
Assembly factors and ATP-dependent proteases in cytochrome c oxidase biogenesis
10
Review Assembly factors and ATP-dependent proteases in cytochrome c oxidase biogenesis Lukas Stiburek, Jiri Zeman ⁎ Charles University in Prague, First Faculty of Medicine, Department of Pediatrics, Prague, Czech Republic abstract article info Article history: Received 4 November 2009 Received in revised form 14 March 2010 Accepted 7 April 2010 Available online 14 April 2010 Keywords: Mitochondria Cytochrome c oxidase Assembly factor SCO1 SCO2 SURF1 TACO1 OXA1L ATP-dependent protease YME1L LON i-AAA m-AAA Eukaryotic cytochrome c oxidase (CcO), the terminal enzyme of the energy-transducing mitochondrial electron transport chain is a hetero-oligomeric, heme–copper oxidase complex composed of both mitochondrially and nuclear-encoded subunits. It is embedded in the inner mitochondrial membrane where it couples the transfer of electrons from reduced cytochrome c to molecular oxygen with vectorial proton translocation across the membrane. The biogenesis of CcO is a complicated sequential process that requires numerous specific accessory proteins, so-called assembly factors, which include translational activators, translocases, molecular chaperones, copper metallochaperones and heme a biosynthetic enzymes. Besides these CcO-specific protein factors, the correct biogenesis of CcO requires an even greater number of proteins with much broader substrate specificities. Indeed, growing evidence indicates that mitochondrial ATP-dependent proteases might play an important role in CcO biogenesis. Out of the four identified energy- dependent mitochondrial proteases, three were shown to be directly involved in proteolysis of CcO subunits. In addition to their well-established protein-quality control function these oligomeric proteolytic complexes with chaperone-like activities may function as molecular chaperones promoting productive folding and assembly of subunit proteins. In this review, we summarize the current knowledge of the functional involvement of eukaryotic CcO-specific assembly factors and highlight the possible significance for CcO biogenesis of mitochondrial ATP-dependent proteases. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Mitochondria are highly dynamic semiautonomous organelles of endosymbiotic, α-proteobacterial descent found in virtually all eukary- otic cells. They harbor several essential metabolic pathways, produce the bulk of cellular ATP and reactive oxygen species (ROS), serve as calcium (Ca 2+ ) stores, and play a central role in programmed cell death [1]. They are surrounded by two biological membranes that delimit two major aqueous subcompartments, the intermembrane space (IMS) and the mitochondrial matrix. Mitochondria contain numerous copies of their own DNA (mtDNA) that codes for a limited number of protein products, almost exclusively subunits of the OXPHOS (oxidative phosphorylation system) machinery and rRNA and tRNA components of a specific mitochondrial translation apparatus. In mammals, 13 protein products are synthesized on mitochondrial ribosomes, all of which are evolutionarily conserved hydrophobic OXPHOS subunits. The mammalian OXPHOS machinery is composed of five multi-subunit inner membrane-embedded enzyme complexes, the respiratory chain and ATP synthase and is built up of more than 90 protein subunits containing numerous prosthetic groups. Thus, the vast majority of the various ∼ 1500 mammalian mitochondrial proteins is encoded in the nucleus, synthesized on cytoplasmic ribosomes and subsequently sorted and imported into the mitochondria by means of a specialized transport machinery [2–4]. Eukaryotic cytochrome c oxidase (CcO) is the terminal enzyme of the mitochondrial electron transport chain. It is responsible for electron transfer from soluble mobile carrier cytochrome c to molecular oxygen as well as for proton translocation across the inner membrane. Mammalian CcO is a hetero-oligomeric inner membrane-embedded complex with a combined molecular weight of 205 kDa and is composed of 13 structural subunits encoded by both mitochondrial and nuclear genes. The core of the enzyme, which incorporates all redox-active cofactors, is composed of three mito- chondrially encoded subunits, Cox1, Cox2 and Cox3 that show high evolutionary conservation. The remaining 10 evolutionarily younger peripheral subunits that associate with the complex core are encoded on nuclear DNA and synthesized in cytoplasm. They are required for stability/protection of the enzyme core as well as for the regulation of its activity. Besides the protein subunits, CcO contains several redox- active prosthetic groups that are responsible for electron transfer and dioxygen reduction. These include two copper centers (Cu A and Cu B ) located on subunits Cox2 and Cox1, respectively, and two heme a moieties (heme a and heme a 3 ) coordinated within the interior of subunit Cox1 [5–7]. Either under mild detergent conditions or when crystallized or reconstituted in phospholipid vesicles, the majority of Biochimica et Biophysica Acta 1797 (2010) 1149–1158 ⁎ Corresponding author. Department of Pediatrics, First Faculty of Medicine, Charles University, Ke Karlovu 2, 12808, Prague 2, Czech Republic. E-mail addresses: [email protected] (L. Stiburek), [email protected] (J. Zeman). 0005-2728/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.bbabio.2010.04.006 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbabio
Transcript of Assembly factors and ATP-dependent proteases in cytochrome c oxidase biogenesis

Biochimica et Biophysica Acta 1797 (2010) 1149–1158
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbab io
Review
Assembly factors and ATP-dependent proteases in cytochrome c oxidase biogenesis
Lukas Stiburek, Jiri Zeman ⁎Charles University in Prague, First Faculty of Medicine, Department of Pediatrics, Prague, Czech Republic
⁎ Corresponding author. Department of Pediatrics, FirUniversity, Ke Karlovu 2, 12808, Prague 2, Czech Repub
E-mail addresses: [email protected] (L. Stib(J. Zeman).
0005-2728/$ – see front matter © 2010 Elsevier B.V. Adoi:10.1016/j.bbabio.2010.04.006
a b s t r a c t
a r t i c l e i n f oArticle history:Received 4 November 2009Received in revised form 14 March 2010Accepted 7 April 2010Available online 14 April 2010
Keywords:MitochondriaCytochrome c oxidaseAssembly factorSCO1SCO2SURF1TACO1OXA1LATP-dependent proteaseYME1LLONi-AAAm-AAA
Eukaryotic cytochrome c oxidase (CcO), the terminal enzyme of the energy-transducing mitochondrialelectron transport chain is a hetero-oligomeric, heme–copper oxidase complex composed of bothmitochondrially and nuclear-encoded subunits. It is embedded in the inner mitochondrial membranewhere it couples the transfer of electrons from reduced cytochrome c to molecular oxygen with vectorialproton translocation across the membrane. The biogenesis of CcO is a complicated sequential process thatrequires numerous specific accessory proteins, so-called assembly factors, which include translationalactivators, translocases, molecular chaperones, copper metallochaperones and heme a biosynthetic enzymes.Besides these CcO-specific protein factors, the correct biogenesis of CcO requires an even greater number ofproteins with much broader substrate specificities. Indeed, growing evidence indicates that mitochondrialATP-dependent proteases might play an important role in CcO biogenesis. Out of the four identified energy-dependent mitochondrial proteases, three were shown to be directly involved in proteolysis of CcO subunits.In addition to their well-established protein-quality control function these oligomeric proteolytic complexeswith chaperone-like activities may function as molecular chaperones promoting productive folding andassembly of subunit proteins. In this review, we summarize the current knowledge of the functionalinvolvement of eukaryotic CcO-specific assembly factors and highlight the possible significance for CcObiogenesis of mitochondrial ATP-dependent proteases.
st Faculty of Medicine, Charleslic.urek), [email protected]
ll rights reserved.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Mitochondria are highly dynamic semiautonomous organelles ofendosymbiotic, α-proteobacterial descent found in virtually all eukary-otic cells. They harbor several essential metabolic pathways, producethe bulk of cellular ATP and reactive oxygen species (ROS), serve ascalcium (Ca2+) stores, and play a central role in programmed cell death[1]. They are surrounded by two biological membranes that delimit twomajor aqueous subcompartments, the intermembrane space (IMS) andthe mitochondrial matrix. Mitochondria contain numerous copies oftheir own DNA (mtDNA) that codes for a limited number of proteinproducts, almost exclusively subunits of the OXPHOS (oxidativephosphorylation system) machinery and rRNA and tRNA componentsof a specific mitochondrial translation apparatus. In mammals, 13protein products are synthesized on mitochondrial ribosomes, all ofwhich are evolutionarily conserved hydrophobic OXPHOS subunits. Themammalian OXPHOS machinery is composed of five multi-subunitinner membrane-embedded enzyme complexes, the respiratory chainand ATP synthase and is built up of more than 90 protein subunitscontaining numerous prosthetic groups. Thus, the vast majority of the
various ∼1500 mammalian mitochondrial proteins is encoded in thenucleus, synthesized on cytoplasmic ribosomes and subsequentlysorted and imported into the mitochondria by means of a specializedtransport machinery [2–4].
Eukaryotic cytochrome c oxidase (CcO) is the terminal enzyme ofthe mitochondrial electron transport chain. It is responsible forelectron transfer from soluble mobile carrier cytochrome c tomolecular oxygen as well as for proton translocation across theinner membrane. Mammalian CcO is a hetero-oligomeric innermembrane-embedded complex with a combined molecular weightof 205 kDa and is composed of 13 structural subunits encoded by bothmitochondrial and nuclear genes. The core of the enzyme, whichincorporates all redox-active cofactors, is composed of three mito-chondrially encoded subunits, Cox1, Cox2 and Cox3 that show highevolutionary conservation. The remaining 10 evolutionarily youngerperipheral subunits that associate with the complex core are encodedon nuclear DNA and synthesized in cytoplasm. They are required forstability/protection of the enzyme core as well as for the regulation ofits activity. Besides the protein subunits, CcO contains several redox-active prosthetic groups that are responsible for electron transfer anddioxygen reduction. These include two copper centers (CuA and CuB)located on subunits Cox2 and Cox1, respectively, and two heme amoieties (heme a and heme a3) coordinated within the interior ofsubunit Cox1 [5–7]. Either under mild detergent conditions or whencrystallized or reconstituted in phospholipid vesicles, the majority of

1150 L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
mammalian CcO exists as a dimer of two thirteen-subunit assemblieswith contacts between monomers mediated merely by subunitsCox6a and Cox6b [8,9]. CcO dimerization likely plays a key structural–functional role, conferring maximal structural stability for thecomplex [10]. In addition to protein subunits and prosthetic groups,crystalline preparations of bovine heart CcOwere shown to contain 13lipids per enzyme dimer, with several of them likely important in O2-transfer process or dimer stabilization [11].
The biogenesis of CcO is a complicated, sequential process thatrequires coordinated expression of mitochondrial and nuclear genes,mitochondrial import and membrane integration of subunit polypep-tides, synthesis/import and incorporation of prosthetic groups, andfinalassembly of matured subunits. A number of specific nuclear geneproducts thatmediate the distinct functions required for CcObiogenesiswere identified. However, the correct biogenesis of the CcO complexrequires an even greater number of protein factors with much broadersubstrate specificities. Indeed, accumulating evidence suggests thatmitochondrial ATP-dependent proteases, particularly the inner mem-brane-bound FtsH/AAA family proteases and the soluble matrixprotease Lon, might also play an important role in this process. Besidesthe well-established role of these proteolytic complexes in protein-quality control, thesemultimeric assemblieswith additional chaperone-like and translocase activities were shown to function in proteolyticprocessing of regulatory proteins, degradation of native subunitpolypeptides and promotion of respiratory chain assembly.
2. CcO assembly factors
The vast majority of the over 30 gene products known to berequired exclusively for proper biogenesis of the eukaryotic CcOcomplex have been identified through functional complementationstudies of respiratory-deficient yeast mutants [12]. Subsequently, anumber of these so-called CcO assembly factors were shown to havehuman homologues, while several of them have been studied at theprotein level [6]. These include copper metallochaperones Sco1, Sco2,Cox11 and Cox17, which are involved in trafficking/insertion of Cu(I/II) ions into CuA and CuB copper centers in Cox2 and Cox1 subunits,respectively; inner membrane proteins Cox10, Cox15 and Surf1,which are required for synthesis or incorporation of heme a moietiesinto Cox1; and Oxa1 and Cox18 translocases, which are involved inthe export/translocation of transmembrane segments of integralinner membrane proteins, including CcO subunits. Although severalpathogenic mutations in each of the three mitochondrially encodedCcO subunits [13] and one mutation in nuclear-encoded COX6B1subunit [14] have been reported, the majority of fatal infantile CcOdeficiencies identified so far result from autosomal recessive muta-tions in genes encoding CcO assembly factors. Indeed, two of thehuman CcO assembly factors, LRPPRC and TACO1, both of which areinvolved in mitochondrial CcO mRNA translation, were identifiedprimarily on the basis of their mutant phenotypes in human patientsusing an integrative genomics approach or functional complementa-tion, respectively [15,16].
2.1. LRPPRC and TACO1
Membrane-bound translational activator proteins that mediatethe membrane-recruitment of translating mitochondrial ribosomeshave been identified in yeast mitochondria [17,18]. They bind to 5′untranslated leader sequences of mitochondrial transcripts andphysically interact with mitochondrial ribosome subunits [19–21].The translational activators are mRNA-specific and, thus, regulatemitochondrially encoded protein synthesis in a gene-specific manner.This mechanism was not expected to be conserved in mammalsbecause mammalian mitochondrial mRNAs lack significant 5′ un-translated region (UTR) sequences, and themajority of genes involvedin translation of mitochondrially encoded proteins lack mammalian
homologues. The only exception was thought to be LRPPRC (leucine-rich pentatricopeptide repeat cassette), the human homologue ofyeast Pet309. Pet309 is a mitochondrial translational activator thatbinds specifically to the 5′ UTR of the COX1 mRNA. LRPPRC wasimplicated in translation and stabilization of COX1 and COX3 mRNAs[16,22]. Mutations in LRPPRC lead to French-Canadian form of CcO-deficient Leigh syndrome, which is characterized by a less severeneurological involvement, including slower progression, when com-pared to the classical Leigh syndrome clinical picture [16,22]. BothPet309 and LRPPRC contain several PPR motifs, consisting ofdegenerated 35-amino acid sequences proposed to form twoantiparallel alpha helices [23]. These repeats characterize a largeprotein family in plants with only a few examples found among fungi,animals and protists. PPR proteins mostly participate in different stepsof sequence-specific RNAmetabolism [24]. Very recently, LRPPRC wasshown to interact with SLIRP (SRA-stem loop interacting RNA-bindingprotein) as part of a high-molecular weight ribonucleoproteincomplex [25]. The disruption of this complex due to knockdown ofLRPPRC was shown to lead to reduction in steady-state levels of allmitochondrial mRNAs. Based on these results LRPPRC was postulatedto act in posttranscriptional mitochondrial gene expression, beinginvolved in regulation of stability and handling of maturemRNAs [25].
Recently, a protein product of the gene CCDC44, subsequentlyrenamed as TACO1 (Translational Activator of COX I), was identified byShoubridge's group as a specific mammalianmitochondrial translation-al activator [15]. Thematurepolypeptideof roughly 30 kDawas found inthe mitochondrial matrix as an oligomer or as part of a ∼74-kDacomplex. Loss of CCDC44/TACO1 results in late-onset Leigh syndromewith CcO deficiency, very likely due to a specific defect in the synthesisofmitochondrially encoded subunit Cox1. The reduced amount of newlysynthesized Cox1 in TACO1 patient fibroblasts is accompanied by com-pensatory increase in some of the mitochondrially encoded subunits ofcomplex I and cytochrome b. The marked decrease in the level of fullyassembled CcO in TACO1-deficient cells was not accompanied byappearance of any of the previously identified subcomplexes, confirm-ing the role of Cox1as the initial seed for CcOassembly. Interestingly, theinactivation of Saccharomyces cerevisiae TACO1 orthologue, YGR021w,which exhibits 29% sequence identity, resulted in almost normal CcOactivity in cells grown on respiratory media [15].
2.2. Oxa1 and Cox18
Oxa1 belongs to the Alb3/Oxa1/YidC family of proteins, membersof which are involved in insertion of membrane proteins inmitochondria, chloroplasts and bacteria. Oxa1 is an integral proteinof the inner mitochondrial membrane that facilitates the cotransla-tional membrane integration of mitochondrial translation products aswell as the insertion of conservatively sorted nuclear gene productsinto the inner membrane [26,27]. Oxa1 attains a Nout–Cin membranetopology and possesses a hydrophobic core domain composed of fivetransmembrane helices that ensures the membrane export of itsprotein substrates [28,29]. Oxa1 proteins were not found inunicellular eukaryotic parasites Giardia lamblia or Trichomonasvaginalis that contain double-membranous mitochondria-like orga-nelles devoid of respiratory complexes and organelle genome [30].Several lines of evidence indicate that Oxa1 interacts with mitochon-drial ribosome [31,32]. Unlike the bacterial Oxa1 homologue, YidC,mitochondrial Oxa1 proteins contain a C-terminalα-helical domain ofroughly 100 residues that protrudes into the matrix [31,33]. Thisdomain is thought to mediate the interaction of Oxa1 withmitochondrial ribosomal subunit Mrp20. This interaction is thoughtto recruit the mitochondrial translation apparatus to the membrane-embedded Oxa1 translocation complex [34]. Chemical crosslinking ofin organello-labeled mitochondrial translation products followed byimmunoprecipitation with anti-Oxa1 antibody has demonstrated thatOxa1 interacts in a transient manner with nascent mitochondrially

1151L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
synthesized polypeptides, including CcO subunits Cox1, Cox2 andCox3 [35]. Whereas the yeast Oxa1 represents a rather general exportmachinery of the inner membrane, the membrane translocation ofboth hydrophilic tails of Cox2 precursor is strictly contingent upon thefunction of Oxa1 [36]. The other substrates of Oxa1, including Oxa1itself, can be inserted independently of its function, albeit withsignificantly reduced efficiencies. The yeast Oxa1-null mutant isrespiratory-deficient, has no detectable CcO activity and showsmarkedly reduced levels of the cytochrome bc1 complex and F1FO-ATP synthase [37,38]. Two distinct Oxa1 orthologues were identifiedin Schizosaccharomyces pombe and both proteins can complement therespiratory defect of yeast Oxa1-null cells [39]. Interestingly, thedouble inactivation of these genes is lethal to this petite-negativeyeast. Depletion of Oxa1 in Neurospora crassa results in a slow-growthphenotype accompanied by reduced subunit levels of CcO and NADH:ubiquinone oxidoreductase (complex I). The N. crassa Oxa1 forms a170–180-kDa homo-oligomeric complex, most likely containing fourOxa1 monomers [40]. In Podospora anserina, Oxa1 appears to berequired for biogenesis of complex I and IV. Furthermore, decreasedproduction of ROS accompanied by increased lifespan was found inP. anserina thermosensitive oxa1 mutant [41]. In addition to Oxa1,mitochondria contain another homologous inner membrane protein,Cox18, that defines the second Alb3/Oxa1/YidC subfamily. Becausedeletion of COX18 in S. cerevisiae, Kluyveromyces lactis and S. pombeleads to complete lack of cytochrome aa3 spectra and normal levels ofthe remaining OXPHOS complexes, the function of Cox18 appearsconfined to CcO in lower eukaryotes [42–44]. In the absence of Cox18,the steady-state levels of Cox2 are severely attenuated due toincreased degradation of the subunit [45,46]. The lack of Cox18 inN. crassa results in partial reduction in cytochrome aa3 accompaniedby substantial induction of the alternative oxidase [47]. In contrast tothe transient interaction of Oxa1, the N. crassa Cox18 interacts withCox2 and Cox3 in a long-lived manner [47].
The human Oxa1 orthologue, referred to as Oxa1l, shares 33%sequence identity with the corresponding yeast polypeptide [48]. Thehuman OXA1L cDNA was initially cloned by partial functionalcomplementation of the respiratory growth defect of the yeastoxa1-79 mutant. It consists of an open reading frame (ORF) predictedto encode a 435-amino acid protein [37]. The human OXA1L mRNAwas found to be enriched in mitochondria-bound polysomes fromHeLa cells, and the 3′ UTR of the OXA1L transcript was functionallyimportant when expressed in yeast cells [49]. We showed that humanOXA1L protein product is a mitochondrial integral membrane proteinthat exists as part of a 600–700-kDa complex not appearing to containany of the OXPHOS complexes [50]. We further demonstrated that thestable short hairpin RNA (shRNA)-mediated knockdown of humanOxa1l in HEK293 cells leads to markedly decreased protein levels andATP hydrolytic activity of the F1FO-ATP synthase and moderatelyreduced levels and activity of NADH:ubiquinone oxidoreductase(complex I). This RNAi phenotype clearly suggests functionalinvolvement of the human protein in the assembly/stability of thesetwo OXPHOS complexes [50]. Similarly, yeast Oxa1 was shown tomediate the assembly of the ATP synthase FO-subunit c into theholoenzyme complex [51]. In sharp contrast to yeast oxa1mutant, theassembly/stability of both CcO and the cytochrome bc1 complex(complex III) was not negatively affected by knockdown of humanOXA1L. Indeed, up-regulated levels of CcO, in terms of both individualsubunits and holoenzyme complex, were found in these cells. In linewith this result, high-resolution respirometry revealed markedlyincreased sodium azide-sensitive oxygen consumption of these cells.
2.3. Surf1
Surf1 is an integral protein of the inner mitochondrial membranerequired for the assembly of the CcO complex. The mature form ofhuman Surf1 has amolecularmass of∼30 kDa and is composed of two
transmembrane domains with a central loop region facing the IMS[52]. Both transmembrane domains and the central loop are requiredfor proper insertion of the human protein into the inner membrane,but the C-terminal tail of the protein is dispensable in this regard. Incontrast to the yeast homologue Shy1 [53], separately expressed N-and C-terminal transmembrane domains of human Surf1 cannot forma functional protein [52]. Although the precise molecular role ofhuman Surf1 in CcO biogenesis remains unknown, several lines ofevidence indicate that theprotein plays a role in someof theearly eventsof CcO assembly, ranging from insertion of heme a/a3 into Cox1 topromotion/stabilization of early subunits' assembly. Human cellslacking Surf1 accumulate CcO subcomplexes composed of merelyCox1, Cox4 and Cox5a, suggesting that the assembly is stalled at arather early stage [54,55]. Surf1 orthologues are found in terminaloxidase operons of several prokaryote species in which the mature CcOconsists of only three core subunits that associate early in the assemblyprocess in eukaryotes [56,57]. Bacterial Surf1 orthologues haverepeatedly been implicated in the insertion and/or stabilization ofheme a/a3, which is thought to occur concurrently with or immediatelyafter membrane integration of Cox1 [58,59]. Because the yeast Surf1homologue Shy1 was found in high-molecular weight CcO-containingcomplexes, the protein is likely to have an additional role in some of thelate CcOassembly events aswell [60]. Recently itwasdemonstrated thatShy1 associates with different inner membrane protein modules,includingCcO subcomplexes aswell as supercomplex species composedof partially and fully assembled forms of CcO and the cytochrome bc1complex. The association of CcO subcomplexes with complex III wasobserved even in the absence of the core subunit Cox2 [61]. Based onthese findings, Shy1, together with another yeast CcO assembly factor,Cox14, was proposed to accompany transient forms of CcO, therebymaintaining their competence for the incorporation of additionalsubunits. This appears consistent with the finding that Cox13, theyeast homologue of human Cox6a, is assembled after formation ofrespiratory supercomplexes [62].
Yeast Shy1 genetically and/or physically interacts with severalfactors involved in translational and early posttranslational steps ofCox1 biogenesis. In S. cerevisiae, Cox1 synthesis is controlled bytranslational activators Mss51 and Pet309. The high-copy expressionof Mss51, which has an additional role in posttranslational events ofCox1 biogenesis, can suppress the CcO-deficient phenotype of shy1Δcells. Mss51 and Cox1 form a transient complex that is stabilized byCox14. The accumulation of this complex is postulated to down-regulate the translation of Cox1 under conditions of impaired CcOassembly by sequestering Mss51 [46,61,63]. Shy1 is thought tofacilitate the release of Mss51 from the ternary complex, making itavailable for Cox1 translation [64]. Recently, Coa1 and Coa2 wereidentified as new yeast CcO assembly factors that act in early steps ofenzyme assembly [61,65,66]. Coa1 is an inner membrane-associatedprotein that exists as part of the Mss51.Cox14.Cox1 ternary complex,as well as in a complex with Shy1 alone. Coa1 may be involved in thetransition of newly synthesized Cox1 from the Mss51 complex to adownstream intermediate involving Shy1 [65]. The respiration in bothcoa1Δ and shy1Δ cells is enhanced by coexpression of Mss51 andCox10, suggesting that Coa1 may link heme a incorporation andcotranslational insertion of Cox1. Coa2, which may possess achaperone function, appears to act downstream of Coa1, and probablystabilizes Cox1 during the heme a3 insertion step [66]. Thisstabilization may be accomplished by promoting Cox5a/Cox6(human Cox4/Cox5a) association with Cox1, which may increasethe stability of the subunit by stabilizing the loops connecting the 12transmembrane helices [66]. This is consistent with the fact that theHAP4-induced upregulation of subunits Cox5 and Cox6 suppressesthe respiratory-deficient phenotype of shy1 cells [67]. Several lines ofevidence indicate that Surf1/Shy1 may be responsible for theinsertion of heme a3 into Cox1. The Rhodobacter sphaeroides Surf1homologue is required for the insertion/stabilization of heme a3

1152 L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
within Cox1, as the majority of CcO lacks heme a3 in R. sphaeroidesSurf1-null mutant [58]. When coexpressed in E. coliwith enzymes forheme a synthesis, the Paracoccus denitrificans Surf1 homologuesSurf1q and Surf1c bind heme a in vivo and probably provide a protein-bound heme a pool for the insertion into Cox1 [59]. cox11Δ cells areperoxide sensitive, showing an almost 2-fold increase in proteincarbonylation, consistent with enhanced ROS production [68]. Theincreased peroxide sensitivity of these cells stem from accumulationof a transient pro-oxidant assembly intermediate composed of Cox1with bound heme a3 moiety. Depletion of Shy1 in cox11Δ backgroundabrogates the peroxide sensitivity of these cells. Furthermore, shy1Δcells remain peroxide resistant even upon overexpression of Cox15,known to markedly increase heme a levels in CcO assembly mutants.The identity of the pro-oxidant heme is substantiated by the fact thatonly heme a3 has an open coordination site that can catalyzeformation of the hydroxyl radical pro-oxidant when solvent-exposed[68]. Mss51, Cox14, Coa1 and Coa2 are found exclusively in fungi,lacking homologues in both higher eukaryotes and bacteria [64].Furthermore, yeast Shy1 possesses additional C-terminal segmentsthat are not conserved in Surf1 proteins of other species. Thus, it is notclear whether similar mechanisms mediated by functional homo-logues of these proteins exist also in higher eukaryotes.
Zordan et al. have shown that ubiquitous posttranscriptionalsilencing of SURF1 in Drosophila melanogaster is associated with 100%egg-to-adult lethality, underdevelopment of the central nervoussystem (CNS), particularly of the optic lobes, and impaired locomotorbehavior in larvae. The altered motor patterns were likely not due tostructural and/or functional abnormalities of muscle fibers orreduction in contractile efficiency; rather they probably resultedfrom defective energy provision due to CcO deficiency. In contrast, apan-neuronal SURF1 knockdown led to slight impairment in locomo-tor behavior and photoreactivity and strikingly increased longevitywhen compared to controls. However, histological analysis failed toreveal any marked neurodegeneration in the brains of these flies [69].
In vertebrates, the SURF1 gene is part of the very tightly organizedand highly conserved surfeit gene cluster, which contains six house-keeping genes (SURF1–6) that encodeboth structurally and functionallyunrelatedproteins [70]. The reason for the conservation of this structureover 250 million years of divergent evolution between birds andmammals remains obscure. Baden et al. reported that posttranscrip-tional silencing of SURF1 in zebrafish (Danio rerio) by morpholinoantisense oligonucleotides resulted in 50% reduction in CcO activity aswell as developmental defects in endodermal tissues, cardiac functionand swimming behavior. The hindbrain and neuronal tube exhibiteddramatically increased apoptosis and secondary motor neurons wereabsent or abnormal. In contrast, the cardiac dysfunction was likely dueto impaired energy metabolism because the heart was devoid ofapoptotic cells, exhibiting increasingly poor performance over time [71].The described phenotype of Surf1-deficient zebrafish was almostidentical to that of the COX5 knockdown animals, suggesting that thedescribed Surf1-deficient phenotype can be readily attributed tosecondary CcO deficiency rather than to a specific lack of Surf1 protein[71]. Two SURF1 knockout mouse models have been generated by theZeviani's group using either replacement of exons 5–7 by neomycin-resistance (NEO)cassette [72]or insertionof a loxP sequence in exon7ofthegene [73]. Theprominent characteristic of the SURF1NEOKOmicewashigh embryonic lethality, which was subsequently attributed todeleterious effects of the presence of the NEO cassette. In contrast,SURF1loxP knockouts showed, in addition to mild CcO deficiency, alteredneuronal Ca2+ homeostasis, moderate functional and morphologicalabnormalities in both skeletal muscle and liver, and substantiallyprolonged lifespan [73]. In sharp contrast to severe CNS involvement ofhuman Surf1-deficient patients, neither mouse SURF1 KO modelexhibited spontaneous neurodegeneration at any age. The positiveeffects of murine SURF1 knockout on the lifespan of (−/−) animals isstrikingly similar to that of the D. melanogaster CNS-specific SURF1
knockdown. On this account, it was suggested that the partialsuppression of respiratory chain activity may positively affect lifespanof (−/−) animals, possibly also due to attenuated ROS production [56].Consistent with these findings, CcO deficiency was associated withreduced oxidative stress in CNS of COX10 knockout mice [74].
Mutations in SURF1, which account for the majority of nuclear-encoded, isolated CcO deficiencies in humans, are characterized by thedevelopment of Leigh syndrome, a subacute necrotizing encephalo-myopathy [75]. The prominent pathology of Leigh syndrome isrepresented by bilateral symmetric necrotic lesions in subcorticalareas of the brain (brain stem, basal ganglia and thalamus) thatconsist of neuronal loss, demyelination and vascular proliferation[76]. Most of the identified SURF1 mutations are predicted to lead toloss of the protein. Human as well as yeast cells lacking Surf1/Shy1retain approximately 10–20% of CcO activity, indicating that thefunction of the protein is partially dispensable in both organisms [52].In contrast to human Surf1, the lack of Shy1 leads to partiallypleiotropic effects including increased levels of cytochrome c andelevated NADH-cytochrome c reductase activity [53]. Furthermore,the human and yeast proteins fail to complement each other.However, the proteins exhibit striking sequence similarity in someconserved domains, and the elevated cytochrome c concentration andincreased complex III activity may reflect a yeast-specific compensa-tory response [60]. Analogous to yeast, overexpression of NF-YA, thecatalytic subunit of the human homologue (NF-Y) of the yeast HAPcomplex increases CcO activity of human SURF1 fibroblasts. Unlike theyeast HAP complex, the human NF-Y complex is not directly involvedin regulatingmitochondrial biogenesis, and themolecular mechanismresponsible for the suppression of the CcO defect in human SURF1fibroblasts is not known [67]. The severe CcO deficiency of SURF1fibroblasts is accompanied by barely detectable changes in cellularrespiratory rates under normoxic conditions [77]. Measuring CcOoxygen kinetics by the partial oxygen pressure at half-maximalrespiration rate revealed markedly attenuated affinity for oxygen ofthe residual enzyme. This phenomenon could exacerbate therespiratory defect in tissues where high energy demand meets upwith very low oxygen pressure, such as in the brain, a prominentpathology site of Leigh syndrome SURF1 patients [78]. The fatalneurological phenotype of CcO-deficient Leigh syndrome is associatedwith a remarkable tissue pattern of CcO assembly impairment,pointing to a profoundly tissue-specific characteristic of CcO biogen-esis [79]. Intriguingly, various tissue samples carrying mutations inSURF1 exhibit, similar to SCO1 and SCO2 tissues, marked tissue-dependent copper deficiency. This suggests that Surf1 is required in atissue-specific manner to maintain the cellular copper levels [55].Interestingly, yeast cells lacking Shy1 are deficient in mitochondrialcopper, whereas their total cellular copper content remains normal[65]. Supplementation of shy1Δ cultures with exogenous copperpartially rescues the respiratory capacity of these cells [67].
2.4. Cox17
The small hydrophilic copper-binding protein Cox17 localizes inyeast to both the cytoplasm and the mitochondrial IMS and was thefirst factor implicated in copper ion provision to CcO [80]. It deliversCu(I) to two downstream CcO copper-chaperones Sco1 and Cox11.The interaction of Cox17 with Sco1 and Cox11 is thought to betransient. Based on its dual localization, Cox17 was initially proposedto act as a copper shuttle between the cytoplasm and the IMS [81].Tethering Cox17 to the inner membrane by a heterologous trans-membrane domain renders the protein fully functional, suggestingthat migration between the cytoplasm and IMS is not essential for itsfunction [82]. Deletion of COX17 in S. cerevisiae does not affectmitochondrial copper content [83]. However, in vitro studies withpurified proteins and the yeast cytoplasm assay have demonstratedthat Cox17 can deliver Cu(I) to both Sco1 and Cox11 [84]. In addition

1153L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
to the Cx9C copper-binding motif, Cox17 possesses additional twoconserved Cys residues upstream of the twin Cys motif. The protein isfunctional without either of the two disulfides in the twin Cx9C motif.Two Cu(I) conformers of Cox17 were described so far. One is amononuclear Cu(I)–Cox17 monomer in the form of helical hairpinthat is stabilized by two disulfide bonds with Cys residues in the twinCx9C motif. The second Cu(I) conformer is an oligomeric proteincomplex containing reduced thiolates that is capable of binding apolycopper–thiolate cluster. Purifying Cox17 from yeast IMS yields aprotein devoid of bound copper [80].
Knockout of COX17 in mice leads to lethality of (−/−) individualsbetween embryonic days E8.5 and E10, confirming the essentialcharacter of Cox17 [85]. The human Cox17 orthologue has beenidentified that shares 48% sequence identity with yeast counterpart[86]. Overexpression of the human Cox17 rescues the CcO activitydefect of human SCO2 but not SCO1-deficient cells [87]. HeLa cells inwhich the expression of Cox17 was downregulated by the use ofsiRNA showed diminished levels of CcO and CcO-containing super-complexes [88]. The accumulation of a 158-kDa Cox1-containingsubcomplex in these cells that was devoid of Cox2 suggested a role forhuman Cox17 in the maturation of Cox2 but not Cox1.
2.5. Sco1 and Sco2
Human Sco1 and Sco2 are closely related inner mitochondrialmembrane copper-binding proteins encoded by paralogous genes.They exert nonoverlapping, cooperative roles in copper delivery toCcO [87]. In addition, they are involved in the maintenance of cellularcopper homeostasis, presumably by controlling cellular copper export[89,90]. Very recently, human SCO proteins were reported to carry outdistinct, stage-specific roles during Cox2 synthesis and CuA sitematuration [91]. Interestingly, the tumor suppressor p53 directlyregulates mitochondrial respiration through transactivation of humanSCO2 transcription [92]. Mutations in both SCO1 and SCO2 causesevere tissue-specific CcO assembly impairment accompanied bymarked cellular copper deficiency [55,79,90,93,94]. However, bothgenes are ubiquitously expressed and display a similar expressionpattern across human tissues. SCO1 mutations have originally beenreported in only a single pedigree, where the two patients, presentingwith fatal infantile encephalomyopathy and hepatopathy, werecompound heterozygotes carrying a nonsense mutation on one alleleand a P174Lmissensemutation on the second allele [94]. Recently, wehave studied the consequences of a novel homozygous SCO1missensemutation (G132S) found in a patient with CcO deficiency associatedwith early onset hypertrophic cardiomyopathy, hypotonia, encepha-lopathy and hepatopathy [55]. Thus, the lack of an apparent cardiacinvolvement in the previously published SCO1 cases, which was insharp contrast to SCO2mutations, very likely resulted either from theconsiderably reduced survival time of both siblings or the distinctnature of the missense allele expressed in these patients. Indeed, theP174L mutant Sco1 exhibits markedly altered functional propertiesand almost normal polypeptide levels [95], whereas the G132S alleleappears to lead to a simple, yet almost complete loss of protein andfunction [55]. In contrast to SCO1, SCO2mutations are more common,with the vast majority of reported patients carrying at least one E140Kmissense allele [96,97]. SCO2mutations cause fatal infantile encepha-lomyopathy and hypertrophic cardiomyopathy. SCO2 patients homo-zygous for the E140K substitution have a delayed onset and slightlyprolonged course of the disease compared with compound hetero-zygotes [96]. Recently,Winge's group demonstrated that yeast cox11Δcells accumulate a CcO subcomplex composed merely of Cox1 subunitwith bound heme a3 that exhibitsmarked pro-oxidant properties [68].Indeed, besides the CcO holoenzyme defect, mitochondria of clinicallyaffected tissues harboring SCO2 mutations are characterized bymarkedly increased levels of Cox1-containing subcomplexes withapparent molecular weight corresponding with that of free Cox1
subunit [79]. Thus, it appears plausible that increased ROS productionmay, in addition to energy deprivation, represent another factorresponsible for the severe clinical involvement of SCO2 tissues withprofound CcO assembly defect.
SCOproteins are integral innermembrane components consisting ofa globular copper-binding domain that protrudes into the IMS [98]. Thisdomain exhibits a thioredoxin fold composed of a central four-strandedβ sheet surrounded by four α helices [99]. A single Cu(I) binding site isformed by cysteinyl residues of the Cx3C motif and a histidyl residuefound within the globular domain. The structures of the metal-freehuman Sco1 conformer and Cu1Sco1 complex are similar with only oneloop showing significant rearrangements [100]. Sco1 and Sco2 aretethered to the innermembrane by a single N-terminal transmembranehelix that was shown to be functionally important in Sco1 [101]. Thehuman Sco2 conformer resembles human Sco1 with the exception ofexhibiting greater conformational dynamics [102]. SCO proteins arethought to act downstream of Cox17 in the copper delivery pathway toCuA site in Cox2. Sco1 is copper-metallated by Cox17 in vitro. It is notknown whether Sco1 delivers both Cu(I) and Cu(II) ions to build thebinuclear, mixed valent CuA center in Cox2. Consistent with thecomposition of CuA center, SCO proteins can bind either Cu(I) or Cu(II) ions [103]. SCO proteinsmay form a complex in order to deliver twocopper ions to Cox2 simultaneously [87]. The involvement of humanSCO proteins in copper delivery to CcO is further supported by the factthat the missense mutations in human SCO1 (P174L) and SCO2 (E140Kand S240F) are located in the vicinity of the conserved Cx3C copper-binding motif [94,104]. Additionally, the CcO defect of both SCO1 andSCO2fibroblasts andmyoblasts is at least partially rescuedbyexogenouscopper supplementation [87,93]. Finally, overexpressing of human SCOproteins with conserved cysteinyl and histidyl residues substituted byalanines fails to rescue the CcO deficiency of either SCO1 or SCO2fibroblasts [103].
Yeast cells lacking Sco1 are devoid of CcO activity and showmarkedly diminished levels of Cox2. Although yeast also encodes aSco2 protein that is capable of binding copper ions [105], this proteinhas no apparent function in CcO assembly [106]. When over-expressed, both yeast Sco1 and Sco2 were shown to physicallyinteracted with Cox2 [107]. Recently, using immunoprecipitation andtwo-dimensional western blotting, we have demonstrated thathuman Sco1 physically interacts with the fully assembled CcOcomplex in both skeletal muscle and HEK293 cell mitochondria [55].Originally, based on sequence similarity of Sco1 with the peroxir-edoxin protein family, the protein was proposed to be involved in themaintenance of CuA site cysteines in reduced state [108]. Furthermore,on the basis of high-resolution structural data, human Sco1 has beenimplicated to function as a redox switch in the IMS [99].
We have shown that human Sco2 acts in a highly tissue-specificmanner at an early stage of CcO assembly, very likely during thematuration of Cox2 subunit [79]. Furthermore, we demonstrated thatthe E140K substitution leads to severely diminished Sco2 levels in apatient heart, liver, brain and fibroblasts, suggesting severelyimpaired stability of the mutant protein. Based on the fact that thisamino acid substitution slightly perturbed copper-binding of Sco2, itwas speculated that the stability of Sco2 may depend on it beingcopper-loaded [91]. The previously identified SCO1 and SCO2missense mutations are located near the conserved copper-bindingmotif, suggesting that the loss-of-function outcome may relate toperturbed copper-binding of the protein. However, SCO2 missensemutations are associated with severely impaired stability of theprotein, and the E140K mutant retains rather appreciable residual Cu(I)-binding properties [109]. Importantly, overexpressing the E140Kmutant Sco2 in the corresponding mutant background led to rescue ofthe CcO defect [90]. In contrast, the P174Lmutation does not affect theability of Sco1 to bind and retain copper ions; however, its ability to becopper-loaded by Cox17 is severely compromised [95]. Based on theinability to detect the residual G132Smutant Sco1 in the dimeric form

1154 L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
on blue-native gels and the fact that the respective mutation lies in aprotein region previously shown to be required for dimerization, wehave proposed that dimerization is required to stabilize Sco1.Whereas the G132S Sco1 skeletal muscle mitochondria accumulatedtwo Cox2-containing subcomplexes, the corresponding Sco2-defi-cient samples are characterized by a complete absence of such species.This result suggests that Sco1 is very likely responsible for a differentposttranslational aspect of Cox2 biogenesis than Sco2 [55]. Thisconclusion is further supported by the fact that the steady-state levelof Sco2 was virtually unaffected in the Sco1-deficient background.Recently, Leary et al. have reported that Cox2 synthesis is diminishedin human SCO2 but not SCO1 cells [91]. The authors concluded thatSco2 is required for the synthesis of Cox2 in a manner that depends onits ability to bind copper, and that it acts upstream of Sco1 during thebiogenesis of Cox2 [91]. It was further demonstrated that a fraction oftotal Sco2 acts as a thiol-disulfide oxidoreductase, oxidizing the copper-coordinating cysteines in Sco1 during Cox2 maturation [91]. Underphysiological conditions, the cysteines in the Cx3Cmotif of Sco1 exist asamixedpopulation of oxidizeddisulfides and reduced thiols. In contrast,Sco1 molecules from either SCO background exhibited altered ratio ofoxidized to reduced cysteines. This ratio was shifted towards disulfidesupon overexpression of wild-type Sco2 and towards thiols uponknockdown of mutant Sco2 in the SCO2 background [91].
Leary et al. showed that tissues and/or fibroblasts culturesharboring mutations in SCO1, SCO2, COX10 or COX15 exhibit markedcopper-deficient phenotype, consistent with the involvement of thecorresponding gene products in cellular copper homeostasis [90].Importantly, the copper deficiency phenotypes of SCO1, SCO2 andCOX15 mutant fibroblasts were fully dissociable from the respectiveCcO defects. Kinetic labeling studies using 64Cu indicated that thecopper defect of SCO1 and SCO2 patient fibroblasts is caused by adefect in cellular copper retention rather than copper uptake.Recently, we have demonstrated that tissues harboring mutations inSURF1 exhibit marked tissue-dependent copper deficiency, furtherexpanding the list of CcO assembly factors with possible roles in themaintenance of cellular copper homeostasis [55]. The observedassociation of human Sco1 with the fully assembled CcO promptedus to hypothesize that the regulation of cellular copper homeostasismay involve CcO as an important cellular copper recipient [55]. Veryrecently, Schon's group generated SCO2 knockout as well as SCO2E140K knock-in mouse models. Whereas the both surviving micemodels, homozygous knock-in mice and knock-in/knockout hetero-zygotes, recapitulate many of the characteristics of human SCO2deficiency, they present with only very mild CcO defect. Indeed, theyhave normal tissue copper levels, with only mitochondrial coppercontent being significantly reduced [110].
3. Involvement of ATP-dependent proteases
A number of more or less specific proteases, including processingpeptidases, ATP-dependent proteases and oligopeptidases, are foundwithin the various subcompartments of mitochondria that mediatethe selective proteolysis of mitochondrial proteins. Two soluble ATP-dependent proteases, the Lon protease and the ClpXP protease, arefound within the mitochondrial matrix. In addition, two ATP-dependent proteolytic complexes, them- and i-AAAmetalloproteases,are anchored within the inner mitochondrial membrane, exposingtheir catalytic domains to the opposite membrane surfaces [111].Three out of these four identified proteolytic complexes were shownto be involved in degradation of unassembled CcO subunits.Mitochondrial ATP-dependent proteases belong to the AAA+ familyof proteins (ATPases Associated with a variety of cellular Activities),which is characterized by the presence of a conserved P-loop ATPasemodule [112]. They assemble into multimeric protein complexes witha central proteolytic chamber and, in addition to proteolytic function,exhibit chaperone-like and translocase activities [113]. They are
implicated in the surveillance of protein-quality control allowing forthe selective degradation of unassembled and damaged proteinswithin the various mitochondrial subcompartments [111,114,115]. Incontrast tomostmammalian cellular proteins that exhibit half-lives ofup to several hours [116], the majority of mitochondrial proteins arequite stable, with half-lives of several days [117–119]. Indeed, thehalf-life of the CcO complex was estimated to be about three days[120]. However, if severely damaged, mitochondria can be removedby a non-selective process, so-called mitochondrial autophagy(mitophagy) [121,122].
Lon is a homo-oligomeric complex with serine protease activity thatforms soluble heptameric ring-shaped assemblieswithin themitochon-drial matrix [114]. It has been shown to recognize, unfold and degrademisfolded, unassembled and oxidatively damaged matrix proteinsthereby preventing their deleterious accumulation [123]. Among theseveral identified endogenous substrates of mammalian Lon are thenuclear-encoded CcO subunits Cox4 and Cox5a (Fig. 1) [114].Furthermore, overexpression of both wild-type and proteolyticallyinactive Lon in cells under ER stress was suggested to promote theassembly of subunit Cox2 into Cox1-containing complexes [124].Subunit Cox4 is thought to associate with the membrane-embeddedsubunit Cox1 at the very beginning of CcO assembly [125,126]. Itsassociation with Cox1 is thought to occur in the form of a Cox4·Cox5aheterodimer [79]. The unassembled Cox4 appears relatively stable, assubstantial levels of the free subunit are found within mitochondriaunder various conditions. On the other hand, excess levels lead to itsrapid proteolytic degradation [126]. Cox4 contains a single transmem-brane helix and, when assembled, exposes a large soluble N-terminaldomain into the mitochondrial matrix [8]. In mammals, subunit Cox4exists as two isoforms; Cox4-1 and Cox4-2 expressed ubiquitously andat high levels in the lungs and trachea, respectively [127]. Expression ofCox4 isoforms was shown to be regulated by oxygen availability inmammalian cells [127]. This regulatory circuit involves HIF-1α, an O2-regulated subunit of hypoxia-inducible factor 1 (HIF-1), and the Lonprotease. HIF-1 is a transcriptional activator that plays a key role inregulating oxygen homeostasis in metazoan species [128]. Underaerobic conditions, the O2-regulated HIF-1α subunit is prolyl hydrox-ylated, and targeted to ubiquitination and proteasomal degradation[129,130]. Under hypoxic conditions, the α-subunit accumulates, andHIF-1 eventually triggers the transcriptional upregulation of both COX4-2 isoform and LON. Lon very likely mediates the degradation of Cox4-1and may also facilitate the assembly of newly imported Cox4-2 into theCcO complex (Fig. 1) [114,127].
m-AAA protease complex is one of the two homologous FtsH/AAAfamily membrane-bound ATP-dependent proteases found within theinner mitochondrial membrane [115]. Subunits of AAA proteasescontain the highly conservedWalker A (P-loop) andWalker B ATPasemodules within their AAA domains. The energy derived from ATPhydrolysis is utilized to unfold substrate proteins and transport theminto the internal proteolytic cavity [131]. The mitochondrial AAAproteases recognize misfolded, solvent-exposed domains of substrateproteins, short terminal protein tails projecting from the membranebilayer, and accessible interhelical loops of polytopic transmembraneproteins [111,115,132].m-AAA protease is active on the matrix side ofthe inner membrane and exists in human mitochondria as twoisoenzyme complexes build up of Spg7 and/or Afg3l2 subunits [133].The yeast m-AAA complex is implicated in the degradation ofunassembled mitochondrially encoded CcO subunit Cox3 (Fig. 1)[134,135]. Cox3 is a highly hydrophobic subunit of the CcO corespanning the inner membrane with seven transmembrane helices. Incontrast to the remaining two mitochondrially encoded CcO subunits,Cox3 does not contain any prosthetic groups and is not directlyinvolved in catalysis [8]. Although a long loop between helices III andIV of the subunit is exposed into the IMS, it is likely the soluble N-terminus protruding into the matrix that is recognized by the m-AAAprotease. Subunit Cox3 is thought to have an important role in CcO
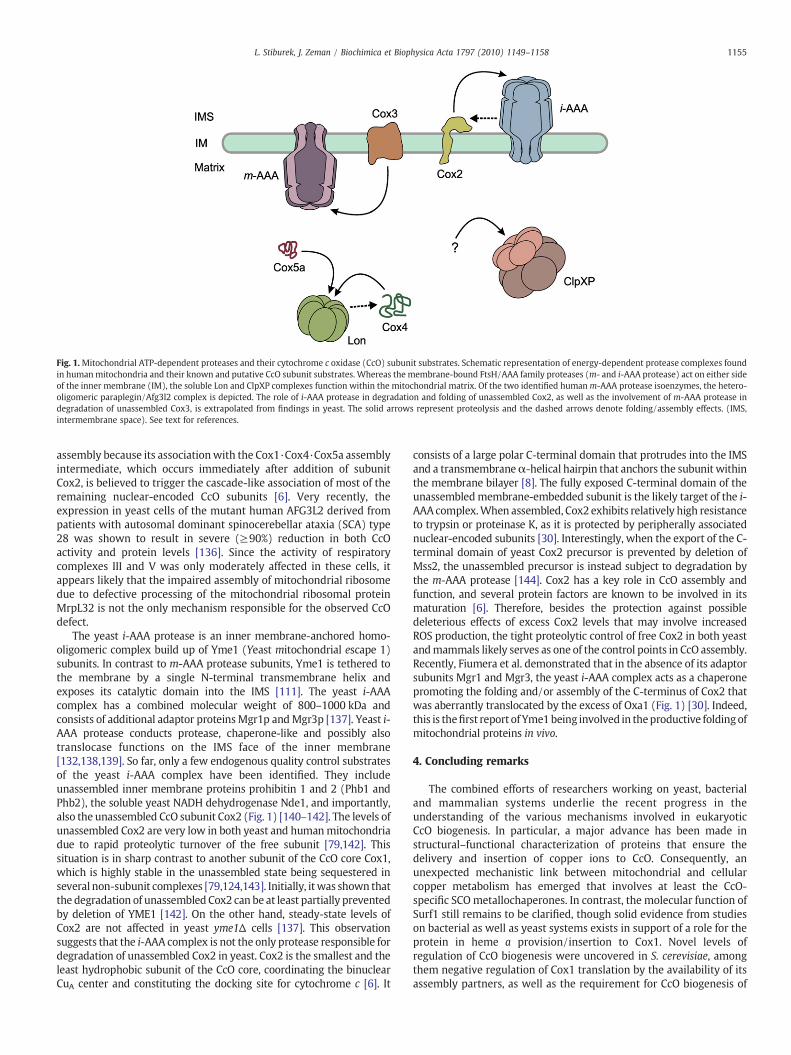
Fig. 1.Mitochondrial ATP-dependent proteases and their cytochrome c oxidase (CcO) subunit substrates. Schematic representation of energy-dependent protease complexes foundin humanmitochondria and their known and putative CcO subunit substrates. Whereas the membrane-bound FtsH/AAA family proteases (m- and i-AAA protease) act on either sideof the inner membrane (IM), the soluble Lon and ClpXP complexes function within the mitochondrial matrix. Of the two identified humanm-AAA protease isoenzymes, the hetero-oligomeric paraplegin/Afg3l2 complex is depicted. The role of i-AAA protease in degradation and folding of unassembled Cox2, as well as the involvement of m-AAA protease indegradation of unassembled Cox3, is extrapolated from findings in yeast. The solid arrows represent proteolysis and the dashed arrows denote folding/assembly effects. (IMS,intermembrane space). See text for references.
1155L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
assembly because its associationwith the Cox1·Cox4·Cox5a assemblyintermediate, which occurs immediately after addition of subunitCox2, is believed to trigger the cascade-like association of most of theremaining nuclear-encoded CcO subunits [6]. Very recently, theexpression in yeast cells of the mutant human AFG3L2 derived frompatients with autosomal dominant spinocerebellar ataxia (SCA) type28 was shown to result in severe (≥90%) reduction in both CcOactivity and protein levels [136]. Since the activity of respiratorycomplexes III and V was only moderately affected in these cells, itappears likely that the impaired assembly of mitochondrial ribosomedue to defective processing of the mitochondrial ribosomal proteinMrpL32 is not the only mechanism responsible for the observed CcOdefect.
The yeast i-AAA protease is an inner membrane-anchored homo-oligomeric complex build up of Yme1 (Yeast mitochondrial escape 1)subunits. In contrast to m-AAA protease subunits, Yme1 is tethered tothe membrane by a single N-terminal transmembrane helix andexposes its catalytic domain into the IMS [111]. The yeast i-AAAcomplex has a combined molecular weight of 800–1000 kDa andconsists of additional adaptor proteinsMgr1p andMgr3p [137]. Yeast i-AAA protease conducts protease, chaperone-like and possibly alsotranslocase functions on the IMS face of the inner membrane[132,138,139]. So far, only a few endogenous quality control substratesof the yeast i-AAA complex have been identified. They includeunassembled inner membrane proteins prohibitin 1 and 2 (Phb1 andPhb2), the soluble yeast NADH dehydrogenase Nde1, and importantly,also the unassembled CcO subunit Cox2 (Fig. 1) [140–142]. The levels ofunassembled Cox2 are very low in both yeast and humanmitochondriadue to rapid proteolytic turnover of the free subunit [79,142]. Thissituation is in sharp contrast to another subunit of the CcO core Cox1,which is highly stable in the unassembled state being sequestered inseveral non-subunit complexes [79,124,143]. Initially, itwas shown thatthe degradation of unassembled Cox2 can be at least partially preventedby deletion of YME1 [142]. On the other hand, steady-state levels ofCox2 are not affected in yeast yme1Δ cells [137]. This observationsuggests that the i-AAA complex is not the only protease responsible fordegradation of unassembled Cox2 in yeast. Cox2 is the smallest and theleast hydrophobic subunit of the CcO core, coordinating the binuclearCuA center and constituting the docking site for cytochrome c [6]. It
consists of a large polar C-terminal domain that protrudes into the IMSand a transmembraneα-helical hairpin that anchors the subunit withinthe membrane bilayer [8]. The fully exposed C-terminal domain of theunassembledmembrane-embedded subunit is the likely target of the i-AAA complex.When assembled, Cox2 exhibits relatively high resistanceto trypsin or proteinase K, as it is protected by peripherally associatednuclear-encoded subunits [30]. Interestingly, when the export of the C-terminal domain of yeast Cox2 precursor is prevented by deletion ofMss2, the unassembled precursor is instead subject to degradation bythe m-AAA protease [144]. Cox2 has a key role in CcO assembly andfunction, and several protein factors are known to be involved in itsmaturation [6]. Therefore, besides the protection against possibledeleterious effects of excess Cox2 levels that may involve increasedROS production, the tight proteolytic control of free Cox2 in both yeastandmammals likely serves as one of the control points in CcO assembly.Recently, Fiumera et al. demonstrated that in the absence of its adaptorsubunits Mgr1 and Mgr3, the yeast i-AAA complex acts as a chaperonepromoting the folding and/or assembly of the C-terminus of Cox2 thatwas aberrantly translocated by the excess of Oxa1 (Fig. 1) [30]. Indeed,this is thefirst report of Yme1being involved in theproductive foldingofmitochondrial proteins in vivo.
4. Concluding remarks
The combined efforts of researchers working on yeast, bacterialand mammalian systems underlie the recent progress in theunderstanding of the various mechanisms involved in eukaryoticCcO biogenesis. In particular, a major advance has been made instructural–functional characterization of proteins that ensure thedelivery and insertion of copper ions to CcO. Consequently, anunexpected mechanistic link between mitochondrial and cellularcopper metabolism has emerged that involves at least the CcO-specific SCOmetallochaperones. In contrast, the molecular function ofSurf1 still remains to be clarified, though solid evidence from studieson bacterial as well as yeast systems exists in support of a role for theprotein in heme a provision/insertion to Cox1. Novel levels ofregulation of CcO biogenesis were uncovered in S. cerevisiae, amongthem negative regulation of Cox1 translation by the availability of itsassembly partners, as well as the requirement for CcO biogenesis of

1156 L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
the fully assembled ATP synthase [145]. The identification of TACO1 as amammalian Cox1-specific translational activator suggests the existenceof an unexpected regulatory level of mammalian mitochondrial geneexpression. Finally, results of recent work on components of mitochon-drial protein-quality control system suggests that, besides theirproteolytic protein-quality control function and involvement in proteinprocessing, mitochondrial ATP-dependent proteases may facilitate theproductive folding and assembly of proteins, including CcO subunits.
Acknowledgements
Work in the authors' laboratory was supported by the GrantAgency of the Czech Republic project GACR P305/10/P414, the GrantAgency of the Ministry of Health of the Czech Republic project NS10581/3 and the institutional project MSM 0021620806.
References
[1] G. Voet, D.J. Voet, Biochemistry, Willey, 1995.[2] R. Lill, F.E. Nargang,W. Neupert, Biogenesis of mitochondrial proteins, Curr. Opin.
Cell Biol. 8 (1996) 505–512.[3] D.J. Pagliarini, S.E. Calvo, B. Chang, S.A. Sheth, S.B. Vafai, S.E. Ong, G.A. Walford, C.
Sugiana, A. Boneh, W.K. Chen, D.E. Hill, M. Vidal, J.G. Evans, D.R. Thorburn, S.A.Carr, V.K. Mootha, A mitochondrial protein compendium elucidates complex Idisease biology, Cell 134 (2008) 112–123.
[4] W. Neupert, J.M. Herrmann, Translocation of proteins into mitochondria, Annu.Rev. Biochem. 76 (2007) 723–749.
[5] H.S. Carr, D.R. Winge, Assembly of cytochrome c oxidase within the mitochon-drion, Acc. Chem. Res. 36 (2003) 309–316.
[6] L. Stiburek, H. Hansikova, M. Tesarova, L. Cerna, J. Zeman, Biogenesis ofeukaryotic cytochrome c oxidase, Physiol. Res. 55 (Suppl 2) (2006) S27–S41.
[7] R.A. Capaldi, Structure and assembly of cytochrome c oxidase, Arch. Biochem.Biophys. 280 (1990) 252–262.
[8] T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, S. Yoshikawa, The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A, Science 272 (1996) 1136–1144.
[9] A. Musatov, N.C. Robinson, Cholate-induced dimerization of detergent- orphospholipid-solubilized bovine cytochrome C oxidase, Biochemistry 41 (2002)4371–4376.
[10] J. Stanicova, E. Sedlak, A. Musatov, N.C. Robinson, Differential stability of dimericand monomeric cytochrome c oxidase exposed to elevated hydrostatic pressure,Biochemistry 46 (2007) 7146–7152.
[11] K. Shinzawa-Itoh, H. Aoyama, K. Muramoto, H. Terada, T. Kurauchi, Y. Tadehara,A. Yamasaki, T. Sugimura, S. Kurono, K. Tsujimoto, T. Mizushima, E. Yamashita, T.Tsukihara, S. Yoshikawa, Structures and physiological roles of 13 integral lipidsof bovine heart cytochrome c oxidase, EMBO J. 26 (2007) 1713–1725.
[12] A. Barrientos, M.H. Barros, I. Valnot, A. Rotig, P. Rustin, A. Tzagoloff, Cytochromeoxidase in health and disease, Gene 286 (2002) 53–63.
[13] E.A. Shoubridge, Cytochrome c oxidase deficiency, Am. J. Med. Genet. 106 (2001)46–52.
[14] V. Massa, E. Fernandez-Vizarra, S. Alshahwan, E. Bakhsh, P. Goffrini, I. Ferrero, P.Mereghetti, P. D'Adamo, P. Gasparini, M. Zeviani, Severe infantile encephalo-myopathy caused by a mutation in COX6B1, a nucleus-encoded subunit ofcytochrome c oxidase, Am. J. Hum. Genet. 82 (2008) 1281–1289.
[15] W. Weraarpachai, H. Antonicka, F. Sasarman, J. Seeger, B. Schrank, J.E. Kolesar, H.Lochmuller, M. Chevrette, B.A. Kaufman, R. Horvath, E.A. Shoubridge, Mutation inTACO1, encoding a translational activator of COX I, results in cytochrome coxidase deficiency and late-onset Leigh syndrome, Nat. Genet. 41 (2009)833–837.
[16] V.K. Mootha, P. Lepage, K. Miller, J. Bunkenborg, M. Reich, M. Hjerrild, T.Delmonte, A. Villeneuve, R. Sladek, F. Xu, G.A. Mitchell, C. Morin, M. Mann, T.J.Hudson, B. Robinson, J.D. Rioux, E.S. Lander, Identification of a gene causinghuman cytochrome c oxidase deficiency by integrative genomics, Proc. Natl.Acad. Sci. U. S. A. 100 (2003) 605–610.
[17] M.E. Sanchirico, T.D. Fox, T.L. Mason, Accumulation of mitochondrially synthesizedSaccharomyces cerevisiae Cox2p and Cox3p depends on targeting information inuntranslated portions of their mRNAs, EMBO J. 17 (1998) 5796–5804.
[18] S. Naithani, S.A. Saracco, C.A. Butler, T.D. Fox, Interactions among COX1, COX2,and COX3 mRNA-specific translational activator proteins on the inner surface ofthemitochondrial innermembrane of Saccharomyces cerevisiae, Mol. Biol. Cell 14(2003) 324–333.
[19] T.W.McMullin, T.D. Fox, COX3mRNA-specific translational activator proteins areassociated with the inner mitochondrial membrane in Saccharomyces cerevisiae,J. Biol. Chem. 268 (1993) 11737–11741.
[20] G.M. Manthey, B.D. Przybyla-Zawislak, J.E. McEwen, The Saccharomycescerevisiae Pet309 protein is embedded in the mitochondrial inner membrane,Eur. J. Biochem. 255 (1998) 156–161.
[21] N.S. Green-Willms, C.A. Butler, H.M. Dunstan, T.D. Fox, Pet111p, an innermembrane-bound translational activator that limits expression of the Saccharo-myces cerevisiae mitochondrial gene COX2, J. Biol. Chem. 276 (2001) 6392–6397.
[22] F. Xu, C. Morin, G. Mitchell, C. Ackerley, B.H. Robinson, The role of the LRPPRC(leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidaseassembly: mutation causes lowered levels of COX (cytochrome c oxidase) I andCOX III mRNA, Biochem. J. 382 (2004) 331–336.
[23] E. Delannoy, W.A. Stanley, C.S. Bond, I.D. Small, Pentatricopeptide repeat (PPR)proteins as sequence-specificity factors in post-transcriptional processes inorganelles, Biochem. Soc. Trans. 35 (2007) 1643–1647.
[24] F. Tavares-Carreon, Y. Camacho-Villasana, A. Zamudio-Ochoa, M. Shingu-Vazquez, A. Torres-Larios, X. Perez-Martinez, The pentatricopeptide repeatspresent in Pet309 are necessary for translation but not for stability of themitochondrial COX1 mRNA in yeast, J. Biol. Chem. 283 (2008) 1472–1479.
[25] F. Sasarman, C. Brunel-Guitton, H. Antonicka, T. Wai, E.A. Shoubridge, L.Consortium, LRPPRC and SLIRP interact in a ribonucleoprotein complex thatregulates posttranscriptional gene expression in mitochondria, Mol. Biol. Cell. 21(8) (2010 April) 1315–1323.
[26] J.M. Herrmann, W. Neupert, Protein insertion into the inner membrane ofmitochondria, IUBMB Life 55 (2003) 219–225.
[27] M. Hildenbeutel, S.J. Habib, J.M. Herrmann, D. Rapaport, New insights into themechanism of precursor protein insertion into the mitochondrial membranes,Int. Rev. Cell. Mol. Biol. 268 (2008) 147–190.
[28] J.M. Herrmann, W. Neupert, R.A. Stuart, Insertion into the mitochondrial innermembrane of a polytopic protein, the nuclear-encoded Oxa1p, EMBO J. 16(1997) 2217–2226.
[29] A. Kuhn, R. Stuart, R. Henry, R.E. Dalbey, The Alb3/Oxa1/YidC protein family:membrane-localized chaperones facilitating membrane protein insertion?Trends Cell Biol. 13 (2003) 510–516.
[30] H.L. Fiumera,M.J. Dunham, S.A. Saracco, C.A. Butler, J.A. Kelly, T.D. Fox, Translocationand assembly of mitochondrially coded Saccharomyces cerevisiae cytochrome coxidase subunit Cox2 byOxa1 and Yme1, in the absence of Cox18, Genetics (2009).
[31] L. Jia, M. Dienhart, M. Schramp, M. McCauley, K. Hell, R.A. Stuart, Yeast Oxa1interacts with mitochondrial ribosomes: the importance of the C-terminalregion of Oxa1, EMBO J. 22 (2003) 6438–6447.
[32] G. Szyrach, M. Ott, N. Bonnefoy,W. Neupert, J.M. Herrmann, Ribosome binding tothe Oxa1 complex facilitates co-translational protein insertion in mitochondria,EMBO J. 22 (2003) 6448–6457.
[33] M. Preuss, M. Ott, S. Funes, J. Luirink, J.M. Herrmann, Evolution of mitochondrialoxa proteins from bacterial YidC. Inherited and acquired functions of a conservedprotein insertion machinery, J. Biol. Chem. 280 (2005) 13004–13011.
[34] L. Jia, J. Kaur, R.A. Stuart, Mapping the yeast Oxa1-mitochondrial ribosomeinterface: identification of MrpL40, a ribosomal protein in close proximity toOxa1 and critical for OXPHOS complex assembly, Eukaryot. Cell 8 (11) (2009November) 1792–1802.
[35] K. Hell, W. Neupert, R.A. Stuart, Oxa1p acts as a general membrane insertionmachinery for proteins encoded by mitochondrial DNA, EMBO J. 20 (2001)1281–1288.
[36] N. Bonnefoy, H.L. Fiumera, G. Dujardin, T.D. Fox, Roles of Oxa1-related inner-membrane translocases in assembly of respiratory chain complexes, Biochim.Biophys. Acta 1793 (2009) 60–70.
[37] N. Bonnefoy, M. Kermorgant, O. Groudinsky, M. Minet, P.P. Slonimski, G.Dujardin, Cloning of a human gene involved in cytochrome oxidase assembly byfunctional complementation of an oxa1− mutation in Saccharomyces cerevisiae,Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 11978–11982.
[38] N. Altamura, N. Capitanio, N. Bonnefoy, S. Papa, G. Dujardin, The Saccharomycescerevisiae OXA1 gene is required for the correct assembly of cytochrome coxidase and oligomycin-sensitive ATP synthase, FEBS Lett. 382 (1996) 111–115.
[39] N. Bonnefoy, M. Kermorgant, O. Groudinsky, G. Dujardin, The respiratory geneOXA1 has two fission yeast orthologues which together encode a functionessential for cellular viability, Mol. Microbiol. 35 (2000) 1135–1145.
[40] F.E. Nargang, M. Preuss, W. Neupert, J.M. Herrmann, The Oxa1 protein forms ahomooligomeric complex and is an essential part of the mitochondrial exporttranslocase in Neurospora crassa, J. Biol. Chem. 277 (2002) 12846–12853.
[41] C.H. Sellem, C. Lemaire, S. Lorin, G. Dujardin, A. Sainsard-Chanet, Interactionbetween the oxa1 and rmp1 genes modulates respiratory complex assembly andlife span in Podospora anserina, Genetics 169 (2005) 1379–1389.
[42] R.L. Souza, N.S. Green-Willms, T.D. Fox, A. Tzagoloff, F.G. Nobrega, Cloning andcharacterization of COX18, a Saccharomyces cerevisiae PET gene required for theassembly of cytochrome oxidase, J. Biol. Chem. 275 (2000) 14898–14902.
[43] M. Gaisne, N. Bonnefoy, The COX18 gene, involved in mitochondrial biogenesis,is functionally conserved and tightly regulated in humans and fission yeast,FEMS Yeast Res. 6 (2006) 869–882.
[44] I. Hikkel, Y. Gbelska, Q.J. van der Aart, G. Lubecu, J. Subik, Cloning andcharacterization of KlCOX18, a gene required for activity of cytochrome oxidasein Kluyveromyces lactis, Curr. Genet. 32 (1997) 267–272.
[45] S.A. Saracco, T.D. Fox, Cox18p is required for export of the mitochondriallyencoded Saccharomyces cerevisiae Cox2p C-tail and interacts with Pnt1p andMss2p in the inner membrane, Mol. Biol. Cell 13 (2002) 1122–1131.
[46] A. Barrientos, A. Zambrano, A. Tzagoloff, Mss51p and Cox14p jointly regulatemitochondrial Cox1p expression in Saccharomyces cerevisiae, EMBO J. 23 (2004)3472–3482.
[47] S. Funes, F.E. Nargang, W. Neupert, J.M. Herrmann, The Oxa2 protein ofNeurospora crassa plays a critical role in the biogenesis of cytochrome oxidaseand defines a ubiquitous subbranch of the Oxa1/YidC/Alb3 protein family, Mol.Biol. Cell 15 (2004) 1853–1861.
[48] A. Rotig, B. Parfait, L. Heidet, G. Dujardin, P. Rustin, A. Munnich, Sequence andstructure of the human OXA1L gene and its upstream elements, Biochim.Biophys. Acta 1361 (1997) 6–10.

1157L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
[49] J. Sylvestre, A. Margeot, C. Jacq, G. Dujardin, M. Corral-Debrinski, The role of the3′ untranslated region in mRNA sorting to the vicinity of mitochondria isconserved from yeast to human cells, Mol. Biol. Cell 14 (2003) 3848–3856.
[50] L. Stiburek, D. Fornuskova, L. Wenchich, M. Pejznochova, H. Hansikova, J. Zeman,Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase andNADH:ubiquinone oxidoreductase, J. Mol. Biol. 374 (2007) 506–516.
[51] L. Jia, M.K. Dienhart, R.A. Stuart, Oxa1 directly interacts with Atp9 and mediatesits assembly into the mitochondrial F1Fo-ATP synthase complex, Mol. Biol. Cell18 (2007) 1897–1908.
[52] J. Yao, E.A. Shoubridge, Expression and functional analysis of SURF1 in Leighsyndrome patients with cytochrome c oxidase deficiency, Hum. Mol. Genet.8 (1999) 2541–2549.
[53] G. Mashkevich, B. Repetto, D.M. Glerum, C. Jin, A. Tzagoloff, SHY1, the yeasthomolog of the mammalian SURF-1 gene, encodes a mitochondrial proteinrequired for respiration, J. Biol. Chem. 272 (1997) 14356–14364.
[54] S.L. Williams, I. Valnot, P. Rustin, J.W. Taanman, Cytochrome c oxidasesubassemblies in fibroblast cultures from patients carrying mutations inCOX10, SCO1, or SURF1, J. Biol. Chem. 279 (2004) 7462–7469.
[55] L. Stiburek, K. Vesela, H. Hansikova, H. Hulkova, J. Zeman, Loss of function of Sco1and its interaction with cytochrome c oxidase, Am. J. Physiol.Cell Physiol. 296 (5)(2009 May) C1218–C1226.
[56] E. Fernandez-Vizarra, V. Tiranti, M. Zeviani, Assembly of the oxidativephosphorylation system in humans: what we have learned by studying itsdefects, Biochim. Biophys. Acta 1793 (2009) 200–211.
[57] F.A. Bundschuh, K. Hoffmeier, B. Ludwig, Two variants of the assembly factorSurf1 target specific terminal oxidases in Paracoccus denitrificans, Biochim.Biophys. Acta 1777 (2008) 1336–1343.
[58] D. Smith, J. Gray, L. Mitchell, W.E. Antholine, J.P. Hosler, Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active siteheme, J. Biol. Chem. 280 (2005) 17652–17656.
[59] F.A. Bundschuh, A. Hannappel, O. Anderka, B. Ludwig, SURF1, associated withLeigh syndrome in humans, is a heme-binding protein in bacterial oxidasebiogenesis, J. Biol. Chem. 284 (38) (2009 September 18) 25735–25741.
[60] L.G. Nijtmans, M. Artal Sanz, M. Bucko, M.H. Farhoud, M. Feenstra, G.A. Hakkaart,M. Zeviani, L.A. Grivell, Shy1p occurs in a high molecular weight complex and isrequired for efficient assembly of cytochrome c oxidase in yeast, FEBS Lett. 498(2001) 46–51.
[61] D.U. Mick, K. Wagner, M. van der Laan, A.E. Frazier, I. Perschil, M. Pawlas, H.E.Meyer, B. Warscheid, P. Rehling, Shy1 couples Cox1 translational regulation tocytochrome c oxidase assembly, EMBO J. 26 (2007) 4347–4358.
[62] K. Brandner, D.U. Mick, A.E. Frazier, R.D. Taylor, C. Meisinger, P. Rehling, Taz1, anouter mitochondrial membrane protein, affects stability and assembly of innermembrane protein complexes: implications for Barth syndrome, Mol. Biol. Cell16 (2005) 5202–5214.
[63] X. Perez-Martinez, S.A. Broadley, T.D. Fox, Mss51p promotes mitochondrialCox1p synthesis and interacts with newly synthesized Cox1p, EMBO J. 22 (2003)5951–5961.
[64] A. Barrientos, K. Gouget, D. Horn, I.C. Soto, F. Fontanesi, Suppression mechanismsof COX assembly defects in yeast and human: insights into the COX assemblyprocess, Biochim. Biophys. Acta 1793 (2009) 97–107.
[65] F. Pierrel, M.L. Bestwick, P.A. Cobine, O. Khalimonchuk, J.A. Cricco, D.R. Winge,Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion incytochrome c oxidase assembly, EMBO J. 26 (2007) 4335–4346.
[66] F. Pierrel, O. Khalimonchuk, P.A. Cobine, M. Bestwick, D.R. Winge, Coa2 is anassembly factor for yeast cytochrome c oxidase biogenesis that facilitates thematuration of Cox1, Mol. Cell. Biol. 28 (2008) 4927–4939.
[67] F. Fontanesi, C. Jin, A. Tzagoloff, A. Barrientos, Transcriptional activators HAP/NF-Y rescue a cytochrome c oxidase defect in yeast and human cells, Hum. Mol.Genet. 17 (2008) 775–788.
[68] O. Khalimonchuk, A. Bird, D.R. Winge, Evidence for a pro-oxidant intermediate inthe assembly of cytochrome oxidase, J. Biol. Chem. 282 (2007) 17442–17449.
[69] M.A. Zordan, P. Cisotto, C. Benna, A. Agostino, G. Rizzo, A. Piccin, M. Pegoraro, F.Sandrelli, G. Perini, G. Tognon, R. De Caro, S. Peron, T.T. Kronnie, A. Megighian, C.Reggiani, M. Zeviani, R. Costa, Post-transcriptional silencing and functionalcharacterization of the Drosophila melanogaster homolog of human Surf1,Genetics 172 (2006) 229–241.
[70] T. Duhig, C. Ruhrberg, O. Mor, M. Fried, The human Surfeit locus, Genomics 52(1998) 72–78.
[71] K.N. Baden, J. Murray, R.A. Capaldi, K. Guillemin, Early developmental pathologydue to cytochrome c oxidase deficiency is revealed by a new zebrafish model,J. Biol. Chem. 282 (2007) 34839–34849.
[72] A. Agostino, F. Invernizzi, C. Tiveron, G. Fagiolari, A. Prelle, E. Lamantea, A.Giavazzi, G. Battaglia, L. Tatangelo, V. Tiranti, M. Zeviani, Constitutive knockoutof Surf1 is associated with high embryonic lethality, mitochondrial diseaseand cytochrome c oxidase deficiency in mice, Hum. Mol. Genet. 12 (2003)399–413.
[73] C. Dell'agnello, S. Leo, A. Agostino, G. Szabadkai, C. Tiveron, A. Zulian, A. Prelle, P.Roubertoux, R. Rizzuto, M. Zeviani, Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice, Hum. Mol. Genet.16 (2007) 431–444.
[74] H. Fukui, F. Diaz, S. Garcia, C.T. Moraes, Cytochrome c oxidase deficiency inneurons decreases both oxidative stress and amyloid formation in a mousemodel of Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A. 104 (2007)14163–14168.
[75] Z. Zhu, J. Yao, T. Johns, K. Fu, I. De Bie, C. Macmillan, A.P. Cuthbert, R.F. Newbold, J.Wang, M. Chevrette, G.K. Brown, R.M. Brown, E.A. Shoubridge, SURF1, encoding a
factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leighsyndrome, Nat. Genet. 20 (1998) 337–343.
[76] D. Leigh, Subacute necrotizing encephalomyelopathy in an infant, J. Neurol.Neurosurg. Psychiatry 14 (1951) 216–221.
[77] P. Pecina, M. Capkova, S.K. Chowdhury, Z. Drahota, A. Dubot, A. Vojtiskova, H.Hansikova, H. Houst'kova, J. Zeman, C. Godinot, J. Houstek, Functional alterationof cytochrome c oxidase by SURF1 mutations in Leigh syndrome, Biochim.Biophys. Acta 1639 (2003) 53–63.
[78] P. Pecina, E. Gnaiger, J. Zeman, E. Pronicka, J. Houstek, Decreased affinity foroxygen of cytochrome-c oxidase in Leigh syndrome caused by SURF1 mutations,Am. J. Physiol. Cell Physiol. 287 (2004) C1384–C1388.
[79] L. Stiburek, K. Vesela, H. Hansikova, P. Pecina, M. Tesarova, L. Cerna, J. Houstek, J.Zeman, Tissue-specific cytochrome c oxidase assembly defects due to mutationsin SCO2 and SURF1, Biochem. J. 392 (2005) 625–632.
[80] O. Khalimonchuk, D.R. Winge, Function and redox state of mitochondriallocalized cysteine-rich proteins important in the assembly of cytochrome coxidase, Biochim. Biophys. Acta 1783 (2008) 618–628.
[81] J. Beers, D.M. Glerum, A. Tzagoloff, Purification, characterization, and localizationof yeast Cox17p, a mitochondrial copper shuttle, J. Biol. Chem. 272 (1997)33191–33196.
[82] A.B. Maxfield, D.N. Heaton, D.R. Winge, Cox17 is functional when tethered to themitochondrial inner membrane, J. Biol. Chem. 279 (2004) 5072–5080.
[83] P.A. Cobine, L.D. Ojeda, K.M. Rigby, D.R. Winge, Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix, J. Biol. Chem. 279(2004) 14447–14455.
[84] Y.C. Horng, P.A. Cobine, A.B. Maxfield, H.S. Carr, D.R. Winge, Specific coppertransfer from the Cox17 metallochaperone to both Sco1 and Cox11 in theassembly of yeast cytochrome C oxidase, J. Biol. Chem. 279 (2004) 35334–35340.
[85] Y. Takahashi, K. Kako, S. Kashiwabara, A. Takehara, Y. Inada, H. Arai, K. Nakada, H.Kodama, J. Hayashi, T. Baba, E. Munekata, Mammalian copper chaperone Cox17phas an essential role in activation of cytochrome C oxidase and embryonicdevelopment, Mol. Cell. Biol. 22 (2002) 7614–7621.
[86] R. Amaravadi, D.M. Glerum, A. Tzagoloff, Isolation of a cDNA encoding the humanhomolog of COX17, a yeast gene essential for mitochondrial copper recruitment,Hum. Genet. 99 (1997) 329–333.
[87] S.C. Leary, B.A. Kaufman, G. Pellecchia, G.H. Guercin, A. Mattman, M. Jaksch, E.A.Shoubridge, Human SCO1 and SCO2 have independent, cooperative functionsin copper delivery to cytochrome c oxidase, Hum. Mol. Genet. 13 (2004)1839–1848.
[88] C. Oswald, U. Krause-Buchholz, G. Rodel, Knockdown of human COX17 affectsassembly and supramolecular organization of cytochrome c oxidase, J. Mol. Biol.389 (2009) 470–479.
[89] S.C. Leary, D.R. Winge, P.A. Cobine, Pulling the plug on cellular copper: the role ofmitochondria in copper export, Biochim. Biophys. Acta 1793 (2009) 146–153.
[90] S.C. Leary, P.A. Cobine, B.A. Kaufman, G.H. Guercin, A. Mattman, J. Palaty, G.Lockitch, D.R. Winge, P. Rustin, R. Horvath, E.A. Shoubridge, The humancytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory rolesin the maintenance of cellular copper homeostasis, Cell Metab. 5 (2007) 9–20.
[91] S.C. Leary, F. Sasarman, T. Nishimura, E.A. Shoubridge, Human SCO2 is requiredfor the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1, Hum.Mol. Genet. 18 (2009) 2230–2240.
[92] S. Matoba, J.G. Kang,W.D. Patino, A.Wragg, M. Boehm, O. Gavrilova, P.J. Hurley, F.Bunz, P.M. Hwang, p53 regulates mitochondrial respiration, Science 312 (2006)1650–1653.
[93] M. Jaksch, C. Paret, R. Stucka, N. Horn, J. Muller-Hocker, R. Horvath, N. Trepesch,G. Stecker, P. Freisinger, C. Thirion, J. Muller, R. Lunkwitz, G. Rodel, E.A.Shoubridge, H. Lochmuller, Cytochrome c oxidase deficiency due to mutations inSCO2, encoding a mitochondrial copper-binding protein, is rescued by copper inhuman myoblasts, Hum. Mol. Genet. 10 (2001) 3025–3035.
[94] I. Valnot, S. Osmond, N. Gigarel, B. Mehaye, J. Amiel, V. Cormier-Daire, A.Munnich, J.P. Bonnefont, P. Rustin, A. Rotig, Mutations of the SCO1 gene inmitochondrial cytochrome c oxidase deficiency with neonatal-onset hepaticfailure and encephalopathy, Am. J. Hum. Genet. 67 (2000) 1104–1109.
[95] P.A. Cobine, F. Pierrel, S.C. Leary, F. Sasarman, Y.C. Horng, E.A. Shoubridge, D.R.Winge, The P174L mutation in human Sco1 severely compromises Cox17-dependent metallation but does not impair copper binding, J. Biol. Chem. 281(2006) 12270–12276.
[96] K. Vesela, H. Hansikova, M. Tesarova, P. Martasek, M. Elleder, J. Houstek, J. Zeman,Clinical, biochemical and molecular analyses of six patients with isolatedcytochrome c oxidase deficiency due to mutations in the SCO2 gene, ActaPaediatr. 93 (2004) 1312–1317.
[97] B.C. Mobley, G.M. Enns, L.J. Wong, H. Vogel, A novel homozygous SCO2 mutation,p.G193S, causing fatal infantile cardioencephalomyopathy, Clin. Neuropathol. 28(2009) 143–149.
[98] P. Buchwald, G. Krummeck, G. Rodel, Immunological identification of yeast SCO1protein as a component of the inner mitochondrial membrane, Mol. Gen. Genet.229 (1991) 413–420.
[99] J.C. Williams, C. Sue, G.S. Banting, H. Yang, D.M. Glerum, W.A. Hendrickson, E.A.Schon, Crystal structure of human SCO1: implications for redox signaling by amitochondrial cytochrome c oxidase “assembly” protein, J. Biol. Chem. 280(2005) 15202–15211.
[100] L. Banci, I. Bertini, V. Calderone, S. Ciofi-Baffoni, S. Mangani, M. Martinelli, P.Palumaa, S. Wang, A hint for the function of human Sco1 from differentstructures, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 8595–8600.
[101] J. Beers, D.M. Glerum, A. Tzagoloff, Purification and characterization of yeastSco1p, a mitochondrial copper protein, J. Biol. Chem. 277 (2002) 22185–22190.

1158 L. Stiburek, J. Zeman / Biochimica et Biophysica Acta 1797 (2010) 1149–1158
[102] L. Banci, I. Bertini, S. Ciofi-Baffoni, I. Leontari, M. Martinelli, P. Palumaa, R. Sillard,S. Wang, Human Sco1 functional studies and pathological implications of theP174L mutant, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 15–20.
[103] Y.C. Horng, S.C. Leary, P.A. Cobine, F.B. Young, G.N. George, E.A. Shoubridge, D.R.Winge, Human Sco1 and Sco2 function as copper-binding proteins, J. Biol. Chem.280 (2005) 34113–34122.
[104] M. Jaksch, I. Ogilvie, J. Yao, G. Kortenhaus, H.G. Bresser, K.D. Gerbitz, E.A.Shoubridge, Mutations in SCO2 are associated with a distinct form ofhypertrophic cardiomyopathy and cytochrome c oxidase deficiency, Hum. Mol.Genet. 9 (2000) 795–801.
[105] P.A. Cobine, F. Pierrel, D.R. Winge, Copper trafficking to the mitochondrion andassembly of coppermetalloenzymes, Biochim. Biophys. Acta 1763 (2006) 759–772.
[106] D.M. Glerum, A. Shtanko, A. Tzagoloff, SCO1 and SCO2 act as high copysuppressors of a mitochondrial copper recruitment defect in Saccharomycescerevisiae, J. Biol. Chem. 271 (1996) 20531–20535.
[107] A. Lode, M. Kuschel, C. Paret, G. Rodel, Mitochondrial copper metabolism inyeast: interaction between Sco1p and Cox2p, FEBS Lett. 485 (2000) 19–24.
[108] Y.V. Chinenov, Cytochrome c oxidase assembly factors with a thioredoxin fold areconserved among prokaryotes and eukaryotes, J. Mol. Med. 78 (2000) 239–242.
[109] P.F. Foltopoulou, G.A. Zachariadis, A.S. Politou, A.S. Tsiftsoglou, L.C. Papadopou-lou, Human recombinant mutated forms of the mitochondrial COX assemblySco2 protein differ from wild-type in physical state and copper binding capacity,Mol. Genet. Metab. 81 (2004) 225–236.
[110] H. Yang, S. Brosel, R. Acin-Perez, V. Slavkovich, I. Nishino, R. Khan, I.J. Goldberg, J.Graziano, G. Manfredi, E.A. Schon, Analysis of mouse models of cytochrome coxidase deficiency due to mutations in Sco2, Hum. Mol. Genet. (2009).
[111] M. Koppen, T. Langer, Protein degradation within mitochondria: versatileactivities of AAA proteases and other peptidases, Crit. Rev. Biochem. Mol. Biol.42 (2007) 221–242.
[112] P.I. Hanson, S.W. Whiteheart, AAA+ proteins: have engine, will work, Nat. Rev.Mol. Cell. Biol. 6 (2005) 519–529.
[113] R.T. Sauer, D.N. Bolon, B.M. Burton, R.E. Burton, J.M. Flynn, R.A. Grant, G.L. Hersch,S.A. Joshi, J.A. Kenniston, I. Levchenko, S.B. Neher, E.S. Oakes, S.M. Siddiqui, D.A.Wah, T.A. Baker, Sculpting the proteome with AAA(+) proteases anddisassembly machines, Cell 119 (2004) 9–18.
[114] I. Lee, C.K. Suzuki, Functional mechanics of the ATP-dependent Lon protease —
lessons from endogenous protein and synthetic peptide substrates, Biochim.Biophys. Acta 1784 (2008) 727–735.
[115] T. Tatsuta, T. Langer, AAA proteases in mitochondria: diverse functions ofmembrane-bound proteolytic machines, Res. Microbiol. 160 (9) (2009 November)711–717.
[116] H.C. Yen, Q. Xu, D.M. Chou, Z. Zhao, S.J. Elledge, Global protein stability profilingin mammalian cells, Science 322 (2008) 918–923.
[117] B. Aschenbrenner, R. Druyan, R. Albin, M. Rabinowitz, Haem a, cytochrome c andtotal protein turnover in mitochondria from rat heart and liver, Biochem. J. 119(1970) 157–160.
[118] R. Druyan, B. DeBernard, M. Rabinowitz, Turnover of cytochromes labeled withdelta-aminolevulinic acid-3H in rat liver, J. Biol. Chem. 244 (1969) 5874–5878.
[119] M.M. Ip, P.Y. Chee, R.W. Swick, Turnover of hepatic mitochondrial ornithineaminotransferase and cytochrome oxidase using (14C)carbonate as tracer,Biochim. Biophys. Acta 354 (1974) 29–38.
[120] S.C. Leary, B.C. Hill, C.N. Lyons, C.G. Carlson, D. Michaud, C.S. Kraft, K. Ko, D.M.Glerum, C.D. Moyes, Chronic treatment with azide in situ leads to an irreversibleloss of cytochrome c oxidase activity via holoenzyme dissociation, J. Biol. Chem.277 (2002) 11321–11328.
[121] T. Yorimitsu, D.J. Klionsky, Autophagy: molecular machinery for self-eating, CellDeath Differ. 12 (Suppl 2) (2005) 1542–1552.
[122] D. Mijaljica, M. Prescott, R.J. Devenish, Different fates of mitochondria:alternative ways for degradation? Autophagy 3 (2007) 4–9.
[123] J.K. Ngo, K.J. Davies, Importance of the lon protease in mitochondrialmaintenance and the significance of declining lon in aging, Ann. N. Y. Acad.Sci. 1119 (2007) 78–87.
[124] O. Hori, F. Ichinoda, T. Tamatani, A. Yamaguchi, N. Sato, K. Ozawa, Y. Kitao, M.Miyazaki, H.P. Harding, D. Ron, M. Tohyama, D. M.S., O. Ogawa, Transmission ofcell stress from endoplasmic reticulum to mitochondria: enhanced expression ofLon protease, J. Cell Biol. 157 (2002) 1151–1160.
[125] L.G. Nijtmans, J.W. Taanman, A.O. Muijsers, D. Speijer, C. Van den Bogert,Assembly of cytochrome-c oxidase in cultured human cells, Eur. J. Biochem. 254(1998) 389–394.
[126] C. Ugalde, M.J. Coenen, M.H. Farhoud, S. Gilinsky, W.J. Koopman, L.P. van denHeuvel, J.A. Smeitink, L.G. Nijtmans, New perspectives on the assembly processof mitochondrial respiratory chain complex cytochrome c oxidase, Mitochon-drion 2 (2002) 117–128.
[127] R. Fukuda, H. Zhang, J.W. Kim, L. Shimoda, C.V. Dang, G.L. Semenza, HIF-1regulates cytochrome oxidase subunits to optimize efficiency of respiration inhypoxic cells, Cell 129 (2007) 111–122.
[128] N.V. Iyer, L.E. Kotch, F. Agani, S.W. Leung, E. Laughner, R.H. Wenger, M.Gassmann, J.D. Gearhart, A.M. Lawler, A.Y. Yu, G.L. Semenza, Cellular anddevelopmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha,Genes Dev. 12 (1998) 149–162.
[129] P.H. Maxwell, M.S. Wiesener, G.W. Chang, S.C. Clifford, E.C. Vaux, M.E. Cockman,C.C. Wykoff, C.W. Pugh, E.R. Maher, P.J. Ratcliffe, The tumour suppressor proteinVHL targets hypoxia-inducible factors for oxygen-dependent proteolysis, Nature399 (1999) 271–275.
[130] P. Jaakkola, D.R. Mole, Y.M. Tian, M.I. Wilson, J. Gielbert, S.J. Gaskell, A.Kriegsheim, H.F. Hebestreit, M. Mukherji, C.J. Schofield, P.H. Maxwell, C.W.Pugh, P.J. Ratcliffe, Targeting of HIF-alpha to the von Hippel–Lindau ubiquityla-tion complex by O2-regulated prolyl hydroxylation, Science 292 (2001)468–472.
[131] T.A. Baker, R.T. Sauer, ATP-dependent proteases of bacteria: recognition logic andoperating principles, Trends Biochem. Sci. 31 (2006) 647–653.
[132] K. Leonhard, A. Stiegler, W. Neupert, T. Langer, Chaperone-like activity of theAAA domain of the yeast Yme1 AAA protease, Nature 398 (1999) 348–351.
[133] M. Koppen, M.D. Metodiev, G. Casari, E.I. Rugarli, T. Langer, Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexeslinked to hereditary spastic paraplegia, Mol. Cell. Biol. 27 (2007) 758–767.
[134] H. Arlt, R. Tauer, H. Feldmann, W. Neupert, T. Langer, The YTA10–12 complex, anAAA protease with chaperone-like activity in the inner membrane ofmitochondria, Cell 85 (1996) 875–885.
[135] G. Steglich, W. Neupert, T. Langer, Prohibitins regulate membrane proteindegradation by the m-AAA protease in mitochondria, Mol. Cell. Biol. 19 (1999)3435–3442.
[136] D. Di Bella, F. Lazzaro, A. Brusco, M. Plumari, G. Battaglia, A. Pastore, A. Finardi, C.Cagnoli, F. Tempia, M. Frontali, L. Veneziano, T. Sacco, E. Boda, A. Brussino, F.Bonn, B. Castellotti, S. Baratta, C. Mariotti, C. Gellera, V. Fracasso, S. Magri, T.Langer, P. Plevani, S. Di Donato, M. Muzi-Falconi, F. Taroni, Mutations in themitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28,Nat. Genet. 42 (4) (2010 April) 313–321.
[137] C.D. Dunn, Y. Tamura, H. Sesaki, R.E. Jensen, Mgr3p and Mgr1p are adaptors forthe mitochondrial i-AAA protease complex, Mol. Biol. Cell 19 (2008) 5387–5397.
[138] K. Leonhard, J.M. Herrmann, R.A. Stuart, G. Mannhaupt, W. Neupert, T. Langer,AAA proteases with catalytic sites on opposite membrane surfaces comprise aproteolytic system for the ATP-dependent degradation of inner membraneproteins in mitochondria, EMBO J. 15 (1996) 4218–4229.
[139] R.N. Rainey, J.D. Glavin, H.W. Chen, S.W. French, M.A. Teitell, C.M. Koehler, A newfunction in translocation for the mitochondrial i-AAA protease Yme1: import ofpolynucleotide phosphorylase into the intermembrane space, Mol. Cell. Biol. 26(2006) 8488–8497.
[140] E.R. Weber, T. Hanekamp, P.E. Thorsness, Biochemical and functional analysis ofthe YME1 gene product, an ATP and zinc-dependent mitochondrial proteasefrom S. cerevisiae, Mol. Biol. Cell 7 (1996) 307–317.
[141] M. Kambacheld, S. Augustin, T. Tatsuta, S. Muller, T. Langer, Role of the novelmetallopeptidase Mop112 and saccharolysin for the complete degradation ofproteins residing in different subcompartments of mitochondria, J. Biol. Chem.280 (2005) 20132–20139.
[142] D.A. Pearce, F. Sherman, Degradation of cytochrome oxidase subunits in mutantsof yeast lacking cytochrome c and suppression of the degradation by mutation ofyme1, J. Biol. Chem. 270 (1995) 20879–20882.
[143] O. Khalimonchuk, M. Bestwick, B. Meunier, T.C. Watts, D.R. Winge, Formation ofthe redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase,Mol. Cell. Biol. 30 1004–1017.
[144] S.A. Broadley, C.M. Demlow, T.D. Fox, Peripheral mitochondrial inner membraneprotein, Mss2p, required for export of the mitochondrially coded Cox2p C tail inSaccharomyces cerevisiae, Mol. Cell. Biol. 21 (2001) 7663–7672.
[145] I.C. Soto, F. Fontanesi, M. Valledor, D. Horn, R. Singh, A. Barrientos, Synthesisof cytochrome c oxidase subunit 1 is translationally downregulated in theabsence of functional F1F0-ATP synthase, Biochim. Biophys. Acta 1793 (2009)1776–1786.




















