
application of hadamard transform ion mobility mass
246
APPLICATION OF HADAMARD TRANSFORM ION MOBILITY MASS SPECTROMETRY TO GLOBAL METABOLOMICS By XING ZHANG A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY WASHINGTON STATE UNIVERSITY Department of Chemistry AUGUST 2014 © Copyright by XING ZHANG, 2014 All Rights Reserved
-
Upload
khangminh22 -
Category
Documents
-
view
5 -
download
0
Transcript of application of hadamard transform ion mobility mass

APPLICATION OF HADAMARD TRANSFORM ION MOBILITY MASS
SPECTROMETRY TO GLOBAL METABOLOMICS
By
XING ZHANG
A dissertation submitted in partial fulfillment of the requirements for the degree of
DOCTOR OF PHILOSOPHY
WASHINGTON STATE UNIVERSITY Department of Chemistry
AUGUST 2014
© Copyright by XING ZHANG, 2014 All Rights Reserved

ii
To the Faculty of Washington State University: The members of the Committee appointed to examine the dissertation of Xing Zhang find it satisfactory and recommend that it be accepted.
Herbert H. Hill, Ph.D., Chair
William F. Siems, Ph.D.
Peter T. A. Reilly, Ph.D.
Brian H. Clowers, Ph.D.
Nairanjana Dasgupta, Ph.D.

iii
ACKNOWLEDGEMENTS
I would like to first express my gratitude to my advisor Dr. Herbert. H. Hill, for accepting me
to the big family of Hill’s group, and for his continual support and guidance throughout the
past five years. I am very lucky to have a great advisor who gives me encouragement all the
time. I would also want to thank Dr. William F. Siems for his advice and help both in lab and
in life. He brings ideas, laugh, and stories that can always cheer me up. I wish to
acknowledge the contributions of Dr. Peter Reilly and Dr. Brian Clowers, for their valuable
time and expertise in Chemistry. I also want to thank Dr. Nairanjana Dasgupta for her
guidance and help in Statistics.
A special thanks to Dr. Kimberly Kaplan for her exceptional help at the beginning of my
graduate career. And I also want to thank all past and present group members, who made my
graduate school life happy and cheerful. I would like to acknowledge our collaborators Dr.
Richard Knochenmuss, Dr. Stephan Graf, Dr. James Schenk, Dr. George Stoica, Dr. Barbara
Sorg, and Dr. Patrick Tso, for their great support.
None of this work would have been possible without the constant support from my parents
and my dear husband Rui Zhu, whose love and support made the life in Pullman sweet and
joyful. Finally, I would like to thank the Department of Chemistry for offering me the chance
to come to USA and pursue my PhD. It has been a wonderful journey!

iv
APPLICATION OF HADAMARD TRANSFORM ION MOBILITY MASS
SPECTROMETRY TO GLOBAL METABOLOMICS
Abstract
by Xing Zhang, Ph.D. Washington State University
August 2014
Chair: Herbert H. Hill, Jr.
Conventional Ion mobility mass spectrometry (IMMS) provides rapid separation and
detection of complex mixtures. It is merging as a powerful analytical platform for the field of
metabolomics but is limited in throughput. Global metabolomics aims at comprehensive
measurement for all metabolites and presents challenges in analytical technique. The work
describe herein evaluates the capability of hadamard transform ion mobility mass
spectrometry (HT-IMMS) for comprehensive metabolomics analysis with high throughput
and high resolving power. The work also presents a number of applications of global
metabolomics regarding to biological systems including human blood, rat brain tissue and
mice plasma.
HT-IMMS has been developed by superimposing a Hadamard transform sequence on the ion
gate. This development provides a 50% duty cycle while retaining high IMS resolving power.

v
Coupling rapid chromatographic separation prior to HT-IMMS enables the detection of more
metabolite features compared to conventional direct infusion IMMS.
Global metabolomic applications of HT-IMMS were extended in this work by developing
general procedure for sample analysis, metabolite identification, data processing and
statistical analysis. The major findings from this work include: 1) HPLC couple with
HT-IMMS provides comprehensive metabolomics analysis within 2 - 3 minutes; 2) ambient
pressure IMMS has high resolving power and allows isomeric separations, providing accurate
assessment of critical biomarkers without the interference of their isomers; 3) the structural
information generated from IMMS analysis complements the MS detection and helps
metabolite identifications; 4) principle component analysis yields pattern recognition that can
reveal the differences between different metabolic states; 5) biomarkers selection requires the
combination of multivariate analysis and univariate analysis; 6) IMMS data pre-processing,
including normalization and the evaluation of censored data, improves statistical analysis.
HT-IMMS is a natural fit for analyzing complex mixtures.

vi
TABLE OF CONTENTS Page
ACKNOWLEDGEMENTS ..................................................................................................... iii
ABSTRACT .............................................................................................................................. iv
TABLE OF CONTENTS .......................................................................................................... vi
LIST OF TABLES .................................................................................................................... ix
LIST OF FIGURES .................................................................................................................. xi
Chapter 1 .................................................................................................................................. 1
Introduction ................................................................................................................................ 1
1.1 Ion Mobility Spectrometry ............................................................................................ 1
1.2 Types of IMS Devices .................................................................................................. 9
1.3 Ion Mobility Mass Spectrometry: Multidimensional Separation ............................... 13
1.4 Application of IMMS to Complex Mixture Analyses ................................................ 18
1.5 Future Improvements .................................................................................................. 21
1.6 Specific Aims .............................................................................................................. 22
1.7 Attribution ................................................................................................................... 24
Chapter 2 ................................................................................................................................ 30
Metabolic Analysis of Striatum Tissues from Parkinson’s Disease-like Rats by Electrospray
Ionization Ion Mobility Mass Spectrometry ............................................................................ 30
2.1 Introduction ................................................................................................................. 32
2.2 Experimental Design ................................................................................................... 36

vii
2.3 Results and Discussion ............................................................................................... 42
2.4 Conclusions ................................................................................................................. 47
Chapter 3 ................................................................................................................................ 59
Evaluation of Hadamard Transform Atmospheric Pressure Ion Mobility Time-of-Flight Mass
Spectrometry (HT-APIMS-TOFMS) for Complex Mixture Analysis ..................................... 59
3.1 Introduction ................................................................................................................. 60
3.2 Theoretical Background .............................................................................................. 65
3.3 Experimental Section .................................................................................................. 66
3.4 Results and Discussion ............................................................................................... 71
3.5 Conclusions ................................................................................................................. 78
3.6 Supplementary Materials ............................................................................................ 90
Chapter 4 ................................................................................................................................ 91
Strategies for Metabolite Identification in Human Blood Metabolome using Electrospray
Ionization Hadamard Transform Ion Mobility Time-of-Flight Mass Spectrometry ............... 91
4.1 Introduction ................................................................................................................. 93
4.2 Experimental Section .................................................................................................. 96
4.3 Results and Discussion ............................................................................................. 101
4.4 Conclusions ............................................................................................................... 107
Chapter 5 .............................................................................................................................. 124
Neuronal Metabolomics by Ion Mobility Mass Spectrometry in Cocaine Self-administering
Rats after Early and Late Withdrawal .................................................................................... 124

viii
5.1 Introduction ............................................................................................................... 125
5.2 Experimental Design ................................................................................................. 128
5.3 Results and Discussion ............................................................................................. 134
5.4 Conclusions ............................................................................................................... 140
Chapter 6 .............................................................................................................................. 157
Metabolomics of Plasma Fluids from Apolipoprotein AV Knockout Mice by Hadamard
Transform Ambient Pressure Ion Mobility Time-of-Flight Mass Spectrometry ................... 157
6.1 Introduction ............................................................................................................... 159
6.2 Experimental Section ................................................................................................ 163
6.3 Results and Discussion ............................................................................................. 167
6.4 Conclusions ............................................................................................................... 174
Chapter 7 .............................................................................................................................. 188
Conclusions ............................................................................................................................ 188
Appendix ............................................................................................................................... 194

ix
LIST OF TABLES Page
Chapter 2
Table 2.1. Number of reproducible ions detected in each sample for principal component
analysis. .................................................................................................................................... 51
Table 2.2. Major ion peaks (ion counts > 100) detected in PD-like samples, BD-IV and SD
healthy controls. m/z ranges between 200 and 900. ................................................................ 52
Table 2.3. Tentative metabolite identifications based on exact m/z using the Human
Metabolome Database, including molecular formula and adducts. ......................................... 53
Chapter 3
Table 3.1 A table of the m/z, drift time, reduced mobility (K0) value and calibration curves
for each metabolite ion. Obtained from the analysis of metabolite standard mixture solutions.
.................................................................................................................................................. 82
Table 3.2 A summary of the absence/presence of six metabolite ions during the analysis of
standard mixture solutions in both HT-IMMS mode and pulsed-IMMS mode. ...................... 83
Chapter 4
Table 4.1 Metabolite identification of 185 major metabolite features detected by
HT-IMtofMS. Measured m/z, drift time and K0 are included. .............................................. 111
Table 4.2 Isotopic ratio analysis results. ................................................................................ 116
Table 4.3 Isomer/isobar separation and analysis results. ....................................................... 116
Table 4.4 Mobility-mass correlation trend lines. ................................................................... 117

x
Chapter 5
Table 5.1 Sample details and cocaine self-administration results including the total number of
active lever presses and the total number of rewards accumulated. ...................................... 145
Table 5.2 Sample list with number of reproducible metabolite features (counts>50) detected
by HT-IMMS in each sample group. ..................................................................................... 145
Table 5.3 Lists of potential biomarkers generated from loadings plots and univariate analysis.
Information includes measured m/z, p-value of t-test, up/down regulation caused by cocaine
administration, adduct form, and identified metabolite name, is listed in table. ................... 146
Chapter 6
Table 6.1 Summary of experimental details. ......................................................................... 178
Table 6.2 Summary of the identifications of the first 40 major metabolite features detected in
all 11 samples. The exact m/z and K0 (cm2V-1s-1) were measured in IMMS analysis, and the
identifications were matched with existing databases. .......................................................... 179

xi
LIST OF FIGURES Page
Chapter 1
Figure 1.1 Illustration of ions separation in flat electrodes DIMS. ……………………….11
Figure 1.2 Schematic picture of electrospray ion mobility time-of-flight mass spectrometer,
with major components: electrospray, DT IMS, IMMS interface and reflectron time-of-flight
mass spectrometer……………………………………………………………………………14
Figure 1.3 IMMS 2-D plot showing fragmentation pattern of [Dopamine + H]+………….15
Chapter 2
Figure 2.1 Schematic diagram of an electrospray ion mobility time-of-flight mass
spectrometer. ............................................................................................................................ 53
Figure 2.2 Multidimensional IMMS spectra of (a) the electrospray background, (b) SD
healthy control metabolome from striatal tissue, and (c) an expanded view of (b). ................ 54
Figure 2.3 MS and IMS spectra for BD-IV and SD healthy control striatal metabolomes are
shown in (a) and (b); MS and IMS spectra for PD affected 20 dpn and 15 dpn strital
metabolomes are shown in (c) and (d).. ................................................................................... 55
Figure 2.4 Principle component analysis results for all reproducible metabolite ions.. .......... 56
Figure 2.5 Selected m/z 154 mobility spectrums abstracted from the overall IMMS spectrums
for ESI background, BD-IV healthy control striatal metabolomes and BD-IV 20dpn affected
striatal metabolomes. ............................................................................................................... 57
Figure 2.6 Scheme of proposed dopamine and 2-(2,4-dihydroxyphenyl) ethylamine path. .... 58

xii
Chapter 3
Figure 3.1 Schematic diagram of the prototype: Electrospray ionization Hadamard transform
atmospheric pressure ion mobility time-of-flight mass spectrometer.. .................................... 84
Figure 3.2 IMMS 3-dimensional spectra of NIST SRM 1950 sample. ................................... 85
Figure 3.3 Twelve calibration curves of the six metabolite ion species under HT-IMMS mode
and pulsed-IMMS mode.. ........................................................................................................ 86
Figure 3.4 IMS spectra of NIST SRM 1950 sample obtained in pulsed-IMMS mode,
HT-IMMS mode, and from Synapt G2 TWIMMS system. ..................................................... 87
Figure 3.5 Comparison of mobility traces of the MS peak m/z 317.12, in pulsed-IMMS mode
(upper) and HT-IMMS mode (lower).. .................................................................................... 88
Figure 3.6 (a) and (b) are the 3-D IMMS-Intensity plots of striatum tissue extract fluid
analyzed by HPLC coupled HT-IMMS and direct infuse HT-IMMS, respectively ................ 89
Figure 3.7 A portion of a typical HT code sequence (top trace), along with multiplexed data
(middle trace) and decoded spectrum (down trace). ................................................................ 90
Chapter 4
Figure 4.1 (a) Schematic diagram of electrospray ionization hadamard transform atmospheric
pressure ion mobility time-of-flight mass spectrometer; (b) major metabolites detected in
human blood metabolome; (c) major metabolites detected in NIST SRM 1950 ................... 118
Figure 4.2 Global metabolomics results from HT-IMtofMS analysis. .................................. 119
Figure 4.3 An example of accurate isotopic ratio analysis. ................................................... 120
Figure 4.4 An example of isomer/isobar separation.. ............................................................ 121

xiii
Figure 4.5 IMMS two-dimensional spectrum of hemoglobin with charge states from +13 to
+17. ........................................................................................................................................ 122
Figure 4.6 IMMS two-dimensional spectrum illustrating the metabolomes of human blood,
with six trend lines identified for different compound classifications. .................................. 123
Chapter 5
Figure 5.1 Schematic diagram electrospray hadamard transform ion mobility time-of-flight
mass spectrometer (HT-IMMS) coupled with HPLC ............................................................ 150
Figure 5.2 IMMS 2-D spectrum of the metabolomes of striatal tissue is displayed in (a). IMS
spectra illustrating the global metabolomes of PFC samples obtained from saline treatment
and cocaine treatment are shown in (b) ................................................................................. 151
Figure 5.3 PCA score plots of six comparisons ..................................................................... 153
Figure 5.4 Dysregulation of creatine and creatinine .............................................................. 154
Figure 5.5 Intensity profiles of GHS, showing its dysregultaion. ......................................... 155
Figure 5.6 Dysregulation of adenosine in STR/PFC/NAC after 1 day withdrawal and after 3
wks withdrawal. ..................................................................................................................... 156
Chapter 6
Figure 6.1 (a) Schematic diagram of electrospray ionization coupled with hadamard
transform ambient pressure ion mobility time-of-flight mass spectrometry. (b) Illustrative
IMMS 3D spectrum of a plasma sample ................................................................................ 181
Figure 6.2 Representative mass spectra (top) and ion mobility spectra (bottom) for four
different groups of samples, including plasma fluids from apoAV KO mice fasted for 5 hrs,

xiv
WT mice fasted for 5 hrs, apoAV KO mice ad lib fed and WT mice ad lib fed. ................. 182
Figure 6.3 PCA results including score plot (top) and loadings plot (bottom).. .................... 184
Figure 6.4 Specific metabolic alternations for lysophospholipid class of metabolites. ......... 185
Figure 6.5 Intensity profiles for glucose (a) and MG (18:0) (b) for four groups of samples. 186
Figure 6.6 Selected-mass ion mobility spectra for monosaccharide ion (m/z = 203.06). ..... 187

1
Chapter 1
Introduction
1.1 Ion Mobility Spectrometry
Ion mobility spectrometry (IMS)1 is an analytical separation technique that separates
gas-phase ions based on their size-to-charge ratio and ion-neutral interactions as they travel
through a drift tube filled with a drift gas. Separation in IMS occurs rapidly in millisecond
with high resolving power2. As an efficient separation with low detection limits, IMS is
applied in separating and detecting illicit drugs3,4, chemical warfare agents5 explosives
detection6, and biomolecules. The advent of soft ionization sources such as electrospray
(ESI)7 and matrix assisted laser desorption ionization (MALDI)8 have extended IMS
application to not only vapor samples, but also aqueous and solid phase samples. In recent
years, IMS has shown great potential for separating complex mixtures, including
pharmaceutical drugs9, biomolecules analysis10, metabolomics11, and proteomics12.
1.1.1 History
Ion mobility spectrometry was first introduced as an analytical technique known as
Plasma Chromatography (PC)13, which produced plasmagrams for ultratrace analysis of
organic compounds. The original PC tube consisted of four parts: 63Ni foil reactor complex to
generate charged particles; ion-injection grid to inject a pulse of ions; ion-drift tube for ion
separation and electrometer as the detector. The drifting action of the charged particles was
initially analogous to that of a time-of-flight mass spectrometer and the drift time was
analogous to the retention time in chromatography. PC showed potential of being a practical

2
and rapid analytical separation technique with very low detection limits, and those properties
made it attractive to military and security use for detecting explosives and chemical warfare
agents. To date, IMS is still a popular choice for security and military applications14. The
performance of IMS device has been improved in both industrial and academic laboratories
on IMS over the past few decades.
1.1.2 Theoretical Background
In the classical case, IMS separates ions based on their mobility through a drift tube in
which an electrostatic field propels the ions through the tube filled with a buffer gas. After
entering the drift tube through an ion gate/pulser, ions are accelerated in the electric field and
decelerated by the collisions with the buffer gas until they reach a constant ion velocity (vd),
which is proportional to the electric field (E). The mobility (K) of an ion is then characterized
as the ratio of its ion velocity to electric field, as shown in Equation 1.
! = !!! = !!
!!! !!!!!!!!!"#$%&'(!1
Where E is the electric field (volts/cm), V (volts) is the voltage drop across the drift tube with
length L (cm), td is the time an ion takes to migrate through the drift tube.
Fundamental information about ionic size under specific conditions can be derived
from mobility measurements. Revercomb and Mason et al. have given the relationship
between ion mobility and collision cross section (Ω) during the collision processes15, as
shown in Equation 2.
! = 3!16!
2!!!!
!/! ! +!!"
!/! 1Ω
!!!!!!!!!!!!"#$%&'(!2

3
Where q is the charge on an electron, N is the number density of the drift gas, kb is
Boltzmann’s constant, T is the temperature, M is the mass of the drift gas and m is the mass
of the ion.
Since IMS devices are operated at a variety of temperatures and that ambient pressure
also changes among geographical regions, and ion’s mobility is different for different
operating parameters. However, the reduced mobility constant (K0) for an ion species remains
constant for a given drift gas after standardized to standard temperature and pressure, as
shown in Equation 3. Thus K0 allows mobility comparisons among laboratories1,16.
!! =!!!!!×
273.15! × !
760 !!!!!!!!!!!!!"#$%&'(!3
1.1.3 IMS Figure of Merits
Resolving power and the number of theoretical plates:
As one of the most important factors for analytical separation techniques, resolving
power (Rp) is the parameter for quantifying the separation. It is commonly defined in terms of
single-peak-based equation, as
!! =!!!! !!!!!!!!!!!!!!!"#$%&'(!4
Where td is the drift time of the ion of interest and wh is the peak width measured at
half-height. As shown in this equation, the resolving power can be increased with narrower
peak width.
The study on peak width in IMS started in 1970’s, researchers first developed an
expression for peak width in IMS using several assumptions15. The first assumption was that

4
the peak shape in IMS is primarily dependent on two factors, which are initial gate pulse
width of the ion gate and the broadening due to diffusion. A pack of ions, after entering the
drift region, will experience diffusional broadening as it travels in the IMS tube. The
second assumption is that the initial ion pack is Gaussian in shape. Thus the final peak width
at half height can be expressed as Equation 5.
!! = !!! + !!"##!!!!!!!!!!!!!!!!!"#$%&'(!5
Where wh is the final peak width at half-height, tg is the initial ion pulse width, tdiff is the
width at half-height of an ion peak which is produced by an infinitely narrow gate pulse.
The diffusion term in Equation 6 was further extended based on the definition of
Brownian diffusion coefficient and Nernst-Einstein Equation, giving the following
expression in for the final peak width at half-height.
!! = !!! +16!!!"#2!"# !!!!!!!!!!!!!"#$%&'(!6
Where kb is the Boltzmann’s constant, T is the temperature, V is the voltage drop on the drift
tube, e is te charge on an electron, z is the number of charges on the ion, and td is the drift
time of the ion. Therefore, the resolving power17 can be written as the following equation:
!! =!!
!!! + 16!"#$2!"# !!!
!!!!!!!!!!!"#$%&'(!7
As shown in Equation 7, in order to increase the resolving power, one must increase
the voltage applied across the IMS, decrease the temperature, or decrease the initial gate
pulse width.

5
In chromatography, another measurement of separation capability is the theoretical
number of plates (N):
! = 16 !!!
!!!!!!!!!!!!!!!"#$%&'(!8
Where tr as retention time, w as the peak width at the base. N can be calculated in IMS with
an easy substitution of retention time with drift time. For a drift time ion mobility
spectrometer device with gate pulse width at 0.2 milliseconds and a drift time of 20
milliseconds, and operation at typical instrumental parameters (temperature at 473 K and
8000 V voltage drop across the tube), the predicting resolving power and number of plate
would be approximately 23,800.
Resolution, separation factor (α) and selectivity:
Resolution in chromatography is another representation of the efficiency of separation,
and in IMS, it is expressed by the commonly used two-peak definition. As shown in Equation
9:
! = !!! − !!!!! + !! 2
!!!!!!!!!!!!!!"#$%&'(!9
Where td1 is the drift time of the ion that drifts faster and td2 is the drift time of the ion that
drifts slower, w is the peak width at the base.
The separation factor (α) in IMS is defined as18 the Equation 10:
! = !!!!!! =
!!"!!" !!!!!!!!!!!!!!"#$%&'(!10
Similar as in chromatography, one would prefer higher α value and any α value of 1 indicates
that the two compounds cannot be separated. Different from chromatography, capacity factor

6
(k’) is very large in IMS. In the definition of capacity factor (k’=(td-t0)/t0), t0 is the time it
takes for an ion to drift down to the drift tube without interaction with the drift gas and it is
negligible when compared to an ion’s drift time in atmospheric pressure condition. The large
capacity factor indicates that in IMS the theoretical number of plates is very close to the
effective number of theoretical plates.
Unlike in chromatography, where the separation factor can be altered by optimizing
mobile/stationary phase, there are only limited factors can be changed in IMS. Several
methods of altering α were previously reported18-20. One method described by M. Tabrizchi et
al. differentiated the declustering/dehydration rates of the analytes by changing temperature;
another idea for altering α was by changing the polarizability and mass of the drift gas on
drift time ion mobility spectrometer, or alternating the composite of drift gas by adding
modifiers21. Studies showed the separation effects of several different drift gases on
structurally similar classes of compounds22,23, and the results showed that alteration of drift
gas changed the drift times of ions, however, the percentages of change were different from
one ion to another, hence the separation factor was altered. It was demonstrated that
separation factor is largely dependent on the polarizability of the drift gas, and optimal
separation can be achieved by optimizing the drift gas.
Analysis speed and sensitivity:
Another attribute of IMS is the rapid analysis time. Each ion mobility separation can
be achieved within 20 – 100 milliseconds. Thus, even through it is common to average
several of these separations for a single analysis; IMS has shortened the analysis time to a
few minutes. High sensitivity in detection also makes it suitable for quantifying trace level
concentrations. The limit of detection (LOD) of IMS is usually below microgram (µg) or

7
even nanogram (ng).
1.1.4 IMS Fundamental Separation Mechanism
Different from chromatography, IMS provides a piece of “hard” information called
collision cross section (Ω)24. Ω is directly related to the ionic structure; therefore, it is where
the ion mobility separation originates. At a given charge state, smaller ions encounter fewer
ion-neutral collisions compared with bigger ions. Although obtaining Ω information is
restricted to drift tube IMS, it is applicable to other types of IMS devices with calibration.
As shown in Equation 11, the average collision cross section can be predicted from
the drift time data after a simple rearrangement of Equation 2. And the obtained Ω can then
be correlated with the gas-phase ion conformation and be a fairly accurate estimate of the
ion’s size25.
Ω = 3!"16!
2!!"#
!/! !"!!!
!273.15
760! !!!!!!!!!!!"#$%&'(!11
Where these parameters include the charge of the ion (z), the drift gas number density (N),
Boltzmann’s constant (k), temperature (T), pressure of the drift gas (P), the voltage drop
across the drift tube (V), drift time (td), length of the drift tube (L), and reduced mass of the
ion-neutral collision pair µ, (µ = mM/(m+M). m and M are the ion and neutral masses,
respectively). In cases where temperature, pressure and gas phase conditions (number density
and gas purity) cannot be measured accurately, calibration procedure using ions with known
Ω is preferred26.
Collision cross section is particularly important with regard to the separation of

8
isomeric compounds27. Isomeric compounds cannot be separated by mass spectrometry,
although they have minor difference in their collision cross sections due to structural
differences; their mass is the same. With the high resolving power of IMS, separation of
isomers can be achieved. The ability to determine collision cross sections and to separate
isomers has assigned IMS enormous potential for the analysis of complex mixtures. When
coupled with MS, conformation information can be obtained. Use of collision cross sections
has been widely applied in proteomics, metabolomics and lipidomics28,29.
The above figure of merits has enabled IMS for complex sample analysis. Efficient
separations can be achieved because of the high resolving power of IMS; the milliseconds
separation in IMS makes it the top choice for degradable complex biological samples and
organic reaction monitoring; the collision cross section information provided by IMS can be
used for class identification of unknown components in complex samples.

9
1.2 Types of IMS Devices
The IMS technique has been improved for decades to provide customized applications.
IMS devices can now be operated under a wide temperature/pressure range using different
electric field conditions. Each type of IMS device has unique advances and properties,
enabling many analytical applications.
1.2.1 Ambient Pressure Drift-Time IMS (DTIMS)
DTIMS employs a simple and conventional design consisting of a stacked series of
metal ring electrodes isolated by ceramic ring insulations but electrically connected using a
resistor chain. This design creates a smooth electric field, driving ions to transmit through the
drift tube. Ion gate, buffer gas and detector are included to complete an IMS device. More
details about the fundamental theory were discussed in the previous section. Other materials
including resistive glass and polymer materials have been assessed for replacing the
conventional metal and ceramic ring components.
Most DTIMS devices operate under ambient pressure to 1) avoid the necessity of
vacuum pumping; 2) provide enough ion-neutral collisions using a miniature size drift tube.
Ambient pressure DTIMS can often achieves high resolving power (>100) separation in
millisecond time scale. Early DTIMS devices typically have a low duty cycle in which only
about 1% of the ion current is used for detection, however, this situation has improved in
recent years by implementing multiplexing methods30,31, in which the duty cycle can be
increased up to about 50%.
1.2.2 Low Pressure Drift-Time IMS
Low pressure DTIMS has been widely used in the analysis of biomolecules. It enables

10
easy coupling with mass spectrometer without complicated pressure interface, therefore, gain
better sensitivity when compared with ambient pressure DTIMS. With the recent
development of ion funnel trap (IFT) technique32,33, low pressure DTIMS is reported with
LODs of picomoles. However, limited Rp of low-pressure IMS systems is inevitable unless
extremely long drift tubes are employed.
1.2.3 Differential Mobility spectrometry (DMS)
Differential ion mobility spectrometry is also called high field asymmetric waveform
ion mobility spectrometry (FAIMS)34. The basic difference between DMS and DTIMS is that
instead of using electrostatic fields, DMS employs a periodic asymmetric electric field in a
gap between two electrodes and in a direction perpendicular to the buffer gas flow. Ions in
DMS will experience alternately strong and weak orthogonal electric fields (or E/N). If the
mobility of an ion swarm is greater in one direction than in the other, the ion will have an
unstable path through the spectrometer and neutralize on the side of the spectrometer. As the
drift gas pushes the ions through the spectrometer, a compensation voltage can be used to
offset the ion swarm’s trajectory toward the side of the gap and enables the ions to migrate
through the spectrometer and be collected and detected on the terminal electrode. The
mobility difference (ΔK) of a given ion species under high and electric fields is expressed by
the compensation field, because only ions with a certain ΔK can be successfully transported
under the present condition. The separation procedure of DMS is shown in Figure 1.1. By
scanning a range of compensation voltages, ions with different ΔK can be transported
through the spectrometer separately.

11
Figure 1.1: Illustration of ions separation in flat electrodes DIMS.
Unlike DTIMS, DMS has an advantage of low-cost construction since most of the
devices work under ambient pressure and are designed in a smaller scale (a few square
centimeters). In addition, DMS is an ion-filtering technique, which provides good
transmission for specific ions. However, it can’t transmit ions simultaneously, limiting its
application to relatively simple mixtures. Recent studies have shown improved resolving
power by increasing the electric field and modification of the drift gas, aiding the separation
of conformers35,36.
1.2.4 Traveling Wave IMS (TWIMS)
TWIMS37 is another recently developed method for mobility measurement.
Structurally similar to DTIMS, TWIMS also consists of stacked ring electrodes. However,
TWIMS operates under low pressure and employs a radio-frequency guided electric field by
applying a DC voltage to one electrode after another, forming a continuous wave along the
TWIMS cell, therefore, transmits ions through the drift cell with different drift times. An ion
will be on top of the wave during the transmission if its mobility matches with the wave,
while faster ions move ahead of the wave and slower ions lag behind.
TWIMS has many advantages, among which high sensitivity is primary. Therefore, it
has been applied on various complex sample analyses. However, the Rp of TWIMS is much

12
lower compared with DTIMS and it does not allow for measurement of collision cross
section values without proper calibration26.

13
1.3 Ion Mobility Mass Spectrometry: Multidimensional Separation
Being simple and inexpensive, the faraday plate is employed by most commercial
IMS stand-alone devices. However, it does not provide further information except for
reduced mobility values. When the interest in IMS blossomed in 1970s, the first commercial
ion mobility mass spectrometer (IMMS) was developed by the Franklin GNO Corporation by
coupling a quadrupole mass spectrometer with ambient pressure DTIMS. In the 1980s, work
continued in the field of IMMS mostly using commercially available instruments for drug
detection. From 1990s, IMMS design has been modified and improved by a number of
research laboratories38 and IMMS instruments were constructed in house by interfacing it
with different types of mass spectrometers such as time-of-flight mass spectrometer,
quadrupole mass spectrometer and ion trap mass spectrometer. Currently, there are a variety
of commercially available IMMS systems, including ion mobility orthogonal time-of-flight
mass spectrometer from Ionwerks (TX, USA)39, resistive glass ion mobility time-of-light
mass spectrometer from Tofwerks (AG, Switzerland)40, Synapt G2 travelling wave ion
mobility mass spectrometer from Waters (Manchester, UK)41 and the ion mobility Q-TOF
mass spectrometer from Agilent (CA, USA)42. Synapt G2 from Waters and ion mobility
Q-TOF from Agilent are low-pressure ion mobility mass spectrometry (LP-IMMS) systems,
they advance in throughput but limited in resolving power; IMMS systems from Ionwerks
and Tofwerks are ambient pressure IMMS (AP-IMMS) systems, which have high resolving
power but low throughput (<1%). Commercially available systems are either limited in
sensitivity or resolving power43.
The schematic diagram of an ambient pressure ion mobility time-of-flight mass
spectrometer (AP-IMMS from Tofwerk) is shown in Figure 2. Major components include: an
electrospray ionization source; an stacked ring drift time ion mobility spectrometer made up

14
by a desolvation region, a BN gate and a drift region; an IMMS interface; and a reflectron
time-of-flight mass spectrometer. This IMMS system was employed in most of the studies in
this dissertation.
Figure 1.2: schematic picture of electrospray ion mobility time-of-flight mass spectrometer,
with major components: electrospray, DT IMS, IMMS interface and reflectron time-of-flight
mass spectrometer.
Coupling ion mobility with mass spectrometry offers a number of advantages,
including 1) High speed IMS separation prior to MS detection to keep mass spectrometer
clean; 2) Increasing signal to noise ratio; 3) Increasing confidence of fragmentation (MS2 or
MSn) analysis; 4) High resolution separation enables isomeric separation; 5) Providing ion
density profiles with collision cross section values; and 6) Providing charge state information.
!

15
1.3.1 Increasing signal to noise ratio (S/N)
The first advantage of interfacing ion mobility spectrometer to mass spectrometer is
that IMS separation before MS detection can prevent neural molecules and contaminates
from entering mass spectrometer to keep the mass spectrometer clean. Therefore, the
two-dimensional IMMS separation spreads the noise, therefore, increases signal to noise ratio
(S/N).
1.3.2 Increasing Confidence of isotopic analysis
Fragmentation patterns can be detected in an IMMS analysis44. Due to the fact that
fragmentation often happens in the IMMS interface region, precursor ions share the same
drift time with the product ions. Figure 3 shows an example of the fragmentation pattern for
[Dopamine + H]+ ions analyzed by ambient pressure drift-time ion mobility time-of-flight
mass spectrometer with quadrupole interface. As stated above, fragmentation happened in the
quadrupole interface after the ion mobility measurement. With precursor ions m/z = 154.08
almost completely fragmented, major product ions with m/z 91.05, 119.05 and 137.06 were
observed.
Figure 1.3: IMMS 2-D plot showing fragmentation pattern of [Dopamine + H]+, with mass
spectrum on x-axis (m/z ranges from 50 to 300) and ion mobility spectrum on y-axis (drift

16
time ranges from 10 ms to 45 ms).
1.3.3 Increasing Confidence of Isotopic Ratio Analysis
Isotope patterns can be recognized by mass spectrometry for specific compound45,
however, for complex mixture analysis, mass peaks can be detected at all mass units and
sometimes multiple mass peaks overlap within one mass unitf. With prior ion mobility
separation, isotope peaks of a specific ion species will be detected at a same drift time,
therefore, simplify the isotopic analysis.
1.3.4 Isomers and Isobars Separation
Ions share the same nominal mass/exact mass are common in complex mixtures
especially in biological samples. These ions have vastly different functions and properties,
and they are a big challenge for mass spectrometry. IMS has emerged as a rapid and effective
approach for isomer separations with its unique capability of ion size separation. IMS has
been widely used in isomeric separations since 1990s. 46,47 A number of isomeric species that
has been successfully separated using IMMS methods include carbohydrates, peptides, lipids
and drug metabolites.
1.3.5 Chiral Separation
Chiral analysis using IMMS was firstly reported by Wu et al. with separation of
peptide diastereomers47. Dwivedi et al. reported that selective interactions between
enantiomer ions and chiral modifier neural molecules altered the collision cross sections of
the enantiomer ions, therefore, provided separation in drift time48. Chiral analysis using
FAIMS-MS has also been reported with amino acid enantiomers separated as metal-bound
complexes49. Campuzano et al. reported another example of epimer separation achieved for

17
betamethasone and dexamethasone, where the diastereomers differed by only one chiral
carbon50. Holness et al. investigated gas-phase separation of chiral molecules found in
amphetamine-type substances by introducing modifier through IMS, demonstrating the
capability of IMS in chiral separation51.
1.3.6 Ion Density Profiles
Identification based on m/z information alone can lead to significant complications
especially for complex mixtures such as biological samples (tissue extracts, fluids). Multiple
matches for each m/z create challenge for unknown compounds identification. In an IMMS
analysis, signals for each class of compounds are highly correlated by forming a
mobility-mass correlation curve (MMCC), and different classes of homologous compounds
occupies the conformation space in IMMS52,53. MMCCs can predict the increase of collision
cross section as a function of increasing mass as shown in Equation 12. In complex mixture
analysis, MMCCs are assigned with the potential of class identifying of unknown
compounds.
Ω! = ! !
! + !!!!!!!!!!!!!!!"#$%&'(!12
Where a is the slope of the MMCC and b is the y-intercept. Mobility-mass correlations of
many classes of homologous compounds have been studied including carbohydrates, lipid,
peptides, fatty acid, etc. More classifications using ion density profiles and MMCCs are
necessary for building a database to aid rapid unknown compounds identification by IMMS54.

18
1.4 Application of IMMS to Complex Mixture Analyses
IMMS have been demonstrated as an efficient analytical tool for the separation and
analysis of complex mixtures from areas such as life science research, pharmaceutical
analysis, forensics and drugs detection. IMMS provides rapid analysis, detecting and
characterizing ion species simultaneously, with minimal sample preparation and purification.
Structural information interpreted by collision cross sections in gas phase is also possible.
1.4.1 Proteomic and Genomic
Characterizations of complex biological systems including genomics, proteomics,
glycomics, and metabolomics, are undergoing for decades. Performing by themselves or the
interactions between them has revealed the assemblies to help understand systems biology.
Consequently, obtaining structural information for these macro and small biomolecules using
analytical tools becomes important. For proteins, many protein complexes can be classified
into structural classes that can be described with purely geometric measurements. Therefore,
the ability of IMS to distinguish the structural differences is helpful. Ruotolo et al. has proved
and optimized the utility of TWIMMS for the separation and characterization of large protein
assemblies55. Other proteomics applications related to top-down proteomics and DMS
method were also reported from Russell et al.38 and Smith et al.56.
1.4.2 Metabolomics
Metabolomics is a fast developing area where large amount of information can be
obtained to fulfill the goal of better understanding system biology. The study of
metabolomics involves the identities, quantization and fluxes of hundreds of thousands of
low molecular weight metabolites, which show wide variations in chemical and physical
properties. Besides the diversity of the metabolome, specific properties of biological samples

19
such as degradability and high sensitivity to environmental changes also present challenges
for analytical measurement. Analytical techniques that required complicated sample
preparation and long analysis time would be biased on monitoring the temporal nature of the
metabolome. IMMS has shown increased popularity in metabolomics because that it allows
efficient and sensitive analysis of all metabolites with minimal sample preparation. It has
been applied on a number of metabolomics studies including metabolic profiling of human
blood57, the metabolic study of human breath58 and cancer tissue samples59.
Due to the facts that massive amount of data generated from metabolomics by IMMS
and that a large portion of the data is unknown, the lack of IMMS database60 and standard
statistical analysis protocol67,68 limits the future applications. The current strategies for
unknown feature identification66 involves matching with public databases, which is tentative
and effort-intensive. In addition, it is difficult to perform pattern recognition and biomarker
selection without the appropriate statistical approach.
1.4.3 Drug Detection/Pharmaceutical Application
The applications of IMS methods including DTIMS and DMS to pharmaceutical
industry have been burgeoning. Ion mobility related techniques have been used mostly in
quality control (QC), quality assurance (QA), process monitoring and cleaning verification,
as quick and efficient alternatives for chromatographic methods61. In cleaning verification
using IMS devices, volatile samples are introduced directly and less volatile samples are
introduced by swiping surface and thermal desorption, the following IMS analysis require
only less than 1 minute per sample. IMS techniques substantially shortened the on line
analyses time, not only for cleaning verification, but also for drug detection. Direct
pharmaceutical compounds detection/identification has been reported with high accuracy

20
good reproducibility and low limit of detection. Recent application of IMMS techniques to
monitoring drug synthesis reaction9 has further demonstrated IMMS to be an appropriate
analytical technique for pharmaceutical industry.
1.4.4 Other Applications to Complex Mixtures
The advantages of IMMS methods in sensitivity and selectivity have made it highly
suitable for various types of complex mixture analyses, including food quality and safety
control, real-time environmental analysis and petroleomics study61,62. The complex nature of
mixture analysis has challenged the detection of one or a class of compounds existing in the
complex matrix. IMMS has the analytical potential to overcome this problem and it has
strongly emerged in other analytical field beyond explosives and chemical warfare agent.
Inspection of scientific literatures proves the successful applications of IMMS including meat
analysis by determining biogenic metabolites produced during spoilage63, clinical analysis by
detecting thiocyanate to distinguish between smokers and non-smokers64, and petroleum
analysis by fingerprinting complex oil mixture for characterization65.

21
1.5 Future Improvements Needed for IMMS
The simplicity, high efficiency and high resolving power of IMS have turned it into a
widely used analytical tool. Coupling with mass spectrometers offers more advantages and
value-added structural information not possible from mass spectrometer alone. Expanding
applications are supported by a growing number of commercially available IMMS systems
from instrumental companies. However, limitations exist especially for the application of
IMMS to the analysis of complex mixtures: 1) As described earlier, the ambient pressure
IMMS systems advance in high Rp but have low throughput, therefore, an IMMS system
with both high Rp and high throughput is needed; 2) With regard to metabolomics,
establishing IMMS database and proper statistical approach for data processing require
significant effort.

22
1.6 Specific Aims
The work described herein aims to improve the current status of applying IMMS to
metabolomics through a variety of technique improvements and to demonstrate the mobility
advantages for MS through specific applications cases. In technique improvement, this work
focuses on increase the duty cycle of DTIMMS device and increase the ionization efficiency.
With regard to metabolomics applications, the add-on values of IMMS can help build a
metabolite database, and the statistical analysis should be further developed to a standard
protocol. The goals will be addressed through the following specific aims:
1. Instrumentation Development. By coupling multiplexing technique Hadamard
transfrom to DTIMMS gating sequence, IMMS analysis can achieve a much higher
duty cycle. Combining IMMS analysis with prior chromatographic separation
provides a solution by adding additional separation to minimize ionization
suppression.
2. IMMS Aiding Metabolite Identification. The add-on values of IMMS including
collision cross section information, isomeric separation and isotopic separation can
help the identification of unknown metabolites. Along with the existing metabolome
databases, we can generate an IMMS database for metaboloites.
3. More Metabolomic Applications. IMMS is emerging in recent years as a technique
for metabolomics. Therefore, more applications are needed to solidify the feasibility
and advances of IMMS in analyzing complex mixtures such as metabolomes from
fluids and tissues. In addition, applying IMMS to studies that lack mechanism
explanation can provide metabolic insight for physiological studies.
4. Improving Statistical Approach. Currently, IMMS data generated from
metabolomics studies are processed with the lack of standard protocal. A processing

23
approach including pre-processing and statistical analysis can provide more
reproducible and reliable metabolic information from the IMMS analysis.

24
1.7 Attribution
The work described in Chapter 2 was conducted by Zhang and samples were provided by
Stoica, the manuscript was prepared in the style required for Analytical Chemistry (Zhang, X.;
Chiu, V. M.; Stocia, G.; Lungu, G.; Schenk, J. O.; Hill, H. H., Jr. Anal. Chem. 2014, 86,
3075-3083). The software in Chapter 3 was provided by Tofwerk (AG, Thun, Switzerland)
and experiments were conducted by Zhang and Liu. The manuscript was prepared according
to the requirement from Analytical Chemistry (Zhang, X.; Knochenmuss, R.; Siems, W. F.;
Liu, W.; Graf, S.; Hill, H. H., Jr. Anal. Chem. 2014, 86 (3), 1661-1670). The experiments in
Chapter 4 were performed by Zhang and Li, and SRM 1950 sample was provided by
National Institute of Standards and Technology (NIST, MD, USA). The manuscript was
written in the format required by Analytical Chemistry. Samples in Chapter 5 were provided
by Sorg and Todd from Department of Neurosciences, Washington State University.
Experiments were conducted by Zhang and Chiu. Manuscript was prepared in the style
required by Analytical Chemistry. The experiments in Chapter 5 were conducted by Zhang,
and the samples were provided by Tso and Xu from University of Cincinnati. The manuscript
was written according to the requirement from Analytical Chemistry. The statistical analysis
in Appendix A was directed by Dasgupta and performed by Zhang, using Minitab (Minitab
Inc., PA, USA) and R (R core team, USA). All manuscripts were prepared by Zhang.
William F. Siems provided advice through all experiments. Hebert H. Hill, Jr. provided
direction throughout all aspects of this work.

25
Reference
(1) Hill, H. H., Jr.; Siems, W. F.; Louis, R. H. S.; McMinn, D. G. Anal. Chem. 1990, 62,
1201A–1209A.
(2) Davis, E. J.; Dwivedi, P.; Tam, M.; Siems, W. F.; H, H. H. Anal. Chem. 2009, 81,
3270–3275.
(3) Wu, C.; Siems, W. F.; H, H. H. Anal. Chem. 2000, 72, 396–403.
(4) Lawrence, A. H. Anal. Chem. 1989, 61, 343–349.
(5) Steiner, W. E.; Klopsch, S. J.; English, W. A.; Clowers, B. H.; H, H. H. Anal. Chem.
2005, 77, 4792–4799.
(6) Ewing, R. G.; Atkinson, D. A.; Eiceman, G. A.; Ewing, G. J. Talanta 2001.
(7) Shumate, C. B.; H, H. H. Anal. Chem. 1989, 61, 601–606.
(8) Gillig, K. J.; Ruotolo, B.; Stone, E. G.; Russell, D. H.; Fuhrer, K.; Gonin, M.;
Schultz, A. J. Anal. Chem. 2000, 72, 3965–3971.
(9) Roscioli, K. M.; Zhang, X.; Li, S. X.; Goetz, G. H. International Journal of Mass
Spectrometry 2012.
(10) Bohrer, B. C.; Merenbloom, S. I.; Koeniger, S. L.; Hilderbrand, A. E.; Clemmer, D.
E. Annual Review of Analytical Chemistry 2008, 1, 293–327.
(11) Dwivedi, P.; Wu, P.; Klopsch, S. J.; Puzon, G. J.; Xun, L.; Hill, H. H., Jr.
Metabolomics 2007, 4, 63–80.
(12) Ruotolo, B. T.; Benesch, J. L. P.; Sandercock, A. M.; Hyung, S.-J.; Robinson, C. V.
Nat Protoc 2008, 3, 1139–1152.
(13) Karasek, F. W. Anal. Chem. 1974, 46, 710A–720a.
(14) Eiceman, G. A.; Karpas Z; Hill. H. H Jr. 2013 Ion Mobility Spectrometry.
(15) Revercomb, H. E.; Mason, E. A. Anal. Chem. 1975, 47, 970–983.
(16) Siems, W. F.; Viehland, L. A.; Herbert H Hill, J. Anal. Chem. 2012, 84, 9782–9791.

26
(17) Siems, W. F.; Wu, C.; Tarver, E. E.; Hill, H. H. J.; Larsen, P. R.; McMinn, D. G.
Anal. Chem. 1994, 66, 4195–4201.
(18) Asbury, G. R.; H, H. H. Anal. Chem. 2000, 72, 580–584.
(19) Tabrizchi, M.; Rouholahnejad, F. Talanta 2006, 69, 87–90.
(20) Tabrizchi, M. Talanta 2004, 62, 65–70.
(21) Eiceman, G. A.; Yuan-Feng, W.; Garcia-Gonzalez, L.; Harden, C. S.; Shoff, D. B.
Analytica Chimica Acta 1995, 306, 21–33.
(22) Ruotolo, B. T.; McLean, J. A.; Gillig, K. J.; Russell, D. H. J. Mass Spectrom. 2004,
39, 361–367.
(23) Matz, L. M.; Hill, H. H.; Beegle, L. W.; Kanik, I. J Am Soc Mass Spectrom 2002, 13,
300–307.
(24) Clemmer, D. E.; Jarrold, M. F. J. Mass Spectrom. 1997, 32, 577–592.
(25) Wu, C.; Siems, W. F.; Asbury, G. R.; H, H. H. Anal. Chem. 1998, 70, 4929–4938.
(26) Bush, M. F.; Hall, Z.; Giles, K.; Hoyes, J.; Robinson, C. V.; Ruotolo, B. T. Anal.
Chem. 2010, 82, 9557–9565.
(27) Li, H.; Giles, K.; Bendiak, B.; Kaplan, K.; Siems, W. F.; Herbert H Hill, J. Anal.
Chem. 2012, 84, 3231–3239.
(28) Pagel, K.; Harvey, D. J. Anal. Chem. 2013, 85, 5138–5145.
(29) Wyttenbach, T.; Bleiholder, C.; Bowers, M. T. Anal. Chem. 2013, 85, 2191–2199.
(30) Knorr, F. J.; Eatherton, R. L.; Siems, W. F.; Hill, H. H. Anal. Chem. 1985, 57, 402–
406.
(31) Clowers, B. H.; Siems, W. F.; H, H. H.; Massick, S. M. Anal. Chem. 2006, 78, 44–
51.
(32) Myung, S.; Lee, Y. J.; Moon, M. H.; Taraszka, M. A.; Sporleder, C. R.; Clemmer, D.
E. Anal. Chem. 2003, 75, 5137–5145.

27
(33) Clowers, B. H.; Ibrahim, Y. M.; Prior, D. C.; Danielson, W. F.; Belov, M. E.; Smith,
R. D. Anal. Chem. 2008, 80, 612–623.
(34) Kolakowski, B. M.; Mester, Z. Analyst 2007, 132, 842–864.
(35) Shvartsburg, A. A.; Smith, R. D. Anal. Chem. 2013, 85, 6967–6973.
(36) Tsai, C.-W.; Yost, R. A.; Garrett, T. J. Bioanalysis 2012, 4, 1363–1375.
(37) Shvartsburg, A. A.; Smith, R. D. Anal. Chem. 2008, 80, 9689–9699.
(38) McLean, J. A.; Ruotolo, B. T.; Gillig, K. J.; Russell, D. H. International Journal of
Mass Spectrometry 2005.
(39) Collins, D.; Lee, M. Anal Bioanal Chem 2001, 372, 66–73.
(40) Kaplan, K.; Graf, S.; Tanner, C.; Gonin, M.; Fuhrer, K.; Knochenmuss, R.; Dwivedi,
P.; Hill, H. H., Jr. Anal. Chem. 2010, 82, 9336–9343.
(41) Giles, K.; Williams, J. P.; Campuzano, I. Rapid Commun. Mass Spectrom. 2011, 25,
1559–1566.
(42) May, J. C.; Goodwin, C. R.; Lareau, N. M.; Leaptrot, K. L.; Morris, C. B.;
Kurulugama, R. T.; Mordehai, A.; Klein, C.; Barry, W.; Darland, E.; Overney, G.;
Imatani, K.; Stafford, G. C.; Fjeldsted, J. C.; McLean, J. A. Anal. Chem. 2014, 86,
2107–2116.
(43) Collins, D.; Lee, M. Anal Bioanal Chem 2001, 372, 66–73.
(44) Castro-Perez, J.; Roddy, T. P.; Nibbering, N. M. M.; Shah, V.; McLaren, D. G.;
Previs, S.; Attygalle, A. B.; Herath, K.; Chen, Z.; Wang, S.-P.; Mitnaul, L.; Hubbard,
B. K.; Vreeken, R. J.; Johns, D. G.; Hankemeier, T. J Am Soc Mass Spectrom 2011,
22, 1552–1567.
(45) Hofstetter, T. B.; Berg, M. TrAC Trends in Analytical Chemistry 2011, 30, 618–627.
(46) Cox, K. A.; Julian, R. K.; Cooks, R. G.; Kaiser, R. E. J Am Soc Mass Spectrom 1994,
5, 127–136.

28
(47) Wu, C.; Siems, W. F.; Klasmeier, J.; H, H. H. Anal. Chem. 2000, 72, 391–395.
(48) Dwivedi, P.; Wu, C.; Matz, L. M.; Clowers, B. H.; Siems, W. F.; H, H. H. Anal.
Chem. 2006, 78, 8200–8206.
(49) Axel Mie; Magnus Jörntén-Karlsson; Bengt-Olof Axelsson; Andrew Ray, A.;
Reimann, C. T. American Chemical Society, 2007; Vol. 79, pp. 2850–2858.
(50) Campuzano, I.; Bush, M. F.; Robinson, C. V.; Beaumont, C.; Richardson, K.; Kim,
H.; Kim, H. I. Anal. Chem. 2011, 84, 1026–1033.
(51) Holness, H. K.; Jamal, A.; Mebel, A.; Almirall, J. R. Anal Bioanal Chem 2012, 404,
2407–2416.
(52) Woods, A. S.; Ugarov, M.; Egan, T.; Koomen, J.; Gillig, K. J.; Fuhrer, K.; Gonin,
M.; Schultz, J. A. Anal. Chem. 2004, 76, 2187–2195.
(53) Kaplan, K.; Dwivedi, P.; Davidson, S.; Yang, Q.; Tso, P.; Siems, W.; Hill, H. H., Jr.
Anal. Chem. 2009, 81, 7944–7953.
(54) Tao, L.; McLean, J. R.; McLean, J. A.; Russell, D. H. J Am Soc Mass Spectrom
2007, 18, 1232–1238.
(55) Ruotolo, B. T.; Gillig, K. J.; Stone, E. G.; Russell, D. H. … of Mass Spectrometry
2002.
(56) Baker, E. S.; Burnum-Johnson, K. E.; Jacobs, J. M. Molecular & Cellular … 2014.
(57) Dwivedi, P.; Schultz, A. J.; Jr, H. H. H. International Journal of Mass Spectrometry
2010, 298, 78–90.
(58) Ruzsanyi, V.; Baumbach, J. I.; Sielemann, S.; Litterst, P.; Westhoff, M.; Freitag, L.
Journal of Chromatography A 2005, 1084, 145–151.
(59) Williams, M. D.; Reeves, R.; Resar, L. S. Analytical and bioanalytical 2013, 405,
5013-5030.
(60) Oldiges, M.; Lütz, S.; Pflug, S.; Schroer, K.; Stein, N.; Wiendahl, C. Appl Microbiol

29
Biotechnol 2007, 76, 495–511.
(61) Armenta, S.; Alcala, M.; Blanco, M. Analytica Chimica Acta 2011, 703, 114–123.
(62) Fasciotti, M.; Lalli, P. M.; Klitzke, C. F.; Corilo, Y. E.; Pudenzi, M. A.; Pereira, R.
C. L.; Bastos, W.; Daroda, R. J.; Eberlin, M. N. Energy Fuels 2013, 27, 7277–7286.
(63) Bota, G. M.; Harrington, P. B. Talanta 2006, 68, 629–635.
(64) Jafari, M. T.; Javaheri, M. Anal. Chem. 2010, 82, 6721–6725.
(65) Fernandez-Lima, F. A.; Becker, C.; McKenna, A. M.; Rodgers, R. P.; Marshall, A.
G.; Russell, D. H. Anal. Chem. 2009, 81, 9941–9947.
(66) Bowen, B. P.; Northen, T. R. J Am Soc Mass Spectrom, 2010, 21, 1471-1476.
(67) Issaq, H. J.; Van, Q. N.; Waybright, T. J.; Muschik, G. M.; Veenstra, T. D. J. Sep.
Science 2009, 32, 2183–2199.
(68) Broadhurst, D. I.; Kell, D. B. Metabolomics 2006, 2, 171–196.

30
Chapter 2
Metabolic Analysis of Striatum Tissues from Parkinson’s
Disease-like Rats by Electrospray Ionization Ion Mobility Mass
Spectrometry
Adapted with permission from “Metabolic analysis of striatum tissues from Parkinson’s disease-like
rats by electrospray ionization ion mobility mass spectrometry” Analytical Chemistry, 2014, 86(6),
3075-3083. Copyright 2014 by Analytical Chemistry
Abstract
Electrospray ionization ion mobility mass spectrometry (ESI-IMMS) was used to
study the striatal metabolomes in a Parkinson’s like disease (PD-like) rat model. Striatum
tissue samples from BD-IV with PD-like disease 20dpn-affected and 15dpn-affected rats (dpn:
days post natal) were investigated and compared with age-matched controls. Ion mobility
mass spectrometer (IMMS) produced multidimensional spectra with mass to charge ratio
(m/z), ion mobility drift time, and intensity information for each individual metabolite.
Principle component analysis (PCA) was applied in this study for pattern recognition and
significant metabolites selection (68% data was modeled in PCA). Both IMMS spectra and
PCA results showed that there were clear global metabolic differences between PD-like
samples and healthy controls. Nine metabolites were selected by PCA and identified as
potential biomarkers using the Human Metabolome Database (HMDB). One targeted
metabolite in this study was dopamine. Selected-mass mobility analysis indicated the absence
of dopamine in PD-like striatal metabolomes. A major discovery of this work, however, was

31
the existence of an isomer of dopamine. By using ion mobility spectrometry, the dopamine
isomer, which has not previously been reported, was separated from dopamine.

32
2.1 Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder of
the central nervous system (CNS)1. It is clinically characterized with symptoms such as
bradykinesia, tremor, rigidity and postural instability2. In the early stage of PD,
movement-related symptoms are most commonly observed, while in the late stage, cognitive
problems and other symptoms may arise, such as sensory and sleep difficulty3. PD affects
over 1% of the population above the age of 654.
PD is partially correlated with dopamine loss in the putamen and caudate nucleus of
the striatum5,6. The disease mechanism, however, remains an undefined question7. Diagnosis
of PD is difficult due to the complicated and gradual progress of symptoms among different
stages8, and there are numbers of other CNS disorders that present similar symptoms9.
Currently, most diagnoses are either symptom-based or brain image assisted, and no clinical
biomarker has been fully validated. Hence, the accuracy of clinical diagnosis is less than
90%10. Therefore, the discovery of biomarkers characteristic for PD is of considerable
interest. New biomarkers are desired, for the purpose of further exploring the mechanisms
involved in PD and in aiding clinical diagnosis.
One approach commonly used for the identification of novel biomarkers is
metabolomics. Metabolomics is a comprehensive measurement of metabolomes in a tissue or
biofluid, providing an overview of the metabolic status. The change of metabolic status
displays observable metabolites changes, and provides the access to the originated generic
variants11. Therefore, given that the health/disease status is captured by metabolic states and
alternations, the idea of using metabolomic analysis for monitoring disease progression and
developing biomarkers has been encouraged. In the past two decades, metabolomics studies

33
have been conducted on a number of CNS and psychiatric disorders, including PD12,
Huntington’s disease13, and depression14.
The application of metabolomics in the 20th century was limited due to the
complicated biological system and less-developed analytical technologies15. In recent years,
technological improvements in both analytical hardware and data-analysis software have
enabled metabolic analysis to become a powerful tool for understanding disease mechanisms
and identifying biomarkers16,17.
Metabolomics approaches for identifying PD biomarkers have been conducted on a
variety of different tissue and fluid samples and a number of potential biomarkers have been
reported. Scatton et al. discovered that the concentration of a series of neurotransmitters, such
as 3,4-dihydroxyphenylacetic acid, homovanillic acid, noradrenaline, and serotonin, changed
from control subjects to parkinsonian patients in brain cortical areas18. Bogdanov et al.
utilized metabolic profiling with high performance liquid chromatography coupled with
electrochemical coulometric array detection to look for biomarkers in plasma that could be
used for diagnosis. They found lower uric acid level and increased glutathione level were
detected in PD patients12. Michell et al. investigated the metabolic profile of serum and urine
samples from 23 PD patients using age and sex-matched controls with gas chromatography
mass spectrometry. They were able to observe subtle separations between PD patients and
healthy controls after principle component analysis as well as partial least-square
discriminate analysis19. More recent work showed that the cholinergic system was closely
related to PD20; iron metabolism was also reported to be linked in many neurodegenerative
diseases including PD21. These cases have demonstrated an understanding that PD is likely to
induce multi-system alternations besides the loss of dopaminergic nigral neurons. They also

34
proved that no single test suffices given the complexity and heterogeneity of PD.
Metabolomics related investigations have been limited to two major analytical
platforms: mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectrometry.
Both platforms have advantages and disadvantages22. MS-based techniques have high
sensitivity and they generate uncomplicated spectra, however, the complexity of a
metabololomic sample has made a pre-separation step necessary, hence, both liquid
chromatography mass spectrometry (LC/MS) and gas chromatography mass spectrometry
(GC/MS) are often required. The addition of chromatographic methods to mass spectrometry
considerably lengthens analysis time, but without them, MS is blind to the structural
information of many isomers that may exist in the sample. On the other hand, NMR
techniques require minimal sample preparation. They are capable of structurally elucidating
molecules and perform quantitative analysis, but suffer from high detection limits, and
relatively complicated spectra. The ideal analytical approach for metabolomics should be
capable of measuring hundreds of metabolites simultaneously, efficiently, and sensitively.
Ion mobility spectrometry has been reported as a potential pre-separation method for mass
spectrometry.
When coupled to mass spectrometry, ion mobility spectrometry (IMS) has been
demonstrated as a novel and efficient analytical platform for metabolomics studies23,24. Ion
mobility mass spectrometry (IMMS) can rapidly generate multi-dimensional information
including ion mobility information based on size-to-charge ratio (Ω/z), ion mass information
based on mass-to-charge (m/z) ratio, and intensity information based on concentration.
IMMS has been demonstrated for application to metabolomics through several metabolic
studies, including the metabolic profiling of human blood, monitoring changes in the lymph

35
metabolomes of fasting and fed rats and characterizing various metabolites23,25,26. IMMS has
evolved into a high-throughput technique with sophisticated data analysis methods.
In this study, the metabolomes of striatum brain region from PD-like rats were
analyzed by electrospray ion mobility mass spectrometry (ESI-IMMS). Our hypothesis in this
study was that the homeostatic perturbation of PD will alter multiple metabolic pathways and
that a global metabolomics approach will derive a more comprehensive picture of the
metabolites and metabolic pathways involved. These metabolites that are affected by PD may
serve in concert or individually as candidate biomarkers, which can be linked to disease
progression and mechanism. The first goal of this study was to differentiate the metabolic
status between PD and healthy control, and also between two PD progression states
(15dpn-affected and 20dpn-affected); the second goal of this study was to selectively analyze
potential biomarkers (influential metabolites) using appropriate analytical and statistical
methods.

36
2.2 Experimental Design
2.2.1 Sample preparation and metabolite extraction:
All animal procedures were performed in Texas A&M University according to the
protocol approved by the Texas A&M University Institutional Animal Care and Use
Committee27. Striatal tissue samples from Berlin Druckrey IV (BD-IV) 15dpn-affected (dpn:
days post natal) rats with PD-like disease, 20dpn-affected rats with PD-like disease and
BD-IV healthy control rats (20dpn) were used. Striatal tissue samples from Sprague Dawley
(SD) healthy control rats (20dpn) were also used as another set of control. Tissues were
frozen and stored at -80°C until used. Right before analysis, each tissue sample was weighed
and placed in eppendorf tubes individually, where the tissue sample was sonicated in 600 µL
of electrospray solvent (methanol: water: formic acid 49.95:49.95:0.1(v/v/v)) for metabolite
extraction28. Cellular debris was separated from metabolites after centrifugation for 30
minutes at 13K rpm by a desktop centrifuge (Model#: 5415D) (Eppendorf AG, Hamburg,
Germany). The supernatant was stored on ice until used for IMMS analysis.
The total number of striatal tissue samples in this study was n = 8, with 1 tissue
sample/animal subject. BD-IV 20dpn-affected samples (n = 2) and BD-IV healthy control
samples (n = 2) were prepared and IMMS analysis was conducted twice for each sample.
BD-IV 15dpn-affected samples (n = 2) and SD healthy control samples (n = 2) were prepared
and IMMS analysis was conducted once each sample. This experimental design guaranteed
both biological replication and instrumental replication, with an emphasis made on the
BD-IV 20dpn-affected samples and BD-IV healthy control samples.
Note: There were 2 sets of PD-like rats, which were all affected but at different ages, 15dpn
and 20dpn. Clinical symptoms and movement disorders were more severe at 20dpn than
15dpn. The affected rats were born normal and showed clinical motor signs characterized by

37
tremor, rigidity, spasticity and postural instability around 15 dpn. The severity of
neurological movement disorder increased with time27. There were also 2 sets of healthy
controls (BD-IV controls and SD controls). In the later descriptions, “PD samples” was
referred to both 15dpn-affected and 20dpn-affected samples.
2.2.2 Electrospray ion mobility time-of-flight mass spectrometer (ESI-IM-TOFMS):
In this study, an ion mobility mass spectrometer from TofWerk (Thun, Switzerland)
was used to ionize, separates, detect, and analyse the complex metabolic samples. Figure
2.1 shows a schematic diagram of the IMMS instrument, which consisted of an electrospray
nebulization and ionization source, an atmospheric drift tube ion mobility spectrometer, a
vacuum interface, a time-of-flight mass spectrometer, and a data collection method. The
design and operating conditions of these instrument components are described in more detail
below.
Electrospray Ionization Source (ESI): Sample solutions were introduced into the
IMMS by an electrospray apparatus with a syringe pump (Model#: Fusion 200) (Chemyx
INC. Stanfford, TX). A 5.0 cm long fused silica capillary with internal diameter of 150 µm
(Polymicro, Phoenix, AZ) was used as the electrospray source and the same capillary was
used as a sample transfer line for the sample solution from syringe to the ESI source. A
biased voltage of 3000 volts and a sample solution flow rate of 5 µL/min were held constant
during the experiment. ESI background was collected before each sample analysis by
electrospraying blank ESI solvent; mass calibration and mobility calibration were conducted
every day during using 2,6-di-tert-butylpyridine.
Ion mobility spectrometer (IMS): A conventional stacked ring IMS constructed at

38
Washington State University was used as the ion mobility spectrometer. It consisted of an
8cm-long dissolvation region, a 21 cm long drift region and a Bradbury-Nielsen (BN) ion
gate. Both dissolvation region and drift region were made up by alternating stainless steel
conducting rings and ceramic insulating rings, where the conducting rings were connected
with resistors (500 kΩ resistors for dissolvation region and 1-MΩ for drift region) to create a
uniform electric field (E = 315 V/cm in drift region). A heated (200°C) steady nitrogen
counter current gas flow (2.5 L/min) was introduced into the IMS to keep the tube clean and
induce ion-neutral collisions. Solvated ion droplets from the electrospray process were
reduced to solvent free analytic ions in the dissolvation region of the spectrometer. These
solvent-free ions were then gated into the drift region of the ion mobility spectrometer with a
gate pulse width of 0.2 ms and a frequency of 20.8 Hz. The IMS tube was held at a constant
temperature of 200oC and operated at ambient pressure (690 - 705 Torr). Metabolite ions
were stable and did not dissociate during the analysis under this condition.
Vacuum interface and time-of-flight mass spectrometer (TOFMS): The interface to
the mass spectrometer was a 300 µm orifice. After the ions passed through the orifice they
were focused and transferred to the mass spectrometer by lenses, nozzles and two segmented
quadrupole ion guides. The operational details have been described elsewhere29. Briefly, the
first segmented quadrupole was operated with a RF of 2.07 MHz at ~3 mbar. The second
segmented quadrupole had a RF of 1.6 MHz at ~1×10-2 mbar. The frequency of the
quadrupole could be optimized for the desired mass range, and the voltage on the lenses and
nozzle could be optimized for desired fragmentation. The DC ion lenses were operated at
~1×10-5 mbar for further transmission.
The TOFMS consisted of a high-resolution time-of-flight mass spectrometer with a

39
multichannel plate (MCP) detector (both were operated at ~4×10-7 mbar). The timing
generator that controlled the IMS gate, the TOFMS extraction and the TDC (operated at 800
ps time resolution) was also from Tofwerk.
2.2.3 IMMS data acquisition and data analysis:
Data acquisition: Tofwerk (AG, Switzerland) developed the data acquisition software
named Tofdaq, which was capable of showing multidimensional data information including
mass spectra, mobility spectra and the total ion intensity (counts per second). This data
acquisition software could be performed at mass spectrometry mode (MS mode) with BN
gate open, as well as ion mobility mass spectrometry mode (IMMS mode) with BN gate
pulsing at 0.2 ms gate pulse width30. The ion mobility mass spectra and ion mobility spectra
were obtained in IMMS mode. In IMMS mode, an IMS experiment cycle was 60 ms, within
which, 1000 MS experiment cycles (60 µs/cycle) were collected and averaged. Multiple IMS
experiment cycles were collected for 15 minutes to achieve one IMMS analysis in this study.
Data Explanation: In the multidimensional data sets obtained from the IMMS, each
ion species was characterized by its ion mobility, m/z ratio and ion intensity (counts). The
mass data and intensity information were generated by TOF mass spectrometer. Ion mobility
data were generated by the time each ion species spent in the ion mobility drift tube and
reported as reduced mobility (K0) in cm2V-1s-1.
(Equation 1)
Where V is the voltage drop over the drift region with length L, td is the time that the
ion takes to migrate through the drift tube. The reduced mobility constant (K0) for an ion
remains constant for a given drift gas, since it corrects for operating pressure (P in Torr) and
Ko =L2
tdV×273.15T
×P760

40
temperature (T in Kelvin) of the drift region.
Initial data analysis: After the multidimensional data acquired by Tofdaq software,
data extraction software named Ionwerks_IMMS_Viewer developed by Exelis Visual
Information Solutions (McLean, VA) was used for generating Microsoft adaptable peaklist
(contained all peaks with each peak assigned with reduced mobility, m/z, and intensity).
Statistical analysis: Two major software approaches were used for further statistical
analysis. The first one was Merge Table Wizard for Microsoft Excel (Ablebits, Homel,
Belarus), which was used for peak alignment among replicates. The second software was
Unscrambler X 10.1 from CAMO Software (Oslo, Norway), which was employed for
principle component analysis. Principal component analysis (PCA) has been extensively
applied in multivariate data analysis, such as MS data analysis and NMR data analysis31,32; it
was utilized for statistical analysis through this study. It is a comprehensive and unsupervised
multivariate analysis method, with principle components (PCs) representing the linear
transformations of the dataset (in this case, the dataset referred to the metabolites that were
assigned with m/z, K0 and intensity). In the score plot, patterns and clusters within the dataset
become apparent without knowing the classification of the samples. In the loadings plot,
metabolites in the dataset are viewed with loading values assigned in specified PCs.
Metabolites that have a large impact on the patterns will have relatively large loading values,
while metabolites that contribute the least have small loading values (absolute values).
Therefore, metabolite concentrations that were altered between parkinsonian samples and
healthy controls could be selected as potential biomarkers based on their loading values.

41
Exact mass analysis for potential biomarkers: Exact m/z information can be obtained
from the IMMS analysis, and biomarkers that show high loading values in PCA loadings’
plot can be expressed and identified using the Human Metabolite Database (HMDB)33.
Selected mass ion mobility analysis: Isomeric analysis is difficult by mass
spectrometry because isomers share the same exact mass but have different molecular
structures. Ion mobility, however, can separate isomeric compounds based on the difference
in their size34,35. Multidimensional data obtained from the IMMS instrument were extracted
by selecting a mass and the mobility spectrum of the corresponding mass appeared in the
IMS window. The time it took for obtaining selected-mass ion mobility spectrum was within
a few seconds. This analysis was used for selected-mass ion mobility analysis and isomeric
separation.

42
2.3 Results and Discussion
2.3.1 Metabolomes of Striatum from Healthy Controls and PD Rats:
Two-dimensional (2D) IMMS spectra for background and the striatal metabolomes of
a healthy control sample are shown in Figure 2.2. In the 2D spectra, the x-axis is assigned
with m/z, the y-axis is assigned with drift time (millisecond (ms)). Every dot displayed in the
2D spectra represents a detected ion. Figure 2.2a is a spectrum of electrospray background
solvent without any sample present. As shown, multiple ions are present, mainly produced by
the ESI solvent. Background spectra were constant throughout all experiments. In practice,
background ions were masked from those found in the sample so that only the ions detected
uniquely in the sample were used for analysis. Figure 2.2b is the spectrum of striatal
metabolomes from a SD healthy control rat. It is clearly different from the ESI background
spectrum. Detailed information (m/z, drift time and intensity) of metabolite ions was
extracted from this spectrum. Ions with high intensities showed up between m/z = 100 and
200, but visible metabolite ions spread out up to an m/z of 600. The limit of detection (LOD)
of IMMS analysis was previously studied using multiple metabolite standards and measured
to range from 13 to 67 nM for different classes of metabolites29,36. Figure 2.2c is a
zoomed-in 2D IMMS spectrum generated from 2.2b (with m/z range of 300 - 400 and drift
time range of 15 - 30 ms), ~40 major metabolite ions were observed from this spectrum,
demonstrating the complexity of striatal metabolomes. The numbers of reproducible
metabolite ions detected in SD healthy control, BD-IV healthy control, 20dpn-affected and
15dpn-affected were 134, 129, 138, and 142, respectively (as shown in Table 2.1).
Global metabolic comparisons between the metabolomes of SD healthy control and
PD samples are shown in Figure 2.3. All spectra in Figure 2.3 range in m/z from 200 to 900
and in drift time from 0 ms to 45 ms. Figure 2.3a and 2.3b represent the MS and IMS

43
spectra for BD-IV healthy control sample and SD healthy control sample, respectively. As
shown in the figures, there were 12 - 13 major ion peaks (with intensities over 100 counts) in
both MS spectra, and their IMS spectra were also similar. Table 2.2 provides the m/z and K0
values for each of these peaks as well as their intensity values. Despite the fact that the
intensities of these major peaks were not exactly the same, there was still considerable
similarity between the BD-IV and SD healthy control samples. Moreover, the intensity
difference was largely caused by variation in tissue weights, and intensity was normalized
upon tissue weight for statistical analysis. Figure 2.3c and 2.3d represent MS and IMS
spectra for PD 20dpn-affected sample and PD 15dpn-affected sample. Responses of the
major ion peaks found in the healthy control samples were different in the PD samples, in
fact, there were only 5 - 6 major ion peaks (with intensity over 100 counts) found in PD
samples. As shown in these figures and more detailed data in the Table 2.2, the MS spectra
and the IMS spectra were similar for the PD samples. However, they were clearly different
from the spectra of the healthy controls. Global metabolic comparisons by spectra indicated a
substantial metabolic difference between healthy controls and PD affected rats. Between
different PD samples (20dpn-affected and 15dpn-affected), however, metabolic patterns were
considerably similar.
2.3.2 Global Metabolic Analysis by Principle Component Analysis (PCA):
Score plots and loadings plots were obtained using Unscramber X10.1. With 36%, 16%
and 15% of the data being explained by PC1, PC2, PC3, respectively, all samples were
plotted in 3D PCA score plot as shown in Figure 2.4. Clear metabolic patterns including
15dpn-affected, 20dpn-affected, BD-IV and SD healthy controls, were observed and assigned.
As expected from the global metabolic comparisons, PCA score plots were able to
distinguish PD metabolomes and healthy control metabolomes. Moreover, even though the

44
two groups of PD affected samples only showed subtle difference in their MS and IMS
spectra, differences between them were observed in PCA score plots.
2.3.3 Biomarkers Identification:
PCA loadings plot shown in Figure 2.4b was used for targeting potential biomarkers.
A loadings plot is a summary of the metabolite ions with regard to their influence on pattern
recognition. Metabolite ions locate away from the center of the loadings plot have higher
loadings values and they have more significant influence on pattern recognition. Therefore,
they have a great potential to serve as biomarkers for PD. Nine metabolite ions were selected
as potential biomarkers for PD. The Human Metabolome Database was used for identifying
potential biomarkers by their exact m/z values with an m/z tolerance of ± 0.010. Table 2.3 is
the identification table. As shown, 6 metabolites including Alanine and Arginine were down
regulated in PD samples, indicating their concentration levels were lower when compared
with healthy controls. The other 3 metabolites including cholesterol were up regulated in PD
samples. Although dopamine is a known biomarker for PD, it was not selected here as a
major potential biomarker by PCA because of its low abundance.
From the previous studies reviewed earlier, levels of multiple metabolites were
altered in PD patients. Our analysis was able to detect neurotransmitters such as
norepinephrine and 3,4-dihydroxyphenylacetic acid, which were found to be altered by
Scatton et al., however, they were not selected as potential biomarkers in statistic analysis
due to the fact that, in these studies, their levels were not significantly changed. It should be
noted, however, that the Scatton studies were of metabolites in the cortical area of the brain
while our studies were from the striatum. Also, other metabolites such as uric acid that have
been found in plasma and urine but were not detected in the brain tissue investigated here.

45
However, nine potential biomarkers were discovered in the striatal metabolomes of PD-like
rats. These potential biomarkers are listed in Table 2.3.
2.3.4 Selected-mass Mobility Analysis of Dopamine:
As noted in the introduction, reduction in dopamine concentration is a common
indicator of PD, Figure 2.5 shows the selected-mass mobility spectra of [Dopamine+H] +
(selected m/z = 154). All figures have x-axis assigned with drift time (ms), y-axis assigned
with relative intensity (arbitrary unit). Figure 2.5a is the selected-mass mobility spectrum of
the ESI solvent without sample. As expected, no peak was detected and the spectrum was
essentially blank. Figure 2.5b and 2.5c compare the selected-mass (m/z = 154) mobility
spectra of a 20dpn-affected sample and a healthy control. Surprisingly, two distinguishable
ion mobility peaks at m/z 154 were observed in healthy control metabolomes, demonstrating
an existing isomer pair for m/z 154 presented in the healthy control metabolomes (exact m/z
was checked to confirm that these two mobility peaks had same exact m/z). A standard
solution of dopamine (3 µM) was analyzed using IMMS; and it was determined that the later
mobility peak had a K0 of 1.52 cm2V-1s-1, matching well with the K0 of dopamine standard
(K0 = 1.51 cm2V-1s-1); and the unknown mobility peak had a higher mobility with a K0 of
1.68 cm2V-1s-1.
For the selected-mass (m/z = 154) mobility spectrum of the 20dpn-affected sample
(Figure 2.5c), however, only one mobility peak was observed at m/z of 154. The missing
peak represented the absence of one isomer. The single peak observed had a K0 of 1.68
cm2V-1s-1, matching the K0 of the unknown mobility peak observed in healthy control
(Figure 2.5b). In order to verify that the missing isomer in 20dpn-affected sample was
dopamine, a dopamine standard was spiked into the 20dpn-affected sample so that the final

46
concentration of dopamine in the sample was 15 µM. The result for this standard addition
showed that a peak was observed at the appropriate mobility of the missing peak, confirming
the absence of dopamine in 20dpn-affected metabolomes while an isomer of dopamine
remained. In summary, dopamine was detected in healthy control samples with a consistent
intensity, however, it was essentially eliminated in 15dpn-affected samples and remained
absent in 20dpn-affected samples. In previous studies, dopamine has been reported as
reduced in concentration, but the present of this unknown dopamine isomer may have
masked the fact that dopamine was at an even lower concentration level in the PD striatal
metabolomes than what people expected.
A proposed structure for the dopamine isomer and the corresponding metabolic
pathway are shown in Figure 2.6. Originating from tyrosine, the proposed metabolic pathway
has a hydroxylation step that may have taken place at a different position compared with the
dopaminergic pathway. Hence, the proposed structure for the unknown isomer is
2-(2,4-dihydroxylphenyl) ethylamine. To confirm this structure, a standard would need to be
analyzed with IMMS match its mobility. Unfortunately, a standard of this metabolite was not
available for purchase and will require specific synthesis. Nevertheless, the presence of an
isomer of dopamine was novel and the fact that the concentration of this isomer was not
affected while the presence of dopamine was reduced below its detection limit was a major
finding of this work.

47
2.4 Conclusions
Ion mobility mass spectrometry (IMMS) is an efficient and sensitive method for the
application of metabolomics to the investigation of Parkinson’s disease. IMMS provides
multidimensional spectra containing hundreds of detected metabolites in minutes, enabling
rapid global comparison of metabolic status. Healthy control striatal tissues have different
global metabolic patterns from those of PD striatal tissues. There are at least 9 metabolites
that are significantly altered by PD and which may be biomarkers for PD. The most
significant finding of this study was the discovery of an isomer, tentatively identified as
2-(2,4-dihydroxylphenyl) ethylamine, of dopamine. While dopamine is depleted in the
striatum of PD affected rats, the isomer is not. The isomer has a higher mobility than
dopamine, 1.68 cm2V-1s-1 vs 1.51 cm2V-1s-1, indicating that it has a small collision cross
section than dopamine. This analysis is consistent with the tentatively identification the
dopamine isomer as 2-(2,4-dihydroxylphenyl) ethylamine. The implication of these
findings is that the reduction of dopamine in PD may be underestimated due to the presence
of its isomer.
Acknowledgements
This article is dedicated to the memory of one of our co-authors, Prof. James Schenk, who
passed away in February of 2013. This project was supported in part by funds provided for
medical and biological research by the State of Washington Initiative Measure No.171.

48
Reference (1) Tanner, C. M.; Goldman, S. M. Neurologic Clinics 1996, 14, 317–335.
(2) Braak, H.; Tredici, K. D.; Rüb, U.; de Vos, R. A. I.; Jansen Steur, E. N. H.; Braak, E.
International journal of mass spectrometry 2003, 24, 197–211.
(3) Henchcliffe, C.; Dodel, R.; Beal, M. F. Progress in Neurobiology 2011, 95, 601–613.
(4) De Rijk, M. C.; Launer, L. J.; Berger, K.; Breteler, M. M.; Dartigues, J. F.;
Baldereschi, M.; Fratiglioni, L.; Lobo, A.; Martinez-Lage, J.; Trenkwalder, C.;
Hofman, A. Neurologic Clinics 2000, 54, S21–S23.
(5) Kish, S. J.; Shannak, K.; Hornykiewicz, O. N Engl J Med 1988, 318, 876–880.
(6) Lotharius, J.; Brundin, P. Nature Reviews Neuroscience 2002, 3, 932–942.
(7) Caudle, W. M.; Bammler, T. K.; Lin, Y.; Pan, S.; Zhang, J. Expert Rev
Neurotherapeutics 2010, 10, 925–942.
(8) PD Study Group, N Engl J Med 2004, 351, 2498–2508.
(9) Aarsland, D.; Larsen, J. P.; Lim, N. G.; Janvin, C.; Karlsen, K.; Tandberg, E.;
Cummings, J. L. Journal of Neurology, Neurosurgery & Psychiatry 1999, 67, 492–
496.
(10) Hughes, A. J. Brain 2002, 125, 861–870.
(11) Nordström, A.; Lewensohn, R. J Neuroimmune Pharmacol 2009, 5, 4–17.
(12) Bogdanov, M.; Matson, W. R.; Wang, L.; Matson, T.; Saunders-Pullman, R.;
Bressman, S. S.; Beal, M. F. Brain 2008, 131, 389–396.
(13) Underwood, B. R. Brain 2006, 129, 877–886.
(14) Paige, L. A.; Mitchell, M. W.; Krishnan, K. R. R.; Kaddurah-Daouk, R.; Steffens, D.
C. Int. J. Geriat. Psychiatry 2007, 22, 418–423.
(15) C E Dalgliesh, E. C. H. M. G. H. K. L. K. K. Y. Biochemical Journal 1966, 101,
792.

49
(16) Nicholson, J. K.; Lindon, J. C. Nature 2008, 455, 1054–1056.
(17) Quinones, M. P.; Kaddurah-Daouk, R. Neurobiology of Disease 2009, 35, 165–176.
(18) Scatton, B.; Javoy-Agid, F.; Rouquier, L.; Dubois, B.; Agid, Y. Brain Research 1983,
275, 321–328.
(19) Michell, A. W.; Mosedale, D.; Grainger, D. J.; Barker, R. A. Metabolomics 2008, 4,
191–201.
(20) Bohnen, N. I.; Albin, R. L. Behavioural Brain Research 2011, 221, 564–573.
(21) Crichton, R. R.; Dexter, D. T.; Ward, R. J. J Neural Transm 2010, 118, 301–314.
(22) Dunn, W. B.; Ellis, D. I. TrAC Trends in Analytical Chemistry 2005, 24, 285–294.
(23) Woods, A. S.; Ugarov, M.; Egan, T.; Koomen, J.; Gillig, K. J.; Fuhrer, K.; Gonin, M.;
Schultz, J. A. Anal. Chem. 2004, 76, 2187–2195.
(24) Dwivedi, P.; Wu, P.; Klopsch, S. J.; Puzon, G. J.; Xun, L.; Hill, H. H., Jr.
Metabolomics 2007, 4, 63–80.
(25) Kaplan, K.; Dwivedi, P.; Davidson, S.; Yang, Q.; Tso, P.; Siems, W.; Hill, H. H., Jr.
Anal. Chem. 2009, 81, 7944–7953.
(26) Dwivedi, P.; Schultz, A. J.; Hill, H. H., Jr. International Journal of Mass
Spectrometry 2010, 1–13.
(27) Stoica, G.; Lungu, G.; Bjorklund, N. L.; Taglialatela, G.; Zhang, X.; Chiu, V.; H, H.
H.; Schenk, J. O.; Murray, I. Journal of Neurochemistry 2012, 122, 812–822.
(28) Villas-Bôas, S. G.; Mas, S.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. Mass Spectrom.
Rev. 2005, 24, 613–646.
(29) Kaplan, K.; Graf, S.; Tanner, C.; Gonin, M.; Fuhrer, K.; Knochenmuss, R.; Dwivedi,
P.; Hill, H. H., Jr. Anal. Chem. 2010, 82, 9336–9343.
(30) Siems, W. F.; Wu, C.; Tarver, E. E.; Hill, H. H. J.; Larsen, P. R.; McMinn, D. G.
Anal. Chem. 1994, 66, 4195–4201.

50
(31) Wagner, M. S.; Castner, D. G. Langmuir 2001.
(32) Pan, Z.; Gu, H.; Talaty, N.; Chen, H.; Shanaiah, N.; Hainline, B. E.; Cooks, R. G.;
Raftery, D. Anal Bioanal Chem 2006, 387, 539–549.
(33) Wishart, D. S.; Knox, C.; Guo, A. C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D. D.;
Psychogios, N.; Dong, E.; Bouatra, S.; Mandal, R.; Sinelnikov, I.; Xia, J.; Jia, L.;
Cruz, J. A.; Lim, E.; Sobsey, C. A.; Shrivastava, S.; Huang, P.; Liu, P.; Fang, L.;
Peng, J.; Fradette, R.; Cheng, D.; Tzur, D.; Clements, M.; Lewis, A.; De Souza, A.;
Zuniga, A.; Dawe, M.; Xiong, Y.; Clive, D.; Greiner, R.; Nazyrova, A.;
Shaykhutdinov, R.; Li, L.; Vogel, H. J.; Forsythe, I. Nucleic Acids Research 2009, 37,
D603–D610.
(34) Creaser, C. S.; Griffiths, J. R.; Bramwell, C. J.; Noreen, S.; Hill, C. A.; Thomas, C. L.
P. Analyst 2004, 129, 984–994.
(35) Borsdorf, H.; Rudolph, M. International Journal of Mass Spectrometry 2001, 208,
67–72.
(36) Zhang, X.; Knochenmuss, R.; Siems, W. F.; Liu, W.; Graf, S.; Herbert H Hill, J.
Anal. Chem. 2014, in press.

51
Table 2.1 Number of reproducible ions detected in each sample for principal component
analysis.
Number of Reproducible Metabolites
SD controls BD-IV controls BD-IV 20dpn-affected BD-IV 15dpn-affected
134 129 138 142
Note: each ion was assigned with m/z, reduced mobility K0 (in cm2V-1s-1) and intensity (in counts
per second) for statistical analysis.

52
Table 2.2 Major ion peaks (ion counts > 100) detected in PD-like samples, BD-IV and SD healthy controls. M/z ranges between 200 and 900.
BD-IV Affected 20dpn
BD-IV Affected 15dpn
m/z K0 Intensity (counts)
m/z K0 Intensity (counts) 268.11 1.34 1492
227.12 1.42 711
227.12 1.42 866
268.1 1.34 548 269.11 1.34 118
228.12 1.42 400
228.13 1.42 109
369.33 0.96 253 225.2 1.38 106
269.1 1.34 178
298.12 1.25 141
BD-IV Healthy Control
SD Healthy Control m/z K0 Intensity (counts)
m/z K0 Intensity (counts)
268.11 1.34 223
268.11 1.34 477 227.12 1.42 217
369.33 0.96 406
307.06 1.21 213
307.06 1.21 250 296.05 1.25 171
296.05 1.25 205
369.33 0.96 161
269.1 1.34 196 453.13 0.93 141
227.12 1.42 194
222.02 1.42 131
308.04 1.21 190 204.1 1.45 128
279.13 1.18 181
269.11 1.34 125
291.05 1.14 167 298.11 1.26 117
222.02 1.42 153
203.23 1.4 114
204.1 1.45 134 228.13 1.42 112
453.12 0.93 127
298.05 1.25 108

53
Table 2.3 Tentative metabolites identification based on exact m/z using the Human
Metabolome Database, including molecular formula and adducts. The changes of metabolites
concentration levels are explained as “up/down regulated” in PD samples when compared
with healthy controls.
Ion # m/z K0 In PD Samples Metabolite
Identification Chemical Formula Adduct
1 90.048 1.82 Down-Regulated Alanine C3H7NO2 [M+H]+ 2 97.034 1.95 Up-Regulated Lactaldehyde C3H6O2 [M+Na]+ 3 114.061 1.89 Down-Regulated Creatinine C4H7N3O [M+H]+
4 123.051 1.82 Down-Regulated Niacinamide/Vitamin
B3 C6H6N2O [M+H]+
5 127.065 1.85 Down-Regulated 5-Aminoimidazole-4-ca
rboxamide C4H6N4O [M+H]+ 6 132.072 1.82 Down-Regulated L-Leucine/L-Isoleucine C6H13NO2 [M+H]+ 7 137.044 1.77 Up-Regulated Hypoxanthine C5H4N4O [M+H]+ 8 175.115 1.59 Down-Regulated Arginine C6H14N4O2 [M+H]+ 9 369.358 0.95 Up-Regulated Cholesterol C27H46O [M-H2O+H]+

53
Figure 2.1 Schematic diagram of an electrospray ion mobility time-of-flight mass
spectrometer (TofWerk AG, Thun, Switzerland), with major components: electrospray
ionization (ESI) source, ion mobility spectrometer, IMMS interface, reflectron time-of-flight
mass spectrometer.

54
Figure 2.2 Multidimensional IMMS spectra of (a) the electrospray background, (b) SD
healthy control metabolome from striatal tissue, and (c) an expanded view of b in the m/z
range from 300 to 400 and drift time range from 15 ms to 30 ms for SD healthy control
striatal metabolome.

55
Figure 2.3 MS and IMS spectra for BD-IV and SD healthy control striatal metabolomes are
shown in (a) and (b); MS and IMS spectra for PD affected 20 dpn and 15 dpn strital
metabolomes are shown in (c) and (d). All spectra have m/z range of 200-900 and drift time
range of 0 ms - 40 ms.

56
Figure 2.4 Principle component analysis results for all reproducible metabolite ions. (a)
shows the 3-D score plot results with PC1 (36%), PC2 (16%) and PC3 (15%). Different
patterns are observed for different samples. (b) is the loadings plot with metabolites plotted
according to their loading values; metabolites highlighted by red circles were identified as
potential biomarkers.
BD-IV 20dpn
Affected
BD-IV Healthy Controls
SD Healthy Controls
BD-IV 15dpn
Affected
a
b

57
Figure 2.5. (a), (b), (c) are the selected m/z 154 mobility spectrums abstracted from the
overall IMMS spectrums for ESI background, BD-IV healthy control striatal metabolomes
and BD-IV 20dpn affected striatal metabolomes. Dopamine and 2-(2,4-dihydroxyphenyl)
ethylamine (proposed structure) were detected as an isomer pair. Both compound ions are
observed in (b), only 2-(2,4-dihydroxyphenyl) ethylamine is observed in (c).
BD-IV Healthy Control
BD-IV 20dpn-Affected
Dopamin
e
2-(2,4-dihydroxyp
henyl) ethylamine
a
b
c

58
Figure 2.6 Scheme of proposed dopamine and 2-(2,4-dihydroxyphenyl) ethylamine path.

59
Chapter 3
Evaluation of Hadamard Transform Atmospheric Pressure Ion
Mobility Time-of-Flight Mass Spectrometry
(HT-APIMS-TOFMS) for Complex Mixture Analysis
Adapted with permission from “Evaluation of hadamard transform atmospheric pressure ion mobility
time-of-flight mass spectrometry (HT-APIMS-TOFMS) for complex mixture analysis” Analytical
Chemistry, 2014, 86(3), 1661-1670. Copyright 2014 by Analytical Chemistry Abstract
Ion mobility mass spectrometry (IMMS) has gained popularity in the analysis of
complex mixtures such as those encountered in metabolomics and proteomics. However, the
challenge that exists in conventional pulsed IMMS is its inherent low duty cycle. The first
application of Hadamard transform (HT)-type signal coupled with atmospheric pressure
IMMS to complex mixtures is presented. Performance of the prototype was assessed by the
analysis of metabolite standard mixture. With 200 times increased IMS duty cycle in HT
mode compared with conventional pulsed mode, the limit of detection (LOD) was decreased
by ~10 times. Evaluation for application to complex mixtures was achieved using the NIST
Standard Reference Material 1950 Metabolites in Human Plasma. Approximately 180
metabolite ions were detected within 1 minute with an IMS resolving power (Rp) of ~100.
Rapid chromatographic separation prior to IMMS analysis was also demonstrated for
improving the response of metabolite ions in rat brain tissue extract.

60
3.1 Introduction
Ion mobility spectrometry (IMS)1 is a gas-phase separation technique, which has been
widely used for rapid detection of explosives, drugs and chemical warfare agents as a
screening method in homeland security2,3. It has gained increased popularity mainly because
of its portability and reasonable low detection limit in field. Internal ionizations sources such
as 63Ni are efficient and stable4, which have been most commonly used in IMS for gas phase
target detection. In the past decades, the development of alternative ionization methods such
as electrospray ionization (ESI)5 and matrix-assisted laser desorption ionization (MALDI)6
has enabled the use of IMS devices for liquid and solid phase targets analysis. In addition, the
success of coupling ion mobility spectrometers to various types of mass spectrometers has
further extended the IMS capabilities to a wide range of analytes, especially complex
mixtures7. The most recent applications of IMMS include proteomics8, metabolomics9 and
organic reaction monitoring10. Interfacing of an ion mobility spectrometer to a mass
spectrometer has not only expanded the application fields, but also provided value-added data
with reduced chemical noise11. Rapid sample analysis on the minute time scale, separation of
isomers and isobars, and two-dimensional identifications based on ion size-to-charge ratio
and ion mass-to-charge ratio (m/z) have made IMMS a natural fit for complex mixture
analysis. However, the inherent high Rp of atmospheric pressure IMS has been limited due to
low duty-cycle operation.
As a pulsed analytical technique, the traditional way of initiating an IMS experiment
is by pulsing open an ion gate for a short time (100 µs-200 µs), admitting a spatially confined
packet of ions into the drift region, where the ions disperse in time based on their
collision-cross-section-to-charge ratios (Ω/z)12. This “pulse-and-wait” approach only pulses
the ion gate one time within an experimental cycle, then an average of multiple experimental

61
cycles yield an ion mobility spectrum. The simplicity of the experiment provides accurate
and reproducible measurement of ion signals. However, with continuous ionization sources
such as ESI, 63Ni and corona discharge, this pulsed mode of operation results in low ion
usage because ion gate cannot be open during the time when the previous ion packet
disperses in the drift region. For a traditional IMS experiment, the duty cycle is usually <1%.
Experiments that require high Rp separation can be achieved by shortening the gate pulsed
open time to further confine the ion packet, producing an even lower duty cycle. Averaging
repetitions is commonly used to obtain a reasonable ion signal; however, the speed of data
collection is sacrificed.
This pulsed-mode constraint of conventional IMS operation does not significantly
affect its performance for target analysis since IMS devices have good selectivity for the
analyte of interest13. However, for analysis of complex mixtures, improvement in the duty
cycle would enable higher sample throughput with better IMMS sensitivity. One method to
improve the duty cycle is the use of an ion trap device before each IMS experiment to
accumulate the ions that are produced by the ionization source while the last ion packet is
disperse in the drift region; then the stored ions are pulsed into the drift region for the next
IMS separation cycle. This approach only works in laboratories that utilize low pressure IMS
devices. Hoaglud et al. 14 and Myung et al. 15 reported methods of interfacing a Paul
geometry ion trap and linear octopole ion trap between the ESI source and the IMS
experiments. With mass spectrometers employed as detector, both Paul trap and linear trap
were demonstrated to improve the duty cycle of ~200 times, at the same time, fewer
collisions resulted in low Rp for IMS separations16. Tang et al.17 and Clowers et al.18 utilized
the ion funnel trap (IFT), which is capable of operating in a higher pressure region (~1 Torr),
to accumulate ions prior to an IMS or IMMS experiment much more efficiently compared

62
with the Paul trap and linear trap, however, they also employed low pressure IMS device
(~2-4 Torr), hence a limitation for Rp of IMS separations still existed. The above studies
illustrate the feasibility of using an ion trap to improve the duty cycle and S/N of IMS or
IMMS experiment in low pressure IMS or IMMS devices. However, the technique for
continuously accumulating ions is not straightforward and it adds to costs for instrumentation.
Moreover, these above designs are not compatible with atmospheric pressure IMS since ions
cannot be effectively transferred across the high-pressure differential of the ion trap-IMS
junction.
Multiplex techniques such as Fourier Transform and Hadamard Transform can
contribute to instrument efficiency as well, with regard to sensitivity, S/N and duty cycle, and
they have been applied to a range of analytical techniques, such as spectroscopy19, mass
spectrometry20 and nuclear magnetic resonance21. Multiplexing has also been used in a
number of low-pressure IMS and IMMS devices. Mclean and Russell et al. 22 employed a
multi-pulse coding to rapidly inject ion packets into the IMS drift tube at a frequency faster
than the conventional pulsed IMS operation mode. Despite the improved IMS efficiency, the
Rp remained a problem. Koeniger et al.23 and Belov et al.24 utilized multiplexing methods to
better confine the ion packet in the IFT to improve the ion transmission and overcome the
low-Rp limitation, but the effect of thermal diffusion and Coulomb repulsion in the
high-charge-density ion packet still caused peak broadening. A commercial IMMS system
offered by Waters has increase the duty cycle to nearly 100% with a low-Rp “travelling wave”
IMMS (TWIMMS)25, and it has been applied to a various types of studies on complex
mixtures such as proteomics26. Although efforts have been made to improve the system
through three instrumentation generations27, the IMS Rp is still about ~40-50, lower than that
of the traditional apIMS (~100).

63
The application of multiplexing method to atmospheric pressure IMS (apIMS) device
was first accomplished by Knorr et al. who implemented FT into apIMS using a two-gate
IMS device28. By controlling both of the ion gates with a frequency sweeping square wave
generator, FT-apIMS interferograms can then be recorded and transformed to normal ion
mobility spectra. This implementation achieved a 3-5 times improvement for S/N and duty
cycle, but the improvement was limited because of apodization in FT. HT29 encoding with a
pseudo-random code has also been applied to apIMS by Clowers et al.30 and Szumlas et al.31
HT-apIMS reported a ~2-10-fold increase in S/N through a 50% duty cycle while still
maintain high Rp. The significance of applying multiplexing methods in apIMS is that it not
only improves duty cycle and S/N, but also maintains high Rp. However, up to now, most of
the related studies were accomplished in apIMS standalone devices using faraday plate as
detector.
While it is clear that duty cycle and S/N can be improved in low pressure IMS devices,
limitations such as inherently low Rp, peak broadening for confined ion packets, and added
instrumentation costs remain problems, which are potentially affecting the application of IMS
or IMMS devices to complex mixtures, especially when it comes to the Rp limitation. High
Rp is desirable in complex mixture analysis due to the existence of isomers and isobars, and
the demand of accurate collision-cross-section measurement for identifications12,32. The
above gaps are the driving forces for developing an analytical platform that allows rapid and
accurate detection for complex mixtures with high duty cycle and high Rp.
In this study, we evaluate an HT multiplexed atmospheric pressure ion mobility
time-of-flight mass spectrometer for complex mixtures analysis. Our hypothesis is that we
can achieve comparable sensitivity and higher Rp when compared with low-pressure IMS

64
systems. With this device, complex mixture analysis can be performed within 1-2 minutes,
with ~1-2 orders magnitudes lower LOD and 2 orders of magnitude higher sensitivity when
compared with pulsed mode analysis.

65
3.2 Theoretical Background
The Hadamard transform is an orthogonal non-sinusoidal transform with low
computational complexity. It can be thought of as a discrete sampling analog of the Fourier
transform. In some cases, such as drift tube IMS, it is better adapted to real-world
multiplexing than the Fourier transform, because it uses a finite number of basis functions of
finite length. The principles and fundamental methods are well known and will not be
repeated here29,33,34.
In HT-IMMS mode, a binary (on/off) sequence is applied to the ion gate. Because of
the nature of the HT sequences, a duty cycle of 50% is achieved. In the ideal case, the signal
increases with the duty cycle, correspondingly improving sensitivity and LODs. However,
this assumes ideal behavior of all components of the experiment. One requirement is that the
ion source be uniform and constant during the modulation period. Real sources often suffer
from fluctuations or instability, but this can usually be brought under control. More
fundamentally, any modulation errors, either due to gating or later degradation of the ion
pulses in the instrument, causes the HT signal to decrease and the baseline noise to increase.
Further degradation occurs if the signal is so low that ion counts are not uniform across all
the gate openings of the sequence. These and other factors can cause real HT data to suffer
from low signal to noise ratios, in spite of the high duty cycle.
As will be shown below, it is now feasible to keep random and systematic HT errors low
enough that the duty cycle advantage is realized in practice. In addition, HT data can be
postprocessed, much as in Fourier transform methods, to improve the quality of the final
result. This allows real improvement of both noise levels and resolution. We report results
with some postprocessing, while the details of the techniques used are presented elsewhere35.

66
3.3 Experimental Section
3.3.1 Chemicals and Reagents and Sample Preparations:
All metabolite standards were purchased from Sigma-Aldrich. Metabolite standards
include amino acids (serine, lysine), peptides (His-Ser, Gly-His-Gly) and carbohydrates
(sucrose, raffinose). Electrospray solvent comprising a 49.75:49.75:0.5 v/v/v mixture of
water, methanol and acetic acid was used as background ESI solvent and to make all standard
sample solutions. Nine standard mixture solutions were made with standard mixture solution
#1 as the initial stock solution, which contained 60 µM of serine, 50 µM of lysine, 150 µM of
His-Ser, 180 µM of Gly-His-Gly, 40 µM of sucrose and 40 µM of raffinose. Standard mixture
solutions #2-#9 were obtained by 3 times, 10 times, 30 times, 60 times, 300 times, 600 times,
3000 times and 10000 times dilution from solution #1.
NIST SRM 1950 Metabolites in Human Plasma was employed as complex mixture. It
consists of a plasma pool collected from an equal number of men and women and with a
racial distribution that reflects the U.S. population. SRM 1950 was completely sealed and
stored at -80°C until before analysis. Extraction solution was made in a glass vial with 800 µl
methanol, 50 µl water and 1 µl acetic acid and it was maintained at 60°C in a water bath. 50
µl of SRM 1950 was added into the extraction solution and kept at 60°C for 30 minutes. The
sample was then transferred to a plastic vial for centrifugation, which was operated in a
desktop centrifuge at 13k RMP and lasted for 30 minutes. All supernatant was removed into a
glass vial and evaporated by a stream of clean nitrogen to a volume of 150 µl. Striatum brain
tissue extract sample from Sprague-Dawley rat was employed as another complex mixture,
the metabolite extraction method was described previously36.

67
3.3.2 Sample Introduction:
ESI was applied through the entire study with two sample introduction methods used.
A biased voltage of 3000 volts was held constant in both sample introduction methods.
The first method was direct infusion. A syringe pump (Model#: Fusion 200) (Chemyx
Inc. TX, USA) was used to control the sample solution flow rate. Fused silica capillary with
150 µm internal diameter (Polymicro, Phoenix, AZ) was used as sample introduction loop
and electrospray source. This direct infusion method was applied to the analysis of metabolite
standard mixture and NIST 1950 SRM complex sample.
The second sample introduction method utilized an Ultra-Plus ΙΙ high performance
liquid chromatography (Micro-Tech Scientific Inc. CA, USA). Rough chromatographic
separation was performed using a C-8 (15 mm × 3 mm) 7-µm guard column (PerkinElmer
Inc. MA, USA) prior to IMMS analysis. Solvent gradient from a “more polar” and “less polar”
mobile phases consisted of 10:90 or 80:20 mixtures (v/v) of methanol and water containing
0.5% acetic acid, respectively, were applied through solvent gradient in 3 minutes, and
delivered at a flow rate of 0.2 mL/min. A 49:1 solution split was applied prior to electrospray
to maintain 4 µL/min solution flow rate for ESI. This HPLC sample introduction method was
applied in the analysis of striatum brain tissue extract sample.
3.3.3 Sample Analysis:
In the analysis of metabolite standard solutions, each solution was analyzed in both
HT-IMMS mode and pulsed-IMMS mode for three times, using sample solution flow rate of
3 µL/min, with 90 seconds data acquisition time for each analysis. SRM 1950 was also

68
analyzed in HT-IMMS mode and pulsed-IMMS mode with sample solution flow rate of 3
µL/min and data acquisition time of 2 minutes. For the analysis of striatum tissue extract, 50
µL sample solution was loaded for HPLC-HT-IMMS analysis and data was collected for 3
minutes; corresponding HT-IMMS analysis using direct infusion method was performed
afterwards for comparison purpose.
IMS standard solution of 2,6-di-tert-butyl pyridine (2,6-DtBp) was prepared and
analyzed at concentration of 2 µM for drift time calibration through the entire study.
3.3.4 HT-apIMtofMS:
As shown in Figure 3.1, a conventional stacked ring IMS constructed at Washington
State University was used as the ion mobility spectrometer. Its main parts included an
8cm-long desolvation region, a 22 cm long drift region and a Bradbury Nielson (BN) ion gate.
The BN gate was constructed using parallel Alloy 46 wires (75µm in diameter) with 0.25 mm
spacing37. Desolvation region and drift region were made up by alternating stainless steel
rings and ceramic rings, where the stainless steel rings were connected with 1 MΩ resistors to
create a uniform electric field (305 V/cm in drift region). A steady nitrogen counter current
gas flow (3 L/min) was introduced into the IMS to induce ion-neutral collisions. Ions were
desolvated in the desolvation region and gated into the drift region by the BN gate. The IMS
tube was held at a constant temperature of 200oC and operated at atmospheric pressure (920 -
940 hPa).
The BN gate pulsing signal was generated using developmental software supplied by
Tofwerk and transferred to the BN gate through a gate pulser made at Washington State
University (also shown in Figure 3.1). The gate pulsing can be easily switched between two

69
different modes: 1. Pulsed mode, when the gate only pulsed open once for 180 µs within one
IMS cycle (72 ms); 2. HT mode, when the gate pulsed open was controlled by a HT sequence,
and the gate pulsed open multiple times with different gate pulse widths within one IMS
cycle (depend on the HT sequence), in this scenario, the smallest gate pulse width is
controlled to 180 µs. The modulation sequence of HT mode was of length 1023 (210-1),
chosen to cover the desired drift time range, taking into account the mass spectrometer
repetition rate. A portion of a typical HT code sequence is shown in Figure 3.7 in the
Supplemental Material, along with typical examples of the multiplexed data and a decoded
spectrum. Neither duty cycle nor Rp depend directly on the sequence length.
IMS separated ions were then transmitted through an IMMS interface, composed of
lenses, nozzles and two segmented quadrupole ion guides, to a TOF mass spectrometer for
mass analysis, as described elsewhere38. In this study, the first segmented quadrupole was
operated with a RF of 2.05 MHz at ~3 mbar, and the second segmented quadrupole had a RF
of 1.55 MHz at ~1×10-2 mbar. TOF mass spectrometer was operated at ~3.7×10-7 mbar in
V-mode. The IMMS interface and TOF mass spectrometer were purchased from Tofwerk
(AG, Switzerland).
3.3.5 Data Acquisition and Processing:
TofDAQ Version 1.92b and IMSviewer Version 1_6d, developed by Tofwerk (AG,
Switzerland), were used for IMMS data collecting and processing. Peak lists that contain drift
time, mass/charge and intensity information for each ion species can be obtained using this
software. They are also capable of generating IMS one-dimensional plots, IMMS
two-dimensional spectra and IMMS-Intensity three-dimensional spectra.

70
Hadamard mode data is processed and inverted using developmental software from
Tofwerk35. The software uses adaptive, complementary algorithms for simultaneous
denoising and sharpening. Both functionalities operate on multiplex domain data, prior to
recovery of the normal spectra. Denoising reduces both random noise and systematic noise
arising from the non-ideal response of the gated ion signal to the encoding sequence; it is
controlled by a user-selected parameter to choose the tradeoff level between noise reduction
in noise and the accompanying loss of resolving power. Sharpening is also controlled by a
user-selected parameter that chooses the tradeoff level between sharpening and the
accompanying loss of S/N.

71
3.4 Results and Discussion
3.4.1 Improvement of HT Multiplexing:
Multiplexing of an IMS signal is expected to improve S/N relative to pulse mode by a
factor of the square root of the ratio of the multiplex duty cycle (in this case 50%) to the pulse
mode duty cycle (in this case 0.25%), or about a factor of 14 in our case. An improvement
of this magnitude has been essentially realized in the present work, but judgment of the
analytical effectiveness of HT IMS should finally be based on number of components
identified, verified, and quantified in complex mixtures. This judgment will be forthcoming
after further studies. Earlier work with HT IMS reported S/N improvements of 2-10,
compared to an expected factor of 930, and it is possible that results from the lower end of the
improvement range suffered from the type non-ideal response of a gated ion stream to
encoding that can be mitigated with the present denoise processing. Earlier work with
Fourier transform (FT) multiplexing and decoding showed S/N improvements of about half
the expected factors of 5-7, with the shortfall being the result mainly of the necessity for
apodization of the multiplex signal28,39. While improvements in S/N from multiplexing have
roughly matched expectations in the past, superior performance in the present case appears to
be the result partially of IMS improvements (especially closer gate wire spacing, longer drift
tube, and less mass dependence of ion transmission to vacuum compared to earlier
experiments) and partially of the adaptive denoise employed here35. Some of the resolving
power values we report are greater than the corresponding values for pulse mode, due mainly
to the sharpening algorithm applied to the multiplex data35. Peak positions and relative
intensities are retained in this processing, but as with other sharpening strategies, there is
always a price of reduced S/N to be paid for sharper peaks.

72
3.4.2 NIST SRM 1950 Metabolites from Human Plasma:
SRM 1950 was employed for illustrating the capability of ESI-HT-apIMtofMS for
analyzing complex mixture. Figure 3.2a shows the IMMS three-dimensional spectrum of
SRM 1950, with x-axis representing m/z (80 to 820), y-axis representing drift time (10 ms to
60 ms) and x-axis representing intensity (arbitrary unit). This spectrum was obtained in
HT-IMMS mode with 1 minute direct infusion acquisition. Figure 3.2b is the IMMS
three-dimensional spectrum (same dimensions as 3.2a) of the same sample, obtained in
conventional pulsed-IMMS mode with 1 minute direct infusion acquisition. Due to the short
data acquisition time and the inherent low duty cycle, the overall response is much lower in
2b. In order to better illustrate detections in the two modes, Figure 3.2c and 3.2d are zoomed
regions of 3.2a and 3.2b, respectively. The higher signal quality and increased sensitivity to
small peaks of HT-IMMS were evident. We were able to obtain ~180 metabolite ions in
HT-IMMS analysis while the pulsed-IMMS analysis only yielded ~80 metabolite ions.
3.4.3 Limit of Detection (LOD) and Calibration Curve:
LOD is an important feature for complex mixture analysis. It was compared between
HT-IMMS mode and pulsed-IMMS mode in this study using metabolite standard mixture
solutions. The detailed information with regard to m/z, drift time, Ko value for each
metabolite ion is listed in Table 3.1.
Table 3.2 is a summary of the absence/presence of the six metabolites ions
([Serine+H]+, [Lysine+H]+, [His-Ser+H]+, [Gly-His-Gly+H]+, [Sucrose+Na]+,
[Raffinose+Na]+) in each analysis. Ion peaks shown as “presence” had intensity (peak height)
greater than three times the noise level. With “û” representing absence and “ü” representing
presence, we can clearly see that in HT-IMMS mode analysis, [Sucrose+Na]+ ion, as the first

73
ion been detected, was firstly observed at 4 nM while the other ions were not observed at this
level. However, in pulsed-IMMS mode analysis, [Sucrose+Na]+ ion was undetected until the
concentration was increased to 67 nM. [Serine+H]+ started to show up when the
concentration level was 20 nM in HT-IMMS mode and 1 µM in pulsed-IMMS mode.
Generally speaking, all six metabolite showed the same trend in this study, they were
detected at lower concentration levels in HT-IMMS mode and at relatively higher
concentration levels in pulsed-IMMS mode. Based on these data, the LODs appeared to be
decreased ~10-50 times by the implementation of HT multiplexing signal into IMMS
analysis. The decrease in LOD could be directly related to the improved duty cycle of the
HT-IMMS mode. As described previously, in pulsed-IMMS mode, duty cycle of 0.25% was
produced; while in HT mode, the overall duty cycle was 50%. Theoretically, the sensitivity
for HT mode analysis would be improved ~200 times compared with pulsed mode; the
lowered LODs in HT-IMMS mode matched with this expectation.
Figure 3.3 includes twelve calibration curves of six metabolite ion species obtained
under HT-IMMS mode and pulsed-IMMS mode. These calibration curves were generated
using the same experimental results that correspond to Table 3.2, and plotted with x-axis as
Log(concentration) and y-axis as Log(intensity). In this plot, each data point represents an
average of three replicate analyses with error bars showing the 95% confident interval. In
each analysis, intensity values were obtained by selecting the target metabolite ion with a
mass range of m/z ± 0.1 and with a drift time range of dt ± 0.1 ms. Data points that reflect the
same metabolite ion species were labeled with the same marker type (big size markers
represent HT-IMMS mode results and small size markers represent pulsed-IMMS mode
results), calibration curves that belong to the same metabolite ion species were also plotted
with the same line style (heavy weight lines represent HT-IMMS mode results and light

74
weight lines represent pulsed-IMMS mode results). Several facts were illustrated in the figure:
1) all calibration curves showed good linearity over a 3-4 orders of magnitude of
concentration levels; 2) the overall y-value of HT mode analysis was approximately 2 units
higher than that of the pulsed mode analysis, since the y-axis represents Log(Intensity), we
can further conclude that the intensity response in HT-IMMS mode was ~2 orders of
magnitude higher than the intensity response in pulsed-IMMS mode; 3) lower LODs were
shown in HT-IMMS calibration curves. Moreover, solutions at higher concentration levels
were not applied in this study to avoid possible “overload” problem for the instrument. Hence,
the linear range in the calibration curve could be even larger than 3-4 orders of magnitude,
for both HT-IMMS mode and Pulsed-IMMS mode.
Table 3.1 also summarizes the information of the calibration curves showing in
Figure 3.2. We can observe from the table that most of the twelve calibration curves had
linear R2 value greater than 98%. The imperfect linear relationships might be due to the error
during sample preparation, moreover, these analyses were performed by electrospraying
metabolite standard mixture, hence the existence of ionization competition among different
ion species might also result in slightly non-linear intensity increase. The results also present
that [Sucrose+Na]+ and [Raffinose+Na]+ had relatively smaller slope values, indicating that
the sensitivity of these two carbohydrate ion species were slightly lower compare to the
sensitivity of amino acid and peptide ion species.
3.4.4 Comparison among Pulsed-IMMS, HT-IMMS and TWIMMS:
Figure 3.4a and 3.4b are the corresponding IMS spectra of SRM 1950 in
pulsed-IMMS mode and HT-IMMS modes, respectively, with m/z assigned to x-axis and
intensity assigned to y-axis. From these two spectra, increased number of IMS features can

75
be observed in HT-IMMS mode when compared with pulsed-IMMS mode. For example,
there are a few low abundant IMS peaks in pulsed-IMMS mode between drift time of 50 ms
and 60 ms, probably due to the low ion intensities. It is highly possible that rather than
providing IMS related information, these peaks would be ignored during data process.
However, the corresponding analysis in HT-IMMS mode clearly showed more detectable
IMS features. In addition, in spite of the higher sensitivity, higher resolution was obtained
with suitable processing. The improved performance might due to two facts. Firstly, the
decreased LOD in HT mode allowed more low concentration ions to be detected; secondly,
S/N was also increased in HT mode as a result of the increased duty cycle, and it also helped
with detecting low concentration ions that responded at a level close to noise.
The same NIST SRM 1950 sample was also analyzed using Synapt G2 TWIMMS
system (Waters, MA, USA). Sample was direct infused by ESI and analyzed for 1 minute.
Figure 4c is the corresponding TW-IMS spectrum, showing that the IMS Rp of Synapt G2
was much lower than both pulsed-IMMS and HT-IMMS mode. Note that the overall IMS
spectra obtained from the two instruments are not exactly same, this was mainly due to the
differences in ionization, IMS separation and ion transmission between these two platforms.
Figure 3.5 is the comparison of IMS spectra of the MS peak at m/z 317.12 in pulsed
mode and HT mode. Both spectra have drift time assigned as x-axis and intensity assigned as
y-axis. As can be seen, this example shows two distinguished IMS peaks for the same m/z
value, illustrating the capability of IMS for isomeric separation. In order to obtain reasonable
signal, the pulsed-IMMS mode was collected for 977 seconds while the HT-IMMS mode was
collected for only 50 seconds. With ~20 times longer collection time, pulsed-IMMS mode
still yielded spectrum with higher noise level and lower peak intensity, further proved the

76
~200 boosted sensitivity in HT-IMMS mode. Moreover, the Rp for the two mobility peaks of
m/z 317.12 were 99 and 127 in HT-IMMS mode, well improved when compared with the
result in pulsed mode. The improvement in Rp in HT-IMMS mode was primarily due to the
effect of sharpening in postprocessing35. The postprocessing allowed improvement of Rp,
while peak positions and relative intensities were retained.
3.4.5 Striatum Tissue Extract Analysis using HPLC coupled HT-IMMS:
With the aim of improving the ability of HT-IMMS for application to complex
mixtures, HPLC was employed for an additional low-resolution chromatographic separation
prior to HT-IMMS analysis. Figure 3.6a shows the 3-D IMMS-Intensity plot of striatum
tissue extract fluid analyzed by HPLC coupled HT IMMS method. 3-D IMMS-Intensity plot
has m/z assigned on x-axis, drift time assigned on y-axis and intensity assigned on z axis,
illustrating a clear overall data presentation. Data in this plot was obtained by a three-minute
HT-IMMS analysis of a continuous HPLC elution of sample fluid. Figure 3.6b is the
corresponding 3-D IMMS-Intensity plot generated by HT-IMMS method alone, without prior
chromatographic separation. Data in this plot was obtained by a three-minute direct infusion
HT-IMMS analysis of the same sample fluid after 16 times dilution by ESI solvent. The 16
times dilution factor was calculated and applied with the consideration of a fair comparison
between two analyses. Figure 3.6c and 3.6d are the 2-D IMMS plots generated from 3.6a and
6b, respectively. As shown by the highlighted red circles in Figure 3.6a and 3.6c, ~20 more
metabolites ions, mostly between m/z 600 to 1000, can be observed with the extended
application of HPLC when compared with Figure 3.6b and 3.6d. Moreover, the HPLC
coupled HT-IMMS analysis results showed less interference from noise and possible
contamination.

77
Overall speaking, adding a pre-chromatographic separation by HPLC further
improved the performance of the HT-IMMS analysis for complex mixtures, mainly due to
two factors: 1) with polar metabolites eluting faster than nonpolar metabolites through the
C-8 column, a rough HPLC separation was achieved, hence the HT-IMMS response for
nonpolar metabolites was improved without the presence of polar metabolites; 2) with the
prior HPLC separation, the metabolites in the sample fluid were batch-separated and
well-distributed in retention time, hence the overall HT-IMMS response was stronger with
regard to the metabolite ions with wide concentration levels, and weaker with regard to noise
and possibly existed contaminations.

78
3.5 Conclusions
HT-apIMtofMS has been demonstrated on complex mixtures. The 2 orders
improvement of duty cycle compared to conventional pulsed mode IMMS was achieved,
while retaining high Rp. Initial results using human plasma standard SRM-1950 showed that
the increased sensitivity allowed substantially better coverage of compounds in the mixture,
in a short measurement time. The HT-IMMS mode was shown to provide a linear response
over at least 4 orders of magnitude, for six representative metabolites. The LODs of these
analytes were also estimated to be 1-2 orders of magnitude lower in multiplexed mode,
compared to conventional. Adding a fast chromatography separation before the IMS aided in
reducing ion suppression effects, which resulted in better coverage of metabolites in a
striatum extract, particularly at high m/z range. The developed HPLC coupled
HT-apIMtofMS instrument represents a high sensitivity, high throughput analytical platform,
that can be employed for various types of applications.
Acknowledgement
This project was supported in part by funds provided for medical and biological
research by the State of Washington Initiative Measure No.171.

79
Reference
(1) Hill, H. H., Jr.; Siems, W. F.; Louis, R. H. S.; McMinn, D. G. Anal. Chem. 1990, 62,
1201A–1209A.
(2) Ewing, R. G.; Atkinson, D. A.; Eiceman, G. A.; Ewing, G. J. Talanta 2001.
(3) Fernández-Maestre, R.; H, H. H. Int. J. Ion Mobil. Spec. 2009, 12, 91–102.
(4) Steiner, W. E.; Clowers, B. H.; Haigh, P. E.; H, H. H. Anal. Chem. 2003, 75, 6068–
6076.
(5) Shumate, C. B.; H, H. H. Anal. Chem. 1989, 61, 601–606.
(6) Gillig, K. J.; Ruotolo, B.; Stone, E. G.; Russell, D. H.; Fuhrer, K.; Gonin, M.;
Schultz, A. J. Anal. Chem. 2000, 72, 3965–3971.
(7) Kanu, A. B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H. H., Jr. J. Mass Spectrom. 2008,
43, 1–22.
(8) McLean, J. A.; Ruotolo, B. T.; Gillig, K. J.; Russell, D. H. Experimental Neurology
2005, 240, 301–315.
(9) Dwivedi, P.; Wu, P.; Klopsch, S. J.; Puzon, G. J.; Xun, L.; Hill, H. H., Jr.
Metabolomics 2007, 4, 63–80.
(10) Roscioli, K. M.; Zhang, X.; Li, S. X.; Goetz, G. H. International Journal of Mass
Spectrometry 2012.
(11) Kanu, A. B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H. H., Jr. J. Mass Spectrom. 2008,
43, 1–22.
(12) Wu, C.; Siems, W. F.; Klasmeier, J.; H, H. H. Anal. Chem. 2000, 72, 391–395.
(13) Eiceman, G. A.; Yuan-Feng, W.; Garcia-Gonzalez, L.; Harden, C. S.; Shoff, D. B.
Analytica Chimica Acta 1995, 306, 21–33.
(14) Hoaglund, C. S.; Valentine, S. J.; Clemmer, D. E. Anal. Chem. 1997, 69, 4156–4161.
(15) Myung, S.; Lee, Y. J.; Moon, M. H.; Taraszka, J.; Sowell, R.; Koeniger, S.;

80
Hilderbrand, A. E.; Valentine, S. J.; Cherbas, L.; Cherbas, P.; Kaufmann, T. C.;
Miller, D. F.; Mechref, Y.; Novotny, M. V.; Ewing, M. A.; Sporleder, C. R.;
Clemmer, D. E. Anal. Chem. 2003, 75, 5137–5145.
(16) Eiceman, G. A.; Karpas, Z. Ion Mobility Spectrometry. Book. 2010.
(17) Tang, K.; Shvartsburg, A. A.; Lee, H. N.; Prior, D. C.; Buschbach, M. A.; Li, F.;
Tolmachev, A. V.; Anderson, G. A.; Smith, R. D. Anal. Chem. 2005, 77, 3330–3339.
(18) Clowers, B. H.; Ibrahim, Y. M.; Prior, D. C.; Danielson, W. F.; Belov, M. E.; Smith,
R. D. Anal. Chem. 2008, 80, 612–623.
(19) Chase, D. B. Anal. Chem. 1986, 108, 7485–7488.
(20) Zare, R. N.; Fernández, F. M.; Kimmel, J. R. Angew. Chem. Int. Ed. 2003, 42, 30–35.
(21) Morris, G. A.; Freeman, R. Journal of Magnetic Resonance (1969) 1978.
(22) McLean, J. A.; Russell, D. H. Int J Ion Mobil Spectrom 2005, 8, 66–71.
(23) Koeniger, S. L.; Bohrer, B. C.; Valentine, S. J.; Clemmer, D. E. Anal. Chem. 2008,
80, 1918–1927.
(24) Belov, M. E.; Clowers, B. H.; Prior, D. C.; Danielson, W. F., III; Liyu, A. V.; Petritis,
B. O.; Smith, R. D. Anal. Chem. 2008, 80, 5873–5883.
(25) Shvartsburg, A. A.; Smith, R. D. Anal. Chem. 2008, 80, 9689–9699.
(26) Thalassinos, K.; Grabenauer, M.; Slade, S. E.; Hilton, G. R.; Bowers, M. T.; Scrivens,
J. H. Anal. Chem. 2008, 81, 248–254.
(27) Giles, K.; Williams, J. P.; Campuzano, I. Rapid Commun. Mass Spectrom. 2011, 25,
1559–1566.
(28) Knorr, F. J.; Eatherton, R. L.; Siems, W. F.; Hill, H. H. Anal. Chem. 1985, 57, 402–
406.
(29) Treado, P. J.; Morris, M. D. Anal. Chem. 1989, 61, 723A–734A.
(30) Clowers, B. H.; Siems, W. F.; H, H. H.; Massick, S. M. Anal. Chem. 2006, 78, 44–

81
51.
(31) Szumlas, A. W.; Ray, S. J.; Hieftje, G. M. Anal. Chem. 2006, 78, 4474–4481.
(32) Wu, C.; Siems, W. F.; Asbury, G. R.; H, H. H. Anal. Chem. 1998, 70, 4929–4938.
(33) Kaneta, T.; Yamaguchi, Y.; Imasaka, T. Anal. Chem. 1999, 71, 5444–5446.
(34) Decker, J. A. Anal. Chem. 1972, 44, 127A–134a.
(35) Knochenmuss, R;, Graf, S.; Fuhrer, K.; Gonin, M. 61th ASMS Conference on Mass
Spectrometry and Allied Topics. 2013, WP 745
(36) Kaplan, K. A.; Chiu, V. M.; Lukus, P. A.; Zhang, X.; Siems, W. F.; Schenk, J. O.; H,
H. H. Anal Bioanal Chem 2013, 405, 1959–1968.
(37) Kimmel, J. R.; Engelke, F.; Zare, R. N. Review of scientific Instruments, 2001.
(38) Kaplan, K.; Graf, S.; Tanner, C.; Gonin, M.; Fuhrer, K.; Knochenmuss, R.; Dwivedi,
P.; Hill, H. H., Jr. Anal. Chem. 2010, 82, 9336–9343.
(39) Louis, R. S.; Siems, W. F.; Hill, H. H., Jr Anal. Chem. 1992, 64, 171-177.

82
Table 3.1 A table of the m/z, drift time, reduced mobility (Ko) value and calibration curves
for each metabolite ion. Obtained from the analysis of metabolite standard mixture solutions.
Metabolite Ions m/z Drift time (ms) Ko (cm2V-1s-1) HT Mode Calibration Curve Pulsed Mode Calibration Curve
[Serine+H]+ 106.05 18.82 1.89y = 0.79x + 0.25
R² = 0.998y = 0.77x - 1.81
R² = 0.972
[Lysine+H]+ 147.11 21.32 1.67y = 0.86x + 0.09
R² = 0.999y = 0.97x - 2.56
R² = 0.994
[His-Ser+H]+ 243.11 26.81 1.33y = 0.89x + 0.43
R² = 0.995y = 0.87x - 1.69
R² = 0.997
[Gly-His-Gly+H]+ 270.12 28.40 1.25y = 0.80x + 0.32
R² = 0.994y = 0.94x - 2.53
R² = 0.989
[Sucrose+Na]+ 365.11 31.24 1.14y = 0.57x + 1.37
R² = 0.992y = 0.66x - 1.03
R² = 0.966
[Raffinose+Na]+ 527.16 39.30 0.91y = 0.76x + 0.60
R² = 0.991y = 0.81x - 1.73
R² = 0.975

83
Table 3.2 A summary of the absence/presence of six metabolite ions during the analysis of
standard mixture solutions in both HT-IMMS mode and pulsed-IMMS mode. The
concentration of each metabolite is shown. “û” represents absence and “ü” represents
presence.
Note: Concentration values are not accurate due to the fact that the initial stock solutions were prepared
with approximate concentration levels (±5%).
[Serine+H]+ HT-IMMS Pulsed-IMMS [Lysine+H]+ HT-IMMS Pulsed-IMMS6 nM û û 5 nM û û
20 nM ü û 17 nM û û
100 nM ü û 83 nM ü û
200 nM ü û 170 nM ü û
1 µM ü ü 830 nM ü ü
2 µM ü ü 1.7 µM ü ü
6 µM ü ü 5 µM ü ü
20 µM ü ü 17 µM ü ü
60 µM ü ü 50 µM ü ü
[His-Ser+H]+ HT-IMMS Pulsed-IMMS [Gly-His-Gly+H]+ HT-IMMS Pulsed-IMMS15 nM û û 18 nM û û50 nM ü û 60 nM ü û
250 nM ü ü 300 nM ü û500 nM ü ü 600 nM ü ü2.5 µM ü ü 3 µM ü ü5 µM ü ü 6 µM ü ü
15 µM ü ü 18 µM ü ü50 µM ü ü 60 µM ü ü
150 µM ü ü 180 µM ü ü
[Sucrose+Na]+ HT-IMMS Pulsed-IMMS [Raffinose+Na]+ HT-IMMS Pulsed-IMMS4 nM ü û 4 nM û û
13 nM ü û 13 nM û û67 nM ü ü 67 nM ü û
130 nM ü ü 130 nM ü û670 nM ü ü 670 nM ü ü1.3 µM ü ü 1.3 µM ü ü4 µM ü ü 4 µM ü ü
13 µM ü ü 13 µM ü ü40 µM ü ü 40 µM ü ü

84
Figure 3.1 Schematic diagram of the prototype: Electrospray ionization Hadamard transform
atmospheric pressure ion mobility time-of-flight mass spectrometer. Stacked ring IMS was
coupled to the TOFMS by an IMMS interface.

85
Figure 3.2 IMMS 3-dimensional spectra of NIST SRM 1950 sample. (a) and (b) show the 3D
spectra obtained in pulsed-IMMS mode and HT-IMMS mode, respectively, with x-axis
representing m/z (100 - 800) and y-axis representing drift time (10 ms – 60 ms). (c) and (d)
are the zoomed-in 3D spectra generated from (a) and (b), with m/z range from 500 to 700 and
drift time range from 42 ms to 58 ms. More components can be observed in HT mode
analysis when compared with pulsed mode analysis.

86
Figure 3.3 Twelve calibration curves of the six metabolite ion species under HT-IMMS mode
and pulsed-IMMS mode. Calibration curves are plotted with x-axis assigned with
Log(concentration) (nM) and y-axis assigned with Log(Intensity) (a.u.). Each data point is an
average of three replicate analysis and the error bars were obtained with 95% confidence
interval.
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
1 1.5 2 2.5 3 3.5 4 4.5 5 5.5 6
HT [Serine+H]+
HT [Lysine+H]+
HT [His-Ser+H]+
HT [Gly-His-Gly+H]+
HT [Sucrose+Na]+
HT [Raffinose+Na]+
Pulsed [Serine+H]+
Pulsed [Lysine+H]+
Pulsed [His-Ser+H]+
Pulsed [Gly-His-Gly+H]+
Pulsed [Sucrose+Na]+
Pulsed [Raffinose+Na]+
Linear (HT [Serine+H]+)
Linear (HT [Lysine+H]+)
Linear (HT [His-Ser+H]+)
Linear (HT [Gly-His-Gly+H]+)
Linear (HT [Sucrose+Na]+)
Linear (HT [Raffinose+Na]+)
Linear (Pulsed [Serine+H]+ )
Linear (Pulsed [Lysine+H]+)
Linear (Pulsed [His-Ser+H]+)
Linear (Pulsed [Gly-His-Gly+H]+)
Linear (Pulsed [Sucrose+Na]+)
Linear (Pulsed [Raffinose+Na]+)
Log
(Int
ensit
y), a
.u.
Log (Concentration), nM

87
Figure 3.4 IMS spectra of NIST SRM 1950 sample. (a) and (b) are the IMS spectra obtained
in pulsed-IMMS mode and HT-IMMS mode, respectively. With x-axis assigned with drift
time and y-axis assigned with intensity in arbitrary unit. (c) presents the IMS spectrum
obtained from Synapt G2 TWIMMS system.

88
Figure 3.5 Comparison of mobility traces of the MS peak m/z 317.12, in pulsed-IMMS mode
(upper) and HT-IMMS mode (lower). X-axis represents drift time in ms and y-axis represents
peak intensity in counts. To obtain reasonable signal, the pulsed mode measurement was
collected for 977 seconds and the HT mode measurement was collected for 50 seconds.
Resolving power for each mobility peak is also labeled in the figure.

89
Figure 3.6 (a) and (b) are the 3-D IMMS-Intensity plots of striatum tissue extract fluid
analyzed by HPLC coupled HT-IMMS and direct infuse HT-IMMS, respectively. They have
m/z (80 - 1000) assigned on x-axis, drift time (10 ms - 60 ms) assigned on y-axis and
intensity assigned on z-axis. (c) and (d) are the corresponding 2-D IMMS plots generated
from (a) and (b). Red circles in (a) and (c) represent that ~20 metabolite ions that were
successfully detected with the help of prior chromatographic separation by HPLC.

90
3.6 Supplementary Materials
Figure 3.7 A portion of a typical HT code sequence (top trace), extracted from HT-IMMS
mode analysis of NIST 1950 sample at m/z = 132.10, along with multiplexed data (middle
trace) and decoded spectrum (down trace).

91
Chapter 4
Strategies for Metabolite Identification in Human Blood
Metabolome using Electrospray Ionization Hadamard Transform
Ion Mobility Time-of-Flight Mass Spectrometry (HT-IMtofMS)
Abstract
With the development of Hadamard transform ion mobility time-of-flight mass spectrometry
(HT-IMtofMS), it is now possible to rapidly measure the metabolites in complex biological
mixtures with high ion mobility resolving powers. However, known metabolites are typically
a small portion of the data. This study investigated various ion mobility mass spectrometric
strategies for the identification of metabolites in human blood metabolome. This study
demonstrates that IMMS can provide useful structural information not possible by other
analytical methods, therefore, increased confidence in metabolite identification. Hadamard
Transform ion mobility mass spectrometry (HT-IMMS) was employed with its capability of
high throughput analysis in 3 minutes and high resolving power separation. Analytical
strategies used for identification in this work include: accurate mass analysis, accurate
mobility analysis, isotopic ration analysis, charge-state analysis, compound class
determination by mobility-mas correlations and isomer/isobar analysis. NIST SRM 1950
(metabolites in human plasma) sample and human whole blood samples were analyzed
individually, with over one thousand metabolite features detected for each sample.
Approximately 250 of the major metabolites from these blood samples were processed for
identification and 185 of them were identified. The reduced mobility (K0) values and adduct

92
forms for each of these identified metabolites are reported and can be used to improve current
metabolome databases.

93
4.1 Introduction
Metabolomics1 is the science of characterizing metabolites in biological samples
(such as fluids, cell and tissue extracts), offering direct measurement of the metabolic state.
As one of the “omics” platform, it is an important and integral part of systems biology2.
Metabolite detection in complex biological mixtures is a difficult and time consuming
process, usually requiring comprehensive extractions, high-resolution chromatographic
separations, and accurate mass and/or MSMS spectrometry. Comprehensive metabolite
identification in complex biological samples is also a challenging process due to the large
number of possible candidates for each metabolite feature. In addition, the chemical and
physical diversity of metabolites complicate the analysis3.
The recent development of ultra-high resolution mass spectrometers such as FTICR4
and Orbitrap5 has helped exact-mass analysis since isobars can be generally distinguished. By
coupling with chromatographic separations6 (mostly gas chromatography (GC) and liquid
chromatography (LC)), MS-based methods yield fairly comprehensive analysis. However,
one major limitations is low separation efficiency due to pre-column sample derivatization7
and long chromatographic separation time8; another problem is the lack of structural analysis
for isomers by mass spectrometer alone, and with the help with chromatographic separation,
the time it takes for isomeric separation decreases sample throughput. A high throughput
analytical technique capable of rapid analysis with structural information would significantly
improve current analytical methodology for comprehensive metabolite detection.
Chromatography has limited capability for metabolite identifications9,10. Its major
advantage is in the pre-separation of complex mixtures prior to mass spectrometry analysis.

94
Although the distribution of analytes between the stationary and mobile phases do reflect
physical and chemical properties such as polarity in liquid chromatography, vapor pressure in
gas chromatography, and charge density in electrophoresis, the retention behavior of a certain
compound does not directly reflect intrinsic structural information that can be used for
identification. Moreover, any slight change in chromatographic conditions such as
temperature, mobility phase flow, or stationary phase activity, can induce shift in the
analyte’s retention time that cannot be calibrated11,12.
Recently there has been considerable interest in using ion mobility spectrometry (IMS)
as a separation technique prior to mass spectrometry. While IMS has a number of advantages
for interfacing with mass spectrometry such as speed, sensitivity, and selectivity, its primary
advantage is that its separation mechanism relies on the intrinsic structure of the analyte ion
(collision cross section) that can be used to aid in identification of unknown metabolites.
Today there are three commercial platforms using ion mobility mass spectrometry (IMMS):
the traveling wave ion mobility–TOF mass spectrometer (TWIMMS)13 by Waters
(Manchester, UK), the low pressure drift tube ion mobility–TOF mass spectrometer
(LP-DTIMMS) by Agilent (CA, USA)14, and the ambient pressure drift tube ion mobility–
TOF mass spectrometer (AP-DTIMMS) by TofWerk15 (AG, Switzerland). All of these
instruments are capable of measuring collision cross sections of metabolite ions. With the
advent of these measurement technologies, IMMS database is needed along with analytical
strategies for using both mass and mobility data to identify metabolites.
Applications of IMMS have been demonstrated for a number of complex biological
samples including metabolomics samples16 and samples containing large protein complexes17.

95
Human blood, in particular, offers an obvious target for the diagnoses of diseases through
metabolomics. Inborn errors in the genetic code have been diagnosed using metabolomics
since the 1980’s18. Other diseases such as type II diabetes19 and cardiovascular disease20 have
been diagnosed using metabolic profiling of blood samples. Recent interests are developing
on a wide range of other diseases including digestive21, metal,22 and pregnancy disorders23
have been investigated by metabolomics of blood samples.
Metabolite identifications are mainly based on the m/z information using metabolite
databases. Public databases have been developed over the past decade, among which, the
Human Metabolome Database (HMDB)24 and METLIN25 are two major databases containing
analytical and molecular information about human metabolites. HMDB aids metabolite
identification based on NMR and mass spectra. It provides information including metabolite
abundance, biological functions, and spectral viewing tools for a large number of metabolites.
METLIN is another online database focused on MS-based data, including comprehensive
MS/MS information, an annotated list of known metabolites and their chemical structures.
These databases remain under development due to the fact that whole human blood
metabolome is not complete. In addition, these databases lack structural information such as
reduced mobility values or collision cross section values, therefore, cannot distinguish
isomers.
The goal of this work is to produce mobility data for metabolites in human blood that
can be used in establishing IMMS database for metabolites, and to demonstrate analytical
strategies in which mobility and mass information can be combined to facilitate metabolite
identifications.

96
4.2 Experimental Section
4.2.1 Sample Preparation:
SRM 1950 Metabolites in Human Plasma was provide by National Institute of
Standards and Technology (NIST). It consisted a plasma pool collected from an equal
number of men and women and with a racial distribution that reflects the U.S. population.
NIST SRM 1950 was sealed and stored at -80°C. A volume of 50 µL of SRM 1950 was
added into extraction solution (800 µL of HPLC grade methanol and 50 µL of HPLC grade
water). The mixture was kept at 60°C in water bath for 30 minutes, and then centrifuged at
13k RMP for 30 minutes. All supernatant was removed into a glass vial and evaporated by a
stream of clean nitrogen to a volume of 150 µL.
Human blood sample was collected from fingertip cleaned with a 70% alcohol pad
and air-dried. A sterile lancet was then used to prick the finger pad. Blood drops were
collected in a glass vial26. 900 µL HPLC grade methanol was used as extraction solution for
metabolite extraction. 50 µL blood was mixed with the extraction solution and kept at 60°C
in water bath for 30 minutes. The sample was then centrifuged at 13k RMP for 30 minutes.
Supernatant was further diluted with 1:4 v/v ratio of supernatant and electrospray solvent
(49.95:49.95:0.1 v/v/v of HPLC grade water, methanol and formic acid).
4.2.2 ESI-HT-IMtofMS Analysis:
Electrospray (ESI): Sample solution was introduced for IMMS analysis by a syringe
pump (model Fusion 200) (Chemyx Inc. TX). Fused silica capillary (Polymicro Inc. AZ) with
internal diameter of 150 µM was used as the electrospray source and sample introduction
loop. A biased voltage of 3000 volts and a sample solution flow rate of 3 µL/min were held

97
constant during the experiment.
Hadamard Transform ion mobility time-of-flight mass spectrometry (HT-IMtofMS)27:
A conventional stacked ring ion mobility spectrometer constructed in-house at Washington
State University, consisting of an 8cm long dissolvation region, a 22 cm long drift region and
a Bradbury-Nielsen (BN) ion gate. Hadamard transform signal was imposed on the BN ion
gate through a gate controller, allowing ion gate to open multiple times during an IMS cycle
to increase the throughput of the IMMS analysis. Both the dissolvation region and the drift
region were constructed using alternating stainless steel conducting rings and ceramic
insulating rings, where the conducting rings were connected with 1 MΩ resistors to create a
uniform electric field (305 V/cm in drift region). A nitrogen drift gas flow was introduced
into the IMS with a flow rate of 2.5 L/min. The IMS tube was held at a constant temperature
of 220°C and operated at ambient pressure (690-705 Torr). Ions were transmitted into a
time-of-flight mass spectrometer (tofMS) through an IMMS interface after IMS separation.
Both IMMS interface and tofMS were purchased from Tofwerk (Thun, Switzerland) and
operational details were descried elsewhere15. Note that the time-of-flight mass spectrometer
was operated in V-mode with a resolution of 5000-7000. The data acquisition software
(TofDaq) and processing software (IMSveiwer1_6d) were also provided by Tofwerk (Thun,
Switzerland).
ESI background spectra were collected using electrospray solvent before each sample
analysis and used for background extraction. Mobility calibration was conducted using
2,6-di-tert-butylpyridine (Sigma-Aldrich). Sodium ion and polydimethylsiloxane ion were
used for internal mass calibration. Each ESI-HT-IMMS analysis lasted for 2 - 3 minutes.

98
4.2.3 Multidimensional Data from ESI-HT-IMtofMS Analysis:
Data collected from ESI-HT-IMtofMS analysis produced a peak list containing
thousands of features characterized by drift time (td), mass-to-charge ratio (m/z) and intensity.
Drift time was recorded as the time required for an ion to travel through the IMS drift region.
It was then used to generate the mobility (K) of an ion species as shown in (1). Reduced
mobility constant shown in (2) remains constant for a given drift gas and E/N, since it
incorporates the pressure and temperature of the drift region.
Where V (volts) is the voltage drop across the IMS drift region with length L (cm), td is drift
time (s). P is the pressure (Torr) and T is the absolute temperature.
Besides reduced mobility values, collision cross-sections (Ω)14 were also obtained.
Collision cross section value reflects ionic size, which complements m/z information
provided by mass spectrometer. By combining m/z and Ω, we were able to assess ionic
density and compound classification. The derivitization of Ω is shown in (3), where e is the
charge on the ion, N is the drift gas number density, K is the mobility, k is Boltzmann’s
constant, T is the absolute temperature, and µ is the reduced mass (4) for the ion (m) and
neutral drift gas (M).
! = ! !!
!!! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!(1)
!!!!!!!!!! =!!!!! !× !
273.15! !× ! !760 !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!(2)
Ω! = !3!
16!"2!!"# !!!!!!!!!!!!!!!!!!!!!(3)

99
Mobility mass correlation is based on a linear relationship between ionic mass (m/z) and
ionic size (Ω/z) 28,29. A specific class of compounds will provide a mobility mass correlation
with unique slope (a) and intercept (b), as shown in (5).
4.2.4 Data Processing for Metabolite Identifications:
Matching with NIST SRM 1950: Peak list generated from NIST SRM 1950 was used
to match with the peak list generated from human blood sample. The metabolite features
detected reproducibly in both samples were identified by matching with the NIST certificate
of analysis (COA) database30.
Matching with public metabolome databases: Human Metabolomes Database and
METLIN Metabolomics Database were applied in this study. The criteria for metabolite
identification included exact m/z obtained from the peak list, a mass accuracy of 10 – 30 ppm,
and adduct forms of [M+H]+, [M+Na]+, [M+K]+ and [M+H-H2O]+. By the above simple and
tentative identification procedure based on m/z, there could be multiple metabolite candidates
matched for one m/z. Therefore, further identification verification was necessary.
Identification based on mobility information: 1) isomeric separations were displayed
!! = ! !"! +! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!(4)
Ω! = ! !
! !+ !!!!!!!!!!!!!!!!!!!(5)

100
using selected-mass mobility analysis by showing multiple mobility peaks for the same m/z;
2) accurate isotopic ratio analysis was performed by measuring the peak areas of the ion
mobility peaks for targeted metabolite and its isotopes; 3) mobility mass correlation curves
were used to identify the classifications of unknown metabolite features; 4) protein ions with
different charge states formed a trend line which was used for calculating the molecular
weight of the protein, therefore, facilitated identification.

101
4.3 Results and Discussion
4.3.1 Metabolite Detection and Separation by ESI-HT-IMMS:
Figure 4.2 illustrates the mass and mobility information obtained from human blood
metabolome. 4.2a shows a typical 3-dimensional spectrum with x-axis representing m/z 100 -
1100, y-axis as drift time ranges from 15 ms to 85 ms, and z-axis as intensity; 4.2b and 4.2c
are the corresponding mass spectrum and ion mobility spectrum. Around ~1000 IMMS
features were detected and assigned with multidimensional information including m/z, drift
time and intensity. The 5000 - 7000 resolution of the TOF mass spectrometer allowed it to
distinguish metabolite features with m/z difference > 0.1. The resolving power of IMS was
80 - 120, sufficient to distinguish isomers and isobars that have small structural difference.
IMMS combined the separation power from both MS and IMS, allowing more metabolite
features to be detected when compared to MS analysis alone. Besides acting as size
separation apparatus, ion mobility spectrometer also prevented unwanted neutral molecules
and containments from entering the mass spectrometer. Neutral molecules especially solvent
molecules generated from electrospray ionization were kept away from the mass
spectrometer by the N2 buffer gas in IMS, therefore, MS can detect metabolite ions that are
completely desolvated. Moreover, IMS spread the noise from MS, efficiently reduced the
noise and increased the signal to noise ratio. Therefore, the overall analysis by
ESI-HT-IMMS was performed with improved signal to noise ratio when compared to using
MS alone.
4.3.2 Metabolite Identification using Databases:
Figure 4.1b and 4.1c display the major metabolite features detected in human blood
sample (250 metabolite features with m/z range from 100 to 1200) and NIST Plasma sample

102
(200 metabolite features with m/z range from 100 to 800), respectively. ~60 metabolite
features were reproducibly detected in both samples. These metabolite features were firstly
matched with the NIST COA database for identification. However, only 12 metabolite
features were identified. The reasons for the low yield of identification are: 1) The NIST
COA database30 contains only 110 metabolites, therefore, there are hundreds of metabolite
features remain unknown and/or not included in the NIST COA database; 2) The database
collected results from 52 different studies, therefore, it is not possible for one single study to
yield all the information. The results described above demonstrated that HT-IMMS analysis
was able to detected hundreds of metabolites in a few minutes, however, the identification
using NIST COA database alone was not sufficient.
Public metabolome databases HMDB and METLIN were then used for the
identifications of the metabolite features in human blood metabolome that were not identified
using the NIST COA database. Since the identification using HMDB and METLIN was
based on m/z only, there were often multiple candidate metabolites for each metabolite
feature. Therefore, reduced mobility information provided by HT-IMMS analysis was
coupled with m/z information to improve the identification procedure. Table 4.1 provides the
identification of 185 metabolite features using the above three databases, as well as using the
reduced mobility information. The measured m/z values, reduced mobility (K0) values, peak
heights, adduct forms, and identifications were reported.
4.3.3 Metabolite Identification using Mass and Mobility information -- Isotopic ratio
Analysis:
Natural isotopic pattern provides important information for compound identification.

103
Isotopic ratio analysis can be operated by mass spectrometer alone if targeted compounds
were known and without interferences. However, for the comprehensive detection of a whole
metabolome, it is difficult to obtain isotope measurement because the frequent occurrence of
multiple peaks within one mass unit. When ion mobility separation applied, isotopes that
belong to the same metabolite feature appear at same drift time because their size differences
are two small to be separated by IMS. Therefore, selected-mass mobility analysis can rapidly
distinguish the isotopes of a given metabolite feature by filtering out interference. Figure 4.3
shows an example of isotopic ratio analysis using IMMS. 4.3a is the mass spectra with x-axis
as mass range from 103.5 to 106.5 and y-axis as intensity. Three peaks with different m/z
values (104.11, 105.00 and 105.11) were shown in the mass spectra, however, it is difficult to
determine the existence of isotope pattern only by mass spectrometry because that each
individual mass peak could represent different metabolite features in the complex human
blood metabolome. 4.3b is the mass selected mobility spectra of m/z = 104.11 (blue), m/z =
105.00 (red) and m/z = 105.11 (green). The mobility peak of m/z 104.11 and m/z 105.11
showed up at the same drift time of 22.52 ms, demonstrating that m/z 105.11 was an isotope
of 104.11. Therefore, the peak area ratio of 104.11 and 105.11 (calculated to be 100: 5.5) was
used for the identification of metabolite feature with m/z 104.11. This metabolite feature was
identified as choline ion [C5H14NO]+. Isotope ratio analysis was observed and applied for
the identification of 8 metabolite features, including amino acid, peptides, fatty acids and
lipid. Table 4.2 shows the isotopic ratio analysis results including measured m/z, drift time,
K0, isotopic ratios, identified chemical formula proposed metabolite identification, and
adduct form.

104
4.3.4 Metabolite Identification using Mass and Mobility information – Isomers and Isobars
separation:
The existence of isomers and isobars in metabolomes has been a challenge for
metabolite detection and identification. Isomeric separation can be achieved easily and
rapidly with IMS. Isobars can be separated using high-resolution mass spectrometer alone,
however, with the application of IMS, they can be separated with low-resolution mass
spectrometer as well. The TOF mass spectrometer employed in this study was operated a
resolution of ~500 - 7000, which was not capable of separating isomers and isobars with m/z
differences less than 0.01. Figure 4.4 illustrates an example of IMS facilitating MS for
isobars separation. 4.4a is the zoomed-in mass spectrum with m/z ranges from 115.0 to 115.6.
It clearly detected one mass peak with broad peak width, which indicated the possibility of
isobars existence, however, the mass spectrometer was unable to distinguish between the
isobars. 4.4b shows the selected mass ion mobility spectrum of the targeted mass peak. With
two mobility peaks detected by IMS, confirming the existence of isobars. The identification
of the mobility peak at 22.6 ms was [N-mononitrosopiperazine+H]+ with m/z of 115.08 K0 of
2.08, and the later mobility peak at 23.6 ms was identified as [Gamma-caprolactone+H]+ with
m/z of 115.07 and K0 of 1.99.
5 pairs of isomers/isobars were detected by IMMS, and Table 4.3 summarized the
information for each metabolite feature including measured m/z, reduced drift time, K0,
metabolite identification, adduct form. The enhanced separation power using IMS also
demonstrated that the addition of IMS can increase peak capacity.

105
4.3.5 Metabolite Identification using Mass and Mobility information – Charge States
Separation:
Ion charge states separation has been reported as another unique merit from IMMS
analysis. Mass spectrometry can differentiate charge states based on the m/z values, while
IMMS can separate and present them into the two dimensional IMMS space. Figure 4.5 is
the IMMS two-dimensional spectra of a protein detected with charge states from +13 to +17.
As shown in the spectra, a trend line was formed with good linearity and increasing m/z
difference (Δm/z) between two adjunct charge states. The different Δm/z values were used
for the identification of hemoglobin and its charge states.
The detailed information of these protein features is listed in Table 4.1, from m/z
895.51 to 1173.91. It is clear that the overall reduced mobility values for hemoglobin ion
species were smaller when compared with the other metabolite features, demonstrating its
compact ionic structure. Moreover, multiple mass peaks were detected within each charge
state, showing the complex isotopic pattern of protein.
4.3.6 Metabolite Identification using Mass and Mobility information – Mobility-mass
Correlations:
The collision cross section to charge ratio (Ω/z) is a property measured by IMS and
directly related to the ionic size and structure. Ω/z can be helpful in identifying an ion
analogously to m/z. Moreover, in the IMMS 2-dimensional space, Specific compound class
tends to form unique mobility-mass correlation, providing class identifications not possible
by any other methods14,31.

106
In the IMMS spectrum, these mobility-mass correlations appear as trend lines with
specific slopes and intercepts that can also be used to provide qualitative information for the
identification of unknowns. Figure 4.6 shows the IMMS 2-dimensional spectra, illustrating
the classification trend lines detected in human blood metabolome. The x-axis represents m/z
ranged from 100 to 1100 and y-axis represents drift time ranged from 15 ms to 90 ms. Five
classification trend lines for amino acids, peptides, and phospholipids were identified and
plotted. Metabolite features fell along a certain trend line were either belonged to that class or
shared structural similarities to that classification of compounds.
In order to better explain the mobility-mass correlations, linear relationships between
1/K0 and m/z were obtained and shown in Table 4.4. With R2 values ranged from 0.89 to
0.99, the correlations were confirmed with good linearity. Since Ω/z is directly proportional
to 1/K0, the results well explained that the relationship between ion’s size-to-charge ratio and
mass-to charge ratio is classification specific. The most recent work by May et al.14
investigated four chemically distinct classes of compounds including quaternary ammonium
salts, lipids, peptides, and carbohydrates, showing near-linear polynomial fits between m/z
and Ω/z. The difference between the result by May and the result here is mainly due to the
different m/z ranges. May et al. studied the correlation in m/z ranging from 200 to near 2000.
However, in this study, a smaller m/z range was investigated.

107
4.4 Conclusions
HT-IMMS analysis enables high throughput and comprehensive metabolite detection
in both NIST SRM 1950 sample and human blood sample, with both ion mobility separation
and m/z detection within 2-3 minutes. It is clear that NIST SRM 1950 and its COA database
are insufficient for metabolite identification for human blood metabolome. With the help of
HMDB and METLIN, metabolite identification is improved, however, still has problems
such as multiple candidates for each metabolite feature. IMMS technique provides reduced
mobility information associated with ionic structure, facilitating metabolite identification. It
allows accurate isotopic ratio analysis without interference; isomers/isobars separation to
increases the peak capacity. It also provides mobility-mass correlations that add qualitative
information regarding to compound classifications. With over a thousand metabolite features
detected and 185 metabolite features identified, it is evident that IMMS is an efficient method
for comprehensive metabolite detection, and the strategies for metabolite identification using
both mobility and mass information yields more reliable identifications. However, generating
IMMS database for metabolome is necessary to further aid metabolite identification.

108
Reference (1) Nordström, A.; Lewensohn, R. J Neuroimmune Pharmacol 2009, 5, 4–17.
(2) Nicholson, J. K.; Lindon, J. C. Nature 2008, 455, 1054–1056.
(3) Dunn, W. B.; Ellis, D. I. TrAC Trends in Analytical Chemistry 2005, 24, 285–294.
(4) Brown, S. C.; Kruppa, G.; Dasseux, J. L. Mass Spectrom. Rev. 2005, 24, 223–231.
(5) Hu, Q.; Noll, R. J.; Li, H.; Makarov, A.; Hardman, M.; Graham Cooks, R. J. Mass
Spectrom. 2005, 40, 430–443.
(6) Wilson, I. D.; Nicholson, J. K.; Castro-Perez, J.; Granger, J. H.; Johnson, K. A.;
Smith, B. W.; Plumb, R. S. J. Proteome Res. 2005, 4, 591–598.
(7) Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.;
Lundgren, K.; Roessner-Tunali, U.; Forbes, M. G.; Willmitzer, L.; Fernie, A. R.;
Kopka, J. FEBS Letters 2005, 579, 1332–1337.
(8) Theodoridis, G. A.; Gika, H. G.; Want, E. J.; Wilson, I. D. Analytica Chimica Acta
2012, 711, 7–16.
(9) Dunn, W. B.; Erban, A.; Weber, R.; Creek, D. J.; Brown, M. Metabolomics 2013.
(10) Prasad, B.; Garg, A.; Takwani, H.; Singh, S. Trends in Analytical Chemistry 2011,
30, 360–387.
(11) Creek, D. J.; Jankevics, A.; Breitling, R.; Watson, D. G.; Barrett, M. P.; Burgess, K.
E. V. Anal. Chem. 2011, 83, 8703–8710.
(12) Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Orešič, M. Chemometrics and
Intelligent Laboratory Systems 2011, 108, 23–32.
(13) Giles, K.; Williams, J. P.; Campuzano, I. Rapid Commun. Mass Spectrom. 2011, 25,
1559–1566.
(14) May, J. C.; Goodwin, C. R.; Lareau, N. M.; Leaptrot, K. L.; Morris, C. B.;
Kurulugama, R. T.; Mordehai, A.; Klein, C.; Barry, W.; Darland, E.; Overney, G.;

109
Imatani, K.; Stafford, G. C.; Fjeldsted, J. C.; McLean, J. A. Anal. Chem. 2014, 86,
2107–2116.
(15) Kaplan, K.; Graf, S.; Tanner, C.; Gonin, M.; Fuhrer, K.; Knochenmuss, R.; Dwivedi,
P.; Hill, H. H., Jr. Anal. Chem. 2010, 82, 9336–9343.
(16) Kaplan, K.; Dwivedi, P.; Davidson, S.; Yang, Q.; Tso, P.; Siems, W.; Hill, H. H., Jr.
Anal. Chem. 2009, 81, 7944–7953.
(17) Ruotolo, B. T.; Benesch, J. L. P.; Sandercock, A. M.; Hyung, S.-J.; Robinson, C. V.
Nat Protoc 2008, 3, 1139–1152.
(18) Ramautar, R.; Berger, R.; van der Greef, J.; Hankemeier, T. Current Opinion in
Chemical Biology 2013, 17, 841–846.
(19) Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A. C.; Peters, A.; Meitinger, T.;
Roden, M.; Wichmann, H.-E.; Pischon, T.; Adamski, J.; Illig, T. Mol. Syst. Biol.
2012, 8, 615.
(20) Shah, S. H.; Bain, J. R.; Muehlbauer, M. J.; Stevens, R. D.; Crosslin, D. R.; Haynes,
C.; Dungan, J.; Newby, L. K.; Hauser, E. R.; Ginsburg, G. S.; Newgard, C. B.;
Kraus, W. E. Circ Cardiovasc Genet 2010, 3, 109.
(21) Schicho, R.; Shaykhutdinov, R.; Ngo, J.; Nazyrova, A.; Schneider, C.; Panaccione,
R.; Kaplan, G. G.; Vogel, H. J.; Storr, M. J. Proteome Res. 2012, 11, 3344–3357.
(22) Bogdanov, M.; Matson, W. R.; Wang, L.; Matson, T.; Saunders-Pullman, R.;
Bressman, S. S.; Beal, M. F.; Flint Beal, M. Brain 2008, 131, 389–396.
(23) Diaz, S. O.; Pinto, J.; Graça, G.; Duarte, I. F.; Barros, A. S.; Galhano, E.; Pita, C.; do
Céu Almeida, M.; Goodfellow, B. J.; Carreira, I. M.; Gil, A. M. J. Proteome Res.
2011, 10, 3732–3742.
(24) Wishart, D. S.; Knox, C.; Guo, A. C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D. D.;
Psychogios, N.; Dong, E.; Bouatra, S.; Mandal, R.; Sinelnikov, I.; Xia, J.; Jia, L.;

110
Cruz, J. A.; Lim, E.; Sobsey, C. A.; Shrivastava, S.; Huang, P.; Liu, P.; Fang, L.;
Peng, J.; Fradette, R.; Cheng, D.; Tzur, D.; Clements, M.; Lewis, A.; De Souza, A.;
Zuniga, A.; Dawe, M.; Xiong, Y.; Clive, D.; Greiner, R.; Nazyrova, A.;
Shaykhutdinov, R.; Li, L.; Vogel, H. J.; Forsythe, I. Nucleic Acids Research 2009,
37, D603–D610.
(25) Smith, C. A.; O'Maille, G.; Want, E. J.; Qin, C. Therapeutic drug … 2005.
(26) Bruce, S. J.; Jonsson, P.; Antti, H.; Cloarec, O.; Trygg, J.; Marklund, S. L.; Moritz,
T. Analytical Biochemistry 2008, 372, 237–249.
(27) Zhang, X.; Knochenmuss, R.; Siems, W. F.; Liu, W.; Graf, S.; Hill, H. H. Anal.
Chem. 2014, 86, 1661–1670.
(28) Fenn, L. S.; Kliman, M.; Mahsut, A.; Zhao, S. R.; McLean, J. A. Anal Bioanal Chem
2009, 394, 235–244.
(29) McLean, J. A.; Ruotolo, B. T.; Gillig, K. J.; Russell, D. H. International Journal of
Mass Spectrometry 2005.
(30) Simón-Manso, Y.; Lowenthal, M. S.; Kilpatrick, L. E.; Sampson, M. L.; Telu, K. H.;
Rudnick, P. A.; Mallard, W. G.; Bearden, D. W.; Schock, T. B.; Tchekhovskoi, D.
V.; Blonder, N.; Yan, X.; Liang, Y.; Zheng, Y.; Wallace, W. E.; Neta, P.; Phinney,
K. W.; Remaley, A. T.; Stein, S. E. Anal. Chem. 2013, 85, 11725–11731.
(31) Woods, A. S.; Ugarov, M.; Egan, T.; Koomen, J.; Gillig, K. J.; Fuhrer, K.; Gonin,
M.; Schultz, J. A. Anal. Chem. 2004, 76, 2187–2195.

111
Table 4.1 Metabolite identification of 185 major metabolite features detected by
HT-IMtofMS. Measured m/z, drift time and K0 are included.
m/z Drift time Ko
Peak Height Metabolite Identification Adduct
100.083 22.20 2.11 447.067 5-aminopentanoic acid [M+H-H2O]+ 102.099 23.21 2.02 735.271 Betaine aldehyde [M+H]+ 104.115 22.51 2.08 7993.72 Choline [M+H]+ 105.000 24.82 1.89 1365.29 Mercaptolactic acid [M+H-H2O]+ 108.052 24.55 1.91 484.306 3-Pyridinaldehyde [M+H]+ 114.075 23.14 2.03 836.098 Creatinine [M+H]+ 115.083 22.55 2.08 115.857 N-Mononitrosopiperazine [M+H]+ 115.083 23.60 1.99 106.525 gamma-caprolactone [M+H]+ 116.081 23.55 1.99 86.3621 Proline [M+H]+ 119.029 23.46 2.00 208.848 Succinic acid [M+H]+ 121.045 23.39 2.01 112.681 Purine [M+H]+ 121.079 23.40 2.01 260.592 4-Dexyerythronic [M+H]+ 122.067 25.98 1.81 270.43 Benzamide [M+H]+ 123.059 23.82 1.97 413.057 Niacinamide/Vitamine B3 [M+H]+ 124.079 26.73 1.76 162.874 5-aminopentanal [M+H]+ 129.067 23.86 1.97 116.565 Dihydrothymine [M+H]+ 132.097 24.27 1.93 214.551 Leucine/Isoleucine [M+H]+ 133.047 24.45 1.92 236.101 Methylsuccinic acid/glutaric acid [M+H]+ 135.060 25.19 1.86 119.312 Homocysteine thiolactone [M+NH4]+ 136.058 25.50 1.84 775.11 Homocysteine [M+H]+ 137.073 25.52 1.84 270.92 N-Mononitrosopiperazine (not sure) [M+Na]+ 138.062 25.85 1.82 175.774 PABA (p-Aminobenzoic acid) [M+H]+ 139.063 26.02 1.80 104.819 8-Hydroxypurine [M+H]+ 139.075 24.81 1.89 101.507 Isocapronic acid [M+Na]+ 140.076 26.04 1.80 471.938 Valine [M+Na]+ 145.042 25.50 1.84 149.583 3-Methylglutaconic acid [M+H]+ 146.070 25.51 1.84 174.778 N/A N/A 147.123 26.06 1.80 124.781 Lysine [M+H]+ 148.060 25.40 1.85 89.8962 Glutamic acid [M+H]+ 152.033 25.41 1.85 401.62 Pyruglutamic acid [M+Na]+ 154.071 26.61 1.76 229.604 Fapy-adenine [M+H]+ 156.051 26.06 1.80 964.567 Histidine [M+H]+ 157.091 26.29 1.79 377.044 Cymene [M+Na]+ 158.066 26.63 1.76 794.719 2-Phenylacetamide [M+Na]+ 159.017 28.50 1.65 82.1021 Erythronic acid [M+Na]+ 163.049 27.29 1.72 216.752 N/A N/A 166.092 27.89 1.68 653.803 Phenylalanine [M+H]+ 169.069 26.52 1.77 84.1626 Uric acid [M+H]+ 170.041 26.61 1.76 243.095 Glutamate [M+Na]+

112
175.144 27.77 1.69 95.3766 Arginine [M+H]+ 182.069 27.92 1.68 815.838 Tyrosine [M+H]+ 184.085 29.75 1.58 133.138 Vitamin B6 [M+H]+ 187.004 28.28 1.66 1590.47 3-phosphoglyceric acid (3PG) [M+H]+ 200.075 29.87 1.57 219.577 N/A N/A 203.060 29.70 1.58 860.53 Glucose [M+Na]+ 203.060 28.47 1.65 263.581 Fructose [M+Na]+ 214.182 31.75 1.48 163.819 N/A N/A 219.034 29.62 1.58 182.375 Tyrosol 4-sulfate [M+H]+ 228.201 33.07 1.42 1031.76 N/A N/A 235.017 29.52 1.59 586.36 N/A N/A 236.164 33.58 1.40 900.538 N,O-Didesmethyltramadol [M+H]+ 238.176 34.05 1.38 227.167 12-amino-dodecanoic acid [M+Na]+ 245.131 32.82 1.43 147.86 Phe-Pho [M+H]+ 250.181 34.94 1.34 2930.05 Lophocerine [M+H]+ 252.151 34.19 1.37 96.4383 N-Decanoylglycine [M+Na]+ 254.153 34.74 1.35 159.059 Gamma-Aminobutyryl-lysine [M+Na]+ 256.268 40.85 1.15 732.798 Palmitic amide [M+H]+ 266.156 35.65 1.32 1166.71 Proline-lysine [M+H]+ 267.127 34.80 1.35 129.335 Phe-Thr [M+H]+ 268.069 33.19 1.41 112.562 Asp-Pro [M+K]+ 268.121 35.63 1.32 587.795 Adenosine [M+H]+ 269.003 33.94 1.38 1057.21 Dimethylallyl pyrophosphate [M+Na]+ 278.241 40.70 1.15 961.488 Palmiti amide [M+Na]+ 279.163 37.17 1.26 230.189 leu-phe [M+H]+ 282.206 36.82 1.27 547.689 Colestipol [M+H]+ 284.296 43.34 1.08 327.502 Sphinganine [M+H-H2O]+ 289.156 39.96 1.17 1640.88 Asparaginyl-arginine [M+H]+ 298.175 37.59 1.25 474.856 Ser-Leu-Pro [M+H-H2O]+ 301.142 39.76 1.18 4428.7 Gly-Gly-Pro-Ala [M+H]+ 302.246 39.70 1.18 747.329 linoleamides [M+Na]+ 303.229 39.57 1.19 899.109 Linoleic acid [M+Na]+ 304.257 41.31 1.14 366.859 Oleamide/Elaidamide [M+Na]+ 305.132 39.80 1.18 818.769 YC-1 [M+H]+ 306.278 42.98 1.09 256.441 Linoleoyl ethanolamide [M+H-H2O]+ 317.114 39.92 1.18 2123.1 glutamy-phenylalanine [M+Na]+ 318.212 40.70 1.15 123.949 N/A N/A 319.121 39.99 1.17 107.902 N/A N/A 325.205 42.30 1.11 129.806 Dinor-PGD2 [M+H]+ 326.234 41.19 1.14 404.939 10-Nitrolinoleic acid [M+H]+ 327.229 41.02 1.14 178.514 Eicosatetraenoic acid (C20:4n) [M+H]+ 332.291 43.56 1.08 522.628 Oleol Ethyl Amide [M+H]+ 335.144 39.70 1.18 240.563 Met-Ala-Asn [M+H]+ 338.337 47.10 1.00 1424.69 N-cyclohexanecarbonylpentadecylamine [M+H]+ 339.338 46.58 1.01 119.882 4,6-Docosanedione [M+H]+

113
342.221 42.06 1.12 159.748 Arachidonoyl amine [M+K]+ 342.328 47.50 0.99 122.611 N,N,N-trimethyl-spingosine [M+H]+ 354.403 50.79 0.92 2089.75 Tetracosene [M+NH4]+ 359.248 42.67 1.10 336.586 2-Hydroxydesogestrel [M+Na]+ 360.318 45.84 1.02 7979.99 13-Docosenamide [M+Na]+ 369.344 46.26 1.01 2251.7 Cholesterol [M+H-H2O]+ 376.294 47.07 1.00 2363.97 13-Docosenamide [M+K]+ 383.322 46.35 1.01 547.388 4,6-cholestadienone [M+H]+ 385.342 48.28 0.97 248.419 PreVitamine D3 [M+H]+ 385.342 47.43 0.99 98.2378 Dehydrocholesterol [M+H]+ 387.190 44.55 1.05 135.668 Pro-Glu-Ala-Ala [M+H]+ 388.344 48.12 0.98 220.69 Aminopentol [M+H-H2O]+ 392.289 48.20 0.97 220.051 12-HETE-Ala [M+H]+ 392.289 47.73 0.98 167.352 15-HEYE-Ala [M+H]+ 398.237 47.20 0.99 176.486 Lys-Pro-Gly-Pro [M+H]+ 405.309 50.81 0.92 374.007 Cholesta-4,6-dien-3-one [M+Na]+ 409.164 45.92 1.02 955.715 Pro-Glu-Ala-Ala [M+Na]+ 413.258 49.67 0.94 710.818 Lyso PC(10:0) [M+H]+ 414.221 47.74 0.98 222.726 Arg-Trp-Ala [M+H-H2O]+ 425.142 45.87 1.02 812.838 His-Phe-OH [M+H]+ 426.327 52.13 0.90 175.69 Oleoylcarnitine [M+H]+ 429.231 49.76 0.94 280.347 Pro-Glu-Ala-Leu [M+H]+ 429.353 52.03 0.90 145.373 N/A N/A 452.273 49.19 0.95 102.959 Arg-Val-Arg [M+Na]+ 469.340 54.05 0.87 547.633 PC(P-14:0/0:0) [M+NH4]+ 496.319 55.98 0.84 276.148 His-Ile-Lys-Val [M+H]+ 518.289 57.35 0.82 661.48 PC(15:0) [M+Na]+ 524.330 58.45 0.80 103.811 Val-Arg-Leu-His [M+H]+ 534.268 57.68 0.81 1115.28 Arg-Cys-Gln-Lys [M+H]+ 542.310 56.36 0.83 107.895 Asn-Pro-Arg-Arg [M+H]+ 544.299 57.95 0.81 232.752 His-Arg-Leu-Pro [M+H]+ 546.320 59.58 0.79 215.682 PE(20:0) [M+Na]+ 547.315 59.67 0.79 176.969 Cys-Arg-Ile-Arg [M+H]+ 558.258 57.21 0.82 169.582 Tyr Thr Phe Gln [M+Na]+ 560.266 58.57 0.80 168.845 Tyr Lys Tyr Ser [M+H]+ 562.284 59.95 0.78 271.205 Arg Gln Gln Met [M+H]+ 617.468 65.48 0.72 103.518 PA(31:1) [M+H]+ 637.263 63.21 0.74 1313.86 Ser-His-Trp-Trp [M+Na]+ 659.241 66.93 0.70 3006.61 Try-Try-Try-Glu [M+Na]+ 671.526 70.46 0.67 3748.32 PA(36:1) [M+H-H2O]+ 673.548 71.00 0.66 730.614 PE-Cer(d14:2(4E,6E)/21:0) [M+H]+ 675.214 66.95 0.70 2732.84 Try-Try-Try-Glu [M+K]+ 687.500 71.28 0.66 272.554 PA(35:2) [M+H]+ 688.506 71.29 0.66 244.606 PC(29:2) [M+H]+ 695.518 71.36 0.66 613.483 PA(35:1) [M+Na]+

114
696.502 71.41 0.66 133.634 PE(34:4) [M+H]+ 697.503 71.47 0.66 107.713 PA(35:0) [M+Na]+ 697.503 72.03 0.65 102.148 PA(P-35:0) [M+Na]+ 711.474 72.56 0.65 83.1371 PA(35:1) [M+Na]+ 725.495 74.18 0.63 309.569 PA(38:4) [M+H]+ 726.495 74.31 0.63 291.408 PE(35:4) [M+H]+ 741.416 74.85 0.63 82.4017 PG(31:3) [M+K]+ 741.475 74.75 0.63 88.7288 PG(32:2) [M+Na]+ 758.510 74.41 0.63 466.089 PC(31:0) [M+K]+ 780.487 74.86 0.63 932.707 PS(36:6) [M+H]+ 782.497 75.04 0.63 497.505 PS (34:2) [M+Na]+ 782.497 76.17 0.62 366.676 PS (36:5) [M+H]+ 796.459 75.70 0.62 1297.31 PS(34:3) [M+K]+ 797.438 76.18 0.62 169.417 PI(30:4) [M+Na]+ 798.454 76.81 0.61 156.723 PS(34:2) [M+K]+ 805.435 75.56 0.62 91.568 PG(36:6) [M+K]+ 806.508 75.76 0.62 219.982 PS (38:7) [M+H]+ 810.502 76.99 0.61 89.1405 PE(42:11) [M+H]+ 820.460 75.74 0.62 126.834 PS(36:5) [M+K]+ 822.471 76.54 0.61 115.621 PS(36:4) [M+K]+ 823.490 76.33 0.61 84.0074 PG(37:4) [M+K]+ 824.480 77.70 0.60 228.71 PS(36:3) [M+K]+ 825.500 77.64 0.60 150.98 PG(37:3) [M+K]+ 830.479 76.32 0.61 124.587 PS(40:9) [M+H]+ 832.529 77.35 0.61 108.391 PC(37:5) [M+K]+ 844.434 76.85 0.61 102.216 PS(38:7) [M+K]+ 895.507 62.86 0.75 98.7142 Hemoglobin [M+17H]17+ 947.831 64.59 0.73 121.932 Hemoglobin [M+16H]16+
1009.090 66.76 0.70 135.644
Hemoglobin [M+15H]15+
1009.140 66.74 0.70 85.9929 1009.220 66.76 0.70 156.075 1009.330 66.71 0.70 164.246 1009.400 66.68 0.70 145.386 1010.550 66.53 0.71 171.284 1010.610 66.67 0.70 100.171 1012.170 66.62 0.70 108.139 1012.400 66.52 0.71 86.9916 1014.670 66.61 0.70 108.922 1081.230 68.87 0.68 392.524
Hemoglobin [M+14H]14+
1081.380 69.08 0.68 83.1947 1084.120 68.60 0.68 178.559 1085.990 68.55 0.68 136.906 1087.090 68.53 0.68 133.202 1088.630 68.70 0.68 93.0488 1090.040 68.69 0.68 118.392

115
1091.350 68.71 0.68 95.4981 1095.170 68.83 0.68 82.4147 1164.180 71.54 0.66 80.4191
Hemoglobin [M+13H]13+
1164.440 71.61 0.66 274.278 1167.280 71.47 0.66 122.149 1167.710 71.31 0.66 102.511 1169.160 71.43 0.66 139.595 1169.300 71.34 0.66 87.5275 1169.300 71.48 0.66 85.6067 1170.970 71.39 0.66 101.411 1173.910 71.41 0.66 101.638

116
Table 4.2 Isotopic ratio analysis results.
# m/z Drift Time Ko Isotopic
Ratio Chemical Formula Metabolite Adduct
Form
1 104.115 22.51 2.08 100%
C5H14NO Choline [M]+ 105.117 22.53 2.08 6%
2 301.142 39.76 1.18 100%
C18H20O4 5-O-Methyllatifolin [M+H]+ 302.145 39.74 1.18 21%
3 317.114 39.92 1.18 100%
C11H18N4O8 Asp Gly Ser Gly [M+H-H2O]+ 318.119 39.96 1.18 13%
4 360.318 45.84 1.02 100%
C22H43NO 13-Docosenamide [M+Na]+ 361.320 45.87 1.02 24% 362.319 45.88 1.02 4%
5 376.294 47.07 1.00 100%
C22H43NO 13-Docosenamide [M+K]+ 377.291 47.10 1.00 24%
6 534.268 57.68 0.81 100%
C20H39N9O6S Arg Cys Gln Lys [M+H]+ 535.283 57.65 0.81 26%
7 659.241 66.93 0.70 100%
C32H36N4O10 Try Try Try Glu [M+Na]+ 660.244 66.95 0.70 32%
8 675.214 66.95 0.70 100%
C32H36N4O10 Try Try Try Glu [M+K]+ 676.218 66.93 0.70 32%
Table 4.3 Isomer/isobar separation and analysis results.
Metabolite Features m/z Drift
Time Ko Chemical Formula Metabolite Adduct
Form 1 115.087 22.55 2.08 C5H10N2O N-Mononitrosopiperazine [M+H]+ 2 115.076 23.60 1.99 C6H10O2 gamma-caprolactone [M+H]+
3 203.053 29.70 1.58 C6H12O6 Glucose [M+Na]+ 4 203.053 28.47 1.65 C6H12O6 Fructose [M+Na]+
5 383.331 46.35 1.01 C27H42O 24-Dehydroprovitamin D3 [M+H]+ 6 383.331 47.20 0.99 C27H42O 4,6-cholestadienone [M+H]+
7 385.347 48.28 0.97 C27H44O 4,6-cholestadienol [M+H]+ 8 385.347 47.43 0.99 C27H44O Vitamin D3 [M+H]+
9 782.494 75.04 0.63 C40H74NO10P PS 34:2 [M+Na]+
10 782.497 76.17 0.62 C42H72NO10P PS 36:5 [M+H]+

117
Table 4.4 Mobility-mass correlation trend lines.
Compound Classification Mobility-mass Correlation
Trend Line R2 Glycerophospholipid (PI) y = 0.0005x + 1.3805 0.89
Glycerophospholipid (PC, PS, PE) y = 0.0008x + 0.9727 0.90 Lyso-phospholipid y = 0.0014x + 0.5089 0.97
Tetrapeptide y = 0.0011x + 0.6337 0.86 Amino acid and Dipeptide y = 0.0018x + 0.2908 0.99

118
Figure 4.1 (a) Schematic diagram of the instrument: Electrospray ionization hadamard
transform atmospheric pressure ion mobility time-of-flight mass spectrometer; (b) major
metabolite features detected in human blood metabolome; (c) major metabolite features
detected in NIST SRM 1950.
a
b c

119
Figure 4.2 Global metabolomics results from HT-IMtofMS analysis. (a) shows the
multidimensional information of the metabolite features detected from human blood, with
x-axis as m/z (ranges from 100 to 1100), y-axis as drift time (ranges from 15 ms to 85 ms)
and z-axis as intensity. (b) and (c) are the corresponding mass spectrum and mobility
spectrum. Over a thousand metabolite features were detected during the analysis.

120
Figure 4.3 An example of accurate isotopic ratio analysis. (a) is the mass spectrum with
zoomed-in mass range from 103.5 to 106.5. Three m/z features were detected with m/z =
104.11, 105.00 and 105.11. (b) is the ion mobility spectra for the above three m/z features.
The mobility peak of m/z 105.11 overlapped with the mobility peak of m/z 104.11, indicating
its identity as an isotope of m/z 104.11.

121
Figure 4.4 An example of isomer/isobar separation. The mass spectrum with zoomed-in mass
range from 115.0 to 115.6 is shown in (a), with one m/z feature detected. (b) is the
corresponding ion mobility spectrum with two ion mobility peaks detected with drift time of
22.6 ms and 23.6 ms, demonstrating the existence of two different metabolite features
identified as [N-mononitrosopiperazine+H]+ and [Gamma-caprolactone+H]+.

122
Figure 4.5 IMMS two-dimensional spectrum of hemoglobin with charge states from +13 to
+17. Good linearity was observed with increasing m/z difference between two adjunct charge
states. This trend lines is also shown in Figure 4.6.

123
Figure 4.6 IMMS two-dimensional spectrum illustrating the metabolomes of human blood.
Each dot observed in this 2D space represents a metabolite feature. Six trend lines were
identified for different compound classification.

124
Chapter 5
Neuronal Metabolomics by Ion Mobility Mass Spectrometry in
Cocaine Self-administering Rats after Early and Late Withdrawal
Abstract
The neuronal metabolomes in rat striatum (STR), prefrontal cortex (PFC) and nucleus
accumbens (NAC) were analyzed by hadamard transform ion mobility mass spectrometry
(HT-IMMS) in order to reveal global and specific metabolic changes induced by cocaine
self-administration after 1-day or 3-weeks withdrawal. Metabolites were comprehensively
separated and detected with the employment of HPLC-IMMS within minutes. Global
metabolic differences were observed using PCA for comparisons between cocaine and saline
treatments after 1-day withdrawal. Potential biomarkers were selected using PCA loadings
plot and unpaired t-test, yielding a complete profile of metabolic changes induced by cocaine
self-administration. Metabolites associated with oxidative stress and energy metabolism were
also specifically investigated. We found out that the dysregulation of creatine/creatinine was
different between the STR and NAC, demonstrated that metabolic alterations are brain
regionally specific. Glutathione and adenosine were also changed in their concentrations, and
the results agreed with previous studies. In general, our findings support the multi-event
mechanism of cocaine-induced disorders proposed by previous investigations, and reveal
additional metabolic targets of cocaine-induced changes after early and extended withdrawal
times.

125
5.1 Introduction
As the second most commonly used illicit drug in the United States, cocaine affects
the lives and health of millions of people. The adverse health consequences to the individual
and the cost to society of cocaine abuse are substantial. Long term users are at greatly
increased risk for neurologic and cardiovascular toxicities as well as other complications1.
The mental elements in play with cocaine withdrawal include craving, exhaustion,
restlessness and depression, making cocaine one of the most addictive drugs2,3.
Classic mechanisms involved in cocaine addiction are increases in dopaminergic
transmission within striatal regions and altered glutamate transmission4,5. Previous
investigations have mainly centered on receptor binding and blocking of enzymes6. However,
additional studies7,8 suggest that the brain states caused by cocaine addiction are produced by
multi-events problems including oxidative stress, aberrant metabolism and
neuroinflammation, leaving a fuller explanation of the mechanisms unsolved. Moreover,
although cocaine withdrawal often has no visible physical symptoms, a severe level of
craving coupled with the lack of effective medications for reducing the craving, causes high
rates of relapse9,10. To better understand the addiction and withdrawal mechanism, it is
necessary to have a comprehensive interpretation of the concert of biochemical events
occurring after cocaine exposure and withdrawal from cocaine exposure.
Global metabolomics, as an exploratory investigation approach, simultaneously and

126
comprehensively measures metabolites to reveal changes in the metabolome. It not only
complements genomics and proteomics data, but also identifies phenotypic changes caused
by stimuli more predicatively than other “omics” approaches11. By measuring all metabolites
in a biological system, global metabolomics provides information for global metabolic
profiling and for the interrogation of specific metabolites of interest. Recent advances in
analytical techniques and data processing software has assisted the relatively new field of
metabolomics. The practice of metabolomics has grown rapidly, being applied to a wide
range of central nervous system diseases including schizophrenia and Parkinson’s disease12,13.
Effects on the metabolome from drug abuse such as cocaine, methamphetamine and
morphine have also been studied14,15.
Previous studies on drug abuse using metabolomics approaches have demonstrated
the potential of global metabolomics and the concept of the multi-event mechanism for
cocaine addiction. Patkar et al.15 performed metabolomics analysis on plasma samples from
cocaine-dependent individuals and drug-free controls using a liquid chromatography
electrochemical array platform, and discovered significant up-regulation in
n-methylserotonine and guanine, while hypoxanthine and xanthine were down regulated.
Metabolomics in brain tissues from cocaine treated rats was investigated by Li et al. using
1H-NMR16. Changes in the tissue concentrations of creatine, taurine, and N-acetylasparate
after cocaine treatment were observed, providing metabolic alterations associated with
neuotransmitters, oxidative stress and mitochondria dysregulation.

127
Comprehensive detection of metabolites with different concentrations, volatilities
and polarities is critical for understanding changes in the metabolome of different brain
regions caused by cocaine addiction. However, previous investigations have the common
problem of lacking comprehensive metabolomics analysis due to limited sensitivity.
IMMS17-19 has been well characterized as an efficient method for metabolomics by a number
of studies in tissues, biofluids, and cell lines. In this study, we employed hadamard transform
ion mobility mass spectrometry (HT-IMMS)20 for metabolomics analysis with its capability
of rapid analysis with nano-molar limit of detection, isomeric separation and generation of
two-dimensional information. We performed global metabolomics on multiple brain tissues
(striatum (STR), prefrontal cortex (PFC) and nucleus accumbens (NAC)) from rats under
cocaine administration followed by different withdrawal times (1 day and 3 weeks).
We predicted that cocaine self-administration would increase or decrease the levels of
multiple metabolites, and that these effects would be both brain region dependent and
withdrawal time dependent. The aim of this study was to investigate the addiction and
withdrawal effects of cocaine on the metabolomes of multiple brain regions globally and
specifically. Our goal was to select regionally specific potential biomarkers, and specifically
exam those biomarkers involved in energy metabolism and oxidative stress for demonstrating
the proposed multi-event mechanism of cocaine addiction.

128
5.2 Experimental Design
5.2.1 Subjects, Cocaine Self-administration and Brain Dissection:
Male Sprague-Dawley rats (300-330 gram) were singly housed in a temperature and
humidity-controlled room with a 12 hr light/dark cycle, with food and water provided ad
libitum except for when animals were engaged in experiments. Experiments were conducted
according to the National Institutes of Health Guide for the Care and Use of Laboratory
Animals (National Research Council, 1996), and experimental protocols were approved by
the University Animal Care and Use Committee.
A total of 13 rats were used, divided into four treatment groups: saline or cocaine,
1-day withdrawal, or saline or cocaine, 3-wks withdrawal. Sample details and cocaine
self-administration results are listed in Table 5.1. All studies were conducted during the same
time of day for each group of animals. Self-administration surgery was conducted according
to a modification of Brown et al.21. Cocaine self-administration training began 5-7 days after
surgery. The acquisition criteria for cocaine self-administration consisted of 3 consecutive
days of 10 rewards during the fixed ratio 1 (FR1) schedule. After FR1 maintenance was
stabilized, animals were switched to an FR3 schedule of reinforcement until met a criterion of
7 consecutive days of at least 10 rewards.
After 1-day or 3-wks of withdrawal time, rats were sacrificed by decapitation and
brains were rapidly removed and dissected on an ice-cold plate. The medial portion of the

129
PFC was dissected from tissue rostral to +2.2 mm, and included the infralimbic, prelimbic,
cingulate cortex area 1 and secondary motor cortex. The NAC included regions between +0.7
to +1.7 mm from bregma, and included both core and shell regions. The STR included the
same rostral to caudal regions dorsal to the NAC, medial to the lateral ventricle and the
corpus callosum.
5.2.2 Metabolite Extraction:
Each tissue sample was weighed individually and sonicated in 600 µL ice-cold ESI
solvent (methanol: water: formic acid 49.95%: 49.95%: 0.1% (v/v/v)). The supernatant was
collected and separated from cellular debris after centrifugation for 30 min at 13K rpm by a
desktop centrifuge. The supernatant was stored at -80 °C before IMMS analysis.
2,6-ditert-butylpyridine was used as internal mobility calibration standard throughout the
study.
5.2.3 IMMS Analysis:
The ion mobility mass spectrometry analysis was performed by hadamard transform
ion mobility time-of-flight mass spectrometry. The components of this instrument include an
electrospray ionization source, a stacked ring ion mobility spectrometer, an IMMS interface
and a time-of-flight mass spectrometer (shown in Figure 5.1). The stack ring ion mobility
spectrometer was built at Washington State University, and the IMMS interface and TOF
mass spectrometer were provided by Tofwerk (AG, Switzerland). The hadamard transform

130
implementation was developed by Tofwerk and evaluated at Washington State University.
The operation details were described elsewhere20,22. Briefly, the electrospray ionization
source ionized the metabolites and created ions for IMMS analysis. Stacked ring ion mobility
spectrometer separated ions based on size-to-charge ratio with an electric field of 305 V/cm
and a drift gas (nitrogen) at a flow rate of 2.5 L/min. The IMS tube was held at ~200 °C and
operated at ambient pressure (690 – 705 Torr). The hadmard transform signal was generated
using software developed and supplied by Tofwerk. The signal was implemented on the
Bradbury Neilson (BN) ion gate with the smallest gate pulse width (GPW) controlled at 180
µs. IMS separated ions were then transferred into the TOF mass spectrometer by IMMS
interface.
Two sample introduction methods before ESI were employed in this study: HPLC
pre-separation and direct infusion. Both methods were described in detail elsewhere20. Briefly,
STR and PFC samples were introduced by HPLC pre-separation using a C-8 guard column to
reduce the ionization suppression problem common in ESI direct infusion. Nucleus
accumbens samples were introduced by direct infusion due to the fact that the limited tissue
size (6 - 10 mg) yielded insufficient metabolite concentration level for HPLC pre-separation.
The ESI ionization was applied with a biased voltage of 3000 V and a flow rate of 4 µL/min.
Each IMMS analysis was completed within 10 minutes.

131
5.2.4 Data Handling and Statistical Analysis:
TofDAQ1.92b and IMSviewer1_6d were developed by Tofwerk and used for IMMS
data acquisition and processing, respectively. The IMMS analysis of each sample generated a
unique data set, containing multidimensional information for detected metabolite features.
Each metabolite feature was characterized by its ion mobility, m/z ratio and ion intensity
(counts). The ion mobility data were generated by the stacked ring ion mobility spectrometer
and represented by reduced mobility K0 (cm2V-1s-1), which is defined as shown in the
following Equation.
!! = !!!!!! !× !
273.15! !× ! !760
Where V is the voltage drop over the drift region with length L, td is the time that the ion
takes to migrate through the drift tube. The reduced mobility constant (K0) for an ion remains
constant for a given drift gas, since it incorporates the pressure (P in Torr) and temperature of
the drift region (T in °K).
Microsoft adaptable peak list for each sample (containing all detected metabolite
features assigned with reduced mobility, m/z, and intensity (counts)) was generated by
IMSviewer1_6d with ~1000 metabolite features detected (counts > 5) for each sample, and
~200 selected by setting a threshold at 50 (only metabolite features with counts > 50 were
selected) for further analysis. Peak lists for replicate samples were then merged using Merge

132
Table Wizard for Microsoft Excel (Ablebits, Homel, Belarus). Also, left censoring23 occurred
in this step due to the limit of detection of the instrument and the biological variation between
replicates. Metabolite features detected in two out of three (or three out of four) replicates
were kept, along with the common metabolite features detected in all replicates, and the
left-censored data24 were replaced by threshold 50.
Principle component analysis (PCA) was applied as an unsupervised statistical tool
for multivariate data, providing pattern recognition for global metabolic analysis. The score
plot provided the patterns among samples without knowing the classification of the samples,
and the loadings plot displayed the metabolite features based on their impact on the pattern
recognition. Metabolites that significantly influenced the pattern recognition had large
absolute loading values, showing as outliers in the loadings’ plot. In addition to PCA,
univariate analysis (unpaired T-test) was also applied on each metabolite feature in order to
select the ones that varied significantly. Metabolite features with a p-value less than 0.05
were considered as significantly different between cocaine samples and the corresponding
control samples. Combining the results from loading’s plot and univariate analysis completed
the potential biomarker selection, where the univariate analysis was applied in all
comparisons, and the loading’s plot was only valid when the score plot differentiated between
cocaine samples and the corresponding controls.

133
Metabolite Identification:
Once the biomarker selection procedure was completed, we identified these
biomarkers based on their accurate m/z using Human Metabolome Database and METLIN
Metabolite Database. Metabolites were identified with an m/z tolerance of ± 0.01 and with
the adduct forms refined to [M+H]+ and [M+Na]+.

134
5.3 Results and Discussion
5.3.1 IMMS Spectra and Global Metabolic Analysis by PCA:
Figure 5.2a illustrates the two-dimensional IMMS spectrum using the metabolomes
of a striatal tissue sample as an example. Metabolite ions detected during the analysis were
displayed in the 2D spectrum with x-axis representing m/z (ranges from 100 to 950) and
y-axis representing drift time (ranges from 15 ms to 75 ms). The mass spectrum and ion
mobility spectrum were also included to explain the complexity of the tissue sample and the
rich information generated from IMMS analysis. Metabolite ions including amino acids, short
peptides and carbohydrates were detected with relatively high peak intensities between m/z
=100 to 400 because of their high abundance and easily ionized nature. However, in general,
higher molecular weight metabolites such as fatty acids and phospholipids had lower
ionization efficiency when compared with lower molecular weight metabolites due to
ionization suppression and were not sensitively detected by direct infusion ESI. By coupling
a rapid but low resolution chromatographic separation prior to ESI ionization, the ionization
suppression problem was significantly reduced20, and metabolite ions with higher m/z (close
to 1000) were detected as well as the low molecular weight metabolites. Notably, the analysis
was improved with HPLC and IMS when compared to MS alone, with regard to the number
of metabolite ions detected, as well as the signal to noise ratio. During the IMMS analyses,
ESI background spectra were collected before each sample analysis and used to mask out the
background ions detected in the sample runs. Table 5.2 summarizes the reproducible major
metabolite ions (counts > 50) detected in the three different brain regions under cocaine

135
self-administration after 1-day or 3-wks withdrawal with their corresponding saline controls.
These metabolite ions were further analyzed using statistical approaches.
An example of the global metabolic difference between cocaine self-administration
after 1-day withdrawal and the corresponding saline control is illustrated by ion mobility
spectra shown in Figure 5.2b. The IMS spectra of metabolites in PFC saline control #1 and
#2 are shown in b1 and b2, respectively. The high degree of similarity between these two
IMS spectra demonstrated the small variation between biological replicates. By comparing
spectra b1 and b2 with b3, which displays the IMS spectra of metabolites in PFC cocaine
sample, significant differences in peak position and intensities can be observed, especially at
drift time of 21 ms and 29 ms (circled in red). Although it is possible to view the global
metabolic patterns by comparing spectra visually, statistical analysis provides a more refined
approach for pattern recognition and potential biomarker selection.
PCA, as the unsupervised multivariate method, was applied to analyze the peak lists
generated from IMMS analysis and to compare between cocaine treatments and their
corresponding saline controls. Six analyses were performed, including STR/PFC/NAC from
cocaine self-administering rats after 1-day withdrawal vs. their corresponding saline controls,
and STR/PFC/NAC from cocaine self-administering rats after 3-wks withdrawal vs. their
corresponding saline controls. The score plots are shown in Figure 5.3 with the 1st PC as
x-axis and 2nd PC as y-axis. Saline samples (marked in green) and cocaine samples (marked

136
in red) are listed in the score plots. Note that at 60% - 80% of the variation was explained by
each score plot. Differences were detected for all the pairwise comparisons between cocaine
and saline treatments after 1-day withdrawal, with the saline samples forming a tight
grouping that was different from the grouping of cocaine samples. This result revealed that
significant global metabolic changes induced by cocaine self-administration after early
withdrawal. In contrast, it appears that there were nearly no global metabolic differences
between saline and cocaine treatments for NAC and PFC after 3-wks withdrawal. The score
plot for STR at 3-wks withdrawal does show global metabolic difference, although the
samples are not tightly grouped in the plot. The results for the 3-wks withdrawal
demonstrated less global metabolic differences compared with the control than did the 1-day
withdrawal samples. It appears that although the metabolome was significantly altered after
cocaine self-administration, it was nearly restored to control conditions after late withdrawal.
5.3.2 Potential Biomarkers:
Potential biomarkers were selected by combining the multivariate and univariate
approaches. As shown in Figure 5.3, comparisons of STR/PFC/STR after 1-day withdrawal,
as well as STR after 3-wks withdrawal, displayed a different grouping compared with their
saline controls. Therefore, the loadings’ plots from the above comparisons were valid in
biomarker selection and the outliers with relatively large loadings’ values were selected as
potential biomarkers. Moreover, for all the six comparisons, metabolites altered due to
cocaine self-administration were selected by unpaired t-test with p-value less than 0.05.

137
Table 5.3 displays all potential biomarkers selected from the six comparisons, with
biomarkers selected by loadings’ plots marked in bold. In this table, information including
measure m/z, K0, p-value from t-test, up/down regulation induced by cocaine
self-administration, adduct, and identified metabolite name are displayed for all selected
biomarkers. The last column contains the final identifications, among which the ones labeled
with “*” were identified in the metabolite databases25 as common metabolites with large
abundance in fluid and tissue, the ones labeled with “**” were identified in both metabolite
databases and previous studies26,27, and the ones without label were identified in metabolite
databases as metabolites with low abundance. Note that there are some metabolite features
without identifications because that no matched identification was found. Metabolite
databases are still under development. As databases become more comprehensive, a more
complete identification may be possible. In general, a wide range of metabolites including the
neurotransmitters (GABA and glutamate), amino acids (serine and proline), and peptides
appeared to be altered due to cocaine self-administration, indicating that multiple metabolic
pathways were altered. Thus, the effects of cocaine self-administration are expressed as a
multi-event mechanism.
5.3.3 Oxidative Stress-related Metabolites Changes:
Evidence was discovered of linkage between oxidative stress and the cocaine-induced
disorder. Creatine and creatinine, as energy supply-related metabolites that increase oxidative
stress, appeared repeatedly as potential biomarkers in Table 5.3 for the 1-day withdrawal

138
comparisons. Thus, these two compounds were studied specifically. The dysregulation of
creatine and its metabolic product creatinine are displayed using intensity profiles in Figure
5.4. Both creatine and creatinine were slightly up-regulated by cocaine self-administration in
the STR. However, they were down-regulated in the NAC (significant for creatine). This
finding matched previous studies showing differences in the response to cocaine addiction
between these two brain regions28. It appears that after 1-day withdrawal, cocaine
self-administration increased their metabolic activity in the STR and decreased it in the NAC.
With regard to the PFC, the creatine level remained relatively unchanged; however, the
creatinine level was significantly elevated by the cocaine self-administration. This result may
reflect an increase amount of creatine involved in the biosynthesis of phosphocreatine
catalyzed by creatine kinase.
Glutathione (GSH) is also widely studied as an important metabolite in oxidative
metabolism. In this study, it was detected as a major metabolite in the STR and PFC,
however, it was not detected in the NAC, perhaps due to the limited tissue size of the NAC.
Its dysregulation is shown in Figure 5.5 by an intensity profile. The intensity of GHS in the
STR was significantly decreased in cocaine self-administering rats. Previous evidence29,30
showed that cocaine treatment over expressed glutathione-peroxidase, which consumed GHS
during the biosynthesis. Our findings of a decrease in the concentration of GHS in the STR
supported the concept that glutathione-peroxidase is over-expressed with cocaine treatment.

139
5.3.4 Energy Metabolism-related Metabolites Changes:
In addition to creatine and creatinine, adenosine was also identified as a potential
biomarker for both the 1-day and 3-wks withdrawal. Adenosine is directly linked to energy
metabolism and enhances energy consumption. The dysregulation of adenosine in all six
comparisons are shown in Figure 5.6. It appears that adenosine levels were increased by
cocaine self-administration in most cases, indicating that neuronal activity was enhanced by
cocaine self-administration. However, the increase was significant only in STR 3-wks and
NAC 3-wks comparisons, indicating that although the global metabolic changes were not
significant after 3-wks withdrawal, there were still specific metabolites alterations. Changes
in adenosine regulation is consistent with previous reports31,32 that cocaine abstinence
decreased total sleep time and sleep efficiency.
5.3.5 Other Metabolites Changes:
As listed in Table 3, there were multiple metabolites alterations induced by cocaine
self-administration in addition to those associated with oxidative stress and energy
metabolism. For example, phospholipids PC (34:1) and PC (34:0) were up-regulated in STR
after 1 day withdrawal, which could contribute to the membrane disruption28 induced by
cocaine treatment. Another example is that increased GABA and glutamate were observed in
STR and NAC, which is also consistent with previous studies33.

140
5.4 Conclusions
HT-IMMS is a novel and powerful analytical method for measuring the dynamic and
complex metabolomes of rat brain tissues. By coupling HPLC and ion mobility spectrometry
with mass spectrometry, global metabolomics detection is achieved less than 10 minutes with
improved metabolite response and less noise when compared with MS alone. Global
metabolic differences occur with cocaine self-administration after 1-day withdrawal, but after
3-wks withdrawal, the metabolomes return to near normal levels. Several metabolites show
brain-region specific changes. Dysregulation of creatine, creatinine, glutathione and
adenosine support a multi-event mechanism of cocaine addiction. The metabolomics
approach using ion mobility mass spectrometry has potential as a powerful analytical method
to help elucidate the complex and molecular mechanisms of cocaine addiction.
Acknowledgement
This project was supported in part by funds provided for medical and biological
research by the State of Washington Initiative Measure No.171.

141
Reference
(1) Nnadi, C. U.; Mimiko, O. A.; McCurtis, H. L.; Cadet, J. L. J Natl Med Assoc 2005,
97, 1504–1515.
(2) Goeders, N. E. J. Pharmacol. Exp. Ther. 2002, 301, 785–789.
(3) Shaham, Y.; Erb, S.; Stewart, J. Brain Res. Brain Res. Rev. 2000, 33, 13–33.
(4) Robbins, T. W.; Everitt, B. J. Neurobiol Learn Mem 2002, 78, 625–636.
(5) Kalivas, P. W.; O'Brien, C. Neuropsychopharmacology 2008, 33, 166–180.
(6) Kovacic, P. Medical Hypotheses 2005, 64, 350–356.
(7) Ng, F.; Berk, M.; Dean, O.; Bush, A. I. The International Journal of
Neuropsychopharmacology 2008, 11, 851–876.
(8) Volkow, N. D.; Wang, G.-J.; Fowler, J. S.; Tomasi, D.; Telang, F. Proc. Natl. Acad.
Sci. U.S.A. 2011, 108, 15037–15042.
(9) Bossert, J. M.; Ghitza, U. E.; Lu, L.; Epstein, D. H.; Shaham, Y. European Journal
of Pharmacology 2005, 526, 36–50.
(10) Weiss, F. Current Opinion in Pharmacology 2005, 5, 9–19.
(11) Zaitsu, K.; Miyawaki, I.; Bando, K.; Horie, H.; Shima, N.; Katagi, M.; Tatsuno, M.;
Bamba, T.; Sato, T.; Ishii, A.; Tsuchihashi, H.; Suzuki, K.; Fukusaki, E. Anal
Bioanal Chem 2013, 406, 1339–1354.
(12) Nordström, A.; Lewensohn, R. J Neuroimmune Pharmacol 2009, 5, 4–17.
(13) Michell, A. W.; Mosedale, D.; Grainger, D. J.; Barker, R. A. Metabolomics 2008, 4,
191–201.

142
(14) Hu, Z.; Deng, Y.; Hu, C.; Deng, P.; Bu, Q.; Yan, G.; Zhou, J.; Shao, X.; Zhao, J.; Li,
Y.; Zhu, R.; Xu, Y.; Zhao, Y.; Cen, X. Behavioural Brain Research 2012, 231, 11–
19.
(15) Patkar, A. A.; Rozen, S.; Mannelli, P.; Matson, W.; Pae, C.-U.; Krishnan, K. R.;
Kaddurah-Daouk, R. Psychopharmacology 2009, 206, 479–489.
(16) Li, Y.; Yan, G. Y.; Zhou, J. Q.; Bu, Q.; Deng, P. C.; Yang, Y. Z.; Lv, L.; Deng, Y.;
Zhao, J. X.; Shao, X.; Zhu, R. M.; Huang, Y. N.; Zhao, Y. L.; Cen, X. B.
Neuroscience 2012, 218, 196–205.
(17) Kaplan, K. A.; Chiu, V. M.; Lukus, P. A.; Zhang, X.; Siems, W. F.; Schenk, J. O.;
Hill, H. H. Anal Bioanal Chem 2013, 405, 1959–1968.
(18) Dwivedi, P.; Schultz, A. J.; Jr, H. H. H. International Journal of Mass Spectrometry
2010, 298, 78–90.
(19) Harry, E. L.; Weston, D. J.; Bristow, A. W. T.; Wilson, I. D.; Creaser, C. S. Journal
of Chromatography B 2008, 871, 357–361.
(20) Zhang, X.; Knochenmuss, R.; Siems, W. F.; Liu, W.; Graf, S.; Hill, H. H. Anal.
Chem. 2014, 86, 1661–1670.
(21) Brown, T. E.; Lee, B. R.; Sorg, B. A. Learn. Mem. 2008, 15, 857–865.
(22) Kaplan, K.; Graf, S.; Tanner, C.; Gonin, M.; Fuhrer, K.; Knochenmuss, R.; Dwivedi,
P.; Hill, H. H., Jr. Anal. Chem. 2010, 82, 9336–9343.
(23) Antweiler, R. C.; Taylor, H. E. Environ. Sci. Technol. 2008, 42, 3732–3738.
(24) Tenori, L.; Oakman, C.; Claudino, W. M.; Bernini, P.; Cappadona, S.; Nepi, S.;

143
Biganzoli, L.; Arbushites, M. C.; Luchinat, C.; Bertini, I.; Di Leo, A. Molecular
Oncology 2012, 6, 437–444.
(25) Wishart, D. S.; Knox, C.; Guo, A. C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D. D.;
Psychogios, N.; Dong, E.; Bouatra, S.; Mandal, R.; Sinelnikov, I.; Xia, J.; Jia, L.;
Cruz, J. A.; Lim, E.; Sobsey, C. A.; Shrivastava, S.; Huang, P.; Liu, P.; Fang, L.;
Peng, J.; Fradette, R.; Cheng, D.; Tzur, D.; Clements, M.; Lewis, A.; De Souza, A.;
Zuniga, A.; Dawe, M.; Xiong, Y.; Clive, D.; Greiner, R.; Nazyrova, A.;
Shaykhutdinov, R.; Li, L.; Vogel, H. J.; Forsythe, I. Nucleic Acids Research 2009,
37, D603–D610.
(26) Lebon, V.; Petersen, K. F.; Cline, G. W.; Shen, J.; Mason, G. F.; Dufour, S.; Behar,
K. L.; Shulman, G. I.; Rothman, D. L. J. Neurosci. 2002, 22, 1523–1531.
(27) Shanta, S. R.; Choi, C. S.; Lee, J. H.; Shin, C. Y.; Kim, Y. J.; Kim, K. H.; Kim, K. P.
The Journal of Lipid Research 2012, 53, 1823–1831.
(28) Li, Y.; Yan, G. Y.; Zhou, J. Q.; Bu, Q.; Deng, P. C.; Yang, Y. Z.; Lv, L.; Deng, Y.;
Zhao, J. X.; Shao, X.; Zhu, R. M.; Huang, Y. N.; Zhao, Y. L.; Cen, X. B.
Neuroscience 2012, 218, 196–205.
(29) Dringen, R. Progress in Neurobiology 2000, 62, 649–671.
(30) Dietrich, J.-B.; Mangeol, A.; Revel, M.-O.; Burgun, C.; Aunis, D.; Zwiller, J.
Neuropharmacology 2005, 48, 965–974.
(31) Matuskey, D.; Pittman, B.; Forselius, E.; Malison, R. T.; Morgan, P. T. Drug and
Alcohol Dependence 2011, 115, 62–66.

144
(32) Yang, S.-L.; Han, J.-Y.; Kim, Y.-B.; Nam, S.-Y.; Song, S.; Hong, J. T.; Oh, K.-W.
Arch. Pharm. Res. 2011, 34, 281–287.
(33) Kalivas, P. Current Opinion in Pharmacology 2004, 4, 23–29.

145
Table 5.1 Sample details and cocaine self-administration results including the total number of
active lever presses and the total number of rewards accumulated.
Treatment Sample size (n) Active lever presses Rewards accumulated 1 day_Cocaine 3 897 ± 37 321 ± 15 1 day_Saline 3 200 ± 68 68 ± 28
3 wks_Cocaine 4 513 ± 67 194 ± 15 3 wks_Saline 3 278 ± 32 118 ± 11
Table 5.2 Sample list with number of reproducible major metabolite features (counts>50)
detected by HT-IMMS in each sample group.
STR_1 day withdrawal STR_3 wks withdrawal Saline Cocaine Saline Cocaine 128 135 98 109
PFC_1 day withdrawal PFC_3 wks withdrawal Saline Cocaine Saline Cocaine 125 115 119 120
NAC_1 day withdrawal NAC_3 wks withdrawal Saline Cocaine Saline Cocaine 125 121 105 114

146
Table 5.3 Lists of potential biomarkers generated from loadings plots and univariate analysis.
Information includes measured m/z, p-value of t-test, up/down regulation caused by cocaine
self-administration, adduct form, and identified metabolite name, are listed in table.
Biomarkers labeled in bold were selected from loadings’ plots. Metabolite names labeled
with “*” are identified to be common metabolites in tissues with large abundance, and the
ones labeled with “**” matched with previously studies.
Striatum (1 day withdrawal) Metabolite m/z Ko P value In Cocaine Adduct Metabolite Name
1 90.08 2.08 0.006 UP N/A N/A
2 91.05 1.76 0.025 UP [M+H-15]+ Serine*
3 104.10 1.96 0.388 DOWN [M+H]+ Choline*
4 114.06 1.86 0.201 UP [M+H]+ Creatinine*
5 127.06 1.84 0.015 UP [M+H]+ 5-Aminoimidazole-4-carboxamide*
7 136.05 1.67 0.060 DOWN [M+H]+ Homocysteine*
8 146.11 1.69 0.034 DOWN [M+H]+ Acetylcholine**
9 152.02 1.72 0.240 UP [M+Na]+ Pyroglutamic acid (5-Oxoproline)*
10 158.03 1.62 0.009 DOWN [M+Na]+ Homocysteine*
11 162.08 1.64 0.041 DOWM [M+H]+ Aminoadipic acid**
12 162.11 1.63 0.024 DOWN [M+H]+ Carnitine**
13 176.05 1.61 0.003 DOWN [M+H]+ N-Acetyl-L-aspartic acid*
14 223.08 1.44 0.046 UP [M+H]+ Cystathionine**
15 268.11 1.34 0.430 UP [M+H]+ Adenosine**
16 280.16 1.28 0.033 UP N/A N/A
17 313.28 1.00 0.034 UP [M+H-H2O]+ MAG(16:0)**
18 331.11 0.96 0.034 UP [M+K]+ Phenylbutyrylglutamine
19 339.29 0.97 0.037 UP [M+H-H2O]+ MAG(18:1)**
20 392.31 0.88 0.031 UP N/A N/A
21 574.93 0.92 0.044 DOWN N/A N/A
22 731.61 0.58 0.007 UP [M+H]+ SM(d18:0/18:1(9Z))**
23 798.55 0.58 0.043 UP [M+K]+ PC(34:1)**
24 800.57 0.57 0.016 UP [M+K]+ PC(34:0)**

147
Striatum (3 wks withdrawal) Metabolite m/z Ko P value In Cociane Adduct Metabolite Name
1 102.06 1.95 0.038 UP N/A N/A
2 114.06 1.87 0.223 UP [M+H]+ Creatinine*
3 146.16 1.66 0.278 UP [M+H]+ Spermidine**
4 189.06 1.61 0.034 DOWN [M+Na]+ 4-Methoxyphenylacetic acid*
5 199.01 1.41 0.037 DOWN [M+H]+ 1-acylglycerone 3-phosphate*
6 202.15 1.47 0.007 UP [M+H]+ Capryloylglycine*
7 212.12 1.38 0.021 DOWN N/A N/A
8 227.12 1.41 0.340 UP [M+H]+ Carnosine**
9 268.11 1.34 0.177 UP [M+H]+ Adenosine**
10 277.10 1.22 0.017 DOWN [M+H]+ Glu-Glu
11 308.10 1.21 0.042 DOWN [M+H]+ Glutathione*
12 341.31 0.94 0.008 DOWN [M+H-H2O]+ MAG(18:0)**
13 365.17 1.14 0.003 DOWN N/A N/A
14 409.20 1.07 0.015 DOWN [2M+H]+ Tryptophan*
Prefrontal Cortex (1 day withdrawal) Metabolite m/z Ko P value In Cociane Adduct Metabolite Name
1 87.06 1.91 0.002 UP [M+H]+ But-2-enoic acid
2 102.07 1.98 0.002 UP [M+H]+ 1-Amino-1-cyclopropane**
3 104.08 1.91 0.679 UP [M+H]+ GABA**
4 114.07 1.89 0.216 UP [M+H]+ Creatinine*
5 123.05 1.82 0.007 UP [M+H]+ Niacinamide (Vitamin B3)*
6 132.08 1.81 0.556 DOWN [M+H]+ Creatine*
7 136.07 1.81 0.003 UP [M+H]+ Adenine*
8 148.07 1.71 0.170 DOWN [M+H]+ Glutamate*
9 166.96 1.70 0.004 DOWN N/A N/A
10 169.06 1.64 0.024 DOWN [M+Na]+ Glutamine*
11 170.04 1.66 0.054 DOWN [M+Na]+ Glutamate*
12 170.09 1.66 0.016 UP [M+H]+ 6-Hydroxydopamine**
13 171.10 1.57 0.030 DOWN N/A N/A
14 176.07 1.63 0.023 DOWN [M+H]+ N-Acetyl-L-aspartic acid*
14 194.05 1.57 0.003 DOWN [M+H]+ Dopachrome**
16 223.00 1.51 0.004 DOWN [M+Na]+ D-Erythrose 4-phosphate*
17 268.11 1.34 0.661 UP [M+H]+ Adenosine**
18 275.06 1.38 0.023 DOWN N/A N/A
19 296.07 1.26 0.025 DOWN [M+H]+ 5-Aminoimidazole Ribonucleotide

148
Prefrontal Cortex (3 wks withdrawal) Metabolite m/z Ko P value In Cociane Adduct Metabolite Name
1 116.08 1.84 0.020 UP [M+H]+ Proline*
2 141.10 1.77 0.030 UP N/A N/A
3 153.14 1.65 0.022 UP N/A N/A
4 166.10 1.55 0.024 UP [M+H]+ Phenylalanine*
5 227.13 1.42 0.000 DOWN [M+H]+ Carnosine**
6 251.98 1.39 0.001 DOWN [M+K]+ L-Aspartyl-4-phosphate
7 369.36 0.95 0.029 UP [M-H2O+H]+ Cholesterol**
Nucleus Accumbens (1 day withdrawal) Metabolite m/z Ko P value In Cociane Adduct Metabolite Name
1 96.04 2.04 0.037 Down [M+Na]+ Aminoacetone
2 106.05 1.88 0.035 Down [M+H]+ Serine*
3 120.07 1.81 0.048 Down [M+H]+ Threonine*
4 123.05 1.83 0.455 Down [M+H]+ Niacinamide (Vitamin B3)*
5 130.05 1.72 0.02 Down [M+H]+ Pyroglutamic acid (5-Oxoproline)*
6 132.08 1.81 0.038 Down [M+H]+ Creatine*
7 147.08 1.72 0.16 Down [M+H]+ Glutamine*
8 157.09 1.66 0.016 Up N/A N/A
9 168.05 1.37 0.023 Up [M+H]+ 8-hydroxyguanine (8-OHG)*
10 170.04 1.65 0.56 Up [M+Na]+ Glutamate*
11 173.06 1.65 0.003 Up [M+H]+ Menadione (Vitamine K3)*
12 187.07 1.57 0.011 Up [M+H]+ AMPA**
13 204.13 1.46 0.006 Down [M+H]+ Succinylmonocholine**
14 237.12 1.33 0.036 Down [M+H]+ Pro-Val/Val-Pro
15 268.11 1.34 0.76 Down [M+H]+ Adenosine**
16 385.36 0.94 0.039 Down [M+H]+ cholestadienol
Nucleus Accumbens (3 wks withdrawal) Metabolite m/z Ko P value In Cociane Adduct Metabolite Name
1 105.03 1.94 0.008 DOWN [M+H]+ Urea-1-carboxylate
2 111.05 1.93 0.001 UP [M+Na]+ Butyric acid*
3 114.06 1.89 0.033 UP [M+H]+ Creatinine*
4 119.03 1.80 0.050 UP [M+H]+ Succinic acid*
5 126.05 1.85 0.028 DOWN N/A N/A
6 130.05 1.73 0.021 UP [M+H]+ Pyroglutamic acid (5-Oxoproline)**
7 136.06 1.80 0.046 UP [M+H]+ Adenine*
8 147.07 1.73 0.045 UP [M+H]+ Glutamine*

149
9 150.06 1.74 0.038 UP [M+H]+ Methionine*
10 161.12 1.63 0.041 DOWN [M+H]+ Tryptamine**
11 173.07 1.64 0.046 UP [M+H]+ Menadione (Vitamine K3)*
12 209.08 1.50 0.023 DOWN [M+H]+ Kynurenine**
13 268.10 1.34 0.047 UP [M+H]+ Adenosine**
14 298.10 1.26 0.042 DOWN [M+Na]+ Gamma-Glutamylglutamine*
15 453.19 0.94 0.033 DOWN [M+H]+ Derrichalcone
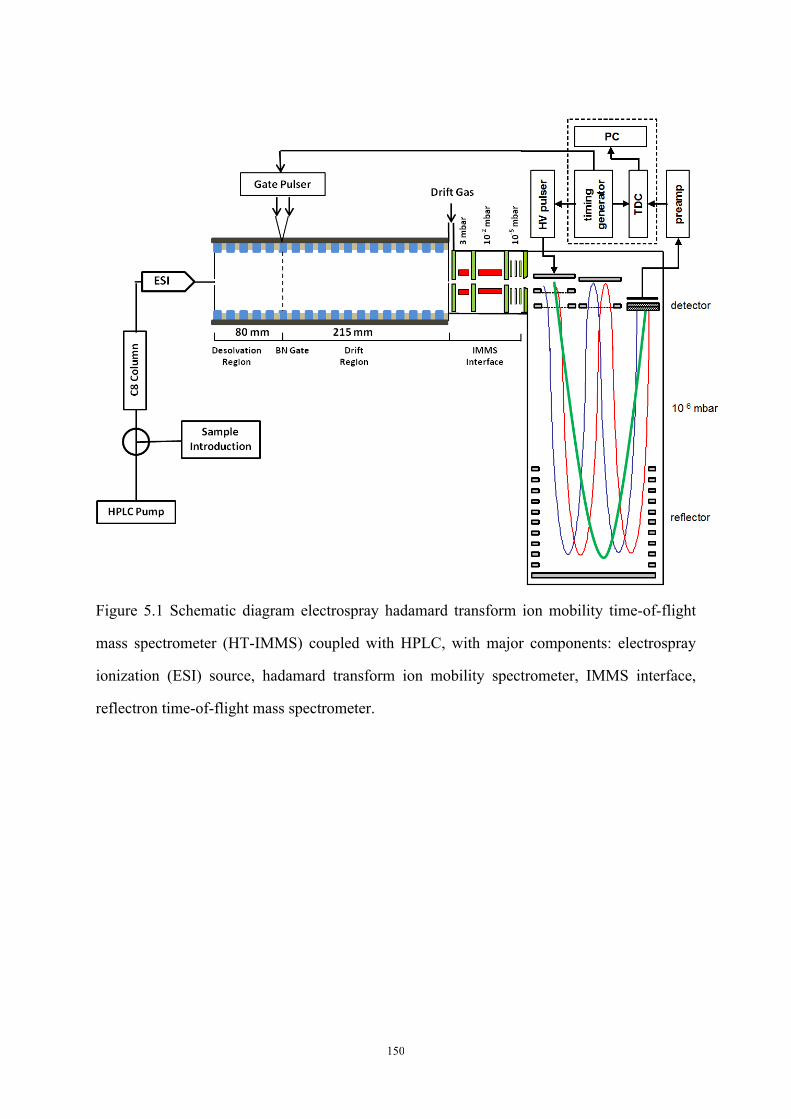
150
Figure 5.1 Schematic diagram electrospray hadamard transform ion mobility time-of-flight
mass spectrometer (HT-IMMS) coupled with HPLC, with major components: electrospray
ionization (ESI) source, hadamard transform ion mobility spectrometer, IMMS interface,
reflectron time-of-flight mass spectrometer.

151
Figure 5.2 IMMS 2-D spectrum of the metabolomes of striatal tissue is displayed in (a), with
x-axis representing m/z (ranges from 100 to 950) and y-axis representing drift time (ranges
from 15 ms to 75ms). The corresponding mass spectrum and ion mobility spectrum are also

152
shown in the figure to illustrate the improvement of 2-D detection when compared with the
1-D spectra. (b) illustrates the global metabolomes of two PFC samples obtained from
repeated saline treated rats with 1-day withdrawal (b1 and b2) and PFC sample obtained from
cocaine treated rats with 1-day withdrawal (b3) using ion mobility spectra.

153
Figure 5.3 PCA score plots of six comparisons: global metabolomes of STR/PFC/NAC
obtained from cocaine treated rats with 1 day withdrawal vs. their corresponding saline
controls, and global metabolomes of STR/PFC/NAC obtained from cocaine
self-administering rats with 3 wks withdrawal vs. their corresponding saline controls. PC-1
and PC-2 represent x-axis and y-axis, and 60% - 80% of the variation is explained in each
score plot.

154
Figure 5.4 Dysregulation of creatine and creatinine in STR/PFC/NAC with 1-day withdrawal.
The magnitudes of the bars represent the average relative intensity value, error bars represent
±SD and asterisk indicates statistical difference (p value < 0.05).
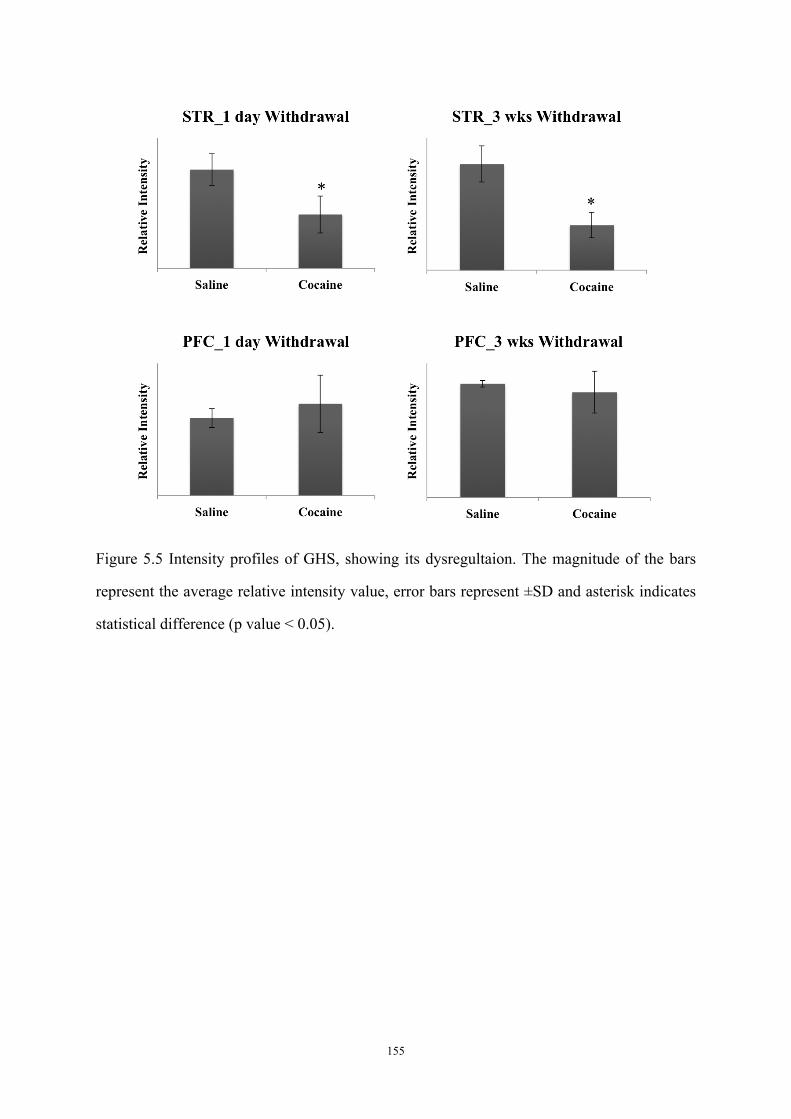
155
Figure 5.5 Intensity profiles of GHS, showing its dysregultaion. The magnitude of the bars
represent the average relative intensity value, error bars represent ±SD and asterisk indicates
statistical difference (p value < 0.05).

156
Figure 5.6 Dysregulation of adenosine in STR/PFC/NAC with 1-day withdrawal and 3-wks
withdrawal. The magnitude of the bars represent the average relative intensity value, error
bars represent ±SD and asterisk indicates statistical difference (p value < 0.05).

157
Chapter 6
Metabolomics of Plasma Fluids from Apolipoprotein AV
Knockout Mice by Hadamard Transform Ambient Pressure Ion
Mobility Time-of-Flight Mass Spectrometry
Abstract
The critical role of apolipoprotein AV (apoAV) in regulating plasma triglyceride level
has been demonstrated but the mechanism remains unresolved. Metabolomics of plasma
fluids from apoAV knockout (KO) mice and wild type (WT) mice under fasted and ad lib fed
dietary treatments was investigated to provide metabolic alternations associated with apoAV.
This is the first comprehensive metabolomics study on apoAV. Hadamard transform ambient
pressure ion mobility time-of-flight mass spectrometry (HT-apIMtofMS) was employed to
perform rapid metabolomics analysis with high resolving power (Rp) separation based on
ion’s size to charge ratio and accurate detection of ion’s mass to charge ratio (m/z). ~1000
metabolite features were detected within 3 minutes, and 40 major metabolite features were
reported and identified based on their accurate m/z values and reduce mobility (K0) values.
Global metabolic analysis by principle component analysis (PCA) on biologically
reproducible metabolite features demonstrated significantly different metabolic patterns for
the four groups of samples. During the specific metabolic analysis, we discovered a “mirror
effect” of dietary treatments on the effect of apoAV on lysophosphatidylcholine related

158
metabolites. Increase of monoacylglyceride class of metabolites caused by the absence of
apoAV was also observed. The separation of glucose and fructose enabled us to monitor the
metabolic alternation of glucose without the interference of its isomer. We found that apoAV
KO subjects have 2-fold increased glucose level when compared with WT, while fructose
level seemed to remain unchanged.

159
6.1 Introduction
Apolipoproteins bind proteins and lipids, playing an important role in lipid and
lipoprotein metabolism. Apolipoprotein AV (ApoAV)1, a 39-kDa protein with 343 residues,
was discovered in 2001 and found to significantly affect plasma triglyceride level with its low
abundance (<1 ug/ml in plasma). The physiological role of apoAV was evaluated by
Pennacchio et al. using transgenic mice that overexpress apoAV protein as well as mice
lacking apoAV2. The 4-fold increased triacylglyceride level in apoAV knockout mice
demonstrated the critical feature of apoAV in modulating plasma lipid homeostasis.
Epidemiological studies further proved that mutations in apoAV have been associated with
hyperglyceridemia3, coronary artery disease4 and type 2 diabetes5 in human and mice.
Knowing that apoAV is located in the apolipoprotein gene cluster in chromosome 11,
and that it’s exclusively synthesized in liver, researchers have successfully developed human
apoAV transgenic mice, allowing specific physiological studies of apoAV. Mechanisms have
been proposed, with regard to its intracellular function of associating with lipid droplets in
the hepatocyte6 and its extracellular function in accelerating the removal of TG-rich
lipoproteins7. More recent investigations associated apoAV with non-alcoholic fatty liver
disease8 and intestinal lipid absorption9. However, the fact that apoAV is present at very low
abundance in plasma leads to the question of its profoundly role in lipid metabolism. The
precise mechanism of apoAV is not completely understood despite the convincing evidence
of its effects. A careful search of the literatures has failed to yield any information linking

160
apoAV with metabolomes other than lipids.
Global metabolomics reveals the identities, quantities of a wide range of diverse
metabolites, providing complementary information to the physiological studies on apoAV.
Metabolomics of plasma has been extensively studied10,11, therefore, the metabolic profiling
as well as the target metabolites including glucose, amino acids, cholesterol and fatty acids
are partly known. The global metabolomics on plasma from apoAV gene modified mice and
wild type (WT) mice monitors the whole metabolome under the designed conditions,
providing metabolic information to reveal more metabolic effects of apoAV in plasma and
enhance our knowledge for understanding the mechanism.
Conventional instrumentations12 used in global metabolomics include nuclear
magnetic resonance (NMR)13, liquid chromatography-mass spectrometry (LC-MS)14 and gas
chromatography-mass spectrometry (GC-MS)15, these methods provide valuable metabolic
information but suffer from either low sensitivity or long analysis time. Hadamard Transform
atmospheric pressure ion mobility time-of-flight mass spectrometry (HT-apIMtofMS)16, a
technique capable of detecting hundreds of metabolites within a few minutes, has been
developed and validated using tissue and fluid extracts.
The general method of ion mobility mass spectrometry (IMMS) has been applied on
various metabolomics studies17-19 and showed its power of sensitive and efficient analysis.

161
Ion mobility spectrometry (IMS) is a rapid gas phase separation technique, originally used for
detecting explosives and drugs. Its capability of differencing ions in millisecond time scale
has made this technique a popular stand-alone system. IMS separation is based on the ion’s
size to charge ratio, making separation of isomers and isobars possible. Ions produced by the
ionization source can quickly attain a constant velocity (νd) under the influences of the
driving force from homogeneous electric field and ion–neutral collisions induced by buffer
gas. Reduced mobility K0 is then derived from νd according to the following equation, can
used to characterize an ion’s identity.
𝐾! = !!
!!! × !"#.!"
! × !
!"#
Where V is the voltage drop across the drift region (length L) of IMS, td is the drift time that
the ion takes to migrate through he drift region, P (in Torr) and T (in Kelvin) are the
operating pressure and temperature.
By coupling IMS to mass spectrometer, the power of separation and detection is
largely increased20. There are several advantages that make IMMS a natural fit for global
metabolomics: 1) rapid pre-MS separation when compared with chromatography methods; 2)
high resolving power IMS separation enabling the differentiation of isomers18; 3) correlation
of mass and mobility (MMCCs) 17,21 providing class identifications not possible by other
methods. Last but not least, HT-apIMtofMS increased the sensitivity by 2 orders of
magnitude when compared with traditional IMMS16, leading to strong responses from the
small amount of plasma samples.

162
The purpose of this work is to apply HT-apIMtofMS to metabolomics on plasma
samples generated from apoAV knockout (KO) mice and WT mice for determining global
and specific metabolic changes associated with apoAV. Moreover, the effects of fasted and
fed on plasma metabolome were also studied in order to associate apoAV with dietary stress.
We believe our work will reveal metabolic effects of apoAV on plasma metabolome, filling
the gap of lacking metabolomics study of apoAV. Meanwhile, the metabolic changes induced
by apoAV and dietary stress could potentially help to understand its mechanism.

163
6.2 Experimental Section
6.2.1 Subjects and Plasma Collection:
Adult male WT and apo AV KO mice (15-17 months of age) were used. These WT
and apo AV KO mice were bred in house in a facility accredited by the American Association
for the Accreditation of Laboratory Animal Care and were housed with corn cob bedding
under conditions of controlled illumination (12:12-h light-dark cycle, lights from 0600 to
1800). All mice had free access to standard rodent chow (5% fat, Harlan Laboratories,
Indianapolis, IN) and water unless otherwise stated. All procedures were approved by the
University of Cincinnati Institutional Animal Care and Use Committee.
Apo AV KO and WT mice (n = 2 - 4/group) were randomly assigned into four groups.
For fasting group, mice were fasted for 5 h (from 0900 to 1400) with continuous access to
water. Blood samples (120 µL for 50 µL of plasma) for plasma separation were collected into
heparinized capillary tubes from tails. The plasma was separated from whole blood by
centrifugation for 10 minutes (4000 x g). Plasma fluids were frozen at -20°C within one hour
from sampling and stored at -70°C until analyzed.
A total number of 11 plasma samples were analyzed in this study (n = 4 for apoAV
KO mice that were fasted for 5 hrs; n = 3 for apoAV KO mice that were ad lib fed; n = 2 for
WT mice that were fasted for 5 hrs; and n = 2 for WT mice that were ad lib fed).

164
6.2.2 Metabolite Extraction:
An evaluation on the extraction solvent of plasma metabolites for global
metabolomics was conducted using methanol, acetone and acetonitrile. Acetonitrile was
chosen to be the optimal extraction solvent22. Plasma samples were delivered on dry ice and
stored at -80ºC until further use. A volume of 200 µL of iced acetonitrile was added to each
50 µL plasma sample, and vigorously mixed for 2 minutes before stored in 4ºC for overnight
extraction. The samples were centrifuged for 30 min at 13 kRPM, and supernatant containing
extracted metabolites was collected for each sample. ESI solvent comprising a
49.75:49.75:0.5 v/v/v mixture of water, methanol and acetic acid was added into each
supernatant using a 2:1 v/v ratio (ESI solvent: supernatant) for further dilution.
6.2.3 HT-apIMtofMS Analysis:
Prepared samples were introduced for IMMS analysis by electrospray ionization. A
stacked ring IMS was constructed at Washington State University using alternating stainless
steel rings and ceramic rings, where the stainless steel rings were connected by 1 MΩ
resistors. The desolvation region was 8 cm-long and the drift region was 22 cm-long.
Metabolite ions were desolvated in the desolvation region and pulsed into the drift region by
a Bradbury Nielson (BN) ion gate. The HT gate pulsing sequence was generated from
software developed by Tofwerk (AG, Switzerland) and transferred to the BN gate by a gate
pulser made at Washington State University. The smallest gate pulse width was controlled to
180 µs. The IMS was held in ambient pressure (690 – 705 torr) and at 200ºC.

165
The time-of-flight mass spectrometer (tofMS) was coupled to the IMS by an IMMS
interface composing of lenses, nozzles and two segmented quadrupole ion guides. The IMMS
interface provided high transmission efficiency of the ions separated in IMS to tofMS. In this
study, the IMMS analysis was operated under positive mode, and the tofMS was operated in
V-mode with ~5000 – 7000 resolution. A summary of operational details can be found in
Table 6.1 and the schematic picture of the instrument is shown in Figure 6.1a.
During the analysis, 2,6-ditert-butylpyridine was used for internal calibration at a
concentration of 2 µM in all samples. Each sample was analyzed by duplicate runs to ensure
reproducibility. Each IMMS analysis was completed in 3 minutes.
6.2.4 Data Processing:
IMMS data was collected by TofDAQ version 1.92b and processed by IMSviewer
version 1_6d, both developed by Tofwerk (AG, Switzerland). The IMMS data format is
multidimensional, consisting of mass-to-charge ratio (m/z), reduced mobility (K0) and
intensity (counts) for each metabolite feature. Microsoft adaptable peak list for each sample,
as well as all the spectra, were also generated by IMSviewer.
Multivariate analysis approach called principle component analysis (PCA), a
commonly used statistical method for multidimensional data, was employed in this study for
global metabolomics to yield pattern recognition among the 11 samples. PCA was performed

166
on the peak list that merged the biologically reproducible metabolite features from all 11
individual peak lists. Because PCA is an unsupervised method, the analysis was
accomplished without knowing the sample group information. Score plot displayed sample
grouping based on the global metabolic difference contained in the peak lists, and loadings
plot selected metabolite features that significantly affected the pattern recognition. Selected
metabolite features were found to be altered in intensity among different sample groups,
therefore, were considered as potential biomarkers.
Major metabolite features as well as potential biomarkers were identified based on
their exact m/z with mass tolerance of 0.01 and K0 with standard deviation of 2%.
Identifications based on exact m/z were matched with Human Metabolome Database
(HMDB), METLIN Metabolite and Tandem MS Database, and related literatures;
identifications based on K0 were matched with previous studies.

167
6.3 Results and Discussion
6.3.1 IMMS Multidimensional Spectrum:
Similar spectra from duplicate runs demonstrated the reproducibility of IMMS
analysis. The whole metabolome of plasma was displayed in the IMMS 3D plot shown in
Figure 6.1b, which has m/z (ranges from 100 to 900) as x-axis, drift time (ranges from 15 ms
to 75 ms) as y-axis, and intensity as z-axis. The corresponding mass spectrum and ion
mobility spectrum are also included to explain the complexity of the plasma metabolome.
Moreover, with the separation power combine from both IMS and MS, the analysis noise was
reduced and the number of detected metabolite features was increased. There were 900-1000
metabolite features detected with ion counts >10 and ~300 with ion counts >50 after masking
out the background ions. Only major metabolite features that did not vary biologically in all
the plasma samples were used in PCA analysis for pattern recognition.
Table 6.2 summarized the identifications of 40 major metabolite features (ranked by
their intensities) commonly detected in all samples. The accurate m/z and K0 values were
obtained from IMMS analysis; the metabolite identifications including metabolite name,
chemical formula and adduct form, were obtained by matching the m/z and K0 information
with existing databases and previous studies. A wide range of metabolites including amino
acids, carbohydrates, lipids, steroids, and peptides were simultaneously detected,
demonstrating the capability of the 3 minutes IMMS analysis for comprehensive
metabolomics.

168
6.3.2 Global Metabolic Analysis:
Global metabolic alternations were observed in the spectra collected from different
groups of samples. Figure 6.2 contains representative mass spectra and ion mobility spectra
of plasma samples collected from apoAV KO mice fasted for 5 hrs, WT mice fasted for 5 hrs,
apoAV KO mice ad lib fed and WT mice ad lib fed. The mass spectra with m/z ranging from
100 to 900 shared similar major mass peaks, due to the fact that the major metabolite ions
were consistent despite different treatments. In the meanwhile, differences can be observed in
relative intensities for multiple mass peaks. For example, boxed area in the mass spectra was
the m/z region where phospholipids were detected. Decrease in intensity for phospholipids
was observed when comparing fasted apoAV KO mice plasma with fasted WT mice plasma.
In contrast, an increase trend was evident in the ad lib fed mice plasma. In the meanwhile, the
ion mobility spectra collected from the 4 groups of samples showed the same intensity
differences for phospholipids. In general, ion mobility spectra differentiated global metabolic
patterns more easily and clearly when compared with the mass spectra because of the screen
nature of IMS. In practice, it is possible to perform the IMMS analysis of a specific plasma
sample, and identify its classification simply based on its ion mobility spectrum.
Despite observable global metabolic alternation demonstrated by visual inspection of
spectra, further analysis showed the difference statistically. As mentioned earlier, PCA was
performed before knowing the sample group information. Figure 6.3 shows the PCA results
including score plot and loadings plot after assigning the sample group information. Both

169
plots were 2D plots with the first principle component (PC-1) and the second principle
component (PC-2) explaining 45% and 20% of the metabolic variation, respectively. Tight
clustering of WT samples demonstrated good biological reproducibility of WT mice within
the same dietary treatment. However, the grouping of apoAV KO samples was not as tight as
WT samples, indicating more biological variation within the same dietary treatment of
apoAV mice. Despite the existence of biological variance, the score plot still yielded a clear
metabolic pattern that distinguished the four sample groups. Loadings plot, instead of
investigating sample clustering, looked into the metabolite features and determined the ones
that significantly influenced the clustering pattern. Considering outliers as influential
metabolite features, we were able to target seven outliers and identify six of them. Their
identifications were PC (18:0), PC (16:0), PE (16:0), monoradylglycerol (18:0),
phosphocreatinine and choline. As influential metabolites, they were bound to be associated
with either apoAV KO or dietary treatment. PCA allowed visualizing the sample grouping
truly based on the metabolic information, which in this case, was the peak list of metabolite
features generated from IMMS analysis. Therefore, it is evident that both dietary treatment
and apoAV significantly changed plasma metabolome.
6.3.3 Specific Metabolic Alternations:
In order to examine the specific metabolic alternations of the potential biomarkers
selected by PCA loadings plot. ANOVA was used (with 95% confidence interval) to
determine the statistical difference in the relative intensities of a specific metabolite among

170
the four groups of samples. As shown in Figure 6.4 and Figure 6.5, the magnitudes of the
bars represent the average relative intensity values, and the error bars represent ± SD. The
asterisk indicates that statistical difference (p value < 0.05) exited between different sample
groups for the target metabolite. An interesting finding from the loading’s plot and bar graphs
is that the intensities of potential biomarkers (outliers in loading’s plot) changed similarly if
they are near one another in the loading’s plot, examples were shown below.
Lysophospholipids: Figure 6.4 illustrates the metabolic alternations for phospholipid
class of metabolites. In PCA loadings plot, choline and lysophosphatidylcholine (lysoPC)
(16:0) were targeted as potential biomarkers and they appeared in the same area (lower right
corner) in the plot. We found out that they shared the same metabolic alternation trend as
shown in 6.4a. In addition to choline and lysoPC (16:0), more lysoPC metabolites appeared
to have the same alternation trend. The bar graph in 6.4a revealed an increase in the lysoPC
for apoAV KO mice plasma when compared with WT mice plasma under fasted dietary
treatment. However, decrease in intensities of lysoPC metabolites was evident in the KO
mice plasma under the ad lib fed dietary treatment. The above results also matched with the
previous discussions on MS and IMS spectra. The effect of the apoAV protein on plasma
lysoPC level was obvious, however, the effect for subjects under ad lib fed was opposite with
the effect for fasted subjects. This “mirror effect” of dietary treatment on the function of
apoAV protein was firstly discovered and it proved that environment stress such as dietary is
not negligible for the investigation of apoAV.

171
As shown in Figure 6.4b, another two potential biomarkers (lysoPC (18:0) and lyso
PE (16:0)) that appeared to be in the same area (upper right corner) in the loadings plot, also
displayed similar metabolic alternation trend. Different from lysoPC (16:0) and choline, their
intensity changes caused by apoAV was not affected by dietary treatment. Decreased
intensities for the above two biomarkers were discovered for apoAV KO mice plasma when
compared with WT mice plasma. It is well known that lysophospholipid class of compounds
can be involved in lipid signaling by binding protein targets, and their functions in lipid
signaling can be vastly different. This partly explained the difference in alternation trend
displayed in 6.4a and 6.4b. Correlation between apoAV and the biological functions of
lysoPC (18:0) and lyso PE (16:0) could lead to a better understanding of their alteration
trends.
Carbohydrates: As one of the most important metabolites in plasma, glucose was
detected and specifically investigated using bar graph as shown in Figure 6.5a. Its metabolic
alternation was clearly affected by both apoAV and dietary treatment. First of all, it is evident
that the average intensity of glucose was significantly higher in the ad lib fed subjects than
the fasted subjects, indicating that 5 hrs fasting treatment had effectively decreased the
glucose level. Secondly, the glucose level was consistently higher in the apoAV KO subjects
than WT subjects, in both fasted and fed treatments. This result suggested that apoAV
disrupted the metabolic homeostatic of glucose, and lack of apoAV induced the up regulation
of glucose. Therefore, the result supported previous studies indicating that apoAV could be

172
associated with diabetes.
There are many isomers existing as monosaccharides, and glucose is known for the
most common one. To further exam their existence in plasma, we performed selected-mass
mobility analysis on monosaccharide. Figure 6.6 shows the selected-mass mobility spectra of
[C6H12O6+Na]+ (selected m/z = 203.06), with x-axis assigned with drift time (ms) and y-axis
assigned with absolute intensity (counts). The spectra generated from apoAV KO plasma
samples were shown on the top with red trace representing ad lib fed sample and blue trace
representing fasted sample, and the spectra for WT plasma samples were on the bottom. It is
evident that two distinguishable ion mobility peaks under the exact same m/z were observed,
demonstrating the capability of IMMS analysis for distinguishing isomers with high Rp (Rp =
~90 for the early peak and Rp = ~60 for the later peak). To identify these two
monosaccharides, their K0 values were calculated to be 1.53 cm2V-1s-1 for the earlier peak and
1.46 cm2V-1s-1 for the later peak. By matching the K0 values with previous studies on
monosaccharide standards by Dwivedi et al., we were able to identify these two peaks as
[Glucose+Na]+ (K0 = 1.53 cm2V-1s-1), and [Fructose+Na]+ (K0 = 1.46 cm2V-1s-1).
Separation of monosaccharide isomers enabled analysis of the metabolic alternation
for glucose without the interference of its isomers. The absolute intensity values of glucose
indicated a 2-fold increase in plasma caused by the absence of apoAV, and a 3-fold decrease
caused by fasted dietary treatment. Moreover, a close look at the later mobility peak revealed
that the fructose level in plasma was not significantly affected by apoAV, indicating the

173
mechanism of apoAV in affecting “sugar level” might be different for different
monosaccharide compounds.
Monoacylglyceride: MG (18:0) was also selected in PCA as a potential biomarker. Its
metabolic alternation was shown in Figure 6.5b. We found that the alternation trend of MG
(18:0) was similar compared with glucose, demonstrating the similar up regulation caused by
the absence of apoAV and down regulation caused by fasted treatment. Beside MG (18:0),
another two MG metabolites were detected as major peaks in Table 6.2 and they were MG
(16:0) and MG (18:2). As expected, the alternation trend of MG (16:0) and MG (18:2)
matched with MG (18:0), indicating that apoAV decreased the MG level in plasma. With the
previous knowledge of the triglyceride lowering function of apoAV, this finding partly
supported previous investigations because that MG and TG have similarity in both structure
and biological activity. The metabolic change of triglyceride is already known from previous
study and can be more effectively measured in negative ion mode. However, our study was
performed in positive ion mode in order to reveal metabolic changes not yet known.

174
6.4 Conclusions
HT-apIMS-tofMS provides three minutes metabolomics analysis and generates
multidimensional spectra containing ~1000 detected metabolites from plasma fluids. PCA
yields significant pattern differences among different sample groups. A “mirror effect” of
dietary treatments on the function of apoAV on certain lysoPC compounds is discovered,
indicating the dietary stress could have affected some functions of apoAV. Another finding
from this study is the detection and separation of glucose and fructose, allowing us to
investigate the metabolic alternation of these isomers individually. While apoAV KO subjects
have higher glucose level than the WT, the fructose level is not affected. Last but not least,
MG class of metabolites is up regulated due to the absence of apoAV, partly supporting the
TG lowering effect of apoAV. The metabolomics study on plasma fluids from designed
apoAV KO mice model generated rich metabolic information that could potentially be used
to resolve the puzzlement of apoAV. Further studies including analyzing the corresponding
lymph fluid metabolomes, as well as performing IMMS analysis in negative mode, could
yield more complementary information about the mechanism of apoAV

175
Reference
(1) Beckstead, J. A.; Oda, M. N.; Martin, D. D. O.; Forte, T. M.; Bielicki, J. K.;
Berger, T.; Luty, R.; Kay, C. M.; Ryan, R. O. Biochemistry 2003, 42, 9416–9423.
(2) Pennacchio, L. A. Science 2001, 294, 169–173.
(3) Priore Oliva, C.; Pisciotta, L.; Li Volti, G.; Sambataro, M. P.; Cantafora, A.;
Bellocchio, A.; Catapano, A.; Tarugi, P.; Bertolini, S.; Calandra, S. Arterioscler
Thromb Vasc Biol 2005, 25, 411–417.
(4) Vaessen, S. F. C.; Schaap, F. G.; Kuivenhoven, J.-A.; Groen, A. K.; Hutten, B.
A.; Boekholdt, S. M.; Hattori, H.; Sandhu, M. S.; Bingham, S. A.; Luben, R.; Palmen,
J. A.; Wareham, N. J.; Humphries, S. E.; Kastelein, J. J. P.; Talmud, P. J.; Khaw,
K.-T. The Journal of Lipid Research 2006, 47, 2064–2070.
(5) Talmud, P. J.; Cooper, J. A.; Hattori, H.; Miller, I. P.; Miller, G. J.; Humphries,
S. E. Diabetologia 2006, 49, 2337–2340.
(6) Shu, X.; Chan, J.; Ryan, R. O.; Forte, T. M. The Journal of Lipid Research 2007,
48, 1445–1450.
(7) Merkel, M. Journal of Biological Chemistry 2005, 280, 21553–21560.
(8) Sharma, V.; Ryan, R. O.; Forte, T. M. … et Biophysica Acta (BBA)-Molecular
and … 2012.
(9) Yoshino, Y.; Okada, T.; Abe, Y.; Odaka, M.; Kuromori, Y.; Yonezawa, R.;
Iwata, F.; Mugishima, H. Obesity Research & Clinical Practice 2013, 7, e415–e419.
(10) Li, W.; Tse, F. L. S. Biomed. Chromatogr. 24, 49–65.

176
(11) Bruce, S. J.; Jonsson, P.; Antti, H.; Cloarec, O.; Trygg, J.; Marklund, S. L.;
Moritz, T. Analytical Biochemistry 2008, 372, 237–249.
(12) Dunn, W. B.; Ellis, D. I. TrAC Trends in Analytical Chemistry 2005, 24, 285–
294.
(13)Hu, Z.; Deng, Y.; Hu, C.; Deng, P.; Bu, Q.; Yan, G.; Zhou, J.; Shao, X.; Zhao, J.;
Li, Y.; Zhu, R.; Xu, Y.; Zhao, Y.; Cen, X. Behavioural Brain Research 2012, 231,
11–19.
(14) Lu, W.; Bennett, B. D.; Rabinowitz, J. D. Journal of Chromatography B 2008,
871, 236–242.
(15) Koek, M. M.; Muilwijk, B.; van der Werf, M. J.; Hankemeier, T. Anal. Chem.
2006, 78, 1272–1281.
(16) Zhang, X.; Knochenmuss, R.; Siems, W. F.; Liu, W.; Graf, S.; Hill, H. H. Anal.
Chem. 2014, 86, 1661–1670.
(17) Kaplan, K.; Dwivedi, P.; Davidson, S.; Yang, Q.; Tso, P.; Siems, W.; Hill, H. H.,
Jr. Anal. Chem. 2009, 81, 7944–7953.
(18) Lapthorn, C.; Pullen, F.; Chowdhry, B. Z. Mass Spectrom. Rev. 2012, 32, 43–
71.
(19) Armenta, S.; Alcala, M.; Blanco, M. Analytica Chimica Acta 2011, 703, 114–
123.
(20) Kanu, A. B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H. H., Jr. J. Mass Spectrom.
2008, 43, 1–22.

177
(21) Fenn, L. S.; Kliman, M.; Mahsut, A.; Zhao, S. R.; McLean, J. A. Anal Bioanal
Chem 2009, 394, 235–244.
(22) Bruce, S. J.; Tavazzi, I.; Parisod, V.; Rezzi, S.; Kochhar, S.; Guy, P. A. Anal.
Chem. 2009, 81, 3285–3296.

178
Table 6.1 Summary of experimental details.
Parameters Details ESI needle bias voltage 3000 V
Sample flow rate 3 ul/min Voltage on the ion gate 7170 V
Electric field 303 V/cm 1st segmented quadruole pressure 3 mbar 2nd segmented quadruole pressure 1 × 10-2 mbar
tofMS chamber pressure 3.8 × 10-7 mbar

179
Table 6.2 Summary of the identifications of the first 40 major metabolite features detected in
all 11 samples. The exact m/z and K0 (cm2V-1s-1) were measured in IMMS analysis, and the
identifications were matched with existing databases.
Number m/z K0 Metabolite Chemical Fomula Adduct
1 104.12 1.97 Choline C5H14NO [M]+
2 111.96 1.53 Methyl isothiocyanate C2H3NS [M+K]+
3 114.96 1.53 Thioacetate C2H3OS [M+K]+
4 155.99 1.54 Homocysteine thiolactone C4H7NOS [[M+K]+
5 157.09 1.67 N/A N/A N/A
6 174.99 1.52 Oxidized dithiothreitol C4H8O2S2 [M+Na]+
7 197.01 1.54 Aconitic acid C6H6O6 [M+Na]+
8 199.01 1.56 Ascorbic acid C6H8O6 [M+Na]+
9 203.06 1.54 Glucose C6H12O6 [M+Na]+
10 204.13 1.48 Acetylcarnitine C8H17N3O3 [M+H]+
11 211.00 1.42 p-Cresol sulfate C7H8O4S [M+Na]+
12 216.01 1.53 Phosphocreatinine C4H8N3O4P [M+Na]+
13 235.02 1.51 5-Carboxyvanillic acid C9H8O6 [M+Na]+
14 250.18 1.27 N/A N/A N/A
15 252.03 1.42 Phosphoribosylamine C5H12NO7P [M+Na]+
16 296.15 1.32 Gly Val Val C12H23N3O4 [M+Na]+
17 313.28 1.00 LysoPE(16:0) C21H44NO7P [M+H-141]+
18 341.31 0.95 LysoPC(18:0) C26H54NO7P [M+H-182]+
19 353.27 0.97 MG(16:0) C19H38O4 [M+Na]+
20 377.25 1.01 MG(18:2) C21H38O4 [M+Na]+
21 381.29 0.92 MG(18:0) C21H42O4 [M+Na]+
22 401.33 0.93 5,6-trans-25-Hydroxyvitamin D3 C27H44O2 [M+H]+
23 437.18 0.96 Ala Met Cys Leu C17H32N4O5S2 [M+H]+
24 445.21 0.89 Asp Thr Ile Pro C19H32N4O8 [M+H]+
25 473.24 0.85 Lys Tyr Tyr C24H32N4O6 [M+H]+
26 496.32 0.79 LysoPC(16:0) C24H50NO7P [M+H]+
27 518.30 0.78 LysoPC(16:0) C24H50NO7P [M+Na]+
28 520.32 0.80 LysoPC(18:2) C26H50NO7P [M+H]+
29 522.33 0.78 LysoPC(18:1) C26H52NO7P [M+H]+
30 524.34 0.76 LysoPC(18:0) C26H54NO7P [M+H]+
31 534.28 0.77 PS(17:0) C23H46NO9P [M+Na]+
32 542.30 0.79 LysoPC(20:5) C28H48NO7P [M+H]+
33 544.31 0.77 LysoPC(20:4) C28H46NO7P [M+H]+
34 566.30 0.78 LysoPC(20:4) C28H50NO7P [M+Na]+
35 568.31 0.77 LysoPC(20:3) C28H52NO7P [M+Na]+

180
36 590.29 0.77 Arg Met His Phe C26H39N9O5S [M+H]+
37 659.25 0.66 Gln Trp Tyr Tyr C34H38N6O8 [M+H]+
38 780.47 0.59 PS(36:6) C42H70NO10P [M+H]+
39 806.47 0.59 PS(38:7) C44H72NO10P [M+H]+
40 828.46 0.58 PS(40:10) C46H70NO10P [M+H]+
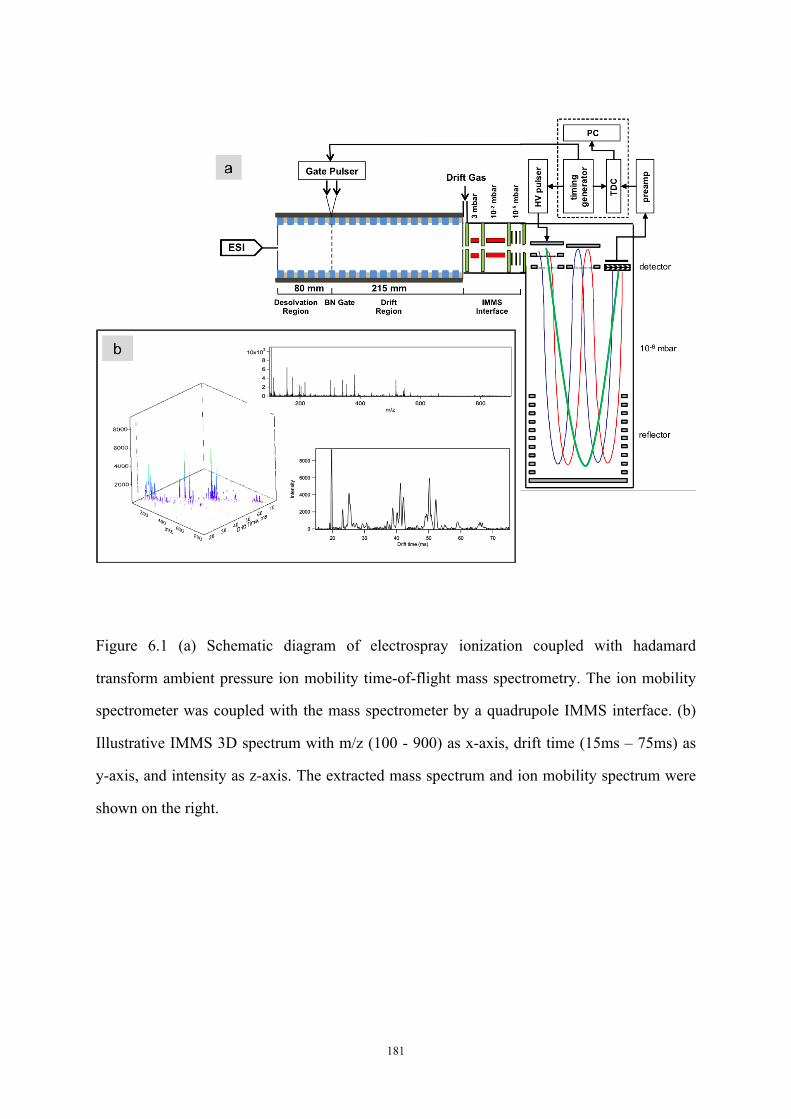
181
Figure 6.1 (a) Schematic diagram of electrospray ionization coupled with hadamard
transform ambient pressure ion mobility time-of-flight mass spectrometry. The ion mobility
spectrometer was coupled with the mass spectrometer by a quadrupole IMMS interface. (b)
Illustrative IMMS 3D spectrum with m/z (100 - 900) as x-axis, drift time (15ms – 75ms) as
y-axis, and intensity as z-axis. The extracted mass spectrum and ion mobility spectrum were
shown on the right.

182
Figure 6.2 Representative mass spectra (top) and ion mobility spectra (bottom) for four
different groups of samples, including plasma fluids from apoAV KO mice fasted for 5 hrs,
WT mice fasted for 5 hrs, apoAV KO mice ad lib fed and WT mice ad lib fed. Mass spectra
have x-axis assigned with m/z (100 - 900) and ion mobility spectra have x-axis assigned with

183
drift time (15ms- 75ms). Both spectra have y-axis as relative intensity. Different patterns can
be observed from the spectra. Areas circled with blue boxes represent the m/z (drift time)
range where the lysophospholipid class of compounds was detected.

184
Figure 6.3 PCA results including score plot (top) and loadings plot (bottom). The first
principle component explained 45% of the variance among 11 samples, and the second
principle component explained 20% of the variance. Clear sample grouping patterns can be
observed in score plot and metabolite features located away from the center of loadings’ plot
were selected as potential biomarkers.

185
Figure 6.4 Specific metabolic alternations for lysophospholipid class of metabolites. (a)
shows the intensity profiles for five metabolites that share the similar metabolic alternation,
and (b) shows the intensity profiles for another two lysophospholipid metabolites displaying
different alternation compared with (4). In both Figure 6.4 and 6.5, error bars represent ±SD
and asterisk indicates statistical difference (p value < 0.05).

186
Figure 6.5 Intensity profiles for glucose (a) and MG (18:0) (b) for four groups of samples.

187
Figure 6.6 Selected-mass ion mobility spectra for monosaccharide ion (m/z = 203.06). Ion
mobility spectra extracted from apoAV KO samples were listed on top with blue trace
representing fasted sample and red trace representing ad lib fed sample. Bottom spectra were
extracted from WT samples. Two ion mobility peaks were observed and identified
[Glucose+Na]+ with K0 = 1.53 cm2V-1s-1 and [Fructose+Na]+ with K0 = 1.46 cm2V-1s-1.

188
Chapter 7
Conclusions
I. Overall Conclusion
Complex mixture analysis has been a great challenge on for analytical chemistry.
Metabolome, as an example of a complex mixture, was specifically investigated in this work
using metabolomics by ion mobility mass spectrometry (IMMS). IMMS showed great
potential for the comprehensive analysis of complex samples with two-dimensional
separation, high sensitivity and enhanced capability for structure elucidation. This work
implemented the hadamard transform multiplexing technique and its related data processing
for metabolomics.
The unique advantages provided by IMMS allow sensitive and comprehensive
metabolomics analysis with the following capabilities: 1) high throughput analyses; 2) easy
coupling with chromatographic separation instruments to achieve a wider detection range; 3)
rapid isomer separation; 4) measurement of collision cross section (Ω) values; 5) isotopic
ratio analysis for metabolite identifications; 6) mobility-mass correlation curves (MMCCs)
for class identifications.

189
II. Specific Conclusions
The metabolomics study in Chapter 2 demonstrated the application of conventional
drift tube ion mobility mass spectrometry for the metabolic analysis of Parkinson’s
disease-like (PD-like) rat striatal tissues. PCA was use as the primary statistical analysis and
displayed significant patterns between PD-like samples and healthy controls. The high IMS
resolving power enabled separation between dopamine and its isomer (proposed structure
2-(2,4-dihydroxylphenyl) ethylamine). The co-existence of these two compounds in healthy
control samples along with the absence of dopamine in PD-like samples demonstrated that
the reduction of dopamine in PD might be underestimated. Potential biomarkers were
selected using the same PCA model, and identified as cholesterol, vitamin B3, lactaldehyde
and amino acids.
The metabolomics study in Chapter 2 employed the conventional pulse mode IMMS
that had a limited duty cycle (0.25%). Therefore, the IMMS analysis time was between 15 –
20 minutes. In order to improve the duty cycle, Chapter 3 employed multiplexing technique
by superimposing hadamard transform (HT) sequence on the ion gate. We obtained ~200
times improvement of duty cycle while retaining high IMS resolving power. The
performance of HT-IMMS was assessed using metabolite standard mixture at a wide
concentration range, and the limit of detection in HT-mode was demonstrated to be ~10 times
lower than conventional pulse mode. NIST Standard Reference Material 1950 Metabolites in
Human Plasma was also analyzed in both modes with more metabolite features detected in

190
HT mode when compared with pulse mode. In order to increase the response of complex
mixture analysis, rapid chromatographic separation prior to IMMS analysis was also
evaluated using a striatal tissue sample. It was evident that the ionization suppression caused
by direct infusion was largely reduced, and more metabolite features were observed in high
m/z region (m/z >800). Therefore, IMMS technique coupled with HT sequence and prior
chromatographic separation was developed and evaluated, allowing metabolomics analysis
within 2 – 3 minutes with high sensitivity.
The study shown in Chapter 4 was the first application of HT-IMMS on
metabolomics, which specifically investigated the metabolomes of human blood. The focus
of this work was to apply the unique merit of HT-IMMS to help metabolomics analysis and
metabolite identifications. Multiple approaches were applied including accurate isotopic ratio
analysis, isomer/isobar separations, charge states separation and mobility-mass correlation
curves classifications. We were able to identify 185 metabolite features after de-isotope using
the exact mass information and unique structural information provided by IMMS. Among the
185 metabolite features, five pairs of isomers and isobars were separated and assigned with
identifications. Moreover, five MMCCs with good linearity were observed and they provided
qualitative information regarding to compound classifications. HT-IMMS provided
comprehensive metabolomics analysis of blood samples within 3 minutes with
multidimensional information generated, demonstrating HT-IMMS as a natural fit for
metabolomics and metabolite identifications.

191
To further extend the applications of HT-IMMS to metabolomics, Chapter 5 described
a neuronal metabolic study, revealing the global and specific neuronal metabolic changes
caused by cocaine self-administration followed by 1 day or 3 weeks withdrawal. With regard
to analytical platform, this study employed chromatographic separation prior to HT-IMMS
analysis and achieved good metabolite response and wide detection range. With regard to
data processing, we improved the biomarker selection model from Chapter 2, and used both
multivariate analysis (PCA) and univariate analysis approaches to target potential biomarkers,
generating a complete profile of metabolic changes induced by cocaine self-administration.
Among the selected potential biomarkers, oxidative stress and energy related metabolites
were specifically investigated. The dyregulation of creatine, creatinine, adenosine and
glutathione matched with previous studies and demonstrated the brain regionally specific
metabolic changes. In general, these findings supported the multi-event mechanism of
cocaine abuse.
Work in extending metabolomics applications was continued in Chapter 6, where the
metabolic function of apolipoprotein AV (apoAV) was investigated using the plasma fluids
collected from apoAV knockout mice and wild type mice under fasted and ad lib fed dietary
treatments. Different metabolic patterns were noted for different sample groups after PCA,
and the biomarkers selected by PCA were extended to their corresponding classes including
lysophosphatidycholine, monoacylglyceride and carbohydrate. A “mirror effect” of dietary

192
treatments on the effect of apoAV on lysophosphatidylcholine related metabolites was
observed, and the increase of monoacyglyceride class of metabolites was noted in the apoAV
KO samples for both dietary treatments. In addition, high resolving power of IMS allowed
separation between glucose and fructose, providing specific metabolic changes without the
interference of each other. It was conclude that the absence of apoAV caused 2-fold increase
in glucose level, while appeared to have little to no influence on fructose level.
Data processing using statistical approach was an important part in Chapter 2, Chapter
5, and Chapter 6. And it was concluded that there’s no standardized procedure for statistical
analysis of metabolomics. Appendix provided the optimized comprehensive data processing
procedure for IMMS type of data, including the estimation of censored values (also known as
missing values), multivariate analysis, univariate analysis and cluster analysis. Censored
values were estimated using distribution analysis by Minitab, enabling us to keep metabolic
information intact. Univariate analysis was optimized using adjusted t-test in linear models
for microarray data (limma), which selected the “real” significant changes using adjusted
variance instead of metabolite feature variance. This univariate analysis approach generated a
fewer number of significant changes when compared to the conventional t-test, allowing us to
focus on a fewer number of biomarkers. Finally, cluster analysis was employed to analyze the
selected biomarkers, providing insights in metabolic pathways.
Chapter 2 showed an example of metabolomics study on PD-like striatal tissue using

193
conventional IMMS approach, advanced in high resolving power but limited in throughput.
The work in Chapter 3 evaluated HT-IMMS by a number of metabolite standards and various
types of complex mixtures, proving the 2 orders of magnitude increase in sensitivity while
maintaining high resolving power. The application study in Chapter 4 illustrated the unique
merit of HT-IMMS in global metabolomics and metabolite identifications. Based on the
above improvements, applications were extended to global metabolic study on cocaine abuse
and apoAV in Chapter 5 and Chapter 6, respectively. The findings in the dysregulation of
potential biomarkers and proposed metabolic pathways may provide complementary
information to the pathological studies. The optimization in data processing was further
addressed in Appendix, illustrating the critical role of statistical analysis in global
metabolomics. With the continued development in HT-IMMS techniques and optimization in
data processing, a multitude of applications should be extended, not only for metabolomics,
but also for all types of complex mixture analyses.

194
Appendix A
Statistical Analysis for Metabolomics using Multivariate
and Univariate Approaches
Introduction
Metabolomic1,2, an important part of integrate “omics”, is the downstream of
genomics and proteomics. Specifically, metabolomics is the scientific study by profiling and
measuring thousands of metabolites. The result of metabolomics represents the collection of
all metabolites in a cell, tissue, or organ. Metabolites are the end products of cellular
processes, including amino acids, glucose, cholesterols, lipids and peptides. And their levels
can be regard as the ultimate response of biological systems. There are a few particular
purposes of metabolomics study. The first aim is to differentiate metabolic states/pattern
recognition, for example, healthy state vs. disease state, early disease state vs. late disease
state. And the second one is to find biomarkers3 that can be used for diagnosis or cure. These
biomarkers refer to the variables that change significantly among different metabolic states,
and can often times be related to the disease mechanism with certain biological functions.
The third one aims to yield significant biological insights by correlating the targeted
biomarkers4 with other variables/metabolites, therefore, generate certain metabolic pathways
that can further be related to proteomics and genomics.

195
Common to all analytical methods5 for metabolomics is that they all produce a
massive amount of data, impossible to be handled completely manually and without the use
of statistics. The number of variables (also called metabolite features) can be hundreds or
even thousands, much larger than the number of experiments (usually 3-20); therefore the
metabolomics data are high dimensional. This situation has made multivariate statistics a
natural fit because that it works well with “long and lean” data sets in general scope.
Multivariate statistics includes dimension reduction methods such as principle component
analysis, partial least square analysis, and linear discriminate analysis. Such multivariate
approaches are also robust to random variation and experimental error.
Nowadays, nuclear magnetic resonance (NMR)6 and mass spectrometry (MS)7 are
two predominant platforms utilized in metabolomics, and both methods perform
metabolomics analysis by detecting the chemical formula and molecular structure of
metabolite ions. MS-based technique is preferred by its high sensitivity, low cost and
straightforward data format. The basis of MS technique is to measure the metabolite ion’s
mass (also called molecular weight). However, due to the complex nature of metabolomics,
pre-separation is often times required before MS analysis for a wider detection range. The
pre-separation step not only increases the number of detectable metabolite features, but also
provides another dimension of information regarding to the structure of the metabolite. Ion
mobility spectrometry (IMS)8 is one of the popular pre-separation techniques and has been
widely applied to metabolomics. IMS separate metabolite ions based on their size, and when

196
coupled with MS technique, ion mobility mass spectrometry (IMMS) can generate
multidimensional information for each metabolite feature.
MS generates a mass to charge ratio (mz) for each metabolite feature, while IMS
generates mobility value (K0) related to ionic size. They can be combined as mzK0 feature9,
which is utilized for charactering each metabolite feature. Every mzK0 feature is also
assigned with an intensity value, which represents the concentration of the corresponding
metabolite feature in a particular sample. The final metabolomics data set for a certain sample
contains hundreds of mzKo features with assigned intensity values. When multiple data sets
generated for multiple samples, one can combine these data sets by merging mzKo features,
and in the end, a complex data matrix presents.
Even though the analytical approach is efficient and reproducible, there are
difficulties along with the data processing using appropriate statistical analysis. Unlike
genomics data and proteomics data, the metabolomics data is more versatile and
hard-to-predict10. Among the statistical approaches utilized in metabolomics, there’s still no
defined standard procedure, therefore, challenges and concerns appeared during recent years.
The major challenges are how to select biomarkers from the massive data matrix, and how to
relate these biomarkers with other metabolites to form a metabolic pathway. This “fishing”
procedure is critical and needs to be handled with caution.

197
The general steps involved in the statistical analysis of metabolomics are usually
followed as data pre-treatment11 (including raw data normalization and mzKo feature
merging), exploratory analysis (e.g. principle component analysis (PCA)12 and partial least
square-discriminant analysis (PLS-DA)13). The exploratory analysis is developed to distill the
large amounts of data by reducing the dimensionality of the raw data set. The result can
elucidate the relations between samples and their classification. For example, by utilizing
PCA, it can simplify large datasets into a new coordinate system (components), and these
new components are derived from the original data in such a way that the greatest variation in
the date is captured in the first group (first component PC1), the second greatest variation in
the second (PC2) and so on. This method of viewing large amounts of data in a simplified
manner provides scientists a means to visualize and detect variations between sample groups.
PCA loadings’ plot shows where the components found their variation and thereby which
metabolites have affected in the relations and groupings. Therefore, potential biomarkers are
defined as the metabolite features that significantly affected the relations. The above example
shows the general approach of statistical analysis on metabolomics type of data, and it indeed
effectively accomplished the global pattern recognition, and also selected some potential
biomarkers for the second aim.
However, improvements are still needed at each stage of data processing, and there
are a few problems/concerns that have been overlooked. One of them is how to appropriately
deal with censored data generated during the analytical experiment. Due to the biological

198
variation among samples and the limit of detection (also called threshold) for the analytical
technique, censored data can appear among biological replicates. It is possible that certain
metabolite features can be detected in the first and second replicates, but not in the third one.
When encountered with this situation, researchers often times mask out metabolite features
that have censored data. However, this is clearly not the ideal solution since the discarded
metabolite features could be important in biological sense. Another problem is biomarker
selection. As described earlier, multivariate statistical models can generate certain biomarkers
with regard to their contribution in pattern recognition. However, the selection procedure was
based on variance, which can be dominated by metabolite features that have high intensity
values. Under this situation, the contribution of metabolite features that have small intensity
values can often times be underestimated or even ignored. The third challenge in
metabolomics is how to relate the selected biomarkers with metabolic pathways, and till
today, this procedure has been realized fully dependent on the biological knowledge.
With the gaps described above as the aims of this study, we developed new data
processing algorithm using Minitab and R. By re-processing the raw data using distribution
analysis on left-censoring data, we were able to avoid discarding information. And by
applying univariate analysis14 on all metabolite features, more potential biomarkers were
selected including the ones with small intensity values. Moreover, cluster analysis was
performed on the selected biomarkers, in order to sort the metabolic pathways from statistical
prospective.

199
Experiments
Sample Preparation:
Six male Sprague Dawley rats were obtained from Washington State University
breeding colony and were housed two or three per cage in a university vivarium with a 12-h
light/dark cycle, at 22 to 24 °C, and with ad libitum access to food and water. Animal
procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory
Animals and were approved by the University Animal Care Committee15. Three animals were
used as saline-treated controls, and three were given an acute dose of cocaine at 30 mg/kg i.p.
45 min every day until maximal locomotor activity was achieved (cocaine addicted). Rats
were given 24 hr withdrawal after the last dose of drug/saline before decapitated and their
brain dissected on an ice-cold watch glass with an ice-cold razor blade. Left and right striatal
samples were pooled from each rat, and stored on dry ice. Samples were weighed and
individually transferred to a sample container where the sample was sonicated in 500 µL of
electrospray solution made up with 50:50 methanol/water, to extract metabolites. Denatured
proteins and cellular debris were separated by centrifugation for 10 min in a desktop
centrifuge. The supernatant was collected and stored in -80°C before analyzed.
Ion Mobility Mass Spectrometry (IMMS) Analysis and Data Format:
Electrospray ionization was applied on the supernatant to ionize the metabolite
molecules into metabolite ions. IMMS analysis lasted for 5 minutes for each sample with a
sample flow rate of 3 ul/min. Multidimensional information was exported and organized for

200
each sample using mzKo representing metabolite features, and intensity values were assigned
to complete one data set. When six data sets were generated, mzKo features were merged into
one data set with all information included.
Table 1 is part of the merged data set, illustrating the data format. As shown in the
table, the data set contains 6 columns with each column corresponding to each sample, and
~150 rows with each row corresponding to each metabolite feature, the number in each cell
represents the intensity value of the corresponding metabolite feature in the corresponding
sample. The samples were named as STR C1, C2, C3 (representing cocaine abused samples)
and S1, S2, S3 (representing saline controls). It is important to note that there are a few cells
with 0 intensity value, which actually indicate the left censoring data not detected by IMMS.

201
Statistical Analysis Results
Evaluate Left-censoring Data using Distribution Analysis:
The prediction of left-censoring data undoubtedly assigned more reasonable intensity
value compared to simply using 0 or discarding the metabolite feature. In this study, we used
the distribution analysis function in Minitab. By setting the instrument analysis threshold at
50, the distribution was selected with the most linear probability plot and the distribution
parameters were estimated by MLE. The prediction of left-censoring data was then
determined by estimating percentiles for certain percent (determined by the number ratio of
left-censoring metabolite features and total metabolite features). The results were shown in
Table 2, with all left-censoring data replaced with the prediction values. To be more specific,
Weibull distribution was found to have the most linear probability, and the left-censoring data
for STR C1, C2, C3, S1, S2, S3 were replaced with predicted values 46, 53, 53, 67, 69, 59,
respectively.
Statistical Analysis using Multivariate Approach:
Principle component analysis (PCA) was utilized as dimension reduction approach
after normalization the raw data and replacing left-censoring data with prediction values. The
unsupervised nature of PCA enables it for pattern recognition by score plot and biomarker
selection by loadings’ plot. PCA was performed using Table 2, and PCA results were shown
in Figure 1 with both score plot and loading’s plot. As shown in the score plot, cocaine
samples (circled in red) are well separated from the saline controls (circled in green). The

202
loadings’ plot has a few outliers representing the metabolite features significantly affected the
pattern recognition. These outliers were extracted further explained by cluster analysis.
Selection of Biomarkers using Univariate Approach:
Unpaired t-test was used initially to obtain biomarkers in addition to the biomarkers
generated from PCA. However, it yielded 31 metabolite features with P-value less than 0.05.
Large amount of potential biomarkers can make the later analysis very difficult because that
each metabolite feature considered as potential biomarker has to be verified, identified and
understood. The adjusted t-test in linear models for microarray data (limma) resolved this
problem by reducing the number of significant features using adjusted variance. The rationale
for adjusted t-test is the fact that the sample variance is not an efficient statistics due to the
small number of observations (3 in this study), and adjusted t-test computes an expected
variance and adjusts the observed ones towards this expected value. By computing the
P-value from adjusted t-test, the number of metabolite features with significant P-value was
effectively reduced to 7. Table 3 contains top 20 metabolite features (ranked by P-value),
showing the significant improvement from adjusted t-test.
Cluster Analysis on Biomarkers for pathway Identification:
The purpose of cluster analysis is to sort metabolite features into respective categories
in a way that the degree of association between metabolite features is maximal if they belong
to the same group and minimal otherwise. Hierarchical clustering analysis was applied on 12

203
metabolite features selected as biomarkers (7 with significant P-values from the adjust t-test,
5 were identified as outliers from PCA), and result was displayed in Figure 2. It shows that
two metabolite features with mzKo 104.10:1.96 and 268.11:1.34 fell into the same category.
These two metabolite features were identified as Choline and Adenosine, which were both
involved in the energy metabolism in a system of biology, proving the feasibility of cluster
analysis.

204
Conclusions
Metabolomics data generated from global metabolomics analysis contains large
amount of information, and often times has more metabolite features than sample size.
Metabolite features characterized by m/z and Ko were obtained from ion mobility mass
spectrometry analysis in a cocaine-abused rats study, and statistical approaches were
developed for pre-processing, pattern recognition and biomarkers selection. Left-censoring
data was evaluated and predicted using distribution analysis. Principle component analysis
was employed as a dimensional reduction method with pattern recognition yielded for
cocaine abused samples and saline control samples. Moreover, it generated 5 verified outliers
(identified as biomarkers) from the loadings’ plot. Univariate analysis approaches were also
evaluated for more biomarkers selection. Plain unpaired t-test was applied with 31 significant
biomarkers appeared to be significant. The number of biomarkers was reduced to 7 by
applying adjusted t-test in linear models for microarray data. Clustering the biomarkers based
their degree of association has further helped for metabolic pathways analysis, which lead to
the final goal of metabolomics study.

205
Reference
(1) Creek, D. J.; Jankevics, A.; Breitling, R.; Watson, D. G.; Barrett, M. P.; Burgess, K.
E. V. Anal. Chem. 2011, 83, 8703–8710.
(2) Bruce, S. J.; Jonsson, P.; Antti, H.; Cloarec, O.; Trygg, J.; Marklund, S. L.; Moritz, T.
Analytical Biochemistry 2008, 372, 237–249.
(3) Griffiths, W. J.; Koal, T.; Wang, Y.; Kohl, M.; Enot, D. P.; Deigner, H. P. Angew.
Chem. Int. Ed. 2010, 49, 5426–5445.
(4) Smith, R. D. Clinical Chemistry 2012, 58, 528–530.
(5) Dunn, W. B.; Ellis, D. I. TrAC Trends in Analytical Chemistry 2005, 24, 285–294.
(6) Aranı bar, N.; Ott, K.-H.; Roongta, V.; Mueller, L. Analytical Biochemistry 2006,
355, 62–70.
(7) Katajamaa, M.; Orešič, M. Journal of Chromatography A 2007, 1158, 318–328.
(8) Kanu, A. B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H. H., Jr. J. Mass Spectrom. 2008,
43, 1–22.
(9) Smith, C. A.; Want, E. J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. Anal. Chem. 2006,
78, 779–787.
(10) Issaq, H. J.; Van, Q. N.; Waybright, T. J.; Muschik, G. M.; Veenstra, T. D. J. Sep.
Science 2009, 32, 2183–2199.
(11) Bijlsma, S.; Bobeldijk, I.; Verheij, E. R.; Ramaker, R.; Kochhar, S.; Macdonald, I. A.;
van Ommen, B.; Smilde, A. K. Anal. Chem. 2006, 78, 567–574.
(12) Pan, Z.; Gu, H.; Talaty, N.; Chen, H.; Shanaiah, N.; Hainline, B. E.; Cooks, R. G.;

206
Raftery, D. Anal Bioanal Chem 2006, 387, 539–549.
(13) Karp, N. A.; Griffin, J. L.; Lilley, K. S. Proteomics 2005, 5, 81–90.
(14) Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J. J. Metabolites 2012.
(15) Kaplan, K. A.; Chiu, V. M.; Lukus, P. A.; Zhang, X.; Siems, W. F.; Schenk, J. O.;
Hill, H. H. Anal Bioanal Chem 2013, 405, 1959–1968.

207
Figure A1: PCA results including Score plot and Loadings’ plot, with 62% of variance
explained.

208
Figure A2: Hierarchical Clustering analysis result on 12 biomarkers.
130.05:1.72
114.06:1.86
176.05:1.61
137.05:1.75
104.07:1.96
162.11:1.63
761.60:0.59
798.55:0.58
142.03:1.73
127.06:1.84
104.10:1.96
268.11:1.34
010000
20000
30000
40000
50000
Cluster Dendrogram
hclust (*, "complete")d
Height

209
Table A1: Part of the raw data set generated from IMMS experiments, illustrating the data
format.
m/z: K0 STR C1 STR C2 STR C3 STR S1 STR S2 STR S3
104.06:1.91 4862 1995 1643 2384 3126 3556
104.10:1.96 8904 11503 7191 10561 12288 19962
105.10:1.96 577 889 528 773 1016 1493
106.04:1.75 357 193 206 466 429 290
112.89:1.95 79 154 0 131 141 235
114.06:1.89 13095 4243 9165 9101 7269 6781
115.06:1.88 1064 517 616 684 560 575
119.03:1.34 553 515 0 456 472 173
120.06:1.80 279 0 0 429 478 376
123.05:1.80 3349 1840 3359 2845 2947 2714
124.04:1.80 541 388 10 1006 0 442
127.06:1.84 546 514 373 0 0 0
129.06:1.80 732 536 980 852 511 557

210
Table A2: Result of censored value estimation, “0” values were replaced by estimations.
m/z: Ko STR C1 STR C2 STR C3 STR S1 STR S2 STR S3
80.04:2.08 608 727 53 1874 927 1234
84.07:1.69 375 53 123 310 163 59
86.05:1.87 6857 9880 4874 4218 3408 4367
87.05:1.92 551 544 53 444 592 419
90.05:1.79 389 452 463 405 623 308
90.08:2.08 5185 4372 4558 3195 2921 1657
90.50:1.96 210 53 166 278 288 67
91.05:1.76 205 214 335 67 69 59
91.09:2.08 304 188 199 227 121 113
99.05:1.81 492 53 851 502 238 59
104.06:1.91 4862 1995 1643 2384 3126 3556
104.10:1.96 8904 11503 7191 10561 12288 19962
105.10:1.96 577 889 528 773 1016 1493
106.04:1.75 357 193 206 466 429 290
112.89:1.95 79 154 53 131 141 235
114.06:1.89 13095 4243 9165 9101 7269 6781
115.06:1.88 1064 517 616 684 560 575
119.03:1.34 553 515 53 456 472 173
120.06:1.80 279 53 53 429 478 376
123.05:1.80 3349 1840 3359 2845 2947 2714
124.04:1.80 541 388 53 1006 69 442
127.06:1.84 546 514 373 67 69 59
129.06:1.80 732 536 980 852 511 557
130.05:1.72 2097 1833 2239 2853 2887 2947
131.05:1.72 330 192 53 171 212 232
132.07:1.80 23766 23893 29439 18474 30974 23224
133.08:1.79 1506 1477 1664 1360 2076 1625
134.04:1.65 367 53 417 553 69 737
136.06:1.79 5811 4760 5012 6253 69 2468
146.11:1.69 139 167 252 420 356 275
146.16:1.67 639 620 729 470 246 644
147.07:1.72 6110 5427 4852 8399 9585 7660
148.06:1.66 4503 2981 3157 7771 9741 5041
150.98:1.68 81 211 114 67 69 59
152.02:1.73 5176 5740 5689 4526 4892 6864
153.02:1.73 255 350 377 402 282 332
154.06:1.63 2509 2220 1911 1857 1967 2974
156.07:1.69 452 566 570 552 554 671

211
159.11:1.67 1628 689 625 1004 567 264
161.12:1.63 368 218 366 404 302 154
162.08:1.64 1836 1909 2485 2840 3065 3286
166.95:1.71 182 419 103 251 446 1018
169.06:1.64 1616 3151 2162 3001 2389 5219
169.10:1.60 2165 1194 1444 1617 473 59
170.03:1.65 10459 17258 16367 13899 15392 21326
171.04:1.65 842 1253 1099 853 1079 1594
172.03:1.65 979 1671 1470 1420 1534 1839
173.06:1.65 475 907 827 344 436 469
174.89:1.70 244 204 216 270 291 598
175.00:1.68 510 800 296 645 797 2623
175.12:1.59 1438 1796 1395 1429 1687 1531
176.05:1.61 516 746 840 2345 2405 2030
184.07:1.58 2364 2239 2729 2570 3397 1477
185.03:1.63 3294 7570 4911 5967 5374 10847
186.04:1.63 363 776 341 529 342 815
187.04:1.63 229 710 482 548 520 904
192.18:1.42 393 336 588 541 814 212
194.03:1.56 87 269 53 233 170 297
201.10:1.25 948 1882 958 1225 1649 209
201.94:1.57 323 332 523 384 509 765
202.12:1.50 949 358 509 905 332 235
203.96:1.56 215 53 246 188 69 210
204.12:1.47 1057 893 233 67 69 59
207.02:1.54 261 904 318 532 448 1040
208.00:1.52 311 342 349 431 495 546
212.85:1.66 135 176 150 67 69 59
222.03:1.45 1045 1666 1261 1093 1572 1318
222.99:1.51 247 860 749 587 729 1462
223.08:1.44 123 254 198 67 69 59
223.98:1.48 201 289 225 145 374 325
227.12:1.41 2158 746 1071 1238 342 601
236.00:1.39 319 230 653 451 423 521
250.10:1.40 1761 776 1245 2044 1193 283
251.10:1.40 417 808 162 283 205 59
251.98:1.39 619 53 982 742 854 825
258.11:1.32 1364 1618 2637 1444 1639 643
268.11:1.34 28870 18573 18319 21564 16163 8807
269.11:1.34 4166 2812 2978 3430 2559 1358
270.11:1.34 1095 825 712 907 457 266

212
276.13:1.28 173 53 288 170 598 330
277.10:1.22 211 350 151 193 182 242
280.10:1.28 715 951 1110 788 547 59
280.16:1.28 226 184 348 67 69 59
282.06:1.29 260 473 353 508 296 576
290.09:1.25 1341 1275 951 549 69 820
296.07:1.26 3871 4529 6100 3212 2947 883
297.08:1.26 661 884 1062 441 484 59
298.09:1.26 957 1454 1060 1286 69 91
307.05:1.21 1588 2731 1397 2279 3205 6495
308.09:1.21 777 53 1087 1334 1688 2600
313.28:1.00 621 746 398 67 69 59
322.06:1.18 412 687 564 591 832 59
324.12:1.19 81 53 112 67 69 59
331.11:0.96 170 268 169 67 69 59
339.29:0.97 132 234 162 67 69 59
341.31:0.94 475 309 267 134 158 59
348.08:1.19 591 608 1082 847 734 156
369.36:0.94 1581 1709 2850 909 489 850
369.36:0.97 1123 245 2710 1044 249 843
370.36:0.97 352 779 1271 365 269 427
392.31:0.88 128 228 208 67 69 59
475.22:0.90 271 378 156 67 69 59
546.53:0.68 455 651 53 67 69 59
548.56:0.67 2062 2632 1944 449 2100 1314
549.56:0.67 1030 877 53 67 69 59
731.61:0.58 313 363 427 67 69 59
732.60:0.58 106 132 164 67 69 59
734.58:0.60 1235 2043 1591 668 1121 784
735.59:0.60 669 953 759 377 770 59
736.58:0.59 74 386 68 67 69 59
760.60:0.59 1607 2890 2802 738 1007 870
761.60:0.59 827 1322 1203 507 660 197
762.61:0.58 167 578 302 67 69 59
772.54:0.58 1375 630 929 141 565 59
773.52:0.58 287 92 122 67 69 59
782.58:0.59 340 164 1044 189 626 358
788.64:0.57 84 582 835 67 69 59
798.55:0.58 1066 756 1154 542 637 227
799.54:0.58 597 197 53 67 69 59
800.57:0.57 114 130 174 67 69 59
213
806.56:0.58 195 804 314 67 69 59
114.06:1.86 46 2096 3414 67 69 59
128.04:1.67 46 1097 973 790 548 552
189.06:1.62 46 356 374 67 69 59
206.05:1.44 46 285 159 67 364 327
233.08:1.45 46 310 211 218 189 59
756.55:0.59 46 129 205 67 69 59
783.58:0.59 46 357 581 67 69 59
810.62:0.57 46 298 316 67 69 59
826.59:0.56 46 344 311 67 69 59
309.11:1.21 46 53 232 530 69 442
158.03:1.62 46 111 53 428 360 252
291.07:1.17 46 313 53 314 654 551
309.04:1.21 46 326 53 218 490 743
84.04:1.72 46 53 53 1164 632 1810
104.07:1.96 46 53 53 1544 1550 1619
105.07:1.91 46 53 53 79 193 220
122.02:1.72 46 53 53 152 179 90
134.04:1.71 46 53 53 659 827 59
136.05:1.67 46 53 53 4286 2000 2442
137.05:1.75 46 53 53 1338 2247 2191
142.03:1.73 46 53 53 462 388 408
212.12:1.38 46 53 53 2036 1124 70
268.24:1.34 46 53 53 1063 774 466
369.24:1.38 46 53 53 443 951 996
574.93:0.92 46 53 53 408 359 305
162.11:1.63 46 53 53 1785 1939 1432

214
Table A3: Top 20 metabolite features with significant P-values.
Metabolite Feature logFC AveExpr t P. Value
adj. P. Value
104.07:1.96 1520.4516 810.8925 27.654404 1.16E-06 0.000170932 162.11:1.63 1667.9802 884.6568 11.614453 8.33E-05 0.005760181 176.05:1.61 1559.64 1480.4657 10.815728 1.18E-04 0.005760181 137.05:1.75 1874.8311 988.0822 6.999526 9.19E-04 0.025983784 130.05:1.72 838.9134 2476.0347 6.970562 9.36E-04 0.025983784 142.03:1.73 368.5252 234.9293 6.783939 1.06E-03 0.025983784 127.06:1.84 -412.7134 271.3567 -5.937015 1.94E-03 0.040705022 574.93:0.92 306.7802 204.0567 5.363985 3.03E-03 0.053414035 731.61:0.58 -302.4004 216.2002 -5.163905 3.58E-03 0.053414035 147.07:1.72 3084.9897 7005.6386 5.142872 3.64E-03 0.053414035 313.28:1.00 -523.4178 326.7089 -5.032628 4.00E-03 0.053414035 136.05:1.67 2858.654 1479.9937 4.549604 6.12E-03 0.064337289 369.24:1.38 746.2683 423.8008 4.481284 6.52E-03 0.064337289 162.08:1.64 987.0945 2570.0676 4.43253 6.82E-03 0.064337289 268.24:1.34 716.9438 409.1386 4.411864 6.95E-03 0.064337289 90.08:2.08 -2114.1302 3648.0152 -4.404063 7.00E-03 0.064337289 760.60:0.59 -1561.5327 1652.2808 -4.107784 9.29E-03 0.080355881 158.03:1.62 276.5577 208.3144 3.909564 1.13E-02 0.092365201 84.04:1.72 1151.1519 626.2426 3.724861 1.37E-02 0.105643351 761.60:0.59 -662.953 786.1506 -3.532025 1.67E-02 0.122865437

215
R Code
#Data input and adjusted-t test dat <- read.csv("/Users/Nancy/Dropbox/A-STAT/xing.csv",header = TRUE, row.names = 1) targ <- read.csv("/Users/Nancy/Dropbox/A-STAT/targxing.csv") head(targ) f <- factor(targ$target, levels=c("O","E")) design <- model.matrix(~0+f) colnames(design) <- c("O","E") contrastNames=c("O-E") contrastMatrix=matrix(c( -1,1),nrow=ncol(design)) colnames(contrastMatrix)=contrastNames contrastMatrix library(limma) fit=lmFit(dat,design=design) fit2=contrasts.fit(fit,contrasts=contrastMatrix) names(fit2) fit2=eBayes(fit2) F.stat=fit2$F p.value=fit2$F.p.value results=classifyTestsF(fit2,p.value=1.0E-1) write.csv(fit2,file="/Users/Nancy/Dropbox/A-STAT/xingresults.csv") topTable(fit2,n=20,coef="O-E") #Cluster analysis (Hierarchical) biomarker<-read.csv("/Users/Nancy/Dropbox/A-STAT/biomarker.csv",header = TRUE, row.names = 1)
d<-dist(biomarker, method ="euclidean") fit<-hclust(d) plot(fit)
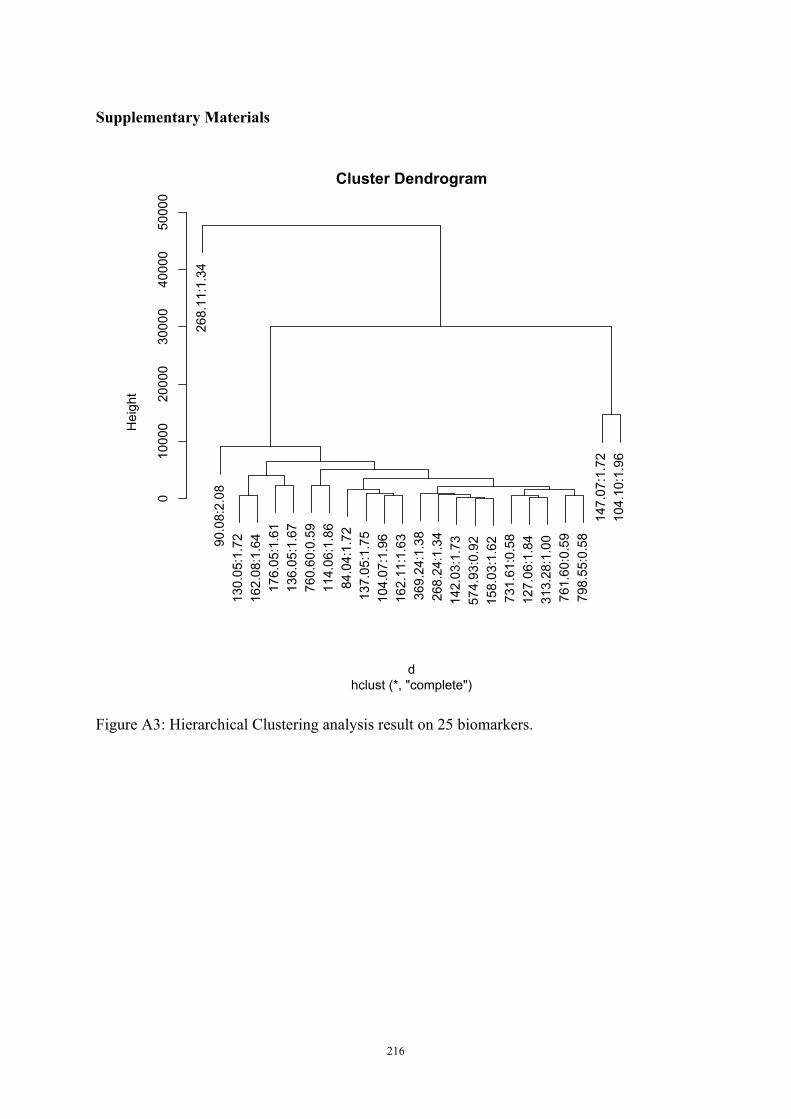
216
Supplementary Materials
Figure A3: Hierarchical Clustering analysis result on 25 biomarkers.
268.11:1.34
90.08:2.08
130.05:1.72
162.08:1.64
176.05:1.61
136.05:1.67
760.60:0.59
114.06:1.86
84.04:1.72
137.05:1.75
104.07:1.96
162.11:1.63
369.24:1.38
268.24:1.34
142.03:1.73
574.93:0.92
158.03:1.62
731.61:0.58
127.06:1.84
313.28:1.00
761.60:0.59
798.55:0.58147.07:1.72
104.10:1.96
010000
20000
30000
40000
50000
Cluster Dendrogram
hclust (*, "complete")d
Height

217
Appendix B
Cleaning Procedure for Resistive Glass Ion Mobility Mass
Spectrometer
1. Turn off the heater, let instrument completely cool down.
2. Turn off the turbo pump.
3. After 30 minutes to 1 hour, turn off the side pump aside the instrument by
double turning the red handle.
4. After another 30 minutes, turn off the rough pump at the back of the instrument
by disconnecting the power.
5. Start dissembling the IMS from the MS.
a. Take off ESI.
b. Take out the gate and store it in aluminum foil.
c. Take off the heat jacket.
d. Unscrew all the screws that connect the IMS and MS.
e. Undo all the leads connection and remove the grey box.
f. Flip the IMS straight up. (Only move with the base. No twist on the resistive
glass tube)

218
Front view of Quadruple 1 as part of the interface:
Cover it with aluminum foil:
Clean dusts on the bottom

219
Dissemble the resistive glass IMS piece by piece:
Ceramic piece between dissolvation tube and drift tube is cracked seriously:
Dirty inside the
tube

220
The IMS end, nozzle and lens:
In order to separate the parts from the metal frame, slightly push the metal showerhead. After
separating the parts, undo the leads and unscrew the metal leads.
Front view of IMS end, nozzle and lens:
Back view of IMS end, nozzle and lens:

221
Unscrew the metal leads and take apart the IMS end, nozzle and lens separately:
Please note that the white piece is an insulator between nozzle and lens.

222
Here are all the parts:

223
6. Cleaning procedure.
a. Rinse the parts with DI water.
b. Use soap solution to clean the resistive glass tubes througoutly.
c. Rinse the resistive glass tubes by DI water for about 10 minutes.
d. Soak the resistive glass tubes using a series of cleaning solvents including
methanol, methylene chloride, acetone and methanol.

224
Put the metal pieces and ceramic pieces into a large beaker and sonicate them in a series of
solutions including water, methanol, methylene chloride, acetone and methanol.
7. Reassembling.
a. Put together the IMS end, nozzle and lens.
b. Assemble IMS end, nozzle and lens into the metal frame.
c. Put the resistive glass tubes and IMS gate back on piece by piece.
d. Couple the IMS with the MS.

225
Put together the IMS end, nozzle and lens.
Matching up the two ceramic pieces by lining up the 2 holes on top.

226
Complete assembling IMS end, nozzle and lens by putting 4 springs in place.
Put the flat showerhead metal piece on top of the springs while match the gas holes.

227
Put the resistive glass tubes and ion gate piece by piece.

228

229

230
Make sure all the springs, nuts and screws are in the right place.
Then flip the IMS to its original position. Clean and put the O ring back on the interface for vacuum seal

231
Tight the screws against the rest of the interface.
Slide in the gate to make sure ceramic piece is in the right place.
8. Put on heating case.
9. Turn on the rough pump behind the instrument.
10. After 30 minutes, turn on the side pump and switch on the red handle.
11. After 20 minutes, turn on the turbo pump and wait till the pressure stabilized.




















