
Analytical Methods of Biological Monitoring for Exposure to Pesticides: Recent Update
14
REVIEW ARTICLE Analytical Methods of Biological Monitoring for Exposure to Pesticides: Recent Update Maria G. Margariti, MSc,* Andreas K. Tsakalof, PhD,† and Aristidis M. Tsatsakis, PhD, DSc* Abstract: Extensive use of synthetic pesticides for agricultural and nonagricultural purposes began in the past 50 years. As a result of their wide and extensive application, exposure to hazardous pesticides is a concern to the general population and occupationally exposed persons. Robust methods are therefore needed for measuring markers of pesticide exposure. This article presents a review of the most recently published analytical methodologies and instrumentations developed for and applied to biological monitoring of exposure to pesticides of various classes. Most of the methods reviewed here are based on chromatography combined with mass spectrometry de- tection. This work clearly demonstrates that although gas chroma- tography still appears to be the most widely employed technique for pesticide analysis in various biological samples, recently a trend has been observed toward the use of liquid chromatography coupled with tandem mass spectrometry. Key Words: biological monitoring, analytical methods, pesticides, chromatography, mass spectrometry (Ther Drug Monit 2007;29:150–163) INTRODUCTION Pesticides are substances used to prevent, destroy, repel, or mitigate any pests, ranging from insects, animals, and weeds to microorganisms such as fungi, molds, bacteria, viruses, and other organisms that compete with humans for food, destroy property, spread disease, or are considered a nuisance. 1,2 Extensive use of synthetic pesticides for agricultural and nonagricultural purposes began in the past 50 years. Pesticide application has increased and improved food supply and also has assisted in the control and reduction of food-borne and vector-borne diseases, which affect and kill many children and adults annually. 3,4 On the other hand, pesticides also pose substantial human health concerns because they are usually, but not always, poisonous to humans and are widely released into environment. As a consequence of their wide and exten- sive application in agricultural and residential settings and their physicochemical properties, these compounds have been found in all compartments of the environment. Consequently, exposure of the general population at some level to several different pesticide residues is almost inevitable. 5 The risks of extended pesticide usage are mainly related to the acute and chronic toxicity of these compounds. Unintentional exposure is a common cause of acute poisoning, particularly among young children. 6 Furthermore, these compounds are sometimes used for suicidal reasons. 7,8 The literature contains a number of cases of acute pesticide poisoning due to ingestion or inhalation of pesticides. 9–14 There also have been a number of studies dealing with chronic pesticide exposures. 15–18 Chronic toxicity can be the conse- quence of occupational exposure or continual consumption of contaminated food and drinking water. It has been shown that pesticides can cause adverse human health effects due to their ability to accumulate in environmental media (air, water, sediments, soil) and food products. Separate occupational risk assessment research is conducted for farmers and other professional pesticide handlers or applicators. Recent studies have indicated that individuals employed in agriculture, as well as their children, may be at higher risk of pesticide exposure than the general population. 19–23 For risk assessment of human exposure to these substances and their metabolites, constant biological monitoring is required. Detection of pesticide residues and/or their metabolites in biologic samples from the general population and occu- pationally exposed persons is in rapid development and has recently been the subject of many articles. 1–118 Current re- search is focusing more on those pesticides that are considered to be endocrine-disrupting and potential risk factors for the development of non-Hodgkin’s lymphoma, leukemia, brain can- cer, uterine cancer, soft-tissue sarcoma, Hodgkin’s disease, and low sperm concentration. More specifically, researchers have focused on organophosphate 30–63,77,98,100 and organochlorine pesticides. 73–83,98,100,117,118 Furthermore, much work has been done on synthetic pyrethroid insecticides, 35,50,51,56,57,77,87–90,100 triazines, 35,50–52,56,77,94 chloroacetanilides, 35,51,52,56,57,94,100 phen- oxyacetic acid herbicides, 35,45,50–52,94,95 chlorophenols, 35,45,55 and neurotoxic carbamates. 35,45,53–60,77,100 All these compounds are of scientific interest because of their potential toxicological risk for human health and their widespread use. Concerning pesticide metabolism, the most widely studied metabolites comprise the six nonspecific dialkyl phosphate (DAP) metabolites of organophosphate pesticides and metabolites specific to chlorpyrifos, malathion, diazinon, and methyl parathion. Other pesticide metabolites frequently monitored in biological samples are dichlorodiphenyldichloro- ethylene [(p,p’-DDE), a very biostable metabolite of Received for publication July 13, 2006; accepted January 17, 2007. From the Departments of Medicine, *Centre of Toxicology Science and Research, School of Health Sciences, University of Crete, Crete; and †University of Thessaly, Larisa, Greece. Correspondence: Prof. Aristidis Tsatsakis, Department of Medicine, Centre of Toxicology Science and Research, School of Health Sciences, University of Crete, Voutes, Heraklion, 71409 Crete, Greece (e-mail: aris@med. uoc.gr). Copyright Ó 2007 by Lippincott Williams & Wilkins 150 Ther Drug Monit Volume 29, Number 2, April 2007
Transcript of Analytical Methods of Biological Monitoring for Exposure to Pesticides: Recent Update

REVIEW ARTICLE
Analytical Methods of Biological Monitoring forExposure to Pesticides: Recent Update
Maria G. Margariti, MSc,* Andreas K. Tsakalof, PhD,† and Aristidis M. Tsatsakis, PhD, DSc*
Abstract: Extensive use of synthetic pesticides for agricultural and
nonagricultural purposes began in the past 50 years. As a result of their
wide and extensive application, exposure to hazardous pesticides is
a concern to the general population and occupationally exposed
persons. Robust methods are therefore needed for measuring markers
of pesticide exposure. This article presents a review of the most
recently published analytical methodologies and instrumentations
developed for and applied to biological monitoring of exposure to
pesticides of various classes. Most of the methods reviewed here are
based on chromatography combined with mass spectrometry de-
tection. This work clearly demonstrates that although gas chroma-
tography still appears to be the most widely employed technique for
pesticide analysis in various biological samples, recently a trend has
been observed toward the use of liquid chromatography coupled with
tandem mass spectrometry.
Key Words: biological monitoring, analytical methods, pesticides,
chromatography, mass spectrometry
(Ther Drug Monit 2007;29:150–163)
INTRODUCTIONPesticides are substances used to prevent, destroy, repel,
or mitigate any pests, ranging from insects, animals, and weedsto microorganisms such as fungi, molds, bacteria, viruses, andother organisms that compete with humans for food, destroyproperty, spread disease, or are considered a nuisance.1,2
Extensive use of synthetic pesticides for agricultural andnonagricultural purposes began in the past 50 years. Pesticideapplication has increased and improved food supply and alsohas assisted in the control and reduction of food-borne andvector-borne diseases, which affect and kill many children andadults annually.3,4 On the other hand, pesticides also posesubstantial human health concerns because they are usually,but not always, poisonous to humans and are widely releasedinto environment. As a consequence of their wide and exten-sive application in agricultural and residential settings andtheir physicochemical properties, these compounds have been
found in all compartments of the environment. Consequently,exposure of the general population at some level to severaldifferent pesticide residues is almost inevitable.5
The risks of extended pesticide usage are mainly relatedto the acute and chronic toxicity of these compounds.Unintentional exposure is a common cause of acute poisoning,particularly among young children.6 Furthermore, thesecompounds are sometimes used for suicidal reasons.7,8 Theliterature contains a number of cases of acute pesticidepoisoning due to ingestion or inhalation of pesticides.9–14
There also have been a number of studies dealing with chronicpesticide exposures.15–18 Chronic toxicity can be the conse-quence of occupational exposure or continual consumption ofcontaminated food and drinking water. It has been shown thatpesticides can cause adverse human health effects due to theirability to accumulate in environmental media (air, water,sediments, soil) and food products. Separate occupational riskassessment research is conducted for farmers and otherprofessional pesticide handlers or applicators. Recent studieshave indicated that individuals employed in agriculture, as wellas their children, may be at higher risk of pesticide exposurethan the general population.19–23 For risk assessment of humanexposure to these substances and their metabolites, constantbiological monitoring is required.
Detection of pesticide residues and/or their metabolitesin biologic samples from the general population and occu-pationally exposed persons is in rapid development and hasrecently been the subject of many articles.1–118 Current re-search is focusing more on those pesticides that are consideredto be endocrine-disrupting and potential risk factors for thedevelopment of non-Hodgkin’s lymphoma, leukemia, brain can-cer, uterine cancer, soft-tissue sarcoma, Hodgkin’s disease, andlow sperm concentration. More specifically, researchers havefocused on organophosphate30–63,77,98,100 and organochlorinepesticides.73–83,98,100,117,118 Furthermore, much work has beendone on synthetic pyrethroid insecticides,35,50,51,56,57,77,87–90,100
triazines,35,50–52,56,77,94 chloroacetanilides,35,51,52,56,57,94,100 phen-oxyacetic acid herbicides,35,45,50–52,94,95 chlorophenols,35,45,55
and neurotoxic carbamates.35,45,53–60,77,100 All these compoundsare of scientific interest because of their potential toxicologicalrisk for human health and their widespread use.
Concerning pesticide metabolism, the most widelystudied metabolites comprise the six nonspecific dialkylphosphate (DAP) metabolites of organophosphate pesticidesand metabolites specific to chlorpyrifos, malathion, diazinon,and methyl parathion. Other pesticide metabolites frequentlymonitored in biological samples are dichlorodiphenyldichloro-ethylene [(p,p’-DDE), a very biostable metabolite of
Received for publication July 13, 2006; accepted January 17, 2007.From the Departments of Medicine, *Centre of Toxicology Science and
Research, School of Health Sciences, University of Crete, Crete; and†University of Thessaly, Larisa, Greece.
Correspondence: Prof. Aristidis Tsatsakis, Department of Medicine, Centre ofToxicology Science and Research, School of Health Sciences, Universityof Crete, Voutes, Heraklion, 71409 Crete, Greece (e-mail: [email protected]).
Copyright � 2007 by Lippincott Williams & Wilkins
150 Ther Drug Monit � Volume 29, Number 2, April 2007

dichlorodiphenyltrichloroethane (p,p�-DDT)], atrazine mer-capturate (metabolite of atrazine), 2-isopropoxyphenol (me-tabolite of propoxur), and 3-phenoxybenzoic acid (metaboliteof the pyrethroids). Table 1 lists pesticides of particularconcern because of their toxicity and/or the extensiveness oftheir application, as well as their metabolites, that nowadaysare widely monitored in biological samples.
A variety of techniques have been applied for theanalysis of pesticides and their major metabolites in biologicalsamples. In general, all the methods reviewed here sharea common scheme, which comprises three main steps: (1)sample pretreatment with the aim of separation, preconcentra-tion, and sometimes derivatization of component(s) of interest;(2) instrumental analysis of the treated sample; and (3)treatment of the acquired chromatographic data and deducinga conclusion. The vast majority of the methods discussed inthis review were developed for forensic and clinicalapplications or for biological monitoring of high-leveloccupational exposures to pesticides, as well as for epidemi-ologic studies of nonoccupationally exposed persons. Thesemethodologies have been applied to diverse biological samples
such as serum, plasma, whole blood, umbilical cord, urine,breast milk, meconium, and amniotic fluid.
Barr and Needham24 published in 2002 a comprehensivereview of the existing analytical methods of biologicalmonitoring for pesticides, covering developments from 1975to 2001. The scope of our article is to present advances inanalytical methodologies and instrumentations and alsosignificant studies of biological monitoring of exposure topesticides of various classes that have occurred primarily in thepast 6 years. The intent is not to provide an exhaustive list ofanalytical methods but to identify recent reliable methods thatare used in occupational and environmental exposure studies.The main attention will be focused on chromatography-basedtechniques with mass spectrometry (MS) detection.
RECENT ANALYTICAL ACHIEVEMENTS INPESTICIDE MONITORING IN BIOLOGICALSAMPLES AND FUTURE RESEARCH GOALS
In recent years, the analysis of pesticide residues ortheir metabolites in biological matrices has incorporated
TABLE 1. Pesticides and Metabolites of Particular Concern and Their Classes
Analyte Name Parent Pesticide Name (Indicator of Exposure)
Organophosphate pesticides: dialkyl phosphate metabolites
Dimethylphosphate (DMP) Azinphos methyl, dichlorvos, dicrotophos, dimethoate, fenitrothion,fenthion, malathion, methyl parathion, trichlorfon
Dimethylthiophosphate (DMTP) Azinphos methyl, dimethoate, fenchlorphos, fenitrothion, fenthion,malathion, methyl parathion
Dimethyldithiophosphate (DMDTP) Azinphos methyl, dimethoate, malathion
Diethylphosphate (DEP) Chlorpyrifos, coumaphos, diazinon, disulfoton, ethion, parathion, phorate
Diethylthiophosphate (DETP) Chlorpyrifos, coumaphos, diazinon, disulfoton, ethion, parathion, phorate
Diethyldithiophosphate (DEDTP) Disulfoton, phorate
Organophosphate pesticides: specific metabolites
2-Isopropyl-4-methyl-6-hydroxypyrimidine (IMPY) Diazinon
Malathion dicarboxylic acid (MDA) Malathion
Para-nitrophenol (PNP) Parathion, methyl parathion
3,5,6-Trichloro-2-pyridinol (TCPY) Chlorpyrifos, chlorpyrifos methyl
Carbamates
2-Isopropoxyphenol (2IPP) Propoxur
1-Naphthol (1N) Carbaryl, naphthalene
Carbofuranphenol Carbofuran, carbosulfan
Pyrethroid pesticides
cis- and trans-3-(2,2-dichlorovinyl)-2,2-dimethyl-cyclopropane-1-carboxylic acids (cis-/trans-DCCA)
Cyfluthrin, cypermethrin, permethrin
cis-3-(2,2-dibromovinyl)-2,2-dimethylcyclopropane-1-carboxylicacid (DBCA)
Deltamethrin
4-Fluoro-3-phenoxybenzoic acid (4F3PBA) Cyfluthrin
3-Phenoxybenzoic acid (3PBA) At least 10 different synthetic pyrethroids
Herbicides
Chloroacetanilide herbicides
Acetochlor mercapturate Acetochlor
Alachlor mercapturate Alachlor
Metolachlor mercapturate Metolachlor
Triazine herbicides
Atrazine mercapturate (AM) Atrazine
Organochlorine pesticides
Dichlorodiphenyldichloroethylene (p,p’-DDE) Dichlorodiphenyltrichloroethane (p,p’-DDT)
q 2007 Lippincott Williams & Wilkins 151
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure

new technologies aiming to develop and apply more efficientprocedures, with the goal of increasing accuracy andminimizing time, labor costs, environmental pollution, andanalysts’ exposure to toxic chemicals. Hence, a variety ofanalytical techniques have been applied, including mainly gaschromatography–tandem mass spectrometry (GC-MS/MS),liquid chromatography–mass spectrometry (LC-MS), andliquid chromatography–tandem mass spectrometry (LC-MS/MS). Usually, the key step in analytical procedureshas been not the instrumental analysis itself but samplepretreatment for isolation of studied compounds from thematrix. Over the years, several procedures have beendeveloped with this aim, comprising liquid–liquid extraction,solid-phase extraction, and microextraction. In the analyticalmethods based on the above-mentioned techniques, mainattention has been paid to simplification, miniaturization, andimprovement of the sample extraction and clean-up steps,which are usually the most time- and labor-expensive partsof the analysis.
Among the recent approaches for measuring pesticidesin biological matrices, multiresidue procedures have been themost common. However, multianalyte method developmentfor the detection of pesticides of various classes isa challenging problem, because compounds of differentpolarities, solubilities, volatilities, and pKa values must besimultaneously extracted and analyzed.25 MS is a verysensitive, universal, yet selective detection technique, mostsuitable for both multiresidue detection and trace levelidentification of a wide range of pesticides.50,51,55,56,77,94,98,100
Chronologically, the gas chromatography–mass spec-trometry (GC-MS)-based methods were among the firstinstrumental techniques introduced for pesticide monitoring,followed by GC-MS/MS. The tandem MS technique (MS/MS)allows highly specific MS analysis26,27 and thereby canimprove detection limits by avoiding most of the inter-ferences, especially when analyzing complex matrices such asbiological samples. However, the number of compounds orbreakdown products of concern that cannot be analyzeddirectly by GC owing to their poor volatility, high polarity,and/or thermal instability has grown dramatically in the pastfew years. Among the analytical approaches used in pesticideanalysis, LC is effective in separating nonvolatile andthermally labile compounds, as well as GC-compatiblepesticides.28
The use of LC in combination with MS in the analysis ofbiomarkers of exposure has shown sustained growth in recentyears. Analytical techniques relying on LC-MS/MS withatmospheric pressure ionization (API) interfaces, mainlyatmospheric pressure chemical ionization (APCI) and elec-trospray (ESI), although (still) relatively expensive, seem tooffer a high degree of selectivity and sensitivity, which arerequired for trace- and ultratrace-level analysis of biologicalsamples.38,39,46,48–51,64,94,95 These techniques have the facility,in some cases, for direct analysis without extensive samplepretreatment and clean-up procedures.38,39 In a recent reviewarticle, Hernandez et al29 demonstrated the advantages ofLC-MS/MS methods for identification, quantification, andconfirmation of pesticides and their metabolites in biologicalsamples.
Organophosphate PesticidesOrganophosphate (OP) pesticides inhibit the cholines-
terase enzymes, including acetylcholinesterase, and can bevery harmful to human health.65 For this reason, occupationalhuman exposure to these pesticides is usually evaluated bymeasuring the reduction in cholinesterase enzyme activity inblood between predose and post-dose exposure.66 However,this indicator lacks selectivity and also sensitivity for low-levelexposure, and it requires the establishment of baseline activityin nonexposed subjects.
OP pesticides are rapidly metabolized in the humanbody,67 suggesting that measurement of intact pesticides inblood or urine is not useful in most cases. Once entering thebody, OPs can be enzymatically converted to their oxon form,which then reacts with available cholinesterase.40 The oxonform also can be enzymatically or spontaneously hydrolyzedto form a specific metabolite moiety and a DAP metabolite,which is a common metabolite of this family of compounds. Ifthe pesticide is not converted to its oxon form, it can undergohydrolysis to its specific metabolite and dialkylthionatemetabolites (dialkylthiophosphate and/or dialkyldithiophos-phate). These metabolites and/or their glucuronide or sulfateconjugates are excreted in urine40 and can be measured asbiomarkers of exposure to OP pesticides.
Dialkyl Phosphate MetabolitesUrinary DAP metabolites have been used to assess
human exposure to OP pesticides.32,33,40–44 The six commonDAP metabolites measured are dimethylphosphate (DMP),diethylphosphate (DEP), dimethylthiophosphate (DMTP),dimethyldithiophosphate (DMDTP), diethylthiophosphate(DETP), and diethyldithiophosphate (DEDTP). The detectionof urinary alkyl phosphates indicates OP intoxication but doesnot verify the presence of a particular OP compound.
In most cases, the methods of analysis that have beendeveloped in recent years enable the simultaneous detection ofall six DAPs.30–35,37 A number of these methods are outlined inTable 2. The application of GC-MS-based methods usuallyrequires metabolite isolation from the matrix, followed by theirchemical derivatization prior to instrumental analysis. Iso-lation of DAPs from the matrix constitutes one of the mostchallenging parts of DAP analysis. Typical isolation techni-ques reported in the literature include liquid–liquid extractionwith use of polar solvents,30,36,37 azeotropic distillation withuse of acetonitrile,32 and solid–liquid extraction of lyophilizedurine samples.31,33 The use of isotopically labeled analogues asinternal standards for each of these analytes (isotope dilution)allows for sample-specific adjustment for recovery and thuspermits a high degree of accuracy and precision. Morerecently, LC-MS/MS methods have also been developed37–39
in order to detect DAPs in urine, and these powerful analyticaltools have inherent advantages (speed of analysis and highselectivity and sensitivity). The substantial simplification ofsample pretreatment is the major advantage of LC-MS/MS-based methods. However, this advantage is partially counter-balanced by the increased instrument cost in comparison withGC-MS systems.
Hardt and Angerer30 have used GC-MS with electronimpact ionization (EI) for the detection of DAPs in human
152 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007
urine. The compounds were extracted from acidified urine intoa mixture of diethylether and acetonitrile. Dibutylphosphatewas used as internal standard (IS). Derivatization was per-formed with pentafluorobenzylbromide (PFBBr). The limit ofdetection (LOD) for DMP was 5 mg/L, and that for the otherfive metabolites was 1 mg/L. The absolute analyte recoveriesranged from 68% to 114%. The method was successfullyapplied to determine the DAPs in urine samples collected fromthe general population.
Significant improvement in sensitivity was obtainedby operating with GC-MS/MS in a negative ion chemicalionization (NCI) mode.31 The combination of NCI andMS/MS allowed a high degree of selectivity and sensitivity,avoiding most of the interferences present in this complexbiological matrix. Before analyses, urine samples werelyophilized and derivatized with PFBBr. The LODs of themethod were 0.5 mg/L for DMP, 0.1 mg/L for DEP and DMTP,0.04 mg/L for DMDTP and DETP, and 0.02 mg/L for DEDTP.With this method, urinary levels of DAPs were measured innonoccupationally exposed persons.
Bravo et al32 also developed an analytical method forthe simultaneous detection of the six DAPs, using azeotropiccodistillation with acetonitrile and derivatization with1-chloro-3-iodopropane, followed by analysis with isotopedilution gas chromatography–positive ion chemical ionization(PCI)–tandem mass spectrometry. The detection limits were inthe low- to mid-pg/mL range. The method was used tomeasure DAP concentrations in approximately 2000 urinesamples collected from the U.S. general population.40 How-ever, according to the authors, recoveries in azeotropicdistillation were typically low (range, 60–80%), probablybecause of incomplete dissolution of the residue or binding ofDAPs to glass; the residues were extremely dirty; and theresidues still contained residual water. Hence, the samplepreparation technique should be further improved.
The same group, Bravo et al33 has recently proposed analternative method for determining urinary DAPs, based onlyophilization of urine samples instead of azeotropic distilla-tion, followed by GC-PCI-MS/MS (triple-quadrupole) analy-sis and isotope dilution quantification. Lyophilization was
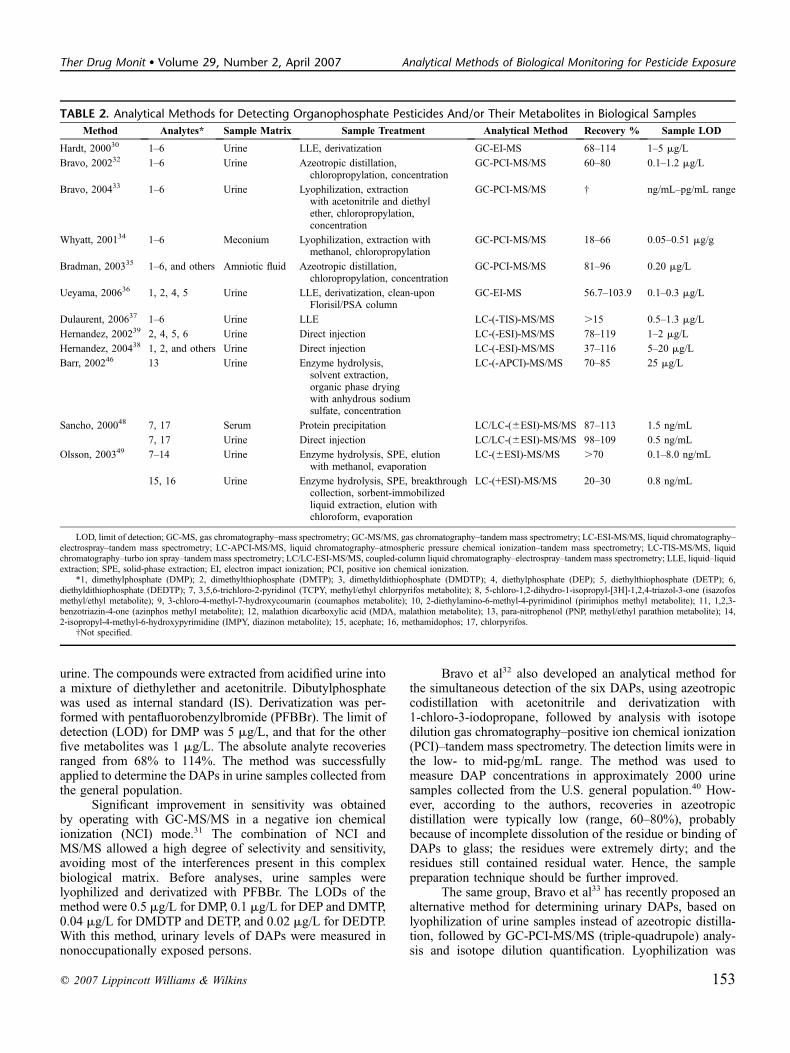
TABLE 2. Analytical Methods for Detecting Organophosphate Pesticides And/or Their Metabolites in Biological Samples
Method Analytes* Sample Matrix Sample Treatment Analytical Method Recovery % Sample LOD
Hardt, 200030 1–6 Urine LLE, derivatization GC-EI-MS 68–114 1–5 mg/L
Bravo, 200232 1–6 Urine Azeotropic distillation,chloropropylation, concentration
GC-PCI-MS/MS 60–80 0.1–1.2 mg/L
Bravo, 200433 1–6 Urine Lyophilization, extractionwith acetonitrile and diethylether, chloropropylation,concentration
GC-PCI-MS/MS † ng/mL–pg/mL range
Whyatt, 200134 1–6 Meconium Lyophilization, extraction withmethanol, chloropropylation
GC-PCI-MS/MS 18–66 0.05–0.51 mg/g
Bradman, 200335 1–6, and others Amniotic fluid Azeotropic distillation,chloropropylation, concentration
GC-PCI-MS/MS 81–96 0.20 mg/L
Ueyama, 200636 1, 2, 4, 5 Urine LLE, derivatization, clean-uponFlorisil/PSA column
GC-EI-MS 56.7–103.9 0.1–0.3 mg/L
Dulaurent, 200637 1–6 Urine LLE LC-(-TIS)-MS/MS .15 0.5–1.3 mg/L
Hernandez, 200239 2, 4, 5, 6 Urine Direct injection LC-(-ESI)-MS/MS 78–119 1–2 mg/L
Hernandez, 200438 1, 2, and others Urine Direct injection LC-(-ESI)-MS/MS 37–116 5–20 mg/L
Barr, 200246 13 Urine Enzyme hydrolysis,solvent extraction,organic phase dryingwith anhydrous sodiumsulfate, concentration
LC-(-APCI)-MS/MS 70–85 25 mg/L
Sancho, 200048 7, 17 Serum Protein precipitation LC/LC-(6ESI)-MS/MS 87–113 1.5 ng/mL
7, 17 Urine Direct injection LC/LC-(6ESI)-MS/MS 98–109 0.5 ng/mL
Olsson, 200349 7–14 Urine Enzyme hydrolysis, SPE, elutionwith methanol, evaporation
LC-(6ESI)-MS/MS .70 0.1–8.0 ng/mL
15, 16 Urine Enzyme hydrolysis, SPE, breakthroughcollection, sorbent-immobilizedliquid extraction, elution withchloroform, evaporation
LC-(+ESI)-MS/MS 20–30 0.8 ng/mL
LOD, limit of detection; GC-MS, gas chromatography–mass spectrometry; GC-MS/MS, gas chromatography–tandem mass spectrometry; LC-ESI-MS/MS, liquid chromatography–electrospray–tandem mass spectrometry; LC-APCI-MS/MS, liquid chromatography–atmospheric pressure chemical ionization–tandem mass spectrometry; LC-TIS-MS/MS, liquidchromatography–turbo ion spray–tandem mass spectrometry; LC/LC-ESI-MS/MS, coupled-column liquid chromatography–electrospray–tandem mass spectrometry; LLE, liquid–liquidextraction; SPE, solid-phase extraction; EI, electron impact ionization; PCI, positive ion chemical ionization.
*1, dimethylphosphate (DMP); 2, dimethylthiophosphate (DMTP); 3, dimethyldithiophosphate (DMDTP); 4, diethylphosphate (DEP); 5, diethylthiophosphate (DETP); 6,diethyldithiophosphate (DEDTP); 7, 3,5,6-trichloro-2-pyridinol (TCPY, methyl/ethyl chlorpyrifos metabolite); 8, 5-chloro-1,2-dihydro-1-isopropyl-[3H]-1,2,4-triazol-3-one (isazofosmethyl/ethyl metabolite); 9, 3-chloro-4-methyl-7-hydroxycoumarin (coumaphos metabolite); 10, 2-diethylamino-6-methyl-4-pyrimidinol (pirimiphos methyl metabolite); 11, 1,2,3-benzotriazin-4-one (azinphos methyl metabolite); 12, malathion dicarboxylic acid (MDA, malathion metabolite); 13, para-nitrophenol (PNP, methyl/ethyl parathion metabolite); 14,2-isopropyl-4-methyl-6-hydroxypyrimidine (IMPY, diazinon metabolite); 15, acephate; 16, methamidophos; 17, chlorpyrifos.
†Not specified.
q 2007 Lippincott Williams & Wilkins 153
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure

chosen as a simpler and robust technique. The multiplereaction monitoring (MRM) technique employed is based onthe simultaneous monitoring of characteristic reactions (pre-cursor ion!product ion transitions), such as the fragmentationof an ion. With the MRM technique, because pairs of ionsare being selected for each compound, interferences fromcoextracted components can be reduced significantly, pro-viding higher selectivity and sensitivity in comparison withselected ion monitoring (SIM), which is a less-specific scanmode. Using this method, urinary levels of DAPs weremeasured in approximately 1100 samples collected fromchildren and pregnant women.
In another study, Whyatt and Barr34 presented aninteresting and promising method for measuring the six DAPsin postpartum meconium using lyophilization, simple meth-anol extraction, and chemical derivatization. Samples wereanalyzed with GC-MS/MS and quantified by isotope dilution.Meconium samples were collected from 20 newborns. LODswere low (0.05–0.51 mg/g) and comparable to or lower thanpreviously reported urine levels (Table 2). The recoveries ofDAPs from 0.5 g spiked meconium samples ranged from 18%to 66%. All samples contained some level of OP metabolites.In particular, DEP was detected in 19 of 20 samples (95%),and DETP in 20 of 20 (100%). DMP and DEDTP were eachdetected in 1 of 20 (5%). DMTP and DMDTP were notdetected. This work is particularly valuable because it provesthat OP metabolites in meconium hold great promise asbiomarkers of prenatal exposure.
In a more recent study, Bradman et al35 demonstratedthat analytical procedures for measuring DAPs and othertoxicants in urine could be effectively transferred to amnioticfluid with minor modifications. They used the methodologyof Bravo et al32 to analyze amniotic fluid samples forDAPs. The method was completely validated, and the re-coveries, detection limits, and precision were similar to thoseof the urine method.32 It is interesting that the researchersreported the detection of DEP and DMP in 2 samples (10%)collected from 20 women and the detection of DMTP in 1sample (5%).
Recently, Ueyama et al36 detected DMP, DEP, DMTP,and DETP in urine by GC-MS with EI, using liquid–liquidextraction (LLE) and PFBBr derivatization. The use ofdibutylphosphate as IS instead of stable isotope analoguesof the DAPs allowed reduction of the analysis cost. Increasedsensitivity in the detection of DAPs was obtained by applyinga clean-up step after derivatization, with the use of a three-layer column of Florisil, Bondesil-Primary/Secondary Amine(PSA), and anhydrous sodium sulfate. Column clean-up wassufficiently effective to remove matrix components and theunreacted PFBBr. As stated by the authors, excess PFBBr ininjected samples would damage GC-MS systems, includingthe column and the detector. These authors markedly reducedthe specimen preparation time to only about 3 hours, andthe LODs of the method were estimated to be approximately0.3 mg/L for DMP and 0.1 mg/L for DEP, DMTP, and DETPfor a 5 mL urine sample. This sensitivity allowed the detectionof DAPs in 48 urine samples collected from a Japanesepopulation with and without occupational exposure to OPs.As pointed out by the authors, the main advantages of this
approach were high sensitivity, reduction of sample prepara-tion time, and use of relatively simple equipment.
LC-MS/MS methods37–39 with ESI and turbo ion spray(TIS) sources have also been developed for measuring DAPsin urine. The main advantage of these approaches was the lackof a tedious derivatization step. In particular, Dulaurent et al37
applied LC-MS/MS for simultaneous detection of the sixDAPs in human urine. Metabolite isolation was performedwith LLE in acidic conditions and the successive use ofdiethylether and ethyl acetate. Dibutylphosphate was used asIS. These authors used a triple-quadrupole mass spectrometerequipped with a TIS source in the negative ionization (NI)mode. The limit of quantification (LOQ) of the method was2 mg/L for all six metabolites in the MRM mode. The methodproved useful for the determination of urine levels of DAPs innonexposed volunteers. They detected concentrations of DEPin all donors, and almost every sample contained DMP andDMTP. DMDTP, DETP, and DEDTP were found in lowerconcentrations. According to the authors, the inconsistent andlow recoveries (.50% except for DMP, which was approx-imately 15%) probably were due to an inefficient extractionprocedure. Therefore, for quantitative determination of DAPs,the extraction step should be improved or even eliminated.
Hernandez et al proposed liquid chromatography–negative ion electrospray–tandem mass spectrometry (LC-ESI-NI-MS/MS) for the direct analysis of four DAPs (DMTP,DEP, DETP, DEDTP)39 and for direct analysis of two DAPs(DMP, DMTP) together with methyl parathion conjugates38 inhuman urine. In these cases of direct sample injection, no ISwas used. In the first method,39 prior to injection, samples werecentrifuged and tetrabutylammonium acetate (TBA) wasadded. MRM was performed after chromatographic separationon a Discovery C18 column. The use of TBA as an ion-pairingreagent enabled the complete separation of the DAPs by LC. Inorder to obtain adequate chromatographic peaks, TBA wasalso added to the sample. However, as stated by the authors,this high content of TBA inhibited the MS response,preventing the detection of DMP. The direct injection of urinesamples into the LC-MS/MS system resulted in betterrecoveries (range, 78–119%) than those obtained with doubleLLE extraction.37 The LODs of this method were 1 mg/L forDEP, DETP, and DEDTP and 2 mg/L for DMTP. Its applicationto the analysis of urine samples from farmers exposed to theOP pesticide chlorpyrifos proved the method’s practical use, aswell as its robustness. Following application of chlorpyrifosonto a field, results showed good correlation betweenapplication of chlorpyrifos and concentration levels of DEPand DETP in urine samples, whereas no relation betweenapplication and presence of DMTP was observed.
In the other approach,38 TBA (ion-pairing agent) was notadded to the mobile phase, as in the previous article,39 in orderto avoid the suppression of MS response and to quantify DMP.Prior to injection, samples were diluted 3-fold with 0.5%HCOOH and TBA was added. Limitations due to the obtainedrecoveries (range, 37–116%) and sensitivity (LODs were 5and 20 mg/L for DMTP and DMP, respectively) reducedthe applicability of this method for quantitative analysis, butaccording to the authors, the method is suitable for semi-quantitative screening. By this semiquantitative method,
154 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007

urinary levels of alkyl phosphates were measured in an unex-posed population and in a grower who applied the OP pesticidemethyl parathion. Hernandez et al concluded that theirmethod, although not being useful for quantification becauseof matrix differences between urine samples, could providevery useful information with regard to exposed populations.
From the above-reviewed published findings, it is clearthat LC-MS/MS provides a valuable alternative to GC-MS-based analytical methods for detection of the polar alkylphosphates, especially in terms of sample preparation (notedious clean-up or derivatization step) and, as a result, overallspeed of analysis. In particular, the use of LC-MS/MS allowsthe direct injection of urine samples into the LC system andrapid detection of DAPs.38,39 Selection of the correctionization mode, such as negative ion TIS, and the optimaleluent system, such as acetonitrile/2 mM ammonium formatepH 3.0 (gradient), for LC-MS/MS analysis influences sub-stantially the LOD of the method.
Specific Organophosphate MetabolitesPesticide-specific metabolites of OPs have also been the
subject of recent research. Whereas DAPs are nonspecificbiomarkers, because they are potential metabolites of most ofthe OPs, detection of the specific metabolite (if it exists) isusually chosen for biological monitoring in order to indicatelow-level exposure to some OP pesticides. For instance,methyl parathion exposure has been estimated on the basis ofurinary measurements of para-nitrophenol (PNP), its mostabundant metabolite. Although it is also a metabolite ofparathion (ethyl parathion), nitrobenzene, and a few similarcompounds, PNP is considered a more specific biomarker ofexposure to methyl parathion than are the DAPs.24,72
Barr et al46 developed a method for quantifying urinaryPNP, using isotope dilution liquid chromatography–negativeion atmospheric pressure chemical ionization–tandem massspectrometry (LC-APCI-NI-MS/MS) on a triple-quadrupoleinstrument. The method involved enzyme hydrolysis andincubation at 37�C to liberate the PNP from the glucuronide orsulfate esters. The samples were then acidified and extracted.The LOD was 25 mg/L, whereas the authors claimed thattheir subsequent studies resulted in a detection limit of only81 ng/L, as calculated in spiked urine samples. The almost300-fold increase in sensitivity was achieved by the applica-tion of a higher (up to 50�C) vaporizer and capillary tem-perature. However, it must be noted that increased sensitivity(0.1 ng/mL) in the detection of PNP was obtained in a morerecent study49 by applying ESI. As compared with formermethods,45 the main advantage of the present approach46 wasthe high specificity of the instrumental part of the analysis, aswell as elimination of the derivatization step. The method wasapplied to analysis of urine samples collected from residentsexposed to methyl parathion and/or PNP in their homes, whichwere contaminated by the illegal application of methylparathion. It was also used by Hryhorczuk et al62 to analyzefor PNP in urine samples collected from 75 individuals whowere members of methyl parathion–contaminated households.
Sancho et al48 used coupled-column liquid chromatog-raphy–electrospray–tandem mass spectrometry (LC/LC-ESI-MS/MS) to detect chlorpyrifos and its main metabolite,
3,5,6-trichloro-2-pyridinol (TCPY), in human serum and urine.Serum samples were protein-precipitated before injection,whereas urine was directly introduced into the LC/LC system.The triple-quadrupole system was operated in positive ion (PI)and NI mode for the analysis of chlorpyrifos and TCPY,respectively. The LOD for both analytes was 1.5 ng/mL inserum and 0.5 ng/mL in urine. The on-line sample clean-upwas sufficiently effective to remove matrix components,compensating for ESI signal suppression and allowing accu-rate quantification of analytes without the use of any IS. Themethod combines sensitivity, specificity, simplicity, speed, andprecision. Direct analysis of urine samples or simplifiedtreatment of serum samples, together with short instrumentalrun time (8 minutes), ensures high throughput of thedeveloped method and renders it suitable for the monitoringof population exposure to chlorpyrifos.
In another study, Olsson et al49 developed a sensitivemethod to quantify specific biomarkers of 10 OP pesticidesor their O,O-dimethyl analogues in human urine, as theirselective metabolites or as the intact pesticide. Urine wasspiked with isotopically labeled analogues of 8 of the 10analytes, enzymatically hydrolyzed and extracted with solid-phase extraction (SPE) on Oasis hydrophily and lypophilybalanced matrix (HLB) cartridges. The analytes were sepa-rated on a narrow-bore column and detected by a triple-quadrupole mass spectrometer in PI or NI ESI mode. LODsfor most of the analytes, related to 2 mL of urine, ranged from0.1 to 1 ng/mL, which are slightly better or about the samein comparison with those in other methods.45,48,50,64,68–71
However, the lack of labeled IS for 2 of the compoundsresulted in higher variation. By this method, urinary levelsof the target analytes were measured in 140 persons withunknown exposure status, and detectable concentrations werefound of 8 of the 10 analytes. The most frequently detectedcompound was PNP, followed by malathion dicarboxylic acid(MDA, metabolite of malathion). These 2 compounds werepresent in 99% and 74% of the samples analyzed.
Organochlorine PesticidesBecause of their persistence in the environment and
potential adverse long-term human health effects, research hasalso been conducted on human exposure to organochlorine(OC) pesticides, including measurements of intact pesticidesor their metabolites in serum, breast milk, meconium, andumbilical cord. The identification and quantification of OCsand their metabolites in biological matrices require applicationof effective extraction and sample purification proceduresand detection by GC, mostly by means of electron capturedetection (ECD), MS, and high-resolution mass spectrometry(HRMS). A powerful development in this field is the intro-duction of Oasis HLB sorbent cartridges for extraction of OCpesticides from serum, which improves extraction recoveriesand eliminates the need for additional laborious clean-upprocedures for removing interferences.77,79
Eleven OC pesticides or their metabolites, together with38 polychlorinated biphenyls (PCBs), have been simultaneouslyseparated with a DB-5MS column and analyzed by isotopedilution of GC-HRMS in human serum.73 The method incor-porated the SPE procedure previously developed by Sandau
q 2007 Lippincott Williams & Wilkins 155
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure
et al74 (Table 3) and achieved LODs in the low-pg/mL range.With an instrumental analysis time of less than 30 minutes, theproposed multicomponent approach provides high throughputfor detection of OC and polychlorinated biphenyl residues. TheGC-HRMS results for OC and PCB analysis in a singleanalytical run73 were in good agreement with data obtained bytwo separate analyses for PCBs and OC pesticides.75 In anotherstudy,76 a novel sample preparation procedure was applied, basedon accelerated solvent extraction and gel permeation chroma-tography (GPC) purification. Despite successful applications, theGPC employed in the methods discussed above73,76 is a time-consuming and expensive clean-up procedure; thus, alternativeclean-up techniques must be adopted.
Lacassie et al77 reported a GC-MS multiresidue methodfor the detection of 12 OCs and other volatile pesticides inserum. The method involved rapid SPE with Oasis HLBcartridges (recovery, 40–99%). For quantitation, SIM proce-dures were applied with use of the cyproheptadine as IS, andthe LOD of the method, related to 1 mL of serum, was 5 ng/mLfor all the compounds. This sensitivity allowed the analysis ofreal samples in cases of poisoning.
GC-ECD-based methods have also been successfullyapplied for the analysis of OCs. Increased sensitivity in thedetection of selected OC pesticides and PCBs in human serumwas obtained by Conka et al.78 Their procedure includesdenaturation of serum proteins, off-line SPE (C18), and elutionwith n-hexane/dichloromethane. The extracts were purifiedwith a florisil–silica gel column and analyzed by GC withmicro-electron capture detection (mECD). The method was
validated by using PCB-174 as IS, and the LODs were 1.5–5.0 pg/mL for OC pesticides.
Very recently, Sundberg et al79 proposed a method forrapid quantitation of OC pesticides and PCBs in avian serumsamples (0.1 or 1.0 mL), by using an Oasis HLB SPE followedby GC-ECD analysis. As reported by the authors, the use ofOasis HLB cartridges (mean recoveries of 74–107% for OCpesticides) eliminated the need for additional lipid clean-up,reducing also the sample-preparation time.
To assess in utero exposure to OC pesticides and PCBs,Burse et al80 developed a method for the detection ofhexachlorobenzene (HCB), p,p’-DDE, and 32 PCB congenersin umbilical cord samples collected from women of the FaroeIslands. The method involved homogenization of the cords,partitioning, and microsilica gel column chromatographypurification. The extracts were analyzed with dual-columncapillary GC (DB-5 and DB-1701) with ECD. The substance1,2-dichloronaphthalene was used as IS, although it was addedonly prior to GC. The compounds HCB and p,p’-DDE werepresent in 100% of the samples analyzed.
The detection of OC pesticides in breast milk has beenproposed as an indicator of exposure of mother and child.Campoy et al81 applied GC-ECD to measure various OCpesticides in milk from lactating mothers. After an LLE, thesamples were purified and analyzed. GC-MS was used forconfirmation of the residues detected in samples. Most of thesamples contained residues of dichlorodiphenyldichloroethane(p,p’-DDD, metabolite of DDT), o,p’-dichlorodiphenyltri-chloroethane (o,p’-DDT), p,p’-DDT, and HCB, whereas all
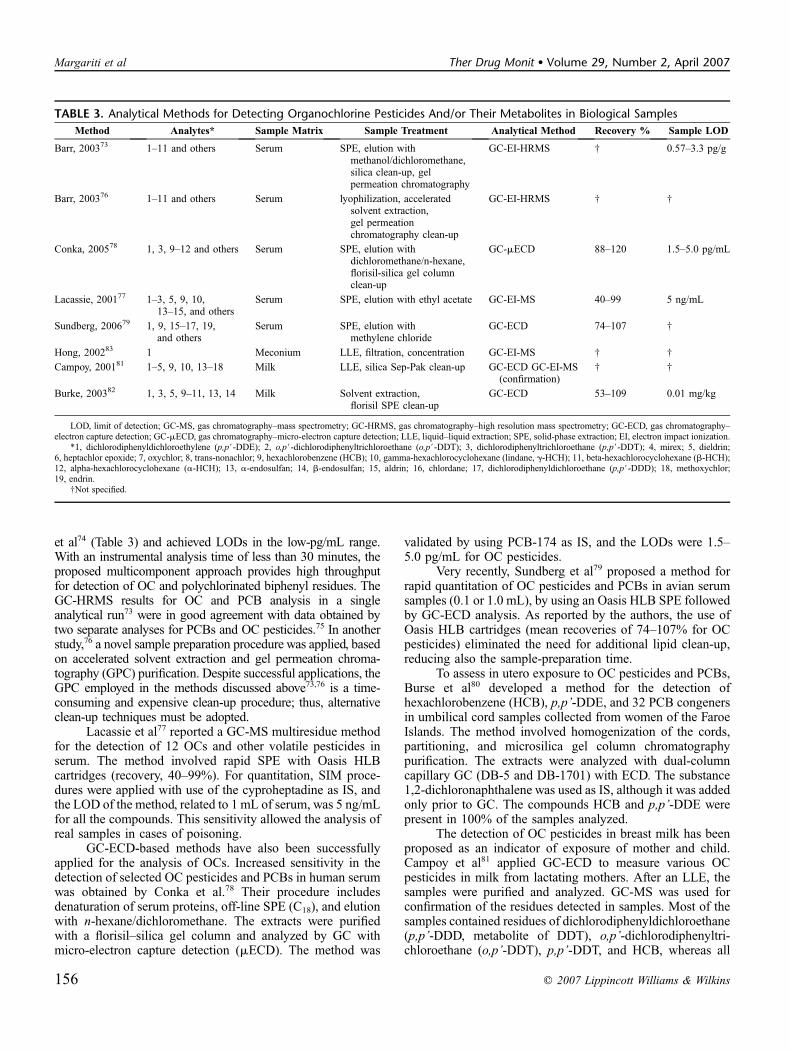
TABLE 3. Analytical Methods for Detecting Organochlorine Pesticides And/or Their Metabolites in Biological Samples
Method Analytes* Sample Matrix Sample Treatment Analytical Method Recovery % Sample LOD
Barr, 200373 1–11 and others Serum SPE, elution withmethanol/dichloromethane,silica clean-up, gelpermeation chromatography
GC-EI-HRMS † 0.57–3.3 pg/g
Barr, 200376 1–11 and others Serum lyophilization, acceleratedsolvent extraction,gel permeationchromatography clean-up
GC-EI-HRMS † †
Conka, 200578 1, 3, 9–12 and others Serum SPE, elution withdichloromethane/n-hexane,florisil-silica gel columnclean-up
GC-mECD 88–120 1.5–5.0 pg/mL
Lacassie, 200177 1–3, 5, 9, 10,13–15, and others
Serum SPE, elution with ethyl acetate GC-EI-MS 40–99 5 ng/mL
Sundberg, 200679 1, 9, 15–17, 19,and others
Serum SPE, elution withmethylene chloride
GC-ECD 74–107 †
Hong, 200283 1 Meconium LLE, filtration, concentration GC-EI-MS † †
Campoy, 200181 1–5, 9, 10, 13–18 Milk LLE, silica Sep-Pak clean-up GC-ECD GC-EI-MS(confirmation)
† †
Burke, 200382 1, 3, 5, 9–11, 13, 14 Milk Solvent extraction,florisil SPE clean-up
GC-ECD 53–109 0.01 mg/kg
LOD, limit of detection; GC-MS, gas chromatography–mass spectrometry; GC-HRMS, gas chromatography–high resolution mass spectrometry; GC-ECD, gas chromatography–electron capture detection; GC-mECD, gas chromatography–micro-electron capture detection; LLE, liquid–liquid extraction; SPE, solid-phase extraction; EI, electron impact ionization.
*1, dichlorodiphenyldichloroethylene (p,p#-DDE); 2, o,p#-dichlorodiphenyltrichloroethane (o,p#-DDT); 3, dichlorodiphenyltrichloroethane (p,p#-DDT); 4, mirex; 5, dieldrin;6, heptachlor epoxide; 7, oxychlor; 8, trans-nonachlor; 9, hexachlorobenzene (HCB); 10, gamma-hexachlorocyclohexane (lindane, g-HCH); 11, beta-hexachlorocyclohexane (b-HCH);12, alpha-hexachlorocyclohexane (a-HCH); 13, a-endosulfan; 14, b-endosulfan; 15, aldrin; 16, chlordane; 17, dichlorodiphenyldichloroethane (p,p#-DDD); 18, methoxychlor;19, endrin.
†Not specified.
156 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007
samples contained the DDT metabolite p,p’-DDE. The methodproposed by Burke et al82 involved solvent extraction, florisilSPE clean-up, and GC-ECD detection. The sensitivityachieved with the latter approach was good (about 0.01mg/kg). Residues of p,p’-DDE and p,p’-DDTwere found in allsamples collected from urban and rural areas of Indonesia.
In another study, Hong et al83 used GC-MS to measurep,p’-DDE in meconium. LLE and filtration were applied to60 meconium samples, and 3 were found to be positive forp,p’-DDE. Chromatographic analysis was performed on aglass column packed with 2% OV-17 on Gaschrom Q. This
study is particularly important because it shows thatmeasurement of persistent OC pesticides or their metabolitesin meconium holds promise as a biomarker of prenatalexposure.
Pyrethroid PesticidesPyrethroid insecticides, which are widely used in
agriculture and in households to control pests, as well as inpublic health to control diseases caused by vectors orintermediate hosts,84,85 have also been the focus of currentresearch. The extensive use of pyrethroid insecticides during
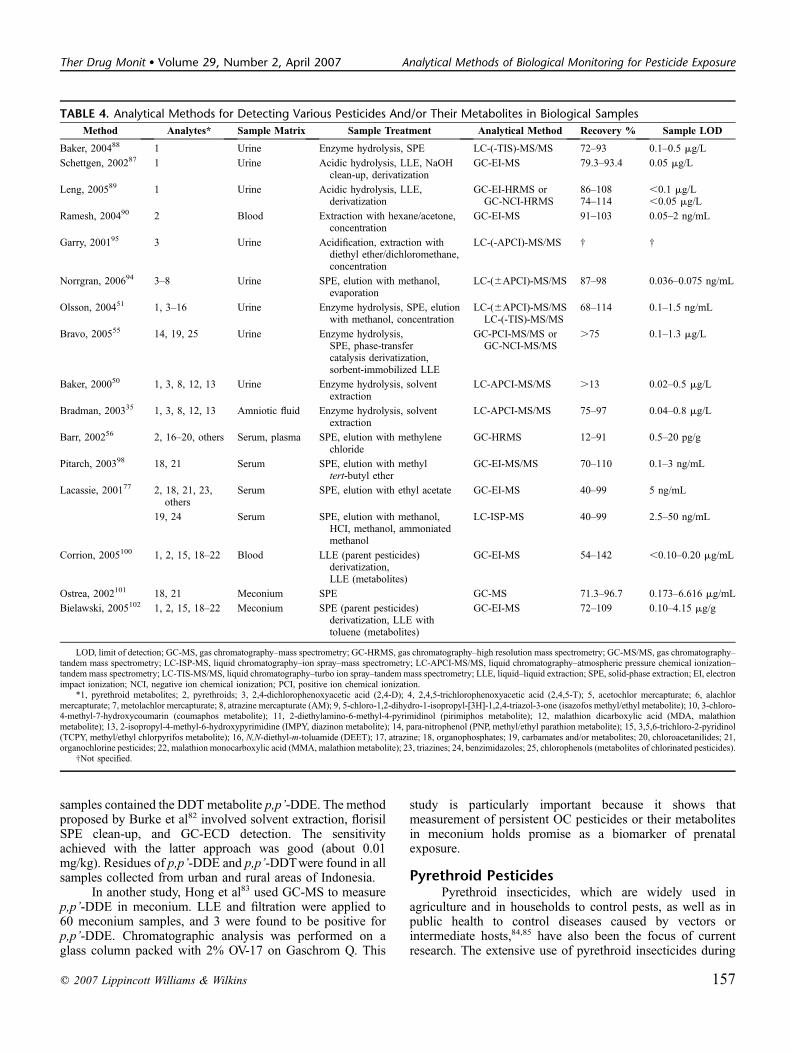
TABLE 4. Analytical Methods for Detecting Various Pesticides And/or Their Metabolites in Biological Samples
Method Analytes* Sample Matrix Sample Treatment Analytical Method Recovery % Sample LOD
Baker, 200488 1 Urine Enzyme hydrolysis, SPE LC-(-TIS)-MS/MS 72–93 0.1–0.5 mg/L
Schettgen, 200287 1 Urine Acidic hydrolysis, LLE, NaOHclean-up, derivatization
GC-EI-MS 79.3–93.4 0.05 mg/L
Leng, 200589 1 Urine Acidic hydrolysis, LLE,derivatization
GC-EI-HRMS orGC-NCI-HRMS
86–10874–114
,0.1 mg/L,0.05 mg/L
Ramesh, 200490 2 Blood Extraction with hexane/acetone,concentration
GC-EI-MS 91–103 0.05–2 ng/mL
Garry, 200195 3 Urine Acidification, extraction withdiethyl ether/dichloromethane,concentration
LC-(-APCI)-MS/MS † †
Norrgran, 200694 3–8 Urine SPE, elution with methanol,evaporation
LC-(6APCI)-MS/MS 87–98 0.036–0.075 ng/mL
Olsson, 200451 1, 3–16 Urine Enzyme hydrolysis, SPE, elutionwith methanol, concentration
LC-(6APCI)-MS/MSLC-(-TIS)-MS/MS
68–114 0.1–1.5 ng/mL
Bravo, 200555 14, 19, 25 Urine Enzyme hydrolysis,SPE, phase-transfercatalysis derivatization,sorbent-immobilized LLE
GC-PCI-MS/MS orGC-NCI-MS/MS
.75 0.1–1.3 mg/L
Baker, 200050 1, 3, 8, 12, 13 Urine Enzyme hydrolysis, solventextraction
LC-APCI-MS/MS .13 0.02–0.5 mg/L
Bradman, 200335 1, 3, 8, 12, 13 Amniotic fluid Enzyme hydrolysis, solventextraction
LC-APCI-MS/MS 75–97 0.04–0.8 mg/L
Barr, 200256 2, 16–20, others Serum, plasma SPE, elution with methylenechloride
GC-HRMS 12–91 0.5–20 pg/g
Pitarch, 200398 18, 21 Serum SPE, elution with methyltert-butyl ether
GC-EI-MS/MS 70–110 0.1–3 ng/mL
Lacassie, 200177 2, 18, 21, 23,others
Serum SPE, elution with ethyl acetate GC-EI-MS 40–99 5 ng/mL
19, 24 Serum SPE, elution with methanol,HCI, methanol, ammoniatedmethanol
LC-ISP-MS 40–99 2.5–50 ng/mL
Corrion, 2005100 1, 2, 15, 18–22 Blood LLE (parent pesticides)derivatization,LLE (metabolites)
GC-EI-MS 54–142 ,0.10–0.20 mg/mL
Ostrea, 2002101 18, 21 Meconium SPE GC-MS 71.3–96.7 0.173–6.616 mg/mL
Bielawski, 2005102 1, 2, 15, 18–22 Meconium SPE (parent pesticides)derivatization, LLE withtoluene (metabolites)
GC-EI-MS 72–109 0.10–4.15 mg/g
LOD, limit of detection; GC-MS, gas chromatography–mass spectrometry; GC-HRMS, gas chromatography–high resolution mass spectrometry; GC-MS/MS, gas chromatography–tandem mass spectrometry; LC-ISP-MS, liquid chromatography–ion spray–mass spectrometry; LC-APCI-MS/MS, liquid chromatography–atmospheric pressure chemical ionization–tandem mass spectrometry; LC-TIS-MS/MS, liquid chromatography–turbo ion spray–tandem mass spectrometry; LLE, liquid–liquid extraction; SPE, solid-phase extraction; EI, electronimpact ionization; NCI, negative ion chemical ionization; PCI, positive ion chemical ionization.
*1, pyrethroid metabolites; 2, pyrethroids; 3, 2,4-dichlorophenoxyacetic acid (2,4-D); 4, 2,4,5-trichlorophenoxyacetic acid (2,4,5-T); 5, acetochlor mercapturate; 6, alachlormercapturate; 7, metolachlor mercapturate; 8, atrazine mercapturate (AM); 9, 5-chloro-1,2-dihydro-1-isopropyl-[3H]-1,2,4-triazol-3-one (isazofos methyl/ethyl metabolite); 10, 3-chloro-4-methyl-7-hydroxycoumarin (coumaphos metabolite); 11, 2-diethylamino-6-methyl-4-pyrimidinol (pirimiphos metabolite); 12, malathion dicarboxylic acid (MDA, malathionmetabolite); 13, 2-isopropyl-4-methyl-6-hydroxypyrimidine (IMPY, diazinon metabolite); 14, para-nitrophenol (PNP, methyl/ethyl parathion metabolite); 15, 3,5,6-trichloro-2-pyridinol(TCPY, methyl/ethyl chlorpyrifos metabolite); 16, N,N-diethyl-m-toluamide (DEET); 17, atrazine; 18, organophosphates; 19, carbamates and/or metabolites; 20, chloroacetanilides; 21,organochlorine pesticides; 22, malathion monocarboxylic acid (MMA, malathion metabolite); 23, triazines; 24, benzimidazoles; 25, chlorophenols (metabolites of chlorinated pesticides).
†Not specified.
q 2007 Lippincott Williams & Wilkins 157
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure

the past 20 years has necessitated the development of robustmethods for detection of these compounds in biologicalsamples. In mammals, pyrethroids are rapidly metabolized intocorresponding carboxylic acids by hydrolytic cleavage of theester bond, followed by oxidation and mainly glucuronization,being eliminated in urine as conjugates.84,86 Because of thisrapid metabolism, the concentrations of intact pyrethroids inblood and serum are much lower than urinary metaboliteconcentrations, and therefore the detection of urinarymetabolites is preferred to blood analysis for the monitoringof pyrethroid exposure.
Schettgen et al87 have used GC-EI-MS for thesimultaneous quantitation of 5 pyrethroid metabolites inurine: cis- and trans-3-(2,2-dichlorovinyl)-2,2-dimethylcyclo-propane-1-carboxylic acids (cis-DCCA and trans-DCCA), cis-3-(2,2-dibromovinyl)-2,2-dimethylcyclopropane-1-carboxylicacid (DBCA), 3-phenoxybenzoic acid (3PBA), and 4-fluoro-3-phenoxybenzoic acid (4F3PBA). After acidic hydrolysis, thecompounds were extracted from urine by LLE with n-hexaneunder acidic conditions, cleaned by NaOH, and derivatized totheir respective volatile esters with N-tert.-butyldimethylsilyl-N-methyltrifluoroacetamide. The substance 2-phenoxybenzoicacid (2PBA) was used as IS. The detection limit (Table 4) was0.05 mg/L for all 5 metabolites in the SIM mode, and thus themethod is sufficiently sensitive for the determination of envi-ronmental exposure levels. By this method, the metabolitestrans-DCCA, 3PBA, and cis-DCCA were the most frequentlydetected in 72%, 70%, and 52% of the samples collected from46 nonoccupationally exposed persons, with median values of0.11, 0.16, and 0.06 mg/L, respectively. Residues of DBCAand 4F3PBA were also detected in some samples.
Baker et al.88 proposed an alternative method forsimultaneous quantitation of the above-mentioned urinarymetabolites of the most common pyrethroids, involving theuse of LC-MS/MS with a TIS source. Before analyses,samples were spiked with isotopically labeled analogues oftrans-DCCA and 3PBA, enzymatically hydrolyzed, and ex-tracted with Oasis HLB SPE cartridges (recovery, 72–93%).The analytes were separated on a Betasil C18 column anddetected in NI mode. Although this approach was not assensitive as the previous one87 (LODs were between 0.1 and0.5 mg/L), the use of LC-TIS-MS/MS instead of GC-MSallowed analysis of urinary pyrethroid metabolites withoutderivatization, reducing also the chromatographic separationtime (total run time of less than 8 minutes). The method wasapplied to measure concentrations of these metabolites in urinefrom persons not occupationally exposed to pyrethroids andsome with suspected residential exposure. Target compoundswere present in 74% of the samples analyzed, and the mostfrequently detected metabolites were 3PBA, trans-DCCA, andcis-DCCA.
A GC-HRMS method that enables for the first timethe simultaneous detection of the 5 pyrethroid metabolitesreported above, together with the pyrethrum metabolite trans-chrysanthemumdicarboxylic acid (trans-CDCA), in humanurine has been developed by Leng et al.89 A particular featureof HRMS detection that was exploited in this study was thecapability to eliminate interfering components even in difficultsamples, such as urine, thus allowing the development of more
sensitive and selective methods for accurately measuring thetarget biomarkers. The method involved acidic hydrolysis,extraction with tert-butyl-methyl-ether, derivatization with1,1,1,3,3,3-hexafluoroisopropanol, and analysis in both EI andNCI modes. The substance 2PBA was used as IS. The analyteswere separated on an Rtx-65 capillary column and detected inSIM mode. The LODs were ,0.1 mg/L in EI mode, whereasthose in NCI mode were estimated to be even lower (,0.05mg/L). The utility of the method is demonstrated by theanalysis of urine samples from 30 persons exposed topyrethroids, as well as from some unexposed subjects. Ascompared with previously developed methods87,88,91 for thequantification of pyrethroid and pyrethrin metabolites inhuman urine, the main advantage of this approach is thepossibility of sensitive and selective detection of all pyrethroidand pyrethrum metabolites in one analytical run with LODs upto 0.05 mg/L in urine.
A rapid and sensitive GC-EI-MS method was developedby Ramesh et al90 for the detection in whole blood of 13pyrethroid insecticides. Detection is difficult because of thetrouble involved in elimination of lipids associated with wholeblood samples and the inherently low concentrations of parentpesticides in blood, in comparison with urinary metaboliteconcentrations. The method involved extraction of pyrethroidswith a hexane/acetone mixture and subsequent evaporation ofthe solvent. Lindane was used as IS and LODs varied between0.05 and 2 ng/mL. When the method was applied to 45 bloodsamples from persons continuously exposed to pyrethroids,none of the samples was found to contain detectable levels of thepyrethroids. The authors attributed their results to the rapidexcretion of these compounds or limitations of the experiment.
Phenoxyacetic Acid HerbicidesPhenoxyacetic acids were synthesized during World War
and were widely and increasingly used as herbicides from theearly 1950s. Previously conducted epidemiologic studies havesuggested an association between chlorophenoxy herbicidesuse and non-Hodgkin’s lymphoma.92,93 Phenoxy herbicides arescarcely biotransformed in mammals.94 The detection in urineof unmodified compounds has been used to monitor humanexposure to these herbicides. Owing to their polarity, acidicherbicides are usually less volatile and thermally instable.Therefore, their analysis with GC is difficult and necessitatesthe use of a complex derivatization procedure.45,96,97 How-ever, in recent years, some studies relying on LC-MS/MShave been proposed for the analysis of these polar pesticideswithout a preceding derivatization step (see Table 4).
Olsson et al51 developed an LC-MS/MS method formeasuring 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), as well as 17 otherurinary markers of human exposure to commonly used pesti-cides. Before analyses, samples were spiked with isotopicallylabeled 13C6-2,4-D and 13C6-2,4,5-T, enzymatically hydro-lyzed, and then extracted with SPE (Oasis HLB). For theanalysis, the researchers used a triple-quadrupole mass spec-trometer equipped with an APCI source in the NI mode. OneMS-MS transition each was monitored for 2,4-D and 2,4,5-T.Limits of detection were 0.2 and 0.1 ng/mL for 2,4-D and2,4,5-T, respectively.
158 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007

In another study, Garry et al95 also applied LC-APCI-NI-MS/MS for determining 2,4-D in urine samples from forestersat the time of estimated maximum 2,4-D exposure. Afteraddition of the IS (C6-ring 2,4-D), samples were acidified andthen extracted with dichloromethane/diethyl ether to eliminatethe interfering compounds. Chromatographic separation wasachieved on a Partisil 5 ODS-3 column. One quantificationand one confirmation ion were observed for both the native2,4-D and the IS. The method was used to elucidate whetherexposure to this herbicide class could contribute to endocrinedisruption and to genotoxicity observed in previous research.Results showed that herbicide applicators with high urinarylevels of 2,4-D exhibited elevated serum luteinizing hormonelevels and also altered genomic stability, which appearsreversible months after peak exposure.
Very recently, Norrgran and coworkers94 presenteda sensitive and selective method for simultaneous detectionof 6 herbicide metabolites, including 2,4-D and 2,4,5-T, inhuman urine. This approach, based on the previously reportedmethod,51 employs automated liquid delivery of ISs andacetate buffer and a mixed-polarity polymeric-phase SPE(Oasis HLB). Samples were analyzed with LC-APCI-NI-MS/MS and quantified by isotope dilution. Chromatographicseparation was achieved on a Betasil Hexylphenyl column. Infact, as reported by the authors, the use of an additional MS-MS transition for each compound enhanced the selectivity ofthe analysis, improving also the sensitivity (LODs for 2,4-Dand 2,4,5-T were 0.054 and 0.075 ng/mL, respectively).Finally, this new methodology has been validated against theprevious, less selective multianalyte method51 by analysis ofhuman specimens with both methods. A good correlation (r =0.9700) was found between concentrations determined by thetwo methods.
Multipesticide Class MethodsBroad-spectrum pesticide screening is frequently re-
quired in forensic and clinical toxicology. Hence, someremarkable multiresidue procedures employing different iso-lation techniques and a variety of different analytical massspectrometry methods have been proposed in the past 6 yearsfor the detection of pesticide residues and/or their metabolitesin biological samples (see Table 4).
In particular, a method for the simultaneous detection of12 urinary phenolic metabolites of pesticides or related chem-icals has been developed by Bravo et al55 combining SPE,chemical derivatization, and isotope dilution GC-MS/MS. Incomparison with existing methods,45,97 this approach im-proved recovery, precision, analytical throughput (3-fold),and sensitivity (LODs were between 0.1 and 1.3 mg/L). Thissensitivity allowed the detection of the target analytes in urinesamples from the general population. However, the authorsnoted that despite the valuable results, this analytical approachneeds further improvement for elimination of the derivatiza-tion step by using LC-MS/MS instead of GC-MS/MS.
An LC-APCI-MS/MS method50 was developed forsimultaneous detection of the urinary metabolites of atrazine,diazinon, malathion, 2,4-D, and certain synthetic pyrethroids,using enzyme hydrolysis followed by extraction with organicsolvents. LODs were shown to be sufficiently low to permit
monitoring of nonexposed persons. In an article already men-tioned in the DAPs section, Bradman et al35 applied the sameapproach50 for analysis of amniotic fluid. Validation experi-ments performed on amniotic fluid revealed that detectionlimits, precision, and analytical recovery of the target analyteswere similar to those in the urine method.50 That article isvaluable because it demonstrates that analytical approachesdeveloped for urine can be successfully applied to amnioticfluid samples.
Olsson et al51 developed an LC-MS/MS multiresiduemethod for high-throughput analysis of 19 markers of com-monly used pesticides in human urine. The approach is basedon 3 previous methods49,50,88 using enzyme hydrolysis fol-lowed by SPE (recovery, 68–114%). All analytes, except thepyrethroid metabolites, were analyzed with APCI interface.TIS interface was also employed because, according to theauthors, the pyrethroid metabolites DBCA and cis- and trans-DCCA could not be measured with the APCI source. The useof a 3 mL SPE cartridge (Oasis HLB) considerably reducedthe clean-up time, as compared with other methods, whichmake use of LLE50 or of a larger SPE cartridge.88 Thisapproach also reduced significantly the sample volume foranalysis (2 mL instead of 17 mL), thus accommodatingstudies involving children, and with an instrument time ofonly 28 minutes (instead of 41 minutes with the 3 formermethods), it provided very high throughput. Despite the favor-able features, the use of two different analyses (an APCI anda TIS analysis) after one common SPE inevitably complicatesthe method. The authors also noted that the matrix effect issuefor some compounds still needs to be addressed.
As has already been mentioned, Norrgran et al94
proposed a multiresidue method for quantitation of 6 herbicidemetabolites in urine, using SPE followed by isotope dilutionLC-APCI-MS/MS analysis. This procedure improves othercurrent methodologies50,51,71,103–106 for the analysis of herbi-cides in biological fluids, especially in specificity andsensitivity. The LOD is ,0.1 ng/mL for all 6 analytes inthe MRM mode, and thus the method is sufficient formeasuring the body burden of the general population but alsoof occupationally exposed subjects. The authors simulta-neously used the previously discussed less-selective method51
for the certification process, by analyzing human specimens,and found good agreement between the methods.
In another study, Pitarch et al98 described a serum assayfor several OC and OP pesticides and PCBs, based on SPE(C18) with GC-MS/MS (ion trap). As stated by the authors,the use of a glass liner packed with CarboFrit in the GCinjection port reduced the background noise by adsorption ofthe sample matrix (eg, lipids) and, consequently, increasedthe signal-to-noise ratio for most of the target analytes. Thisapproach was successfully applied to the detection of thesechemicals in samples from exposed and nonexposed farmers,as well as from the general population. The authors con-cluded that their method was more sensitive and much fasterthan a comparable previously published method based onSPE and GC-ECD/nitrogen phosphorus detection (NPD).107
Lacassie et al77 used GC-MS to determine 47 volatile(OP, OC, phtalimide, uracil, triazine, pyrethroid) pesticidesand ion spray LC-MS for 14 thermolabile and polar pesticides
q 2007 Lippincott Williams & Wilkins 159
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure

(carbamates and benzimidazoles) in serum after an SPE step.These methods were applied to measure pesticide levels insamples collected from intoxication cases. Beside the advan-tage of including various classes of pesticides, these ap-proaches offer simplicity and sensitivity (LODs, 2.5–50 mg/L)in comparison with the existing methods,108,111,114–116 whichoften required laborious sample pretreatment, involvingderivatization or clean-up steps, or employment of NCI.108
Barr et al56 performed multiresidue analysis for 29contemporary pesticides in human serum or plasma, by usingsimple SPE (Oasis cartridge) with isotope dilution GC-HRMS.The LODs were in the low-pg/g range for a 4 g sample, whichare quite low in comparison with LODs in existing analyticalmethodologies.48,99,109–113 This sensitivity of the GC-HRMSsystem is suited for detecting incidental exposures to severalpesticides. In fact, as reported by the authors, HRMS iseffective for reducing matrix interferences and provides higherspecificity than with single quadrupoles or other common low-resolution mass spectrometers. The method was applied todetection of these pesticides in the plasma of urban women. Bythe same approach, contemporary pesticides were detected forthe first time in maternal and/or umbilical cord plasma samplescollected at delivery from 230 mother and newborn pairs.57
The latter study is particularly important because it reveals thatthe pesticides are readily transferred to the developing fetusduring pregnancy.
New methods continue to be published for the assess-ment of prenatal exposure to pesticides. Corrion et al100
published a GC-EI-MS method to measure pesticides ofvarious classes and their metabolites in maternal and umbilicalcord blood. This method using LLE provided LODs of lessthan 0.10 and 0.20 mg/mL for parent pesticides and theirmetabolites, respectively. The method was applied to theanalysis of maternal and umbilical cord blood collected frommother and newborn pairs living in a rural area. It was con-cluded that in terms of sensitivity and recovery, the perfor-mance of the present method is comparable with that of othercurrent methodologies.90,114 Another important feature to men-tion is that the proposed multiclass method reduces consid-erably the chromatographic analysis time (34 minutes versus75 minutes) and, at the same time, allows the detection ofmore target analytes in comparison with former methods.90,114
There are also studies being conducted to assess fetalexposure to environmental toxins, utilizing meconium asa suitable biological matrix. In particular, Ostrea et al101
published a GC-MS multiresidue procedure for detectingselected OC and OP pesticides in meconium. This method,using SPE (C18) (recovery, 71.3–96.7%), provided LODsranging from 0.173 to 6.616 mg/mL. More recently, Bielawskialso published an article102 about meconium analysis fordetermination of fetal exposure to several classes of pesticides(OCs, OPs, pyrethroids, carbamate, and chloroacetanilide), inwhich a modified version of the method of Ostrea et al101 wasapplied for detecting parent pesticides; in addition, a GC-EI-MS method based on methanolic/hydrochloric acid methylester derivatization, followed by LLE, was developed for theanalysis of their metabolites. LODs for parent and metabolitecompounds ranged, respectively, from 0.10 to 1.56 mg/g andfrom 0.31 to 4.15 mg/g in the SIM mode. It was concluded that
the modified method was more sensitive and much faster thanthe existing procedure101 The application of each method wasinvestigated by analysis of meconium samples from infantsborn in a rural area.
CONCLUSIONSQuantification of pesticide metabolites and parent com-
pounds in different biological samples is an establishedapproach for the monitoring of occupational or environmentalexposure in humans and is an integral part of pesticide appli-cation risk assessment and management. These studies requirethe development of sensitive, selective, reliable analyticalmethods with high sample throughput and, at the same time,acceptable costs. As is demonstrated in this review, most of themethods applied nowadays have LODs in the low-ng/mLrange or even lower, and this enables monitoring of bothoccupational and occasional/environmental human exposure.These detection limits are mainly achieved by the wide appli-cation of sensitive and selective detection techniques, such asMS and especially MS/MS, but also by recent developments insample preparation/sample pretreatment methods.
Proper sample pretreatment can contribute to theimprovement of detection limits and method reliability byremoving matrix interferences. At the same time, it remains themost time-consuming, labor-consuming, error-prone part ofanalysis. Trends to simplify or even eliminate sample pre-treatment can be followed in the reviewed literature. Thislatter goal is accomplished by the application of selectivedetection techniques, such as MS/MS, and by the selection ofa cleaner matrix for biomonitoring, such as urine or salivainstead of whole blood. Direct injection of untreated urinesamples into an LC-MS system is often practiced.38,39
However, proper selection of a biological matrix for moni-toring should be ruled mainly by how representative it is forthe toxicant of interest. The ability to detect pesticides andother chemicals in new matrices such as meconium, amnioticfluid, and umbilical cord results in the demonstration thatsuch biologic media are useful specimens for biomonitoringof environmental pollutants. Measurements of pesticides andtheir metabolites in meconium and amniotic fluid, which areproduced by the fetus, have recently been used to estimatedirect fetal pesticide exposure.
Sample treatment can be simplified and accelerated bythe application of coupled-column LC or on-line SPE-LC,48
which are now more widely applied. At the same time, thisreview demonstrates that off-line sample pretreatment stillremains popular, and solvent extraction and off-line SPE arewidely applied.
In this review, we also demonstrated that notwithstand-ing the trend toward wider application of LC-MS/(MS), theGC-MS- and GC-MS/MS-based methodologies can still bemethods of choice. The latter methodologies are well-established,and the increased labor and time consumption, in most cases,can be compensated for by a substantially lower instrumentcost in comparison with LC-MS/MS systems. Moreover,because (unlike GC) the fragmentation pathway of eachanalyte depends on the chromatographic conditions and theLC-MS system, no spectral libraries are available for these
160 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007

instruments. This is probably the main limitation to applicationof LC-MS in the screening of unknowns in biologicalmatrices.
For some classes of pesticides, and especially fororganochlorine pesticides, the GC-ECD still remains themethod of choice, providing very good detection limitscomparable with those of GC-MS.
In conclusion, it should be noted that the developmentof multiresidue methodologies, which may screen simulta-neously (in one run) an increased number of different classesof pesticides, is of great value for monitoring populationexposure to pesticides, and a trend in the development ofsuch methodologies in the past few years can beobserved.51,56,77,98,100,101
REFERENCES1. Laws ER, Hayes WJ. Handbook of Pesticide Toxicology. San Diego, CA:
Academic Press; 1991.2. U.S. Environmental Protection Agency. Available at http://www.epa.
gov./ebtpages/pesticides.html.3. Centers for Disease Control and Prevention. Disease Information: Food-
Borne Illness Technical Information. Atlanta, GA: US Department ofHealth and Human Services, Centers for Disease Control andPrevention, National Center for Infectious Diseases, Division ofBacterial and Mycotic Diseases; December 2002. Available at http://www.cdc.gov/ncidod/dbmd/diseaseinfo/foodborneinfections_t.htm.
4. Gubler DJ. Resurgent vector-borne diseases as a global health problem.Emerg Infect Dis. 1998;4:442–450.
5. Morgan D. Exposure of general population to pesticides. PesticideOutlook. 1992;3:24.
6. Lifshitz M, Shahak E, Sofer S. Carbamate and organophosphatepoisoning in young children. Pediatr Emerg Care. 1999;15:102–103.
7. Michalodimitrakis MN, Tsatsakis AM, Christakis-Hampsas MG, et al.Death following intentional methyl bromide poisoning: toxicologicaldata and literature review. Vet Hum Toxicol. 1997;39:30–34.
8. Tsatsakis AM, Aguridakis P, Michalodimitrakis MN, et al. Experienceswith acute organophosphate poisonings in Crete. Vet Hum Toxicol. 1996;38:101–107.
9. Tsatsakis AM, Tsakalof AK, Siatitsas Y, et al. Acute poisoning withcarbamate pesticides: the Cretan experience. Sci Justice. 1996;36:35–39.
10. Tsatsakis AM, Manousakis A, Anastasaki M. Clinical and toxicologicaldata in fenthion and omethoate acute poisoning. J Environ Sci Health B.1998;33:657–670.
11. Tsatsakis AM. More fatal methomyl poisonings in Crete. Sci Justice.1998;38:282–283.
12. Tsatsakis AM, Tsakalof AK, Michalodimitrakis EN. The analysis ofmethomyl, a carbamate pesticide, in post-mortem samples. Sci Justice.1996;36:41–45.
13. Tsatsakis AM, Perakis K, Koumantakis E. Experience with acuteparaquat poisoning in Crete. Vet Hum Toxicol. 1996;38:113–117.
14. Tsatsakis AM, Bertsias GK, Mammas IN, et al. Acute fatal poisoning bymethomyl caused by inhalation and transdermal absorption. Bull EnvironContam Toxicol. 2001;66:415–420.
15. Tsatsakis AM, Tutudaki MI, Tzatzarakis MN, et al. Pesticide depositionin hair: preliminary results of a model study of methomyl incorporationinto rabbit hair. Vet Hum Toxicol. 1998;40:200–203.
16. Covaci A, Tutudaki M, Tsatsakis AM, et al. Hair analysis: anotherapproach for the assessment of human exposure to selected persistentorganochlorine pollutants. Chemosphere. 2002;46:413–418.
17. Tutudaki M, Tsakalof A, Tsatsakis AM. Hair analysis used to assesschronic exposure to the organophosphate diazinon: a model study withrabbits. Hum Exp Toxicol. 2003;22:159–164.
18. Tutudaki M, Tsatsakis AM. Pesticide hair analysis: Development ofa GC-NCI-MS method to assess chronic exposure to diazinon in rats.J Anal Toxicol. 2005;29:805–809.
19. Dolapsakis G, Vlachonikolis IG, Varveris C, et al. Mammographicfindings and occupational exposure to pesticides currently in use onCrete. Eur J Cancer. 2001;37:1531–1536.
20. Fenske RA, Lu C, Barr D, et al. Children’s exposure to chlorpyrifos andparathion in an agricultural community in central Washington State.Environ Health Persp. 2002;110:549–553.
21. Curl CL, Fenske RA, Kissel JC, et al. Evaluation of take-homeorganophosphorus pesticide exposure among agricultural workers andtheir children. Environ Health Persp. 2002;110:787–792.
22. Lu C, Fenske RA, Simcox NJ, et al. Pesticide exposure of children in anagricultural community: evidence of household proximity to farmlandand take home exposure pathways. Environ Res. 2000;84:290–302.
23. Curwin BD, Hein MJ, Sanderson WT, et al. Urinary and hand wipepesticide levels among farmers and nonfarmers in Iowa. J Expo AnalEnviron Epidemiol. 2005:(in press).
24. Barr DB, Needham LL. Analytical methods for biological monitoring ofexposure to pesticides: a review. J Chromatogr B. 2002;778:5–29.
25. Stajnbaher D, Zupancic-Kralj L. Multiresidue method for determinationof 90 pesticides in fresh fruits and vegetables using solid-phaseextraction and gas chromatography–mass spectrometry. J Chromatogr A.2003;1015:185–198.
26. Hoffmann E. Tandem mass spectrometry: a primer. J Mass Spectrom.1996;31:129–137.
27. Frias MM, Frenich AG, Martinez Vidal JL, et al. Analyses of lindane,vinclozolin, aldrin, p,p’-DDE, o,p’-DDT and p,p’-DDT in human serumusing gas chromatography with electron capture detection and tandemmass spectrometry. J Chromatogr B. 2001;760:1–15.
28. Hogendoorn E, Zoonen P. Recent and future developments of liquidchromatography in pesticide trace analysis. J Chromatogr A. 2000;892:435–453.
29. Hernandez F, Sancho JV, Pozo OJ. Critical review of the application ofliquid chromatography/mass spectrometry to the determination ofpesticide residues in biological samples. Anal Bioanal Chem. 2005;382:934–946.
30. Hardt J, Angerer J. Determination of dialkyl phosphates in human urineusing gas chromatography–mass spectrometry. J Anal Toxicol. 2000;24:678–684.
31. Oglobline AN, Elimelakh H, Tattam B, et al. Negative ion chemicalionization GC/MS-MS analysis of dialkylphosphate metabolites oforganophosphate pesticides in urine of nonoccupationally exposedsubjects. Analyst. 2001;126:1037–1041.
32. Bravo R, Driskell WJ, Whitehead RD Jr, et al. Quantitation of dialkylphosphate metabolites of organophosphate pesticides in human urineusing GC-MS-MS with isotopic internal standards. J Anal Toxicol. 2002;26:245–252.
33. Bravo R, Caltabiano LM, Weerasekera G, et al. Measurement of dialkylphosphate metabolites of organophosphorus pesticides in human urineusing lyophilization with gas chromatography–tandem mass spectrom-etry and isotope dilution quantification. J Expo Anal Environ Epidemiol.2004;14:249–259.
34. Whyatt RM, Barr DB. Measurement of organophosphate metabolites inpostpartum meconium as a potential biomarker of prenatal exposure:a validation study. Environ Health Persp. 2001;109:417–420.
35. Bradman A, Barr DB, Claus Henn BG, et al. Measurement of pesticidesand other toxicants in amniotic fluid as a potential biomarker of prenatalexposure: a validation study. Environ Health Persp. 2003;111:1779–1782.
36. Ueyama J, Saito I, Kamijima M, et al. Simultaneous determination ofurinary dialkylphosphate metabolites of organophosphorus pesticidesusing gas chromatography–mass spectrometry. J Chromatogr B. 2006:(in press).
37. Dulaurent S, Saint-Marcoux F, Marquet P, et al. Simultaneousdetermination of six dialkylphosphates in urine by liquid chromatog-raphy tandem mass spectrometry. J Chromatogr B. 2006;831:223–229.
38. Hernandez F, Sancho JV, Pozo OJ. An estimation of the exposure toorganophosphorus pesticides through the simultaneous determination oftheir main metabolites in urine by liquid chromatography–tandem massspectrometry. J Chromatogr B. 2004;808:229–239.
39. Hernandez F, Sancho JV, Pozo OJ. Direct determination of alkylphosphates in human urine by liquid chromatography/electrospraytandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:1766–1773.
40. Barr DB, Bravo R, Weerasekera G, et al. Concentrations of dialkylphosphate metabolites of organophosphorus pesticides in the U.S.population. Environ Health Persp. 2004;112:186–200.
q 2007 Lippincott Williams & Wilkins 161
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure

41. Castorina R, Bradman A, McKone TE, et al. Cumulative organophos-phate pesticide exposure and risk assessment among pregnant womenliving in an agricultural community: a case study from the CHAMACOScohort. Environ Health Persp. 2003;111:1640–1648.
42. Wessels D, Barr DB, Mendola P. Use of biomarkers to indicate exposureof children to organophosphate pesticides: implications for a longitudinalstudy of children’s environmental health. Environ Health Persp. 2003;111:1939–1946.
43. Balluz L, Philen R, Ortega L, et al. Investigation of systemic lupuserythematosus in Nogales, Arizona. Am J Epidemiol. 2001;154:1029–1036.
44. Eskenazi B, Harley K, Bradman A, et al. Association of in uteroorganophosphate pesticide exposure and fetal growth and length ofgestation in an agricultural population. Environ Health Persp. 2004;112:1116–1124.
45. Hill RH, Shealy DB, Head SL, et al. Determination of pesticidemetabolites in human urine using isotope dilution technique and tandemmass spectrometry. J Anal Toxicol. 1995b;19:323–329.
46. Barr DB, Turner WE, DiPietro E, et al. Measurement of p-nitrophenol inthe urine of residents whose homes were contaminated with methylparathion. Environ Health Persp. 2002;110(Suppl 6):1085–1091.
47. Hore P, Robson M, Freeman N, et al. Chlorpyrifos accumulation patternsfor child-accessible surfaces and objects and urinary metaboliteexcretion by children for 2 weeks after crack-and-crevice application.Environ Health Persp. 2005;113:211–219.
48. Sancho JV, Pozo OJ, Hernandez F. Direct determination of chlorpyrifosand its main metabolite 3,5,6-trichloro-2-pyridinol in human serumand urine by coupled-column liquid chromatography/electrospray–tandem mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:1485–1490.
49. Olsson AO, Nguyen JV, Sadowski MA, et al. A liquid chromatography/electrospray ionization–tandem mass spectrometry method for quanti-fication of specific organophosphorus pesticide biomarkers in humanurine. Anal Bioanal Chem. 2003;376:808–815.
50. Baker SE, Barr DB, Driskell WJ, et al. Quantification of selectedpesticide metabolites in human urine using isotope dilution high-performance liquid chromatography/tandem mass spectrometry. J ExpoAnal Environ Epidemiol. 2000;10:789–798.
51. Olsson AO, Baker SE, Nguyen JV, et al. A liquid chromatography–tandem mass spectrometry multiresidue method for quantification ofspecific metabolites of organophosphorus pesticides, synthetic pyreth-roids, selected herbicides, and DEET in human urine. Anal Chem. 2004;76:2453–2461.
52. Swan SH, Kruse RL, Liu F, et al. Semen quality in relation to biomarkersof pesticide exposure. Environ Health Persp. 2003;111:1478–1484.
53. Meeker JD, Ryan L, Barr DB, et al. The relationship of urinarymetabolites of carbaryl/naphthalene and chlorpyrifos with human semenquality. Environ Health Persp. 2004;112:1665–1670.
54. Meeker JD, Singh NP, Ryan L, et al. Urinary levels of insecticidemetabolites and DNA damage in human sperm. Hum Reprod. 2004;19:2573–2580.
55. Bravo R, Caltabiano LM, Fernandez C, et al. Quantification of phenolicmetabolites of environmental chemicals in human urine using gaschromatography–tandem mass spectrometry and isotope dilutionquantification. J Chromatogr B. 2005;820:229–236.
56. Barr DB, Barr JR, Maggio VL, et al. A multi-analyte method for thequantification of contemporary pesticides in human serum and plasmausing high-resolution mass spectrometry. J Chromatogr B. 2002;778:99–111.
57. Whyatt RM, Barr DB, Camann DE, et al. Contemporary-use pesticidesin personal air samples during pregnancy and blood samples at deliveryamong urban minority mothers and newborns. Environ Health Persp.2003;111:749–756.
58. Whyatt RM, Rauh V, Barr DB, et al. Prenatal insecticide exposures andbirth weight and length among an urban minority cohort. Environ HealthPersp. 2004;112:1125–1132.
59. Meeker JD, Barr DB, Ryan L, et al. Temporal variability of urinary levelsof nonpersistent insecticides in adult men. J Expo Anal EnvironEpidemiol. 2005;15:271–281.
60. Whyatt RM, Camann D, Perera FP, et al. Biomarkers in assessingresidential insecticide exposures during pregnancy and effects on fetalgrowth. Toxicol Appl Pharm. 2005;206:246–254.
61. Hill RH Jr, Head S, Barr DB, et al. Public health decisions: thelaboratory’s role in the Lorain County, Ohio, investigation. EnvironHealth Persp. 2002;110(Suppl 6):1057–1059.
62. Hryhorczuk DO, Moomey M, Burton A, et al. Urinary p-nitrophenol asa biomarker of household exposure to methyl parathion. Environ HealthPersp. 2002;110(Suppl 6):1041–1046.
63. Kissel JC, Curl CL, Kedan G, et al. Comparison of organophosphoruspesticide metabolite levels in single and multiple daily urine samplescollected from preschool children in Washington State. J Expo AnalEnviron Epidemiol. 2005;15:164–171.
64. Sancho JV, Pozo OJ, Lopez FJ, et al. Different quantitation approachesfor xenobiotics in human urine samples by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom.2002;16:639–645.
65. Costa LG, Cole TB, Vitalone A, et al. Measurement of paraoxonase(PON1) status as a potential biomarker of susceptibility to organophos-phate toxicity. Clin Chim Acta. 2005;352:37–47.
66. He F, Chen S, Tang X, et al. Biological monitoring of combined exposureto organophosphates and pyrethroids. Toxicol Lett. 2002;134:119–124.
67. Gallo MA, Lawryk NJ. Organic phosphorus pesticides. In: Hayes WJ Jr,Laws ER Jr, eds. Handbook of pesticide toxicology: classes of pesticides.Vol. 2. San Diego: Academic Press; 1991:917–1123.
68. Aprea C, Betta A, Catenacci G, et al. Reference values of urinary 3,5,6-trichloro-2-pyridinol in the Italian population-validation of analyticalmethod and preliminary results (multicentric study). J AOAC Internat.1999;82:305–312.
69. Koch HM, Angerer J. Analysis of 3,5,6-trichloro-2-pyridinol in urinesamples from the general population using gas chromatography–massspectrometry after steam distillation and solid-phase extraction.J Chromatogr B. 2001;759:43–49.
70. Yokley RA, Shen N, Cheung MW. Determination of two oxy-pyrimidinemetabolites of diazinon in urine by gas chromatography/mass selectivedetection and liquid chromatography/electrospray ionization/massspectrometry/mass spectrometry. J AOAC Internat. 2000;83:1229–1238.
71. Beeson MD, Driskell WJ, Barr DB. Isotope dilution high-performanceliquid chromatography/tandem mass spectrometry method for quantify-ing urinary metabolites of atrazine, malathion, and 2,4-dichlorophenoxy-acetic acid. Anal Chem. 1999;71:3526–3530.
72. Esteban E, Rubin C, Hill R, et al. Association between indoor residentialcontamination with methyl parathion and urinary para-nitrophenol.J Expo Anal Environ Epidemiol. 1996;6:375–387.
73. Barr JR, Maggio VL, Barr DB, et al. New high-resolution massspectrometric approach for the measurement of polychlorinated bi-phenyls and organochlorine pesticides in human serum. J Chromatogr B.2003;794:137–148.
74. Sandau CD, Sjodin A, Davis MD, et al. Comprehensive solid-phaseextraction method for persistent organic pollutants. Validation andapplication to the analysis of persistent chlorinated pesticides. AnalChem. 2003;75:71–77.
75. DiPietro ES, Lapeza CR, Turner WE, et al. A fast universal automatedcleanup system for the isotope-dilution high resolution mass spectro-metric analysis of PCDDs, PCDFs, coplanar-PCBs, PCB congeners, andpersistent pesticides from the same serum sample. OrganohalogenComp. 1997;31:26–31.
76. Barr DB, Weihe P, Needham LL, et al. PCBs and organochlorinepesticide concentrations in a Faroe Island 14-year old cohort: measure-ment using new methodology and evaluation of correlations andpatterns. Organohalogen Comp. 2003;63:5–8.
77. Lacassie E, Marquet P, Gaulier J, et al. Sensitive and specificmultiresidue methods for the determination of pesticides of variousclasses in clinical and forensic toxicology. Forensic Sci Int. 2001;121:116–125.
78. Conka K, Drobna B, Kocan A, et al. Simple solid-phase extractionmethod for determination of polychlorinated biphenyls and selectedorganochlorine pesticides in human serum. J Chromatogr A. 2005;1084:33–38.
79. Sundberg SE, Ellington JJ, Evans JJ. A simple and fast extraction methodfor organochlorine pesticides and polychlorinated biphenyls in smallvolumes of avian serum. J Chromatogr B. 2006;831:99–104.
80. Burse VW, Najam AR, Williams CC, et al. Utilization of umbilical cordsto assess in utero exposure to persistent pesticides and polychlorinatedbiphenyls. J Expo Anal Environ Epidemiol. 2000;10:776–788.
162 q 2007 Lippincott Williams & Wilkins
Margariti et al Ther Drug Monit � Volume 29, Number 2, April 2007

81. Campoy C, Jimenez M, Olea-Serrano MF, et al. Analysis of organo-chlorine pesticides in human milk: preliminary results. Early Hum Dev.2001;65(Suppl):S183–S190.
82. Burke ER, Holden AJ, Shaw IC. A method to determine residue levels ofpersistent organochlorine pesticides in human milk from Indonesianwomen. Chemosphere. 2003;50:529–535.
83. Hong Z, Gunter M, Randow FE. Meconium: a matrix reflecting potentialfetal exposure to organochlorine pesticides and its metabolites.Ecotoxicol Environ Saf. 2002;51:60–64.
84. Soderlund DM, Clark JM, Sheets LP, et al. Mechanisms of pyrethroidneurotoxicity: implications for cumulative risk assessment. Toxicology.2002;171:3–59.
85. International Programme on Chemical Safety (IPCS). EnvironmentalHealth Criteria 97: Deltamethrin. Oxford, UK: World HealthOrganization; 1990.
86. Dorman DC, Beasley VR. Neurotoxicology of pyrethrin and thepyrethroid insecticides. Vet Hum Toxicol. 1991;33:238–243.
87. Schettgen T, Koch HM, Drexler H, et al. New gas chromatographic–massspectrometric method for the determination of urinary pyrethroid metabolitesin environmental medicine. J Chromatogr B. 2002;778:121–130.
88. Baker SE, Olsson AO, Barr DB. Isotope dilution high-performanceliquid chromatography–tandem mass spectrometry method for quanti-fying urinary metabolites of synthetic pyrethroid insecticides. ArchEnviron Contam Toxicol. 2004;46:281–288.
89. Leng G, Gries W. Simultaneous determination of pyrethroid andpyrethrin metabolites in human urine by gas chromatography–highresolution mass spectrometry. J Chromatogr B. 2005;814:285–294.
90. Ramesh A, Ravi PE. Electron ionization gas chromatography–massspectrometric determination of residues of thirteen pyrethroid insecti-cides in whole blood. J Chromatogr B. 2004;802:371–376.
91. Arrebola FJ, Martinez-Vidal JL, Fernandez-Gutierrez A, et al. Moni-toring of pyrethroid metabolites in human urine using solid-phaseextraction followed by gas chromatography–tandem mass spectrometry.Anal Chim Acta. 1999;401:45–54.
92. Hoar SK, Blair A, Holmes FF, et al. Agricultural herbicide use and risk oflymphoma and soft-tissue sarcoma. JAMA. 1986;256:1141–1147.
93. Jahm SH, Waisenburger DD, Babbitt PA, et al. A case-control study ofnon-Hodgkin’s lymphoma and the herbicide 2,4-dichlorophenoxyaceticacid (2,4-D) in eastern Nebraska. Epidemiology. 1990;1:349–356.
94. Norrgran J, Bravo R, Bishop AM, et al. Quantification of six herbicidemetabolites in human urine. J Chromatogr B. 2006;830:185–195.
95. Garry VF, Tarone RE, Kirsch IR, et al. Biomarker correlations of urinary2,4-D levels in foresters: genomic instability and endocrine disruption.Environ Health Persp. 2001;109:495–500.
96. Thompson TS, Treble RG. Solid phase extraction of 2,4-D from humanurine. Chemosphere. 1996;33:1515–1522.
97. Shealy DB, Bonin MA, Wooten JV, et al. Application of an improvedmethod for the analysis of pesticides and their metabolites in the urine offarmer applicators and their families. Environ Int. 1996;22:661–675.
98. Pitarch E, Serrano R, Lopez FJ, et al. Rapid multiresidue determinationof organochlorine and organophosphorus compounds in human serumby solid-phase extraction and gas chromatography coupled to tandemmass spectrometry. Anal Bioanal Chem. 2003;376:189–197.
99. Qui H, Jun HW. Solid-phase extraction and liquid chromatographicquantitation of insect repellent N,N-diethyl-m-toluamide in plasma.J Pharm Biomed Anal. 1996;15:241–250.
100. Corrion ML, Ostrea EM Jr, Bielawski DM, et al. Detection of prenatalexposure to several classes of environmental toxicants and theirmetabolites by gas chromatography–mass spectrometry in maternaland umbilical cord blood. J Chromatogr B. 2005;822:221–229.
101. Ostrea EM, Morales V, Ngoumgna E, et al. Prevalence of fetal exposureto environmental toxins as determined by meconium analysis. Neuro-toxicology. 2002;23:329–339.
102. Bielawski D, Ostrea E Jr, Posecion N Jr, et al. Detection of several classesof pesticides and metabolites in meconium by gas chromatography–massspectrometry. Chromatographia. 2005;62:623–629.
103. Aprea C, Sciarra G, Bozzi N. Analytical methods for the determinationof urinary 2,4-dichlorophenoxyacetic acid and 2-methyl-4-chlorophe-noxyacetic acid in occupationally exposed subjects and in the generalpopulation. J Anal Toxicol. 1997;21:262–267.
104. Lyubimov AV, Garry VF, Carlson RE, et al. Simplified urinaryimmunoassay for 2,4-D: Validation and exposure assessment. J LabClin Med. 2000;136:116–124.
105. Jaeger LL, Jones AD, Hammock BD. Development of an enzyme-linkedimmunosorbent assay for atrazine mercapturic acid in human urine.Chem Res Toxicol. 1998;11:342–352.
106. Barr JR, Barr DB, Patterson DG Jr, et al. Quantification of non-persistentpesticides in human samples by isotope dilution mass spectrometry.Toxicol Environ Chem. 1998;66:3–10.
107. Pitarch E, Lopez FJ, Serrano R, et al. Multiresidue determination oforganophosphorus and organochlorine pesticides in human biologicalfluids by capillary gas chromatography. Fresenius J Anal Chem. 2001;369:502–509.
108. Mariani G, Benfenati E, Fanelli R. A NICI-GC-MS method to analyseendosulfan in biological samples. Int J Environ Anal Chem. 1995;58:67–72.
109. Lee XP, Kumazawa T, Sato K. Rapid extraction and capillary gaschromatography for diazine herbicides in human body fluids. ForensicSci Int. 1995;72:199–207.
110. Cho Y, Matsuoka N, Kamiya A. Determination of organophosphoruspesticides in biological samples of acute poisoning by HPLC with diode-array detector. Chem Pharm Bull. 1997;45:737–740.
111. Futagami K, Narazaki C, Kataoka Y. Application of high-performancethin-layer chromatography for the detection of organophosphorusinsecticides in human serum after acute poisoning. J Chromatogr BBiomed Sci Appl. 1997;704:369–373.
112. Lee HS, Kim K, Kim JH, et al. On-line sample preparation of paraquat inhuman serum samples using high-performance liquid chromatographywith column switching. J Chromatogr B Biomed Sci Appl. 1998;716:371–374.
113. Frenzel T, Sochor H, Speer K, et al. Rapid multimethod for verificationand determination of toxic pesticides in whole blood by means ofcapillary GC-MS. J Anal Toxicol. 2000;24:365–371.
114. Waliiszewski SM, Szymczynski GA. Persistent organochlorine pesti-cides in blood serum and whole blood. Bull Environ Contam Toxicol.1991;46:803–809.
115. Kawasaki S, Ueda H, Itoh H, et al. Screening of organophosphoruspesticides using liquid-chromatography/mass spectrometry. J Chromatogr.1992;595:193–202.
116. Itoh H, Kawasaki S, Tadano J. Application of liquid-chromatography/mass spectrometry to pesticide analysis. J Chromatogr A. 1996;754:61–76.
117. Hardell E, Eriksson M, Lindstrom G, et al. Case-control study onconcentrations of organohalogen compounds and titers of antibodies toEpstein-Barr virus antigens in the etiology of non-Hodgkin lymphoma.Leuk Lymphoma. 2001;42:619–629.
118. Nordstrom M, Hardell L, Lindstrom G, et al. Concentrations oforganochlorines related to titers to Epstein-Barr virus early antigen IgGas risk factors for hairy cell leukemia. Environ Health Persp. 2000;108:441–445.
q 2007 Lippincott Williams & Wilkins 163
Ther Drug Monit � Volume 29, Number 2, April 2007 Analytical Methods of Biological Monitoring for Pesticide Exposure




















