
Analysis of SM22 Deficient Mice Reveals Unanticipated Insights into Smooth Muscle Cell...
9
MOLECULAR AND CELLULAR BIOLOGY, 0270-7306/01/$04.0010 DOI: 10.1128/MCB.2001.21.4.1336–1344.2001 Feb. 2001, p. 1336–1344 Vol. 21, No. 4 Copyright © 2001, American Society for Microbiology. All Rights Reserved. Analysis of SM22a-Deficient Mice Reveals Unanticipated Insights into Smooth Muscle Cell Differentiation and Function JANET C. L. ZHANG, 1 STEVEN KIM, 2 BRIAN P. HELMKE, 3 WILLIAM W. YU, 1 KEVIN L. DU, 1 MIN MIN LU, 1 MARK STROBECK, 1 QIAN-CHUN YU, 4 AND MICHAEL S. PARMACEK 1 * Departments of Medicine, 1 Bioengineering, 3 and Cell and Molecular Engineering, 4 University of Pennsylvania, Philadelphia, Pennsylvania 19104, and Department of Medicine, University of Chicago, Chicago, Illinois 60637 2 Received 17 October 2000/Accepted 7 November 2000 SM22a is a 22-kDa smooth muscle cell (SMC) lineage-restricted protein that physically associates with cytoskeletal actin filament bundles in contractile SMCs. To examine the function of SM22a, gene targeting was used to generate SM22a-deficient (SM22 2/2LacZ ) mice. The gene targeting strategy employed resulted in insertion of the bacterial lacZ reporter gene at the SM22a initiation codon, permitting precise analysis of the temporal and spatial pattern of SM22a transcriptional activation in the developing mouse. Northern and Western blot analyses confirmed that the gene targeting strategy resulted in a null mutation. Histological analysis of SM22 1/2LacZ embryos revealed detectable b-galactosidase activity in the unturned embryonic day 8.0 embryo in the layer of cells surrounding the paired dorsal aortae concomitant with its expression in the primitive heart tube, cephalic mesenchyme, and yolk sac vasculature. Subsequently, during postnatal devel- opment, b-galactosidase activity was observed exclusively in arterial, venous, and visceral SMCs. SM22a- deficient mice are viable and fertile. Their blood pressure and heart rate do not differ significantly from their control SM22a 1/2 and SM22a 1/1 littermates. The vasculature and SMC-containing tissues of SM22a- deficient mice develop normally and appear to be histologically and ultrastructurally similar to those of their control littermates. Taken together, these data demonstrate that SM22a is not required for basal homeostatic functions mediated by vascular and visceral SMCs in the developing mouse. These data also suggest that signaling pathways that regulate SMC specification and differentiation from local mesenchyme are activated earlier in the angiogenic program than previously recognized. Smooth muscle cells (SMCs) subserve a variety of functions in higher vertebrates, including the regulation of arterial tone, the control of airway resistance, and the modulation of gastro- intestinal and genitourinary tract contractility and basal tone. Despite the myriad of functions mediated by SMCs, relatively little is understood about the developmental programs that regulate SMC specification and differentiation. This is due, in part, to the complex embryological origins of the SMC line- age(s) and the lack of definitive SMC markers (27, 28). In contrast to striated muscle cells which terminally differentiate, SMCs retain their capacity to proliferate and modulate their phenotype during postnatal development (27, 34, 36, 37). This characteristic presumably evolved to facilitate reparative pro- cesses such as wound healing. However, it has also been im- plicated in the pathophysiology of a number of disease processes, including hypertension, restenosis following angioplasty, post- transplant arteriopathy, and asthma (13, 25, 32, 34). Cytoskeletal dynamics and organization play an important role in regulating SMC morphology and phenotype (for a re- view, see reference 41). The SMC cytoskeleton is composed of actin filaments, as well as intermediate filaments and their associated proteins. The insoluble network of intermediate filaments has been implicated in the maintenance of SMC shape (42). The intermediate filaments colocalize with a sub- population of F-actin filament bundles that is distinct from the contractile apparatus (26). The cytoskeletal actin filaments and smooth muscle actin converge upon a-actinin-containing dense bodies that serve as a possible coupling point between the SMC contractile apparatus and the cytoskeleton (26). Cy- toskeletal actin filaments are anchored within focal adhesions which form rib-like arrays over the entire SMC surface (40). The geometric organization of these arrays assures that con- tractile tension is distributed uniformly over the SMCs to the extracellular matrix. SM22a is a 22-kDa cytoskeletal protein that is expressed abundantly and exclusively in visceral and vascular SMCs dur- ing postnatal development. SM22a has been variably desig- nated SM22a (17, 37), transgelin (16), WS3-10 (45), and p27 (1). SM22a mRNA has been detected in the dorsal aorta of the mouse embryo as early as embryonic day 9.5 (E9.5) (18). Like most other markers of the SMC lineage, SM22a is also ex- pressed in embryonic cardiac and skeletal muscle through mid- gestation (6, 17, 18, 37). SM22a is downregulated in concert with other SMC-restricted myofibrillar proteins in late-passage primary aortic SMCs and in neointimal SMCs that arise in response to arterial injury (35, 36). In human atherosclerotic lesions, SM22a is downregulated within neointimal SMCs but is expressed in SMCs that form the fibrous caps of complicated atherosclerotic lesions (35, 36). Our group, and others, re- ported that the mouse SM22a promoter restricts transgene expression to arterial SMCs, suggesting that distinct transcrip- tional programs may distinguish previously unrecognized SMC sublineages (14, 19, 22, 43). The activity of the mouse SM22a promoter is critically dependent upon two CArG box-contain- * Corresponding author. Mailing address: Department of Medicine, University of Pennsylvania, 91234 Founders Pavilion, 3400 Spruce St., Philadelphia, PA 19104-4283. Phone: (215) 662-3140. Fax: (215) 349- 8017. E-mail: [email protected]. 1336 on December 1, 2014 by guest http://mcb.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Analysis of SM22 Deficient Mice Reveals Unanticipated Insights into Smooth Muscle Cell...

MOLECULAR AND CELLULAR BIOLOGY,0270-7306/01/$04.0010 DOI: 10.1128/MCB.2001.21.4.1336–1344.2001
Feb. 2001, p. 1336–1344 Vol. 21, No. 4
Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Analysis of SM22a-Deficient Mice Reveals UnanticipatedInsights into Smooth Muscle Cell Differentiation and Function
JANET C. L. ZHANG,1 STEVEN KIM,2 BRIAN P. HELMKE,3 WILLIAM W. YU,1 KEVIN L. DU,1
MIN MIN LU,1 MARK STROBECK,1 QIAN-CHUN YU,4 AND MICHAEL S. PARMACEK1*
Departments of Medicine,1 Bioengineering,3 and Cell and Molecular Engineering,4 University of Pennsylvania,Philadelphia, Pennsylvania 19104, and Department of Medicine, University of Chicago, Chicago, Illinois 606372
Received 17 October 2000/Accepted 7 November 2000
SM22a is a 22-kDa smooth muscle cell (SMC) lineage-restricted protein that physically associates withcytoskeletal actin filament bundles in contractile SMCs. To examine the function of SM22a, gene targeting wasused to generate SM22a-deficient (SM222/2LacZ) mice. The gene targeting strategy employed resulted ininsertion of the bacterial lacZ reporter gene at the SM22a initiation codon, permitting precise analysis of thetemporal and spatial pattern of SM22a transcriptional activation in the developing mouse. Northern andWestern blot analyses confirmed that the gene targeting strategy resulted in a null mutation. Histologicalanalysis of SM221/2LacZ embryos revealed detectable b-galactosidase activity in the unturned embryonic day8.0 embryo in the layer of cells surrounding the paired dorsal aortae concomitant with its expression in theprimitive heart tube, cephalic mesenchyme, and yolk sac vasculature. Subsequently, during postnatal devel-opment, b-galactosidase activity was observed exclusively in arterial, venous, and visceral SMCs. SM22a-deficient mice are viable and fertile. Their blood pressure and heart rate do not differ significantly from theircontrol SM22a1/2 and SM22a1/1 littermates. The vasculature and SMC-containing tissues of SM22a-deficient mice develop normally and appear to be histologically and ultrastructurally similar to those of theircontrol littermates. Taken together, these data demonstrate that SM22a is not required for basal homeostaticfunctions mediated by vascular and visceral SMCs in the developing mouse. These data also suggest thatsignaling pathways that regulate SMC specification and differentiation from local mesenchyme are activatedearlier in the angiogenic program than previously recognized.
Smooth muscle cells (SMCs) subserve a variety of functionsin higher vertebrates, including the regulation of arterial tone,the control of airway resistance, and the modulation of gastro-intestinal and genitourinary tract contractility and basal tone.Despite the myriad of functions mediated by SMCs, relativelylittle is understood about the developmental programs thatregulate SMC specification and differentiation. This is due, inpart, to the complex embryological origins of the SMC line-age(s) and the lack of definitive SMC markers (27, 28). Incontrast to striated muscle cells which terminally differentiate,SMCs retain their capacity to proliferate and modulate theirphenotype during postnatal development (27, 34, 36, 37). Thischaracteristic presumably evolved to facilitate reparative pro-cesses such as wound healing. However, it has also been im-plicated in the pathophysiology of a number of disease processes,including hypertension, restenosis following angioplasty, post-transplant arteriopathy, and asthma (13, 25, 32, 34).
Cytoskeletal dynamics and organization play an importantrole in regulating SMC morphology and phenotype (for a re-view, see reference 41). The SMC cytoskeleton is composed ofactin filaments, as well as intermediate filaments and theirassociated proteins. The insoluble network of intermediatefilaments has been implicated in the maintenance of SMCshape (42). The intermediate filaments colocalize with a sub-population of F-actin filament bundles that is distinct from the
contractile apparatus (26). The cytoskeletal actin filaments andsmooth muscle actin converge upon a-actinin-containing densebodies that serve as a possible coupling point between theSMC contractile apparatus and the cytoskeleton (26). Cy-toskeletal actin filaments are anchored within focal adhesionswhich form rib-like arrays over the entire SMC surface (40).The geometric organization of these arrays assures that con-tractile tension is distributed uniformly over the SMCs to theextracellular matrix.
SM22a is a 22-kDa cytoskeletal protein that is expressedabundantly and exclusively in visceral and vascular SMCs dur-ing postnatal development. SM22a has been variably desig-nated SM22a (17, 37), transgelin (16), WS3-10 (45), and p27(1). SM22a mRNA has been detected in the dorsal aorta of themouse embryo as early as embryonic day 9.5 (E9.5) (18). Likemost other markers of the SMC lineage, SM22a is also ex-pressed in embryonic cardiac and skeletal muscle through mid-gestation (6, 17, 18, 37). SM22a is downregulated in concertwith other SMC-restricted myofibrillar proteins in late-passageprimary aortic SMCs and in neointimal SMCs that arise inresponse to arterial injury (35, 36). In human atheroscleroticlesions, SM22a is downregulated within neointimal SMCs butis expressed in SMCs that form the fibrous caps of complicatedatherosclerotic lesions (35, 36). Our group, and others, re-ported that the mouse SM22a promoter restricts transgeneexpression to arterial SMCs, suggesting that distinct transcrip-tional programs may distinguish previously unrecognized SMCsublineages (14, 19, 22, 43). The activity of the mouse SM22apromoter is critically dependent upon two CArG box-contain-
* Corresponding author. Mailing address: Department of Medicine,University of Pennsylvania, 91234 Founders Pavilion, 3400 Spruce St.,Philadelphia, PA 19104-4283. Phone: (215) 662-3140. Fax: (215) 349-8017. E-mail: [email protected].
1336
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

ing elements that bind the MADS box transcription factor SRF(14, 19, 22, 43).
Despite the fact that SM22a is expressed abundantly inSMCs and has been localized within the cytoskeletal apparatus,relatively little is understood about its function. SM22a shareshigh-level amino acid sequence identity with several other pro-teins, including the thin filament SMC-restricted myofibrillarregulatory protein calponin (33, 44), the Caenorhabditis elegansbody wall muscle protein Unc-87 (8), the Drosophila muscleprotein Mp20 (2), and the neuronal-restricted protein NP25(31). Mutation of the unc-87 and mp20 genes results in paral-ysis of the C. elegans body wall and of the Drosophila flightmuscle, respectively (2, 8). SM22a colocalizes to actin filamentbundles and stress fibers (41). Purified SM22a protein bindsdirectly to actin filaments at a ratio of 1:6 actin monomers (38,39). These data suggest that SM22a may serve to organize thespatial relationships of actin filaments in SMCs (38, 39, 41). Inthis regard, it is also noteworthy that SM22a gene expression isdownregulated when vascular SMCs assume a synthetic phe-notype and by cellular transformation processes that involvemajor cytoskeletal rearrangements (35, 37). Taken together,these data suggest that SM22a may play a role in organizationof the cytoskeleton and directly, or indirectly, regulate SMCmorphology or other processes involving the cytoskeleton.
In the studies described here, gene targeting was used todefine precisely the spatial and temporal patterns of SM22atranscriptional activation during vascular development and toexamine the function of the SM22a protein in SMCs. Surpris-ingly, in E8.0 SM221/2LacZ embryos b-galactosidase activitywas observed in the dorsal aorta. This is 24 to 36 h prior to thedocumented expression of genes encoding vascular SMCmarkers in the embryonic mouse. During postnatal develop-ment, SM22a gene was activated exclusively in visceral andvascular SMCs. SM22a-deficient embryos (SM222/2) are via-ble and fertile and develop normally. SMC-containing tissuesfrom SM22a-deficient mice are histologically indistinguishablefrom the tissues of their control littermates, and only subtledefects in the arrangement of actin filaments are observed inSM22a-deficient SMCs. These data demonstrate that SM22ais not required for the normal development of the mouseembryo and basal homeostatic functions mediated by SMCs. Inaddition, these data demonstrate that the devleopmental pro-gram leading to vascular stabilization and patterning duringembryonic development is initiated earlier than was suggestedby previous studies.
MATERIALS AND METHODS
Molecular cloning and generation of SM22a mutant embryonic stem (ES)cells and mice. A genomic clone including the 59 end of the murine SM22a genewas isolated from an SV129 mouse library (43). The targeting vector was gen-erated in the pPNT plasmid (46), which contains the PGK-neo-poly(A) andPGK-tk-poly(A) cassettes for positive and negative selection, respectively. Asschematically represented in Fig. 1A, the pPNTSM22.LacZ targeting vectorcontains a genomic subfragment that includes approximately 5-kb of the SM22a59 flanking sequence, exon 1, intron 1, and exon 2 sequence through the initiationcodon (denoted as ATG). This was cloned in frame to the bacterial lacZ reportergene (denoted as LacZ), which in turn was subcloned into NotI/XhoI-digestedpPNT. The targeting vector also includes the 2.6-kb BamHI/NcoI SM22agenomic subfragment that spans exon 4 through intron 5 sequences subclonedinto the EcoRI site of pPNT. In pPNTSM22.LacZ, sequences 39 of the initiationcodon of exon 2 are replaced by the lacZ reporter gene and the PGK-neocassette.
The SM22a targeting construct was linearized with NotI and electroporatedinto RW ES cells as described previously (24). After 24 h, neomycin-resistanttransfectants were selected in 250 mg of G418 per ml and 1 mM gancyclovir for8 to 10 days. DNA from resistant ES cell clones was analyzed by Southern blotanalysis after EcoRI digestion with a radiolabeled probe derived from genomicsequences located 39 of the targeting vector (see Fig. 1A). To generate SM22a-deficient mice, ES cells from two independently derived SM221/2LacZ cloneswere microinjected into C57BL/6 donor blastocysts that were implanted intopseudopregnant CD1 females (3, 12). The resulting male chimeras were matedwith C57BL/6 females, and agouti offspring were genotyped by Southern blotanalysis as described previously (24). SM221/2LacZ mice were then outbred withC57BL/6 and CD1 mice to generate heterozygous SM222/2LacZ mice that wereinterbred for phenotype analysis. All animal experimentation was performedaccording to National Institutes of Health guidelines in the University of Penn-sylvania Animal Care Facility.
Preparation of SM22a polyclonal antiserum. The pGEX4TSM22a expressionplasmid encoding the glutathione S-transferase (GST)–mouse SM22a (aminoacids 1 to 201) fusion protein was transformed into bacteria, and GST-SM22afusion protein was induced and purified from bacterial extracts as describedpreviously (23). Rabbits were immunized with the purified GST-SM22a fusionprotein according to the standard protocol of Cocalico Biologicals, Inc. (Reams-town, Pa.). Preimmune immunoglobulin G (IgG) and anti-SM22a polyclonalIgG (no. 1387-4) were isolated from the serum by GST-SM22a affinity chroma-tography and protein A-Sepharose chromatography.
Northern and Western blot analyses. RNA was isolated from tissues of wild-type, heterozygous SM221/2LacZ, and homozygous SM222/2LacZ mice as de-scribed elsewhere (24). Northern blot analyses were performed using 10 mg ofRNA/sample, and the radiolabeled 754-bp (bp 29 to 811) mouse SM22a cDNAprobe as described previously (29). Western blot analyses were performed asdescribed earlier using the affinity-purified rabbit polyclonal a-SM22a antiserumand control rabbit preimmune IgG (17).
Cardiovascular physiological assessment. The systolic blood pressure (SBP)and heart rate (HR) of conscious wild-type, heterozygous (SM221/2LacZ), andhomozygous-null (SM222/2LacZ) adult male mice were compared using a mousephysiological recording system according to the manufacturer’s instructions (Vis-itech System, Apex, N.C.). Each measurement was performed in triplicate andwas repeated the following day to ensure reproducibility. To assess cardiacfunction in wild-type, heterozygous (SM221/2LacZ), and homozygous-null(SM222/2LacZ) adult male mice, echocardiography was performed using an S12transducer (5 to 12 MHz) and a HP5500 Sonos system as described earlier (7).M-mode tracings were made at the level of the papillary muscle in the leftventricular (LV) short-axis view. The heart rate was determined from the M-mode and pulsed-Doppler tracings of the peak aortic flow velocity in the ascend-ing aorta. On-line measurement of LV dimensions was made using a Tom TecImaging System. End-systolic and diastolic dimensions were determined by plain-imetry. The percent left ventricular fractional shortening was calculated using astandard formula: [(LVEDd 2 LVESd)/LVEDd] 3 100. The percent ejectionfraction was estimated from the fractional area change (FAC) in the LV short-axis view as follows: FAC 5 [(LVEDa 2 LVESa)/LVEDa] 3 100. Color flowmapping and pulsed Doppler wave forms were obtained across the pulmonaryartery to evaluate patent ductus arteriosus. The statistical significance was cal-culated by analysis of variance ANOVA and by two-tailed unpaired Student t test(a P value of ,0.05 was considered significant).
Cell biology, histology, and ultrastructural analyses. Tissues from adult wild-type and SM222/2LacZ mice were fixed, embedded in paraffin, sectioned, andstained with hematoxylin and eosin as described previously (24). Embryos andtissue sections from adult mice were fixed and stained for b-galactosidase activityand counterstained with hematoxylin and eosin as described previously (5). A7r5SMCs were stably transfected with an expression plasmid encoding Ds-REDepitope-tagged mouse SM22a as described previously (15). Three-dimensional(3-D) fluorescence images of A7r5 cells and primary SMCs were acquired usinga Delta Vision microscopy system (Applied Precision, Issaquah, Wash.)equipped with an appropriate barrier filter set (Chroma, Battleboro, Vt.) forDs-Red or rhodamine (lex 5 555 nm, lem 5 617 nm). Spatial and temporalnormalization of the illumination intensity allowed quantitative analysis of thefluorescence intensity (11). A constrained iterative deconvolution algorithm (11)using an experimentally measured point spread function was applied to 3-Darrays of optical sections to yield a high-resolution spatial distribution of fluo-rescence intensity that accurately represented the 3-D cytoskeletal structure (9).Image restoration and volume projections were computed using SoftWoRx soft-ware (Applied Precision). For electron microscopy, the aorta and bladder fromwild-type and SM222/2LacZ mice were fixed in 2.5% glutaraldehyde and 2%paraformaldehyde and then postfixed with 2% aqueous osmium tetroxide as
VOL. 21, 2001 TARGETED MUTATION OF SM22a 1337
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
described previously (4). The samples were washed and dehydrated with ethanoland embedded in LX-112 medium that was polymerized at 70°C for 48 h.Ultrathin sections (80 nm) were cut with a diamond knife and stained with uranylacetate. Immunogold labeling of tissue sections was performed as describedpreviously (4). Samples were fixed with 2% paraformaldehyde and 0.05% glu-taraldehyde in phosphate-buffered saline for 1 h, treated with 10 mM NH4Cl for30 min, dehydrated at 220°C, and embedded in Lowicryl K4M medium. Thesamples were polymerized with UV light (365 nm), and ultrathin sections (90nm) were cut, mounted on Formvar-coated nickel grids, blocked with 1% bovineserum albumin and 2% goat serum, and incubated with affinity-purified anti-SM22a polyclonal antiserum or control rabbit IgG at a 1:100 dilution. Thesections were then incubated with 15-nm gold-conjugated anti-rabbit IgG andstained with uranyl acetate, and the images were visualized with a Philips CM100electron microscope at 60 kV.
RESULTS
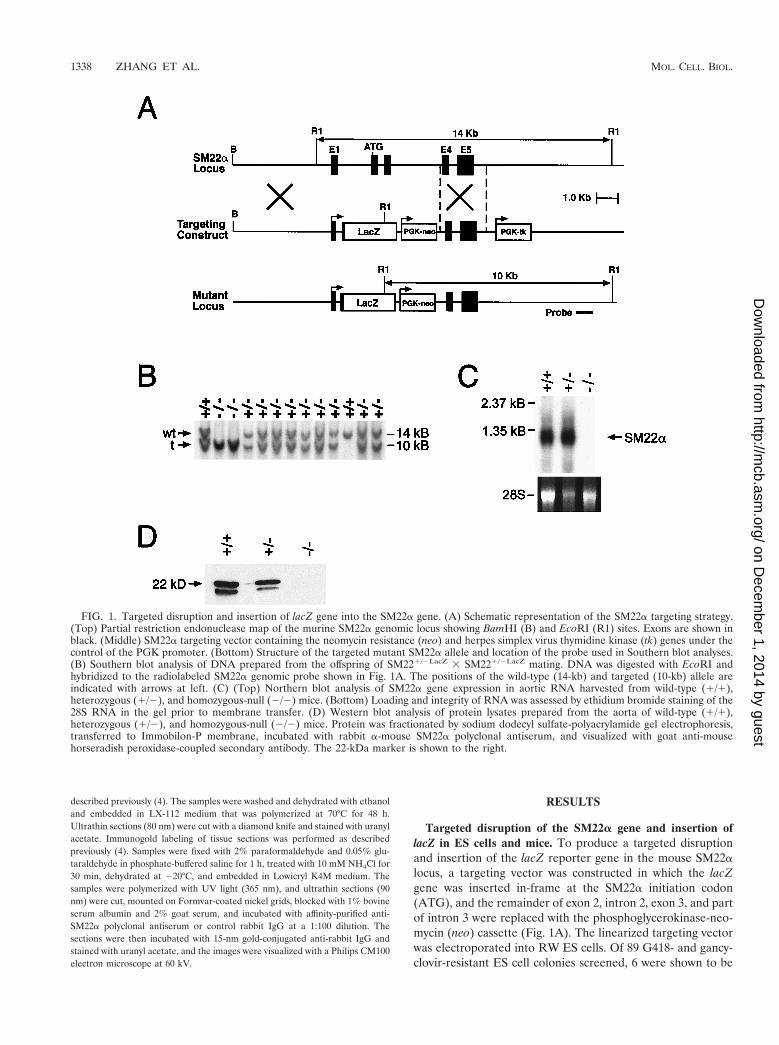
Targeted disruption of the SM22a gene and insertion oflacZ in ES cells and mice. To produce a targeted disruptionand insertion of the lacZ reporter gene in the mouse SM22alocus, a targeting vector was constructed in which the lacZgene was inserted in-frame at the SM22a initiation codon(ATG), and the remainder of exon 2, intron 2, exon 3, and partof intron 3 were replaced with the phosphoglycerokinase-neo-mycin (neo) cassette (Fig. 1A). The linearized targeting vectorwas electroporated into RW ES cells. Of 89 G418- and gancy-clovir-resistant ES cell colonies screened, 6 were shown to be
FIG. 1. Targeted disruption and insertion of lacZ gene into the SM22a gene. (A) Schematic representation of the SM22a targeting strategy.(Top) Partial restriction endonuclease map of the murine SM22a genomic locus showing BamHI (B) and EcoRI (R1) sites. Exons are shown inblack. (Middle) SM22a targeting vector containing the neomycin resistance (neo) and herpes simplex virus thymidine kinase (tk) genes under thecontrol of the PGK promoter. (Bottom) Structure of the targeted mutant SM22a allele and location of the probe used in Southern blot analyses.(B) Southern blot analysis of DNA prepared from the offspring of SM221/2LacZ 3 SM221/2LacZ mating. DNA was digested with EcoRI andhybridized to the radiolabeled SM22a genomic probe shown in Fig. 1A. The positions of the wild-type (14-kb) and targeted (10-kb) allele areindicated with arrows at left. (C) (Top) Northern blot analysis of SM22a gene expression in aortic RNA harvested from wild-type (1/1),heterozygous (1/2), and homozygous-null (2/2) mice. (Bottom) Loading and integrity of RNA was assessed by ethidium bromide staining of the28S RNA in the gel prior to membrane transfer. (D) Western blot analysis of protein lysates prepared from the aorta of wild-type (1/1),heterozygous (1/2), and homozygous-null (2/2) mice. Protein was fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis,transferred to Immobilon-P membrane, incubated with rabbit a-mouse SM22a polyclonal antiserum, and visualized with goat anti-mousehorseradish peroxidase-coupled secondary antibody. The 22-kDa marker is shown to the right.
1338 ZHANG ET AL. MOL. CELL. BIOL.
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

homologous recombinants by Southern blot analyses. Each ofthese SM221/2LacZ clones contained a single site of integrationof the vector in the host genome as determined by Southernblot analyses performed with a neo probe (data not shown).
Two independently derived SM221/2LacZ ES cell lines wereused to generate chimeric mice, and these mice were crossed toC57BL/6 mice to transmit the targeted allele through the germline as determined by Southern blot analysis (Fig. 1B). Het-erozygous SM221/2LacZ mice were phenotypically normal andfertile. These mice were outbred to the BL/6 and CD1 back-ground to produce heterozygotes that were intercrossed togenerate homozygous SM222/2LacZ mice. To confirm that anull mutation was created with this targeting strategy, North-ern blot analyses were performed on RNA isolated from SMC-containing tissues (aorta and uterus) harvested from wild-type(SM221/1), heterozygous (SM221/2LacZ), and homozygous(SM222/2LacZ) null mice. As anticipated, a 1.3-kb transcriptcorresponding to the expected size of SM22a mRNA was ob-served in samples of RNA prepared from the aorta (Fig. 1C,lanes 1 and 2) and uterus (data not shown) of wild-type andheterozygous mice. In contrast, hybridization of the radiola-beled SM22a cDNA probe to RNA samples prepared from the
tissues of SM222/2LacZ mice was not detected (Fig. 1C, lane3). Western blot analyses performed with SM22a-specific poly-clonal antiserum and tissue lysates prepared from the aorta ofwild-type (SM221/1), heterozygous (SM221/2LacZ), and ho-mozygous (SM222/2LacZ) null mice revealed the expected 22-kDa band (Fig. 1D, lanes 1 and 2). In addition, a 20-kDa signalwas reproducibly observed in lysates prepared from the aortaof wild-type (SM221/1) and heterozygous (SM221/2LacZ)mice. In contrast, no hybridization signal was detected in tissuelysates prepared from the aorta (Fig. 1D, lane 3) and uterus(data not shown) of homozygous SM222/2LacZ null mice.Taken together, these data demonstrate that the gene target-ing strategy utilized generated a null mutation in the geneencoding SM22a.
Transcriptional activation of the SM22a gene in the embry-onic and adult mouse. The gene targeting strategy effectivelyknocked in the bacterial lacZ reporter gene under the tran-scriptional control of the endogenous SM22a promoter, as wellas other unidentified elements flanking and within the SM22agene. To determine when and where in the postgastrulationembryo the SM22a gene is transcribed, E8.0 SM221/2LacZ
embryos were harvested from staged matings of SM221/2LacZ 3
FIG. 2. The SM22a gene is activated in the dorsal aorta of the E8.0 embryo. E8.0 SM221/2LacZ embryos harvested from staged matings ofSM221/2LacZ 3 SM221/2LacZ mice genotyped by Southern blot analyses and stained for b-galactosidase activity as described in Materials andMethods. (A) Whole-mount b-galactosidase activity in an E8.0 SM221/2LacZ embryo. b-Galactosidase activity (blue staining) is observed in theembryonic heart, aorta, cephalic mesenchyme (CM), and yolk sac. The planes of the sections shown in panels B, C, and D are indicated by whitelines. (B) Cross-section through the head of an E8.0 SM221/2LacZ embryo showing b-galactosidase activity restricted to cells within the cephalicmesenchyme (CM). Staining was not observed in the surface ectoderm (ECT) or neuroepithelium (NE). (C) Cross-section through the thoraciccavity of an E8.0 SM221/2LacZ embryo demonstrating b-galactosidase activity in the myocardium (Myo), pericardium (P), aorta (Ao), andnotochord (NC). (D) High-power view of a section through the thoracic cavity of an E8.0 SM221/2LacZ embryo demonstrating b-galactosidaseactivity in cells surrounding the paired dorsal aorta (Ao). In addition, faint blue staining is observed in scattered cells within the paraxial mesoderm(PM), which gives rise to the somites.
VOL. 21, 2001 TARGETED MUTATION OF SM22a 1339
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
SM221/2LacZ mice and stained for b-galactosidase activity. AtE8.0, the process of “embryonic turning” is initiated, the heartconsists of common atrial and ventricular chambers, and theunfused dorsal aortae are easily recognizable. In the E8.0SM221/2LacZ embryo, b-galactosidase activity (blue staining)was obvious within the head, heart, cardiac outflow tract, dor-sal aorta, and yolk sac membranes (Fig. 2A). Sections obtainedthrough the headfold revealed that lacZ-positive cells wererestricted to the cephalic mesenchyme (Fig. 2B). No stainingwas observed within the neuroepithelium or surface ectoderm(Fig. 2B). Sections through the superior thoracic cavity re-vealed blue-stained pericardial and myocardial cells. In con-trast, the single layer of endocardial cells did not express b-ga-lactosidase (Fig. 2C). In addition, blue-stained cells werereproducibly observed surrounding the paired dorsal aorta(Fig. 2C and D, A.). Of note, this is 24 to 36 h prior to thereported expression of genes encoding SMC markers in themouse embryo (27). Finally, scattered blue stained cells wereobserved within the paraxial mesoderm (Fig. 2D), which givesrise to the somites and in the notochord (Fig. 2C).
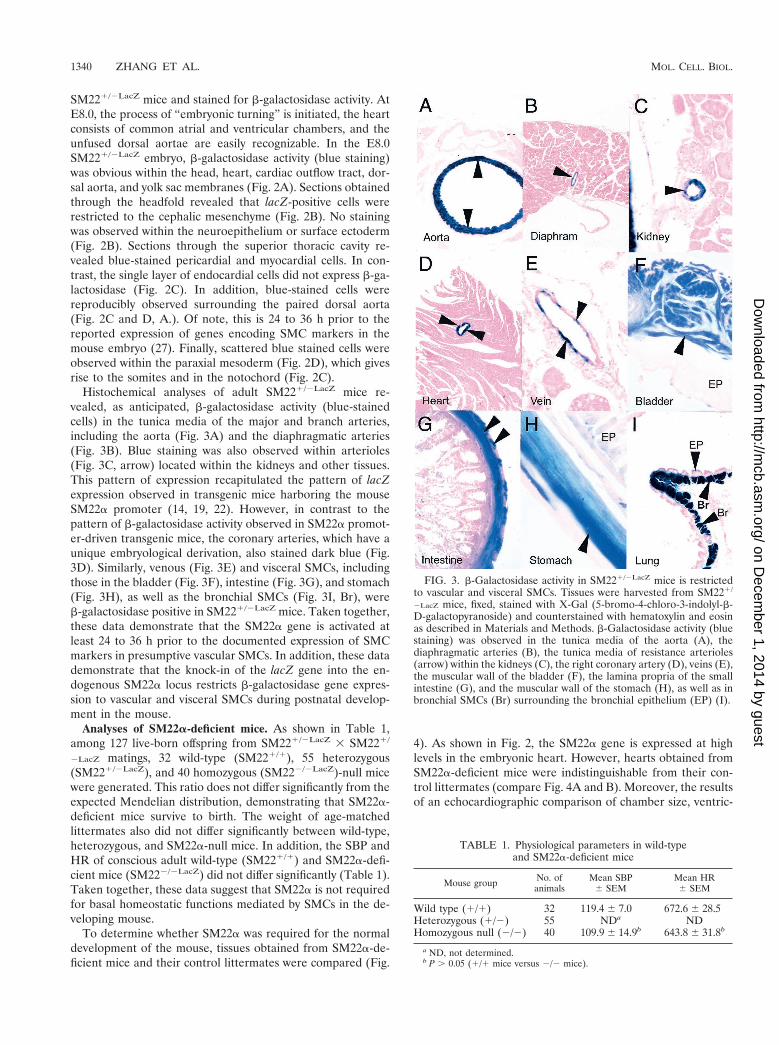
Histochemical analyses of adult SM221/2LacZ mice re-vealed, as anticipated, b-galactosidase activity (blue-stainedcells) in the tunica media of the major and branch arteries,including the aorta (Fig. 3A) and the diaphragmatic arteries(Fig. 3B). Blue staining was also observed within arterioles(Fig. 3C, arrow) located within the kidneys and other tissues.This pattern of expression recapitulated the pattern of lacZexpression observed in transgenic mice harboring the mouseSM22a promoter (14, 19, 22). However, in contrast to thepattern of b-galactosidase activity observed in SM22a promot-er-driven transgenic mice, the coronary arteries, which have aunique embryological derivation, also stained dark blue (Fig.3D). Similarly, venous (Fig. 3E) and visceral SMCs, includingthose in the bladder (Fig. 3F), intestine (Fig. 3G), and stomach(Fig. 3H), as well as the bronchial SMCs (Fig. 3I, Br), wereb-galactosidase positive in SM221/2LacZ mice. Taken together,these data demonstrate that the SM22a gene is activated atleast 24 to 36 h prior to the documented expression of SMCmarkers in presumptive vascular SMCs. In addition, these datademonstrate that the knock-in of the lacZ gene into the en-dogenous SM22a locus restricts b-galactosidase gene expres-sion to vascular and visceral SMCs during postnatal develop-ment in the mouse.
Analyses of SM22a-deficient mice. As shown in Table 1,among 127 live-born offspring from SM221/2LacZ 3 SM221/
2LacZ matings, 32 wild-type (SM221/1), 55 heterozygous(SM221/2LacZ), and 40 homozygous (SM222/2LacZ)-null micewere generated. This ratio does not differ significantly from theexpected Mendelian distribution, demonstrating that SM22a-deficient mice survive to birth. The weight of age-matchedlittermates also did not differ significantly between wild-type,heterozygous, and SM22a-null mice. In addition, the SBP andHR of conscious adult wild-type (SM221/1) and SM22a-defi-cient mice (SM222/2LacZ) did not differ significantly (Table 1).Taken together, these data suggest that SM22a is not requiredfor basal homeostatic functions mediated by SMCs in the de-veloping mouse.
To determine whether SM22a was required for the normaldevelopment of the mouse, tissues obtained from SM22a-de-ficient mice and their control littermates were compared (Fig.
4). As shown in Fig. 2, the SM22a gene is expressed at highlevels in the embryonic heart. However, hearts obtained fromSM22a-deficient mice were indistinguishable from their con-trol littermates (compare Fig. 4A and B). Moreover, the resultsof an echocardiographic comparison of chamber size, ventric-
FIG. 3. b-Galactosidase activity in SM221/2LacZ mice is restrictedto vascular and visceral SMCs. Tissues were harvested from SM221/
2LacZ mice, fixed, stained with X-Gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside) and counterstained with hematoxylin and eosinas described in Materials and Methods. b-Galactosidase activity (bluestaining) was observed in the tunica media of the aorta (A), thediaphragmatic arteries (B), the tunica media of resistance arterioles(arrow) within the kidneys (C), the right coronary artery (D), veins (E),the muscular wall of the bladder (F), the lamina propria of the smallintestine (G), and the muscular wall of the stomach (H), as well as inbronchial SMCs (Br) surrounding the bronchial epithelium (EP) (I).
TABLE 1. Physiological parameters in wild-typeand SM22a-deficient mice
Mouse group No. ofanimals
Mean SBP6 SEM
Mean HR6 SEM
Wild type (1/1) 32 119.4 6 7.0 672.6 6 28.5Heterozygous (1/2) 55 NDa NDHomozygous null (2/2) 40 109.9 6 14.9b 643.8 6 31.8b
a ND, not determined.b P . 0.05 (1/1 mice versus 2/2 mice).
1340 ZHANG ET AL. MOL. CELL. BIOL.
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

ular wall thickness, and indices of contractile function, includ-ing left ventricular fractional shortening and ejection fraction,did not differ significantly (data not shown). Similarly, thearteries and veins of wild-type and SM22a-deficient adult micewere virtually indistinguishable, including quantitative mor-phometric analyses of the arterial intima to media ratios (com-pare Fig. 4C and D). In addition, visceral organs obtained fromwild-type and SM22a-deficient mice, including the small intes-tine (compare Fig. 4E and F) and uterus (compare Fig. 4G andH), developed normally and were histologically indistinguish-able.
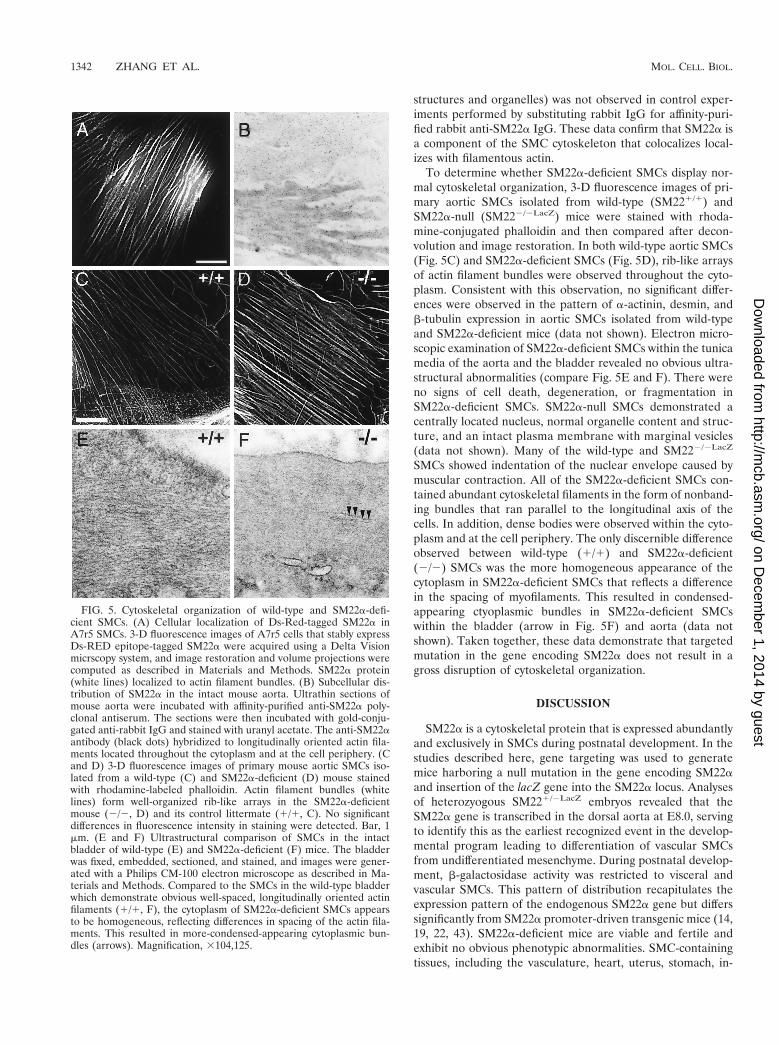
Ultrastructural analyses of SM22a-deficient SMCs. SM22acolocalizes to the cytoskeleton of SMCs (38, 39, 41). There-fore, it was of interest to define the subcellular localization ofSM22a in SMCs and to determine whether SM22a-deficient
SMCs exhibited alterations in cytoskeletal architecture. A 3-Dreconstruction of A7r5 SMCs that stably expressed a Ds-Red-tagged SM22a protein revealed localization to the F-actin fil-ament bundles (Fig. 5A). This pattern of expression was iden-tical to that observed when these cells were stained with theactin-binding protein phalloidin (data not shown). Immuno-gold localization of SM22a protein performed using affinity-purified SM22a polyclonal antiserum revealed gold particles(black dots) localizing to longitudinally oriented actin fila-ments in the cytoplasm and patches at the cell periphery (Fig.5B). In addition, gold-labeled SM22a protein was also ob-served on the surface of some myosin bundles (dark patches inFig. 5B). Signal (black dots in Fig. 5B) was not detected in theextracellular space, fibroblasts, or endothelial cells (data notshown). Hybridization to actin filaments (or other subcellular
FIG. 4. SMC-containing tissues develop normally in SM22a-deficient mice. Tissues harvested from wild-type (wt) and SM22a-deficient (2/2)mice were fixed and stained with hematoxylin and eosin as described in Materials and Methods. (A and B) Comparison of hearts from wild-type(A) and 2/2 (B) mice demonstrating normal left ventricular (LV) and right ventricular (RV) chamber dimensions and morphology. (C and D)Comparison of the aorta harvested from a wild-type (C) and 2/2 (D) adult mouse demonstrating normal tunica intima (End), tunica media (M),and adventitia (AD). (E and F) Comparison of small intestine harvested from wild-type (E) and 2/2 (F) mice demonstrating intact lamina propriaand intestinal epithelium (EP). (G and H) Comparison of uterus harvested from wild-type (G) and 2/2 (H) mice demonstrating normal SMCarrangement within the muscular layers (SMCs) of the uterus.
VOL. 21, 2001 TARGETED MUTATION OF SM22a 1341
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
structures and organelles) was not observed in control exper-iments performed by substituting rabbit IgG for affinity-puri-fied rabbit anti-SM22a IgG. These data confirm that SM22a isa component of the SMC cytoskeleton that colocalizes local-izes with filamentous actin.
To determine whether SM22a-deficient SMCs display nor-mal cytoskeletal organization, 3-D fluorescence images of pri-mary aortic SMCs isolated from wild-type (SM221/1) andSM22a-null (SM222/2LacZ) mice were stained with rhoda-mine-conjugated phalloidin and then compared after decon-volution and image restoration. In both wild-type aortic SMCs(Fig. 5C) and SM22a-deficient SMCs (Fig. 5D), rib-like arraysof actin filament bundles were observed throughout the cyto-plasm. Consistent with this observation, no significant differ-ences were observed in the pattern of a-actinin, desmin, andb-tubulin expression in aortic SMCs isolated from wild-typeand SM22a-deficient mice (data not shown). Electron micro-scopic examination of SM22a-deficient SMCs within the tunicamedia of the aorta and the bladder revealed no obvious ultra-structural abnormalities (compare Fig. 5E and F). There wereno signs of cell death, degeneration, or fragmentation inSM22a-deficient SMCs. SM22a-null SMCs demonstrated acentrally located nucleus, normal organelle content and struc-ture, and an intact plasma membrane with marginal vesicles(data not shown). Many of the wild-type and SM222/2LacZ
SMCs showed indentation of the nuclear envelope caused bymuscular contraction. All of the SM22a-deficient SMCs con-tained abundant cytoskeletal filaments in the form of nonband-ing bundles that ran parallel to the longitudinal axis of thecells. In addition, dense bodies were observed within the cyto-plasm and at the cell periphery. The only discernible differenceobserved between wild-type (1/1) and SM22a-deficient(2/2) SMCs was the more homogeneous appearance of thecytoplasm in SM22a-deficient SMCs that reflects a differencein the spacing of myofilaments. This resulted in condensed-appearing ctyoplasmic bundles in SM22a-deficient SMCswithin the bladder (arrow in Fig. 5F) and aorta (data notshown). Taken together, these data demonstrate that targetedmutation in the gene encoding SM22a does not result in agross disruption of cytoskeletal organization.
DISCUSSION
SM22a is a cytoskeletal protein that is expressed abundantlyand exclusively in SMCs during postnatal development. In thestudies described here, gene targeting was used to generatemice harboring a null mutation in the gene encoding SM22aand insertion of the lacZ gene into the SM22a locus. Analysesof heterozyogous SM221/2LacZ embryos revealed that theSM22a gene is transcribed in the dorsal aorta at E8.0, servingto identify this as the earliest recognized event in the develop-mental program leading to differentiation of vascular SMCsfrom undifferentiated mesenchyme. During postnatal develop-ment, b-galactosidase activity was restricted to visceral andvascular SMCs. This pattern of distribution recapitulates theexpression pattern of the endogenous SM22a gene but differssignificantly from SM22a promoter-driven transgenic mice (14,19, 22, 43). SM22a-deficient mice are viable and fertile andexhibit no obvious phenotypic abnormalities. SMC-containingtissues, including the vasculature, heart, uterus, stomach, in-
FIG. 5. Cytoskeletal organization of wild-type and SM22a-defi-cient SMCs. (A) Cellular localization of Ds-Red-tagged SM22a inA7r5 SMCs. 3-D fluorescence images of A7r5 cells that stably expressDs-RED epitope-tagged SM22a were acquired using a Delta Visionmicrscopy system, and image restoration and volume projections werecomputed as described in Materials and Methods. SM22a protein(white lines) localized to actin filament bundles. (B) Subcellular dis-tribution of SM22a in the intact mouse aorta. Ultrathin sections ofmouse aorta were incubated with affinity-purified anti-SM22a poly-clonal antiserum. The sections were then incubated with gold-conju-gated anti-rabbit IgG and stained with uranyl acetate. The anti-SM22aantibody (black dots) hybridized to longitudinally oriented actin fila-ments located throughout the cytoplasm and at the cell periphery. (Cand D) 3-D fluorescence images of primary mouse aortic SMCs iso-lated from a wild-type (C) and SM22a-deficient (D) mouse stainedwith rhodamine-labeled phalloidin. Actin filament bundles (whitelines) form well-organized rib-like arrays in the SM22a-deficientmouse (2/2, D) and its control littermate (1/1, C). No significantdifferences in fluorescence intensity in staining were detected. Bar, 1mm. (E and F) Ultrastructural comparison of SMCs in the intactbladder of wild-type (E) and SM22a-deficient (F) mice. The bladderwas fixed, embedded, sectioned, and stained, and images were gener-ated with a Philips CM-100 electron microscope as described in Ma-terials and Methods. Compared to the SMCs in the wild-type bladderwhich demonstrate obvious well-spaced, longitudinally oriented actinfilaments (1/1, F), the cytoplasm of SM22a-deficient SMCs appearsto be homogeneous, reflecting differences in spacing of the actin fila-ments. This resulted in more-condensed-appearing cytoplasmic bun-dles (arrows). Magnification, 3104,125.
1342 ZHANG ET AL. MOL. CELL. BIOL.
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

testine, and bladder, all developed normally. Consistent withthese findings, the cytoskeletal organization of SM22a-defi-cient and wild-type vascular and visceral SMCs is virtuallyindistinguishable.
Our group and others have reported that the SM22a pro-moter restricts expression of a lacZ reporter gene to arterialSMCs in transgenic mice (14, 19, 22, 43). In SM22a promoter-driven transgenic mice, b-galactosidase activity is not observedin venous, coronary arterial, or visceral SMCs despite the factthat the endogenous SM22a gene is expressed in these cells.This led to the hypothesis that distinct transcriptional pro-grams distinguish tissue-restricted subsets of SMCs or SMCsublineages. This hypothesis was supported by the finding thatthe smooth muscle a-actin promoter and intragenic enhancer(20), the smooth muscle myosin heavy-chain promoter andintragenic enhancer (21), the telokin promoter (10), and theg-enteric actin promoter (30) restrict transgene expression tounique tissue-restricted subsets of SMCs in transgenic mice. Itis noteworthy that each of these transcriptional regulatory el-ements also contains a functionally important CArG box thatbinds directly to the MADS box transcription factor SRF. Theobserved b-galactosidase activity in visceral and vascular SMCsof SM221/2LacZ mice is consistent with this model and leads usto predict that additional, as-yet-unidentified, transcriptionalregulatory elements required for expression in venous, coro-nary arterial, and visceral SMCs exist and are located in distantregions flanking the SM22a gene. Identification and charac-terization of these regulatory elements should provide funda-mental insights into the transcriptional programs that distin-guish SMC sublineages.
Relatively little is currently understood about the transcrip-tional programs and signaling pathways that regulate the spec-ification of SMCs from undifferentiated mesenchyme duringembryonic development (for a review, see references 27 and28). The differentiation of SMCs from mesenchyme is a criticalevent in angiogenic patterning that ultimately distinguishesarteries, veins, and capillary beds. In the mouse, SMC markers,including SM–a-actin, calponin, and SM22a, had been de-tected as early as E9.5 in the dorsal aorta (27). Therefore, wewere surprised to detect b-galactosidase activity in the dorsalaorta of E8.0 unturned SM221/2LacZ embryos. These datasuggest that the signaling pathways that regulate the specifica-tion and the differentiation of vascular SMCs from the mesen-chyme are activated earlier in the developing mouse embryothan was recognized previously. As such, the SM221/2LacZ
mouse and lacZ-tagged SMCs isolated from these mice mayserve as valuable experimental reagents for studying the mo-lecular basis of SMC differentiation. Moreover, becauseSM22a is downregulated in concert with other contractile pro-teins in atherosclerotic lesions (35–37), these mice may be usedto examine the molecular mechanisms that modulate SMCphenotype and to examine the pathological mechanisms un-derlying vascular proliferative syndromes.
What then is the function of the cytoskeletal protein SM22ain SMCs and other embryonic tissues, including the developingheart and skeletal muscle? SM22a colocalizes with actin fila-ments and is abundantly expressed in contractile SMCs. It hasbeen conserved through evolution and shows high-level se-quence identity to the myofibrillar regulatory protein calponin.However, SM22a-deficient mice are viable and fertile and ex-
hibit no overt phenotype. SMC-containing tissues, includingarteries, veins, heart, gut, uterus, and bladder, develop nor-mally. Basal homeostatic functions regulated in whole or inpart by SMCs, including the regulation of blood pressure,bladder control, uterine contraction, and gastrointestinal mo-tility, are not grossly altered in SM22a-deficient mice. Consis-tent with these data, the SMC ultrastructure and cytoskeletalorganization in SM22a-deficient mice is virtually indistinguish-able from their control littermates. It remains theoreticallypossible that SM22a is partially redundant with another struc-turally related protein such as calponin. In this regard, it isnoteworthy that we recently cloned a novel mouse cDNA en-coding a protein with greater than 70% sequence identity toSM22a that is also expressed in some SMC containing tissues(J. Zhang and M. Parmacek, unpublished data). Alternatively,SM22a may not be required for basal homeostatic functionsbut may be required for stress responses such as wound heal-ing. The availability of SM22a-deficient mice allows these andother possibilities to be examined.
ACKNOWLEDGMENTS
We thank Lisa Gottschalk for her help preparing the figures, AngieBoyce for expert secretarial assistance, and Theodore J. Plappert fortechnical assistance in performing echocardiography.
This project was supported in part by NIH NRSA grants HL10058to B.P.H. and NIH-R0156915 to M.S.P. M.S.P. is an Established In-vestigator of the American Heart Association.
REFERENCES
1. Almendral, J. M., J. F. Santaren, J. Perera, M. Zerial, and R. Bravo. 1989.Expression, cloning and cDNA sequence of a fibroblast serum-regulatedgene encoding a putative actin-associated protein (p27). Exp. Cell Res.181:518–530.
2. Ayme-Southgate, A., P. Lasko, C. French, and M. L. Pardue. 1989. Charac-terization of the gene for mp20: a Drosophila muscle protein that is not foundin asynchronous oscillatory flight muscle. J. Cell Biol. 108:521–531.
3. Bradley, A. 1987. Production and analysis of chimaeric mice, p. 113–151. InE. J. Robertson (ed.), Teratocarcinomas and embryonic stem cells: a prac-tical approach. IRL Press, Oxford, England.
4. Chan, Y. M., Q. C. Yu, J. D. Fine, and E. Fuchs. 1993. The genetic basis ofWeber-Cockayne epidermolysis bullosa simplex. Proc. Natl. Acad. Sci. USA90:7414–7418.
5. Chang, M. W., E. Barr, J. Seltzer, Y.-Q. Jiang, G. J. Nabel, E. G. Nabel, M. S.Parmacek, and J. M. Leiden. 1995. Cytostatic gene therapy for vascularproliferative disorders with a constitutively active form of the retinoblastomagene product. Science 267:518–522.
6. Duband, J. L., M. Gimona, M. Scatena, S. Sartore, and J. V. Small. 1993.Calponin and SM22 as differentiation markers of smooth muscle: spatiotem-poral distribution during avian embryonic development. Differentiation 55:1–11.
7. Fentzke, R. C., C. E. Korcarz, R. M. Lang, H. Lin, and J. M. Leiden. 1998.Dilated cardiomyopathy in transgenic mice expressing a dominant-negativeCREB transcription factor in the heart. J. Clin. Investig. 101:2415–2426.
8. Goetinck, S., and R. H. Waterston. 1994. The Caenorhabditis elegans muscle-affecting gene unc-87 encodes a novel thin filament associated protein.J. Cell Biol. 127:79–93.
9. Helmke, B. P., R. D. Goldman, and P. F. Davies. 2000. Rapid displacementof vimentin intermediate filaments in living cells exposed to flow. Circ. Res.86:745–752.
10. Herring, B. P., and A. F. Smith. 1996. Telokin expression is mediated by asmooth muscle cell-specific promoter. Am J. Physiol. 270:C1656–C1665.
11. Hiraoka, Y., J. W. Sedat, and D. A. Agard. 1990. Determination of three-dimensional properties of a light microscope system: partial confocal behav-ior in epifluorescence microscopy. Biophys. J. 57:325–333.
12. Hogan, B., R. Beddington, F. Constantini, and E. Lacy (ed.). 1994. Manip-ulating the mouse embryo, 2nd ed. Cold Spring Harbor Laboratory Press,Plainview, N.Y.
13. James, A. L., P. D. Pare, and J. C. Hogg. 1989. The mechanics of airwaynarrowing in asthma. Am. Rev. Respir. Dis. 139:242–246.
14. Kim, S., H. S. Ip, M. M. Lu, C. Clendenin, and M. S. Parmacek. 1997. Aserum response factor-dependent transcriptional regulatory program iden-tifies distinct smooth muscle cell sublineages. Mol. Cell. Biol. 17:2266–2278.
15. Kuo, C. T., E. E. Morrisey, R. Anandappa, K. Sigrist, M. M. Lu, M. S.
VOL. 21, 2001 TARGETED MUTATION OF SM22a 1343
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

Parmacek, C. Soudais, and J. M. Leiden. 1997. GATA4 transcription factoris required for ventral morphogenesis and heart tube formation. Genes Dev.11:1048–1060.
16. Lawson, D., M. Harrison, and C. Shapland. 1997. Fibroblast transgelin andsmooth muscle SM22a are the same protein, the expression of which isdown-regulated in many cell lines. Cell Motil. Cytoskel. 38:250–257.
17. Lees-Miller, J. P., D. H. Heeley, and L. B. Smillie. 1987. An abundant andnovel protein of 22 kDa (SM22) is widely distributed in smooth muscles.Purification form bovine aorta. Biochem. J. 244:705–709.
18. Li, L., J. M. Miano, P. Cserjesi, and E. N. Olson. 1996. SM22a, a marker ofadult smooth muscle, is expressed in multiple myogenic lineages duringembryogenesis. Circ. Res. 78:188–195.
19. Li, L., J. M. Miano, B. Mercer, and E. N. Olson. 1996. Expression of theSM22a promoter in transgenic mice provides evidence for distinct transcrip-tional regulatory programs in vascular and visceral smooth muscle cells.J. Cell Biol. 132:849–859.
20. Mack, C. P., and G. K. Owens. 1999. Regulation of smooth muscle a-actinexpression in vivo is dependent on CArG elements within the 59 and firstintron promoter regions. Circ. Res. 84:852–861.
21. Madsen, C. S., R. C. P., J. E. Hungerford, S. L. White, I. Manabe, and G. K.Owens. 1998. Smooth muscle-specific expression of the smooth muscle my-osin heavy chain gene in transgenic mice requires 59-flanking and first in-tronic DNA sequence. Circ. Res. 82:908–917.
22. Moessler, H., M. Mericskay, Z. Li, S. Nagl, D. Paulin, and J. V. Small. 1996.The SM22 promoter directs tissue-specific expression in arterial but not invenous or visceral smooth muscle cells in transgenic mice. Development122:2415–2425.
23. Morrisey, E. E., H. S. Ip, Z. Tang, and M. S. Parmacek. 1997. GATA-4activates transcription via two novel domains that are conserved within theGATA-4/5/6 subfamily. J. Biol. Chem. 272:8515–8524.
24. Morrisey, E. E., Z. Tang, K. Sigrist, M. M. Lu, F. Jiang, H. S. Ip, and M. S.Parmacek. 1998. GATA6 regulates HNF4 and is required for differentiationof visceral endoderm in the mouse embryo. Genes Dev. 12:3579–3590.
25. Newby, A. C., and A. B. Zaltsman. 1999. Fibrous cap formation or destruc-tion—the critical importance of vascular smooth muscle cell proliferation,migration and matrix formation. Cardiovasc. Res. 41:345–360.
26. North, A. J., M. Gimona, Z. Lando, and J. V. Small. 1994. Actin isoformcompartments in chicken gizzard smooth muscle cells. J. Cell. Sci. 107:445–455.
27. Owens, G. K. 1998. Molecular control of vascular smooth muscle cell differ-entiation. Acta Physiol. Scand. 164:623–635.
28. Parmacek, M. S. Transcriptional programs regulating vascular smooth mus-cle cell development and differentiation. Curr. Top. Dev. Biol., in press.
29. Parmacek, M. S., and J. M. Leiden. 1989. Structure and expression of themurine slow/cardiac troponin C gene. J. Biol. Chem. 264:13217–13225.
30. Qian, J., A. Kumar, J. C. Szuscik, and J. L. Lessard. 1996. Tissue anddevelopmental specific expression of murine smooth muscle g-actin fusion
genes in transgenic mice. Dev. Dyn. 207:135–144.31. Ren, W., G. Y. K. Ng, R.-X. Wang, P. H. Wu, B. F. O’Dowd, S. George, D. H.
Osmond, and C. Liew. 1994. The identification of NP25: a novel protein thatis differentially expressed by neuronal subpopulations. Mol. Brain Res. 22:173–185.
32. Ross, R. 1999. Mechanisms of disease: atherosclerosis—an inflammatorydisease. N. Engl. J. Med. 340:115–126.
33. Samaha, F. F., H. S. Ip, E. E. Morrisey, J. Seltzer, Z. Tang, J. Solway, andM. S. Parmacek. 1996. Developmental pattern of expression and genomicorganization of the calponin-h1 gene; a contractile smooth muscle cellmarker. J. Biol. Chem. 271:395–403.
34. Schwartz, S. M., and C. E. Murry. 1998. Proliferation and the monoclonalorigins of atherosclerotic lesions. Annu. Rev. Med. 49:437–460.
35. Shanahan, C. M., N. R. B. Cary, J. C. Metcalfe, and P. L. Weissberg. 1994.High expression of genes for calcification-regulating proteins in human ath-erosclerotic plaques. J. Clin. Investig. 93:2393–2402.
36. Shanahan, C. M., and P. L. Weissberg. 1997. Smooth muscle cell heteroge-neity: Patterns of gene expression in vascular smooth muscle cells in vitroand in vivo. Arterioscler. Thromb. Vasc. Biol. 183:333–338.
37. Shanahan, C. M., P. L. Weissberg, and J. C. Metcalfe. 1993. Isolation of genemarkers of differentiated and proliferating vascular smooth muscle cells.Circ. Res. 73:193–204.
38. Shapland, C., J. J. Hsuan, N. F. Totty, and D. Lawson. 1993. Purification andproperties of transgelin: a transformation and shape change sensitive actin-gelling protein. J. Cell Biol. 121:1065–1073.
39. Shapland, C., P. Lowings, and D. Lawson. 1988. Identification of new actin-associated polypeptides that are modified by viral transformation andchanges in cell shape. J. Cell Biol. 107:153–161.
40. Small, J. V. 1985. Geometry of actin-membrane attachments in the smoothmuscle cell: the localisations of vinculin and a-actinin. EMBO J. 4:45–49.
41. Small, J. V., and M. Gimona. 1998. The cytoskeleton of the vertebratesmooth muscle cell. Acta Physiol. Scand. 164:341–348.
42. Small, J. V., and A. Sobieszek. 1977. Studies on the function and compositionof the 10-nm (100-A) filaments of vertebrate smooth muscle. J. Cell Sci.23:243–268.
43. Solway, J., J. Seltzer, F. F. Samaha, S. Kim, L. E. Alger, Q. Niu, E. E.Morrisey, H. S. Ip, and M. S. Parmacek. 1995. Structure and expression ofa smooth muscle cell-specific gene, SM22a. J. Biol. Chem. 270:13460–13469.
44. Takahashi, K., and B. Nadal-Ginard. 1991. Molecular cloning and sequenceanalysis of smooth muscle calponin. J. Biol. Chem. 266:13284–13288.
45. Thweatt, R., C. K. Lumpkin, and S. Goldstein. 1992. A novel gene encodinga smooth muscle protein is overexpressed in senescent human fibroblasts.Biochem. Biophys. Commun. 187:1–7.
46. Tybulewicz, V. L. J., C. E. Crawford, P. K. Jackson, R. T. Bronson, and R. C.Mulligan. 1991. Neonatal lethality and lymphopenia in mice with a homozy-gous disruption of the c-abl proto-oncogene. Cell 65:1153–1163.
1344 ZHANG ET AL. MOL. CELL. BIOL.
on Decem
ber 1, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from




















