
Analysis of nucleotide diversity among alleles of the major bacterial blight resistance gene Xa27 in...
13
ORIGINAL ARTICLE Analysis of nucleotide diversity among alleles of the major bacterial blight resistance gene Xa27 in cultivars of rice (Oryza sativa) and its wild relatives Waikhom Bimolata • Anirudh Kumar • Raman Meenakshi Sundaram • Gouri Shankar Laha • Insaf Ahmed Qureshi • Gajjala Ashok Reddy • Irfan Ahmad Ghazi Received: 29 January 2013 / Accepted: 23 April 2013 / Published online: 8 May 2013 Ó Springer-Verlag Berlin Heidelberg 2013 Abstract Xa27 is one of the important R-genes, effective against bacterial blight disease of rice caused by Xantho- monas oryzae pv. oryzae (Xoo). Using natural population of Oryza, we analyzed the sequence variation in the functionally important domains of Xa27 across the Oryza species. DNA sequences of Xa27 alleles from 27 rice accessions revealed higher nucleotide diversity among the reported R-genes of rice. Sequence polymorphism analysis revealed synonymous and non-synonymous mutations in addition to a number of InDels in non-coding regions of the gene. High sequence variation was observed in the promoter region including the 5 0 UTR with ‘p’ value 0.00916 and ‘h w ’ = 0.01785. Comparative analysis of the identified Xa27 alleles with that of IRBB27 and IR24 indicated the operation of both positive selection (Ka/Ks [ 1) and neutral selection (Ka/Ks & 0). The genetic distances of alleles of the gene from Oryza nivara were nearer to IRBB27 as compared to IR24. We also found the presence of conserved and null UPT (upregulated by transcriptional activator) box in the isolated alleles. Considerable amino acid polymorphism was localized in the trans-membrane domain for which the functional sig- nificance is yet to be elucidated. However, the absence of functional UPT box in all the alleles except IRBB27 sug- gests the maintenance of single resistant allele throughout the natural population. Keywords Allele mining Allelic diversity Bacterial blight Synonymous mutation Nonsynonymous mutation Trans-membrane domain Abbreviations BB Bacterial blight NBS-LRR Nucleotide binding site leucine rich repeats RLL Relative lesion length TSS Transcription start site UTR Untranslated region UPT Upregulated by transcriptional activator NJ Neighbor joining Introduction Plants are constantly subjected to attacks from various biotic stresses such as different diseases. Both the plants and the pathogens have been co-evolving in response to Electronic supplementary material The online version of this article (doi:10.1007/s00425-013-1891-3) contains supplementary material, which is available to authorized users. W. Bimolata A. Kumar I. A. Ghazi (&) Department of Plant Sciences, School of Life Sciences, University of Hyderabad, Prof. C. R. Rao Road, Gachibowli, Hyderabad 500046, India e-mail: [email protected] W. Bimolata e-mail: [email protected] R. M. Sundaram G. A. Reddy Crop Improvement Section, Directorate of Rice Research, Rajendranagar, Hyderabad 500030, India e-mail: [email protected] G. S. Laha Crop Protection Section, Directorate of Rice Research, Rajendranagar, Hyderabad 500030, India e-mail: [email protected] I. A. Qureshi Department of Biotechnology, School of Life Sciences, University of Hyderabad, Prof. C. R. Rao Road, Gachibowli, Hyderabad 500046, India e-mail: [email protected] 123 Planta (2013) 238:293–305 DOI 10.1007/s00425-013-1891-3
Transcript of Analysis of nucleotide diversity among alleles of the major bacterial blight resistance gene Xa27 in...

ORIGINAL ARTICLE
Analysis of nucleotide diversity among alleles of the majorbacterial blight resistance gene Xa27 in cultivars of rice(Oryza sativa) and its wild relatives
Waikhom Bimolata • Anirudh Kumar • Raman Meenakshi Sundaram •
Gouri Shankar Laha • Insaf Ahmed Qureshi • Gajjala Ashok Reddy •
Irfan Ahmad Ghazi
Received: 29 January 2013 / Accepted: 23 April 2013 / Published online: 8 May 2013
� Springer-Verlag Berlin Heidelberg 2013
Abstract Xa27 is one of the important R-genes, effective
against bacterial blight disease of rice caused by Xantho-
monas oryzae pv. oryzae (Xoo). Using natural population
of Oryza, we analyzed the sequence variation in the
functionally important domains of Xa27 across the Oryza
species. DNA sequences of Xa27 alleles from 27 rice
accessions revealed higher nucleotide diversity among the
reported R-genes of rice. Sequence polymorphism analysis
revealed synonymous and non-synonymous mutations in
addition to a number of InDels in non-coding regions of the
gene. High sequence variation was observed in the
promoter region including the 50UTR with ‘p’ value
0.00916 and ‘hw’ = 0.01785. Comparative analysis of
the identified Xa27 alleles with that of IRBB27 and
IR24 indicated the operation of both positive selection
(Ka/Ks [ 1) and neutral selection (Ka/Ks & 0). The
genetic distances of alleles of the gene from Oryza nivara
were nearer to IRBB27 as compared to IR24. We also
found the presence of conserved and null UPT (upregulated
by transcriptional activator) box in the isolated alleles.
Considerable amino acid polymorphism was localized in
the trans-membrane domain for which the functional sig-
nificance is yet to be elucidated. However, the absence of
functional UPT box in all the alleles except IRBB27 sug-
gests the maintenance of single resistant allele throughout
the natural population.
Keywords Allele mining � Allelic diversity � Bacterial
blight � Synonymous mutation � Nonsynonymous mutation �Trans-membrane domain
Abbreviations
BB Bacterial blight
NBS-LRR Nucleotide binding site leucine rich repeats
RLL Relative lesion length
TSS Transcription start site
UTR Untranslated region
UPT Upregulated by transcriptional activator
NJ Neighbor joining
Introduction
Plants are constantly subjected to attacks from various
biotic stresses such as different diseases. Both the plants
and the pathogens have been co-evolving in response to
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00425-013-1891-3) contains supplementarymaterial, which is available to authorized users.
W. Bimolata � A. Kumar � I. A. Ghazi (&)
Department of Plant Sciences, School of Life Sciences,
University of Hyderabad, Prof. C. R. Rao Road,
Gachibowli, Hyderabad 500046, India
e-mail: [email protected]
W. Bimolata
e-mail: [email protected]
R. M. Sundaram � G. A. Reddy
Crop Improvement Section, Directorate of Rice Research,
Rajendranagar, Hyderabad 500030, India
e-mail: [email protected]
G. S. Laha
Crop Protection Section, Directorate of Rice Research,
Rajendranagar, Hyderabad 500030, India
e-mail: [email protected]
I. A. Qureshi
Department of Biotechnology, School of Life Sciences,
University of Hyderabad, Prof. C. R. Rao Road,
Gachibowli, Hyderabad 500046, India
e-mail: [email protected]
123
Planta (2013) 238:293–305
DOI 10.1007/s00425-013-1891-3

each other’s modulation. Resistance (R)-genes play a major
role in the first line of defense against pathogens. R-genes
interact with their cognate avirulent (avr) genes encoded by
the pathogens and mediate host plant resistance. This
interaction between the host and its pathogen is believed to
play a major role in evolution of genetic variations in the
R-genes of plants and virulence/avirulence genes of
pathogens. The loss of R-gene mediated resistance is
attributed to mutation in effector gene leading to genetic
changes during the course of evolution which alter the
effector’s structure and function. An in-depth analysis of
molecular evolution and maintenance mechanism of
resistance and susceptibility alleles of R-gene may unravel
the role of pathogen-imposed selection of R-genes (Tiffin
and Moeller 2006).
In rice, the molecular evolution and genetic diversity of
genes related to disease response have been widely studied
for the major blast resistance gene, Pi-ta (Huang et al.
2008; Yoshida et al. 2009; Lee et al. 2011). Other R-genes
which have been extensively studied in the context of
sequence diversity are ‘Pm3’ locus in wheat conferring
resistance against powdery mildew (Bhullar et al. 2009),
Rpm1 (Stahl et al. 1999), RPS2 (Caicedo et al. 1999) and
RPP13 (Rose et al. 2004) in Arabidopsis, Cf2 in tomato
(Caicedo et al. 2004) and RGC2 in lettuce (Kuang et al.
2004). Similar studies have been done for disease response
genes in higher plants such as loblolly pine (Ersoz et al.
2010); however, till date, genetic diversity analysis of any
of the rice bacterial blight (BB) resistance genes has not
been reported. Bacterial blight caused by different races of
the bacterium Xanthomonas oryzae pv. oryzae (Xoo) is one
of the oldest known and ravaging diseases of rice. This
disease hampers rice production in Asia up to 60 % (Re-
issig 1986) and is predominantly rampant in humid tropical
and temperate regions. Till date, more than 30 BB resis-
tance genes have been identified and six of these have been
cloned and functionally characterized (Chun et al. 2012).
The existence of natural variation for a particular gene
provides the platform to identify multiple allelic variations
contributing different phenotypic patterns of resistance
(Iyer-Pascuzzi et al. 2007).
Xa27, a major BB R-gene identified from Oryza minuta
(BBCC), is one of the six BB resistance genes, which have
been cloned. Its function and mechanism of action has been
well characterized (Gu et al. 2005, 2009; Romer et al.
2009). It is a dosage-dependent, differentially expressed
R-gene which confers resistance against Xoo PXO99A. It
also shows broad-spectrum resistance against diverse
strains of Xoo obtained from different Asian countries
including two Indian strains A3842 and A3857 (Gu et al.
2004). Plants with Xa27 gene show resistance only against
incompatible strains of Xoo, harboring the avirulence gene
avrXa27. Xa27 is an intronless gene encoding a protein of
113 amino acids (Gu et al. 2005). Its recessive allele xa27
from IR24 also codes for the same protein without any
change in the protein sequence. The main polymorphic
regions between these two alleles are localized upstream of
transcription start site (TSS) and TATA box. There is a
deletion of three nucleotides ‘AGA’ at -51 position in the
promoter of the recessive allele of the gene (i.e., xa27, Gu
et al. 2005). Unlike other well-known R-genes, Xa27 does
not code any known conserved domains like NBS-LRR or
LRR receptor kinase and has been predicted to encode a
protein with a trans-membrane domain (Gu et al. 2005).
With the availability of Xa27 sequence in public domain,
allelic diversity analysis of wild relatives and landraces
may help in unlocking new and wide spectrum resistant
alleles. Analysis of DNA polymorphism for Xa27 in Indian
accessions of two major wild species of rice, O. nivara and
O. sativa may offer a prospect to divulge the process of
fixation of alleles and haplotypes responsible for adaptive
variation during the course of evolution. It will also reveal
the level to which the genes and other regulatory systems
are conserved across the species (Koornneef et al. 2004).
In the present study, we analyzed inter and intraspecific
polymorphism in Xa27 locus of Asian cultivated rice O.
sativa and its ancestor O. nivara from 27 different wild rice
accessions, introgression lines and landraces. The major
objectives of this study were: (1) to determine if the Xa27
alleles of O. sativa and its wild relative share identity; (2)
to analyze the level of polymorphism of Xa27 at the
sequence level and explain its molecular evolution; and (3)
to analyze the possibility of selection at Xa27 locus.
Materials and methods
Plant materials and growth conditions
A total of 27 accessions of different Oryza species were
analyzed for sequence diversity of Xa27 locus (Online
Resource 1). IR24, PB1 and TN1 were also used as disease-
sensitive control. The germplasms were obtained from
Directorate of Rice Research, Hyderabad and NBPGR,
New Delhi (India). Rice seeds were germinated in dark at
28 �C in germination box layered with pre-soaked germi-
nation paper. After a week, the germinated seeds were
shifted to 10-cm diameter pots containing soil. The seed
beds were uniformly watered and fertilized with a half-
strength Hoagland nutrient solution. After 30 days, healthy
seedlings were transplanted to 20-l earthen pots. Five pots
were kept for each genotype with three plants in each pot
filled with a mixture of clay and peat (1:1, v/v). The wild
accessions were propagated as tillers.
294 Planta (2013) 238:293–305
123

Bacterial strains, plant inoculation and disease
phenotyping
After 45 days of transplantation, each accession was
challenged with five Indian virulent isolates of Xoo. The
isolates used for screening were DX011 (Pantnagar, Utta-
rakhand), DX127 (Cuttack, Orissa), DX020 (Hyderabad,
Andhra Pradesh), DX015 (Aduthurai, Tamil Nadu) and
DX133 (Raipur, Chhattisgarh). These strains were obtained
from Department of Plant Pathology, DRR, Hyderabad,
and cultured on modified Wakimoto’s medium at 28 �C for
72 h. The bacteria were then scraped and suspended in
sterile distilled water and the concentration was adjusted to
0.1–0.2 OD (1 9 108–1 9 109 CFU/ml). Using this bac-
terial suspension, 5–6 uppermost leaves of plants at the
booting stage were inoculated following leaf clipping
method (Kauffman et al. 1973). Three plants were used for
each isolate. The control plants were clipped with scissors
dipped in sterile water. After 15 days of inoculation, lesion
lengths were measured and the ratios of lesion length to
leaf length (RLL) were calculated. Scoring of disease was
done following standard evaluation system for rice (SES
scale; IRRI 2002). RLLs\25 % were scored as resistant or
tolerant genotypes and [25 % were scored as susceptible.
During the process of screening for bacterial blight resis-
tance, seedlings were uniformly irrigated and fertilized
using Hoagland nutrient solution.
DNA isolation, gene amplification and sequencing
Genomic DNA was isolated from young leaves following
CTAB method (Doyle and Doyle 1990) with minor mod-
ifications (Rajendrakumar et al. 2007). The entire Xa27
gene including the promoter and 30UTRs were amplified
from 27 genotypes using overlapping gene-specific primers
designed from GenBank accession AY986492 (Fig. 1).
Genotypes which were either highly resistant or moder-
ately resistant to most of the strains were selected along
with few susceptible ones. Whole gene amplification was
performed using high-fidelity jump start AccuTaq poly-
merase. PCR was carried out in 30 lL reaction volume
containing 50 ng of genomic DNA, 0.3 lM of each primer,
0.2 mM of each dNTPs, 109 PCR buffer (10 mM Tris–
HCl, pH 8.0, 2.5 mM MgCl2) and 0.75 U of AccuTaq
DNA polymerase (Sigma-Aldrich). The PCR profile con-
sisting of 3 min initial denaturation at 96 �C, 30 cycles of
amplification with 30 s DNA denaturation at 94 �C, 45 s
annealing at 60 �C and 2 min 15 s elongation at 68 �C was
adopted. Subsequently, the amplified products were visu-
alized on 1 % agarose gels. The PCR products were then
purified using gel extraction kit (Sigma-Aldrich), cloned
into pTZ57R/T (Fermentas) and sequenced. The sequenced
alleles were deposited in NCBI GenBank.
Allelic diversity and sequence data analysis
Based on the sequences obtained from the genotypes under
study, the coding regions and the UTR regions of the new
alleles were predicted through the software utility, FGE-
NESH (http://www.softberry.com) (Salamov and Solovyer
2000). Xa27 CDS of IRBB27 (GenBank accession
AY986492) and its recessive allele from IR24 (GenBank
accession AY986491) were used as reference for compar-
ative analysis. The percentage identity of each alleles and
their deduced protein sequence with that of Xa27 (IRBB27)
and xa27 (IR24) were determined by pairwise alignment.
Pairwise alignment was performed in BLASTN and
BLASTP (Altschul et al. 1990), respectively. Intraspecific
and interspecific polymorphisms were analyzed using
DnaSP program version 5.0 (Librado and Rozas 2009).
Sequences were assembled and subjected to multiple
alignments using ClustalX version 1.81 (Thompson et al.
1997) and Multalin version 5.4.1 (Corpet 1988). The
aligned file was used as an input for analysis in DnaSP
(http://www.ub.edu/dnasp). The numbers of polymorphic
sites, including SNPs, InDel polymorphism, synonymous
and non-synonymous substitutions in coding and non-
coding region (Nei and Gojobori 1986) were determined
according to DnaSP. All the analysis was done in sliding
window for 50 bp per window. Alignment gaps were
excluded from comparative analysis. Nucleotide diversity
was analyzed by estimating p (average number of nucle-
otide diversity per site Nei and Li 1979); and hw, (number
of segregating sites, Watterson 1975). Initially, each allele
was compared to Xa27 (IRBB27) and xa27 (IR24),
respectively. Later on, overall comparative analysis for all
the sequenced alleles was performed. Different test selec-
tion analysis such as Tajima’s D test (Tajima 1989) and Fu
Fig. 1 A schematic presentation of Xa27 gene. Overlapping primers were designed covering the entire gene. The arrows represent the regions
and the orientations of the primers designed
Planta (2013) 238:293–305 295
123

and Li’s D test (Fu and Li 1993) were conducted separately
for O. nivara and O. sativa accessions using DnaSP pro-
gram to determine departures from neutrality. A dendro-
gram predicting the evolutionary relationship among 26
alleles was generated by developing bootstrapped unrooted
linear neighbour joining (NJ) tree in MEGA 4 (Tamura
et al. 2007). We also used maximum parsimony and
maximum likelihood methods to construct the tree; how-
ever, there was not much difference found from NJ plot.
The robustness of the tree was determined by taking
bootstrap value up to 100,000. To analyze the level of
diversity in regulatory region, the alleles were divided into
three parts; promoter with 50UTR, 30UTR and coding
region. Sequence diversity analysis was also done sepa-
rately for O. nivara and O. sativa accessions. The presence
of trans-membrane helices in the coded protein were pre-
dicted using TOPCONS (Bernsel et al. 2009) and signal
peptide prediction was done with the help of SignalP 4.0
(Petersen et al. 2011). SIFT analysis was done for the
deduced protein sequence to determine the deleterious
nature of the mutation (Kumar et al. 2009).
Results
Phenotypic evaluation
Out of the 27 accessions selected for allelic diversity
analysis, 10 O. nivara accessions were resistant to all the
five strains and three of them were susceptible to either one
of the strain; both O. alta and O. officinalis were resistant
to all the five strains, whereas among the O. sativa acces-
sions TN1, IR24 and IR20 were susceptible to all the five
Xoo strains and remaining accessions were resistant or
susceptible to one or the other strain (Online resource 1).
Gene amplification and sequencing
The sequence encompassing the entire Xa27 gene was
amplified from 27 rice accessions using overlapping gene-
specific primers. Sequencing was done for all the cloned
alleles. The size of the sequenced alleles ranged from 2,056
to 2,733 bp in length. Overall sequence similarity of the
entire gene ranged from 95 to 99 %. The deduced proteins
of Xa27 alleles from WR132, WR11, WR14, WR82,
WR110 and ON15 (O. nivara); PAU933 and R9 (O. sativa)
coded for 112 amino acids while the remaining alleles
encoded proteins of 113 amino acids, with few differences
in the protein sequence (Tables 1, 2). In WR14, WR82,
WR110, ON15, PAU933, R9 and WR132, three basepairs
(TAC) deletion (Online Resource 2) was observed in the
coding region which lead to deletion of threonine
(T) (Fig. 2a). However, deletion of nucleotide ‘G’ in
WR11 at 90th position (Online Resource 2) resulted in a
frameshift mutation as shown in Fig. 2a. All the sequences
were deposited in NCBI GenBank database and the
following accession numbers JF304301–JF304303,
HQ888852–HQ888857, JN016505–JN016521 and JN601064
were obtained.
Polymorphism of Xa27 alleles
The alleles from 29 genotypes including IR24 and IRBB27
were aligned. The total alignment length ranged from 2,056
to 2,780 bp, including sites with alignment gaps, promoter,
50 and 30UTR. Comparative analysis in DnaSP, excluding
sites with gaps, showed 103 polymorphic sites (in total
1,999 available sites), 74 singleton variable sites, 40 par-
simony informative site and 32 InDel events ranging from
1 to 50 bp in length. The average number of nucleotide
difference, ‘K’ of the entire gene was estimated to be
15.892. The genetic diversity, ‘p’ of the tested gene was
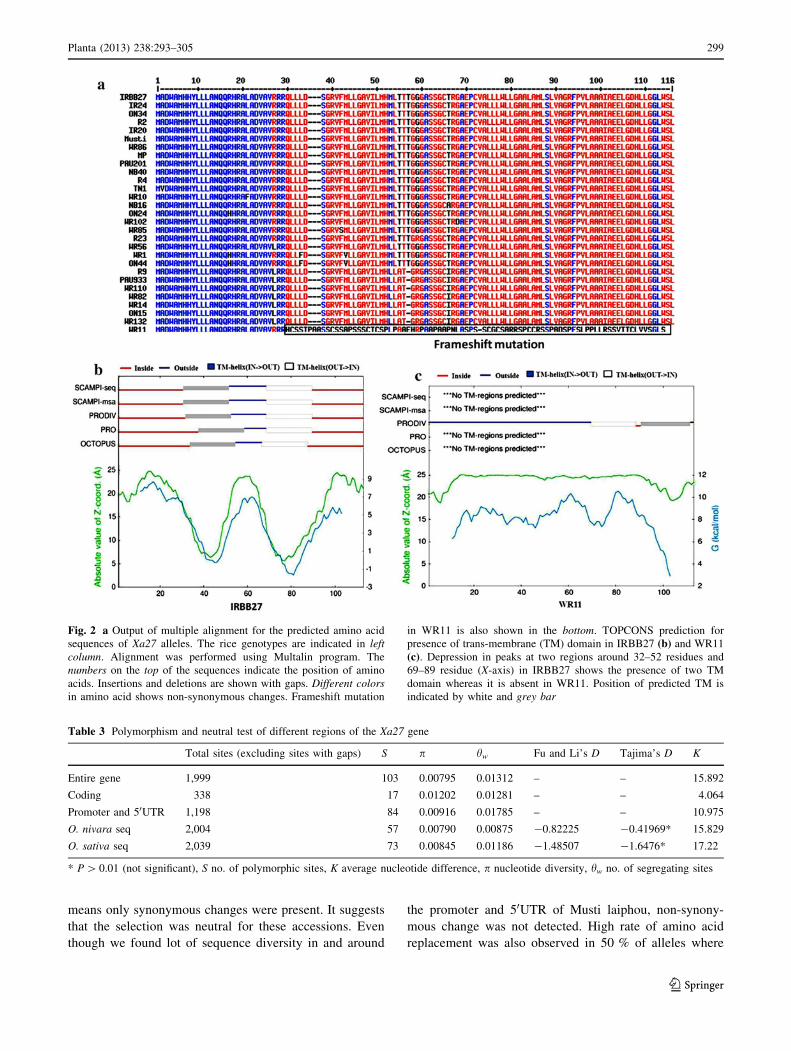
0.00795 and ‘hw’ equal to 0.01312 (Table 3). Among all
accessions, Mustilaiphou was found to be highly diverse
from IRBB27 (p = 0.02291) than IR24 (p = 0.0163)
(Tables 4, 5). The highest sequence variability was
observed in the 50 flanking region of the alleles with ‘p’
value 0.00916 and least variability in 30UTR. The test of
neutrality failed to give significant Tajima’s and Fu and
Li’s D (P [ 0.01) in O. nivara and O. sativa species, even
though hw value was greater than p resulting in negative
Tajima’s D (Table 3). InDel polymorphism was also
detected predominantly in promoter region with few cases
in exonic part. Some of the InDel sites were consistent
between the species. Fixed differences of nucleotide pat-
tern were detected among O. sativa and O. nivara alleles
with few cases of variation within the species too. Intra-
specific polymorphism was lower in O. nivara
(p = 0.00790) than O. sativa (p = 0.00845) (Table 3).
Deletion of &400 bp from the upstream region of Xa27
alleles was noticed in WR132, WR56, WR82, PAU933,
ON15, and WR110. Details of InDel polymorphisms and
SNPs detected in comparison to IR24 and IRBB27 was
summarized in Tables 1 and 2. The level of amino acid
polymorphism was highest in O. nivara. Nine alleles from
species O. nivara had single amino acid replacement,
whereas WR56 (two changes), WR1 and ON44 (three
changes) had more than one. Single amino acid replace-
ment was seen only in three accessions of O. sativa
(Fig. 2a). Both conservative and non-conservative amino
acid polymorphisms were present in the alleles, out of
which, two conservative and three non-conservative amino
acid polymorphism lie in the first trans-membrane domain;
however, these mutations did not make any major changes
in the trans-membrane structure. SIFT analysis also
showed that the amino acid mutations in the alleles were
296 Planta (2013) 238:293–305
123
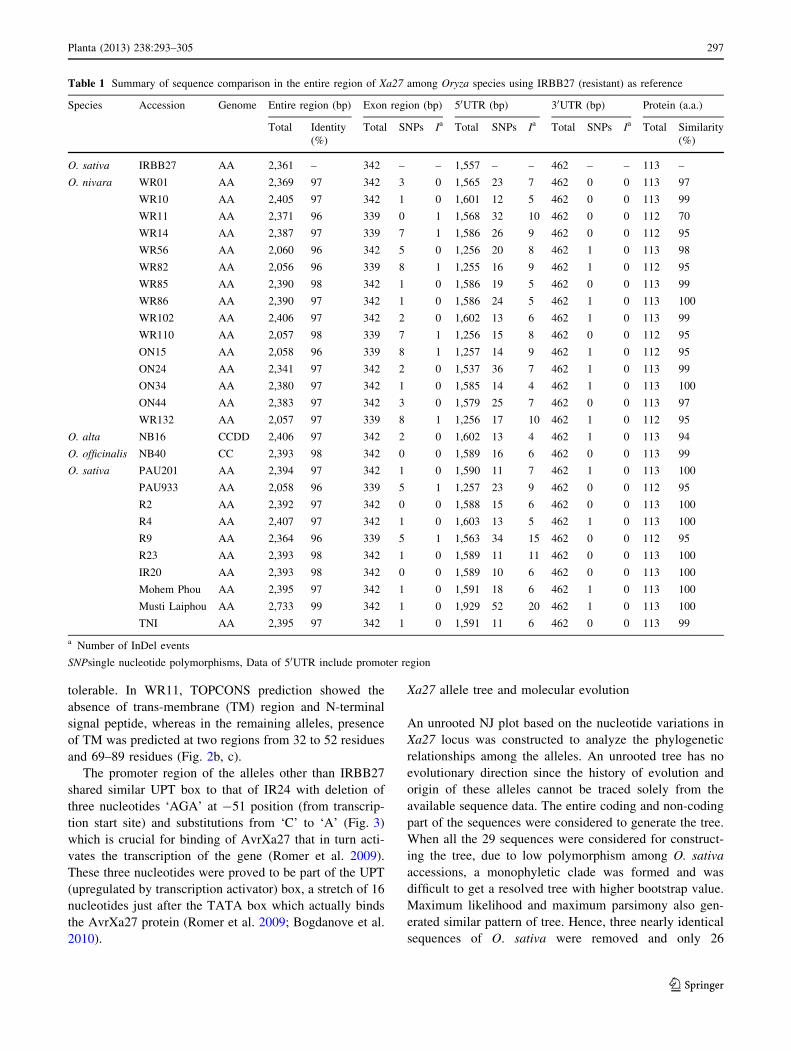
tolerable. In WR11, TOPCONS prediction showed the
absence of trans-membrane (TM) region and N-terminal
signal peptide, whereas in the remaining alleles, presence
of TM was predicted at two regions from 32 to 52 residues
and 69–89 residues (Fig. 2b, c).
The promoter region of the alleles other than IRBB27
shared similar UPT box to that of IR24 with deletion of
three nucleotides ‘AGA’ at -51 position (from transcrip-
tion start site) and substitutions from ‘C’ to ‘A’ (Fig. 3)
which is crucial for binding of AvrXa27 that in turn acti-
vates the transcription of the gene (Romer et al. 2009).
These three nucleotides were proved to be part of the UPT
(upregulated by transcription activator) box, a stretch of 16
nucleotides just after the TATA box which actually binds
the AvrXa27 protein (Romer et al. 2009; Bogdanove et al.
2010).
Xa27 allele tree and molecular evolution
An unrooted NJ plot based on the nucleotide variations in
Xa27 locus was constructed to analyze the phylogenetic
relationships among the alleles. An unrooted tree has no
evolutionary direction since the history of evolution and
origin of these alleles cannot be traced solely from the
available sequence data. The entire coding and non-coding
part of the sequences were considered to generate the tree.
When all the 29 sequences were considered for construct-
ing the tree, due to low polymorphism among O. sativa
accessions, a monophyletic clade was formed and was
difficult to get a resolved tree with higher bootstrap value.
Maximum likelihood and maximum parsimony also gen-
erated similar pattern of tree. Hence, three nearly identical
sequences of O. sativa were removed and only 26
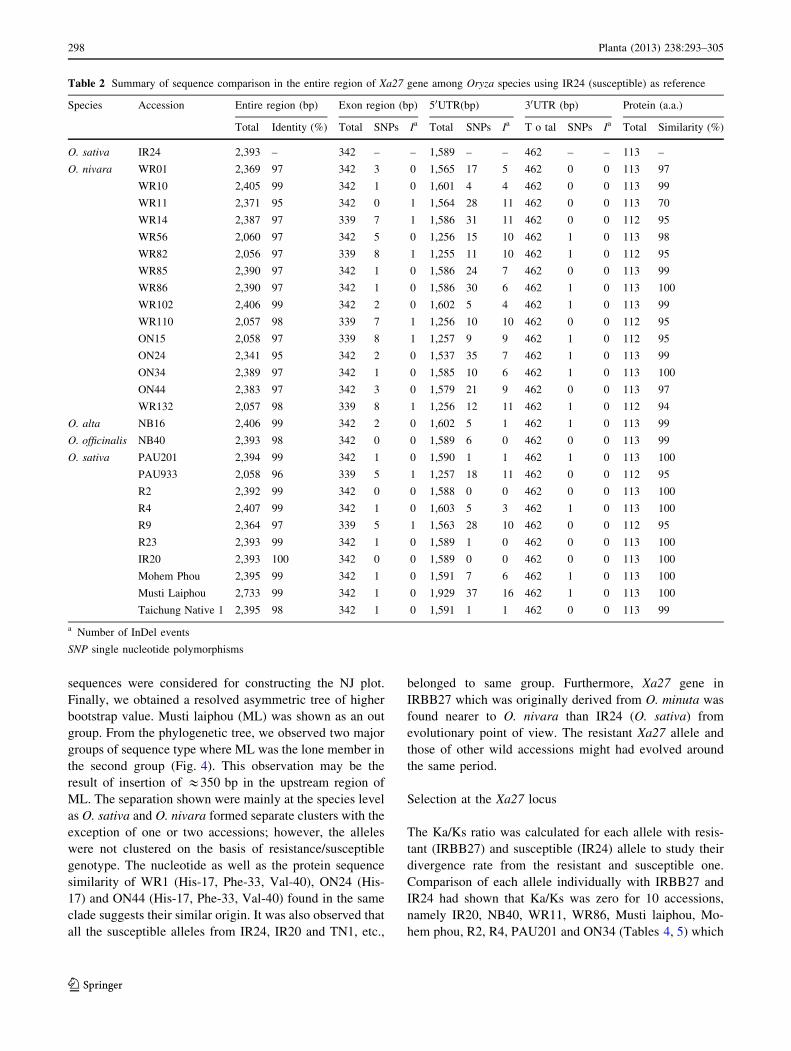
Table 1 Summary of sequence comparison in the entire region of Xa27 among Oryza species using IRBB27 (resistant) as reference
Species Accession Genome Entire region (bp) Exon region (bp) 50UTR (bp) 30UTR (bp) Protein (a.a.)
Total Identity
(%)
Total SNPs Ia Total SNPs Ia Total SNPs Ia Total Similarity
(%)
O. sativa IRBB27 AA 2,361 – 342 – – 1,557 – – 462 – – 113 –
O. nivara WR01 AA 2,369 97 342 3 0 1,565 23 7 462 0 0 113 97
WR10 AA 2,405 97 342 1 0 1,601 12 5 462 0 0 113 99
WR11 AA 2,371 96 339 0 1 1,568 32 10 462 0 0 112 70
WR14 AA 2,387 97 339 7 1 1,586 26 9 462 0 0 112 95
WR56 AA 2,060 96 342 5 0 1,256 20 8 462 1 0 113 98
WR82 AA 2,056 96 339 8 1 1,255 16 9 462 1 0 112 95
WR85 AA 2,390 98 342 1 0 1,586 19 5 462 0 0 113 99
WR86 AA 2,390 97 342 1 0 1,586 24 5 462 1 0 113 100
WR102 AA 2,406 97 342 2 0 1,602 13 6 462 1 0 113 99
WR110 AA 2,057 98 339 7 1 1,256 15 8 462 0 0 112 95
ON15 AA 2,058 96 339 8 1 1,257 14 9 462 1 0 112 95
ON24 AA 2,341 97 342 2 0 1,537 36 7 462 1 0 113 99
ON34 AA 2,380 97 342 1 0 1,585 14 4 462 1 0 113 100
ON44 AA 2,383 97 342 3 0 1,579 25 7 462 0 0 113 97
WR132 AA 2,057 97 339 8 1 1,256 17 10 462 1 0 112 95
O. alta NB16 CCDD 2,406 97 342 2 0 1,602 13 4 462 1 0 113 94
O. officinalis NB40 CC 2,393 98 342 0 0 1,589 16 6 462 0 0 113 99
O. sativa PAU201 AA 2,394 97 342 1 0 1,590 11 7 462 1 0 113 100
PAU933 AA 2,058 96 339 5 1 1,257 23 9 462 0 0 112 95
R2 AA 2,392 97 342 0 0 1,588 15 6 462 0 0 113 100
R4 AA 2,407 97 342 1 0 1,603 13 5 462 1 0 113 100
R9 AA 2,364 96 339 5 1 1,563 34 15 462 0 0 112 95
R23 AA 2,393 98 342 1 0 1,589 11 11 462 0 0 113 100
IR20 AA 2,393 98 342 0 0 1,589 10 6 462 0 0 113 100
Mohem Phou AA 2,395 97 342 1 0 1,591 18 6 462 1 0 113 100
Musti Laiphou AA 2,733 99 342 1 0 1,929 52 20 462 1 0 113 100
TNI AA 2,395 97 342 1 0 1,591 11 6 462 0 0 113 99
a Number of InDel events
SNPsingle nucleotide polymorphisms, Data of 50UTR include promoter region
Planta (2013) 238:293–305 297
123
sequences were considered for constructing the NJ plot.
Finally, we obtained a resolved asymmetric tree of higher
bootstrap value. Musti laiphou (ML) was shown as an out
group. From the phylogenetic tree, we observed two major
groups of sequence type where ML was the lone member in
the second group (Fig. 4). This observation may be the
result of insertion of &350 bp in the upstream region of
ML. The separation shown were mainly at the species level
as O. sativa and O. nivara formed separate clusters with the
exception of one or two accessions; however, the alleles
were not clustered on the basis of resistance/susceptible
genotype. The nucleotide as well as the protein sequence
similarity of WR1 (His-17, Phe-33, Val-40), ON24 (His-
17) and ON44 (His-17, Phe-33, Val-40) found in the same
clade suggests their similar origin. It was also observed that
all the susceptible alleles from IR24, IR20 and TN1, etc.,
belonged to same group. Furthermore, Xa27 gene in
IRBB27 which was originally derived from O. minuta was
found nearer to O. nivara than IR24 (O. sativa) from
evolutionary point of view. The resistant Xa27 allele and
those of other wild accessions might had evolved around
the same period.
Selection at the Xa27 locus
The Ka/Ks ratio was calculated for each allele with resis-
tant (IRBB27) and susceptible (IR24) allele to study their
divergence rate from the resistant and susceptible one.
Comparison of each allele individually with IRBB27 and
IR24 had shown that Ka/Ks was zero for 10 accessions,
namely IR20, NB40, WR11, WR86, Musti laiphou, Mo-
hem phou, R2, R4, PAU201 and ON34 (Tables 4, 5) which
Table 2 Summary of sequence comparison in the entire region of Xa27 gene among Oryza species using IR24 (susceptible) as reference
Species Accession Entire region (bp) Exon region (bp) 50UTR(bp) 30UTR (bp) Protein (a.a.)
Total Identity (%) Total SNPs Ia Total SNPs Ia T o tal SNPs Ia Total Similarity (%)
O. sativa IR24 2,393 – 342 – – 1,589 – – 462 – – 113 –
O. nivara WR01 2,369 97 342 3 0 1,565 17 5 462 0 0 113 97
WR10 2,405 99 342 1 0 1,601 4 4 462 0 0 113 99
WR11 2,371 95 342 0 1 1,564 28 11 462 0 0 113 70
WR14 2,387 97 339 7 1 1,586 31 11 462 0 0 112 95
WR56 2,060 97 342 5 0 1,256 15 10 462 1 0 113 98
WR82 2,056 97 339 8 1 1,255 11 10 462 1 0 112 95
WR85 2,390 97 342 1 0 1,586 24 7 462 0 0 113 99
WR86 2,390 97 342 1 0 1,586 30 6 462 1 0 113 100
WR102 2,406 99 342 2 0 1,602 5 4 462 1 0 113 99
WR110 2,057 98 339 7 1 1,256 10 10 462 0 0 112 95
ON15 2,058 97 339 8 1 1,257 9 9 462 1 0 112 95
ON24 2,341 95 342 2 0 1,537 35 7 462 1 0 113 99
ON34 2,389 97 342 1 0 1,585 10 6 462 1 0 113 100
ON44 2,383 97 342 3 0 1,579 21 9 462 0 0 113 97
WR132 2,057 98 339 8 1 1,256 12 11 462 1 0 112 94
O. alta NB16 2,406 99 342 2 0 1,602 5 1 462 1 0 113 99
O. officinalis NB40 2,393 98 342 0 0 1,589 6 0 462 0 0 113 99
O. sativa PAU201 2,394 99 342 1 0 1,590 1 1 462 1 0 113 100
PAU933 2,058 96 339 5 1 1,257 18 11 462 0 0 112 95
R2 2,392 99 342 0 0 1,588 0 0 462 0 0 113 100
R4 2,407 99 342 1 0 1,603 5 3 462 1 0 113 100
R9 2,364 97 339 5 1 1,563 28 10 462 0 0 112 95
R23 2,393 99 342 1 0 1,589 1 0 462 0 0 113 100
IR20 2,393 100 342 0 0 1,589 0 0 462 0 0 113 100
Mohem Phou 2,395 99 342 1 0 1,591 7 6 462 1 0 113 100
Musti Laiphou 2,733 99 342 1 0 1,929 37 16 462 1 0 113 100
Taichung Native 1 2,395 98 342 1 0 1,591 1 1 462 0 0 113 99
a Number of InDel events
SNP single nucleotide polymorphisms
298 Planta (2013) 238:293–305
123
means only synonymous changes were present. It suggests
that the selection was neutral for these accessions. Even
though we found lot of sequence diversity in and around
the promoter and 50UTR of Musti laiphou, non-synony-
mous change was not detected. High rate of amino acid
replacement was also observed in 50 % of alleles where
Fig. 2 a Output of multiple alignment for the predicted amino acid
sequences of Xa27 alleles. The rice genotypes are indicated in left
column. Alignment was performed using Multalin program. The
numbers on the top of the sequences indicate the position of amino
acids. Insertions and deletions are shown with gaps. Different colors
in amino acid shows non-synonymous changes. Frameshift mutation
in WR11 is also shown in the bottom. TOPCONS prediction for
presence of trans-membrane (TM) domain in IRBB27 (b) and WR11
(c). Depression in peaks at two regions around 32–52 residues and
69–89 residue (X-axis) in IRBB27 shows the presence of two TM
domain whereas it is absent in WR11. Position of predicted TM is
indicated by white and grey bar
Table 3 Polymorphism and neutral test of different regions of the Xa27 gene
Total sites (excluding sites with gaps) S p hw Fu and Li’s D Tajima’s D K
Entire gene 1,999 103 0.00795 0.01312 – – 15.892
Coding 338 17 0.01202 0.01281 – – 4.064
Promoter and 50UTR 1,198 84 0.00916 0.01785 – – 10.975
O. nivara seq 2,004 57 0.00790 0.00875 -0.82225 -0.41969* 15.829
O. sativa seq 2,039 73 0.00845 0.01186 -1.48507 -1.6476* 17.22
* P [ 0.01 (not significant), S no. of polymorphic sites, K average nucleotide difference, p nucleotide diversity, hw no. of segregating sites
Planta (2013) 238:293–305 299
123
rate of non-synonymous changes by rate of synonymous
changes (Ka/Ks) was [1. Alleles of TN1 and R23 when
compared to IR24 showed very high Ka/Ks & 8.00, where
the rate of amino acid replacement was occurring at a faster
rate than the synonymous changes which suggest rapid
adaptive evolution (Bergelson et al. 2001). However, when
compared with IRBB27, the rate of amino acid changes
was substantially lower with Ka/Ks = 0.788. Along with
positive selection, we could also see purifying selection in
some of the alleles, ON24, WR85, NB16, WR102 and
WR56 where Ka/Ks \ 1 (Tables 4, 5).
Discussion
Natural variation in the alleles of R-genes due to SNPs,
InDels and insertion or deletions of large DNA fragments
plays an important role in providing phenotypic diversity
or variability with respect to disease resistance. Consid-
ering the existence and emergence of virulent pathotypes
of the bacterial blight pathogen, there is a need to exploit
the potential of allele mining by identification of novel
alleles of R-genes. Study on molecular evolution and
understanding genetic diversity of known R-gene(s) will
also help us in finding key region(s) controlling disease
resistance. Maintenance of allelic diversity at R-gene in
natural population occurs as a result of co-evolution of
host and pathogen (May and Anderson 1983). Pathogens
are believed to play an important role as a selective agent
during the course of R-gene evolution (Kover and Schaal
2002). In the present study, we initially identified a set of
rice genotypes possessing resistance against multiple
virulent strains of Xoo and analyzed the nucleotide
and amino acid sequence diversity with respect to Xa27,
an R-gene involved in gene-for-gene interaction with
AvrXa27.
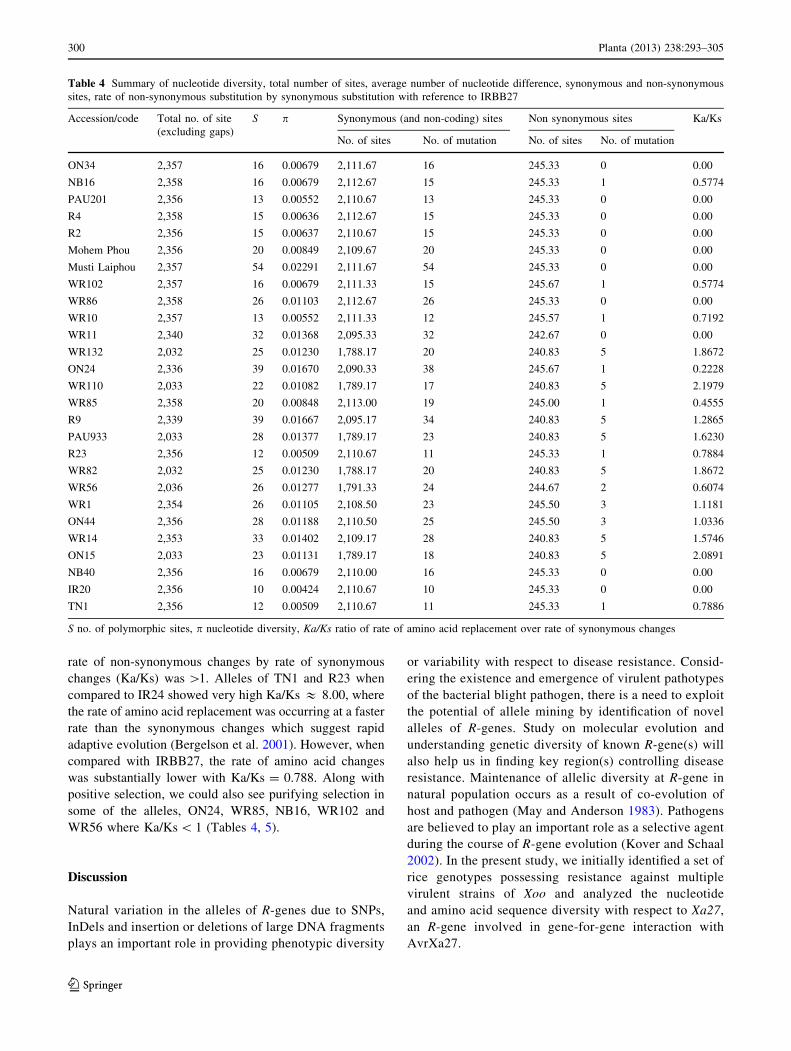
Table 4 Summary of nucleotide diversity, total number of sites, average number of nucleotide difference, synonymous and non-synonymous
sites, rate of non-synonymous substitution by synonymous substitution with reference to IRBB27
Accession/code Total no. of site
(excluding gaps)
S p Synonymous (and non-coding) sites Non synonymous sites Ka/Ks
No. of sites No. of mutation No. of sites No. of mutation
ON34 2,357 16 0.00679 2,111.67 16 245.33 0 0.00
NB16 2,358 16 0.00679 2,112.67 15 245.33 1 0.5774
PAU201 2,356 13 0.00552 2,110.67 13 245.33 0 0.00
R4 2,358 15 0.00636 2,112.67 15 245.33 0 0.00
R2 2,356 15 0.00637 2,110.67 15 245.33 0 0.00
Mohem Phou 2,356 20 0.00849 2,109.67 20 245.33 0 0.00
Musti Laiphou 2,357 54 0.02291 2,111.67 54 245.33 0 0.00
WR102 2,357 16 0.00679 2,111.33 15 245.67 1 0.5774
WR86 2,358 26 0.01103 2,112.67 26 245.33 0 0.00
WR10 2,357 13 0.00552 2,111.33 12 245.57 1 0.7192
WR11 2,340 32 0.01368 2,095.33 32 242.67 0 0.00
WR132 2,032 25 0.01230 1,788.17 20 240.83 5 1.8672
ON24 2,336 39 0.01670 2,090.33 38 245.67 1 0.2228
WR110 2,033 22 0.01082 1,789.17 17 240.83 5 2.1979
WR85 2,358 20 0.00848 2,113.00 19 245.00 1 0.4555
R9 2,339 39 0.01667 2,095.17 34 240.83 5 1.2865
PAU933 2,033 28 0.01377 1,789.17 23 240.83 5 1.6230
R23 2,356 12 0.00509 2,110.67 11 245.33 1 0.7884
WR82 2,032 25 0.01230 1,788.17 20 240.83 5 1.8672
WR56 2,036 26 0.01277 1,791.33 24 244.67 2 0.6074
WR1 2,354 26 0.01105 2,108.50 23 245.50 3 1.1181
ON44 2,356 28 0.01188 2,110.50 25 245.50 3 1.0336
WR14 2,353 33 0.01402 2,109.17 28 240.83 5 1.5746
ON15 2,033 23 0.01131 1,789.17 18 240.83 5 2.0891
NB40 2,356 16 0.00679 2,110.00 16 245.33 0 0.00
IR20 2,356 10 0.00424 2,110.67 10 245.33 0 0.00
TN1 2,356 12 0.00509 2,110.67 11 245.33 1 0.7886
S no. of polymorphic sites, p nucleotide diversity, Ka/Ks ratio of rate of amino acid replacement over rate of synonymous changes
300 Planta (2013) 238:293–305
123
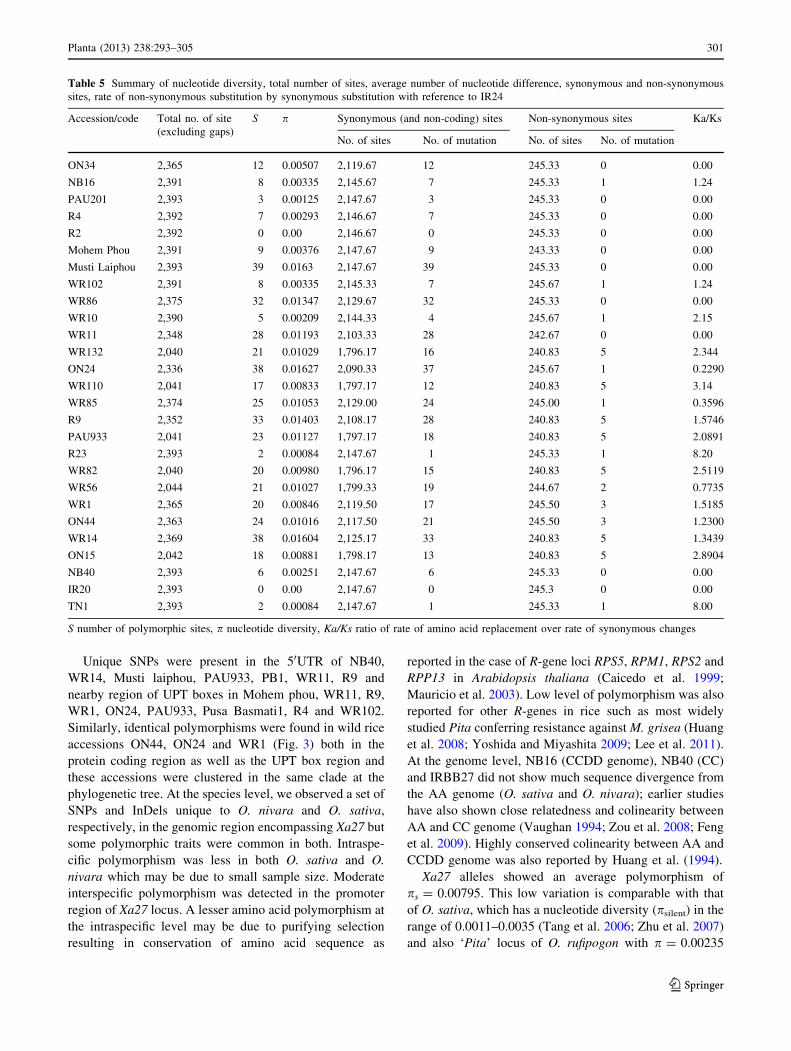
Unique SNPs were present in the 50UTR of NB40,
WR14, Musti laiphou, PAU933, PB1, WR11, R9 and
nearby region of UPT boxes in Mohem phou, WR11, R9,
WR1, ON24, PAU933, Pusa Basmati1, R4 and WR102.
Similarly, identical polymorphisms were found in wild rice
accessions ON44, ON24 and WR1 (Fig. 3) both in the
protein coding region as well as the UPT box region and
these accessions were clustered in the same clade at the
phylogenetic tree. At the species level, we observed a set of
SNPs and InDels unique to O. nivara and O. sativa,
respectively, in the genomic region encompassing Xa27 but
some polymorphic traits were common in both. Intraspe-
cific polymorphism was less in both O. sativa and O.
nivara which may be due to small sample size. Moderate
interspecific polymorphism was detected in the promoter
region of Xa27 locus. A lesser amino acid polymorphism at
the intraspecific level may be due to purifying selection
resulting in conservation of amino acid sequence as
reported in the case of R-gene loci RPS5, RPM1, RPS2 and
RPP13 in Arabidopsis thaliana (Caicedo et al. 1999;
Mauricio et al. 2003). Low level of polymorphism was also
reported for other R-genes in rice such as most widely
studied Pita conferring resistance against M. grisea (Huang
et al. 2008; Yoshida and Miyashita 2009; Lee et al. 2011).
At the genome level, NB16 (CCDD genome), NB40 (CC)
and IRBB27 did not show much sequence divergence from
the AA genome (O. sativa and O. nivara); earlier studies
have also shown close relatedness and colinearity between
AA and CC genome (Vaughan 1994; Zou et al. 2008; Feng
et al. 2009). Highly conserved colinearity between AA and
CCDD genome was also reported by Huang et al. (1994).
Xa27 alleles showed an average polymorphism of
ps = 0.00795. This low variation is comparable with that
of O. sativa, which has a nucleotide diversity (psilent) in the
range of 0.0011–0.0035 (Tang et al. 2006; Zhu et al. 2007)
and also ‘Pita’ locus of O. rufipogon with p = 0.00235
Table 5 Summary of nucleotide diversity, total number of sites, average number of nucleotide difference, synonymous and non-synonymous
sites, rate of non-synonymous substitution by synonymous substitution with reference to IR24
Accession/code Total no. of site
(excluding gaps)
S p Synonymous (and non-coding) sites Non-synonymous sites Ka/Ks
No. of sites No. of mutation No. of sites No. of mutation
ON34 2,365 12 0.00507 2,119.67 12 245.33 0 0.00
NB16 2,391 8 0.00335 2,145.67 7 245.33 1 1.24
PAU201 2,393 3 0.00125 2,147.67 3 245.33 0 0.00
R4 2,392 7 0.00293 2,146.67 7 245.33 0 0.00
R2 2,392 0 0.00 2,146.67 0 245.33 0 0.00
Mohem Phou 2,391 9 0.00376 2,147.67 9 243.33 0 0.00
Musti Laiphou 2,393 39 0.0163 2,147.67 39 245.33 0 0.00
WR102 2,391 8 0.00335 2,145.33 7 245.67 1 1.24
WR86 2,375 32 0.01347 2,129.67 32 245.33 0 0.00
WR10 2,390 5 0.00209 2,144.33 4 245.67 1 2.15
WR11 2,348 28 0.01193 2,103.33 28 242.67 0 0.00
WR132 2,040 21 0.01029 1,796.17 16 240.83 5 2.344
ON24 2,336 38 0.01627 2,090.33 37 245.67 1 0.2290
WR110 2,041 17 0.00833 1,797.17 12 240.83 5 3.14
WR85 2,374 25 0.01053 2,129.00 24 245.00 1 0.3596
R9 2,352 33 0.01403 2,108.17 28 240.83 5 1.5746
PAU933 2,041 23 0.01127 1,797.17 18 240.83 5 2.0891
R23 2,393 2 0.00084 2,147.67 1 245.33 1 8.20
WR82 2,040 20 0.00980 1,796.17 15 240.83 5 2.5119
WR56 2,044 21 0.01027 1,799.33 19 244.67 2 0.7735
WR1 2,365 20 0.00846 2,119.50 17 245.50 3 1.5185
ON44 2,363 24 0.01016 2,117.50 21 245.50 3 1.2300
WR14 2,369 38 0.01604 2,125.17 33 240.83 5 1.3439
ON15 2,042 18 0.00881 1,798.17 13 240.83 5 2.8904
NB40 2,393 6 0.00251 2,147.67 6 245.33 0 0.00
IR20 2,393 0 0.00 2,147.67 0 245.3 0 0.00
TN1 2,393 2 0.00084 2,147.67 1 245.33 1 8.00
S number of polymorphic sites, p nucleotide diversity, Ka/Ks ratio of rate of amino acid replacement over rate of synonymous changes
Planta (2013) 238:293–305 301
123

(Huang et al. 2008). Nucleotide divergence for O. nivara
accessions also were much lower with ‘p’ = 0.0079
(Table 3). Yoshida and Miyashita (2009) also reported
very low diversity at silent site of ‘Pita’, ps = 0.0101.
However, we may get higher allelic variability if we
include more number of species such as O. rufipogon, O.
longistaminata or O. minuta, etc., in the study. We may
also conduct this study in different sets of Oryza species for
better understanding of Xa27 molecular evolution. The
nucleotide polymorphism in 50 non-coding region
(p = 0.00916) was shown to be much higher than that of
the entire region (Table 3). This may be due to the low
polymorphisms of the 30UTR and exclusion of sites with
gaps from the analysis. We found that the genetic diversity
of Xa27 alleles was in and around the range of those
reported defense-related genes. Nucleotide diversity of
other defense-related genes such as Adh3 locus in wild
barley has p value 0.0219 (Lin et al. 2001); Adh loci of
allogamous species depicted p values from 0.00204 to
0.01742 (Cummings and Clegg 1998). In A. thaliana, a
chitinase-encoding gene has p = 0.0104 (Kawabe et al.
1997) and Pto, disease resistance gene of wild tomato
(Lycopersicon) was reported with p = 0.012 (Rose et al.
2007). Bakker et al. (2008) also reported that seven genes
encoding pathogen-related proteins, had ps = 0.00183 and
pa = 0.00126.
The divergence rate ratios for most of the O. nivara
accessions from IRBB27 were high with Ka/Ks [ 1 which
suggest the occurrence of positive selection or adaptive
evolution (Bergelson et al. 2001). Most of the O. sativa
accessions exhibited neutral selection with Ka/Ks = 0,
while few O. nivara and ILs have Ka/Ks \ 1 illustrating
the presence of selection constraint. Even though, WR11
(O. nivara) showed zero divergence rate from IRBB27, at
the translation level, it encoded a non-functional protein
due to frame shift mutation. This miscalculation arose due
to the exclusion of single base pair deletion from the
analysis by the software. We also observed similar pattern
of divergence rate between the susceptible allele xa27
(IR24) and the new alleles. The rate of diversity was very
much higher than those observed between Xa27 (IRBB27)
and the alleles. A strong positive selection was shown in
TN1, R23 and WR110 against IR24. This observation was
due to very low polymorphism at silent sites (ps) of the said
alleles. In general, adaptive evolution is most commonly
observed in regions involved in host pathogen recognition
(Endo et al. 1996) such as in the case of Xa21 and Xa21D,
occurrence of non-synonymous mutation was higher than
synonymous mutation in LRR domain (Wang et al. 1998).
For thorough analysis of adaptive and positive selection of
R-genes, we should consider only those sites predicted to
play an important role in recognition (Wang et al. 1998).
Amino acids evolve at a faster rate in functionally impor-
tant regions of R-proteins than the corresponding rate of
synonymous changes (Meyers et al. 1998). Similar pattern
was observed in this case with majority of the amino acid
replacement taking place in the TM domain despite the fact
that this substitution did not cause major changes in the
pattern of the TM. Probably, Xa27 was evolving under
relaxed constraint. The role of these TMs in pathogen
recognition is yet to be explored in order to understand the
resistance mechanism of Xa27 completely.
Fig. 3 Multiple alignment of the promoter region of Xa27 covering
the predicted UPTAvrXa27. The rice genotypes are indicated in left
column. Multiple alignment was constructed in Multalin. Deletion of
three bp ‘AGA’ and substitution of ‘C’ by ‘A’ in the UPT box is
indicated by black bar. It is at -51 position of the gene (from TSS)
302 Planta (2013) 238:293–305
123

When Tajima’s D was calculated for O. nivara acces-
sions and O. sativa accessions, the difference between pand h was very less which failed to give a significant
D value. The non-significant Tajima’s D and Fu and Li’s
F may be due to small sample size and random sampling.
Simultaneously, we cannot overrule the possibility of
selection occurring at this locus: firstly, as the samples
under study were selected randomly from a natural popu-
lation and not from an interbreeding population; secondly,
Xa27 locus interacts directly with the pathogen where the
possibility of co-evolution of both plant and pathogen exist.
So, a population-level study of R-gene locus will yield
accurate measurement of selection (Caicedo et al. 1999).
Other than looking only at the coding region for selection,
we found high level of nucleotide polymorphism at the
promoter and 50UTR among the accessions which pos-
sessed many regulatory sites. The roles of these polymor-
phisms in the gene function are yet to be explored and this
region may also be under natural selection. More impor-
tantly, the deletion and substitution in the UPTAvrXa27 box
in all alleles except IRBB27 will make these alleles unable
to signal AvrXa27 recognition (Romer et al. 2009), which
suggest the selective maintenance of dominant Xa27 or we
can say the gain of function of this allele during the course
of evolution among the natural population. This might also
be the possible case of an ‘‘evolutionary arms race’’ or a
negative frequency-dependent selection. A more detailed
study with more number of natural populations may pro-
vide crucial insights to understand the exact evolutionary
dynamics of Xa27 locus.
Twenty-seven alleles of Xa27 showing numerous
sequence diversity have been identified. Other than the few
changes in the coded protein sequences, the UPT box was
similar to that of IR24. We presume that the binding
affinity of AvrXa27 to the UPT box of these alleles will be
weak due to presence of defective UPT box and hence
binding of AvrXa27 will be unable to induce the expres-
sion of the alleles. It may be interesting to study the dif-
ferent alleles where amino acid changes had occurred, after
fusing these newly found coding sequences with a func-
tional UPTAvrxa27 by promoter engineering as described by
Romer et al. (2009) and Hummel et al. (2012). Clear
understanding of functional difference among alleles will
help in deciphering its structure and function (Bergelson
et al. 2001) as well as studying the molecular diversity of
avrxa27 will clear the co-evolutionary process of this R-avr
gene. The available sequence data of different alleles and
the level of diversity found at the Xa27 locus may provide
great opportunity for studying the evolution of plant–
pathogen interactions as well as a chance to study both
evolutionary and genetic aspects of this unique R-gene.
Acknowledgments The authors are grateful to two anonymous
reviewers for their critical review and valuable suggestions to
improve the manuscript. The research is supported by Department of
Science and Technology, New Delhi, India, Grant no. SR/SO/PS-21/
09. The authors also thank DRR, Hyderabad, Rajendra Agricultural
College, Pusa (Bihar) and NBPGR, New Delhi, for providing the
germplasms. Financial supports from XI plan seed money, DBT-
CREBB, DST-FIST, UGC-SAP-CAS-I and maintenance grant from
University of Hyderabad is gratefully acknowledged. W.B. is also
grateful to CSIR for the fellowship (Award no. 09/414/(0832)/2008-
EMR-I).
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic
local alignment search tool. J Mol Biol 215:403–410
Bakker EG, Traw MB, Toomajian C, Kreitman M, Bergelson J (2008)
Low levels of polymorphism in genes that control the activation
of defence response in Arabidopsis thaliana. Genetics
178:2031–2043
Fig. 4 The evolutionary relationship of 26 Xa27 alleles was inferred
using the neighbor-joining method. The optimal tree with the sum of
branch length = 0.07944762 is shown. The bootstrap consensus tree
which is inferred from 100,000 replicates is taken to represent the
evolutionary history of the alleles analyzed. The percentage of
replicate trees in which the alleles clustered together in the bootstrap
test (100,000 replicates) are shown next to the branches. The tree is
drawn to scale, with branch lengths. All positions containing gaps and
missing data were eliminated from the dataset (complete deletion
option). There were a total of 1,998 positions in the final dataset.
Phylogenetic analyses were conducted in MEGA4 (Tamura et al.
2007)
Planta (2013) 238:293–305 303
123

Bergelson J, Kreitman M, Stahl EA, Tian D (2001) Evolutionary
dynamics of plant R genes. Science 292:2281–2285
Bernsel A, Viklund H, Hennerdal A, Elofsson A (2009) TOPCONS:
consensus prediction of membrane protein topology. Nucleic
Acids Res 37 (Webserver issue):W465–W468
Bhullar NK, Street K, Mackay M, Keller NYB (2009) Unlocking
wheat genetic resources for the molecular identification of
previously undescribed functional alleles at the Pm3 resistance
locus. Proc Natl Acad Sci USA 106:9519–9524
Bogdanove AJ, Schornack S, Lahaye T (2010) TAL effectors: finding
plant genes for disease and defense. Curr Opin Plant Biol
13:394–401
Caicedo AL, Schaal BA (2004) Heterogenous evolutionary processes
affect R gene diversity in natural populations of Solanum
pimpinellifolium. Proc Natl Acad Sci USA 101:17444–17449
Caicedo AL, Schaal BA, Kunkel BN (1999) Diversity and molecular
evolution of the RPS2 resistance gene in Arabidopsis thaliana.
Proc Natl Acad Sci USA 96:302–306
Chun X, Hongqi C, Xudong Z (2012) Identification, mapping,
isolation of the genes resisting to bacterial blight and breeding
application in rice. Mol Plant Breed 3:121–131
Corpet F (1988) Multiple sequence alignment with hierarchical
clustering. Nucl Acids Res 16:10881–10890
Cummings MP, Clegg MT (1998) Nucleotide sequence diversity at
the alcohol dehydrogenase 1 locus in wild barley (Hordeum
vulgare ssp. spontaneum): an evaluation of the background
selection hypothesis. Proc Natl Acad Sci USA 95:5637–5642
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue.
Focus 12:13–15
Endo T, Ikeo K, Gojobori T (1996) Large-scale search for genes on
which positive selection may operate. Mol Biol Evol 13:685–690
Ersoz ES, Wright MH, Gonzalez-Martınez SC, Langley CH, Neale
DB (2010) Evolution of disease response genes in loblolly pine:
insights from candidate genes. PLoS One 5:e14234
Feng Q, Huang T, Zhao Q, Zhu J, Lin Z, Han B (2009) Analysis of
collinear regions of Oryza AA and CC genomes. J Genet
Genomics 36:667–677
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations.
Genetics 133:693–709
Gu K, Tian D, Yang F, Wu L, Sreekala C, Wang D, Wang GL, Yin Z
(2004) High-resolution genetic mapping of Xa27(t), a new
bacterial blight resistance gene in rice, Oryza sativa L. Theor
Appl Genet 108:800–807
Gu K, Yang B, Tian D, Wang D, Wu L, Sreekala C, Yang F, Chu Z,
Wang GL, White FF, Yin Z (2005) R-gene expression induced
by a type-III effector triggers disease resistance in rice. Nature
435:1122–1125
Gu K, Tian D, Qiu C, Yin Z (2009) Transcription activator-like type
III effector AvrXa27 depends on OsTFIIAg5 for the activation of
Xa27 transcription in rice that triggers disease resistance to
Xanthomonas oryzae pv. oryzae. Mol Plant Pathol 10:829–835
Huang H, Kochert G (1994) Comparative RFLP mapping of an
allotetraploid wild rice species (Oryza latifolia) and cultivated
rice (O. sativa). Plant Mol Biol 25:633–648
Huang CL, Hwang SY, Chiang YC, Lin TP (2008) Molecular
evolution of the Pi-ta gene resistant to rice blast in wild rice
(Oryza rufipogon). Genetics 179:1527–1538
Hummel AW, Doyle EL, Bogdanove AJ (2012) Addition of
transcription activator-like effector binding sites to a pathogen
strain-specific rice bacterial blight resistance gene makes it
effective against additional strains and against bacterial leaf
streak. New Phytol 195:883–893
IRRI (2002) Standard evaluation system for rice (SES). International
Rice Research Institute, Manila
Iyer-Pascuzzi AS, Sweeney MT, Sarla N, McCouch SR (2007) Use of
naturally occurring alleles for crop improvement. In: Upadhyaya
NM (ed) Rice functional genomics-challenges, progress and
prospects. Springer, New York, pp 113–143
Kauffman HE, Reddy APK, Hsieh SPY, Merca SD (1973) An
improved technique for evaluating resistance of rice varieties to
Xanthomonas oryzae. Plant Dis Rep 57:537–541
Kawabe A, Innan H, Terauchi R, Miyashita NT (1997) Nucleotide
polymorphism in the acidic chitinase locus (ChiA) region of the
wild plant Arabidopsis thaliana. Mol Biol Evol 14:1303–1315
Koornneef M, Alonso-Blanco C, Vreugdenhil D (2004) Naturally
occurring genetic variation in Arabidopsis thaliana. Annu Rev
Plant Biol 55:141–172
Kover PX, Schaal BA (2002) Genetic variation for disease resistance
and tolerance among Arabidopsis thaliana accessions Proc Natl
Acad Sci USA 99:11270–11274
Kuang H, Woo SS, Meyers BC, Nevo E, Michelmore RW (2004)
Multiple genetic processes result in heterogeneous rates of
evolution within the major cluster disease resistance genes in
lettuce. Plant Cell 16:2870–2894
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding
non-synonymous variants on protein function using the SIFT
algorithm. Nat Protoc 4:1073–1081
Lee S, Jia Y, Jia M, Gealy DR, Olsen KM, Caicedo AL (2011)
Molecular evolution of the rice blast resistance gene Pi-ta in
invasive weedy rice in the USA. PLoS One 6(10):e26260
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive
analysis of DNA polymorphism data. Bioinformatics
25:1451–1452
Lin JZ, Brown AHD, Clegg MT (2001) Heterogeneous geographic
patterns of nucleotide sequence diversity between two alcohol
dehydrogenase genes in wild barley (Hordeum vulgare subspe-
cies spontaneum). Proc Natl Acad Sci USA 98:531–536
Mauricio R, Stahl EA, Korves T, Tian D, Kreitman M, Bergelson J (2003)
Natural selection for polymorphism in the disease resistance gene
Rps2 of Arabidopsis thaliana. Genetics 163:735–746
May RM, Anderson RM (1983) Parasite-host coevolution. In:
Futuyama DJ, Slatkin M (eds) Coevolution. Sinauer Associates,
Sunderland, pp 186–206
Meyers BC, Shen KA, Rohani R, Gaut BS, Michelmore RW (1998)
Receptor-like genes in the major resistance locus of lettuce are
subject to divergent selection. Plant Cell 10:1833–1846
Nei M, Gojobori T (1986) Simple methods for estimating the numbers
of synonymous and nonsynonymous nucleotide substitutions.
Mol Biol Evol 3:418–426
Nei M, Li WH (1979) Mathematical model for studying genetic
variation in terms of restriction endonucleases. Proc Natl Acad
Sci USA 76:5269–5273
Petersen TN, Brunak S, Heijne GV, Nielsen H (2011) SignalP 4.0:
discriminating signal peptides from transmembrane regions. Nat
Method 8:785–786
Rajendrakumar P, Sujatha K, Rao KS, Natarajkumar P, Viraktamath
BC, Balachandran SM, Biswal AK, Sundaram RM (2007) A
protocol for isolation of DNA suitable for rapid seed and grain
purity assessments in rice. Rice Genet Newsl 23:92–95
Reissig WH, Heinrichs EA, Litsinger JA, Moody K, Fiedler L, Mew
TW, Barrion AT (1986) Illustrated guide to integrated pest
management in rice in tropical Asia. International Rice Research
Institute, Manila (Philippines)
Romer P, Recht S, Lahaye T (2009) A single plant resistance gene
promoter engineered to recognize multiple TAL effectors from
disparate pathogens. Proc Natl Acad Sci USA 106:20526–20531
Rose LE, Bittner-Eddy PD, Langley CH, Holub EB, Michelmore RW,
Beynon JL (2004) The maintenance of extreme amino acid
diversity at the disease resistance gene, RPP13, in Arabidopsis
thaliana. Genetics 166:1517–1527
Rose LE, Michelmore RW, Langley CH (2007) Natural variation in
the Pto disease resistance gene within species of wild tomato
304 Planta (2013) 238:293–305
123

(Lycopersicon) II population genetics of Pto. Genetics
175:1307–1319
Salamov AA, Solovyer VV (2000) Ab initio gene finding in
Drosophila genomic DNA. Genome Res 10:516–522
Stahl EA, Dwyer G, Mauricio R, Kreitman M, Bergelson J (1999)
Dynamics of disease resistance polymorphism at the Rpm1 locus
of Arabidopsis. Nature 400:667–671
Tajima F (1989) Statistical method for testing the neutral mutation
hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular
evolutionary genetics analysis (MEGA) software version 4.0.
Mol Biol Evol 24:1596–1599
Tang T, Lu J, Huang JZ, He JH, McCouch SR, Shen Y (2006)
Genomic variation in rice: genesis of highly polymorphic
linkage blocks during domestication. PLoS Genet 2(11):e199
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG
(1997) The CLUSTAL_X Windows interface: flexible strategies
for multiple sequence alignment aided by quality analysis tools.
Nucleic Acids Res 25:4876–4882
Tiffin P, Moeller DA (2006) Molecular evolution of plant immune
system genes. Trends Genet 22:662–670
Vaughan DA (1994) Wild relatives of rice: genetic resources
handbook. International Rice Research Institute, Manila
Wang GL, Ruan DL, Song WY, Sideris S, Chen L, Pi LY, Zhang S,
Zhang Z, Fauquet C, Gaut BS, Whalen MC, Ronald PC (1998)
Xa21D encodes a receptor-like molecular with a leucine-rich
repeat domain that determines race-specific recognition and is
subject to adaptive evolution. Plant Cell 10:765–779
Watterson GA (1975) On the number of segregating sites in genetical
models without recombination. Theoret Pop Biol 7:256–276
Yoshida K, Miyashita NT (2009) DNA polymorphism in the blast
resistance gene Pita of wild rice Oryza rufipogon and its related
species. Genes Genet Syst 84:121–126
Zhu Q, Zheng X, Luo J, Gaut BS, Ge S (2007) Multilocus analysis of
nucleotide variation of Oryza sativa and its wild relatives: severe
bottleneck during domestication of rice. Mol Biol Evol
24:875–882
Zou XH, Zhang FM, Zhang JG, Zang LL, Tang L, Wang J, Sang T,
Ge S (2008) Analysis of 142 genes resolves the rapid diversi-
fication of the rice genus. Genome Biol 9:R49.1–R49.13
Planta (2013) 238:293–305 305
123




















