
Axial D3-trishomocubane derivatives with potential: Dreams or reality?
29
Send Reprints Orders on [email protected] 2632 Current Organic Chemistry, 2012, 16, 2632-2660 1385-2728/12 $58.00+.00 © 2012 Bentham Science Publishers Axial D 3 -trishomocubane Derivatives with Potential: Dreams or Reality? Dmitry I. Sharapa, 1 Igor A. Levandovskiy 2 and Tatyana E. Shubina *,1 1 Computer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Nägelsbachstr. 25, 91052 Erlangen, Germany 2 Department of Organic Chemistry, Kiev Polytechnic Institute, pr. Pobedy 37, 03056 Kiev, Ukraine Abstract: The D3-trishomocubane is a unique high symmetrical chiral stabilomeric compound. Its derivatives have great potential to be used as scaffolds for drugs, in structure-oriented design, asymmetric catalysis, light-driven systems and much more. Axial substitution is the most useful method for this purpose but the least studied. This paper critically analyzes various synthetic strategies aiming at intro- ducing substituents into C 2 /C 9 positions of D3-trishomocubane. Herein we cover formation of trishomocubane skeleton and rearrange- ment of Cs-trishomocubanes. Based on a comprehensive retrosynthetic analysis a general synthetic scheme is proposed. Keywords: C s -trishomocubane, D 3 -trishomocubane, tertiary derivatives, axial derivatives, retrosynthetic analysis, cyclopentadienes, quinones. INTRODUCTION D 3 -trishomocubane (pentacyclo[6.3.0.0 2,6 .0 3,10 .0 5,9 ]undecane, THC) is one of the few molecules that has D 3 symmetry, yet it is chiral and exists in the form of two optical isomers. Moreover, D 3 - trishomocubane is the most stable molecule among the cage struc- tures with the empirical formula C 11 H 14 . This hydrocarbon, like adamantane, possesses specific proper- ties, viz., a relatively large cage size (its diameter is 5.5Å), high lipophilicity and conformational rigidity. The two latter properties are especially valuable for novel drug design. Additionally, its axially substituted derivatives (2-, and/or 2,9-) retain three-fold symmetry and can be potentially used as nanode- vices. Analysis of the two existing reviews on the chemistry of D 3 - trishomocubane [1,2] shows that these derivatives remain virtually unexplored. This review aims at revealing perspectives of such compounds and possible synthetic pathways (with retrosynthetic analysis) to obtain them and intends to challenge and stimulate interest to this topic. (±)-D 3 -trishomocubane was first synthesized in 1970 [3], the pure R-(+)- and S-(–)- forms were synthesized few years later [4-8]. The rotation angle [] D 165° is one of the largest among chiral cage hydrocarbons [much larger than for C 2 -bishomocubane ([] D 58°) [9], but smaller than for twistane ([] D 414-440°) [10- 13] and tribblatane ([] D 617-621°) [14]. Resolution of racemates of various D 3 -trishomocubane derivatives can be via different methods (Table 1). Additionally, monoketone and alcohol can be converted from one to another by microbiological methods stereoselectively (but not stereospecifically, the ee depends on the system) [16-18]. Thus, it seems that it is possible to find appropriate conditions for the resolution of any synthesized compound. Also it has been found 5 9 10 3 6 8 1 2 11 4 7 (–) (+) Fig. (1). Different graphical representations of the D3-trishomocubane isomers. * Address correspondence to this authors at the Computer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Nägelsbachstr. 25, 91052 Erlangen, Germany; Tel: +49(0)9131-85 26580; Fax: +49(0)9131-85 26565; E-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Axial D3-trishomocubane derivatives with potential: Dreams or reality?

Send Reprints Orders on [email protected]
2632 Current Organic Chemistry, 2012, 16, 2632-2660
1385-2728/12 $58.00+.00 © 2012 Bentham Science Publishers
Axial D3-trishomocubane Derivatives with Potential: Dreams or Reality?
Dmitry I. Sharapa,1 Igor A. Levandovskiy2 and Tatyana E. Shubina*,1
1Computer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Nägelsbachstr. 25, 91052 Erlangen, Germany
2Department of Organic Chemistry, Kiev Polytechnic Institute, pr. Pobedy 37, 03056 Kiev, Ukraine
Abstract: The D3-trishomocubane is a unique high symmetrical chiral stabilomeric compound. Its derivatives have great potential to be
used as scaffolds for drugs, in structure-oriented design, asymmetric catalysis, light-driven systems and much more. Axial substitution is
the most useful method for this purpose but the least studied. This paper critically analyzes various synthetic strategies aiming at intro-
ducing substituents into C2/C9 positions of D3-trishomocubane. Herein we cover formation of trishomocubane skeleton and rearrange-
ment of Cs-trishomocubanes. Based on a comprehensive retrosynthetic analysis a general synthetic scheme is proposed.
Keywords: Cs-trishomocubane, D3-trishomocubane, tertiary derivatives, axial derivatives, retrosynthetic analysis, cyclopentadienes, quinones.
INTRODUCTION
D3-trishomocubane (pentacyclo[6.3.0.02,6.03,10.05,9]undecane, THC) is one of the few molecules that has D3 symmetry, yet it is chiral and exists in the form of two optical isomers. Moreover, D3-trishomocubane is the most stable molecule among the cage struc-tures with the empirical formula C11H14.
This hydrocarbon, like adamantane, possesses specific proper-ties, viz., a relatively large cage size (its diameter is 5.5Å), high lipophilicity and conformational rigidity. The two latter properties are especially valuable for novel drug design.
Additionally, its axially substituted derivatives (2-, and/or 2,9-) retain three-fold symmetry and can be potentially used as nanode-vices.
Analysis of the two existing reviews on the chemistry of D3-trishomocubane [1,2] shows that these derivatives remain virtually unexplored. This review aims at revealing perspectives of such
compounds and possible synthetic pathways (with retrosynthetic analysis) to obtain them and intends to challenge and stimulate interest to this topic.
(±)-D3-trishomocubane was first synthesized in 1970 [3], the pure R-(+)- and S-(–)- forms were synthesized few years later [4-8]. The rotation angle [�]D�165° is one of the largest among chiral cage hydrocarbons [much larger than for C2-bishomocubane ([�]D�58°) [9], but smaller than for twistane ([�]D�414-440°) [10-13] and tribblatane ([�]D�617-621°) [14]. Resolution of racemates of various D3-trishomocubane derivatives can be via different methods (Table 1).
Additionally, monoketone and alcohol can be converted from one to another by microbiological methods stereoselectively (but not stereospecifically, the ee depends on the system) [16-18]. Thus, it seems that it is possible to find appropriate conditions for the resolution of any synthesized compound. Also it has been found
5
9 10
3
6
8 1
2
11
4
7
(–)
(+)
Fig. (1). Different graphical representations of the D3-trishomocubane isomers.
* Address correspondence to this authors at the Computer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Nägelsbachstr. 25, 91052 Erlangen, Germany; Tel: +49(0)9131-85 26580; Fax: +49(0)9131-85 26565; E-mail: [email protected]
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2633
that the use of enantiomerically pure substrates allow one enanti-omer of D3-trishomocubane derivative to be obtained [19].
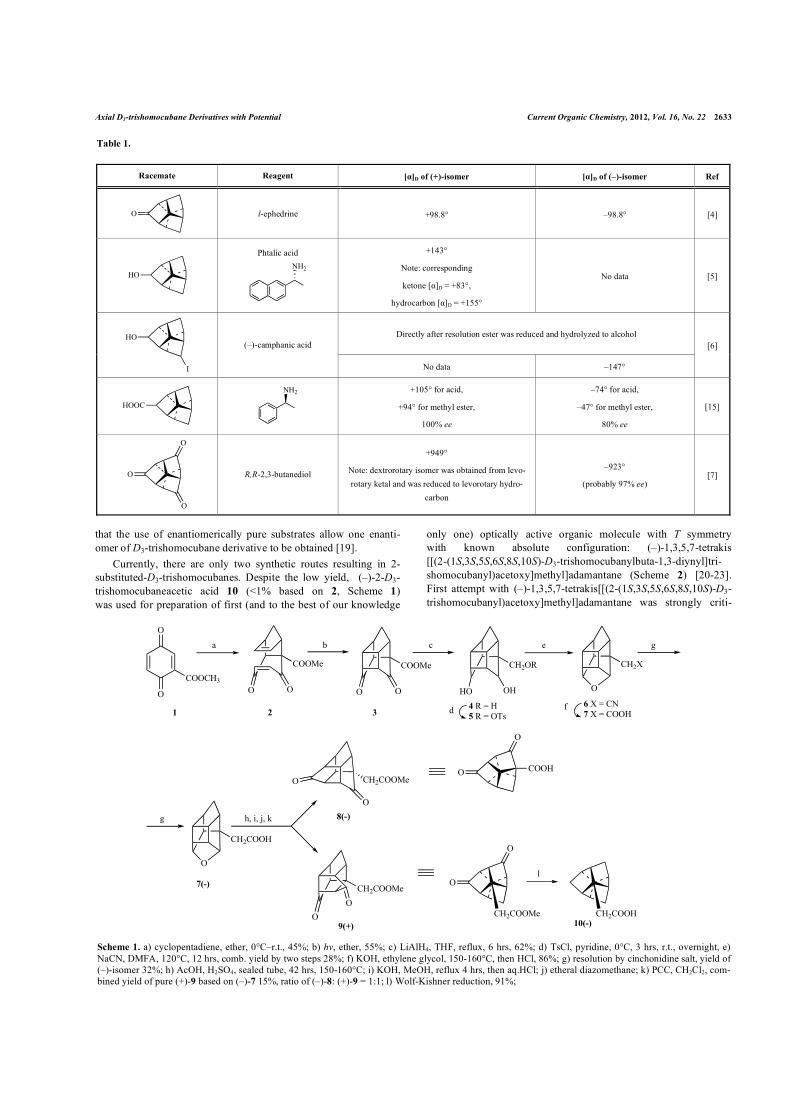
Currently, there are only two synthetic routes resulting in 2-substituted-D3-trishomocubanes. Despite the low yield, (–)-2-D3-trishomocubaneacetic acid 10 (<1% based on 2, Scheme 1) was used for preparation of first (and to the best of our knowledge
only one) optically active organic molecule with T symmetry with known absolute configuration: (–)-1,3,5,7-tetrakis [[(2-(1S,3S,5S,6S,8S,10S)-D3-trishomocubanylbuta-1,3-diynyl]tri-shomocubanyl)acetoxy]methyl]adamantane (Scheme 2) [20-23]. First attempt with (–)-1,3,5,7-tetrakis[[(2-(1S,3S,5S,6S,8S,10S)-D3-trishomocubanyl)acetoxy]methyl]adamantane was strongly criti-
Table 1.
Racemate Reagent [�]D of (+)-isomer [�]D of (–)-isomer Ref
O
l-ephedrine +98.8° –98.8° [4]
HO
Phtalic acid
NH2
+143°
Note: corresponding
ketone [�]D = +83°,
hydrocarbon [�]D = +155°
No data [5]
Directly after resolution ester was reduced and hydrolyzed to alcohol HO
I
(–)-camphanic acid
No data –147°
[6]
HOOC
NH2
+105° for acid,
+94° for methyl ester,
100% ee
–74° for acid,
–47° for methyl ester,
80% ee
[15]
O
O
O
R,R-2,3-butanediol
+949°
Note: dextrorotary isomer was obtained from levo-
rotary ketal and was reduced to levorotary hydro-
carbon
–923°
(probably 97% ee) [7]
O
O
COOCH3
OO
COOMe
OO
COOMe
OHHO
CH2OR
O
CH2X
6 X = CN
7 X = COOH
a b c e g
f
O
CH2COOH
O
CH2COOMeO
CH2COOMe
O
O CH2COOMe
COOH
O
O
O
O
CH2COOH
g h, i, j, k
l
4 R = H
5 R = OTsd321
7(-)
8(-)
9(+) 10(-)
Scheme 1. a) cyclopentadiene, ether, 0°C–r.t., 45%; b) hv, ether, 55%; c) LiAlH4, THF, reflux, 6 hrs, 62%; d) TsCl, pyridine, 0°C, 3 hrs, r.t., overnight, e) NaCN, DMFA, 120°C, 12 hrs, comb. yield by two steps 28%; f) KOH, ethylene glycol, 150-160°C, then HCl, 86%; g) resolution by cinchonidine salt, yield of (–)-isomer 32%; h) AcOH, H2SO4, sealed tube, 42 hrs, 150-160°C; i) KOH, MeOH, reflux 4 hrs, then aq.HCl; j) etheral diazomethane; k) PCC, CH2Cl2, com-bined yield of pure (+)-9 based on (–)-7 15%, ratio of (–)-8: (+)-9 = 1:1; l) Wolf-Kishner reduction, 91%;

2634 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
cized [24], also in both papers 2-derivatives of (1S,3S,5S,6S, 8S,10S)-D3-trishomocubane were referred as (1S,3S,5R,6S,8R,10R)-D3-trishomocubane [24].
Also 2-substituted-D3-trishomocubanes (25 - 27, Scheme 3) were used for NMR relaxation studies, as they are 'pseudo-symmetric tops' ”since complete characterization of the dynamic behaviour of completely asymmetric molecules in solution is very
difficult” [25]. The pathway depicted on Scheme 3 results in much better yield (<7% based on 1, can be improved up to �14%), but the procedure still looks rather complicated and some yields are poorly reproduced (see below) [25].
Until now, no synthetic methods which obtain 2,9-substituted-D3-trishomocubane were reported, or concepts on how to synthesize it. Yet such compounds can be used as:
CH2OH
HOH2C
HOH2C
CH2OH
C CH
HC C
HC C
C CH
C C-Br
R
R
R
R
CH2COOH
-C C-C CR=
11 ([�]D = +19.6°)
unstable
8-step procedure
9-step procedure
a
R+
10 ([�]D = –68.2°)
12 13
14 15
Scheme 2. a) CuCl, aq. EtNH2, THF, mixture contains unreacted 11:15:14 in ratio � 2:4,5:3, respectively; yield of pure 14 (after recrystallization) - 13%
OO OHHO
X X
OHHO
X
OHO
OHO
X
X
+
HO
OH
X
X
+
8%
66%
18
19 21
20
X = COOMe
a b c d
2 16 17
OH
COOMe COOH
OHOH
COOH
OH
COOMe
O
COOMe COOH COOMe CH2OH
e f g
g h i j
20 22 23
24 25 26 27
Scheme 3. a) NaBH4, CeCl3·7H2O, MeOH, –10°C – r.t., 12 hrs., 61%); b) hv, acetone, 81%; c) PDC, CH2Cl2, r.t., overnight, 88%; d) Wolff-Kishner reduc-tion, then CH2N2, Et2O; e) glacial AcOH, conc. H2SO4, 150°C, 36 hrs., then KOH/EtOH, reflux, 3.5 hrs., 85%; f) (MeO)2SO2, K2CO3, acetone, reflux, 4 hrs, 95%; g) PCC, CH2Cl2, r.t., 5 hrs., 80%; h) Wolff-Kishner reduction, 91%; i) (MeO)2SO2, K2CO3, acetone, reflux, 4 hrs, 96%; j) LiAlH4, THF, 0°C, then reflux, 4 hrs, 88%.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2643
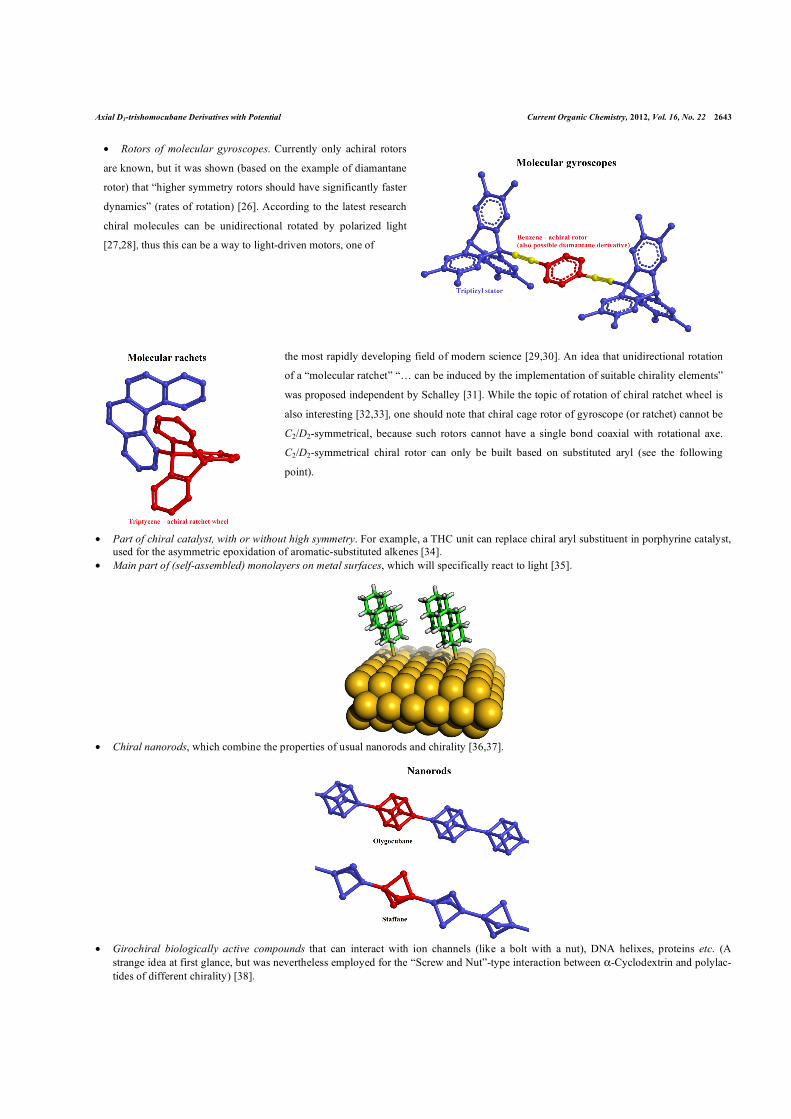
• Rotors of molecular gyroscopes. Currently only achiral rotors
are known, but it was shown (based on the example of diamantane
rotor) that “higher symmetry rotors should have significantly faster
dynamics” (rates of rotation) [26]. According to the latest research
chiral molecules can be unidirectional rotated by polarized light
[27,28], thus this can be a way to light-driven motors, one of
the most rapidly developing field of modern science [29,30]. An idea that unidirectional rotation
of a “molecular ratchet” “… can be induced by the implementation of suitable chirality elements”
was proposed independent by Schalley [31]. While the topic of rotation of chiral ratchet wheel is
also interesting [32,33], one should note that chiral cage rotor of gyroscope (or ratchet) cannot be
C2/D2-symmetrical, because such rotors cannot have a single bond coaxial with rotational axe.
C2/D2-symmetrical chiral rotor can only be built based on substituted aryl (see the following
point).
• Part of chiral catalyst, with or without high symmetry. For example, a THC unit can replace chiral aryl substituent in porphyrine catalyst,
used for the asymmetric epoxidation of aromatic-substituted alkenes [34].
• Main part of (self-assembled) monolayers on metal surfaces, which will specifically react to light [35].
• Chiral nanorods, which combine the properties of usual nanorods and chirality [36,37].
• Girochiral biologically active compounds that can interact with ion channels (like a bolt with a nut), DNA helixes, proteins etc. (A
strange idea at first glance, but was nevertheless employed for the “Screw and Nut”-type interaction between �-Cyclodextrin and polylac-
tides of different chirality) [38].
2644 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
PLLA - poly(L-lactide); PDLA - poly(D-lactide)
Table 2. Relative stabilities of D3-trishomocubane +C
1,
+C
2 and
+C
4 cations and �C
1, �C
2 and
�C4
radicals (�E + ZPE, kcal/mol, relative to +C
1 and �C
4,
respectively).
Cations Radicals
+
+C1=+C3=+C6
+
+C2=+C9
+
+C4=+C7=+C11
�C1=�C3=�C6
�C2=�C9
�C4=�C7=�C11
B3PW91/6–31G(d) 0.0 5.6 1.2 4.0 2.6 0.0
B3PW91/6–311+G(d,p) 0.0 6.1 0.9 4.7 3.5 0.0
MP2/cc–pVDZ 0.0 7.9 0.8 2.4 2.4 0.0
I. RETROSYNTHETIC ANALYSIS
How can one introduce functional groups in 2 and/or 2,9 posi-tions in D3-trishomocubane? The seemingly obvious answer would be – by standard chemical reactions (i.e. halogenation). Unfortu-nately, in most cases the direct methods of functionalization lead to formation of 4-derivatives. The reason for that is preferred forma-tion of either +C1/+C4 cations (in electrophilic reactions) or �C1/�C4 radicals (in radical transformations). As implied by our calcula-tions, Table 2) [39], the D3-trishomocubane +C1 cation and �C4 radi-cal are most stable. The other D3-trishomocubane cations and radi-
cals are less stable [MP2/cc-pVDZ]: +C1 (0.0 kcal/mol) < +C4 (0.8
kcal/mol) < +C2 (7.9 kcal/mol) and �C1 (0.0 kcal/mol) < �C4 (2.4 kcal/mol) � �C2 (2.4 kcal/mol), respectively. Hence, under electro-philic conditions one would expect formation of 4-derivatives and under radical conditions - 1-derivatives, which was shown experi-mentally [40,41] (Scheme 4).
E
EE E
2829 30
Scheme 4. Radical pathway: (COCl)2, (PhCOO)2, 80-90°C, 24 hrs, then MeOH, 5 hrs, 56% (E = COOMe); electrophilic pathway: Br2, AlBr3, r.t., 1.5 hrs, 41% (E = Br).
Another approach – is the formation of the D3-trishomocubane skeleton via the expansion of the C2-bishomocubane due to the reaction with chlorodicarbonylrhodium dimer (Scheme 5) [42,43]. This rearrangement was reported only twice by the same group of authors and resulted in low yields for several substituted substrates.
It is unclear whether it is possible to use this method for the synthesis of the target compound (Scheme 6).
R1 R2 Yield,
%
H H 100
OH H 100
O H 65
OAc H 34
Cl H 0
R1 R1
O
a
31 32
R2 R2
Me Me 0
Scheme 5. a) Rh2(CO)4Cl2, benzene, in a sealed pressure tube under N, at 68°C.
OY
YX X
?
33
Scheme 6.
Hence, we will only focus on the rearrangement of Cs-trishomocubane skeleton to D3-trishomocubane catalyzed by H+ or Lewis acids with already existing substituents which will be at positions 2 and 2,9 in D3-trishomocubane.
The chemistry of Cs-THC derivatives is much more rich com-pared to D3-trishomocubane, but no suitable direct methods for the functionalization of the positions 2,6,9,10 (that would become axial after the rearrangement) exist (Scheme 7).
5
9 10
3
6
7 1
2
8 11
4
A
B
H
H
A
B
Y
A
B
XH
H
Scheme 7.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2637
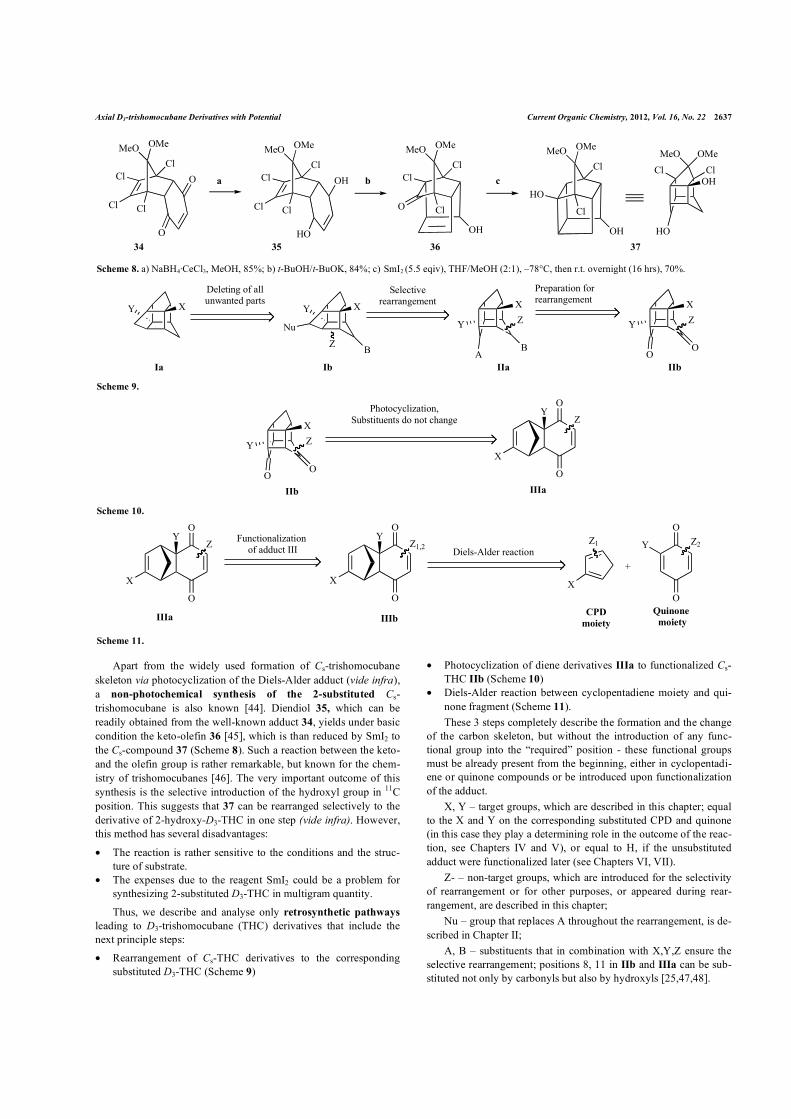
Apart from the widely used formation of Cs-trishomocubane skeleton via photocyclization of the Diels-Alder adduct (vide infra), a non-photochemical synthesis of the 2-substituted Cs-trishomocubane is also known [44]. Diendiol 35, which can be readily obtained from the well-known adduct 34, yields under basic condition the keto-olefin 36 [45], which is than reduced by SmI2 to the Cs-compound 37 (Scheme 8). Such a reaction between the keto- and the olefin group is rather remarkable, but known for the chem-istry of trishomocubanes [46]. The very important outcome of this synthesis is the selective introduction of the hydroxyl group in 11C position. This suggests that 37 can be rearranged selectively to the derivative of 2-hydroxy-D3-THC in one step (vide infra). However, this method has several disadvantages:
• The reaction is rather sensitive to the conditions and the struc-ture of substrate.
• The expenses due to the reagent SmI2 could be a problem for synthesizing 2-substituted D3-THC in multigram quantity.
Thus, we describe and analyse only retrosynthetic pathways leading to D3-trishomocubane (THC) derivatives that include the next principle steps:
• Rearrangement of Cs-THC derivatives to the corresponding substituted D3-THC (Scheme 9)
• Photocyclization of diene derivatives IIIa to functionalized Cs-THC IIb (Scheme 10)
• Diels-Alder reaction between cyclopentadiene moiety and qui-none fragment (Scheme 11).
These 3 steps completely describe the formation and the change of the carbon skeleton, but without the introduction of any func-tional group into the “required” position - these functional groups must be already present from the beginning, either in cyclopentadi-ene or quinone compounds or be introduced upon functionalization of the adduct.
X, Y – target groups, which are described in this chapter; equal to the X and Y on the corresponding substituted CPD and quinone (in this case they play a determining role in the outcome of the reac-tion, see Chapters IV and V), or equal to H, if the unsubstituted adduct were functionalized later (see Chapters VI, VII).
Z- – non-target groups, which are introduced for the selectivity of rearrangement or for other purposes, or appeared during rear-rangement, are described in this chapter;
Nu – group that replaces A throughout the rearrangement, is de-scribed in Chapter II;
A, B – substituents that in combination with X,Y,Z ensure the selective rearrangement; positions 8, 11 in IIb and IIIa can be sub-stituted not only by carbonyls but also by hydroxyls [25,47,48].
O
O
OMeMeO
Cl
ClCl
ClOH
HO
OMeMeO
Cl
ClCl
Cl
OH
OMeMeO
Cl
ClO
Cl
OH
OMeMeO
Cl
Cl
HO
OH
ClCl
MeO OMe
HO
34 35 36 37
a b c
Scheme 8. a) NaBH4·CeCl3, MeOH, 85%; b) t-BuOH/t-BuOK, 84%; c) SmI2 (5.5 eqiv), THF/MeOH (2:1), –78°C, then r.t. overnight (16 hrs), 70%.
Z
XYZ
Preparation for
rearrangementSelective
rearrangement
Deleting of all
unwanted parts
IbIa IIa
AB
XY
Nu
X
Y Z
IIb
OO
X
Y
B
Scheme 9.
O
O
ZPhotocyclization,
Substituents do not change
IIIa
Z
IIb
OO
X
Y
Y
X
Scheme 10.
O
O
Z1,2
O
O
Z2Z1
Diels-Alder reaction
Functionalization
of adduct III
CPD
moiety
Quinone
moietyIIIa IIIb
O
O
ZY
X X
Y
X
Y
+
Scheme 11.

2638 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
According to the expected application, one has to choose which substituents can be in axial positions. Evidently, targeted substitu-ents X and Y can be alkyl or aryl groups (although they can be barely converted further or used in previously mentioned systems, see Introduction), as well as halogen or hydroxyl (thiols) groups (it was shown that OH group is non-reactive in tertiary position of THC systems, Scheme 12) [49,50]. This leads to to the assumption that groups such as –COOAlk, –C(O)Me, –C=CH2, –CH2OH, –CH2Hal, –CH2COOAlk, etc might be more suitable.
HO HOOC
Br
38 40
39
a
b
c
Scheme 12. a) HCOOH, H2SO4, 20°C, 30 min, 27%; b) HBr, reflux, 15 hrs, 50%; c) HCOOH, H2SO4, 3.5 hrs, 52%.
Since the substituents A and B in IIb originate from adduct IIIb (A=B=O) and take part in the rearrangement (see chapter Re-arrangement), these groups (in forms of ketones or reduced to hy-droxyls) should exist after photocyclization and must be removed after selective rearrangement, together with all the other unwanted substituents. These substituents can also be defined at this point: since the removal of functional group from the tertiary positions of the cage is an extremely complicated task, only halogens should be present on the tertiary positions of structures Ib, IIa, IIb (Scheme
13) [51]. Possible substitutions on the 4C- atom (secondary posi-tion) are described in chapter IV.
Br
O
O
O
O
41 42
Scheme 13. Li/tBuOH, 84%.
II. SELECTIVE REARRANGEMENT OF THE CS-CAGE TO
D3
The problem of selectively rearranging Cs-trishomocubane is obvious: if the functional groups in the positions 8 and 11 are the same (usually after photocyclization they are either keto- or hy-droxyl- groups), there are two possible ways for the rearrangement to occur (Scheme 14). Thus, from a molecule with a plane of sym-metry the two enantiomers (R and S) are always obtained while a chiral Cs-substrate yields two different isomers.
Nu is the nucleophile that attacks the rearranged cation, A is the group that causes the rearrangement (either a hydroxyl or a car-bonyl), note that A=B in accordance with the numbering on the previous schemes.
To obtain the single product one of the two strategies can be applied:
• The rearrangement must be selective for the substituents (A=B),
• The substrate must have only one possibility for rearrange-ment (A�B). The synthesis of such compounds requires selective changing of the A or B groups, which can be done only based on the presence of specific target groups.
5
9 10
3
6
8 1
2
11
4
7 10
3
1
2
11
4
9
5
7
6
8
front sideback side
(R) (S)A
A AA
NuNu
5
9 10
3
6
8 1
2
11
4
710
3
1
2
11
4
9
5
7
6
8
A
AAA
NuNu Y
Y
Y
axial isomernonaxial isomer
Chiral substrate
Achiral substrate
Scheme 14.

Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2639
The influence of the substituents on the reactivity of groups A and B plays a major role and is different for diols and diketones.
Mild oxidation was found to be the one method, which can dif-ferentiate the two hydroxyl groups according to the substituents on the “frontside” or the “backside” (the less hindered hydroxyl is more reactive) of the Cs-trishomocubane (Scheme 15 [52] and Scheme 16 steps a-c). The rearrangement of the ether 7 was non-selective, therefore it is expected that the situation with the diol would be the same. Unfortunately, no attempts on the rearrange-ment of 17 were made. Yet substituted diketo- compounds have high selectivity in various reactions. For example, the hindrance of carbonyl group decreases its reactivity Scheme 17 [53,54], Scheme
16 steps d-f [55]. Even when the frontside and the backside sub-stituents are the same their influence is quite different (Scheme 18)
[56,57]. Both pathways were used to synthesize 18 and its corresponding ketal 46 (Scheme 16).
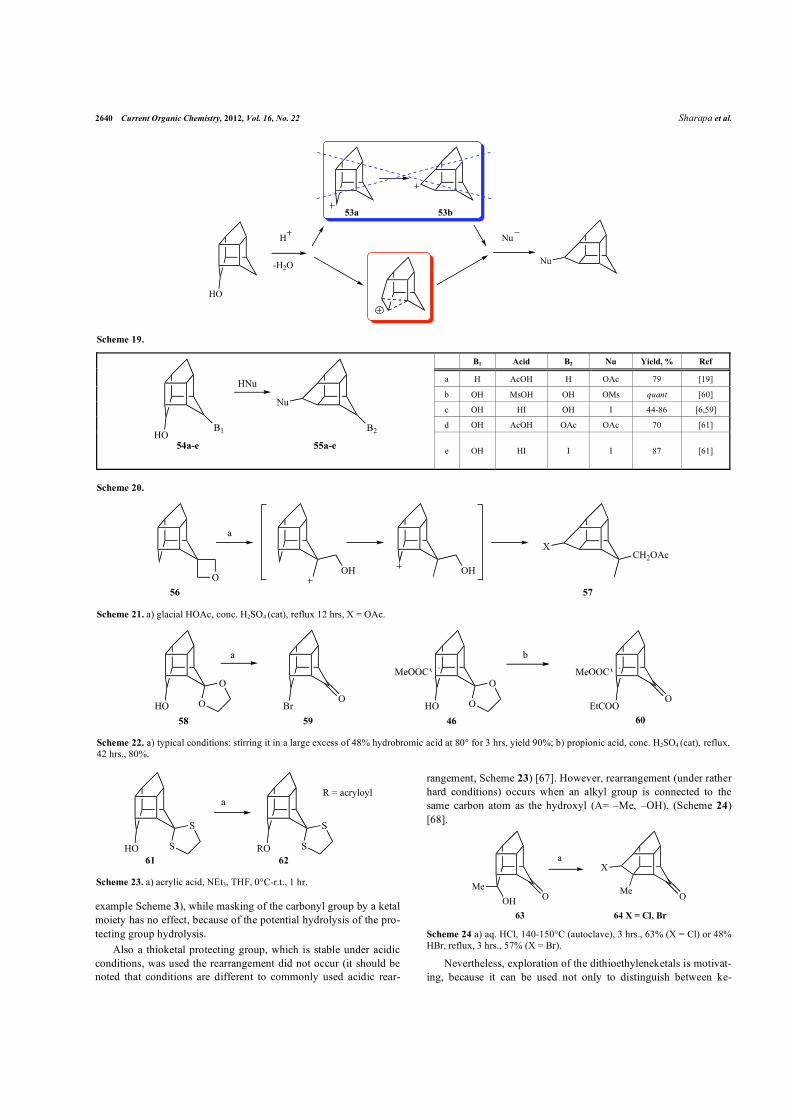
Rearrangement of the Cs-THC-alcohol in acidic media is the most popular method for rearranging this cage. According to the classical idea, 8Cs-THC-cation 53a undergoes rearrangement to 4D3-THC-cation 53b, followed by a reaction with a nucleophile [59]. Our ab initio and DFT study showed that the reaction proceeds via the formation of only one D3-nonclassical cation instead (Scheme 19) [39]. Hence, this method seems to be suitable to form substi-
tuted D3-skeleton based on Cs-THC. However, the rearrangement of Cs-THC-alcohol in acidic media only yields a single isomer (vide
supra), when the second group is not a hydroxyl (A=OH, B�OH).
The effect of the B-substituent on this rearrangement was found to be essential. The cation, which is formed on a first step by the removal of the hydroxyl group (Scheme 20), successfully rear-ranges when there is no substituent in position 11 (B1 = H,H), or the substituent is another hydroxyl group (B1 = OH, H).
It was also found that complex carbon substituent (B = Me, CH2OH) does not affect the rearrangement (Scheme 21) [62].
However, the presence of a keto group in this position com-pletely prevents rearrangement (different substrates and acids were tried by several authors, Scheme 22) [58,62-66]. The reduction of the carbonyl group is a possible solution for this problem (see for
HOOH
Ph
HOO
Ph
Me Me
43 44
a
Scheme 15. a) PCC, CH2Cl2, 0–25°C.
OO
MeOOC
O O
OMeOOC
OO
MeOOC
HOOH
MeOOC
HOOH
MeOOC
a
b c (selective)
d
e f
selective
HOO
MeOOC
HO O
OMeOOC
2
16 17 18
3 45 46
Scheme 16. a, b, c – see Scheme 3; d) hv, EtOAc, 88%[55] (compare with step b, Scheme 1); e) ethylene glycol, TsOH, 85%;[55] f) NaBH4, MeOH, r.t., 48 hrs., 95% [58].
OO
O
R R
a
OH
O
OEt
b
OCH2
R
4748 49
R = Me
Ph
R = Ph
Scheme 17. a) Ph3P–CH3-Br+, n-BuLi, 0°-25°C, 48%[53]; b) ethyl diazoacetate (EDA), BF3·Et2O, 0°C–r.t., 2 hrs., 25% for both substituents [54].
OOO
Br
BrBr
HOOH
Br
a b
O
Br
Br
O
Br
Br
O OO
O
+
5051 52a 52b
Scheme 18. a) moist air;[56] b) ethylene glycol, TsOH · H2O, ratio 52a:52b = 1:5, also bisethylenketal formed, which can be converted back to a mixture 52a+52b by hydrolysis 10% H2SO4, THF, total yield � quant.
2640 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
example Scheme 3), while masking of the carbonyl group by a ketal moiety has no effect, because of the potential hydrolysis of the pro-tecting group hydrolysis.
Also a thioketal protecting group, which is stable under acidic conditions, was used the rearrangement did not occur (it should be noted that conditions are different to commonly used acidic rear-
rangement, Scheme 23) [67]. However, rearrangement (under rather hard conditions) occurs when an alkyl group is connected to the same carbon atom as the hydroxyl (A= –Me, –OH), (Scheme 24) [68].
OH
a
O O
MeMe
X
63 64 X = Cl, Br
Scheme 24 a) aq. HCl, 140-150°C (autoclave), 3 hrs., 63% (X = Cl) or 48% HBr, reflux, 3 hrs., 57% (X = Br).
Nevertheless, exploration of the dithioethyleneketals is motivat-ing, because it can be used not only to distinguish between ke-
HO
H Nu
-H2ONu
53a 53b
Scheme 19.
B1 Acid B2 Nu Yield, % Ref
a H AcOH H OAc 79 [19]
b OH MsOH OH OMs quant [60]
c OH HI OH I 44-86 [6,59]
d OH AcOH OAc OAc 70 [61] HO
HNu
B1 B2
Nu
54a-e 55a-e
e OH HI I I 87 [61]
Scheme 20.
OOH
XCH2OAc
56 57
a
OH
Scheme 21. a) glacial HOAc, conc. H2SO4 (cat), reflux 12 hrs, X = OAc.
HO
a
HO
b
O
O
MeOOC
BrO
EtCOOO
MeOOC
58 59 6046
O
O
Scheme 22. a) typical conditions: stirring it in a large excess of 48% hydrobromic acid at 80° for 3 hrs, yield 90%; b) propionic acid, conc. H2SO4 (cat), reflux, 42 hrs., 80%.
HO S
S
RO S
S
61 62
aR = acryloyl
Scheme 23. a) acrylic acid, NEt3, THF, 0°C-r.t., 1 hr.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2641
togroups (like ethyleneketals), but also for the reduction of the non-targeted keton in one step under mild condition (compared to widely used Wolf-Kishner reaction), Scheme 25.
Some methods allow the formation of some “unwanted” keto-alcohols (Scheme 26) [69].
This result, however, can also be used: by converting such compounds into bromo-ketones without rearrangement (Scheme
22), with the possibility that after reduction to bromo-alcohols these compounds can successfully undergo rearrangement (Scheme 27).
Since dienetriol 70 (obtained from acid 16 by multistep synthe-
sis involving reductions and resolutions) can be converted to ace-
tonide 71 (Scheme 28) [70], we can expect that the same can be
done with the cage-triol 4.
OO
X
HO
X
HO
X
S
S
O
X
S
S
X
HO
Ni/Ra
Scheme 25.
O
O
O
TBSOH2CO
OH
O
TBSOH2C
81%
H H
O
O
MeOOCO
OH
MeOOC
H H
O
OH
TBSOH2C
H
OOH
MeOOC
65 662
68 6967
a b
c
Scheme 26. a) NaBH4 (1.1 equiv), CeCl3·7H2O, -30°–0°C, 30 min; b) hv, acetone, 90%; c) NaBH4, MeOH, 15°C, 81%.
O
OH
X
OOH
X
OBr
X
HOBr
X
X
HO
Br
Scheme 27.
OH
H
HOH
CH2OH
O
H
HOH
O
O
H
O
O
OH
H
HOH
COOMe
OH
H
HOH
CH2OH
O
H
HOH
O
O
H
O
O
16 70 71 72
4
Scheme 28. a) LiAlH4, THF, 0°C, 1 hr, r.t. 2 hrs.; b) acetone, PPTS (cat), r.t., 12 hrs., overall yield from 16 51%; c) PDC, CH2Cl2, 0°C–r.t. 2 hrs.

2642 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
Based on the selectivity of the reactions with diketones it is an-
ticipated that these compounds can undergo rearrangement in one
step to form a single product. The previously reported rearrange-
ment of �-diketone by chlorosulphonic acid [39,71-73] is studied
much less than the rearrangement of alcohols, but is characterized
by rather promising features:
• The rearrangement occurs under low temperature (this usually
corresponds to higher selectivity)
• The rearrangement is selective towards “frontside” substitution
(reaction with “backside”-substituted diketones was never at-
tempted, Scheme 29).
OO
Br
Br
HSO3Cl
?
50
We propose that this rearrangement (Scheme 29) and the rear-
rangement shown on Scheme 24 occur via the formation of the
analogous intermediates 77b-d, which reactivity differ from the
unsubstituted 77a (Table 3).
We also expect that polysubstituted Cookson diketones, (for
example 50) will rearrange in only one possible way (probably by
the less hindered carbonyl). Contrary to the first method (rear-
rangement of alcohols), chlorosulfation does not require removal of
a substituent near 11
C, so it can be used for the synthesis of 4,7,11-
substituted-THC with functional groups in position 2/9. Thus, this
method can be useful for diketones, which are obtained from halo-
substituted quinones (see chapter V).
In the case of synthesis of mono-axial-substituted D3-THC the
problem of the selective rearrangement could be omitted by using
type II compounds, which have same functional group in positions
9 and 10 (for example 116) or 2 and 7 (based on dicarboxyfulvene
120b), Scheme 30.
Also this method solves the problem of the selective synthesis
of 2-X,9-Y-substituted III. No compounds of this type are known at
the moment, since the selectivity of functionalization of these two
positions is in general independent from each other and in the case
of high sensitivity for the individual reaction – unpredictable (thus
its possible to form IIIa or IIIc by functionalization, while using a
“trisubstituted strategy” suggests the synthesis of 2,9-substituted
compounds of type IIId, IIIe, Scheme 31).
OO
HO
ClO
OO
HO
ClO
MeMe
HSO3Cl HSO3Cl
73 74 75 76
Scheme 29. a) HSO3Cl, CH3Cl, 0°C, overnight, 75%;[39] b) HSO3Cl, 0°C, 57% [73].
Table 3.
X Rearrangement Conditions Ref.
H – Any acid [58,62-66]
Me + HBr, (or HCl), reflux, P [68] X
O
77 Cl + HSO3Cl [39,71-73]
2
7
A
B
X
X
10
9
A
B
H
Y
Y
HA B
X X
AB
YYZZ
nonselective rearrangament
Scheme 30.
2
7
6
5
4
31
10
9
8
A
B
X
Y
IIIa
H
H
2
7
10
9
A
B
X
H
IIIc
Y
H
210
9
A
B
X
Y
IIId
Y
H
2
7
10
A
B
X
H
IIIe
Y
X
Scheme 31.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2643
However such simplification causes another problem: the re-gioselective reduction of a functional group that appears on a non-axial position. This problem would be even more complicated than searching for a pathway for compound IIIa.
III. CYCLOPENTADIENE DERIVATIVES (9-
SUBSTITUTED III)
In general several types of starting compounds with CPD-fragment can be used:
• 2-Substituted cyclopentadienes • 2-Substituted cyclopentadienone or its ketal • 2-Substituted fulvenes • 2-Substituted cyclopentadienes with two substituents at the
atom C5
3 2
1
5
4
RO OR R1 R2
X X X X
The main problem of substituted cyclopentadiens is the equi-librium of several isomers. The structure of a product depends on the substituent pattern in CPD, the structure of the dienophile, and the presence of a Lewis acid catalyst and is generally unpredictable (e.g. Scheme 32) [74-80].
Very often the product is a mixture of several adducts (Scheme 33). In some cases it is a mixture of endo- and exo- adducts, but more often a mixture of regioisomers (1 or 9) [81,82].
Cyclopentadiene derivatives usually exist in their dimer form, and the respective monomer (that participates in the DA reaction) can be obtained by distillation, whereby the yields are often low (e.g. monomerization of Thiele ester (Carbomethoxycyclopentadi-ene dimer) to methylcyclopenadienecarboxylate 85 – 50%[81]).
The presence of heterosubstituents poses additional problems, because possible structures are in equilibrium with each other and undergo very fast rearrangement. For example, bistrimethylsylil-CPD has 7 equilibrium structures, with 5,5-isomer being major one (�95% in mixture). But its reactivity in the DA reaction is rather low, and in all reported cases the 2,5-isomer is the reactive one [83]. The adduct with quinone was then readily photocyclized into
O
O
O
O
O
O
O
O
O
O
O
O
Ph
O
O
Ph
Me
Me
Me
Me
Me
Me
Me
78
79 80
81 82
83 84
a
b
c
Scheme 32. a) benzene, sealed tube, 90°C, 5 hrs, 77%; [74] b) benzene, 0-5°C, 4 hrs., 40%; [76] c) no experimental data is available [75].
41
8
O
O
O
O
+
MeOOC
2
59
O
O
MeOOC
MeOOC
+
O
O
+
(MeO)3C
O
O
MeOOC
+
O
O
MeOOC
H
H
H
H
85 86 87 88
89 86 88 90
a
b
Scheme 33. a) CH2Cl2/benzene, -75°C-r.t., overnight, ratio 87:88 = 3:1, total yield unknown, 87 was isolated in 30% yield;[81] b) no experimental data is available; ratio 88:90 = 3:2 [82].
2644 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
2,4-bis-TMS-derivative of the Cookson diketone [84] (Scheme 34),
however attempts to control the DA-reaction by a TMS group was
in general unsuccessful (Scheme 35) [85].
The derivatives of cyclopentadienone and their ketals exist
as one isomer, but are usually very reactive (ethyl ketal of cy-
clopentadienone dimerizes about 465 times more rapidly than cy-
clopentadiene; the ethylene ketal dimerizes nearly 5�105
times
faster than cyclopentadiene) [86]. The dimerization rate can be
decreased by introducing steric or electron-accepting substituents:
while ketals of nonsubstituted cyclopentadienone are usually used
without isolation [87,88], e.g. dimethyl ketal of tetrachlorocy-
clopentadienone is stable [89], and ethylene ketal of 3-t-butyl-
cyclopentadienone 101 dimerizes slowly enough to obtain adducts
with p-benzoquinone [90] (Scheme 36). Annulated 2,5-
bis(trimethylsilyl)cyclopentadienone 104 is stable even without any
ketal protection [91]. However, the problem of low yield still exists:
dehydrobromination of 2,5-dibromo-3-t-butyl-cyclopentanone ketal
100 produces only 50% of 3-t-butyl-cyclopentadienone ketal 101
(after distillation) and 40% are lost to dimerisation (conditions for
the monomerization are unknown) [92]. The yield of the adduct 109
is even lower – 30% [93].
Me3Si SiMe3SiMe3
Me3Si
COOMe
COOMe
Me3Si
SiMe3
SiMe3
Me3SiCN
CN
CN
CN
O
Me3Si
O
Me3Si
H
H
SiMe3
SiMe3
OO
O
O
MeOOC COOMe
CN
NC CN
NC91
92
93 94
95
a
d
b
c
Scheme 34. a) CCl4, 20°C, 24 hrs (or xylene, reflux, 2 hrs); b) MeOH, r.t., 89%; c) hv, EtOAc, quant.; d) CH2Cl2, 20°C, 24 hrs., 94%
SiMe3
MeN
O
O
Ph
SiMe3
N
O
O
Ph
SiMe3
N
O
O
Ph
++
96 97a 97b
a
Scheme 35. a) Et2O, r.t., 1 hr., 97a:97b = 9:11 (the same result obtained for methylCPD).
X
X
X
X
OMe
OMe
ButO
O
ButO
O
Br
BrO
O
But
O
O
endo/exo
stereochemistry
is unknown
OMeMeO
X
O
O
X
X
X Cl
Cl ClCl
MeOOMe
OO
98 X=H
34 X=Cl
99
100 101 102
a b
dc
5,5-dimethoxycyclopentadiene X=H
tetrachloro-5,5-dimethoxyCPD X=Cl
Scheme 36. a) toluene, 0-70°C, 12 hrs., 74% (X = H) [94], toluene, reflux, 24 hrs., 83% (X = Cl); [89] b) hv, acetone, 90%; [89] c) MeONa, DMSO, 25°C, 40
hrs., 50%; [92] d) benzoquinone, MeOH, r.t., 12 hrs., quant [90].
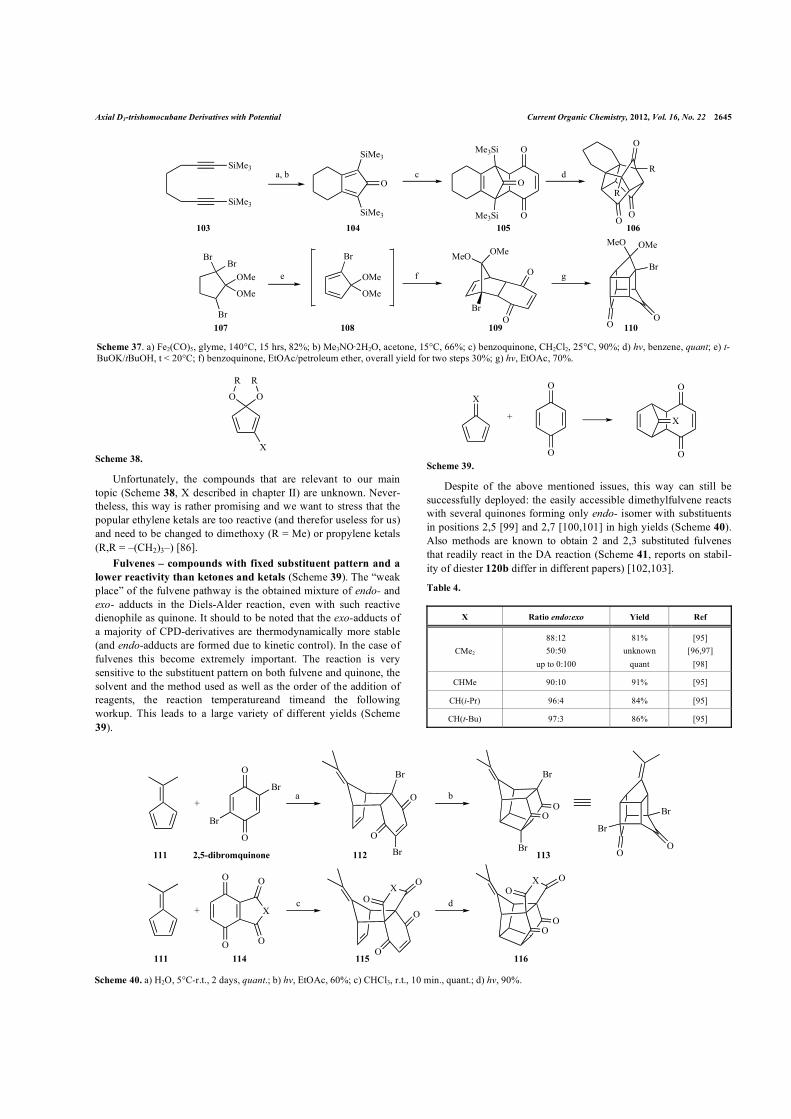
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2645
O
RR
O
X Scheme 38.
Unfortunately, the compounds that are relevant to our main topic (Scheme 38, X described in chapter II) are unknown. Never-theless, this way is rather promising and we want to stress that the popular ethylene ketals are too reactive (and therefor useless for us) and need to be changed to dimethoxy (R = Me) or propylene ketals (R,R = –(CH2)3–) [86].
Fulvenes – compounds with fixed substituent pattern and a
lower reactivity than ketones and ketals (Scheme 39). The “weak place” of the fulvene pathway is the obtained mixture of endo- and exo- adducts in the Diels-Alder reaction, even with such reactive dienophile as quinone. It should to be noted that the exo-adducts of a majority of CPD-derivatives are thermodynamically more stable (and endo-adducts are formed due to kinetic control). In the case of fulvenes this become extremely important. The reaction is very sensitive to the substituent pattern on both fulvene and quinone, the solvent and the method used as well as the order of the addition of reagents, the reaction temperatureand timeand the following workup. This leads to a large variety of different yields (Scheme 39).
X
+
O
O
X
O
O Scheme 39.
Despite of the above mentioned issues, this way can still be successfully deployed: the easily accessible dimethylfulvene reacts with several quinones forming only endo- isomer with substituents in positions 2,5 [99] and 2,7 [100,101] in high yields (Scheme 40). Also methods are known to obtain 2 and 2,3 substituted fulvenes that readily react in the DA reaction (Scheme 41, reports on stabil-ity of diester 120b differ in different papers) [102,103].
Table 4.
X Ratio endo:exo Yield Ref
CMe2
88:12
50:50
up to 0:100
81%
unknown
quant
[95]
[96,97]
[98]
CHMe 90:10 91% [95]
CH(i-Pr) 96:4 84% [95]
CH(t-Bu) 97:3 86% [95]
SiMe3
SiMe3
O
SiMe3
SiMe3
Me3Si
Me3Si
O
O
OR
R
OO
O
OMe
OMe
OMe
OMe
Br
Br
OMeMeO
O
O
Br Br
Br
Br
MeO OMe
OO
104103 105 106
107 108 109 110
a, b
e
c
f
d
g
Scheme 37. a) Fe2(CO)5, glyme, 140°C, 15 hrs, 82%; b) Me3NO·2H2O, acetone, 15°C, 66%; c) benzoquinone, CH2Cl2, 25°C, 90%; d) hv, benzene, quant; e) t-BuOK/tBuOH, t < 20°C; f) benzoquinone, EtOAc/petroleum ether, overall yield for two steps 30%; g) hv, EtOAc, 70%.
+
O
O
X
O
O
O
O
X
O
O
O
O
X
O
O
+
O
O
Br
Br
O
O
Br
O
O
Br
BrBr O
O
Br
Br
111 2,5-dibromquinone 112 113
111 114 115 116
a b
c d
Scheme 40. a) H2O, 5°C-r.t., 2 days, quant.; b) hv, EtOAc, 60%; c) CHCl3, r.t., 10 min., quant.; d) hv, 90%.
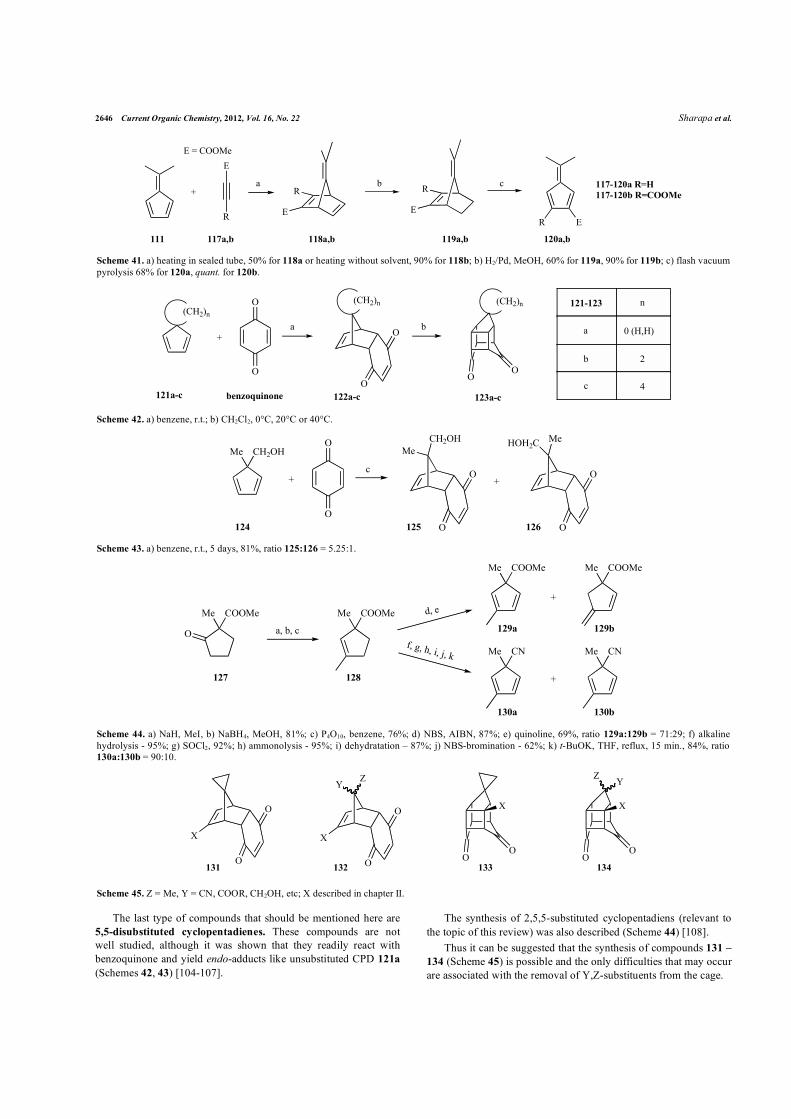
2646 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
The last type of compounds that should be mentioned here are
5,5-disubstituted cyclopentadienes. These compounds are not
well studied, although it was shown that they readily react with
benzoquinone and yield endo-adducts like unsubstituted CPD 121a
(Schemes 42, 43) [104-107].
The synthesis of 2,5,5-substituted cyclopentadiens (relevant to
the topic of this review) was also described (Scheme 44) [108].
Thus it can be suggested that the synthesis of compounds 131 –
134 (Scheme 45) is possible and the only difficulties that may occur
are associated with the removal of Y,Z-substituents from the cage.
R
E
R R
EE
R E
+
E = COOMe
111 117a,b 118a,b 119a,b 120a,b
a b c 117-120a R=H
117-120b R=COOMe
Scheme 41. a) heating in sealed tube, 50% for 118a or heating without solvent, 90% for 118b; b) H2/Pd, MeOH, 60% for 119a, 90% for 119b; c) flash vacuum
pyrolysis 68% for 120a, quant. for 120b.
121-123 n
a 0 (H,H)
b 2
c 4
+
(CH2)n
O
O
O
OO
O
(CH2)n (CH2)n
122a-c 123a-c121a-c benzoquinone
a b
Scheme 42. a) benzene, r.t.; b) CH2Cl2, 0°C, 20°C or 40°C.
+
Me CH2OH
O
O
O
O
CH2OH
Me
O
O
HOH2CMe
+
124 125 126
c
Scheme 43. a) benzene, r.t., 5 days, 81%, ratio 125:126 = 5.25:1.
O
Me COOMe Me COOMe
Me CN
Me COOMe
127 128
129a
130a
a, b, c
d, e
f, g, h, i, j, k
Me COOMe
129b
+
+
Me CN
130b
Scheme 44. a) NaH, MeI, b) NaBH4, MeOH, 81%; c) P4O10, benzene, 76%; d) NBS, AIBN, 87%; e) quinoline, 69%, ratio 129a:129b = 71:29; f) alkaline
hydrolysis - 95%; g) SOCl2, 92%; h) ammonolysis - 95%; i) dehydratation – 87%; j) NBS-bromination - 62%; k) t-BuOK, THF, reflux, 15 min., 84%, ratio
130a:130b = 90:10.
O
O OO
O
O
ZY
OO
YZ
X X
X X
131 132 133 134
Scheme 45. Z = Me, Y = CN, COOR, CH2OH, etc; X described in chapter II.
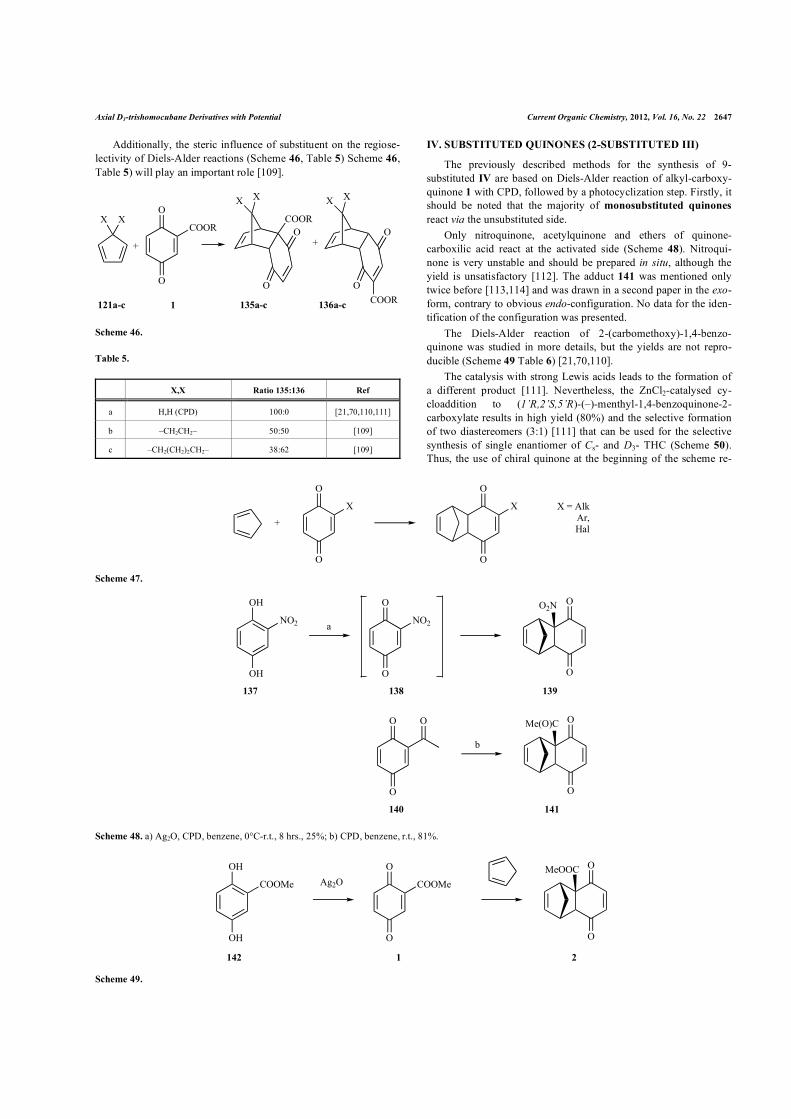
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2647
Additionally, the steric influence of substituent on the regiose-lectivity of Diels-Alder reactions (Scheme 46, Table 5) Scheme 46, Table 5) will play an important role [109].
O
O
XX
COOR
+
X X
O
O
O
O
XX
COOR
+
COOR
121a-c 1 135a-c 136a-c
Scheme 46.
Table 5.
X,X Ratio 135:136 Ref
a H,H (CPD) 100:0 [21,70,110,111]
b –CH2CH2– 50:50 [109]
c –CH2(CH2)2CH2– 38:62 [109]
IV. SUBSTITUTED QUINONES (2-SUBSTITUTED III)
The previously described methods for the synthesis of 9-substituted IV are based on Diels-Alder reaction of alkyl-carboxy-quinone 1 with CPD, followed by a photocyclization step. Firstly, it should be noted that the majority of monosubstituted quinones react via the unsubstituted side.
Only nitroquinone, acetylquinone and ethers of quinone-carboxilic acid react at the activated side (Scheme 48). Nitroqui-none is very unstable and should be prepared in situ, although the yield is unsatisfactory [112]. The adduct 141 was mentioned only twice before [113,114] and was drawn in a second paper in the exo-form, contrary to obvious endo-configuration. No data for the iden-tification of the configuration was presented.
The Diels-Alder reaction of 2-(carbomethoxy)-1,4-benzo-quinone was studied in more details, but the yields are not repro-ducible (Scheme 49 Table 6) [21,70,110].
The catalysis with strong Lewis acids leads to the formation of a different product [111]. Nevertheless, the ZnCl2-catalysed cy-cloaddition to (1’R,2’S,5’R)-(–)-menthyl-1,4-benzoquinone-2-carboxylate results in high yield (80%) and the selective formation of two diastereomers (3:1) [111] that can be used for the selective synthesis of single enantiomer of Cs- and D3- THC (Scheme 50). Thus, the use of chiral quinone at the beginning of the scheme re-
O
O
O
O
+
X X X = Alk
Ar,
Hal
Scheme 47.
O
O
O
O
NO2
O2NOH
OH
NO2 a
O
O
O
O
Me(O)CO
137 138 139
140 141
b
Scheme 48. a) Ag2O, CPD, benzene, 0°C-r.t., 8 hrs., 25%; b) CPD, benzene, r.t., 81%.
O
O
O
O
COOMe
MeOOCOH
OH
COOMe Ag2O
142 1 2
Scheme 49.
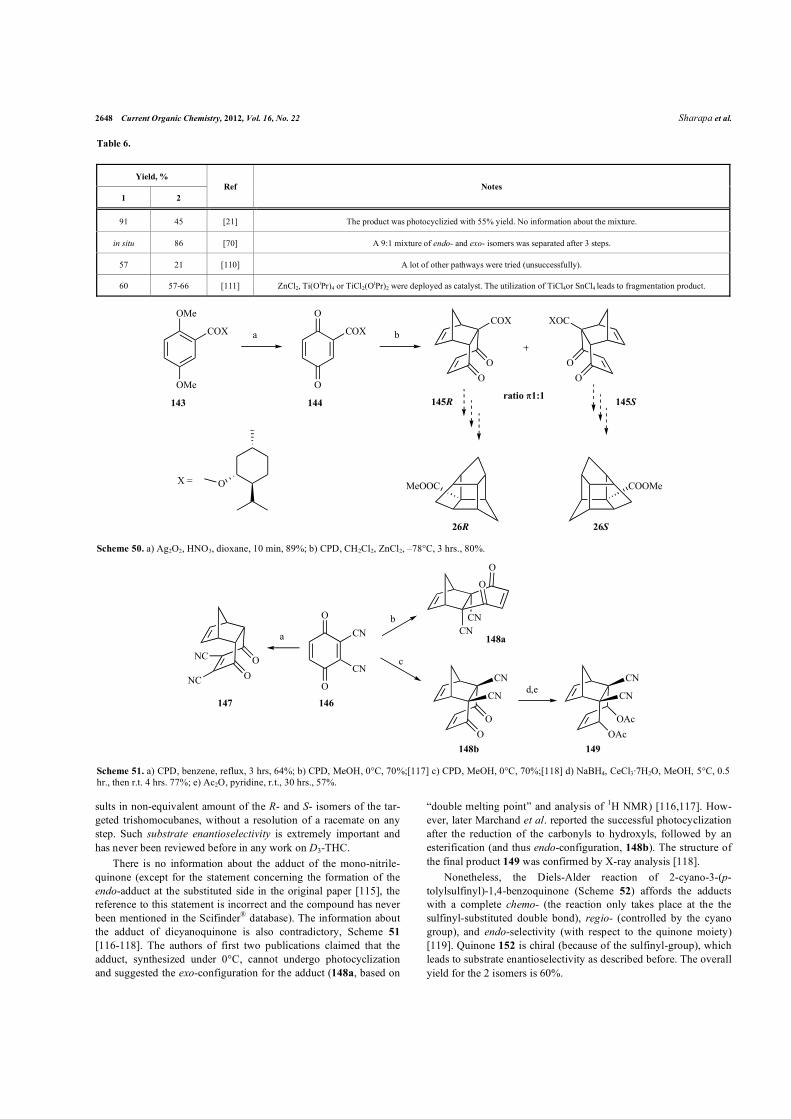
2648 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
sults in non-equivalent amount of the R- and S- isomers of the tar-geted trishomocubanes, without a resolution of a racemate on any step. Such substrate enantioselectivity is extremely important and has never been reviewed before in any work on D3-THC.
There is no information about the adduct of the mono-nitrile-quinone (except for the statement concerning the formation of the endo-adduct at the substituted side in the original paper [115], the reference to this statement is incorrect and the compound has never been mentioned in the Scifinder® database). The information about the adduct of dicyanoquinone is also contradictory, Scheme 51
[116-118]. The authors of first two publications claimed that the adduct, synthesized under 0°C, cannot undergo photocyclization and suggested the exo-configuration for the adduct (148a, based on
“double melting point” and analysis of 1H NMR) [116,117]. How-ever, later Marchand et al. reported the successful photocyclization after the reduction of the carbonyls to hydroxyls, followed by an esterification (and thus endo-configuration, 148b). The structure of the final product 149 was confirmed by X-ray analysis [118].
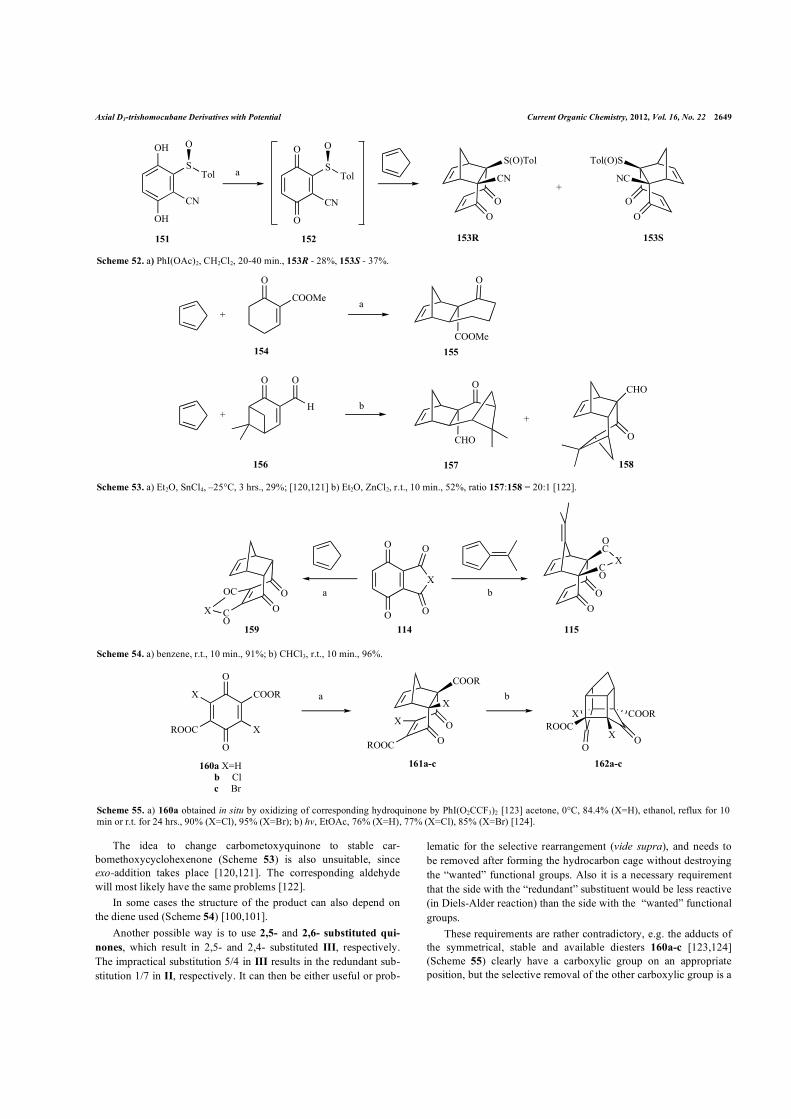
Nonetheless, the Diels-Alder reaction of 2-�yano-3-(p-tolylsulfinyl)-1,4-benzoquinone (Scheme 52) affords the adducts with a complete chemo- (the reaction only takes place at the the sulfinyl-substituted double bond), regio- (controlled by the cyano group), and endo-selectivity (with respect to the quinone moiety) [119]. Quinone 152 is chiral (because of the sulfinyl-group), which leads to substrate enantioselectivity as described before. The overall yield for the 2 isomers is 60%.
Table 6.
Yield, %
1 2
Ref Notes
91 45 [21] The product was photocyclizied with 55% yield. No information about the mixture.
in situ 86 [70] A 9:1 mixture of endo- and exo- isomers was separated after 3 steps.
57 21 [110] A lot of other pathways were tried (unsuccessfully).
60 57-66 [111] ZnCl2, Ti(OiPr)4 or TiCl2(OiPr)2 were deployed as catalyst. The utilization of TiCl4or SnCl4 leads to fragmentation product.
O
O
COX
OMe
OMe
COX
O
O
COX
O
O
XOC
MeOOC COOMe
ratio �1:1
X = O
143 144
a b
145R 145S
26R 26S
Scheme 50. a) Ag2O2, HNO3, dioxane, 10 min, 89%; b) CPD, CH2Cl2, ZnCl2, –78°C, 3 hrs., 80%.
O
O
CN
CN
O
O
CN
CN
O
O
CN
CN
O
O
NC
NC
a
b
c
d,e
OAc
OAc
CN
CN
146147
148a
148b 149
Scheme 51. a) CPD, benzene, reflux, 3 hrs, 64%; b) CPD, MeOH, 0°C, 70%;[117] c) CPD, MeOH, 0°C, 70%;[118] d) NaBH4, CeCl3·7H2O, MeOH, 5°C, 0.5 hr., then r.t. 4 hrs. 77%; e) Ac2O, pyridine, r.t., 30 hrs., 57%.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2649
The idea to change carbometoxyquinone to stable car-bomethoxycyclohexenone (Scheme 53) is also unsuitable, since exo-addition takes place [120,121]. The corresponding aldehyde will most likely have the same problems [122].
In some cases the structure of the product can also depend on the diene used (Scheme 54) [100,101].
Another possible way is to use 2,5- and 2,6- substituted qui-
nones, which result in 2,5- and 2,4- substituted III, respectively. The impractical substitution 5/4 in III results in the redundant sub-stitution 1/7 in II, respectively. It can then be either useful or prob-
lematic for the selective rearrangement (vide supra), and needs to be removed after forming the hydrocarbon cage without destroying the “wanted” functional groups. Also it is a necessary requirement that the side with the “redundant” substituent would be less reactive (in Diels-Alder reaction) than the side with the “wanted” functional
groups.
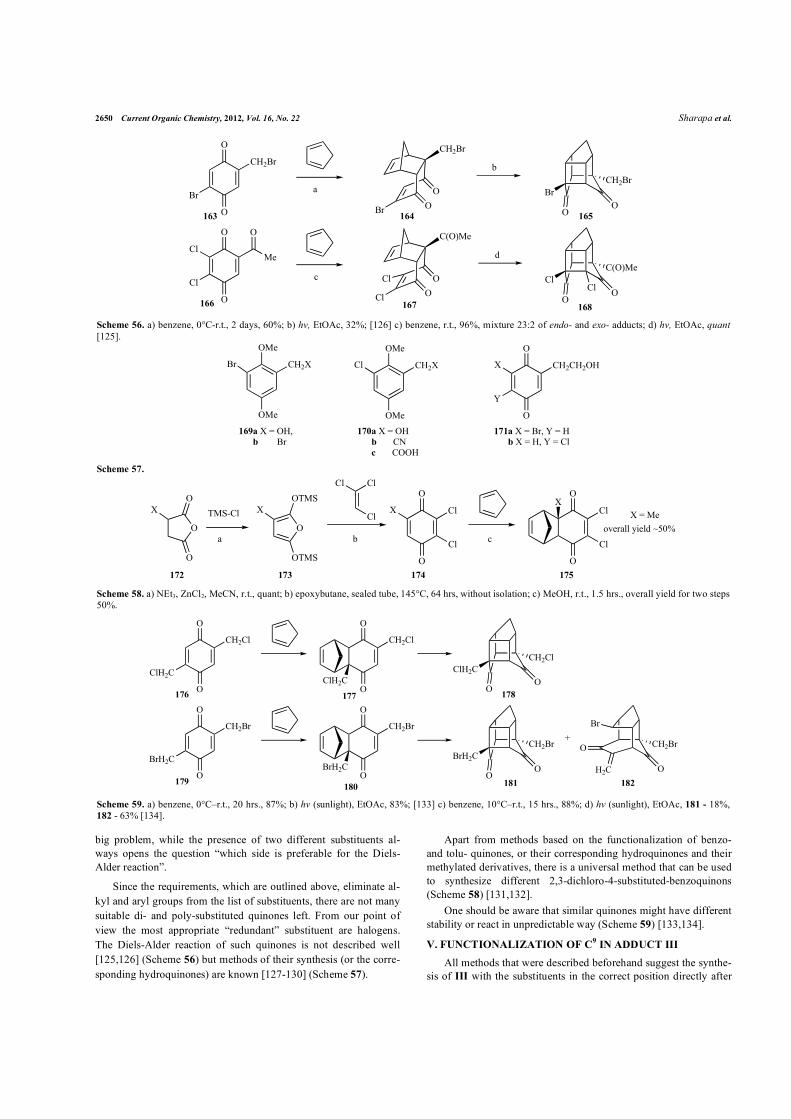
These requirements are rather contradictory, e.g. the adducts of the symmetrical, stable and available diesters 160a-c [123,124] (Scheme 55) clearly have a carboxylic group on an appropriate position, but the selective removal of the other carboxylic group is a
O
O
S
CN O
O
CN
S(O)Tol
Tol
O
O
O
NC
Tol(O)S
+
OH
OH
S
CN
Tol
O
a
151 152 153R 153S
Scheme 52. a) PhI(OAc)2, CH2Cl2, 20-40 min., 153R - 28%, 153S - 37%.
O
+
COOMe
COOMe
O
O
+
O
H
CHO
OCHO
O
+
154 155
156 157 158
a
b
Scheme 53. a) Et2O, SnCl4, –25°C, 3 hrs., 29%; [120,121] b) Et2O, ZnCl2, r.t., 10 min., 52%, ratio 157:158 = 20:1 [122].
O
O
X
O
O
O
O
CO
OC
X
O
OCO
OC
X
114 115159
a b
Scheme 54. a) benzene, r.t., 10 min., 91%; b) CHCl3, r.t., 10 min., 96%.
O
O
O
O
COOR
COORX
X
ROOC
X
X
COORX
ROOCX
OO
ROOC
160a X=H
b Cl
c Br
161a-c 162a-c
a b
Scheme 55. a) 160a obtained in situ by oxidizing of corresponding hydroquinone by PhI(O2CCF3)2 [123] acetone, 0°C, 84.4% (X=H), ethanol, reflux for 10 min or r.t. for 24 hrs., 90% (X=Cl), 95% (X=Br); b) hv, EtOAc, 76% (X=H), 77% (X=Cl), 85% (X=Br) [124].
2650 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
big problem, while the presence of two different substituents al-ways opens the question “which side is preferable for the Diels-Alder reaction”.
Since the requirements, which are outlined above, eliminate al-
kyl and aryl groups from the list of substituents, there are not many
suitable di- and poly-substituted quinones left. From our point of
view the most appropriate “redundant” substituent are halogens.
The Diels-Alder reaction of such quinones is not described well
[125,126] (Scheme 56) but methods of their synthesis (or the corre-
sponding hydroquinones) are known [127-130] (Scheme 57).
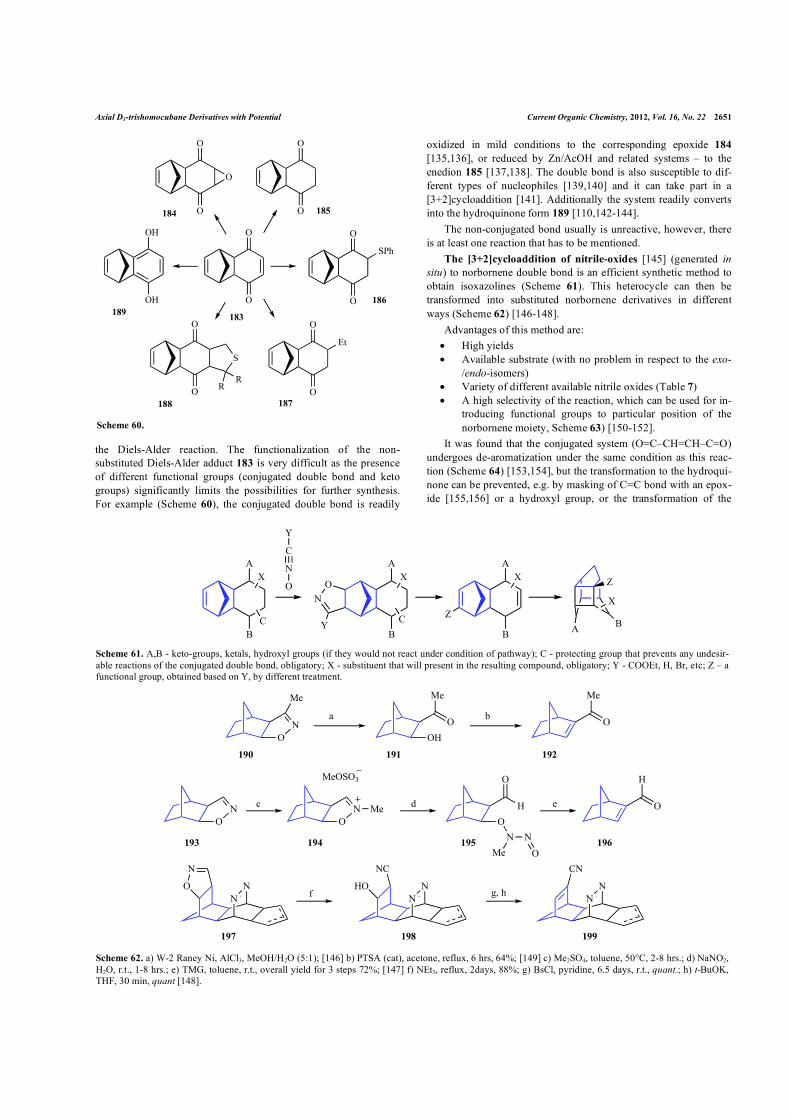
Apart from methods based on the functionalization of benzo- and tolu- quinones, or their corresponding hydroquinones and their methylated derivatives, there is a universal method that can be used to synthesize different 2,3-dichloro-4-substituted-benzoquinons (Scheme 58) [131,132].
One should be aware that similar quinones might have different stability or react in unpredictable way (Scheme 59) [133,134].
V. FUNCTIONALIZATION OF C9 IN ADDUCT III
All methods that were described beforehand suggest the synthe-sis of III with the substituents in the correct position directly after
O
O
O
O
CH2Br
CH2Br
Br
CH2Br
Br
OO
Br
O
O
O
O
C(O)Me
Cl
C(O)Me
Cl
OO
Cl
Me
O
Cl
ClCl
163 164 165
166 167 168
a
b
c
d
Scheme 56. a) benzene, 0°C-r.t., 2 days, 60%; b) hv, EtOAc, 32%; [126] c) benzene, r.t., 96%, mixture 23:2 of endo- and exo- adducts; d) hv, EtOAc, quant
[125]. OMe
OMe
CH2XBr
169a X = OH,
b Br
OMe
OMe
CH2XCl
170a X = OH
b CN
c COOH
O
O
CH2CH2OHX
Y
171a X = Br, Y = H
b X = H, Y = Cl
Scheme 57.
O
O
ClXX = Me
O
O
X
O
O
OTMS
X
OTMS
Cl
O
O
ClX
Cl
Cl
Cl Cl
TMS-Cl
overall yield ~50%
172 173 174 175
a b c
Scheme 58. a) NEt3, ZnCl2, MeCN, r.t., quant; b) epoxybutane, sealed tube, 145°C, 64 hrs, without isolation; c) MeOH, r.t., 1.5 hrs., overall yield for two steps 50%.
O
O
CH2Cl
O
O
CH2Cl
ClH2CClH2C
CH2Cl
ClH2C
OO
CH2Br
BrH2C
OO
CH2Br
H2C
O
O
Br
O
O
CH2Br
O
O
CH2Br
BrH2CBrH2C
176 177 178
179180
181 182
+
Scheme 59. a) benzene, 0°C–r.t., 20 hrs., 87%; b) hv (sunlight), EtOAc, 83%; [133] c) benzene, 10°C–r.t., 15 hrs., 88%; d) hv (sunlight), EtOAc, 181 - 18%, 182 - 63% [134].
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2651
the Diels-Alder reaction. The functionalization of the non-substituted Diels-Alder adduct 183 is very difficult as the presence of different functional groups (conjugated double bond and keto groups) significantly limits the possibilities for further synthesis. For example (Scheme 60), the conjugated double bond is readily
oxidized in mild conditions to the corresponding epoxide 184
[135,136], or reduced by Zn/AcOH and related systems – to the enedion 185 [137,138]. The double bond is also susceptible to dif-ferent types of nucleophiles [139,140] and it can take part in a [3+2]cycloaddition [141]. Additionally the system readily converts into the hydroquinone form 189 [110,142-144].
The non-conjugated bond usually is unreactive, however, there is at least one reaction that has to be mentioned.
The [3+2]cycloaddition of nitrile-oxides [145] (generated in
situ) to norbornene double bond is an efficient synthetic method to obtain isoxazolines (Scheme 61). This heterocycle can then be transformed into substituted norbornene derivatives in different ways (Scheme 62) [146-148].
Advantages of this method are:
• High yields • Available substrate (with no problem in respect to the exo-
/endo-isomers) • Variety of different available nitrile oxides (Table 7) • A high selectivity of the reaction, which can be used for in-
troducing functional groups to particular position of the norbornene moiety, Scheme 63) [150-152].
It was found that the conjugated system (O=C–CH=CH–C=O) undergoes de-aromatization under the same condition as this reac-tion (Scheme 64) [153,154], but the transformation to the hydroqui-none can be prevented, e.g. by masking of C=C bond with an epox-ide [155,156] or a hydroxyl group, or the transformation of the
O
O
O
O
O
O
O
O
O
O
Et
SPh
OH
OH
O
O
S
RR
184 185
186
187188
189183
O
Scheme 60.
A
BA
B
X
C
A
B
XO
N
Y
A
B
X
ZC
Z
X
C
Y
N
O
Scheme 61. A,B - keto-groups, ketals, hydroxyl groups (if they would not react under condition of pathway); C - protecting group that prevents any undesir-able reactions of the conjugated double bond, obligatory; X - substituent that will present in the resulting compound, obligatory; Y - COOEt, H, Br, etc; Z – a functional group, obtained based on Y, by different treatment.
O
N O
H
N
NO
N
N
NHO
NC
N
N
CN
O
N
O
N
Me
O
H
Me
N
O
c d e
f g, h
O
N
Me
OH
O
Me
a
190 191
O
Me
b
192
193 194 195 196
197 198 199
MeOSO3
Scheme 62. a) W-2 Raney Ni, AlCl3, MeOH/H2O (5:1); [146] b) PTSA (cat), acetone, reflux, 6 hrs, 64%; [149] c) Me2SO4, toluene, 50°C, 2-8 hrs.; d) NaNO2, H2O, r.t., 1-8 hrs.; e) TMG, toluene, r.t., overall yield for 3 steps 72%; [147] f) NEt3, reflux, 2days, 88%; g) BsCl, pyridine, 6.5 days, r.t., quant.; h) t-BuOK, THF, 30 min, quant [148].

2652 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
keto-groups to ketals or hydroxyls or the use of a 2-substituted ad-duct (Scheme 65).
Additionally, it was found that nitroalkanes can yield the same isoxazolines [168], but this method is more complicated due to reactivity of the nitroalkanes towards other functional groups in the adduct.
VI. FUNCTIONALIZATION OF C2 IN ADDUCT III
Treatment of epoxide 184 by a mixture of acetic and sulfuric
acid produces the acetoxy-adduct 205 [169]. Since the configura-
tion of the adduct does not change, it can undergo photocyclization to form the acetate of 9-hydroxy-Cookson diketone (Scheme 67). This is, perhaps, the simplest possible way to synthesis 9-substituted Cs-THC. The disadvantage of this scheme is similar to that of Scheme 8: the substituents of the THC-cage hardly change from one to another.
Different nucleophiles can react with trienedione 209, resulting
in the endo- adducts (Scheme 68) [170].
The question of the individual reactivity for adducts with dif-ferent substituents is still open. It was shown that the addition of
an arylsulfonyl group to the phenyl-substituted adduct 212 can successfully take place (Scheme 69) [172,173]. However, such addition is very sensitive to the solvent – the formation of the hy-
droquinone product (210, 213) is a concurrent process.
Despite the fact that the arylsulphuric substituents are probably not the best one, they can be useful for retrieving the endo- configu-
CH2OH CH2OHON CH2OH
O
N
Ph
Ph
+CH2OH
O
N
(EtO)2P
200201 202 203
O
a b
Scheme 63. a) (i-PrO)2P(O)C(Cl)NOH, NEt3, CH2Cl2, 12 hrs, 75%; b) PhC(Cl)NOH, NEt3, DMF, 1 min, 82%, 202:203=3:2.
Table 7.
N-oxide Precursor Ref
carbethoxyformonitrile oxide (CEFNO) EtO2C–C�N+–O-
O
O
N
Cl
OH
[157]
Cyanogens N-oxide (CNO). N�C–C�N+–O- NC N
Cl
OH
[158]
2-oxopropanenitrile oxide CH3–C�N+–O- Acetone + CAN [159,160]
trifluoroacetonitrile oxide CF3–C�N+–O- F3C N
Cl
OH
[161]
1,3-Dithiane of 2-Oxopropanenitrile Oxide
N
S S
Me C O N OH
S S
Me C
H
[162]
bromonitrile oxide Br–C�N+–O- Br N
Br
OH
[163]
benzenesulfonylcarbonitrile oxide PhSO2–C�N+–O- O
S
O
N
Br
OHPh
[164]
Me3Si C N O from Hg(CNO)2 and TMS-Br [165,166] fulminic acid HCNO
H N
I
OH
from Hg(CNO)2 and HI, KI
[167]
O
N
C
R
C
N
R
O
+
O
O OH
OH183 204
a
Scheme 64. a) Ethyl chloroximidoacetate, NaHCO3, [bmim]BF4, ionic liquid, r.t., 3-4 hrs., 72% (R = COOEt).
Table 8.
R Ref
Ph [153]
COOEt [154]
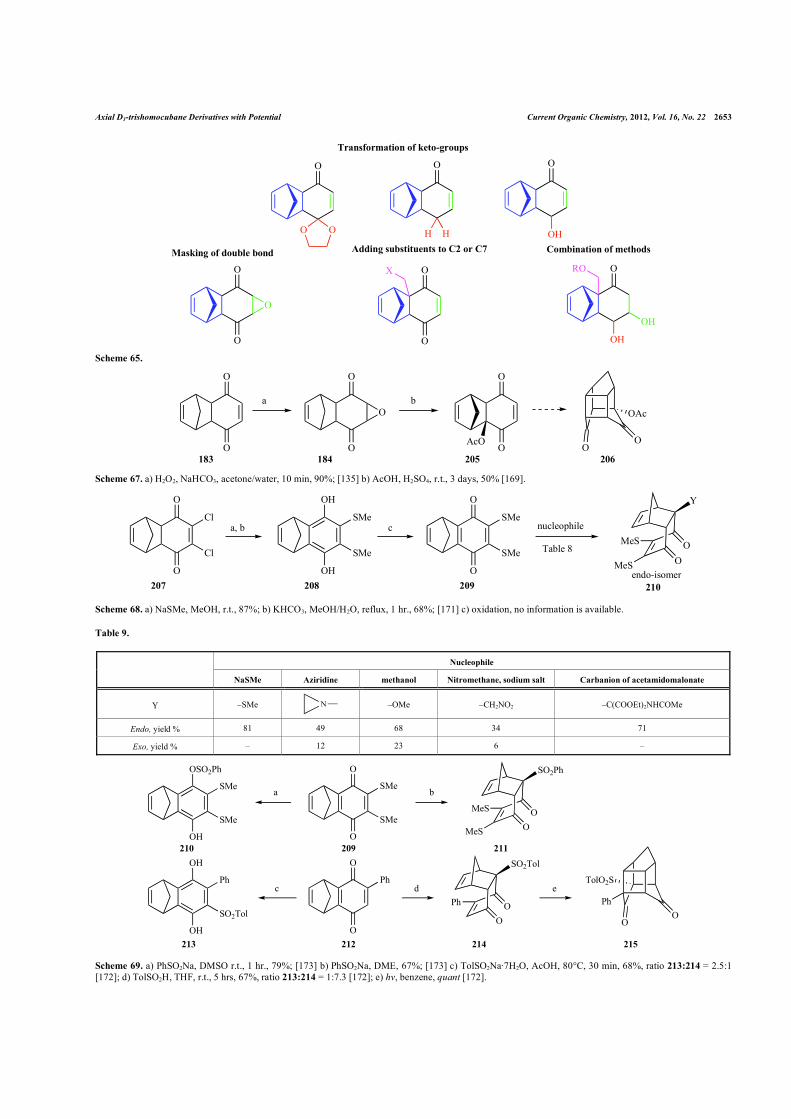
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2653
O
OX
OH
O
O
O
O
OO
O
OH
O
OH
RO
Adding substituents to C2 or C7
Transformation of keto-groups
O
Masking of double bond Combination of methods
H H
Scheme 65.
O
O
O
OAcO
OAc
OO
O
O
O
183 184 205 206
a b
Scheme 67. a) H2O2, NaHCO3, acetone/water, 10 min, 90%; [135] b) AcOH, H2SO4, r.t., 3 days, 50% [169].
O
O
Y
MeS
MeS
O
O
SMe
SMe
OH
OH
SMe
SMe
O
O
Cl
Cl
nucleophile
endo-isomer207 208 209 210
a, b c
Table 8
Scheme 68. a) NaSMe, MeOH, r.t., 87%; b) KHCO3, MeOH/H2O, reflux, 1 hr., 68%; [171] c) oxidation, no information is available.
Table 9.
Nucleophile
NaSMe Aziridine methanol Nitromethane, sodium salt Carbanion of acetamidomalonate
Y –SMe N
–OMe –CH2NO2 –C(COOEt)2NHCOMe
Endo, yield % 81 49 68 34 71
Exo, yield % – 12 23 6 –
OSO2Ph
OH
O
O
SO2Ph
SMe
SMe
MeS
MeS
O
O
SMe
SMe
OH
OH
O
O
SO2Tol
Ph
Ph
O
O
Ph
SO2Tol
a b
209210 211
212 214213
c d
TolO2S
OO
Ph
215
e
Scheme 69. a) PhSO2Na, DMSO r.t., 1 hr., 79%; [173] b) PhSO2Na, DME, 67%; [173] c) TolSO2Na·7H2O, AcOH, 80°C, 30 min, 68%, ratio 213:214 = 2.5:1 [172]; d) TolSO2H, THF, r.t., 5 hrs, 67%, ratio 213:214 = 1:7.3 [172]; e) hv, benzene, quant [172].
2654 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
O
O
O
O OO
OO
O
O
HOH2C HOH2C
O
O
O
O
CH2OH
OO
O
HOH2C HOH2C
HOH2C
HOH2C
HOH2C HOH2C
H
H
95%
quant
184
216a 216b
217
Scheme 70. a) DBU (0.1 equiv), 40% formalin, THF, 0°C, 95% [178]; b) DBU (2.1 equiv.), 40% formalin (excess), 0°C, quant.; compare 70 and 216b.
O
O
O
O
HOH2C
O
O
O
HOH2C
H
H
O
O
O
H
H
O
O
O
OTES
O
HOH2C
O
O
H
H
O
O
H
R R
OTESXY XY
O O O O
HOH2C
218 219 220 221
222 223 224 225
a
b
c
d
Scheme 71. a) DBU, 40% formalin, THF, 0°C, 65% (X,Y=O), 86% (X=OH, Y=H), 91% (X=H, Y=OH); b) DBU, 35% formalin, THF, 0 °C, 2 h, 92%; [183]
c) HCHO (3 equiv), DBU (0.5 equiv), THF, rt, 5 h, 94% [187]; also see [188] d) DBU, 37% formalin, THF, 0°C-25°C, 98% [181].
O
O
O
HOH2C
O
OH
O
TBSOH2C
O
OH
O
TBSOH2C
OAc
O
O
TBSOH2C
45%
46%
H
H
H
H
OH
O
O
HOH2C
H
215
226
228
(–)-227
(+)-227
a, b
c
d
Scheme 72. a) TBSCl, imid. DMAP, DMF, rt, 92%; b) NaBH4, MeOH, 15°C, 81%; c) Lipase PS-D (Amano), vinyl acetate, rt, 28h. [178]; d) DIBAL-H, THF,
–78°C, 65% [189].
ration of adduct once it is turned into the hydroquinone form (by
functionalization in the intended position). Another way is to use
the triene 209, followed by the removal of the methylsulfanyl sub-
stituents. For example, this can be done by changing the groups to
halogens [174] or by oxidizing and removing them after the forma-
tion of the cage [175-177]. Anyway, this pathway seems to be too
complicated and long.
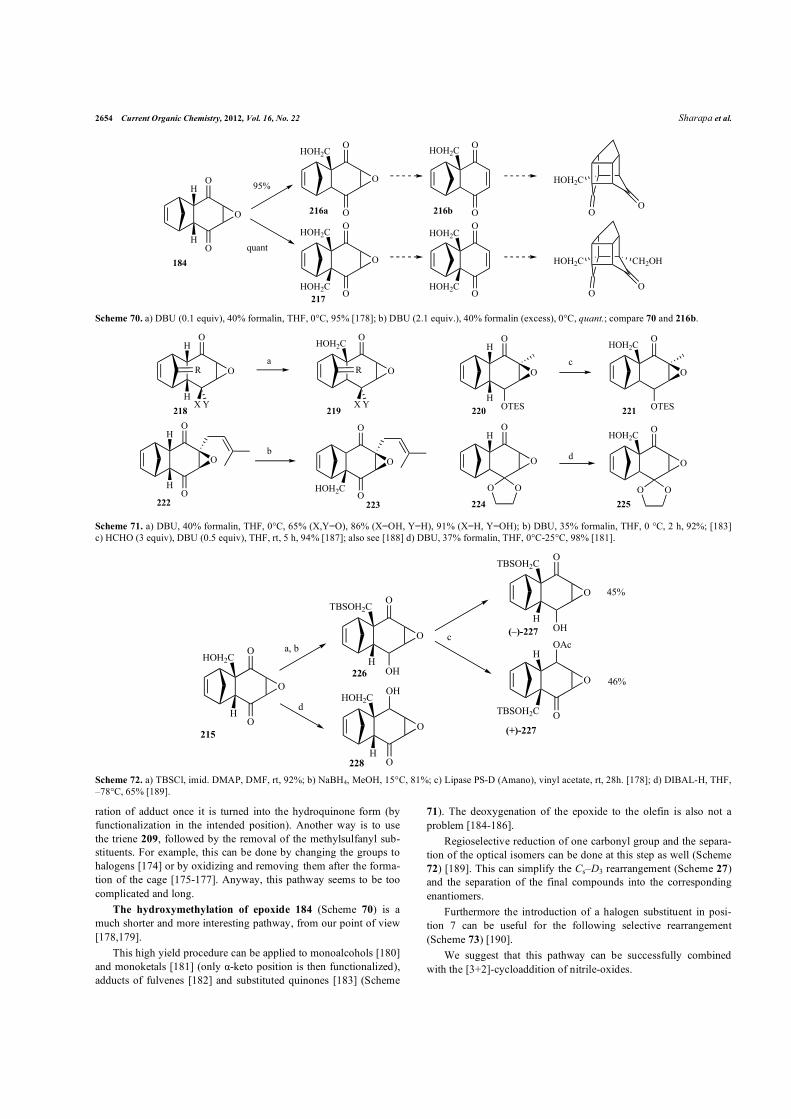
The hydroxymethylation of epoxide 184 (Scheme 70) is a
much shorter and more interesting pathway, from our point of view
[178,179].
This high yield procedure can be applied to monoalcohols [180]
and monoketals [181] (only �-keto position is then functionalized),
adducts of fulvenes [182] and substituted quinones [183] (Scheme
71). The deoxygenation of the epoxide to the olefin is also not a
problem [184-186].
Regioselective reduction of one carbonyl group and the separa-
tion of the optical isomers can be done at this step as well (Scheme
72) [189]. This can simplify the Cs–D3 rearrangement (Scheme 27)
and the separation of the final compounds into the corresponding
enantiomers.
Furthermore the introduction of a halogen substituent in posi-
tion 7 can be useful for the following selective rearrangement
(Scheme 73) [190].
We suggest that this pathway can be successfully combined
with the [3+2]-cycloaddition of nitrile-oxides.
Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2655
OH
O
O
TBSOH2C
H
228
OH
O
O
TBSOH2C
BrR R
229
a
OO
Br
HO
OCl
HO
BrHOH2C
Scheme 73. a) 1,2-dibromotetrachloroethane (2.0 equiv) DBU, CH2Cl2, r.t., 10 hrs, quant. R = allyl.
O
O
O
O
O
O
CH2
RO
O
OH
O
CH2
RO
O
OH
O
CH2
RO
O
N
C
Z
O
OH
OH
CH2
RO
HO
Y
O
OH
CH2
RO
Y
O
O
O
CH2
RO
O
N
C
Z
O
O
CH2
RO
Y
OH
OH
O
CH2
RO
OH
OH
CH2
RO
O
N
C
Z
OH
OH
CH2
RO
Y
OH
OH
CH2
RO
O
O
CH2
RO
CH2OR
O
O
Y
COOMe
O
Y O
O
COOMe
OH
Y
H2C
YOH
OH
OR
H2C
Y
O
O
O
CH2OH
OH
Y
CH2OR
OH
O
Y
CH2OR
Br
O
Y
CH2OR
Br
OH
Y
YX
Nu
YX
photocyclization
rearrangement
introduction of first target group
introduction
of
second
target
group
V
VI
VII
IX
VIII
XII
XIII
XIX
XIV
XV XVI XVII
XVIII XIX XX
XXVXXIIIXXI
XXII XXIV XXVI
XXVII
XXVIII
Scheme 74.
2656 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
O
O
HOH2C
H
OH
O
HOH2C
BrO
O
Br
HO
OCl
HOBr
HOH2C
O
O
O
H
H
O
N
Z
O
O
O
HOH2C
H
O
N
Z
Y
Y
Y
Ias it was described on previous
scheme
Y
XI XVIXXIX
XXX XXXI XXXII Scheme 75.
O
OR
R
Br
Br
O
OR
RO
O
R R
O
O
hindered
positionhindered
position
OO
R R
O
O
Y
CH2OHR = Me, Ph, etc XXXIII
XXXIV
OEt
OEt
OEtEtO
O
O
O
O
O
XXXV Scheme 76.
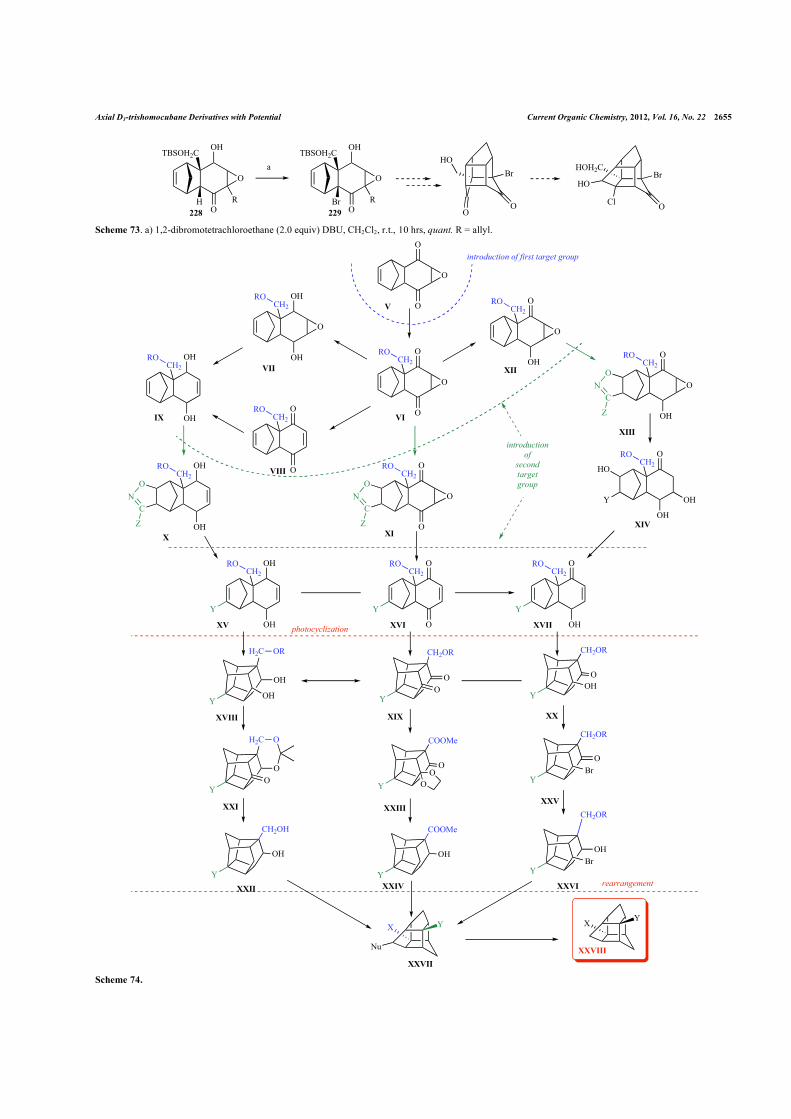
VII. GENERAL SYNTHESIS SCHEME
Hence, based on the reviewed methods and pathways above, the general pathways to 2,9-D3-trishomocubane can be drawn (Scheme 74). As a starting compound the readily available epoxide V can be used. First the substituent can be introduced into position 2 via hydroxymethylation. After that several options are possible: the [3+2]cycloaddition of nitrile-oxides can be done directly, either after the reduction of one carbonyl or after the deoxygenation of epoxide and the reduction of both carbonyls. The opening of the heterocycles followed by a dehydration of the alcohols should result in disubstituted adducts (XV-XVII), and adduct XVI, which can be converted into the other two (XV and XVII).
The Cs-trishomocubane derivatives XVII-XX (obtained by pho-tocyclization of adducts XV-XVI, respectively), can be converted into one another by reduction-oxidation steps. Thus, independently from the first synthetic steps, at least three independent pathways for the formation of suitable monoalcohol derivative for the rear-rangement can be proposed:
1) Selective ketalization or thioketalization of 9-substituted diketone XIX, followed by reduction of the second carbonyl group and removal of the protecting one (via Ni-Ra for thioketal or via deprotection and Wolf-Kishner reduction);
2) Changing of the hydroxyl group into ketoalcohol XX to bromine followed by reduction of the carbonyl;
3) Protection of one hydroxyl group in XVIII by the formation of an acetonide XXI, followed by the oxidation of the second hy-droxyl group and the reduction of the carbonyl as it was described before.
The monoalcohols XXII, XXIV, XXVI can undergo rear-rangement to form the D3-THC-derivative XXVII, which after removal of all useless halo- and hydroxy- groups gives the targeted “diaxial-substituted D3-THC” XXVIII.
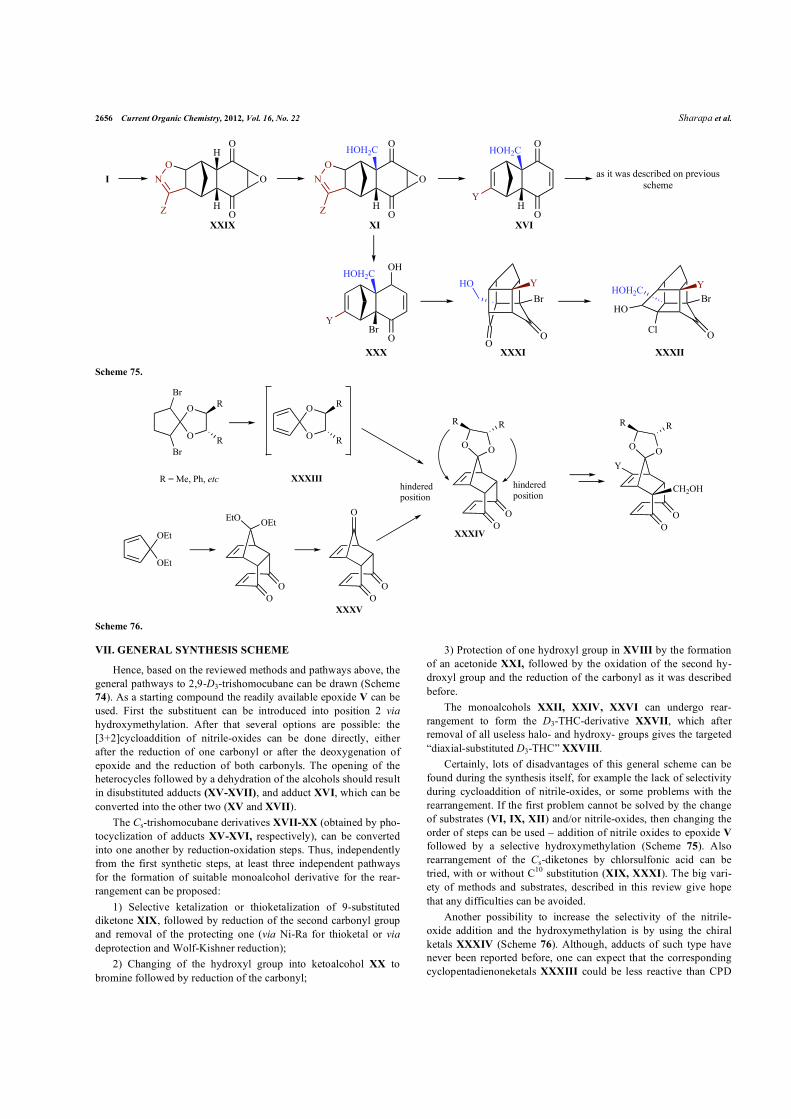
Certainly, lots of disadvantages of this general scheme can be found during the synthesis itself, for example the lack of selectivity during cycloaddition of nitrile-oxides, or some problems with the rearrangement. If the first problem cannot be solved by the change of substrates (VI, IX, XII) and/or nitrile-oxides, then changing the order of steps can be used – addition of nitrile oxides to epoxide V followed by a selective hydroxymethylation (Scheme 75). Also rearrangement of the Cs-diketones by chlorsulfonic acid can be tried, with or without C10 substitution (XIX, XXXI). The big vari-ety of methods and substrates, described in this review give hope that any difficulties can be avoided.
Another possibility to increase the selectivity of the nitrile-oxide addition and the hydroxymethylation is by using the chiral ketals XXXIV (Scheme 76). Although, adducts of such type have never been reported before, one can expect that the corresponding cyclopentadienoneketals XXXIII could be less reactive than CPD

Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2657
ethylene ketal because of steric hindrance. Also the ketal XXXIV can possibly be synthesized from the triketone XXXV.
VIII. CONCLUSIONS AND FUTURE PERSPECTIVES
The D3-trishomocubane is one of the few molecules belonging to the D3 point symmetry group and despite the high symmetry has a chiral cage structure. C3-symmetry axe is also retained in 2- and 2,9- derivatives of D3-trishomocubane. While such derivatives have great potential to be used as scaffolds for drugs, in structure-oriented design, asymmetric catalysis, rigid axial rods, light-driven systems etc., they still remain to be elusive. The direct functionali-zation of D3-trishomocubane usually leads to formation of 4-derivatives. As an alternative methodology rearrangement of Cs-trishomocubane derivatives to corresponding D3-trishomocubane can be used. However, in such case the major problem is also func-tionalization of Cs-trishomocubane. Consequently, functional groups should be already present from the beginning, either in cy-clopentadiene or quinone or to be introduced upon functionalization of the adduct.
Based on the extensive retro-synthetic analysis a general scheme for the synthesis of 2- and 2,9- derivatives of D3-trishomocubane is suggested. All steps of the proposed scheme are reviewed and discussed.
Although D3-trishomocubane is known for more than four dec-ades, its chemistry is still insufficiently studied. Further investiga-tions on the chemistry, reactivity and biological properties of such compounds are required. We hope that this review will instigate such studies.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
This work was supported by the State Basic Research Fund of Ukraine (grants from the President of Ukraine to young scientists 2005-2011). We thank “Macrochem” and “UkrOrgSynthesis” for their support.
REFERENCES
[1] Marchand, A. P. Synthesis and chemistry of homocubanes, bishomocubanes, and trishomocubanes Chem. Rev. 1989, 89, 1011-1033.
[2] Levandovsky, I. A.; Sharapa, D. I.; Cherenkova, O. A.; Gaidai, A. V.; Shubina, T. E. The chemistry of D3-trishomocubane Russ. Chem. Rev. 2010, 79, 1005-1026.
[3] Underwood, G. R.; Ramamoorthy, B. Chemical studies of caged compounds. II. Synthesis of pentacyclo[6.3.0.02,6.03,10.05,9]undecane: trishomocubane Tetrahedron Lett. 1970, 4125-4127.
[4] Eaton, P. E.; Leipzig, B. Resolution of trishomocubanone: the enantiomeric (D3)-trishomocubanes J. Org. Chem. 1978, 43, 2483-2484.
[5] Nakazaki, M.; Naemura, K.; Arashiba, N. Syntheses and chiroptical properties of (-)-ditwist-brendane and (+)-D3-trishomocubane J. Org. Chem. 1978, 43, 689-692.
[6] Helmchen, G.; Staiger, G. Synthesis and absolute configuration of the enantiomers of pure D3-tris(homocubanes) (pentacyclo[6.3.0.02,6.03,10.05,9]undecane) and tris(homocubanones) Angew. Chem. 1977, 89, 119-120.
[7] Fessner, W. D.; Prinzbach, H. D3-Trishomocubanetrione. Synthesis and optical resolution Tetrahedron 1986, 42, 1797-1803.
[8] Nakazaki, M.; Naemura, K.; Arashiba, N.; Iwasaki, M. Syntheses of novel gyrochiral pentacyclic systems with C2 symmetry. (-)-C2-Bismethanotwistane and (±)-C2-methanoditwistane J. Org. Chem. 1979, 44, 2433-2438.
[9] Nakazaki, M.; Naemura, K.; Chikamatsu, H.; Iwasaki, M.; Hashimoto, M. Synthesis and absolute configuration of optically active D3-tritwistane; the gyrochiral prototype of "twist" diamond J. Org. Chem. 1981, 46, 2300-2306.
[10] Adachi, K.; Naemura, K.; Nakazaki, M. Synthesis and absolute configuration of optically active tricyclo[4.4.0.03,8]decane Tetrahedron Lett. 1968, 5467-5470.
[11] Tichy, M.; Sicher, J. Synthesis and absolute configuration of tricyclo [4.4.0.0.3,8]dec-4-ene (twistene) Tetrahedron Lett. 1969, 4609-4613.
[12] Nakazaki, M.; Naemura, K.; Nakahara, S. Absolute configurations and absolute rotations of C2-bishomocubane, ditwist-brendane, and twistane J.
Org. Chem. 1978, 43, 4745-4750. [13] McCann, D. M.; Stephens, P. J.; Cheeseman, J. R. Determination of Absolute
Configuration Using Density Functional Theory Calculation of Optical Rotation: Chiral Alkanes J. Org. Chem. 2004, 69, 8709-8717.
[14] Mueller-Boetticher, H.; Fessner, W. D.; Melder, J. P.; Prinzbach, H.; Gries, S.; Irngartinger, H. Functionalized enantiomerically pure [1.1.1]-, [2.1.1.]-, [2.2.1]-, and [2.2.2]triblattanes Chem. Ber. 1993, 126, 2275-2297.
[15] Gaidai, A. V.; Volochnyuk, D. M.; Shishkin, O. V.; Fokin, A. A.; Levandovskiy, I. A.; Shubina, T. E. D3-trishomocubane-4-carboxylic acid as a new chiral building block: synthesis and absolute configuration Synthesis 2012, 44, 810-816.
[16] Nakazaki, M.; Chikamatsu, H.; Naemura, K.; Nishino, M.; Murakami, H.; Asao, M. Microbial stereodifferentiating reduction of the carbonyl groups located on the C2 axes of gyrochiral molecules J. Org. Chem. 1979, 44, 4588-4593.
[17] Nakazaki, M.; Chikamatsu, H.; Naemura, K.; Sasaki, Y.; Fujii, T. The C2-ketone rule in horse liver alcohol dehydrogenase (HLADH)-catalyzed oxidation and reduction J. Chem. Soc., Chem. Commun. 1980, 626-627.
[18] Nakazaki, M.; Chikamatsu, H.; Naemura, K.; Suzuki, T.; Iwasaki, M.; Sasaki, Y.; Fujii, T. C2-ketone rule in horse liver alcohol dehydrogenase (HLADH) mediated oxidoreduction J. Org. Chem. 1981, 46, 2726-2730.
[19] Naemura, K.; Fujii, T.; Chikamatsu, H. Selective and stereospecific horse liver alcohol dehydrogenase-catalyzed reduction of cage-shaped meso-diketone. An efficient access to optically active D3-trishomocubane derivative Chem. Lett. 1986, 923-926.
[20] Nakazaki, M.; Naemura, K. Synthesis and absolute configuration of the first optically active organic molecule with T symmetry. (-)-1,3,5,7-Tetrakis[2-(1S,3S,5R,6S,8R,10R)-D3-trishomocubanylacetoxymethyl]adamantane J.
Chem. Soc., Chem. Commun. 1980, 911-912. [21] Nakazaki, M.; Naemura, K. Synthesis and absolute configuration of the first
optically active organic molecule with T symmetry: (-)-1,3,5,7-tetrakis[[(2-(1S,3S,5R,6S,8R,10R)-D3-trishomocubanyl)acetoxy]methyl]adamantane J.
Org. Chem. 1981, 46, 106-111. [22] Nakazaki, M.; Naemura, K.; Hokura, Y. Synthesis of (+)-1,3,5,7-tetrakis[2-
(1S,3S,5R,6S,8R,10R)-D3-trishomocubanylbuta-1,3-diynyl]adamantane. An optically active organic molecule with T symmetry of known absolute configuration J. Chem. Soc., Chem. Commun. 1982, 1245-1246.
[23] Naemura, K.; Hokura, Y.; Nakazaki, M. Synthesis of (+)-1,3,5,7-tetrakis[2-(1S,3S,5R,6S,8R,10R)-D3-trishomocubanylbuta-1,3-diynyl]adamantane, the first optically active organic molecule with T symmetry and of known absolute configuration Tetrahedron 1986, 42, 1763-1768.
[24] Mislow, K. On the symmetry of (-)-1,3,5,7-tetrakis[2-(1S,3S,5R,6S,8R,10R)-D3-trishomocubanylacetoxymethyl]adamantane J. Chem. Soc., Chem.
Commun. 1981, 234. [25] Schwartz, M.; Marchand, A. P.; Wang, K. S.; Reddy, S. P.; Redda, G. M.;
Gadgil, V. R.; Watson, W. H.; Kashyap, R. P.; Krawiec, M. Synthesis of 2-substituted pentacyclo[6.3.0.02,6.03,10.05,9]undecanes (D3-trishomocubanes) and a study of their carbon-13 spin-lattice relaxation times in solution J. Chem. Soc., Perkin Trans. 2 1993, 1829-1836.
[26] Karlen, S. D.; Ortiz, R.; Chapman, O. L.; Garcia-Garibay, M. A. Effects of Rotational Symmetry Order on the Solid State Dynamics of Phenylene and Diamantane Rotators J. Am. Chem. Soc. 2005, 127, 6554-6555.
[27] Hoki, K.; Yamaki, M.; Fujimura, Y. Optical control of chiral molecular motors Springer Ser. Chem. Phys. 2008, 89, 92-112.
[28] Yamaki, M.; Nakayama, S.-i.; Hoki, K.; Kono, H.; Fujimura, Y. Quantum dynamics of light-driven chiral molecular motors Phys. Chem. Chem. Phys. 2009, 11, 1662-1678.
[29] Kottas, G. S.; Clarke, L. I.; Horinek, D.; Michl, J. Artificial Molecular Rotors Chem. Rev. (Washington, DC, U. S.) 2005, 105, 1281-1376.
[30] Kelly, T. R.; Editor Molecular Machines. [In: Top. Curr. Chem., 2005; 262], 2005.
[31] Schalley, C. A. Of molecular gyroscopes, Matroshka dolls, and other "nano"-toys Angew. Chem., Int. Ed. 2002, 41, 1513-1515.
[32] Kelly, T. R.; Sestelo, J. P.; Tellitu, I. New Molecular Devices: In Search of a Molecular Ratchet J. Org. Chem. 1998, 63, 3655-3665.
[33] Sebastian, K. L. Molecular ratchets: Verification of the principle of detailed balance and the second law of dynamics Phys. Rev. E Stat. Phys., Plasmas,
Fluids, Relat. Interdiscip. Top. 2000, 61, 937-939. [34] Halterman, R. L.; Jan, S. T. Catalytic asymmetric epoxidation of
unfunctionalized alkenes using the first D4-symmetric metallotetraphenylporphyrin J. Org. Chem. 1991, 56, 5253-5254.
[35] Zheng, Y. B.; Hao, Q.; Yang, Y.-W.; Kiraly, B.; Chiang, I. K.; Huang, T. J. Light-driven artificial molecular machines J. Nanophotonics 2010, 4, No pp given.
[36] Schwab, P. F. H.; Levin, M. D.; Michl, J. Molecular Rods. 1. Simple Axial Rods Chem. Rev. (Washington, D. C.) 1999, 99, 1863-1933.

2658 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
[37] Pati, R.; Karna, S. P. Length-dependence of intramolecular electron transfer in �-bonded rigid molecular rods: an ab initio molecular orbital study Chem.
Phys. Lett. 2002, 351, 302-310. [38] Ohya, Y.; Takamido, S.; Nagahama, K.; Ouchi, T.; Ooya, T.; Katoono, R.;
Yui, N. Molecular "Screw and Nut": �-Cyclodextrin Recognizes Polylactide Chirality Macromolecules (Washington, DC, U. S.) 2007, 40, 6441-6444.
[39] Sharapa, D. I.; Gayday, A. V.; Mitlenko, A. G.; Levandovskiy, I. A.; Shubina, T. E. A Convenient Road to 1-Chloropentacycloundecanes - A Joint Experimental and Computational Investigation Eur. J. Org. Chem. 2011, 2554-2561, S2554/2551-S2554/2537.
[40] Sorochinskii, A. E.; Aleksandrov, A. M.; Petrenko, A. E.; Kukhar, V. P. Bromination of pentacyclo[6.3.0.02,6.03,10.05,9]undecane Zh. Org. Khim. 1987, 23, 2247.
[41] Petrenko, A. E.; Aleksandrov, A. M.; Sorochinskii, A. E.; Kukhar, V. P. Chlorocarbonylation of pentacyclo[6.3.0.02,6.03,10.05,9]undecane Zh. Org.
Khim. 1987, 23, 2248. [42] Blum, J.; Zlotogorski, C.; Zoran, A. Simple route from dicyclopentadiene to
trishomocubanone Tetrahedron Lett. 1975, 1117-1120. [43] Zlotogorski, C.; Blum, J.; Osawa, E.; Schwarz, H.; Hoehne, G.
Carbonylation and valence isomerization of 1,3-dihomocubane derivatives by chlorodicarbonylrhodium dimer J. Org. Chem. 1984, 49, 971-976.
[44] Suri, S. C.; Hardcastle, K. I. A non-photochemical approach for synthesizing functionalized pentacyclo[5.4.0.02,6.03,10.05,9]undecanes using samarium (II) iodide J. Org. Chem. 1992, 57, 6357-6360.
[45] Suri, S. C. Base-promoted eliminative cyclization: novel synthesis of functionalized tetracyclo[6.2.1.02,7.04,10]undecane and tetracyclo[5.3.0.02, 6.O5,9]decane systems Tetrahedron Lett. 1990, 31, 3695-3698.
[46] Aleksandrov, A. M.; Sorochinskii, A. E.; Kukhar, V. P. Reaction of 12-oxahexacyclo[5.4.1.02,6.03,10.05,9.08,11]dodecane with bromine Zh. Org.
Khim. 1986, 22, 2233-2234. [47] Axt, M.; Oulyadi, H.; Pannecoucke, X.; Quirion, J.-C.; Pohlmann, A. R.;
Costa, V. E. U. Peptide analogs containing the pentacyclo[5,4,0,02,6,03,6, 05,9]undecane scaffold: conformational analysis in solution J. Mol. Struct. 2004, 689, 49-60.
[48] Marchand, A. P.; LaRoe, W. D.; Sharma, G. V. M.; Suri, S. C.; Reddy, D. S. Facile stereoselective reductions of enediones and cage diketones suing sodium borohydride-cerium(III) chloride J. Org. Chem. 1986, 51, 1622-1625.
[49] Aleksandrov, A. M.; Sorochinskii, A. E.; Petrenko, A. E.; Kukhar, V. P. Synthesis of 1-substituted pentacyclo[6.3.0.02,6.03,10.05,9]undecane (symmetrical trishomocubane) Zh. Org. Khim. 1987, 23, 756-761.
[50] Aleksandrov, A. M.; Sorochinskii, A. E.; Petrenko, A. E.; Kukhar, V. P. Fluorination of pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione with sulfur tetrafluoride Zh. Org. Khim. 1988, 24, 149-152.
[51] Petrenko, A. E.; Aleksandrov, A. M.; Sorochinskii, A. E.; Kukhar, V. P. Reactions of tetracyclo[6.3.0.04,11.05,9]undecane-2,7-dione with electro-philic halogenating reagents Zh. Org. Khim. 1990, 26, 2361-2365.
[52] Watson, W. H.; Kashyap, R. P.; Krawiec, M.; Marchand, A. P.; Tsay, F. R. Structures of three substituted pentacyclo[5.4.0.02,6.03,10.05,9]undecanes and a computational analysis of bond lengths Struct. Chem. 1994, 5, 21-35.
[53] Watson, W. H.; Kashyap, R. P.; Marchand, A. P.; Vidyasagar, V. Structure of a pentacyclic cage enone Acta Crystallogr., Sect. C Cryst. Struct.
Commun. 1990, C46, 926-928. [54] Marchand, A. P.; Annapurna, P.; Reddy, S. P.; Watson, W. H.; Nagl, A.
Lewis acid-promoted reactions of substituted pentacyclo[5.4.0.02,6.03, 1005,9]undecane-8,11-diones with ethyl diazoacetate J. Org. Chem. 1989, 54, 187-193.
[55] Watson, W. H.; Nagl, A.; Marchand, A. P.; Reddy, G. M.; Reddy, S. P. Regiospecific formation of a methyl pentacyclo[5.4.0.02,6.03,10.05,9]unde-cane-8,11-dione-9-carboxylate monoketal Acta Crystallogr., Sect. C Cryst.
Struct. Commun. 1989, C45, 659-661. [56] Watson, W. H.; Nagl, A.; Kashyap, R. P.; Marchand, A. P.; Lu, S. P.; Dave,
P. R.; Annapurna, P. Structures of two hydrated cage diketones Acta
Crystallogr., Sect. C Cryst. Struct. Commun. 1990, C46, 1875-1879. [57] Rodionov, V. N.; Sklyarova, A. S.; Shamota, T. V.; Schreiner, P. R.; Fokin,
A. A. Selective reductive dimerization of homocubane series oximes Russ. J.
Org. Chem. 2011, 47, 1695-1702. [58] Watson, W. H.; Nagl, A.; Marchand, A. P.; Reddy, G. M. Structure of an
open-ended cage compound Acta Crystallogr., Sect. C Cryst. Struct.
Commun. 1989, C45, 1600-1602. [59] Kent, G. J.; Godleski, S. A.; Osawa, E.; Schleyer, P. v. R. Syntheses and
relative stability of (D3)-trishomocubane (pentacyclo[6.3.0.02,6.03,10.05,9] undecane), the pentacycloundecane stabilomer J. Org. Chem. 1977, 42, 3852-3859.
[60] Mehta, G.; Chaudhuri, B. An efficient preparative route to functionalized (D3)-trishomocubanes Indian J. Chem., Sect. B 1979, 17B, 421-422.
[61] Smith, E. C.; Barborak, J. C. Syntheses of the pentacyclo[6.3.0.02,6.03, 10.05,9]undecyl (trishomocubyl) and tetracyclo[6.3.0.04,11.05,9]undeca-2,6-dienyl (homohypostrophenyl) systems J. Org. Chem. 1976, 41, 1433-1437.
[62] Marchand, A. P.; Wang, Y.; Ren, C.-t.; Vidyasagar, V.; Wang, D. On the mechanism of acid promoted ring opening of a pentacyclo [5.4.0.02,6.03,10.05,9]undecane-spiroannulated oxetane Tetrahedron 1996, 52, 6063-6072.
[63] Eaton, P. E.; Cassar, L.; Hudson, R. A.; Hwang, D. R. Synthesis of homopentaprismane and homohypostrophene and some comments on the
mechanism of metal ion catalyzed rearrangements of polycyclic compounds J. Org. Chem. 1976, 41, 1445-1448.
[64] Mehta, G.; Reddy, M. S. A strategy for the construction of novel tetracyclic Lycopodium alkaloids of paniculatine- and magellanine-type Tetrahedron
Lett. 1990, 31, 2039-2042. [65] Martins, F. J. C.; Viljoen, A. M.; Kruger, H. G.; Fourie, L.; Roscher, J.;
Joubert, A. J.; Wessels, P. L. Enantioselective synthesis of amino acids from pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione Tetrahedron 2001, 57, 1601-1607.
[66] Mehta, G.; Sreenivasa Reddy, M.; Thomas, A. Synthetic studies towards novel tetracyclic lycopodium alkaloids: a synthesis of deoxymagellaninone Tetrahedron 1998, 54, 7865-7882.
[67] Furuya, A.; Arai, H.; (DIC Corporation, Japan). Application: JP, 2008, p 12pp.
[68] Aleksandrov, A. M.; Turov, A. V.; Kornilov, M. Y.; Kukhar, V. P. Hydroxy derivatives of pentacyclo[5.4.0.02,6.03,10.05,9]undecane Zh. Org. Khim. 1991, 27, 2566-2572.
[69] Watson, W. H.; Kashyap, R. P.; Marchand, A. P.; Reddy, S. P. Formation of methyl 11-hydroxy-8-oxopentacyclo[5.4.0.02,6.03,10.05,9]undecanecar-boxylate by a regiospecific and stereospecific reduction Acta Crystallogr.,
Sect. C Cryst. Struct. Commun. 1991, C47, 376-378. [70] Yoshida, N.; Konno, H.; Kamikubo, T.; Takahashi, M.; Ogasawara, K.
Preparation of a synthetic equivalent of chiral methyl 2,5-dihydroxycyclo-hexane-1,4-dienecarboxylate Tetrahedron Asymmetry 1999, 10, 3849-3857.
[71] Tolstikov, G. A.; Lerman, B. M.; Galin, F. Z. Skeletal rearrangement during reaction of skeletal gamma -diketones with chlorosulfonic acid Zh. Org.
Khim. 1976, 12, 1133-1134. [72] Tolstikov, G. A.; Lerman, B. M.; Galin, F. Z. Synthesis and some reactions
of chlorooxochlorosulfates of trishomocubane and dihomobasketane Zh.
Org. Khim. 1977, 13, 1634-1638. [73] Tolstikov, G. A.; Lerman, B. M.; Galin, F. Z.; Struchkov, Y. T.; Andrianov,
V. G. Synthesis of trishomocubane and dihomobasketane derivatives via the skeletal rearrangement under the action of chlorosulfonic acid Tetrahedron
Lett. 1978, 4145-4148. [74] Watson, W. H.; Nagl, A.; Kashyap, R. P.; Marchand, A. P.; Zhao, D. A
Diels-Alder cycloadduct of methylcyclopentadiene with 2,6-dimethyl-p-benzoquinone and the intramolecular photocyclization product of this cycloadduct Acta Crystallogr., Sect. C Cryst. Struct. Commun. 1989, C45, 1342-1346.
[75] Watson, W. H.; Kashyap, R. P.; Krawiec, M.; Marchand, A. P.; Lu, S. P. Three substituted hexahydro-5,8-dimethanonaphthalenes Acta Crystallogr.,
Sect. C Cryst. Struct. Commun. 1992, C48, 1290-1294. [76] Minter, D. E.; Marchand, A. P.; Tsay, F.-R. Assignment of the 1H and 13C
NMR spectra of 1-methyl-6-phenyl-1�,4�,4a�,5�,8�,8a�-hexahydro-1,4-methanonaphthalene-5,8-diol ARKIVOC (Gainesville, FL, U. S.) 2003, 8-17.
[77] Goering, H. L.; Chang, C.-S. Regioselectivity of Lewis acid catalyzed Diels-Alder reactions of methylcyclopentadiene J. Org. Chem. 1975, 40, 2565.
[78] Kresze, G.; Schulz, G.; Waltz, H. Substituted cyclopentadienes and their Diels-Alder reactions Justus Liebigs Ann. Chem. 1963, 666, 45-53.
[79] Reymond, S.; Cossy, J. Copper-Catalyzed Diels-Alder Reactions Chem. Rev.
(Washington, DC, U. S.) 2008, 108, 5359-5406. [80] Marchand, A. P.; Vidyasagar, V.; Buckner, M. B.; Holman, P. O. Lewis acid
catalysis of a Diels-Alder cycloaddition. An undergraduate organic experiment J. Chem. Educ. 1987, 64, 642-644.
[81] Kyba, E. P.; Hudson, C. W. 1,2-H shifts in carbenes. The benzonorbornenylidene system J. Org. Chem. 1977, 42, 1935-1939.
[82] Yates, P.; Gupta, I. Diels-Alder reactions of (trimethoxymethyl)cyclo-pentadienes. Formation of adducts derived from the 2-substituted tautomer J.
Chem. Soc., Chem. Commun. 1981, 449-451. [83] Ustynyuk, Y. A.; Kisin, A. V.; Pribytkova, I. M.; Zenkin, A. A.; Antonova,
N. D. Nuclear magnetic resonance spectroscopy of metal cyclopentadienyls. X. Proton magnetic resonance spectra of, and dynamic behavior in, bis(trimethylsilyl)cyclopentadiene J. Organometal. Chem. 1972, 42, 47-63.
[84] Tolstikov, G. A.; Lerman, B. M.; Galin, F. Z.; Danilova, N. A. Synthesis of silicon-containing cage diketones Zh. Obshch. Khim. 1977, 47, 1656-1659.
[85] Fleming, I.; Williams, R. V. The reaction between bis(trimethylsilyl)cyclo-pentadiene and dichloroketene, and the Diels-Alder reactions between N-phenylmaleimide and two silylated methylcyclopentadienes J. Chem. Soc.,
Perkin Trans. 1 1981, 684-688. [86] Eaton, P. E.; Hudson, R. A. Cyclopentadienone ketals J. Am. Chem. Soc.
1965, 87, 2769-2771. [87] Allred, E. L.; Anderson, C. L. 2,3-Diazabicyclo[2.2.1]heptyl ring system. III.
Synthesis and characterization of some 7-substituted derivatives J. Org.
Chem. 1967, 32, 1874-1877. [88] Hoffmann, G. G.; Klein, H. New derivatives of 2,4-diphenylbicyclo[3.2.1]
oct-6-ene Chem. Ber. 1986, 119, 514-523. [89] Mehta, G.; Nair, M. S. Experiments in pursuit of pentagonal dodecahedrane:
model synthesis of convex polyquinanes J. Am. Chem. Soc. 1985, 107, 7519-7524.
[90] Kosman, D.; Stock, L. M. Electron paramagnetic resonance spectra of semiquinones. VI. Spin and electron density distributions in bicyclo [2.2.1]heptyl and bicyclo[2.2.2]octyl derivatives of semiquinone J. Amer.
Chem. Soc. 1969, 91, 2011-2021. [91] Knoelker, H.-J.; Baum, E.; Heber, J. Transition metal-diene complexes in
organic synthesis. part 25. Cycloadditions of annulated 2,5-

Axial D3-trishomocubane Derivatives with Potential Current Organic Chemistry, 2012, Vol. 16, No. 22 2659
bis(trimethylsilyl)cyclopentadienones Tetrahedron Lett. 1995, 36, 7647-7650.
[92] Garbisch, E. W., Jr.; Sprecher, R. F. tert-Butyl-substituted cyclopentadi-enones J. Amer. Chem. Soc. 1969, 91, 6785-6800.
[93] Dao Cong, D.; Edward, J. T. Synthesis of some brominated cage molecules as possible precursors to pentaprismane J. Chem. Eng. Data 1982, 27, 211-213.
[94] Ganji, P.; Ibrahim, H. Deoxygenation of Hydroquinones as a General Route to Norbornane-Fused Aromatic Systems: An Entry into Substituted and Functionalized Dimethano- and Methanoanthracenes J. Org. Chem. 2012, 77, 511-518.
[95] Griesbeck, A. G. Synthesis of 7-alkylidenetricyclo[6.3.0.02,6]undeca-4,9-diene-3,11-diones J. Prakt. Chem./Chem.-Ztg. 1992, 334, 558-562.
[96] Ichihara, A.; Kobayashi, M.; Oda, K.; Sakamura, S. Synthetic studies of highly oxygenated cyclohexane derivatives. V. Facile synthesis of quinone epoxides and epoxycyclohexenones by retro-Diels-Alder reaction Tetrahedron Lett. 1974, 4231-4234.
[97] Ichihara, A.; Kobayashi, M.; Oda, K.; Sakamura, S.; Sakai, R. Synthetic studies of highly oxygenated cyclohexane derivatives. Part XII. Facile synthesis of quinone epoxides and 5,6-epoxy-4-hydroxy-2-cyclohexenones via the retro-Diels-Alder reaction Tetrahedron 1979, 35, 2861-2866.
[98] Buttrus, N. H.; Cornforth, J.; Hitchcock, P. B.; Kumar, A.; Stuart, A. S. Synthesis of substituted dibenzophospholes. Part 5. Synthesis of intermediates for 4- and 6-aryl substituents J. Chem. Soc., Perkin Trans. 1 1987, 851-857.
[99] Guan, X.-P.; Su, Z.; Sun, J.-G.; Yu, Y.-Z. Synthesis of halogenated 4-isopropylidenepentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-diones Molecules 1996, 1, 46-50.
[100] Morita, S.; Fukushima, S.; Kanematsu, K. p-Benzoquinone-2,3-dicarboxylic anhydride as highly reactive and versatile electron-accepting dienophile Tetrahedron Lett. 1979, 2151-2154.
[101] Kanematsu, K.; Morita, S.; Fukushima, S.; Osawa, E. Reagent design and study of p-benzoquinone derivatives as highly reactive electron-attracting dienophiles. A promising class of reagents (synthons) for cycloaddition J.
Am. Chem. Soc. 1981, 103, 5211-5215. [102] Wilson, W. S.; Warrener, R. N. Mild retro-[�4s+�2s] cleavage route to
furans and fulvenes J. Chem. Soc., Chem. Commun. 1972, 211-212. [103] Herges, R.; Reif, W. Synthesis of methyl 6,6-dimethylfulvene-2-carboxylate
and dimethyl 6,6-dimethylfulvene-2,3-dicarboxylate as well as their conversion to 2,3,5,6-substituent norbornadienes Chem. Ber. 1994, 127, 1143-1145.
[104] Singh, V. K.; Raju, B. N. S.; Deota, P. T. A convenient synthesis of novel pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione-4-spiro-1-cyclopropane Synth. Commun. 1986, 16, 1731-1735.
[105] Paquette, L. A.; Vanucci, C.; Rogers, R. D. Isodicyclopentadienes and related molecules. Part 41. Stereochemical course of Diels-Alder cycloadditions to (hydroxymethyl)-substituted plane-nonsymmetric cyclopentadienes J. Am. Chem. Soc. 1989, 111, 5792-5800.
[106] Marchand, A. P.; Sorokin, V. D.; Watson, W. H.; Carlson, T. F.; Krawiec, M. Structure of a C26H28 alkene formed via titanium-promoted reductive dimerization of 4-(spirocyclopropyl)pentacyclo[5.4.0.02,6.03,10.05,9] undecan-8-one Struct. Chem. 1994, 5, 367-373.
[107] Singh, V. K.; Raju, B. N. S.; Deota, P. T. Micellar catalysis. �4s + �2s Cycloaddition in aqueous media Synth. Commun. 1988, 18, 567-574.
[108] Klaerner, F. G.; Adamsky, F. Synthesis and rearrangement of substituted bicyclo[2.1.0]pent-2-enes. Thermal walk rearrangement Chem. Ber. 1983, 116, 299-322.
[109] Ashnagar, A.; Bruce, J. M. Michael adducts of 2-methoxycarbonyl-1,4-benzoquinone with different donor molecules Asian J. Chem. 2010, 22, 2058-2072.
[110] Mamaghani, M.; Pourali, A. Synthesis of Methyl 3,6-Dioxo-endo-tricyclo[6.2.1.02,7]undeca- 4,9-diene-2-carboxylate as Synthetic Intermediate for Conduritol Derivatives Russ. J. Org. Chem. 2002, 38, 347-349.
[111] Brimble, M. A.; Duncalf, L. J.; Reid, D. C. W.; Roberts, T. R. Addition of cyclopentadiene and 2-trimethylsilyloxyfuran to quinones bearing a menthyl ester chiral auxiliary Tetrahedron 1998, 54, 5363-5374.
[112] Marchand, A. P.; Suri, S. C.; Earlywine, A. D.; Powell, D. R.; Van der Helm, D. Synthesis of methyl- and nitro-substituted pentacyclo[5.4.0.02,6.03, 10.05,9]undecane-8,11-diones J. Org. Chem. 1984, 49, 670-675.
[113] Cooper, S. C.; Sammes, P. G. (1,5)-Acetyl shifts in cycloadducts derived from 2-acetyl-1,4-benzoquinones J. Chem. Soc., Chem. Commun. 1980, 633-634.
[114] Cooper, S. C.; Sammes, P. G. Acyl rearrangements in acylbenzoquinone cycloadducts J. Chem. Soc., Perkin Trans. 1 1984, 2407-2413.
[115] Ashnagar, A.; Bruce, J. M. Synthesis of Diels-Alder mono-adducts of methoxycarbonyl-1,4-benzoquinone Int. J. ChemTech Res. 2010, 2, 224-232.
[116] Perry, G. J.; Sutherland, M. D. The hydrocyanation of free and polymer-bound benzoquinone Tetrahedron 1982, 38, 1471-1476.
[117] Bott, S. G.; Marchand, A. P.; Kumar, K. A. Thermodynamic vs. kinetic control in the Diels-Alder cycloaddition of cyclopentadiene to 2,3-dicyano-p-benzoquinone J. Chem. Crystallogr. 1996, 26, 281-286.
[118] Marchand, A. P.; Ganguly, B.; Watson, W. H.; Bodige, S. G. Thermodynamic vs. kinetic control in the Diels-Alder cycloaddition of
cyclopentadiene to 2,3-dicyano-p-benzoquinone: kinetic control revisited Tetrahedron 1998, 54, 10967-10972.
[119] Ruano, J. L. G.; Alemparte, C. Synthesis and Dienophilic Behavior of (S)-2-Cyano-3-(p-tolylsulfinyl)-1,4- benzoquinone J. Org. Chem. 2004, 69, 1405-1408.
[120] Liu, H. J.; Ngooi, T. K. A direct approach to the cis-1-octalone system. 2-Carbomethoxy-2-cyclohexenone as a dienophile Synth. Commun. 1982, 12, 715-722.
[121] Liu, H. J.; Ngooi, T. K.; Browne, E. N. C. Diels-Alder reactions of 2-carbomethoxy-2-cyclohexen-1-one Can. J. Chem. 1988, 66, 3143-3152.
[122] Liu, H.-J.; Li, Y.; Browne, E. N. C. Face-selective Diels-Alder reactions of (1R,5R)-3-formyl-6,6-dimethylbicyclo[3.1.1]hept-3-en-2-one Can. J. Chem. 1994, 72, 1883-1893.
[123] Guan, X.; Yu, Y. Synthesis of benzoquinone-2,5-dicarboxylates Youji
Huaxue 1994, 14, 80-84. [124] Guan, X.-P.; Yu, Y.-Z. Synthesis of substituted Cs-trishomocubane
derivatives. Part 1. Synthesis of Cs-trishomocubane-8,11-dione-1,9-dicarboxylate derivatives Youji Huaxue 1995, 15, 263-267.
[125] Beddoes, R. L.; Bruce, J. M.; Finch, H.; Heelam, L. M. J.; Hunt, I. D.; Mills, O. S. Benzoquinones and related compounds. Part 4. Thermolysis of the Diels-Alder adduct of 2-acetyl-5,6-dichloro-1,4-benzoquinone and cyclopentadiene: evidence for a partial retro-diene reaction J. Chem. Soc.,
Perkin Trans. 1 1981, 2670-2676. [126] Sudhir, U.; James, B.; Joly, S.; Nair, M. S. Diels-Alder reactivity of 2-
(bromomethyl)-1,4-quinone and 2-bromo-5-(bromomethyl)-1,4-quinone with cyclopentadiene and the synthesis of new substituted pentacyclic systems Res. Chem. Intermed. 2003, 29, 523-532.
[127] Benbow, J. W.; Katoch-Rouse, R. A Biomimetic Approach to Dihydrobenzofuran Synthesis J. Org. Chem. 2001, 66, 4965-4972.
[128] Evano, G.; Schaus, J. V.; Panek, J. S. A Convergent Synthesis of the Macrocyclic Core of Cytotrienins: Application of RCM for Macrocyclization Org. Lett. 2004, 6, 525-528.
[129] Nguyen, V. T.; Verniest, G.; Claessens, S.; De Kimpe, N. Total synthesis of four naturally occurring 2-azaanthraquinone antibiotics, 6-deoxy-8-methylbostrycoidin, 6-deoxybostrycoidin, 7-O-demethyl-6-deoxybostry-coidin and scorpinone Tetrahedron 2005, 61, 2295-2300.
[130] Veldhuizen, E. J. A.; Vaillancourt, F. H.; Whiting, C. J.; Hsiao, M. M. Y.; Gingras, G.; Xiao, Y.; Tanguay, R. M.; Boukouvalas, J.; Eltis, L. D. Steady-state kinetics and inhibition of anaerobically purified human homogentisate 1,2-dioxygenase Biochem. J. 2005, 386, 305-314.
[131] Ruettimann, A.; Lorenz, P. A new synthetic route to ubiquinones Helv.
Chim. Acta 1990, 73, 790-796. [132] Falcou, A.; Boullais, C. Synthesis of [2,3-13C2-2,5-cyclohexadienyl]
ubiquinone 3 J. Labelled Compd. Radiopharm. 1998, 41, 657-668. [133] Sudhir, U.; Rath, N. P.; Nair, M. S. Synthesis of novel tetra- and pentacyclic
aza-cage systems Tetrahedron 2001, 57, 7749-7753. [134] Nair, M. S.; Sudhir, U.; Joly, S.; Rath, N. P. Two fascinating rearrangements
through selective placement of bromine substituents. Photochemical synthesis of 3-bromo-7-(bromomethyl)tetracyclo[5.3.1.02,6.04,8]undec-10(12)-ene-9,11-dione and its rearrangement with amines Tetrahedron 1999, 55, 7653-7660.
[135] Alder, K.; Flock, F. H.; Beumling, H. Preparation of p-quinone epoxides Chem. Ber. 1960, 93, 1896-1899.
[136] Genski, T.; Taylor, R. J. K. The synthesis of epi-epoxydon utilizing the Baylis-Hillman reaction Tetrahedron Lett. 2002, 43, 3573-3576.
[137] Vankar, Y. D.; Kumaravel, G.; Mukherjee, N.; Rao, C. T. Sodium iodide-chlorotrimethylsilane (or boron trifluoride etherate) and zinc-chlorotrimethylsilane: mild reagent systems for the conversion of enediones into 1,4-diketones Synth. Commun. 1987, 17, 181-187.
[138] Marchand, A. P.; Ngooi, T. K.; Watson, W. H.; Kashyap, R. P. Endo-Tricyclo[6.2.1.0.2,7]undec-9-ene-3,6-dione: a versatile synthetic intermediate Tetrahedron 1991, 47, 961-974.
[139] Asaoka, M.; Naito, S.; Takei, H. Total synthesis of (±)-pyrenolide B Tetrahedron Lett. 1985, 26, 2103-2106.
[140] Ghandi, M.; Shahidzadeh, M. Experimental and semiempirical studies of chemical reactivity of dialkylcadmium reagents addition to �,�-enones J.
Organomet. Chem. 2006, 691, 4918-4925. [141] Mloston, G.; Celeda, M.; Heimgartner, H. Chemo- and stereoselectivity in
1,3-dipolar cyclo-additions of thiocarbonyl ylides with a 1,4-methano-naphthalene-5,8-dione derivative Heterocycles 2003, 59, 767-777.
[142] Marchand, A. P.; Alihodzic, S.; Shukla, R. Simple procedure for preparing annulated p-benzoquinones. Improved synthesis of 1,4-dihydro-1,4-methanonaphthalene-5,8-dione Synth. Commun. 1998, 28, 541-546.
[143] Mitra, A. K.; Gawandi, V. B.; George, K.; Mohan, H.; Mukherjee, T. Antioxidant activity of 5,8-dihydroxy-1,4-dihydro-1,4-methanonaphthalene Res. Chem. Intermed. 2008, 34, 85-92.
[144] Mitra, A. K.; Gawandi, V. B.; George, K.; Mohan, H.; Mukherjee, T. Investigations on the antioxidant activity of 5,8-dihydroxy-1,4-dihydro-1,4-methanonaphthalene (DDMN) Res. Chem. Intermed. 2009, 35, 13-20.
[145] Feuer, H.; Editor Nitrile Oxides, Nitrones, and Nitronates in Organic
Synthesis; Volume 2, 2008. [146] Kozikowski, A. P.; Adamczyk, M. Methods for the conversion of
isoxazolines to �-hydroxy ketones Tetrahedron Lett. 1982, 23, 3123-3126.

2660 Current Organic Chemistry, 2012, Vol. 16, No. 22 Sharapa et al.
[147] Kwiatkowski, S. A new, efficient approach to �,�-unsaturated ketones and �-hydroxy ketones from 4,5-dihydroisoxazoles J. Chem. Soc., Chem. Commun. 1987, 1496-1498.
[148] Brand, U.; Huenig, S. Azo bridges from Azines. Part XX. Parallel cyanovinylene and azo groups - synthesis and chemistry Liebigs Ann. 1996, 585-592.
[149] Gottschalk, F. J.; Weyerstahl, P. Simple synthesis of 3-acetyl-2-norbornanone via Diels-Alder reaction and reactivity of its derivatives Chem.
Ber. 1980, 113, 555-565. [150] Gutsmiedl, K.; Wirges, C. T.; Ehmke, V.; Carell, T. Copper-Free "Click"
Modification of DNA via Nitrile Oxide-Norbornene 1,3-Dipolar Cycloaddition Org. Lett. 2009, 11, 2405-2408.
[151] Mayo, P.; Hecnar, T.; Tam, W. 1,3-Dipolar cycloaddition of nitrile oxides with unsymmetrically substituted norbornenes Tetrahedron 2001, 57, 5931-5941.
[152] Pavlov, V. A.; Kurdyukov, A. I.; Aristova, N. V.; Gorin, B. I.; Zyablikova, T. A.; Moskva, V. V. Phosphorylnitrile oxides. III. Synthesis of phosphorylated isoxazoles and isoxazolines Zh. Obshch. Khim. 1994, 64, 1373-1387.
[153] Fernandez, M. P.; Gonzalez, B.; Pardo, M.; Soto, J. L. 1,3-Dipolar cycloaddition of nitrile oxides with Diels-Alder adducts of p-benzoquinone and 1,4-naphthoquinone An. Quim. 1994, 90, 477-482.
[154] Darvatkar, N. B.; Wankhede, K. S.; Bhilare, S. V.; Deorukhkar, A. R.; Raut, D. G.; Vaidya, V. V.; Trivedi, G. K.; Salunkhe, M. M. 1,3-Dipolar cycloaddition of nitrile oxides with symmetrical tri- and polycyclic strained olefins J. Heterocycl. Chem. 2010, 47, 1004-1010.
[155] Bardhan, S.; Schmitt, D. C.; Porco, J. A., Jr. Total Synthesis and Stereochemical Assignment of the Spiroisoxazoline Natural Product (+)-Calafianin Org. Lett. 2006, 8, 927-930.
[156] Barlow, M. G.; Haszeldine, R.; Peck, C. J. Hexafluoro-3-oxatricyclo[3.2.0.02,4]hept-6-ene (hexafluoro-Dewar-benzene oxide) from the photochemical oxidation of hexafluorobenzene J. Chem. Soc., Chem.
Commun. 1980, 158-159. [157] Caldirola, P.; De Amici, M.; De Micheli, C.; Wade, P. A.; Price, D. T.;
Bereznak, J. F. Metal hydride reduction of isoxazoline-3-carboxylate esters Tetrahedron 1986, 42, 5267-5272.
[158] Kozikowski, A. P.; Adamcz, M. Methods for the stereoselective cis-cyanohydroxylation and -carboxyhydroxylation of olefins J. Org. Chem. 1983, 48, 366-372.
[159] Itoh, K.-i.; Takahashi, S.; Ueki, T.; Sugiyama, T.; Takahashi, T. T.; Horiuchi, C. A. A novel one-pot synthesis of 3-acetyl- and 3-benzoylisoxazole derivatives using ammonium cerium nitrate (CAN) Tetrahedron Lett. 2002, 43, 7035-7037.
[160] Itoh, K.-i.; Horiuchi, C. A. Formation of isoxazole derivatives via nitrile oxide using ammonium cerium nitrate (CAN): a novel one-pot synthesis of 3-acetyl- and 3-benzoylisoxazole derivatives Tetrahedron 2004, 60, 1671-1681.
[161] Tanaka, K.; Masuda, H.; Mitsuhashi, K. Applications of fluorinated 1,3-dipolar compounds as the building blocks of the heterocycles with fluorine groups. Part X. Exo/endo stereoselectivity in 1,3-dipolar cycloaddition of trifluoroacetonitrile oxide and -nitrilimine with bicyclic olefins Bull. Chem.
Soc. Jpn. 1986, 59, 3901-3904. [162] Barrow, S. J.; Easton, C. J.; Savage, G. P.; Simpson, G. W. Exploiting the
1,3-dithiane of 2-oxopropanenitrile oxide to limit competing dimerization in 1,3-dipolar cycloaddition reactions Tetrahedron Lett. 1997, 38, 2175-2178.
[163] Vyas, D. M.; Chiang, Y.; Doyle, T. W. A short, efficient total synthesis of (±) acivicin and (±) bromoacivicin Tetrahedron Lett. 1984, 25, 487-490.
[164] Wade, P. A.; Bereznak, J. F. Sulfonylisoxazolines: reliable intermediates for the preparation of �-hydroxy nitriles J. Org. Chem. 1987, 52, 2973-2977.
[165] De Sarlo, F.; Brandi, A.; Guarna, A.; Goti, A.; Corezzi, S. Simple in situ preparation of fulminic acid Tetrahedron Lett. 1983, 24, 1815-1816.
[166] De Sarlo, F.; Guarna, A.; Brandi, A.; Goti, A. The chemistry of fulminic acid revised Tetrahedron 1985, 41, 5181-5185.
[167] Huisgen, R.; Christl, M. 1,3-Dipolar cycloadditions. 72. Reactions of fulminic acid with unsaturated compounds Chem. Ber. 1973, 106, 3291-3311.
[168] Cecchi, L.; De Sarlo, F.; Machetti, F. 4,5-Dihydroisoxazoles by copper(II)-catalysed condensation of primary nitroalkanes with dipolarophiles Synlett 2007, 2451-2453.
[169] Ichihara, A.; Kobayashi, M.; Oda, K.; Sakamura, S. Regioselective, abnormal ring opening of the epoxide of quinone epoxide-cyclopentadiene adducts Bull. Chem. Soc. Jpn. 1978, 51, 826-829.
[170] Wladislaw, B.; Di Vitta, C.; Marzorati, L.; De Arruda Campos, I. P.; Lucchini, V. Novel stereoselective addition of some nucleophiles to 2,3-bis(methylsulfanyl)norbornenobenzoquinone Tetrahedron Lett. 1997, 38, 2625-2628.
[171] Wladislaw, B.; Marzorati, L.; Di Vitta, C. Some new 2,3-dialkylthio-1,4-benzoquinones Synthesis 1983, 464-466.
[172] Lockshin, M. P.; Filosa, M. P.; Zuraw, M. J.; Carlier, P. R. Formation of a Novel Sulfonated Enedione J. Org. Chem. 1996, 61, 2556-2558.
[173] Di Vitta, C.; de Arruda Campos, I. P.; Farah, J. P. S.; Zukerman-Schpector, J. New reductive addition of hard nucleophiles to 6,7-bis(methylsulfanyl)-1,4-dihydro-1,4-methanonaphthalene-5,8-dione Perkin 1 2000, 3692-3694.
[174] Wladislaw, B.; Marzorati, L.; Lima Netto, S. Some sulfur- and nitrogen-substituted xyloquinones Phosphorus Sulfur 1987, 33, 173-177.
[175] Carreno, M. C.; Ruano, J. L. G.; Urbano, A. Synthesis and asymmetric Diels-Alder reactions of (S)-2-p-tolylsulfinyl-1,4-benzoquinone Tetrahedron Lett. 1989, 30, 4003-4006.
[176] Wladislaw, B.; Marzorati, L.; Campos, I. P. d. A.; Viertler, H. The importance for cage compound formation of the oxidation potential of the enedionic system in benzoquinone-cyclopentadiene adducts J. Chem. Soc.,
Perkin Trans. 2 1992, 475-477. [177] Carreno, M. C.; Ruano, J. L. G.; Urbano, A.; Lopez-Solera, M. I. (SS)-2-(p-
Tolylsulfinyl)norborneno-p-benzoquinones: A New Type of Facially Perturbed Enantiopure Quinones J. Org. Chem. 1997, 62, 976-981.
[178] Mehta, G.; Islam, K. Enantioselective total synthesis of epoxyquinone natural products (-)-phyllostine, (+)-epoxydon, (+)-epiepoxydon and (-)-panepophenanthrin: access to versatile chiral building blocks through enzymatic kinetic resolution Tetrahedron Lett. 2004, 45, 7683-7687.
[179] Mehta, G.; Islam, K. Total synthesis of the novel angiogenesis inhibitors epoxyquinols A and B Tetrahedron Lett. 2003, 44, 3569-3572.
[180] Mehta, G.; Ramesh, S. S. Enantioselective total synthesis of (+)-panepophenanthrin, a novel inhibitor of the ubiquitin-activating enzyme Tetrahedron Lett. 2004, 45, 1985-1987.
[181] Chae, H. I.; Hwang, G.-S.; Jin, M. Y.; Ryu, D. H. Efficient asymmetric synthesis of chiral monomer of epoxyquinols and (-)-phyllostine Bull.
Korean Chem. Soc. 2010, 31, 1047-1050. [182] Ichihara, A.; Kimura, R.; Oda, K.; Moriyasu, K.; Sakamura, S. Synthetic
studies of highly oxygenated cyclohexane derivatives. Part XV. Synthesis of (±)-epoxydon and related natural compounds Agric. Biol. Chem. 1982, 46, 1879-1883.
[183] Mehta, G.; Pan, S. C. Total Synthesis of the Novel Antifungal Agent (±)-Jesterone Org. Lett. 2004, 6, 811-813.
[184] Wong, H. N. C.; Fok, C. C. M.; Wong, T. Stereospecific deoxygenation of epoxides to olefins Heterocycles 1987, 26, 1345-1382.
[185] Takano, S.; Moriya, M.; Ogasawara, K. Enantiocontrolled route to optically pure carvone using chiral cyclohexa-2,5-dienone synthon Synlett 1993, 601-602.
[186] Lei, X.; Zaarur, N.; Sherman, M. Y.; Porco, J. A., Jr. Stereocontrolled Synthesis of a Complex Library via Elaboration of Angular Epoxyquinol Scaffolds J. Org. Chem. 2005, 70, 6474-6483.
[187] Mehta, G.; Sunil Kumar, Y. C.; Das, M. A de novo Diels-Alder strategy toward the novel pentacyclic natural product fluostatin C: a concise synthesis of 6-deoxyfluostatin C Tetrahedron Lett. 2011, 52, 3505-3508.
[188] Jin, M. Y.; Hwang, G.-S.; Chae, H. I.; Jung, S. H.; Ryu, D. H. Highly efficient synthesis of (+)-bromoxone, (+)-epiepoxydon and (+)-epiepoformin Bull. Korean Chem. Soc. 2010, 31, 727-730.
[189] Mehta, G.; Pujar, S. R.; Ramesh, S. S.; Islam, K. Enantioselective total synthesis of polyoxygenated cyclohexanoids: (+)-streptol, ent-RKTS-33 and putative '(+)-parasitenone'. Identity of parasitenone with (+)-epoxydon Tetrahedron Lett. 2005, 46, 3373-3376.
[190] Jung, S. H.; Hwang, G.-S.; Lee, S. I.; Ryu, D. H. Total Synthesis of (+)-Ambuic Acid: �-Bromination with 1,2-Dibromotetrachloroethane J. Org.
Chem. 2012, 77, 2513-2518.




















