) metastases.[117] Simi-lar to integrin αvβ3, this integrin is a receptor for vitronectin, a mediator of cell ad-hesion and spreading,[115] among others, by internalizing conformationally-modifiedvitronectin.[118] Furthermore, it is a receptor for osteopontin.[116] Very often, theaction of αvβ5 is described in alignment with αvβ3.[79, 119, 120] For example, theyboth regulate human cardiac myofibroblast differentiation via activation of TGF-β1,albeit via different pathways.[121]
Integrin αvβ6
Integrin αvβ6 is expressed in a restricted set of epithelial cells, the expression levelsof which are low in healthy tissue, but significantly upregulated during progressionof e.g. epithelial malignancies.[79, 122] Furthermore, αvβ6 is highly expressed in lungadenocarinoma, but could not be detected in e.g. melanoma.[117] This integrin bindsligands such as fibronectin, osteopontin and LAP-TGF-β.[105, 123] Moreover, it pro-motes migration of keratinocytes by binding fibronectin and vitronectin,[124] and itsextracellular and transmembrane domains activate and adhere to TGF-β,[122, 125]
whereas the intracellular β6 subunit is suggested to be a mediator of cell prolifera-tion and MMP production.[126, 127]
Integrin αvβ8
This heterodimer is strongly expressed in brain tumors[117, 128] and as a fibrin recep-tor on Schwann cells.[129] It is further expressed on rat and mouse primary astrocytesand promotes their migration.[130] It functions as an activator of TGF-β, a cytokinewith multiple functions in homeostasis, through coordinated interactions with met-alloproteases (MMPs).[131] In murine intestinal dendritic cells, it activates TGF-β
21

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
in order to prevent gut inflammation.[132] Moreover, it binds and mediates adhesionto LAP-TGF-β.[133]
Integrin α5β1
This integrin is highly expressed on adipose-derived and bone marrow-derived stemcells[116] and is a regulator of angiogenesis.[134] Moreover, it was reported that α5β1controls αvβ3 during in vitro migration and in vivo angiogenesis,[135] and that itincreases the invasiveness of breast cancer cells.[136] It efficiently mediates fibronectinfibrillogenesis,[137] but also binds osteopontin, fibrillin and thrombospondin.[105]
Integrin α8β1
As compared to other integrins (e.g. αvβ3 or α5β1) this αβ heterodimer is notthoroughly investigated yet. It is particularly expressed in kidney and mesenchymalstem cells adjacent to epithelial cells.[138] It binds tenascin, osteopontin, fibronectin,vitronectin, nephronectin and LAP-TGF-β.[105, 139, 140]
2.2.4. RGD peptides: ECM protein mimics with broad integrinselectivities
More than 30 years ago, Pierschbacher and Ruoslahti found that the tripeptideArg-Gly-Asp (RGD) forms the core of the cell recognition motif in the ECM pro-tein fibronectin,[141] and that it is also present in various other ECM components,such as vitronectin, osteopontin, collagens, fibrinogen, thromospondin and vWF.[142]
Through binding to cell surface integrins, RGD either promotes cell adhesion andprovides traction for migration when immobilized onto a surface, or it inhibits celladhesion when present in solution.[142, 143] Linear RGD peptides of different lengthshave been applied as fibronectin/vitronectin mimics, including RGD, RGDS, GRGD,GRGDS, GRGDSP, GRGDNP, GRGDTP and GRGDSPK.[144] All these peptidesexhibit similar, but very low affinities to integrins αvβ3, αvβ5 and α5β1.[144] Lin-ear RGD peptides such as GRGDS are accessible via standard solid-phase synthe-sis approaches, however, they lack integrin selectivity and are prone to proteolyticdegradation (Table 2.1). A major contribution to the improvement of integrin se-lectivity and proteolytic stability of RGD peptides has been made by Kessler andcoworkers. Starting in 1991 with the development of cyclic RGD peptides as in-hibitors of cell adhesion to vitronectin,[145] Kessler et al. gradually optimized the
22

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
selectivity and affinity of these peptides for the integrins αvβ3 and α5β1,[146, 147] andeventually developed the potent, but non-selective αvβ3/αvβ5 antagonist cilengi-tide (cyclo-[V(N -Me)RGDf]),[29] which was recently reported to also bind with highaffinity to integrin α5β1.[144] Cyclic peptides such as cyclo-[KRGDf] and cilengitideexhibit significantly higher integrin affinities, and their chemical synthesis (involvinga C-→N-term cyclization step) is relatively straightforward (Table 2.1). Moreover,Kessler et al. also developed RGD-containing cyclic peptides with high selectivitiesfor the integrins αvβ6[148] and α5β1.[149] Based on these cyclic RGD peptides, alsopeptidomimetics with high selectivities for integrins αvβ3[150, 151] and α5β1[152, 153]
were developed. These compounds combine high proteolytic stabilities with higherselectivities as compared to the previously reported cyclic RGD peptides. However,their synthesis is much more complicated and does not allow for easy post-peptide-synthesis modifications (Table 2.1).
Table 2.1.: Qualitative evaluation of previously reported integrin-binding com-pounds in terms of integrin affinity and selectivity, proteolytic stability,and synthetic effort.
Protein engineering is a different approach to develop high affinity RGD-basedintegrin binders. Cochran and coworkers developed a family of high-affinity integrinαvβ3/αvβ5/α5β1-binding ‘cysteine-knot’ (knottin) RGD peptides which are con-sidered great candidates for drug development.[154, 155] Recently, a dimeric versionwas reported to comprise a 3650-fold stronger tumor cell migration and proliferationas compared to cilengitide.[156] It is slightly advantageous that knottin RGD pep-tides can be synthesized both chemically and recombinantly, however, they entirelylack selectivity for any of the integrins αvβ3, αvβ5 and α5β1 (Table 2.1).
23

22
2.2. INTEGRINS AND THE ROLE OF ARG-GLY-ASP (RGD) AS AN INTEGRIN-BINDINGECM PROTEIN MIMIC
Non-RGD-based compounds could also represent an alternative to the common RGDpeptide-based integrin inhibitors. For example, integrin-selective antibodies com-prise both very high affinities and selectivities, but they are costly and cannot easilybe functionalized in a directed manner, for example, in order to modify surfaces forimproved cell adhesion (Table 2.1).
2.2.5. Methods for measuring RGD peptide-integrin interactions
The most commonly applied experimental approaches to access binding affinitiesinvolve ELISA (Enzyme Linked Immunosorbent Assay) techniques.[157] Kessler andco-workers determined affinities of RGD peptides using competition ELISA exper-iments with extracellular matrix (ECM) proteins as competitors, for example, fi-bronectin and vitronectin, either by applying the ECM proteins in solution andsurface-immobilizing the integrin[29, 146, 158] or vice versa.[144, 150, 159–161] In contrast,Piras et al. used biotinylated cyclic RGD peptides to determine integrin αvβ3affinities of RGD peptidomimetics.[162] More recently, the same group analyzed thebinding of bioconjugates of 5-fluoro-5-desoxyribose (FDR) and cyclic RGD peptidesto both immobilized and cellular integrin αvβ3 to conclude that [18F]FDR can beefficiently conjugated to RGD peptides.[163] An alternative method involves the 125I-Echistatin assay, which was applied by Alsibai et al. to determine binding affinitiesof RGD mimics to integrins αvβ3 and αIIbβ3.[164]
Recently, Cho et al. used a high-throughput one-bead–one-compound method toscreen RGD peptides for their binding to αvβ3-expressing human cancer cells. [165]
In order to identify peptides with high-affinity to integrins, microarrays that are suit-able for high-throughput screening and displaying the αvβ3 protein immobilized onchips were developed by Lee et al.[166] Gagnon et al. used a high-throughput in vivoscreening approach to identify potentially new imaging molecules with high affinityand selectivity to integrin αvβ6.[167] Other high-throughput methods involve phagedisplay[168] and yeast display screening, the latter of which led to the selection of thehigh integrin affinity knottin-RGD peptides described in the previous section.[154]
In addition to these indirect methods, there are also various techniques to accessbinding affinities in a direct manner. A relatively straightforward method was re-ported by Wang et al.[169] who applied fluorescence polarization to measure bindingaffinities of cyclic peptides to integrin αvβ3. However, the applied integrin concen-tration of 290 nM (roughly corresponding to 35 µg/mL) in combination with the
24

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
time effort per experiment precludes its use as a cost-effective screening method.Liu et al. investigated binding of various cyclic RGD peptides to integrin αvβ3using a surface plasmon resonance (SPR)-based binding assay.[170] Finally, surfaceplasmon-enhanced fluorescence spectroscopy (SPFS), an extended variant of SPR,has been applied for monitoring binding of fluorescently labeled RGD-based peptidesto integrins αvβ3 and αvβ5.[171]
2.3. Applications of RGD peptides in biomaterials
The selection of materials that are described in the following sections is based on aWeb of Science search on “RGD biomaterials” and considers publications as of 2004.These sections all focus on the most relevant publications in each field, i.e. nature-inspired and ii) synthetic polymers and hydrogels, respectively, and iii) inorganicmaterials. For literature published before 2004 the reader is kindly referred to areview by Hersel et al.[172] An overview of relevant RGD-functionalized biomaterials,as well as chemical structures of selected biomaterials is given in Figure 2.5.
Figure 2.5.: (A) Overview of RGD-functionalized biomimetic materials, divided ininorganic materials, nature-inspired, and synthetic polymers and hy-drogels. Materials discussed in this chapter are highlighted in boldprint; (B) Chemical structures of selected nature-inspired and syn-thetic polymers.
25

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
2.3.1. Nature-inspired polymers and hydrogels
Alginate
Alginate (Figure 2.5B), a natural polymer collected from brown seaweed, can begelated via bivalent cations, and possesses low toxicity and high biocompatibilityin combination with a relatively cost-efficient production.[173, 174] It is of particularinterest to tissue engineering and drug delivery research, and the covalent function-alization with RGD motifs provides an anchor for integrin-mediated cell adhesion.The RGD organization and stiffness of alginate hydrogels controls the cell growthrate.[175] For RGD peptides, it was found that a spacer length of at least four glycineunits is required for improved in vitro fibroblast growth.[176]
As microcapsules, RGD-modified alginates were also investigated as drug deliverysystems.[177, 178] Moreover, various research groups investigated the differentiationof adipose-derived stromal cells and mesenchymal stem cells,[179–181], and differentcell-alginate systems have been reported for cardiac,[182] retinal[183] and bone tissueengineering research.[184, 185] It was also shown that chondrogenesis of bone marrowstromal cells (BMSCs) in GGGGRGDSY-functionalized alginate hydrogels can besignificantly inhibited by soluble GRGDSP.[186]
In addition to RGD, other integrin-binding sequences,[187, 188] or enzymatically cleav-able sequences,[189] were applied to further control biocompatibility.
Silk(protein)
The natural protein silk fibroin produced by wild silkworm or domestic Bombyx moriconsists of recurrent sequences of [GSGAGA]n and comprises great biocompatibil-ity and -degradibility.[190] Silk modification with RGD peptides improved humancornea fibroblast adhesion and proliferation as well as expression of collagens andproteoglycans in the fibroblasts,[191] suggesting a possible strategy for engineeringof human cornea tissue.[192, 193] In a different study, recombinant spider silk proteinsthat were recombinantly modified with a linear RGD peptide (GGSGGRGDSPG)comprised similar or even slightly better mouse fibroblast adhesion than proteinsthat were chemically modified with cyclo-[KRGDf].[194] Widhe et al. presented thecyclic disulfide RGD peptide cyclo[CS-STGRGDSPACS-S] as a turn motif in silkmimicking the integrin α5β1-binding site of fibronectin, and reported improvedkeratinocyte proliferation and migration.[195] Moreover, transgenic modification ofsilk fibroin with RGD peptides (TGRGDSPAS and RGD) or vascular endothelial
26

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
growth factor (VEGF)-binding peptide supported cellularization of human umbilicalendothelial cells (HUVECs).[196]
The weak mechanical properties of regenerated silk fibroin fibers was successfully op-timized by blending with other components,[190, 197] for example, hydroxyapatite.[198]
RGD-modified silk protein was also applied as a graft with titanium and showedstrong adherence of fibroblast cells, suggesting possible applications in implantablemedical devices.[199]
Self-assembling molecules
Molecules comprising hydrophobic and hydrophilic sequences (amphiphiles) undergoself-assembly via a combination of electrostatic interactions, hydrogen bonding andhydrophobic interactions to form physical hydrogels able to support cell growth.[200]
Functionalization of these amphiphiles with RGD sequences significantly influencesthe biological and mechanical behavior of these hydrogels. For example, King et al.described stiff hydrogels (G’ up to 100 kPa) formed by self-assembly of two hexapep-tides, EEFKWKFKEE and KKFEWEFEKK. The addition of RGD-functionalizedEEFKWKFKEE significantly increased the survival and spreading of 3T3 mousefibroblasts.[201] In another study, it was shown that the self-assembly properties ofdifferent RGD sequences (G6KRGDY, A6KRGDY, V6KRGDY) significantly influ-ence the rheological properties of peptide-alginate hybrids.[202] Interesting work byRamakers et al. describes RGD- and diacetylene-containing peptide amphiphilesthat change color upon cell adhesion, which is potentially useful in tracking cell mi-gration in 2D and 3D.[203] Moreover, the biological properties of RGD-functionalizedself-assembling peptides modified with other cell adhesion-promoting sequences werealso investigated. For example, nanofibrous β-sheet peptide hydrogels presentingcyclic RGDS and PHSRN, separated by 3.2 nm, showed upregulation of α5 integrinsubunit after encapsulation of HUVECs.[204] Furthermore, self-assembling peptidescomprising RGD in combination with laminin-derived IKVAV were reported, for ex-ample, in the form of nanofibers that support growth of cardiac fibroblasts,[205] or inthe form of mechanically-tunable, cross-linked hydrogels promoting human neuralstem and progenitor cell (hNSC) differentiation into neurons.[206]
Stephanopoulos and co-workers described the relatively new concept of nanotubesconsisting of co-assemblies of DNA and RGD-functionalized DNA that guide neuralstem cell growth and differentiation into neurons.[207]
27

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
Elastin-like proteins & elastin-like recombinamers
Elastin-like proteins (ELPs) consist of repeating sequences found in elastin,[208] themost commonly observed motif of which is VPGXG (X: any amino acid, exceptP).[209] In aqueous solutions, ELPs undergo a temperature-dependent transition froma soluble state, characterized by fully hydrated random polymer coils (hydrophobichydration), to a self-assembled and phase-separated state, characterized by foldedpolymer chains that have completely lost the ordered water structures.[209] Incorpo-ration of the RGD motif into ELPs was either realized recombinantly or via covalentmodification.[210] When ELPs are produced using recombinant techniques, they areusually referred to as elastin-like recombinamers (ELRs).[211] Soft ELR hydrogels (G’:1–10 kPa) were fabricated by copper-free click reaction of an azide-functionalized,RGD-bearing ELR with a cyclooctyne-functionalized ELR.[212] Recently, hydrogelscross-linked via Staudinger ligation were reported to have potential applications intherapeutic cell delivery and bioprinting.[213]
Furthermore, elastin-like proteins were investigated as surface coatings to improvethe cell adhesion properties of e.g. chitosan,[214] silicon-doped hydroxyapatite[215]
and poly(lactic) acid (PLA).[216]
Chitosan
Chitosan, a de-acetylated derivative of chitin, is a linear polysaccharide consisting ofD-glycosamine residues with a structure comparable to various glycosaminoglycans,structural components of the extracellular matrix.[217] Grafting of the GRGDS pep-tide onto carboxymethyl-functionalized chitosan improved the biocompatibility andresulted in 3-5 times faster human dermal fibroblast cell adhesion.[218] Yang et al.showed that RGD-modified chitosan-poly(methacrylic acid) exhibits excellent HU-VEC adhesion, spreading and proliferation properties.[219] Moreover, RGD-modifiedchitosan scaffolds also showed improved adhesion and proliferation of ATDC5 chon-drocytes.[220] Furthermore, Park et al. described stiff chitosan-pluronic composites(G’ > 100 kPa at 37 ℃) with superior chondrocyte viability over alginate hydrogels,suggesting application in articular cartilage tissue engineering.[221] Chitosan was alsoapplied as surface coating for titanium, leading to improved osteoblast adhesion andslower bacterial growth.[222]
28

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
Hyaluronic acid
Hyaluronic acid (or hyaluronan, HA, Figure 2.5B, p.25) is a nature-derived gly-cosaminoglycan and one of the main components of the extracellular matrix andresponsible, among others, for the release of growth factors.[223, 224] Kim and cowork-ers reported that mesenchymal stem cell proliferation and focal adhesion formation,as well as the mechanical properties of electrospun HA fibers can be controlled bythe density of conjugated RGD peptides (GCGYGRGDSPG).[225] Wang et al. syn-thesized injectable RGD-functionalized HA-hydrogels that supported HUVEC andfibroblast cell growth, and as a result of this, in vitro and in vivo vascularizationafter 2 weeks.[226] Wade et al. described the synthesis of RGD- and fluorescently-patterned HA-hydrogels that allowed controlled 3T3 fibroblast growth,[227] whileTarus et al. observed vertical neurite outgrowth in soft HA-hydrogels (G’ = 400 Pa)as a function of GRGDS peptide density.[228]
In a recent paper, photo-crosslinkable 3D-printed HA hydrogel filaments with long-term mechanical stability (> 1 month) were reported to support 3T3 fibroblastadhesion upon RGD-modification.[229]
Pectin
Pectins are mostly used as jellifying agents in food industry, but were also investi-gated for drug delivery applications.[230] For example, injectable RGD-pectin conju-gates promoted osteogenic differentiation of pre-osteoblasts, while maintaining highviability after 29 days.[230] Another injectable hydrogel that gelates upon reacting ofRGD-functionalized pectin aldehyde groups with hydrazide-functionalized HA wasreported to show controllable mechanical properties, degradation, and excellent cellcompatibility.[231]
2.3.2. Synthetic polymers and hydrogels
Polyethylene glycol (PEG)
Polyethylene glycole (PEG)-based hydrogels are of interest for a broad range of tissueengineering applications due to their tuneable mechanical[232, 233], cell adhesion[234]
and biological[235, 236] properties. The formation of these hydrogels is usually re-alized via crosslinking of PEG derivatives, such as PEG methacrylate,[237] PEGdiacrylate,[238–240] PEG sebacate diacrylate,[241, 242] or PEG vinylsulfone.[243, 244] The
29

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
cell response of PEG hydrogels can be tuned by external stimuli. For example,the aspartic acid (D)-residue in cyclo-[KRGDf]-functionalized PEG hydrogels wasprotected with a UV-cleavable group, which allowed for controlled and non-invasivedeprotection in vivo upon exposure to UV light (350/365 nm, 10 min), thereby con-trolling cell adhesion, inflammation and vascularization of the hydrogel.[236] More-over, injectable PEG hydrogels covalently functionalized with thiol containing GC-GYGRGDSPG were reported to show much better cell viability as compared to thenon-functionalized hydrogels,[245] while starPEG-heparin hydrogels functionalizedwith an RGD peptide (sequence was not reported) promoted fibroblast adhesion,spreading and proliferation as compared to non-modified hydrogels.[246]
As for the nature-inspired hydrogels described earlier, other cell-adhesion promot-ing peptides or proteins can be applied to modify the properties. Combined co-valent functionalization of PEG acrylate hydrogels with a thiolated RGD peptide(GCGGGRGDSPG) and a thiolated, laminin-derived YIGSR peptide (GCGGGYI-GSRG) led to 25% improved endothelial cell migration as compared to hydrogelssolely functionalized with the RGD peptide.[247] Another sequence that was appliedin combination with RGD is the integrin α5β1-binding PHSRN motif. For ex-ample, PEG hydrogels covalently functionalized with the peptide RGDG13PHSRNshowed improved osteoblast adhesion, spreading and formation of focal contactsas compared to hydrogels solely functionalized with the peptide RGDS.[248] In an-other study, PEG hydrogels functionalized with both collagen-binding sequence CK-LERG and integrin-binding sequence CRGDSG via thiol-acrylate reaction promotedchondrogenic differentiation of mesenchymal stem cells to a much higher extent ascompared to hydogels solely functionalized with CRGDSG, indicated, among oth-ers, by a 2.5-fold higher amount of glycosaminoglycan production per cell after14 days.[249] Moreover, PEG hydrogels containing immobilized fibroblast growthfactor (bFGF) were found to migrate towards increasing bFGF concentrations.[250]
More recently, Kolodziej et al. created patterned PEG surfaces with well-definedspatial distributions of covalently-functionalized RGD peptides (Lev-GRGDSPG,Lev: 5-aminolevulinic acid) and non-covalently bound bFGF, resulting in confine-ments of HUVEC focal adhesions to the RGD- and bFGF-modified parts of thesurface.[251] In a different study, VEGF was covalently linked to RGD-PEG gels, butthis did not significantly improve bone regeneration.[252]
Various RGD-modified PEG composites were also reported to promote or mod-ulate cellular responses, for example, in combination with poly(lactic acid),[253–255]
30

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
poly(glycolic acid),[256], poly(glutamic acid),[257] as well as extracellular glycosamino-glycans such as heparin.[224, 246, 258, 259]
Polyethylene terephthalate (PET)
Owing to its biocompatibility and mechanical strength, polyethylene terephthalate(PET, Figure 2.5B, p.25) is broadly used for application in cell-culturing, surgi-cal sutures and cross-band engineering.[260] As for the previous materials, it canbe modified with RGD peptides in order to promote cell adhesion and differen-tiation. For efficient endothelial cell and osteoblast adhesion onto PET surfaces,RGD surface densities of at least 1 pmol/mm2 seem to be required.[261] Further-more, it was shown that other ECM protein-binding sequences, such as YIGSR,REDV or SVVYGLR, were significantly less efficient in mediating cell adhesionthan RGD, whereas PET-functionalization with a combination of RGD peptide andthe pro-angiogenic SVVYGLR peptide resulted in significantly improved migrationas compared with RGD-functionalization alone.[262]
Poly-(lactide-co-glycotide) (PLGA)
Poly-(lactide-co-glycotide) (PLGA, Figure 2.5B, p.25) is one of the most widelyapplied polymers in biomedicine and of particular interest for tissue engineeringpurposes, since lactic acid, the degradation product can be resorbed via metabolicpathways.[263] In order to allow functionalization of PLGA with RGD peptides, ad-ditional functionalities need to be introduced. For example, Gu et al. synthesizedpoly(lactic-co-glycolic acid-co-lysine) (PLGAL), and covalently bound RGD pep-tides via the lysine side-chain of the terpolymer.[264]
PLGA composite materials have recently been investigated for tissue engineeringapplications. For example, porous nanocomposite scaffolds of RGD-modified PLGAand hydroxyapatite promoted osteoblast and 3T3 endothelial cell adhesion and pro-liferation in vitro[265] as well as bone formation in vivo.[265, 266]
Poly(2-hydroxyethyl methacrylate) (PHEMA)/Poly(ethylene glycolmethacrylate) (PEGMA)
Poly(2-hydroxyethyl methacrylate) (PHEMA, Figure 2.5B, p.25) and poly(ethyleneglycol methacrylate) (PEGMA) represent a class of polymers that are used to pre-pare non-fouling polymer surfaces with a lack of cell adhesivity.[267–269] In order to
31

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
induce cell adhesion onto these materials, functionalization with cell-adhesive pep-tides, ECM proteins or growth factors is usually required.[270] This was achieved, forexample, via in situ copolymerization with RGD-containing monomers,[271] or viacopper catalyzed 1,3-dipolar cycloaddition of azide-bearing polymers with alkyne-functionalized RGD peptides.[270] Santander-Borrego et al. reported that the in-corporation of RGD residues into PHEMA based hydrogels significantly improvedepithelial cell adhesion, growth and proliferation, in particular at the lowest pep-tide density tested (~0.14 peptide chains/nm3).[270] More recently, Karimi et al.described fast endothelial cell adhesion and migration in PEGMA-based hydrogelswith exposed nano-clusters of RGD peptides, which is considered important for nextgeneration cardiovascular biomaterials.[272]
2.3.3. Inorganic materials
Titanium alloys
RGD-functionalized titanium alloys have thoroughly been investigated for their useas bone and dental implants. Prior to functionalization with RGD peptides, thesurfaces were physically or chemically treated. Linker molecules such as thiolsor silanes can be applied to support RGD immobilization.[273] For example, sur-faces modified with acrylic acid (plasma treatment) and RGD showed an increasedalkaline phosphatase activity of pre-osteoblasts, suggesting their potential use asosteo-conductive bone implants.[274] Grafting of titanium with chitosan and subse-quent coating with RGD was reported to be a promising route to improve tissueintegration of implants by promoting osteoblast functions as well as reducing bac-terial adhesion.[222] In another study, titanium bone implants functionalized withRGD-containing PEO-nanoparticles showed enhanced pre-osteoblast adhesion andfilopodia-like structure formation among the nanoparticles.[275] It was also shownthat the topography of sandblasted Ti6Al4V disks functionalized with cyclic RGDpeptides particularly supports early osteoblast adhesion and spreading.[276] Recently,titanium implants that were chemically functionalized with RGDS in combinationwith its fibronectin-derived, α5β1-binding synergy motif PHSRN were applied tostimulate human mesenchymal stem cell (hMSC) differentiation in vitro, and thuspromote bone growth in a rat calvarial defect.[277] Furthermore, titanium surfacesfunctionalized with a cyclo-[KRGDf]/heparin-binding peptide conjugate showed im-proved adhesion, proliferation and viability of osteoblast-like cells as compared to
32

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
the surface functionalized with heparin-binding peptide alone.[278]
Gold
Gold surfaces functionalized with RGD peptides are generally part of differentanalaytical techniques, for example, AFM[279, 280] or SPR/SPFS,[171] but are alsocommonly used as model materials to study processes like cell adhesion and prolif-eration. For example, it was reported that application of a negative electric potentialon stimuli-responsive RGD-modified gold surfaces induces a conformational changeof the RGD ligands, which inhibits cell adhesion.[281] Cell adhesion and growth canalso be controlled via formation of ordered or disordered nanopatterns, as shownfor osteoblasts on gold nanoparticles functionalized with cyclo-[KRGDf].[282] Fur-thermore, RGD-functionalized gold nanodots can regulate the spreading and dif-ferentiation of mesenchymal stem cells on PEG hydrogels.[283] Very recently, it wasreported that the RGD density on gold nanoparticles regulates osteogenic and adi-pogenic differentiation of mesenchymal stem cells.[284]
Due to the high costs, gold might not be suitable as an implant material, however,in the future smart hydrogels containing gold nanoparticles could represent a potentplatform for guided cell adhesion, proliferation and growth.
Hydroxyapatite
Hydroxyapatite (HAp),¶ a major component in bone and tooth, has been investi-gated as implant material for bone tissue engineering. When coated with RGD, HApstimulates the adhesion, but not the spreading, of mesenchymal stem cells.[285] Theuse of various sera seems to be more effective for spreading.[286] In 2008, it was re-ported that RGD peptides alone even have a detrimental influence on HAp implantperformance.[287] This issue probably led to the development of hybrid materials thatrecently raised interest as bone tissue regeneration materials. For example, Zhanget al. described porous scaffolds of RGD-conjugated copolymers and HAp surface-grafted with poly(L-lactide) to promote adhesion and growth of 3T3 fibroblasts andosteoblasts as well as in vivo bone formation.[288] More recently, a nanocomposite ofnatural silk protein fibroin (exhibits RGD in the sequence) and HAp, deposited onfibroin scaffolds, was reported to improve the mechanical strength and cytocompat-ibility of these scaffolds for osteoblasts.[289]
¶The common abbreviation for hydroxyapatite is “HA”. In order to prevent ambiguity errorwith hyaluronic acid, “HAp” is chosen as an abbreviation for this thesis.
33

22
2.3. APPLICATIONS OF RGD PEPTIDES IN BIOMATERIALS
It can be concluded that RGD-based peptides are applied in a wide range ofnature-inspired, synthetic and inorganic materials with the aim to control or evenimprove their cell adhesion and proliferation properties. Remarkably, the vast major-ity of literature on RGD-functionalized biomaterials involves non-selective integrin-binding peptides such as GRGDS or cyclo-[KRGDf]. One of the aims of this thesiswas to investigate if more selective integrin-binders (i.e. the bicyclic RGD peptidesthat will be introduced in Chapters 4 and 5) are more efficient promoters of cellularadhesion and proliferation in biomaterials. Within the scope of the thesis, theseinvestigations were performed on elastin-like recombinamers as well as in polyiso-cyanopeptide hydrogels, the results of which will be presented in Chapters 7 and 8,respectively.
34

22
2.4. REFERENCES
2.4. References[1] R. B. Merrifield, J. Am. Chem. Soc. 1963, 85, 2149.[2] NobelPrize.org, The Nobel Prize in Chemistry 1984, https://www.nobel-
prize.org/prizes/chemistry/1984/summary/, 2018.[3] D. J. Craik, D. P. Fairlie, S. Liras, et al., Chem. Biol. Drug Des. 2013, 81,
136–147.[4] W. Pytlik, The growing significance of peptide therapeutics, https://www.ge-
sundheitsindustrie-bw.de/en/article/news/the-growing-significance-of-peptide-therapeutics, 2014.
[5] A. Ellert-Miklaszewska, K. Poleszak, B. Kaminska, Future Med. Chem. 2017,9, 199–221.
[6] K. Fosgerau, T. Hoffmann, Drug Discov. Today 2015, 20, 122–128.[7] P. Vlieghe, V. Lisowski, J. Martinez, et al., Drug Discov. Today 2010, 15,
40–56.[8] L. Diao, B. Meibohm, Clin. Pharmacokinet. 2013, 52, 855–868.[9] K. A. Witt, T. J. Gillespie, J. D. Huber, et al., Peptides 2001, 22, 2329–2343.[10] I. W. Hamley, Angew. Chem. Int. Ed. 2007, 46, 8128–8147.[11] D. P. McGregor, Curr. Opin. Pharmacol. 2008, 8, 616–619.[12] P. Chames, M. van Regenmortel, E. Weiss, et al., Br. J. Pharmacol. 2009,
157, 220–233.[13] M. V. Carriero, I. Longanesi-Cattani, K. Bifulco, et al., Mol. Cancer Ther.
2009, 8, 2708–2717.[14] R. Liu, X. Li, W. Gao, et al., Clin. Cancer Res. 2013, 19, 6802–6811.[15] L. Rasche, J. Duell, C. Morgner, et al., PLoS ONE 2013, 8, e63414.[16] M. A. Arap, J. Lahdenranta, P. J. Mintz, et al., Cancer Cell 2004, 6, 275–
284.[17] Y. R. Miao, B. L. Eckhardt, Y. Cao, et al., Clinical Cancer Research 2013,
19, 2107–2116.[18] Q. Liu, J. Wang, B. J. Boyd, Talanta 2015, 136, 114–127.[19] F. Guida, A. Battisti, I. Gladich, et al., Biosens. Bioelectron. 2018, 100,
298–303.[20] K. E. Sapsford, J. B. Blanco-Canosa, P. E. Dawson, et al., Bioconjugate
Chem. 2010, 21, 393–398.[21] H. J. Hwang, M. Y. Ryu, J. P. Park, RSC Adv. 2015, 5, 55300–55302.[22] M. Pelay-Gimeno, A. Glas, O. Koch, et al., Angew. Chem. Int. Ed. 2015, 54,
8896–8927.
35

22
2.4. REFERENCES
[23] R. J. Simon, R. S. Kania, R. N. Zuckermann, et al., Proc. Natl. Acad. Sci.USA 1992, 89, 9367–9371.
[24] M. T. Dohm, R Kapoor, A. E. Barron, Curr. Pharm. Des. 2011, 17, 2732–2747.
[25] B. Mojsoska, R. N. Zuckermann, H. Jenssen, Antimicrob. Agents Chemother.2015, 59, 4112–4120.
[26] A. Rüegger, M. Kuhn, H. Lichti, et al., Helv. Chim. Acta 1976, 59, 1075–1092.
[27] B. D. Kahan, N. Engl. J. Med. 1989, 321, 1725–1738.[28] S. Gibaud, D. Attivi, Expert Opin. Drug Deliv. 2012, 9, 937–951.[29] M. A. Dechantsreiter, E. Planker, B. Matha, et al., J. Med. Chem. 1999, 42,
3033–3040.[30] R. Stupp, M. E. Hegi, T. Gorlia, et al., Lancet Oncol. 2014, 15, 1100–1108.[31] T. R. White, C. M. Renzelman, A. C. Rand, et al., Nat. Chem. Biol. 2012,
7, 810–817.[32] J. Chatterjee, F. Rechenmacher, H. Kessler, Angew. Chem. Int. Ed. 2013,
52, 254–269.[33] L. L. Ling, T. Schneider, A. J. Peoples, et al., Nature 2015, 517, 455–459.[34] A. Rembratt, C. Graugaard-Jensen, T. Senderovitz, et al., Eur. J. Clin. Phar-
macol. 2004, 60, 397–402.[35] W. L. Robson, A. K. Leung, J. P. Norgaard, J. Urol. 2007, 178, 24–30.[36] M. Kojima, H. Hosoda, Y. Date, et al., Nature 1999, 402, 656–660.[37] B. D. Welch, A. P. VanDemark, A. Heroux, et al., Proc. Natl. Acad. Sci. USA
2007, 104, 16828–16833.[38] K. Hamamoto, Y. Kida, Y. Zhang, et al., Microbiol. Immunol. 2002, 46,
741–749.[39] S. Chen, D. Gfeller, S. A. Buth, et al., ChemBioChem 2013, 14, 1316–1322.[40] L. Marinelli, A. Lavecchia, K.-E. Gottschalk, et al., J. Med. Chem. 2003, 46,
4393–4404.[41] C. Garcia-Echeverria, P. Chene, M. J. Blommers, et al., J. Med. Chem. 2000,
43, 3205–3208.[42] Z. Zhang, Q. Luo, X. Yan, et al., Anal. Chem. 2012, 84, 8946–8951.[43] T. A. Stone, C. M. Deber, Biochim. Biophys. Acta 2017, 1859, 577–585.[44] A. R. Nelson, L. Borland, N. L. Allbritton, et al., Biochemistry 2007, 46,
14771–14781.[45] W. A. Banks, M. Tschöp, S. M. Robinson, et al., J. Pharmacol. Exp. Ther.
2002, 302, 822–827.
36

22
2.4. REFERENCES
[46] K. Bellmann- Sickert, A. G. Beck-Sickinger, ChemMedChem 2011, 6, 193–200.
[47] E Yodoya, K Uemura, T Tenma, et al., J. Pharmacol. Exp. Ther. 1994, 271,1509–1513.
[48] P Dasgupta, R Mukherjee, Br. J. Pharmacol. 2000, 129, 101–109.[49] M. A. Amon, M. Ali, V. Bender, et al., Biochim. Biophys. Acta 2006, 1763,
879–888.[50] A. Zorzi, K. Deyle, C. Heinis, Curr. Opin. Chem. Biol. 2017, 38, 24–29.[51] C. J. White, A. K. Yudin, Nature Chem. 2011, 3, 509–524.[52] E. Koivunen, D. A. Gay, E. Ruoslahti, J. Biol. Chem. 1993, 268, 20205–
20210.[53] R Rink, A Arkema-Meter, I Baudoin, et al., J. Pharmacol. Tox. Met. 2010,
61, 210–218.[54] C. A. Lipinski, F. Lombardo, B. W. Dominy, et al., Adv. Drug Delivery Rev.
1997, 23, 3–25.[55] C. A. Lipinski, Drug Discov. Today 2004, 1, 337–341.[56] D. S. Nielsen, N. E. Shepherd, W. Xu, et al., Chem. Rev. 2017, 117, 8094–
8128.[57] I. Namatame, H. Tomoda, S. Ishibashi, et al., Proc. Natl. Acad. Sci. USA
2004, 101, 737–742.[58] C. T. Wong, D. K. Rowlands, C. H. Wong, et al., Angew. Chem. Int. Ed.
2012, 51, 5620–5624.[59] V. Baeriswyl, C. Heinis, ChemMedChem 2013, 8, 377–384.[60] W. Liu, C. Wu, Chinese Chem. Lett. 2018, in press, DOI 10.1016/j.cclet.
2018.03.015.[61] E. M. Driggers, S. P. Hale, J. Lee, et al., Nature Rev. Drug Discov. 2008, 7,
608–624.[62] C. K. Wang, D. J. Craik, Biopolymers 2016, 106, 901–909.[63] A. Holmgren, M. Bjornstedt in Methods Enzymol. Vol. 252, Academic Press,
1995, pp. 199–208.[64] G. Powis, W. R. Montfort, Annu. Rev. Pharmacol. Toxicol. 2001, 41, 261–
295.[65] M. Bartoloni, X. Jin, M. J. Marcaida, et al., Chem. Sci. 2015, 6, 5473–5490.[66] C. M.B. K. Kourra, N. Cramer, Chem. Sci. 2016, 7, 7007–7012.[67] V. J. Thombare, C. A. Hutton, Peptide Sci. 2018, e24057.[68] D. Kemp, P. McNamara, Tetrahedron Letters 1981, 22, 4571–4574.
37

22
2.4. REFERENCES
[69] P. Timmerman, J. Beld, W. C. Puijk, et al., ChemBioChem 2005, 6, 821–824.
[70] P. Timmerman, W. C. Puijk, R. H. Meloen, J. Mol. Recognit. 2007, 20, 283–299.
[71] C. Heinis, T. Rutherford, S. Freund, et al., Nat. Chem. Biol. 2009, 5, 502–507.
[72] H. Jo, N. Meinhardt, Y. Wu, et al., J. Am. Chem. Soc. 2012, 134, 17704–17713.
[73] R. Wallbrecher, L. Depré, W. P. Verdurmen, et al., Bioconjugate Chem. 2014,25, 955–964.
[74] N. K. Bashiruddin, M. Nagano, H. Suga, Bioorg. Chem. 2015, 61, 45–50.[75] G. J. Richelle, S. Ori, H. Hiemstra, et al., Angew. Chem. Int. Ed. 2018, 57,
501–505.[76] S. Chen, J. Morales-Sanfrutos, A. Angelini, et al., ChemBioChem 2012, 13,
1032–1038.[77] K. Chua, E. Fung, E. D. Micewicz, et al., Bioorg. Med. Chem. Lett. 2015,
25, 4961–4969.[78] M. A. Schwartz, M. D. Schaller, M. H. Ginsberg, Annu. Rev. Cell Dev. Biol.
1995, 11, 549–599.[79] R. O. Hynes, Nat. Med. 2002, 8, 918–921.[80] M. Barczyk, S. Carracedo, D. Gullberg, Cell Tissue Res. 2010, 339, 269–280.[81] B.-H. Luo, C. V. Carman, T. A. Springer, Annu. Rev. Immunol. 2007, 25,
619–647.[82] M. A. Arnaout, S. L. Goodman, J.-P. Xiong, Curr. Opin. Cell Biol. 2007,
19, 495–507.[83] I. D. Campbell, M. J. Humphries, Cold Spring Harb. Perspect. Biol. 2011,
3, a004994.[84] F. G. Giancotti, E. Ruoslahti, Science 1999, 285, 1028–1032.[85] R. Zaidel-Bar, S. Itzkovitz, A. Ma’ayan, et al., Nat. Cell Biol. 2007, 9, 858–
867.[86] G. Baneyx, L. Baugh, V. Vogel, Proc. Natl. Acad. Sci. USA 2002, 99, 5139–
5143.[87] V. P. Hytönen, B. Wehrle-Haller, Exp. Cell Res. 2016, 343, 35–41.[88] S. J. Shattil, C. Kim, M. H. Ginsberg, Nat. Rev. Mol. Cell Biol. 2010, 11,
288–300.[89] H. Hamidi, M. Pietilä, J. Ivaska, Brit. J. Cancer 2016, 115, 1017–1023.
38

22
2.4. REFERENCES
[90] B. Geiger, J. P. Spatz, A. D. Bershadsky, Nat. Rev. Mol. Cell Biol. 2009,10, 21–33.
[91] V. Schaufler, H. Czichos-Medda, V. Hirschfeld-Warnecken, et al., Cell Adhe-sion and Migration 2016, 6918, 1–11.
[92] J. D. Humphries, J. Cell Sci. 2006, 119, 3901–3903.[93] S. Hu, D. Cui, X. Yang, et al., Molecular vision 2011, 17, 1334–42.[94] J. Heino in I Domain Integrins, (Ed.: D. Gullberg), Springer Netherlands,
Dordrecht, 2014, pp. 143–155.[95] R. Nishiuchi, J. Takagi, M. Hayashi, et al., Matrix Biol. 2006, 25, 189–197.[96] C. S. Stipp, Exp. Rev. Mol. Med. 2010, 12, 1–24.[97] V. Ramovs, L. te Molder, A. Sonnenberg, Matrix Biol. 2017, 57-58, 213–243.[98] E. S. Harris, T. M. McIntyre, S. M. Prescott, et al., J. Biol. Chem. 2000,
275, 23409–23412.[99] D. M. Rose, R. Alon, M. H. Ginsberg, Immunol. Rev. 2007, 218, 126–134.[100] I. Mitroulis, V. I. Alexaki, I. Kourtzelis, et al., Pharmacol. Ther. 2015, 14,
123–135.[101] T. E. O’Toole, D. Mandelman, J. Forsyth, et al., Science 1991, 254, 845–847.[102] B. S. Coller, S. J. Shattil, Blood 2010, 112, 3011–3025.[103] P. Hermand, P. Gane, M. Huet, et al., J. Biol. Chem. 2003, 278, 4892–4898.[104] N. I. Reed, H. Jo, C. Chen, et al., Science Trans. Med. 2015, 7, 330–343.[105] Y. Takada, X. Ye, S. Simon, Genome Biol. 2007, 8, 215.[106] C. L. Gladson, D. A. Cheresh, J. Clin. Invest. 1991, 88, 1924–1932.[107] C. L. Gladson, J. Neuropath. Exp. Neur. 1996, 55, 1143–1149.[108] I. Pecheur, O. Peyruchaud, C.-M. Serre, et al., FASEB J. 2002, 16, 1266–
1268.[109] P. C. Brooks, R. A. F. Clark, D. A. Cheresh, Science 1994, 264, 569–571.[110] F. P. Ross, J Chappel, J. I. Alvarez, et al., J. Biol. Chem. 1993, 268, 9901–
9907.[111] R. B. Brem, S. G. Robbins, D. J. Wilson, et al., Invest. Ophthalmol. Vis. Sci.
1994, 35, 3466–3474.[112] M. Rolli, E. Fransvea, J. Pilch, et al., Proc. Natl. Acad. Sci. USA 2003, 100,
9482–9487.[113] P. C. Brooks, S Strömblad, R Klemke, et al., J. Clin. Invest. 1995, 96, 1815–
1822.[114] Z. Liu, F. Wang, X. Chen, Drug Dev. Res. 2008, 69, 329–339.
39

22
2.4. REFERENCES
[115] Y. Asano, H. Ihn, K. Yamane, et al., Am. J. Pathol. 2004, 164, 1275–1292.[116] U. R. Goessler, P Bugert, K Bieback, et al., Int. J. Mol. Med. 2008, 21,
271–279.[117] J. Schittenhelm, A. Klein, M. S. Tatagiba, et al., Int. J. Clin. Exp. Pathol.
2013, 6, 2719–2732.[118] T. S. Panetti, P. J. McKeown-Longo, J. Biol. Chem. 1993, 268, 11492–11495.[119] M. Friedlander, C. L. Theesfeld, M. Sugita, et al., Proc. Natl. Acad. Sci. USA
1996, 93, 9764–9769.[120] K. P. Conroy, L. J. Kitto, N. C. Henderson, Cell Tissue Res. 2016, 365,
511–519.[121] V. Sarrazy, A. Koehler, M. L. Chow, et al., Cardiovasc. Res. 2014, 102, 407–
417.[122] L. A. Koopman Van Aarsen, D. R. Leone, S. Ho, et al., Cancer Res. 2008,
68, 561–570.[123] J. S. Munger, X. Huang, H. Kawakatsu, et al., Cell 1999, 96, 319–328.[124] X Huang, J Wu, S Spong, et al., J. Cell Sci. 1998, 111, 2189–2195.[125] A. Weinacker, A. Chen, M. Agrez, et al., J. Biol. Chem. 1994, 269, 6940–
6948.[126] M Agrez, A Chen, R. I. Cone, et al., J. Cell Biol. 1994, 127, 547–56.[127] M. R. Morgan, G. J. Thomas, A. Russell, et al., J. Biol. Chem. 2004, 279,
26533–26539.[128] J. Schittenhelm, E. I. Schwab, J. Sperveslage, et al., J. Neuropath. Exp. Neur.
2013, 72, 194–210.[129] M. A. Chernousov, D. J. Carey, Exp. Cell Res. 2003, 291, 514–524.[130] R. Milner, X. Huang, J. Wu, et al., J. Cell Sci. 1999, 112, 4271–4279.[131] D. Mu, S. Cambier, L. Fjellbirkeland, et al., J. Cell Biol. 2002, 157, 493–507.[132] T. M. Fenton, A. Kelly, E. E. Shuttleworth, et al., Mucosal Immunol. 2017,
10, 624–634.[133] S. Cambier, S. Gline, D. Mu, et al., Am. J. Pathol. 2005, 166, 1883–1894.[134] C. J. Avraamides, B. Garmy-Susini, J. A. Varner, Nat. Rev. Cancer 2008,
8, 604–617.[135] S. Kim, M. Harris, J. A. Varner, J. Biol. Chem. 2000, 275, 33920–33928.[136] C. T. Mierke, B. Frey, M. Fellner, et al., J. Cell Sci. 2011, 124, 369–383.[137] E. H. J. Danen, P. Sonneveld, C. Brakebusch, et al., J. Cell Biol. 2002, 159,
1071–1086.[138] U. Müller, D. Wang, S. Denda, et al., Cell 1997, 88, 603–613.
40

22
2.4. REFERENCES
[139] U Müller, B Bossy, K Venstrom, et al., Mol. Biol. Cell 1995, 6, 433–48.[140] L. M. Schnapp, N. Hatch, D. M. Ramos, et al., J. Biol. Chem. 1995, 270,
23196–23202.[141] M. D. Pierschbacher, E. Ruoslahti, Proc. Natl. Acad. Sci. USA 1984, 81,
5985–5988.[142] E. Ruoslahti, Annu. Rev. Biochem. 1988, 57, 375–413.[143] E. Ruoslahti, M. D. Pierschbacher, Science 1987, 238, 491–497.[144] T. G. Kapp, F. Rechenmacher, S. Neubauer, et al., Sci. Rep. 2017, 7, 39805.[145] M. Aumailley, M. Gurrath, J. Calvete, et al., FEBS J. 1991, 291, 50–54.[146] M Pfaff, K Tangemann, B Müller, et al., J. Biol. Chem. 1994, 269, 20233–
20238.[147] R Haubner, R Gratias, B Diefenbach, et al., J. Am. Chem. Soc. 1996, 118,
7461–7472.[148] O. V. Maltsev, U. K. Marelli, T. G. Kapp, et al., Angew. Chem. Int. Ed.
2016, 55, 1535–1539.[149] T. G. Kapp, F. S. Di Leva, J. Notni, et al., J. Med. Chem. 2018, 61, 2490–
2499.[150] J. D. Hegemann, M. De Simone, M. Zimmermann, et al., J. Med. Chem.
2014, 57, 5829–5834.[151] S. Neubauer, F. Rechenmacher, R. Brimioulle, et al., J. Med. Chem. 2014,
57, 3410–3417.[152] D. Heckmann, A. Meyer, L. Marinelli, et al., Angew. Chem. Int. Ed. 2007,
46, 3571–3574.[153] F. Rechenmacher, S. Neubauer, J. Polleux, et al., Angew. Chem. Int. Ed.
2013, 52, 1572–1575.[154] R. H. Kimura, A. M. Levin, F. V. Cochran, et al., Proteins 2009, 77, 359–
369.[155] R. H. Kimura, R. Teed, B. J. Hackel, et al., Clin. Cancer Res. 2012, 18,
839–849.[156] J. W. Kim, F. V. Cochran, J. R. Cochran, J. Am. Chem. Soc. 2015, 137,
6–9.[157] E. Engvall, P. Perlmann, Immunochemistry 1971, 8, 871–874.[158] S. L. Goodman, G. Hoelzemann, G. A. G. Sulyok, et al., J. Med. Chem.
2002, 45, 1045–1051.[159] A. O. Frank, E. Otto, C. Mas-Moruno, et al., Angew. Chem. Int. Ed. 2010,
49, 9278–9281.
41

22
2.4. REFERENCES
[160] T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, et al., Angew. Chem. Int.Ed. 2011, 50, 8714–8717.
[161] A. Bochen, U. K. Marelli, E. Otto, et al., J. Med. Chem. 2013, 56, 1509–1519.
[162] M. Piras, I. N. Fleming, W. T. A. Harrison, et al., Synlett 2012, 23, 2899–2902.
[163] S. Dall’Angelo, Q. Zhang, I. N. Fleming, et al., Org. Biomol. Chem. 2013,11, 4551–8.
[164] W. Alsibai, A. Hahnenkamp, M. Eisenblätter, et al., J. Med. Chem. 2014,57, 9971–9982.
[165] C.-F. Cho, B. Behnam Azad, L. G. Luyt, et al., ACS Comb. Sci. 2013, 15,393–400.
[166] Y. Lee, D.-K. Kang, S.-i. Chang, et al., J. Biomol. Screen. 2004, 9, 687–694.[167] M. K. J. Gagnon, S. H. Hausner, J. Marik, et al., Proc. Natl. Acad. Sci. U.
S. A. 2009, 106, 17904–17909.[168] J. Richards, M. Miller, J. Abend, et al., J. Mol. Biol. 2003, 326, 1475–1488.[169] W. Wang, Q. Wu, M. Pasuelo, et al., Bioconjugate Chem. 2005, 16, 729–734.[170] Y. Liu, Y. Pan, Y. Xu, J. Biomol. Screen. 2010, 15, 131–137.[171] D. Lössner, H. Kessler, G. Thumshirn, et al., Anal. Chem. 2006, 78, 4524–
4533.[172] U. Hersel, C. Dahmen, H. Kessler, Biomaterials 2003, 24, 4385–4415.[173] K. Y. Lee, D. J. Mooney, Progr. Polym. Sci. 2012, 37, 106–126.[174] E. S. Place, L. Rojo, E. Gentleman, et al., Tissue Eng. Part A 2011, 17,
2713–2722.[175] S. X. Hsiong, P. Carampin, H.-J. Kong, et al., J. Biomed. Mater. Res. Part
A 2008, 85A, 145–156.[176] J. W. Lee, Y. J. Park, S. J. Lee, et al., Biomaterials 2010, 31, 5545–5551.[177] G. Orive, M. De Castro, H.-J. Kong, et al., J. Control. Release 2009, 135,
203–210.[178] A. Garate, E. Santos, J. L. Pedraz, et al., J. Drug Target. 2015, 23, 806–812.[179] S.-W. Kang, B.-H. Cha, H. Park, et al., Macromol. Biosci. 2011, 11, 673–
679.[180] O. Jeon, E. Alsberg, Tissue Eng. Part A 2013, 19, 1424–1432.[181] H. R. Caires, M. Gomez-Lazaro, C. M. Oliveira, et al., Sci. Rep. 2015, 5,
1–13.[182] M. Shachar, O. Tsur-Gang, T. Dvir, et al., Acta Biomater. 2011, 7, 152–162.
42

22
2.4. REFERENCES
[183] N. C. Hunt, D. Hallam, A. Karimi, et al., Acta Biomater. 2017, 49, 329–343.[184] M. Grellier, P. L. Granja, J. C. Fricain, et al., Biomaterials 2009, 30, 3271–
3278.[185] S. S. Ho, A. T. Keown, B. Addison, et al., Biomacromolecules 2017, 18,
4331–4340.[186] J. T. Connelly, A. J. García, M. E. Levenston, Biomaterials 2007, 28, 1071–
1083.[187] R. Nakaoka, Y. Hirano, D. J. Mooney, et al., J. Artif. Org. 2013, 16, 284–93.[188] O. Tsur-Gang, E. Ruvinov, N. Landa, et al., Biomaterials 2009, 30, 189–195.[189] K. B. Fonseca, S. J. Bidarra, M. J. Oliveira, et al., Acta Biomater. 2011, 7,
1674–1682.[190] X. Li, Q. Zhang, D. Ye, et al., Polym. Eng. Sci. 2007, 57, 206–213.[191] E. S. Gil, B. B. Mandal, S.-H. Park, et al., Biomaterials 2010, 31, 8953–8963.[192] L. Wang, R. Ma, G. Du, et al., J. Biomed. Mater. Res. B. Appl. Biomater.
2015, 103, 204–211.[193] L. Jia, C. E. Ghezzi, D. L. Kaplan, J. Biomed. Mater. Res. B. Appl. Biomater.
2016, 104, 431–441.[194] S. Wohlrab, S. Müller, A. Schmidt, et al., Biomaterials 2012, 33, 6650–6659.[195] M. Widhe, N. Dekki, M. Hedhammar, Biomaterials 2016, 74, 256–266.[196] T. Saotome, H. Hayashi, R. Tanaka, et al., J. Mater. Chem. B 2015, 3,
7109–7116.[197] A. W. Morgan, K. E. Roskov, S. Lin-Gibson, et al., Biomaterials 2008, 29,
2556–2563.[198] S. Behera, D. Naskar, S. Sapru, et al., Nanomed. Nanotechnol. 2017, 13,
1745–1759.[199] G. Vidal, T. Blanchi, A. J. Mieszawska, et al., Acta Biomater. 2013, 9, 4935–
4943.[200] R. J. Mart, R. D. Osborne, M. M. Stevens, et al., Soft Matter 2006, 2, 822.[201] P. J. S. King, M. Giovanna Lizio, A. Booth, et al., Soft Matter 2016, 12,
1915–1923.[202] G. Ochbaum, R. Bitton, Polymer 2017, 108, 87–96.[203] B. E. I. Ramakers, S. A. Bode, A. R. Killaars, et al., J. Mater. Chem. B
2015, 3, 2954–2961.[204] E. T. Pashuck, B. J. Duchet, C. S. Hansel, et al., ACS Nano 2016, 10, 11096–
11104.[205] K. Luder, K. Kulkarni, H. W. Lee, et al., Chem. Commun. 2016, 52, 4549–
4552.
43

22
2.4. REFERENCES
[206] W. Liyanage, H. A. M. Ardoña, H. Q. Mao, et al., Bioconjugate Chem. 2017,28, 751–759.
[207] N. Stephanopoulos, R. Freeman, H. A. North, et al., Nano Lett. 2015, 15,603–609.
[208] J. F. Almine, D. V. Bax, S. M. Mithieux, et al., Chem. Soc. Rev. 2010, 39,3371–3379.
[209] J. C. Rodríguez-Cabello, J. Reguera, A. Girotti, et al., Adv. Polym. Sci.2006, 200, 119–167.
[210] D. Kaufmann, A. Fiedler, A. Junger, et al., Macromol. Biosci. 2008, 8, 577–588.
[211] A. Girotti, D. Orbanic, A. Ibáñez-Fonseca, et al., Adv. Healthc. Mater. 2015,4, 2423–2455.
[212] I. González De Torre, M. Santos, L. Quintanilla, et al., Acta Biomater. 2014,10, 2495–2505.
[213] C. M. Madl, L. M. Katz, S. C. Heilshorn, Adv. Funct. Mater. 2016, 26, 3612–3620.
[214] B. R. R. Costa, C. A. Custódio, A. M. Testera, et al., Adv. Funct. Mater.2009, 19, 3210–3218.
[215] M. Vila, A. García, A. Girotti, et al., Acta Biomater. 2016, 45, 349–356.[216] X. Punet, R. Mauchauffé, M. I. Giannotti, et al., Biomacromolecules 2013,
14, 2690–2702.[217] A. Di Martino, M. Sittinger, M. V. Risbud, Biomaterials 2005, 26, 5983–
5990.[218] A. Hansson, N. Hashom, F. Falson, et al., Carbohydr. Polym. 2012, 90, 1494–
1500.[219] Z. Yang, S. Yuan, B. Liang, et al., Macromol. Biosci. 2014, 14, 1299–1311.[220] R. S. Tigli, M. Gümüşderelioglu, Int. J. Biol. Macromol. 2008, 43, 121–128.[221] K. M. Park, Y. K. Joung, K. D. Park, et al., Macromol. Res. 2008, 16, 517–
523.[222] Z. Shi, K. G. Neoh, E. T. Kang, et al., J. Biomed. Mater. Res. A 2008, 86,
865–872.[223] J. Kim, Y. Park, G. Tae, et al., J. Biomed. Mater. Res. A 2009, 88, 967–975.[224] M. V. Tsurkan, K. Chwalek, M. Schoder, et al., Bioconjugate Chem. 2014,
25, 1942–1950.[225] I. L. Kim, S. Khetan, B. M. Baker, et al., Biomaterials 2013, 34, 5571–5580.[226] L. S. Wang, F. Lee, J. Lim, et al., Acta Biomater. 2014, 10, 2539–2550.
44

22
2.4. REFERENCES
[227] R. J. Wade, E. J. Bassin, W. M. Gramlich, et al., Adv. Mater. 2015, 27,1356–1362.
[228] D. Tarus, L. Hamard, F. Caraguel, et al., ACS Appl. Mater. Inter 2016, 8,25051–25059.
[229] L. Ouyang, C. B. Highley, C. B. Rodell, et al., ACS Biomater. Sci. Eng.2016, 2, 1743–1751.
[230] F. Munarin, S. G. Guerreiro, M. A. Grellier, et al., Biomacromolecules 2011,12, 568–577.
[231] F. Chen, Y. Ni, B. Liu, et al., Carbohydr. Polym. 2017, 166, 31–44.[232] A. K. Blakney, M. D. Swartzlander, S. J. Bryant, J. Biomed. Mater. Res. A
2012, 100 A, 1375–1386.[233] K. Ye, X. Wang, L. Cao, et al., Nano Lett. 2015, 15, 4720–4729.[234] M. S. Weiss, B. P. Bernabé, A. Shikanov, et al., Biomaterials 2012, 33, 3548–
3559.[235] M. D. Swartzlander, C. A. Barnes, A. K. Blakney, et al., Biomaterials 2015,
41, 26–36.[236] T. T. Lee, J. R. García, J. I. Paez, et al., Nat. Mater. 2015, 14, 352–360.[237] B. M. Müller, R. Loth, P. G. Hoffmeister, et al., Acta Biomater. 2017, 51,
148–160.[238] J. Zhu, C. Tang, K. Kotte-Marchant, et al., Bioconjugate Chem. 2010, 20,
333–339.[239] A. D. Lynn, A. K. Blakney, T. R. Kyriakides, et al., J. Biomed. Mater. Res.
A 2011, 96 A, 621–631.[240] B. Yañez-Soto, S. J. Liliensiek, C. J. Murphy, et al., J. Biomed. Mater. Res.
A 2013, 101 A, 1184–1194.[241] J. Kim, K.-W. Lee, T. E. Hefferan, et al., Biomacromolecules 2008, 9, 149–
157.[242] J. Kim, M. Dadsetan, S. Ameenuddin, et al., J. Biomed. Mater. Res. A 2010,
95, 191–197.[243] B. K. Wacker, S. K. Alford, E. A. Scott, et al., Biophys. J. 2008, 94, 273–285.[244] J. Kim, Y. P. Kong, S. M. Niedzielski, et al., Soft Matter 2016, 12, 2076–
2085.[245] S. Qiong Liu, Q. Tian, L. Wang, et al., Macromol. Rapid Commun. 2010,
31, 1148–1154.[246] A. Watarai, L. Schirmer, S. Thönes, et al., Acta Biomater. 2015, 25, 65–75.[247] M. H. Fittkau, P. Zilla, D. Bezuidenhout, et al., Biomaterials 2005, 26, 167–
174.
45

22
2.4. REFERENCES
[248] D. S. W. Benoit, K. S. Anseth, Biomaterials 2005, 26, 5209–5220.[249] C. N. Salinas, K. S. Anseth, J. Biomed. Mater. Res. A 2009, 90, 456–464.[250] S. A. DeLong, J. J. Moon, J. L. West, Biomaterials 2005, 26, 3227–34.[251] C. M. Kolodziej, S. H. Kim, R. M. Broyer, et al., J. Am. Chem. Soc. 2012,
134, 247–255.[252] J. R. García, A. Y. Clark, A. J. García, J. Biomed. Mater. Res. A 2016, 104,
889–900.[253] E. Lieb, M. Hacker, J. Tessmar, et al., Biomaterials 2005, 26, 2333–2341.[254] D. Grafahrend, J. L. Calvet, K. Klinkhammer, et al., Biotechnol. Bioeng.
2008, 101, 609–621.[255] N. Orgovan, R. Ungai-Salánki, S. Lukácsi, et al., Biointerphases 2016, 11,
031001.[256] S. A. Bencherif, A. Srinivasan, J. A. Sheehan, et al., Acta Biomater. 2009,
5, 1872–1883.[257] Q. Xu, Z. Zhang, C. Xiao, et al., Biomacromolecules 2017, 18, 1411–1418.[258] T. Nie, R. E. Akins, K. L. Kiick, Acta Biomater. 2009, 5, 865–875.[259] E. Hesse, U. Freudenberg, T. Niemietz, et al., J. Tissue Eng. Reg. Med. 2018,
12, 229–239.[260] C. Chollet, S. Lazare, F. Guillemot, et al., Colloid Surface B 2010, 75, 107–
114.[261] C. Chollet, C. Chanseau, M. Remy, et al., Biomaterials 2009, 30, 711–720.[262] Y. Lei, M. Remy, C. Labrugere, et al., J. Mater. Sci.: Mater. Med. 2012, 23,
2761–2772.[263] E. Jabbari, X. He, M. T. Valarmathi, et al., J. Biomed. Mater. Res. A 2009,
89, 124–137.[264] S. Y. Gu, Z. M. Wang, C. Y. Zhang, et al., Carbohydr. Polym. 2008, 74,
572–578.[265] P. Zhang, H. Wu, H. Wu, et al., Biomacromolecules 2011, 12, 2667–2680.[266] Y. Huang, J. Ren, T. Ren, et al., J. Biomed. Mater. Res. A 2010, 95, 993–
1003.[267] R. R. Bhat, B. N. Chaney, J. Rowley, et al., Adv. Mater. 2005, 17, 2802–
2807.[268] S. Tugulu, P. Silacci, N. Stergiopulos, et al., Biomaterials 2007, 28, 2536–
2546.[269] S. Desseaux, J. P. Hinestrosa, N. Schüwer, et al., Macromolecules 2016, 49,
4609–4618.
46

22
2.4. REFERENCES
[270] M. Santander-Borrego, D. W. Green, T. V. Chirila, et al., J. Polym. Sci. PartA: Polym. Chem. 2014, 52, 1781–1789.
[271] S. M. Paterson, A. M. Shadforth, J. A. Shaw, et al., Mater. Sci. Eng. C2013, 33, 4917–4922.
[272] F. Karimi, T. G. McKenzie, A. J. O’Connor, et al., J. Mater. Chem. B 2017,5, 5942–5953.
[273] D. Khatayevich, M. Gungormus, H. Yazici, et al., Acta Biomater. 2010, 6,4634–4641.
[274] H. S. Seo, Y. M. Ko, J. W. Shim, et al., Appl. Surf. Sci. 2010, 257, 596–602.[275] M. N. Nguyen, T. Lebarbe, O. F. Zouani, et al., Biomacromolecules 2012,
13, 896–904.[276] C. Mas-Moruno, P. M. Dorfner, F. Manzenrieder, et al., J. Biomed. Mat. Res.
A 2013, 101 A, 87–97.[277] R. Fraioli, K. Dashnyam, J. H. Kim, et al., Acta Biomater. 2016, 43, 269–
281.[278] M. Pagel, R. Hassert, T. John, et al., Angew. Chem. Int. Ed. 2016, 55, 4826–
4830.[279] M. A. Hussain, A. Agnihotri, C. A. Siedlecki, Langmuir 2005, 21, 6979–6986.[280] C. H. Yea, B. Lee, H. Kim, et al., Ultramicroscopy 2008, 108, 1144–1147.[281] M. Lashkor, F. J. Rawson, A. Stephenson-Brown, et al., Chem. Commun.
2014, 50, 15589–15592.[282] J. Huang, J. Ding, Soft Matter 2010, 6, 3395.[283] K. Ye, X. Wang, L. Cao, et al., Nano Lett. 2015, 15, 4720–4729.[284] J. Li, Y. Chen, N. Kawazoe, et al., Nano Res. 2018, 11, 1247–1261.[285] A. A. Sawyer, D. M. Weeks, S. S. Kelpke, et al., Biomaterials 2005, 26,
7046–7056.[286] A. A. Sawyer, K. M. Hennessy, S. L. Bellis, Biomaterials 2007, 28, 383–392.[287] K. M. Hennessy, W. C. Clem, M. C. Phipps, et al., Biomaterials 2008, 29,
3075–3083.[288] P. Zhang, H. Wu, H. Wu, et al., Biomacromolecules 2011, 12, 2667–2680.[289] S. Behera, D. Naskar, S. Sapru, et al., Nanomed. Nanotechnol. 2017, 13,
1745–1759.
47

22

333
3. Development of an ELISA Setupfor the Detection of RGD-bindingto Various Integrins
Parts of the work described in this chapter were published: D.Bernhagen, L. De Laporte, P.Timmerman, Anal. Chem. 2017, 89, 5991–5997.

333
3.1. ABSTRACT
3.1. Abstract
In this chapter, the development of a highly sensitive competition ELISA to mea-sure integrin-binding of RGD peptides in high-throughput without using cells, ECM-proteins or antibodies, is described. The assay measures (non-labeled) RGD-peptides’ability to inhibit binding of a biotinylated knottin-RGD peptide to surface-immobilizedintegrins αvβ3, αvβ5, and α5β1, and thus enables quantification of the bindingstrength of high-, medium- and low-affinity RGD-binders. A biotinylated knottin-RGD peptide instead of biotinylated cyclo-[KRGDf] (as reported by Piras et al.)was introduced, as integrin-binding was much stronger and clearly detectable forall three integrins. In order to maximize sensitivity and cost-efficiency, several pa-rameters were first optimized, such as integrin-immobilization levels, knottin-RGDconcentration, buffer compositions, type of detection tag (biotin, His- or cMyc-tag),and spacer length. In this way, two key factors were identified, i.e. i) the criticalspacer length (longer than Gly) and ii) the presence of Ca2+ and/or Mg2+ in allincubation and washing buffers. Binding of knottin-RGD peptide was strongest forαvβ3, but also detectable for both αvβ5 and α5β1, while binding of biotinylatedcyclo-[KRGDf] was very weak and only detectable for αvβ3. For assay validation,IC50 values for three unlabeled peptides were determined, i.e. i) linear GRGDS,ii) cyclo-[KRGDf], and iii) the knottin-RGD itself for binding to abovementionedintegrins. Major benefits of the novel assay are: i) the extremely low consump-tion of integrin (50 ng/peptide), ii) the fact that neither antibodies/ECM-proteins,nor integrin-expressing cells are required for detection, and iii) its suitability forhigh-throughput screening of (RGD-)peptide libraries (→ Chapters 4 & 5).
3.2. Introduction
Integrins, a family of cell adhesion receptors composed of 24 αβ heterodimers, me-diate interactions between different cells and the extracellular matrix.[1, 2] In thesearch for high-affinity and high-selectivity integrin binders the evaluation of bind-ing affinities is a key step.[3–5]
Different high-throughput screening methods have been developed to achieve this.[6, 7]
Recently, Cho et al. used a high-throughput one-bead-one-compound method toscreen RGD peptides for their binding to αvβ3-expressing human cancer cells.[8]
Wang et al. determined IC50 values for various cyclic RGD peptides by measuring
50

333
3.2. INTRODUCTION
change in fluorescence polarization of an αvβ3 integrin in complex with a fluo-rescently labeled peptide upon addition of the unlabeled peptides.[9] However, theintegrin concentrations used here (290 nM, roughly corresponding to 35 µg/mL)would not allow for cost-effective screening of peptide libraries.Also, several ELISA-based techniques[10–12] have been used to evaluate binding affini-ties of RGD peptides to integrins. For example, Piras et al. used biotinylated, cyclicRGD peptides[13] and, more recently, 5-fluoro-5-desoxyribose (FDR)-conjugates ofcyclic RGD peptides, to determine affinities to both immobilized and cellular in-tegrin αvβ3.[14] This method requires modification of the RGD peptides with afluorescent label, which may significantly affect their binding affinities to integrins.Kessler and coworkers showed the detection of RGD peptide binding to integrins inELISA using extracellular matrix (ECM) proteins such as fibronectin or vitronectin,either by surface-immobilizing and applying the ECM proteins in solution,[3, 15, 16]
or vice versa.[17–21]
As such, many analytical techniques reported in literature give reliable and preciseresults, but are not broadly applicable due to high acquisition costs,[6, 22] compli-cated experimental setups,[6, 7, 12–14, 23] the requirement to use integrin-expressingcells,[4, 24, 25] or milligram quantities of integrins.[19, 26] This precludes their generalapplicability for the screening of large compound libraries. Therefore, novel high-throughput screening assays are required. Ideally, the method is highly sensitive,delivers reproducible results, is cheap in terms of material effort, quick and easy touse, and does not need highly-specialized devices or facilities.In this chapter, the development of an easy-to-use ELISA for the screening of RGD-peptide binding to various integrins is described. Instead of using ECM proteins andantibodies reported in literature, a high-affinity knottin-RGD peptide equipped witha biotin-tag (1a) was applied as the key component because of its reported high-affinity to integrins αvβ3, αvβ5 and α5β1.[27] Candidate peptides were screenedfor their ability to inhibit binding of 1a to integrins αvβ3, αvβ5 and α5β1. Itwas found that both the presence of a long spacer between the knottin sequenceand the biotin-label, as well as the presence of divalent cations (i.e. Ca2+/Mg2+)in both washing and incubation buffers is crucial to integrin-binding of 1a. Forvalidation of the assay, IC50 values for three literature-annotated RGD-peptides(1d, 2c, GRGDS) were determined with respect to their binding to αvβ3, αvβ5and α5β1, for which binding affinities were also determined previously in variousother assays.[3, 9, 13–21] Hypothetically, the use of chemically synthesizable peptides
51

333
3.3. RESULTS & DISCUSSION
(instead of antibodies, integrin-expressing cells or ECM-proteins) should be optimalfor assay-standardization, and should further reduce the costs associated with thistype of screening-assays to an acceptable level.
3.3. Results & discussion
3.3.1. Preliminary experiments
With the ultimate objective to setup a simple ELISA experiment for detecting thebinding of a broad range of RGD peptides to various integrin receptors, the bind-ing of a common set of biotinylated RGD peptides, i.e. linear GRGDS and twocyclo-[KRGDf] peptides (2a and 2b, Figure 3.1) with different spacer length, tosurface-immobilized integrin αvβ3 was studied using the recently described methodby Piras et al.[13]. RGD peptides are more straightforward to work with in ELISA ascompared to the full ECM-proteins fibronectin and vitronectin, and standardizationof an RGD peptide based assay will be easier to handle and encounter less develop-ment issues (i.e. variability, reproducibility, etc.). However, despite several efforts,it was not possible to observe measurable binding, even though the peptides testedwere reported to bind αvβ3 with very decent affinities.[9, 13] Furthermore, it wasdemonstrated that the observed absence of integrin-binding for these peptides wasnot attributable to incomplete or inadequate immobilization levels of αvβ3, sincestrong binding of anti-αvβ3 mAbs using the same ELISA-plates could be detected(Appendix, Figure 3.8, p.68).
3.3.2. Binding of knottin-RGD peptide to integrin αvβ3
Subsequently, the integrin-binding of knottin-RGD peptides 1, first reported byKimura et al. and known to bind with nanomolar affinities to various integrin recep-tors (αvβ3, αvβ5 and α5β1), was studied in ELISA.[27] Therefore, the biotinylatedvariant 1a was synthesized to enable detection using a streptavidin-HRP conjugate.Binding of 1a to αvβ3 was tested over a wide range of different concentrations(0.1-1000 nM) and also in combination with a set of different concentrations of thesurface-immobilized integrin αvβ3 (0.1-1.5 µg/mL). Even though the binding of 1a(OD405>1.0 A.U.) was detectable, it was only observed at the highest concentrationsof surface-immobilized αvβ3 (1.5 µg/mL) and knottin-RGD peptide 1a (1000 nM)investigated (Figure 3.2A).
52

333
3.3. RESULTS & DISCUSSION
Figure 3.1.: Overview of (A) knottin-RGD peptides, and (B) cyclo-[KRGDf] pep-tides. (C) Molecular weights (calculated and experimental) of purifiedpeptides as observed by UPLC/ESI-MS analysis.
Based on a report by Legler et al. in 2001, stating that the affinity of αvβ3 toligands such as fibronectin or vitronectin can be modulated using bivalent cations,[28]
the binding studies of 1a with αvβ3 were then repeated in the presence of 1.0 mMCaCl2 and 0.5 mM MgCl2 in all buffers. In the presence of these cations, bindingof peptide 1a to αvβ3 was drastically improved and could already be detected(OD405~1.0 A.U.) at a concentration of 0.5 µg/mL of surface-immobilized αvβ3and 0.1 nM of 1a, respectively, which is equivalent to an approximately 10000-fold reduction as compared to the measurements in the absence of Ca2+ and Mg2+
(Figure 3.2A). Furthermore, the absorbance increased almost 10-fold (from OD~0.3to 2.9 A.U.) when using αvβ3 at 1.0 µg/mL and 1a at 100 nM.
3.3.3. Role of bivalent cations on integrin binding
Next, it was investigated whether Ca2+ and Mg2+ exert their binding-promotingeffect exclusively in the incubation step, or whether the presence of these cations in
53

333
3.3. RESULTS & DISCUSSION
Figure 3.2.: Absorbance (405 nm) as a function of knottin-RGD 1a concentra-tion. (A) Concentration of integrin αvβ3 (blue circles: 1.5 µg/mL;green squares: 1 µg/mL; red triangles: 0.5 µg/mL; black diamonds:0.1 µg/mL) in the overall presence (plain lines) and absence (dashedlines) of Ca2+/Mg2+ was varied. (B) Integrin αvβ3 concentration:0.5 µg/mL. Influence of Ca2+/Mg2+ present in only coating- and in-cubation steps (dashed lines) and, additionally, washing steps (plainlines) were studied. All experiments were carried out in triplicate.Error bars represent standard deviations.
the washing buffer is part of the overall binding-promoting effect. In these exper-iments, 0.5 µg/mL of αvβ3 was immobilized in the ELISA plates and a standardconcentration of 1.0 mM Ca2+ and 0.5 mM Mg2+ in all incubation and blockingsteps was applied, while varying the Ca2+ and 0.5 mM Mg2+ in the washing bufferfrom zero to 1.0 and 0.5 mM, respectively (Figure 3.2B). At 0.1 µM, binding wasimproved by 33% (OD405 1.11 iso. 0.84 A.U.), while at higher concentrations of1a (1.0 and 10 µM), the effect was even further enhanced to 43% (OD405 1.24 iso.0.86 A.U.) and 52% (OD 1.43 iso. 0.94 A.U.), respectively.
Moreover, it was briefly studied how the use of different ratios (2:1/1:1 iso. 1:2)and concentrations (2.0 iso. 1.0 mM) of Ca2+/Mg2+, or the use of cations other thanCa2+ and Mg2+ (i.e. Mn2+) would affect the binding of knottin-RGD peptide 1a(Figure 3.3). To better analyze the effect of the cations, only 0.25 µg/mL of integrinαvβ3 was immobilized. Even though the effects were marginal, the use of 0.5 mMof Mn2+ (in combination with 1 mM Ca2+) instead of, or in addition to 0.5 mMMg2+ (also tested by Legler et al.[28]) led to slightly higher binding (0.01 µM of 1a:OD405 0.45 and 0.43 A.U., respectively iso. 0.38 A.U.), and the use of 1.0 mM Mg2+
alone also slightly increased the binding of 1a (OD~0.41 iso. 0.38 A.U.). Similarly,
54

333
3.3. RESULTS & DISCUSSION
Figure 3.3.: Effect of cations in the ELISA buffers. Absorbance (background-subtracted, 405 nm) as a function of knottin-RGD 1a concentrationand the bivalent cations present in the washing buffers. Integrin αvβ3concentration: 0.25 µg/mL in coating buffer; blocking: 1% I-Block inPBSTC; washing: PBST including depicted bivalent ions. All exper-iments were carried out in triplicate. Error bars represent standarddeviations.
doubling the concentrations of both Ca2+ and Mg2+ (each 2.0 mM) also increasedbinding to some extent (0.01 µM of 1a: OD 0.44 iso. 0.38 A.U.). Similar trendswere observed for the other concentrations of 1a tested (0.1 and 1 µM). Hence, itseems that both Mg2+ and Mn2+ are slightly more effective as compared to Ca2+ inmoderating the binding properties of αvβ3.
3.3.4. Role of spacer length between Biotin and RGD moietyNext to this, it was found that the linker connecting the knottin-RGD peptide in 1ato the biotin tag is also of crucial importance to binding and should be of sufficientlength in order to be able to detect integrin binding. In the absence of Ca2+ andMg2+, the binding of knottin-RGD peptides 1b (Gly-spacer) and 1c (no spacer)was undetectable (OD405<0.2 A.U.) at or below 10 µM, while 1a (PEG2 spacer)showed clear and distinct binding to αvβ3 (OD ~1.0 A.U.) at this concentration(Figure 3.4A). The binding levels of 1b and 1c did hardly improve when bivalentcations were added. Presumably, the longer spacer in 1a enhances the accessibilityof the biotin-tag to the deeply embedded binding pocket of streptavidin, thus en-abling the visualization of 1a-binding.
55

333
3.3. RESULTS & DISCUSSION
Figure 3.4.: Effect of (i) spacer length of biotinylated knottin-RGD peptides 1, and(ii) the presence of cations. Absorbance (background-subtracted, 405nm) as a function of peptide 1a-c concentration in the absence (opencolumns), presence only in incubation buffers (checkered columns),or presence in all buffers (filled columns) of Ca2+/Mg2+. (A) Integrinαvβ3; (B) αvβ5; (C) α5β1 (for absorbances, see Appendix, p.70, Fig-ure 3.10A). Integrin concentration: 0.5 µg/mL; blocking: 1% I-Block.All experiments were carried out in triplicate; those marked with anasterisk represent average values of two independent experiments. Er-ror bars represent standard deviations.
56

333
3.3. RESULTS & DISCUSSION
3.3.5. Binding of knottin-RGD peptide to integrins αvβ5, andα5β1
Following the significant enhancement of 1a-binding to αvβ3, it was then evaluatedwhether the presence of Ca2+ and Mg2+ (both in binding and washing buffers) couldalso promote binding to other integrins, like αvβ5 and α5β1.Binding of 1a to surface-immobilized αvβ5 was significantly enhanced (0.1 µM of1a: OD405 0.52 iso. <0.2 A.U., Figure 3.4B), even though the binding levels wereup to 3-fold lower than for αvβ3 (OD405 1.31 A.U.). Similarly, binding of 1a toα5β1 was also improved (Figure 3.4C). In the absence of Ca2+ and Mg2+ bindingwas basically undetectable (OD405<0.2 A.U.) even at 1.0 µM, whereas addition ofCa2+/Mg2+ made it visible even at 0.1 µM (OD405 0.45 A.U.).
3.3.6. Role of detection tag
It was also investigated if the use of tags other than biotin, such as FLAG-, His-or cMyc-tag (knottin peptides 1e-g, Figure 3.1, p.53),[29] could be used to detectintegrin-binding of knottin 1 in ELISA. In fact, not much known is known in liter-ature about using these ‘protein-tags’ in combination with medium-sized peptides,like 1. Surprisingly, binding was not observed when incubating surface-immobilizedαvβ3 with knottin-peptides 1f or 1g (Figure 3.5A), even not at the highest con-centration (10 µM) tested. Only the binding of knottin peptide 1e exhibiting theFLAG-tag was observed, albeit with low intensity and at very high concentrationscompared to knottin-RGD peptide 1a. In order to investigate the role of pep-tide sequence attached to a detection tag, two other non-related His-tagged pep-tides CGSGRIVRIHRKIQRIVNLIQHIGSGH6 (3a) and CGSGRIVSIDRKIQRIV-NLIQHIGSGH6 (3b) were tested for their binding to an anti-His polyclonal Ab-serum. Only one of these bound the antibodies reasonably well at 10 µM (OD405
~1.7 A.U.), while binding to the other was not detectable at all (OD405<0.2 A.U.,Figure 3.5B). This leads to the assumption that the highly-charged His-tag prefer-ably interacts with the peptide sequence to which it is attached, thus losing itsaffinity for the anti-His antibody. The results exemplify the drawbacks of usingFLAG, His or cMyc as detection tags for visualizing peptide binding in ELISA, asantibody-binding may be highly dependent on the nature of the peptide to whichit is connected. Therefore, it was considered that biotin is the ideal tool to detect
57

333
3.3. RESULTS & DISCUSSION
binding of RGD-based peptides in the present ELISA setup.
Figure 3.5.: Absorbance (background-subtracted, 405 nm) as a function of pep-tide concentration. (A) Effect of detection tags on knottin-RGDpeptide binding to αvβ3. Peptides were detected using Strep-HRP,anti-FLAG-HRP, anti-His6-HRP, and anti-cMyc-HRP. For 1f and 1gno trendline was determined; (B) Detection of surface-coated His-tagged peptides 3a and 3b using anti-His6 in combination with rab-bit anti-mouse-HRP. Integrin concentration: 0.5 µg/mL in coatingbuffer; blocking: 1% I-Block. All coating and washing buffers con-tained 1 mM Ca2+ and 0.5 mM Mg2+. All experiments were carriedout in triplicate. Error bars represent standard deviations.
3.3.7. Binding of cyclo-[RGD] peptides to integrins αvβ3, αvβ5,and α5β1
Finally, it was studied whether the binding-promoting effect of Ca2+ and Mg2+
could also be used to improve the integrin-binding of the cyclo-[KRGDf] peptides 2a(PEG2 spacer) and 2b (no spacer) in ELISA. As mentioned, binding of both 2a and2b to αvβ3 was not detectable in ELISA in the absence of Ca2+ and Mg2+, but sur-prisingly, only a weak enhancement of 2a-binding in the presence of those ions in allbuffers compared to the presence only in incubation buffers (i.e., from OD405 0.30to 0.44 A.U. for 2a at 10 µM, or from OD405 0.21 to 0.27 A.U. at 0.1 µM) wasobserved, whereas binding of 2b remained undetectable (OD405<0.1 A.U.), even inthe presence of Ca2+ and Mg2+ in all buffers (Figure 3.6A).Similar as for integrin αvβ3, biotinylated peptide 2b neither showed binding to
58

333
3.3. RESULTS & DISCUSSION
αvβ5 nor to α5β1 in this assay, independent of whether or not Ca2+- and Mg2+-ions were present in the incubation buffer (Figure 3.6B and C). As for knottin-RGDpeptide 1a, the biotinylated peptide 2a showed weaker binding levels to α5β1 com-pared to αvβ3, independent of whether or not Ca2+- and Mg2+-ions were presentin the incubation and washing buffers (Figures 3.6B and C).
Figure 3.6.: Effect of cations on direct detection biotinylated cyclic RGD binding.Absorbance (background-subtracted, 405 nm) as a function of peptide2a/2b concentration in the absence (open columns), presence onlyin incubation buffers (checkered columns), or presence in all buffers(filled columns) of Ca2+/Mg2+. (A) Integrin αvβ3; (B) αvβ5; (C)α5β1 (for absorbances, see Appendix, p.70, Figure 3.10A). Integrinconcentration: 0.5 µg/mL; blocking: 1% I-Block. All experimentswere carried out in triplicate; those marked with an asterisk representaverage values of two independent experiments. Error bars representstandard deviations.
59

333
3.3. RESULTS & DISCUSSION
3.3.8. Determination of IC50 values via competition ELISA
In order to take advantage of the exclusive binding properties of 1a, a competitionELISA setup was developed that is capable of quantifying the binding of unlabeledpeptides to the abovementioned integrins. This assay measures the peptides’ abilityto inhibit binding of 1a to surface-immobilized integrins and could serve to ranka large library of unmodified peptides’ binding capacity to different integrins. Inorder to validate this assay, binding levels of well-described RGD peptides to dif-ferent integrins were determined by incubating a fixed concentration of 1a togetherwith varying concentrations of the non-biotinylated peptides 1d, 2c, GRGDS, andcyclo-[FYFDLRK] as a negative control.The competition ELISA data for αvβ3 showed that all tested RGD peptides inhibit1a-binding, albeit at very different concentrations (Figure 3.7A). Both knottin-RGDpeptide 1d and cyclo-[KRGDf] peptide 2c showed full inhibition at 10 µM in con-trast to the linear GRGDS peptide, the integrin-binding of which is reported to bemuch weaker. For the latter, an IC50 value of approximately 5 µM was estimated(in agreement with reported binding strength[13]), while that for the negative con-trol peptide cyclo-[FYFDLRK] was >100 µM (Figure 3.7D). Interestingly, the IC50
value determined for 2c (0.16 µM) was slightly lower as for 1d (0.25 µM), which wasunexpected based on the observed differences in the direct binding of their labeledcounterparts (1a/2a) in the binding ELISA (Figures 3.4A and 3.6A). This couldsuggest that biotinylation of 2c has lowered its affinity for αvβ3, but other reportsconfirm the much higher αvβ3-affinity of knottins 1.[27] Moreover, the much steepercurvature of the titration curve for 1d suggests a difference in binding kinetics for1d and 2c and so the results may be time- and temperature-dependent.
Subsequently, RGD peptide-binding to integrin αvβ5 was investigated using thiscompetition ELISA (Figure 3.7B). The data revealed that 1d and 2c both showeddetectable inhibition already at low concentrations (~0.05 µM), while for GRGDSthis started only around 1.0 µM (20-fold higher) and for cyclo-[FYFDLRK] inhi-bition was not detectable at all. The latter confirms that the competition ELISAis able to correctly predict integrin binding strengths.[21] However, the binding ofcyclo-[KRGDf] peptide 2c in this assay is much stronger as compared to the valuesrecently reported by Kapp et al.[21] Also, the data from the binding ELISA exper-iments presented in this chapter (Figures 3.4B, p.56 and 3.6B, p.59) suggested amuch lower integrin-binding for 2c as compared to 1d.
60

333
3.3. RESULTS & DISCUSSION
Figure 3.7.: Validation of the assay. Absorbance (405 nm) as a function of con-centration of different RGD-peptides for (A) integrins αvβ3 (for ab-sorbances: Appendix, p.70, Figure 3.10B), (B) αvβ5, and (C) α5β1;(D) IC50 values determined via nonlinear regression analysis using thesoftware GraphPad Prism. The IC50 values marked with an aster-isk were based on a non-linear analysis using absorbance from 1d at30 µM as the lower plateau. All experiments were carried out in trip-licate; those marked with an asterisk represent average values of twoindependent experiments. Error bars represent standard deviations.
Finally, competition ELISA experiments applying integrin α5β1 (Figure 3.7C)were carried out. Here, only knottin-RGD peptide 1d showed detectable inhibitionbetween 0.1 and 10 µM, and in comparison to integrins αvβ3 and αvβ5, the IC50 for1d was approximately half the value (0.10 µM), which is in agreement with earlierreports by Kim et al.[30] Interestingly, peptide 2c did not show a measurable compe-tition, which is consistent with the almost 100-fold lower affinity of this peptide forα5β1, as reported by Kapp et al.[21] Most notably, these results emphasize the bene-fit of the presented competition ELISA, as the IC50 values for 2c could convenientlybe determined, even though the direct binding levels of biotinylated cyclo-[KRGDf]
61

333
3.4. CONCLUSION & OUTLOOK
peptides were undetectable (see Figure 3.6, p.59).The much lower IC50 values, as observed by Kapp et al. (~100-fold for αvβ3 and~1000-fold for α5β1) can be, at least partly, explained by the use of the exctracellu-lar matrix (ECM) proteins vitronectin and fibronectin, for which integrin-binding ismuch weaker than that for knottin-RGD peptide 1a.[12, 21, 27] Hence, much lower con-centrations of RGD-peptides are required to inhibit ligand–integrin binding, leadingto overall lower IC50 values. In this respect, Kapp et al. propose denaturation ofα5β1 upon coating in ELISA to explain the lower binding that they observed (whencompared to coating of ECM proteins).[21] In the present assay, however, the positivebinding data observed with knottin-RGD peptide 1a preclude this option. In ad-dition, the present assay exposes a mixture of peptide and ligand (i.e., biotinylatedknottin-RGD peptide 1a) to the surface-immobilized integrin, while other groupspreincubated peptide and integrin before exposing this to surface-immobilized ligand(i.e., vitronectin or fibronectin).[21] This could lead to non-reversible preoccupationof integrins’ RGD-binding pocket with peptide and eventually lead to overestima-tion of the peptide’s affinity for the integrin due to slow off-rate kinetics of thepeptide–integrin interaction. Finally, other assays used different concentrations foreach integrin assay (αvβ3: 2 µg/mL, vitronectin: 1 µg/mL; αvβ5: 3 µg/mL, vit-ronectin: 5 µg/mL; α5β1: 2 µg/mL, fibronectin: 0.5 µg/mL),[21] while the presentassay always applied identical conditions for each integrin. This leads to betterstandardization and enables direct comparison of resulting IC50 values.
3.4. Conclusion & outlook
Despite low detection levels in direct binding ELISA experiments, the IC50 valuesof non-labeled cyclic and linear RGD peptides were conveniently determined witha competition ELISA using the high-affinity biotinylated knottin-RGD peptide 1a.The results clearly indicate that the binding affinity of knottin, and thus, the inhi-bition levels for the selected RGD peptides, strongly depends on the presence of asufficiently long spacer between the peptide and the biotin tag, as well as the pres-ence of Ca2+ and Mg2+ in all buffers used. Furthermore, it was demonstrated thatthis novel method has the potential to serve as a cheaper, highly standardized, andreproducible alternative to similar assays involving antibodies or integrin-expressingcells. Moreover, since this assay does not need any detection antibodies, cells, orexpensive analytical devices, and provides reliable results with very low levels of
62

333
3.5. MATERIALS & METHODS
integrins used, it can be applied in almost any facility. Another advantage of apply-ing the knottin-RGD peptide is its highly stable structure, compared to the weaklong-term stability of antibodies.∗ However, it needs to be mentioned that the newmethod can only be used for integrin-receptors that bind knottin-RGD peptide 1a.For integrin-receptors that do not bind 1a, a surrogate binder needs to be developedfirst.The described ELISA method is suitable to be applied for the high-throughputscreening of libraries of high-affinity, RGD-containing bicyclic peptide binders tointegrins αvβ3, αvβ5, and α5β1, the results of which are thoroughly described inthe following chapters 4 and 5.
3.5. Materials & methods
Reagents & chemicals
Incubation and washing buffers were prepared using standard protocols. Recom-binant human integrins were purchased from R&D Systems (Minneapolis, USA).Strep-HRP (Streptavidin-Horseradish Peroxidase conjugate, Southern-Biotech, Birm-ingham, USA), anti-Flag-HRP, anti-c-Myc-HRP (both Sigma, Missouri, USA), anti-His6-HRP (Roche, Mannheim, Germany), His·Tag mAb (Merck, Darmstadt, Ger-many) and rabbit anti-mouse-HRP (Southern-Biotech, Birmingham, USA) were di-luted 1:200-1:1000 for ELISA experiments. Amino acids were purchased from IrisBiotech (Marktredwitz, Germany) and Matrix Innovation (Quebec, Canada). Resinswere purchased from Rapp Polymere (Tübingen, Germany) and Merck (Darmstadt,Germany). MnCl2·4H2O was purchased from Sigma-Aldrich (Steinheim, Germany).CaCl2·2H2O and MgCl2·6H2O were purchased from Merck (Darmstadt, Germany).Tween80 was purchased from Faryon (Capelle, The Netherlands) and I-Block™ waspurchased from Tropix (Bedford, USA).
Peptide synthesis
All synthesized peptides are reported in Figure 3.1 (p.53). Knottin-RGD peptides1a-g were based on the knottin structure as published by Cochran et al.[27] andwere synthesized via Fmoc-based solid-phase peptide synthesis (SPPS) on a Rink-
∗Knottin-RGD peptide 1a was still stable after more than six months regardless of severalthaw-freeze cycles.
63

333
3.5. MATERIALS & METHODS
amide resin using standard protocols. Subsequent oxidative folding was performedin a cysteine/cystine-buffer according to published protocols.[31] Cyclo-[RGD] pep-tides 2a-c were synthesized on a 2-chlorotrityl resin, followed by cleavage with 2%TFA/DCM as a fully side-chain protected peptide and backbone cyclization and sub-sequent full deprotection using 95% TFA. Linear GRGDS amide was synthesized ona Rink resin and finally acetylated at the N-terminus.
Peptide analysis and purification
UPLC analysis was performed on a Waters Acquity Ultra Performance LC System,equipped with a Waters Acquity UPLC BEH130 C18 1.7 µm column. A lineargradient of 5–55% MeCN (0.05% TFA) in H2O (0.05% TFA) was used. All pep-tides were purified by preparative HPLC (Waters Prep LC) on an RP-C18 column(Reprosil-Pur 120 C18-AQ 150x20 mm, Dr. Maisch GmbH, Ammerbuch, Germany)using a MeCN/milliQ H2O gradient including 0.05% TFA followed by lyophilizationon a Christ Alpha 2-4 LDplus lyophilizer.
Binding ELISA
The ELISA setup was established as follows: Plates were coated with 100 µL ofan 0.5-1.5 µg/mL integrin solution in coating buffer (0.1 M Na2HPO4 adjusted topH 8 using 12 M HCl, with or without cations (i.e. 1.0/0.5 mM Ca2+/Mg2+)),onto 96-well NUNC Polysorp© plates at 4 ◦C overnight, followed by blocking with150 µL 1% I-Block™ in washing buffer (PBS/Tween80, 0.05%) with (PBSTC) orwithout (PBST) cations (1.0/0.5 mM Ca2+/Mg2+) for 1 hour at room temperature.After 3x washing with 400 µL of PBST/PBSTC, plates were incubated with 100 µLof biotinylated peptide solution in PBSC (containing 1.0 mM CaCl2 and 0.5 mMMgCl2) for 90 min at room temperature. After washing 3x with PBST/PBSTC,the plates were incubated with 100 µL of 1:1000 Strep-HRP in PBSC (1 hour atroom temperature). After washing the plates 4x with PBST/PBSTC, they wereincubated with 150 µL substrate buffer containing 0.91 mM ABTS (2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) and 0.006% H2O2 in substrate buffer (0.2 MNa2HPO4 adjusted to pH 4 using 0.2 M citric acid). Absorbance was measured after45 min using a Molecular Devices Spectramax M2 plate reader. In the experimentsinvolving different cations, absorbance was corrected for background absorption inview of the strong differences in absorption for different cations (background refers
64

333
3.5. MATERIALS & METHODS
to absorbance without biotinylated peptide present). All experiments were carriedout in triplicate.
Competition ELISAThe protocols for coating, blocking and washing in the competition ELISA weresimilar to that for the binding ELISA. Coating of the integrins was performed usingPBSC. Peptides, whose integrin binding strength was to be determined, were mixedin 12 different concentrations (each threefold dilutions) with a fixed concentrationof 1a (both in PBSC, 15 min, r.t.) followed by incubation of the plates with pep-tide/1a solutions. Substrate buffer incubation and absorbance measurements wereidentical to that for binding ELISA. All experiments were carried out in triplicate.IC50 values were calculated via non-linear regression analysis using the GraphPadPrism software and represent the peptide concentration at which 50% inhibition ofbiotinylated knottin binding is observed.Schematic representations of the applied binding and competition ELISA setups aredepicted in Figure 3.11 (Appendix, p.71).
65
333

333
3.6. REFERENCES
3.6. References[1] M. Barczyk, S. Carracedo, D. Gullberg, Cell Tissue Res. 2010, 339, 269–280.[2] Y. Takada, X. Ye, S. Simon, Genome Biol. 2007, 8, 215.[3] M. A. Dechantsreiter, E. Planker, B. Matha, et al., J. Med. Chem. 1999, 42,
3033–3040.[4] A. P. Silverman, A. M. Levin, J. L. Lahti, et al., J. Mol. Biol. 2009, 385,
1064–1075.[5] W. Alsibai, A. Hahnenkamp, M. Eisenblätter, et al., J. Med. Chem. 2014,
57, 9971–9982.[6] Y. Lee, D.-K. Kang, S.-i. Chang, et al., J. Biomol. Screen. 2004, 9, 687–694.[7] M. K. J. Gagnon, S. H. Hausner, J. Marik, et al., Proc. Natl. Acad. Sci. U.
S. A. 2009, 106, 17904–17909.[8] C.-F. Cho, B. Behnam Azad, L. G. Luyt, et al., ACS Comb. Sci. 2013, 15,
393–400.[9] W. Wang, Q. Wu, M. Pasuelo, et al., Bioconjugate Chem. 2005, 16, 729–734.[10] E. Engvall, P. Perlmann, Immunochemistry 1971, 8, 871–874.[11] J. Chatterjee, O. Ovadia, G. Zahn, et al., J. Med. Chem. 2007, 50, 5878–
5881.[12] D. Arosio, L. Manzoni, E. M. V. Araldi, et al., Bioconjugate Chem. 2009,
20, 1611–1617.[13] M. Piras, I. N. Fleming, W. T. A. Harrison, et al., Synlett 2012, 23, 2899–
2902.[14] S. Dall’Angelo, Q. Zhang, I. N. Fleming, et al., Org. Biomol. Chem. 2013,
11, 4551–8.[15] M Pfaff, K Tangemann, B Müller, et al., J. Biol. Chem. 1994, 269, 20233–
20238.[16] S. L. Goodman, G. Hoelzemann, G. A. G. Sulyok, et al., J. Med. Chem.
2002, 45, 1045–1051.[17] A. O. Frank, E. Otto, C. Mas-Moruno, et al., Angew. Chem. Int. Ed. 2010,
49, 9278–9281.[18] T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, et al., Angew. Chem. Int.
Ed. 2011, 50, 8714–8717.[19] A. Bochen, U. K. Marelli, E. Otto, et al., J. Med. Chem. 2013, 56, 1509–
1519.[20] J. D. Hegemann, M. De Simone, M. Zimmermann, et al., J. Med. Chem.
2014, 57, 5829–5834.
67

333
3.7. APPENDIX
[21] T. G. Kapp, F. Rechenmacher, S. Neubauer, et al., Sci. Rep. 2017, 7, 39805.[22] Y. Liu, Y. Pan, Y. Xu, J. Biomol. Screen. 2010, 15, 131–137.[23] D. Lössner, H. Kessler, G. Thumshirn, et al., Anal. Chem. 2006, 78, 4524–
4533.[24] S. Cressman, Y. Sun, E. J. Maxwell, et al., Int. J. Pept. Res. Ther. 2009,
15, 49–59.[25] Z.-b. Li, W. Cai, Q. Cao, et al., J. Nucl. Med. 2007, 48, 1162–1171.[26] J. Richards, M. Miller, J. Abend, et al., J. Mol. Biol. 2003, 326, 1475–1488.[27] R. H. Kimura, A. M. Levin, F. V. Cochran, et al., Proteins 2009, 77, 359–
369.[28] D. F. Legler, G Wiedle, F. P. Ross, et al., J. Cell Sci. 2001, 114, 1545–1553.[29] K Terpe, Appl. Microbiol. Biotechnol. 2003, 60, 523–533.[30] J. W. Kim, F. V. Cochran, J. R. Cochran, J. Am. Chem. Soc. 2015, 137,
6–9.[31] Y. Sohma, B. L. Pentelute, J. Whittaker, et al., Angew. Chem. Int. Ed. 2008,
47, 1102–1106.
3.7. Appendix
Figure 3.8.: Absorbances determined in binding ELISA experiments with immo-bilized integrin αvβ3 and soluble αvβ3 monoclonal antibody (αvβ3mAb). Bound αvβ3 mAb was detected with rat anti-mouse lgG-HRPand ABTS.
68

333
3.7. APPENDIX
Figure 3.9.: UPLC/ESI-MS spectra of A) purified knottin-RGD peptide 1a, andB) purified cyclo-[RGD] peptide 2a (top: UV detection at 215 nm,middle/bottom: ESI-MS detection).
69

333
3.7. APPENDIX
Figure 3.10.: Absorbances determined in (A) binding ELISA experiments of pep-tides 1a-c and 2a+b to integrin α5β1, and (B) competition ELISAexperiments of peptides 1d, 2c, GRGDS and cyclo-[FYFDLRK] tointegrin αvβ3. Values marked with an asterisk were not consideredfor IC50 calculation.
70

333
3.7. APPENDIX
Figure 3.11.: Schematic representation of the applied binding ELISA and compe-tition ELISA.
71

333

4444
4. Screening for High Affinity andSelectivity Bicyclic RGD-Bindersto Integrin αvβ3
Parts of the work described in this chapter were published: D. Bernhagen, V. Jungbluth, N.Gisbert Quilis, J. Dostalek, P. B. White, K. Jalink, P. Timmerman, ACS Comb. Sci. 2019, 21,198–206.

4444
4.1. ABSTRACT
4.1. Abstract
In this chapter, the identification of high-affinity and high-selectivity integrin αvβ3-binding bicyclic RGD peptides via high-throughput screening of partly random-ized peptide libraries is described. Peptide libraries (~700 different compounds)comprising the universal integrin-binding sequence Arg-Gly-Asp (RGD) in the firstloop, and a randomized sequence XXX (where X represents one of 18 canonicalL-amino acids except Cys and Met) in the second loop, both enclosed by eitheran L- or D-Cys residue, were converted to bicyclic peptides via reaction with 1,3,5-tris(bromomethyl)benzene (T3). Screening of 1st generation libraries yielded sub-micromolar lead bicyclic inhibitors for integrin αvβ3, e.g. CT3HEQcT3RGDcT3
(IC50: 195 nM). Next generation (2nd and 3rd) libraries were designed by partiallyvarying the structure of the strongest 1st generation lead inhibitors and screenedfor improved affinities and selectivities. In this way, three highly selective αvβ3-binders (CT3HPQcT3RGDcT3, IC50: 30 nM; CT3HPQCT3RGDcT3, IC50: 31 nM;CT3HSQCT3RGDcT3, IC50: 42 nM) were identified, comprising affinities compara-ble to that of a high-affinity knottin-RGD peptide (32 amino acids, IC50: 38 nM)and superior selectivities over integrins αvβ5 (IC50 >10,000 nM) and α5β1 (IC50
>10,000 nM). The affinity data were confirmed by surface plasmon-enhanced flu-orescence spectroscopy (SPFS), yielding a Kd of 1.1 nM for Cy5-labeled bicycleCT3HPQcT3RGDcT3, while a Kd of 0.6 nM for Cy5-labeled knottin-RGD peptidewas measured. Moreover, it is briefly demonstrated that biotinylated versions ofthese bicyclic peptides may replace biotinylated knottin-RGD peptide as a ligandin competition ELISA experiments.
4.2. Introduction
(Multi-)cyclic peptides represent an important platform in drug development owingto their unique properties, such as conformational restriction and low toxicity. Pep-tides produced by nature, for example, romidepsin, vancomycin and ciclosporin, aswell as semisynthetic peptides such as dalbavancin, are established peptide-baseddrugs.[1–3] Over the past years, the bicyclic CLIPS -peptide platform (Chemical Link-age of Peptides onto Scaffolds) has attracted considerable interest by combining hightarget binding affinity and selectivity with an appreciable proteolytic stability.[4–6]
In particular, the cyclization of cysteine-containing peptide loops using synthetic
74

4444
4.2. INTRODUCTION
scaffolds, first described by Timmerman et al.,[7] has been applied to explore vari-ous bicyclic peptide inhibitors. For example, Heinis et al. combined cyclization ofcysteine-containing peptides with phage-display screening and identified a bicyclicpeptide, ACT3’SDRFRNCT3’PADEALCT3’G (T3’ stands for scaffold derived from1,3,5-trisbromomesitylene), exhibiting nanomolar affinity to plasma kallikrein (Ki =1.5 nM).[8] Here, a consensus motif ‘SDRFR’ was identified in the first selections,followed by sequential optimization of the second loop. Notably, the kallikrein in-hibitory activities of the linear peptides were at least 250-fold lower as comparedto the corresponding T3-bicycles. The same group reported the bicyclic peptideACT3SRYEVDCT3RGRGSACT3G that inhibits urokinase-type plasminogen acti-vator with a Ki of 53 nM,[9] and, most recently, ACT3HSRCT3PQLPPCT3G asan inhibitor for sortase A (Ki: 1.1 µM).[10] Lian et al. developed a one-beadtwo-compound screening-technology to identify bicyclic peptide inhibitors, withdouble-digit nanomolar Ki values (10-100 nM), for protein-tyrosine phosphatase 1B(PTP1B), a target for treatment of type II diabetes.[11] These peptides both containa target-binding sequence in the first loop, and a cell-penetrating peptide FΦRRRQ(Φ: L-naphthylalanine) in the second loop. Recently, the same group reported asub-micromolar bicyclic inhibitor for K-Ras, combining a cell-permeable peptide se-quence in the first loop with the K-Ras binding motif AJFRnΨID (J: D-Leu, Ψ:L-propargylglycine, n: D-asparagine) in the second loop.[12] Finally, Luzi et al. iden-tified a bicyclic CLIPS -peptide binder (ACT3PPCT3LWQVLCT3, Kd: 10 nM) totumor necrosis factor-alpha (TNFα), a mediator of inflammatory disorders.[13] Allof the above described bicyclic peptide binders and inhibitors fully rely on the high-throughput potential of random phage-display library technology.In contrast to this fully random-based screening approach, Timmerman et al. pre-viously reported a design-based approach to identify bicyclic peptides with goodtumor-cell activities that were derived from CDR-loop sequences of anti-gastrinmonoclonal antibodies (mAbs) via microarray screening.[5] In this approach, an es-sential part of the sequence of the protein to be mimicked is taken as a lead sequencefor development.In this chapter, a similar approach is described to identify bicyclic peptides thatbind integrin αvβ3 with high affinity and selectivity. These bicycles consist of anRGD-loop in combination with a second loop (XXX) that further supports the RGD-integrin affinity, and also brings selectivity into the peptide. Integrins represent afamily of cell adhesion receptors[14, 15] that are potential targets for novel therapeutic
75

4444
4.3. RESULTS & DISCUSSION
agents resulting from their significant role in pathological processes. A major con-tribution to the investigation of integrin-binding peptides was made by Kessler andcoworkers, who developed the potent αvβ3 antagonist cilengitide,[16] and other cy-clopeptides with decent affinities for integrins, such as αvβ3, α5β1 and α6β1.[17–23]
Recently, Cochran and coworkers described a family of high-affinity integrin-binding‘cysteine-knot’ (knottin)-RGD peptides which are considered great candidates fordrug development.[24–26] However, these peptides express basically no selectivity inbinding to integrins αvβ3, α5β1, and αvβ5. The bicyclic RGD peptide platformdescribed in this chapter therefore provides an attractive alternative to these high-affinity integrin-binders. Moreover, the rational-design-screening approach can beapplied to identify high-affinity binders to other integrins (→ Chapter 5, p.111), andmight in principle also be suitable to develop bicyclic peptide-mimics of other ECM-proteins, for example, based on the laminin-derived YIGSR or IKVAV motifs.[27–29]
4.3. Results & discussion
The first important factor to be considered for the peptide library design is themethod to be used to screen the peptides for high-affinity integrin binding. Amongthe various labeled and label-free detection methods to measure peptide-integrininteractions that are available, the ELISA setup described in Chapter 3 representsa reliable medium-throughput screening method with an acceptable practical andfinancial burden. The binding ELISA experiment measures the direct interactionbetween a labeled peptide and the surface-immobilized integrin, whereas the com-petitive ELISA experiment measures a non-labeled peptide’s ability to inhibit theinteraction between the surface-immobilized integrin and a labeled high-affinity in-tegrin binding peptide. Since the attachment of a label might substantially influencethe integrin affinity, it was decided to design ‘label-free’ peptide libraries compris-ing acetylated N-termini and C-terminal amides, the binding properties of whichwere evaluated by means of measuring the level of inhibition of the knottin-integrininteraction in competitive ELISA experiments.
4.3.1. General procedure for library screening
The approach involves the design of partially randomized libraries of small, RGD-containing bicyclic CLIPS [30] peptides to achieve an iterative affinity and selectivity
76

4444
4.3. RESULTS & DISCUSSION
optimization process for the integrin receptor αvβ3. All 672 bicyclic RGD peptideswere screened for inhibition of binding of the biotinylated knottin-RGD peptide tointegrin αvβ3 using the ELISA setup as described in Chapter 3.∗ Occasionally, asecond screening for the best 30 hits from the first screening, which gave at least 90%inhibition, was performed at a lower concentration (2.5-10 µM) to determine the dif-ferences in their affinities for the integrin more precisely. Following screening of thevarious crude bicyclic RGD peptide libraries, the best binders were re-synthesizedand HPLC-purified, followed by determination of IC50 values.
4.3.2. Design & synthesis of RGD peptide librariesWith the ultimate goal to determine high-affinity and selectivity integrin binders,linear peptide libraries were then designed, consisting of two separate binding mo-tifs surrounded by three cysteines. The first motif contains the well-known RGDsequence that should provide the basic integrin affinity, while the second motif con-tains a random sequence intended to provide integrin-selectivity to the bicycle. Themotifs are enclosed by three cysteines that allow for the double CLIPS -cyclizationusing the trivalent tris(bromomethyl)benzene scaffold T3, and hence the formationof a bicyclic peptide comprising two different loops (for a schematic representation,see Figure 4.1).
Figure 4.1.: Formation of a bicyclic CLIPS peptide via a triple nucleophilic sub-stitution reaction (SN2-type) of the thiol groups of the three cysteineresidues with trivalent scaffold 1,3,5-tris(bromomethyl) benzene (T3).
The first challenge was to determine the proper size of both the ‘RGD’ and the∗Detailed experimental conditions are depicted in Table 4.2, p.104, Appendix.
77

4444
4.3. RESULTS & DISCUSSION
random ‘X(X)nX’ loop. The well-known integrin-binder cilengitide (cyclo-[V(N -Me)RGDf])[16] consists of five amino acids. In a bicyclic peptide, a 5-mer loop canbe created by enclosing the 3-mer ‘RGD’ sequence with two cysteines, hence theminimal binding motif ‘CRGDC’ was selected. At this point it was decided tokeep the ‘RGD’ loop size constant in the 1st generation library and to optimize thisfurther at a later stage. For the second loop containing the ‘X(X)nX’ sequence,the right level of structural diversity had to be created that should maximize thechance to achieve a largely improved integrin affinity and selectivity as comparedto the monocyclic CRGDC-peptide. Considering all natural amino acids (exceptcysteine and methionine), herein represented by ‘X’, there are 324 (182) variants ofa dimer motif ‘XX’, 5,832 possible trimer sequences ‘XXX’, and 104,976 variantsfor the tetramer motif ‘XXXX’. A trimer sequence ‘XXX’ was considered suitableto provide reasonable structural and conformational diversity in this loop. Since 96different ‘XXX’ motifs would represent 1.6% of the total possible tripeptide space,the chance of missing a high-affinity integrin binder after several rounds of structuraloptimization was considered relatively low, compared to an ‘XXXX’ motif, where96 sequences would only represent 0.09% of the total variant structural space.Thereby, it was decided to locate the ‘RGD’ motif either in the C-terminal (“right”)loop, and consequently the ‘XXX’ sequence in the N-terminal (“left”) loop, orvice versa. 96 randomly generated ‘XXX’ sequences were generated for each loopby using the software program “R”. Because of the strong impact of D-aminoacids on the integrin binding-affinity,[16, 31] additional sub-libraries comprising dif-ferent combinations of L- and D-cysteines were also designed. Hence, the entire1st generation library consisted of four sub-libraries showing 96 different randomsequences in the left loop (CXXXCRGDC, CXXXcRGDC, CXXXCRGDc, andCXXXcRGDc), and three sub-libraries comprising 96 different random sequencesin the right loop (cRGDCXXXC, CRGDcXXXC, cRGDcXXXC), leading to a to-tal amount of 672 different peptides that were then converted to bicyclic pep-tides via reaction with scaffold T3 (CT3XXXCT3RGDCT3, CT3XXXcT3RGDCT3,CT3XXXCT3RGDcT3, and CT3XXXcT3RGDcT3, cT3RGDCT3XXXCT3, CT3RG-DcT3XXXCT3, cT3RGDcT3XXXCT3, Figure 4.2). After screening of these librariesfor binding integrin αvβ3 (method described below), different lead motifs for nextgeneration libraries were chosen based on the IC50 values of the best binders. Anoverview of all applied amino acids is shown in the Appendix (p.105, Table 4.3A).
78

4444
4.3. RESULTS & DISCUSSION
Figure 4.2.: Overview of the design process for the determination of high-affinitybicyclic peptides to integrin αvβ3.
For the design of the 2nd generation libraries, derived from several promising leadsfrom 1st generation screening, either the second loop of this lead (e.g. ‘QAD’) wasextended by one amino acid (‘CXQADc’ or ‘CQADXc’), or a full replacement anal-ysis for the ‘E’-residue in the motif CT3HEQcT3RGDcT3/CT3HEQCT3RGDcT3
was performed, since in the 1st generation screening multiple positive hits for the‘HXQ’ motif in the second loop were observed. After screening and determinationof IC50 values for the best 2nd generation αvβ3-binders, 3rd generation libraries weredesigned based either on 1) a full amino acid replacement analysis for the ‘HWQ’motif, or 2) enlarging the ‘RGD’ loops (e.g. ‘CRGDXc’ or ‘CXRGDSc’) while main-taining the sequence of the second loop ‘CHWQC’ constant.
79

4444
4.3. RESULTS & DISCUSSION
4.3.3. Screening for αvβ3-binding peptides
1st generation screening
About 4% of the 672 1st generation bicyclic RGD peptides showed OD405 valuesbelow 0.4 A.U., corresponding to more than 80% inhibition of knottin-RGD bind-ing to αvβ3 (OD405 0.2 A.U. = 100%, OD405 1.2 A.U. = 0%).† For the strongestαvβ3-binding bicycles, we observed that peptides comprising the ‘RGD’ motif inthe right loop generally gave higher binding than peptides from libraries compris-ing the ‘RGD’ motif in the left loop.‡ Moreover, the best binders all had at leastone D-Cys attached to the ‘RGD’ sequence, either C-terminal, or both N- and C-terminal, which is consistent with the works of Kessler et al., who reported enhancedintegrin-binding for (mono)cyclic peptides with a D-amino acid next to the ‘RGD’-motif.[31] Furthermore, the present results revealed that peptides comprising the mo-tif ‘HXQ’ (X: any L-amino acid) in the left loop mostly showed significant inhibition.Surprisingly, approximately 50% of bicyclic RGD peptides showed OD405 values of2.0 A.U. or higher, while the OD405 of pure biotinylated knottin-RGD peptide wasonly ~1.2 A.U. Apparently, those RGD-bicycles do not only lack the ability to blockbinding of knottin-RGD to integrin αvβ3, but somehow even seem to ‘activate’ theRGD-binding pocket of this integrin-receptor for knottin-RGD binding. Currently,there is now explanation for this penomenon, but it was beyond doubt that thosebicycles were not considered of interest as potent and selective αvβ3-binders.Then, the best 96 hits from the first screening (OD405 0.18–0.66) were tested asecond time, now using only a 100-fold excess (10 µM) over the knottin-RGD pep-tide, in which 28% (27 peptides) showed >50% inhibition. Notably, all 96 peptidesshowed a neutral or negative net charge.§ The four strongest binders selected in bothscreenings (CT3QADcT3RGDcT3, CT3HEQcT3RGDcT3, CT3QWGCT3RGDcT3,CT3WGDCT3RGDcT3) were then re-synthesized, purified and their IC50 values de-termined to be between 195 nM for CT3HEQcT3RGDcT3 and 835 nM for CT3WG-DCT3RGDcT3 (Figure 4.3A). These values are still significantly higher than theIC50 values determined for the knottin-RGD peptide itself (38 nM), cyclo-[KRGDf]peptide (35 nM), and finally cilengitide (121 nM) from Kessler et al.[16, 31]
†These OD405 values were determined after the screenings of 1st generation libraries.‡The OD405 values of all 1st, 2nd and 3rd generation library screenings are depicted in the
Supplementary Information, Section A.1, p.207.§The contribution to the net charge is +1 for each K or R, -1 for each E or D, and 0 for all
other amino acids.
80

4444
4.3. RESULTS & DISCUSSION
Figure 4.3.: (A) IC50 values for 1st, 2nd, and 3rd generation bicyclic integrin αvβ3-binders; (B) structures of highest integrin αvβ3 affinity 1st, 2nd and3rd generation bicycles. Each concentration was tested in triplicate.IC50 values were calculated via nonlinear regression analysis using thesoftware GraphPad Prism. Inhibition values were calculated based onabsorbance when no bicyclic competitor (0%, OD405~1.8 A.U.) or non-labeled knottin-RGD at 30 µM (100%, OD405~0.2 A.U.) was applied.
81

4444
4.3. RESULTS & DISCUSSION
2nd generation screening
In order to further improve these affinities, 2nd generation libraries were designedusing the six highest affinity bicycles as a lead. These libraries (Figure 4.2) con-tained peptides comprising 1) a 3-mer to 4-mer extended second loop, for example,‘XQAD’ or ‘QADX’ (total of 12x18=216 peptides), 2) a full replacement analysis forthe ‘E’-residue in the motif ‘HEQ’ (25) following the presence of numerous strongbinders containing this motif in the first screening, 3) a set of 12 non-natural variantsfrom potent lead sequences,¶ and 4) the best 8 hits from the 1st generation screen-ing (as positive controls). From a total set of 261 2nd generation RGD-bicycles 31%(82 peptides) showed >50% inhibition, and 7% (18) showed even >70% inhibitionat 5 µM concentration. To further narrow down the selection, a re-screening of thebest 30 hits was executed at 2.5 µM, in which 6% (15) showed >50% inhibition andonly 2% (6) of the peptides >70% inhibition. None of these six peptides comprisedan extended motif such as ‘XQAD’ or ‘WGDX’ in the left loop, indicating that theinitial selection for the trimer sequence seemed the correct choice. Instead, five pep-tides comprised the ‘HXQ’ motif (CT3HWQCT3RGDcT3, CT3HPQcT3RGDcT3,CT3HWQcT3RGDcT3, CT3HNQcT3RGDcT3, and CT3HFQcT3RGDcT3) and onecomprised the motif ‘Q[Abu]D’ (CT3Q[Abu]DcT3RGDcT3, Abu: 2-aminobutyricacid). The IC50 values of the best six (re-synthesized and HPLC-purified) pep-tides ranged from 30 nM (CT3HPQcT3RGDcT3) to 225 nM (CT3HWQcT3RGDcT3,Figure 4.3A). Five of the six peptides showed an IC50 that was lower than thebest 1st generation bicycle CT3HEQcT3RGDcT3 (195 nM). The bicyclic peptideCT3HPQcT3RGDcT3 displayed an IC50 value (30 nM) comparable to that of theknottin-RGD published by Kimura et al. (38 nM).[24]
3rd generation screening
Following, 3rd generation libraries were designed comprising 1) a full position-replacementanalysis on the 2nd generation lead sequence ‘HWQ’ (86 peptides), 2) RGD-loopsextended by an additional amino acid ‘X’ while keeping the ‘HWQ’-loop constant(57 peptides), and 3) the extended 5-mer loops ‘GRGDX’ and ‘XRGDS’ and a con-stant ‘HWQ’-loop (56 peptides, Figure 4.2). From the total of 199 bicycles that werescreened at 5 µM, 35% (79) showed >50% inhibition, 18% (35) >70% inhibition, and
¶The list of peptides applied in 2nd generation libraries is given in the appendix (p.105, Ta-ble 4.3B).
82

4444
4.3. RESULTS & DISCUSSION
yet 4% of the peptides (8) >90%. In the re-screening of the best 20 hits at 2.5 µM, 19peptides (95%) showed >80% inhibition, yet four peptides (20%) even >90% inhibi-tion. From this screening, the best six hits were re-synthesized and HPLC-purifiedfor IC50 determination: CT3HPQCT3RGDcT3, CT3H[Aib]QCT3RGDcT3 (Aib: 2-aminoisobutyric acid), CT3HRQCT3RGDcT3, CT3HFQCT3RGDcT3, CT3HWE-CT3RGDcT3 andCT3HSQCT3RGDcT3. Amongst these, the best binder wasCT3H-PQCT3RGDcT3 (IC50: 31 nM), and differed only in configuration by one cysteinein comparison to the 2nd generation binder CT3HPQcT3RGDcT3 (Figure 4.3A).The other bicyclic 9-mer peptides showed IC50 values between 42 and 157 nM. TheIC50 values clearly indicate that ‘H’ (histidine) and ‘Q’ (glutamine) are essential forhigh integrin αvβ3 affinity (for structures of the best αvβ3 inhibitors from eachgeneration, see Figure 4.3B).
4.3.4. Amino acid replacement analysis for cysteines
It was also investigated whether replacement of L/D-cysteines by the non-naturalamino acid derivatives L/D-homocysteine (HCy) and L/D-penicillamine (Pen) canfurther improve the αvβ3 inhibition of 2nd generation lead CT3HWQCT3RGDcT3.Therefore, a library of 27 peptides, in which one, two or all cysteines were re-placed was screened at two different cocentrations (10 and 1 µM). The lead peptideshowed 88% inhibition at 10 µM and 55% inhibition at 1 µM (Appendix, p.103,Figure 4.10). A relatively small decrease in inhibitory activity was observed forthe bicycles CT3HWQ(L-HCy)T3RGDcT3 (85% at 10 µM/53% at 1 µM) andCT3HWQCT3RGD(D-HCy)T3 (80%/37%), whereas a huge decrease was observedfor (L-Pen)T3HWQCT3RGDcT3 (31%/8%) and (L-HCy)T3HWQCT3RGD(D-H-Cy)T3 (20%/12%). Generally, replacement of cysteine by Pen led to a much strongerdecrease in αvβ3 affinity than the replacement by HCy. For example, the bicy-cle CT3HWQ(L-HCy)T3RGDcT3 (85% at 10 µM) showed a relatively strong in-hibitory activity compared with CT3HWQ(L-Pen)T3RGDcT3 (0% at 10 µM). Also,replacement of the C-terminal D-Cys by D-Pen, for example, CT3HWQCT3RGD(D-Pen)T3 (0% inhibition at 10 µM) resulted in a complete loss of inhibition, regardlessof whether the other positions comprised Cys, HCy, Pen, or combinations thereof,which suggests that the additional methyl groups in penicillamine entirely block theintegrin-bicycle interaction.
83

4444
4.3. RESULTS & DISCUSSION
4.3.5. Screening of control single-loop peptides
In order to illustrate the importance of the bicyclic-peptide format, we synthesizedsingle-loop variants for some of the 1st generation lead αvβ3 inhibitors (CT3QAD-cT3RGDcT3, CT3HEQcT3RGDcT3, and CT3WGDCT3RGDcT3), in which one ofthe three D/L-cysteines was replaced by D/L-Ala. The peptides were then convertedto single-loop variants using scaffold 1,3-bis(bromomethyl)benzene (mT2), for ex-ample, CmT2QADaRGDcmT2, AQADcmT2RGDcmT2, and CmT2QADcmT2RGDa.This scaffold represents the bivalent equivalent of the trivalent T3 scaffold (see Fig-ure 4.4A). ForCT3QADcT3RGDcT3, the αvβ3 inhibitory activity (Appendix, p.106,Table 4.4) strongly decreased when opening the QAD-loop (AQADcmT2RGDcmT2,from 79% to 7% at 10 µM), the RGD-loop (CmT2QADcmT2RGDa, 0%), or whenone big loop was formed (CmT2QADaRGDcmT2, 32%). Likewise, a vast decrease inaffinity was observed for single-loop variants of bicyclic peptidesCT3HEQcT3RGDcT3
(from 92% to 16%, 22% and 28%, respectively) and CT3WGDCT3RGDcT3 (from75% to 23%, 52% and 8%, respectively), which exemplifies the essence of con-straining both loops for optimal αvβ3 inhibition. For comparison, the single-loop RGD peptides CmT2RGDCmT2 (23% at 10 µM), CmT2RGDcmT2 (57%) andcmT2RGDcmT2 (41%), showed much lower inhibition values as compared to the bestbicyclic peptides.Moreover, it was investigated whether the RGD loop can be further optimized forintegrin αvβ3 affinity by extension of the ‘RGD’ sequence, and/or by applicationof scaffolds comprising different arene substitution patterns of the bromomethylresidues (i.e. ortho, meta, para). Hence, various linear RGD peptides were syn-thesized (CRGDc, CGRGDc, CRGDSC, CVRGDfC and CGRGDSC), and cyclizedeither by S-S-oxidation (S-S), or by using scaffolds 1,2-bis(bromomethyl)benzene(oT2), 1,3-bis(bromomethyl)benzene (mT2), 3,5-bis(bromomethyl)pyridine (mP2)and 1,4-bis(bromomethyl)benzene (pT2, Figure 4.4A). The scaffoldmP2 representsa hydrophilic variant of mT2, and the scaffolds oT2 and pT2 are the ortho- andpara-substitution variants of mT2. Subsequently, the ability of these peptides wasmeasured to inhibit binding of the biotinylated knottin-RGD peptide (0.1 µM) tointegrin αvβ3 (Figure 4.4B).
The monocyclic RGD peptide CmT2RGDcmT2 (51%) showed much weaker in-hibition compared to bicycle CT3HWQCT3RGDcT3 (95%), which underlines theimportance of the second loop ‘HWQ’ for integrin αvβ3 inhibition. Extension
84

4444
4.3. RESULTS & DISCUSSION
Figure 4.4.: (A) Overview of bi- and trivalent scaffolds for the synthesis of mono-and bicyclic peptides; (B) αvβ3-inhibitory capacities of various mono-cyclic peptides. The value marked with an asterisk was taken frominhibition curves of IC50 determination. Inhibition values were cal-culated based on absorbance values with no bicyclic competitor(0%, OD405~1.8 A.U.) or non-labeled knottin-RGD at 30 µM (100%,OD405~0.2 A.U.) applied.
of the RGD loop consecutively led to lower inhibition values, ranging from 45%(CmT2GRGDcmT2) to 11% (CmT2GRGDScmT2). Surprisingly, CS-SRGDcS-S com-prised even slightly higher inhibition at 1 µM (81%) than cyclo-[KRGDf] (78%),however it cannot serve as a lead for bicycles, since there is no possibility to ex-tend the bivalent disulfide bond into the 3rd dimension. Two other monocyclicRGD peptides, CmP2RGDcmP2 (60%) and CpT2GRGDcpT2 (52%) showed morethan 50% inhibition. The slightly higher inhibition of CmP2RGDcmP2 (60%) overCmT2RGDcmT2 (51%) suggests that the use of a more water soluble scaffold, suchas 2,4,6-tris(bromomethyl)pyridine (“mP3”) might also improve the affinities of bi-cyclic αvβ3-binders (please note the formation of three different isomers for thiscompound). However, for the vast majority of the monocyclic peptides, especiallycontaining ‘VRGDf’ and ‘GRGDS’, inhibition rates below 40% were measured il-lustrating that changing the RGD loop size and/or constraint in bicyclic peptideswould probably not lead to overall higher inhibition values.
4.3.6. Testing streptavidin-binding of ‘HPQ’-containing bicycles
As mentioned before, the screening revealed several high-affinity αvβ3-binders com-prising ‘H’ and ‘Q’ in the second loop. Since the tripeptide ‘HPQ’ is known to bind
85

4444
4.3. RESULTS & DISCUSSION
streptavidin under certain circumstances,[32] it was important to exclude the possibil-ity that the bicycles containing ‘HPQ’ or similar motifs would interact with strepta-vidin in this competition ELISA setup. Therefore, selected aminooxy-functionalizedbicyclic and monocyclic peptides were incubated onto glutardialdehyde (GDA)-coated 96-well plates and their affinity to soluble streptavidin-HRP was measured(Appendix, p.106, Table 4.5). Interestingly, only the bicyclic peptide CT3HFQCT3-RGDcT3 and monocyclic control CmT2HPQcmT2 showed weak binding to strepta-vidin, but only at the highest concentration tested (100 µM) indicating that strep-tavidin does not bind the ‘HPQ’-motif in the relevant bicyclic peptides within theconcentration range of the competition ELISA experiments (30–0.005 µM).
4.3.7. Determination of affinity binding constants (Kd)
In addition to measuring the IC50 values in ELISA, the bicycle-integrin interac-tion was also measured in a direct manner using an optical technique that com-bines surface plasmon resonance (SPR) with surface plasmon-enhanced fluores-cence spectroscopy (SPFS).‖ For that purpose, one bicyclic integrin αvβ3-inhibitor(CT3HPQcT3RGDcT3), linear GRGDS, cyclo-[KRGDf] and knottin-RGD were func-tionalized with an N-terminal linker (K-PPPSG[Abz]SG, Abz: 4-aminobenzoic acid)following studies of Pallarola et al., suggesting that the use of this linker shouldnot affect the integrin-affinity of the bicyclic peptides.[33] In a first experiment, thebinding of RGD-bicycles and controls to gold surface-immobilized integrins was de-termined by measuring the change in refractive index induced by changes of surfaceplasmon resonance excitation upon binding of the bicycles (data not shown). How-ever, this approach lacked sensitivity, probably due to the low molecular weight ofthe bicycles (~2 kDa). Therefore, the same selection of peptides was labeled with afluorescent tag (Cy5),∗∗ and the surface plasmon field was applied at the wavelengthcoincident with the absorption band of Cy5 in order to locally excite fluorescencesignal in close proximity to the gold surface. This approach increases the fluores-cence signal originating from the surface, which allowed the performance of kineticmeasurements.[34] A thiol SAM biointerface architecture was employed with bind-
‖SPFS experiments were performed and analyzed by V. Jungbluth, N. Gisbert Quilis and J.Dostalek, AIT Austrian Institute of Technology GmbH, Tulln, Austria.
∗∗K(Cy5)-PPPSG[Abz]SG will be abbreviated before each sequence with the prefix “K(Cy5)-linker“. The syntheis of Cy5-functionalized peptides is described in Chapter 6, p.145.
86

4444
4.3. RESULTS & DISCUSSION
ing distances d<20 nm (Appendix, p.107, Figure 4.11A). Concentration-dependentfluorescence signal curves F(c) were normalized to ∆Fmax (value measured at sat-uration concentration), and fitted via Langmuir isotherms (Figure 4.5A). In thisway, dissociation equilibrium constants (Kd) were determined for K(Cy5)-linker-CT3HPQcT3RGDcT3 (optimized for αvβ3), and controls K(Cy5)-linker-GRGDS,cyclo-[K(K(Cy5)-linker)RGDf] and K(Cy5)-linker-knottin-RGD (Figure 4.5B).
Figure 4.5.: (A) Concentration-dependent, normalized fluorescence signals for se-lected Cy5-labeled peptides’ binding to integrin αvβ3; (B) Overviewof measured equilibrium dissociation constants (Kd). Linker:PPPSG[4-Abz]SG. Values for αvβ3 were obtained applying a thiolSAM surface, values for αvβ3 were obtained applying a 3D hydrogelsurface architecture (for details, see Materials & methods, p.98).
The SPFS analysis revealed that K(Cy5)-linker-CT3HPQcT3RGDcT3 binds αvβ3with a Kd of 0.4 nM (for SPR sensogram, see Appendix, p.107, Figure 4.11B),which is similar to that observed for K(Cy5)-linker-knottin-RGD (Kd=0.6 nM).These results are in line with the ELISA competition data (Figure 4.3A), wheresimilarly strong inhibition was measured for both the bicyclic and knottin-RGDpeptide. For cyclo-[K(K(Cy5)-linker)RGDf], a slightly weaker affinity constant of4.1 nM was determined, suggesting that the Cy5-linker functionalization affectscyclo-[KRGDf] binding to αvβ3 more significantly than for knottin-RGD or the bi-cyclic RGD-peptide (see IC50 values, Figure 4.3). Moreover, linear peptide K(Cy5)-linker-GRGDS did not show measurable binding to αvβ3 in SPFS (Kd >100 nM),which confirms the findings in ELISA that this peptide hardly interacts with inte-grin αvβ3.In order to validate selectivity for αvβ3, the same selection of peptides was testedfor binding to integrin α5β1. However, when applying a thiol SAM surface, the
87

4444
4.3. RESULTS & DISCUSSION
obtained fluorescence signal was too weak (data not shown). In order to reducethe effect of quenching, the average distance from the gold surface was increasedby using a hydrogel binding matrix (d: 100 nm). Interestingly, both Cy5-labeledCT3HPQcT3RGDcT3, cyclo-[KRGDf] and GRGDS (Kd: >100 nM) did not showbinding to α5β1, while Cy5-labeled knottin-RGD showed reasonable binding (Kd:9.0 nM).To additionally investigate the selectivity of unlabeled αvβ3-binding bicycles (andcontrol RGD peptides) competition ELISA experiments were performed.
4.3.8. Selectivity experiments (ELISA)
Finally, the selectivity of binding to three different integrins (αvβ3, αvβ5, α5β1)of the three highest-affinity bicycles (CT3HPQcT3RGDcT3, CT3HPQCT3RGDcT3
and CT3HSQCT3RGDcT3) was tested and compared to knottin-RGD and cyclo-[KRGDf] by measuring IC50 values (Table 4.1).All three bicyclic peptides showed superior selectivity for integrin αvβ3, whichmeans that zero affinity for both αvβ5 and α5β1 integrins was observed (IC50
>10,000 nM). In contrast to this, cyclo-[KRGDf] showed strong binding to αvβ5(IC50: 182 nM), whereas knottin-RGD bound all integrins with almost equally strongaffinity (αvβ5: 76 nM, α5β1: 114 nM) basically not expressing any sort of selec-tivity.†† These results reveal that bicyclic RGD peptides showed by far superiorselectivities for integrin αvβ3 as compared to several other known RGD peptides,e.g. cilengtide that showed high affinity for both αvβ3 and αvβ5.[16]
In a recent publication, Neubauer et al. describe RGD peptidomimetics with highαvβ3-selectivities over α5β1.[22] For the strongest binders, the IC50 ratio for αvβ3and α5β1 was 0.006 and 0.007, respectively. In contrast to this, this ratio is ≤ 0.003for the bicyclic αvβ3-binder CT3HPQcT3RGDcT3. Despite the small differences inmethodology, the integrin selectivity for the RGD-bicycles seems at least comparableif not slightly better than those of the peptidomimetics published by Neubauer etal.
††The IC50 values for cyclo-[KRGDf] and knottin-RGD slightly differ from the values reportedin Chapter 3, which can be explained by different experimental conditions.
88

4444
4.3. RESULTS & DISCUSSION
Table 4.1.: Selectivities for optimized αvβ3-binding bicyclic peptides, knottin-RGD, cyclo-[KRGDf], cilengitide and GRGDS. IC50 values were de-termined using competition ELISA via nonlinear regression analysisusing the software GraphPad Prism. Each concentration was tested intriplicate.
4.3.9. Conformational analysis of αvβ3-binding bicycles
Circular dichroism (CD) spectroscopy
In order to measure the secondary structure in solution, a CD (circular dichroism)analysis for the three highest affinity bicycles CT3HPQcT3RGDcT3, CT3HPQCT3-RGDcT3 and CT3HSQCT3RGDcT3 and two monocyclic RGD peptides (CmT2RG-DcmT2 and cmT2RGDcmT2) was performed (Figure 4.6A). Monocycle CmT2RG-DcmT2 shows a minimum at 203 nm (∆ε~-51,000 cm2dmol-1). For monocyclecmT2RG-DcmT2, only deviating in configuration of the N-terminal cysteine, thespectral shape is similar, but with a higher molar ellipticity ∆ε (~-7,900 cm2dmol-1)at the minimum (205 nm). Compared to typical spectra of secondary protein struc-tures (Figure 4.6B) the spectra of these monocycles reveal the presence of mainlyrandom coil/disordered structures in solution (green spectrum, Figure 4.6B). Thespectra of the bicyclic peptides resemble those for the monocycles, and only vary inmolar ellipticity. They show minima at 205 nm (∆ε ~-24,000– -42,500 cm2dmol-1).Like for the monocyclic peptides, the curve shape reveals the presence of randomcoil/disordered structures. This could be explained by the fact that the peptidesonly consist of nine amino acids, probably not allowing for the formation of definedsecondary structural motifs in solution.
89
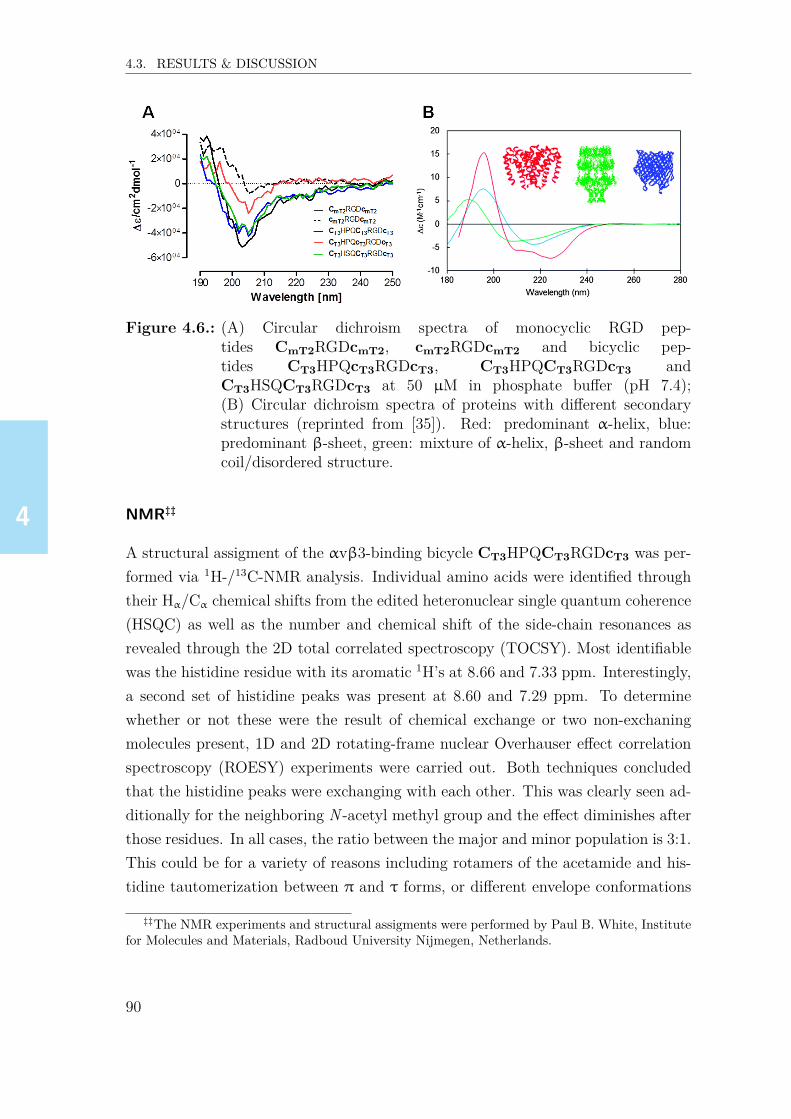
4444
4.3. RESULTS & DISCUSSION
Figure 4.6.: (A) Circular dichroism spectra of monocyclic RGD pep-tides CmT2RGDcmT2, cmT2RGDcmT2 and bicyclic pep-tides CT3HPQcT3RGDcT3, CT3HPQCT3RGDcT3 andCT3HSQCT3RGDcT3 at 50 µM in phosphate buffer (pH 7.4);(B) Circular dichroism spectra of proteins with different secondarystructures (reprinted from [35]). Red: predominant α-helix, blue:predominant β-sheet, green: mixture of α-helix, β-sheet and randomcoil/disordered structure.
NMR‡‡
A structural assigment of the αvβ3-binding bicycle CT3HPQCT3RGDcT3 was per-formed via 1H-/13C-NMR analysis. Individual amino acids were identified throughtheir Hα/Cα chemical shifts from the edited heteronuclear single quantum coherence(HSQC) as well as the number and chemical shift of the side-chain resonances asrevealed through the 2D total correlated spectroscopy (TOCSY). Most identifiablewas the histidine residue with its aromatic 1H’s at 8.66 and 7.33 ppm. Interestingly,a second set of histidine peaks was present at 8.60 and 7.29 ppm. To determinewhether or not these were the result of chemical exchange or two non-exchaningmolecules present, 1D and 2D rotating-frame nuclear Overhauser effect correlationspectroscopy (ROESY) experiments were carried out. Both techniques concludedthat the histidine peaks were exchanging with each other. This was clearly seen ad-ditionally for the neighboring N -acetyl methyl group and the effect diminishes afterthose residues. In all cases, the ratio between the major and minor population is 3:1.This could be for a variety of reasons including rotamers of the acetamide and his-tidine tautomerization between π and τ forms, or different envelope conformations
‡‡The NMR experiments and structural assigments were performed by Paul B. White, Institutefor Molecules and Materials, Radboud University Nijmegen, Netherlands.
90

4444
4.3. RESULTS & DISCUSSION
of the proline side chain. Considering the possibilities, the presence of tautomers ofhistidine is unlikely as the major and minor 1H/13C shifts were similar (Major/Minor1H // Major/Minor 13C: 8.66/8.60 ppm // 136.2/136.0 ppm and 7.33/7.29 ppm //130.8/130.7 ppm). Furthermore, the presence of different rotamers is unlikely as thepeptide contains an N -acetyl group, however the CT3HPQCT3RGDcT3 displayedexchange behavior. Therefore, it is likely the presence of different conformations ofthe proline that influences the chemical shift of neighboring residues and groups.This is evident also in the larger difference in the proline 1Hδ chemical shift: 3.92 vs3.66 ppm and 3.75 vs 3.50 ppm. The 1H and 13C chemical shifts of the major speciesresidues pointed to a similar conclusion that the peptide adopts more of a randomcoil structure rather than an ordered secondary structure (Figure 4.7). Consideringthe sharpness of the resonances, relaxation measurements were not pursued as thestructure is most likely monomeric.
Figure 4.7.: Chemical shift difference plots for Cα and Cβ calculated by ∆δ13Cα =δ13Cα,rc – δ13Cα,i and ∆δ13Cβ = δ13Cβ,i – δ13Cβ,rc (i: measured aminoacid in bicycle, rc: random coil). Positive values reflect more beta-sheet character while negative values represent more alpha-helicalcharacter. Amino acids that are close to the baseline are indicative ofrandom coil structure, or show both α-helical and β-sheet character,or alternatively structured sequences.
91

4444
4.3. RESULTS & DISCUSSION
4.3.10. Binding ELISA studies on biotinylated αvβ3-bindingRGD bicycles
In order to investigate whether the bicycles can serve as a surrogate binder forknottin-RGD in competition ELISA experiments, the highest affinity αvβ3-bindingbicycles and knottin-RGD peptide comprising a peptidic linker K-PPPSG[4-Abz]SG(as described in section 4.3.7, p.86) were functionalized with a biotin-tag, and sub-sequently tested at four different concentrations (1.0, 0.1, 0.01 and 0.001 µM)for binding to surface-immobilized integrin αvβ3. All three biotinylated bicy-cles showed similar absorbances at 1.0 µM (OD405~1.1–1.3 A.U.) and at 0.1 µM(OD405~1.0–1.1 A.U., Figure 4.8). Even at a concentration of 0.01 µM, still sig-nificant binding was measured, e.g. for biotin-functionalized CT3HPQcT3RGDcT3
(OD405~0.75 A.U.). At the lowest concentration tested (0.001 µM), only weak bind-ing was detected (OD405 ≤ 0.3 A.U.).
Figure 4.8.: Binding ELISA results for biotinylated αvβ3-binding bicyclesand biotinylated knottin-RGD. Background-subtracted absorbances(405 nm) are shown as a function of peptide concentration. Experi-ments were carried out in triplicate and error bars represent standarddeviations.
The results also reveal significantly higher binding levels of the biotinylated knottin-RGD at 0.01-1 µM as compared to the biotinylated bicyclic peptides even thoughthe IC50 for unlabeled knottin-RGD and bicycles were very similar (Figure 4.3,p.81). Most likely, covalent coupling of the biotin-linker to the bicyclic peptides ledto a significantly stronger decrease in αvβ3-affinity as compared to knottin-RGD,clearly resulting in lower binding levels. The overall binding levels, however, indicate
92

4444
4.3. RESULTS & DISCUSSION
that biotinylated RGD-bicycles can still serve as a surrogate binder in competitionELISA experiments.
4.3.11. Trimerization of RGD peptides and its effect onαvβ3-affinity
Peptide multimerization is a common approach to achieve higher protein bindinglevels, for example, to improve integrin-affinities of linear,[36] cyclic[37–39], or knottin-RGD peptides.[26] If a high-affinity ligand is needed as a tool to measure inhibitionin competition ELISA, but binding of a monomeric peptide to its target cannot bedetected, as observed in preliminary binding ELISA experiments for linear GRGDSpeptide (Chapter 3, p.52), this approach could result in significantly improved bind-ing. Hence, trimeric versions of each bicyclic αvβ3-binder (CT3HPQcT3RGDcT3),linear GRGDS and knottin-RGD were synthesized.First step was the synthesis of trivalent peptidic scaffold K-[GSGSK(S)]3, compris-ing a free N-terminal lysine for biotinylation, three lysine moieties with serines ontheir side chains, and a GSGS spacer between these lysines. The serines were thenoxidized with sodium periodate to form aldehydes. Subsequently, the N-terminallysine was biotinylated on its side chain, followed by oxime ligation with RGD pep-tides comprising the linker KPPPSG[4-Abz]SG and an aminooxy-group located atthe lysine side-chain (see Appendix, Figure 4.12, p.108).Subsequently, binding ELISA experiments were performed using four different con-centrations (1.0, 0.1, 0.01 and 0.001 µM) of monovalent and trivalent RGD pep-tides (Figure 4.9). As expected, the monomeric and trimeric knottin-RGD pep-tides showed the highest absorbances, followed by monomeric/trimeric RGD-bicycleCT3HPQcT3RGDcT3 and monomeric/trimeric GRGDS.For example, at 1 µM the highest absorbance was measured for monomeric knottin-RGD (OD405 ~1.75 A.U.) followed by trimeric knottin-RGD (OD405 ~1.4 A.U.) andmonomeric and trimeric CT3HPQcT3RGDcT3 (OD405 ~1.3 and 1.25 A.U.). At 0.1and 0.01 µM the absorbances for the bicyclic peptides were again slightly lowercompared with the knottin-RGD peptides. However, at the lowest concentrationtested (0.001 µM) the absorbance of trimeric bicycle CT3HPQcT3RGDcT3 was evenslightly higher than for trimeric knottin-RGD peptide. In sharp contrast to this, theabsorbances measured for monomeric GRGDS were much lower as compared to theother biotinylated peptides (OD405 ~0.31 A.U. at 1 µM). However, trimeric GRGDS
93

4444
4.3. RESULTS & DISCUSSION
showed ~50% higher absorbance (OD405 ~0.48 A.U. at 1 µM) as compared to themonomeric version, suggesting a significant higher αvβ3-affinity for this peptide.Also at lower concentrations (0.1, 0.01 and 0.001 µM) the absorbance for trimericGRGDS was significantly higher, and at 0.01 and 0.001 µM binding of monomericGRGDS was not detectable anymore.
Figure 4.9.: Binding ELISA applying monovalent and trivalent versions of knottin-RGD, GRGDS and CT3HPQcT3RGDcT3. Background-subtractedabsorbances (405 nm) are shown as a function of peptide concen-tration. Experiments were carried out in triplicate and error barsrepresent standard deviations.
Furthermore, the results reveal that the binding affinity of the trimeric as com-pared to the monomeric peptides is dependent on the concentration applied. Forexample, trimeric CT3HPQcT3RGDcT3 showed slightly lower binding as comparedto monomeric at 1 µM (OD405~1.25 vs. ~1.35 A.U.) and 0.1 µM (~1.0 vs. ~1.1 A.U.),whereas at 0.01 µM (~0.9 vs. ~0.75 A.U.) and 0.001 µM (~0.9 vs. ~0.3 A.U.) theabsorbance was significantly higher. The absorbances are probably the result of twoindependent factors, i.e. the molar ratio between RGD moieties and the integrin,and the affinity difference between monomeric and trimeric peptide. Consideringthe molecular weight of the integrin (~130 kDa, 105–150 kDa according to manu-facturer), approximately 0.2 pmol were coated per well. In contrast, the amount ofRGD moieties for monomeric/trimeric peptides lies between 100/300 pmol at 1 µMand 0.1/0.3 pmol at 0.001 µM. This is, for example, applying 0.001 µM of peptideapproximately means a two-fold molar shortfall of RGD moieties for the monomeric,and a 1.5-fold molar excess (0.1 pmol) for the trimeric peptide. At this approxi-mately equimolar concentration compared to integrin trimeric CT3HPQcT3RGDcT3
94

4444
4.4. CONCLUSION & OUTLOOK
binds with much higher affinity to integrin as compared to monomeric CT3HPQcT3-RGDcT3. At 0.1 or 1 µM, however, the RGD moieties were applied with high excessover integrin. It is fair to assume that in this concentration regime all RGD-bindingpockets are bound to an RGD moiety. However, per binding pocket, the trimericpeptide exposes only 1/3 of the biotin amount compared with the monomeric pep-tide, which would lead to a threefold lower signal in binding ELISA (OD405 –66%) ifthe affinities of both peptides were equal. However, the absorbance of the trimericbicycle was only slightly lower at 0.1 µM (OD405 0.98 vs. 1.11 A.U., –13%), whichmight reveal a higher affinity of the trimeric peptide. All these considerations, how-ever, are based on the assumptions that all RGD-moieties of the trimeric peptidesare bound to different integrin molecules, and that no RGD-moeity is unbound. Insummary, these results show that multimerization of RGD peptides represents an in-teresting approach for competition ELISA experiments, in particular when relativelylow biotinylated peptide concentrations (≤ 0.01 µM) are needed.
4.4. Conclusion & outlook
It was shown that bicyclic RGD peptides represent a novel platform for high-affinityand selectivity integrin αvβ3-binders. These peptides offer a straightforward, cost-effective and versatile alternative for established binders, such as knottin-RGD orcyclo-[KRGDf], and the observed high selectivities suggest applications in integrin-mediated in vivo tumor staining, cancer diagnostics, and/or cancer therapeutics.The staining of cell membranes using fluorescently labeled bicycles will be describedin Chapter 6. Moreover, the bicyclic RGD peptides were evaluated for integrin-mediated cell adhesion and proliferation, e.g. via covalent functionalization of hy-drogels, the results of which will be described in Chapters 7 and 8.Finally, the high affinities and selectivities observed for integrin αvβ3 also initiatedthe idea for screening of these bicyclic peptide libaries for novel high-affinity bindersto integrins αvβ5 and α5β1 that mediate cell interactions. The results of thesestudies which will be described in Chapter 5.
95

4444
4.5. MATERIALS & METHODS
4.5. Materials & methods
Reagents & chemicals
Incubation and washing buffers were prepared using standard protocols. Recom-binant human integrins were purchased from R&D Systems (Minneapolis, USA).Strep-HRP (Streptavidin-Horseradish Peroxidase conjugate, Southern-Biotech, Birm-ingham, USA) was diluted 1:1000 for ELISA experiments. Amino acids were pur-chased from Iris Biotech (Marktredwitz, Germany) and Matrix Innovation (Quebec,Canada). Resins were purchased from Rapp Polymere (Tübingen, Germany) andMerck (Darmstadt, Germany). MnCl2·4H2O and 1,3,5-tris(bromomethyl)benzenewere purchased from Sigma-Aldrich (Steinheim, Germany). CaCl2·2H2O and MgCl2·6H2O were purchased from Merck (Darmstadt, Germany). Tween80 was purchasedfrom Faryon (Capelle, the Netherlands) and I-Block™ was purchased from Tropix(Bedford, USA).
Peptide synthesis
Purified knottin-RGD peptide, cyclic and linear RGD peptides and bicyclic pep-tides were synthesized according to published protocols.[24, 40, 41] Random peptidesequences XXX (where X represents any natural amino acid except cysteine or me-thionine) were generated using the software R. Peptide libraries were synthesized(modified peptide synthesizer Syro II, MultiSynTech; cysteine couplings were per-formed manually) via solid-phase peptide synthesis (SPPS) using Fmoc strategy ona TentaGel® Ram resin (2 µmol) followed by acetylation and full deprotection usinga cleavage mixture (TFA/DODT/H2O/thio-anisole/triisopropylsilane volume ratio95:5:5:2.5:2.5). Libraries were lyophilized, dissolved at 4 mM in MeCN and con-verted to bicyclic peptides by adding 1.1 equiv. of 1,3,5-tris-(bromomethyl)benzene(4.1 mM in MeCN) followed by adjusting to pH 8 with 150 mM ammonium bicar-bonate solution. After ~45 min. reactions were quenched with 0.5% ethanethiol inH2O/MeCN (1:1), and the bicyclic libraries were lyophilized. The peptides were dis-solved in 200 µL DMSO to achieve a concentration of approx. 10 mM, and furtherdiluted to 2.5-100 µM using milliQ water.
96

4444
4.5. MATERIALS & METHODS
Peptide analysis and purification
UPLC analysis was performed on a Waters Acquity Ultra Performance LC System,equipped with a Waters Acquity UPLC BEH130 C18 1.7 µm column. A linear gra-dient of 5–55% MeCN (0.05% TFA) in H2O (0.05% TFA) was used. If not stateddifferently, the peptides were purified by preparative HPLC (Waters Prep LC) onan RP-C18 column (Reprosil-Pur 120 C18-AQ 150x20 mm, Dr. Maisch GmbH,Ammerbuch, Germany) using a MeCN/milliQ H2O gradient including 0.05% TFAfollowed by lyophilization on a Christ Alpha 2-4 LDplus lyophilizer.An overview of the UPLC data for the peptides described in this chapter contain-ing non-functionalized, linker-modified, biotinylated, Cy5-functionalized peptides,trimeric scaffolds and multimeric peptides is shown in the appendix (p.109, Ta-ble 4.6).
Competition ELISA
Competition ELISA studies were performed like described in Chapter 3 with sev-eral modifications. Plates were coated with 100 µL of an integrin solution (0.25-0.5 µg/mL, in PBS buffer containing 1 mM CaCl2 and 0.5 mM MgCl2 (PBSMg))onto 96-well Polysorp© plates at 4 ℃ overnight followed by blocking with 150 µL1% I-Block™ in 0.05% Tween80/PBS with 1.0 mM CaCl2 and 0.5 mM MgCl2(PBSTMg) for 1 h at room temperature. After 3x washing with 400 µL 0.05%Tween80/PBS with 1.0 mM CaCl2 and 0.5 mM MnCl2 (PBSTMn), plates were in-cubated with 100 µL of biotinylated peptide solution in (PBSMg) for 90 min at r.t.After washing 3x with PBSTMn the plates were incubated with 100 µL of 1:1000Strep-HRP in PBSMg (1 h, r.t.). After washing the plates 4x with PBSTMn, theywere incubated with 150 µL substrate buffer containing 0.91 mM ABTS (2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) and 0.006% H2O2 in substrate buffer(0.2 M Na2HPO4 adjusted to ~pH 4 using 0.2 M citric acid). Absorbance was mea-sured after 45 min using a Molecular Devices Spectramax M2 plate reader. All exper-iments were carried out in triplicate. IC50 were calculated via non-linear regressionanalysis using the GraphPad Prism software. This value represents the concentra-tion of peptide that is required to inhibit binding of the biotinylated knottin-RGDpeptide 1a (Chapter 3, p.53, figure 3.1A) with 50%.
97

4444
4.5. MATERIALS & METHODS
Surface plasmon-enhanced fluorescence spectroscopy (SPFS)
For measuring the binding affinity of Cy5-labeled peptides to immobilized integrinligands, PBS with 1 mM CaCl2, 0.5 mM MnCl2, 1 mg/mL BSA and 0.05% Tween20was used as running buffer. Different concentrations of the peptide (0.1, 1, 5, 10,50, 100, and 1000 nM) were sequentially flushed over the sensor surface. Each con-centration was allowed to react with the integrin for 30 minutes followed by rinsingthe surface with running buffer solution for 10 min. The binding of target analytewas monitored in real-time by measuring the fluorescence intensity F(t) originat-ing from the close proximity to the sensor surface that was probed by resonantlyexcited surface plasmons (Appendix, p.4.11, Figure 4.11B). The fluorescence signalF gradually increased upon binding of target analyte, and for each concentration,the equilibrium fluorescence signal ∆F was determined as a difference to the fluo-rescence baseline after 10 min rinsing with running buffer. The titration curve wasestablished based on these values and it was fitted with a Langmuir isotherm model[function ∆F = ∆Fmaxc/(Kd+c)] in order to determine the equilibrium dissociationconstant Kd.
Circular dichroism (CD) spectroscopy
CD spectra were recorded applying a JASCO J-810 CD spectrometer equipped withan air-cooled 150 W Xenon arc lamp, a computer-controlled Peltier device and aquartz cuvette with a pathlength of 1 mm. Working concentrations of 50 µM peptidein phosphate buffer pH 7.4 and (optional) 20 µM urea were applied, since at theseconcentrations absorbances ≤ 1.0 were detected over the entire wavelength (250–190 nm) for all peptides. In order to avoid ozone formation the CD spectrometer wasflushed with N2 for ~10 min prior use, whereas during the experiments a continuousflow of ~20 psi was applied.
NMR
All NMR spectra were collected on a Bruker Avance III 500 MHz spectrometerequipped with a Prodigy BB cryoprobe at 298 K. Samples were prepared by dissolv-ing the compounds in D2O and adding a small amount of 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) for internal referencing. 1H spectra were acquired using 32scans and a relaxation delay of 3 s. 2D COSY-DQF spectra with presaturationwere acquired with a 6000 Hz spectral width in both dimensions using 2048 x 512
98

4444
4.5. MATERIALS & METHODS
points and processed using 2048 x 512 points, 4 scans per increment and a relax-ation delay of 1.5 s. 2D gradient TOCSY spectra with presaturation were acquiredwith a 5000 Hz spectral width in both dimensions using 1024 x 512 points andprocessed using 1024 x 1024 points, 8 scans per increment, a relaxation delay of2 s and a TOCSY mix time of 100 ms. A TOCSY spinlock field of 8.3 kHz wasapplied. Multiplicity-edited 1H-13C HSQC spectra were acquired using a 6010 Hzspectral width in F2 and 18868 Hz spectral width in F1 using 1024 x 512 pointsand processed to 1024 x 1024 points, 2 scans per increment, relaxation delay of 1.5 sand 1-bond JCH = 145 Hz. 1H-13C HMBC spectra were acquired using a 5319.1 Hzspectral width in F2 and 22321.4 Hz spectral width in F1 using 2048 x 512 pointsand processed to 2048 x 2048 points, 4 scans per increment, relaxation delay of 1.5 sand a long-range JCH = 8 Hz.T1 measurements were performed by properly calibrating the 90 degree pulse lengthand then performing estimates using the 1D inversion recovery sequence with exci-tation sculpting water suppression. After the longest T1 was determined to be ap-proximately 2 s, a pseudo-2D inversion recovery experiment was performed with 10separate delays of 8 scans each with a total longitudinal relaxation time of 10.3 s. T2
measurements were acquired by first performing estimates using the 1D PROJECT-CMPG sequence with presaturation water suppression.[42] After the longest T2 wasdetermined to be approximately 1 s, a pseudo-2D PROJECT-CPMG sequence ex-periment with presaturation was performed with 12 separate delays of 8 scans each,a cycle time of 4 ms with a total longitudinal relaxation time of 10.3 s.
99
4444

4444
4.6. REFERENCES
4.6. References[1] E. M. Driggers, S. P. Hale, J. Lee, et al., Nature Rev. Drug Discov. 2008, 7,
608–624.[2] D. J. Craik, D. P. Fairlie, S. Liras, et al., Chem. Biol. Drug Des. 2013, 81,
136–147.[3] C. K. Wang, D. J. Craik, Biopolymers 2016, 106, 901–909.[4] H Li, N. S. Sampson, J. Peptide. Res. 2002, 59, 45–54.[5] P. Timmerman, R. Barderas, J. Desmet, et al., J. Biol. Chem. 2009, 284,
34126–34134.[6] V. Baeriswyl, C. Heinis, ChemMedChem 2013, 8, 377–384.[7] P. Timmerman, J. Beld, W. C. Puijk, et al., ChemBioChem 2005, 6, 821–
824.[8] C. Heinis, T. Rutherford, S. Freund, et al., Nat. Chem. Biol. 2009, 5, 502–
507.[9] A. Angelini, L. Cendron, S. Chen, et al., ACS Chem. Biol. 2012, 7, 817–821.[10] I. Rentero Rebollo, S. McCallin, D. Bertoldo, et al., ACS Med. Chem. Lett.
2016, 7, 606–611.[11] W. Lian, B. Jiang, Z. Qian, et al., J. Am. Chem. Soc. 2014, 136, 9830–9833.[12] T. B. Trinh, P. Upadhyaya, Z. Qian, et al., ACS Comb. Sci. 2016, 18, 75–85.[13] S. Luzi, Y. Kondo, E. Bernard, et al., Protein Eng. Des. Sel. 2015, 28, 45–52.[14] M. Barczyk, S. Carracedo, D. Gullberg, Cell Tissue Res. 2010, 339, 269–280.[15] Y. Takada, X. Ye, S. Simon, Genome Biol. 2007, 8, 215.[16] M. A. Dechantsreiter, E. Planker, B. Matha, et al., J. Med. Chem. 1999, 42,
3033–3040.[17] L. Marinelli, A. Lavecchia, K.-E. Gottschalk, et al., J. Med. Chem. 2003, 46,
4393–4404.[18] D. Heckmann, A. Meyer, L. Marinelli, et al., Angew. Chem. Int. Ed. 2007,
46, 3571–3574.[19] A. Bochen, U. K. Marelli, E. Otto, et al., J. Med. Chem. 2013, 56, 1509–
1519.[20] F. Rechenmacher, S. Neubauer, J. Polleux, et al., Angew. Chem. Int. Ed.
2013, 52, 1572–1575.[21] J. D. Hegemann, M. De Simone, M. Zimmermann, et al., J. Med. Chem.
2014, 57, 5829–5834.[22] S. Neubauer, F. Rechenmacher, R. Brimioulle, et al., J. Med. Chem. 2014,
57, 3410–3417.
101

4444
4.6. REFERENCES
[23] O. V. Maltsev, U. K. Marelli, T. G. Kapp, et al., Angew. Chem. Int. Ed.2016, 55, 1535–1539.
[24] R. H. Kimura, A. M. Levin, F. V. Cochran, et al., Proteins 2009, 77, 359–369.
[25] R. H. Kimura, R. Teed, B. J. Hackel, et al., Clin. Cancer Res. 2012, 18,839–849.
[26] J. W. Kim, F. V. Cochran, J. R. Cochran, J. Am. Chem. Soc. 2015, 137,6–9.
[27] L. Aucoin, C. M. Griffith, G. Pleizier, et al., J. Biomater. Sci. Polymer Edn2002, 13, 447–462.
[28] A. Rezania, K. E. Healy, Biotechnol. Progr. 1999, 15, 19–32.[29] M. B. Rahmany, M. Van Dyke, Acta Biomater. 2013, 9, 5431–5437.[30] P Timmerman, J Beld, R. H. Meloen, et al., Method for Selecting a Candidate
Drug Compound, 2004.[31] M. Aumailley, M. Gurrath, J. Calvete, et al., FEBS J. 1991, 291, 50–54.[32] L. B. Giebel, R. T. Cass, D. L. Milligan, et al., Biochemistry 1995, 34, 15430–
15435.[33] D. Pallarola, A. Bochen, H. Boehm, et al., Adv. Funct. Mater. 2014, 24,
943–956.[34] M. Bauch, K. Toma, M. Toma, et al., Plasmonics 2014, 9, 781–799.[35] A. J. Miles, B. A. Wallace, Chem. Soc. Rev. 2016, 45, 4859–4872.[36] Z.-H. Jin, T. Furukawa, A. Waki, et al., Biol. Pharm. Bul. 2010, 33, 370–378.[37] D. Lössner, H. Kessler, G. Thumshirn, et al., Anal. Chem. 2006, 78, 4524–
4533.[38] I. Dijkgraaf, A. Y. Rijnders, A. Soede, et al., Org. Biomol. Chem. 2007, 5,
935–944.[39] C. Wängler, S. Maschauer, O. Prante, et al., ChemBioChem 2010, 11, 2168–
2181.[40] D. Bernhagen, L. De Laporte, P. Timmerman, Anal. Chem. 2017, 89, 5991–
5997.[41] Y. Sohma, B. L. Pentelute, J. Whittaker, et al., Angew. Chem. Int. Ed. 2008,
47, 1102–1106.[42] J. A. Aguilar, M. Nilsson, G. Bodenhausen, et al., Chem. Commun. 2012,
48, 811–813.
102

4444
4.7. APPENDIX
4.7. AppendixThe appendix contains an overview of paramters in the competition ELISA setups(Table 4.2), a list of all amino acids applied in the screenings (Table 4.3), molecu-lar drawings of derivatives of cysteine and inhibition values for various derviativesof a 2nd generation αvβ3-binders (Figure 4.10), a table of inhibibitory activitiesfor selected bicyclic αvβ3-binders and their monocyclic analogues (Table 4.4), abinding ELISA study to measure streptavidin affinity of selected 2nd generationαvβ3-selective bicycles (Table 4.5), an example of an SPR sensogram and an ex-ample of fluorescence signal kinetics acquired in an SPFS experiment (Figure 4.11),a schematic representation of the formation of trivalent peptides (Figure 4.12) anda complete list of UPLC/ESI-MS data obtained for all non-modified and modified,purified RGD peptides (Table 4.6).
Figure 4.10.: (A) Molecular drawings of L/D-cysteine (Cys) and non-natural aminoacids L-homocysteine (HCy), L-penicillamine (Pen), D-homocysteine(1) and D-penicillamine (2); (B) Inhibition values (using setup 2,Table 4.2B) for various derivatives of 2nd generation αvβ3-inhibitorCT3HWQCT3RGDcT3.
103

4444
4.7. APPENDIX
Table 4.2.: Parameters varied in the two different competition ELISA setups (A),and selected setups for library screening and IC50 determination (B).
104

4444
4.7. APPENDIX
Table 4.3.: List of amino acis applied in this study including short codes, 1-lettercodes and protective groups.
105

4444
4.7. APPENDIX
Table 4.4.: Inhibitory activities (determined using setup 1, Table 4.2B) for αvβ3integrin for selected bicyclic peptides (three cysteines, constrained with1,3,5-tris(bromomethyl)benzene, T3) and their monocyclic analogues(two cysteines, constrained with 1,3-bis(bromomethyl)benzene, mT2).
Table 4.5.: Binding ELISA studies to measure streptavidin-affinity of selected2nd generation αvβ3-binding bicyclic peptides and control sequences(monocyclic HPQ and RGD, and biotinylated linear GRGDS). Ab-sorbance values are means of triplicates including standard deviation.
106

4444
4.7. APPENDIX
Figure 4.11.: Example of (A) SPR sensorgram showing covalent immobilizationof integrin αvβ3 ligand into a 3D hydrogel binding matrix, and (B)fluorescence signal kinetics acquired upon titration of K(Cy5)-linker-CT3HPQcT3RGDcT3.
107

4444
4.7. APPENDIX
Figure 4.12.: Formation of trimeric RGD-peptides. In the scaffold K-[GSGSK(S)]3, the serine residues connected to lysine side-chains areoxidized to aldehydes with sodium periodate. After biotinylationof the oxidized scaffold, the RGD-variants R, bearing an aminooxygroup on their lysine side chain, are connected via oxime ligation.
108

4444
4.7. APPENDIX
Table 4.6.: Molecular weights of non-modified and modified, purified RGD pep-tides described in Chapter 4, determined via UPLC/ESI-MS analysis.Weights were calculated using ChemDraw software.
109

4444

55555
5. Screening for High Affinity andSelectivity Bicyclic RGD-Bindersto Integrins α5β1 and αvβ5
Parts of the work described in this chapter were published: D. Bernhagen, V. Jungbluth, N.Gisbert Quilis, J. Dostalek, P. B. White, K. Jalink, P. Timmerman, ACS Comb. Sci. 2019, 21,598–607.

55555
5.1. ABSTRACT
5.1. Abstract
In this chapter, the screening of bicyclic peptide libraries for high-affinity bindersto integrins α5β1 and αvβ5 is described. Screening of 1st generation libraries,comprising the universal integrin-binding sequence RGD in the first loop, and arandomized sequence (XXX) in the second loop, yielded a sub-micromolar α5β1-binder (CT3RGDcT3AYGCT3, IC50: 406 nM), and a low micromolar αvβ5-binder(CT3RGDcT3NWGCT3, IC50: 1457 nM). Next generation libraries were designedby partially varying the structure of the strongest 1st generation lead inhibitors andscreening for improved affinities and selectivities for both receptors. In this way, asingle medium-affinity αvβ5-binding bicycle CT3RGDcT3NWaCT3, IC50: 650 nM),and three high-affinity α5β1-binders (CT3RGDcT3AYJCT3, J: D-Leu, IC50: 90 nM;CT3RGDcT3AYaCT3, IC50: 156 nM; CT3RGDcT3AWGCT3, IC50: 173 nM) wereidentified, one of which showed an even higher α5β1-affinity than the 32 aminoacid benchmark knottin-RGD peptide (IC50: 114 nM). Affinity data were confirmedfor the α5β1-binding bicycle by SPFS analysis showing a Kd of 4.1 nM for Cy5-labeled RGD-bicycle CT3RGDcT3AYJCT3 and a slightly higher Kd (9.0 nM) for theCy5-labeled benchmark knottin-RGD. Ala-replacement analysis data for all threeα5β1-binders did confirm the essential role of both the bicyclic structure for thesebinders and also the presence of the second loop. Furthermore, the α5β1-bindingbicycles showed excellent selectivities over αvβ5 (typical IC50 ratio α5β1/αvβ5<0.009 and <0.017, respectively) and acceptable selectivities over αvβ3 (typicalIC50 ratio α5β1/αvβ3 between 0.090 and 0.157).
5.2. Introduction
Integrins α5β1 and αvβ5 represent two of 24 different heterodimeric transmembranereceptor proteins[1, 2] that are involved in many cellular processes, such as signaling,proliferation, migration and differentiation via interaction with extracellular matrixproteins such as vitronectin and fibronectin.[3, 4] For example, they fulfill a varietyof different functions in angiogenesis next to other integrins like α1β1, α2β1, α4β1,α6β1, α6β4, α9β1 and αvβ3.[5] Furthermore, integrin α5β1, when overexpressedin breast cancer cells, increases its invasiveness into 3D collagen fiber matrices bythreefold as compared to cells with low levels of α5β1.[6] Integrin αvβ5 has beenless thoroughly investigated as compared to integrin α5β1, and its action is gen-
112

55555
5.2. INTRODUCTION
erally described in alignment with αvβ3.[7–9] For example, integrin αvβ5 controlsthe internalization of vitronectin from the extracellular matrix in human foreskinfibroblasts,[10] and improves the vitronectin-dependent activation of plasminogenactivater inhibitor-1 in scleroderma fibroblasts.[11] In general, integrins exhibit in-tensive crosstalk amongst each other.[4] For example, integrin α5β1 regulates the invitro and in vivo function of αvβ3,[12] and both integrins αvβ3 and αvβ5 individu-ally direct human cardiac myofibroblast differentiation via activation of TGF-β1.[9]
However, αvβ5 promotes angiogenesis that is induced by VEGF in in vivo cornealmodels, while αvβ3 supports bFGF-induced angiogenesis.[13]
The expression of integrins is dynamic and depends on the microenvironment anddevelopmental age of a particular cell.[1] For example, undifferentiated epidermal andneural stem cells overexpress β1, while adipose-tissue derived stem cells overexpressα5β1 in undifferentiated state as well as during chondrogenic differentiation.[14] Incontrast to this, integrin αvβ5 is overexpressed in scleroderma fibroblasts.[11] More-over, the expression pattern of integrins depends on the tissue. For example, studiesin brain metastases and their corresponding primary tumors revealed high expres-sion of αvβ6 in lung adenocarcinoma, but no expression in prostate carcinoma ormelanoma,[15] whereas glioblastoma showed high levels of αvβ3 expression.[16, 17]
In order to target the wide variety of cellular integrin receptors, either for biomate-rial, therapeutic or imaging applications, it is strongly preferred to apply selectiveand high-affinity integrin binding molecules, or – more specifically – peptides. So far,several high-affinity, but non-selective αvβ5 and α5β1 integrin-binders have beenreported, including cilengitide,[18] knottin-RGD (αvβ3/αvβ5/α5β1-binders),[19–21]
and echistatin (binds αvβ3, αvβ5, αvβ6, αvβ8, α5β1 and αIIbβ3).[22, 23] More se-lective ligands, for example the α5β1-selective PHSCN (ATN-161)[24, 25] or severalα5β1-binding peptidomimetics[26] recently also raised interest as potential cancertherapeutics. When covalently linking the peptidomimetics to a gold surface, α5β1-transfected fibroblasts showed a much higher level of cell adhesion as compared toαvβ3-transfected cells.[27] However, the manufacturing of peptidomimetics often re-quires complex multistep syntheses and therefore does not yet allow for affordablelarge-scale production. Novel peptidic α5β1-integrin-binders would circumvent thisissue. Very recently, Kapp et al. reported N -methylated, cyclic isoDGR peptideswith high α5β1-selectivity over integrins αvβ6 and αvβ3.[28]
Bicyclic RGD peptides could provide an attractive alternative due to their straight-forward synthesis and relative ease of N-terminal functionalization. Furthermore, a
113

55555
5.3. RESULTS & DISCUSSION
thorough research of relevant literature did not yield any result for selective peptidicαvβ5-binders. Therefore, the excellent integrin αvβ3-affinities and selectivities thatwere identified for several bicyclic RGD-peptides (see Chapter 4) raised interest toalso search for similar selective binders to α5β1 and αvβ5 integrins. This chapterdescribes both the design and the experimental work on the screening and opti-mization of bicyclic RGD-peptide libraries for high-affinity binders to each integrinsα5β1 and αvβ5.
5.3. Results & discussion
5.3.1. Design & synthesis of randomized RGD-peptide libraries
Design and synthesis of the bicyclic RGD peptide libraries was basically identical asdescribed for the αvβ3-binder selections in Chapter 4. Briefly, the libraries consist oftwo separate trimeric peptide motifs, surrounded by three different cysteine residues(in total 9-mer peptides). The first and most important motif comprises the well-known RGD-sequence that should provide the basic integrin affinity to the bicyclicbinder, while the second motif contains a trimeric random peptide sequence (‘XXX’),which is intended to introduce binding selectivity for the different integrin targets,and also to further improve the overall binding affinities. The trimeric motifs are en-closed by three different cysteine residues (either L- or D), which allow for the doubleCLIPS -cyclization via reaction with the scaffold molecule 1,3,5-tris(bromomethyl)benzene (T3, Figure 5.1), and hence formation of a bicyclic peptide comprising twodifferent loops.The RGD sequence was located either in the N-terminal (left) loop and the random‘XXX’ sequence in the C-terminal (right) loop, or vice versa. Seven different lin-ear libraries (C(c)RGDC(c)XXXC), CXXXC(c)RGDC(c), “C(c)” indicates twodifferent libraries with either L- or D-Cys at this position), each containing 96 pep-tides, were synthesized and converted to bicyclic peptides via reaction with T3 atbasic pH under high-dilution conditions (Figure 5.1), followed by quenching with0.5% ethane thiol and 2x lyophilization. An overview of all amino acids used in thisscreening is given in the Appendix (p.138, Table 5.6).
114

55555
5.3. RESULTS & DISCUSSION
5.3.2. General procedure for library screeningThe bicyclic RGD peptide libraries were then screened for integrin-inhibition usingexperimental setups basically identical to that described in Chapter 4 (Appendix,p. 139, Table 5.7). All 672 bicyclic RGD-peptides were screened in a competi-tion ELISA at one single concentration (i.e. 10 µM) for inhibition of biotinylatedknottin-RGD binding to both integrin receptors α5β1 and αvβ5. For each integrin,all peptides were ranked in order of decreasing inhibitory capacity, and from this aselection of the best 1st generation binders (two binders for α5β1, one binder forαvβ5) was determined. Based on the highest affinity 1st generation lead binder foreach integrin, the design of the 2nd generation libraries was performed. These li-braries contained peptides comprising extended RGD loops as well as peptides withnon-extended RGD loops in combination with a position replacement analysis of thesecond loop (Figure 5.1).∗ The 2nd generation libraries were screened in a competi-tion ELISA at 5 µM, followed by a second screening (contrary to the 1st generationscreening) with the best hits at 2.5 µM in order to determine the differences in theiraffinities for the integrins more precisely. Based on this screening, the best hitswere re-synthesized and HPLC-purified, followed by determination of IC50 values,yielding six 2nd generation integrin α5β1-binders and three 2nd generation αvβ5-binders. For the three highest-affinity α5β1-selective bicycles an Ala-replacementanalysis was performed to confirm the essential role of each loops for α5β1-binding.Furthermore, a replacement analysis with non-canonical amino acids was performedwith the intention to further improve integrin affinity, followed by an integrin selec-tivity study using competition ELISA.
5.3.3. Screening for α5β1-binding peptides1st generation screening
The 1st generation bicycle libraries were screened for binding to integrin receptorα5β1 at a concentration of 10 µM in a competition ELISA setup with biotiny-lated knottin-RGD as described for αvβ3 (Chapter 4).† For this receptor onlynine out of 672 peptides (1.3%) showed >70% inhibition, and all of these peptidescomprised the RGD motif in the left loop enclosed by an N-terminal L-Cys and a
∗The design of the 2nd generation libraries was equivalent to the design of the 3rd generationlibraries for the determination of αvβ3-selective bicycles (see Chapter 4).
†The OD405 values of all 1st and 2nd generation library screenings are depicted in the Supple-mentary Information, Section A.2, p.221.
115

55555
5.3. RESULTS & DISCUSSION
Figure 5.1.: Methodology for the design of 1st generation bicyclic RGD peptidelibraries, and the optimization process for the determination of high-affinity bicyclic peptides to integrins α5β1 (A) and αvβ5 (B). ‘X’represents any canonical L-amino acid. L-cysteines are indicated inyellow, D-cysteines in orange, and RGD motifs in green. Lead motifsare shown in light blue (A) and ruby (B), respectively.
“central” D-Cys. Still 29 peptides (4%) showed >50% inhibition, whereas 540 pep-tides (80%) showed inhibition levels <30%. Two bicycles, CT3RGDcT3AYGCT3
(100% inhibition) and CT3RGDcT3NWGCT3 (91% inhibition) showed outstand-ing binding at 10 µM. These two hits were subsequently re-synthesized, purifiedand tested for inhibition at concentration range of 30 µM down to 0.014 µM. ForCT3RGDcT3AYGCT3 an IC50 value of 406 nM was determined, whereas bicycleCT3RGDcT3NWGCT3 exhibited an IC50 >2 µM (Table 5.1A). The unexpected dis-crepancy between percentage inhibition of the latter peptide in the screening (91%at 10 µM) and IC50 (>2 µM) may be partly explained by the fact that peptidesof different purity were applied (screening: crude, >70%; IC50 determination: puri-fied, >95%). Due to oligo-/polymeric impurities, the IC50 value of purified bicyclemay be much higher than the apparent binding ability of the crude bicycle. The
116

55555
5.3. RESULTS & DISCUSSION
discrepancy could also be partly explained by the application of different setups inscreening and IC50 determination (Appendix, p.139, Table 5.7B). Remarkably, theIC50 for CT3RGDcT3AYGCT3 (406 nM) was much lower than observed for cyclo-[KRGDf] and cilengitide (each >10,000 nM), while it was ~3.5-fold higher than forknottin-RGD (114 nM). The much lower affinity of the bicycle as compared to cy-clo-[KRGDf] reveals that the additional loop (AYG) selectively binds parts of α5β1,and/or introduces a conformational change into the peptide that results in a higherα5β1 affinity of the RGD loop. The fact that in the high-affinity knottin-RGDpeptide the RGD motif is presented within a multicyclic structure suggests thatcertain constrained peptides in proximity to RGD stabilize the RGD-α5β1 bindinginteraction.
2nd generation screening
Based on the sequence of CT3RGDcT3AYGCT3, 2nd generation libraries were de-signed including 1) a full position replacement analysis of the lead ‘AYG’, i.e.CT3RGDcT3XYGCT3,CT3RGDcT3AXGCT3,CT3RGDcT3AYXCT3, 2) RGD-loopsextended by an additional amino acid ‘X’ while keeping the ‘AYG’ loop constant,i.e. CT3RGDcT3XYGCT3, CT3XRGDcT3AYGCT3, CT3RGDXcT3AYGCT3, and3) the extended 5-mer loops ‘GRGDX’ and ‘XGRGDS’ and a constant ‘AYG’ loop,i.e. CT3XRGDScT3AYGCT3, CT3GRGDXcT3AYGCT3 (Figure 5.1, p.116).‡ ‘X’represents all canonical L-amino acids and a number of selected non-natural aminoacids.§ All 196 2nd generation bicycles were screened at 5 µM, and 18 bicycles (9%)identified with inhibition rates >70%. In a second screening of the best 20 hits at2.5 µM, the top 15 peptides (75%) showed >70% and the top seven peptides (35%)even showed >85% inhibition. Also, eight of the 20 peptides exhibited higher inhi-bition values than the parent lead sequence CT3RGDcT3AYGCT3 (82% inhibition),for example CT3RGDcT3VYGCT3 (90% inhibition) or CT3RGDcT3AYaCT3 (87%inhibition).Following this, the best six 2nd generation hits were re-synthesized at larger scale(20 µmol), purified by preparative HPLC, and their inhibitory activity analyzedat various concentrations (30 µM down to 0.014 µM) to determine their IC50 val-
‡As a consequence of the successful screening of 3rd generation αvβ3-binders (Chapter 4) therespective design concept was selected for the design of 2nd generation α5β1 libraries.
§A list of all applied amino acids in the library screenings is depicted in the Appendix, Table 5.6
117

55555
5.3. RESULTS & DISCUSSION
ues. Remarkably, the IC50 of the best inhibitor CT3RGDcT3AYJCT3 (90 nM, J:D-Leu, for UPLC/MS spectrum see Appendix, p.137, Figure 5.6) was even slightlylower than that for knottin-RGD peptide (114 nM) and showed 78% higher inhi-bition than 1st generation lead CT3RGDcT3AYGCT3. The other five peptides alldisplayed IC50 values in the range between 150 and 400 nM. For example, for theG/D-Ala-variant bicycle CT3RGDcT3AYaCT3 an IC50 value of 156 nM was deter-mined, which is ~2.5 fold lower than that for parent lead CT3RGDcT3AYGCT3.For the bicycles CT3RGDcT3AWGCT3 and CT3RGDcT3VYGCT3, quite compara-ble values were determined (173 and 211 nM), whereas the inhibitory concentrationsfor CT3RGDcT3AYiCT3 (395 nM) and CT3RGDcT3IYGCT3 (386 nM) were all sig-nificantly higher.
Table 5.1.: (A) Inhibitory activity of crude samples and IC50 values for purified1st and 2nd generation bicyclic integrin α5β1-binders. #Values fromcrude library screening at 10 µM, $values from screening at 2.5 µM,§Per definition 100% (reference); (B) α5β1-inhibitory capacities of var-ious monocyclic variants. The value marked with an asterisk was takenfrom inhibition curves of IC50 determination. IC50 values were deter-mined via nonlinear regression analysis using the software GraphPadPrism based on absorbance when no bicyclic competitor (0%, OD4050.9–1.5 A.U.) or non-labeled knottin-RGD at 30 µM (100%, OD4050.2 A.U.) was applied.
118

55555
5.3. RESULTS & DISCUSSION
5.3.4. α5β1-binding of single-loop variants
As for integrin αvβ3-binders (Chapter 4), it was then investigated whether theRGD-loop size can further be optimized for higher α5β1-affinity by synthesizing var-ious monocyclic peptides comprising the sequences CRGDc, CGRGDc, CRGDSC,CVRGDfC, and CGRGDSC, followed by constraining these using various bis(bromo-methyl)benzene scaffolds or disulfide bond formation. Remarkably, RGD-monocycleCmT2RGDcmT2 entirely lost the inhibitory capacity (<0%) compared to the bicy-cle CT3RGDcT3AYJCT3 (85%), which corresponds to the inactivity of the struc-turally similar monocycle cyclo-[KRGDf] as described earlier, and highlights theessential role of the second loop for α5β1 binding activity (Table 5.1B). MonocycleCmT2GRGDcmT2 showed 10% inhibition, whereas other peptides containing ‘RGD’,‘RGDS’, ‘VRGDf’ or ‘GRGDS’ did not show any competition at all.
5.3.5. Ala-replacement study for selected α5β1-binders
In order to prove the unique binding affinity of the bicycles, linear peptide librariesbased on the sequences of the three highest-affinity α5β1-binding bicycles were syn-thesized, in which each amino acid was sequentially replaced by alanine (Ala), fol-lowed by CLIPS cyclization with mT2 to give the corresponding monocyclic pep-tides (for Cys/Ala replacements), or with T3 to give the corresponding bicyclicpeptides (for all remaining replacements). Subsequently, the crude peptide librarieswere analyzed for α5β1-binding in competition ELISA experiments at 1 µM. Theinhibitory data for the crude peptides revealed that the affinities decrease massivelywhen single amino acids in the best α5β1-binders are substituted by Ala (Table 5.2).Ala-replacement of amino acids of the RGD loop led to significant decreases for eachbicycle CT3RGDcT3AYJCT3 (Table 5.2A), CT3RGDcT3AYaCT3 (Table 5.2B) andCT3RGDcT3AWGCT3 (Table 5.2C). For example, replacing ‘R’ or ‘G’ in bicycleCT3RGDcT3AYJCT3, ‘R’ or ‘D’ in bicycle CT3RGDcT3AYaCT3, and ‘R’, ‘G’ or‘D’ in bicycle CT3RGDcT3AWGCT3 led to a complete loss of α5β1-affinity (inhibi-tion at 1 µM ≤ 1%).Similarly, Ala-replacement of the cysteines (and hence loss of the bicyclic struc-ture) also led to a massive decrease in binding affinity. For example, substitu-tion of either the N-terminal L-cysteine or middle D-cysteine (opening of RGDloop) led to a complete loss of α5β1-affinity for bicycles CT3RGDcT3AYaCT3 and
119
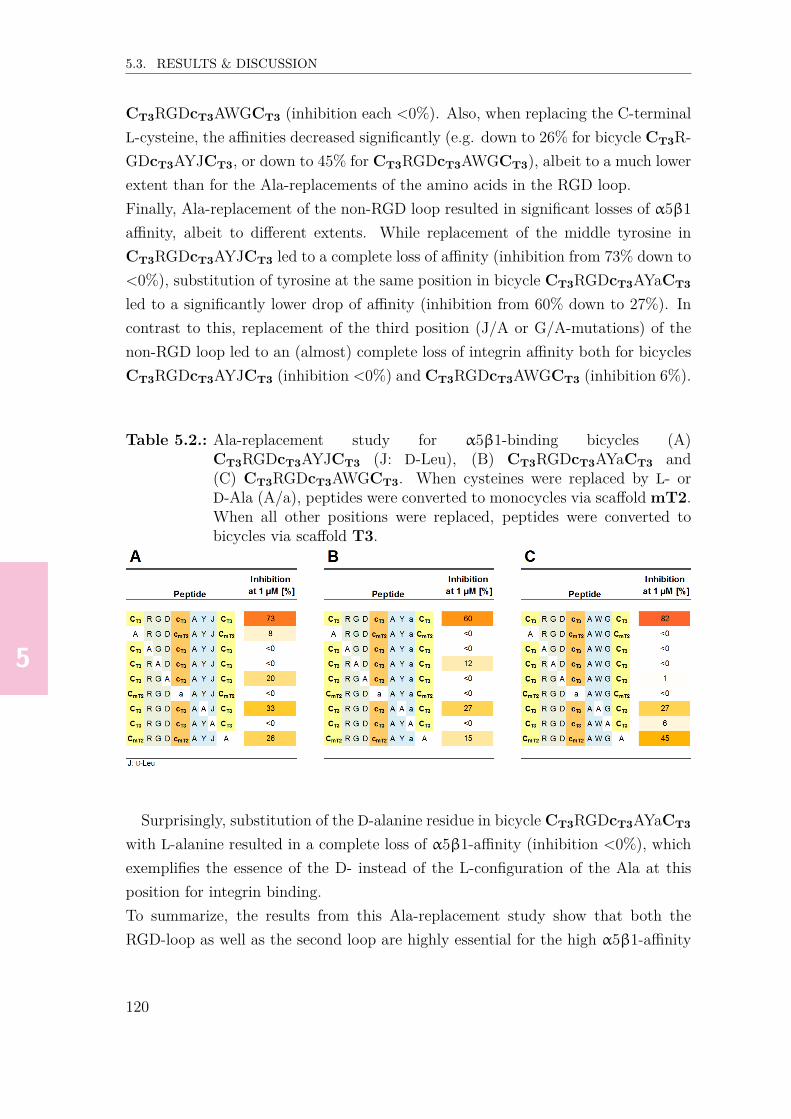
55555
5.3. RESULTS & DISCUSSION
CT3RGDcT3AWGCT3 (inhibition each <0%). Also, when replacing the C-terminalL-cysteine, the affinities decreased significantly (e.g. down to 26% for bicycle CT3R-GDcT3AYJCT3, or down to 45% for CT3RGDcT3AWGCT3), albeit to a much lowerextent than for the Ala-replacements of the amino acids in the RGD loop.Finally, Ala-replacement of the non-RGD loop resulted in significant losses of α5β1affinity, albeit to different extents. While replacement of the middle tyrosine inCT3RGDcT3AYJCT3 led to a complete loss of affinity (inhibition from 73% down to<0%), substitution of tyrosine at the same position in bicycle CT3RGDcT3AYaCT3
led to a significantly lower drop of affinity (inhibition from 60% down to 27%). Incontrast to this, replacement of the third position (J/A or G/A-mutations) of thenon-RGD loop led to an (almost) complete loss of integrin affinity both for bicyclesCT3RGDcT3AYJCT3 (inhibition <0%) and CT3RGDcT3AWGCT3 (inhibition 6%).
Table 5.2.: Ala-replacement study for α5β1-binding bicycles (A)CT3RGDcT3AYJCT3 (J: D-Leu), (B) CT3RGDcT3AYaCT3 and(C) CT3RGDcT3AWGCT3. When cysteines were replaced by L- orD-Ala (A/a), peptides were converted to monocycles via scaffold mT2.When all other positions were replaced, peptides were converted tobicycles via scaffold T3.
Surprisingly, substitution of the D-alanine residue in bicycle CT3RGDcT3AYaCT3
with L-alanine resulted in a complete loss of α5β1-affinity (inhibition <0%), whichexemplifies the essence of the D- instead of the L-configuration of the Ala at thisposition for integrin binding.To summarize, the results from this Ala-replacement study show that both theRGD-loop as well as the second loop are highly essential for the high α5β1-affinity
120

55555
5.3. RESULTS & DISCUSSION
observed. Moreover, high integrin affinity is only expressed when the peptides areconstrained into the bicyclic CLIPS format, and is almost entirely lost on the levelof monocyclic CLIPS peptides.
5.3.6. Replacement of the non-RGD loop of 2nd generationα5β1-binders with non-canonical amino acids
In order to further improve the affinities of the three highest-affinity α5β1-bindingbicycles, small libraries were designed, in which the amino acids of the non-RGDloop were replaced by structurally similar non-canonical amino acids, complementedby a small selection of canonical amino acids. For example, in all three bicy-cles CT3RGDcT3AYJCT3, CT3RGDcT3AWGCT3 and CT3RGDcT3AYaCT3 theL-alanine (A) in the second loop was substituted by either D-alanine (a), nor-leucine (5), norvaline (XN), aminobutyric acid (B) or glycine (G), whereas forCT3RGDcT3AYJCT3 and CT3RGDcT3AYaCT3 the L-tyrosine (Y) residue was re-placed by D-tyrosine (y), phenylalanine (F), D-phenylalanine (f), 4-chlorophenylala-nine (XC) and 4-bromophenylalanine (XP). In total, 15 variants per peptide in-cluding the lead (in total 48 bicycles) were synthesized (Tables 5.3A–C, p.123). Allcrude bicycles were screened for α5β1-affinity at 1 µM.For bicycle CT3RGDcT3AYJCT3 (73% inhibition at 1 µM), five derivatives showedhigher inhibition (Table 5.3A), for exampleCT3RGDcT3BYJCT3 (89%, B: aminobu-tyric acid) or CT3RGDcT3AYXBCT3 (81%, XB: D-norleucine). In contrast, forthree derivatives a strong decrease in inhibitory activity (<30%) was observed,i.e. CT3RGDcT3GYJCT3 (25%), CT3RGDcT3AfJCT3 (16%) and CT3RGDcT3-AYXYCT3 (15%, XY: cyclopropylglycine).Surprisingly, for bicycle CT3RGDcT3AWGCT3 (82% inhibition at 1 µM), not a sin-gle amino acid permutation produced an increased inhibitory activity (Table 5.3B).For example, substitution of tryptophan (W) with D-tryptophan (w) in the ‘AWG’loop resulted in complete loss of inhibition (<0%), while replacement of the glycineresidue (G) with D-alanine (a, 65%) or D-serine (s, 69%) showed only a small de-crease in activity. Overall, 11 out of the 15 variants of CT3RGDcT3AWGCT3 showedlower than 50% inhibition, which reveals that further attempts to replace singleamino acids in the ‘AWG’ loop would most likely not be successful in improving theoverall affinity of this optimized lead.In sharp contrast to this, 5 out of 15 variants of CT3RGDcT3AYaCT3 (60% inhibi-
121

55555
5.3. RESULTS & DISCUSSION
tion, Table 5.3C) showed higher inhibition values (>60%), albeit only one of thoseshowed >66% inhibition, i.e. CT3RGDcT3BYaCT3 (78%, B: aminobutyric acid).On the contrary, inhibition was entirely lost when substituting the “A” in the ‘AYa’motif by D-alanine (a, 5% inhibtion) or glycine (G, <0%).Finally, from the total of 45 derivatives, the best 5 hits were re-synthesized andpurified, followed by IC50 determination as described earlier.¶ The lowest valuewas determined for CT3RGDcT3BYJCT3 (IC50: 574 nM, B: aminobutyric acid,Table 5.3D), followed by CT3RGDcT3BYaCT3 (IC50: 647 nM) and CT3RGDcT3-AYXBCT3 (701 nM). Surprisingly, the IC50 values were significantly higher thanfor the best 2nd generation binders (approx. 100 nM) even though the inhibitionvalues of the crude bicycles suggested the opposite. As for the 1st generation li-brary screening, it may be assumed that the occurrence of these false-positive hitsin the crude library screening might have been caused by polymeric impurities orbe due to inaccurate determinations of peptide concentrations that in fact weresignificantly higher than 1 µM. This hypothesis is supported by the fact that fortwo bicycles that were synthesized two times within the same batch, each two dif-ferent inhibition values were obtained (confer Tables 5.3A and C). That is, forCT3RGDcT3AYXBCT3 (XB: D-Norleucine) inhibitions of 81% and 63% were mea-sured, whereas for CT3RGDcT3AYXYCT3 (XY: Cyclopropylglycine) values of 15%and 6% were determined. Therefore, it would be preferable in the future to includethe determination of IC50 values using peptide concentrations that are corrected fornet peptide content.
¶Since lower IC50 were to be expected as compared to the 2nd generation binders, a concen-tration range from 10 µM to 0.0046 µM (instead of 30 µM to 0.014 µM) was applied.
122

55555
5.3. RESULTS & DISCUSSION
Table 5.3.: Non-canonical amino acid-replacement study for α5β1-binding bicycles(A) CT3RGDcT3AYJCT3 (J: D-Leu), (B) CT3RGDcT3AYaCT3 and(C)CT3RGDcT3AWGCT3. Each two peptides marked with an asteriskor “$” were analyzed for inhibition two times, confer (A); (D) IC50values for the best five binders as determined in competition ELISAexperiments.
123

55555
5.3. RESULTS & DISCUSSION
5.3.7. Determination of affinity binding constants (Kd)
The determination of IC50 values is an indirect method to determine binding affini-ties. In order to be able to measure the bicycle-α5β1 integrin interaction in adirect manner, surface plasmon-enhanced fluorescence spectroscopy (SPFS) was per-formed‖ applying the α5β1-inhibitor CT3RGDcT3AYJCT3, linear GRGDS, cyclo-[KRGDf] and knottin-RGD, each modified with a K-PPPSG[Abz]SG linker∗∗ and afluorescent Cy5 label.†† Dissociation equilibrium constants (Kd) for bicycle K(Cy5)-linker-CT3RGDcT3AYJCT3 and the benchmark peptides were determined for eachα5β1 and αvβ3 integrins in order to be able to measure both affinities and selec-tivities. Concentration-dependent fluorescence signal curves F(c) were normalizedto ∆Fmax (value measured at saturation concentration), and fitted via Langmuirisotherm model (Figure 5.2A). For bicycle K(Cy5)-linker-CT3RGDcT3AYJCT3 aKd of 4.3 nM was determined (Figure 5.2B). Surprisingly, the interaction is sub-stantially stronger as compared to K(Cy5)-linker-knottin-RGD (Kd: 9.0 nM), whichdoes not reflect the observed difference in IC50 values for both binders in ELISA. Thediscrepancy might be explained by a loss of activity as a result of the attachmentof the Cy5-linker. For cyclo-[K(K(Cy5)-linker)RGDf] and K(Cy5)-linker-GRGDS,binding to α5β1 in SPFS was not observed, which is in accordance with the cor-responding IC50 values. Moreover, bicycle K(Cy5)-linker-CT3RGDcT3AYJCT3 didalso not show any interaction with αvβ3 in SPFS. This result confirms that bicycleK(Cy5)-linker-CT3RGDcT3AYJCT3 is indeed able to express complete selectivityfor integrin α5β1 over αvβ3.
5.3.8. Selectivity experiments (ELISA)
Following the αvβ3-binders described in Chapter 4, the selectivity of unlabeledα5β1-binders for different integrin receptors was assessed by determining the IC50
values for integrins αvβ3 and αvβ5 and subsequently calculating the ratio of thesetwo values (Table 5.4).
‖SPFS experiments were performed and analyzed by V. Jungbluth, N. Gisbert Quilis and J.Dostalek, AIT Austrian Institute of Technology GmbH, Tulln, Austria.
∗∗Based on findings of Pallarola et al.,[29] as already described in Chapter 4.††Like in Chapter 4, K(Cy5)-PPPSG[Abz]SG will be abbreviated before each sequence with
the prefix “K(Cy5)-linker“. The synthesis of Cy5-functionalized peptides will be described inChapter 6, p.145.
124

55555
5.3. RESULTS & DISCUSSION
Figure 5.2.: (A) Concentration-dependent, normalized fluorescence signals forselected binding of Cy5-labeled bicycle CT3RGDcT3AYJCT3 andknottin-RGD to integrin α5β1 (triplicate measurement). B) Overviewof measured equilibrium dissociation constants (Kd). Values for αvβ3were obtained by applying a 3D hydrogel surface architecture, val-ues for αvβ3 were obtained by applying a mixed thiol SAM surface.§Values were also described in Chapter 4.
Table 5.4.: Selectivities for optimized α5β1-binding bicyclic peptides. IC50 val-ues were determined using competition ELISA via nonlinear regressionanalysis using the software GraphPad Prism. Each concentration wastested in triplicate.
Bicycles CT3RGDcT3AYJCT3 and CT3RGDcT3AWGCT3 showed excellent se-lectivities over integrin αvβ5 (IC50 of each bicycle >10,000 nM) corresponding toan IC50 ratio α5β1/αvβ5 of <0.009 and <0.017, respectively. The observed selec-tivities for both bicycles over αvβ3 were much lower (IC50: 1019 nM and 1185 nM,respectively), corresponding to an IC50 ratio α5β1/αvβ3 of approximately 0.09.Bicycle CT3RGDcT3AYaCT3, however, exhibited slightly reduced selectivity (IC50
ratio α5β1/αvβ5 = 0.034; α5β1/αvβ3 = 0.157).
125

55555
5.3. RESULTS & DISCUSSION
5.3.9. Conformational analysis of α5β1-binding bicycles
Circular dichroism (CD) spectroscopy
As for the αvβ3-binding bicycles (Chapter 4, p.90) CD (circular dichroism) analysiswas performed for two α5β1-binding bicycles (CT3RGDcT3AYJCT3 and CT3RGD-cT3AYacT3) and two monocyclic RGD peptides (CmT2RGDcmT2 and cmT2RGDcmT2,Figure 5.3A). Monocycle CmT2RGDcmT2 shows a minimum in molar ellipticity at203 nm (∆ε ~ -51,000 cm2dmol-1), while for monocycle cmT2RGDcmT2, only devi-ating in configuration of the N-terminal cysteine, the spectral shape is much dif-ferent, with also a much lower molar ellipticity (∆ε ~ -7,900 cm2dmol-1) at theminimum (205 nm). When comparing with typical spectra of secondary proteinstructures (Figure 5.3B) these monocycles suggest the presence of mainly randomcoil/disordered structure in solution. In sharp contrast to this, CD spectra of bothbicycles do not show any distinct minimum, not revealing any sign of a well-definedsecondary structure in solution (Figure 5.3A). One explanation could be the pres-ence of various types of secondary structures, the spectra of which compensate eachother, resulting in a cumulative spectrum not showing any distinct minimum or max-imum. Another explanation is the formation of structural aggregates, the moleculesof which adopt different secondary structures from which the various spectra alsocompensate each other. However, experiments in buffers containing the chaotropicurea led to very similar CD spectra (data not shown), which precludes this option.The overall conclusion of these results should be that CD spectroscopy is probablynot a suitable technique to gain information about the conformation of the α5β1-binding bicycles in solution.
1D and 2D NMR spectroscopy‡‡
In order to further investigate the secondary structures of the α5β1-binders in solu-tion, a detailed structural assignment using a series of 1D and 2D NMR-spectroscopictechniques was performed for bicycle CT3RGDcT3AYJCT3.Individual amino acids were identified through their Hα/Cα chemical shifts from theedited spectrum obtained by heteronuclear single-quantum correlation spectroscopy(HSQC), as well as via the number and chemical shift of the side-chain resonances
‡‡The NMR experiments and structural assignments were performed by Dr. Paul White, Insti-tute for Molecules and Materials, Radboud University Nijmegen, Netherlands.
126

55555
5.3. RESULTS & DISCUSSION
Figure 5.3.: (A) Circular dichroism spectra of monocyclic RGD pep-tides CmT2RGDcmT2, cmT2RGDcmT2 and bicyclic peptidesCT3RGDcT3AYJCT3 and CT3RGDcT3AYacT3 at 50 µM inphosphate buffer; (B) Circular dichroism spectra of proteins withdifferent secondary structures (reprinted from [30]). Red: predomi-nant α-helix, blue: predominant β-sheet, green: mixture of α-helix,β-sheet and unordered structure.
as revealed through 2D total correlated spectroscopy (TOCSY). The residues moststraightforward to identify belonged to the side chain of tyrosine. The aromaticprotons at 7.11 and 6.83 ppm were assigned ortho and meta to the phenol, respec-tively, and could be connected to the 13C’s at 133.3 and 118.3 ppm, respectively.Using the 1H-13C heteronuclear multiple-bond correlation spectroscopy (HMBC)long-range coupling experiment, Hα and Hβ of tyrosine were identified as 4.44 ppmand 3.03/2.90 ppm. The corresponding Cα and Cβ were identified as 58.9 ppm and38.2 ppm, respectively, via the 1-bond 1H-13C HSQC correlation experiment. Theseshifts were consistent with a random coil conformation as opposed to α helix or βsheet. An identical analysis was performed for all readily identifiable residues, whichconcluded overall a random coil conformation of the peptide (Figure 5.4A).In order to determine the effect of linking the peptide chain to the T3-scaffold viathe cysteine residues, T1 and T2 NMR relaxations were determined for both thelinear peptide and the bicyclic compound. T1 relaxation for the protons arises pri-marily through dipole-dipole interactions with neighboring nuclei that occur at theLarmour precession frequency, usually via molecular motion. T2 relaxation involvesthe loss of spin coherence in the XY plane through variations in local magneticfields of any frequency interacting with the nuclear spin. As both of these parame-ters involve molecular motion, they can be sensitive probes for phenomena such as
127

55555
5.3. RESULTS & DISCUSSION
polymerization, aggregation, and complexation, which affect the rotational correla-tion time of the molecule.
Figure 5.4.: (A) Chemical shift difference plots for Cα and Cβ calculated by ∆δ13Cα
= δ13Cα,rc – δ13Cα,i and ∆δ13Cβ = δ13Cβ,i – δ13Cβ,rc (i: measuredamino acid in bicycle, rc: random coil). Positive values reflect moreβ-sheet character while negative values represent more α-helical char-acter. Amino acids that are close to the baseline are indicative ofrandom coil structure, or show both α-helical and β-sheet character,or alternatively structured sequences; (B) NMR relaxation times T1and T2 as a function of amino acid residue.
The residues that were most readily identifiable in both linear and bicyclic peptidewere Hα,Gly, Hβ,Ala, Hδ,Tyr and Hε,Tyr (Figure 5.4B). In all cases, the T1 values ofthese protons deviated less than 10% between the linear and bicyclic conformation,indicating that overall the molecular rotation of the molecule remains relativelyunchanged, which means that the secondary structure for the bicycle is virtuallyidentical to that of the corresponding linear peptide, i.e. very likely a random coil.The T2 decreased slightly more, reflecting the increase in rigidity of the peptide,however not significant enough to indicate dimerization or aggregation. Therefore,it can be concluded with relative certainty that these compounds exist as monomersin solution.
128

55555
5.3. RESULTS & DISCUSSION
5.3.10. Binding ELISA studies with biotinylated α5β1-bindingRGD bicycles
In order to investigate whether biotinylated versions of bicyclic α5β1-binders couldreplace the biotinylated knottin-RGD peptide in competition ELISA experiments,their binding to integrin α5β1 was determined in a direct binding ELISA setup(Figure 5.5).
Figure 5.5.: Binding ELISA of biotinylated α5β1-binding bicycles and knottin-RGD. Background-subtracted absorbances (OD at 405 nm) are shownas a function of peptide concentration. Experiments were carried outin triplicate and error bars represent standard deviations.
The biotinylated peptideCT3RGDcT3AYacT3 showed the strongest binding amongthe bicyclic peptides at 1 µM (OD405 ~2.2 A.U.) and 0.1 µM (OD405 ~1.7 A.U.),whereas binding levels of biotinylated bicycles CT3RGDcT3AYJCT3 and CT3RGD-cT3AWGcT3 at 1 µM (OD405 ~1.7 A.U.) and 0.1 µM (OD405 ~1.3 A.U.) were verysimilar. Binding of the bicycles could be detected even at 0.01 µM (OD405 ~0.35–0.50 A.U.), while at 0.001 µM integrin binding was not detectable anymore (OD405
<0.2 A.U.) for any of the biotinylated peptides, including the knottin-RGD pep-tide. Moreover, at all concentrations tested, biotinylated knottin-RGD showed thestrongest absorbance, which is not fully in agreement with the very similar IC50
values for the non-labeled versions (p.118, Table 5.1A). As for the αvβ3-bindingbicycles, the functionalization with the linker plus biotinylation might have led to astronger decrease of binding affinity for the bicycles as compared to knottin-RGD.Nonetheless, the data reveal that all bicycles could represent possible surrogate
129

55555
5.3. RESULTS & DISCUSSION
ligands for a competition ELISA experiment to identify novel multicyclic integrinbinders.
5.3.11. Screening for αvβ5-binding peptides
1st generation screening
Screening of 1st generation bicycle libraries for integrin αvβ5 at 10 µM revealed thatonly one out of 672 peptides showed more than 50% inhibition of knottin-RGD-αvβ5binding (CT3RGDcT3NWGCT3, 55% inhibition).§§ Remarkably, this bicycle wasalso determined to be a low-affinity 1st generation binder for α5β1 (IC50,α5β1:2084 nM, Table 5.1A, p.118) despite the fact that both integrin α- and β-subunitsare different. One explanation, which might be verified with computational meth-ods, could be that parts of the integrin subunits (αv/α5 and β5/β1, respectively)that interact with this bicycle are conserved. However, the observation should notbe overestimated in view of the per se low inhibitory activity in case of αvβ5 (55%at 10 µM). Approximately 94% of the RGD-bicycles (631 peptides) showed <25%inhibition, independent of whether the ‘RGD’-motif is located in the left or rightloop. The remaining 6% (except for the abovementioned hit, 40 peptides) showedbetween 25 and 46% inhibition. As a consequence of the overall very low inhibi-tion values (671 out of 672 peptides <50% inhibition at 10 µM), only one hit wasselected (CT3RGDcT3NWGCT3) in contrast to the five leads identified as 1st gen-eration leads for αvβ3 (Chapter 4). Subsequently, this bicycle was purified and itsinhibition for knottin-RGD binding to αvβ5 tested at eight different concentrations(from 30 down to 0.014 µM, with three-fold dilution steps) in order to determinethe corresponding IC50 value. Interestingly, a relatively high IC50 value of 1457 nMwas determined for this bicycle (Table 5.5A), the inhibiting ability of which wasstill much lower than that of the benchmark peptides knottin-RGD (76 nM), cyclo-[KRGDf] (182 nM), and cilengitide (26 nM).
2nd generation screening
Similar to the screening for α5β1-binders, a number of different 2nd generationlibraries (in total 196 peptides) were designed based on 1) a full amino acid replace-
§§The OD405 values of all 1st and 2nd generation library screenings are depicted in the Supple-mentary Information, Section A.3, p.231.
130

55555
5.3. RESULTS & DISCUSSION
ment study for the ‘NWG’ motif (including a set of non-canonical amino acids, seeAppendix, p.138, Table 5.6), and 2) extending the length of the ‘RGD’ loops (forexample, ‘CRGDXc’ or ‘CXRGDSc’) while maintaining the length for the secondloop ‘cNWGC’ constant (Figure 5.1). This time, these libraries were screened forintegrin αvβ5 inhibition at 5 µM. Surprisingly, not a single peptide of the 2nd gener-ation bicycles exhibited any further improved inhibition as compared to that of the1st generation lead bicycle CT3RGDcT3NWGCT3 itself (66% inhibition). In fact,only three of the total of 196 2nd generation bicycles, i.e. CT3RGDcT3NWaCT3
(57%), CT3GRGDacT3NWGCT3 (54%) and CT3RGDcT3NWfCT3 (53%), showedmore than 50% inhibition. One reason for the remarkably low amount of hits couldbe that only seven different non-natural amino acids were tested in the 2nd gen-eration library screening; hence a broader range of variants, including deviationsfrom natural amino acids and N-methylated amino acids, might possibly increasethe probability to detect more high-affinity hits for αvβ5.In order to determine IC50 values for these three optimized leads, they were re-synthesized, purified by preparative HPLC and tested again for integrin αvβ5 in-hibition at a concentration range from 30 to 0.014 µM. Surprisingly, two of these,i.e. CT3GRGDacT3NWGCT3 (1150 nM) and CT3RGDcT3NWaCT3 (650 nM) didexhibit a lower IC50 value than their parent lead (1457 nM), in contradiction to thescreening data for the crude libraries (see Table 5.5A). However, the 3rd optimizedlead, i.e. CT3RGDcT3NWfCT3, exhibited a much higher IC50 value (>3.5 µM,Table 5.5A) than was initially expected based on crude library screening (53% vs.66% inhibition of the parent lead at 5 µM). Apparently, a small but significantdiscrepancy in inhibitory activity between crude and purified bicycles was observed(Table 5.5A). For example, optimized lead peptide CT3RGDcT3NWaCT3 showed aweaker inhibition in crude form as compared to the parent lead (57% vs. 66% at5 µM), whereas their IC50 values (650 nM vs. 1457 nM) determined using HPLC-purified bicycles indicated significantly stronger inhibition for the optimized lead. Ingeneral, the IC50 values obtained with purified bicycles are considered more reliablesince the screening with the crude peptide libraries contains several uncertainties,i.e. i) only a single concentration was tested, ii) concentrations were not determineddirectly, but calculated from amount of resin used for the synthesis, and iii) strongvariations in net bicyclic peptide content due to differences in cyclization efficiencies,and consequently the presence of different amounts of oligo-/polymeric impurities.Since the binding affinities of the 2nd generation αvβ5-binding bicycles were still
131

55555
5.3. RESULTS & DISCUSSION
much lower as compared to the benchmark RGD peptides mentioned above, the bi-cyclic CLIPS RGD peptide platform is maybe not very well suitable for identifyinga high-affinity αvβ5-binder.
Table 5.5.: (A) Percentage inhibition (# values from crude library screening at5 µM) and IC50 values for 1st and 2nd generation bicyclic integrin αvβ5-binders. § Per definition 100% (reference); (B) αvβ5-inhibitory valuesof various monocyclic peptides. The value marked with an asteriskwas taken from inhibition curves for IC50 determination. Inhibitionvalues were obtained via nonlinear regression analysis based on ab-sorbance values with no bicyclic competitor present (0%, OD405 ~0.8–1.3 A.U.) or in presence of 30 µM non-labeled knottin-RGD (100%,OD405 ~0.2 A.U.).
5.3.12. αvβ5-binding of single-loop variants
It was also investigated to what extent the second loop (‘cNWaC’) of the opti-mized lead bicycle contributes to αvβ5-binding, and whether the size and shapeof the RGD-loop can be further optimized or improved for higher αvβ5-affinities.Therefore, various monocyclic RGD peptides were synthesized comprising the se-quences CRGDc, CGRGDc, CRGDSC, CVRGDfC, and CGRGDSC, followed byconstraining these with various bivalent CLIPS-scaffolds or via disulfide formation(for chemical structures of scaffolds, see Chapter 4, p.85, Figure 4.4A), and theirabilities to block binding of knottin-RGD to integrin αvβ5 were studied in com-petition ELISA. In comparison to bicycle CT3RGDcT3NWaCT3 (57% inhibitionat 1 µM), monocyclic CmT2RGDcmT2 showed much lower inhibition (16%), which
132

55555
5.4. CONCLUSION & OUTLOOK
proves that the second loop ‘NWa’ in CT3RGDcT3NWaCT3 is apparently highly es-sential for integrin αvβ5 binding (Table 5.5B). For several extended loop-variants,e.g. the hexacyclic peptide CmT2GRGDcmT2 (18%) or the heptacyclic peptideCmT2GRGDScmT2 (17%) the binding capability did not improve much, suggestingthat elongating the RGD-loop is not the preferred option for optimized binding.Moreover, all other extended monocycles containing ‘RGDS’ or ‘VRGDf’ showedinhibition values of 12% or lower, confirming the fact that further extension of theRGD-loop size is not expected to yield better binding bicycles.In sharp contrast to this, the monocyclic disulfide-bridged peptide CoxRGDcox
showed the highest inhibition value at 1 µM observed for a monocyclic peptide,i.e. 36%, suggesting that a further reduction in size and/or conformational freedomlikely would further improve the RGD-loop’s binding ability. However, there is nostraightforward way to combine a disulfide-bridged RGD-loop with a randomizedsecond loop. One way to achieve this could be to synthesize a linear peptide com-prising a random XXX motif and an RGD motif enclosed by two cysteines. Bythen backbone-cyclizing the peptide, followed by SS-oxidation, a bicyclic structurecould be created. Moreover, the significantly higher inhibition values observed forCmP2RGDcmP2 (24%) over CmT2RGDcmT2 (16%) and CmP2GRGDScmP2 (25%)over CmT2GRGDScmT2 (17%) might suggest that applying the more water-solublescaffold 2,4,6-tris(bromomethyl) pyridine (“mP3”) as a replacement for the muchmore hydrophobic T3-scaffold could potentially improve the αvβ5-inhibition valuesfor the corresponding bicycles to a similar extent. However, it has to be mentionedthat application of non-C3-symmetrical trivalent scaffolds would result in the for-mation of three different isomers.
5.4. Conclusion & outlook
The screening of partially randomized RGD-bicycle libraries was successfully appliedin the search for integrin α5β1-binders, eventually yielding three high-affinity 2nd
generation bicyclic RGD peptides, i.e. CT3RGDcT3AYJCT3 (J: D-leucine, IC50:90 nM), CT3RGDcT3AYaCT3 (IC50: 156 nM) and CT3RGDcT3AWGCT3 (IC50:173 nM), with very good selectivity over αvβ5 (IC50 ratios α5β1/αvβ5 <0.007–<0.034) and moderate selectivity over αvβ3 (IC50 ratios α5β1/αvβ3 0.09–0.157).It was also shown that these bicycles have superior α5β1-affinity over the bench-mark RGD peptides cyclo-[KRGDf] and linear GRGDS as reflected from the data
133

55555
5.5. MATERIALS & METHODS
that were obtained in competition ELISA experiments. Therefore, these bicyclesrepresent an alternative platform to target integrin α5β1, be it in the context oftherapeutic applications, biomaterial functionalization, or in vitro/in vivo tracers.Unfortunately, a similar screening operation to search for αvβ5-specific RGD-bicyclesdid not yield any high-affinity bicycles (best 2nd generation binder: CT3RGDcT3-NWaCT3 (IC50: 650 nM)). Since the initial libraries only represented a small num-ber of all possible bicyclic RGD-peptides (~1.5%), it cannot be excluded that ahigh-affinity ‘3x3’ RGD-bicyclic binder is existent, but was somehow missed in thescreening. Other approaches apart from the bicyclic CLIPS platform could be morepromising, for example the design and screening of tricyclic or tetracyclic peptidelibraries, the chemistry of which was very recently published by Richelle et al.[31]
Another method that is suitable to screen much higher numbers of peptides (>109),and which is also compatible with the CLIPS platform would be phage-displayscreening.[32, 33] The major drawback is, however, that it is limited to natural aminoacids so far. Hence, none of the high-affinity binders described in Chapters 4 and 5would have been detected, since they all comprise at least one non-canonical aminoacid, i.e. a D-cysteine.
5.5. Materials & methodsAll reagents and methods were identical to those used and described in Chapter 4(see p.96) with following deviations:
Peptide synthesis, analysis and purificationAn overview of the UPLC/ESI-MS data for the peptides described in this chaptercontaining non-functionalized, linker-modified, biotinylated, and Cy5-functionalizedpeptides is shown in the appendix (Table 5.8, p.140).
Competition ELISACompetition ELISA experiments were performed as described in Chapter 4 (p.97)applying an integrin αvβ5/α5β1 concentration of 0.5 µM and a biotinylated knottin-RGD concentration of 0.2 µM. Absorbance readout and IC50 determination wasperformed as described in Chapter 4.
134

55555
5.6. REFERENCES
5.6. References[1] M. Barczyk, S. Carracedo, D. Gullberg, Cell Tissue Res. 2010, 339, 269–280.[2] Y. Takada, X. Ye, S. Simon, Genome Biol. 2007, 8, 215.[3] M. A. Schwartz, M. D. Schaller, M. H. Ginsberg, Annu. Rev. Cell Dev. Biol.
1995, 11, 549–599.[4] M. A. Schwartz, M. H. Ginsberg, Nat. Cell Biol. 2002, 4, 65–68.[5] C. J. Avraamides, B. Garmy-Susini, J. A. Varner, Nat. Rev. Cancer 2008,
8, 604–617.[6] C. T. Mierke, B. Frey, M. Fellner, et al., J. Cell Sci. 2011, 124, 369–383.[7] M. Friedlander, C. L. Theesfeld, M. Sugita, et al., Proc. Natl. Acad. Sci. USA
1996, 93, 9764–9769.[8] K. P. Conroy, L. J. Kitto, N. C. Henderson, Cell Tissue Res. 2016, 365,
511–519.[9] V. Sarrazy, A. Koehler, M. L. Chow, et al., Cardiovasc. Res. 2014, 102, 407–
417.[10] T. S. Panetti, P. J. McKeown-Longo, J. Biol. Chem. 1993, 268, 11492–11495.[11] Y. Asano, H. Ihn, K. Yamane, et al., Am. J. Pathol. 2004, 164, 1275–1292.[12] S. Kim, M. Harris, J. A. Varner, J. Biol. Chem. 2000, 275, 33920–33928.[13] M. Friedlander, P. C. Brooks, R. W. Shaffer, et al., Science 1995, 270, 1500–
1502.[14] U. R. Goessler, P Bugert, K Bieback, et al., Int. J. Mol. Med. 2008, 21,
271–279.[15] J. Schittenhelm, A. Klein, M. S. Tatagiba, et al., Int. J. Clin. Exp. Pathol.
2013, 6, 2719–2732.[16] C. L. Gladson, D. A. Cheresh, J. Clin. Invest. 1991, 88, 1924–1932.[17] C. L. Gladson, J. Neuropath. Exp. Neur. 1996, 55, 1143–1149.[18] M. A. Dechantsreiter, E. Planker, B. Matha, et al., J. Med. Chem. 1999, 42,
3033–3040.[19] R. H. Kimura, A. M. Levin, F. V. Cochran, et al., Proteins 2009, 77, 359–
369.[20] R. H. Kimura, R. Teed, B. J. Hackel, et al., Clin. Cancer Res. 2012, 18,
839–849.[21] J. W. Kim, F. V. Cochran, J. R. Cochran, J. Am. Chem. Soc. 2015, 137,
6–9.[22] Z.-R. Gan, R. J. Gould, J. W. Jacobs, et al., J. Biol. Chem. 1988, 263,
19827–19832.
135

55555
5.6. REFERENCES
[23] T. G. Kapp, F. Rechenmacher, S. Neubauer, et al., Sci. Rep. 2017, 7, 39805.[24] O Stoeltzing, W Liu, N Reinmuth, et al., Int. J. Cancer 2003, 104, 496–503.[25] P. Khalili, A. Arakelian, G. Chen, et al., Mol. Cancer Ther. 2006, 5, 2271–
2281.[26] D. Heckmann, A. Meyer, B. Laufer, et al., ChemBioChem 2008, 9, 1397–
1407.[27] F. Rechenmacher, S. Neubauer, J. Polleux, et al., Angew. Chem. Int. Ed.
2013, 52, 1572–1575.[28] T. G. Kapp, F. S. Di Leva, J. Notni, et al., J. Med. Chem. 2018, 61, 2490–
2499.[29] D. Pallarola, A. Bochen, H. Boehm, et al., Adv. Funct. Mater. 2014, 24,
943–956.[30] A. J. Miles, B. A. Wallace, Chem. Soc. Rev. 2016, 45, 4859–4872.[31] G. J. Richelle, S. Ori, H. Hiemstra, et al., Angew. Chem. Int. Ed. 2018, 57,
501–505.[32] C. Heinis, T. Rutherford, S. Freund, et al., Nat. Chem. Biol. 2009, 5, 502–
507.[33] S. Chen, D. Gfeller, S. A. Buth, et al., ChemBioChem 2013, 14, 1316–1322.
136

55555
5.7. APPENDIX
5.7. AppendixThe appendix contains an overview of parameters in the competition ELISA se-tups (Table 5.7), a list of all amino acids applied in the screenings (Table 5.6),a UPLC/ESI-MS spectrum of a purified α5β1-binding bicycle (Figure 5.6) and acomplete list of UPLC/ESI-MS data obtained for all non-modified and modified,purified RGD peptides (Table 5.8).
Figure 5.6.: UPLC/ESI-MS spectrum of purified bicycle CT3RGDcT3AYJCT3 (J:D-Leu, top: UV detection at 215 nm, middle/bottom: ESI-MS detec-tion)
137
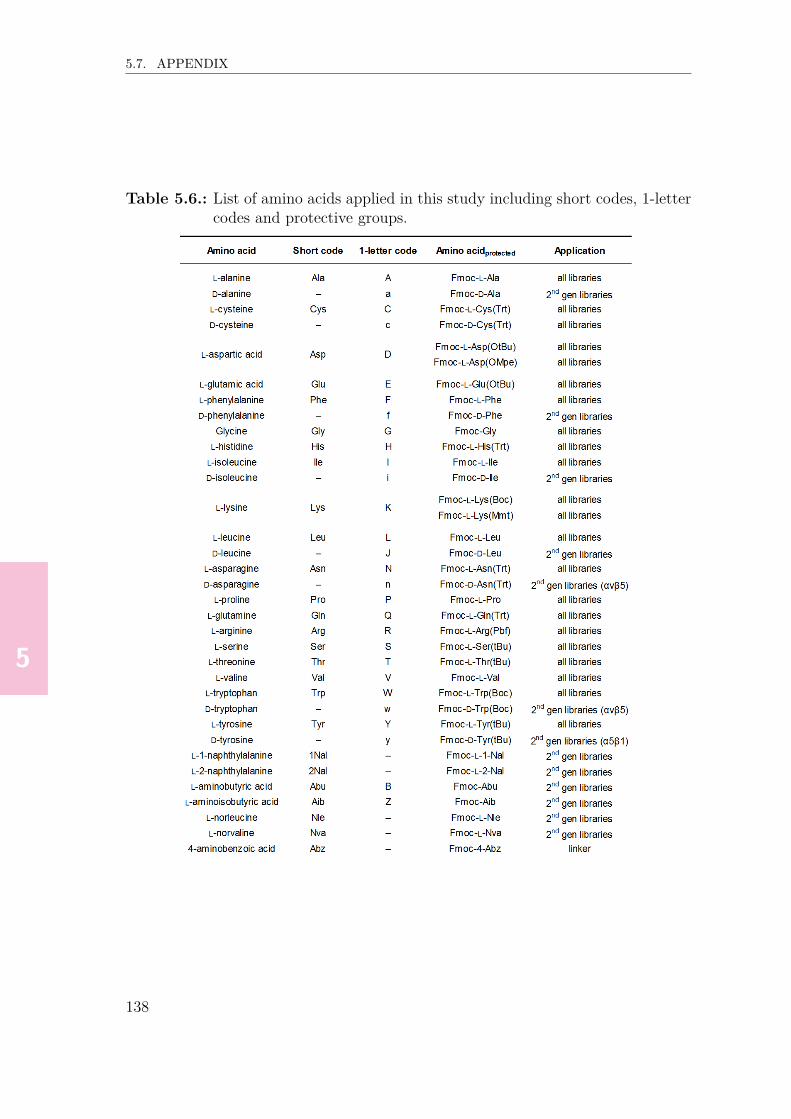
55555
5.7. APPENDIX
Table 5.6.: List of amino acids applied in this study including short codes, 1-lettercodes and protective groups.
138

55555
5.7. APPENDIX
Table 5.7.: Parameters varied in the two different competition ELISA setups (A),and selected setups for library screening, IC50 determination and selec-tivity experiments (B).
139

55555
5.7. APPENDIX
Table 5.8.: Molecular weights of non-modified and modified, purified RGD pep-tides described in Chapter 5, determined via UPLC/ESI-MS analysis.Weights were calculated using ChemDraw software.
140

666666
6. Visualization of Integrin-Bindingon Cells using FluorescentlyLabeled RGD Bicyclic Peptides
The work described in this chapter resulted from a collaboration with the group of Prof. Dr.Kees Jalink from the Netherlands Cancer Institute, Amsterdam, the Netherlands. Parts of thework were published: D. Bernhagen, V. Jungbluth, N. Gisbert Quilis, J. Dostalek, P. B. White,K. Jalink, P. Timmerman, ACS Comb. Sci. 2019, 21, 198–206; D. Bernhagen, V. Jungbluth, N.Gisbert Quilis, J. Dostalek, P. B. White, K. Jalink, P. Timmerman, ACS Comb. Sci. 2019, 21,598–607.

666666
6.1. ABSTRACT
6.1. Abstract
In this chapter, the in vitro staining properties of fluorescently labeled bicyclic pep-tides 2a-2c (αvβ3-selective) and 3a-3c (α5β1-selective) towards human colorectaladenocarcinoma (HT29) cells (express integrin subunits αv- and β1), HeLa cells(express integrins αvβ3 and α5β1) and human adipose-derived stem cells (ASCs,express α5, β1 and β5) are described.Incubation of HT29 cells at 37 ℃ revealed significant extra- as well as intracellularmembrane staining both for benchmark 1a (knottin-RGD, equally binds all threeintegrins αvβ3/αvβ5/α5β1) and αvβ3-selective bicycle 2a, while α5β1-selectivebicycles 3a–3c as well as benchmark 1b (αvβ3- and αvβ5-selective) and negativecontrol 1c (low integrin affinity) only showed very weak staining levels. Interestingly,bicycle 2b, comprising the same affinity to recombinant αvβ3 in immunoassays as2a and varying in only the absolute configuration (L/D-) of the middle cysteine, didnot show any staining. When measuring at 4 ℃ (suppression of internalization),bicycle 2a again bound the HT29 outer cell membrane much stronger than 2b and1b, and even significantly stronger than benchmark 1a or the α5β1-selective bicy-cles 3b and 3c.Similar studies with HeLa cells revealed the highest staining levels for benchmarkpeptide 1a and bicycles 3b and 3c, while 3a showed a significantly lower level ofstaining. Among the αvβ3-selective bicycles, 2c showed slightly stronger stainingthan 2a and 2b, revealing that the more hydrophilic HSQ loop better adapts to theconformation of the integrins’ RGD-binding pocket than HPQ. For α5β1-selectivebicycles 3b and 3c, an even higher staining intensity was observed compared with2c, possibly as a result of superior expression of α5β1 over αvβ3. In contrast,benchmark peptide 1b and negative control 1c did not show any detectable stain-ing.Finally, ASCs showed the highest staining levels when incubated with either α5β1-selective bicycle 3b or benchmark 1a, the staining of which spreads to the entire cellat 37 ℃. The staining intensity of 3b was much higher than for 3a and 3c, indicat-ing that the nature of the non-RGD loop clearly influences binding of the bicyclesto cell surface α5β1 integrin. Moreover, weak membrane staining was observed forαvβ3-selective bicycle 2c, whereas the other αvβ3-selective bicycles 2a and 2b, aswell as benchmark 1b and negative control 1c did not show any significant mem-brane integrin binding.
142

666666
6.2. INTRODUCTION
6.2. IntroductionThe identification of a series of bicyclic ECM protein mimics that bind integrinsαvβ3 or α5β1 with very high affinity and selectivity (Chapters 4 and 5), wasachieved by measuring their binding affinities towards recombinant proteins thatwere expressed outside their natural environment. However, for in vitro or in vivoapplications, it has to be considered that the natural environment of membrane-integrated integrins is a complex and dynamic system involving intracellular, ex-tracellular, outside-in, and inside-out signaling cascades,[1–4] which highlights thequestion whether it is possible to visualize binding of the bicyclic RGD-peptides tonative integrin receptors in their natural environment, i.e. integrated in the mem-brane of intact cells.The aim of this study was to visualize binding of fluorescently labeled RGD-bicyclesto cellular membrane integrins. For this purpose, confocal imaging of two humancancer cell lines (HT29, HeLa)∗ and one human stem cell line (ASC) was stud-ied, using these fluorescently labeled bicycles in combination with monocylic andknottin-RGD peptide benchmarks as well as a linear RGD peptide as negative con-trol.Fluorescence microscopy visualizes microscopic structures in cells or tissue by mea-suring the fluorescence emitted from ligands bound to the specimen. Previous stud-ies already described in vitro and in vivo imaging of cells and tissues via fluores-cently labeled RGD peptides,[5–8] but the majority of articles focuses on the labelingof tumor tissue, which is related to the overexpression of certain integrins duringcancer development.[9–11] For example, 125I-functionliazed y-cyclo-[RGDyV] and Y-cyclo-[RGDfY] were developed by Haubner et al. in order to investigate angiogenesisand metastasis in vivo.[12] Recently, a fluorescent naphthalenediimide cyclo-[KRGDf]conjugate was reported as an in vitro αvβ3 binder in prostate cancer cells (PC-3).[8]
While the conjugate showed labeling mainly in the vesicular cytoplasm regions, theunconjugated naphtalenediimide compound showed more uniform labeling of the cy-toplasm. In yet another in vivo study, a fluorescent, high affinity αvβ3-, αvβ5- andα5β1-binding knottin-RGD peptide was applied to image brain tumors (intracra-nial medulloblastoma).[13] However, to the best of our knowledge, studies on the in
∗Derived from cervical cancer cells by Henrietta Lacks.
143

666666
6.2. INTRODUCTION
vivo or in vitro imaging specifically applying α5β1-selective, fluorescently labeledpeptides have thus far not been reported.The resolution of fluorescence microscopy is limited by light diffraction, as describedby Abbe’s Law:
d = λ
2n · sinθHere, d represents the minimum resolvable distance between two points, λ the wave-length of light, n the refractive index of the medium and θ the opening angle of theobjective.Super resolution microscopy (SRM) techniques aim to overcome this limitation.There are two different approaches to achieve this, i.e. via near field methods suchas Total Internal Reflection Fluorescence (TIRF)[14] or Near field Scanning Opti-cal Microscopy (NSOM),[15] and using far field methods like Confocal Microscopy(CM)[16] (Figure 6.1A+B) or Stimulated Emission Depletion (STED).[17]
Figure 6.1.: (A) Schematic of a confocal microscope including light pathways. Thecrucial component is a detector pinhole that filters out “out-of-focus”fluorescence leading to a higher spatial and lateral resolution. (B)Conventional epifluorescence image (1) in comparison with a confocalimage (2) of a similar region of a whole mount of a butterfly (Pre-cis coenia) pupal wing stained with propidium iodide; scale bar notdefined; reprint from [18].
The studies described in this chapter utilize Confocal Microscopy to analyze thein vitro cell membrane integrin staining properties of fluorescently labeled RGDbicycles as well as benchmark and negative control peptides.
144

666666
6.3. RESULTS & DISCUSSION
6.3. Results & discussion
6.3.1. Selection of a fluorescent label
Visualization of membrane staining via confocal microscopy requires the presenceof a fluorescent label. Factors that need to be considered when selecting a suitabledye include i) the absorbance- and emission spectrum of the dye, ii) the fluorescenceintensity, iii) the water solubility, iv) the availability and costs of the dye, and v)the peptide sequence, which ultimately determines the functionalization strategy.For membrane-labeling of non-transfected cells, the emission spectrum (blue, redor green) is subordinate since this type of cells does not express fluorescent pro-teins, whose emission spectrum would have to be considered. For high fluorescenceintensities, the dye should comprise high absorbance coefficients and fluorescencequantum yields. Cyanine-based dyes, such as Cy2, Cy3, Cy5 and Alexa Fluor 647,as well as rhodamine dyes, such as Alexa Fluor 488 and Atto 488, fulfill thesecriteria.[19, 20] The water solubility of a dye depends on its hydrophilicity and canbe improved, for example, by modification with polar groups such as sulfonates.Moreover, the price of a fluorescent dye depends on factors such as synthesis ef-forts, possible functionalizations, and suitability for microscopy (quantum yield, ab-sorbance coefficient). There are inexpensive compounds†, for example, RhodamineB (~75€/100 g) and 5(6)-Carboxyfluorescein (~100€/5 g), medium-priced dyes, suchas Cy5 or diSulfo-Cy5 NHS ester (each ~1000€/100 mg), and high-end labels, forexample, Alexa Fluor 488 NHS ester (~2100€/5 mg). Moreover, there are variousoptions to modify a peptide, for example, via the N-terminal or side-chain aminesusing NHS-ester- or isothiocyanate-functionalized dyes, or via cysteine thiols usingmaleimide- or iodoacetamido-functionalized dyes.Considering all the above mentioned criteria, the water-soluble label diSulfo-Cy5-NHS ester (for molecular structure, see Figure 6.2A) was selected, comprising highfluorescence intensity, an affordable pricing, and straightforward and selective cou-pling via amine functionalities.
†Approximate prices effective 7 March 2018; providers: Sigma-Aldrich, Lumiprobe, ThermoFisher Scientific).
145

666666
6.3. RESULTS & DISCUSSION
6.3.2. Synthesis of Cy5-labeled peptides
The Cy5-labled RGD-bicycles were composed of i) the bicyclic integrin-binding mo-tif, ii) a peptidic linker and iii) the Cy5-label, and synthesized via solid-phase peptidesynthesis (integrin-binding precursor motif + linker), followed by CLIPS cyclizationand Cy5-functionalization. The linker motif, KPPPSG[Abz]SG (see Figure 6.2A,hereafter referred to as “K-linker”, Abz: 4-aminobenzoic acid), was derived from thehexaPPP-motif reported by Pallarola et al. that previously proved to hardly reducethe αvβ3-inhibitory activity of cyclic RGD peptides.[21] The N-terminal α-amine oflysine was acetylated while the ε-amine (side-chain amine) was functionalized withdiSulfo-Cy5-NHS ester (Figure 6.2A).For this study, each three αvβ3-selective bicycles (2a–2c) and α5β1-selective bicy-cles (3a–3c), and additionally two benchmark RGD-peptides (1a and 1b) and onenegative control (1c) were modified with diSulfo-Cy5-NHS ester (Figure 6.2B).
6.3.3. Experimental setup of membrane-labeling experiments
Two different experimental setups were established to investigate cell labeling (Fig-ure 6.3). In the first setup, freshly trypsinized cells were cooled to ~4 ℃ and mixedwith the labeled peptides. After incubation for 1 min (4 ℃), the peptide-labeledcells were transferred onto glass coverslips and incubated for 3-4 h at 37 ℃, followedby washing and analysis via confocal microscopy. In the second setup, cells werefirst transferred onto glass coverslips, incubated at 37 ℃ until ~40-50% confluency(24 h – 4 d, depending on the cell line) and cooled to 4 ℃, followed by additionof Cy5-functionalized peptide and incubation for a defined period of time at 4 ℃(modified experimental setup 2: 37 ℃). After washing, the cells were fixed withparaformaldehyde (PFA) before confocal microscopy analysis. A more detailed de-scription is given in the Materials & methods section (p.164).The Cy5-labeled benchmark peptide 1a binds with high affinity to integrins αvβ3,α5β1 and αvβ5 (see competition ELISA data, Chapters 3–5). The very similaraffinities as determined for the αvβ3- and α5β1-binding RGD-bicycles 2a-c and3a-c (for IC50 values of non-functionalized peptides, see Chapters 4 and 5) give agood reason to also expect very similar membrane staining properties as observedfor benchmark peptide 1a.
146

666666
6.3. RESULTS & DISCUSSION
Figure 6.2.: (A) Formation of fluorescently labeled RGD-bicycle K(Cy5)-linker-CT3HPQCT3RGDcT3. (B) Integrin selectivities and selected IC50values (integrin αvβ3) of Cy5-functionalized peptides investigated inthis study (linker: PPPSG[Abz]SG, J: D-Leu). The three disulfidebridges in 1a are indicated by “Cox” of identical colors (red, brownand black). *The IC50 values for the non-functionalized RGD-bicycles2a–2c (αvβ3) and 3a–3c (α5β1), and benchmarks 1a+1b as well asnegative control 1c (αvβ3) were already reported in Chapters 4 and5.
6.3.4. HT29 cells
The first cell line tested, HT29, is a colon carcinoma cell line which expresses theintegrin subunits α2, α3, α6, αv and β1, but no α1, α5 or β3.[22, 23]
In a first set of experiments, HT29 cells were treated following experimental setup 1(Figure 6.3) for benchmark peptides 1a–c, as well as αvβ3-selective bicycles 2a+b
147

666666
6.3. RESULTS & DISCUSSION
Figure 6.3.: Schematic illustrations of the different experimental setups applied inthis study. Setup 1: Cells are pre-incubated with peptides at 4 ℃ for1 min and then transferred onto glass coverslips, incubated for 3-4 hat 37 ℃, followed by multiple washing steps and confocal analysis.Setup 2: Cells are transferred onto glass coverslips and incubated forat least 24 h at 37 ℃ and cooled to 4 ℃, followed by addition ofthe peptide, incubation for 10–30 min (4 ℃, modified setup: 37 ℃)and washing (plus optional fixation) prior to confocal analysis (Cellmembranes in blue, fluorescence emission of the Cy5-labels in red).
and α5β1-selective bicycles 3a–c (1 µM). From the two positive (1a/1b) and sin-gle negative (1c) benchmark peptides, only peptide 1a showed significant stainingof the inner cell membrane and intracellular vesicles (Figure 6.4), whereas stainingwith RGD monocycle 1b (positive benchmark) or the linear peptide 1c (negativebenchmark) was hardly observed. Instead, the αvβ3-selective bicycle 2a did showstaining levels and intensities that were very similar to those of benchmark peptide1a, while the αvβ3-selective bicycle 2b hardly showed staining of the HT29 cells.This is remarkable considering that the peptide sequences of both bicycles differ onlyin the chiral configuration of the middle cysteine (i.e. D for 2a, L for 2b). Moreover,the very different staining behavior of 2a and 2b cannot be understood either when
148

666666
6.3. RESULTS & DISCUSSION
considering their almost equal binding to αvβ3 (IC50: 30 and 31 nM) as determinedin competition ELISA (see Chapter 4). A plausible explanation for this could bethat the difference in chirality of the middle cysteine somehow causes a different3D orientation of bicycle 2a, which therefore fits well into the binding pocket ofthe cellular integrin αvβ3, while bicycle 2b does not. A different explanation couldbe that both bicycles do bind the cellular integrin with similar affinities, however,only one (i.e. 2a) gets internalized as a result of the different conformation. Theα5β1-binding bicycles (3a–c) all showed measurable staining of intracellular vesi-cles, albeit weak as compared to the αvβ3-binders.Internalization of fluorescently labeled RGD-peptides at 37 ℃ was previously re-ported, for example, for M21 melanoma cells.[24] With the aim to avoid internal-ization of the RGD-bicycles and to more efficiently promote membrane integrinstaining, HT29 cells were treated with selected peptides (benchmarks 1a and 1b,αvβ3-selective bicycles 2a and 2b, and α5β1-selective bicycles 3b and 3c) followingexperimental setup 2 (Figure 6.3). Surprisingly, αvβ3-selective bicycle 2a showedeven higher staining levels than observed for benchmark peptide 1a. The stainingwith bicycle 2a was basically located at the cell membranes, and therefore revealinghigh levels of efficient integrin αvβ3 staining (Figure 6.5).Remarkably, as in the experiments at 37 ℃, bicycle 2b did not show any staining,suggesting that the second, non-RGD loop of 2b does not fit the integrin bind-ing pocket, contrary to the the second loop in bicycle 2a. For benchmark 1a andα5β1-selective bicycles 3b and 3c, moderate membrane staining was observed, whilemonocycle 1b hardly showed cell staining. Considering the integrin expression pat-tern of HT29 cells (expression of αv and β1; no expression of α5 and β3),[22, 23] thesimilar staining levels for benchmark 1a (binds equally to αvβ3, αvβ5 and α5β1)and α5β1-selective bicycles 3b and 3c might be explained by the possibility thatbicycles 3b and 3c only bind to the β1 subunit while peptide 1a binds to only oneof the subunits αv and β1. It has to be mentioned that this hypothesis, assumingsimiliar expression levels of both αv and β, was not verified experimentally. Follow-ing this line of reasoning, the αv subunit seems to be conformationally advantageousparticularly for binding of αvβ3-selective bicycle 2a. Another interesting observa-tion is that the difference in staining efficiency for benchmark 1a and on the otherhand the α5β1-selective bicycles 3b + 3c is much less pronounced at 4 ℃ than at37 ℃. One possible explanation might involve a temperature-dependent conforma-tional flexibility of the various integrin subunits, allowing membrane internalization
149

666666
6.3. RESULTS & DISCUSSION
Figure 6.4.: Confocal microscopy images of HT29 cells incubated with benchmarkRGD-peptides 1a-c, and bicyclic peptides 2b+c and 3a–c obtainedby applying experimental setup 1. Cells were treated following ex-perimental setup 1 (peptide incubation: 3 h [2a: 4 h] at 37 ℃). Allimages were acquired at identical imaging conditions, and processedvia ImageJ (LUT: Fire). The contrast is shown in arbitrary units(a.u.). 0: No fluorescence, 100: maximum fluorescence. Scale bars:50 µm (1c), 75 µm (2a), 25 µm (rest).
150

666666
6.3. RESULTS & DISCUSSION
Figure 6.5.: Confocal microscopy images of HT29 cells incubated with benchmarkRGD-peptides 1a+b, and bicyclic peptides 2a+b and 3b+c obtainedby applying experimental setup 2 (peptide incubation: 30 min at 4 ℃,fixation with 4% PFA). All images were acquired at identical imagingconditions, and processed via ImageJ (LUT: Fire). The contrast isshown in arbitrary units (a.u.). 0: No fluorescence, 50: maximumfluorescence. Scale bars: 50 µm (1a, 2b, 3b), 75 µm (rest).
of peptides at higher temperatures.Without considering the weak fluorescence intensity for αvβ3-binder 2b, the strongstaining observed for αvβ3-binder 2a compared to the α5β1-binders 3a+b couldalso be explained by the high expression levels of integrin subunit αv and the absenceof α5 in HT29 cells.[22, 23] In order to test this hypothesis, the fluorescently labeledRGD peptides were subsequently tested for efficient membrane integrin staining ofa cell line that is known to also express integrin heterodimer α5β1.
151

666666
6.3. RESULTS & DISCUSSION
6.3.5. HeLa cells
The cervical carcinoma cell line HeLa is known to express both integrins α5β1 andαvβ3.[25–28]
In this set of experiments HeLa cells were treated following experimental setup 2(Figure 6.3) for bicyclic RGD peptides (2b+c and 3b+c) and benchmark sequences1a–c. Contrary to the results for HT29 cells at 4 ℃, the strongest staining intensi-ties of HeLa cell membranes were observed with benchmark peptide 1a (Figure 6.6),presumably as a consequence of the high integrin affinity for both integrins α5β1and αvβ3. The locally higher intensities could be a result of integrin-clustering.In contrast, application of the benchmark peptides monocyclic RGD 1b and linearRGD 1c did not result in cell staining. The surprisingly weak performance of 1b(high αvβ3- and αvβ5-affinity) could be explained by i) an overrepresentation ofα5β1 integrin, ii) a significant decrease of integrin affinity due to the presence ofthe linker and the fluorescent label (IC50: 900 nM, Figure 6.2B, p.147), and/or iii)the possibility that the cell surface integrin’s RGD binding pocket (αvβ3 and/orαvβ5) is conformationally unfavorable for binding of 1b as opposed to the RGDbinding pocket of recombinant integrins αvβ3 and αvβ5 applied in competitionELISA (Chapter 4).Cells that were incubated with αvβ3-binding bicycles 2b and 2c showed weak stain-ing, the latter of which showed slightly higher fluorescence intensities, while cellsincubated with 2a did not show any detectable membrane staining. These resultswere not expected due to the fact that HeLa cells express integrin αvβ3, but mightbe explained by i) a different integrin conformation in vitro compared with the re-combinant form applied in immunoassays, ii) the possibility that the ‘HSQ’ loopof 2c better fits the integrins’ RGD-binding pocket than ‘HPQ’ in 2a/2b, or iii) adecrease in the bicycles’ integrin affinity due to the presence of a linker and a fluo-rescent label (the influence of the linker and the Cy5-label on the integrin affinity ofboth αvβ3-bicycles and benchmark RGD peptides will be discussed in section 6.3.7,p.160). As opposed to this, incubation with α5β1-binding bicycles 3b and 3c led toreasonable HeLa membrane-staining, each showing slightly higher staining localiza-tion on the membrane part located distal from the glass coverslip surface. However,despite its high α5β1-affinity as observed in immunoassays (see Chapter 5), bicycle3a did not show any staining, presumably due to an integrin conformation in vitrothat is not favorable for binding of this peptide, or a significant loss in integrinaffinity as a consequence of Cy5-labeling. The slightly higher staining intensities
152

666666
6.3. RESULTS & DISCUSSION
observed for the α5β1-binding bicycles 3b and 3c over the αvβ3-binding bicycles2a–c could be explained by the facts that HeLa cells express both integrins α5β1and αvβ3,[28] and that the α5β1-binding bicycles do also show a certain extent ofbinding to αvβ3 (IC50 ratio α5β1/αvβ3 ~0.1, see Chapter 5).
153

666666
6.3. RESULTS & DISCUSSION
Figure 6.6.: Confocal microscopy images of HeLa cells incubated with benchmarkRGD-peptides 1a–c, and bicyclic peptides 2a–c and 3a–c obtainedby applying experimental setup 2 (peptide incubation: 10 min at 4 ℃,fixation with 4% PFA). All images were acquired at identical imagingconditions, and processed via ImageJ (LUT: Fire). The contrast isshown in arbitrary units (a.u.). 0: No fluorescence, 50: maximumfluorescence. Scale bars: 50 µm (2a and 3a, 75 µm (rest).
154

666666
6.3. RESULTS & DISCUSSION
Additionally, three-dimensional z-stack images were taken of cells labeled withbenchmark 1a and bicycle 3b (Figure 6.7). This technique allows to detect flu-orescence of thin cross sections throughout the cell, which gives a more detailedinsight into the 3D membrane staining efficiency. In these experiments, the distancebetween each cross section was set to one micrometer. The distances are relativevalues each with the bottom image as a reference value (z = 0 µM).The first z-stack analysis, applying benchmark 1a, reveals that the cell aggregate
Figure 6.7.: Z-stack analysis of HeLa cells incubated with 1a and bicycle 3b (usingexperimental setup 2). Scale bars: 75 µm (1a) and 25 µm (3b).
is more than 12 µm thick. The top part of the membranes can be detected from
155

666666
6.3. RESULTS & DISCUSSION
12 to 9 µm. Below 9 µm the lateral part of the membranes becomes more visiblethrough each section. The strong lateral membrane staining on the bottom imagereveals that the basal membrane probably lies below this section. In contrast, thez-stack analysis of the cell labeled with 3b reveals that this cell is thinner comparedto the cell aggregate stained with 1a. While the top part of the membrane is notvisible, the lateral part becomes visible below 6 µm. Starting at 2 µm, the basalmembrane becomes visible. The images taken at 1 and 2 µm show a slight accu-mulation of 3b on the lateral membrane to the right and on the top part, possiblydue to integrin clustering. As for the other z-stack analysis, the basal membrane isprobably situated below the lowest section (0 µm) analyzed.
6.3.6. Adipose-derived stem cells
So far, two cell lines were investigated for integrin staining, i.e. HT29 overexpressingintegrin subunits αv and β1, and HeLa cells expressing both heterodimers αvβ3 andα5β1. Adipose-derived stem cells (ASCs) overexpress integrin subunits α5, β1 andβ5, while showing low levels of αv and β3 expression.[29, 30] This integrin expressionpattern would suggest effective staining levels when applying the α5β1-selectivebicycles 3a–c, while little to no staining would be expected for the αvβ3-selectivebicycles 2a–c.First of all, it is noteworthy mentioning that ASCs showed relatively slow growthand were therefore cultured for at least 4 d prior to trypsinization.‡ Applyingexperimental setup 2, Cy5-labeled benchmark peptides 1a+1b, negative control1c, αvβ3-selective bicycles 2a–c and α5β1-selective bicycles 3a–c were added tothe adhered cells and incubated at 4 ℃ for 10 min, followed by washing and fixationprior to confocal analysis (Figure 6.8). The strongest membrane fluorescence wasobserved for cells stained with benchmark 1a, the focal adhesion regions of whichlocally even showed higher staining intensities. In contrast, negative control linearpeptide 1c showed only very weak staining, whereas the benchmark monocyclicpeptide 1b did not show any staining at all. Hence, these two benchmarks didhardly show any membrane integrin staining of either of the three cell lines tested.
‡Moreover, the cell surface area of adhered ASCs was much higher than for the other two celllines, which explains the lower cell density that can be seen on the confocal images.
156

666666
6.3. RESULTS & DISCUSSION
Figure 6.8.: Confocal microscopy images of adipose-derived stem cells incubatedwith benchmark RGD-peptides 1a–c, and bicyclic peptides 2a–c and3a–c obtained by applying experimental setup 2 (cell incubation: 4 d,peptide incubation: 10 min at 4 ℃, fixation with 4% PFA). All im-ages were acquired at identical imaging conditions, and processed viaImageJ (LUT: Fire). The contrast is shown in arbitrary units (a.u.).0: No fluorescence, 50: maximum fluorescence. Scale bars: 50 µm(1a) and 75 µm (rest).
157

666666
6.3. RESULTS & DISCUSSION
In contrast, intense ASC membrane staining was observed for α5β1-binder 3b,while hardly any fluorescence was observed for cell membranes stained with theother two α5β1-binders 3a and 3c. An explanation for this observation might bethat the AWG loop in bicycle 3b is essential for the binding to α5β1 integrins ex-posed on ASC membranes at 4 ℃, contrary to the respective AYJ and AYa loopsin bicycles 3a and 3c. For the αvβ3-selective bicycles, weak (2c) or no staining(2a+b) was observed; a result that was expected as a consequence of the integrinexpression pattern (only little αv and β3 expression).[29, 30]
Finally, a selection of four peptides (benchmark peptides 1a+c, αvβ3-selective bicy-cle 2a and α5β1-selective bicycle 3b) was incubated with ASCs at 37 ℃ for 10 min(modified variant of experimental setup 2, Figure 6.9) to validate that membraneintegrin staining is temperature-dependent.Surprisingly, 1a-stained cells showed by far the strongest fluorescence, which waseven higher than at 4 ℃, and located in the entire intracellular environment, whereas1c again did not show any staining. Presumably, the much higher fluorescence of 1aas compared with the experiments at 4 ℃ can be explained by the higher membranepermeability for this peptide that internalizes subsequent to integrin binding. Incontrast, since the integrin affinity for benchmark 1c is per se very low, it cannotbind to integrins and therefore not being internalized, which would explain the lowstaining levels even at 37 ℃. Interestingly, the confocal microscopy images of cellsincubated with αvβ3-selective bicycle 2a and α5β1-selective bicycle 3b reveal sim-ilar staining levels. Remarkably, at 37 ℃ 3b showed much weaker staining levelsthan at 4 ℃. One hypothetical explanation might be that integrin α5β1 exhibits adifferent conformation at 37 ℃ that decreases binding efficiency of 3b.
158

666666
6.3. RESULTS & DISCUSSION
Figure 6.9.: Confocal microscopy images of adipose-derived stem cells incubatedwith benchmark RGD-peptides 1a+c, and bicyclic peptides 2a and3b obtained by applying a modified variant of experimental setup 2(cell incubation: 5 d, peptide incubation: 10 min at 37 ℃, fixationwith 4% PFA). All images were acquired at identical imaging condi-tions, and processed via ImageJ (LUT: Fire). The contrast is shown inarbitrary units (a.u.). 0: No fluorescence, 50: maximum fluorescence.Scale bars: 75 µm (1a+c, 2a) and 50 µm (2b).
In order to validate that both the ‘RGD’ loop and the ‘AWG’ loop in α5β1-selective bicycle 3b are essential for membrane integrin staining, two bicycles weresynthesized, each comprising either one of the two loops with a scrambled sequence,i.e. one bicycle exhibiting a scrambled RGD-loop (GDR-3b) and one bicycle ex-hibiting a scrambled AWG-loop (WGA-3b, Figure 6.10A).The scrambled peptides were tested for their ability to bind to membrane integrinsof ASCs at 4 ℃ applying setup 2 (see caption of Figure 6.10). As expected, al-most no staining was observed for GDR-3b, which confirms the essential role ofRGD for binding to membrane integrins. For the second scrambled peptide WGA-3b mediocre staining levels were observed, albeit much weaker than for 3b (Fig-
159

666666
6.3. RESULTS & DISCUSSION
ure 6.10B). This also indicates an essential role of the ‘AWG’ loop for the bindingof the bicycle to membrane integrin α5β1, however, to a lesser extent than for the‘RGD’ loop.
Figure 6.10.: Confocal microscopy images of adipose-derived stem cells incubatedwith scrambled, bicyclic peptides GDR-3b and WGA-3b in compar-ison with α5β1-binder 3b (image adopted from Figure 6.8) obtainedby applying experimental setup 2 (cell incubation: 4 d, peptide in-cubation: 10 min at 4 ℃, fixation with 4% PFA). All images wereacquired at identical imaging conditions, and processed via ImageJ(LUT: Fire). The contrast is shown in arbitrary units (a.u.). 0: Nofluorescence, 50: maximum fluorescence. Scale bars: 75 µm.
6.3.7. Inhibition properties of linker-modified and Cy5-labeledbicyclic peptides
It was mentioned earlier that the functionalization of RGD bicycles and bench-marks with both the linker and the Cy5-label might have led to reduced integrinaffinities, which could explain the low staining efficiencies observed for, for exam-ple, benchmark 1b and in some cases αvβ3-bicycles 2a–c. In order to addressthis, non-labeled variants of two αvβ3-selective RGD bicycles (2a and 2b), andthree positive/negative benchmark peptides (1a-1c) were functionalized with eitherthe linker only, or with both the linker and Cy5 dye, and tested for inhibition ofbiotinylated knottin-RGD–integrin αvβ3 binding in competition ELISA (IC50 de-termination), followed by comparison of these values with the IC50 values of thenon-functionalized RGD peptides (see Table 6.1A).§
§The determination of IC50 values for non-functionalized RGD peptides was already describedin Chapter 4.
160

666666
6.3. RESULTS & DISCUSSION
When adding the linker, the IC50 values increased ~4–4.5-fold for both bicyclesCT3HPQcT3RGDcT3 and CT3HPQcT3RGDcT3 (from 30 to 165 nM and from 31to 150 nM, respectively, Table 6.1A). Furthermore, for the Cy5-functionalized vari-ant of the first bicycle (corresponds to 2a) an even higher IC50 (~8-fold, 276 nM) wasdetermined. In contrast, the IC50 value of cyclo-[KRGDf] increased ~15-fold (from36 to to 609 nM), and even ~25-fold after funtionalization with Cy5 (corresponds topeptide 1b), which may at least in parts explain the weak staining intensities ob-served in all cell lines tested. In contrast the IC50 value of Cy5-linker-functionalizedknottin-RGD (corresponds to 1a) was only ~4-fold higher (198 nM) as compared tothe non-functionalized variant (38 nM). These data suggest that the (Cy5)-linkerfunctionalization impairs the integrin αvβ3-affinity of cyclo-[KRGDf] to a largerextent than for the bicyclic peptides and the knottin-RGD peptide.Furthermore, the integrin affinities for each one αvβ3-selective and one α5β1-selective bicycle carrying various newly designed linkers were evaluated (see Ta-ble 6.1B). Various linker sequences have been reported to control the distance be-tween the integrin-binding motif and a functional residue, for example, for the pur-pose of surface functionalization.[21]
Following this, non-modified variants of each one αvβ3- and α5β1-selective bicy-cle (2a+2b) were modified with five different linkers to investigate their influenceon integrin affinity, i.e. G, PPPPPPPPP, [PEG]2, [PEG]3 and GGSGGSGGS.¶
Subsequently, these peptides were analyzed for inhibition of binding of a biotiny-lated knottin-RGD peptide to the respective integrins αvβ3 and α5β1 (Table 6.1B).
For the αvβ3-binder, the GGSGGSGGS-modified variant comprised the lowestIC50 value (101 nM), followed by the [PEG]3-modified bicycle (121 nM), that is bothpeptides show higher αvβ3-affinity than the bicycle comprising the KPPPSG[Abz]SGlinker (165 nM, Table 6.1). Surprisingly, the bicycle extended by one glycine evenshowed a higher IC50 than the GGSGGSGGS-modified bicycle, which exemplifiesthe high sensitivity of the integrin-RGD bicycle interaction. A similar trend wasobserved for the α5β1-binding bicyle, the GGSGGSGGS-modification of whichled to a 2-fold increase of IC50 (189 nM) as compared to the non-modified bicy-cle (90 nM, Table 5.1, p.118) whereas modification with glycine resulted in a ~4-fold higher value (388 nM). Moreover, for both bicycles application of a proline
¶The term [PEG] refers to an amino acid comprising two ethylene glycol units (8-amino-3,6-dioxaoctanoic acid).
161

666666
6.3. RESULTS & DISCUSSION
Table 6.1.: (A) IC50 values of non-modified, KPPPSG[Abz]SG- orK(Cy5)PPPSG[Abz]SG-modified bicyclic and benchmark RGDpeptides determined via competition ELISA using surface-immobilizedintegrin αvβ3 and soluble peptides (biotinylated knottin-RGD +respective non-labeled RGD peptide); (B) IC50 values for each oneαvβ3- and α5β1-binding bicycle as a function of linker attached. Eachconcentration (0.014–30 µM) was tested in triplicate and the IC50values were determined via nonlinear regression analysis using thesoftware GraphPad Prism.
linker (PPPPPPPPP) led to the highest loss of integrin affinity (IC50,αvβ3: 302 nMfor PPPPPPPPPCT3HPQcT3RGDcT3, IC50,α5β1: 869 nM for PPPPPPPPPCT3-RGDcT3AYJCT3), probably as a consequence of the low conformational flexibilityof this linker.Interestingly, for both peptides and integrins, the application of the flexible linkerGGSGGSGGS affected the peptide-integrin interaction to the lowest level, whichwould suggest the application of this linker for both αvβ3- and α5β1-selective bi-cycles in future cell membrane staining experiments.
162

666666
6.4. CONCLUSION & OUTLOOK
6.4. Conclusion & outlook
The studies described in this chapter show that the simple translation of resultsobtained in competition ELISA experiments (IC50 values) to in vitro experiments(labeling of cell membrane integrins) is not straightforward. Various other factorsinfluence the staining efficiency in addition to the affinity of the bicyclic RGD pep-tides, i.e. integrin expression pattern of the cell line, size of the integrin-binder(ease of membrane internalization), linker sequence (decrease of integrin affinity)or incubation temperature. For example, very strong staining levels of HT29 cellmembranes were detected for fluorescently labeled αvβ3-selective bicycle 2a, whileαvβ3-selective bicycle 2b, varying in only the absolute configuration of the mid-dle cysteine, did not show any HT29 membrane staining. In contrast, HeLa cellmembranes showed much lower staining levels when applying αvβ3-selective bicy-cle 2a, while incubation with α5β1-selective bicycles 3b and 3c resulted in clearlydetectable membrane staining, albeit to a much lower extent as compared to high-affinity knottin-RGD benchmark peptide 1a. Moreover, the α5β1-selective bicycle3b (and the benchmark peptide 1a) also showed the strongest staining levels of ASCmembranes, while staining was neither detected for αvβ3-selective bicycles 2a and2b nor for monocyclic benchmark peptide 1b and linear negative control peptide1c. The results reveal that fluorescently labeled, integrin-selective bicyclic RGDpeptides could represent a very good starting point for the development of noveldiagnostics on the basis of highly selective integrin targeting.There are various possibilities to further improve the staining efficiency in futureexperiments. The first option is the application of Cy5-functionalized bicycles com-prising a different linker connecting the integrin-binding moiety with the fluorescentlabel, for example, GGSGGSGGS which was shown to affect αvβ3- and α5β1-affinity of the bicycles to a lower extent than the KPPPSG[Abz]SG linker appliedin this study. Another alternative is the application of different buffer systems toreduce the amount of photobleaching, for example, by introducing oxidizing and re-ducing agents that reduce the life time of the triplet state, but also promote the quickrecovery of the triplet state.[31] Earlier in this thesis (Chapter 4, Figure 4.9, p.94) abiotinylated trimeric bicyclic peptide was described to show high affinity to surface-immobilized integrin αvβ3, in particular at low peptide concentrations (≤ 0.01 µM).Perhaps, Cy5-functionalized versions of trimeric bicyclic peptides might representan elegant solution to increase the cell membrane staining efficiency. However, while
163

666666
6.5. MATERIALS & METHODS
the size of such a construct might reduce the probability of cell internalization, itwill also most likely impair the resolution in confocal microscopy, as well as in po-tential SRM experiments.
6.5. Materials & methods
Reagents & chemicalsFor general information about reagents and chemicals, as well as the synthesis ofthe bicyclic peptides, see Chapter 4 (p.96). Disulfo-Cy5-NHS ester was purchasedfrom Cyandye (Sunny Isles Beach, USA).
Synthesis of fluorescently labeled RGD peptidesPeptides were dissolved at 4 mM in DMSO, followed by adding disulfo-Cy5 NHS(1.0 equiv., 20 mg/mL in DMSO) and DIPEA (10 equiv.). After completion (30-60 min), the reaction was quenched with 10% TFA/H2O (2x the volume of DIPEAused), and the product was subsequently purified using preparative RP-HPLC. Alllabeled peptides were isolated with high purities >90%, and stored in the dark at-20 ℃ prior use.
BuffersCBS buffer (Carbonate-Buffered Saline, pH 7.2) contains 140 mM NaCl, 5 mMKCl, and 23 mM NaHCO3. Adjustment of pH was realized using dry ice. The work-ing buffer, HCG buffer (HEPES-CBS-Glucose), consists of CBS containing 10 mMHEPES, 10 mM glucose, 1 mM CaCl2, 0.5 mM MgCl2, and 0.5 mM MnCl2. For theexperiments at 4 ℃ the working buffer was cooled to 4 ℃ before use.
Cell culture & confocal microscopyHuman colorectal adenocarinoma cells (HT29) and human cervical carcinoma (HeLa)cells were obtained from the Netherlands Cancer Institute (NKI). Human adipose-derived stem cells (ASCs) were a kind gift from Dr. Egbert Oosterwijk (Institutefor Molecular Life Sciences, Radboud University Medical Center Nijmegen (Rad-boudumc), the Netherlands).
164

666666
6.5. MATERIALS & METHODS
Experimental setup 1: HT29 cells were mixed together with Cy5-labeled peptide(1 mM in DMSO), ratio 1:1000, in order to achieve a peptide concentration of1 µM, followed by incubation on clean glass coverslips (Fisher Scientific, thickness1.5, d: 24 mm) for 3 h (~30% confluency). Next, the glass coverslips were washedat least five times with HCG buffer in order to reduce background fluorescence.Subsequently, the cells were analyzed via confocal microscopy using a Leica TCSSP8 confocal microscope equipped with a supercontinuum white light laser (NKTPhotonics) and water immersion objectives (63x W PL APO CS2, NA 1.2/40x WPL APO CS2, NA 1.1). The excitation wavelength was set to 633 nm while fluores-cence was detected from 646 to 778 nm.Experimental setup 2: HT29 cells, HeLa cells or ASCs were allowed to adhere onclean glass coverslips for at least 24 h (ASC: at least 4 d) until reaching approx. 40-50% (ASC: ~10-20%) confluency. Then, the glass coverslips were washed two timeswith cold HCG buffer to remove non-adhered cells, followed by adding cold HCGbuffer, and cooling of the glass coverslips to ~4 ℃.‖ Afterwards, the Cy5-labeledpeptides were incubated at 1 µM for 10-30 min at 4 ℃ (modified setup: 37 ℃),followed by at least five washing steps with HCG buffer, fixation with 4% formalde-hyde solution in PBS (20 min) and another four washing steps with HCG bufferprior to confocal analysis.
‖Since the cooling was realized with ice, the exact temperatures of the solution and the glasscoverslips, onto which the cells were immobilized were not determined and therefore estimated tobe 4 ℃.
165

666666

666666
6.6. REFERENCES
6.6. References[1] M. A. Schwartz, M. D. Schaller, M. H. Ginsberg, Annu. Rev. Cell Dev. Biol.
1995, 11, 549–599.[2] M. A. Schwartz, M. H. Ginsberg, Nat. Cell Biol. 2002, 4, 65–68.[3] C. J. Avraamides, B. Garmy-Susini, J. A. Varner, Nat. Rev. Cancer 2008,
8, 604–617.[4] V. P. Hytönen, B. Wehrle-Haller, Exp. Cell Res. 2016, 343, 35–41.[5] Z. Zhang, Q. Luo, X. Yan, et al., Anal. Chem. 2012, 84, 8946–8951.[6] M. H. Lee, J. Y. Kim, J. H. Han, et al., J. Am. Chem. Soc. 2012, 134, 12668–
12674.[7] Y. Zheng, S. Ji, A. Czerwinski, et al., Bioconjugate Chem. 2014, 25, 1925–
1941.[8] Z. Hu, R. L. Arrowsmith, J. A. Tyson, et al., Chem. Commun. 2015, 51,
6901–6904.[9] F. Aoudjit, K. Vuori, Chemoth. Res. Pract. 2012, 2012, 283181.[10] J. Schittenhelm, A. Klein, M. S. Tatagiba, et al., Int. J. Clin. Exp. Pathol.
2013, 6, 2719–2732.[11] M. Nieberler, U. Reuning, F. Reichart, et al., Cancers 2017, 9, 1–33.[12] R. Haubner, H.-J. Wester, U. Reuning, et al., J. Nucl. Med. 1999, 40, 1061–
1071.[13] S. J. Moore, M. G. Hayden Gephart, J. M. Bergen, et al., Proc. Natl. Acad.
Sci. USA 2013, 110, 14598–14603.[14] A. L. Mattheyses, S. M. Simon, J. Z. Rappoport, J. Cell Sci. 2010, 123,
3621–3628.[15] E. Betzig, J. K. Trautman, T. D. Harris, et al., Science 1991, 251, 1468–
1470.[16] P. Davidovits, M. D. Egger, Scanning laser microscope, 1969.[17] S. W. Hell, J. Wichmann, Opt. Lett. 1994, 19, 780–782.[18] S. W. Paddock, Mol. Biotechnol. 2000, 16, 127–149.[19] G. T. Dempsey, J. C. Vaughan, K. H. Chen, et al., Nat. Methods 2011, 8,
1027–1040.[20] L. Nahidiazar, A. V. Agronskaia, J. Broertjes, et al., PLoS ONE 2016, 11,
e0158884.[21] D. Pallarola, A. Bochen, H. Boehm, et al., Adv. Funct. Mater. 2014, 24,
943–956.
167

666666
6.6. REFERENCES
[22] C. Schreiner, J. Bauer, M. Margolis, et al., Clin. Exp. Metastas. 1991, 9,163–178.
[23] J. Haler, M. Nasralla, G. L. Nicolson, Brit. J. Cancer 1999, 80, 1867–1874.[24] S. Castel, R. Pagan, F. Mitjans, et al., Lab Invest. 2001, 81, 1615–1626.[25] T. Riikonen, P. Vihinen, M. Potila, et al., Biochem. Biophys. Res. Commun.
1995, 209, 205–212.[26] M. Oba, S. Fukushima, N. Kanayama, et al., Bioconjugate Chem. 2007, 18,
1415–1423.[27] L. Xiong, M. Yu, M. Cheng, et al., Mol BioSyst. 2009, 5, 241–243.[28] N. Orgovan, B. Peter, S. Bosze, et al., Sci. Rep. 2014, 4, 4034.[29] U. R. Goessler, P Bugert, K Bieback, et al., Int. J. Mol. Med. 2008, 21,
271–279.[30] A. B. J. Prowse, F. Chong, P. P. Gray, et al., Stem Cell Res. 2011, 6, 1–12.[31] J. Vogelsang, R. Kasper, C. Steinhauer, et al., Angew. Chem. Int. Ed. 2008,
47, 5465–5469.
168

7777777
7. Synthesis and BiologicalEvaluation of ELR SurfacesModified with Integrin αvβ3- andα5β1-Selective RGD-Bicycles
Parts of the work described in this chapter were published: F. Cipriani, D. Bernhagen, C.García-Arévalo, I. Gonzàlez de Torre, P. Timmerman, J. Rodríguez-Cabello, Biomed. Mater.2019, 14, 035009.

7777777
7.1. ABSTRACT
7.1. Abstract
In this study, the high affinity integrin αvβ3- and α5β1-selective RGD bicycles de-scribed in Chapters 4 and 5 were covalently bound to elastin-like recombinamers(ELRs) via copper-free click chemistry in order to investigate their HUVEC ad-hesion and proliferation promoting properties. Next to the bicycles, various posi-tive and negative benchmark ELRs (e.g. knottin-RGD-functionalized, fibronectin-coated, linear GRGDS-functionalized, or non-functionalized ELRs) were studiedwhich allows a proper classification of the obtained results. MALDI-TOF-MS anal-ysis revealed covalent functionalization with all bicycles and controls, also indicatingdifferences in effective functionalization degrees. Proliferation studies (PicoGreen®assay) after 30 minutes revealed that all ELRs modified with αvβ3- (P1a-P1c) andα5β1-selective bicycles (P2a-P2c) comprised significantly higher cell counts thanGRGDS control-functionalized (P3c), BSA-coated (P0-BSA) or non-modified ELR(P0). For two αvβ3-selective ELRs (P1a+P1b), even higher cell counts were ob-served than for the RGD-monocycle-functionlized positive benchmark ELR (P3b),while similar cell counts as compared to the knottin-RGD-functionalized positivebenchmark ELR (P3a) were detected. After 4 hours and 1 day, αvβ3-selectivebicycle-functionalized ELRs P1a-P1c comprised much higher cell counts than neg-ative control ELR P3c (GRGDS-functionalized), as well as the benchmark ELRrecombinantly modified with RGD as part of the ELR backbone (P0-RGD), andslightly higher cell counts than α5β1-selective bicycle-functionalized ELRs P2a-P2c. After 3 days, cell counts for RGD-bicycle-functionalized ELRs P1a-P1c andP2a-P2c were on the same level as fibronectin-coated, positive ELR benchmarkP0-FN, and still significantly higher than for the GRGDS-functionalized negativebenchmark ELR P3c and all other negative benchmark ELRs (P0, P0-BSA, P0-RGD). Notably, ELR P1a (modified with bicycle CT3HPQcT3RGDcT3) showed aneven higher cell count than the positive benchmark ELRs P3a and P3b. After 5 and7 days, both αvβ3-selective ELRs P1a and P1b comprised significantly higher cellnumbers than benchmark ELRs P3a-P3c, and yet slightly higher cell counts ascompared to α5β1-selective ELRs P2a-P2c. After 14 days, the differences betweenRGD-bicycle-functionalized and benchmark ELRs were only marginal. Finally, mor-phology studies with nuclei-, actin skeleton- and vinculin-stained HUVECs revealedthat ELR P1a, similarly to benchmark ELR P3a, comprised extended focal contactsalready after 4 hours, whereas formation of focal contacts on bicycle-functionalized
170

7777777
7.2. INTRODUCTION
ELRs P1b, P1c, P2a-P2c as well as on benchmark ELRs P3b, P3c and P0was only observed after 1 day or even later. The overall data suggest that i) highintegrin-affinity RGD-bicycles promote HUVEC cell adhesion and proliferation onELRs much more efficiently than the linear RGD peptide, and that ii) two par-ticular αvβ3-selective RGD-bicycles, depending on the time point, are even moreefficient mediators of cell growth than the benchmark peptides knottin-RGD andcyclo-[KRGDf].
7.2. Introduction
Recombinant elastin-like recombinamers (ELRs) or elastin-like proteins (ELPs) arederived from the extracellular matrix protein (ECM) elastin and represent a groupof ECM-mimicking biomaterials. ELRs are thermo-sensitive polymers, which arecharacterized by a transition temperature Tt, below which the polymers are fully hy-drated (hydrophic hydration) and show water-soluble random-coil structures. Abovethis temperature the polymer chains become insoluble, because they form so-calledβ-spiral structures that are stabilized by hydrophobic interactions, thereby losingtheir hydrophobic hydration.[1–3] The advantages of ELRs for tissue engineering ap-plications are their straightforward synthesis, together with their defined macro-molecular structure, controlled swelling behavior and porosity, degradability, andcontrollable mechanical properties.[4, 5]
First described in 1992 by Nicol et al.,[6] ELRs comprising integrin-binding motifssuch as ‘RGD’[3, 7–9] or ‘REDV’[9, 10] (Figures 7.1A and 7.1B) have been investigatedas possible materials for tissue engineering applications. More recently, ELR com-posite materials have been reported for various applications, for example, ELR-fibringels as artificial heart valves[11], or ELR-coated silicon doped hydroxyapatite for pos-sible application in bone regeneration.[12]
However, the recombinant synthesis of ELRs does neither allow incorporation ofnon-canonical amino acids nor the formation of cyclic peptides. An elegant ap-proach to circumvent this limitation, and an additional lever to control cell ad-hesion and proliferation is the post-functionalization with peptides, for example,via the side-chain amines of lysines (Figures 7.1A–C). For example, ELP hydro-gels covalently functionalized with a peptide mimic of the receptor-binding regionof VEGF (vascular endothelial growth factor) showed enhanced HUVEC prolifera-tion over non-functionalized hydrogels,[13] whereas ELPs functionalized with RGD
171

7777777
7.2. INTRODUCTION
via maleimide-thiol coupling showed improved attachment, spreading, migrationand proliferation of endothelial and mesenchymal stem cells as compared to non-functionalized ELR.[14] In an earlier study, ELRs functionalized with cyclo-[KRGDf]showed ~100% improved mouse osteoblast adhesion over ELRs functionalized withlinear FGRGDS.[15] Herein, the focus laid on the chemical functionalization of ELRsand did not include recombinantly synthesized RGD-ELRs. In contrast, recombi-nant silk spider proteins covalently functionalized with a cyclic RGD peptide showedsimilar cell adhesion behavior as compared to the proteins comprising RGD as part ofthe recombinant backbone.[16] However, the linear RGD sequence was incorporatedrecombinantly, whereas the cyclic RGD sequence was coupled chemically, whichmakes a precise comparison of the cell adhesion-promoting properties of linear ver-sus cyclic RGD rather difficult.
Figure 7.1.: Selection of ELRs applied for tissue engineering applications. ELRscomprising (A) an integrin-binding RGD motif reported by GonzálezDe Torre et al.,[17] and (B) an integrin-binding REDV motif reportedby Girotti et al.[10] (C) ELR applied in this study.
Earlier in this thesis, high affinity bicyclic peptides for both αvβ3 and α5β1 in-tegrins were reported (Chapters 4 and 5), together with their application in cellmembrane staining (Chapter 6). In the following study, the αvβ3- and α5β1-binding RGD bicycles are covalently connected to ELRs to investigate their HU-VEC adhesion- and proliferation-promoting behavior. In addition, several con-trol ELRs, for example, fibronectin-coated or knottin-RGD functionalized ELRswere investigated in order to classify the overall benefit of bicyclic peptides as celladhesion/proliferation-promoting ligands. Moreover, the differences in cell adhe-
172

7777777
7.3. RESULTS & DISCUSSION
sion properties between recombinant ELRs comprising RGD as part of the polymerbackbone and ELRs post-functionalized with different RGD peptides are discussed.
7.3. Results & discussion
7.3.1. Selection of RGD peptides and cyclooctyne conjugation
With respect to the high integrin affinities and selectivities of the RGD-bicyclesas described in Chapters 4 and 5, each three αvβ3- and α5β1-selective bicycleswere selected for investigation of cell adhesion and proliferation-promoting prop-erties on 2D ELR surfaces. Like in the previous chapters, the three peptidesGRGDS, cyclo-[KRGDf] and knottin-RGD were selected as benchmarks. All pep-tides were functionalized with the KPPPSG[Abz]SG linker (hereafter referred to as“K-linker”), as described in the previous chapters, followed by conversion to theircyclooctyne-functionalized variants via reaction of the BCN-NHS activated esterwith the side-chain amine of the N-terminal lysine (Figure 7.2A). A complete listof BCN-functionalized peptides, including nomenclature and integrin selectivities isgiven in Figure 7.2B.
7.3.2. Synthesis of RGD peptide-functionalized ELRs
The ELR applied in this study comprised 24 lysine residues, of which 80% wereazide-functionalized (NMR data not shown). The copper-free click reactions wereperformed in cold water at 4 ℃ to ensure complete dissolution of the ELRs. Aschematic representation of the peptide conjugation reaction is depicted in Fig-ure 7.2A. Six different RGD bicycles, of which three bind integrin αvβ3 and an-other three bind integrin α5β1, and three benchmark RGD peptides (knottin-RGD,cyclo-[KRGDf] and linear GRGDS) were covalently bound to the ELR. Moreover,ELRs with two different degrees of azide functionalization were used, i.e. 5% or 10%,resulting on average in approx. 1 (5%) and 2 (10%) RGD-conjugations per ELRmolecule. Additionally, four benchmark ELRs that were not covalently function-alized were tested, i.e. ELRs bearing RGD in the polymer backbone (P0-RGD),the non-functionalized ELR azide (P0), and ELRs coated with either fibronectin(P0-FN) or bovine serum albumin (P0-BSA). P0-RGD and P0-FN representpositive benchmarks, while P0 and P0-BSA were selected as negative controls.In order to verify the functionalization of ELRs with the RGD peptides, MALDI-
173

7777777
7.3. RESULTS & DISCUSSION
Figure 7.2.: (A) Schematic representation of the functionalization of an ELR withbicyclic peptide CT3HPQCT3RGDcT3. (B) Overview of ELRs in-vestigated in this study, including degree of functionalization, RGDpeptide, and integrin selectivities. ELR labels containing “P0” serveas benchmarks that were not covalently modified via copper-free clickreaction. * Selectivities as described in Chapters 4 and 5. Brack-ets indicate partial selectivity for αvβ3 as determined in selectivityexperiments described in Chapter 5.
TOF-MS analyses were performed. A selection of spectra (P0, P1a, P2a, andP3a–c) is given in Figure 7.3.
The peaks around m/z 62,200 represent non-functionalized ELR, whereas addi-tional peaks reveal ELRs that were mono-, di-, tri- or even tetrafunctionalized withRGD peptides. Even though MALDI-TOF-MS analysis does not allow to quantifythe functionalization degree, comparison of the spectra revealed different degrees offunctionalization depending on the type and the amount of peptide applied. Forexample, for P1a and P3a (each 10%) the peak for monofunctionalized ELR (m/z64,383 and 66,558, respectively) shows the highest intensity, whereas the highest
174

7777777
7.3. RESULTS & DISCUSSION
Figure 7.3.: MALDI-TOF spectra of non-functionalized ELR and ELRs function-alized with 5%/10% of bicyclic RGD peptides 1a, 2a and benchmarkRGD peptides 3a–c.
peak in the spectrum for P2a (10%) results from non-functionalized ELR (m/z62,139), indicating a degree of functionalization which is <10%. One explanationcould be that the conjugation reaction with bicycle 2a was less efficient than withbicycle 1a, possibly as a result of a lower degree of solubilization of 2a. Anotherexplanation might be that the degree of hydrolysis for the BCN active ester groupwas higher in bicycle 2a than in bicycle 1a.As expected, the spectra also confirm the different degrees of functionalization foreach peptide. For example, when comparing the spectra for the ELR functional-ized with bicycle 1a (P1a), the spectrum representing 10% RGD-functionalizationindeed shows peaks with higher intensity for mono- and di-RGD-functionalized rela-tively to the peak representing non-functionalized ELR, as compared to the spectrumrepresenting 5% RGD-functionalization.Following the successful RGD peptide-functionalization of the ELRs, a PicoGreen®assay was performed with the ELRs representing 5% functionalization and bench-mark ELRs in order to study the RGD peptide-dependent HUVEC proliferation.
175

7777777
7.3. RESULTS & DISCUSSION
7.3.3. Time-dependent proliferation studies∗ For HUVEC proliferation studies, cells were fixed at defined time points (30 min,4 h, 1 d, 3 d, 5 d, 7 d, 14 d) and incubated with PicoGreen® in order to quantifythe total amount of DNA, which can be directly related to the number of cells. Flu-orescence intensities were transformed into cell numbers using a calibration curveobtained from measuring fluorescence of defined cell numbers (n = 0, 100, 1000,10,000 and 100,000): n = y+4.4615
0.0157 (n: number of cells, y: fluorescence emission). Forstraightforward comparison, data were subdivided related to time point, i.e. short-term (30 min, 4 h, 1 d), medium-term (3 d, 5 d) and long-term (7 d, 14 d), and typeof ELR, that is ELR functionalized with bicyclic RGD peptides (P1a–c and P2a–c),and benchmark ELRs (ELRs functionalized with 3a–c → P3a–c; ELR comprisingRGD as part of the backbone → P0-RGD; non-functionalized ELR azide → P0;ELR coated with fibronectin → P0-FN; ELR coated with BSA → P0-BSA).Among the bicycle-functionalized ELRs, P1a showed the highest number of cellsafter 30 min (~3,350) followed by P1b (~3,100) and P2a (~2,700, Figure 7.4A).
After 4 hours, P1a, P1b and P2c showed the highest cell numbers (~3,700), whilethe ELRs functionalized with α5β1-binding bicycle CT3RGDcT3AWGCT3 (P2b)comprised significantly lower cell numbers (~3,000). After 1 day, the number of cellshad not increased for the majority of the bicycle-funtionalized ELRs, suggesting thatthe adhered cells did not proliferate yet.Can the very similar cell numbers between ELRs functionalized with αvβ3- (P1a-c)and α5β1-selective bicycles (P2a-c) be explained by the surface integrin expressionof HUVEC cells?Flow cytometry analysis with fluorescently labeled antibodies performed by Aidoudiet al. showed that HUVECs express both integrins αvβ3 and α5β1.[18] Baranska etal. reported that the HUVEC surface expression levels of integrin α5β1 are morethan twice the expression levels of αvβ3,[19] which was more recently confirmed byGimenes et al.[20] From these findings, one could expect that α5β1-selective bicy-cles should promote HUVEC adhesion more efficiently than αvβ3-selective bicy-cles. However, a higher α5β1 expression might be compensated by a slightly lowerfunctionalization degree for the α5β1-selective bicycles as indicated by the MALDIstudies discussed earlier (see spectra for P2a and P1a, Figure 7.3, p.175), which
∗The cell experiments described in the following subsections were performed by Filippo Ciprianiat Technical Proteins Nanobiotechnology Valladolid, Spain.
176

7777777
7.3. RESULTS & DISCUSSION
Figure 7.4.: Time-dependent proliferation assay (30 min, 4 h, 1 d). Number ofcells determined for ELRs containing (A) bicyclic peptides 1a–c (P1a-P1c) and 2a–c (P2a-P2c), and (B) benchmark RGD peptides 3a–c(P3a-P3c), RGD within the ELR-backbone (P0-RGD), no RGDpeptide (P0), fibronectin coating (P0-FN) and coating with BSA(P0-BSA). Grid lines at 2,500 and 4,000 are shown for better com-parison of data. The number of cells was calculated from the fluo-rescence intensities using a calibration curve. All experiments werecarried out in triplicate and error bars show standard deviations.
could at least partly explain the slightly higher cell numbers for ELRs P1a and P1bas compared to ELRs P2a and P2b.Within the group of benchmark ELRs P3a displayed the highest cell numbers after
177

7777777
7.3. RESULTS & DISCUSSION
30 min (each ~3,700, Figure 7.4B) followed by P0-FN(~3,100) and P3b (~2,650).The significantly higher cell count for P3a as compared to P3b also reflects thedifferent integrin-affinities of the respective benchmark peptides (knottin-RGD →P3a, cyclo-[KRGDf] → P3b). Moreover, ELRs functionalized with linear GRGDS(P3c) showed a significantly higher cell count (~2,100) than negative control ELRsP0 (~1,500) and P0-BSA (~1,250), but a significantly lower cell count as comparedto benchmark ELRs P3a and P3b, which is fully in line with the integrin affinitiesas observed in Chapters 4 & 5.When comparing the cell counts for the bicycle-functionalized ELRs (P1a-P1c andP2a-P2c) and benchmark/negative control ELRs P3a-3c after 30 min, they gen-erally reflect the integrin-affinities of the respective peptides as described in Chap-ters 4 & 5. While ELRs functionalized with high-affinity αvβ3-selective bicycles(IC50,αvβ3: ~30 nM, P1a and P1b) showed almost similar cell counts as comparedto knottin-RGD-functionalized ELR P3a (IC50,αvβ3: 38 nM), the cell counts forELRs functionalized with α5β1-selective bicycles (P2a-P2c, IC50,α5β1: 90-173 nM)were comparable to those of cyclo-[KRGDf]-functionalized benchmark ELR P3b(IC50,αvβ3: 182 nM). Additionally, all bicycle-functionalized ELRs showed signifi-cantly higher cell counts than the ELR functionalized with low-affinity integrin-binder GRGDS (IC50,αvβ3: ~5,000 nM, see Chapter 3).After 4 hours, P0-FN (~4,050), P3a (~3,950) and P3b (~3,700) showed the highestcell numbers, whereas the ELR comprising RGD as part of the polymer backbone(P0-RGD) comprised < 1,700 cells. Like observed for the bicycle-functionalizedELRs, the benchmark ELRs generally did not offer higher proliferation levels after1 day compared with 4 hours, except for the non-functionalized ELR P0 (~2,000, 1 dvs. ~1,250, 4 h). Amongst all bicycle-functionalized and benchmark ELRs analyzedafter 30 min and 4 hours, P3a overall showed the highest cell numbers (30 min:~3,700, 4 h: ~3,950), followed by P1a (30 min: ~3,350, 4 h: ~3,700), P3b (30 min:~2,650, 4 h: ~3,700) and P2c (30 min: ~2,550, 4 h: ~3,700). After 1 day, 7 out of 13ELRs showed similar cell numbers (~3,300–3,650), whereas the non-functionalized(P0), backbone RGD-functionalized (P0-RGD) and BSA-coated ELRs (P0-BSA)exhibited much lower cell numbers (≤ 2,000).The next time points analyzed were after 3 days and 5 days (Figure 7.5A). Amongstthe bicycle-functionalized ELRs, P1a comprised the highest cell number (~6,850),followed byP2c (~6,250) andP1b (~6,000). Also for the other bicycle-functionalizedELRs (except P2b) cell numbers >5,700 were detected. After 5 days, P1a again
178

7777777
7.3. RESULTS & DISCUSSION
displayed the highest cell numbers (~12,950) followed by P1b (~12,600) and P2a(~11,600), while ELRs functionalized with the αvβ3-binder 1c (~10,600) or theα5β1-binders 2b+c (~5,900 and 9,650, respectively) showed significantly less pro-liferation levels.The observed cell numbers suggest that ELR functionalization with αvβ3-selectivebicycles 1a and 1b is slightly more efficient as compared to the α5β1-selective bi-cycles 2a and 2b. As discussed earlier, a higher peptide functionalization degreeof the ELR might partly explain this observation. A different explanation could bethe fact that the integrin affinities of αvβ3-selective bicycles 1a and 1b (IC50: 30and 31 nM, respectively, Chapter 4) are slightly higher than the affinities of theα5β1-selective bicycles 2a and 2b (IC50: 90 and 173 nM, respectively, Chapter 5),resulting in more efficient integrin binding. However, it has to be noted that thishypothesis cannot be verified since the conformation of recombinant integrins asapplied in ELISA (Chapters 4 and 5) may strongly differ from the conformation ofintegrins expressed on living HUVECs.The benchmark ELRs varied significantly amongst each other with regard to cellproliferation (Figure 7.5B). Interestingly, after 3 days, proliferation levels for P3b(~6,600 cells) were on average higher compared with P3a (~5,300), and much higherthan the GRGDS-functionalized ELR (P3c) comprising a significantly lower cellnumber (~4,050), which reflects the strong difference in integrin affinities (see Chap-ters 4 & 5. The highest cell number was observed for P0-FN (~6,850), interestinglyresembling the cell number for P1a (~6,850), while the lowest number of cells wasobserved for P0-BSA (~1,100). However, none of the benchmark ELRs showedhigher cell numbers than the best bicycle-functionalized ELR P1a (~6,850).
After 5 days, the cell numbers increased significantly in all ELRs except for P0-BSA. In contrast to the data obtained for benchmarks P3a–c after 3 days, prolif-eration levels for P3c after 5 days (~5,400) were much lower in comparison to P3b(~11,750) or P3a (~9,000). Again, the fibronectin-coated ELR P0-FN comprisedthe highest cell numbers among the positive benchmark ELRs (~12,350) which is,however, still significantly lower compared to the ELR modified with αvβ3-bindingbicycle (P1a, ~12,950). As for the other investigated time points, P0-RGD andP0 showed much lower proliferation levels (cell numbers <4,000 after 3 days and<6,500 after 5 days), clearly indicating that RGD-post-functionalization of ELR,via the side-chain of free lysines, is much more efficient compared to exclusive re-
179

7777777
7.3. RESULTS & DISCUSSION
Figure 7.5.: Time-dependent proliferation assay (3 d, 5 d). Number of cells de-termined for ELRs containing (A) bicyclic peptides 1a–c (P1a-P1c)and 2a–c (P2a-P2c), and (B) benchmark RGD peptides 3a–c (P3a-P3c), RGD within the ELR-backbone (P0-RGD), no RGD peptide(P0), fibronectin coating (P0-FN) and coating with BSA (P0-BSA).Grid lines at 5,000 and 12,500 are shown for better comparison of data.The number of cells was calculated from the fluorescence intensitiesusing a calibration curve. All experiments were carried out in tripli-cate and error bars show standard deviations.
combinant ELR synthesis.Finally, the long-term proliferation (7 d, 14 d) of bicycle-functionalized ELRs was
180

7777777
7.3. RESULTS & DISCUSSION
evaluated (Figure 7.6A). After 7 days, the highest cell numbers were detected onELRs functionalized with αvβ3-bicycles, i.e. P1b (~24,900), P1a (~24,050) andP1c (~21,450). In contrast, a significantly lower number of cells was observed onELRs modified with α5β1-bicycles (P2a–c), for example, for P2c (~19,850 cells) orP2a (~19,400 cells). As for the proliferation data after 3 and 5 days, P2b comprisedthe lowest cell numbers after 7 days (~13,500). At last, cell counts after 14 dayswere approximately doubled as compared to after 7 days for all bicycle-functionalizedELRs. Here, differences between αvβ3- and α5β1-functionalized ELRs were hardlydetectable. For P1a, P1b and P2a cell numbers >62,000 were detected, whereasthe other bicycle-functionalized ELRs comprised cell numbers between ~54,600 and60,350.Proliferation data for the positive control ELRs revealed significantly lower cell num-bers after 7 days as compared to bicycle-functionalized ELRs (~20,000), for example,for P3a (~16,950), P3c (~11,050), or P0-FN (~16,200, Figure 7.6B). The highestproliferation levels among the positive controls were determined for ELRs modifiedwith cyclo-[KRGDf] (P3b, ~18,800), however proliferation levels were still loweras compared to ELR P1a described earlier (~24,900). After 14 days, cell num-bers >60,000 were observed for P3b and P3c, whereas the other benchmark ELRs(except for P0-BSA) showed cell numbers between approx. 45,000 and 58,000.Interestingly, P0-FN comprised the second lowest number of cells, even though atearlier time points (30 min–5 d) relatively strong proliferation was observed for thisELR as compared to the RGD-bicycle-functionalized ELRs P1a-P1c and P2a-P2c,and to the benchmark ELRs, e.g. P3b, P3c and P0-RGD. This observation mightbe explained by nascent degradation of the fibronectin-coated ELR.
In summary, the data from the PicoGreen® assay show that, depending on theincubation time, ELRs functionalized with integrin-selective RGD-bicycles (P1a–cand P3a–c) promote cell adhesion and proliferation to a similar or even higher ex-tent as compared to the various benchmark ELRs (e.g. P3a and P3b).While after 30 minutes, P1a and P3a provided the best cell adhesion properties,the highest cell numbers after 3 and 5 days were detected on ELRs functionalizedwith αvβ3-selective bicycle 1b (P1b) and benchmark cyclo-[KRGDf] (P3b). Gen-erally, the proliferation levels between αvβ3- and α5β1-selective ELRs were similar;at certain time points, αvβ3-binding bicycles supported proliferation to a slightlyhigher extent than α5β1-binding bicycles. This is surprising considering the fact
181

7777777
7.3. RESULTS & DISCUSSION
Figure 7.6.: Time-dependent proliferation assay (7 d, 14 d). Number of cells de-termined for ELRs containing (A) bicyclic peptides 1a–c (P1a-P1c)and 2a–c (P2a-P2c), and (B) benchmark RGD peptides 3a–c (P3a-P3c), RGD within the ELR-backbone (P0-RGD), no RGD peptide(P0), fibronectin coating (P0-FN) and coating with BSA (P0-BSA).Grid lines at 20,000 and 80,000 are shown for better comparison ofdata. The number of cells was calculated from the fluorescence inten-sities using a calibration curve. All experiments were carried out intriplicate and error bars show standard deviations.
182

7777777
7.3. RESULTS & DISCUSSION
that HUVECs were reported to express significantly higher levels of integrin α5β1as compared to αvβ3.[19] However, a lower effective peptide-functionalization de-gree of ELRs modified with α5β1-selective bicycles, as indicated by the differentMALDI-TOF-MS spectra (Figure 7.3, p.175), might have overcompensated the roleof the HUVEC integrin expression pattern. Even though ELRs functionalized withintegrin αvβ3-selective RGD-bicycles (e.g. P1a) very efficiently promote cell ad-hesion and proliferation, it needs to mentioned that certain benchmark ELRs, forexample, P3a and P3b showed similar adhesion and proliferation levels. A pos-sible explanation might be that these positive benchmark peptides are much morepromiscuous than the RGD-bicycles, and therefore also bind to other integrins orproteins involved in cell proliferation.
7.3.4. Morphology studies
In order to investigate the time-dependent cell adhesion, proliferation, and the for-mation of focal adhesions, the cells were stained with rhodamin phalloidin (actinskeleton), DAPI (nuclei) and a fluorescent mAb (vinculin), and examined after de-fined time points via fluorescence microscopy (Figures 7.7 and 7.8).After discarding non-adherent cells after 30 minutes of seeding, the analysis of theadhered cells suggests small and spherically shaped HUVECs with small protru-sions in the periphery of the ring-shaped adhesions at the beginning of cell culture(30 min) on practically all tested ELR surfaces except for P0 and P0-BSA. After4 hours, the RGD-bicycle-functionalized ELR P1a in particular showed already ex-tended focal contacts (Figure 7.7), comparable to those of benchmark ELRs P3aand P0-FN (Figure 7.8). In contrast to this, the focal contacts in the other ELRswere mainly circumscribed to the perinuclear zone, for example, on ELRs P1c, P2a,P2b (Figure 7.7), or benchmark ELRs P3b, P3c and P0-RGD (Figure 7.8). After1 day, also cells incubated on P2a and P3b developed extended focal contacts, whilethe adhesions on P2b, P2c and P0-RGD showed a more contorted shape. After3 days, a big difference in morphology was observed between the two αvβ3-selectiveELRs P1a (cobblestone appearance) and P1b (contorted shape, Figure 7.7). Possi-ble explanations for the more contorted focal contacts observed on ELR P1b couldbe a slightly lower functionalization degree and/or a less homogenous distributionof the RGD bicycle residues. Moreover, after 3 days, overlapping actin and vinculincaptures were found (yellow coloration), for example, on ELRs P1b, P2a, 2b and
183

7777777
7.3. RESULTS & DISCUSSION
P3c.After 5 days, a mix of contorted and cobblestone-like adhesions was observed forall bicycle-functionalized ELRs (P1a–P2c, Figure 7.7) and most of the benchmarkELRs, i.e. P3a, P3b, P3c, P0-RGD and P0-FN (Figure 7.8). Finally, after7 days, all bicycle-functionalized ELRs (except for P2b) almost reached confluence.The morphological differences at that point in time were still very low, be it forthe majority of the bicycle-functionalized ELRs (Figure 7.7) or for the majorityof the benchmark ELRs (Figures 7.8). One exception involves α5β1-selective ELRP2b, as well as negative benchmark ELRs P3c and P0, showing more contorted cellmorphologies. Another exception is the fact that HUVECs incubated on fibronectin-coated ELRs (P0-FN) showed a more uniform cobblestone appearance, while onthe other ELR surfaces the cells adopted a more elongated and contorted shape.
Figure 7.7.: Morphology studies of ELRs functionalized with bicycles 1a–c (P1a-P1c) and 2a–c (P2a-P2c).
184

7777777
7.4. CONCLUSION & OUTLOOK
Figure 7.8.: Morphology studies of ELRs functionalized with benchmark sequences3a–c (P3a-P3c)), and comprising RGD within the ELR-backbone(P0-RGD), no RGD peptide (P0), fibronectin coating (P0-FN) andcoating with BSA (P0-BSA).
7.4. Conclusion & outlook
The in vitro cell adhesion and proliferation studies presented in this chapter showedthat integrin αvβ3- and α5β1-selective RGD-bicycles (1a–c and 2a–c) are capableof promoting fast HUVEC cell adhesion and proliferation on 2D elastin-like recom-binamers (ELRs) to even higher levels than the established linear control GRGDS(3c). Moreover, two out of three αvβ3-selective bicycles (1a and 1b) mostly showedan even better performance in mediating cell adhesion and proliferation than high-integrin affinity benchmarks cyclo-[KRGDf] (3b) and knottin-RGD peptide (3a).Furthermore, covalent RGD-functionalization of ELRs via copper-free click reactionwas shown to be much more efficient to promote cell adhesion and proliferation ascompared to the sole recombinant synthesis of ELRs that exhibit RGD as part oftheir backbone (P0-RGD). Therefore, this RGD-functionalization strategy is pre-ferred for future syntheses of biomaterials, which also allows the tuning of RGDdensities and therefore optimization of the cell-biomaterial interaction.
185

7777777
7.5. MATERIALS & METHODS
7.5. Materials & methods
Reagents & chemicals
Amino acids were purchased from Iris Biotech (Marktredwitz, Germany) and Ma-trix Innovation (Quebec, Canada). Resins were purchased from Rapp Polymere(Tübingen, Germany) and Merck (Darmstadt, Germany). 1,3,5-tris(bromomethyl)benzene (T3) and (1R,8S,9s)-Bicyclo[6.1.0]non-4-yn-9-ylmethyl N -succinimidyl car-bonate (BCN-NHS) were purchased from Sigma-Aldrich (Steinheim, Germany).
Peptide synthesis
Peptide syntheses were carried out on a fully automated peptide synthesizer fromGyros Protein Technologies (Symphony) via Fmoc-based solid-phase peptide synthe-sis on Rink-amide resin using standard protocols. Folding of knottin-RGD peptideand synthesis of cyclo-[KRGDf] were performed as described in previous chapters(for example, see p.63). For the formation of bicyclic peptides, purified linear pep-tides were dissolved at 0.5 mM in 1:3 MeCN/H2O, followed by adding 1.1 equiv.1,3,5-tris(bromomethyl) benzene (T3) dissolved in MeCN (10 mM) and 1.4 equiv.ammonium carbonate (0.2 M in H2O) were added. After completion (30-60 min,monitored by UPLC/MS), reaction was quenched with 10% TFA/H2O to pH < 4,followed by lyophilization. All peptides were purified by preparative HPLC on anRP-C18 column (Reprosil-Pur 120 C18-AQ 150x20 mm, Dr. Maisch GmbH, Am-merbuch, Germany) using a MeCN/miliQ H2O gradient (5-65%) including 0.05%TFA followed by lyophilization (Christ Alpha 2-4 LDplus lyophilizer).
Synthesis of peptide-cyclooctyne conjugates
To the peptides dissolved in DMSO (5 mM, for TEC218 10 mM) was added 1.1 equiv.BCN-NHS ester and 10 equiv. N,N -diisopropylamine. After completion of thereaction (15-30 min, monitored by UPLC/MS), the reaction was quenched with10% TFA/DMSO to pH < 4. The product was subsequently purified by prepara-tive HPLC using a MeCN/H2O gradient (5-65%) including 0.05% TFA followed bylyophilization. The successful conjugation was verified by MALDI-TOF-MS (Voy-ager STR, Applied Biosystems).
186

7777777
7.5. MATERIALS & METHODS
Formation of peptide-functionlized ELRs
The ELRs comprising 5% or 10% peptide-functionalization were based on followingcalculations: Azide-functionalized ELRs (62,809 Da) comprised 24 lysine molecules,of which 80% were functionalized with azides (analyzed in NMR, data not shown),corresponding to 19.2 azide groups per ELR molecule. The application of 1 equiv.BCN-funtionalized peptide would result in the functionalization of 5.21% of theazide groups. Hence, application of 0.96/1.92 equiv. BCN-functionalized peptidewould result in 5%/10% functionalization.The conjugation of ELRs with BCN-functionalized peptides was performed as fol-lows: Azide-functionalized ELRs were dissolved in miliQ water (4℃, 20 mg/mL).BCN-functionalized peptides were dissolved at 5 mM in H2O (TEC213-BCN in 50%MeCN/H2O), and added to the ELR solutions. After shaking the copper-free clickreactions for at least 24 h at 4℃, the ELRs were lyophilized followed by dialysis toremove unreacted cyclooctyne-monomers (3 d).
Cell culture & Cell adhesion assay
HUVEC cells (Cat.# C-015-10C, Gibco) were used in all experiments at passage2. Cells were cultured in Medium 200 (Gibco) containing Low Serum Supplement(LSGS) kit (Gibco) providing following final concentrations: 2% (v/v) fetal bovineserum (FBS), 1 µg/mL hydrocortisone, 10 ng/mL human epidermal growth factor,10 ng/mL basic fibroblast growth factor, 10 µg/mL heparin, 10 µg/mL gentamicinand 0.25 µL amphotericin solution (Gibco). HUVECs were incubated at 37 ℃ and5% CO2, and harvested at 90% confluence via trypsin-EDTA treatment.HUVECs were seeded at a density of 5300/cm2 in serum-free Medium 200 (Gibco)for 30 min on different surfaces (n = 3), and further allowed to adhere for 30 minfollowed by removal of Medium 200 and culturing for 14 days. Cell adhesion andspreading were evaluated at the beginning (30 min) and after 4 h, 1, 3, 5, 7 and 14 dof incubation, during which cultures were daily provided with fresh media.
DNA analysis
DNA content was determined by PicoGreen® assay after 4 h, 1, 3, 5, 7 and 14 daysof incubation. Briefly, the cells were lysed with a solution of 0.1% Triton X-100(Sigma Aldrich) in PBS (v/v), and the PicoGreen® analysis for DNA content wasperformed in 96-well plates applying standard fluorescein wavelengths (excitation:
187

7777777
7.5. MATERIALS & METHODS
480 nm, emission: 520 nm) according to the manufacturer’s instructions (Invitrogen)using an automated plate reader (Bionova Cientifica, Molecular Devices).
2D Immunofluorescence staining
Immunofluorescence staining was performed to visualize the HUVECs on differentELR surfaces. After fixation of cells with 4% (w/v) paraformaldehyde, cells werepermeabilized with 0.1% Triton X-100 and blocked with 1% (w/v) BSA/PBS. Focaladhesion contact formation was evaluated via staining of vinculin (monoclonal rabbitanti-vinculin antibody-Alexa Fluor® 488 conjugate, Abcam, 1:200 v/v), actin skele-ton (rhodamin phallaodin, Invitrogen, 1:80 v/v) and nuclei (DAPI, Lonza, 1:10,000v/v). Cell adhesion and morphological changes were examined with an invertedfluorescence microscope (Nikon Eclipse Ti E) by taking 2–3 representative imagesper sample (10x magnification).
188

7777777
7.6. REFERENCES
7.6. References[1] D. W. Urry, Angew. Chem. Int. Ed. Engl. 1993, 32, 819–841.[2] J. C. Rodríguez-Cabello, J. Reguera, M. Alonso, et al., Chem. Phys. Lett.
2004, 388, 127–131.[3] B. R. R. Costa, C. A. Custódio, A. M. Testera, et al., Adv. Funct. Mater.
2009, 19, 3210–3218.[4] J. F. Almine, D. V. Bax, S. M. Mithieux, et al., Chem. Soc. Rev. 2010, 39,
3371–3379.[5] J. C. Rodríguez-Cabello, F. J. Arias, M. A. Rodrigo, et al., Adv. Drug De-
livery Rev. 2016, 97, 85–100.[6] A. Nicol, D. Channe Gowda, D. W. Urry, J. Biomed. Mater. Res. 1992, 26,
393–413.[7] J. C. Liu, D. A. Tirrell, Biomacromolecules 2008, 9, 2984–2988.[8] K. S. Straley, S. C. Heilshorn, Soft Matter 2009, 5, 114–124.[9] M. Putzu, F. Causa, V. Nele, et al., Biofabrication 2016, 8, 045009.[10] A. Girotti, J. Reguera, J. C. Rodríguez-Cabello, et al., J. Mater. Sci.: Mater.
Med. 2004, 15, 479–484.[11] I. Gonzalez de Torre, M. Weber, L. Quintanilla, et al., Biomater. Sci. 2016,
4, 1361–1370.[12] M. Vila, A. García, A. Girotti, et al., Acta Biomater. 2016, 45, 349–356.[13] L. Cai, C. B. Dinh, S. C. Heilshorn, Biomater. Sci. 2015, 2, 757–765.[14] S. Ravi, V. R. Krishnamurthy, J. M. Caves, et al., Acta Biomater. 2012, 8,
627–635.[15] D. Kaufmann, A. Fiedler, A. Junger, et al., Macromol. Biosci. 2008, 8, 577–
588.[16] S. Wohlrab, S. Müller, A. Schmidt, et al., Biomaterials 2012, 33, 6650–6659.[17] I. González De Torre, M. Santos, L. Quintanilla, et al., Acta Biomater. 2014,
10, 2495–2505.[18] S. Aidoudi, K. Bujakowska, N. Kieffer, et al., Plos ONE 2008, 3, e2657.[19] P. Baranska, H. Jerczynska, Z. Pawlowska, et al., Cancer Genom. Proteom.
2005, 270, 265–269.[20] S. N. C. Gimenes, D. S. Lopes, P. T. Alves, et al., Sci. Rep. 2017, 7, 7077.
189

7777777
7.7. APPENDIX
7.7. Appendix
Table 7.1.: Molecular weights of BCN-functionalized RGD peptides as obtainedfrom UPLC/MS analysis. Weights were calculated using ChemDrawsoftware.
190

88888888
8. In Vitro Evaluation of MechanicalProperties of PolyisocyanopeptideHydrogels Modified with Integrinαvβ3- and α5β1-SelectiveRGD-Bicycles
The work described in this chapter resulted from a collaboration with Kaizheng Liu (groupof Dr. Paul Kouwer, Institute for Molecules and Materials, Radboud University Nijmegen, theNetherlands) and was supported by the group of Dr. Egbert Oosterwijk, Institute for MolecularLife Sciences, Radboud UMC, Nijmegen, the Netherlands. Parts of the work described in thischapter will be published: K. Liu, J. Vandaele, D. Bernhagen, E. Oosterwijk, P. Timmerman, S.Rocha, P. H. J. Kouwer, in preparation.

88888888
8.1. ABSTRACT
8.1. Abstract
In this project, the impact of the presence of high affinity integrin αvβ3- and α5β1-selective RGD-bicycles as well as benchmark integrin-binders knottin-RGD and cy-clo-[KRGDf] on the mechanical stability and stem cell adhesion/proliferation prop-erties of polyisocyanopeptide (PIC) hydrogels was investigated via i) determinationof gel stiffness and critical stress in rheological experiments, and ii) (confocal) mi-croscopy analysis to investigate the shape of non-stained and actin-stained cells.All peptide-functionalized PIC hydrogels exhibited similar mechanical propertieswith respect to gel stiffness, G0, and critical stress, σcrit, ranging from 200 Pa till480 Pa for G0, and 23–31 Pa for σcrit, respectively, suggesting that neither the con-formation nor the integrin-affinity and -selectivity of the RGD peptides significantlyaffects the mechanical stability of the gels.Adhesion studies (brightfield microscopy) with adipose-derived stem cells showedthat the hydrogel functionalized with α5β1-selective bicycle CT3RGDcT3AWGCT3
featured superior cell adhesion and numerous protrusions after 1 day incubation ascompared with hydrogels functionalized with αvβ3-selective bicycles or benchmarkRGD peptides. Moreover, this hydrogel showed incipient degradation, suggestingthat cells efficiently deform this gel via integrin-binding to CT3RGDcT3AWGCT3.Confocal microscopy of actin- and nuclei-stained cells revealed superior cell adhesionand a higher amount of protrusions in hydrogels functionalized with α5β1-selectivebicycle CT3RGDcT3AYJCT3 (J: D-Leu) after 2 days of incubation as comparedto hydrogels exhibiting αvβ3-selective bicycle CT3HPQcT3RGDcT3 or benchmarkpeptides knottin-RGD and cyclo-[KRGDf]. Finally, WST-1 assays revealed thathydrogels functionalized with α5β1-selective bicycle CT3RGDcT3AWGCT3 showedthe highest proliferation levels after 1 and 2 days, whereas all other hydrogels showedlower, but relatively similar proliferation levels.The overall results suggest that CT3RGDcT3AWGCT3 seems the most promisingpeptide to effectively promote fast stem cell adhesion and proliferation in PIC-basedhydrogels.
8.2. Introduction
Polyisocyanopeptide (PIC)-based hydrogels represent a relatively new family of bio-materials, reported to be the first synthetic hydrogels to exhibit strain-stiffening
192

88888888
8.2. INTRODUCTION
properties (gels become stiffer with increasing stress) similar to cytoskeletal proteinhydrogels such as actin and collagen.[1, 2] The precursor polymers, polyisocyanopep-tides are synthesized via nickel-catalyzed polymerization of building blocks thatcomprise isocyanide-groups, and form a β-helical structure via formation of a sta-ble hydrogen-bonded network.[3] Prior to polymerization, these building blocks aregrafted with oligo(ethylene glycol) residues to allow bundle formation (Figure 8.1),and eventually the reversible formation of PIC hydrogels of tunable thermal transi-tion in aqueous solutions and at low polymer concentrations.[1, 4]
Figure 8.1.: (A) Synthesis of oligo(ethylene glycol)-substituted PICs. The for-mation of hydrogels could be observed, for example, for polymerswith m=3 and n=11,000.[1] (B) Schematic representation of the sec-ondary helical structure stabilized by a hydrogen-bond network. Mod-ified reprint with permission from Springer Nature, license number4405330831499.[1]
Strain-stiffening can be controlled via variation of polymer length, concentration,temperature,[2, 5] or the formation of composite materials, e.g. with polyacrylamide,carbon nanotubes or fibrin.[6, 7] Despite the excellent mechanical properties, studiesrevealed that cell adhesion and proliferation properties of hydrogels based on thePIC platform are not optimal yet.[7] The integrin-binding linear peptide RGD andmodifications thereof, e.g. GRGDS, is still very broadly applied to introduce celladhesion and proliferation properties into synthetic polymer matrices.[8, 9] Recently,Das et al. investigated mesenchymal cell adhesion in PIC hydrogels covalently func-
193

88888888
8.3. RESULTS & DISCUSSION
tionalized with GRGDS, showing that mesenchymal stem cells remain viable after36 h.[10] However, the spherical cell shape as observed in brightfield microscopy sug-gests that the cells do not form focal adhesions, which are crucial for cell-ECMsignalling and eventually proliferation. An explanation might be that the interac-tion between cellular integrins and the GRGDS peptides immobilized on the PICbackbone is too weak to induce the signalling cascades that result in the formationof focal adhesions. One option to circumvent this issue might involve the applicationof high affinity integrin αvβ3- and α5β1-selective RGD-bicycles.In Chapter 7, the cell-adhesion promoting properties of RGD-bicycles on 2D ELRsurfaces were discussed, raising the question whether these peptides would promotecell-adhesion in a 3D environment, such as in PIC hydrogels, as well. In this study,the rheological and cell adhesion properties of PIC hydrogels were investigated thatwere functionalized with three αvβ3- and two α5β1-selective RGD-bicycles, andcompared with PIC hydrogels bearing integrin-binding benchmark peptides knottin-RGD (binds αvβ3/αvβ5/α5β1) and cyclo-[KRGDf] (binds αvβ3/αvβ5).
8.3. Results & discussion
PIC polymers, comprising a molecular weight of ~5 kDa and a theoretical isocyanidemonomer/azide-isocyanate monomer ratio of 30:1, were used for conjugation of theRGD peptides. The cyclooctyne-functionalized peptides were conjugated to theazide-containing PIC scaffolds via copper-free click reaction (Figure 8.2A) in a waythat theoretically 1 out of 100 isocyanopeptide monomers is eventually peptide-functionalized (for calculation details, see Materials & Methods). Seven differentpeptide-PIC conjugates were synthesized, three of which comprise αvβ3-selectivebicycles (PIC-1a – PIC-1c), two exhibiting α5β1-selective bicycles (PIC-2a andPIC-2b), and two comprising benchmark sequences knottin-RGD (PIC-3a) andcyclo-[KRGDf] (PIC-3b, for sequences see Figure 8.2B).
8.3.1. Rheological characterization
In order to investigate the impact of the presence of RGD-bicycles and benchmarkRGD-peptides on the mechanical properties of PIC hydrogels, their stress-stiffeningbehavior was tested. For quantification, it is necessary to determine the differentialmodulus K’ = δσ
δγ, which is a function of oscillatory stress, δσ, and oscillatory strain,
194

88888888
8.3. RESULTS & DISCUSSION
Figure 8.2.: (A) Reaction scheme for the formation of peptide-functionalized PICsvia copper-free click reaction of azide-functionalized PICs and cyclooc-tyne (BCN)-functionalized peptides. (B) Nomenclature of PIC hydro-gels investigated in this study, including RGD peptide sequence andintegrin selectivity. Linker: PPPSG[Abz]SG (Abz: 4-aminobenzoicacid). The three disulfide bridges in the peptide corresponding toPIC-3a are illustrated with same-colored “Cox”. * Data from compe-tition ELISA.
δγ. The oscillatory strains, δγ, can be obtained from a set of frequency sweeps(value at f = 1 Hz) as a function of pre-stress, σ, with a range from σ = 0.5 Pa inthe first frequency sweep till σ = 200 Pa in the final frequency sweep.For each hydrogel, the following rheological experiments were implemented consec-
195

88888888
8.3. RESULTS & DISCUSSION
utively:
(i) Temperature sweep (T = 5–37 ℃, f = 1 Hz). This experiment determines thetransition temperature, at which the PIC solution forms a hydrogel.
(ii) Time sweep (T = 37 ℃, f = 1 Hz, t = 30 min). This experiment at a constanttemperature and frequency allows the gel to equilibrate.
(iii) 23 Frequency sweeps at pre-stresses σ ranging from 0.5–200 Pa (T = 37 ℃, f= 10–0.1 Hz with oscillatory stress δσ<0.1 σ). From the frequency sweeps, themeasured oscillatory strains δγ at 1 Hz can be extracted to calculate stress-dependent K’ values.
From the frequency sweeps, 23 different K’ values were obtained and plottedagainst the pre-stress. The resulting curves are depicted in Figure 8.3A. At lowstress values (σ<5 Pa), K’ equals G0, referred to as plateau modulus, which in-dicates the equilibrium bulk stiffness (Figure 8.3A, small graph). At high stressvalues (σ>70 Pa) K’ increases in a power law manner with increasing stress, whichis referred to as “strain stiffening”. The onset of stress stiffening is defined as criticalstress σcrit. The gel stiffness G0 and critical stress σcrit for each hydogel is depictedin Figure 8.3B.
Figure 8.3.: Rheological characterization of PIC hydrogels. (A) Differential mod-ulus, K’, as a function of stress, σ, and (B) gel stiffness, G0, and crit-ical stress, σcrit for PIC hydrogels functionalized with bicyclic RGDpeptides and benchmarks, obtained at 37 ℃ (cPIC = 2 mg/mL inα-MEM).
196

88888888
8.3. RESULTS & DISCUSSION
The gel stiffnesses ranged from ~200 Pa for PIC-1b until ~480 Pa for PIC-2a.Interestingly, for PIC-1b, which was conjugated with an αvβ3-selective bicyclestructurally very similar compared to the bicycle in PIC-1a, the gel stiffness wasmuch lower (G0 <200 Pa). Also, PIC-3a, functionalized with knottin-RGD (32amino acids) even showed a higher stiffness than PIC-1b. Therefore, assumingthat all hydrogels have similar effective functionalization percentages ( 1%), no clearimpact of structure and size of the RGD peptide on the gel stiffness can be notedunder these experimental conditions.Furthermore, critical stress values between ~23 Pa (PIC-1a and PIC-3a) and~31 Pa (PIC-2a) were calculated. Recently, it was reported that the contrac-tile stress that fibroblasts can apply to fibrin hydrogels is equivalent to an externalstress of up to 14 Pa.[11] This range was later reported as a reference for “biologicallyrelevant stress regime”.[2, 10] The critical stress values determined herein all slightlyexceed this range, suggesting that when cells are incubated in these hydrogels, theymight only be able to deform the gels in the linear regime, which would limit theactive control of spreading, migration, proliferation, and differentiation.[11–14]
8.3.2. Imaging of hydrogel-cell scaffolds
In order to estimate the cell adhesion and spreading properties, the cells were en-capsulated in the hydrogels and analyzed via brightfield and confocal microscopy.
Brightfield microscopy
In the hydrogels functionalized with the αvβ3-selective bicycles (PIC-1a–PIC-1c)the majority of the cells was spherically shaped after 24 h of culturing, while in hydro-gel PIC-1a, a small number of cells showed protrusions (Figure 8.4A). In sharp con-trast to this, PIC-2b, functionalized with α5β1-selective bicycle CT3RGDcT3AW-GCT3 showed intense protrusions throughout the entire gel (see also 40-fold magni-fication, Figure 8.4B). The “cloudy” regions, as observed in the top left part of theimage, reveal inceptive degradation of the gel, most likely induced by cell contrac-tile forces via binding of α5β1 integrin receptors to the immobilized RGD-bicycles.The degradation is remarkable considering the fact that this gel comprised a crit-ical stress value that was above the biologically relevant stress regime (see above).The fact that ASCs overexpress integrin subunits α5 and β1 while lacking αv
197

88888888
8.3. RESULTS & DISCUSSION
and β3[15, 16] could at least partly explain why hydrogels comprising αvβ3-selectivebicycles (PIC-1a–PIC1c) and αvβ3/αvβ5-selective monocyclic RGD (PIC-3b)do not promote cell protusion and hydrogel degradation to the same extent. Incontrast to PIC-2b, hydrogel PIC-2a, functionalized with α5β1-selective bicycleCT3RGDcT3AYJCT3 (J: D-Leu), showed a much lower degree of protrusion and nodegradation at all, despite the fact that both RGD-bicycles immobilized in PIC-2aand PIC-2b have very similar integrin affinities (IC50: 90 and 173 nM, Chapter 5).One explanation could be that the recombinant integrins (as applied in ELISA)and cellular integrins (as applied here) have different conformations, and bicycle 2bbinds more efficiently to cellular integrins than 2a. Since mechanical properties mayhave an impact on the cell adhesion behavior,[10] another explanation could be anin fact much higher difference in mechanical properties of the hydrogels containingstem cells (to be determined) as compared to the very similar mechanical propertiesdetermined for hydrogels that do not contain stem cells (as described earlier: PIC-2b: G0:332, σcrit: 25.8 Pa; PIC-2a: G0:466 Pa, σcrit: 30.8 Pa).
Confocal microscopy of actin cytoskeleton and nuclei
In order to visualize the adhesion and protrusion properties more clearly, the cellswere stained with DAPI (nuclei) and Texas Red™ X Phalloidin (actin cytoskeleton)in a selected set of hydrogels (PIC-1a, PIC-2a, PIC-2b, PIC-3a and PIC-3b)and analyzed via confocal microscopy after 2 days. It has to be noted that PIC-2bwas too soft for confocal analysis, probably as a result of almost entire degradation.The confocal images for the remaining hydrogels, depicted in Figure 8.5, validatethe images obtained from brightfield microscopy. In PIC-1a (comprising an αvβ3-selective bicycle) and PIC-3b (cyclo-[KRGDf], the vast majority of the cells arespherically shaped and the actin filaments are mainly circumscribed to the perin-uclear zone, suggesting minimal amounts of focal adhesion contacts. In contrast,the cells in PIC-2a (α5β1-selective bicycle) and PIC-3a (knottin-RGD) formedprotrusions, visualized by elongated actin filaments which partly exhibit a multipleof the size of the nuclei. Interestingly, these protrusions were not observed after24 h (see brightfield images, Figure 8.4A), suggesting that the protrusion rate canbe controlled by the cellular integrin-selectivity, size and conformation of the RGDpepitdes.
198

88888888
8.3. RESULTS & DISCUSSION
Figure 8.4.: Brightfield microscopy images for PIC hydrogels functionalized withbicyclic RGD-peptides and benchmarks taken after 24 h of stem cellincubation with (A) 4-fold magnification, and (B) 20-fold magnifica-tion.
199
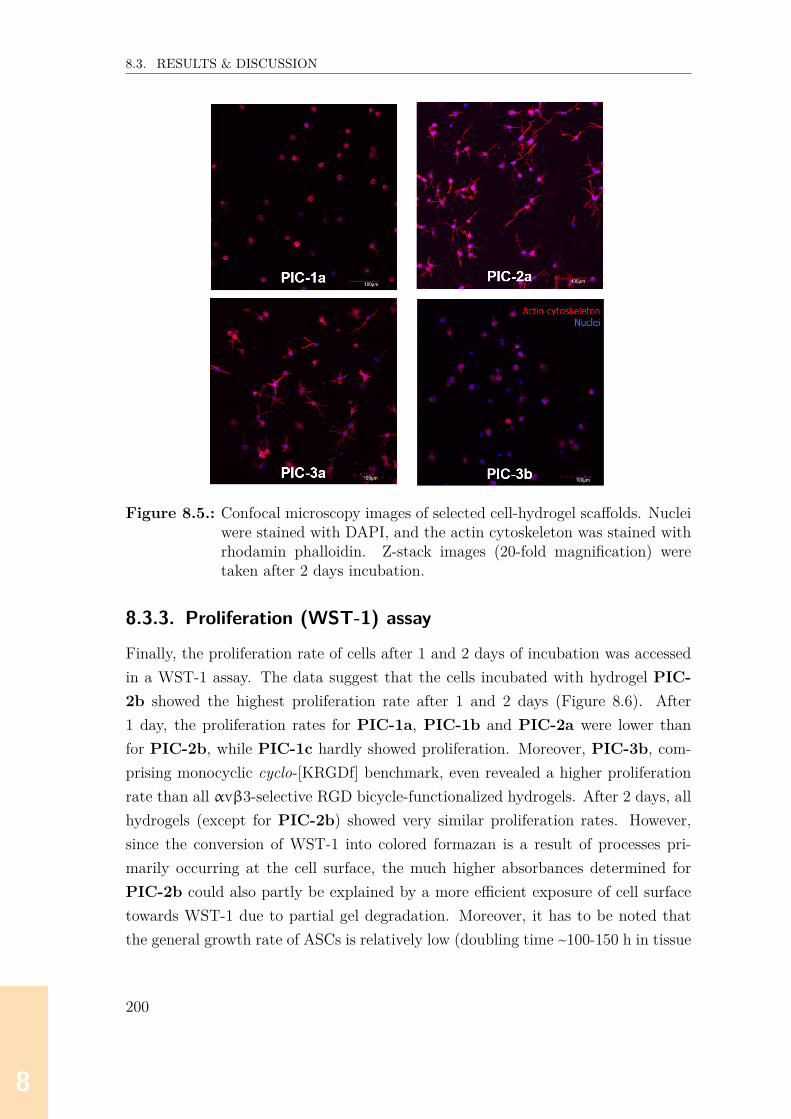
88888888
8.3. RESULTS & DISCUSSION
Figure 8.5.: Confocal microscopy images of selected cell-hydrogel scaffolds. Nucleiwere stained with DAPI, and the actin cytoskeleton was stained withrhodamin phalloidin. Z-stack images (20-fold magnification) weretaken after 2 days incubation.
8.3.3. Proliferation (WST-1) assay
Finally, the proliferation rate of cells after 1 and 2 days of incubation was accessedin a WST-1 assay. The data suggest that the cells incubated with hydrogel PIC-2b showed the highest proliferation rate after 1 and 2 days (Figure 8.6). After1 day, the proliferation rates for PIC-1a, PIC-1b and PIC-2a were lower thanfor PIC-2b, while PIC-1c hardly showed proliferation. Moreover, PIC-3b, com-prising monocyclic cyclo-[KRGDf] benchmark, even revealed a higher proliferationrate than all αvβ3-selective RGD bicycle-functionalized hydrogels. After 2 days, allhydrogels (except for PIC-2b) showed very similar proliferation rates. However,since the conversion of WST-1 into colored formazan is a result of processes pri-marily occurring at the cell surface, the much higher absorbances determined forPIC-2b could also partly be explained by a more efficient exposure of cell surfacetowards WST-1 due to partial gel degradation. Moreover, it has to be noted thatthe general growth rate of ASCs is relatively low (doubling time ~100-150 h in tissue
200

88888888
8.4. CONCLUSION & OUTLOOK
culture flasks[17]), hence the influence of integrin selectivity and affinity of the RGDsequence for proliferation should not be overinterpreted.
Figure 8.6.: Proliferation assay after 1 and 2 days of incubation. The absorbancedirectly correlates with the number of cells.
8.4. Conclusion & outlookThe results described in this chapter reveal that integrin α5β1-selective bicycleCT3RGDcT3AWGCT3 has the best potential to significantly improve the stem celladhesion and growth properties in soft materials. However, further questions needto be addressed, for example:1. When do the cells start to form the first protrusions (in particular in hydrogelPIC-2b which was almost entirely degraded after 2 d)?2. When does the formation of focal adhesion complexes start and how can thesebe quantified?3. Do the cells migrate or even differentiate?In order to answer these questions, further studies need to be performed. For ex-ample, the formation of protrusions, degradation of gels or cell migration can beobserved in live-cell imaging experiments, while in order to visualize focal adhesioncomplexes, the membrane-cytoskeletal protein vinculin is a suitable target since itcontrols focal adhesion formation.[18]
Finally, it can be concluded that the stem cell adhesion/proliferation behavior ina PIC hydrogel is not only influenced by the integrin affinity and selectivity of the
201

88888888
8.5. MATERIALS & METHODS
conjugated RGD peptide, but also by the mechanical properties of the hydrogel.Moreover, it is likely that the capability of the RGD peptide to bind to the rele-vant, cellular conformation of integrins (and not only to the conformation presentin recombinant integrins applied in ELISA experiments) is another important factorthat potentially explains why cell adhesion properties can still be very different, evenwhen two peptides have similar (recombinant) integrin α5β1-affinities and selectiv-ities (compare PIC-2a and PIC-2b, Figure 8.4, p.199).
8.5. Materials & methods
Synthesis of polyisocyanopeptides
Polyisocyanopeptide (PIC) polymers were synthesized as previously reported.[10] Inshort, the methoxy-terminated isocyanide monomer (a) was dissolved in freshlydistilled toluene at 50 mg/mL, and azide-terminated isocyanide monomer (b) wassubsequently added in a mole ratio of a:b = 30:1. Following, a catalyst solution(c) of Ni(ClO4)2·6 H2O (0.37 mg/mL) in freshly distilled toluene/ethanol (4:1) wasadded, and the mole ratio between catalyst and monomers was 1:5000. The mixturewas stirred at room temperature and the reaction progress was monitored by IR-ATR. The resulting polymer (PIC azide 5 kDa) was precipitated in diisopropyl etherunder vigorous stirring, collected by filtration or centrifugation and air-dried.
Synthesis of peptide-cyclooctyne conjugates
The synthesis of peptide-cyclooctyne conjugates (BCN-peptides) is described inChapter 7 (p.186).
Conjugation of RGD peptides
PIC azide 5 kDa was dissolved at 2.5 mg/mL in MeCN for 8 h at 4 ℃. BCN-peptide(calculation of amounts described below), dissolved at 6 mg/mL in DMSO, wasadded and reaction was allowed to stir overnight at room temperature. Following,the peptide-functionalized polymer was precipitated in isopropylether (1:10 v/v),washed with isopropylether, and allowed to dry overnight at room temperature.The amount of peptide-cyclooctyne conjugate (BCN-peptide) to be added was cal-culated as follows: The total amount of azide residues nAzide residues in a defined
202

88888888
8.5. MATERIALS & METHODS
amount of PIC azide is: nAzide residues = m·xa(xa·Ma)+(xb·Mb) , where xa (96.67%) and Ma
(316.35 Da) represent the percentage and molecular weight of methoxy-terminatedmonomer, and xb (3.33%) and Mb (371.39 Da) the percentage and molecular weightof the azide-terminated monomer. The amount of BCN-peptide nBCN-peptide tobe added to achieve a 1% PIC polymer functionalization is nBCN-peptide = 1
xb·
nAzide residues.
Cell culture and encapsulation into hydrogels
Human adipose-derived stem cells (ASCs, obtained from Radboud biobank) werecultured in α-MEM (Invitrogen) supplemented with 10% FCS and 1% penicillin/streptomycin at 37 ℃ with 5% CO2. Dry PIC polymers were sterilized under UVlight for 20 min and dissolved in medium for 24 h at 4 ℃. After reaching confluency,cells were trypsinized and resuspended with fresh medium. Cell suspension andpolymer solution were mixed on ice applying a polymer concentration of 2 mg/mLand a cell density of 200,000/mL, subsequently followed by transferring 200 µL ofthe cell-gel constructs into 48 well plates (for bright field and confocal microscopy)and 8 well chambers (for proliferation assay). After addition of an extra portionof warm culture medium (200 µL) all samples were incubated under standard cellculture conditions (37 ℃, 5% CO2).
Rheological analysis of polymer hydrogels
Dried polymer was dissolved in α-MEM (without serum) at 2 mg/mL overnight at4 ℃. Following, rheological measurements were performed as previously described.[10]
In short, rheological tests were carried out on a stress-controlled rheometer (Dis-covery HR-1 or HR-2, TA Instruments) using an aluminum or steel parallel plategeometry with a diameter of 40 mm and a gap of 500 µm. Following a nonlinearpre-stress measurement, a temperature sweep from 5 to 37 ℃ was performed, fol-lowed by a time sweep for 30 min at 37 ℃ to ensure that samples were fully stable.Then, 23 frequency sweeps ranging from σ values of 0.5 till 200 Pa (f = 10–0.1 Hzwith oscillatory stress δσ<0.1 σ) were performed at 37 ℃, from which strains at f= 1 Hz were extracted to calculate stress-dependent differential moduli K’.
203

88888888
8.5. MATERIALS & METHODS
Brightfield microscopyBright field images of cells in hydrogels were acquired after 24 h using a Leica DC200 microscope.
Staining of cytoskeleton and nucleiFollowing procedures were all performed at 37 ℃. The cell-gel constructs werewashed with phosphate-buffered saline (PBS, pH 7.4) and fixed with 4% formalde-hyde in PBS (v/v) for 40 min, followed by membrane permeabilization with 0.1%Triton X-100 in PBS for (10 min) and blocking with 1% BSA/PBS (w/v) for 30 min.Next, the gel-encapsulated cells were incubated with Texas Red™ X Phalloidin(1:40 in 1% BSA/PBS [v/v], Thermo Fischer Scientific) for 1 h, followed by incu-bation with 4’,6-diamidin-2-phenylindol (DAPI, Invitrogen, 1:200 in PBS [v/v]) for10 min. Imaging of cyctoskeleton (Texas Red™ X Phalloidin, excitation/emissionwavelength: 559/612 nm) and nuclei (DAPI, 405/461 nm) was performed using anOlympus FLUOVIEW FV1000 confocal laser scanning microscope and a Leica SP8.
Cell proliferation assayCulture media and non-encapsulated cells were gently removed and fresh mediumsupplemented with cell proliferation reagent WST-1 (Roche), at a final concentration(1:10, v/v) was added, followed by incubation for 2 h at 37 ℃. Absorbance wasmeasured at λ = 450 nm with a Perkin Elmer plate reader (Perkin Elmer 1420Multilabel Counter).
204

88888888
8.6. REFERENCES
8.6. References[1] P. H. Kouwer, M. Koepf, V. A. Le Sage, et al., Nature 2013, 493, 651–655.[2] M. Jaspers, M. Dennison, M. F. Mabesoone, et al., Nat. Commun. 2014, 5,
1–8.[3] J. J. Cornelissen, J. J. Donners, R. De Gelder, et al., Science 2001, 293,
676–680.[4] M. Jaspers, A. C. Pape, I. K. Voets, et al., Biomacromolecules 2016, 17,
2642–2649.[5] M. Jaspers, A. E. Rowan, P. H. Kouwer, Adv. Funct. Mater. 2015, 25, 6503–
6510.[6] M. Jaspers, S. L. Vaessen, P. Van Schayik, et al., Nat. Commun. 2017, 8,
1–10.[7] S. M. Bruekers, M. Jaspers, J. M. Hendriks, et al., Cell Adhes. Migr. 2016,
10, 495–504.[8] U. Hersel, C. Dahmen, H. Kessler, Biomaterials 2003, 24, 4385–4415.[9] E. Mauri, A. Sacchetti, N. Vicario, et al., Biomater. Sci. 2018, 6, 501–510.[10] R. K. Das, V. Gocheva, R. Hammink, et al., Nat. Mater. 2016, 15, 318–325.[11] K. A. Jansen, R. G. Bacabac, I. K. Piechocka, et al., Biophys. J. 2013, 105,
2240–2251.[12] R. J. Pelham, Y.-L. Wang, Proc. Natl. Acad. Sci. USA 1997, 94, 13661–
13665.[13] R McBeath, D. M. Pirone, C. M. Nelson, et al., Dev. Cell 2004, 6, 483–495.[14] A. J. Engler, S. Sen, H. L. Sweeney, et al., Cell 2006, 126, 677–689.[15] U. R. Goessler, P Bugert, K Bieback, et al., Int. J. Mol. Med. 2008, 21,
271–279.[16] A. B. J. Prowse, F. Chong, P. P. Gray, et al., Stem Cell Res. 2011, 6, 1–12.[17] H. E. Gruber, S Somayaji, F Riley, et al., Biotech. Histochem. 2011, 87,
303–311.[18] J. D. Humphries, P. Wang, C. Streuli, et al., J. Cell Biol. 2007, 179, 1043–
1057.
205

88888888

999999999
A. Supplementary Information
A.1. Absorbance data (OD405 values) for all libraryscreenings for αvβ3-binders
A.1.1. First generation screening of 672 random-diversitypeptides
207

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.1.: Absolute absorbances of library screening for 1st generation of αvβ3-binders. Motif: CT3XXXCT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 1.14 C1 PNV 0.60 E1 PHI 1.22 G1 LPD 1.30A2 AIP 1.99 C2 PYI 0.85 E2 YPS 0.82 G2 RLG 4.00A3 DGY 0.73 C3 TPT 1.33 E3 TRV 3.44 G3 YIY 1.28A4 ILP 1.09 C4 NWG 0.55 E4 VVR 4.00 G4 FRA 2.44A5 VSL 1.21 C5 FTQ 0.93 E5 NST 1.17 G5 LTI 1.16A6 DEW 0.56 C6 QYL 1.01 E6 FLW 0.87 G6 APS 1.09A7 YEE 1.03 C7 WGD 0.39 E7 NSY 0.96 G7 RDP 0.81A8 PLE 1.04 C8 ISY 0.74 E8 ILK 1.56 G8 DHL 0.72A9 KKP 0.83 C9 EIG 1.25 E9 SDQ 1.18 G9 NDA 1.95
A10 QGS 0.91 C10 WFH 0.73 E10 WQY 0.74 G10 DAD 1.65A11 HVK 3.84 C11 NKP 0.48 E11 YYT 1.43 G11 NYA 1.20A12 RNS 1.41 C12 PNE 0.91 E12 NVA 1.05 G12 EDT 1.27B1 DTI 1.26 D1 KTN 1.36 F1 NWQ 0.73 H1 WYV 1.19B2 QAK 1.07 D2 GYE 0.64 F2 TFI 0.66 H2 QSL 1.86B3 WSL 0.67 D3 SYD 1.14 F3 QRG 0.90 H3 TIH 1.09B4 RAA 0.97 D4 IEE 0.86 F4 LDW 0.86 H4 GLA 1.35B5 KHI 3.54 D5 HLQ 0.81 F5 PHL 0.94 H5 LWI 1.22B6 RGS 0.88 D6 GNS 0.68 F6 KID 0.56 H6 GLY 1.07B7 IIV 2.39 D7 IRW 1.30 F7 RQV 3.30 H7 SSG 1.41B8 WFT 1.06 D8 IIG 1.25 F8 KRW 4.00 H8 GWN 0.80B9 RSK 4.00 D9 DFP 1.16 F9 QAD 1.48 H9 SWS 1.49B10 GHF 0.59 D10 PPG 0.74 F10 ERV 1.21 H10 QIH 0.74B11 QDH 1.00 D11 QGI 1.43 F11 YLT 0.95 H11 KQL 0.78B12 HEQ 0.49 D12 FSH 1.05 F12 RWD 0.54 H12 GYG 1.16
208
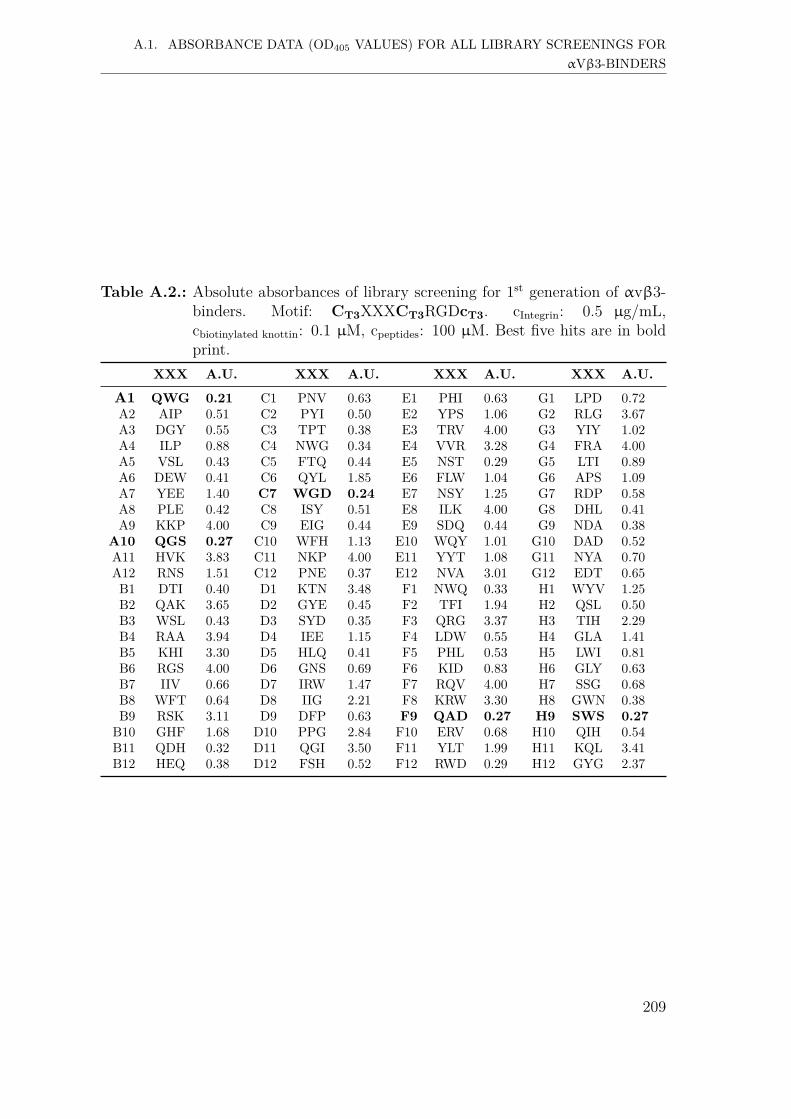
999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.2.: Absolute absorbances of library screening for 1st generation of αvβ3-binders. Motif: CT3XXXCT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.21 C1 PNV 0.63 E1 PHI 0.63 G1 LPD 0.72A2 AIP 0.51 C2 PYI 0.50 E2 YPS 1.06 G2 RLG 3.67A3 DGY 0.55 C3 TPT 0.38 E3 TRV 4.00 G3 YIY 1.02A4 ILP 0.88 C4 NWG 0.34 E4 VVR 3.28 G4 FRA 4.00A5 VSL 0.43 C5 FTQ 0.44 E5 NST 0.29 G5 LTI 0.89A6 DEW 0.41 C6 QYL 1.85 E6 FLW 1.04 G6 APS 1.09A7 YEE 1.40 C7 WGD 0.24 E7 NSY 1.25 G7 RDP 0.58A8 PLE 0.42 C8 ISY 0.51 E8 ILK 4.00 G8 DHL 0.41A9 KKP 4.00 C9 EIG 0.44 E9 SDQ 0.44 G9 NDA 0.38
A10 QGS 0.27 C10 WFH 1.13 E10 WQY 1.01 G10 DAD 0.52A11 HVK 3.83 C11 NKP 4.00 E11 YYT 1.08 G11 NYA 0.70A12 RNS 1.51 C12 PNE 0.37 E12 NVA 3.01 G12 EDT 0.65B1 DTI 0.40 D1 KTN 3.48 F1 NWQ 0.33 H1 WYV 1.25B2 QAK 3.65 D2 GYE 0.45 F2 TFI 1.94 H2 QSL 0.50B3 WSL 0.43 D3 SYD 0.35 F3 QRG 3.37 H3 TIH 2.29B4 RAA 3.94 D4 IEE 1.15 F4 LDW 0.55 H4 GLA 1.41B5 KHI 3.30 D5 HLQ 0.41 F5 PHL 0.53 H5 LWI 0.81B6 RGS 4.00 D6 GNS 0.69 F6 KID 0.83 H6 GLY 0.63B7 IIV 0.66 D7 IRW 1.47 F7 RQV 4.00 H7 SSG 0.68B8 WFT 0.64 D8 IIG 2.21 F8 KRW 3.30 H8 GWN 0.38B9 RSK 3.11 D9 DFP 0.63 F9 QAD 0.27 H9 SWS 0.27B10 GHF 1.68 D10 PPG 2.84 F10 ERV 0.68 H10 QIH 0.54B11 QDH 0.32 D11 QGI 3.50 F11 YLT 1.99 H11 KQL 3.41B12 HEQ 0.38 D12 FSH 0.52 F12 RWD 0.29 H12 GYG 2.37
209

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.3.: Absolute absorbances of library screening for 1st generation of αvβ3-binders. Motif: CT3XXXcT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.98 C1 PNV 0.99 E1 PHI 1.31 G1 LPD 0.86A2 AIP 0.91 C2 PYI 3.88 E2 YPS 0.72 G2 RLG 3.89A3 DGY 0.69 C3 TPT 0.58 E3 TRV 3.87 G3 YIY 4.00A4 ILP 1.20 C4 NWG 0.52 E4 VVR 4.00 G4 FRA 4.00A5 VSL 1.07 C5 FTQ 0.53 E5 NST 0.63 G5 LTI 2.10A6 DEW 0.76 C6 QYL 2.21 E6 FLW 4.00 G6 APS 1.19A7 YEE 2.93 C7 WGD 1.48 E7 NSY 0.61 G7 RDP 4.00A8 PLE 1.13 C8 ISY 2.08 E8 ILK 3.93 G8 DHL 1.06A9 KKP 4.00 C9 EIG 0.79 E9 SDQ 0.67 G9 NDA 0.81A10 QGS 0.93 C10 WFH 4.00 E10 WQY 4.00 G10 DAD 0.69A11 HVK 4.00 C11 NKP 4.00 E11 YYT 4.00 G11 NYA 0.52A12 RNS 4.00 C12 PNE 1.19 E12 NVA 4.00 G12 EDT 1.26B1 DTI 0.82 D1 KTN 4.00 F1 NWQ 0.43 H1 WYV 2.83B2 QAK 3.27 D2 GYE 2.34 F2 TFI 2.39 H2 QSL 0.88B3 WSL 3.32 D3 SYD 0.99 F3 QRG 3.70 H3 TIH 2.63B4 RAA 4.00 D4 IEE 2.29 F4 LDW 1.16 H4 GLA 1.78B5 KHI 4.00 D5 HLQ 0.66 F5 PHL 4.00 H5 LWI 4.00B6 RGS 4.00 D6 GNS 0.95 F6 KID 0.91 H6 GLY 2.46B7 IIV 1.32 D7 IRW 4.00 F7 RQV 4.00 H7 SSG 0.65B8 WFT 2.88 D8 IIG 3.62 F8 KRW 4.00 H8 GWN 0.74B9 RSK 3.22 D9 DFP 1.42 F9 QAD 0.69 H9 SWS 0.74
B10 GHF 1.27 D10 PPG 1.42 F10 ERV 0.76 H10 QIH 0.99B11 QDH 1.17 D11 QGI 0.55 F11 YLT 3.85 H11 KQL 4.00B12 HEQ 1.39 D12 FSH 1.78 F12 RWD 4.00 H12 GYG 2.18
210
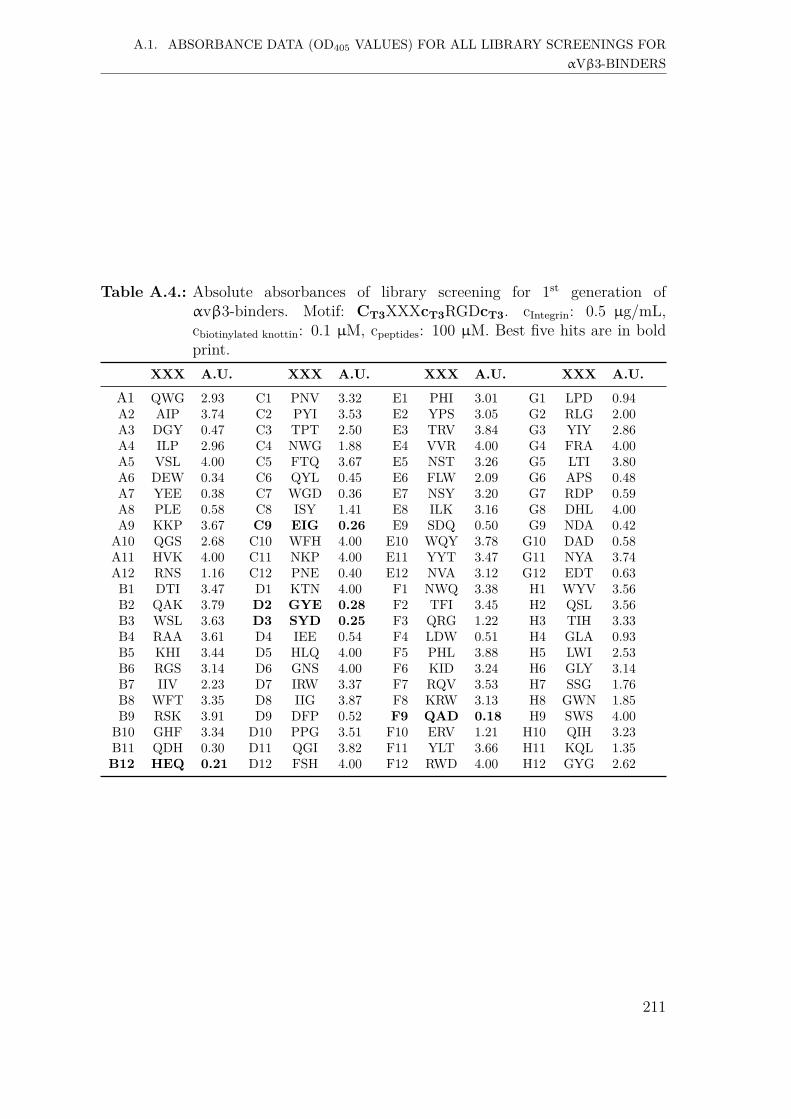
999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.4.: Absolute absorbances of library screening for 1st generation ofαvβ3-binders. Motif: CT3XXXcT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 2.93 C1 PNV 3.32 E1 PHI 3.01 G1 LPD 0.94A2 AIP 3.74 C2 PYI 3.53 E2 YPS 3.05 G2 RLG 2.00A3 DGY 0.47 C3 TPT 2.50 E3 TRV 3.84 G3 YIY 2.86A4 ILP 2.96 C4 NWG 1.88 E4 VVR 4.00 G4 FRA 4.00A5 VSL 4.00 C5 FTQ 3.67 E5 NST 3.26 G5 LTI 3.80A6 DEW 0.34 C6 QYL 0.45 E6 FLW 2.09 G6 APS 0.48A7 YEE 0.38 C7 WGD 0.36 E7 NSY 3.20 G7 RDP 0.59A8 PLE 0.58 C8 ISY 1.41 E8 ILK 3.16 G8 DHL 4.00A9 KKP 3.67 C9 EIG 0.26 E9 SDQ 0.50 G9 NDA 0.42A10 QGS 2.68 C10 WFH 4.00 E10 WQY 3.78 G10 DAD 0.58A11 HVK 4.00 C11 NKP 4.00 E11 YYT 3.47 G11 NYA 3.74A12 RNS 1.16 C12 PNE 0.40 E12 NVA 3.12 G12 EDT 0.63B1 DTI 3.47 D1 KTN 4.00 F1 NWQ 3.38 H1 WYV 3.56B2 QAK 3.79 D2 GYE 0.28 F2 TFI 3.45 H2 QSL 3.56B3 WSL 3.63 D3 SYD 0.25 F3 QRG 1.22 H3 TIH 3.33B4 RAA 3.61 D4 IEE 0.54 F4 LDW 0.51 H4 GLA 0.93B5 KHI 3.44 D5 HLQ 4.00 F5 PHL 3.88 H5 LWI 2.53B6 RGS 3.14 D6 GNS 4.00 F6 KID 3.24 H6 GLY 3.14B7 IIV 2.23 D7 IRW 3.37 F7 RQV 3.53 H7 SSG 1.76B8 WFT 3.35 D8 IIG 3.87 F8 KRW 3.13 H8 GWN 1.85B9 RSK 3.91 D9 DFP 0.52 F9 QAD 0.18 H9 SWS 4.00B10 GHF 3.34 D10 PPG 3.51 F10 ERV 1.21 H10 QIH 3.23B11 QDH 0.30 D11 QGI 3.82 F11 YLT 3.66 H11 KQL 1.35B12 HEQ 0.21 D12 FSH 4.00 F12 RWD 4.00 H12 GYG 2.62
211
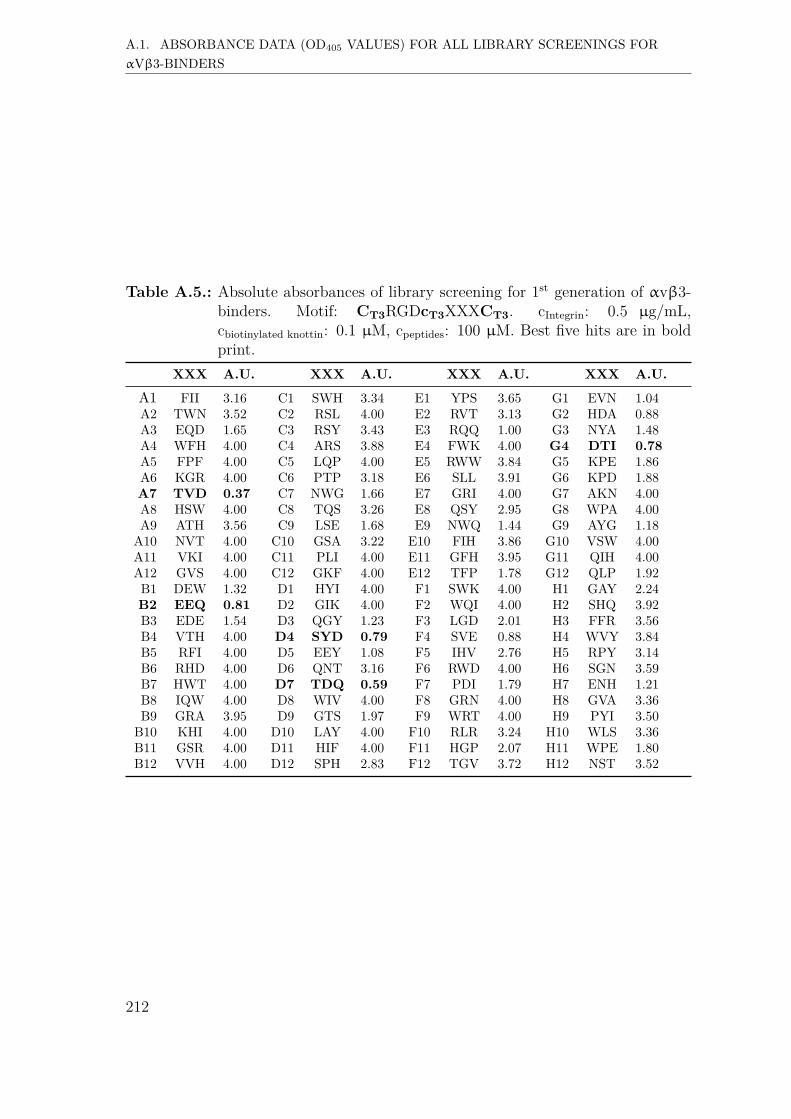
999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.5.: Absolute absorbances of library screening for 1st generation of αvβ3-binders. Motif: CT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 3.16 C1 SWH 3.34 E1 YPS 3.65 G1 EVN 1.04A2 TWN 3.52 C2 RSL 4.00 E2 RVT 3.13 G2 HDA 0.88A3 EQD 1.65 C3 RSY 3.43 E3 RQQ 1.00 G3 NYA 1.48A4 WFH 4.00 C4 ARS 3.88 E4 FWK 4.00 G4 DTI 0.78A5 FPF 4.00 C5 LQP 4.00 E5 RWW 3.84 G5 KPE 1.86A6 KGR 4.00 C6 PTP 3.18 E6 SLL 3.91 G6 KPD 1.88A7 TVD 0.37 C7 NWG 1.66 E7 GRI 4.00 G7 AKN 4.00A8 HSW 4.00 C8 TQS 3.26 E8 QSY 2.95 G8 WPA 4.00A9 ATH 3.56 C9 LSE 1.68 E9 NWQ 1.44 G9 AYG 1.18A10 NVT 4.00 C10 GSA 3.22 E10 FIH 3.86 G10 VSW 4.00A11 VKI 4.00 C11 PLI 4.00 E11 GFH 3.95 G11 QIH 4.00A12 GVS 4.00 C12 GKF 4.00 E12 TFP 1.78 G12 QLP 1.92B1 DEW 1.32 D1 HYI 4.00 F1 SWK 4.00 H1 GAY 2.24B2 EEQ 0.81 D2 GIK 4.00 F2 WQI 4.00 H2 SHQ 3.92B3 EDE 1.54 D3 QGY 1.23 F3 LGD 2.01 H3 FFR 3.56B4 VTH 4.00 D4 SYD 0.79 F4 SVE 0.88 H4 WVY 3.84B5 RFI 4.00 D5 EEY 1.08 F5 IHV 2.76 H5 RPY 3.14B6 RHD 4.00 D6 QNT 3.16 F6 RWD 4.00 H6 SGN 3.59B7 HWT 4.00 D7 TDQ 0.59 F7 PDI 1.79 H7 ENH 1.21B8 IQW 4.00 D8 WIV 4.00 F8 GRN 4.00 H8 GVA 3.36B9 GRA 3.95 D9 GTS 1.97 F9 WRT 4.00 H9 PYI 3.50
B10 KHI 4.00 D10 LAY 4.00 F10 RLR 3.24 H10 WLS 3.36B11 GSR 4.00 D11 HIF 4.00 F11 HGP 2.07 H11 WPE 1.80B12 VVH 4.00 D12 SPH 2.83 F12 TGV 3.72 H12 NST 3.52
212

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.6.: Absolute absorbances of library screening for 1st generation of αvβ3-binders. Motif: cT3RGDCT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 2.24 C1 SWH 4.00 E1 YPS 1.87 G1 EVN 1.35A2 TWN 1.21 C2 RSL 4.00 E2 RVT 3.39 G2 HDA 0.74A3 EQD 1.25 C3 RSY 3.45 E3 RQQ 3.66 G3 NYA 0.77A4 WFH 3.82 C4 ARS 3.62 E4 FWK 3.59 G4 DTI 0.52A5 FPF 3.41 C5 LQP 1.61 E5 RWW 3.47 G5 KPE 0.97A6 KGR 4.00 C6 PTP 0.88 E6 SLL 3.39 G6 KPD 1.08A7 TVD 1.07 C7 NWG 1.10 E7 GRI 3.53 G7 AKN 4.00A8 HSW 1.65 C8 TQS 1.36 E8 QSY 2.30 G8 WPA 1.88A9 ATH 2.02 C9 LSE 1.11 E9 NWQ 0.98 G9 AYG 1.04
A10 NVT 1.46 C10 GSA 0.75 E10 FIH 2.92 G10 VSW 4.00A11 VKI 3.98 C11 PLI 3.39 E11 GFH 3.46 G11 QIH 3.20A12 GVS 2.71 C12 GKF 4.00 E12 TFP 3.39 G12 QLP 1.80B1 DEW 0.61 D1 HYI 3.49 F1 SWK 4.00 H1 GAY 1.66B2 EEQ 0.97 D2 GIK 3.77 F2 WQI 4.00 H2 SHQ 1.10B3 EDE 0.95 D3 QGY 1.83 F3 LGD 0.97 H3 FFR 3.39B4 VTH 4.00 D4 SYD 0.76 F4 SVE 0.73 H4 WVY 3.27B5 RFI 4.00 D5 EEY 1.41 F5 IHV 1.65 H5 RPY 2.41B6 RHD 4.00 D6 QNT 1.62 F6 RWD 3.67 H6 SGN 0.93B7 HWT 3.72 D7 TDQ 0.83 F7 PDI 0.36 H7 ENH 1.10B8 IQW 3.05 D8 WIV 2.91 F8 GRN 1.93 H8 GVA 3.50B9 GRA 2.72 D9 GTS 1.03 F9 WRT 4.00 H9 PYI 2.42B10 KHI 4.00 D10 LAY 3.71 F10 RLR 3.30 H10 WLS 3.14B11 GSR 1.97 D11 HIF 3.39 F11 HGP 3.35 H11 WPE 1.06B12 VVH 4.00 D12 SPH 3.36 F12 TGV 4.00 H12 NST 3.62
213

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.7.: Absolute absorbances of library screening for 1st generation ofαvβ3-binders. Motif: cT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.1 µM, cpeptides: 100 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 1.63 C1 SWH 4.00 E1 YPS 2.23 G1 EVN 1.34A2 TWN 2.26 C2 RSL 3.38 E2 RVT 4.00 G2 HDA 1.15A3 EQD 1.54 C3 RSY 3.57 E3 RQQ 3.08 G3 NYA 1.45A4 WFH 3.98 C4 ARS 2.09 E4 FWK 4.00 G4 DTI 0.35A5 FPF 2.43 C5 LQP 1.79 E5 RWW 4.00 G5 KPE 1.42A6 KGR 3.78 C6 PTP 1.88 E6 SLL 1.83 G6 KPD 1.33A7 TVD 0.65 C7 NWG 0.90 E7 GRI 4.00 G7 AKN 4.00A8 HSW 1.17 C8 TQS 1.12 E8 QSY 0.80 G8 WPA 3.24A9 ATH 4.00 C9 LSE 1.10 E9 NWQ 0.80 G9 AYG 2.01A10 NVT 3.08 C10 GSA 1.10 E10 FIH 4.00 G10 VSW 3.31A11 VKI 3.68 C11 PLI 2.41 E11 GFH 3.58 G11 QIH 4.00A12 GVS 4.00 C12 GKF 1.71 E12 TFP 1.78 G12 QLP 1.60B1 DEW 1.38 D1 HYI 3.84 F1 SWK 3.37 H1 GAY 1.90B2 EEQ 1.07 D2 GIK 4.00 F2 WQI 4.00 H2 SHQ 2.04B3 EDE 1.41 D3 QGY 1.31 F3 LGD 1.37 H3 FFR 3.65B4 VTH 2.43 D4 SYD 0.83 F4 SVE 0.61 H4 WVY 3.03B5 RFI 3.85 D5 EEY 2.56 F5 IHV 2.35 H5 RPY 2.47B6 RHD 3.33 D6 QNT 2.68 F6 RWD 1.69 H6 SGN 2.19B7 HWT 3.35 D7 TDQ 1.43 F7 PDI 1.54 H7 ENH 2.27B8 IQW 3.02 D8 WIV 4.00 F8 GRN 3.52 H8 GVA 3.48B9 GRA 3.17 D9 GTS 1.07 F9 WRT 3.37 H9 PYI 4.00
B10 KHI 3.38 D10 LAY 2.19 F10 RLR 3.85 H10 WLS 3.52B11 GSR 3.46 D11 HIF 4.00 F11 HGP 1.32 H11 WPE 1.25B12 VVH 3.60 D12 SPH 2.11 F12 TGV 3.22 H12 NST 3.24
214

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
A.1.2. Second generation screening of 260 peptides
Table A.8.: Absolute absorbances of library screening for 2nd generation of αvβ3-binders. Motif: CT3XXXXZT3RGDcT3. A1-C12: Z=c, D1-H12:Z=C. cIntegrin: 0.25 µg/mL, cbiotinylated knottin: 0.1 µM, cpeptides: 5 µM.Best five hits are in bold print.
XXXX A.U. XXXX A.U. XXXX A.U. XXXX A.U.
A1 AQAD 0.97 C1 QADI 0.95 E1 TQWG 0.64 G1 GWGD 0.29A2 DQAD 0.91 C2 QADK 0.70 E2 VQWG 0.60 G2 HWGD 0.54A3 EQAD 1.09 C3 QADL 0.67 E3 WQWG 1.31 G3 IWGD 0.52A4 FQAD 0.90 C4 QADN 0.69 E4 YQWG 0.52 G4 KWGD 0.52A5 GQAD 0.79 C5 QADP 0.78 E5 QWGA 0.55 G5 LWGD 0.49A6 HQAD 0.68 C6 QADQ 0.61 E6 QWGD 0.48 G6 NWGD 0.50A7 IQAD 0.86 C7 QADR 0.63 E7 QWGE 0.47 G7 PWGD 0.49A8 KQAD 0.76 C8 QADS 0.67 E8 QWGF 1.03 G8 RWGD 0.67A9 LQAD 0.50 C9 QADT 0.66 E9 QWGG 0.50 G9 SWGD 0.47
A10 NQAD 0.77 C10 QADV 0.85 E10 QWGH 0.44 G10 TWGD 0.54A11 PQAD 0.81 C11 QADW 0.72 E11 QWGI 0.77 G11 VWGD 0.59A12 QQAD 0.72 C12 QADY 0.69 E12 QWGL 0.93 G12 WWGD 0.64B1 RQAD 1.29 D1 AQWG 0.80 F1 QWGN 0.69 H1 YWGD 0.82B2 SQAD 0.94 D2 DQWG 0.54 F2 QWGP 0.75 H2 WGDA 0.65B3 TQAD 1.51 D3 EQWG 0.56 F3 QWGQ 0.61 H3 WGDD 0.56B4 VQAD 1.07 D4 FQWG 0.72 F4 QWGS 0.61 H4 WGDE 0.66B5 WQAD 1.14 D5 GQWG 0.69 F5 QWGT 0.79 H5 WGDF 0.71B6 YQAD 0.88 D6 HQWG 0.52 F6 QWGV 1.18 H6 WGDG 0.48B7 QADA 1.00 D7 IQWG 0.86 F7 QWGW 1.24 H7 WGDH 0.44B8 QADD 1.19 D8 LQWG 0.70 F8 QWGY 1.64 H8 WGDI 0.62B9 QADE 1.01 D9 NQWG 0.61 F9 AWGD 0.57 H9 WGDK 0.53B10 QADF 1.33 D10 PQWG 0.68 F10 DWGD 0.50 H10 WGDL 0.59B11 QADG 0.80 D11 QQWG 1.01 F11 EWGD 0.57 H11 WGDN 0.48B12 QADH 0.65 D12 SQWG 0.88 F12 FWGD 0.58 H12 WGDP 0.61
reference: no peptide: 1.17
215

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.9.: Absolute absorbances of library screening for 2nd generation of αvβ3-binders. Motif: CT3XXXXZT3RGDcT3. A1-A7: Z=C, A8-H12:Z=c. cIntegrin: 0.25 µg/mL, cbiotinylated knottin: 0.1 µM, cpeptides: 5 µM.Best five hits are in bold print.
XXXX A.U. XXXX A.U. XXXX A.U. XXXX A.U.
A1 WGDQ 0.69 C1 YHEQ 0.96 E1 HEIG 0.85 G1 EIGQ 1.90A2 WGDR 0.69 C2 HEQA 0.86 E2 IEIG 2.13 G2 EIGR 3.54A3 WGDS 0.69 C3 HEQD 0.64 E3 KEIG 2.11 G3 EIGS 0.96A4 WGDT 0.80 C4 HEQE 0.75 E4 LEIG 1.04 G4 EIGT 0.97A5 WGDV 0.70 C5 HEQF 0.86 E5 NEIG 0.75 G5 EIGV 0.98A6 WGDW 0.88 C6 HEQG 0.73 E6 PEIG 0.71 G6 EIGW 0.90A7 WGDY 0.97 C7 HEQH 0.71 E7 QEIG 0.71 G7 EIGY 0.99A8 AHEQ 0.87 C8 HEQI 0.81 E8 REIG 1.68 G8 ASYD 0.98A9 DHEQ 0.84 C9 HEQK 1.44 E9 SEIG 0.76 G9 DSYD 0.91
A10 EHEQ 0.75 C10 HEQL 0.70 E10 TEIG 0.72 G10 ESYD 0.91A11 FHEQ 0.83 C11 HEQN 0.71 E11 VEIG 0.91 G11 FSYD 1.35A12 GHEQ 0.81 C12 HEQP 0.83 E12 WEIG 0.82 G12 GSYD 0.79B1 HHEQ 0.59 D1 HEQQ 0.50 F1 YEIG 1.30 H1 HSYD 1.14B2 IHEQ 0.86 D2 HEQR 0.91 F2 EIGA 0.95 H2 ISYD 1.26B3 KHEQ 0.84 D3 HEQS 0.72 F3 EIGD 0.86 H3 KSYD 1.82B4 LHEQ 0.74 D4 HEQT 0.72 F4 EIGE 0.78 H4 LSYD 0.93B5 NHEQ 0.83 D5 HEQV 0.73 F5 EIGF 1.15 H5 NSYD 0.96B6 PHEQ 0.74 D6 HEQW 0.73 F6 EIGG 0.69 H6 PSYD 1.00B7 QHEQ 0.61 D7 HEQY 0.77 F7 EIGH 0.66 H7 QSYD 0.98B8 RHEQ 0.64 D8 AEIG 0.71 F8 EIGI 0.79 H8 RSYD 1.05B9 SHEQ 0.68 D9 DEIG 0.83 F9 EIGK 1.55 H9 SSYD 0.97B10 THEQ 0.63 D10 EEIG 0.71 F10 EIGL 0.93 H10 TSYD 0.87B11 VHEQ 0.51 D11 FEIG 1.14 F11 EIGN 0.77 H11 VSYD 0.95B12 WHEQ 0.79 D12 GEIG 0.77 F12 EIGP 0.76 H12 WSYD 1.29
216

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.10.: Absolute absorbances of library screening for 2nd generation of αvβ3-binders. Motif: CT3XXX(X)ZT3RGDcT3. A1-B7, C10-D11, E5,E8-E11, F3, F6-F8: Z=c, B8-C9, D12-E4, E6, E7, E12-F2, F4, F5:Z=C. cIntegrin: 0.25 µg/mL, cbiotinylated knottin: 0.1 µM, cpeptides: 5 µM.Best five hits are in bold print.
XXXX A.U. XXX(X) A.U. XXX A.U. XXX A.U.
A1 YSYD 0.96 B7 SYDY 0.80 D1 HGQ 1.14 E7 H5Q 0.75A2 SYDA 1.01 B8 HAQ 0.56 D2 HHQ 1.47 E8 E4G 0.73A3 SYDD 1.18 B9 HDQ 0.43 D3 HIQ 1.28 E9 E5G 0.72A4 SYDE 0.85 B10 HFQ 0.64 D4 HNQ 0.52 E10 S1D 0.56A5 SYDF 0.96 B11 HGQ 0.55 D5 HPQ 0.41 E11 S2D 0.96A6 SYDG 0.71 B12 HHQ 0.53 D6 HQQ 0.99 E12 WGD 0.36A7 SYDH 0.78 C1 HIQ 1.12 D7 HSQ 0.62 F1 QWG 1.43A8 SYDI 0.96 C2 HNQ 0.64 D8 HTQ 1.11 F2 QAD 0.75A9 SYDK 0.89 C3 HPQ 0.64 D9 HVQ 1.61 F3 QAD 0.74
A10 SYDL 1.07 C4 HQQ 0.52 D10 HWQ 0.43 F4 HLQ 1.45A11 SYDN 0.82 C5 HSQ 0.82 D11 HYQ 0.72 F5 HEQ 0.51A12 SYDP 0.98 C6 HTQ 0.60 D12 1GD 0.75 F6 HEQ 0.47B1 SYDQ 0.88 C7 HVQ 0.66 E1 2GD 0.62 F7 EIG 0.77B2 SYDR 1.22 C8 HWQ 0.44 E2 Q1G 1.02 F8 SYD 0.77B3 SYDS 0.84 C9 HYQ 0.81 E3 Q2G 1.01B4 SYDT 0.82 C10 HAQ 0.99 E4 Q3D 0.43B5 SYDV 0.79 C11 HDQ 0.58 E5 Q3D 0.40B6 SYDW 0.86 C12 HFQ 0.51 E6 H4Q 0.92
reference: no peptide: 1.17
217

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
A.1.3. Third generation screening of 199 peptides
Table A.11.: Absolute absorbances of library screening for 3rd generation ofαvβ3-binders. Motif: CT3XXXCT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.2 µM, cpeptides: 5 µM. Best five hits are in boldprint.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 AWQ 2.30 C1 iWQ 3.46 E1 H4Q 0.82 G1 HWV 3.55A2 DWQ 1.11 C2 9WQ 1.29 E2 H5Q 3.28 G2 HWW 1.81A3 EWQ 1.78 C3 aWQ 1.06 E3 H6Q 0.55 G3 HWY 1.35A4 FWQ 2.44 C4 hWQ 0.75 E4 H7Q 0.37 G4 HW3 1.83A5 GWQ 1.03 C5 RGD 0.61 E5 H8Q 0.30 G5 HW4 1.54A6 IWQ 1.81 C6 HAQ 0.82 E6 HfQ 0.79 G6 HW5 0.98A7 KWQ 2.03 C7 HDQ 0.54 E7 HiQ 1.00 G7 HW6 2.10A8 LWQ 2.11 C8 HEQ 0.48 E8 H9Q 0.87 G8 HW7 1.50A9 NWQ 0.77 C9 HFQ 0.42 E9 HaQ 0.92 G9 HW8 1.34A10 PWQ 1.41 C10 HGQ 0.94 E10 HwQ 0.95 G10 HWf 2.37A11 QWQ 2.63 C11 HHQ 0.71 E11 HWA 1.10 G11 HWi 1.60A12 RWQ 1.97 C12 HIQ 1.21 E12 HWD 0.95 G12 HW9 3.16B1 SWQ 2.25 D1 HKQ 2.21 F1 HWE 0.41 H1 HWa 2.84B2 TWQ 1.43 D2 HLQ 1.50 F2 HWF 1.79 H2 HWq 0.51B3 VWQ 1.48 D3 HNQ 0.49 F3 HWG 0.69B4 WWQ 1.83 D4 HPQ 0.26 F4 HWH 0.58B5 YWQ 1.39 D5 HQQ 2.31 F5 HWI 2.17B6 3WQ 1.16 D6 HRQ 0.44 F6 HWK 0.96B7 4WQ 1.65 D7 HSQ 0.52 F7 HWL 1.05B8 5WQ 1.31 D8 HTQ 0.60 F8 HWN 0.56B9 6WQ 2.13 D9 HVQ 0.45 F9 HWP 1.47
B10 7WQ 2.75 D10 HWQ 0.41 F10 HWR 0.77B11 8WQ 1.03 D11 HYQ 0.56 F11 HWS 0.82B12 fWQ 3.45 D12 H3Q 1.57 F12 HWT 2.25
references: knottin-RGD (5µM): 0.23, no peptide: 2.46
218

999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.12.: Absolute absorbances of library screening for 3rd generation of αvβ3-binders. Motif: CT3HWQCT3XXXXcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.2 µM, cpeptides: 5 µM. Best five hits are in boldprint.
XXXX A.U. XXXX A.U. XXXX A.U.
A1 RGDA 0.98 C1 RGDf 3.32 E1 4RGD 1.51A2 RGDD 0.79 C2 RGDi 2.21 E2 5RGD 1.37A3 RGDE 1.03 C3 RGD9 3.00 E3 6RGD 3.29A4 RGDF 1.44 C4 RGDa 0.78 E4 7RGD 2.87A5 RGDG 1.31 C5 ARGD 1.46 E5 8RGD 1.56A6 RGDH 1.25 C6 DRGD 1.49 E6 fRGD 2.68A7 RGDI 1.36 C7 ERGD 1.37 E7 iRGD 1.58A8 RGDK 2.36 C8 FRGD 3.45 E8 9RGD 1.68A9 RGDL 1.52 C9 GRGD 1.29 E9 aRGD 1.68
A10 RGDN 1.45 C10 HRGD 1.81A11 RGDP 2.49 C11 IRGD 2.73A12 RDGQ 2.30 C12 KRGD 2.42B1 RGDR 2.01 D1 LRGD 1.68B2 RGDS 0.78 D2 NRGD 1.33B3 RGDT 0.82 D3 PRGD 1.17B4 RGDV 0.89 D4 QRGD 1.66B5 RGDW 2.79 D5 RRGD 3.24B6 RGDY 2.29 D6 SRGD 1.56B7 RGD3 0.90 D7 TRGD 1.51B8 RGD4 1.22 D8 VRGD 1.95B9 RGD5 1.31 D9 WRGD 3.41B10 RGD6 3.27 D10 YRGD 3.82B11 RGD7 3.46 D11 2RGD 2.36B12 RGD8 1.50 D12 3RGD 1.41
references: knottin-RGD (5µM): 0.23, no peptide: 2.46
219
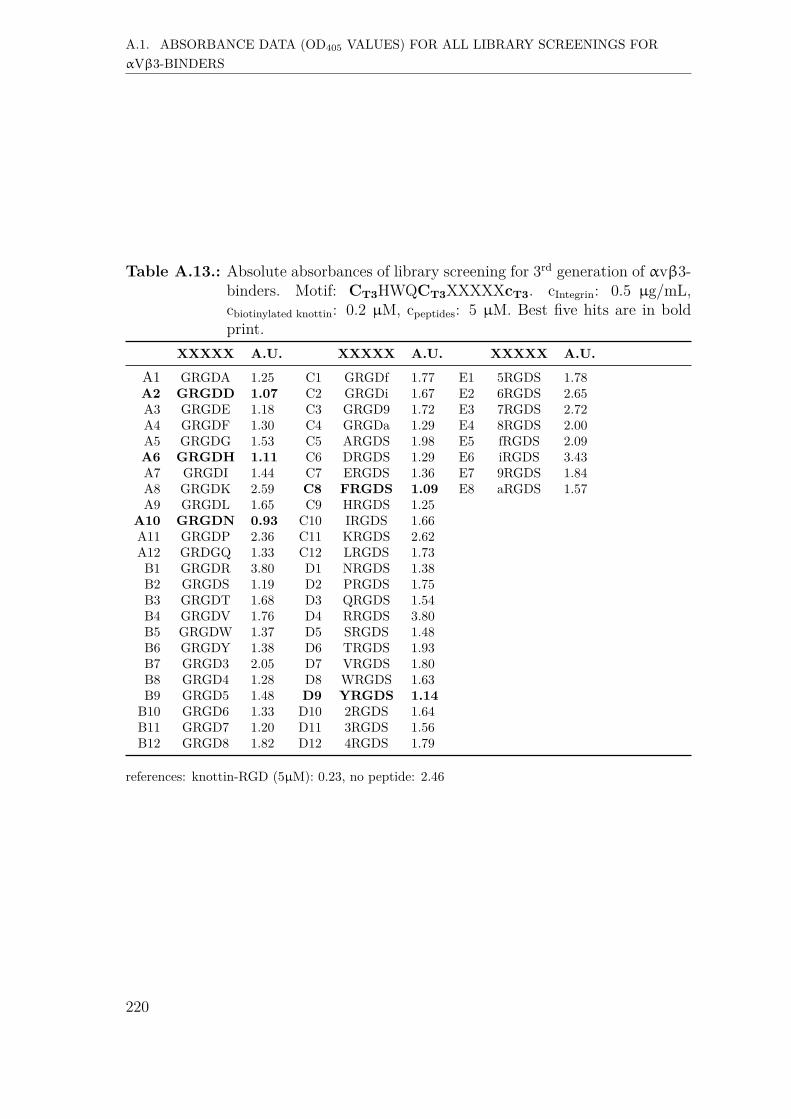
999999999
A.1. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ3-BINDERS
Table A.13.: Absolute absorbances of library screening for 3rd generation of αvβ3-binders. Motif: CT3HWQCT3XXXXXcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin: 0.2 µM, cpeptides: 5 µM. Best five hits are in boldprint.
XXXXX A.U. XXXXX A.U. XXXXX A.U.
A1 GRGDA 1.25 C1 GRGDf 1.77 E1 5RGDS 1.78A2 GRGDD 1.07 C2 GRGDi 1.67 E2 6RGDS 2.65A3 GRGDE 1.18 C3 GRGD9 1.72 E3 7RGDS 2.72A4 GRGDF 1.30 C4 GRGDa 1.29 E4 8RGDS 2.00A5 GRGDG 1.53 C5 ARGDS 1.98 E5 fRGDS 2.09A6 GRGDH 1.11 C6 DRGDS 1.29 E6 iRGDS 3.43A7 GRGDI 1.44 C7 ERGDS 1.36 E7 9RGDS 1.84A8 GRGDK 2.59 C8 FRGDS 1.09 E8 aRGDS 1.57A9 GRGDL 1.65 C9 HRGDS 1.25
A10 GRGDN 0.93 C10 IRGDS 1.66A11 GRGDP 2.36 C11 KRGDS 2.62A12 GRDGQ 1.33 C12 LRGDS 1.73B1 GRGDR 3.80 D1 NRGDS 1.38B2 GRGDS 1.19 D2 PRGDS 1.75B3 GRGDT 1.68 D3 QRGDS 1.54B4 GRGDV 1.76 D4 RRGDS 3.80B5 GRGDW 1.37 D5 SRGDS 1.48B6 GRGDY 1.38 D6 TRGDS 1.93B7 GRGD3 2.05 D7 VRGDS 1.80B8 GRGD4 1.28 D8 WRGDS 1.63B9 GRGD5 1.48 D9 YRGDS 1.14
B10 GRGD6 1.33 D10 2RGDS 1.64B11 GRGD7 1.20 D11 3RGDS 1.56B12 GRGD8 1.82 D12 4RGDS 1.79
references: knottin-RGD (5µM): 0.23, no peptide: 2.46
220

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
A.2. Absorbance data (OD405 values) for all libraryscreenings for α5β1-binders
A.2.1. First generation screening of 672 random-diversitypeptides
Table A.14.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: CT3XXXCT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.81 C1 PNV 0.86 E1 PHI 1.05 G1 LPD 1.01A2 AIP 0.81 C2 PYI 0.87 E2 YPS 0.97 G2 RLG 0.81A3 DGY 0.79 C3 TPT 0.83 E3 TRV 0.95 G3 YIY 1.04A4 ILP 0.95 C4 NWG 0.79 E4 VVR 1.61 G4 FRA 0.88A5 VSL 0.75 C5 FTQ 0.85 E5 NST 1.11 G5 LTI 1.06A6 DEW 0.76 C6 QYL 0.87 E6 FLW 0.98 G6 APS 0.84A7 YEE 0.66 C7 WGD 1.00 E7 NSY 0.99 G7 RDP 0.99A8 PLE 0.63 C8 ISY 0.88 E8 ILK 1.89 G8 DHL 0.83A9 KKP 0.72 C9 EIG 0.80 E9 SDQ 0.91 G9 NDA 0.86A10 QGS 0.77 C10 WFH 0.84 E10 WQY 1.47 G10 DAD 0.84A11 HVK 0.93 C11 NKP 0.78 E11 YYT 1.02 G11 NYA 0.89A12 RNS 1.01 C12 PNE 0.79 E12 NVA 1.06 G12 EDT 0.93B1 DTI 0.86 D1 KTN 0.70 F1 NWQ 0.95 H1 WYV 1.12B2 QAK 0.90 D2 GYE 0.86 F2 TFI 1.40 H2 QSL 0.92B3 WSL 0.85 D3 SYD 0.99 F3 QRG 0.94 H3 TIH 1.17B4 RAA 0.88 D4 IEE 1.03 F4 LDW 0.95 H4 GLA 1.00B5 KHI 0.88 D5 HLQ 1.09 F5 PHL 0.92 H5 LWI 1.67B6 RGS 0.78 D6 GNS 0.89 F6 KID 0.90 H6 GLY 1.01B7 IIV 1.04 D7 IRW 0.85 F7 RQV 0.94 H7 SSG 1.09B8 WFT 0.78 D8 IIG 0.86 F8 KRW 3.05 H8 GWN 1.05B9 RSK 1.11 D9 DFP 0.81 F9 QAD 0.96 H9 SWS 0.98B10 GHF 0.77 D10 PPG 0.81 F10 ERV 0.91 H10 QIH 0.99B11 QDH 0.74 D11 QGI 0.88 F11 YLT 1.07 H11 KQL 0.94B12 HEQ 0.76 D12 FSH 1.39 F12 RWD 0.86 H12 GYG 1.06
references: knottin-RGD (5µM): 0.19, no peptide: 1.12.
221

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.15.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: CT3XXXCT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 1.50 C1 PNV 1.56 E1 PHI 1.64 G1 LPD 1.70A2 AIP 2.00 C2 PYI 1.76 E2 YPS 1.36 G2 RLG 3.81A3 DGY 1.99 C3 TPT 1.63 E3 TRV 2.40 G3 YIY 1.95A4 ILP 1.81 C4 NWG 1.38 E4 VVR 1.99 G4 FRA 1.79A5 VSL 1.46 C5 FTQ 1.84 E5 NST 2.06 G5 LTI 1.99A6 DEW 1.57 C6 QYL 2.23 E6 FLW 2.19 G6 APS 1.97A7 YEE 2.24 C7 WGD 1.74 E7 NSY 1.73 G7 RDP 1.84A8 PLE 1.49 C8 ISY 2.24 E8 ILK 3.56 G8 DHL 1.79A9 KKP 2.31 C9 EIG 1.80 E9 SDQ 1.92 G9 NDA 1.90
A10 QGS 1.33 C10 WFH 1.88 E10 WQY 1.77 G10 DAD 2.11A11 HVK 3.20 C11 NKP 1.69 E11 YYT 2.01 G11 NYA 2.06A12 RNS 1.73 C12 PNE 1.64 E12 NVA 2.00 G12 EDT 2.00B1 DTI 2.07 D1 KTN 2.44 F1 NWQ 1.68 H1 WYV 2.53B2 QAK 3.18 D2 GYE 1.90 F2 TFI 2.09 H2 QSL 1.79B3 WSL 1.61 D3 SYD 2.06 F3 QRG 1.94 H3 TIH 2.18B4 RAA 2.33 D4 IEE 1.88 F4 LDW 2.07 H4 GLA 1.98B5 KHI 2.13 D5 HLQ 1.64 F5 PHL 1.74 H5 LWI 2.67B6 RGS 1.74 D6 GNS 1.85 F6 KID 1.66 H6 GLY 1.66B7 IIV 1.74 D7 IRW 1.91 F7 RQV 3.03 H7 SSG 1.79B8 WFT 1.89 D8 IIG 1.80 F8 KRW 3.82 H8 GWN 1.87B9 RSK 1.96 D9 DFP 1.93 F9 QAD 2.21 H9 SWS 1.74
B10 GHF 1.78 D10 PPG 2.02 F10 ERV 1.93 H10 QIH 2.15B11 QDH 1.65 D11 QGI 1.57 F11 YLT 2.04 H11 KQL 2.07B12 HEQ 1.53 D12 FSH 1.84 F12 RWD 1.81 H12 GYG 1.94
references: knottin-RGD (5µM): 0.25, no peptide: 2.08.
222
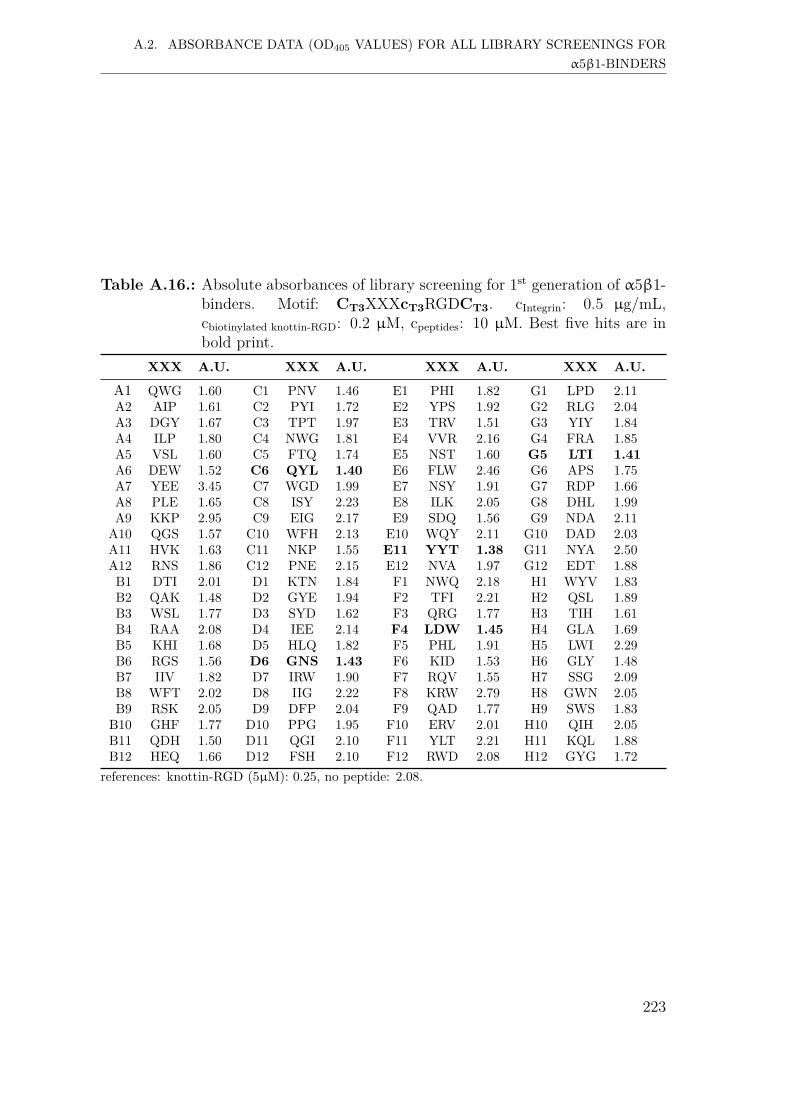
999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.16.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: CT3XXXcT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 1.60 C1 PNV 1.46 E1 PHI 1.82 G1 LPD 2.11A2 AIP 1.61 C2 PYI 1.72 E2 YPS 1.92 G2 RLG 2.04A3 DGY 1.67 C3 TPT 1.97 E3 TRV 1.51 G3 YIY 1.84A4 ILP 1.80 C4 NWG 1.81 E4 VVR 2.16 G4 FRA 1.85A5 VSL 1.60 C5 FTQ 1.74 E5 NST 1.60 G5 LTI 1.41A6 DEW 1.52 C6 QYL 1.40 E6 FLW 2.46 G6 APS 1.75A7 YEE 3.45 C7 WGD 1.99 E7 NSY 1.91 G7 RDP 1.66A8 PLE 1.65 C8 ISY 2.23 E8 ILK 2.05 G8 DHL 1.99A9 KKP 2.95 C9 EIG 2.17 E9 SDQ 1.56 G9 NDA 2.11
A10 QGS 1.57 C10 WFH 2.13 E10 WQY 2.11 G10 DAD 2.03A11 HVK 1.63 C11 NKP 1.55 E11 YYT 1.38 G11 NYA 2.50A12 RNS 1.86 C12 PNE 2.15 E12 NVA 1.97 G12 EDT 1.88B1 DTI 2.01 D1 KTN 1.84 F1 NWQ 2.18 H1 WYV 1.83B2 QAK 1.48 D2 GYE 1.94 F2 TFI 2.21 H2 QSL 1.89B3 WSL 1.77 D3 SYD 1.62 F3 QRG 1.77 H3 TIH 1.61B4 RAA 2.08 D4 IEE 2.14 F4 LDW 1.45 H4 GLA 1.69B5 KHI 1.68 D5 HLQ 1.82 F5 PHL 1.91 H5 LWI 2.29B6 RGS 1.56 D6 GNS 1.43 F6 KID 1.53 H6 GLY 1.48B7 IIV 1.82 D7 IRW 1.90 F7 RQV 1.55 H7 SSG 2.09B8 WFT 2.02 D8 IIG 2.22 F8 KRW 2.79 H8 GWN 2.05B9 RSK 2.05 D9 DFP 2.04 F9 QAD 1.77 H9 SWS 1.83B10 GHF 1.77 D10 PPG 1.95 F10 ERV 2.01 H10 QIH 2.05B11 QDH 1.50 D11 QGI 2.10 F11 YLT 2.21 H11 KQL 1.88B12 HEQ 1.66 D12 FSH 2.10 F12 RWD 2.08 H12 GYG 1.72
references: knottin-RGD (5µM): 0.25, no peptide: 2.08.
223

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.17.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: CT3XXXCT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.86 C1 PNV 1.23 E1 PHI 1.21 G1 LPD 1.22A2 AIP 0.97 C2 PYI 1.03 E2 YPS 1.04 G2 RLG 0.99A3 DGY 0.82 C3 TPT 1.02 E3 TRV 1.97 G3 YIY 2.01A4 ILP 1.72 C4 NWG 0.93 E4 VVR 3.68 G4 FRA 1.06A5 VSL 2.56 C5 FTQ 1.02 E5 NST 0.77 G5 LTI 2.64A6 DEW 0.86 C6 QYL 0.74 E6 FLW 1.41 G6 APS 1.05A7 YEE 0.81 C7 WGD 0.85 E7 NSY 1.08 G7 RDP 0.97A8 PLE 0.97 C8 ISY 0.87 E8 ILK 1.82 G8 DHL 1.35A9 KKP 1.02 C9 EIG 0.82 E9 SDQ 0.84 G9 NDA 0.83A10 QGS 0.77 C10 WFH 2.82 E10 WQY 0.85 G10 DAD 0.96A11 HVK 2.57 C11 NKP 1.16 E11 YYT 0.87 G11 NYA 0.89A12 RNS 0.89 C12 PNE 0.87 E12 NVA 1.41 G12 EDT 1.06B1 DTI 1.27 D1 KTN 1.27 F1 NWQ 1.47 H1 WYV 1.05B2 QAK 0.85 D2 GYE 0.63 F2 TFI 1.22 H2 QSL 2.26B3 WSL 0.93 D3 SYD 0.80 F3 QRG 0.87 H3 TIH 1.41B4 RAA 0.93 D4 IEE 0.81 F4 LDW 0.76 H4 GLA 0.99B5 KHI 2.65 D5 HLQ 0.91 F5 PHL 0.94 H5 LWI 3.51B6 RGS 1.09 D6 GNS 0.71 F6 KID 1.03 H6 GLY 1.05B7 IIV 1.51 D7 IRW 1.41 F7 RQV 1.22 H7 SSG 1.01B8 WFT 3.76 D8 IIG 1.20 F8 KRW 1.60 H8 GWN 0.97B9 RSK 2.82 D9 DFP 0.63 F9 QAD 0.96 H9 SWS 1.16
B10 GHF 0.85 D10 PPG 0.72 F10 ERV 1.11 H10 QIH 2.88B11 QDH 0.84 D11 QGI 1.90 F11 YLT 1.54 H11 KQL 0.84B12 HEQ 0.81 D12 FSH 0.86 F12 RWD 1.85 H12 GYG 1.27
references: knottin-RGD (5µM): 0.19, no peptide: 1.12.
224

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.18.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: CT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 0.49 C1 SWH 0.54 E1 YPS 0.89 G1 EVN 0.25A2 TWN 0.29 C2 RSL 0.29 E2 RVT 0.30 G2 HDA 0.26A3 EQD 0.31 C3 RSY 0.27 E3 RQQ 0.59 G3 NYA 0.26A4 WFH 0.35 C4 ARS 0.29 E4 FWK 1.97 G4 DTI 0.26A5 FPF 1.38 C5 LQP 0.26 E5 RWW 1.66 G5 KPE 0.28A6 KGR 3.65 C6 PTP 0.26 E6 SLL 0.25 G6 KPD 0.28A7 TVD 0.25 C7 NWG 0.17 E7 GRI 0.30 G7 AKN 0.64A8 HSW 0.34 C8 TQS 0.24 E8 QSY 0.22 G8 WPA 1.72A9 ATH 0.58 C9 LSE 0.21 E9 NWQ 0.23 G9 AYG 0.15
A10 NVT 0.28 C10 GSA 0.25 E10 FIH 0.25 G10 VSW 0.31A11 VKI 3.35 C11 PLI 1.62 E11 GFH 0.32 G11 QIH 0.23A12 GVS 0.24 C12 GKF 3.41 E12 TFP 0.27 G12 QLP 0.24B1 DEW 0.28 D1 HYI 0.27 F1 SWK 0.83 H1 GAY 0.28B2 EEQ 0.24 D2 GIK 0.51 F2 WQI 0.20 H2 SHQ 0.28B3 EDE 0.28 D3 QGY 0.20 F3 LGD 0.23 H3 FFR 0.61B4 VTH 0.20 D4 SYD 0.21 F4 SVE 0.22 H4 WVY 0.33B5 RFI 0.92 D5 EEY 0.30 F5 IHV 0.49 H5 RPY 0.30B6 RHD 0.32 D6 QNT 0.20 F6 RWD 0.24 H6 SGN 0.31B7 HWT 0.28 D7 TDQ 0.21 F7 PDI 0.22 H7 ENH 0.29B8 IQW 0.30 D8 WIV 0.72 F8 GRN 0.27 H8 GVA 0.28B9 GRA 0.28 D9 GTS 0.22 F9 WRT 0.22 H9 PYI 1.51B10 KHI 0.57 D10 LAY 0.34 F10 RLR 3.56 H10 WLS 0.81B11 GSR 1.47 D11 HIF 0.28 F11 HGP 0.21 H11 WPE 0.28B12 VVH 0.30 D12 SPH 0.20 F12 TGV 0.22 H12 NST 0.27
references: knottin-RGD (5µM): 0.15, no peptide: 0.33.
225
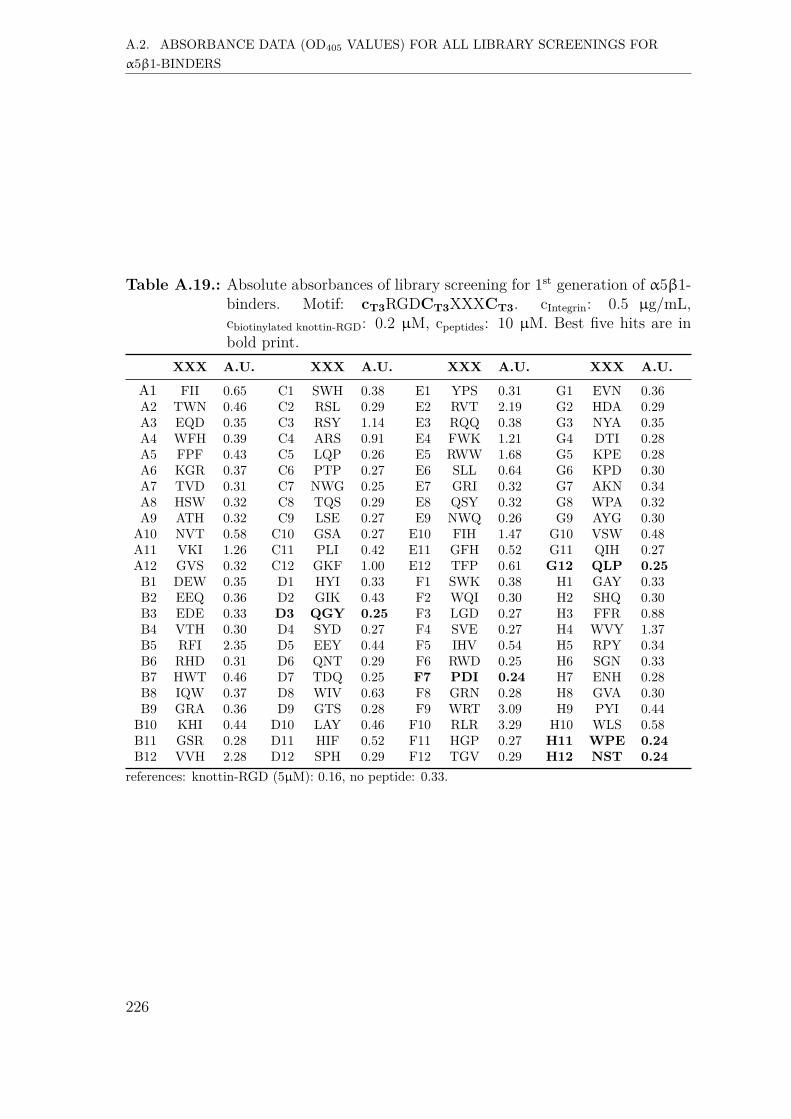
999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.19.: Absolute absorbances of library screening for 1st generation of α5β1-binders. Motif: cT3RGDCT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 0.65 C1 SWH 0.38 E1 YPS 0.31 G1 EVN 0.36A2 TWN 0.46 C2 RSL 0.29 E2 RVT 2.19 G2 HDA 0.29A3 EQD 0.35 C3 RSY 1.14 E3 RQQ 0.38 G3 NYA 0.35A4 WFH 0.39 C4 ARS 0.91 E4 FWK 1.21 G4 DTI 0.28A5 FPF 0.43 C5 LQP 0.26 E5 RWW 1.68 G5 KPE 0.28A6 KGR 0.37 C6 PTP 0.27 E6 SLL 0.64 G6 KPD 0.30A7 TVD 0.31 C7 NWG 0.25 E7 GRI 0.32 G7 AKN 0.34A8 HSW 0.32 C8 TQS 0.29 E8 QSY 0.32 G8 WPA 0.32A9 ATH 0.32 C9 LSE 0.27 E9 NWQ 0.26 G9 AYG 0.30A10 NVT 0.58 C10 GSA 0.27 E10 FIH 1.47 G10 VSW 0.48A11 VKI 1.26 C11 PLI 0.42 E11 GFH 0.52 G11 QIH 0.27A12 GVS 0.32 C12 GKF 1.00 E12 TFP 0.61 G12 QLP 0.25B1 DEW 0.35 D1 HYI 0.33 F1 SWK 0.38 H1 GAY 0.33B2 EEQ 0.36 D2 GIK 0.43 F2 WQI 0.30 H2 SHQ 0.30B3 EDE 0.33 D3 QGY 0.25 F3 LGD 0.27 H3 FFR 0.88B4 VTH 0.30 D4 SYD 0.27 F4 SVE 0.27 H4 WVY 1.37B5 RFI 2.35 D5 EEY 0.44 F5 IHV 0.54 H5 RPY 0.34B6 RHD 0.31 D6 QNT 0.29 F6 RWD 0.25 H6 SGN 0.33B7 HWT 0.46 D7 TDQ 0.25 F7 PDI 0.24 H7 ENH 0.28B8 IQW 0.37 D8 WIV 0.63 F8 GRN 0.28 H8 GVA 0.30B9 GRA 0.36 D9 GTS 0.28 F9 WRT 3.09 H9 PYI 0.44
B10 KHI 0.44 D10 LAY 0.46 F10 RLR 3.29 H10 WLS 0.58B11 GSR 0.28 D11 HIF 0.52 F11 HGP 0.27 H11 WPE 0.24B12 VVH 2.28 D12 SPH 0.29 F12 TGV 0.29 H12 NST 0.24
references: knottin-RGD (5µM): 0.16, no peptide: 0.33.
226
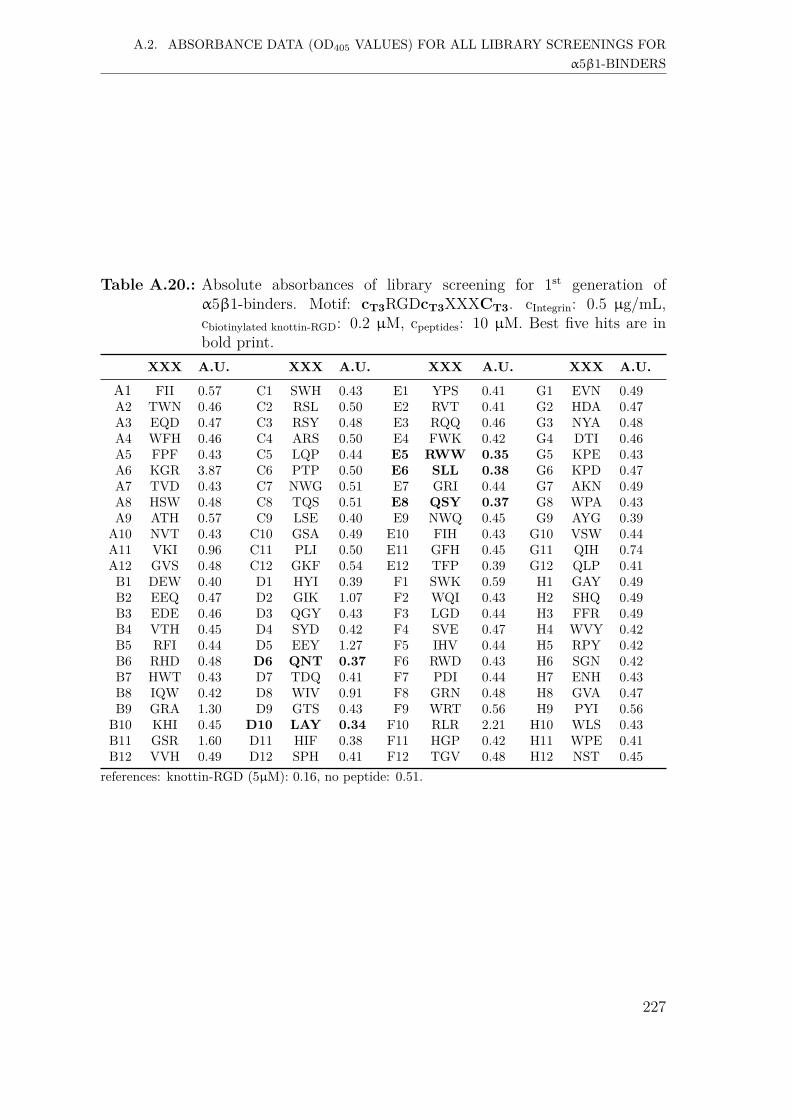
999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.20.: Absolute absorbances of library screening for 1st generation ofα5β1-binders. Motif: cT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 0.57 C1 SWH 0.43 E1 YPS 0.41 G1 EVN 0.49A2 TWN 0.46 C2 RSL 0.50 E2 RVT 0.41 G2 HDA 0.47A3 EQD 0.47 C3 RSY 0.48 E3 RQQ 0.46 G3 NYA 0.48A4 WFH 0.46 C4 ARS 0.50 E4 FWK 0.42 G4 DTI 0.46A5 FPF 0.43 C5 LQP 0.44 E5 RWW 0.35 G5 KPE 0.43A6 KGR 3.87 C6 PTP 0.50 E6 SLL 0.38 G6 KPD 0.47A7 TVD 0.43 C7 NWG 0.51 E7 GRI 0.44 G7 AKN 0.49A8 HSW 0.48 C8 TQS 0.51 E8 QSY 0.37 G8 WPA 0.43A9 ATH 0.57 C9 LSE 0.40 E9 NWQ 0.45 G9 AYG 0.39
A10 NVT 0.43 C10 GSA 0.49 E10 FIH 0.43 G10 VSW 0.44A11 VKI 0.96 C11 PLI 0.50 E11 GFH 0.45 G11 QIH 0.74A12 GVS 0.48 C12 GKF 0.54 E12 TFP 0.39 G12 QLP 0.41B1 DEW 0.40 D1 HYI 0.39 F1 SWK 0.59 H1 GAY 0.49B2 EEQ 0.47 D2 GIK 1.07 F2 WQI 0.43 H2 SHQ 0.49B3 EDE 0.46 D3 QGY 0.43 F3 LGD 0.44 H3 FFR 0.49B4 VTH 0.45 D4 SYD 0.42 F4 SVE 0.47 H4 WVY 0.42B5 RFI 0.44 D5 EEY 1.27 F5 IHV 0.44 H5 RPY 0.42B6 RHD 0.48 D6 QNT 0.37 F6 RWD 0.43 H6 SGN 0.42B7 HWT 0.43 D7 TDQ 0.41 F7 PDI 0.44 H7 ENH 0.43B8 IQW 0.42 D8 WIV 0.91 F8 GRN 0.48 H8 GVA 0.47B9 GRA 1.30 D9 GTS 0.43 F9 WRT 0.56 H9 PYI 0.56B10 KHI 0.45 D10 LAY 0.34 F10 RLR 2.21 H10 WLS 0.43B11 GSR 1.60 D11 HIF 0.38 F11 HGP 0.42 H11 WPE 0.41B12 VVH 0.49 D12 SPH 0.41 F12 TGV 0.48 H12 NST 0.45
references: knottin-RGD (5µM): 0.16, no peptide: 0.51.
227

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
A.2.2. Second generation screening of 197 peptides
Table A.21.: Absolute absorbances of library screening for 2nd generation of α5β1-binders. Motif: CT3RGDcT3XXXCT3. cIntegrin: 0.49 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 DYG 1.82 C1 iYG 1.75 E1 A5G 0.81 G1 AYW 2.45A2 EYG 1.53 C2 9YG 1.41 E2 A6G 0.74 G2 AYY 2.33A3 FYG 1.23 C3 aYG 0.92 E3 A7G 0.64 G3 AY3 1.54A4 GYG 1.51 C4 RGD 1.50 E4 A8G 0.98 G4 AY4 2.21A5 HYG 1.47 C5 AAG 0.66 E5 AfG 1.33 G5 AY5 1.49A6 IYG 0.52 C6 ADG 0.91 E6 AiG 1.05 G6 AY6 2.10A7 KYG 1.54 C7 AEG 0.68 E7 A9G 1.15 G7 AY7 2.79A8 LYG 0.89 C8 AFG 0.66 E8 AaG 1.13 G8 AY8 1.07A9 NYG 0.91 C9 AGG 1.97 E9 AyG 1.04 G9 AYf 1.58A10 PYG 1.35 C10 AGG 1.19 E10 AYA 0.93 G10 AYi 0.56A11 QYG 0.61 C11 AIG 0.71 E11 AYD 0.94 G11 AY9 0.64A12 RYG 1.95 C12 AKG 1.17 E12 AYE 0.82 G12 AYa 0.54B1 SYG 0.97 D1 ALG 0.81 F1 AYF 1.65B2 TYG 0.95 D2 ANG 1.13 F2 AYH 1.61B3 VYG 0.45 D3 APG 1.29 F3 AYI 1.80B4 WYG 1.33 D4 AQG 1.09 F4 AYK 3.02B5 YYG 1.55 D5 ARG 1.11 F5 AYL 1.90B6 3YG 0.47 D6 ASG 0.92 F6 AYN 1.06B7 4YG 0.54 D7 ATG 0.54 F7 AYP 0.87B8 5YG 0.54 D8 AVG 0.54 F8 AYQ 1.21B9 6YG 1.22 D9 AWG 0.52 F9 AYR 3.21
B10 7YG 2.12 D10 AYG 0.65 F10 AYS 1.32B11 8YG 1.14 D11 A3G 1.31 F11 AYT 1.24B12 fYG 2.14 D12 A4G 0.93 F12 AYV 1.62
references: knottin-RGD (5µM): 0.37, no peptide: 1.54.
228
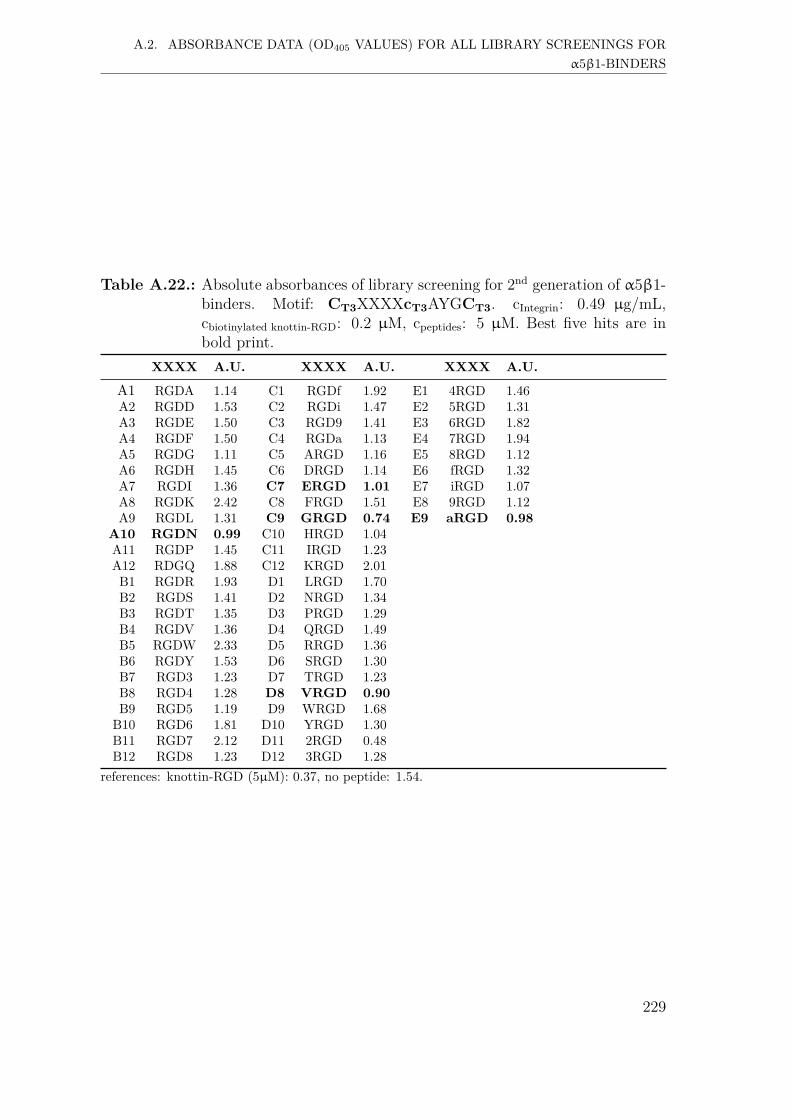
999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.22.: Absolute absorbances of library screening for 2nd generation of α5β1-binders. Motif: CT3XXXXcT3AYGCT3. cIntegrin: 0.49 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXXX A.U. XXXX A.U. XXXX A.U.
A1 RGDA 1.14 C1 RGDf 1.92 E1 4RGD 1.46A2 RGDD 1.53 C2 RGDi 1.47 E2 5RGD 1.31A3 RGDE 1.50 C3 RGD9 1.41 E3 6RGD 1.82A4 RGDF 1.50 C4 RGDa 1.13 E4 7RGD 1.94A5 RGDG 1.11 C5 ARGD 1.16 E5 8RGD 1.12A6 RGDH 1.45 C6 DRGD 1.14 E6 fRGD 1.32A7 RGDI 1.36 C7 ERGD 1.01 E7 iRGD 1.07A8 RGDK 2.42 C8 FRGD 1.51 E8 9RGD 1.12A9 RGDL 1.31 C9 GRGD 0.74 E9 aRGD 0.98
A10 RGDN 0.99 C10 HRGD 1.04A11 RGDP 1.45 C11 IRGD 1.23A12 RDGQ 1.88 C12 KRGD 2.01B1 RGDR 1.93 D1 LRGD 1.70B2 RGDS 1.41 D2 NRGD 1.34B3 RGDT 1.35 D3 PRGD 1.29B4 RGDV 1.36 D4 QRGD 1.49B5 RGDW 2.33 D5 RRGD 1.36B6 RGDY 1.53 D6 SRGD 1.30B7 RGD3 1.23 D7 TRGD 1.23B8 RGD4 1.28 D8 VRGD 0.90B9 RGD5 1.19 D9 WRGD 1.68B10 RGD6 1.81 D10 YRGD 1.30B11 RGD7 2.12 D11 2RGD 0.48B12 RGD8 1.23 D12 3RGD 1.28
references: knottin-RGD (5µM): 0.37, no peptide: 1.54.
229

999999999
A.2. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORα5β1-BINDERS
Table A.23.: Absolute absorbances of library screening for 2nd generation of α5β1-binders. Motif: CT3XXXXXcT3AYGCT3. cIntegrin: 0.49 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXXXX A.U. XXXXX A.U. XXXXX A.U.
A1 GRGDA 0.99 C1 GRGDf 1.09 E1 5RGDS 1.34A2 GRGDD 1.11 C2 GRGDi 1.05 E2 6RGDS 2.48A3 GRGDE 1.31 C3 GRGD9 1.79 E3 7RGDS 2.08A4 GRGDF 1.31 C4 GRGDa 2.60 E4 8RGDS 3.40A5 GRGDG 1.12 C5 ARGDS 1.59 E5 fRGDS 2.00A6 GRGDH 1.17 C6 DRGDS 1.33 E6 iRGDS 1.80A7 GRGDI 1.11 C7 ERGDS 1.58 E7 9RGDS 1.67A8 GRGDK 1.35 C8 FRGDS 1.38 E8 aRGDS 1.53A9 GRGDL 1.54 C9 HRGDS 1.58A10 GRGDN 1.35 C10 IRGDS 1.45A11 GRGDP 1.46 C11 KRGDS 1.38A12 GRDGQ 1.18 C12 LRGDS 1.62B1 GRGDR 1.04 D1 NRGDS 1.29B2 GRGDS 1.05 D2 PRGDS 1.68B3 GRGDT 1.57 D3 QRGDS 2.54B4 GRGDV 1.61 D4 RRGDS 3.82B5 GRGDW 1.70 D5 SRGDS 3.41B6 GRGDY 1.32 D6 TRGDS 3.12B7 GRGD3 1.17 D7 VRGDS 2.48B8 GRGD4 1.38 D8 WRGDS 3.12B9 GRGD5 1.27 D9 YRGDS 1.63
B10 GRGD6 1.94 D10 2RGDS 1.34B11 GRGD7 2.09 D11 3RGDS 1.38B12 GRGD8 1.16 D12 4RGDS 1.59
references: knottin-RGD (5µM): 0.37, no peptide: 1.54.
230

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
A.3. Absorbance data (OD405 values) for all libraryscreenings for αvβ5-binders
A.3.1. First generation screening of 672 random-diversitypeptides
Table A.24.: Absolute absorbances of library screening for 1st generation of αvβ5-binders. Motif: CT3XXXCT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.69 C1 PNV 0.61 E1 PHI 0.64 G1 LPD 0.72A2 AIP 0.70 C2 PYI 0.67 E2 YPS 0.71 G2 RLG 0.85A3 DGY 0.67 C3 TPT 0.66 E3 TRV 0.63 G3 YIY 0.69A4 ILP 0.72 C4 NWG 0.61 E4 VVR 1.98 G4 FRA 0.72A5 VSL 0.75 C5 FTQ 0.62 E5 NST 0.75 G5 LTI 0.78A6 DEW 0.56 C6 QYL 0.81 E6 FLW 0.73 G6 APS 0.64A7 YEE 0.70 C7 WGD 0.60 E7 NSY 0.73 G7 RDP 0.69A8 PLE 0.72 C8 ISY 0.66 E8 ILK 1.09 G8 DHL 0.68A9 KKP 0.78 C9 EIG 0.62 E9 SDQ 0.70 G9 NDA 0.74
A10 QGS 0.73 C10 WFH 0.74 E10 WQY 0.69 G10 DAD 0.72A11 HVK 0.79 C11 NKP 0.64 E11 YYT 0.75 G11 NYA 0.72A12 RNS 0.71 C12 PNE 0.71 E12 NVA 0.75 G12 EDT 0.76B1 DTI 0.71 D1 KTN 0.63 F1 NWQ 0.70 H1 WYV 0.80B2 QAK 0.71 D2 GYE 0.58 F2 TFI 0.74 H2 QSL 0.75B3 WSL 0.69 D3 SYD 0.64 F3 QRG 0.69 H3 TIH 0.83B4 RAA 0.71 D4 IEE 0.54 F4 LDW 0.69 H4 GLA 0.71B5 KHI 0.71 D5 HLQ 0.58 F5 PHL 0.71 H5 LWI 0.96B6 RGS 0.66 D6 GNS 0.58 F6 KID 0.74 H6 GLY 1.02B7 IIV 0.72 D7 IRW 0.57 F7 RQV 0.70 H7 SSG 0.71B8 WFT 0.70 D8 IIG 0.58 F8 KRW 1.86 H8 GWN 0.73B9 RSK 0.75 D9 DFP 0.55 F9 QAD 0.75 H9 SWS 0.76B10 GHF 0.70 D10 PPG 0.60 F10 ERV 0.73 H10 QIH 0.77B11 QDH 0.76 D11 QGI 0.57 F11 YLT 0.81 H11 KQL 0.74B12 HEQ 0.66 D12 FSH 0.62 F12 RWD 0.78 H12 GYG 0.75
231

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.25.: Absolute absorbances of library screening for 1st generation of αvβ5-binders. Motif: CT3XXXCT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.79 C1 PNV 0.73 E1 PHI 0.66 G1 LPD 0.74A2 AIP 1.34 C2 PYI 0.68 E2 YPS 0.68 G2 RLG 3.20A3 DGY 0.67 C3 TPT 0.74 E3 TRV 1.16 G3 YIY 0.79A4 ILP 0.73 C4 NWG 0.80 E4 VVR 1.50 G4 FRA 1.09A5 VSL 0.87 C5 FTQ 0.78 E5 NST 0.64 G5 LTI 1.03A6 DEW 0.74 C6 QYL 0.88 E6 FLW 1.03 G6 APS 0.73A7 YEE 0.74 C7 WGD 0.77 E7 NSY 0.68 G7 RDP 0.71A8 PLE 0.62 C8 ISY 0.76 E8 ILK 2.13 G8 DHL 0.74A9 KKP 3.73 C9 EIG 0.75 E9 SDQ 0.66 G9 NDA 0.95
A10 QGS 0.65 C10 WFH 0.68 E10 WQY 0.70 G10 DAD 0.80A11 HVK 2.68 C11 NKP 1.11 E11 YYT 0.67 G11 NYA 0.84A12 RNS 0.87 C12 PNE 0.68 E12 NVA 1.39 G12 EDT 1.23B1 DTI 0.79 D1 KTN 1.42 F1 NWQ 0.67 H1 WYV 1.02B2 QAK 1.41 D2 GYE 0.69 F2 TFI 0.82 H2 QSL 0.82B3 WSL 0.77 D3 SYD 0.74 F3 QRG 0.73 H3 TIH 1.03B4 RAA 1.30 D4 IEE 0.87 F4 LDW 0.77 H4 GLA 0.83B5 KHI 0.82 D5 HLQ 0.67 F5 PHL 0.61 H5 LWI 1.17B6 RGS 0.97 D6 GNS 0.76 F6 KID 0.72 H6 GLY 0.89B7 IIV 0.82 D7 IRW 1.06 F7 RQV 1.03 H7 SSG 1.04B8 WFT 1.05 D8 IIG 1.35 F8 KRW 3.80 H8 GWN 0.81B9 RSK 1.86 D9 DFP 0.64 F9 QAD 0.72 H9 SWS 0.91
B10 GHF 0.69 D10 PPG 0.72 F10 ERV 0.70 H10 QIH 1.36B11 QDH 0.69 D11 QGI 0.96 F11 YLT 1.08 H11 KQL 0.95B12 HEQ 0.73 D12 FSH 0.70 F12 RWD 0.73 H12 GYG 0.91
references: knottin-RGD (5µM): 0.27, no peptide: 0.77.
232

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.26.: Absolute absorbances of library screening for 1st generation of αvβ5-binders. Motif: CT3XXXcT3RGDCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.80 C1 PNV 0.86 E1 PHI 0.71 G1 LPD 0.75A2 AIP 0.74 C2 PYI 1.22 E2 YPS 0.68 G2 RLG 0.74A3 DGY 0.77 C3 TPT 0.73 E3 TRV 0.96 G3 YIY 1.36A4 ILP 0.79 C4 NWG 0.70 E4 VVR 0.74 G4 FRA 1.68A5 VSL 0.78 C5 FTQ 0.68 E5 NST 0.72 G5 LTI 0.80A6 DEW 0.73 C6 QYL 0.80 E6 FLW 3.30 G6 APS 0.70A7 YEE 2.90 C7 WGD 0.73 E7 NSY 0.70 G7 RDP 0.67A8 PLE 0.77 C8 ISY 0.74 E8 ILK 0.77 G8 DHL 0.80A9 KKP 1.18 C9 EIG 0.66 E9 SDQ 0.70 G9 NDA 0.88
A10 QGS 0.72 C10 WFH 2.22 E10 WQY 0.84 G10 DAD 0.89A11 HVK 0.82 C11 NKP 0.73 E11 YYT 0.92 G11 NYA 1.45A12 RNS 0.75 C12 PNE 0.83 E12 NVA 0.72 G12 EDT 0.88B1 DTI 0.84 D1 KTN 0.80 F1 NWQ 0.73 H1 WYV 2.25B2 QAK 0.79 D2 GYE 0.75 F2 TFI 1.04 H2 QSL 0.79B3 WSL 1.66 D3 SYD 0.74 F3 QRG 0.66 H3 TIH 0.96B4 RAA 0.90 D4 IEE 0.99 F4 LDW 0.76 H4 GLA 0.75B5 KHI 0.86 D5 HLQ 0.70 F5 PHL 0.70 H5 LWI 2.31B6 RGS 0.91 D6 GNS 0.72 F6 KID 0.73 H6 GLY 0.81B7 IIV 0.81 D7 IRW 1.72 F7 RQV 0.75 H7 SSG 0.85B8 WFT 1.74 D8 IIG 1.15 F8 KRW 3.45 H8 GWN 0.79B9 RSK 1.54 D9 DFP 0.67 F9 QAD 0.72 H9 SWS 0.79B10 GHF 0.78 D10 PPG 0.70 F10 ERV 0.72 H10 QIH 0.80B11 QDH 0.74 D11 QGI 0.74 F11 YLT 1.19 H11 KQL 1.03B12 HEQ 0.74 D12 FSH 0.73 F12 RWD 0.80 H12 GYG 0.75
references: knottin-RGD (5µM): 0.27, no peptide: 0.77.
233
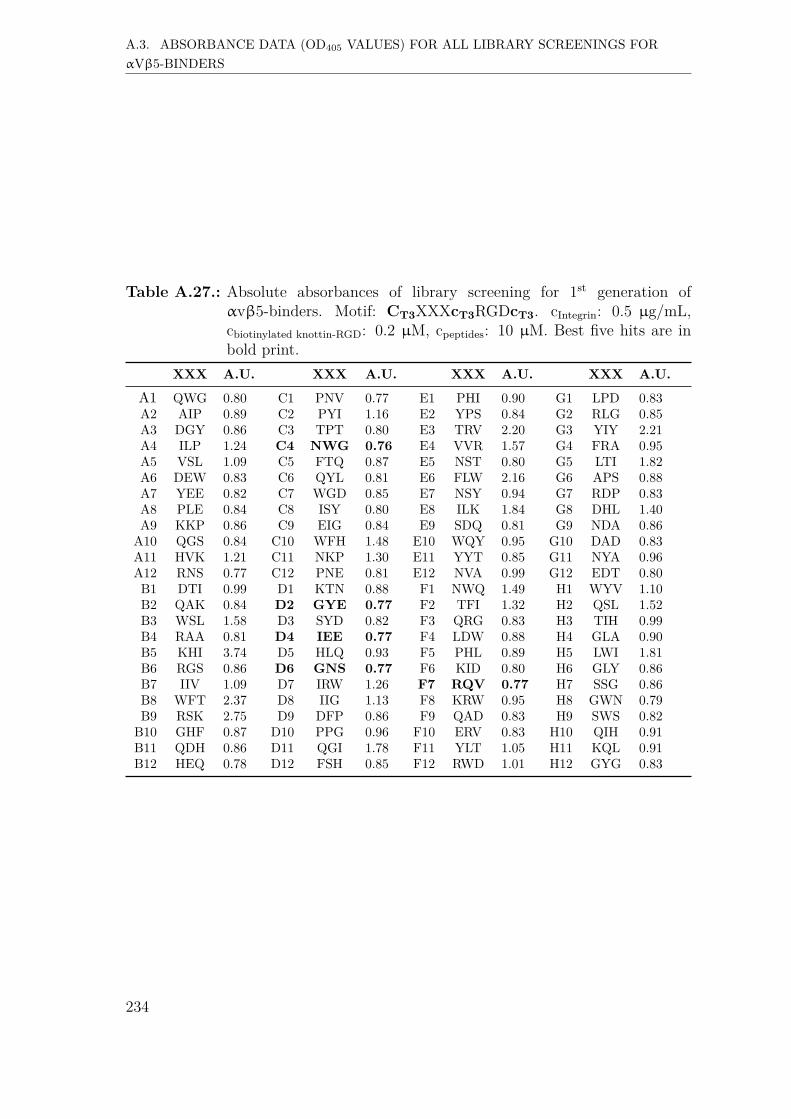
999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.27.: Absolute absorbances of library screening for 1st generation ofαvβ5-binders. Motif: CT3XXXcT3RGDcT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 QWG 0.80 C1 PNV 0.77 E1 PHI 0.90 G1 LPD 0.83A2 AIP 0.89 C2 PYI 1.16 E2 YPS 0.84 G2 RLG 0.85A3 DGY 0.86 C3 TPT 0.80 E3 TRV 2.20 G3 YIY 2.21A4 ILP 1.24 C4 NWG 0.76 E4 VVR 1.57 G4 FRA 0.95A5 VSL 1.09 C5 FTQ 0.87 E5 NST 0.80 G5 LTI 1.82A6 DEW 0.83 C6 QYL 0.81 E6 FLW 2.16 G6 APS 0.88A7 YEE 0.82 C7 WGD 0.85 E7 NSY 0.94 G7 RDP 0.83A8 PLE 0.84 C8 ISY 0.80 E8 ILK 1.84 G8 DHL 1.40A9 KKP 0.86 C9 EIG 0.84 E9 SDQ 0.81 G9 NDA 0.86A10 QGS 0.84 C10 WFH 1.48 E10 WQY 0.95 G10 DAD 0.83A11 HVK 1.21 C11 NKP 1.30 E11 YYT 0.85 G11 NYA 0.96A12 RNS 0.77 C12 PNE 0.81 E12 NVA 0.99 G12 EDT 0.80B1 DTI 0.99 D1 KTN 0.88 F1 NWQ 1.49 H1 WYV 1.10B2 QAK 0.84 D2 GYE 0.77 F2 TFI 1.32 H2 QSL 1.52B3 WSL 1.58 D3 SYD 0.82 F3 QRG 0.83 H3 TIH 0.99B4 RAA 0.81 D4 IEE 0.77 F4 LDW 0.88 H4 GLA 0.90B5 KHI 3.74 D5 HLQ 0.93 F5 PHL 0.89 H5 LWI 1.81B6 RGS 0.86 D6 GNS 0.77 F6 KID 0.80 H6 GLY 0.86B7 IIV 1.09 D7 IRW 1.26 F7 RQV 0.77 H7 SSG 0.86B8 WFT 2.37 D8 IIG 1.13 F8 KRW 0.95 H8 GWN 0.79B9 RSK 2.75 D9 DFP 0.86 F9 QAD 0.83 H9 SWS 0.82
B10 GHF 0.87 D10 PPG 0.96 F10 ERV 0.83 H10 QIH 0.91B11 QDH 0.86 D11 QGI 1.78 F11 YLT 1.05 H11 KQL 0.91B12 HEQ 0.78 D12 FSH 0.85 F12 RWD 1.01 H12 GYG 0.83
234

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.28.: Absolute absorbances of library screening for 1st generation of αvβ5-binders. Motif: CT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 1.92 C1 SWH 1.75 E1 YPS 1.17 G1 EVN 0.65A2 TWN 0.93 C2 RSL 0.75 E2 RVT 0.76 G2 HDA 0.62A3 EQD 0.67 C3 RSY 0.73 E3 RQQ 1.02 G3 NYA 0.60A4 WFH 1.90 C4 ARS 0.76 E4 FWK 2.51 G4 DTI 0.63A5 FPF 2.78 C5 LQP 0.78 E5 RWW 2.74 G5 KPE 0.69A6 KGR 3.93 C6 PTP 0.74 E6 SLL 0.78 G6 KPD 0.66A7 TVD 0.61 C7 NWG 0.46 E7 GRI 0.73 G7 AKN 1.08A8 HSW 0.78 C8 TQS 0.65 E8 QSY 0.70 G8 WPA 1.15A9 ATH 1.27 C9 LSE 0.70 E9 NWQ 0.66 G9 AYG 0.57
A10 NVT 0.62 C10 GSA 0.66 E10 FIH 0.81 G10 VSW 0.74A11 VKI 3.57 C11 PLI 1.78 E11 GFH 0.83 G11 QIH 0.73A12 GVS 0.70 C12 GKF 3.83 E12 TFP 0.76 G12 QLP 0.66B1 DEW 0.61 D1 HYI 1.24 F1 SWK 2.70 H1 GAY 0.71B2 EEQ 0.64 D2 GIK 1.47 F2 WQI 0.81 H2 SHQ 0.75B3 EDE 0.63 D3 QGY 0.68 F3 LGD 0.66 H3 FFR 2.35B4 VTH 0.67 D4 SYD 0.70 F4 SVE 0.67 H4 WVY 0.97B5 RFI 2.38 D5 EEY 0.74 F5 IHV 1.48 H5 RPY 0.68B6 RHD 0.75 D6 QNT 0.74 F6 RWD 0.70 H6 SGN 0.66B7 HWT 0.85 D7 TDQ 0.70 F7 PDI 0.67 H7 ENH 0.65B8 IQW 0.79 D8 WIV 1.61 F8 GRN 0.72 H8 GVA 0.67B9 GRA 0.60 D9 GTS 0.68 F9 WRT 0.72 H9 PYI 1.62B10 KHI 0.78 D10 LAY 0.82 F10 RLR 3.77 H10 WLS 1.08B11 GSR 2.73 D11 HIF 1.52 F11 HGP 0.60 H11 WPE 0.65B12 VVH 1.18 D12 SPH 0.69 F12 TGV 0.67 H12 NST 0.73
references: knottin-RGD (5µM): 0.23, no peptide: 0.74.
235

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.29.: Absolute absorbances of library screening for 1st generation of αvβ5-binders. Motif: cT3RGDCT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 1.04 C1 SWH 1.14 E1 YPS 0.64 G1 EVN 0.57A2 TWN 1.41 C2 RSL 0.73 E2 RVT 3.56 G2 HDA 0.58A3 EQD 0.65 C3 RSY 1.98 E3 RQQ 0.67 G3 NYA 0.59A4 WFH 1.14 C4 ARS 1.22 E4 FWK 2.45 G4 DTI 0.60A5 FPF 0.78 C5 LQP 0.66 E5 RWW 2.86 G5 KPE 0.60A6 KGR 0.76 C6 PTP 0.73 E6 SLL 1.22 G6 KPD 0.64A7 TVD 0.67 C7 NWG 0.64 E7 GRI 0.92 G7 AKN 0.64A8 HSW 0.62 C8 TQS 0.71 E8 QSY 0.77 G8 WPA 0.81A9 ATH 0.71 C9 LSE 0.63 E9 NWQ 0.78 G9 AYG 0.69A10 NVT 0.75 C10 GSA 1.40 E10 FIH 1.82 G10 VSW 1.71A11 VKI 2.00 C11 PLI 0.90 E11 GFH 1.06 G11 QIH 0.65A12 GVS 0.73 C12 GKF 1.11 E12 TFP 1.26 G12 QLP 0.60B1 DEW 0.80 D1 HYI 1.58 F1 SWK 0.66 H1 GAY 0.69B2 EEQ 0.64 D2 GIK 0.80 F2 WQI 0.68 H2 SHQ 0.66B3 EDE 0.63 D3 QGY 0.64 F3 LGD 0.55 H3 FFR 1.46B4 VTH 0.74 D4 SYD 0.64 F4 SVE 0.66 H4 WVY 2.42B5 RFI 3.31 D5 EEY 0.86 F5 IHV 0.83 H5 RPY 0.69B6 RHD 0.74 D6 QNT 0.71 F6 RWD 0.72 H6 SGN 0.66B7 HWT 1.56 D7 TDQ 0.69 F7 PDI 0.67 H7 ENH 0.89B8 IQW 0.93 D8 WIV 1.36 F8 GRN 0.67 H8 GVA 0.78B9 GRA 0.68 D9 GTS 0.65 F9 WRT 3.30 H9 PYI 0.79
B10 KHI 0.87 D10 LAY 0.82 F10 RLR 3.81 H10 WLS 1.58B11 GSR 0.69 D11 HIF 0.78 F11 HGP 0.63 H11 WPE 0.65B12 VVH 2.47 D12 SPH 0.67 F12 TGV 0.89 H12 NST 0.65
references: knottin-RGD (5µM): 0.23, no peptide: 0.74.
236
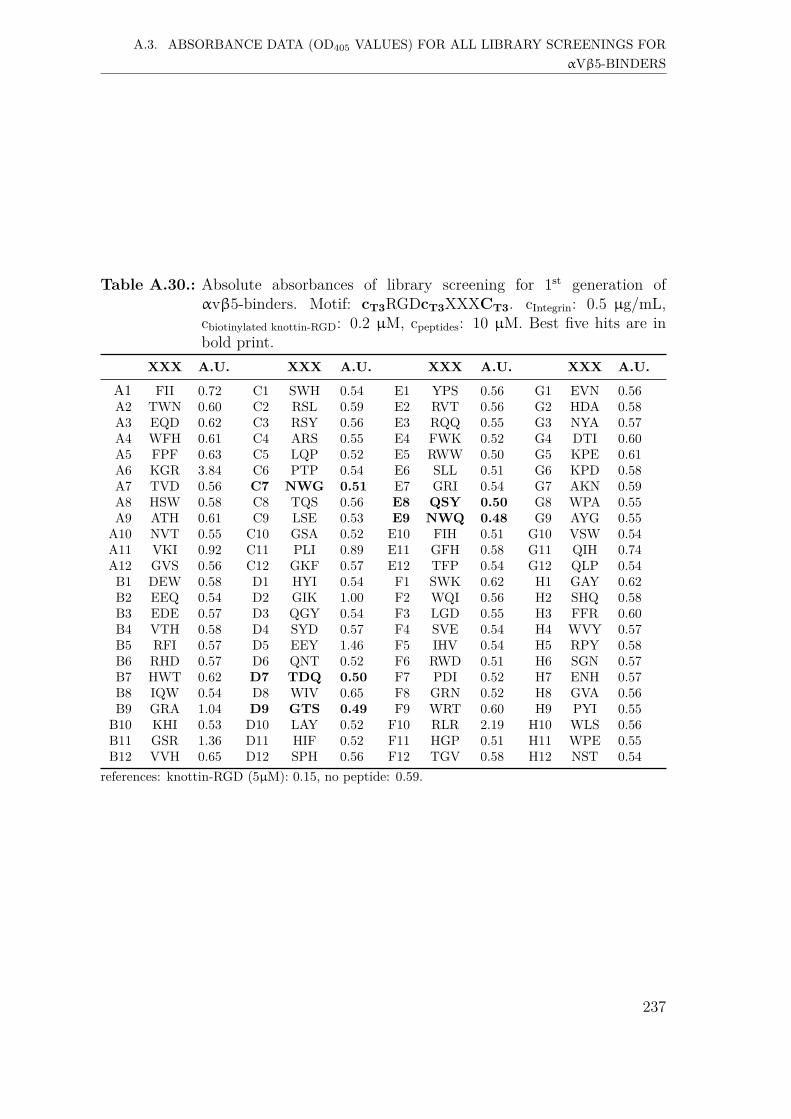
999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.30.: Absolute absorbances of library screening for 1st generation ofαvβ5-binders. Motif: cT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 10 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 FII 0.72 C1 SWH 0.54 E1 YPS 0.56 G1 EVN 0.56A2 TWN 0.60 C2 RSL 0.59 E2 RVT 0.56 G2 HDA 0.58A3 EQD 0.62 C3 RSY 0.56 E3 RQQ 0.55 G3 NYA 0.57A4 WFH 0.61 C4 ARS 0.55 E4 FWK 0.52 G4 DTI 0.60A5 FPF 0.63 C5 LQP 0.52 E5 RWW 0.50 G5 KPE 0.61A6 KGR 3.84 C6 PTP 0.54 E6 SLL 0.51 G6 KPD 0.58A7 TVD 0.56 C7 NWG 0.51 E7 GRI 0.54 G7 AKN 0.59A8 HSW 0.58 C8 TQS 0.56 E8 QSY 0.50 G8 WPA 0.55A9 ATH 0.61 C9 LSE 0.53 E9 NWQ 0.48 G9 AYG 0.55
A10 NVT 0.55 C10 GSA 0.52 E10 FIH 0.51 G10 VSW 0.54A11 VKI 0.92 C11 PLI 0.89 E11 GFH 0.58 G11 QIH 0.74A12 GVS 0.56 C12 GKF 0.57 E12 TFP 0.54 G12 QLP 0.54B1 DEW 0.58 D1 HYI 0.54 F1 SWK 0.62 H1 GAY 0.62B2 EEQ 0.54 D2 GIK 1.00 F2 WQI 0.56 H2 SHQ 0.58B3 EDE 0.57 D3 QGY 0.54 F3 LGD 0.55 H3 FFR 0.60B4 VTH 0.58 D4 SYD 0.57 F4 SVE 0.54 H4 WVY 0.57B5 RFI 0.57 D5 EEY 1.46 F5 IHV 0.54 H5 RPY 0.58B6 RHD 0.57 D6 QNT 0.52 F6 RWD 0.51 H6 SGN 0.57B7 HWT 0.62 D7 TDQ 0.50 F7 PDI 0.52 H7 ENH 0.57B8 IQW 0.54 D8 WIV 0.65 F8 GRN 0.52 H8 GVA 0.56B9 GRA 1.04 D9 GTS 0.49 F9 WRT 0.60 H9 PYI 0.55B10 KHI 0.53 D10 LAY 0.52 F10 RLR 2.19 H10 WLS 0.56B11 GSR 1.36 D11 HIF 0.52 F11 HGP 0.51 H11 WPE 0.55B12 VVH 0.65 D12 SPH 0.56 F12 TGV 0.58 H12 NST 0.54
references: knottin-RGD (5µM): 0.15, no peptide: 0.59.
237

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
A.3.2. Second generation screening of 198 peptides
Table A.31.: Absolute absorbances of library screening for 2nd generation ofαvβ5-binders. Motif: CT3RGDcT3XXXCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXX A.U. XXX A.U. XXX A.U. XXX A.U.
A1 AWG 1.70 C1 iWG 1.80 E1 N4G 1.34 G1 NWV 1.38A2 DWG 1.53 C2 9WG 1.80 E2 N5G 1.25 G2 NWW 1.62A3 EWG 1.73 C3 aWG 1.63 E3 N6G 1.29 G3 NWY 1.61A4 FWG 2.05 C4 nWG 1.53 E4 N7G 1.27 G4 NW3 1.37A5 GWG 1.37 C5 RGD 1.61 E5 N8G 1.65 G5 NW4 1.68A6 HWG 1.31 C6 NAG 1.28 E6 NfG 1.65 G6 NW5 1.59A7 IWG 1.66 C7 NDG 1.54 E7 NiG 1.44 G7 NW6 1.63A8 KWG 1.74 C8 NEG 1.33 E8 N9G 1.48 G8 NW7 1.35A9 LWG 1.65 C9 NFG 1.15 E9 NaG 1.59 G9 NW8 1.37A10 PWG 1.82 C10 NGG 1.38 E10 NwG 1.63 G10 NWf 1.01A11 QWG 1.71 C11 NHG 1.72 E11 NWA 1.57 G11 NWi 1.21A12 RWG 1.81 C12 NIG 1.31 E12 NWD 1.47 G12 NW9 1.21B1 SWG 1.60 D1 NKG 1.44 F1 NWE 1.38 H1 NWa 0.96B2 TWG 1.70 D2 NLG 1.44 F2 NWF 1.84B3 VWG 1.75 D3 NNG 1.10 F3 NWH 1.56B4 WWG 1.86 D4 NPG 1.71 F4 NWI 1.21B5 YWG 1.79 D5 NQG 1.40 F5 NWK 1.70B6 3WG 1.67 D6 NRG 1.43 F6 NWL 1.55B7 4WG 1.53 D7 NSG 1.14 F7 NWN 1.39B8 5WG 1.45 D8 NTG 1.17 F8 NWP 1.22B9 6WG 1.82 D9 NVG 1.45 F9 NWQ 1.42
B10 7WG 1.83 D10 NWG 0.86 F10 NWR 2.24B11 8WG 1.61 D11 NYG 1.16 F11 NWS 1.40B12 fWG 2.03 D12 N3G 1.20 F12 NWT 1.51
references: knottin-RGD (5µM): 0.43, no peptide: 1.68.
238

999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.32.: Absolute absorbances of library screening for 2nd generation of αvβ5-binders. Motif: CT3XXXXcT3NWGCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXXX A.U. XXXX A.U. XXXX A.U.
A1 RGDA 1.58 C1 RGDf 1.90 E1 4RGD 1.44A2 RGDD 1.52 C2 RGDi 1.66 E2 5RGD 1.47A3 RGDE 1.62 C3 RGD9 1.64 E3 6RGD 1.91A4 RGDF 1.83 C4 RGDa 1.45 E4 7RGD 1.94A5 RGDG 1.49 C5 ARGD 1.41 E5 8RGD 1.79A6 RGDH 1.58 C6 DRGD 1.39 E6 fRGD 1.58A7 RGDI 1.64 C7 ERGD 1.54 E7 iRGD 1.55A8 RGDK 1.70 C8 FRGD 1.72 E8 9RGD 1.56A9 RGDL 1.55 C9 GRGD 1.22 E9 aRGD 1.56
A10 RGDN 1.46 C10 HRGD 1.50A11 RGDP 1.70 C11 IRGD 1.85A12 RDGQ 1.50 C12 KRGD 1.93B1 RGDR 1.89 D1 LRGD 1.41B2 RGDS 1.45 D2 NRGD 1.59B3 RGDT 1.48 D3 PRGD 1.68B4 RGDV 1.54 D4 QRGD 1.70B5 RGDW 2.11 D5 RRGD 1.76B6 RGDY 1.90 D6 SRGD 1.52B7 RGD3 1.66 D7 TRGD 1.56B8 RGD4 1.63 D8 VRGD 1.54B9 RGD5 1.62 D9 WRGD 1.72B10 RGD6 2.07 D10 YRGD 1.61B11 RGD7 2.37 D11 2RGD 1.16B12 RGD8 1.52 D12 3RGD 1.43
references: knottin-RGD (5µM): 0.43, no peptide: 1.68.
239
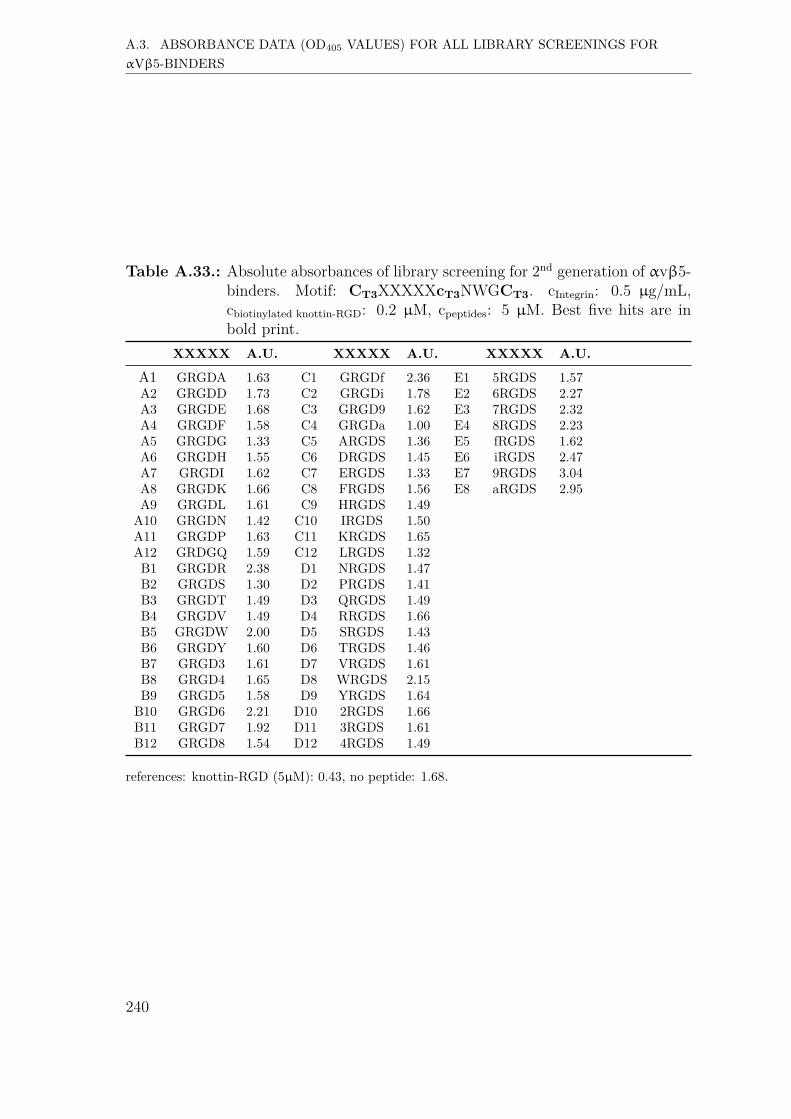
999999999
A.3. ABSORBANCE DATA (OD405 VALUES) FOR ALL LIBRARY SCREENINGS FORαVβ5-BINDERS
Table A.33.: Absolute absorbances of library screening for 2nd generation of αvβ5-binders. Motif: CT3XXXXXcT3NWGCT3. cIntegrin: 0.5 µg/mL,cbiotinylated knottin-RGD: 0.2 µM, cpeptides: 5 µM. Best five hits are inbold print.
XXXXX A.U. XXXXX A.U. XXXXX A.U.
A1 GRGDA 1.63 C1 GRGDf 2.36 E1 5RGDS 1.57A2 GRGDD 1.73 C2 GRGDi 1.78 E2 6RGDS 2.27A3 GRGDE 1.68 C3 GRGD9 1.62 E3 7RGDS 2.32A4 GRGDF 1.58 C4 GRGDa 1.00 E4 8RGDS 2.23A5 GRGDG 1.33 C5 ARGDS 1.36 E5 fRGDS 1.62A6 GRGDH 1.55 C6 DRGDS 1.45 E6 iRGDS 2.47A7 GRGDI 1.62 C7 ERGDS 1.33 E7 9RGDS 3.04A8 GRGDK 1.66 C8 FRGDS 1.56 E8 aRGDS 2.95A9 GRGDL 1.61 C9 HRGDS 1.49A10 GRGDN 1.42 C10 IRGDS 1.50A11 GRGDP 1.63 C11 KRGDS 1.65A12 GRDGQ 1.59 C12 LRGDS 1.32B1 GRGDR 2.38 D1 NRGDS 1.47B2 GRGDS 1.30 D2 PRGDS 1.41B3 GRGDT 1.49 D3 QRGDS 1.49B4 GRGDV 1.49 D4 RRGDS 1.66B5 GRGDW 2.00 D5 SRGDS 1.43B6 GRGDY 1.60 D6 TRGDS 1.46B7 GRGD3 1.61 D7 VRGDS 1.61B8 GRGD4 1.65 D8 WRGDS 2.15B9 GRGD5 1.58 D9 YRGDS 1.64
B10 GRGD6 2.21 D10 2RGDS 1.66B11 GRGD7 1.92 D11 3RGDS 1.61B12 GRGD8 1.54 D12 4RGDS 1.49
references: knottin-RGD (5µM): 0.43, no peptide: 1.68.
240

10101010101010101010
B. List of abbreviations
1Nal L-1-naphthylalanine2Nal L-2-naphtyhlalanineA, Ala L-Alaninea D-AlanineABTS 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)Abu → BAbz 4-Aminobenzoic acidAc AcetylAib → ZAla → Aapprox. approximatelyArg → RASC Adipose-derived stem cell(s)Asn → NAsp → DA.U. Absorption unitsB, Abu L-Aminobutyric acidBCN-NHS Bicyclo[6.1.0]non-4-yn-9-ylmethyl N -succinimidyl carbonatebFGF Basic fibroblast growth factorBMSCs Bone marrow stromal cellsBoc tert-ButyloxycarbonylBSA Bovine serum albuminC, Cys L-Cysteinec D-CysteineCBS Carbonate-buffered salineCD Circular dichroismCDR Complementarity-determining regionCLIPS Chemical linkage of peptides to scaffolds
241

10101010101010101010
CM Confocal microscopyCNS Central nervous systemCOSY-DQF Correlation spectroscopy with double quantum filterCys → CD, Asp L-Aspartic acidDAPI 4’,6-diamidino-2-phenylindoleDCM DichloromethaneD-HCy D-HomocysteineDIPEA N,N -DiisopropylethylamineDMSO Dimethyl sulfoxideD-Pen D-PenicillamineE, Glu L-Glutamic acidECM Extracellular matrixe.g. for example (exempli gratia)ELISA Enzyme-linked immunosorbent assayELP(s) Elastin-like protein(s)ELR(s) Elastin-like recombinamer(s)ESI Electrospray ionisationF, Phe L-Phenylalaninef D-PhenylalanineFITC Fluorescein isothiocyanateFmoc FluorenylmethoxycarbonylFN FibronectinG, Gly GlycineGDA GlutardialdehydeGln → QGlu → EGly → GGSDIM Ground state depletion (followed by) individual molecule (return)H, His L-Histidineh D-HistidineHA Hyaluronic acidHAp HydroxyapatiteHCG HEPES-CBS-GlucoseHEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
242

10101010101010101010
His → HHMBC Heteronuclear multiple-bond correlation spectroscopyHPLC High performance liquid chromatographyHRP → strep-HRPHSQC Heteronuclear single-quantum correlation spectroscopyHUVEC(s) Human umbilical vein endothelial cell(s)I, Ile L-Isoleucinei D-IsoleucineIC50 Half maximal inhibitory concentrationi.e. that is (id est)Ile → IK, Lys L-LysineL, Leu L-LeucineL-HCy L-HomocysteineL-Pen L-PenicillamineLys → KM, Met L-MethioninemAb(s) monoclonal antibody(ies)MALDI-TOF Matrix-assisted laser desorption/ionization time-of-flightMeCN Acetonitrile(α-)MEM Mimimum Essential Medium EagleMet → MMmt Monomethoxytrityl (ε-4-Methoxytrityl)mP2 Scaffold derived from 3,5-bis(bromomethyl)pyridineMpe 3-methyl-pent-3-ylMS Mass spectrometrymT2 Scaffold derived from 1,3-bis(bromomethyl)benzeneN, Asn L-AsparagineNHS N -HydroxysuccinimideNle L-NorleucineNMR Nuclear magnetic resonanceNSOM Near field scanning optical microscopyNva L-NorvalineOD Optical densityoT2 Scaffold derived from 1,2-bis(bromomethyl)benzene
243

10101010101010101010
P, Pro L-ProlinePALM Photoactivated localization microscopyPB Phosphate bufferPbf 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonylPBS Phosphate-buffered salinePEG Polyethylene glycolPCL PolycaprolactonePFA ParaformaldehydePHEMA Poly(2-hydroxyethyl methacrylate)PIC(s) Polyisocyanopeptide(s)PLL PolylysinePLLA Poly-small L-lactidePPEGMA Poly(poly(ethylene glycol) methyl ether monomethacrylate)PPI Protein-protein interactionsPro → PPS PolystyrenepT2 Scaffold derived from 1,4-bis(bromomethyl)benzenePU PolyurethaneQ, Gln L-Glutamineq D-GlutamineR, Arg L-ArginineRGD Trieptide of L-argininge, glycine and L-aspartic acidROESY Rotating frame nuclear Overhauser effect spectroscopyS, Ser L-SerineSAM Self-assembled monolayerSer → SSMLM Single molecule localization based microscopySPFS Surface plasmon-enhanced fluorescence spectroscopySPPS Solid-phase peptide synthesisSPR Surface plasmon resonanceSRM Super resolution microscopyS-S Index for disulfideSTED Stimulated emission depletionSTORM Stochastic optical reconstruction microscopyStrep-HRP Streptavidin-horseradish peroxidase
244

10101010101010101010
T, Trp L-ThreonineT3 Scaffold derived from 1,3,5-tris(bromomethyl)benzenetBu tert-ButylTFA Trifluoroacetic acidTGF Transforming growth factorThr → TTIRF Total internal reflection fluorescenceTOCSY Total correlation spectroscopyTrp → WTrt Triphenylmethyl (Trityl)UPLC Ultra high performance liquid chromatographyUV UltravioletV, Val L-ValineVEGF Vascular endothelial growth factorvs. versusvWF von Willebrand factorW, Trp L-Tryptophanw D-TryptophanZ (Aib) L-Aminoisobutyric acid
245

10101010101010101010

10101010101010101010
C. Acknowledgements
People told me that this is the best and easiest part to write, since you are at theend of your thesis. You do not need to do literature research for this chapter. Youjust have to think of all the people that supported you during your PhD project,professionally and privately. Being happy making it to this part of the thesis, I hopeI will acknowledge everyone who had a share in making this work possible in anappropriate way.
My first gratitudes belong to my supervisors Prof. Dr. Martin Möller and Prof.Dr. Peter Timmerman.Herr Prof. Möller, ich bedanke mich sehr herzlich bei Ihnen für die Übernahmeder Erstbetreuung. Vor dem Hintergrund Ihrer vielfältigen Aufgaben und Verpflich-tungen als Direktor des DWI’s schätze ich es sehr, dass Sie mir die Chance zu einerPromotion an der RWTH gegeben haben. Peter, you gave me the chance to per-form my PhD studies in a biotech company and made it possible to work on aninteresting and innovative research topic. Your supervision was great. Not only didyou provide very valuable input from your long-term experience in peptide-basedprotein mimicry, but also managed to find motivating words when times were hard.I cannot thank you enough for all the work you invested with respect to my thesis,my papers and the BIOGEL administration. Let us hope the RGD bicycles will finda useful application someday. Herr Prof. Weinhold, vielen Dank, dass Sie sichdie Zeit genommen haben, und sich als Prüfer zur Verfügung gestellt haben. HerrProf. Richtering, Ihnen möchte ich für die Übernahme des Prüfungsvorsitzes her-zlich danken.
I would like to thank my great colleagues at Pepscan for the trouble-free collabo-ration and all the things I could learn during the entire 3 years.Wim M, thank you for hiring me and for fruitful discussions about future careerperspectives. Sylvia, thank you a lot for your support, be it related to administra-
247

10101010101010101010
tive questions especially at the beginning of the three years or organizing peptidedeliveries to my collaborators. Dropathie, Joop en Joshua, van harte bedanktvoor jullie hulp met het introduceeren van ELISA en andere dingen, bijvoorbeeldhet maken van PBST buffer en aminozuuranalyse. Richard and Thomas, thankyou guys for introducing me to the HPLC and UPLC systems, and the rest of theAnalytics team for the really nice atmosphere when working in your lab. WimS, Nicole, Jeroen and Dana, thank you all so much sharing your experience inpeptide chemistry, which I really appreciate. Sonja, thank you for teaching me howto operate the Syro machines and your help finding accommodation close to work.Anneloes, thank you your support in operating the Syro’s and ordering compoundsfor my research. Nicolas, thank you for sharing your experience in chemistry andprogramming, and being a good friend. Hope you will be happy in Southern France.Katja and JW, thank you for all your great scientific support, be it related tobiological questions or generating peptide libraries. Katja, thank you for beinga good friend. All the best for your future in Amsterdam and for finalizing yourdream appartment. Mike, vielen Dank für wisschenschaftliche Diskussionen undfür das ein oder andere Bier. Bin froh, einen neuen Freund gewonnen zu haben.Mal schauen, welche wilden Tiere uns auf der nächsten Reise begegnen... Pelin,hope you are doing great. It was great to meet you. All the best for your future!Gaston, similar science, similar taste of music. We should have met earlier. Hope,your tricycles and tetracycles will conquer the peptide therapeutics world. Thankyou Evert, Steffen, Jordi and Remy for the good time when we shortly workedtogether.
I was very lucky and pleased having the opportunity to work on projects in dif-ferent fields of science. Various very commited and smart people had a share inmaking this possible.
I received great support from my colleagues at DWI Aachen, where I spent thefirst of my secondments.Laura, thank you very much for suggesting Pepscan as an institution to performthe PhD studies within the BIOGEL network. I am impressed of the level of yourcommitment to your work and your students, and I really hope your hydrogelswill regenerate the spinal chord of a human being someday. Sheila, vielen Dank,dass du mir das Kultivieren von Zellen gezeigt hast und für all deine praktische
248

10101010101010101010
Hilfe während meiner Zeit bei euch. Christopher, du hast mich sehr unterstütztwährend meiner Zeit am DWI, auch privat. Ich danke dir dafür und wünsche diralles Gute mit Maggie. Du bist ein cooler Typ! Sitara and Winnie, thank youfor the cooperation, and for making the RGD bicycles a part of your research. Ly-dia, vielen Dank für deinen Support bei administrativen Fragen. Du hilfst vielenStudenten und das ist großartig. Frau Fuge, danke, dass Sie sich so souverän umVersicherungsfragen gekümmert haben und sich nicht von meiner komplizierten Sit-uation haben abschrecken lassen.
I spent my next secondment at Technical Protein Nanobiotechnology in Valladolid.Israel, thank you for your help in organizing this secondment and your supervisionduring my time in Valladolid. Filippo and Leander, you supported me a lot in thelab, be it by providing material, introducing me into different analytical methodsor facilitating communiation with your excellent knowledge of Spanish. Leander,I will never forget our trip to Porto. Remember, not every car fits every parkinggarage. I wish you happiness and joy with your family. Filippo, we had a goodtime in Northern Spain. All the best for you and Sara! Rocío and Alicia, manythanks for your lab and administrative support. Nora, thank you for performingthe MALDI-TOF MS analysis and providing the spectra.
During my third secondment at the National Cancer Institute in Amsterdam, Ireceived great support as well.Kees, I met you as a straightforward and passionate scientist. Thank you so muchfor the opportunity to learn confocal microscopy in your group and for sharing yourlong-year experience in scientific discussions. I really appreciate it. Jeffrey, youintroduced me into the lab facilities, took care of the cell culture various times, andalways helped if I had any questions or challenges. Thank you for everything. Leilaand Bram, thank you for your support and the scientific discussions.
My final secondment was in the group of molecular materials at RU Nijmegen.Paul Kouwers, I really appreciate the great cooperation and supervision during theproject. Thank you for this. You are one of the most “normal” scientists I have met,smart, with a good sense of humor, and able to give great scientific presentations.Max (Kaizheng), thank you so much for the good cooperation in our project,and all your support, e.g. during the rheological experiments or in the creation of
249

10101010101010101010
confocal images. I hope you will gain many more interesting insights into the prop-erties of bicycle-hydrogel conjugates. Good luck, my friend. Paula W, thank youfor all the administrative support during my project at RU. Egbert Oosterwijkand Dorien (Radboud UMC), thank you for providing the space and support ourexperiments with the adipose-derived stem cells. Dennis Löwik, thank you verymuch for unselfishly taking the time to show me the use of the CD spectrometer.
I would also express my gratitude to the co-authors of my papers that were notmentioned yet, and without whom the quality of the work would not be the same.Paul B. White, thank you for performing and analyzing NMR experiments. It is apleasure to have an NMR-expert like you part of the collaboration. Moreover, thankyou, Jakub and Vanessa, for performing and analyzing the SPFS experiments thatvalidated that the bicycles are indeed strong and selective integrin binders.
I would like to thank the European Union and all European tax payers for fi-nancing the Marie Curie Training Network, and for giving me the opportunity toperform innovative research in a foreign country. Long live the peace!
To all my colleagues in the BIOGEL network, Filippo, Leander, Max, Sitara,Arturo, Nestor, Evgenia, Luis, Jenny, Marcel, Daria, Paula, Melanie andDelphine, thank you all for your contributions to this network. The team was aperfect fit and I really enjoyed cooperating with you. I will always remember thegood times during workshops and meetings, and hope to meet you guys again soon.
Esther, Marcel, Edo en Jasper, ik had echt een heel mooie tijd met jullie bijde LTTC. Spannende wedstrijden, goed tafeltennissen en daarna lekker biertjes engezelligheid. Bedankt daarvoor en veel success met de volgende competities!
Finally, I would like to express my gratitudes to my dearest and nearest.Ela, du warst in den gut 3 Jahren der wichtigste Mensch und beste Freundin fürmich. Unsere Gespräche über unsere Arbeitstage haben mir geholfen, die positivenErlebnisse zu feiern und die negativen zu verarbeiten. Du hast mir Halt gegeben,wenn es mir nicht gut ging, hast mir Mut gemacht, wenn ich aufgeben wollte, hastmich auf Kurs gebracht, wenn es notwendig wurde, und mich oft bestärkt in meinenEntscheidungen, auch wenn sie teilweise hart waren. Ich bin dankbar, dass ich dich
250

10101010101010101010
habe.Mama & Papa, danke, dass ihr immer für mich da wart. Papa, du warst mitdeinem Fleiß und deinem uneigennützigen Einsatz Vorbild für uns viele. Wo auchimmer du bist, wünsche ich dir Frieden und gute Gesellschaft bis wir uns wiederse-hen. Mama, wir konnten in all dieser Zeit immer miteinander reden, auch wenn wiruns selten gesehen haben. Ich bin beeindruckt von deiner Stärke und auch wenn eskomisch klingt, es als Sohn zu schreiben, aber ich bin stolz auf dich. Danke, dass esdich gibt! Maleen & Alex, ich bin froh euch als Geschwister zu haben. Bleibt sowie ihr seid, verfolgt eure Ziele und werdet glücklich. Ich habe euch sehr lieb. Oma& Opa, ich habe höchsten Respekt vor eurer Lebensleistung. Bleibt gesund, munterund noch lange glücklich miteinander. Oma & Opa Bayern, für eure offene undstreitbare Art respektiere ich euch. Ich wünsche euch Glück und alles Gute.Julian, ich bin froh dich als besten Kumpel zu haben und das schon seit fast 20Jahren (Billigcola. Jo Jakko, ik kom voor de printer. Footswing. PES.). Mann,werden wir alt. Das Highlight in den letzten Jahren war sicherlich deine Hochzeitin Kairo. Dass wir jetzt wieder in der Nähe wohnen, finde ich klasse, auch wenn essicherlich nicht für ewig anhält. Ich hoffe, dass wir noch lange Freunde bleiben undwünsche dir und deiner Fayrouz alles erdenklich Gute.Nils, du bist seit meiner Zeit bei den Brünnis ein sehr guter Freund. Auch wennwir uns nicht oft sehen, waren und sind mir die Treffen und Gespräche mit dirund Michael sehr wichtig. Heja BVB! (Entschuldige bitte, irgendwo musste ich dieBorussia erwähnen).Norbert, hattest du mir als Lehrer noch Beharrlichkeit vermittelt, sind du und Pe-tra nach den Ereignissen Mitte 2015 gefühlt zu einem Teil meiner Familie geworden.Dass ihr beide für unsere Familie da wart, hat mir geholfen, die Flinte nicht ins Kornzu werfen. Dass wir dazu auch noch auf einer Wellenlänge sind, ist ein Glücksfallfür mich.
I thank everyone who contributed to the success of this work by whatevermeans, especially those kind people I did not acknowledge personally.
251

10101010101010101010

10101010101010101010
Eidesstattliche Erklärung
Dominik Bernhagen erklärt hiermit, dass diese Dissertation und die darin dargelegtenInhalte die eigenen sind und selbstständig, als Ergebnis der eigenen originärenForschung, generiert wurden.
Hiermit erkläre ich an Eides statt
1. Diese Arbeit wurde vollständig oder größtenteils in der Phase als Doktoranddieser Fakultät und Universität angefertigt;
2. Sofern irgendein Bestandteil dieser Dissertation zuvor für einen akademischenAbschluss oder eine andere Qualifikation an dieser oder einer anderen Institutionverwendet wurde, wurde dies klar angezeigt;
3. Wenn immer andere eigene- oder Veröffentlichungen Dritter herangezogen wur-den, wurden diese klar benannt;
4. Wenn aus anderen eigenen- oder Veröffentlichungen Dritter zitiert wurde, wurdestets die Quelle hierfür angegeben. Diese Dissertation ist vollständig meine eigeneArbeit, mit der Ausnahme solcher Zitate;
5. Alle wesentlichen Quellen von Unterstützung wurden benannt;
6. Wenn immer ein Teil dieser Dissertation auf der Zusammenarbeit mit anderenbasiert, wurde von mir klar gekennzeichnet, was von anderen und was von mir selbsterarbeitet wurde;
7. Kein Teil dieser Arbeit wurde vor deren Einreichung veröffentlicht.
253
Copyright © 2022 FDOKUMEN








![Preparation of [5 + 6]-, [6 + 6]-, and [6 + 7]Bicyclic Guanidines fromC,C'Bis(iminophosphoranes](https://static.fdokumen.com/doc/165x107/631f2da3d10f1687490fada7/preparation-of-5-6-6-6-and-6-7bicyclic-guanidines-fromccbisiminophosphoranes.jpg)











