
An investigation of structural motifs in gold complexes - The ...
240
An investigation of structural motifs in gold complexes A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Science and Engineering. 2017 Arij Taher Mohamed Addaraidi School of Chemistry
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of An investigation of structural motifs in gold complexes - The ...

An investigation of structural motifs in gold
complexes
A thesis submitted to the University of Manchester for the degree
of Doctor of Philosophy in the Faculty of Science and Engineering.
2017
Arij Taher Mohamed Addaraidi
School of Chemistry

2
Table of contents
Abstract ............................................................................................................................ 8
Declaration: ..................................................................................................................... 9
Copyright: ...................................................................................................................... 10
Acknowledgements ......................................................................................................... 11
List of Abbreviations ...................................................................................................... 7
1.0 Thesis structure ....................................................................................................... 13
1.1 Gold and phosphine chemistry .............................................................................. 14
1.1.1 General introduction of gold .................................................................................. 14
1.1.2 Physical and chemical properties of gold .............................................................. 14
1.1.3 Applications of gold ............................................................................................... 15
1.1.3.1 Usage of gold in medicine .................................................................................. 16
1.1.4 Co-ordination chemistry of gold ............................................................................ 17
1.1.5 Aurophilicity of gold(I) .......................................................................................... 18
1.1.6 Phosphine ligands systems ..................................................................................... 21
1.1.6.1 PR3 oordination chemistry .................................................................................. 22
1.1.6.2 Properties of PR3 ligands .................................................................................... 24
1.1.6.2.1 Steric parameter and cone angle ...................................................................... 25
1.1.6.2.2 Electronic parameter υ ..................................................................................... 26
1.1.7 Phosphine-gold (I) complexes ............................................................................... 27
1.1.8 Understanding aurophilic interactions ................................................................... 29
1.2 X-ray Crystallography ............................................................................................ 31
1.2.1 Basic concepts in crystallography .......................................................................... 31
1.2.1.1 Crystal structure .................................................................................................. 31
1.2.1.2 The crystal lattice and unit cell ........................................................................... 32
1.2.1.3 Crystal systems and the 14 Bravais Lattices ....................................................... 34
1.2.1.4 Symmetry and space group ................................................................................. 35
1.2.2 X-ray diffraction..................................................................................................... 38

3
1.2.2.1 Braggs Law ......................................................................................................... 39
1.2.2.2 Characterization techniques ................................................................................ 40
1.2.2.2.1 Single crystal X-ray diffractometer and method of analysis ............................ 40
1.2.2.2.1.1 Obtaining and growing crystals .................................................................... 41
1.2.2.2.1.2 Choosing a suitable crystal ........................................................................... 43
1.2.2.2.1.3 Data collection .............................................................................................. 44
1.2.2.2.1.4 Data reduction and correction ....................................................................... 45
1.2.2.2.1.5 Structure solution .......................................................................................... 45
1.2.2.2.1.6 Refining the structure .................................................................................... 46
1.3 References ................................................................................................................ 48
Chapter 2 ....................................................................................................................... 54
2. The systematic introduction of fluorine into the phenyl rings of phosphine
ligands and its influence on Au···Au distances in the solid state structures of
[AuX(PR3)] complexes. ................................................................................................. 55
2.1 Abstract .................................................................................................................... 55
2.2 Introduction ............................................................................................................. 55
2.3 Experimental ........................................................................................................... 57
2.3.1 Synthetic procedure ................................................................................................ 57
2.3.2 Synthesis of gold (I) phosphine complexes............................................................. 58
2.3.3 X-ray data collection and structures refinement .................................................... 59
2.4 Results and discussion ............................................................................................ 60
2.4.1 Synthesis and structural characterization of the complexes .................................. 60
2.4.2 Structure determination of complexes [AuX{P(4-C6H4F)3}], X = Cl, 1a, X = Br,
1b. .................................................................................................................................... 63
2.4.3 Structure determination of complexes [AuX{P(3-C6H4F)3}], X = Cl, 2a, X = Br,
2b and X = I, 2c. ............................................................................................................. 65
2.4.4 Structure determination of complexes [AuX{P(3,5-C6H3F2)3}], X = Cl, 3a, X = Br,
3b and X = I, 3c. ............................................................................................................. 69
2.4.5 Structure determination of complexes [AuX{P(3, 4, 5-C6H2F3)3}], X = Cl, 4a, X =
Br, 4b and X = I, 4c. ....................................................................................................... 71
2.5 Conclusion ................................................................................................................ 76

4
Acknowledgement ......................................................................................................... 77
References ...................................................................................................................... 78
Chapter 3 ....................................................................................................................... 81
3. Crystal structures of [AuCl{PPh2(C6F5)}] and [AuBr{PPh2(C6F5)}] ................... 82
3.1 Abstract .................................................................................................................... 82
3.2 Chemical context ..................................................................................................... 82
3.3 Structural commentary .......................................................................................... 83
3.4 Supramolecular features ........................................................................................ 85
3.5 Database survey ....................................................................................................... 89
3.6 Synthesis and crystallization .................................................................................. 89
3.6.1 Synthesis of [AuCl{PPh2(C6F5)}] (I)...................................................................... 89
3.6.2 Synthesis of [AuBr{PPh2(C6F5)}] (II) .................................................................... 89
3.7 Refinement ............................................................................................................... 90
References ...................................................................................................................... 91
Supporting information ................................................................................................ 92
Chapter 4 ..................................................................................................................... 110
4. Crystal structure of [AuI{PPh2(i-C3F7)}] .............................................................. 111
4.1 Structure description ............................................................................................ 111
4.2 Synthesis and crystallization ................................................................................ 114
4.3 Refinement ............................................................................................................. 115
Acknowledgement ....................................................................................................... 115
References .................................................................................................................... 116
Full crystallographic data........................................................................................... 117
Chapter 5 ..................................................................................................................... 123

5
5. The single crystal structures of gold(I) halide complexes containing the P(3,5-
(CF3)2C6H3)3 or P(3,5-(CF3)2C6H3)(C6H5)2 ligands-An unusual case of decreasing
Au···Au distances in the chloride, bromide and iodide complexes. ....................... 124
5.1 Abstract .................................................................................................................. 124
5.2 Introduction ........................................................................................................... 124
5.3 Experimental ......................................................................................................... 126
5.3.1 General procedure ............................................................................................... 126
5.3.2 Synthesis of R3P–Au–Cl complexes ...................................................................... 126
5.3.3 Synthesis of R3P–Au–Br and R3P–Au–I complexes ............................................. 127
5.3.4 Crystallographic details ....................................................................................... 127
5.4 Results and discussion .......................................................................................... 127
5.4.1 Synthesis and characterization ............................................................................ 127
5.4.2 Crystal structures of [AuX{PPh2(3,5-(CF3)2C6H3)}], X= Cl (1a), Br (1b), I (1c).130
5.4.3 Crystal structure of complexes [AuX{P(3,5-(CF3)2C6H3)3}], X = Cl, 2a, X = Br,
2b, X=I, 2c. ................................................................................................................... 133
5.5 Conclusions ............................................................................................................ 140
Reference ...................................................................................................................... 142
Chapter 6 ..................................................................................................................... 144
6. The single crystal structures of gold(I) halide complexes containing the P(4-
MeOC6H4)3, PPh(4-MeOC6H4)2 and PPh2(4-MeOC6H4) ligands............................ 145
6.1 Abstract .................................................................................................................. 145
6.2 Introduction ........................................................................................................... 145
6.3 Experimental ......................................................................................................... 146
6.3.1 Synthesis and crystallization ................................................................................ 146
6.4 Refinement ............................................................................................................. 147
6.5 Results and discussion .......................................................................................... 148
6.6 Summary ................................................................................................................ 157
References .................................................................................................................... 158

6
Supporting information .............................................................................................. 160
Chapter 7 ..................................................................................................................... 185
7. The single crystal structures of some gold(I) halide complexes containing the
PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and P(4-C6H4NMe2)3 ligands. ............... 186
7.1 Abstract .................................................................................................................. 186
7.2 Introduction ........................................................................................................... 186
7.3 Experimental ......................................................................................................... 187
7.3.1 Synthesis and crystallization ................................................................................ 187
7.3.1.1 Synthesis of [AuCl{PPh2(4-C6H4NMe2)}] (1a), [AuCl{PPh(4-C6H4NMe2)2}] (2a)
and [AuCl{P(4-C6H4NMe2)3}] (3a). ............................................................................. 187
7.3.1.2 Synthesis of [AuI{PPh2(4-C6H4NMe2)}] (1c) .................................................... 188
7.3.1.3 Synthesis of [AuBr{PPh(4-C6H4NMe2)2}] (2b) and [AuBr{P(4-C6H4NMe2)3}]
(3b). ............................................................................................................................... 188
7.4 Refinement ............................................................................................................. 188
7.5 Results and discussion .......................................................................................... 190
7.6 Summary ................................................................................................................ 198
References .................................................................................................................... 200
Supporting information .............................................................................................. 202
Chapter 8 ..................................................................................................................... 233
8.1 Conclusions ............................................................................................................ 234
8.2 References .............................................................................................................. 239
Appendix ……………………………………………………………………………..240

7
List of abbreviations
Abbreviation Definition
α Alpha
β Beta
γ Gamma
θ Theta
λ Wavelength
Å Angstrom (10-10
m)
Chemical shift
CIF Crystallographic Information Files
CSD Cambridge Structural Database
NHCs N-heterocyclic carbenes
Me Methyl
MRSA Methicillin resistant Staphylococcus aureus
m.wt Molecular weight
NMR Nuclear Magnetic Resonance
OMe Methoxy
NMe2 Dimethyl-amine
ptol (4-methylphenyl)
CDCl3 Deuterated chloroform
tht Tetrahydrothiophene
PPh3 Triphenylphosphine
CH2Cl2 Dichloromethane
EtOH Ethanol
R Alkyl, aryl or hydrogen group
Z Number of formula units per unit cell
ppm Parts per million
Ar Aryl

8
An investigation of structural motifs in gold
complexes
Arij Taher Mohamed Addaraidi
A thesis submitted to the University of Manchester for the degree of Doctor of
Philosophy in the Faculty of Sciences and Engineering 2017.
Abstract
A series of gold(I) phosphine halides complexes of the type [AuX(PAr3)] have been
synthesized. All of the complexes have been investigated and structurally characterised
by means of NMR spectroscopy and single crystal X-ray crystallography. In the solid
state all of the complexes adopt an approximately linear geometry at gold with the bond
angles ranging from 170.50(12)° to 179.19(6)° for Cl–Au–P, 169.32(6)° to 179.10(6)°
for Br–Au–P and 165.35(7)° to 178.62(7)° for I–Au–P. The Au–X bond lengths ranged
from 2.277(2) Å to 2.319(3) Å (X = Cl), 2.3470(4) Å to 2.4248(10) Å (X = Br) and
2.5513(6) Å to 2.5787(7) Å (X = I), while the Au–P distances were between 2.219(2) Å
and 2.261(2) Å.
The presence, or otherwise, of short Au···Au interactions as a function of halide and
phosphine steric and electronic parameters has been investigated. The Au···Au
distances range from 3.1273(8) Å in [AuCl{P(3,4,5-C6H2F3)3}] to 9.0059(6) Å in
[AuCl(3,5-(CF3)2C6H3)3]. For all complexes the Au···Au distances increase as the
halide changes from Cl, Br, I, except for [AuX{P(3,5-(CF3)2C6H3)3}], X = Cl, Br, I,
where the reverse trend is observed. The secondary C–H···X, C–H···F and F···F
interactions are also investigated.

9
Declaration: This thesis is a presentation of my original research work. No portion of the work
referred to in the thesis has been submitted in support of an application for another
degree or qualification of this or any other university or other institute of learning.
Signed
……………………………………………………………………………………………

10
Copyright:
i. The author of this thesis (including any appendices and/or schedules to this thesis)
owns certain copyright or related rights in it (the “Copyright”) and s/he has given
The University of Manchester certain rights to use such Copyright, including for
administrative purposes.
ii. Copies of this thesis, either in full or in extracts and whether in hard or electronic
copy, may be made only in accordance with the Copyright, Designs and Patents Act
1988 (as amended) and regulations issued under it or, where appropriate, in
accordance with licensing agreements which the University has from time to time.
This page must form part of any such copies made.
iii. The ownership of certain Copyright, patents, designs, trademarks and other
intellectual property (the “Intellectual Property”) and any reproductions of
copyright works in the thesis, for example graphs and tables (“Reproductions”),
which may be described in this thesis, may not be owned by the author and may be
owned by third parties. Such Intellectual Property and Reproductions cannot and
must not be made available for use without the prior written permission of the
owner(s) of the relevant Intellectual Property and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property
University IP Policy
(seehttp://documents.manchester.ac.uk/display.aspx?DocID=24420), in any relevant
Thesis restriction declarations deposited in the University Library, The University
Library’s regulations (see http://www.library.manchester.ac.uk/about/regulations/)
and in The University’s policy on Presentation of Theses.

11
Acknowledgements
I would like to express my special thanks and grateful to my supervisor, Dr Alan
Brisdon, for his continuous help and patience throughout my PhD research.
Special and Particular thanks goes to my Co- supervisor Dr Robin Pritchard for his
great help, patience, guidance in all areas throughout this work including in solving
crystal structures.
I would like to express my deepest gratitude to my parents for their un-ending
supporting, prayers and love, my words are not enough to express my thanks to you; I
would like to dedicate my thesis to you. My warm thanks go to my husband (Sami), for
his material and spiritual support and encouragement in all aspects of my life.
I cannot finish without thanking you, my little sons (Mwaid and Islam) for being a
powerful source of energy and happiness during this research.
Thanks to all my colleagues in Brisdon and Pritchard group and my friend (Amina
Ejgandi) who has shown lots of concern, support and encouragement at the time of
difficulties.
Special thanks go to my sisters and brothers, for being supportive of my graduate
studies.
Thanks to the Libyan government for the funding.
The Author
The author completed her first degree in the University of Zawia/Libya 2004/2005, and
MSc degree in Analytical chemistry 2009/2010 from the University of Huddersfield.

12
Chapter 1
Introduction

13
1.0 Thesis structure
The thesis has been prepared in alternative format, and contains 6 research papers that
have been prepared for publication. This thesis consists of 8 chapters. Chapter 1
provides an introduction to gold in connection with phosphine ligands, followed by
describing the detailed background into X-ray crystallography. In all of the research
papers described, complex formation, crystal growth, X-ray data collection and solution
was performed by the thesis author. Manuscript preparation was primarily by the thesis
author, with assistance from supervisor and co-supervisor.
Chapter 2 contains “The systematic introduction of fluorine into the phenyl rings of
phosphine ligands and its influence on Au···Au distances in the solid state structures of
[AuX(PR3)] complexes”, this has been prepared for submission to CrystEngComm.
Chapter 3 contains “Crystal structures of [AuCl{PPh2(C6F5)}] and
[AuBr{PPh2(C6F5)}]”, this has been prepared for submission to Acta Crystallographic
Section E.
Chapter 4 contains “Crystal structure of [AuI{PPh2(i-C3F7)}]”, this has been prepared
for submission to IUCrData.
Chapter 5 contains “The single crystal structures of gold(I) halide complexes
containing the P(3,5-(CF3)2C6H3)3 or P(3,5-(CF3)2C6H3)(C6H5)2 ligands – An unusual
case of decreasing Au···Au distances in the chloride, bromide and iodide complexes”,
this has been prepared for submission to CrystEngComm.
Chapter 6 contains “The single crystal structures of gold(I) halide complexes
containing the P(4-MeOC6H4)3, PPh(4-MeOC6H4)2 and PPh2(4-MeOC6H4) ligands”,
this has been prepared for submission to Acta Crystallographic Section C.
Chapter 7 contains “The single crystal structures of some gold(I) halide complexes
containing the PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and PPh(4-C6H4NMe2)3
ligands”, this has been prepared for submission to Acta Crystallographic Section C.
Chapter 8 contains a conclusion to the work presented in this thesis.

14
1.1 Gold and phosphine chemistry
1.1.1 General introduction of gold
Gold is the king of the elements, and one of the most noble metals, which has a unique
position in the Periodic Table. The scientific symbol for gold i.e. Au, is derived from
the name of the Greek goddess of dawn, called ‘Aura’. Aurum, the Latin name of gold,
itself describes the appreciation of the physical properties of the metal, e.g. shine,
glittering yellow colour and worth, by ancient people. Gold is found in alluvial deposits
and in seawater in its pure form and as alloys and tellurides. Found in concentrations on
average of 0.004 ppm, gold is one of the 23 trace elements that form only 0.0003 % of
the earth’s crust. Some other, rather unlikely, places for gold deposition are human
lungs and horns of animals like deer, rhinos and antelopes. It is also found in some
herbs, and is associated with the curing effect of those herbs. The very little that was
known to the ancient people about the medicinal use and therapeutic effect of gold has,
overtime, bloomed into a field of scientific research.1
1.1.2 Physical and chemical properties of gold
Gold is a d-block transition metal placed in period 6 and group 11 of the periodic table.
The only one naturally occurring stable isotope has an atomic number of 79, atomic
weight of 196.9665 g mol-1
, and atomic volume of 10.2 cm3 mol
-1.2 In its pure form,
gold is found as isomeric face-centred cubic crystals, with an inter-atomic distance of
135 pm. The high density of gold makes it easily extractable from residual rocks. In
gaseous form, gold is found as diatomic molecules with a bond length of 2.30-4.80 Ǻ.3,4
Furthermore, gold exhibits the aurophilicity phenomenon in the solid state, where gold
complexes tend to form an Au···Au interaction. This bond length measures about 300-
350 pm and is comparable in bond strength to the gas phase dimer.5
The noble nature of
gold, as evident from its low reactivity with oxygen, sulphur and aqueous bases, is due
to the low oxidation potential and small radius of metal.6
However, it has been found to
react with all halogens such as chlorine, bromine and iodine to form Au2Cl6, Au2Br6 and
AuI, respectively, and dissolves in hydrochloric acid and cyanide solution, in the
presence of a strong oxidant.2
The reactivity of atomic gold is higher than its molecular
form. The key features that confer gold its unique character are its inertness,

15
“aurophilicity”, low oxidative potential, high electronegativity and its metallic yellow
colour.5
The uniqueness of gold is mainly due to the so-called relativistic effect, which is more
pronounced in gold than the other elements of its group.7
Both, the physical and
chemical properties of gold, are significantly different from other members of its group
in the periodic table (Table 1).
Table 1: Physical properties of gold in comparison with silver and copper.
Properties/Elements Cu Ag Au
Group, Period, Block 11,4,d 11,5,d 11,6,d
Colour bronze silver yellow
Atomic weight (g mol−1
) 63.546 107.868 196.966
Electronic configuration [Ar]3d10
5s1 [Kr]4d
105s
1 [Xe]4f
145d
10 6s
1
Density (g cm-3
) 48.9 510.49 19.3
Melting point 1084.62°C 961.78°C 1064°C
Boiling point 2562°C 2162°C 2856°C
Heat of fusion (kJ mol-1
) 13.26 11.28 12.55
Heat of vaporization (kJ mol-1
) 300.4 250.58 324
Specific heat capacity (J mol-1
.K-1
) 24.44 25.35 25.418
Oxidation states 1, 2, 3, 4 1, 2, 3 -1, 1, 2, 3, 4, 5
Electronegativity (Pauling scale) 1.9 1.93 2.54
First ionization energy (kJ mol-1
) 745.5 731.0 890.1
Atomic radius (pm) 128 160 135
Covalent radius (pm) 138 153 144
Van der Waals radius (pm) 140 172 166
Thermal conductivity (300 K),
(W m-1
K-1
) 401 429 318
1.1.3 Applications of gold
Due to its unique physical properties, gold has also earned the famous title of ‘the king
of the metals’, and has been used in jewellery and decorative pieces.1
Other, relatively

16
less traditional, applications of gold include its use in the electronic and electrical
industry, e.g. microelectronics, solar energy collectors, gold plating in aerospace
equipment.6,8,9
Photography is another important area of gold applications.10
In recent
times the chemistry of gold is one of the fastest growing fields of chemistry due to its
linkage to a large number of topics, involving catalysis11
material science1 and
medicine.12
Occasional historical medicinal use of gold suggests awareness of its role as
a therapeutic agent, therefore there are a large number of gold based complexes have
been developed and tested for their therapeutic role.13,14
Moreover, the complexes of
gold(I) combining phosphines have been developed, particularly for their use as
effective and selective homogeneous catalysts in organic transformations.15
A brief
review of this historic journey is described as follow.
1.1.3.1 Usage of gold in medicine
The historic use of gold in medicines to cure various diseases dates back to 2500 BC in
Arabia, China and India. In medieval Europe, alchemists used gold powder and flakes
to make an elixir known as ‘aurum potabile’. The 17th
century witnessed the use of
‘gold cordial’, a medicine used in treatment of ailments caused by “vital spirits” such as
fainting and fever etc.16
The modern use of gold in medicinal application did not begin
until the use of gold sodium chloride for treatment of syphilis in the early 19th
century,
by Chrestien and Figuier. Later a German physician, Robert Koch, discovered the
antibacterial effects of Au(CN)2-, which led to the landmark discovery of gold-based
anti-arthritis drugs (RA rheumatoid arthritis). The word coined for the treatment of RA
with gold complexes is ‘chrysotherapy’ and has been in use for last 70 years.1,17,18
The first generation of chrysotherapy drugs, e.g. aurothioglucose, di-sodium
aurothiomalate, aurothiopropanal sulphonate and gold(I) 4-amino-2-mercaptobenzoate,
were mostly Au(I) based. The second generation of RA treatment drugs include
Auranofin [tetra-O-acetyl-β-D-(glucopyranosyl)thio]-(triethylphosphine)gold(I).
Introduction of a phosphine as the ligand of this second-generation drug produced a
lipophilic character, which increased solubility and bio-distribution of the drug and
made its oral administration possible.19
Following the footsteps of Robert Koch, Fricker and co-workers synthesised a range of
Au(I) and Au(III) based cyanide and related complexes, e.g. [Au(SCN)(PMe3)], and
proved their anti-microbial activity for Gram-positive bacteria including MRSA. In their

17
recent work, Ghosh et al. found that the gold complex, [1-benzyl-3-tertbutylimidazol-2-
ylidene]AuCl, has significant antimicrobial properties.20
Another important medicinal area where gold complexes can play an important role is
in chronic asthma. The only treatment for this disease currently in use, is the continuous
use of corticosteroids. Although still in need of through investigation, some side-effects
associated with the use of gold complexes have also been reported.21
Gold compounds
have also been investigated as a method for treating various disorders involving
inflammation of skin, such as psoriasis 22
pemphigus 23
and urticarial.24
1.1.4 Co-ordination chemistry of gold
Gold is a soft Lewis acid with a soft metal centre and reacts with a range of generally
soft π-acceptor ligands, e.g. CN−, PR3, RS
-, R2S, RN
-, RCO2
-, in the presence of strong
oxidising agents. Although gold is found in multiple oxidation states i.e. -1 to V, its
coordination chemistry is dominated by Au(-1) – (Au)III based complexes. The toxic
nature of Au(III)-based complexes undermines its use in most of its applications,
leaving Au(I) as the most important candidate for gold coordination chemistry. The
most common coordination number of gold compounds in the oxidation state (I) is two-
coordinate with a linear geometry. These complexes can be formed as neutral, anionic
or cationic molecules such as [Ph3PAuCl], [Au(CN)2]- or [Au(PPh3)2]
+, respectively.
25
However, a large number of gold (I) complexes with higher coordination numbers are
also known, they are usually 3-coordinate trigonal-planar and tetrahedral 4-coordinate
complexes.25
Examples are given in Figure 1.
Figure 1: Examples of typical Au(I) complexes.
Trigonal Au(I)
Tetrahedral Au(I)
Linear Au(I)

18
Au(I), although thermodynamically stable, in aqueous solutions, disproportionate into
atomic gold and Au(III) in the absence of a stabilising ligand.6
3Au(I) (aq) 2Au (s) + Au(III) (aq)
While studying various Au-L coordination, Schröder et al. determined the ligand bond
strength for a range of ligand i.e L (ligand) = Xe < C6F6 < H2O < CO < H2S < CH3CN ≈
C2H4 ≈ NH3 < CH3SCH3 < PH3.26
Unlike Cu(I) and Ag(I), Au(I) preferably forms complexes with two coordinate, rather
than three and four coordinate geometry. Moreover, the low coordination numbers
arising from the rod-like geometry for the linear complexes allows for the formation of
weak additional bonds in the solid state. The so-called aurophillic bonding between the
gold atoms make them align in opposite directions in firstly, a long zigzag chain of
molecules in a head to tail (Figure 2). Such complexes of gold have a simple linear
geometry and pronounced presence of aurophilic forces with a typical bond length of
350 pm and bond energy of 21-46 kJmol-1
.27
Figure 2: Examples of gold (I) complexes [C3H7NCAuCl] with linear geometry.
1.1.5 Aurophilicity of gold(I)
Aurophilic interactions28
are an important factor of many two-coordinate
gold(I)-containing compounds. The term aurophilicity is now broadly used to describe
many types of Au···Au interactions within and between gold complexes. Consequently,
it is essential to have a clear definition of the effect.29

19
Recent development in crystallographic and spectroscopic studies of gold complexes
has revealed the presence of closer-than-normal Au···Au distances in gold complexes,
these were called aurophilic forces by H.Schmidbaur. These forces have been revealed
to be non-covalent interatomic forces of strength comparable with that of hydrogen
bonding and twice as strong as Van der Waals forces.29,30
The Au···Au interactions are
characterised by, a typical, interatomic distance ranging from 2.9 to 3.60 Å (the
estimated Van der Waals distance being 3.60 Å), and a bond strength, as measured by
temperature- dependent NMR spectroscopy, in the range of 20 to 50 k J mol-1
. The
exact bond length and bond energy of an aurophilic bond however, tends to change with
the physical state of gold and chemical nature of the ligand in a gold-based complex.
For example, in gaseous gold atoms such Au···Au attractions have a bond length of
2.47Å, and bond energy of 288 kJ mol-1
. Similarly, in bulk metallic gold, a bond length
of 2.88 Å and bond energy of 100 k J mol-1
is found. Although more pronounced for
Au(I), aurophilic forces are found in all oxidation states of gold, both in their pure form
and in an aggregate of various oxidation states together.31
Interesting physical properties
such as: enhanced luminescence can be shown in complexes which have aurophilic
interactions.32
Aurophilicity, in the standard case, appears to be operative in between the closed-shell
gold centres in the oxidation state Au+1
and in the linearly two-coordinate state. The low
coordination number is an important condition because it reduces the steric repulsions
between the ligands in the aggregates. Therefore the attraction is hardly observed for
coordination numbers >2. For independent ions or parts of multinuclear complexes or
mononuclear molecules, in the case of a flexible skeleton, the metal centres approach
takes place vertically to the molecular axis to reach an equilibrium Au–Au distance of
ca. 3 Å as shown in Scheme 1. (a–c).33

20
Scheme 1: (a-c) some typical arrangements resulting in aurophilic interactions.
The conformation of the aggregates may be crossed or parallel with, where applicable,
like or the opposite directions of the individual units as shown in scheme 2. (d-f).
Scheme 2: (d-f) crossed head-to-head or head-to-tail aurophilic interactions.
The gold (I) centre can deliver only one or numerous aurophilic contacts. Dimers and
chainlike polymers are the most common arrangements (helical linear, or zigzag), but
oligomer arrangements are also known (scheme 3. g-i).33
(e)
(f)
(d)
(a) (b) (c)

21
Scheme 3: (g-i) some oligomers arising from aurophilic interactions.
In L–Au–X complexes the geometry of each unit shows only slight deviations from
linearity in the aggregates, but the minor bindings which have been observed are all in
the direction that brings the gold atoms closer together, indicating attraction, but not
repulsion.33
Despite of their key role in gold chemistry, the existence of aurophilic forces in gold
have been intriguing researchers for a long time, and could not be explained with
classical bonding theories. It was not until the discovery of relativistic effects34
that a
realistic explanation for the existence of aurophilic forces could be given (see Section:
1.1.8).
One way in which an understanding of aurophilic interactions may be studied is by
systematically changing the ligands. One class of suitable ligands for such an
investigation are phosphines.
1.1.6 Phosphine ligands systems
Phosphines (PR3) are soft and strong σ-donor ligands used in transition-metal
complexes. The electronic, steric, and stereo-chemical properties of a phosphine ligand
are based on the substituents attached to the phosphorus atom. Selection of a phosphine
with an appropriate group of substituents, allows control over the steric and electronic
environment of a complex, which is of considerable interest in applications like
(g) (h)
(i)

22
tunability/optimization of catalysts. Because of this a large number of
phosphine-containing homogeneous catalysts have been developed for a wide variety of
organic reactions including hydrogenation, hydroformylation, hydration, hydrolysis,
cross-couplings, and carbon-heteroatom bond formation.35
Primary hydrogen derivatives of the heavy elements of group 15 (phosphorus, arsenic
and antimony) i.e., also known as pnictanes, are important starting materials and
intermediates for many useful chemical reactions. They have been controversial as
laboratory reagents due to their toxicity and reactive chemical nature. Development of
their secondary and tertiary derivatives, especially with heavy substituents, however,
have helped to overcome the toxicity and reactivity issue to a large extent, and increased
the use of pnictanes more than ever.
Phosphine, in its simplest form as PH3, is known to be an extremely toxic and volatile
gaseous substance, which is spontaneously flammable in an oxygen atmosphere.
Phosphine finds its real position in coordination chemistry in the form of its
derivatives.35
Due to relevance with the current project, only the complexes of tertiary
phosphines with elements of group 11, especially gold, are discussed further here.
1.1.6.1 PR3 coordination chemistry
Organophosphorus compounds, such as the tertiary phosphines which have three
hydrocarbon substituents, are the most commonly found ligands. Other compounds
which are well documented are primary (RPH2), secondary (R2PH) and phosphines with
P–ER linkages (E = heteroatom such as O/N, R = alkyl/aryl). As a progenitor, usually
one or more P–X bonds (X= halogen) are used to form phosphines by the reaction with
Grignard or organolithium reagents, although a few, such as PF3, act as ligands on their
own.36
In the 1950s, Dewar 37
Chatt and Duncansen 38
proposed a model, on the basis of
which the bonding of a phosphine to a metal is usually broken-down into two
components, 𝜎-donation and π-acceptance. When the lone pair of the phosphorus centre
donates into a metal d-orbital of suitable symmetry, a 𝜎-bond is produced. The orbitals
involved in the π-bonding component are not as well defined. Initially it was thought
that low lying 3d orbitals of phosphorus were used in 𝜋-back bonding.39
Later on, this idea became obsolete after suggestions were given that the anti-bonding
orbitals of the P-R bonds can be the π-accepting orbitals.40

23
Figure 3: (a) The σ-donation and π-back donation for CO ligand and a metal, (b) the
σ -donation and π-back donation for a phosphine and a metal (adapted from 40
).
The following order shows the π-acceptor ability of some phosphines:
PMe3 = P(NR2)3 < PAr3 < P(OMe)3 < P(OAr)3 < PCl3 < CO = PF3,
The order shown above illustrates that P(NR2)3 is a less good 𝜋-donor than was
expected because the lone electron pairs of nitrogen compete with metal dπ to donate
into the PR σ* orbitals. PF3 is the only phosphine ligand that has similar π-acceptor
properties as CO.
Two contrasting effects are brought about by the interactions between metal and
phosphine ligands. The P-R bond length is increased when dπ orbital of the metal
donates electron density to the vacant σ* orbital of P–R. While a reduction in the P-R
bond length can be brought about by donation of the lone pair from phosphorus to the
metal, which in turn causes a decrease in P (lone pair)–R (bonding pair) repulsion. An
assessment of the two effects can be made by making a comparison of the crystal
structures of pairs of phosphine-metal complexes, such as [(η3-C8H13) Fe{P(OMe)3}3]
n+,
where n = 0 or 1. A reduction in the ability of Fe to donate electron density to the
phosphite is made by the oxidation of this complex, which in turn causes an increase in
the Fe–P bond length and a decrease in the P–O bond length.40
Phosphine based gold complexes have been the focus of interest due to their therapeutic
effects in anticancer and anti-arthritics drugs, their chemiluminescent properties, and
their catalytic role, e.g. hydration of alkynes. Au(I) phosphine complexes are also
known for evidence of their pronounced aurophilic interactions.41

24
1.1.6.2 Properties of PR3 ligands
Both the steric and the electronic properties of the ligand influence the metal-ligand
bonding that is why advances have been made in many areas of measurement of these
properties. Through this it was though that it would be possible to rationalise the
reactivity of transition metal-phosphine complexes.42 The influence of a P(III) ligand
was explained in terms of electronic effects before 1970, but only a few references to
steric effects were made. Tolman made a proposal in the following year concerning
electronic and steric parameters based on the ligand cone angles (θ) of space-filling
CPK molecular models and the A1 carbonyl stretching frequencies (ν) of the carbonyl
group of Ni(CO)3L complexes, this is why these measures are named after him even
today.43
Changes in electron donor or acceptor properties that are usually brought about due to
differences in electron density, through transmission along chemical bonds by addition
of an electron-donating or -withdrawing atom or group, are termed as electronic effects;
for instance, these might be adjusting the methyl group in P(p-C6H4CH3)3 to a chlorine
atom, as in P(p-C6H4Cl)3, which leads to significant electronic effects. Non-bonding
forces emerging from changes in the size or shape of a molecule are termed as steric
effects; for instance, adjusting the substituent orientation from P(p-C6H4CH3)3 to
P(o-C6H4CH3)3 has a steric impact.44
An important impact is made by the electronic effects on steric properties and vice
versa. For example, the percentage of s-character of the phosphorus lone pair will
decrease when there is an increase in the angle between substituents. An influence on
the bond distances and angles is made by the presence of electronegative atoms or
groups. That is why there is a significant relation between electronic and steric effects,
and individually, each effect is difficult to separate.45

25
Figure 4: Represent a schematic of steric and electronic effects.
1.1.6.2.1 Steric parameter and Cone angle
When considering the coordination chemistry of phosphines, it is of utmost importance
to also consider the steric demand. The cone angle concept made by Tolman in the late
1970s, is a very common method of quantifying the size of a phosphine.45
But the
Tolman cone angle concept has a few limitations and can strictly only be applied to
nickel systems, and does not include the prospect that ligand substituents can
‘intermesh’. Furthermore, it initially applied only to phosphines in which all three
substituents were the same. When a phosphine is composed of different groups, the
average of the sum of half angles is need.
A half angle 𝜃i/2 i the angle between one M-P bond and the vector dividing the P-M-P
angle for chelating diphosphines.
The original cone angles were revised on the basis of the calculated structures and
crystallographic data, however the original values of Tolman are still extensively used
in their original form. But suggestions have been made for other steric parameters, such
as the solid angle, which are founded on the “shadow” cast by asymmetric ligands
amongst other possible measures.46

26
Figure 5: Show the measurement of the Tolman cone angle, (a) for symmetric PR3 ligands
and (b) for asymmetric PR3 ligands (adapted from47
).
1.1.6.2.2 Electronic parameter 𝝊
If a change is made to a part of a molecule that results in a different electronic
distribution within the molecule, then an electronic effect is produced.48
This effect is
carried through transmission over the chemical bonds. Tolman, in 1977, used a
parameter grounded on the vibrational spectra of [Ni(CO)3L] complexes as a measure of
the electronic properties of a ligand – he suggested using the highest CO stretching
frequency, υ, of the phosphine-substituted transition metal carbonyl complex. IR
frequencies can be determined with sufficient precision to provide distinct values for
common substituents. He identified a group electronic parameter χi by determining the
difference in the CO frequencies of the complexes with different phosphines.49
The electronic parameter (χi) of PR3 ligands can be accurately obtained by measuring
the v(CO) of different types of [Ni(CO)3L] complexes and identifying the three attached
groups phosphines by Equation 1.
The (χi) values are in the range of 0 to 20, compared to the tBu group (χ of
tBu = 0); for
instance, the χ value of the phenyl group is 4.3, and that of (CF3)2CHO is 17.43,50
𝒗 = 𝟐𝟎𝟓𝟔. 𝟏 + ∑ 𝝌𝟑𝒊=𝟏 i
Equation 1: Tolman equation for calculating the electronic parameter, v, of phosphine
ligand.
Suggestions have been made for several substitute metal complexes, such as
[Rh(CO)Cl(PR3)2] and [Mo(CO)5PR3], because some nickel carbonyl [Ni(CO)3PR3]
(a) (b)

27
complexes, and the precursor, have a high toxicity. The v(CO) stretching frequencies of
a series of [Cr(CO)5L] complexes were found by Stohmeier and Horrocks so a
comparison could be made for the properties of phosphine ligands.51
1.1.7 Phosphine-gold (I) complexes
Phosphine gold(I) complexes of the type (R3P)AuX are among the key reagents in gold
chemistry and have been the subject of considerable interest owing to their therapeutic
value as anti-arthritic and anti-cancer drugs 52
and their chemiluminescent properties.53
Gold(I) phosphine complexes are also important for homogeneous catalysis involving
four coordinated diphosphane-diorgano complexes as the intermediates in the process.
Another important feature of high coordination gold (I) complexes i.e., with
three-coordinate species, is their luminescent character. This particular property can be
very useful for its applications in light-emitting diodes.54
Due to the occurrence of gold-
gold interactions that are displayed in the solid state, the supramolecular chemistry of
gold(I) phosphine complexes is a major source of attraction.55
In the presence of bulky trialkyl phosphines in compounds like [AuCl(Pi-Pr3)], the
aurophilic interactions are lost.56
Irrespective of this fact, according to the CSD
database, the size of a phosphine is not a predominant factor for deduction of aurophilic
interactions as for the complex of P(p-CH3C6H4)3 (cone angle = 145°) an aurophilic
interaction is observed, d(Au–Au) = 3.375 Å, whilst for triphenyl phosphine ligand
(cone angle also 145◦) the Au–Au contacts are over 6.9 Å.57
It can be said that the occurrence of aurophilic interactions in triaryl phosphine
complexes is dependent on the capability of these interactions to challenge other weaker
intermolecular forces. Moreover, there are cases like that of [AuCl{P(p-CH3C6H4)3}],
where there are two known polymorphs.58
One has an aurophilic interaction 3.375(1)
Å,59
and the other with no specifically close gold–gold approach.60
Additionally, bulky
triaryl phosphines do not always prevent the occurrence of gold-gold contacts, as
observed in crystallographic data. For instance, the complexes [AuCl{P(m–CF3C6H4)3}]
and [AuCl{P(3,5–(CF3)2C6H3)3}] show aurophilic interactions of 3.0738(9) and
3.341(1) Å, respectively.61
Irrespective of the fact that countless research into the crystallographic nature on gold
(I) phosphine complexes of aryl and alkyl phosphines have taken place, there are a

28
limited number of systems making use of unsaturated alkenyl or alkenyl fragments. A
gold-gold interaction of 3.0934(5) Å62
is displayed by the structure of
[AuCl{P(CH=CH2)3}], whereas [AuCl{PPh2(C≡CH)}] does not show such bonds, but
exhibits links into dimers through hydrogen bonding that takes place between the
acetylenic protons and the metal bound chlorides.63
The aurophilic interactions are no
longer observed when the phenyl groups are substituted for more bulky substituents, for
example in [AuCl{Pi-Pr2(C≡CH)}].64
Incomparison, the iodo-analogue
[AuI{PPh2(C≡CH)}] shows an Au···Au contact of 3.0625(9) Å. But Brisdon and
co-workers, have previously reported the gold(I) complexes of fluorovinyl substituted
phosphines, [AuCl{PPh(CF=CF2)2}] and [AuCl{PPh2(CF=CF2)}] however only the
first formed crystals suitable for diffraction work, which showed that the complex
aggregates as dimers with a short Au···Au distance of 3.1945(5) Å.65
The nature of the anionic ligand X in the complexes [X–Au–L] has an effect upon the
strength of the Au–Au interaction. Theoretical and experimental data has suggested that
the softer the ligand the stronger the gold–gold interaction.66
In addition, searches of the CSD for structures contain P–Au–X (Figure 6) have been
performed in order to determine Au···Au interactions where the distance is less than
twice the Van der Walls radius (3.60 Å).31
As a result, there were found 114 hits having
an Au···Au interaction which are less than twice the Van der Waals radius, as shown in
the histogram below (Figure 7).
Figure 6: Show how the search has been performed.

29
Figure 7: Histogram of the Au···Au distance of structures in the CSD.
1.1.8 Understanding aurophilic interactions
There have been a number of attempts to describe and understand aurophilic
interactions. One of these is based on the ‘relativistic effect’, the aurophilic attraction
between gold nuclei result from a transfer of electrons from a filled ‘d’ orbital of one
gold atom to an empty ‘p’ orbital of another gold atom, under the influence of relativity
and correlation effects existing in atoms with heavy nuclei.67
The phenomenon of a
relativistic effect is based on the fact that an increase in velocity of a moving object
affects its mass in such way that at a speed equal to the speed of light, the mass of the
object becomes infinite. The increase in objects mass by the relativistic effect can be
calculated with the equation shown below.68
𝒎ᵣ =𝒎˳
√𝟏 − (𝒗/𝒄)²
Equation 2: Calculating objects mass by the relativistic effect.
Where mo is the rest mass, v is the velocity of the object and c is the velocity of light.
Under the large positive charge of nuclei of heavy metals like gold, electrons undergo
an increase in their velocity and hence a relative change in their mass. Such changes
have consequences for the orbitals of the metal, dividing them into relativistic and
non-relativistic radii. The ratio of relativistic and non-relativistic radii has been found to
have a direct relation with the atomic number of atoms. The ratio in the case of gold

30
tends to deviate from the generally observed unity value, and therefore, is considered
important in determining the unique chemical behaviour of the metal.67
The relativistic increase in electron mass has three effects on the atomic orbitals, firstly:
s-orbitals and also p-orbitals, but to a lesser extent, become smaller and energetically
stabilized. Secondly: outer d and f orbitals expand and become energetic destabilized.
Thirdly: spin-orbit coupling occurs, for example splitting of p, d, and f orbitals
energies.68
Such simultaneous contraction and expansion of orbitals increases the energy gap
between them and pushes the otherwise closed shell d orbital into an outward position.
The contracted s and p orbitals also shield the d orbital of the metal from the nuclear
charge making its electrons loosely held by the nucleus and more readily available for
chemical interactions.67
There have also been many attempts made to either predict when aurophilic interactions
will occur, or to explain their occurrence. One of those by Anderson et al. reported that
compounds with Au···Au interactions have a greater a tendency to produce structures
that have more than one molecule in the asymmetric unit (Zʹ > 1). The calculations of
molecular volume, have shown that Zʹ = 1 is favoured if the two ligands have a variance
in size, ligands which have equivalent size can form structures with Zʹ > 1 as a non-
crystallographic twist is needed to maximize the strength and, hence, minimize the
length of the Au···Au interaction. Another factor that differs exhibiting for the Zʹ
behaviour is the type of packing arrangement; with Zʹ > 1 structures being molecules
rather than infinite chains. In the case of Zʹ = 1 structures are in some cases evenly split
between these two possible arrangements and don’t normally show aggregation beyond
dimers.69
Flowers and colleagues proposed that in some cases the structures of the dihalogen
adducts of phosphines, i.e. I-I-PR3 and the equivalent gold(I) complexes I-Au-PR3 are
isomorphous indicating that ligand packing, rather than aurophilic interactions, are
most significant in determining the crystallographic structure. Where the structures
digress, it was proposed that this is due to the greater ability of the dihalogen adduct to
be involved in hydrogen bonding or because of changes in the nature of the ring
embraces exhibited by the aromatic rings of the phosphine ligands.70

31
1.2 X-ray crystallography
1.2.1 Basic concepts in crystallography
Some understanding of the physical and chemical properties of materials in the solid
state can be determined from the arrangement of atoms or molecules in crystals, and
that will lead to obtaining information related to the structure of both molecular and
non-molecular materials.
A crystal is a solid which has long-range three-dimensional internal order of the atoms,
molecules or ions. The structure of a crystal can be understood as the continuous
three-dimensional translational repetition of a basic structural unit, which can contain
one or more types of atoms, and is described starting in section 1.2.1.1.
The scientific study of determining the arrangement of atoms in crystalline solids is
termed as crystallography. The development of this field is behind the improvement of
the X-ray diffraction methods and also the understanding of three-dimensional
structures.71,72
The technique of crystal structure determination by means of X-ray diffraction and will
be explained in detail in section 1.2.2.
1.2.1.1 Crystal structure
A crystal is a solid object consisting of a large number of identical units, which may be
atoms, molecules, or ions that repeat in three-dimensional space.71
There are two kinds
of solid; crystalline and non-crystalline (amorphous) solids. A perfectly crystalline solid
is a highly ordered structure made up of a large number of identical units/molecules,
which are arranged in a precisely regular way repeated in all directions. On the other
hand, the non-crystalline or amorphous state has only a short-range order of the atoms
or unit cells. These two possibilities can usually be determined from their X-ray
diffraction patterns, amorphous solids result in very wide humps in the diffraction
pattern rather than clear diffraction peaks found for crystalline materials.71,73
Figure 8
below shows the difference between crystalline and amorphous solids.

32
Figure 8: Molecular order within (a) a crystalline solid and (b) an amorphous solid.73
1.2.1.2 The crystal lattice and unit cell
A crystal lattice is the representation of the three-dimensional arrangement of atoms,
molecules or ions in the crystalline state as points.74
The lattice point is an imaginary
point which aids to determine these positions and that can be selected by ensuring the
environment surrounding each point is similar.73
An array of possible lattice points
giving rise to a crystal is shown in Figure 9.
Figure 9: Crystal lattice.75
The structure of a crystal consists of a recurring element in three dimensions. This
repeated element is termed as the unit cell. It is held as the foundation of the crystal
structure.
There are two main types of unit cell: primitive and non-primitive. Primitive unit cells
consist of only one lattice point, which arises from the lattice point at each of the
Lattice point

33
corners. Non-primitive unit cells consist of additional lattice points, either on a face of
the unit cell or within the unit cell, in addition to the corner lattice points, and therefore,
have more than one lattice point for every unit cell.76,77
Unit Cell Parameters
A unit cell is characterized by three vectors a, b, and c, which lie along the crystal axes
x, y and z, respectively, with angles between the vectors labelled as α, β and γ. Figure
10 shows the parameters of the unit cell. The faces of the unit cells are explained below:
The unit cell faces are defined as:78
A: edges defined by lattice vectors b and c
B: edges defined by lattice vectors a and c
C: edges defined by lattice vectors a and b
In a similar manner, the inter-facial angles of the unit cell are labelled as:
α: angle between edges b and c
β: angle between edges a and c
γ: angle between edges a and b
Figure 10: Schematic representation of unit cell parameters.
However, the smallest unique part of a crystal structure known as the asymmetric unit
which can be grown to a complete crystalline structure through space group symmetry
operation (including translation), and is often a single molecule but could be a number
of molecules or a fraction of a molecule if the molecule itself possesses symmetry
elements.73

34
1.2.1.3 Crystal systems and the 14 Bravais Lattices
For the large range of crystalline materials known in solids, many choices of unit cell
are possible for any one lattice. There are conventions that are used to guide the choice
of a unit cell. In the absence of any rotational and reflection symmetry, for example, a
conventional unit cell has the shortest possible sides, and angles as close as possible as
to 90°. Presence of rotational and reflection symmetry, on the other hand puts some
limitations on the length of the sides and the angles between them; seven crystal
systems are known, and the shapes of these unit cells are shown in Table 2 with their
lattice parameters.79
Auguste Bravais, in 1850, showed that an including centring crystals can be divided into
14 unit cell types (Table 2). His classification of unit cells was based on the
assumptions that:73
The unit cell is the simplest repeating unit in the crystal
The unit cells have parallel opposite faces
The edge of the unit cells connect equivalent points
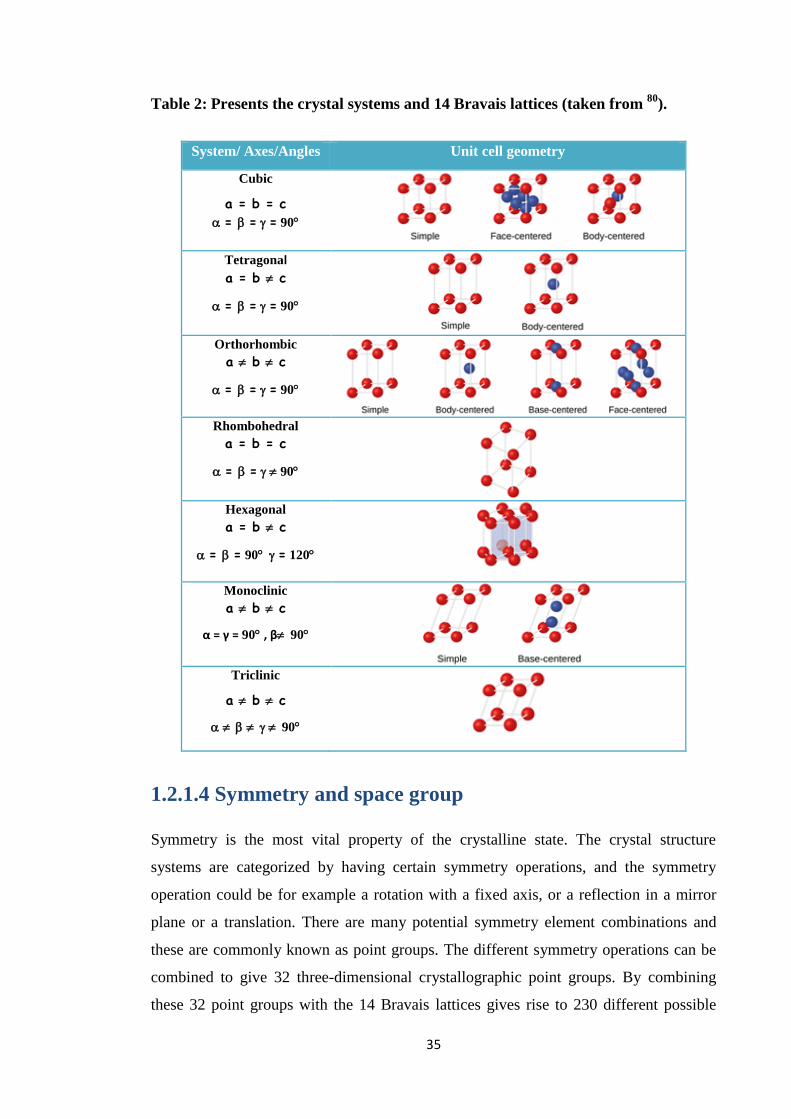
35
Table 2: Presents the crystal systems and 14 Bravais lattices (taken from 80
).
System/ Axes/Angles Unit cell geometry
Cubic
a = b = c
= ==90
Tetragonal
a = b c
= ==90
Orthorhombic
a b c
= ==90
Rhombohedral
a = b = c
= =90
Hexagonal
a = b c
= =90 =120
Monoclinic
a b c
α = γ = 90 , β 90
Triclinic
a b c
90
1.2.1.4 Symmetry and space group
Symmetry is the most vital property of the crystalline state. The crystal structure
systems are categorized by having certain symmetry operations, and the symmetry
operation could be for example a rotation with a fixed axis, or a reflection in a mirror
plane or a translation. There are many potential symmetry element combinations and
these are commonly known as point groups. The different symmetry operations can be
combined to give 32 three-dimensional crystallographic point groups. By combining
these 32 point groups with the 14 Bravais lattices gives rise to 230 different possible

36
arrangements, and each of these arrangements is called a space group that can be
described as a mathematical representation of symmetry operations that are applied to
the asymmetric unit.72
The space groups and their related symmetry operations are characterised in Volume A
of the International Table for Crystallography.81
Table 3 shows the number of space
groups that can be found in each crystal system.
Table 3: Number of space groups in each crystal system.73
Crystal systems Number of space groups
Triclinic 2
Monoclinic 13
Orthorhombic 59
Tetragonal 68
Trigonal 25
Hexagonal 27
Cubic 36
Total 230
Defining the space group of a crystal is important as an operation may reveal some
symmetry in the unit cell’s contents. Furthermore, the space group aids greatly in
simplifying the analysis of a diffraction pattern, since different regions of the pattern
may be identified as identical. The symmetry elements that are found in crystal lattices
can be categorized into two groups, non-translational and translational symmetry
elements.

37
Figure 11: Shows the two symmetry element groups.
The non-translational symmetry operation includes rotation (specifically one, two,
three, four, or six-fold), reflection, inversion and rotary-inversion. All of these elements
do not cause systematic absences in the diffraction data.73
The detail of these symmetry
elements will described in the following.
Inversion centre: An inversion centre or a centre of symmetry is usually recognized by
a darkened or bold point. After an inversion operation, the coordinates of a specific
object at (x, y, z) will change to (-x, -y, -z). In writing, it is also symbolized as a bar
across the top of a number, e.g., 1̅. 73
Reflection: Is a process that takes place across a mirror plane (expressed by the letter
m).73
Rotation: Rotation in crystallography occurs in an anticlockwise direction motion and
takes place on only a fraction of a circle. Expressed in the form of an integer n (n-fold),
where the angle of rotation is 360°/n, there are five types of rotations namely one, two,
three, four, or six fold.73
Rotation-inversion: A rotation-inversion consists of a combination of two symmetry
elements, where a rotation is followed by an inversion that is represented as a bar across
the top of the rotation number, n, e.g., 2̅, 3̅, 4̅, or 6̅. 73
Translation symmetry elements can be specified by a translation, where the translation
of an object is defined as the moving of an object in a particular direction (a, b, c) In
crystallography, there are two types of translation symmetry, namely the glide plane and
symmetry elements
Tranlslation elements
Screw axis Glide
planes
Non translation elements
Inversion centre
Relflection Rotation Rotation-inversion

38
screw axis. These types of symmetry elements can be identified by absences in
diffraction data.
Screw Axis: A screw axis results from a combination of two symmetry operations, a
rotation followed by translation along the direction of axis. It is denoted by two
integers, the first representing the rotation value and the second written as a subscript,
the translation for example, a 21 screw axis is a translation of ½ way along an axis
followed by a two-fold rotation about that axis.73
Glide planes: Similar to a screw axis, glide planes also consist of two symmetry
processes except in this case, after a reflection, there is translation. They are expressed
in the form of the plane along which the glide occurs usually in the form of a, b, c or n
which is used to express the diagonal direction in the unit cell.73
1.2.2 X-ray diffraction
X-ray diffraction (XRD) is an important technique used for structural analysis of
crystalline materials.82
The basic principle behind this technique is similar in some
respects to a microscope. In a microscope a lens collects a portion of the light scattered
by a micro object and refracts it back to the observers’ eye as an image. The diffraction
patterns generated by objects are characteristic of objects and carry key features about
their shape. Unfortunately, the viewing power of a microscope is limited by the
wavelength of light (400-700 nm), and is unable to generate images of objects at a
molecular level. Light waves do not undergo any scattering by individual molecules and
hence are not capable of generating an image at that scale.83
The mismatch between the wavelength of light and the size of crystals in matter was
resolved with the discovery of X-rays. Although discovered in 1895, the nature of
X-rays as waves was not ascertained until 1912. Soon after that, Walter Freidrich and
Paul Knipping, using crystals of copper sulphate, performed the pioneering experiment
of X-ray diffraction.84,85
A year later, Lawrence Bragg successfully determined the
crystal structures of sodium and potassium chloride using XRD.86
For five decades,
crystal structure determination was a slow process taking up-to one year to solve the
structure of a single crystal. The advent of computer technology in 1960s brought a
revolution in the determination of crystal structures by XRD at a pace unprecedented
before. Today, crystal structures containing hundreds of atoms can be analysed within
39
the matter of a few hours. The key features that distinguish this technique from typically
used spectroscopic techniques are that it utilizes a monochromatic beam of X-rays, and
measures any change in the intensity of the beam as it interacts with a solid. Unlike
absorption spectroscopies, XRD is based on the phenomenon of interference, also
known as diffraction, and is capable of providing complete details of structures.83,87
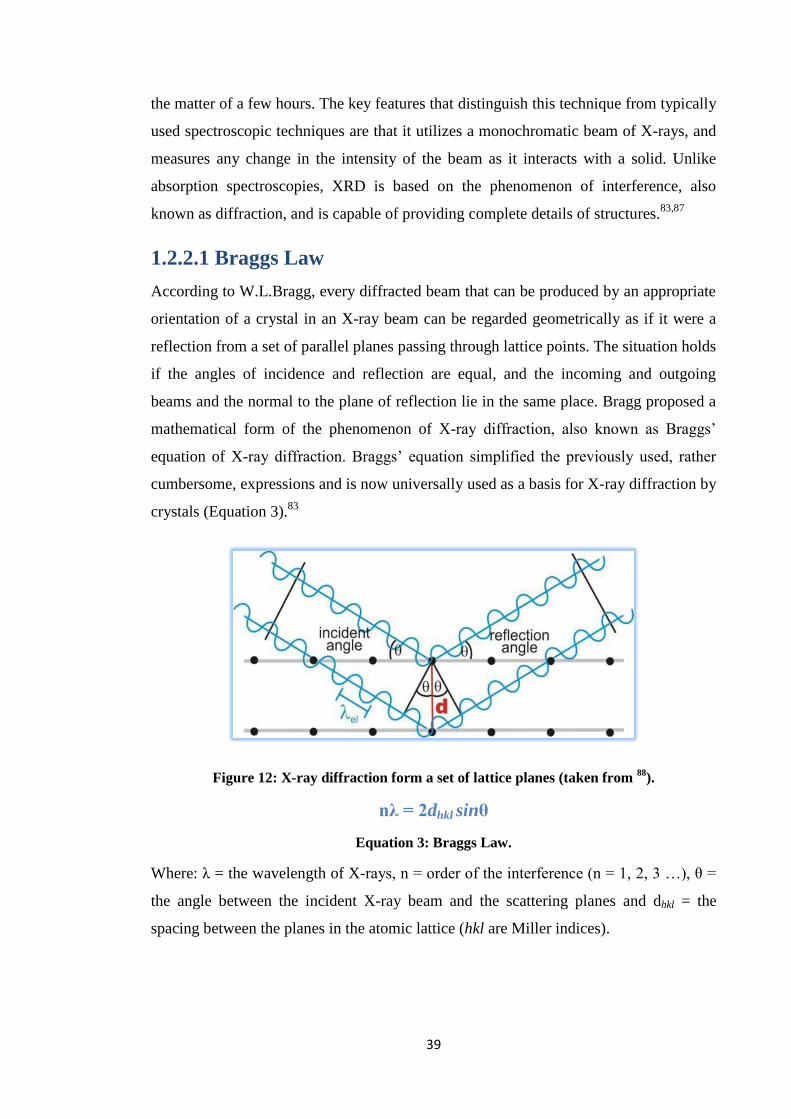
1.2.2.1 Braggs Law
According to W.L.Bragg, every diffracted beam that can be produced by an appropriate
orientation of a crystal in an X-ray beam can be regarded geometrically as if it were a
reflection from a set of parallel planes passing through lattice points. The situation holds
if the angles of incidence and reflection are equal, and the incoming and outgoing
beams and the normal to the plane of reflection lie in the same place. Bragg proposed a
mathematical form of the phenomenon of X-ray diffraction, also known as Braggs’
equation of X-ray diffraction. Braggs’ equation simplified the previously used, rather
cumbersome, expressions and is now universally used as a basis for X-ray diffraction by
crystals (Equation 3).83
Figure 12: X-ray diffraction form a set of lattice planes (taken from 88
).
nλ = 2dhkl sinθ
Equation 3: Braggs Law.
Where: λ = the wavelength of X-rays, n = order of the interference (n = 1, 2, 3 …), θ =
the angle between the incident X-ray beam and the scattering planes and dhkl = the
spacing between the planes in the atomic lattice (hkl are Miller indices).

40
1.2.2.2 Characterization techniques
In practice two types of X-ray diffraction are in common use, these are powder and
single crystal X-ray diffraction.89
Powder X-ray diffraction is a more convenient method
as it does not require a material of good crystallinity, and involves simple sample
preparation. This method is mostly used for the identification of known substances or to
test the purity of a mixture by its powder XRD "fingerprint". However, for
poly-crystalline samples, powder X-ray diffraction is incapable of producing structural
data of high quality.90
Unlike powder X-ray diffraction, single crystal X-ray diffraction requires a good quality
single crystal. Although more time consuming, this technique is capable of generating
detailed structure data about the crystal, e.g. spatial positions of atoms, bond-length,
bond angles, conformations, and three dimensional geometries etc., with high precision
and accuracy. The non-destructive nature of the technique, along with many other
advantageous features, has made this technique an essential tool in many scientific
fields for structure determination of the micro and macromolecules. Single crystal X-ray
diffraction utilises the fact that most solids cool down into crystals of specific geometry.
The idea behind single crystal X-ray diffraction is that when a monochromatic X-ray
beam is passed through a single crystal, the radiation interacts with the electrons in the
atoms, resulting in scattering of the radiation to produce a unique diffraction pattern. By
rotating the crystal in the path of the X-ray beam, multiple images can be recorded on a
detector, and these can be used to build a three dimensional image of the crystal after a
computationally intensive analysis.89-92
1.2.2.2.1 Single crystal X-ray diffractometer and method of analysis
A single crystal X-ray diffractometer consists of an X-ray source, a collimator for beam
focusing, a goniometer to hold and rotate the crystal, and a detector. In addition to these
primary parts, an XRD instrument has a beam-stop to halt the primary X-ray beam from
hitting the detector, and a video microscope to help with positioning the crystal. A
liquid-nitrogen based cryostat is used for data collection at low temperature.89
A schematic flowchart (Figure 13) adopted from71
provides a simple and step-wise
description of the method typically used for XRD analysis of solid crystals.

41
Figure 13: A flowchart describing the procedure of crystal structure determination
(adopted from 71
).
The details of each step will be explained in the following sections.
1.2.2.2.1.1 Obtaining and growing crystals
The quality of a crystal is vital for correct determination of its structure. Although, there
are no hard-and-fast rules for good crystal growth, there are some important measures
than can be useful. Theoretically speaking, the crystallisation process of a compound
should start as soon as its concentration in a solvent gets higher than its solubility
product. The kinetic hindrances involved in the process however, require the solution to
be in supersaturated state. The crystallisation process begins with the phenomenon of
nucleation, which either begins spontaneously or is induced by vibration of particles. A
good control of nucleation is vital to control the quality of the consequently formed
Obtaining and growing crystals
Selection of a suitable Crystal and mounting it for X-ray
study
Measurement of intensity data
Obtain the unit cell geometry and preliminary symmetry
information
Data reduction by applying necessary corrections
Refine the structure model
Solving the structure by: direct methods or Patterson
synthesis
Interpreting the results

42
crystals. A speedy nucleation process, for example, generates too many crystals of small
size, which are not appropriate for a good XRD analysis. Good quality large crystals of
XRD standard are typically 0.1-0.3 mm in each dimension.93,4
The various methods
used for growing crystals are shown in Figure 14 and will be discussed below.
Figure 14: Schematic descriptions of crystallisation methods.
Slow evaporation
The simplest method to grow crystals is slow evaporation. In this method, a saturated
solution of a given material is prepared in a suitable solvent. A few millilitres of the
solution are transferred to another container and left, loosely covered for evaporation of
the solvent. Although easy to perform, the slow evaporation process requires a lot of
material to prepare a saturated solution. Further, a saturated solution can also result in
extensive nucleation and is not suitable for air-sensitive compounds.93,94
Slow cooling
In this method, similar to slow-evaporation, a saturated solution of the solid-to-be-
studied is prepared. The saturated solution is allowed to cool down slowly, either by
leaving it immersed in a water bath or leaving it to cool down to room temperature.
Liquid nitrogen has also been used in variations of the method. The slow-cooling
method works best for moderately soluble substances, but requires a lot of material to
prepare a saturated solution. Amongst the disadvantages of this method is the formation
of twined and disordered crystals from the saturated solution used in this method.93,94
Vapour-diffusion
This method is based on a binary solvent system i.e. typically two solvents that mix
well. One of the solvents with high boiling point and better solubility for the compound

43
to be crystallised, is known as ‘solvent’, while the other, with comparatively low boiling
point and low solubility for the compound-to-be-analysed is known as ‘the precipitant’.
In a typical experimental protocol for this method, a solution of the compound is
prepared in a small open container. The contained is then placed inside a larger
container containing some precipitant. The whole set-up is sealed and allowed to stand.
Over time the precipitant, which is more volatile, will diffuse through the gas phase into
the solvent, leading to oversaturation, nucleation and, if all goes well, finally
crystallization. The diffusion speed can be regulated by varying the temperature. The
challenge of this method lies in finding the correct solvents for the binary mix for this
experiment.93,94
Liquid-liquid diffusion
Similar to the vapour-diffusion method, a liquid-liquid diffusion method is based on a
binary mix. Unlike the former, which utilises boiling points of the solvents as key
selection criteria, the latter requires that the specific densities of the two liquids are
different. In this method, a concentrated solution of the concerned compound is
prepared. A small volume of the liquid with higher specific density is transferred in a
narrow receptacle and carefully layered with the other liquid (this works best with a
syringe and hypodermic needle). Over time, diffusion of the two solvents results in
formation of crystals. In a variation of this method, to promote a clean separation
between the two layers, the lower layer is frozen before adding the second liquid. The
main advantages of this method are that: it works well with small amounts, and involves
easy to controll parameters. Again, as in the vapour-diffusion method, it is a little
complicated to find the solvents with optimum characteristics.95,96
1.2.2.2.1.2 Choosing a suitable crystal
A single crystal of perfect quality is required for XRD analysis. The selection procedure
can be performed by a careful examination of the product under a microscope, and any
crystals with curved, deformed or fragmented structure can be discarded at this stage.
Apparent crystal-like, amorphous structures can be identified with the help of a
polariser mounted on a microscope. A carefully chosen perfect crystal is mounted onto a
glass fibre loop in a film of fomblin oil for X-ray examination.71-73

44
1.2.2.2.1.3 Data collection
In the data collection stage of X-ray analysis, using calculations based on Braggs’ law
the computer determines the crystal orientation, unit cell parameters, and reflection
indices.71
According to a rearranged form of Bragg’s equation (Equation 4), the distance of each
spot from the centre of an X-ray diffraction pattern is proportional to sinθ and 1/dhkl.
Sinθ = (λ/2). (1/ dhkl)
Equation 4: Bragg equation
Figure 15: Show a schematic of four-circle diffractometer (taken from 97
).
At this stage, systematic absences in the diffraction pattern provide useful clues that
help toward assignment of the space group of a crystal. A space group contains
information about the symmetry, including screw axes and glide planes, of a crystal and
position of lattice points in its unit cell. A unit cell centring can also be determined from
an associated pattern of systematic absences. A primitive (P) centred unit cell, for
example, does not have any conditions for observed intensity, and therefore, does not
result in any systematic absences in the diffraction pattern. In a C-centred unit cell on
the other hand, for a reflection to be seen the sum of its indices (h+k) must be an even
number (h + k =2n). All reflections with (h+k) equalling an odd number will be
systematically absent.

45
Similarly, for an F-centred cell, a pattern will be visible if all indices are either all even
or all odd, e.g. hkl = will not appear in the diffraction pattern. In a body centred cell (I),
a reflection will be observed if the sum of its indices is even (h + k +1 =2n). Similar to
centring of a unit cell, crystal symmetry such as screw axis and glides also produces
associated systematic absences, which are observed in the selected parts of the
diffraction pattern.98
Sometimes, several space groups have the same systematic absences and cannot be
distinguished from each other. In such a situation, a trial and error method of
assignment is adopted. In such cases, at the data collection stage, the best practice is to
start with an assumption based on lowest possible symmetry. In structure refinement
however, preference should be given to highest possible symmetry. The primarily
collected data consists of long lists of reflections each with hkl indices and measured
intensity, and requires corrections before further interpretation.98
1.2.2.2.1.4 Data reduction and correction
The data correction step involves conversion of the measured intensities (I) to the
‘observed structure amplitudes’ Fo2, and the corresponding standard uncertainties σ
(Fo2). Important corrections that are made include Lorentz correction, polarization
correction, absorption correction and decay corrections. Lorentz correction is applied
when diffraction from some planes have been recorded for longer than others.
Polarization corrections are applied to allow for partial polarization of the X-ray
reflected beam. Some materials have the capacity to absorb X-rays and show relatively
poor diffraction, absorption corrections are applied to correct for errors resulting from
such situations. In crystals showing signs of degradation, a decay correction is
applied.71,73
1.2.2.2.1.5 Structure solution
The step of structure solution is the process in which a molecular model for a crystal
structure can be calculated and obtained. This process known as phasing, which is
involves solving the phase problem. The process of transforming the measured
diffraction pattern into a picture of the crystal structure which produced it is known as
reverse Fourier transformation. A reverse Fourier transform is used to obtain the

46
electron density from the diffraction pattern which is mathematically represented by the
equation (5):
ρ(xyz) = 1/V Σhkl |F(hkl)| . exp[iΦ(hkl)] . exp[-2πi(hx + ky +lz)]
Equation 5: Constructing an electron density map from the diffraction pattern.71
Where: p(xyz) is the electron density at location (x, y, z) in the unit cell, V is the volume
of the unt cell, F(hkl) are the structure factors and hkl are the Miller indeces.
Based on this equation, the amplitudes |F(hkl)| for the diffracted X-rays have been
measured, and the final exponential term for the contribution of hkl can be calculated
and converted into atomic positions xyz, however, the phases of the reflection are
unknown, so the calculation of the electron density map cannot be directly carried out as
some of the information is missing. This is known as the “phase problem”. There are
two main methods used in order to overcome this problem: direct methods and
Patterson synthesis. The uses of these methods depend on the kinds of molecule being
studied.71,73
The Patterson synthesis is used for structures containing heavy atoms, and for inorganic
and organometallic crystal structures. In contrast to the heavy atom method, direct
methods are used for organic molecules and those that do not involve a heavy atom in
the crystal. This method is a numerical technique based on simple facts such as the
electron density in a unit-cell does not drop below zero at any point.
After using either direct methods or Patterson synthesis it is possible to produce a
Fourier Map of electron density ρ(xyz) within the unit-cell, which allows for an initial
structure to be produced.71,73,94
1.2.2.2.1.6 Refining the structure
Once a part of the molecular structure has been determined, the rest of the atoms in the
molecule can be located. Therefore, the development of a complete crystal structure
model needs the initial structure estimated to be refined. This can be performed based
on a statistical approach known as least squares.

47
The obtained structure model should be compared against the experimental reflection
data in order to observe how well it fits, and to obtain the best agreement between the
observed structure factor and the calculated structure factor, this given by the residual
(R) factor (Equation 6).
R= Σ (ǀ Foǀ– ǀ Fcǀ)/ Σ ǀ Foǀ
Equation 6: Residual (R) Factor.71
Where: |Fc| is the calculated structure factor; |Fo| is the observed structure factor. If the
values of |Fc| and |Fo| are similar, the trial structure should be approximately correct. The
R factor for the complete refinement of the structure is typically around 0.02 and 0.07.
After the model structure is improved, the crystallographic information file (CIF) is
written, which is the standard archive file for crystallographic data.99
In the final step the crystallographer can present and interpret the results, which contain
the geometrical results, for instance: bond lengths, bond angles, torsion angles and
intermolecular geometry, and the CIF can be submitted to the Cambridge
Crystallographic Database when publishing the structure.71,73,97

48
1.3 References
1. G. Morteani and H. Schmidbaur, (ed.): Gold-Progress in Chemistry,
Biochemistry and Technology, Wiley & Sons, Chichester, 1999, p.40.
2. http://en.wikipedia.org/wiki/Gold - (Last assessed date 2017).
3. C. Q. Sun, H. L. Bai, B. K. Tay, S. Li, T.P. Chen, C. Li and E.Y. Jiang, J.
Phys.Chem., 2004, 108, 2162-2167.
4. H. Schmidbaur, Angew. Chem. Int. Ed., 1976, 15, 728-740.
5. P. Schwerdtfeger, P. D. Boyd, A. K. Burrell, W. T. Robinson and M. J.
Taylor, Inorg. Chem., 1990, 29, 3593-3607; P. Pyykkö, Chem. Rev., 1997,
97, 597-636.
6. R. J. Puddephatt, The Chemistry of Gold, Elsevier Scientific Publishing
Company, New York, 1980, p1.
7. P. Schwerdtfeger, Hetero Chem., 2002, 13, 578-584.
8. C. Corti and R. Holliday, Gold: science and applications, 2009: CRC Press.
9. R. D. Rakhimov, K. P. Butin and K. I. Grandberg, J. Organomet., Chem.,
1994, 464, 253-260.
10. J. H. Yip, R. Feng and J. J. Vittal, Inorg. Chem., 1999, 38, 3586-3589.
11. A. S. K. Hashmi and D. F.Toste, Modern Gold Catalyzed Synthesis; Ed.;
Wiley-VCH: Weinheim, Germany, 2012.
12. S. J. Berners-Price and A. Filipovska, Metallomics 2011, 3, 863-873.
13. C. F. Shaw, Chem. Rev., 1999, 99, 2589-2600.
14. K. Nomiya, R. Noguchi, K. Ohsawa and K. Tsuda, J. Chem. Soc., Dalton
Trans., 1998, 4101-4108.
15. A. Gomez-Suarez and S. P. Nolan, Angew. Chem., Int. Ed. 2012, 51,
8156-8159; A. S. K. Hashmi, Angew. Chem., Int. Ed. 2010, 49, 5232-5241.
16. E. R. T. Tiekink and M. W. Whitehouse, Chem. Aust., 1990, 57, 346-348.
17. G. J. Higby, Gold Bull. 1982, 15, 130-140.
18. W. F. Kean and I. R. F. Kean, Inflammo, 2008, 16, 112-125.
19. S. P. Fricker, Gold Bull., 1996, 29, 53-60.
20. S. P. Fricker, Medicinal chemistry of gold compounds, in The Chemistry of
Organic Derivatives of Gold & Silver, John Wiley & Sons Ltd, New York,
1999, pp 641.

49
21. G. Nierop, W. P. Gijzel, E. H. Bel, A. H. Zwinderman and J. H. Dijkman,
Thorax, 1992, 47, 349-354.
22. R. E. Thomas and R. A. Papandrea, Med. J. Aust., 1993, 158, 720.
23. B. A. Becker and A. A. Gaspari, Dermatol. Clin., 1993, 11, 429-452.
24. A. D. Ormerod, Drugs, 1994, 48, 717-730.
25. R. E. Sue and P. J. Sadler, Metal-Based Drugs, 1994, 1, 107-144.
26. D. Schröder, H. Schwarz, J. Hrusak and P. Pyykkö, Inorg. Chem., 1998,
37, 624-632.
27. H. Schmidbaur, Nature (London), 2001, 413, 31-33; M. J. Katz, K. Sakai
and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884-1895.
28. H. Schmidbaur, K. Dziwok, A. Grohmann and G. Müller, Chem. Ber.,
1989, 122, 893-895; H. Schmidbaur, Gold Bull, 1990, 23, 11-21; H.
Schmidbaur, Chem. Soc. Rev., 1995, 24, 391-400.
29. F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur,
Angew. Chem. Int. Ed. Engl., 1988, 27, 1544-1546.
30. S. S., Pathaneni and G. R., Desiraju, J. Chem. Soc., Dalton Trans., 1993,
319-322; R. V. Parish, Hyperfine Interact. 1988, 40, 159-169; M. Melnik,
R. V. Parish, Coor, Chem. Rev., 1986, 70, 157-257; P. G. Jones, Gold
Bull., 1981, 14, 102-118; P. G. Jones, Gold Bull., 1983, 16, 114-124.
31. T. G. Spiro, Progr. Inorg. Chem., 1970, 11, 1; K. A. Gingerich, J. Cryst.
Growth, 1971, 9, 31-45.
32. J. C. Vickery and A. L. Balch, Inorg. Chem., 1997, 36, 5978-5983.
33. H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931-1951.
34. W. Morris, R.L., M.M. Olmstead, and A.L. Balch, J. Am. Chem. Soc.,
2003, 125, 1033-1040.
35. M. Brynda, Coord, Chem., 2005, 249, 2013-2034.
36. J. F. Nixon, Adv. Inorg. Chem., 1985, 29, 41-141.
37. J. S. Dewar, Bull. Soc. Chem. Fr., 1951, 18, C71-C79.
38. J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939-2947.
39. J. Chatt and A. A. Williams, J. Chem. Soc., 1951, 3061-3067.
40. A. G. Orpen and N.G. Connelly, Organomet., 1990, 9, 1206-1210; D.
Gilheany, Chem. Rev., 1994, 94, 1339-1374.
41. S. J. Berners-Price and P. J. Sadler, Inorg. Chem., 1986, 25, 3822-3827.

50
42. C. J. K. Paul and W. N. M. Piet. Van Leeuwen., “Phosphorus (III) Ligands
in Homogeneous Catalysis: Design and Synthesis”. 2012: John Wiley &
Sons. p7.
43. C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953-2956.
44. B. Gumbo, Short-cutting the phosphorus cycle in urban ecosystems,
Balkema, Amsterdam, 2005.
45. C. Tolman, Chem. Rev., 1977, 77, 313-348.
46. A. Immirzi, A. Musco and B.E. Mann, Inorg. Chim. Acta., 1977, 21, L37.
47. C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953-2956; C.A. Tolman,
W.C. Seidel and L. W. Gosser, J. Am. Chem. Soc., 1974, 96, 53-60.
48. J. Mathew, T. Thomas and C.H. Suresh, Inorg. Chem, 2007, 46, 10800-
10809.
49. P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the
Art, Springer, 2004, p11-12.
50. C. Flener Lovitt, G. Frenking and G. Girolami, Organomet., 2012, 31,
4122-4132.
51. P. Leeuwen, Homogeneous catalysis, Kluwer Academic Publishers,
Dordrecht, 2004.
52. M. J. McKeage, L. Maharaj and S. J. Berners-Price, Coord. Chem. Rev.,
2002, 232, 127-135.
53. Z. Assefa, B. G. McBurnett, R. J. Staples, J. P. Fackler, B. Assmann, K.
Angermaier and H.Schmidbaur, Inorg.Chem.,1995, 34, 75-83; Z. Assefa,
B. G. McBurnett, R. J. Staples and J. P. Fackler, Inorg. Chem., 1995, 34,
4965-4972.
54. W. Henderson, B. K. Nicholson and E.R. Tiekink, Inorg. Chim. Acta.,
2006, 359, 204-214.
55. H. Fang, X. Gang. Zhang and S. G. Wang, Phys. Chem. Chem. Phys.,
2009, 11, 5796-5804; T. Tunyogi, A. Deák, G. Tárkányi, P. Király, and G.
Pálinkás, Inorg. Chem. 2008, 47, 2049-2055.
56. K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem.,
1994, 472, 371-376.
57. N. C. Baenziger, W. E. Bennett and D. M. Soboroff, Acta Cryst., Sect. B:
Struct. Crystallogr. Cryst. Chem., 1976, 32, 962-963.

51
58. I. Dance and M. Scudder, J. Chem. Soc., Chem. Commun., 1995, 1039-
1040.
59. R. C. Bott, P. C. Healey and G. Smith, Aust. J. Chem., 2004, 57, 213-218.
60. P. D. Cookson and E. R. T. Tiekink, Acta Cryst., Sect. C: Cryst. Struct.
Commun., 1994, 50, 1896-1898.
61. K. Nunokawa, S. Onaka, T. Tatematsu, M. Ito and J. Sakai, Inorg. Chim.
Acta, 2001, 322, 56-64.
62. U. Monkowius, S. Nogai and H. Schmidbaur, Organomet., 2003, 22, 145-
152.
63. M. Bardaji, P. G. Jones and A. Laguna, J. Chem. Soc., Dalton Trans.,
2002, 3624.
64. G. Jia, R. J. Puddephatt and J. J. Vittal, J. Organomet, Chem., 1993, 449,
211-217.
65. K. K. Banger, R. P. Banham, A. K. Brisdon,W. I. Cross, G. Damant, S.
Parsons, R. G. Pritchard and A. Sousa-Pedares, J. Chem. Soc., Dalton
Trans., 1999, 427.
66. P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133-138;
P. Pyykkö, N. Runeburg and F. Mendizabal, Chem.–Eur. J., 1997, 3,
1451-1457.
67. H. Schmidbaur, Gold Bulletin, 1990, 23, 11-21; H. Schmidbaur, S. Cronje,
B. Djordjevic and O. Schuster, Chemical Physics., 2005. 311, 151-161; H.
Schmidbaur, Gold Bulletin, 2000, 33, 3-10.
68. P. Pyykkö, Chem. Rev., 1988, 88, 563-594; K. S. Pitzer, Acc.Chem. Res.,
1979, 12, 271-276; P. Pyykkö, Adu. Quantum Chem., 1978, 11, 353-409.
69. K. M. Anderson, A. E. Goeta and J. W. Steed, Inorg. Chem., 2007, 46,
6444-6451.
70. N. A. Barnes, K. R. Flower, S. A. Fyyaz, S. M. Godfrey, A. T. McGown,
P. J. Miles, R. G. Pritchard and J. E. Warren, CrystEngComm, 2010, 12,
784-794.
71. W. Clegg, Crystal structure determination. Oxford Chemistry Primers,
2011. 60(1).
72. J. P. Glusker and K.N. Trueblood, Crystal structure analysis: a primer.
Vol. 14. 2010: Oxford University Press.

52
73. L. Ooi, Principles of X-ray crystallography, United States: Oxford
University Press, New York, 2010.
74. askIITians. Crystal Lattices and Unit Cells 2006 [cited 2017; Available
from: http://www.askiitians.com/iit-jee-Solid-State/crystal-lattices-and-
unit-cells.html.
75. http://eguruchela.com/chemistry/learning/Types_of_unit_cell.ph [cited
2017].
76. http://www.doitpoms.ac.uk/tlplib/crystallography3/unit_cell.php [cited
2017]
77. http://home.iitk.ac.in/~sangals/crystosim/crystaltut.html [cited 2017;
Available from: http://www.askiitians.com/iit-jee-Solid-State/crystal-
lattices-and-unit-cells.html.
78. http://home.iitk.ac.in/~sangals/crystosim/crystaltut.html [cited 2017].
79. W. Clegg, A.J. Blake and J.M. Cole, Crystal structure analysis: principles
and practice. [Second edition]: International Union of Crystallography,
2009.
80. https://opentextbc.ca/chemistry/chapter/10-6-lattice-structures-in-
crystalline-solids [cited 2017].
81. M. M. Woolfson, An Introduction to X-ray Crystallography. 2nd Ed.
Cambridge: Cambridge University Press, 1997.
82. B.E. Warren, X-ray Diffraction, 1969: Courier Dover Publications.
83. W. Clegg, A.J. Blake and J.M. Cole, Crystal structure analysis: principles
and practice: International Union of Crystallography, 2001.
84. L. Bragg, The Development of X-ray Analysis. London: G. Bell and Sons
Ltd, 1975.
85. G. H. Stout and L. H. Jensen, X-ray Structure Determination. 2nd Ed.
New York: John Wiley and Sons, Inc, 1989.
86. E. W. Nuffield, X-ray Diffraction Methods. New York: John Wiley and
Sons, Inc, 1966.
87. H. P. Klug and L.E. Alexander, X-ray diffraction procedures: for
polycrystalline and amorphous materials, 2nd Edition, by Harold P. Klug,
Leroy E. Alexander, 1974, 1, pp. 992.
88. http://www.microscopy.ethz.ch/bragg.htm [cited 2017].

53
89. T. Boyd and A.R. Barron, an Introduction to Single-Crystal X-Ray
Crystallography, 2013.
90. J. P. Glusker and K. N. Trueblood, Crystal structure analysis: a primer,
New ed.; Oxford University Press, 1985.
91. L.V. Azaroff and M.J. Buerger, Powder method in X-ray crystallography,
1958.
92. J. Helliwell and M. Helliwell, Chem. Commun., 1996, 14, 1595-1602.
93. R. H. Powell, Molecular structure from X-ray diffraction, Annual Reports
Section" C"(Physical Chemistry), 2010, 106, 192-210.
94. R. A. Keraliya, T. G. Soni, V. T. Thakkar and T. R. Gandhi, Dissolution
Technol., 2010, 1, 16-21.
95. P. D. Boyle, Growing crystals that will make your crystallographer happy,
Department of Chemistry, North Carolina State University, 2007.
96. S. Ramukutty and E. Ramachandran, Journal of Crystal Growth, 2012,
351, 47-50.
97. U. Shmueli, Theories and Techniques of Crystal Structure Determination.
Oxford: Oxford University Press, 2007.
98. W. Clegg, A.J. Blake and J.M. Cole, Crystal structure analysis: principles
and practice. 2009[secound edition]: International Union of
Crystallography.
99. J. Gasteiger and T. Engel, Chemoinformatics: A Textbook. Weinheim:
WILEY-VCH, 2003.

54
Chapter 2
The systematic introduction of fluorine into the phenyl
rings of phosphine ligands and its influence on Au···Au
distances in the solid state structures of [AuX(PR3)]
complexes.

55
2. The systematic introduction of fluorine into the phenyl rings of
phosphine ligands and its influence on Au···Au distances in the solid
state structures of [AuX(PR3)] complexes.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, University of Manchester, Manchester, M13 9PL, UK.
2.1 Abstract
Gold(I) phosphine halides of the type [AuX(PAr3)] (where Ar = (3-FC6H4), (4-FC6H4),
(3,5-F2C6H3), (3,4,5-F3C6H2), and X = Cl, Br, I) have been prepared with the exception
of [AuI{P(4-FC6H4)3}], and structurally characterised in the solid state by single crystal
X-ray diffraction in order to investigate the presence of short Au···Au (aurophilic)
interactions. The resulting two-coordinate gold complexes exhibit Au–X bond lengths
ranging from 2.277(3) to 2.319(3) Å (X = Cl), 2.3843(13) to 2.4248(10) Å (X = Br) and
2.5513(6) to 2.5787(7) Å (X = I), while the Au–P distances were between 2.224(3) and
2.249(2) Å. For all the complexes containing only meta-fluorines, ie (3-FC6H4) or (3,5-
F2C6H3) the Au–Au distances observed are in the range 6.982(2) – 8.3507(7) Å, while
those containing either (4-FC6H4) or (3,4,5-F3C6H2) are found between 3.1273(8) and
4.3592(5) Å. The presence, or otherwise, of short Au···Au interactions as a function of
halide, phosphine steric and electronic parameters has been probed, and secondary C–
H···X, C–H···F and F···F interactions are investigated.
2.2 Introduction
Gold(I) phosphine complexes of the type [AuX(PAr3)] are among the most common in
gold chemistry and have been the subject of considerable interest for their applications,
such as their therapeutic value as anti-arthritic and anti-cancer drugs,1 and their
chemiluminescent properties.2 The supra-molecular chemistry of these compounds also
attracts much interest, owing to the presence of Au···Au interactions that are shown to
exist in some of these complexes in the solid state,3 although there is still little
understanding of exactly what factors determine the presence, or otherwise, of
aurophilic interactions in a particular complex.

56
The attraction between two gold atoms with closed shell 5d10
configurations, result in
dispersive interactions called “aurophilic” interactions,4 which have been used widely to
explain various kinds of gold-gold interactions, in and among gold complexes.5,6
According to theoretical studies, hybridization between the filled 5d and empty 6s/6p
shells of gold are not the origin of aurophilic interactions, instead they arise from
dispersive, inductive and electrostatic interactions arising from relativistic effects.7-11
The Au···Au distance detected in complexes showing aurophilic interactions commonly
lie between that of the Au2 molecule in the gaseous state (2.47Å) and twice the van der
Waals radius of gold (ca. 3.60 Å).5 The strength of such interactions are greater than
commonly observed for van der Waals forces (approximately 21-46 k J mol-1
),12
and
often equivalent in power to hydrogen bonding. In the domain of crystal engineering,
such relationships are of great importance and have been proposed as a method of
designing particular structural motifs.13
In monomeric gold (I) phosphine halide compounds the aurophilic interaction usually
results in the formation of dimers, where two monomeric [XAuL] units are joined
together, typically in a perpendicular (crossed-swords motif) manner, although
connection in a parallel or antiparallel alignment has also been demonstrated.14,15 In
addition to the observation of dimers, examples are also known where monomeric units
demonstrate extended interactions, for instance [AuCl(PMe3)]n is found as an elongated
infinite chain,16
and [AuCl(PPhMe2)] crystallises in both trimeric and dimeric forms.15
It
has been shown that greater aurophilic interactions exist in complexes that contain the
more polarizable halides, and that neutral strongly donating ligands, such as phosphines
and NHCs result in more significant aurophilic interactions than those that are less
donating.17,18 These interactions are most commonly detected for trialkyl phosphines
with relatively small cone angles,14,15 for example [AuCl(PMe3)],
19-21
[AuBr(PMe3)]19-21
and [AuCl(PEt3)]22
all have short Au···Au contacts, varying from
3.27–3.98 Å. A similar situation arises with the gold(I) halides of tertiary-substituted
arylphosphines of the type [AuX(PAr3)] (where X = Cl, Br, I) in that short Au···Au
distances are considered more likely to arise when the steric impact of the substituents
is low, and a wide variety of crystal arrangements of this type of complex have been
obtained.23–31
According to studies of the CSD database, though, the size of a phosphine per se is not
a fool-proof indicator of the likelihood of aurophilic interactions, for example in the
complex of P(p-CH3C6H4)3 (cone angle = 145º) an aurophilic relation is detected,

57
d(Au···Au) = 3.375Å,28
whilst for PPh3 (cone angle also 145º) the closest Au···Au
contacts are over 6.9 Å.32
Similarly crystallographic studies have revealed that bulky
triaryl phosphines do not necessarily prevent the formation of Au···Au interactions. For
instance, the compounds [AuCl{P(3-CF3C6H4)3}] and [AuCl{P(3,5-(CF3)2C6H3)3}]
demonstrate Au···Au contacts of 3.0738(9) and 3.341(1) Å,33
correspondingly.
We have previously reported that a short Au···Au distance is observed in the solid state
structure of [AuCl{PPh2(CF=CF2)}], d(Au···Au) = 3.1945(6) Å,34
whereas in
[AuCl{PPh2(CF=CHF)}] the shortest Au···Au distance is 8.081 Å.34
It is unlikely that
replacement of a single fluorine of the trifluorovinyl group with hydrogen will have a
very large influence on either the steric or electronic properties of the phosphine,
suggesting that the factors that result in a change in crystal habit, from C2/c to P21/n
and observation of very different Au···Au distances in two otherwise very similar
complexes are more subtle than can be predicted on the basis of the cone angle or
donating properties of the phosphine alone. Similarly, contrary to expectations, is the
observation of a longer Au···Au interaction in [AuCl(PPh3)], d(Au···Au) = 6.904 –
7.004 Å, [CTPPAU]23
compared with that found for the larger and less donating ligand
system, [AuCl{P(C6F5)3}], d(Au···Au) = 3.783 Å [MUVSIF].26
Here we report an
investigation of a systematic introduction of fluorine into the phenyl rings of a
phosphine ligand and the effect this has on the presence, or otherwise, of aurophilic
interactions in the solid state structures.
2.3 Experimental
2.3.1 Synthetic procedure
All syntheses were carried out using standard glassware unless otherwise stated. The
solvents hexane, EtOH and CH2Cl2, were used as supplied. Other chemicals were
obtained from commercial sources and used as received. 31
P{1H} NMR spectra were
recorded as CDCl3 solutions on a Bruker Advance III 400 MHz spectrometer operating
at 161.975 MHz. All [AuX(PAr3)] (X = Cl, Br, I) complexes were synthesized via
literature methods34,35 as shown in scheme 1, and the crystals were grown at room
temperature. All glassware and equipment was carefully cleaned to minimise
contamination and to encourage the formation of larger crystals.

58
2.3.2 Synthesis of gold (I) phosphine complexes
AuCl(tht): K[AuCl4] (0.5 g, 1.3 mmol) was dissolved in ethanol (12.5 cm3) and water
(5 cm3). Tetrahydrothiophene (2.75 cm
3, 0.03 mmol) was added dropwise, and the
reaction mixture was stirred for 15 minutes, during which time the solution mixture
changed from yellow to white in colour. The mixture was filtered and the white solid
was dried in vacuum.
[AuCl{P(4-C6H4F)3}], 1a: A solution of P(4-C6H4F)3 (0.132 g, 0.31 mmol) in
dichloromethane (5 cm3) was added to AuCl(tht) (0.1 g, 0.3 mmol) and the mixture was
allowed to stir overnight, after which time the volatiles were removed in vacuo. This
resulted in a white solid compound.31
P {1H} NMR (CDCl3): δ 30.70 ppm (s).
[AuBr{P(4-C6H4F)3}], 1b: To 1a (0.1 g, 0.18 mmol) dissolved in EtOH (10 cm3) was
added LiBr (0.016 g, 0.2 mmol) and the solution was left to stir under 80 ºC, after that
time the solution was placed under vacuum. The resulting gold complex was extracted
in to dichloromethane, from which crystals were grown. 31
P{1H} NMR (CDCl3): δ
32.82 ppm (s).
[AuCl{P(3-C6H4F)3}], 2a: P(3-C6H4F)3 (0.132 g, 0.31 mmol) was added to AuCl(tht)
(0.1 g, 0.3 mmol) in CH2Cl2 (5 cm3) and the mixture was allowed to stir overnight, then
the volatiles were removed in vacuo, to afford a white solid product. 31
P{1H} NMR
(CDCl3): δ 33.26 ppm (s).
[AuBr{P(3-C6H4F)3}], 2b: To 2a (0.1 g, 0.18 mmol) dissolved in EtOH (10 mL) was
added LiBr (0.016 g, 0.2 mmol) and the solution was left to stir under 80 ºC, then the
solution volatiles were removed in vacuo. The title compound was dissolved in CH2Cl2,
from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 34.75 ppm (s).
[AuI{P(3-C6H4F)3}], 2c: In 50 mL around bottom flask 2a (0.1 g, 0.18 mmol) was
dissolved in EtOH (10 mL) and KI (0.03 g, 0.18) was added. The solution was left to
stir under 80 ºC, after that time the mixture was left under vacuo, to afford the desired
product. 31
P{1H} NMR (CDCl3): δ 36.00 ppm (s).
[AuCl{P(3,5-C6H3F2)3}], 3a: A solution of P(3,5-C6H3F2)3 (0.115 g, 0.31 mmol) in
CH2Cl2 (5 cm3) was added to AuCl(tht) (0.1 g, 0.3 mmol) and the mixture was allowed
to stir overnight, after that the volatiles were removed in vacuo. This resulted in a white
solid product. 31
P{1H} NMR (CDCl3): δ 36.06 ppm (s).

59
[AuBr{P(3,5-C6H3F2)3}], 3b: To 3a (0.1 g, 0.16 mmol) dissolved in EtOH (10 mL) was
added LiBr (0.03 g, 0.18). The solution was left to stir under 80 ºC, after that time the
mixture was left under vacuum. The resulting gold complex was dissolved in CH2Cl2,
from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 37.71 ppm (s).
[AuI{P(3,5-C6H3F2)3}], 3c: To (0.1 g, 0.16 mmol) 3a dissolved in (10 mL) of ethanol
was added KI (0.027 g, 0.16 mmol) and the solution was left to stir under 80 ºC, then
the solution volatiles were removed in vacuo. The resulting gold compound was
dissolved in CH2Cl2, from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 40.72
ppm (s).
[AuCl{P(3,4,5-C6H2F3)3}], 4a: A mixture of (0.13 g, 0.31 mmol) P(3,4,5-C6H2F3)3 in
CH2Cl2 (5 cm3) was added to AuCl(tht) (0.1 g, 0.3 mmol), then the mixture was stirred
overnight. The volatiles were removed in vacuo affording the compound. 31
P{1H} NMR
(CDCl3): δ 36.83 ppm (s).
[AuBr{P(3,4,5-C6H2F3)3}], 4b: To 4a (0.1 g, 0.15 mmol) dissolved in EtOH (10 mL)
was added LiBr (0.013 g, 0.15 mmol), then the mixture was stirred under 80 ºC. The
solution was then removed under vacuo. The title compound was resulted and dissolved
in dichloromethane, from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 38.27
ppm (s).
[AuI{P(3,4,5-C6H2F3)3}], 4c: 4a (0.1 g, 0.15 mmol) was dissolved in ethanol (10 mL),
KI (0.025 g, 0.15 mmol) was added and the mixture was stirred for 15 minutes under 80
ºC, then solution was removed under vacuo, and the title compound was dissolved in
dichloromethane, from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 41.16
ppm (s).
2.3.3 X-ray Data collection and structures Refinement
Crystals of complexes 1b, 3a, 3b and 4b were obtained by using slow evaporation from
saturated solutions in CH2Cl2 and 1a, 2a, 2b, 2c, 3c, 4a and 4c grown by vapour
diffusion of hexane into a saturated CH2Cl2 solution of compounds at room temperature.
The reflection data were collected at 150 k by use of the Supernova for 1a, 1b, 3a, 3b,
3c, 4a and 4c using graphite monochromated Mo Kα radiation ( λ=0.71073 Å) with the
CCD detector, and those for 2a, 2b, 2c and 4a on an Oxford Xcalibur2 diffractometer at
293 K using graphite monochromated Mo Kα radiation ( λ=0.71073 Å) with the CCD
detector. The crystallographic and structural refinement data are summarized in Table 1.

60
All Structural data were solved by direct methods using SHELXT 36
and refined on F2
by full-matrix least squares using SHELXL within the OleX2.37
All non-hydrogen
atoms were refined with anisotropic parameters; hydrogen atoms were added in
idealised locations. The structures were manipulation and displayed using the
MERCURY program.38
Moreover, the Cambridge Crystallography Database Centre
(CCDC) and Conquest program were used to compare the structures with any existing
data, also to check if each structure had been previously submitted. The structure of 3c
was solved with a disordered CH2Cl2 molecule. Also, the three fluorine atoms in one
phenyl ring are disordered over two positions in structures 2a and 2b, with refined site
occupancies of 0.74: 0.26 and 0.615: 0.385 for 2a and 0.60: 0.40 and 0.811: 0.189 for
2b. Additionally, one of the three fluorine atoms in phenyl ring is disordered over two
positions in 2c structure and was fixed at occupancies of 0.5: 0.5.
2.4 Results and discussion
2.4.1 Synthesis and structural characterization of the complexes
A series of gold(I) triarylphosphine halide complexes (halide = Cl, Br, I) were prepared
from four related phosphines, two bearing a single fluorine atom in either the meta or
para position, ie P(3-C6H4F)3 and P(4-C6H4F)3, as well as the difluorinated phosphine,
P(3,5-C6H3F2)3, and the phosphine in which all of the meta and para positions were
fluorinated, ie P(3,4,5-C6H2F3)3. We did not include the phosphines with ortho-fluorine
substituents because this is likely to result in a large change in the steric properties of
the phosphines. Thus, the complexes [AuCl{P(4-C6H4F)3}]1a, [AuBr{P(4-C6H4F)3}]1b,
[AuCl{P(3-C6H4F)3}]2a, [AuBr{P(3-C6H4F)3}]2b, [AuI{P(3-C6H4F)3}]2c,
[AuCl{P(3,5-C6H3F2)3}]3a, [AuBr{P(3,5-C6H3F2)3}]3b, [AuI{P(3,5-C6H3F2)3}]3c,
[AuCl{P(3,4,5-C6H2F3)3}]4a, [AuBr{P(3,4,5-C6H2F3)3}]4b and [AuI{P(3,4,5-
C6H2F3)3}]4c were synthesized according to the literature procedures,34,35
as shown in
scheme 1. For all of the resulting complexes, a shift in the 31
P{1H} NMR resonance of
the phosphine ligand is detected of ca. 30 to 41 ppm to higher frequencies compared
with the starting phosphines, which confirms the coordination of the ligand.

61
Scheme 1 Synthesis of gold(I) halide phosphine complexes.
Crystals of complexes 1b, 3a, 3b and 4b were grown by slow evaporation of a saturated
CH2Cl2 solution of the compounds, and crystals of complexes 1a, 2a, 2b, 2c, 3c, 4a and
4c were grown by vapour diffusion of hexane into saturated CH2Cl2 solution of
compounds. Unfortunately we were unable to obtain X-ray quality crystals of complex
1c. For the remaining complexes single crystal X-ray diffraction data was obtained, and
crystallographic parameters and data are summarised in Table 1. Selected bond lengths
and angles determined for these complexes are given in Table 2.

62
Table 1 Crystal data and structure refinement parameters for complexes
1a 1b 2a 2b 2c 3a 3b 3c 4a 4b 4c
Formula C18H12AuClF3P C18H12AuBrF3P C18H12AuClF3P C18H12AuBrF3P C18H11AuF3IP C18H9AuClF6P C18H9AuBrF6P C18H9AuF6IP·0.5(CH2Cl2)
C36H12Au2Cl2F18P2 C36H12Au2Br2F18P2 C36H12Au2F18I2P2
FW 548.66 593.12 548.48 592.93 639.10 602.64 647.10 736.55 1313.23 1402.15 1496.13
Crystal system Triclinic Triclinic Orthorhombic Orthorhombic Orthorhombic Orthorhombic Orthorhombic Monoclinic Triclinic Triclinic Triclinic
Space group P-1 P-1 P212121 P212121 P212121 P212121 P212121 P21/c P-1 P-1 P-1
a, b, c (A˚ )
8.5103 (5), 8.9794 (5),
11.8046 (7)
8.3356 (5), 9.2497 (5),
11.7918 (7)
10.4024 (6), 12.3014 (7),
13.2226 (9)
10.3318 (6), 12.3731 (7),
13.5000 (9)
10.4203 (8), 12.3403 (9),
14.0931 (13)
10.3597 (5), 12.7160 (5),
13.5327 (5)
10.3197 (5), 12.6200 (5),
13.8667 (5)
9.8077 (5), 13.7626 (5),
16.5593 (9)
10.5641 (11), 14.2052 (17),
14.3111 (15)
10.0678 (3), 14.3463 (7),
15.0508 (7)
10.1524 (8), 14.6698 (11),
15.0844 (14)
α, β, γ (º)
87.074 (5), 71.970 (5),
79.435 (5)
88.174 (5), 72.400 (5),
80.557 (5)
90, 90, 90 90, 90, 90 90, 90, 90 90, 90, 90 90, 90, 90 90, 103.354
(5), 90
62.039 (11), 77.722 (9), 78.744
(9)
116.001 (5), 93.013
(3), 100.855 (3)
118.413 (9), 90.713 (7),
102.081 (7)
V (A˚ 3) 843.22 (9) 854.72 (9) 1692.03 (18) 1725.79 (18) 1812.2 (3) 1782.72 (13) 1805.92 (13) 2174.73 (18) 1842.1 (3) 1896.29 (14) 1915.7 (3)
Z 2 2 4 4 4 4 4 4 2 2 2
Crystal size
(mm3)
0.1 × 0.09 ×
0.06
0.08 × 0.04 ×
0.06
0.16 × 0.14 ×
0.03
0.2 × 0.18 ×
0.09
0.12 × 0.08 ×
0.06
0.19 × 0.12 ×
0.03 0.16 × 0.12 ×
0.08
0.12 × 0.1 ×
0.02 0.14 × 0.06 × 0.04 0.12 × 0.1 × 0.08 0.13 × 0.09 × 0.06
Temperature
(K) 150 150 293 293 293 150 150 150 293 150 100
Dx (Mg m_3) 2.161 2.305 2.138 2.283 2.342 2.245 2.380 2.250 2.368 2.456 2.594
µ (MoKa)
(mm_1) 9.00 11.06 8.97 10.96 9.93 8.55 10.51 8.43 8.31 10.04 9.46
F(000) 516 552 1016 1104 1172 1128 1200 1356 1224 1296 1368
Theta Min-
Max, degrees 3.6°–26.0° 3.6°–29.3° 3.0–26.5° 3.0–26.0° 2.9°, 26.0° 3.4°, 26.0° 3.4°, 26.0° 3.3°, 27.5° 2.9°, 26.5° 3.3°,26.0° 3.1°,26.5°
N (total) 6180 6453 4539 4492 3894 4816 5218 9085 10369 13769 10811
N (unique) 3308 3873 3387 3050 3167 3378 3290 4781 7514 7444 7830
Observed Data 2946 3386 2627 2611 2847 2984 2785 3397 5261 5702 5831
R, wR2 ,S 0.049, 0.085,
1.06
0.041, 0.066,
1.02
0.058, 0.127,
1.07
0.050, 0.125,
1.05
0.045, 0.124,
1.09
0.037, 0.062,
1.04
0.043, 0.057,
0.99
0.052, 0.095,
1.02
0.060, 0.146,
1.07 0.046, 0.080, 1.00
0.053, 0.095,
0.95
Min. and Max.
Resd Density /
e Å -3
-1.80, 1.92 -1.28, 1.33 -2.03, 3.01 2.79, -1.25 -1.07, 2.00 -0.99, 1.30 -0.87, 1.58 -1.75, 1.97 -3.76, 3.11 -1.37,1.50 -1.65, 1.92

63
All the complexes adopt essentially linear coordination at the Au centres with P–Au–X
bond angles ranging from 173.66 (11)° to 177.8 (19)° for P–Au–Cl systems, 171.81(6)°
to 177.37 (12)° for P–Au–Br complexes, and 170.01(7)° to 177.83 (6)° for P–Au–I
systems. The larger deviations from linearity occur where significant intermolecular
Au···Au interactions are observed, e.g. for the series of [AuX{P(3,4,5-C6H2F3)3}]
complexes, X = Cl: Au···Au, 3.1273(7) Å; P–Au–Cl, 173.66(11)°, X = Br: Au···Au,
3.2322(5)Å; P–Au–Br, 171.81(6)° and X = I: Au···Au, 3.2341(7) Å; P–Au–I,
170.01(7)°.
Table 2 Selected bond lengths and angles for complexes 1a– 4c
Complex Au-P (Å) Au-X (Å) P-Au-X (°) Au···Au (Å) H···Xav (Å) (C–H···F)av (Å) F···Fav (Å)
1a 2.227(2) 2.299(2) 176.12(8) 4.3592(5) 2.884 2.5404 -
1b 2.2338(16) 2.4181(7) 175.91(4) 4.3260(4) 2.923 2.659(1) -
2a 2.228(4) 2.280(4) 177.8(19) 6.982(2) 2.846 2.415 -
2b 2.230(4) 2.386(2) 177.37(12) 7.121(1) 2.963 2.43 -
2c 2.253(5) 2.5583(14) 176.93(11) 7.179(1) 3.155 2.405 2.90(9)
3a 2.224(3) 2.277(3) 177.51(10) 7.1873(6) 2.807 2.571 2.86(1)
3b 2.227(3) 2.3843(13) 176.5(9) 7.2895(6) 2.827 2.343 2.86(1)
3c 2.249(2) 2.5513(6) 177.83(6) 8.3507(7) 3.18 2.575 2.76(1)
4a 2.225(3)
/2.228(3)
2.315(3)
/2.319(3) 176.83(11)
/170.50(12) 3.1273(7) 2.624 2.442 2.833(1)
4b 2.232(2)
/2.234(2)
2.4050(10)
/2.4248(10)
174.31(6)
/169.32(6) 3.2322(5) 2.826 2.547 2.835(8)
4c 2.248 (2)
/2.246(3)
2.5787(7)
/2.5769(9) 165.35(7)
/174.67(7) 3.2341(7) 3.043 2.485 2.821(1)
2.4.2 Structure determination of complexes [AuX{P(4-C6H4F)3}], X = Cl, 1a, X = Br,
1b.
Compounds 1a and 1b both crystallize in the triclinic space group P-1 with two
molecules in the unit cell and they adopt isomorphous structures. The molecular
structures of 1a and 1b are shown in Fig. 1(a-b), respectively.

64
Fig. 1 View of the molecular structures with the atom numbering scheme of 1a (a) and 1b (b).
Thermal ellipsoids shown at the 50% probability level.
The crystal structure of 1a has been reported before, [LAQBUB]39
and while the Au–P
distance we determine (2.227(2) Å) is similar to that reported previously, 2.224(2) Å,
the Au–Cl distance of 1a (2.299(2) Å) is 7 longer than the previous determination,
2.285(2) Å. The structure of complex 1b has not been reported before, however the Au–
Br bond length in 1b (2.4181(7) Å) is similar to that of the related complex
[AuBr(PPh3)] d(Au–Br) = 2.407(6) Å,
24 and slightly longer than for [AuP(ptol)3Br]
d(Au–Br) = 2.391(5) Å.28
Both 1a and 1b show very similar Au···Au separations of 4.3592(5) and 4.3260(4) Å,
respectively, which are somewhat longer than twice the sum of the van der Waals radius
of gold (ca. 3.60 Å). However, intermolecular interactions which are less than the sum
of the relevant van der Waals radii are observed in the crystal packing of both 1a and 1b
(Fig. 2(a-b)). The gold-bound halide forms a number of hydrogen bond interactions, for
1a these are to two ortho hydrogens of one phenyl ring (H2 and H6), although one of
these is only just less than the sum of the van der Waals radii, and to one meta hydrogen
of another ring (H15) with dav(H···Cl) = 2.884 Å. In the case of complex 1b, the
bromide makes two contacts, to corresponding ortho C–H protons, dav(H···Br) = 2.923
Å. Both structures also show C–H···F interactions (see later) formed between a fluorine
in the para-position of one phenyl ring and an ortho-hydrogen of an adjacent molecule,
the most significant of which is d(H18···F1) = 2.432 Å (X = Cl), 2.453 Å (X = Br) as
shown in Fig. 3.
(a) (b)

65
Fig. 2 A view of the crystal packing of 1a (a) showing H···Cl contacts and 1b (b) showing
H···Br contacts, along the a-axis.
Fig. 3 A view of the crystal packing of 1a showing H···F contacts, along the a-axis.
2.4.3 Structure determination of complexes [AuX{P(3-C6H4F)3}], X = Cl, 2a, X = Br,
2b and X = I, 2c.
We were able to successfully grow crystals of all three of the gold halide complexes of
P(3-C6H4F)3, 2a, 2b and 2c. These three complexes are isomorphous, and crystallise in
(a) (b)
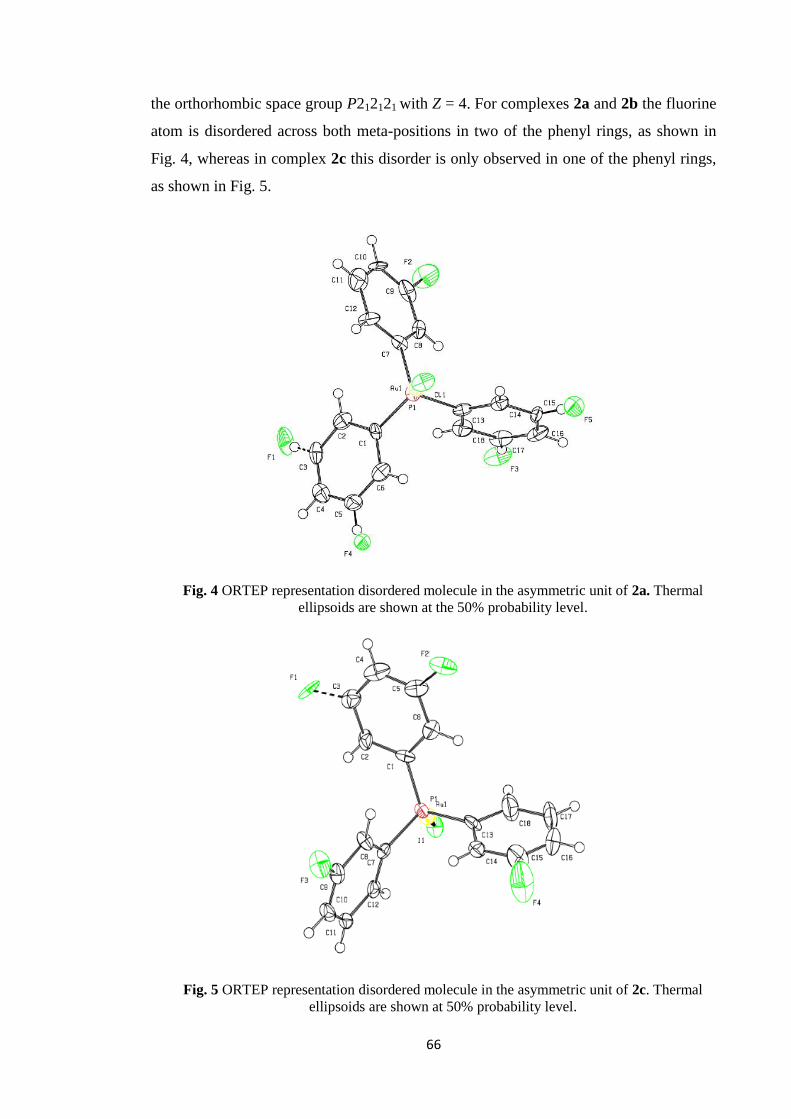
66
the orthorhombic space group P212121 with Z = 4. For complexes 2a and 2b the fluorine
atom is disordered across both meta-positions in two of the phenyl rings, as shown in
Fig. 4, whereas in complex 2c this disorder is only observed in one of the phenyl rings,
as shown in Fig. 5.
Fig. 4 ORTEP representation disordered molecule in the asymmetric unit of 2a. Thermal
ellipsoids are shown at the 50% probability level.
Fig. 5 ORTEP representation disordered molecule in the asymmetric unit of 2c. Thermal
ellipsoids are shown at 50% probability level.

67
The crystal structure of complex 2a has been reported once before, [XAKGOH]40
and
the Au–P bond lengths determined here and reported previously are in good agreement,
as is the shortest Au···Au contact of 6.988 Å observed before,40
and that found here,
6.982(2) Å. On going from complex 2a to 2b and 2c increases are observed in both the
Au–P and Au–X bond lengths, while the closest Au···Au distances are similar at
6.982(2), 7.121(1) and 7.179(1) Å for 2a, 2b and 2c, respectively. These distances
suggest that there are no significant gold-gold interactions in the solid state. However, a
number of intermolecular H···X hydrogen bonds are observed in the extended structures
of these molecules as shown in Fig. 6 (a-b). These involve the gold-bound halide
forming a hydrogen bonding interaction with, in the case of 2a and 2b, one meta and
two para-hydrogens, while 2c only shows a weak interaction with single para hydrogen.
Once again each complex exhibits C–H···F–C interactions that are considerably less
than the sum of the relevant van der Waal’s radii, involving the meta-fluorine and a
para-C–H, as shown in Fig. 7(a-b), with d(H16···F2) = 2.415 in 2a, d(H16···F2) =
2.435 in 2b and d(H4···F2) = 2.443, d(H10···F3) = 2.373 Å in 2c).
Fig. 6 View of the crystal packing of 2a (a) showing H···Cl contacts and 2b (b) showing H···Br
contacts, along the a-axis.
(a) (b)

68
Fig. 7 View of the crystal packing of 2a (a) and 2b (b) showing H···F contacts, along the c-axis.
Fig. 7 (c) A view of the crystal packing of 2c showing H···F contacts, viewed down the b-
axis.
(a) (b)
(c)

69
2.4.4 Structure determination of complexes [AuX{P(3,5-C6H3F2)3}], X = Cl, 3a, X =
Br, 3b and X = I, 3c.
Increasing the amount of fluorination on the aromatic rings, by using the phosphine
ligand P(3,5-C6H3F2)3 resulted in the formation of the three complexes 3a, 3b and 3c as
white solids. Compounds 3a and 3b are isomorphous and crystallise as orthorhombic
crystals in the P212121 space group with Z = 4, whereas compound 3c crystallises in the
monoclinic space group P21/c and has four molecules in the unit cell, along with one
molecule of disordered CH2Cl2. In all three complexes the anticipated approximately
linear-at-gold molecular structures are observed, as shown in Fig. 8 (a-b-c).
Fig. 8 View of the molecular structures of 3a (a), 3b and 3c (c) with the numbering scheme.
Thermal ellipsoids are shown at the 50% probability level. (Solvent of dichloromethane of 3c
omitted for clarity).
(a) (b)
(c)

70
The Au–P bond lengths of complexes 3a and 3b are almost identical to those found in
the related complex [AuCl(PPh3)] d(Au–P) = 2.235(3) Å,
23 whilst the Au–P distance of
3c is slightly longer. The closest Au···Au distances in 3a, 3b and 3c are 7.1873(6),
7.2895(6) and 8.3507(8) Å, respectively, all slightly longer than those found for
complexes 2a, 2b and 2c, and much longer than twice the van der Waals radius of gold,
suggesting that there are no significant Au···Au interactions in any of these complexes.
In the crystal packing of 3a and 3b intermolecular hydrogen bonding interactions
between C–Hortho···F and C–Hpara···X are detected, as shown in Fig. 10 (a-b). For
complex 3c no hydrogen bonding is observed between the iodide and any of the phenyl
ring hydrogens, instead there are interactions with the dichloromethane molecule.
Because of this difference it is probably not sensible to try and draw too many
comparisons between the interactions observed in 3c with those found in 3a and 3b. In
both 3a and 3b there are some distances between the meta- fluorine atoms of
neighbouring molecules, with F5···F6 = 2.878(9), F4···F6 = 2.922(9) Å for 3a, F5···F6
= 2.86(1) Å for 3b, as shown in Fig. 9, which are less than twice the van der Waals’
radius of fluorine (2.94 Å).
Fig. 9 View of the crystal packing of 3a (a) showing H···Cl contacts and 3b (b) showing H···Br
contacts, viewed down a-axis.
(a) (b)

71
Fig. 10 View of the crystal packing of 3a (a) and 3b (b) showing F···F contacts, viewed down
a-axis.
2.4.5 Structure determination of complexes [AuX{P(3, 4, 5-C6H2F3)3}], X = Cl, 4a, X
= Br, 4b and X = I, 4c.
When the P(3,4,5-C6H2F3)3 phosphine ligand was used complexes 4a, 4b and 4c are
formed. These crystallise in the triclinic space group P-1 with two molecules in the unit
cell. The molecular structures of 4a and 4c are illustrated in Fig. 11 (a-b).
(a)
(a) (b)

72
Fig. 11 View of the molecular structures of 4a (a) and 4c (b) as a dimer. Thermal ellipsoids are
shown at the 50% probability level.
The P–Au bond lengths found in 4a (2.224(4) and 2.229(4) Å), 4b (2.232(3) and
2.234(2) Å) and 4c (2.247(3) and 2.246(2) Å) increase slightly as the nature of the
halide changes, but are much as expected based on the Au–P bond lengths found in the
related complexes [AuCl(PPh3)]23
and [AuCl{P(C6F5)3}]41
which have Au–P distances
of 2.235(3) and 2.215(9)/2.206(9) Å, respectively. The Au–Cl bond distances of 4a are
(2.315(3) and 2.320(4) Å), however, slightly longer than those found in both
[AuCl{P(C6F5)3}]41
d(Au–Cl )=2.269(9)/2.271(9) Å and [AuCl(PPh3)]23
d(Au–Cl)=
2.279(3) Å.
The most interesting observation for this series of [AuX{PAr3}] complexes is that an
aurophilic interaction is observed in each complex, with the Au···Au separation
distance in 4a, 4b and 4c being 3.1273(7), 3.2322(5) and 3.2341(7) Å with torsion
angles of P–Au–Au–P are 114.2(1)°, 117.66(9)° and 113.7(1)°, respectively. All of
these distances are less than twice the van der Waals radius of gold, 3.60 Å, and all are
shorter than the Au···Au distances in both [AuCl(PPh3)], d(Au···Au) = 6.904 – 7.004 Å
[CTPPAU]23
, [AuCl{P(C6F5)3}], d(Au···Au) = 3.783 Å [MUVSIF]41
, as well as the
distances found in complexes 1a and 1b (4.3592(5) and 4.3260(4) Å).
The crystal packing of these complexes shows H···X hydrogen bonding interactions
between the gold-bound halide and two ortho protons of the partially fluorinated
(b)

73
aromatic rings of adjacent molecules with the following metrical data: 4a: d(Cl1···H12
= 2.682 Å, Cl1···H12–C12 = 168º and d(Cl2···H36) = 2.566 Å, Cl2···H36–C36 =
174.7º; 4b: d(Br1···H12) = 2.801 Å, Br1···H12–C12=144.4º and d(Br2···H30) = 2.852
Å, Br2···H30–C30 = 162.3º and 4c: d(I1···H12) = 2.952 Å, I1···H12–C12 = 123.9º and
d(I2···H30) = 3.135 Å, I2···H30–30 = 150.3º. All these values are less than the sum of
the van der Waals radii of hydrogen and chlorine, hydrogen and bromine and hydrogen
and iodine respectively, and are shown in Fig. 12 (a-b). In complex 4b a weak
secondary Br···Au interaction is observed (3.294 Å), while in 4c two equivalent I···Au
interactions are obvious (3.471 and 3.501 Å).
In addition to the halide···H hydrogen bonds, there are also a large number of C–H···F
and F···F distances observed in the structures of these complexes that are less than the
sum of the relevant van der Waals radii, these contacts shown in Fig. 13, viewed down
the a-axis. Thus 4a displays five C–H···F and one F···F interactions, whilst 4b displays
three C–H···F and three F···F interactions and 4c has four C–H···F and four F···F
interactions. The F···F interactions tend to be between fluorine atoms in the 3- and
5-positions, while both meta- and para- fluorines are involved in the C–H···F
interactions.
(a)

74
Fig. 12 (a) A view of the crystal packing of 4a showing H···Cl contacts, viewed down the
a-axis.
Fig. 12 (b) A view of the crystal packing of 4c showing H···I contacts, viewed down the
a-axis.
(b)

75
Fig. 13 Crystal packing of 4c showing F···F contacts, viewed down the a-axis.
For the four series of gold(I) triarylphosphine halide complexes, irrespective of the
identity of the halide, those containing phosphines of partially fluorinated aromatic
rings with fluorine in the meta positions, ie P(3-C6H4F)3 and P(3,5-C6H3F2)3 resulted in
complexes which do not exhibit aurophilic interactions. However, all the complexes of
P(4-C6H4F)3 and P(3,4,5-C6H2F3)3 show much shorter Au···Au distances. It is doubtful
that for this set of phosphines these differences in Au···Au distances arise from either
the steric or electronic properties of these phosphines, which are all rather similar. For
example the cone angle of PPh3, P(3-C6H4F)3 and P(4-C6H4F)3 are all the same, 145°,42
yet P(4-C6H4F)3 exhibits a much shorter Au···Au distance than the complexes of the
other two ligands. Although the electron donating properties of the phosphines is likely
to decrease in the order P(4-C6H4F)3> P(3-C6H4F)3 > P(3,5-C6H3F2)3 >
P(3,4,5-C6H2F3)3, it is the complexes of the first and last of these phosphines that give
rise to the shorter Au···Au distances. Furthermore, P(C6F5)3 and P(OPh)3 possess
similar electronic properties ((CO) for their Ni(CO)3P complexes being 2090.9 and
2085.3 cm-1
, respectively), and while the gold chloride of P(C6F5)3 exhibits a short
Au···Au distance, 3.783 Å, [MUVSIF]41
in the analogous P(OPh)3 complex d(Au···Au)
= 8.285 Å.43
Since we are unable to account for the observed differences in terms of the identity of
the halide or the steric or electronic properties of the phosphine we turned our attention
to the secondary interactions in the structures. All of the complexes exhibit a number of
halogen bonds between the gold-bound halide and protons of the partially fluorinated
rings. As the number of fluorines increases on the rings it is anticipated that there will
be an increase in the acidity of the remaining protons. This in turn might be expected to
give rise to a stronger Au–X···H interaction. Indeed the largest average difference
between the observed X···H distances and the sum of the relevant van der Waals radii is
found for the complexes (X = Cl) of P(3,4,5-C6H2F3)3 ( = -0.326 Å) followed by
P(3,5-C6H3F2)3 ( = -0.180 Å), then P(3-C6H4F)3 ( = -0.103 Å) and finally
P(4-C6H4F)3 ( = -0.066 Å). However, this alone, again, does not explain the
observation of short Au···Au distances for complexes 2a-c and 4a-c.
We note that for the complexes containing tris(4-fluorophenyl)phosphine and
particularly tris(3,4,5-trifluorophenyl)phosphine a larger number of C–H···F and F···F
interactions less than the sum of the van der Waal’s radii are observed, compared with

76
the phosphines which possess fluorine atoms only in the meta positions. C–H···F, and
related C–H···X interactions are of topical interest; a recent statistical analysis of a CSD
dataset suggest that H–C···F interactions occur more than three times as often as might
be expected on a purely random basis.44
As such their influence on crystal packing is
expected to lie between that of the strong hydrogen bonds of HO- and HN- donors and
very strong halogen bonds (N···I), but to be greater than that of other moderate
hydrogen-bond interactions. Aurophilic interactions are frequently cited as being of
similar strength to hydrogen bonds,12
and calculations performed on the
[Au(C6F5)(N(H)=CPh2)] system confirmed that in that system the two interactions are
comparable and that the solid state structure, which exhibits both aurophilic and
hydrogen bonding, arises from a competition between these two factors.45
It is likely
that in our systems the observed structures will depend on a balance of all the different
possible interactions.
In the gold chloride complexes of P(3-C6H4F)3 and P(3,5-C6H3F2)3 a single C–H···F
interaction is observed, while for P(4-C6H4F)3 two are seen and for P(3,4,5-C6H2F3)3
four exist. This greater number of interactions, combined with the anticipated enhanced
interaction arising from the more acidic aryl protons of the trifluorophenyl rings taken
together might explain the observation of the shortest Au···Au interaction in the
P(3,4,5-C6H2F3)3 systems. Of course, whether the observed structures are driven by an
aurophilic interaction that result in close approaches of other parts of the molecule, or
the numerous other interactions result in proximity of the gold centres is difficult to
determine, and warrants further investigation both from the structural and computational
standpoints.
2.5 Conclusion
In conclusion for the single X-ray crystal structures of the complexes [AuX(PAr3)] (Ar
= (3-FC6H4), X = Cl, Br and Ar = (4-FC6H4), (3,5-F2C6H3), (3,4,5-F3C6H2), X = Cl, Br,
I) we find that the observed Au–Au distances are a function of the phosphine alone. For
all of the complexes containing phosphines that are fluorinated only at the
meta-position, ie (3-FC6H4) or (3,5-F2C6H3) the observed Au–Au distances are greater
than ca. 7 Å, however, for systems containing either (4-FC6H4) or (3,4,5-F3C6H2) very
much shorter distances are observed in the range ca. 3.2 – 4.5 Å.
It appears unlikely that, in this series of complexes, the presence, or otherwise, of short
Au···Au interactions is a function of the halide or the steric and electronic properties of

77
the phosphines. However, a significant number of secondary C–H···X, C–H···F and
F···F interactions are observed in the complexes containing the P(4-FC6H4)3 and
P(3,4,5-F3C6H2)3, phosphines which taken together may account for the shorter Au···Au
distances found in the solid state structures of these complexes.
Acknowledgement
We thank Dr N. Barnes for preparation of complexes 2a, 2c, 4a and 4c.

78
References
1. See for example M. J. McKeage, L. Maharaj and S. J. Berners-Price, Coord.
Chem. Rev., 2002, 232, 127-135, and references therein.
2. See for example L. J. Larson, E. M. McCauley, B. Weissbart and D. S. Tinti,
J. Phys. Chem., 1995, 99, 7218-7226; B. Weissbart, D. V. Toronto, A. L.
Balch and D. S. Tinti, Inorg. Chem., 1996, 35, 2490-2496; Z. Assefa, B. G.
McBurnett, R. J. Staples, J. P. Fackler, B. Assmann, K. Angermaier and H.
Schmidbaur, Inorg. Chem., 1995, 34, 75-83.
3. P. G. Jones, Gold Bull., 1981, 14, 102-118; P. G. Jones, Gold Bull., 1986, 19,
46-57.
4. F. Scherbaum, A. Grohman, B. Huber, C. Kruger and H. Schmidbaur, Angew.
Chem., Int. Ed. Engl., 1988, 27, 1544-1546; H. Schmidbaur, Gold Bull., 1990,
23, 11-21; H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391-400; H.
Schmidbaur, Nature, 2001, 413, 31-34.
5. H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931-1951; H.
Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370-412.
6. D. V. Toronto, B. Weissbart, D. S. Tinti and A. L. Balch, Inorg. Chem., 1996,
35, 2484-2489; D. T. Walters, K. R. England, K. B. Ghiassi, F. Z. Semma, M.
M. Olmstead, and L. Balch, Polyhdedron, 2016, 117, 535-541.
7. P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412-4456.
8. P. Pyykkö, Chem. Rev., 1997, 97, 597-636.
9. P. Pyykkö and Y. Zhao, Angew. Chem., Int. Ed. Engl., 1991, 30, 604-605.
10. P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133-138.
11. P. Pyykkö, W. Schneider, A. Bauer, A. Bayler and H. Schmidbaur, Chem.
Commun., 1997, 1111-1112.
12. H. Schmidbaur, W. Graf and G. Müller, Angew. Chem., Int. Ed. Engl., 1988,
27, 417-419; H. Schmidbaur, K. Dziwok, A. Grohmann and G. Müller, Chem.
Ber., 1989, 122, 893-895; K. Dziwok, J. Lachmann, D. L. Wilkinson and G.
Müller, Chem. Ber., 1990, 123, 423-431; R. Narayanaswamy, M. A. Young,
E. Parkhurst, M. Ouellette, M. E. Kerr, D. M. Ho, R. C. Elder, A. E. Bruce
and M. R. M. Bruce, Inorg. Chem., 1993, 32, 2506-2517.

79
13. M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884-
1895.
14. D. V. Toronto, B. Weissbart, D. S. Tinti and A. L. Balch, Inorg. Chem., 1996,
35, 2484-2489.
15. P. D. Cookson and E. R. T. Tiekink, Acta Cryst., Sect. C: Cryst. Struct.
Commun., 1993, 49, 1602-1603.
16. K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994,
472, 371-376.
17. R. Pretorius, M. R. Fructos, H. Müller-Bunz, R. A. Gossage, P. J.
Pérez and M. Albrecht, Dalton Trans., 2016, 45, 14591-14602.
18. P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967-1997.
19. K. Angermaier, G.A. Bowmaker, E.N. de Silva, P. C. Healy, B.E. Jones and
H. Schmidbaur, J. Chem. Soc., Dalton Trans, 1996, 3121-3129.
20. K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994,
472, 371-376.
21. S. Ahrland, K. Dreisch, B. Noren and A. Oskarsson, Acta Chem. Scand., Ser.,
1987, A41, 173-177.
22. E. R. T. Tiekink, Acta Cryst., 1989, C45, 1233-1234.
23. N. C. Baenziger, W. E. Bennett and D. M. Soboroff, Acta Cryst., 1976, B32,
962-963.
24. P. F. Barron, L. H. Engelhardt, P.C. Healy, J. Oddy and A. H. White,
Aust.J.Chem., 1987, 40, 1545-1555.
25. L. -J. Baker, R. C. Bott, G. A. Bowmaker, P. C. Healy, B. W. Skelton, P.
Schwerdtfeger and A. H. White, J. Chem. Soc., Dalton Trans., 1995, 1341.
26. H. W. Chen and E. R. T. Tiekink, Acta Cryst., 2003, E59, m50-m53.
27. C. S. W. Harker and E. R. T. Tiekink, Acta Cryst., 1990, C46, 1546.
28. R. C. Bott, P. C. Healy and G. Smith, Aust. J. Chem., 2004, 57, 213-218.
29. P. D. Cookson and E. R. T. Tiekink, Acta Cryst., 1994, C50, 1896-1898.
30. S. Y. Ho and E. R. T. Tiekink, Acta Cryst., 2001, E57, m549-m550.
31. E.C. Alyea, G. Ferguson and S. Kannon, Polyhedron, 2000, 19, 2211-2213.
32. N. C. Baenziger, W. E. Bennett and D. M. Soboroff, Acta Cryst., Sect. B:
Struct. Cryst. Chem., 1976, 32, 962-963.

80
33. K. Nunokawa, S. Onaka, T. Tatematsu, M. Ito and J. Sakai, Inorg. Chim.
Acta, 2001, 322, 56-64.
34. N. A. Barnes, A. K. Brisdon, F. R. W. Brown, W. I. Cross, I. R. Crossley, C.
Fish, C. J. Herbert, R. G. Pritchard and J. E. Warren, J. Chem. Soc., Dalton
Trans., 2011, 40, 1743-1750.
35. C. A. McAuliffe, R. V. Parish and D. Randall, J. Chem. Soc., Dalton Trans.,
1979, 1730-1735.
36. G. M. Sheldrick. Acta Cryst., 2008, A64, 112-122.
37. O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H.
Puschmann, J. Appl. Cryst., 2009, 42, 339-341.
38. C. F. Macrea, P. R. Eddington, P. McCabe, E. Pidcock, G. P. Shields, R.
Taylor, M. Towler and J. van de Streek, J. Appl. Cryst., 2006, 39, 453-457.
39. P. Tasker, D. Coventry, S. Parsons and D. Messenger. (2005) Private
Communication to the Cambridge structure Database, deposition number
CCDC276800.
40. O. B. Shawkataly, A. Tariq, S. S. Ghani, C. S. Yeap and H. K. Fun, Acta
Cryst., 2010, E66, m1217-m1218.
41. H. W. Chen and E. R. T. Tiekink, Acta Cryst., 2003, E59, m50- m52
42. C. A. Tolman, Chem. Rev., 1977, 77, 313-348.
43. P. B. Hitchcock and P. L. Pye, J. Chem. Soc., Dalton Trans., 1977, 1457-
1460.
44. R. Taylor, Crystal Growth & Design, 2016, 16, 4165-4168.
45. A. Codina, E. J. Fernandez, P. G. Jones, A. Laguna, J. M. Lopez-de-
Luzuriaga, M. Monge, M. E. Olmos, J. Perez and M. A. Rodriguez, J. Am.
Chem. Soc., 2002, 124, 6781-6786.

81
Chapter 3
Crystal structures of [AuCl{PPh2(C6F5)}] and
[AuBr{PPh2(C6F5)}]

82
3. Crystal structures of [AuCl{PPh2(C6F5)}] and [AuBr{PPh2(C6F5)}]
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, University of Manchester, Manchester, M13 9PL, UK.
Keywords: Crystal structure; Single crystal; F···F interaction; Aurophilic interaction;
C—H···X interactions.
3.1 Abstract
The two title compounds are analogs of [AuX(PPh3)] (X = Cl, Br) in which one of the
phenyl groups is replaced by C6F5. The crystals grown by vapour diffusion of hexane
into a saturated dichloromethane solution are isomorphous with two independent
molecules in the asymmetric unit, which display near-linear geometries at gold. In the
extended structure of C18H10AuClF5P (I) a head to tail packing motif with C–H···Cl, C–
H···F and π-π stacking interactions is observed. For C18H10AuBrF5P (II) C–H···Br and
π-π stacking interactions are found. In both compounds F···F interactions are observed
between chains resulting in three-dimensional networks.
3.2 Chemical context
Phosphine–containing metal complexes have a wide range of different applications
(Downing & Smith, 2004; Tolman, 1977). In most cases the phosphines contain
hydrocarbon organic fragments; some perfluorinated analogues are known, but
relatively few examples in which the phosphine is partially fluorinated exist. For
example while the structure of [AuCl(PPh3)] (Baenziger et al., 1976), [AuBr(PPh3)]
(Barron et al., 1987) and [AuClP(C6F5)3] (Chen & Tiekink, 2002) are known, in
comparison the analogues containing PPh2(C6F5) are unknown. This set of complexes
are of interest because it has been shown that substituents can influence aurophilic
interactions in a series of Au (I) complexes (Onaka et al., 2001), and while aurophilicity
has been observed in [AuCl{P(C6F5)3}] (Chen & Tiekink, 2002), it was not present in
the structures of either [AuCl(PPh3)] (Baenziger et al., 1976) and [AuBr(PPh3)] (Barron
et al., 1987).

83
The compounds (I) and (II) were synthesized according to a literature procedure
(Brisdon et al., 2010), which involved the reduction of the Au(III) salt K[AuCl4] with
tetrahydrothiophene (tht) in an ethanol/water mixture, to obtain [AuCl(tht)], which was
then reacted with the phosphine in CH2Cl2. This resulted in [AuCl{PPh2(C6F5)}] as a
white solid, which could then be reacted with LiBr in ethanol, to give
[AuBr{PPh2(C6F5)}] (scheme 1).
Scheme 1 Synthesis of Au(I) phosphine complexes.
3.3 Structural commentary
Both [AuCl{PPh2(C6F5)}] (I) (Fig. 1) and [AuBr{PPh2(C6F5)}] (II) (Fig. 2) are
isomorphous, crystallising in the Triclinic P-1 space group and containing two
(I) X= Cl
(II) X= Br
X= Br

84
independent molecules, labelled as A and B, in the unit cell. All molecules exhibit
essentially linear geometry at the Au atom.
Figure 1 A view of the two independent molecules (A) and (B) of (I) with the atom-numbering
scheme. Displacement ellipsoids are shown at the 50% probability level.
Figure 2 A view of the two independent molecules (A) and (B) of (II) with the atom-numbering
scheme. Displacement ellipsoids are shown at the 50% probability level.
(A)
(A)
(B)
(B)

85
The P–Au–Cl and P–Au–Br angles for each pair of molecules are 178.68(7)/178.53(7)°
and 178.48(6)/179.10(6)°. The Au–P and Au–Cl bond lengths of (I) for molecules A
and B are 2.2408(17)/2.2312(18) and 2.2834(16)/ 2.2832(17) Å, respectively, while the
Au–P and Au–Br distances in the bromo analogue (II) for A and B are
2.231(2)/2.243(2) and 2.3984(9)/2.3992(10) Å, respectively. Interestingly, there is a
slightly greater difference in the Au–P values for molecules A and B in both (I) and (II)
than there is between the Au–Cl and Au–Br bond distances.
The Au–P bond lengths in the chloro derivative (I) are significantly longer than those
found in [AuCl{P(C6F5)3}] d(Au–P) = 2.2060(9)/2.2149(9) Å (Chen & Tiekink, 2002),
but not significantly different from [AuCl(PPh3)] d(Au–P) = 2.235(3) Å (Baenziger et
al., 1976). The Au–Cl bond distances of (I), in both molecules, are similar to that
reported in [AuCl(PPh3)] d(Au–Cl) = 2.279(3) Å (Baenziger et al., 1976) and that found
in the related complex [AuCl{P(C6F5)3}] d(Au–Cl) = 2.2688(9)/2.2706(9) Å (Chen &
Tiekink, 2002).
By comparing both Au–P and Au–Br distances of the bromo analog (II) for molecules
A and B, with the analogous distances of 2.252(6) and 2.407(6) Å, respectively, from
the complex [AuBr(PPh3)] (Barron et al., 1987) shows that there is little significant
difference in either of these distances.
The dihedral angles between the fluoro aryl ring planes for compound (I) and (II) are
similar, as indicated by the Au1–P1–C1–C2 and Au2–P2–C19–C20 torsion angles for
(I) which are 144.8(6) and 108.9(7)°, respectively, and in the other complex (II)
112.0(8) and 146.6(8)° for planes Au1–P1–C1–C2 and Au2–P2–C19–C20,
respectively.
3.4 Supramolecular features
In the crystal structure of [AuCl{PPh2(C6F5)}] (I), the molecules pack together via van
der Waals contacts, C–H···Cl, C–H···F and F···F interactions and -stacking. The C–
H···Cl and C–H···F interactions are shown in Fig. 3 and listed in Table 1. There are
short intermolecular interactions between the fluorine atoms F2···F7 of 2.808(7) and
F3···F8 of 2.868(8) Å which are both significantly less than twice the van der Waals’
radius of fluorine (2.94 Å) (Bondi, 1964).

86
In the extended structure of (II) the notable intermolecular interactions detected include
-stacking, C–H···Br and F···F interactions which result in a linked three-dimensional
lattice (Fig. 4 and Table 2). The shortest F···F contacts are F2···F7 of 2.825(7) Å and
F3···F8 of 2.924(9) Å which are less than twice the van der Waals’ radius of fluorine
(2.94 Å) (Bondi, 1964).
The shortest Au···Au interaction distance for complexes (I) and (II) are 4.9795(4) and
5.0178(6) Å, respectively, which are longer than twice the van der Waals radii of gold,
3.60 Å (Bondi, 1964), suggesting that no significant gold-gold interactions occur in
either complex. These distances can be compared with those observed in the PPh3
(Baenziger et al., 1976) and P(C6F5)3 (Chen & Tiekink, 2002) containing analogues of
6.989 and 3.7828(2) Å, respectively.
Offset -stacking is seen in compounds (I) and (II), and is characterized by a centroid-
to-centroid distance of 4.653/5.331 Å for (I) and 5.356 Å for complex (II).
Table 4
Hydrogen-bond geometry (Å, º) for (I).
D—H---A D—H H---A D---A D—H---A
C35—H35···F2i 0.929 2.653 3.30(1) 127.6
C10—H10···Cl2ii 0.930 2.938 3.609(8) 140.7
C12—H12···Cl2 0.929 2.864 3.705(9) 159.2
C18—H18···Cl2 0.930 2.854 3.748(9) 138.6
Symmetry codes: (i) -x,1-y,1-z; (ii) -x,1-y,1-z.
Table 5
Hydrogen-bond geometry (Å, °) for (II).
D—H---A D—H H---A D---A D—H---A
C10—H10···Br2i 0.93 3.013 3.81(1) 144.2
C12—H12···Br2 0.93 2.994 3.86(1) 156.3
C18—H18···Br2ii 0.93 2.9766 3.704(8) 136.2
C30—H30···Br1iii
0.93 3.027 3.83(1) 144.9
Symmetry codes: (i) -x,1-y,1-z; (ii) x,y,z; (iii) -1+x,-1+y,-1+z.

87
Figure 3 A view of the crystal packing of (I) viewed down the c-axis, showing Cl···H (in blue)
and H···F (in red) contacts.
Figure 4 A view of the crystal packing of (I) viewed down the a-axis, showing F···F contacts
(in blue).

88
Figure 5 A view of the crystal packing of (II) viewed down the c-axis, showing Br···H contacts
(in blue).
Figure 6 A view of the crystal packing of (II) viewed down the a-axis, showing F···F contacts
(in blue).

89
3.5 Database survey
A search of the Cambridge Structural Database (Version 5.37, 2016 with updates;
Groom et al., 2016), for complexes of the type [AuX{P(C6H5)n(C6F5)3-n}] (X=Cl, Br, I)
reveals only two are known [AuX(PPh3)] (X=Cl, Br, I) (Baenziger et al., 1976),
(Barron et al., 1987) and [AuX{P(C6F5)3}] (X=Cl) (Chen & Tiekink, 2002); there are no
such reported structures containing PPh2(C6F5).
3.6 Synthesis and crystallization
3.6.1 Synthesis of [AuCl{PPh2(C6F5)}] (I)
The synthesis of [AuCl{PPh2(C6F5)}] (I) is based on a reported literature procedure
(Brisdon et al., 2010). K[AuCl4] (0.2 g, 0.5 mmol) was dissolved in ethanol (5 cm3) and
water (2 cm3), and then tetrahydrothiophene (0.1 ml, 1.1 mmol) was added drop-wise,
and the reaction mixture stirred for 15 minutes, during which time the solution changed
from yellow to white in colour. After filtration the resulting white solid [AuCl(tht)] was
dried in vacuo. A mixture of PPh2(C6F5) (0.11g, 0.31 mmol) in dichloromethane (5 cm3)
was added to [AuCl(tht)] (0.1 g, 0.3 mmol) in dichloromethane (5 cm3) and the mixture
was allowed to stir overnight after which time a white solid resulted. 31
P{1H} NMR
(CDCl3): δ 15.3 ppm (s); 19
F NMR (CDCl3): δ -125.4 ppm (m, 2F), -144.5 (m, 1F), -
157.7 (m, 2F); 1H NMR (CDCl3): δ 7.4-7.6 (m, 10H, ArH). Single crystals of
[AuCl{PPh2(C6F5)}] were obtained by vapour diffusion of hexane into a saturated
dichloromethane solution.
3.6.2 Synthesis of [AuBr{PPh2(C6F5)}] (II)
This complex was prepared using a modified procedure (McAuliffe et al., 1979).
[AuCl{PPh2(C6F5)}] (I) (0.1 g, 0.17 mmol) was dissolved in EtOH (10 cm3) and LiBr
(0.014 g, 0.16 mmol) was added, then the mixture was left to stir for 15 minutes reflux
at 80 ºC, after that time the solvent was removed under vacuo. The resulting gold
complex was dissolved in dichloromethane, from which crystals were grown. 31
P{1H}
NMR (CDCl3): δ 17.6 ppm (s); 19
F NMR (CDCl3): δ -125.5 ppm (m, 2F), -144.6 (m,
1F), -157.7 (m, 2F); 1H NMR (CDCl3): δ 7.4-7.6 (m, 10H, ArH). Single crystals of
[AuBr{PPh2(C6F5)}] were obtained by vapour diffusion of hexane into a saturated
dichloromethane solution.

90
3.7 Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2.
Table 2
Experimental details
Crystal data
Chemical formula C18H10AuClF5P C18H10AuBrF5P
Mr 584.65 629.11
Crystal system, space group Triclinic, P-1 Triclinic, P-1
Temperature (K) 150 150
a, b, c (Å) 11.7939(6), 13.1570(5),
13.4024(6)
11.7733(7), 13.1903(7),
13.6025(8)
, , (°) 107.191(4), 102.074(4),
109.541(4)
107.387(5), 102.314(5),
108.504(5)
V (Å3) 1759.34(14) 1797.44(19)
Z 4 4
Radiation type Mo K Mo K
(mm-1
) 8.65 10.54
Crystal size (mm) 0.16 × 0.12 × 0.08 0.08 × 0.06 × 0.04
Data collection
Diffractometer SuperNova, Single source at offset,
Eos diffractometer
SuperNova, Single source at
offset, Eos diffractometer
Absorption correction Multi-scan (CrysAlis PRO;
Agilent, 2014)
Multi-scan (CrysAlis PRO;
Agilent, 2014)
Tmin, Tmax 0.548, 1.000 0.413, 1.000
No. of measured,
independent and observed
[I > 2(I)] reflections
14408, 6914, 5316 15324, 7071, 5386
Rint 0.043 0.055
(sin /)max (Å-1
) 0.617 0.617
Refinement
R[F2 > 2(F
2)], wR(F
2), S 0.041, 0.071, 0.98 0.047, 0.078, 1.00
No. of reflections 6914 7071
No. of parameters 469 469
H-atom treatment H-atom parameters constrained H-atom parameters
constrained
max, min (e Å-3
) 1.84, -1.12 1.95, -1.88

91
References
Baenziger, N. C., Bennett, W. E. & Soboroff, D. M. (1976). Acta Cryst. B32, 962-963.
Barnes, N. A., Brisdon, A. K., Brown, F. R. W., Cross, W. I., Crossley, I. R., Fish, C.,
Herbert, C. J., Pritchard, R. G. & Warren, J. E. (2011). Dalton Trans. 40, 1743-1750.
Barron, P. F., Engelhardt, L.H., Healy, P.C., Oddy, J. & White, A. H. (1987). Aust. J.
Chem. 40, 1545-1555.
Bondi, A. (1964). J. Phys. Chem. 68, 441-451.
Bourhis, L. J., Dolomanov, O. V., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2015) Acta Cryst. A71, 59-75.
Chen, H. W., Tiekink, E. R. T. (2002). Acta Cryst. E59, m50-m52.
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2009). J. Appl. Cryst. 42, 339-341.
Groom, C. R. & Allen, F. H. (2016). Angew. Chem. Int. Ed. 53, 662-671.
McAuliffe, C. A., Parish, R. V. & Randall, D. (1979). J.Chem Soc.,. Dalton Trans.
1730-1735.
Nunokawa, K., Onaka, S., Tatematsu, T., Ito. M. & Sakai, J. (2001). Inorg. Chim. Acta.
322, 56-64.
Palatinus, L. & Chapuis, G. (2007). J. Appl. Cryst. 40, 786-790; Palatinus, L. & van der
Lee, A. (2008). J. Appl. Cryst. 41, 975-984; Palatinus, L., Prathapa, S. J. & van
Smaalen, S. (2012). J. Appl. Cryst. 45, 575-580.
Sheldrick, G. M. (2008). Acta Cryst. A64, 112-122.
Sheldrick, G. M. (2015). Acta Cryst. C71, 3-8.
Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925.
92
Supporting information
Crystal structures of [AuCl{PPh2(C6F5)}] and [AuBr{PPh2(C6F5)}]
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
Computing details
Data collection: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33 (release 27-03-2014
CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); cell refinement: CrysAlis PRO, Agilent
Technologies, Version 1.171.37.33 (release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27
2014,17:12:48); data reduction: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33
(release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); program(s) used to
solve structure: Superflip (Palatinus & Chapuis, 2007;Palatinus & van der Lee, 2008; Palatinus
et al., 2012); program(s) used to refine structure: SHELXL (Sheldrick, 2015); molecular
graphics: Olex2 (Dolomanov et al., 2009); software used to prepare material for publication:
Olex2 (Dolomanov et al., 2009).
[AuCl{PPh2(C6F5)}] (I)
Crystal data
C18H10AuClF5P Z = 4
Mr = 584.65 F(000) = 1096
Triclinic, P¯1 Dx = 2.207 Mg m-3
a = 11.7939 (6) Å Mo K radiation, = 0.71073 Å
b = 13.1570 (5) Å Cell parameters from 4917 reflections
c = 13.4024 (6) Å = 3.5–28.1°
= 107.191 (4)° = 8.65 mm-1
= 102.074 (4)° T = 150 K
= 109.541 (4)° clear light colourless
V = 1759.34 (14) Å3 0.08 × 0.06 × 0.04 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
6914 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source, Incoatec Is
5316 reflections with I > 2(I)
Mirror monochromator Rint = 0.043
Detector resolution: 8.0714 pixels mm-1
max = 29.0°, min = 3.3°
scans h = -1316
Absorption correction: multi-scan k = -1717

93
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
Tmin = 0.548, Tmax = 1.000 l = -1818
14408 measured reflections
Refinement
Refinement on F2 Primary atom site location: iterative
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.040 H-atom parameters constrained
wR(F2) = 0.071 w = 1/[
2(Fo
2) + (0.015P)
2]
where P = (Fo2 + 2Fc
2)/3
S = 0.98 (/)max = 0.001
6914 reflections max = 1.84 e Å-3
469 parameters min = -1.12 e Å-3
6 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate
(isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au2 0.07792 (3) 0.20009 (2) 0.49841 (2) 0.02279 (8)
Au1 0.60042 (3) 0.90957 (2) 0.85375 (2) 0.02184 (8)
Cl2 0.15209 (18) 0.32893 (15) 0.67943 (14) 0.0283 (4)
Cl1 0.74388 (18) 1.10221 (15) 0.94397 (15) 0.0307 (5)
P1 0.45881 (18) 0.72159 (15) 0.76946 (15) 0.0196 (4)
P2 0.00266 (18) 0.07598 (16) 0.31968 (15) 0.0210 (4)
F1 0.1865 (4) 0.6581 (4) 0.7985 (3) 0.0351 (11)
F10 0.2723 (4) 0.1120 (4) 0.4249 (4) 0.0383 (12)
F5 0.5998 (4) 0.6855 (3) 0.9560 (3) 0.0307 (10)
F6 -0.1211 (4) -0.1875 (3) 0.1550 (3) 0.0334 (11)
F3 0.2755 (5) 0.5496 (4) 1.0975 (4) 0.0435 (13)
F2 0.1115 (4) 0.5817 (4) 0.9470 (4) 0.0414 (12)

94
F4 0.5207 (4) 0.6040 (4) 1.1021 (3) 0.0397 (12)
F8 0.2384 (5) -0.2644 (4) 0.2427 (4) 0.0551 (15)
F9 0.3792 (5) -0.0391 (4) 0.3923 (4) 0.0529 (14)
C12 0.2334 (6) 0.5777 (6) 0.5857 (5) 0.0228 (17)
H12 0.2339 0.5161 0.6060 0.027*
C3 0.2304 (8) 0.6048 (6) 0.9469 (6) 0.030 (2)
C14 0.6090 (6) 0.6504 (6) 0.6581 (5) 0.0221 (17)
H14 0.6384 0.7254 0.6583 0.026*
C32 0.0925 (6) 0.1071 (6) 0.1470 (5) 0.0213 (17)
H32 0.0998 0.0366 0.1344 0.026*
C31 0.0445 (6) 0.1487 (6) 0.2285 (5) 0.0203 (16)
C21 0.0579 (9) -0.2262 (6) 0.2008 (7) 0.035 (2)
C2 0.2721 (7) 0.6447 (6) 0.8710 (6) 0.0251 (18)
C13 0.5211 (6) 0.6173 (6) 0.7108 (5) 0.0196 (16)
F7 -0.0125 (5) -0.3364 (4) 0.1240 (4) 0.0452 (13)
C25 -0.1700 (6) 0.0003 (6) 0.2734 (5) 0.0198 (16)
C19 0.0699 (7) -0.0324 (6) 0.2952 (6) 0.0219 (17)
C34 0.1176 (7) 0.2746 (6) 0.1007 (6) 0.0283 (18)
H34 0.1442 0.3177 0.0594 0.034*
C7 0.3269 (6) 0.6934 (6) 0.6524 (5) 0.0191 (16)
C16 0.6092 (7) 0.4606 (6) 0.6067 (6) 0.0288 (19)
H16 0.6390 0.4079 0.5716 0.035*
C18 0.4763 (7) 0.5058 (6) 0.7103 (5) 0.0233 (17)
H18 0.4160 0.4833 0.7437 0.028*
C8 0.3228 (7) 0.7836 (6) 0.6213 (6) 0.0282 (18)
H8 0.3842 0.8600 0.6648 0.034*
C30 -0.2473 (7) -0.0040 (6) 0.1771 (6) 0.0237 (17)
H30 -0.2111 0.0278 0.1318 0.028*
C29 -0.3786 (7) -0.0562 (6) 0.1496 (6) 0.0279 (19)
H29 -0.4298 -0.0582 0.0854 0.033*
C36 0.0297 (7) 0.2515 (6) 0.2427 (6) 0.0235 (17)
H36 -0.0050 0.2786 0.2956 0.028*
C1 0.3960 (7) 0.6698 (6) 0.8685 (6) 0.0217 (17)
C35 0.0654 (7) 0.3143 (6) 0.1798 (6) 0.0267 (18)
H35 0.0547 0.3831 0.1902 0.032*
C15 0.6519 (6) 0.5719 (6) 0.6061 (5) 0.0234 (17)
H15 0.7098 0.5935 0.5704 0.028*
C11 0.1417 (7) 0.5568 (7) 0.4909 (6) 0.0294 (19)
H11 0.0808 0.4804 0.4467 0.035*
C6 0.4776 (7) 0.6580 (6) 0.9502 (6) 0.0232 (17)

95
C23 0.2550 (8) -0.0754 (7) 0.3368 (7) 0.035 (2)
C20 0.0015 (7) -0.1471 (6) 0.2178 (6) 0.0244 (17)
C24 0.1966 (7) -0.0002 (6) 0.3522 (6) 0.0263 (18)
C28 -0.4373 (7) -0.1061 (6) 0.2142 (6) 0.031 (2)
H28 -0.5260 -0.1403 0.1946 0.037*
C4 0.3137 (8) 0.5905 (6) 1.0241 (6) 0.0271 (19)
C5 0.4385 (8) 0.6165 (6) 1.0262 (6) 0.0282 (19)
C17 0.5224 (7) 0.4271 (6) 0.6591 (6) 0.0282 (18)
H17 0.4949 0.3526 0.6602 0.034*
C26 -0.2266 (7) -0.0502 (6) 0.3389 (6) 0.0243 (17)
H26 -0.1752 -0.0486 0.4028 0.029*
C27 -0.3578 (7) -0.1025 (6) 0.3094 (7) 0.033 (2)
H27 -0.3940 -0.1359 0.3538 0.039*
C10 0.1383 (7) 0.6470 (7) 0.4603 (6) 0.037 (2)
H10 0.0758 0.6312 0.3954 0.044*
C33 0.1294 (7) 0.1708 (6) 0.0843 (6) 0.0285 (19)
H33 0.1625 0.1431 0.0304 0.034*
C9 0.2284 (8) 0.7618 (7) 0.5261 (6) 0.036 (2)
H9 0.2255 0.8233 0.5064 0.043*
C22 0.1831 (9) -0.1908 (7) 0.2604 (7) 0.037 (2)
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au2 0.02255 (17) 0.02470 (16) 0.02079 (16) 0.00964 (13) 0.00725 (13) 0.00911
(13)
Au1 0.02316 (17) 0.01919 (15) 0.02123 (16) 0.00764 (13) 0.00753 (13) 0.00706
(12)
Cl2 0.0286 (11) 0.0286 (10) 0.0209 (9) 0.0074 (8) 0.0066 (8) 0.0075 (8)
Cl1 0.0301 (11) 0.0208 (9) 0.0298 (10) 0.0030 (8) 0.0070 (9) 0.0064 (8)
P1 0.0200 (10) 0.0189 (10) 0.0208 (10) 0.0082 (8) 0.0087 (9) 0.0078 (8)
P2 0.0228 (11) 0.0216 (10) 0.0202 (10) 0.0102 (8) 0.0087 (9) 0.0086 (8)
F1 0.029 (3) 0.056 (3) 0.038 (3) 0.028 (2) 0.020 (2) 0.026 (2)
F10 0.024 (3) 0.041 (3) 0.051 (3) 0.016 (2) 0.009 (2) 0.019 (2)
F5 0.029 (3) 0.042 (3) 0.025 (2) 0.017 (2) 0.009 (2) 0.016 (2)
F6 0.040 (3) 0.027 (2) 0.027 (2) 0.012 (2) 0.010 (2) 0.005 (2)
F3 0.067 (4) 0.042 (3) 0.047 (3) 0.029 (3) 0.038 (3) 0.031 (2)
F2 0.037 (3) 0.049 (3) 0.057 (3) 0.023 (2) 0.035 (3) 0.027 (2)
F4 0.059 (3) 0.052 (3) 0.026 (2) 0.035 (3) 0.017 (2) 0.023 (2)
F8 0.076 (4) 0.057 (3) 0.071 (4) 0.057 (3) 0.042 (3) 0.032 (3)
F9 0.041 (3) 0.076 (4) 0.062 (3) 0.041 (3) 0.019 (3) 0.033 (3)
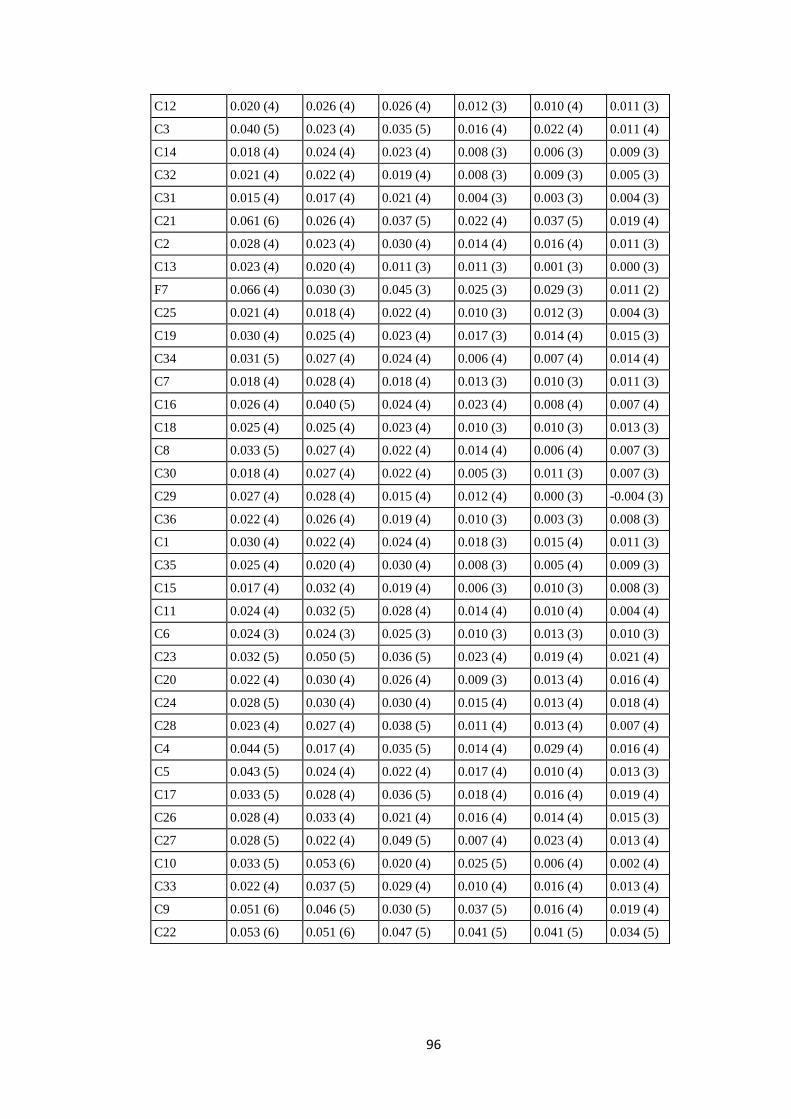
96
C12 0.020 (4) 0.026 (4) 0.026 (4) 0.012 (3) 0.010 (4) 0.011 (3)
C3 0.040 (5) 0.023 (4) 0.035 (5) 0.016 (4) 0.022 (4) 0.011 (4)
C14 0.018 (4) 0.024 (4) 0.023 (4) 0.008 (3) 0.006 (3) 0.009 (3)
C32 0.021 (4) 0.022 (4) 0.019 (4) 0.008 (3) 0.009 (3) 0.005 (3)
C31 0.015 (4) 0.017 (4) 0.021 (4) 0.004 (3) 0.003 (3) 0.004 (3)
C21 0.061 (6) 0.026 (4) 0.037 (5) 0.022 (4) 0.037 (5) 0.019 (4)
C2 0.028 (4) 0.023 (4) 0.030 (4) 0.014 (4) 0.016 (4) 0.011 (3)
C13 0.023 (4) 0.020 (4) 0.011 (3) 0.011 (3) 0.001 (3) 0.000 (3)
F7 0.066 (4) 0.030 (3) 0.045 (3) 0.025 (3) 0.029 (3) 0.011 (2)
C25 0.021 (4) 0.018 (4) 0.022 (4) 0.010 (3) 0.012 (3) 0.004 (3)
C19 0.030 (4) 0.025 (4) 0.023 (4) 0.017 (3) 0.014 (4) 0.015 (3)
C34 0.031 (5) 0.027 (4) 0.024 (4) 0.006 (4) 0.007 (4) 0.014 (4)
C7 0.018 (4) 0.028 (4) 0.018 (4) 0.013 (3) 0.010 (3) 0.011 (3)
C16 0.026 (4) 0.040 (5) 0.024 (4) 0.023 (4) 0.008 (4) 0.007 (4)
C18 0.025 (4) 0.025 (4) 0.023 (4) 0.010 (3) 0.010 (3) 0.013 (3)
C8 0.033 (5) 0.027 (4) 0.022 (4) 0.014 (4) 0.006 (4) 0.007 (3)
C30 0.018 (4) 0.027 (4) 0.022 (4) 0.005 (3) 0.011 (3) 0.007 (3)
C29 0.027 (4) 0.028 (4) 0.015 (4) 0.012 (4) 0.000 (3) -0.004 (3)
C36 0.022 (4) 0.026 (4) 0.019 (4) 0.010 (3) 0.003 (3) 0.008 (3)
C1 0.030 (4) 0.022 (4) 0.024 (4) 0.018 (3) 0.015 (4) 0.011 (3)
C35 0.025 (4) 0.020 (4) 0.030 (4) 0.008 (3) 0.005 (4) 0.009 (3)
C15 0.017 (4) 0.032 (4) 0.019 (4) 0.006 (3) 0.010 (3) 0.008 (3)
C11 0.024 (4) 0.032 (5) 0.028 (4) 0.014 (4) 0.010 (4) 0.004 (4)
C6 0.024 (3) 0.024 (3) 0.025 (3) 0.010 (3) 0.013 (3) 0.010 (3)
C23 0.032 (5) 0.050 (5) 0.036 (5) 0.023 (4) 0.019 (4) 0.021 (4)
C20 0.022 (4) 0.030 (4) 0.026 (4) 0.009 (3) 0.013 (4) 0.016 (4)
C24 0.028 (5) 0.030 (4) 0.030 (4) 0.015 (4) 0.013 (4) 0.018 (4)
C28 0.023 (4) 0.027 (4) 0.038 (5) 0.011 (4) 0.013 (4) 0.007 (4)
C4 0.044 (5) 0.017 (4) 0.035 (5) 0.014 (4) 0.029 (4) 0.016 (4)
C5 0.043 (5) 0.024 (4) 0.022 (4) 0.017 (4) 0.010 (4) 0.013 (3)
C17 0.033 (5) 0.028 (4) 0.036 (5) 0.018 (4) 0.016 (4) 0.019 (4)
C26 0.028 (4) 0.033 (4) 0.021 (4) 0.016 (4) 0.014 (4) 0.015 (3)
C27 0.028 (5) 0.022 (4) 0.049 (5) 0.007 (4) 0.023 (4) 0.013 (4)
C10 0.033 (5) 0.053 (6) 0.020 (4) 0.025 (5) 0.006 (4) 0.002 (4)
C33 0.022 (4) 0.037 (5) 0.029 (4) 0.010 (4) 0.016 (4) 0.013 (4)
C9 0.051 (6) 0.046 (5) 0.030 (5) 0.037 (5) 0.016 (4) 0.019 (4)
C22 0.053 (6) 0.051 (6) 0.047 (5) 0.041 (5) 0.041 (5) 0.034 (5)

97
Geometric parameters (Å, º)
Au2—Cl2 2.2833 (16) C19—C20 1.383 (9)
Au2—P2 2.2408 (17) C19—C24 1.380 (10)
Au1—Cl1 2.2832 (17) C34—H34 0.9300
Au1—P1 2.2312 (18) C34—C35 1.392 (9)
P1—C13 1.822 (6) C34—C33 1.376 (10)
P1—C7 1.808 (7) C7—C8 1.380 (10)
P1—C1 1.839 (7) C16—H16 0.9300
P2—C31 1.811 (7) C16—C15 1.382 (10)
P2—C25 1.809 (7) C16—C17 1.385 (9)
P2—C19 1.833 (7) C18—H18 0.9300
F1—C2 1.342 (9) C18—C17 1.398 (9)
F10—C24 1.353 (8) C8—H8 0.9300
F5—C6 1.343 (8) C8—C9 1.388 (10)
F6—C20 1.339 (8) C30—H30 0.9300
F3—C4 1.339 (8) C30—C29 1.380 (9)
F2—C3 1.332 (8) C29—H29 0.9300
F4—C5 1.339 (9) C29—C28 1.394 (10)
F8—C22 1.331 (8) C36—H36 0.9300
F9—C23 1.339 (9) C36—C35 1.378 (9)
C12—H12 0.9300 C1—C6 1.382 (10)
C12—C7 1.411 (9) C35—H35 0.9300
C12—C11 1.370 (10) C15—H15 0.9300
C3—C2 1.381 (10) C11—H11 0.9300
C3—C4 1.373 (11) C11—C10 1.375 (10)
C14—H14 0.9300 C6—C5 1.381 (9)
C14—C13 1.397 (9) C23—C24 1.374 (10)
C14—C15 1.371 (9) C23—C22 1.390 (10)
C32—H32 0.9300 C28—H28 0.9300
C32—C31 1.388 (8) C28—C27 1.395 (11)
C32—C33 1.387 (9) C4—C5 1.387 (10)
C31—C36 1.384 (9) C17—H17 0.9300
C21—F7 1.344 (8) C26—H26 0.9300
C21—C20 1.401 (10) C26—C27 1.377 (9)
C21—C22 1.365 (11) C27—H27 0.9300
C2—C1 1.397 (9) C10—H10 0.9300
C13—C18 1.380 (9) C10—C9 1.391 (10)
C25—C30 1.388 (10) C33—H33 0.9300
C25—C26 1.397 (9) C9—H9 0.9300

98
P2—Au2—Cl2 178.68 (7) C29—C30—H30 120.5
P1—Au1—Cl1 178.53 (7) C30—C29—H29 118.7
C13—P1—Au1 115.7 (2) C30—C29—C28 122.6 (7)
C13—P1—C1 103.3 (3) C28—C29—H29 118.7
C7—P1—Au1 113.5 (2) C31—C36—H36 119.5
C7—P1—C13 103.3 (3) C35—C36—C31 121.0 (6)
C7—P1—C1 109.4 (3) C35—C36—H36 119.5
C1—P1—Au1 110.9 (2) C2—C1—P1 125.3 (6)
C31—P2—Au2 113.0 (2) C6—C1—P1 118.5 (6)
C31—P2—C19 103.5 (3) C6—C1—C2 116.2 (7)
C25—P2—Au2 109.8 (2) C34—C35—H35 120.0
C25—P2—C31 108.2 (3) C36—C35—C34 119.9 (7)
C25—P2—C19 109.3 (3) C36—C35—H35 120.0
C19—P2—Au2 112.8 (2) C14—C15—C16 120.2 (6)
C7—C12—H12 120.2 C14—C15—H15 119.9
C11—C12—H12 120.2 C16—C15—H15 119.9
C11—C12—C7 119.6 (7) C12—C11—H11 119.5
F2—C3—C2 120.8 (8) C12—C11—C10 121.1 (7)
F2—C3—C4 119.9 (7) C10—C11—H11 119.5
C4—C3—C2 119.2 (8) F5—C6—C1 119.5 (6)
C13—C14—H14 120.1 F5—C6—C5 117.6 (7)
C15—C14—H14 120.1 C5—C6—C1 122.8 (7)
C15—C14—C13 119.8 (7) F9—C23—C24 121.5 (7)
C31—C32—H32 120.0 F9—C23—C22 119.8 (7)
C33—C32—H32 120.0 C24—C23—C22 118.7 (8)
C33—C32—C31 119.9 (7) F6—C20—C21 116.8 (7)
C32—C31—P2 123.0 (5) F6—C20—C19 121.7 (7)
C36—C31—P2 118.0 (5) C19—C20—C21 121.4 (7)
C36—C31—C32 119.0 (7) F10—C24—C19 120.1 (6)
F7—C21—C20 119.1 (8) F10—C24—C23 116.1 (7)
F7—C21—C22 120.7 (7) C23—C24—C19 123.7 (7)
C22—C21—C20 120.2 (7) C29—C28—H28 121.4
F1—C2—C3 116.3 (7) C29—C28—C27 117.3 (7)
F1—C2—C1 121.3 (7) C27—C28—H28 121.4
C3—C2—C1 122.4 (8) F3—C4—C3 120.7 (7)
C14—C13—P1 117.7 (5) F3—C4—C5 118.9 (8)
C18—C13—P1 121.6 (5) C3—C4—C5 120.4 (7)
C18—C13—C14 120.5 (6) F4—C5—C6 120.4 (7)
C30—C25—P2 121.5 (5) F4—C5—C4 120.7 (7)

99
C30—C25—C26 119.4 (7) C6—C5—C4 118.9 (7)
C26—C25—P2 119.0 (6) C16—C17—C18 119.7 (7)
C20—C19—P2 123.6 (6) C16—C17—H17 120.2
C24—C19—P2 120.1 (5) C18—C17—H17 120.2
C24—C19—C20 116.3 (7) C25—C26—H26 119.8
C35—C34—H34 120.4 C27—C26—C25 120.5 (7)
C33—C34—H34 120.4 C27—C26—H26 119.8
C33—C34—C35 119.2 (7) C28—C27—H27 119.4
C12—C7—P1 120.6 (6) C26—C27—C28 121.1 (8)
C8—C7—P1 120.1 (5) C26—C27—H27 119.4
C8—C7—C12 119.0 (7) C11—C10—H10 120.0
C15—C16—H16 119.7 C11—C10—C9 120.1 (8)
C15—C16—C17 120.5 (6) C9—C10—H10 120.0
C17—C16—H16 119.7 C32—C33—H33 119.5
C13—C18—H18 120.3 C34—C33—C32 120.9 (6)
C13—C18—C17 119.4 (7) C34—C33—H33 119.5
C17—C18—H18 120.3 C8—C9—C10 119.3 (8)
C7—C8—H8 119.5 C8—C9—H9 120.4
C7—C8—C9 120.9 (7) C10—C9—H9 120.4
C9—C8—H8 119.5 F8—C22—C21 120.6 (8)
C25—C30—H30 120.5 F8—C22—C23 119.6 (8)
C29—C30—C25 119.1 (7) C21—C22—C23 119.6 (7)
Au2—P2—C31—C32 133.7 (5) C13—P1—C1—C6 55.7 (6)
Au2—P2—C31—C36 -45.2 (6) C13—C14—C15—C16 -0.6 (11)
Au2—P2—C25—C30 128.9 (5) C13—C18—C17—C16 -2.0 (11)
Au2—P2—C25—C26 -48.2 (6) F7—C21—C20—F6 -0.1 (10)
Au2—P2—C19—C20 144.9 (5) F7—C21—C20—C19 -179.6 (6)
Au2—P2—C19—C24 -37.9 (6) F7—C21—C22—F8 0.8 (12)
Au1—P1—C13—C14 -39.8 (6) F7—C21—C22—C23 177.9 (6)
Au1—P1—C13—C18 145.2 (5) C25—P2—C31—C32 -104.5 (6)
Au1—P1—C7—C12 176.0 (4) C25—P2—C31—C36 76.6 (6)
Au1—P1—C7—C8 1.6 (6) C25—P2—C19—C20 22.5 (7)
Au1—P1—C1—C2 108.8 (6) C25—P2—C19—C24 -160.2 (6)
Au1—P1—C1—C6 -68.8 (6) C25—C30—C29—C28 -0.6 (10)
P1—C13—C18—C17 176.7 (5) C25—C26—C27—C28 -0.1 (11)
P1—C7—C8—C9 174.4 (5) C19—P2—C31—C32 11.4 (7)
P1—C1—C6—F5 0.7 (9) C19—P2—C31—C36 -167.5 (6)
P1—C1—C6—C5 -178.0 (5) C19—P2—C25—C30 -106.9 (6)
P2—C31—C36—C35 176.9 (5) C19—P2—C25—C26 75.9 (6)

100
P2—C25—C30—C29 -175.8 (5) C7—P1—C13—C14 84.9 (6)
P2—C25—C26—C27 176.2 (5) C7—P1—C13—C18 -90.2 (6)
P2—C19—C20—F6 -0.6 (10) C7—P1—C1—C2 -17.2 (7)
P2—C19—C20—C21 178.9 (6) C7—P1—C1—C6 165.2 (5)
P2—C19—C24—F10 -1.1 (10) C7—C12—C11—C10 -0.8 (10)
P2—C19—C24—C23 -178.4 (6) C7—C8—C9—C10 -1.1 (11)
F1—C2—C1—P1 -0.5 (10) C30—C25—C26—C27 -1.0 (10)
F1—C2—C1—C6 177.2 (6) C30—C29—C28—C27 -0.5 (10)
F5—C6—C5—F4 -0.5 (10) C29—C28—C27—C26 0.9 (10)
F5—C6—C5—C4 178.2 (6) C1—P1—C13—C14 -161.1 (5)
F3—C4—C5—F4 -1.7 (10) C1—P1—C13—C18 23.9 (7)
F3—C4—C5—C6 179.6 (6) C1—P1—C7—C12 -59.6 (6)
F2—C3—C2—F1 -0.3 (10) C1—P1—C7—C8 126.0 (6)
F2—C3—C2—C1 180.0 (6) C1—C6—C5—F4 178.3 (6)
F2—C3—C4—F3 2.3 (10) C1—C6—C5—C4 -3.0 (11)
F2—C3—C4—C5 -178.7 (6) C35—C34—C33—C32 -1.3 (11)
F9—C23—C24—F10 1.0 (11) C15—C14—C13—P1 -175.5 (5)
F9—C23—C24—C19 178.4 (7) C15—C14—C13—C18 -0.4 (10)
F9—C23—C22—F8 -0.2 (12) C15—C16—C17—C18 1.0 (11)
F9—C23—C22—C21 -177.4 (7) C11—C12—C7—P1 -173.4 (5)
C12—C7—C8—C9 0.0 (10) C11—C12—C7—C8 1.0 (10)
C12—C11—C10—C9 -0.4 (11) C11—C10—C9—C8 1.3 (11)
C3—C2—C1—P1 179.2 (5) C20—C21—C22—F8 -178.2 (7)
C3—C2—C1—C6 -3.1 (10) C20—C21—C22—C23 -1.1 (12)
C3—C4—C5—F4 179.3 (6) C20—C19—C24—F10 176.3 (6)
C3—C4—C5—C6 0.6 (11) C20—C19—C24—C23 -1.0 (11)
C14—C13—C18—C17 1.7 (11) C24—C19—C20—F6 -177.9 (6)
C32—C31—C36—C35 -2.1 (10) C24—C19—C20—C21 1.5 (11)
C31—P2—C25—C30 5.1 (6) C24—C23—C22—F8 178.7 (7)
C31—P2—C25—C26 -172.0 (5) C24—C23—C22—C21 1.6 (12)
C31—P2—C19—C20 -92.6 (6) C4—C3—C2—F1 -179.3 (6)
C31—P2—C19—C24 84.6 (6) C4—C3—C2—C1 1.0 (11)
101
C31—C32—C33—C34 -0.9 (11) C17—C16—C15—C14 0.4 (11)
C31—C36—C35—C34 -0.1 (11) C26—C25—C30—C29 1.3 (10)
C2—C3—C4—F3 -178.6 (6) C33—C32—C31—P2 -176.3 (5)
C2—C3—C4—C5 0.3 (11) C33—C32—C31—C36 2.6 (10)
C2—C1—C6—F5 -177.1 (5) C33—C34—C35—C36 1.9 (11)
C2—C1—C6—C5 4.2 (10) C22—C21—C20—F6 178.9 (7)
C13—P1—C7—C12 49.9 (6) C22—C21—C20—C19 -0.5 (12)
C13—P1—C7—C8 -124.5 (6) C22—C23—C24—F10 -178.0 (6)
C13—P1—C1—C2 -126.7 (6) C22—C23—C24—C19 -0.5 (12)
[AuBr{PPh2(C6F5)}] (II)
Crystal data
2(C18H10AuBrF5P) Z = 2
Mr = 1258.21 F(000) = 1168
Triclinic, P¯1 Dx = 2.325 Mg m-3
a = 11.7733 (7) Å Mo K radiation, = 0.71073 Å
b = 13.1903 (7) Å Cell parameters from 4549 reflections
c = 13.6025 (8) Å = 3.9–27.8°
= 107.387 (5)° = 10.54 mm-1
= 102.314 (5)° T = 150 K
= 108.504 (5)° , clear light colourless
V = 1797.44 (19) Å3 0.08 × 0.06 × 0.04 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
7071 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source, Incoatec Is
5386 reflections with I > 2(I)
Mirror monochromator Rint = 0.055
Detector resolution: 8.0714 pixels mm-1
max = 26.0°, min = 3.3°
scans h = -1316
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1715

102
Tmin = 0.413, Tmax = 1.000 l = -1717
15324 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-
invariant direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.047 H-atom parameters constrained
wR(F2) = 0.078 w = 1/[
2(Fo
2)]
where P = (Fo2 + 2Fc
2)/3
S = 1.00 (/)max = 0.001
7071 reflections max = 1.95 e Å-3
469 parameters min = -1.88 e Å-3
6 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate
(isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au2 0.08279 (3) 0.19907 (3) 0.49728 (3) 0.02346 (10)
Au1 0.59486 (3) 0.90740 (3) 0.85491 (3) 0.02384 (10)
Br2 0.15841 (8) 0.32813 (7) 0.68648 (7) 0.0279 (2)
Br1 0.74183 (8) 1.10779 (7) 0.94996 (7) 0.0302 (2)
P1 0.45792 (19) 0.72081 (19) 0.77074 (18) 0.0214 (5)
P2 0.00900 (19) 0.07815 (19) 0.32024 (18) 0.0219 (5)
F1 0.1829 (4) 0.6488 (4) 0.7922 (4) 0.0358 (13)
F10 0.2770 (4) 0.1112 (5) 0.4280 (4) 0.0407 (14)
F5 0.6013 (4) 0.6887 (4) 0.9572 (4) 0.0350 (13)
F6 -0.1078 (4) -0.1786 (4) 0.1520 (4) 0.0347 (13)
F3 0.2754 (5) 0.5468 (5) 1.0909 (4) 0.0481 (15)
F2 0.1097 (4) 0.5725 (4) 0.9386 (4) 0.0402 (14)
F4 0.5225 (5) 0.6070 (5) 1.0989 (4) 0.0446 (14)
F8 0.2462 (5) -0.2637 (5) 0.2467 (5) 0.0581 (17)
F9 0.3830 (5) -0.0426 (5) 0.3981 (5) 0.0602 (18)

103
C12 0.2381 (7) 0.5807 (7) 0.5889 (6) 0.023 (2)
H12 0.2421 0.5195 0.6080 0.027*
C3 0.2299 (8) 0.5987 (7) 0.9415 (8) 0.030 (2)
C14 0.6055 (7) 0.6501 (7) 0.6561 (7) 0.028 (2)
H14 0.6309 0.7235 0.6540 0.034*
C32 0.0955 (7) 0.1098 (7) 0.1511 (7) 0.026 (2)
H32 0.1050 0.0402 0.1394 0.032*
C31 0.0474 (7) 0.1503 (7) 0.2308 (7) 0.0220 (19)
C21 0.0681 (9) -0.2208 (8) 0.2027 (8) 0.037 (2)
C2 0.2700 (8) 0.6380 (7) 0.8657 (7) 0.028 (2)
C13 0.5233 (7) 0.6193 (7) 0.7121 (7) 0.0214 (19)
F7 0.0018 (5) -0.3291 (4) 0.1229 (4) 0.0468 (15)
C25 -0.1631 (7) 0.0013 (7) 0.2739 (7) 0.024 (2)
C19 0.0780 (8) -0.0300 (7) 0.2951 (7) 0.026 (2)
C34 0.1170 (7) 0.2740 (8) 0.1040 (7) 0.031 (2)
H34 0.1420 0.3161 0.0626 0.037*
C7 0.3271 (7) 0.6918 (7) 0.6555 (7) 0.0221 (19)
C16 0.6149 (7) 0.4649 (8) 0.6087 (7) 0.031 (2)
H16 0.6466 0.4133 0.5744 0.037*
C18 0.4866 (7) 0.5087 (7) 0.7150 (7) 0.026 (2)
H18 0.4307 0.4864 0.7512 0.031*
C8 0.3192 (8) 0.7824 (7) 0.6247 (7) 0.030 (2)
H8 0.3781 0.8585 0.6676 0.036*
C30 -0.2408 (7) -0.0050 (7) 0.1788 (7) 0.0225 (19)
H30 -0.2060 0.0255 0.1333 0.027*
C29 -0.3726 (7) -0.0579 (7) 0.1520 (7) 0.027 (2)
H29 -0.4255 -0.0618 0.0883 0.033*
C36 0.0307 (7) 0.2537 (7) 0.2462 (7) 0.029 (2)
H36 -0.0037 0.2812 0.2985 0.035*
C1 0.3955 (7) 0.6694 (7) 0.8666 (7) 0.0205 (19)
C35 0.0658 (7) 0.3148 (7) 0.1829 (7) 0.029 (2)
H35 0.0552 0.3837 0.1932 0.034*
C15 0.6490 (8) 0.5728 (8) 0.6043 (7) 0.032 (2)
H15 0.7023 0.5933 0.5655 0.038*
C11 0.1426 (7) 0.5562 (8) 0.4942 (7) 0.032 (2)
H11 0.0838 0.4800 0.4513 0.038*
C6 0.4782 (8) 0.6577 (7) 0.9501 (7) 0.023 (2)
C23 0.2611 (9) -0.0776 (9) 0.3397 (9) 0.039 (3)
C20 0.0129 (8) -0.1427 (8) 0.2160 (7) 0.025 (2)
C24 0.2050 (9) 0.0000 (8) 0.3548 (7) 0.031 (2)

104
C28 -0.4267 (8) -0.1050 (7) 0.2182 (8) 0.032 (2)
H28 -0.5147 -0.1394 0.1995 0.038*
C4 0.3149 (9) 0.5873 (7) 1.0187 (7) 0.032 (2)
C5 0.4388 (8) 0.6168 (7) 1.0217 (8) 0.032 (2)
C17 0.5337 (8) 0.4337 (8) 0.6640 (8) 0.035 (2)
H17 0.5105 0.3606 0.6667 0.042*
C26 -0.2164 (8) -0.0464 (7) 0.3422 (7) 0.030 (2)
H26 -0.1636 -0.0415 0.4065 0.037*
C27 -0.3471 (8) -0.1001 (7) 0.3131 (8) 0.037 (2)
H27 -0.3821 -0.1332 0.3570 0.045*
C10 0.1359 (8) 0.6459 (9) 0.4646 (8) 0.036 (2)
H10 0.0729 0.6306 0.4011 0.043*
C33 0.1301 (7) 0.1715 (8) 0.0878 (7) 0.033 (2)
H33 0.1625 0.1429 0.0342 0.039*
C9 0.2238 (9) 0.7595 (8) 0.5303 (8) 0.041 (3)
H9 0.2189 0.8203 0.5110 0.049*
C22 0.1896 (10) -0.1893 (9) 0.2625 (9) 0.044 (3)
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au2 0.0289 (2) 0.02043 (19) 0.0202 (2) 0.01101 (15) 0.00678 (14) 0.00703
(16)
Au1 0.0311 (2) 0.01499 (18) 0.0230 (2) 0.00933 (15) 0.00786 (15) 0.00532
(15)
Br2 0.0357 (5) 0.0218 (5) 0.0197 (5) 0.0081 (4) 0.0061 (4) 0.0060 (4)
Br1 0.0357 (5) 0.0168 (5) 0.0294 (5) 0.0066 (4) 0.0069 (4) 0.0052 (4)
P1 0.0274 (12) 0.0154 (12) 0.0227 (13) 0.0100 (10) 0.0083 (10) 0.0079
(10)
P2 0.0274 (13) 0.0196 (12) 0.0216 (13) 0.0121 (10) 0.0080 (10) 0.0093
(10)
F1 0.038 (3) 0.048 (3) 0.036 (3) 0.028 (3) 0.019 (2) 0.020 (3)
F10 0.038 (3) 0.041 (3) 0.041 (4) 0.020 (3) 0.009 (3) 0.014 (3)
F5 0.031 (3) 0.037 (3) 0.031 (3) 0.014 (2) 0.003 (2) 0.011 (3)
F6 0.050 (3) 0.024 (3) 0.023 (3) 0.015 (2) 0.006 (2) 0.004 (2)
F3 0.077 (4) 0.041 (4) 0.048 (4) 0.032 (3) 0.038 (3) 0.025 (3)
F2 0.048 (3) 0.039 (3) 0.054 (4) 0.024 (3) 0.035 (3) 0.026 (3)
F4 0.068 (4) 0.046 (4) 0.036 (3) 0.035 (3) 0.018 (3) 0.025 (3)
F8 0.081 (4) 0.065 (4) 0.071 (5) 0.064 (4) 0.041 (4) 0.036 (4)
F9 0.048 (4) 0.080 (5) 0.080 (5) 0.046 (3) 0.028 (3) 0.043 (4)
C12 0.027 (5) 0.027 (5) 0.017 (5) 0.013 (4) 0.009 (4) 0.008 (4)
C3 0.043 (6) 0.025 (5) 0.038 (6) 0.022 (4) 0.030 (5) 0.014 (5)

105
C14 0.027 (5) 0.019 (5) 0.032 (6) 0.005 (4) 0.009 (4) 0.008 (4)
C32 0.021 (5) 0.023 (5) 0.028 (6) 0.001 (4) 0.006 (4) 0.010 (4)
C31 0.022 (5) 0.014 (4) 0.028 (5) 0.008 (4) 0.009 (4) 0.004 (4)
C21 0.055 (7) 0.027 (6) 0.038 (7) 0.017 (5) 0.024 (5) 0.019 (5)
C2 0.038 (5) 0.028 (5) 0.029 (6) 0.022 (4) 0.017 (4) 0.013 (5)
C13 0.019 (4) 0.020 (5) 0.024 (5) 0.012 (4) 0.001 (4) 0.007 (4)
F7 0.080 (4) 0.029 (3) 0.042 (4) 0.031 (3) 0.032 (3) 0.011 (3)
C25 0.035 (5) 0.016 (4) 0.024 (5) 0.019 (4) 0.011 (4) 0.003 (4)
C19 0.043 (6) 0.027 (5) 0.022 (5) 0.021 (4) 0.016 (4) 0.017 (4)
C34 0.025 (5) 0.028 (6) 0.035 (6) 0.004 (4) 0.006 (4) 0.017 (5)
C7 0.029 (5) 0.022 (5) 0.020 (5) 0.016 (4) 0.011 (4) 0.007 (4)
C16 0.033 (5) 0.033 (6) 0.025 (6) 0.024 (4) 0.009 (4) -0.001 (5)
C18 0.032 (5) 0.022 (5) 0.033 (6) 0.016 (4) 0.018 (4) 0.013 (4)
C8 0.047 (6) 0.025 (5) 0.021 (5) 0.018 (4) 0.017 (4) 0.006 (4)
C30 0.031 (5) 0.020 (5) 0.022 (5) 0.016 (4) 0.014 (4) 0.008 (4)
C29 0.034 (5) 0.026 (5) 0.023 (5) 0.019 (4) 0.004 (4) 0.007 (4)
C36 0.036 (5) 0.020 (5) 0.030 (6) 0.012 (4) 0.009 (4) 0.008 (4)
C1 0.022 (5) 0.014 (4) 0.031 (5) 0.010 (4) 0.013 (4) 0.009 (4)
C35 0.040 (5) 0.010 (4) 0.028 (6) 0.010 (4) 0.003 (4) 0.004 (4)
C15 0.037 (5) 0.029 (5) 0.035 (6) 0.021 (4) 0.015 (4) 0.010 (5)
C11 0.023 (5) 0.025 (5) 0.036 (6) 0.007 (4) 0.012 (4) -0.001 (5)
C6 0.033 (4) 0.017 (4) 0.022 (4) 0.015 (3) 0.009 (3) 0.006 (3)
C23 0.032 (6) 0.052 (7) 0.061 (8) 0.033 (5) 0.025 (5) 0.036 (6)
C20 0.038 (5) 0.026 (5) 0.025 (5) 0.022 (4) 0.018 (4) 0.013 (4)
C24 0.050 (6) 0.032 (6) 0.028 (6) 0.026 (5) 0.023 (5) 0.019 (5)
C28 0.036 (6) 0.013 (5) 0.044 (7) 0.011 (4) 0.017 (5) 0.005 (5)
C4 0.062 (7) 0.022 (5) 0.026 (6) 0.023 (5) 0.028 (5) 0.013 (5)
C5 0.043 (6) 0.016 (5) 0.039 (6) 0.016 (4) 0.011 (5) 0.012 (5)
C17 0.044 (6) 0.028 (6) 0.041 (6) 0.019 (5) 0.016 (5) 0.018 (5)
C26 0.040 (6) 0.029 (5) 0.034 (6) 0.022 (4) 0.015 (4) 0.018 (5)
C27 0.039 (6) 0.017 (5) 0.051 (7) 0.008 (4) 0.023 (5) 0.007 (5)
C10 0.041 (6) 0.056 (7) 0.027 (6) 0.035 (5) 0.021 (5) 0.014 (5)
C33 0.034 (5) 0.036 (6) 0.027 (6) 0.012 (5) 0.010 (4) 0.014 (5)
C9 0.062 (7) 0.039 (6) 0.034 (6) 0.036 (5) 0.013 (5) 0.017 (5)
C22 0.063 (7) 0.040 (7) 0.061 (8) 0.037 (6) 0.030 (6) 0.037 (6)
Geometric parameters (Å, º)
Au2—Br2 2.3992 (10) C19—C20 1.388 (11)
Au2—P2 2.243 (2) C19—C24 1.402 (11)

106
Au1—Br1 2.3984 (9) C34—H34 0.9300
Au1—P1 2.231 (2) C34—C35 1.394 (11)
P1—C13 1.816 (8) C34—C33 1.368 (11)
P1—C7 1.793 (8) C7—C8 1.402 (11)
P1—C1 1.823 (8) C16—H16 0.9300
P2—C31 1.801 (8) C16—C15 1.374 (11)
P2—C25 1.816 (8) C16—C17 1.375 (10)
P2—C19 1.837 (9) C18—H18 0.9300
F1—C2 1.346 (9) C18—C17 1.367 (11)
F10—C24 1.356 (10) C8—H8 0.9300
F5—C6 1.348 (8) C8—C9 1.392 (11)
F6—C20 1.342 (9) C30—H30 0.9300
F3—C4 1.347 (9) C30—C29 1.395 (10)
F2—C3 1.334 (9) C29—H29 0.9300
F4—C5 1.346 (9) C29—C28 1.389 (11)
F8—C22 1.342 (10) C36—H36 0.9300
F9—C23 1.334 (9) C36—C35 1.384 (10)
C12—H12 0.9300 C1—C6 1.410 (10)
C12—C7 1.376 (10) C35—H35 0.9300
C12—C11 1.389 (10) C15—H15 0.9300
C3—C2 1.389 (11) C11—H11 0.9300
C3—C4 1.367 (11) C11—C10 1.379 (11)
C14—H14 0.9300 C6—C5 1.343 (11)
C14—C13 1.391 (10) C23—C24 1.374 (12)
C14—C15 1.361 (11) C23—C22 1.376 (13)
C32—H32 0.9300 C28—H28 0.9300
C32—C31 1.375 (11) C28—C27 1.394 (12)
C32—C33 1.388 (10) C4—C5 1.373 (11)
C31—C36 1.400 (11) C17—H17 0.9300
C21—F7 1.354 (10) C26—H26 0.9300
C21—C20 1.372 (12) C26—C27 1.380 (10)
C21—C22 1.344 (12) C27—H27 0.9300
C2—C1 1.398 (10) C10—H10 0.9300
C13—C18 1.399 (10) C10—C9 1.391 (12)
C25—C30 1.378 (10) C33—H33 0.9300
C25—C26 1.414 (10) C9—H9 0.9300
P2—Au2—Br2 179.10 (6) C29—C30—H30 120.6
P1—Au1—Br1 178.48 (6) C30—C29—H29 119.2
C13—P1—Au1 115.6 (3) C28—C29—C30 121.6 (8)

107
C13—P1—C1 104.4 (4) C28—C29—H29 119.2
C7—P1—Au1 113.9 (3) C31—C36—H36 120.2
C7—P1—C13 102.5 (4) C35—C36—C31 119.5 (9)
C7—P1—C1 108.9 (4) C35—C36—H36 120.2
C1—P1—Au1 110.7 (3) C2—C1—P1 126.1 (6)
C31—P2—Au2 113.7 (3) C2—C1—C6 115.1 (7)
C31—P2—C25 108.0 (4) C6—C1—P1 118.8 (6)
C31—P2—C19 103.9 (4) C34—C35—H35 119.7
C25—P2—Au2 110.2 (3) C36—C35—C34 120.6 (8)
C25—P2—C19 108.4 (4) C36—C35—H35 119.7
C19—P2—Au2 112.5 (3) C14—C15—C16 120.8 (9)
C7—C12—H12 118.7 C14—C15—H15 119.6
C7—C12—C11 122.7 (8) C16—C15—H15 119.6
C11—C12—H12 118.7 C12—C11—H11 120.3
F2—C3—C2 120.1 (8) C10—C11—C12 119.3 (8)
F2—C3—C4 120.7 (8) C10—C11—H11 120.3
C4—C3—C2 119.2 (8) F5—C6—C1 118.1 (7)
C13—C14—H14 120.0 C5—C6—F5 119.3 (7)
C15—C14—H14 120.0 C5—C6—C1 122.5 (8)
C15—C14—C13 120.1 (8) F9—C23—C24 119.5 (10)
C31—C32—H32 119.6 F9—C23—C22 121.9 (9)
C31—C32—C33 120.8 (9) C24—C23—C22 118.5 (9)
C33—C32—H32 119.6 F6—C20—C21 118.3 (8)
C32—C31—P2 122.7 (6) F6—C20—C19 120.3 (8)
C32—C31—C36 119.2 (8) C21—C20—C19 121.4 (8)
C36—C31—P2 118.0 (7) F10—C24—C19 118.8 (8)
F7—C21—C20 119.3 (9) F10—C24—C23 118.5 (8)
C22—C21—F7 119.4 (9) C23—C24—C19 122.8 (9)
C22—C21—C20 121.1 (9) C29—C28—H28 120.5
F1—C2—C3 116.8 (7) C29—C28—C27 119.0 (8)
F1—C2—C1 120.7 (7) C27—C28—H28 120.5
C3—C2—C1 122.5 (8) F3—C4—C3 119.3 (8)
C14—C13—P1 118.4 (6) F3—C4—C5 120.9 (8)
C14—C13—C18 119.3 (8) C3—C4—C5 119.8 (8)
C18—C13—P1 122.2 (6) F4—C5—C4 119.6 (8)
C30—C25—P2 121.7 (6) C6—C5—F4 119.5 (8)
C30—C25—C26 120.4 (7) C6—C5—C4 120.9 (8)
C26—C25—P2 117.8 (6) C16—C17—H17 119.5
C20—C19—P2 123.9 (7) C18—C17—C16 121.0 (8)
C20—C19—C24 115.7 (8) C18—C17—H17 119.5

108
C24—C19—P2 120.4 (7) C25—C26—H26 120.1
C35—C34—H34 120.3 C27—C26—C25 119.8 (8)
C33—C34—H34 120.3 C27—C26—H26 120.1
C33—C34—C35 119.4 (8) C28—C27—H27 119.8
C12—C7—P1 122.2 (6) C26—C27—C28 120.3 (8)
C12—C7—C8 117.5 (7) C26—C27—H27 119.8
C8—C7—P1 120.1 (6) C11—C10—H10 120.2
C15—C16—H16 120.2 C11—C10—C9 119.6 (8)
C15—C16—C17 119.6 (8) C9—C10—H10 120.2
C17—C16—H16 120.2 C32—C33—H33 119.8
C13—C18—H18 120.3 C34—C33—C32 120.4 (9)
C17—C18—C13 119.4 (8) C34—C33—H33 119.8
C17—C18—H18 120.3 C8—C9—H9 119.8
C7—C8—H8 119.7 C10—C9—C8 120.3 (9)
C9—C8—C7 120.6 (8) C10—C9—H9 119.8
C9—C8—H8 119.7 F8—C22—C21 121.5 (10)
C25—C30—H30 120.6 F8—C22—C23 118.0 (9)
C25—C30—C29 118.8 (8) C21—C22—C23 120.4 (10)
Au2—P2—C31—C32 133.3 (6) C13—P1—C1—C6 56.3 (7)
Au2—P2—C31—C36 -44.7 (7) C13—C14—C15—C16 -1.5 (13)
Au2—P2—C25—C30 130.7 (6) C13—C18—C17—C16 -1.0 (14)
Au2—P2—C25—C26 -45.4 (7) F7—C21—C20—F6 2.9 (12)
Au2—P2—C19—C20 146.6 (6) F7—C21—C20—C19 -178.7 (7)
Au2—P2—C19—C24 -35.6 (7) F7—C21—C22—F8 -1.5 (14)
Au1—P1—C13—C14 -42.6 (7) F7—C21—C22—C23 176.2 (8)
Au1—P1—C13—C18 142.0 (6) C25—P2—C31—C32 -104.1 (7)
Au1—P1—C7—C12 175.3 (6) C25—P2—C31—C36 77.9 (7)
Au1—P1—C7—C8 1.1 (8) C25—P2—C19—C20 24.6 (8)
Au1—P1—C1—C2 112.0 (7) C25—P2—C19—C24 -157.6 (6)
Au1—P1—C1—C6 -68.8 (7) C25—C30—C29—C28 -0.6 (13)
P1—C13—C18—C17 176.3 (7) C25—C26—C27—C28 -1.7 (13)
P1—C7—C8—C9 174.8 (7) C19—P2—C31—C32 10.8 (8)
P1—C1—C6—F5 2.1 (10) C19—P2—C31—C36 -167.2 (6)
P1—C1—C6—C5 -177.8 (7) C19—P2—C25—C30 -105.9 (7)
P2—C31—C36—C35 176.5 (6) C19—P2—C25—C26 78.0 (7)
P2—C25—C30—C29 -175.3 (6) C7—P1—C13—C14 82.0 (7)
P2—C25—C26—C27 176.6 (7) C7—P1—C13—C18 -93.4 (7)
P2—C19—C20—F6 0.1 (11) C7—P1—C1—C2 -14.0 (9)
P2—C19—C20—C21 -178.3 (6) C7—P1—C1—C6 165.2 (6)

109
P2—C19—C24—F10 -2.6 (10) C7—C12—C11—C10 0.3 (13)
P2—C19—C24—C23 179.7 (7) C7—C8—C9—C10 -0.7 (14)
F1—C2—C1—P1 -2.9 (12) C30—C25—C26—C27 0.4 (13)
F1—C2—C1—C6 177.9 (7) C30—C29—C28—C27 -0.6 (13)
F5—C6—C5—F4 -0.3 (13) C29—C28—C27—C26 1.7 (13)
F5—C6—C5—C4 178.2 (7) C1—P1—C13—C14 -164.4 (6)
F3—C4—C5—F4 -1.1 (13) C1—P1—C13—C18 20.2 (8)
F3—C4—C5—C6 -179.7 (8) C1—P1—C7—C12 -60.6 (8)
F2—C3—C2—F1 0.6 (13) C1—P1—C7—C8 125.2 (7)
F2—C3—C2—C1 178.4 (7) C1—C6—C5—F4 179.5 (7)
F2—C3—C4—F3 1.4 (13) C1—C6—C5—C4 -1.9 (14)
F2—C3—C4—C5 -178.7 (8) C35—C34—C33—C32 -1.4 (12)
F9—C23—C24—F10 1.2 (13) C15—C14—C13—P1 -175.2 (7)
F9—C23—C24—C19 178.9 (8) C15—C14—C13—C18 0.3 (12)
F9—C23—C22—F8 -0.2 (14) C15—C16—C17—C18 -0.2 (13)
F9—C23—C22—C21 -178.0 (8) C11—C12—C7—P1 -174.5 (6)
C12—C7—C8—C9 0.4 (13) C11—C12—C7—C8 -0.2 (13)
C12—C11—C10—C9 -0.6 (13) C11—C10—C9—C8 0.8 (14)
C3—C2—C1—P1 179.5 (7) C20—C21—C22—F8 -177.0 (8)
C3—C2—C1—C6 0.2 (12) C20—C21—C22—C23 0.7 (15)
C3—C4—C5—F4 179.0 (8) C20—C19—C24—F10 175.3 (7)
C3—C4—C5—C6 0.4 (14) C20—C19—C24—C23 -2.4 (12)
C14—C13—C18—C17 0.9 (12) C24—C19—C20—F6 -177.8 (7)
C32—C31—C36—C35 -1.6 (12) C24—C19—C20—C21 3.9 (12)
C31—P2—C25—C30 6.0 (8) C24—C23—C22—F8 178.6 (8)
C31—P2—C25—C26 -170.1 (6) C24—C23—C22—C21 0.8 (15)
C31—P2—C19—C20 -90.0 (7) C4—C3—C2—F1 -179.3 (8)
C31—P2—C19—C24 87.7 (7) C4—C3—C2—C1 -1.6 (14)
C31—C32—C33—C34 0.1 (12) C17—C16—C15—C14 1.5 (13)
C31—C36—C35—C34 0.2 (12) C26—C25—C30—C29 0.7 (12)
C2—C3—C4—F3 -178.6 (8) C33—C32—C31—P2 -176.5 (6)
C2—C3—C4—C5 1.2 (13) C33—C32—C31—C36 1.4 (12)
C2—C1—C6—F5 -178.6 (7) C33—C34—C35—C36 1.3 (12)
C2—C1—C6—C5 1.5 (12) C22—C21—C20—F6 178.4 (8)
C13—P1—C7—C12 49.6 (8) C22—C21—C20—C19 -3.2 (14)
C13—P1—C7—C8 -124.6 (7) C22—C23—C24—F10 -177.6 (8)
C13—P1—C1—C2 -122.9 (8) C22—C23—C24—C19 0.1 (14)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].

110
Chapter 4
Crystal structure of [AuI{PPh2(i-C3F7)}]

111
4. Crystal structure of [AuI{PPh2(i-C3F7)}]
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, University of Manchester, Manchester, M13 9PL, UK.
Keywords: Crystal structure; F···F interaction; hydrogen bonding; Gold (I) Phosphine.
Crystals of the title compound [AuI{PPh2(i-C3F7)}] were grown by slow evaporation of
the solvent from a CH2Cl2 solution of [AuI{PPh2(i-C3F7)}]. The Au atom adopts a
linear geometry; the Au‒P and Au‒I bond lengths are 2.257(2) and 2.5593(7) Å,
respectively, and the P‒Au‒I angle is 178.62(7)º. In the crystal, weak C–H···F
hydrogen bonding (2.636 Å) results in chains which are linked by F···F interactions
(2.78(1) and 2.93(1) Å) into a three-dimensional network.
3D view Chemical scheme
4.1 Structure description
Many linear two-coordinate complexes of gold (I) halides with tertiary-substituted
arylphosphines of the type [Au X( PR3)] (where X = Cl, Br, I) have been studied (Boot
et al., 2007). However a very much more limited number of complexes are known when
considering phosphines with fluoroalkyl substituents (Brisdon et al., 2010).

112
The structure of the title compound derived from single crystal X-ray diffraction data is
shown in Fig. 1. Coordination at the gold centre is essentially linear, the P‒Au‒I angle
is 178.62(7)º, with Au‒P and Au‒I bond lengths of 2.257(2) and 2.5593(7) Å,
respectively. The Au‒I distance is slightly longer than that found for the complex
[AuI(PPh3)] d(Au‒I) = 2.553(1) Å (Ahrland et al., 1987), while the Au‒P distance is
similar to that reported for the non-isomorphous [AuCl{PPh2(i-C3F7)}] d(Au‒P) =
2.2433(51) Å (Brisdon et al., 2010). The P‒C distance to the fluorinated group is longer
at 1.913(11) Å than the distances to the phenyl rings at 1.814(11) Å and 1.819(11) Å.
Figure 1 The molecular structure of [AuI{PPh2(i-C3F7)}], with displacement ellipsoids drawn at
the 50% probability level.
In the extended structure hydrogen bond interactions are observed between molecules
involving a meta proton on an aryl ring and one of the CF3 fluorines of an adjacent
molecule, C12‒H12···F4, d(H···F) = 2.636 Å (Table 1) resulting in the formation of
chains of molecules in the b direction.
Table 1:
Hydrogen-bond geometry (Å, °
).

113
D—H···A D—H H···A D···A D—H···A
C12—H12···F4i 0.93 2.636 3.28(1) 127.1
Symmetry code: (i) x, -1+y, z.
Figure 2 Crystal packing of the title compound showing the C–H···F interactions, viewed down
the a-axis.
There are also F···F interactions which link the hydrogen bonded chains, with F···F
distances of (F2···F2 = 2.93(1) Å and F5···F5 = 2.78(1) Å). Both of these distances are
shorter than twice the sum of the van der Waals’ radius of fluorine (2.94 Å) (Bondi,
1964), but in the case of the F2···F2 interaction only just so. The shortest Au···Au
distance is 4.6975(8) Å which is closer to Au···Au distance in Cl analogue (Brisdon et
al., 2010) at 4.502 Å. These distances are larger than twice the sum of the van der
Waals’ radius of gold (3.60 Å), suggesting that there are no significant aurophilic
(Au···Au) interactions in these complexes.

114
Figure 3 Crystal packing of the title compound showing the F···F interactions between
hydrogen bonded chains, viewed down the a-axis.
4.2 Synthesis and crystallization
The title compound (I) was synthesized based on literature procedures (Brisdon et al.,
2010; McAuliffe et al., 1979) in two steps. K[AuCl4] (0.2 g, 0.5 mmol) of was dissolved
in ethanol (5 cm3) and water (2 cm
3). Tetrahydrothiophene (0.1 g, 1.1 mmol) was added
drop-wise, and the reaction mixture stirred for 15 minutes, during which time the
solution changed from yellow to white in colour. After filtration the resulting AuCl(tht)
white solid was dried in vacuo. In a flask containing [PPh2(i-C3F7)] (0.11g, 0.31 mmol)
dissolved in dichloromethane (5 cm3) was added AuCl(tht) (0.1 g, 0.3 mmol) in
dichloromethane (5 cm3) and the mixture was allowed to stir for 24 h. The resulting
solid [AuCl{PPh2(i-C3F7)}] was dissolved in ethanol (10 ml) and KI (0.02 g, 0.12
mmol) was added. The mixture was left to stir for 15 minutes at 80 ºC, after which time
the solution was removed under vacuo, to give [AuI{PPh2(i-C3F7)}]. Crystals were
obtained by slow evaporation from saturated solutions of CH2Cl2. Single crystals were
analysed and data collected at 100 K on a SuperNova diffractometer using graphite
monochromated Mo Kα radiation (λ = 0.71073 Å) with CCD detector.

115
4.3 Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2.
Table 2:
Experimental details:
Crystal data
Chemical formula C15H10AuF7IP
Mr 678.07
Crystal system, space group Triclinic, P-1
Temperature (K) 100
a, b, c (Å) 7.4332(6), 9.9661(8), 12.8272(9)
, , (°) 79.797(6), 84.130(6), 72.245(7)
V (Å3) 889.48(12)
Z 2
Radiation type Mo K
(mm-1
) 10.16
Crystal size (mm) 0.13 × 0.09 × 0.05
Data collection
Diffractometer SuperNova, Single source at offset, Eos
Absorption correction
Multi-scan CrysAlis PRO, Agilent
Technologies, Version 1.171.36.32 (release 02-
08-2013 CrysAlis171 .NET) (compiled Aug 2
2013,16:46:58)
Tmin, Tmax 0.556, 1.000
No. of measured, independent and
observed [I > 2(I)] reflections 4728, 3110, 2541
Rint 0.058
(sin /)max (Å-1
) 0.595
Refinement
R[F2 > 2(F
2)], wR(F
2), S 0.052, 0.112, 1.02
No. of reflections 3110
No. of parameters 226
No. of restraints 150
H-atom treatment H-atom parameters constrained
max, min (e Å-3
) 2.41, -1.77
Acknowledgement
We thank Dr N. Barnes for preparation of the complex [AuI{PPh2(i-C3F7)}].

116
References
Ahrland, S., Dreisch, K., Norén, B. & Oskarsson, A. (1987). Acta Chem. Scand. A41,
173-177.
Barnes, N. A., Brisdon, A. K., Brown, F. R. W., Cross, W. I., Crossley, I. R., Fish, C.,
Herbert, C. J., Pritchard, R. G. & Warren, J. E. (2010). Dalton Trans. 40, 1743-1750.
Bott, R. C., Healy, P. C. & Smith, G. (2007). Polyhedron. 26, 2803-2809.
Bourhis, L. J., Dolomanov, O. V., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2015). Acta Cryst. 71A, 59-75.
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2009). J. Appl. Cryst. 42, 339-341.
McAuliffe, C. A., Parish, R. V. & Randall, D. (1979). J.Chem. Soc., Dalton Trans.
1730-1735.
Sheldrick, G. M. (2008). Acta Cryst. A64, 112-122.
Sheldrick, G. M. (2015). Acta Cryst. C71, 3-8.
Westrip, S. P. (2010). J. Appl. Cryst., 43, 920-925.

117
Full crystallographic data
Crystal structure of [AuI{PPh2(i-C3F7)}]
Arij T. Addaraidi, Alan K. Brisdon and Robin G. Pritchard
Computing details
Data collection: CrysAlis PRO, Agilent Technologies, Version 1.171.36.32 (release 02-
08-2013 CrysAlis171 .NET) (compiled Aug 2 2013,16:46:58); cell refinement:
CrysAlis PRO, Agilent Technologies, Version 1.171.36.32 (release 02-08-2013
CrysAlis171 .NET) (compiled Aug 2 2013,16:46:58); data reduction: CrysAlis PRO,
Agilent Technologies, Version 1.171.36.32 (release 02-08-2013 CrysAlis171 .NET)
(compiled Aug 2 2013,16:46:58); program(s) used to solve structure: SHELXS
(Sheldrick, 2008); program(s) used to refine structure: SHELXL (Sheldrick, 2015);
molecular graphics: Olex2 (Dolomanov et al., 2009); software used to prepare material
for publication: Olex2 (Dolomanov et al., 2009).
Crystal data
C15H10AuF7IP Z = 2
Mr = 678.07 F(000) = 620
Triclinic, P¯1 Dx = 2.532 Mg m-3
a = 7.4332 (6) Å Mo K radiation, = 0.71073 Å
b = 9.9661 (8) Å Cell parameters from 1948 reflections
c = 12.8272 (9) Å = 3.6–28.1°
= 79.797 (6)° = 10.16 mm-1
= 84.130 (6)° T = 100 K
= 72.245 (7)° clear light colourless
V = 889.48 (12) Å3 0.13 × 0.09 × 0.05 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
3110 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source 2541 reflections with I > 2(I)
Mirror monochromator Rint = 0.058
Detector resolution: 8.0714 pixels mm-1
max = 25.0°, min = 3.1°

118
scans h = -88
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.36.32 (release 02-08-2013 CrysAlis171
.NET) (compiled Aug 2 2013,16:46:58)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1111
Tmin = 0.556, Tmax = 1.000 l = -1515
4728 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.052 H-atom parameters constrained
wR(F2) = 0.112 w = 1/[
2(Fo
2) + (0.0264P)
2]
where P = (Fo2 + 2Fc
2)/3
S = 1.02 (/)max < 0.001
3110 reflections max = 2.41 e Å-3
226 parameters min = -1.77 e Å-3
150 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
C1 0.3710 (17) 0.2398 (12) 0.3651 (10) 0.023 (3)
C2 0.482 (2) 0.3463 (14) 0.3640 (11) 0.033 (3)
C3 0.2078 (18) 0.2601 (13) 0.4498 (10) 0.023 (3)
C4 0.4929 (16) 0.2131 (11) 0.1383 (9) 0.014 (2)
C5 0.5613 (15) 0.3276 (11) 0.0991 (9) 0.014 (2)
H5 0.5063 0.4135 0.1246 0.017*
C6 0.7071 (15) 0.3185 (11) 0.0241 (8) 0.012 (2)
H6 0.7507 0.3971 -0.0003 0.014*
C7 0.7897 (17) 0.1921 (12) -0.0154 (9) 0.022 (3)
119
H7 0.8902 0.1848 -0.0657 0.027*
C8 0.7208 (18) 0.0758 (13) 0.0211 (10) 0.025 (3)
H8 0.7740 -0.0090 -0.0060 0.030*
C9 0.5783 (15) 0.0858 (11) 0.0951 (8) 0.015 (3)
H9 0.5345 0.0071 0.1187 0.018*
C10 0.2296 (16) 0.0647 (11) 0.2580 (9) 0.015 (3)
C11 0.3574 (18) -0.0635 (12) 0.2909 (9) 0.024 (3)
H11 0.4823 -0.0685 0.2991 0.028*
C12 0.3007 (17) -0.1860 (12) 0.3120 (9) 0.022 (3)
H12 0.3883 -0.2723 0.3362 0.026*
C13 0.1213 (17) -0.1838 (12) 0.2985 (9) 0.021 (3)
H13 0.0869 -0.2677 0.3146 0.025*
C14 -0.0121 (17) -0.0562 (11) 0.2606 (9) 0.019 (3)
H14 -0.1348 -0.0542 0.2500 0.023*
C15 0.0436 (16) 0.0703 (12) 0.2386 (9) 0.017 (3)
H15 -0.0416 0.1563 0.2115 0.020*
Au1 0.04773 (6) 0.42910 (4) 0.18216 (4) 0.01753 (16)
F1 0.5031 (11) 0.1056 (6) 0.4043 (5) 0.0289 (18)
F2 0.5028 (12) 0.3687 (8) 0.4625 (6) 0.043 (2)
F3 0.6565 (10) 0.3001 (7) 0.3206 (5) 0.0283 (17)
F4 0.3915 (11) 0.4713 (7) 0.3081 (6) 0.039 (2)
F5 0.1075 (12) 0.3985 (9) 0.4391 (6) 0.050 (2)
F6 0.0850 (12) 0.1908 (10) 0.4426 (6) 0.054 (2)
F7 0.2666 (12) 0.2256 (9) 0.5479 (6) 0.045 (2)
I1 -0.22481 (11) 0.65467 (8) 0.13202 (6) 0.0225 (2)
P1 0.2881 (4) 0.2322 (3) 0.2301 (2) 0.0136 (6)
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
C1 0.022 (3) 0.023 (3) 0.023 (3) -0.0065 (13) -0.0011 (10) -0.0035 (11)
C2 0.033 (3) 0.033 (3) 0.033 (3) -0.0092 (14) -0.0014 (10) -0.0053 (11)
C3 0.023 (3) 0.023 (3) 0.023 (3) -0.0070 (13) -0.0015 (10) -0.0039 (11)
C4 0.014 (3) 0.015 (3) 0.015 (3) -0.0043 (12) -0.0012 (10) -0.0023 (11)
C5 0.014 (3) 0.014 (3) 0.014 (3) -0.0042 (12) -0.0005 (10) -0.0018 (11)
C6 0.012 (3) 0.012 (3) 0.012 (3) -0.0039 (12) -0.0012 (10) -0.0014 (11)
C7 0.022 (3) 0.023 (3) 0.022 (3) -0.0065 (13) -0.0016 (10) -0.0035 (11)
C8 0.025 (3) 0.025 (3) 0.025 (3) -0.0074 (13) -0.0014 (10) -0.0042 (11)
C9 0.015 (3) 0.015 (3) 0.015 (3) -0.0047 (12) -0.0008 (10) -0.0022 (11)
C10 0.015 (3) 0.015 (3) 0.015 (3) -0.0043 (12) -0.0001 (10) -0.0023 (11)
C11 0.024 (3) 0.023 (3) 0.024 (3) -0.0070 (13) -0.0010 (10) -0.0034 (11)

120
C12 0.022 (3) 0.022 (3) 0.022 (3) -0.0064 (12) -0.0012 (10) -0.0029 (11)
C13 0.021 (3) 0.021 (3) 0.020 (3) -0.0061 (12) -0.0010 (10) -0.0032 (11)
C14 0.019 (3) 0.020 (3) 0.019 (3) -0.0058 (12) -0.0010 (10) -0.0030 (11)
C15 0.016 (3) 0.016 (3) 0.016 (3) -0.0048 (12) -0.0001 (10) -0.0022 (11)
Au1 0.0142 (3) 0.0129 (3) 0.0243 (3) -0.00383
(18)
0.00195 (19) -0.00170
(19)
F1 0.050 (5) 0.009 (3) 0.023 (4) -0.004 (3) -0.012 (4) 0.005 (3)
F2 0.059 (6) 0.053 (5) 0.030 (5) -0.030 (4) -0.013 (4) -0.014 (4)
F3 0.030 (4) 0.031 (4) 0.028 (4) -0.015 (3) -0.008 (3) -0.001 (3)
F4 0.055 (6) 0.022 (4) 0.038 (5) -0.002 (4) -0.016 (4) -0.005 (4)
F5 0.044 (5) 0.052 (5) 0.042 (5) 0.000 (4) 0.011 (4) -0.009 (4)
F6 0.044 (5) 0.097 (7) 0.037 (5) -0.043 (5) 0.016 (4) -0.024 (5)
F7 0.052 (6) 0.062 (6) 0.022 (4) -0.023 (5) 0.006 (4) -0.001 (4)
I1 0.0157 (4) 0.0132 (4) 0.0376 (5) -0.0036 (3) -0.0018 (4) -0.0020 (4)
P1 0.0133 (10) 0.0141 (10) 0.0133 (10) -0.0045 (8) -0.0015 (8) -0.0007 (8)
Geometric parameters (Å, º)
C1—C2 1.527 (18) C7—C8 1.395 (17)
C1—C3 1.534 (17) C8—H8 0.9300
C1—F1 1.438 (13) C8—C9 1.340 (16)
C1—P1 1.918 (12) C9—H9 0.9300
C2—F2 1.353 (14) C10—C11 1.365 (15)
C2—F3 1.336 (15) C10—C15 1.412 (15)
C2—F4 1.337 (14) C10—P1 1.818 (11)
C3—F5 1.343 (13) C11—H11 0.9300
C3—F6 1.320 (14) C11—C12 1.384 (16)
C3—F7 1.326 (13) C12—H12 0.9300
C4—C5 1.383 (15) C12—C13 1.356 (16)
C4—C9 1.416 (14) C13—H13 0.9300
C4—P1 1.811 (12) C13—C14 1.395 (15)
C5—H5 0.9300 C14—H14 0.9300
C5—C6 1.366 (15) C14—C15 1.418 (15)
C6—H6 0.9300 C15—H15 0.9300
C6—C7 1.382 (14) Au1—I1 2.5589 (9)
C7—H7 0.9300 Au1—P1 2.254 (3)
C2—C1—C3 111.2 (10) C9—C8—C7 120.3 (12)
C2—C1—P1 115.3 (9) C9—C8—H8 119.8
C3—C1—P1 111.7 (8) C4—C9—H9 119.2
F1—C1—C2 103.1 (10) C8—C9—C4 121.7 (11)

121
F1—C1—C3 104.5 (9) C8—C9—H9 119.2
F1—C1—P1 110.1 (7) C11—C10—C15 119.8 (11)
F2—C2—C1 112.6 (12) C11—C10—P1 123.4 (9)
F3—C2—C1 111.3 (10) C15—C10—P1 116.7 (8)
F3—C2—F2 106.2 (10) C10—C11—H11 120.1
F3—C2—F4 109.0 (12) C10—C11—C12 119.8 (12)
F4—C2—C1 109.3 (11) C12—C11—H11 120.1
F4—C2—F2 108.3 (10) C11—C12—H12 119.0
F5—C3—C1 109.0 (10) C13—C12—C11 121.9 (11)
F6—C3—C1 114.5 (10) C13—C12—H12 119.0
F6—C3—F5 105.4 (10) C12—C13—H13 119.9
F6—C3—F7 108.1 (11) C12—C13—C14 120.3 (11)
F7—C3—C1 112.9 (10) C14—C13—H13 119.9
F7—C3—F5 106.2 (9) C13—C14—H14 120.7
C5—C4—C9 116.8 (11) C13—C14—C15 118.6 (11)
C5—C4—P1 120.9 (8) C15—C14—H14 120.7
C9—C4—P1 121.9 (9) C10—C15—C14 119.5 (10)
C4—C5—H5 119.0 C10—C15—H15 120.3
C6—C5—C4 122.0 (10) C14—C15—H15 120.3
C6—C5—H5 119.0 P1—Au1—I1 178.60 (8)
C5—C6—H6 120.1 C1—P1—Au1 109.6 (4)
C5—C6—C7 119.9 (11) C4—P1—C1 106.7 (5)
C7—C6—H6 120.1 C4—P1—C10 109.0 (5)
C6—C7—H7 120.4 C4—P1—Au1 113.5 (4)
C6—C7—C8 119.3 (12) C10—P1—C1 100.9 (5)
C8—C7—H7 120.4 C10—P1—Au1 116.1 (4)
C7—C8—H8 119.8
C2—C1—C3—F5 50.1 (13) C11—C12—C13—
C14
1.1 (18)
C2—C1—C3—F6 168.0 (11) C12—C13—C14—
C15
-1.1 (17)
C2—C1—C3—F7 -67.7 (13) C13—C14—C15—
C10
-1.6 (17)
C3—C1—C2—F2 38.7 (14) C15—C10—C11—
C12
-4.4 (17)
C3—C1—C2—F3 157.8 (10) C15—C10—P1—C1 123.7 (9)
C3—C1—C2—F4 -81.7 (13) C15—C10—P1—C4 -124.3 (9)
C4—C5—C6—C7 0.5 (17) C15—C10—P1—Au1 5.3 (10)
C5—C4—C9—C8 1.1 (16) F1—C1—C2—F2 -72.8 (12)
C5—C4—P1—C1 -75.4 (10) F1—C1—C2—F3 46.4 (13)

122
C5—C4—P1—C10 176.4 (9) F1—C1—C2—F4 166.8 (10)
C5—C4—P1—Au1 45.4 (10) F1—C1—C3—F5 160.7 (9)
C5—C6—C7—C8 0.9 (16) F1—C1—C3—F6 -81.5 (12)
C6—C7—C8—C9 -1.2 (18) F1—C1—C3—F7 42.9 (13)
C7—C8—C9—C4 0.2 (18) P1—C1—C2—F2 167.2 (8)
C9—C4—C5—C6 -1.5 (16) P1—C1—C2—F3 -73.6 (12)
C9—C4—P1—C1 111.1 (9) P1—C1—C2—F4 46.8 (13)
C9—C4—P1—C10 2.9 (11) P1—C1—C3—F5 -80.3 (11)
C9—C4—P1—Au1 -128.1 (8) P1—C1—C3—F6 37.6 (13)
C10—C11—C12—
C13
1.7 (18) P1—C1—C3—F7 161.9 (8)
C11—C10—C15—
C14
4.4 (17) P1—C4—C5—C6 -175.3 (8)
C11—C10—P1—C1 -59.7 (11) P1—C4—C9—C8 174.8 (9)
C11—C10—P1—C4 52.4 (11) P1—C10—C11—C12 179.0 (9)
C11—C10—P1—Au1 -178.0 (8) P1—C10—C15—C14 -178.8 (8)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].

123
Chapter 5
The single crystal structures of gold(I) halide complexes containing the
P(3,5-(CF3)2C6H3)3 or P(3,5-(CF3)2C6H3)(C6H5)2 ligands–An unusual
case of decreasing Au···Au distances in the chloride, bromide and
iodide complexes.

124
5. The single crystal structures of gold(I) halide complexes containing
the P(3,5-(CF3)2C6H3)3 or P(3,5-(CF3)2C6H3)(C6H5)2 ligands-An unusual
case of decreasing Au···Au distances in the chloride, bromide and
iodide complexes.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, The University of Manchester, Manchester, M13 9PL, UK.
5.1 Abstract
A series of gold(I) phosphine halides of the type [AuX(PAr3)] (where PAr3 = P(3,5-
(CF3)2C6H3)3, P(3,5-(CF3)2C6H3)(C6H5)2 and X = Cl, Br, I) have been synthesized. All
of the complexes have been characterized by means of NMR spectroscopy and single
crystal X-ray crystallography. In the solid state all of the complexes adopt an
approximately linear geometry at gold with average bond lengths of d(Au–P)av =
2.2892(7) Å and d(Au–X)av = 2.2809(9) Å (X = Cl), 2.3779(5) Å (X = Br) and
2.5654(9) Å (X = I). The Au···Au distances range from 3.262(1) Å in [AuI{P(3,5-
(CF3)2C6H3)3}] to 9.0059(6) Å in [AuCl{P(3,5-(CF3)2C6H3)3}]. Unusually, the Au···Au
distances decrease for the complexes containing P(3,5-(CF3)2C6H3)3 as the halide ion
increases in size. The secondary C–H···X, C–H···F and F···F interactions are
investigated.
5.2 Introduction
A number of linear gold (I) phosphine halide complexes of the type [AuX(PAr3)]
(where X = Cl, Br, I) have been shown in the solid state to demonstrate the existence of
short Au···Au distances, which have been termed aurophilic interactions.1,2
These
closed-shell interactions are estimated to be in the range 21–46 kJmol-1
, and in
combination with other intermolecular interactions can give rise to well-defined
extended structures.3,4
Aurophilic interactions are typically recognized from X-ray
diffraction studies, and are defined as occurring when the distance separating two gold
centres is less than twice the van der Waals radius of gold (ca. 3.60 Å).5,6
However,
there is little rationale that satisfactorily explains whether such an interaction is likely to

125
exist in the solid state structure of a particular complex. There is however a dependence
on the identity of the halide, with a clear preference for shorter Au···Au distances to be
observed for a particular phosphine in the order Cl < Br < I. But, attempts to understand
the influence that the phosphine has on the aurophilic distance have been much less
successful.
It has been suggested that steric factors arising from the phosphines have the greatest
effect on the packing environment of the gold centres 7-9
and hence on the existence, or
otherwise, of Au···Au interactions. Thus short gold-gold distances are observed for
complexes of a series of small phosphines, such as PMe3 and PEt3. However, size alone
is not a sufficiently accurate predictor, for example, the solid-state structure of gold (I)
complex of tris(4-methylphenyl)phosphine exhibits a short Au···Au distance of
3.375(1) Å, whereas that of the analogous unsubstituted phosphine, which has the same
size based on its cone angle measurement, [AuCl(PPh3)] has no comparable Au···Au
interactions.10-12
In fact, a longer Au···Au distance is observed in [AuCl(PPh3)],
d(Au···Au) = 6.904 – 7.004 Å, [CTPPAU]10
than in the complex containing the very
much larger P(C6F5)3 based system, [AuCl{P(C6F5)3}], d(Au···Au) = 3.783 Å,
[MUVSIF]13
.
If instead of the size of a phosphine the packing of linear XYPR3 complexes and all the
possible non-bonding interactions are considered, then strong parallels are observed in
the structures of a series of [IAu(PAr3)] complexes and [I-IPAr3] adducts. This has been
interpreted as suggesting that aurophilic interactions might arise as a consequence of the
packing of molecules, rather than vice versa.14
The differences in Au···Au distances observed in [AuCl(PPh3)]10
and
[AuCl{P(C6F5)3}]13
prompted us to investigate the structures of complexes based on the
mixed ligand system PPh2(C6F5).15
The structures of [AuCl{PPh2(C6F5)}] and
[AuBr{PPh2(C6F5)}] displayed extensive hydrogen-bonding interactions, but the
shortest Au···Au distances were 4.9795(4) and 5.0178(6) Å, respectively. We
subsequently investigated the crystal structures of a series of gold(I) halide phosphine
complexes in which the aromatic rings of the phosphine are partially fluorinated and
found that while complexes of the 3-FC6H4- and 3,5-F2C6H3-containing phosphines
resulted in Au···Au distances in the range 6.982(2) – 8.3507(7) Å, those containing
either 4-FC6H4 or 3,4,5-F3C6H2 possessed remarkably shorter gold-gold distances of
between 3.1273(8) and 4.3592(5) Å.16

126
Since the location of fluorine on the aromatic ring gave rise to such dramatic effects on
the observed Au···Au distances in the solid state structures we decided to extend our
study by investigating the gold complexes of phosphines possessing CF3-containing
aromatic groups. The structure of [AuCl{P(2-CF3-C6F4)3}] has been reported before,
and the closest Au···Au distances are 7.947 Å,17
whereas in the corresponding
complexes containing the (3-CF3-C6F4) and (4-CF3-C6F4) substituents only the meta-CF3
containing complex showed an aurophilic interaction (d(Au···Au) = 3.0738(9) Å.18
Here we report our findings on the structures of the gold complexes containing the
ligand systems PPh2(3,5-(CF3)2C6H3) and P(3,5-(CF3)2C6H3)3.
5.3 Experimental
5.3.1 General procedure
All syntheses were carried out using standard glassware unless otherwise stated.
Solvents and materials were obtained from commercial sources and used as received.
31P NMR spectra were recorded as CDCl3 solutions on a Bruker Advance III 400 MHz
NMR spectrometer operating at 161.975 MHz respectively. The phosphine ligands were
prepared through a literature procedure,19
and all [AuX(PAr3)] (X = Cl, Br, I)
complexes were synthesized via literature methods.20,21
The crystals were grown at
room temperature. All glassware and equipment was scrupulously cleaned to minimise
contamination and to encourage the formation of larger crystals.
5.3.2 Synthesis of R3P–Au–Cl complexes
Complexes [AuCl{PPh2(3,5-(CF3)2C6H3)}],1a and [AuCl{P(3,5-(CF3)2C6H3)3}],2a
were prepared via the AuCl(tht) complex (tht = tetrahydrothiophene) which was
prepared as follows: K[AuCl]4 (0.2 g, 0.5 mmol) was dissolved in ethanol (5 cm3) and
water (2 cm3). Tetrahydrothiophene (0.1 ml, 1.1 mmol) was added dropwise, and the
mixture was stirred for 15 min, during which time the solution mixture changed from
yellow to white in colour. The mixture was filtered and the white solid was dried in
vacuo. A solution of the PPh2(3,5-F3CC6H3) (0.1 g, 0.25 mmol) or P(3,5-F3CC6H3)3
(0.24 g, 0.36 mmol) in dichloromethane (5 cm3) was added to AuCl(tht) (0.1 g, 0.3
mmol) and the reaction mixture was stirred for 24 h at room temperature, after which
time the volatiles were removed in vacuo. The gold compounds were isolated as white

127
solid products. (1a): 31
P{1H} NMR (CDCl3): δ 33.82 ppm (s); (2a):
31P{
1H} NMR
(CDCl3): δ 35.20 ppm (s).
5.3.3 Synthesis of R3P–Au–Br and R3P–Au–I complexes
The compounds 1b, 1c, 2b and 2c were synthesized via halide exchange,20
by
dissolving 1a (0.1 g, 0.16 mmol) or 2a (0.1 g, 0.11 mmol) in ethanol (10 mL) then
adding LiBr or KI, followed by and stirring the mixture for 15 minutes at 80 ºC, after
which time the solution was filtered and the solvent was removed under vacuo. The
gold complexes were dissolved in dichloromethane, from which crystals were grown.
(1b): 31
P{1H} NMR (CDCl3): δ 35.68 ppm (s); (1c):
31P{
1H} NMR (CDCl3): δ 37.48
ppm (s); (2b): 31
P{1H} NMR (CDCl3): δ 36.45 ppm (s); (2c):
31P{
1H} NMR (CDCl3): δ
38.38 ppm (s).
5.3.4 Crystallographic details
Crystals of complexes 2a and 2c were obtained by slow evaporation of the solvent from
saturated CH2Cl2 solutions. 1a, 1b, 1c and 2b were grown by vapour diffusion of
hexane into a saturated CH2Cl2 solution of the relevant compound at room temperature.
Reflection data for 1a, 1b, 1c, 2a and 2b were collected at 150 K on Oxford Xcalibur2
diffractometer and reflection data for 2c were collected at 293 K on Supernova
diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) with
the CCD detector. The crystallographic data are presented in Table 1. Structure solution
for all complexes was by direct methods using SHELXS,22
and refined with full- matrix
least squares refinement of F2
using using SHELXL in OleX2.23
All non-hydrogen
atoms were refined with anisotropic parameters; hydrogen atoms were added in
idealised locations. The computer package MERCURY 24
was used to display the
structure and investigate the extended bonding. The Cambridge Crystallography
Database Centre (CCDC) and Conquest program were used to compare the structures
with any existing data. The structures of 2a and 2b were solved with disorder in the
fluorine atoms in one CF3 group with refined site occupancies of 0.89: 0.11 and 0.48:
0.52, respectively.
5.4 Results and discussion
5.4.1 Synthesis and characterization
The gold(I) chloride phosphine complexes were prepared by reaction of AuCl(tht) [tht =

128
tetrahydrothiothene] with the phosphines PPh2(3,5-(CF3)2C6H3) and P(3,5-(CF3)2C6H3)3.
20 Subsequent halide exchange
21 generated the bromide and iodide analogues resulting
in the two series of complexes: [AuX{PPh2(3,5-(CF3)2C6H3)}] [X= Cl (1a), Br (1b) and
I (1c)], and [AuX{P(3,5-(CF3)2C6H3)3}] [X=Cl (2a), Br (2b), I (2c)].
Crystals of complexes 1a, 1b, 1c and 2b were obtained by slow diffusion of hexane into
a saturated CH2Cl2 solution of the compounds, and crystals of complexes 2a and 2c
were grown by slow evaporation of the solvent from a saturated CH2Cl2 solution of the
compounds. For all complexes single crystal X-ray diffraction data was obtained, and
crystallographic parameters and data are summarised in Table 1. Selected bond lengths
and angles for these complexes are given in Table 2.

129
Table 1 Crystal data and structure refinement for compounds 1a–2c
Compound 1a 1b 1c 2a 2b 2c
Formula C20H13AuClF6P C20H13AuBrF6P C20H13AuF6IP C24H9AuClF18P C48H18Au2Br2F36P2 C24H9AuF18IP
FW 630.69 675.15 722.14 902.70 1894.32 994.15
Crystal system Monoclinic Monoclinic Triclinic Monoclinic Monoclinic Triclinic
Space group I2/a I2/a P-1 I2/a P21/n P-1
a, b, c (A˚ )
17.2099(11),
8.9357(7),
27.6160(18)
17.3933(11), 8.9849(7),
27.4888(18)
8.9730(5), 9.9540(7),
14.0671(10)
18.8721(14),
9.0059(5), 33.577(2)
13.7905(3), 17.1274(6),
23.6202(6)
12.8929(7), 30.6929(17),
32.675 (2)
α, β, γ (º) 90, 105.845(7), 90 90, 104.715(7), 90 70.770(6), 74.251(5),
64.338(6) 90, 105.304(7), 90 95.110(3)
117.299(6), 90.882(5),
92.981(5)
V (A˚ 3) 4085.5(5) 4155.0(5) 1057.00(14) 5504.3(7) 5556.8(3) 11463.3(13)
Z 8 8 2 8 4 16
Crystal size (mm3) 0.16 × 0.12 × 0.08 0.18 × 0.16 × 0.08 0.18 × 0.16 × 0.09 0.12 × 0.1 × 0.1 0.15 × 0.15 × 0.04 0.13 × 0.08 × 0.06
Temperature (K) 150 150 150 150 150 293
Dx (Mg m_3
) 2.051 2.159 2.269 2.179 2.264 2.304
µ (MoKa) (mm_1
) 7.47 9.14 8.55 5.64 6.93 6.40
F(000) 2384 2528 668 3408 3552 7392
Theta Min-Max,
degrees
min = 3.2°, max =
25.0° min = 3.2°, max = 26.0°
min = 3.3°, max =
26.0°,
min = 2.8°, max =
26.5° min = 3.3°, max = 26.5° min = 3.0°, max = 26.0°
N (total) 6823 8101 7576 9722 27818 67406
N (unique) 3590 4076 4144 5656 11494 44486
Observed Data 2423 3168 3404 4507 8973 22130
R, wR2 ,S 0.075, 0.148, 1.03 0.043, 0.066, 1.04 0.046, 0.082, 1.00 0.040, 0.100, 1.07 0.050, 0.129, 1.03 0.103, 0.207, 1.03
Min. and Max. Resd
Density / e Å -3 -2.47, 3.48 -0.93, 1.32 -1.58, 1.50 -0.81, 1.61 -3.13, 1.58 -1.76, 3.38

130
Table 2 Selected bond lengths (Å) and bond angles (°) for complexes 1a-2c.
Complex 1a 1b 1c 2a 2b 2c
Au-P (Å)
2.231(3)
2.234(2)
2.258(2)
2.2186(17)
2.2290(6)/
2.2248(6)
av =2.2445(6)
Au-X (Å)
2.285(4)
2.4088(9) 2.5688(7)
2.2769(18)
2.3470(4)/
2.3756(3)
av=2.562(17)
P-Au-X (°)
175.3(1)
175.04(5)
174.68(5) 179.19(6) 174.497(18)/
173.112(17)
av=174(14)
Au···Au (Å) 3.7519(8) 3.8067(6) 4.3393(6) 9.0059(6) 3.3196(1) 3.262(1)
F···Fav (Å) - 2.883 - 2.833(8) 2.839375 2.8522
H···Fav (Å) 2.61 2.602 2.554 2.545 2.5785 2.5654
H···Xav (Å) 2.8975 3.006 - 2.763 2.843 3.102
5.4.2 Crystal structures of [AuX{PPh2(3,5-CF3C6H3)}], X= Cl (1a), Br (1b), I (1c).
The molecular structure of [AuCl{PPh2(3,5-CF3C6H3)}] 1a (Fig. 1-a) and the
corresponding bromide complex [AuBr{PPh2(3,5-CF3C6H3)}] 1b (Fig. 1-b) are
isostructural. Both crystallise in the monoclinic space group I2/a, with 8 molecules in
the unit cell. Whereas the iodide complex [AuI{PPh2(3,5-CF3C6H3)}] 1c (Fig. 1-c)
crystallises in the triclinic space group P-1 with two molecules in the unit cell.
(a) (b)

131
Fig. 1 Molecular structures showing crystallographic numbering schemes for [AuX{PPh2(3,5-
CF3C6H3)}]. Displacement ellipsoids are drawn at the 50% probability level. (a) X = Cl (1a), (b)
X = Br (1b), (c) X = I (1c).
Each of these complexes display approximately linear geometry at the gold atom, with
P–Au–X angles of 175.3(1) (X = Cl), 175.04(5) (X = Br) and 174.68(5)° (X = I). The
Au–X bond lengths of complexes 1a, 1b and 1c are 2.285(4), 2.4088(9) and 2.568(7) Å,
respectively. These bond lengths are similar to those found in related systems, for
example of 1a the Au–Cl bond distance is comparable to those found in [AuCl(PPh3)]10
d(Au–Cl) = 2.279(3) Å and [AuCl{P(3,5-(CF3)2C6H3}3]17
d(Au–Cl) = 2.274(7)/2.284(6)
Å. The Au–P bond lengths in complexes 1a (2.231(3) Å) and 1b (2.234(2) Å) are
similar to that found in the complex [AuCl(PPh3)]10 d(Au–P) = 2.235(3) Å, whereas, the
Au–P distance of 1c (2.258(2) Å) is slightly longer. The closest Au···Au distances in
1a, 1b and 1c are 3.7519(8), 3.8067(6) and 4.3393(6) Å, respectively, while these
distances are all somewhat longer than twice the van der Waals radius of gold (ca. 3.60
Å), they follow the usual trend of observing longer Au···Au interactions on going from
Cl to Br to I.
In the crystal packing of 1a and 1b intermolecular hydrogen bonding interactions
between C–Hortho···X and H(ortho, para)···F are observed, as shown in Figs. 2 and 3,
respectively. While, in complex 1c no hydrogen bonding is detected between the iodide
and any of the phenyl ring hydrogens, instead there are Hmeta···F interactions (Fig. 4).
In 1b there is one distance between the fluorine atoms of adjacent molecules (F5···F5i =
(c)

132
2.883 Å, shown in Fig. 5) which is just less than twice the van der Waals’ radius of
fluorine (2.94 Å).
Fig. 2 Crystal packing of 1a, showing Cl···H contacts, viewed down the b-axis.
Fig. 3 Crystal packing of 1b, showing H···F contacts, viewed down the b-axis.

133
Fig. 4 Crystal packing of 1c, showing H···F contacts, viewed down the a-axis.
Fig. 5 Crystal packing of 1b, showing F···F contacts, viewed down the b-axis.
5.4.3 Crystal structure of complexes [AuX{P(3,5-(CF3)2C6H3)3}], X = Cl, 2a, X = Br,
2b, X=I, 2c.
The gold complex, [AuCl{P(3,5-(CF3)2C6H3)3}] 2a crystallises in the monoclinic space
group I2/a, and the molecular structure was solved and refined as a single molecule.
While that of 2b crystallises in the monoclinic space group P21/n and refined as two
independent molecules in the asymmetric unit. Whereas the complex of 2c crystallise in
the triclinic space group P-1 containing eight independent molecules in the asymmetric

134
unit. All three complexes exhibit the expected linear geometry at the gold atom, with P-
Au-X angles of 179.19(6), 2a, 174.50(6), 2b and 174.00(14)° 2c. The molecular
structures of these complexes are shown in Fig. 6 (a-c).
(a)
(b)

135
Fig. 6 Molecular structures showing crystallographic numbering schemes for [AuX{3,5-
(CF3)2C6H3)3}]. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen
atoms have been omitted for clarity. (a) X= Cl (2a), (b) X= Br (2b), (c) X= I (2c).
A crystal structure of 2a has been published previously [BAVSIB]17
, however in that
case three different crystals were identified, two were monoclinic, while the other was
triclinic. They report data for only one of these crystals with the following parameters:
{a = 23.624(6), b = 17.041(5), c = 13.817(4) Å, β = 95.23(2)°, Z = 8; space group,
P21/n}. Bond lengths and angles are reported for two unique molecules; d(Au–P) =
2.215(6) and 2.216(6) Å, and d(Au–Cl) = 2.274(7) and 2.284(6) Å.
The Au···Au distance in 2a is 9.0059(6) Å, which is very much longer than that of
previously reported (3.341(1) Å) 17
meaning that there is no Au···Au interaction in our
crystals of 2a compared with twice the van der Waals radius of gold, 3.60 Å.
The structures of 2b and 2c have not been reported before. In 2b the average Au–P and
Au–Br distances are 2.2269(6) and 2.3470(4) Å, respectively, while the average Au–P
bond distance of 2c is 2.2445(6) Å, ranging from 2.239(6) Å to 2.250(6) Å, and the
average Au–I bond distance is 2.562(17) Å, ranging from 2.5561(17) Å to 2.5711(17)
Å. These distances are similar to those found in related gold(I) phosphine halide
complexes, e.g. [AuI(PPh3)]8, 2.2499(2) Å and 2.553(1) Å for the Au–P and Au–I
bonds, respectively.
(c)

136
While 2a shows no aurophilic interaction, when the chloride is replaced by bromide or
iodide an Au···Au interaction is observed in 2b and 2c; the shortest Au···Au distances
are 3.3196(1) and 3.262(1) Å with torsion angles of P–Au–Au–P are 118.16(3)° and
119.8(2)°, respectively, both of which are below the upper limit of the aurophilicity
range (ca. 3.60 Å). While 2b exhibits a single Au...Au distance, the unit cell of complex
2c contains four pairs of dimers with Au···Au distances of between 3.262(1) and
3.700(1) Å [Au2···Au4 = 3.700(1), Au1···Au5 = 3.553(1), Au3···Au7 = 3.347(1),
Au6···Au8 = 3.262(1) Å]. Each dimer is connected to an adjacent dimer by somewhat
longer Au···Au distances [Au1···Au3 = 4.436(1), Au5···Au7 = 4.712(1), Au2···Au6 =
4.531(1), Au4···Au8 = 4.530(1) Å]. Fig. 7 shows the zig-zag alternating longer and
shorter gold-gold distances (viewed down the b* direction). It is also apparent from this
picture that the fluorines of the CF3 groups aggregate together to form a fluorous
surface.
Fig. 7 Packing diagram of 2c showing shorter and longer Au···Au distances viewed down b*
direction.
It is in interesting to note that when considering the shortest of the aurophilic
interactions in each of the complexes the reverse trend in Au···Au distance is observed
on changing the halide compared with the previous ligand system, and indeed most
others. That is for tris(3,5-trifluoromethyl)phenyl)phosphine the Au···Au distance is
longest for the gold chloride complex and becomes successively shorter for the bromide

137
and iodide. Although a second aurophilic interaction is observed in 2b between adjacent
gold atoms that is longer Au···Au distance = 4.297(1) Å to the neighbouring gold
atoms, this gives rise to an alternating series of longer and shorter Au···Au distances
through the crystal, and so a longer average aurophilic distance.
The extended packing of complexes 2a, 2b and 2c shows C–H···X hydrogen bonding
interactions between the gold-bound halide and ortho protons of the aromatic rings of
neighbouring molecules with the following data: 2a: d(Cl1···H6) = 2.763 Å, Cl1···H6–
C6 = 172.7º; 2b: d(Br1···H18) = 2.843 Å, Br1···H18–C18=142.1º and 2c: d(I3···H7) =
3.151 Å, I3···H7–C24 = 129°, d(I4···H15) = 3.078 Å, I4···H15–C34 = 157°, d(I5···H1)
= 2.998 Å, I5···H1–C8 = 158°, d(I7···H37) = 3.179 Å, I7···H37–C104 = 135° and
d(I8···H28) = 3.104 Å, I8···H28–C80 = 169º. All these values are less than the sum of
the relevant van der Waals radii of hydrogen and chlorine, hydrogen and bromine and
hydrogen and iodine, and are shown in Fig. 8 (a-b).
Fig. 8 (a) A view of the crystal packing of 2a, showing H···Cl contacts.

138
Fig. 8 (b) A view of the crystal packing of 2b, showing H···Br contacts.
In addition to the halide-hydrogen interactions, there are also a large number of
intermolecular contacts between C–H···F and an F···F contact is observed in the
structures of these complexes that are shorter than the sum of the van der Waals radii of
F···F at (2.94 Å) and H···F at (2.67 Å), these contacts shown in Fig. 9 (a-c) and 10 (a-
b), respectively.
Fig. 9 (a) A view of the crystal packing of 2a, showing H···F contacts.
(a)

139
Fig. 9 (b) A view of the crystal packing of 2b, showing H···F contacts.
Fig. 9 (c) A view of the crystal packing of 2c, showing H···F contacts.
(b)
(c)

140
Fig. 10 (a) A view of the crystal packing of 2a, showing F···F contacts.
Fig. 10 (b) A view of the crystal packing of 2b, showing F···F contacts.
5.5 Conclusions
For the ligands PPh2(3,5-(CF3)2C6H3) and P(3,5-(CF3)2C6H3)3 single X-ray crystal
structures of the phosphine gold(I) chloride, bromide and iodide complexes have been
(a)
(b)

141
obtained. As expected, in each case the Au–X bond distance increases in the order X =
Cl, Br, I. For the Au–P distances a much smaller difference is observed on altering the
halide. The Au···Au distances observed for the complexes containing PPh2(3,5-
(CF3)2C6H3) are longer than twice the van der Waals radius of gold, and increase as the
halide changes from Cl, Br, I (3.7519(8), 3.8067(6) and 4.3393(6) Å, respectively).
For the complexes containing the P(3,5-(CF3)2C6H3)3 ligand the chloride shows a long
gold-gold distance (9.0059(6) Å), whilst the corresponding bromide and iodide yield
much shorter distances, 3.3196 (1) and 3.262(1) Å respectively; this is the reverse of the
usual trends in Au..Au distances with respect to the halide identity. In both of the later
complexes the extended structure shows a sequence of alternating shorter and longer
Au···Au distances. Other non-bonded interactions identified include extensive C–H···X
hydrogen bonding interactions between the gold-bound halide and ortho protons of the
aromatic rings of neighbouring molecules, and C–H···F and F···F intermolecular
contacts which taken together may account for the shorter Au···Au distances found in
the solid state structures of these complexes.

142
Reference
1. H. Schmidbaur, Gold Bull., 1990, 23, 11-21.
2. K. Angermaier, G. A. Bowmaker, E. N. de Silva, P. C. Healy, B. E. Jones and
H. Schmidbaur, J. Chem. Soc., Dalton Trans, 1996, 3121-3129.
3. H. Schmidbaur, K. Dziwok, A. Grohmann and G. Müller, Chem. Ber., 1989,
122, 893-895.
4. J. Vicente, M.-T. Chicote, M. D. Abrisqueta, R. P. Guerrero and G. Jones,
Angew. Chem., Int. Ed. Engl., 1997, 36, 1203.
5. H. Schmidbaur, W. Graf and G. Müller, Angew. Chem., Int. Ed. Engl., 1988, 27,
417-419.
6. C. M. Che, H. L. Kwong, V. W.W. Yam and K. C. J. Cho, Chem. Commun.,
1989, 885.
7. K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472,
371-376.
8. S. Ahrland, K. Dreisch, B. Noren and A. Oskarsson, Acta Chem. Scand., Ser.,
1987, A41, 173-177.
9. E. R. T. Tiekink, Acta Cryst., 1989, C45, 1233-1234.
10. N. C. Baenziger, W. E. Bennett and D. M. Soboroff, Acta Cryst., 1975, B32,
962-963.
11. P. F. Barron, L. H. Engelhardt, P. C. Healy, J. Oddy and A. H. White, Aust. J.
Chem., 1987, 40, 1545-1555.
12. R. C. Bott, P. C. Healy and G. Smith, Aust. J. Chem., 2004, 57, 213-218.
13. H. W. Chen and E. R. T. Tiekink, Acta Cryst., 2002, E59, m50-m52.
14. N. A. Barnes, K. R. Flower, S. A. Fyyaz, S. M. Godfrey, A. T. Mc Gown, P. J.
Miles, R. G. Pritchard and J. E. Warren, CrystEngComm., 2010, 12, 784-794.
15. Chapter 3
16. Chapter 2
17. A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organomet., 2012, 31,
644-661.
18. K. Nunokawa, S. Onaka, T. Tatematsu, M. Ito and J. Sakai, Inorg. Chim. Acta,
2001, 322, 56-64.
19. S. Haji and C. Erkey, Tetrahedron, 2002, 58, 3929-3941.

143
20. N. A. Barnes, A. K. Brisdon, F. R. W. Brown, W. I. Cross, I. R. Crossley, C.
Fish, C. J. Herbert, R. G. Pritchard and J. E. Warren, Dalton Trans., 2011, 40,
1743-1750.
21. C. A. McAuliffe, R.V. Parish and D. Randall, J.C.S. Dalton, 1979, 1730-1735
22. G. M. Sheldrick. Acta Cryst., 2008, A64, 112-122.43.
23. O. V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard and H. Puschmann,
J. Appl. Cryst., 2009, 42, 339-341.
24. C. F. Macrea, P. R. Eddington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor,
M. Towler and J. van de Streek, J. Appl. Cryst., 2006, 39, 453-457.

144
Chapter 6
The single crystal structures of gold(I) halide complexes
containing the P(4-MeOC6H4)3, PPh(4-MeOC6H4)2 and
PPh2(4-MeOC6H4) ligands.

145
6. The single crystal structures of gold(I) halide complexes containing
the P(4-MeOC6H4)3, PPh(4-MeOC6H4)2 and PPh2(4-MeOC6H4) ligands.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, The University of Manchester, Manchester, M13 9PL, UK.
6.1 Abstract
Linear gold(I) complexes of the 4-methoxy-substituted triarylphosphines, of the type
[AuCl{P(4-MeOC6H4)3}](1), [AuCl{PPh(4-MeOC6H4)2}](2) and [AuX{PPh2(4-
MeOC6H4)}] [X = Cl, (3a) and X = Br, (3b)] have been prepared by the reaction of the
appropriate ligand with [AuCl(tht)] in dichloromethane. The structures of all four
complexes have been obtained by single crystal X-ray diffraction revealing near linear,
two coordinate gold(I) complexes. Hydrogen bonding is prominent in complexes 1, 2,
3a and 3b, but none of the complexes show an aurophilic interaction.
6.2 Introduction
Compounds of gold (I) phosphines have potential roles in many areas of chemistry,
ranging from catalysis to the medical and biological field (Fackler Jr et al., 2009; Ott,
2009), including their use as antitumor or anticancer drugs (Tiekink, 2002; Rackham et
al., 2007). Gold (I) complexes of the type [AuX(PAr3)] (where X = Cl, Br, I) with
tertiary arylphosphines are dominated by two-coordinate linear complexes of the type
L-Au-X that may be isomorphous as the halide is varied. Thus, it has been observed that
for a series of gold compounds isomorphous crystals are a common occurrence between
analogous iodide and bromide members, though series that include all three halide
homologues are also known, for example: with PPh3 (Baenzinger et al, 1976; Barron et
al., 1987; Ahrland et al., 1987) and [P(tris(2-methylphenyl)phosphine)3] (Bott et al.,
2004).
A feature of these complexes that has interested chemists is the potential for significant
intermolecular interactions that frequently generate supramolecular structures based on
aurophilic Au···Au bonding (Schmidbaur, 1990; Angermaier et al, 1996) and hydrogen
bonding. These interactions are estimated to be of a similar magnitude and often occur
together to give well-defined extended structures (Schmidbaur et al., 1989; Vicente et

146
al., 1997).
Typically, aurophilic interactions are recognized from X-ray diffraction studies, where
an aurophilic interaction is defined when the separation distance between two gold
centres is less than twice the van der Waals radius of gold. Au–Au distances have been
observed in compounds that lie between the distance observed for the Au2 molecule in
the gaseous state (2.47 Å) up to twice the van der Waals radius of gold (ca. 3.60 Å)
(Schmidbaur et al., 1988; Che et al., 1989).
It has been suggested [for a large number of crystal structures, that steric factors are the
most important effect on the packing environment of the gold centres (Angermaier et
al., 1994; Ahrland et al., 1987; Tiekink, 1989) and hence on the existence, or otherwise,
of Au···Au interactions. Thus it is surprising that the ring-substituted triarylphosphine
gold (I) complex tris(p-methylphenyl)phosphine, results in short Au···Au interactions
of 3.375(1) Å, whereas the analogous unsubstituted phosphine complex [AuCl(PPh3)]
has no comparable Au···Au interactions (Baenzinger et al, 1976; Barron et al., 1987;
Bott et al., 2004). Care must be taken not to introduce very significant modification of a
phosphine, when comparing structures, for example in complexes containing tris(2,4,6-
trimethoxylphenyl)phosphine the methoxy groups affect both the gold environment and
the ligand basicity (Bott et al., 1995; Alyea et al., 2000). However, substitution at the
para-position of the aryl rings is likely to result in very little change in the steric demand
of a triaryl phosphine, whilst generating phosphines that possess a mixture of
substituted- and non-substituted-aryl substituents is likely to result in smaller
perturbations in the steric and electronic properties of the gold centre.
6.3 Experimental
6.3.1 Synthesis and crystallization
The compounds 1, 2, 3a were synthesized based on existing literature methods to
prepare the chloride containing complexes from AuCl(tht) and the relevant phosphine
ligand (Barnes et al, 2011). 1: 31
P{1H} NMR (CDCl3): δ 29.18 ppm (s). 2:
31P{
1H}
NMR (CDCl3): δ 30.58 ppm (s). 3a: 31
P{1H} NMR (CDCl3): δ 31.77 ppm (s).
Complex 3b was prepared by halide exchange (McAuliffe et al., 1979) of 3a. To a
solution of 3a (0.1 g, 0.19 mmol) dissolved in ethanol (10 mL) was added LiBr (0.016
g, 0.18 mmol), and the mixture was left to stir for 15 minutes at 80 ºC, after which time

147
the solvent was removed under vacuo. The gold compound was dissolved in CH2Cl2,
from which crystals were grown. 31
P{1H} NMR (CDCl3): δ 33.96 ppm (s).
Suitable crystals of complexes 2, 3a and 3b were grown by vapour diffusion of hexane
into a saturated dichloromethane solution, while crystals of complex 1 were obtained by
vapour diffusion of diethyl ether into a saturated dichloromethane solution.
6.4 Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1.
Table 1
Experimental details
1 2 3a 3b
Crystal data
Chemical formula C21H21AuClO3P 2(C20H19AuClO2P) C19H17AuClOP C19H17AuBrOP
Mr 584.76 1109.47 524.71 569.17
Crystal system,
space group Monoclinic, C2/c Monoclinic, P21/c Monoclinic, P21/c Monoclinic, P21/c
Temperature (K) 150 150 150 150
a, b, c (Å)
14.2728(4),
14.4854(4),
20.0982(6)
20.085(3),
12.2804(10),
16.376(2)
6.6337(3),
12.6665(5),
21.1922(10)
6.6997(3),
12.7762(5),
21.1763(10)
(°) 101.544(3) 108.693(16) 90.960(5) 90.721(5)
V (Å3) 4071.2(2) 3825.9(9) 1780.45(13) 1812.47(13)
Z 8 8 4 4
Radiation type Mo K Mo K Mo K Mo K
(mm-1
) 7.46 7.92 8.50 10.41
Crystal size (mm) 0.16 × 0.14 × 0.06 0.16 × 0.12 × 0.08 0.04 × 0.02 × 0.02 0.16 × 0.14 × 0.08
Data collection
Diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
Absorption
correction
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Tmin, Tmax 0.517, 1.000 0.605, 1.000 0.776, 1.000 0.611, 1.000
No. of measured,
independent and
observed [I >
2(I)] reflections
7978, 4212, 3560 15768, 7511, 3142 7053, 3677, 3010 4602, 2620, 2296
Rint 0.046 0.178 0.035 0.024
(sin /)max (Å-1
) 0.628 0.617 0.628 0.595

148
Refinement
R[F2 > 2(F
2)],
wR(F2), S
0.039, 0.085, 1.00 0.093, 0.167,
0.93
0.037, 0.064,
1.02 0.028, 0.063, 1.03
No. of reflections 4212 7511 3677 2620
No. of parameters 247 455 209 209
H-atom treatment H-atom parameters
constrained
H-atom
parameters
constrained
H-atom
parameters
constrained
H-atom parameters
constrained
max, min (e Å-3
) 1.39, -3.50 2.50, -1.70 1.47, -1.16 0.64, -1.16
6.5 Results and discussion
The series of 4-methoxy-substituted-phosphine gold(I) halide complexes, [AuCl{P(4-
MeOC6H4)3}](1), [AuCl{PPh(4-MeOC6H4)2}](2), [AuCl{PPh2(4-MeOC6H4)}](3a) and
[AuBr{PPh2(4-MeOC6H4)}](3b) were prepared as described above and confirmation of
complex formation was achieved by 31
P{1H} NMR spectroscopy where a shift in the
phosphorus resonance is observed compared with that of the free phosphine. The
structure of each complex was obtained by single crystal X-ray diffraction. Selected
bond lengths and angles for all of the complexes are given in Table 2.
Table 2
Selected bond lengths (Å) and angles (o) for complexes.
Complex 1 2 3a 3b
Au–P /Å 2.2292(16) 2.231(7) /2.237(6) 2.2323(15) 2.2399(16)
Au–X /Å 2.2871(16) 2.279(7)/ 2.295(6) 2.2881(15) 2.3967(7)
P–Au–X /Å 175.87(6) 174.9(2)/ 176.91(18) 176.28(5) 176.43(5)
Au···Au /Å 6.2246(4) 7.133(1) 6.5239(6) 6.5643(6)
Although a search of the CCD revealed that the structure of [AuCl{P(4-MeOC6H4)3}]
(1) has been published previously (Yei and Tiekink, 2001, ACEZAJ), this was only as a
short structure report. Both our sample, and that previously reported crystallize in the
C2/c monoclinic space group, with 8 molecules in the unit cell and one molecule in the
asymmetric unit {comparative cell data for that previously reported (Yei and Tiekink,
2001): a = 14.2875(4), b = 14.5396(4), c = 20.2212(6) Å, β = 101.504(1)°, Z = 8}.

149
Figure 1 A view of the molecule structure of 1 showing the atom-labelling scheme, with
displacement ellipsoids drawn at the 50% probability level.
The bonded distances determined from our study are within experimental limits the
same as those reported previously. For example, the Au–P and Au–Cl bond lengths
were determined as 2.2292(16) and 2.2871(16) Å, which are comparable to those of Yei
and Tiekink, 2001, where Au–P and Au–Cl distances of 2.233(8) and 2.2885(9) Å are
reported.
The previous report identifies a single C–H···O hydrogen bond between a methyl
hydrogen and the oxygen of an adjacent methoxy group [d(H···O) = 2.58 Å], whereas
our data shows in addition to these types of interaction [H14···O1= 2.548, H21···O2=
2.635 and H···O3= 2.710 Å] (Fig. 2) a C–H···Cl interaction that is 0.11 Å less than the
sum of the H and Cl van der Waals radii, Cl(1)···H(20): 2.839 Å between the chlorine
and an ortho-proton on one of the aryl rings (Fig. 3).

150
Table 3
Hydrogen-bond geometry (Å, °) for complex (1)
D—H···A D—H H···A D---A D—H···A
C20—H20···Cl1i 0.93 2.839 3.697(6) 153.9
C14—H14C···O1ii 0.93 2.548 3.411(8) 149.5
C21—H21B···O2iii
0.96 2.635 3.297(8) 126.4
C5—H5···O3iv 0.93 2.710 3.587(7) 157.4
Symmetry codes: (i) 1/2-x,-1/2+y,1/2-z; (ii) -1/2+x,1.5-y,-1/2+z; (iii) ) x,1-y,-1/2+z; (iv) -1/2+x,- 1/2+y,z.
Figure 2 A view of the crystal packing of 1 showing the C–H···Cl interactions, viewed down
the a-axis.
Figure 3 A view of the crystal packing of 1 showing the C–H···O interactions.

151
Based on twice the van der Waals radius of Au at 3.60 Å there is no significant
gold-gold interaction apparent in this complex, because the shortest Au···Au distance is
6.2246(4) Å.
While the structure of the tris-4-methoxyphenylphosphine-containing gold(I) halide
complex have been prepared and crystallised before there are no reports of the mixed
Ph2(4-MeO-C6H4)P or Ph(4-MeO-C6H4)2P containing complexes. The gold chloride
complex of Ph(4-MeO-C6H4)2P, resulted in crystals of complex 2 found to be in the
monoclinic system, space group P21/c, with 8 molecules in the unit cell, and the
asymmetric unit containing two molecules, labelled A and B (Fig. 4). The Au–P and
Au–Cl distances in both molecules A and B are 2.231(7)/ 2.237(6) Å and 2.279(7)/
2.295(6) Å, respectively. These values are similar, within experimental error, to those
found in the related complexes [AuClP(2-MeOC6H4)3] (Yei & Tiekink, 2001) and
[AuClP(2-MeOC6H4)3] (Bott et al., 2007), for which d(Au–P) = 2.233(1) and 2.250(4)
Å, and d(Au–Cl) = 2.289(1) and 2.294(5) Å, respectively.
Figure 4 View of molecular structures of 2 showing the atom-labelling scheme, with
displacement ellipsoids drawn at the 30% probability level.
As expected, an essentially linear geometry is found for the Au atom in both molecules
with Cl–Au–P angles being 174.9(2)° and 176.91(18)° for molecules A and B,
(A)
(B)

152
respectively. The sequence of (Au–P–C–C) torsion angles for molecule A are
169.8(17)°, 126.0(18)° and 122.0(18)°, and 153.2(17)°, 142.3(16)° and 119.8(16)°
found for molecule B.
In the extended structure of 2, molecules are connected through C–H···Cl hydrogen
bonds to form chains (see Fig. 5 and Table 4 for details and symmetry codes). Cl1 is
involved in a hydrogen bonding interaction with a proton of a methoxy group, while Cl2
is involved in interactions with both a methoxy proton and an ortho aromatic proton.
The packing diagram in figure 6 shows that four different hydrogen bonding
interactions are apparent (Table 4), two of these involve the methoxy oxygens and
protons in the ortho or meta positions of the aromatic rings [O1···H13= 2.42 Å,
O2···H37= 2.59 Å], while a further two involve the methoxy oxygen and protons of
another methoxy group [O3···H14A= 2.62 Å and O4···H7 Å= 2.54] with all of these
distances being less than the sum of van der Waals radii of hydrogen and oxygen at 2.72
Å.
Table 4
Hydrogen-bond geometry (Å , °) for complex (2)
D—H···A D—H H···A D···A D—H···A
C27—H27A···Cl1i 0.96 2.847 3.609(8) 159
C14—H14B···Cl2ii 0.96 2.851 3.82(2) 177
C40—H40···Cl2iii
0.93 2.899 3.65(2) 138
C13—H13···O1iv 0.93 2.42 3.32(2) 161
C37—H37···O2 0.93 2.59 3.48(3) 159
C14—H14B···O3v 0.96 2.62 3.49(3) 151
C7—H7A···O4vi 0.96 2.54 3.23(2) 129
Symmetry codes: (i) -x,-1/2+y,1/2-z; (ii) -x,-1/2+y,1/2-z; (iii) x,1.5-y,-1/2+z; (iv) 1-x,-1/2+y,1/2-z; (v) -1+y,z; (vi)
1+x,-1+y,z.

153
Figure 5 A view of the crystal packing of 2 viewed along the b-axis, showing C–H···Cl
contacts.
Figure 6 A view of the crystal packing of 2 showing C–H···O contacts.
The shortest Au···Au distance is 7.133(1) Å, so there is again no significant gold-gold
interaction apparent in this complex, based on twice the van der Waals radius of gold
being 3.60 Å.
The crystals of [AuCl{PPh2(4-MeOC6H4)}] 3a and of the analogous bromide complex
[AuBr{PPh2(4-MeOC6H4)}] 3b are isostructural; both crystallize in the monoclinic

154
space group P21/c with 4 molecules in the unit cell. These complexes show (Fig. 7 a-b)
near-linear geometry at the gold atom, with P–Au–Cl and P–Au–Br angles of
176.28(5)° and 176.43(5)°, respectively. The Au–Cl and Au–P bond lengths of 3a are
2.2881(15) and 2.2323(15) Å, while the Au–Br and Au–P distances in the bromide
analogue 3b are 2.3967(7) and 2.2399(16) Å, respectively. The values of d(Au–P) for
3a and 3b are significantly different, reflecting a difference in the trans-influence of the
two halides. The values of d(Au–Cl) and d(Au–Br) which can be compared with those
of the previously reported related structures [AuCl{P(4-MeOC6H4)}3] (Yei & Tiekink,
2001), [AuBr{P(4-MeOC6H4)}3] (Bott et al., 2007) and 2, where it is found that there is
no significant difference in d(Au–Cl) between 3a and the related structures (Yei &
Tiekink, 2001) d =2.2885(9) Å, 2 d = 2.237(6)/ 2.295(6) Å, whereas there is small, but
significant difference in d(Au–Br) found in 3b (2.3967(7) Å) and [AuBr{P(4-
MeOC6H4)}3] (Bott et al., 2007) d = 2.385(2) Å).
Figure 7 (a) A view of the molecular structure of (3a), showing the atom-labelling scheme, with
displacement ellipsoids drawn at the 50% probability level.
(a)

155
Figure 7 (b) A view of the molecular structure of (3b), showing the atom-labelling scheme,
with displacement ellipsoids drawn at the 50% probability level.
The Au···Au distances for compounds 3a and 3b are 6.5643(6) and 6.5239(6) Å,
respectively, which are longer than twice the van der Waals radius of Au, 3.60 Å,
suggesting that no significant Au···Au interactions exist in either complex.
In the crystal structures of 3a and 3b, the molecules pair-up via weak hydrogen bonding
C–H···Cl and C–H···Br interactions involving the halogen and one ortho and one para
hydrogen on the two rings not bearing the methoxy group. These interactions are shown
in Fig. 8 (a-b) and listed in Table 5.
Table 5:
Hydrogen-bond geometry (Å , °) for complexes (3a, 3b).
D—H···A D—H H···A D···A D—H···A
C10—H10···Cl1i 0.931 2.896 3.799(7) 164.0
C14—H14···Cl1ii 0.930 2.897 3.514(6) 125.1
C10—H10···Br1iii
0.931 2.9603 3.867(7) 165.0
C14—H14···Br1iv 0.931 3.0166 3.614(7) 123.5
Symmetry codes: (i) 1-x,-y,1-z ; (ii) -1+x,y,z ; (iii)1-x,-y,1-z; (iv) -1+x,y,z.
(b)

156
Figure 8 (a) A view of the crystal packing of 3a viewed along the a-axis, showing Cl···H
contacts.
Figure 8 (b) A view of the crystal packing of 3b, viewed along the a-axis, showing Br···H
contacts.
(a)
(b)

157
6.6 Summary
The single-crystal structures of the complexes [AuCl{P(4-MeOC6H4)3}] (1),
[AuCl{PPh (4-MeOC6H4)2}] (2) and [AuX{PPh2(4-MeOC6H4)}] [X = Cl, (3a) and X =
Br, (3b)] have been obtained. In all cases the gold adopts a near-linear geometry, and
comparison of the Au–P distances suggests that there is little electronic difference
between the ligands P(4-MeOC6H4)3, PPh (4-MeOC6H4)2 and PPh2(4-MeOC6H4). None
of the complexes were found to exhibit an aurophilic interaction, instead hydrogen
bonding involving the oxygen of the methoxy group was found in the complexes that
contain P(4-MeOC6H4)3} and PPh (4-MeOC6H4)2. In all of the complexes interactions
involving the halogen atoms and hydrogens were identified.

158
References
Abdou, H. E., Mohamed, A. A., Fackler Jr, J. P., Burini, A., Galassi, R., López-de-
Luzuriaga, J. M. & Olmos, M. E. (2009). Coord. Chem. Rev. 253, 1661-1669.
Ahrland, S., Dreisch, K., Noren, B. & Oskarsson, A. (1987). Acta Chem. Scand.,
Ser. A41, 173-177.
Alyea, E.C., Ferguson, G. & Kannon, S. (2000). Polyhedron. 19, 2211-2213.
Angermaier, K., Bowmaker, G. A., de Silva, E.N., Healy, P.C., Jones, B.E. &
Schmidbaur, H. (1996). J. Chem. Soc., Dalton Trans. 3121-3129.
Angermaier, K., Zeller, E. & Schmidbaur, H. J. (1994). J. Organomet. Chem. 472,
371-376.
Baenzinger, N.C., Bennett, W.E. & Soboroff, D. M. (1976). Acta Cryst. B32, 962-
963.
Baker, L.-J., Bott, R. C., Bowmaker, G. A., Healy, P. C., Skelton, B. W.,
Schwerdtfeger, P. & White, A. H. (1995). J. Chem. Soc., Dalton Trans. 1341-1347.
Barnes, N. A., Brisdon, A. K., Brown, F. R. W., Cross, W. I, Crossley, I. R., Fish,
C. Herbert, C. J., Pritchard, R. G., Warren, J. E., (2011) Dalton Trans. 40, 1743-
1750.
Barron, P. F., Engelhardt, L. H., Healy, P. C., Oddy, J. & White, A. H. (1987). Aust.
J. Chem. 40, 1545-1555.
Bondi, A. (1964). J. Phys. Chem. 68, 441-451.
Bott, R.C., Healy, P.C. & Smith, G. (2004). Aust. J. Chem. 57, 213.
Bott, R. C., Healy, P. C. & Smith, G. (2007). Polyhedron. 26, 2803-2809.
Che, C. M., Kwong, H. L., Yam, V. W.W. & Cho, K. C. J. (1989). Chem. Commun.
885.
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2009). J. Appl. Cryst. 42, 339-341.
McAuliffe, C. A., Parish, R. V. & Randall, D. (1979). J.C.S.Dalton. 1730-1735.
Ott, I. (2009). Coord. Chem. Rev. 253, 1670-1681.
Rackham, O., Nichols, S. J., Leedman, P. J., Berners-Price, S. J. & Filipovska, A.
(2007). Biochem. Pharm. 74, 992-1002.
Schmidbaur, H. (1990). Gold Bulletin. 23, 11-21.

159
Schmidbaur, H., Dziwok, K., Grohmann, A. & Muller, G. (1989). Chem. Ber. 122,
893-895.
Schmidbaur, H., Graf, W. & Muller, G. (1988). Angew.Chem., Int. Ed. Engl. 27,
417-419.
Schneider, W., Bauer, A. & Schmidbaur, H. (1996). Organomet., 15, 5445-5446.
Sheldrick, G. M. (2008). Acta Cryst. A64, 112-122.
Sheldrick, G. M. (2015). Acta Cryst. A71, 3-8.
Tiekink, E. R. T. (2002). Crit. Rev. Hematol. Oncol. 42(3), 225-248.
Tiekink, E. R. T. (1989). Acta Crystallogr. C45, 1233-1234.
Vicente, J., Chicote, M.-T., Abrisqueta, M. D., Guerrero, R. P. & Jones, G. (1997).
Angew. Chem., Int. Ed. Engl. 36, 1203-1205.
Yei, S. H. & Tiekink, E. R. T. (2001). Acta Cryst. E57, m549-m550.

160
Supporting information
The single crystal structures of gold(I) halide complexes containing the P(4-
MeOC6H4)3, PPh(4-MeOC6H4)2 and PPh2(4-MeOC6H4) ligands.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
Computing details
Data collection: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33 (release 27-03-2014
CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); cell refinement: CrysAlis PRO, Agilent
Technologies, Version 1.171.37.33 (release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27
2014,17:12:48); data reduction: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33
(release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); program(s) used to
solve structure: ShelXT (Sheldrick, 2015); program(s) used to refine structure: SHELXL
(Sheldrick, 2015); molecular graphics: Olex2 (Dolomanov et al., 2009); software used to
prepare material for publication: Olex2 (Dolomanov et al., 2009).
[AuCl{P(4-MeOC6H4)3}] 1 Crystal data
C21H21AuClO3P F(000) = 2256
Mr = 584.76 Dx = 1.908 Mg m-3
Monoclinic, C2/c Mo K radiation, = 0.71073 Å
a = 14.2728 (4) Å Cell parameters from 3667 reflections
b = 14.4854 (4) Å = 3.5–28.0°
c = 20.0982 (6) Å = 7.46 mm-1
= 101.544 (3)° T = 150 K
V = 4071.2 (2) Å3 clear light colourless
Z = 8 0.16 × 0.14 × 0.06 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
4212 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source 3560 reflections with I > 2(I)
Mirror monochromator Rint = 0.046
Detector resolution: 8.0714 pixels mm-1
max = 26.5°, min = 3.0°
scans h = -1715
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version k = -186
161
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
Tmin = 0.517, Tmax = 1.000 l = -2524
7978 measured reflections
Refinement
Refinement on F2 Primary atom site location: dual
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.039 H-atom parameters constrained
wR(F2) = 0.085 w = 1/[
2(Fo
2) + (0.025P)
2]
where P = (Fo2 + 2Fc
2)/3
S = 1.00 (/)max = 0.001
4212 reflections max = 1.39 e Å-3
247 parameters min = -3.50 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Au1 0.28315 (2) 0.29598 (2) 0.18433 (2) 0.01809 (9)
P1 0.25652 (10) 0.40901 (11) 0.25351 (8) 0.0170 (3)
Cl1 0.31265 (11) 0.18803 (11) 0.10805 (8) 0.0272 (4)
O2 0.0903 (3) 0.7238 (3) 0.0789 (2) 0.0288 (11)
O1 0.6010 (3) 0.5217 (3) 0.4553 (2) 0.0279 (10)
O3 -0.0358 (3) 0.3016 (3) 0.4133 (2) 0.0344 (12)
C2 0.4389 (4) 0.3840 (4) 0.3288 (3) 0.0223 (14)
H2 0.4351 0.3263 0.3080 0.027*
C8 0.2084 (4) 0.5100 (4) 0.2053 (3) 0.0149 (12)
C13 0.1145 (4) 0.5399 (4) 0.2020 (3) 0.0244 (14)
H13 0.0767 0.5114 0.2287 0.029*
C15 0.1719 (4) 0.3787 (4) 0.3061 (3) 0.0169 (12)
C9 0.2631 (4) 0.5535 (5) 0.1655 (3) 0.0232 (14)

162
H9 0.3258 0.5343 0.1676 0.028*
C16 0.1270 (4) 0.2940 (4) 0.2970 (3) 0.0224 (14)
H16 0.1420 0.2530 0.2651 0.027*
C3 0.5187 (4) 0.4064 (5) 0.3774 (3) 0.0240 (14)
H3 0.5677 0.3636 0.3899 0.029*
C11 0.1336 (4) 0.6549 (4) 0.1194 (3) 0.0204 (13)
C10 0.2266 (4) 0.6250 (5) 0.1225 (3) 0.0245 (14)
H10 0.2645 0.6530 0.0957 0.029*
C6 0.3726 (4) 0.5341 (4) 0.3400 (3) 0.0213 (13)
H6 0.3243 0.5774 0.3272 0.026*
C1 0.3640 (4) 0.4466 (4) 0.3104 (3) 0.0170 (12)
C20 0.1482 (4) 0.4398 (4) 0.3534 (3) 0.0216 (13)
H20 0.1784 0.4969 0.3600 0.026*
C12 0.0774 (4) 0.6121 (5) 0.1592 (3) 0.0236 (14)
H12 0.0149 0.6316 0.1572 0.028*
C18 0.0349 (4) 0.3309 (5) 0.3809 (3) 0.0226 (14)
C19 0.0801 (4) 0.4165 (5) 0.3911 (3) 0.0235 (14)
H19 0.0647 0.4576 0.4229 0.028*
C7 0.6743 (4) 0.4548 (5) 0.4798 (3) 0.0345 (17)
H7A 0.7044 0.4363 0.4433 0.052*
H7B 0.6463 0.4019 0.4969 0.052*
H7C 0.7212 0.4816 0.5155 0.052*
C5 0.4527 (4) 0.5564 (4) 0.3881 (3) 0.0207 (13)
H5 0.4580 0.6147 0.4079 0.025*
C14 0.1460 (5) 0.7742 (5) 0.0403 (3) 0.0320 (16)
H14A 0.1649 0.7342 0.0073 0.048*
H14B 0.2019 0.7981 0.0700 0.048*
H14C 0.1088 0.8244 0.0176 0.048*
C4 0.5253 (4) 0.4924 (5) 0.4072 (3) 0.0207 (13)
C21 -0.0709 (5) 0.3662 (5) 0.4554 (4) 0.0385 (19)
H21A -0.0961 0.4191 0.4290 0.058*
H21B -0.0198 0.3852 0.4915 0.058*
H21C -0.1206 0.3381 0.4743 0.058*
C17 0.0596 (4) 0.2692 (5) 0.3349 (3) 0.0283 (15)
H17 0.0312 0.2112 0.3294 0.034*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.01889 (14) 0.01789 (14) 0.01842 (13) -0.00117 (9) 0.00600 (9) -0.00130
(10)

163
P1 0.0152 (7) 0.0178 (8) 0.0183 (8) 0.0000 (6) 0.0039 (6) -0.0004 (7)
Cl1 0.0329 (8) 0.0236 (8) 0.0279 (8) -0.0032 (7) 0.0131 (7) -0.0082 (7)
O2 0.032 (2) 0.030 (3) 0.023 (2) 0.004 (2) 0.0019 (19) 0.009 (2)
O1 0.022 (2) 0.037 (3) 0.023 (2) -0.004 (2) 0.0017 (18) -0.004 (2)
O3 0.033 (3) 0.041 (3) 0.035 (3) -0.004 (2) 0.021 (2) 0.008 (2)
C2 0.026 (3) 0.018 (3) 0.025 (3) -0.006 (3) 0.008 (3) -0.005 (3)
C8 0.019 (3) 0.011 (3) 0.014 (3) 0.001 (2) 0.000 (2) 0.001 (2)
C13 0.024 (3) 0.024 (4) 0.027 (3) 0.004 (3) 0.009 (3) 0.002 (3)
C15 0.015 (3) 0.022 (3) 0.014 (3) 0.000 (3) 0.003 (2) -0.004 (3)
C9 0.017 (3) 0.029 (4) 0.024 (3) 0.002 (3) 0.004 (2) 0.000 (3)
C16 0.024 (3) 0.022 (3) 0.022 (3) 0.000 (3) 0.007 (3) -0.003 (3)
C3 0.016 (3) 0.029 (4) 0.027 (3) 0.008 (3) 0.004 (3) 0.004 (3)
C11 0.030 (3) 0.015 (3) 0.016 (3) -0.003 (3) 0.004 (2) -0.001 (3)
C10 0.026 (3) 0.029 (4) 0.019 (3) -0.005 (3) 0.006 (3) -0.002 (3)
C6 0.016 (3) 0.022 (3) 0.026 (3) 0.000 (3) 0.005 (2) -0.001 (3)
C1 0.016 (3) 0.017 (3) 0.018 (3) 0.002 (2) 0.003 (2) 0.000 (3)
C20 0.021 (3) 0.016 (3) 0.027 (3) -0.003 (3) 0.005 (3) 0.001 (3)
C12 0.016 (3) 0.028 (4) 0.026 (3) 0.005 (3) 0.003 (2) -0.001 (3)
C18 0.020 (3) 0.030 (4) 0.018 (3) 0.002 (3) 0.003 (2) 0.013 (3)
C19 0.021 (3) 0.030 (4) 0.020 (3) 0.006 (3) 0.006 (2) 0.003 (3)
C7 0.028 (3) 0.045 (5) 0.026 (4) 0.003 (3) -0.005 (3) 0.000 (4)
C5 0.024 (3) 0.018 (3) 0.023 (3) -0.004 (3) 0.012 (3) -0.005 (3)
C14 0.040 (4) 0.030 (4) 0.024 (4) -0.004 (3) 0.001 (3) 0.008 (3)
C4 0.016 (3) 0.032 (4) 0.015 (3) -0.003 (3) 0.006 (2) 0.001 (3)
C21 0.036 (4) 0.051 (5) 0.033 (4) 0.006 (4) 0.020 (3) 0.008 (4)
C17 0.030 (3) 0.027 (4) 0.029 (4) -0.012 (3) 0.008 (3) 0.000 (3)
Geometric parameters (Å, º)
Au1—P1 2.2292 (16) C3—C4 1.377 (9)
Au1—Cl1 2.2871 (16) C11—C10 1.386 (8)
P1—C8 1.813 (6) C11—C12 1.388 (8)
P1—C15 1.811 (6) C10—H10 0.9300
P1—C1 1.805 (5) C6—H6 0.9300
O2—C11 1.356 (7) C6—C1 1.394 (8)
O2—C14 1.418 (8) C6—C5 1.381 (8)
O1—C7 1.439 (7) C20—H20 0.9300
O1—C4 1.366 (6) C20—C19 1.389 (8)
O3—C18 1.372 (7) C12—H12 0.9300
O3—C21 1.419 (8) C18—C19 1.393 (9)
C2—H2 0.9300 C18—C17 1.381 (9)

164
C2—C3 1.383 (8) C19—H19 0.9300
C2—C1 1.395 (8) C7—H7A 0.9600
C8—C13 1.396 (7) C7—H7B 0.9600
C8—C9 1.376 (8) C7—H7C 0.9600
C13—H13 0.9300 C5—H5 0.9300
C13—C12 1.390 (8) C5—C4 1.387 (8)
C15—C16 1.379 (8) C14—H14A 0.9600
C15—C20 1.390 (8) C14—H14B 0.9600
C9—H9 0.9300 C14—H14C 0.9600
C9—C10 1.383 (8) C21—H21A 0.9600
C16—H16 0.9300 C21—H21B 0.9600
C16—C17 1.388 (8) C21—H21C 0.9600
C3—H3 0.9300 C17—H17 0.9300
P1—Au1—Cl1 175.87 (6) C6—C1—P1 122.4 (4)
C8—P1—Au1 110.68 (19) C6—C1—C2 118.8 (5)
C15—P1—Au1 113.7 (2) C15—C20—H20 119.6
C15—P1—C8 106.4 (3) C19—C20—C15 120.8 (6)
C1—P1—Au1 112.83 (19) C19—C20—H20 119.6
C1—P1—C8 106.2 (3) C13—C12—H12 120.0
C1—P1—C15 106.5 (3) C11—C12—C13 120.1 (5)
C11—O2—C14 118.0 (5) C11—C12—H12 120.0
C4—O1—C7 116.8 (5) O3—C18—C19 124.5 (6)
C18—O3—C21 117.4 (5) O3—C18—C17 115.3 (6)
C3—C2—H2 119.6 C17—C18—C19 120.1 (6)
C3—C2—C1 120.9 (6) C20—C19—C18 119.3 (6)
C1—C2—H2 119.6 C20—C19—H19 120.3
C13—C8—P1 122.2 (4) C18—C19—H19 120.3
C9—C8—P1 118.8 (4) O1—C7—H7A 109.5
C9—C8—C13 118.8 (5) O1—C7—H7B 109.5
C8—C13—H13 119.8 O1—C7—H7C 109.5
C12—C13—C8 120.3 (6) H7A—C7—H7B 109.5
C12—C13—H13 119.8 H7A—C7—H7C 109.5
C16—C15—P1 119.1 (5) H7B—C7—H7C 109.5
C16—C15—C20 119.1 (5) C6—C5—H5 119.8
C20—C15—P1 121.8 (5) C6—C5—C4 120.3 (6)
C8—C9—H9 119.3 C4—C5—H5 119.8
C8—C9—C10 121.3 (5) O2—C14—H14A 109.5
C10—C9—H9 119.3 O2—C14—H14B 109.5
C15—C16—H16 119.6 O2—C14—H14C 109.5

165
C15—C16—C17 120.9 (6) H14A—C14—H14B 109.5
C17—C16—H16 119.6 H14A—C14—H14C 109.5
C2—C3—H3 120.2 H14B—C14—H14C 109.5
C4—C3—C2 119.7 (5) O1—C4—C3 124.4 (5)
C4—C3—H3 120.2 O1—C4—C5 115.4 (6)
O2—C11—C10 125.1 (6) C3—C4—C5 120.2 (5)
O2—C11—C12 115.4 (5) O3—C21—H21A 109.5
C10—C11—C12 119.5 (6) O3—C21—H21B 109.5
C9—C10—C11 120.0 (6) O3—C21—H21C 109.5
C9—C10—H10 120.0 H21A—C21—H21B 109.5
C11—C10—H10 120.0 H21A—C21—H21C 109.5
C1—C6—H6 119.9 H21B—C21—H21C 109.5
C5—C6—H6 119.9 C16—C17—H17 120.1
C5—C6—C1 120.1 (5) C18—C17—C16 119.8 (6)
C2—C1—P1 118.7 (5) C18—C17—H17 120.1
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].
[AuCl{PPh(4-MeOC6H4)2}] 2
Crystal data
2(C20H19AuClO2P) F(000) = 2128
Mr = 1109.47 Dx = 1.926 Mg m-3
Monoclinic, P21/c Mo K radiation, = 0.71073 Å
a = 20.085 (3) Å Cell parameters from 1054 reflections
b = 12.2804 (10) Å = 3.6–23.6°
c = 16.376 (2) Å = 7.92 mm-1
= 108.693 (16)° T = 150 K
V = 3825.9 (9) Å3 clear colourless
Z = 4 0.16 × 0.12 × 0.08 mm
Data collection
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
Rint = 0.178
Tmin = 0.605, Tmax = 1.000 max = 26.0°, min = 3.3°
15768 measured reflections h = -1924
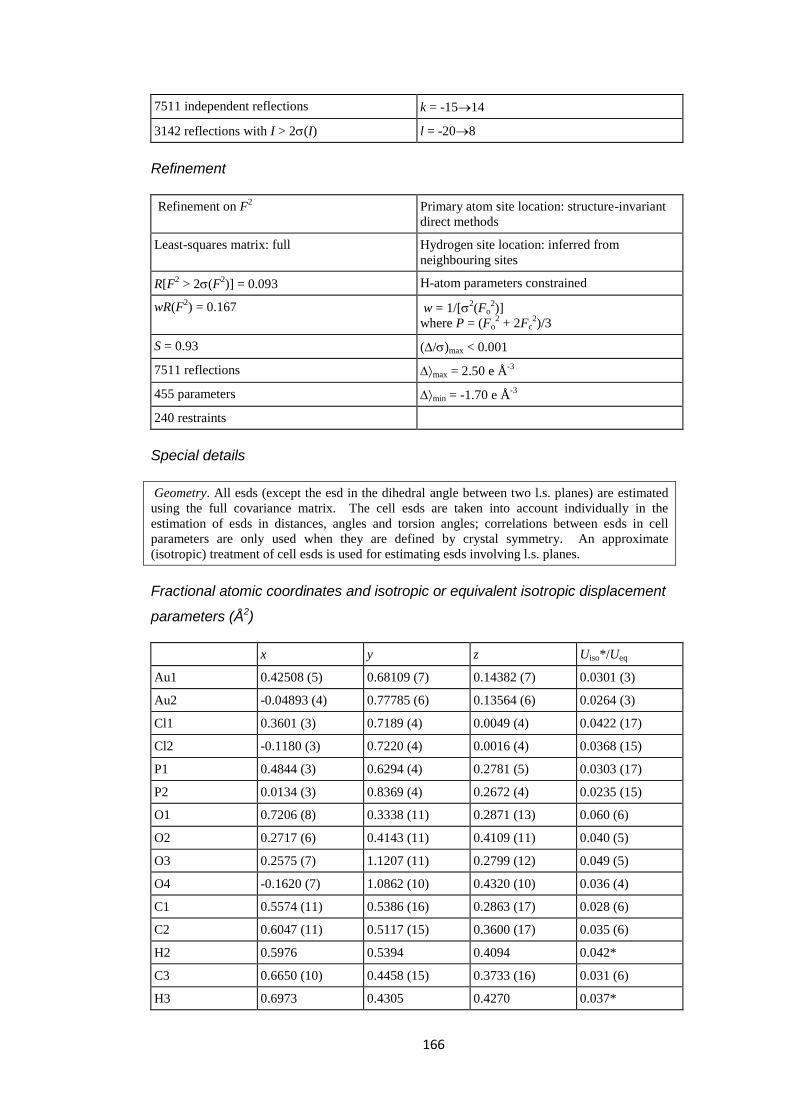
166
7511 independent reflections k = -1514
3142 reflections with I > 2(I) l = -208
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.093 H-atom parameters constrained
wR(F2) = 0.167 w = 1/[
2(Fo
2)]
where P = (Fo2 + 2Fc
2)/3
S = 0.93 (/)max < 0.001
7511 reflections max = 2.50 e Å-3
455 parameters min = -1.70 e Å-3
240 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate
(isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.42508 (5) 0.68109 (7) 0.14382 (7) 0.0301 (3)
Au2 -0.04893 (4) 0.77785 (6) 0.13564 (6) 0.0264 (3)
Cl1 0.3601 (3) 0.7189 (4) 0.0049 (4) 0.0422 (17)
Cl2 -0.1180 (3) 0.7220 (4) 0.0016 (4) 0.0368 (15)
P1 0.4844 (3) 0.6294 (4) 0.2781 (5) 0.0303 (17)
P2 0.0134 (3) 0.8369 (4) 0.2672 (4) 0.0235 (15)
O1 0.7206 (8) 0.3338 (11) 0.2871 (13) 0.060 (6)
O2 0.2717 (6) 0.4143 (11) 0.4109 (11) 0.040 (5)
O3 0.2575 (7) 1.1207 (11) 0.2799 (12) 0.049 (5)
O4 -0.1620 (7) 1.0862 (10) 0.4320 (10) 0.036 (4)
C1 0.5574 (11) 0.5386 (16) 0.2863 (17) 0.028 (6)
C2 0.6047 (11) 0.5117 (15) 0.3600 (17) 0.035 (6)
H2 0.5976 0.5394 0.4094 0.042*
C3 0.6650 (10) 0.4458 (15) 0.3733 (16) 0.031 (6)
H3 0.6973 0.4305 0.4270 0.037*

167
C4 0.6699 (10) 0.4066 (16) 0.2952 (16) 0.026 (5)
C5 0.6198 (11) 0.4288 (16) 0.2115 (18) 0.044 (7)
H5 0.6245 0.3994 0.1613 0.053*
C6 0.5649 (10) 0.4956 (15) 0.2103 (16) 0.029 (6)
H6 0.5317 0.5129 0.1577 0.035*
C7 0.7803 (11) 0.3269 (17) 0.3657 (17) 0.047 (7)
H7A 0.8118 0.2712 0.3595 0.071*
H7B 0.7640 0.3093 0.4132 0.071*
H7C 0.8043 0.3955 0.3763 0.071*
C8 0.4240 (10) 0.5592 (14) 0.3250 (14) 0.020 (5)
C9 0.4366 (10) 0.4567 (14) 0.3605 (14) 0.022 (5)
H9 0.4789 0.4225 0.3651 0.027*
C10 0.3883 (10) 0.4041 (16) 0.3890 (15) 0.034 (6)
H10 0.3969 0.3335 0.4104 0.041*
C11 0.3256 (10) 0.4571 (14) 0.3860 (14) 0.018 (5)
C12 0.3108 (10) 0.5617 (14) 0.3457 (14) 0.023 (5)
H12 0.2685 0.5961 0.3415 0.027*
C13 0.3571 (10) 0.6112 (16) 0.3139 (15) 0.034 (6)
H13 0.3462 0.6776 0.2853 0.041*
C14 0.2831 (10) 0.3071 (16) 0.4483 (17) 0.048 (7)
H14A 0.2946 0.2578 0.4093 0.073*
H14B 0.2412 0.2827 0.4588 0.073*
H14C 0.3211 0.3093 0.5018 0.073*
C15 0.5234 (10) 0.7412 (14) 0.3531 (15) 0.023 (5)
C16 0.5095 (9) 0.7613 (15) 0.4274 (14) 0.023 (5)
H16 0.4769 0.7157 0.4396 0.028*
C17 0.5392 (10) 0.8448 (15) 0.4888 (16) 0.039 (6)
H17 0.5276 0.8564 0.5387 0.046*
C18 0.5899 (10) 0.9092 (16) 0.4636 (16) 0.037 (6)
H18 0.6122 0.9666 0.4989 0.044*
C19 0.6067 (10) 0.8885 (15) 0.3882 (15) 0.030 (6)
H19 0.6404 0.9305 0.3749 0.036*
C20 0.5735 (9) 0.8071 (14) 0.3351 (14) 0.020 (5)
H20 0.5844 0.7947 0.2848 0.024*
C21 -0.0389 (10) 0.9157 (15) 0.3100 (15) 0.026 (5)
C22 -0.0137 (10) 0.9986 (14) 0.3716 (15) 0.026 (5)
H22 0.0339 1.0152 0.3877 0.031*
C23 -0.0545 (10) 1.0576 (15) 0.4103 (14) 0.024 (5)
H23 -0.0352 1.1128 0.4497 0.028*
C24 -0.1255 (11) 1.0309 (16) 0.3877 (16) 0.033 (6)

168
C25 -0.1560 (10) 0.9522 (14) 0.3236 (14) 0.018 (5)
H25 -0.2040 0.9383 0.3065 0.022*
C26 -0.1126 (10) 0.8960 (14) 0.2867 (15) 0.026 (5)
H26 -0.1325 0.8435 0.2451 0.032*
C27 -0.2330 (11) 1.0526 (17) 0.4215 (17) 0.053 (7)
H27A -0.2541 1.1028 0.4509 0.079*
H27B -0.2326 0.9810 0.4453 0.079*
H27C -0.2598 1.0515 0.3613 0.079*
C28 0.0891 (10) 0.9239 (15) 0.2715 (15) 0.022 (5)
C29 0.0786 (10) 0.9945 (14) 0.2000 (16) 0.027 (5)
H29 0.0370 0.9952 0.1537 0.033*
C30 0.1365 (10) 1.0649 (14) 0.2040 (15) 0.023 (5)
H30 0.1332 1.1152 0.1604 0.028*
C31 0.1966 (11) 1.0572 (15) 0.2724 (16) 0.026 (5)
C32 0.2078 (12) 0.9833 (17) 0.3399 (18) 0.051 (7)
H32 0.2509 0.9787 0.3835 0.061*
C33 0.1516 (11) 0.9167 (17) 0.3391 (17) 0.044 (7)
H33 0.1560 0.8675 0.3837 0.053*
C34 0.2544 (11) 1.1828 (18) 0.2043 (17) 0.052 (7)
H34A 0.3007 1.2081 0.2088 0.078*
H34B 0.2237 1.2440 0.1996 0.078*
H34C 0.2369 1.1376 0.1541 0.078*
C35 0.0460 (10) 0.7230 (16) 0.3415 (16) 0.032 (6)
C36 0.0948 (10) 0.6524 (14) 0.3289 (15) 0.030 (6)
H36 0.1105 0.6608 0.2817 0.035*
C37 0.1210 (11) 0.5668 (16) 0.3880 (16) 0.032 (6)
H37 0.1536 0.5175 0.3803 0.038*
C38 0.0967 (12) 0.5587 (18) 0.4578 (18) 0.051 (7)
H38 0.1129 0.5018 0.4966 0.062*
C39 0.0494 (10) 0.6318 (15) 0.4721 (16) 0.033 (6)
H39 0.0343 0.6236 0.5199 0.040*
C40 0.0246 (9) 0.7165 (14) 0.4160 (14) 0.022 (5)
H40 -0.0055 0.7682 0.4265 0.026*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.0389 (6) 0.0300 (5) 0.0262 (7) 0.0042 (4) 0.0171 (5) 0.0081 (5)
Au2 0.0326 (5) 0.0313 (5) 0.0176 (6) -0.0028 (4) 0.0114 (4) -0.0041 (5)
Cl1 0.052 (4) 0.047 (4) 0.035 (5) 0.011 (3) 0.024 (3) 0.009 (4)
Cl2 0.033 (3) 0.058 (4) 0.020 (4) 0.002 (3) 0.009 (3) -0.014 (3)

169
P1 0.028 (4) 0.022 (3) 0.048 (5) -0.003 (3) 0.020 (3) 0.007 (3)
P2 0.017 (3) 0.035 (4) 0.018 (4) -0.001 (2) 0.005 (3) -0.008 (3)
O1 0.048 (12) 0.044 (10) 0.09 (2) -0.016 (9) 0.032 (12) -0.025 (11)
O2 0.012 (9) 0.054 (10) 0.039 (14) -0.002 (7) -0.011 (8) -0.003 (10)
O3 0.044 (11) 0.052 (10) 0.054 (16) -0.024 (8) 0.020 (10) -0.003 (10)
O4 0.036 (10) 0.038 (9) 0.031 (13) 0.008 (7) 0.005 (8) -0.010 (9)
C1 0.030 (7) 0.028 (7) 0.027 (7) -0.002 (4) 0.010 (5) 0.001 (5)
C2 0.037 (7) 0.034 (7) 0.035 (8) 0.003 (5) 0.012 (5) -0.002 (5)
C3 0.032 (7) 0.029 (7) 0.031 (7) 0.002 (5) 0.008 (5) 0.002 (5)
C4 0.024 (7) 0.025 (7) 0.027 (7) -0.002 (4) 0.008 (5) 0.000 (5)
C5 0.044 (8) 0.048 (8) 0.044 (8) 0.000 (5) 0.020 (5) -0.001 (5)
C6 0.027 (7) 0.030 (7) 0.029 (7) -0.004 (5) 0.008 (5) 0.004 (5)
C7 0.047 (10) 0.041 (10) 0.049 (11) 0.008 (8) 0.009 (8) 0.005 (9)
C8 0.021 (6) 0.017 (6) 0.020 (7) -0.002 (4) 0.003 (5) 0.000 (5)
C9 0.020 (6) 0.023 (7) 0.024 (7) 0.000 (4) 0.006 (5) 0.000 (5)
C10 0.036 (7) 0.030 (7) 0.035 (8) -0.002 (5) 0.010 (5) 0.000 (5)
C11 0.019 (6) 0.018 (6) 0.016 (7) -0.004 (4) 0.004 (5) -0.008 (5)
C12 0.021 (6) 0.024 (7) 0.022 (7) -0.002 (4) 0.006 (5) 0.001 (5)
C13 0.036 (7) 0.034 (7) 0.034 (8) -0.001 (5) 0.013 (5) 0.004 (5)
C14 0.047 (10) 0.045 (10) 0.055 (11) -0.001 (8) 0.018 (8) 0.001 (8)
C15 0.023 (6) 0.020 (7) 0.025 (7) -0.001 (4) 0.006 (5) 0.003 (5)
C16 0.025 (6) 0.022 (7) 0.022 (7) 0.003 (4) 0.008 (5) -0.001 (5)
C17 0.039 (7) 0.036 (8) 0.039 (8) 0.002 (5) 0.010 (5) 0.000 (5)
C18 0.034 (7) 0.031 (7) 0.039 (8) 0.003 (5) 0.003 (5) -0.001 (5)
C19 0.031 (7) 0.026 (7) 0.031 (7) -0.001 (4) 0.009 (5) 0.004 (5)
C20 0.018 (6) 0.019 (6) 0.022 (7) 0.002 (4) 0.006 (5) -0.001 (5)
C21 0.028 (7) 0.026 (7) 0.024 (7) -0.004 (4) 0.009 (5) 0.001 (5)
C22 0.025 (7) 0.026 (7) 0.026 (7) -0.007 (4) 0.007 (5) -0.002 (5)
C23 0.023 (6) 0.021 (6) 0.024 (7) -0.004 (4) 0.003 (5) 0.002 (5)
C24 0.036 (7) 0.031 (7) 0.033 (8) 0.001 (5) 0.012 (5) 0.002 (5)
C25 0.017 (6) 0.018 (6) 0.018 (7) -0.001 (4) 0.004 (5) -0.001 (5)
C26 0.028 (7) 0.025 (7) 0.026 (7) 0.000 (4) 0.009 (5) -0.001 (5)
C27 0.055 (10) 0.060 (11) 0.053 (12) 0.015 (8) 0.030 (9) 0.003 (8)
C28 0.020 (6) 0.022 (6) 0.022 (7) 0.001 (4) 0.006 (5) -0.001 (5)
C29 0.028 (7) 0.026 (7) 0.029 (7) 0.000 (4) 0.009 (5) 0.000 (5)
C30 0.025 (6) 0.023 (7) 0.022 (7) 0.003 (4) 0.008 (5) 0.000 (5)
C31 0.028 (7) 0.025 (7) 0.026 (7) -0.001 (4) 0.011 (5) 0.000 (5)
C32 0.051 (8) 0.053 (8) 0.050 (9) -0.003 (5) 0.018 (5) 0.004 (5)
C33 0.045 (8) 0.046 (8) 0.042 (8) -0.005 (5) 0.016 (5) 0.003 (5)
C34 0.049 (10) 0.052 (10) 0.056 (12) -0.006 (8) 0.019 (9) 0.002 (9)

170
C35 0.030 (7) 0.031 (7) 0.033 (7) 0.000 (5) 0.009 (5) 0.000 (5)
C36 0.029 (7) 0.031 (7) 0.031 (7) -0.006 (4) 0.011 (5) -0.004 (5)
C37 0.031 (7) 0.031 (7) 0.033 (7) -0.001 (5) 0.009 (5) -0.002 (5)
C38 0.054 (8) 0.050 (8) 0.051 (9) -0.004 (5) 0.017 (5) -0.001 (5)
C39 0.031 (7) 0.034 (7) 0.032 (7) -0.001 (5) 0.008 (5) -0.001 (5)
C40 0.024 (6) 0.020 (6) 0.019 (7) -0.001 (4) 0.006 (5) -0.004 (5)
Geometric parameters (Å, º)
Au1—Cl1 2.279 (7) C16—H16 0.9300
Au1—P1 2.231 (7) C16—C17 1.43 (3)
Au2—Cl2 2.294 (6) C17—H17 0.9300
Au2—P2 2.237 (6) C17—C18 1.45 (3)
P1—C1 1.81 (2) C18—H18 0.9300
P1—C8 1.843 (18) C18—C19 1.40 (3)
P1—C15 1.84 (2) C19—H19 0.9300
P2—C21 1.733 (19) C19—C20 1.35 (3)
P2—C28 1.841 (18) C20—H20 0.9300
P2—C35 1.83 (2) C21—C22 1.41 (3)
O1—C4 1.39 (2) C21—C26 1.43 (2)
O1—C7 1.45 (3) C22—H22 0.9300
O2—C11 1.38 (2) C22—C23 1.39 (2)
O2—C14 1.44 (2) C23—H23 0.9300
O3—C31 1.42 (2) C23—C24 1.39 (2)
O3—C34 1.44 (3) C24—C25 1.41 (3)
O4—C24 1.37 (2) C25—H25 0.9300
O4—C27 1.44 (2) C25—C26 1.39 (2)
C1—C2 1.32 (3) C26—H26 0.9300
C1—C6 1.40 (3) C27—H27A 0.9600
C2—H2 0.9300 C27—H27B 0.9600
C2—C3 1.41 (2) C27—H27C 0.9600
C3—H3 0.9300 C28—C29 1.42 (3)
C3—C4 1.40 (3) C28—C33 1.38 (3)
C4—C5 1.44 (3) C29—H29 0.9300
C5—H5 0.9300 C29—C30 1.43 (2)
C5—C6 1.37 (2) C30—H30 0.9300
C6—H6 0.9300 C30—C31 1.36 (3)
C7—H7A 0.9600 C31—C32 1.39 (3)
C7—H7B 0.9600 C32—H32 0.9300
C7—H7C 0.9600 C32—C33 1.39 (3)
C8—C9 1.37 (2) C33—H33 0.9300

171
C8—C13 1.45 (2) C34—H34A 0.9600
C9—H9 0.9300 C34—H34B 0.9600
C9—C10 1.37 (2) C34—H34C 0.9600
C10—H10 0.9300 C35—C36 1.37 (2)
C10—C11 1.41 (2) C35—C40 1.42 (3)
C11—C12 1.43 (3) C36—H36 0.9300
C12—H12 0.9300 C36—C37 1.41 (3)
C12—C13 1.35 (2) C37—H37 0.9300
C13—H13 0.9300 C37—C38 1.38 (3)
C14—H14A 0.9600 C38—H38 0.9300
C14—H14B 0.9600 C38—C39 1.38 (3)
C14—H14C 0.9600 C39—H39 0.9300
C15—C16 1.36 (3) C39—C40 1.37 (3)
C15—C20 1.39 (2) C40—H40 0.9300
P1—Au1—Cl1 174.9 (2) C17—C18—H18 118.7
P2—Au2—Cl2 176.93 (18) C19—C18—C17 123 (2)
C1—P1—Au1 114.3 (9) C19—C18—H18 118.7
C1—P1—C8 107.8 (9) C18—C19—H19 120.3
C1—P1—C15 104.3 (10) C20—C19—C18 119 (2)
C8—P1—Au1 109.2 (7) C20—C19—H19 120.3
C15—P1—Au1 115.1 (7) C15—C20—H20 118.9
C15—P1—C8 105.5 (9) C19—C20—C15 122 (2)
C21—P2—Au2 110.3 (8) C19—C20—H20 118.9
C21—P2—C28 105.1 (9) C22—C21—P2 124.7 (15)
C21—P2—C35 107.0 (10) C22—C21—C26 114.7 (17)
C28—P2—Au2 115.2 (8) C26—C21—P2 120.5 (15)
C35—P2—Au2 111.3 (8) C21—C22—H22 117.5
C35—P2—C28 107.6 (9) C23—C22—C21 125.1 (19)
C4—O1—C7 111.9 (19) C23—C22—H22 117.5
C11—O2—C14 116.3 (14) C22—C23—H23 121.3
C31—O3—C34 114.7 (18) C22—C23—C24 117 (2)
C24—O4—C27 118.2 (16) C24—C23—H23 121.3
C2—C1—P1 123.5 (19) O4—C24—C23 115 (2)
C2—C1—C6 118 (2) O4—C24—C25 123.8 (19)
C6—C1—P1 118.8 (19) C23—C24—C25 121.4 (19)
C1—C2—H2 116.0 C24—C25—H25 120.7
C1—C2—C3 128 (2) C26—C25—C24 118.6 (19)
C3—C2—H2 116.0 C26—C25—H25 120.7
C2—C3—H3 124.3 C21—C26—H26 118.7

172
C4—C3—C2 111 (2) C25—C26—C21 122.7 (19)
C4—C3—H3 124.3 C25—C26—H26 118.7
O1—C4—C3 125 (2) O4—C27—H27A 109.5
O1—C4—C5 110 (2) O4—C27—H27B 109.5
C3—C4—C5 125 (2) O4—C27—H27C 109.5
C4—C5—H5 122.0 H27A—C27—H27B 109.5
C6—C5—C4 116 (2) H27A—C27—H27C 109.5
C6—C5—H5 122.0 H27B—C27—H27C 109.5
C1—C6—H6 119.0 C29—C28—P2 114.9 (16)
C5—C6—C1 122 (2) C33—C28—P2 121.3 (17)
C5—C6—H6 119.0 C33—C28—C29 123.8 (19)
O1—C7—H7A 109.5 C28—C29—H29 122.4
O1—C7—H7B 109.5 C28—C29—C30 115 (2)
O1—C7—H7C 109.5 C30—C29—H29 122.4
H7A—C7—H7B 109.5 C29—C30—H30 120.4
H7A—C7—H7C 109.5 C31—C30—C29 119 (2)
H7B—C7—H7C 109.5 C31—C30—H30 120.4
C9—C8—P1 123.2 (14) C30—C31—O3 123 (2)
C9—C8—C13 119.7 (16) C30—C31—C32 125 (2)
C13—C8—P1 116.7 (14) C32—C31—O3 112 (2)
C8—C9—H9 119.2 C31—C32—H32 121.6
C10—C9—C8 121.5 (18) C31—C32—C33 117 (2)
C10—C9—H9 119.2 C33—C32—H32 121.6
C9—C10—H10 120.1 C28—C33—C32 120 (2)
C9—C10—C11 119.7 (18) C28—C33—H33 120.2
C11—C10—H10 120.1 C32—C33—H33 120.2
O2—C11—C10 126.4 (17) O3—C34—H34A 109.5
O2—C11—C12 114.4 (15) O3—C34—H34B 109.5
C10—C11—C12 118.9 (17) O3—C34—H34C 109.5
C11—C12—H12 119.5 H34A—C34—H34B 109.5
C13—C12—C11 121.1 (18) H34A—C34—H34C 109.5
C13—C12—H12 119.5 H34B—C34—H34C 109.5
C8—C13—H13 120.6 C36—C35—P2 120.2 (17)
C12—C13—C8 118.7 (19) C36—C35—C40 121 (2)
C12—C13—H13 120.6 C40—C35—P2 118.1 (15)
O2—C14—H14A 109.5 C35—C36—H36 120.2
O2—C14—H14B 109.5 C35—C36—C37 120 (2)
O2—C14—H14C 109.5 C37—C36—H36 120.2
H14A—C14—H14B 109.5 C36—C37—H37 121.1
H14A—C14—H14C 109.5 C38—C37—C36 118 (2)

173
H14B—C14—H14C 109.5 C38—C37—H37 121.1
C16—C15—P1 124.6 (15) C37—C38—H38 118.7
C16—C15—C20 117.0 (19) C37—C38—C39 123 (2)
C20—C15—P1 118.3 (15) C39—C38—H38 118.7
C15—C16—H16 116.5 C38—C39—H39 120.0
C15—C16—C17 127 (2) C40—C39—C38 120 (2)
C17—C16—H16 116.5 C40—C39—H39 120.0
C16—C17—H17 124.3 C35—C40—H40 120.9
C16—C17—C18 111 (2) C39—C40—C35 118.2 (19)
C18—C17—H17 124.3 C39—C40—H40 120.9
Au1—P1—C1—C2 169.8 (17) C11—C12—C13—C8 -3 (3)
Au1—P1—C1—C6 -9.2 (18) C13—C8—C9—C10 -3 (3)
Au1—P1—C8—C9 126.0 (18) C14—O2—C11—C10 4 (3)
Au1—P1—C8—C13 -46.7 (18) C14—O2—C11—C12 177.4 (19)
Au1—P1—C15—C16 121.8 (18) C15—P1—C1—C2 43 (2)
Au1—P1—C15—C20 -61.4 (17) C15—P1—C1—C6 -135.8 (16)
Au2—P2—C21—C22 -153.2 (17) C15—P1—C8—C9 -109.8 (19)
Au2—P2—C21—C26 29.8 (19) C15—P1—C8—C13 77.5 (19)
Au2—P2—C28—C29 35.9 (16) C15—C16—C17—C18 -1 (3)
Au2—P2—C28—C33 -142.3 (16) C16—C15—C20—C19 -1 (3)
Au2—P2—C35—C36 66.1 (18) C16—C17—C18—C19 -1 (3)
Au2—P2—C35—C40 -119.8 (16) C17—C18—C19—C20 2 (3)
P1—C1—C2—C3 -177.0 (15) C18—C19—C20—C15 -1 (3)
P1—C1—C6—C5 178.3 (15) C20—C15—C16—C17 2 (3)
P1—C8—C9—C10 -175.3 (17) C21—P2—C28—C29 -85.6 (17)
P1—C8—C13—C12 178.8 (17) C21—P2—C28—C33 96.2 (19)
P1—C15—C16—C17 178.4 (15) C21—P2—C35—C36 -173.4 (18)
P1—C15—C20—C19 -177.6 (16) C21—P2—C35—C40 1 (2)
P2—C21—C22—C23 -175.8 (16) C21—C22—C23—C24 2 (3)
P2—C21—C26—C25 175.6 (15) C22—C21—C26—C25 -2 (3)
P2—C28—C29—C30 178.2 (13) C22—C23—C24—O4 175.6 (18)
P2—C28—C33—C32 179.7 (16) C22—C23—C24—C25 -4 (3)
P2—C35—C36—C37 178.1 (16) C23—C24—C25—C26 4 (3)
P2—C35—C40—C39 -179.4 (14) C24—C25—C26—C21 -1 (3)
O1—C4—C5—C6 177.1 (16) C26—C21—C22—C23 1 (3)
O2—C11—C12—C13 -176.5 (19) C27—O4—C24—C23 -171.3 (19)
O3—C31—C32—C33 -179.9 (17) C27—O4—C24—C25 9 (3)
O4—C24—C25—C26 -175.9 (19) C28—P2—C21—C22 -29 (2)
C1—P1—C8—C9 1 (2) C28—P2—C21—C26 154.4 (17)

174
C1—P1—C8—C13 -171.4 (17) C28—P2—C35—C36 -61 (2)
C1—P1—C15—C16 -112.1 (19) C28—P2—C35—C40 113.2 (17)
C1—P1—C15—C20 64.6 (18) C28—C29—C30—C31 2 (3)
C1—C2—C3—C4 -1 (3) C29—C28—C33—C32 2 (3)
C2—C1—C6—C5 -1 (3) C29—C30—C31—O3 177.4 (16)
C2—C3—C4—O1 -175.4 (17) C29—C30—C31—C32 2 (3)
C2—C3—C4—C5 -1 (3) C30—C31—C32—C33 -4 (3)
C3—C4—C5—C6 2 (3) C31—C32—C33—C28 2 (3)
C4—C5—C6—C1 -1 (3) C33—C28—C29—C30 -4 (3)
C6—C1—C2—C3 2 (3) C34—O3—C31—C30 -8 (3)
C7—O1—C4—C3 -17 (3) C34—O3—C31—C32 168.1 (19)
C7—O1—C4—C5 167.6 (18) C35—P2—C21—C22 86 (2)
C8—P1—C1—C2 -69 (2) C35—P2—C21—C26 -91.4 (19)
C8—P1—C1—C6 112.4 (16) C35—P2—C28—C29 160.6 (15)
C8—P1—C15—C16 1 (2) C35—P2—C28—C33 -18 (2)
C8—P1—C15—C20 178.1 (15) C35—C36—C37—C38 -1 (3)
C8—C9—C10—C11 -3 (3) C36—C35—C40—C39 -5 (3)
C9—C8—C13—C12 6 (3) C36—C37—C38—C39 -1 (4)
C9—C10—C11—O2 179 (2) C37—C38—C39—C40 0 (4)
C9—C10—C11—C12 6 (3) C38—C39—C40—C35 3 (3)
C10—C11—C12—C13 -2 (3) C40—C35—C36—C37 4 (3)
[AuCl{PPh2(4-MeOC6H4)}] 3a
Crystal data
C19H17AuClOP F(000) = 1000
Mr = 524.71 Dx = 1.957 Mg m-3
Monoclinic, P21/c Mo K radiation, = 0.71073 Å
a = 6.6337 (3) Å Cell parameters from 2867 reflections
b = 12.6665 (5) Å = 3.9–28.4°
c = 21.1922 (10) Å = 8.50 mm-1
= 90.960 (5)° T = 150 K
V = 1780.45 (13) Å3 colourless
Z = 4 0.04 × 0.02 × 0.02 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
3677 independent reflections
Radiation source: SuperNova (Mo) X-ray 3010 reflections with I > 2(I)

175
Source
Mirror monochromator Rint = 0.035
Detector resolution: 8.0714 pixels mm-1
max = 26.5°, min = 3.4°
scans h = -78
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using
spherical harmonics, implemented in
SCALE3 ABSPACK scaling algorithm.
k = -915
Tmin = 0.776, Tmax = 1.000 l = -2626
7053 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.037 H-atom parameters constrained
wR(F2) = 0.064 w = 1/[
2(Fo
2) + (0.0155P)
2]
where P = (Fo2 + 2Fc
2)/3
S = 1.02 (/)max < 0.001
3677 reflections max = 1.47 e Å-3
209 parameters min = -1.16 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate
(isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.08277 (3) 0.07899 (2) 0.35624 (2) 0.02276 (8)
Cl1 -0.1199 (2) -0.06595 (12) 0.34381 (8) 0.0353 (4)
P1 0.2977 (2) 0.21338 (12) 0.36832 (7) 0.0188 (3)
O1 -0.0971 (6) 0.6300 (3) 0.4067 (2) 0.0334 (11)

176
C2 0.0195 (8) 0.3695 (5) 0.3414 (3) 0.0233 (14)
H2 -0.0255 0.3232 0.3102 0.028*
C18 0.4866 (8) 0.3204 (5) 0.2681 (3) 0.0221 (14)
H18 0.4132 0.3800 0.2791 0.026*
C3 -0.0806 (8) 0.4667 (5) 0.3487 (3) 0.0247 (14)
H3 -0.1899 0.4846 0.3229 0.030*
C12 0.6579 (8) 0.2320 (5) 0.4376 (3) 0.0255 (14)
H12 0.7020 0.2703 0.4031 0.031*
C1 0.1824 (8) 0.3407 (5) 0.3792 (2) 0.0182 (13)
C15 0.7040 (9) 0.1415 (5) 0.2337 (3) 0.0275 (15)
H15 0.7748 0.0816 0.2218 0.033*
C4 -0.0123 (8) 0.5341 (5) 0.3950 (3) 0.0221 (14)
C6 0.2474 (8) 0.4121 (5) 0.4258 (3) 0.0247 (14)
H6 0.3557 0.3946 0.4522 0.030*
C13 0.4634 (8) 0.2264 (5) 0.3006 (2) 0.0171 (12)
C7 0.4697 (8) 0.1949 (4) 0.4354 (2) 0.0197 (13)
C17 0.6233 (8) 0.3239 (5) 0.2181 (3) 0.0258 (15)
H17 0.6416 0.3866 0.1962 0.031*
C16 0.7292 (9) 0.2362 (5) 0.2016 (3) 0.0291 (15)
H16 0.8192 0.2397 0.1685 0.035*
C14 0.5715 (8) 0.1375 (5) 0.2840 (3) 0.0243 (14)
H14 0.5557 0.0750 0.3064 0.029*
C8 0.4033 (9) 0.1390 (5) 0.4881 (3) 0.0269 (15)
H8 0.2729 0.1122 0.4887 0.032*
C11 0.7891 (9) 0.2181 (5) 0.4863 (3) 0.0321 (16)
H11 0.9190 0.2456 0.4849 0.039*
C5 0.1515 (9) 0.5088 (5) 0.4328 (3) 0.0322 (16)
H5 0.1979 0.5565 0.4631 0.039*
C9 0.5303 (10) 0.1239 (5) 0.5385 (3) 0.0344 (17)
H9 0.4857 0.0875 0.5737 0.041*
C10 0.7261 (10) 0.1628 (5) 0.5375 (3) 0.0348 (17)
H10 0.8135 0.1512 0.5716 0.042*
C19 -0.2744 (10) 0.6581 (5) 0.3724 (3) 0.0428 (19)
H19A -0.2432 0.6667 0.3287 0.064*
H19B -0.3265 0.7232 0.3886 0.064*
H19C -0.3735 0.6035 0.3768 0.064*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.01897 (12) 0.01951 (13) 0.02973 (14) -0.00125 -0.00156 (9) 0.00105 (12)

177
(11)
Cl1 0.0260 (8) 0.0231 (9) 0.0563 (11) -0.0038 (7) -0.0096 (7) 0.0061 (9)
P1 0.0192 (7) 0.0182 (8) 0.0189 (8) 0.0010 (7) 0.0003 (6) 0.0013 (7)
O1 0.034 (3) 0.025 (3) 0.041 (3) 0.006 (2) -0.003 (2) -0.004 (2)
C2 0.019 (3) 0.027 (4) 0.023 (3) -0.006 (3) -0.001 (3) -0.001 (3)
C18 0.022 (3) 0.020 (3) 0.024 (3) 0.003 (3) -0.002 (3) 0.004 (3)
C3 0.019 (3) 0.026 (4) 0.029 (3) 0.004 (3) -0.001 (3) -0.001 (3)
C12 0.022 (3) 0.035 (4) 0.019 (3) 0.009 (3) -0.008 (2) -0.011 (3)
C1 0.016 (3) 0.022 (3) 0.017 (3) 0.002 (3) 0.002 (2) 0.000 (3)
C15 0.028 (3) 0.031 (4) 0.024 (3) 0.004 (3) 0.004 (3) 0.000 (3)
C4 0.025 (3) 0.016 (3) 0.026 (3) 0.003 (3) 0.009 (3) 0.000 (3)
C6 0.025 (3) 0.023 (3) 0.026 (3) 0.007 (3) -0.007 (3) 0.000 (3)
C13 0.018 (3) 0.020 (3) 0.013 (3) -0.004 (3) -0.002 (2) 0.000 (3)
C7 0.031 (3) 0.013 (3) 0.015 (3) 0.001 (3) 0.010 (3) -0.004 (3)
C17 0.029 (3) 0.030 (4) 0.018 (3) 0.001 (3) 0.004 (3) 0.011 (3)
C16 0.026 (3) 0.037 (4) 0.024 (3) -0.001 (3) 0.002 (3) 0.001 (3)
C14 0.023 (3) 0.023 (4) 0.026 (3) 0.004 (3) -0.001 (3) 0.002 (3)
C8 0.029 (3) 0.025 (4) 0.027 (3) 0.002 (3) 0.004 (3) -0.004 (3)
C11 0.023 (3) 0.041 (4) 0.032 (4) 0.003 (3) 0.002 (3) -0.019 (4)
C5 0.033 (4) 0.029 (4) 0.035 (4) 0.001 (3) -0.006 (3) -0.015 (3)
C9 0.053 (4) 0.031 (4) 0.019 (3) -0.001 (4) 0.002 (3) 0.002 (3)
C10 0.052 (4) 0.037 (4) 0.015 (3) 0.018 (4) -0.009 (3) -0.006 (3)
C19 0.038 (4) 0.029 (4) 0.061 (5) 0.011 (3) -0.010 (4) -0.006 (4)
Geometric parameters (Å, º)
Au1—Cl1 2.2881 (15) C15—C14 1.393 (7)
Au1—P1 2.2323 (15) C4—C5 1.377 (8)
P1—C1 1.801 (6) C6—H6 0.9300
P1—C13 1.830 (5) C6—C5 1.390 (8)
P1—C7 1.823 (6) C13—C14 1.384 (8)
O1—C4 1.363 (7) C7—C8 1.400 (7)
O1—C19 1.418 (7) C17—H17 0.9300
C2—H2 0.9300 C17—C16 1.363 (8)
C2—C3 1.407 (8) C16—H16 0.9300
C2—C1 1.383 (7) C14—H14 0.9300
C18—H18 0.9300 C8—H8 0.9300
C18—C13 1.385 (7) C8—C9 1.363 (8)
C18—C17 1.407 (7) C11—H11 0.9300
C3—H3 0.9300 C11—C10 1.364 (8)
C3—C4 1.372 (8) C5—H5 0.9300

178
C12—H12 0.9300 C9—H9 0.9300
C12—C7 1.334 (7) C9—C10 1.390 (9)
C12—C11 1.349 (8) C10—H10 0.9300
C1—C6 1.402 (8) C19—H19A 0.9600
C15—H15 0.9300 C19—H19B 0.9600
C15—C16 1.390 (8) C19—H19C 0.9600
P1—Au1—Cl1 176.28 (5) C14—C13—C18 120.8 (5)
C1—P1—Au1 115.18 (18) C12—C7—P1 123.8 (4)
C1—P1—C13 106.4 (3) C12—C7—C8 117.2 (6)
C1—P1—C7 106.1 (3) C8—C7—P1 119.1 (4)
C13—P1—Au1 111.70 (19) C18—C17—H17 119.7
C7—P1—Au1 112.44 (19) C16—C17—C18 120.6 (6)
C7—P1—C13 104.3 (2) C16—C17—H17 119.7
C4—O1—C19 118.1 (5) C15—C16—H16 119.7
C3—C2—H2 118.9 C17—C16—C15 120.7 (5)
C1—C2—H2 118.9 C17—C16—H16 119.7
C1—C2—C3 122.2 (6) C15—C14—H14 120.0
C13—C18—H18 120.7 C13—C14—C15 120.0 (6)
C13—C18—C17 118.6 (5) C13—C14—H14 120.0
C17—C18—H18 120.7 C7—C8—H8 120.1
C2—C3—H3 120.9 C9—C8—C7 119.9 (6)
C4—C3—C2 118.2 (6) C9—C8—H8 120.1
C4—C3—H3 120.9 C12—C11—H11 120.9
C7—C12—H12 117.5 C12—C11—C10 118.2 (6)
C7—C12—C11 124.9 (6) C10—C11—H11 120.9
C11—C12—H12 117.5 C4—C5—C6 120.1 (6)
C2—C1—P1 119.5 (5) C4—C5—H5 120.0
C2—C1—C6 117.7 (5) C6—C5—H5 120.0
C6—C1—P1 122.8 (4) C8—C9—H9 119.9
C16—C15—H15 120.4 C8—C9—C10 120.2 (6)
C16—C15—C14 119.2 (6) C10—C9—H9 119.9
C14—C15—H15 120.4 C11—C10—C9 119.6 (6)
O1—C4—C3 123.6 (6) C11—C10—H10 120.2
O1—C4—C5 115.1 (6) C9—C10—H10 120.2
C3—C4—C5 121.3 (6) O1—C19—H19A 109.5
C1—C6—H6 119.7 O1—C19—H19B 109.5
C5—C6—C1 120.5 (6) O1—C19—H19C 109.5
C5—C6—H6 119.7 H19A—C19—H19B 109.5
C18—C13—P1 122.7 (4) H19A—C19—H19C 109.5

179
C14—C13—P1 116.5 (4) H19B—C19—H19C 109.5
Au1—P1—C1—C2 -44.1 (5) C1—P1—C7—C8 95.2 (5)
Au1—P1—C1—C6 134.2 (4) C1—C2—C3—C4 0.1 (8)
Au1—P1—C13—C18 127.4 (4) C1—C6—C5—C4 1.7 (9)
Au1—P1—C13—C14 -54.5 (5) C13—P1—C1—C2 80.2 (5)
Au1—P1—C7—C12 148.6 (5) C13—P1—C1—C6 -101.4 (5)
Au1—P1—C7—C8 -31.5 (5) C13—P1—C7—C12 27.4 (6)
P1—C1—C6—C5 -179.1 (4) C13—P1—C7—C8 -152.7 (5)
P1—C13—C14—C15 -178.7 (4) C13—C18—C17—C16 0.7 (9)
P1—C7—C8—C9 179.6 (5) C7—P1—C1—C2 -169.2 (4)
O1—C4—C5—C6 178.8 (5) C7—P1—C1—C6 9.2 (5)
C2—C3—C4—O1 -179.7 (5) C7—P1—C13—C18 -110.9 (5)
C2—C3—C4—C5 1.0 (8) C7—P1—C13—C14 67.2 (5)
C2—C1—C6—C5 -0.7 (8) C7—C12—C11—C10 -0.5 (10)
C18—C13—C14—
C15
-0.6 (9) C7—C8—C9—C10 -0.7 (9)
C18—C17—C16—
C15
0.1 (9) C17—C18—C13—P1 177.6 (4)
C3—C2—C1—P1 178.3 (4) C17—C18—C13—C14 -0.4 (8)
C3—C2—C1—C6 -0.2 (8) C16—C15—C14—C13 1.4 (9)
C3—C4—C5—C6 -1.9 (9) C14—C15—C16—C17 -1.2 (9)
C12—C7—C8—C9 -0.5 (9) C8—C9—C10—C11 1.4 (10)
C12—C11—C10—C9 -0.8 (9) C11—C12—C7—P1 -179.0 (5)
C1—P1—C13—C18 0.9 (5) C11—C12—C7—C8 1.1 (9)
C1—P1—C13—C14 179.0 (4) C19—O1—C4—C3 4.9 (8)
C1—P1—C7—C12 -84.7 (5) C19—O1—C4—C5 -175.8 (5)
[AuBr{PPh2(4-MeO C6H4)}] 3b
Crystal data
C19H17AuBrOP F(000) = 1072
Mr = 569.17 Dx = 2.086 Mg m-3
Monoclinic, P21/c Mo K radiation, = 0.71073 Å
a = 6.6997 (3) Å Cell parameters from 2503 reflections
b = 12.7762 (5) Å = 3.7–28.4°
c = 21.1763 (10) Å = 10.41 mm-1
= 90.721 (5)° T = 150 K
V = 1812.47 (13) Å3 , clear light colourless
Z = 4 0.16 × 0.14 × 0.08 mm

180
Data collection
SuperNova, Single source at offset, Eos
diffractometer
2620 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source 2296 reflections with I > 2(I)
Mirror monochromator Rint = 0.024
Detector resolution: 8.0714 pixels mm-1
max = 25.0°, min = 3.3°
scans h = -47
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1415
Tmin = 0.611, Tmax = 1.000 l = -1725
4602 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.028 H-atom parameters constrained
wR(F2) = 0.063 w = 1/[
2(Fo
2) + (0.0209P)
2 + 4.1946P]
where P = (Fo2 + 2Fc
2)/3
S = 1.03 (/)max = 0.001
2620 reflections max = 0.64 e Å-3
209 parameters min = -1.16 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate
(isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq

181
Au1 0.08491 (4) 0.08632 (2) 0.35685 (2) 0.02253 (10)
Br1 -0.12911 (11) -0.06328 (5) 0.34708 (4) 0.0402 (2)
P1 0.3011 (2) 0.21928 (12) 0.36654 (8) 0.0192 (4)
O1 -0.0975 (7) 0.6295 (4) 0.4094 (2) 0.0341 (12)
C2 0.0267 (9) 0.3742 (5) 0.3398 (3) 0.0224 (16)
H2 -0.0151 0.3289 0.3078 0.027*
C18 0.4946 (10) 0.3235 (5) 0.2663 (3) 0.0230 (16)
H18 0.4285 0.3841 0.2784 0.028*
C3 -0.0726 (9) 0.4696 (5) 0.3478 (3) 0.0236 (16)
H3 -0.1784 0.4881 0.3213 0.028*
C12 0.6608 (9) 0.2402 (5) 0.4331 (3) 0.0263 (16)
H12 0.7003 0.2799 0.3986 0.032*
C1 0.1852 (9) 0.3462 (5) 0.3782 (3) 0.0193 (15)
C15 0.6957 (12) 0.1451 (6) 0.2310 (3) 0.0331 (19)
H15 0.7652 0.0852 0.2195 0.040*
C4 -0.0112 (9) 0.5357 (5) 0.3957 (3) 0.0244 (16)
C6 0.2493 (10) 0.4153 (5) 0.4250 (3) 0.0260 (17)
H6 0.3578 0.3976 0.4507 0.031*
C13 0.4610 (9) 0.2308 (5) 0.2987 (3) 0.0171 (14)
C7 0.4729 (10) 0.1998 (4) 0.4325 (3) 0.0182 (15)
C17 0.6247 (10) 0.3268 (5) 0.2164 (3) 0.0267 (17)
H17 0.6436 0.3891 0.1945 0.032*
C16 0.7264 (10) 0.2383 (6) 0.1989 (3) 0.0295 (17)
H16 0.8156 0.2409 0.1656 0.035*
C14 0.5629 (10) 0.1414 (5) 0.2796 (3) 0.0259 (17)
H14 0.5404 0.0782 0.3002 0.031*
C8 0.4134 (11) 0.1417 (5) 0.4844 (3) 0.0288 (17)
H8 0.2861 0.1128 0.4853 0.035*
C11 0.7943 (11) 0.2257 (6) 0.4813 (3) 0.0341 (19)
H11 0.9214 0.2548 0.4796 0.041*
C5 0.1538 (10) 0.5093 (5) 0.4336 (3) 0.0305 (18)
H5 0.1989 0.5554 0.4647 0.037*
C9 0.5467 (12) 0.1269 (6) 0.5354 (3) 0.038 (2)
H9 0.5065 0.0899 0.5709 0.045*
C10 0.7383 (11) 0.1675 (6) 0.5326 (3) 0.0344 (19)
H10 0.8287 0.1552 0.5655 0.041*
C19 -0.2747 (10) 0.6564 (5) 0.3743 (4) 0.039 (2)
H19A -0.2444 0.6606 0.3302 0.058*
H19B -0.3237 0.7228 0.3886 0.058*
H19C -0.3746 0.6037 0.3808 0.058*

182
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.02000 (16) 0.01984 (14) 0.02769 (16) -0.00164
(12)
-0.00216
(13)
0.00021 (10)
Br1 0.0327 (4) 0.0288 (4) 0.0589 (5) -0.0065 (3) -0.0133 (4) 0.0056 (3)
P1 0.0210 (9) 0.0172 (8) 0.0193 (9) 0.0000 (7) -0.0014 (8) 0.0008 (6)
O1 0.034 (3) 0.027 (3) 0.041 (3) 0.010 (2) -0.006 (3) -0.005 (2)
C2 0.023 (4) 0.019 (3) 0.025 (4) -0.002 (3) 0.003 (3) 0.002 (3)
C18 0.023 (4) 0.022 (3) 0.024 (4) 0.003 (3) -0.005 (4) -0.002 (3)
C3 0.021 (4) 0.026 (4) 0.024 (4) 0.000 (3) -0.001 (3) 0.006 (3)
C12 0.022 (4) 0.035 (4) 0.021 (4) 0.009 (3) -0.006 (3) -0.008 (3)
C1 0.020 (4) 0.022 (3) 0.016 (3) -0.001 (3) 0.010 (3) 0.001 (3)
C15 0.040 (5) 0.035 (4) 0.025 (4) 0.014 (4) 0.005 (4) 0.000 (3)
C4 0.022 (4) 0.022 (3) 0.029 (4) 0.004 (3) 0.005 (4) 0.003 (3)
C6 0.022 (4) 0.024 (3) 0.031 (4) 0.010 (3) -0.006 (3) 0.000 (3)
C13 0.016 (3) 0.024 (3) 0.011 (3) -0.001 (3) 0.002 (3) -0.001 (2)
C7 0.024 (4) 0.014 (3) 0.017 (3) 0.006 (3) 0.007 (3) -0.004 (2)
C17 0.029 (4) 0.021 (4) 0.030 (4) -0.003 (3) 0.001 (4) 0.010 (3)
C16 0.030 (4) 0.044 (4) 0.015 (3) -0.006 (4) 0.003 (3) 0.007 (3)
C14 0.031 (4) 0.024 (4) 0.023 (4) 0.002 (3) 0.005 (4) 0.007 (3)
C8 0.029 (4) 0.029 (4) 0.028 (4) -0.005 (3) 0.004 (4) 0.005 (3)
C11 0.028 (4) 0.040 (4) 0.034 (4) 0.008 (4) -0.004 (4) -0.018 (3)
C5 0.032 (4) 0.031 (4) 0.028 (4) 0.001 (4) -0.004 (4) -0.009 (3)
C9 0.059 (6) 0.038 (4) 0.016 (4) 0.008 (4) -0.001 (4) 0.009 (3)
C10 0.041 (5) 0.036 (4) 0.026 (4) 0.016 (4) -0.010 (4) -0.008 (3)
C19 0.033 (4) 0.023 (4) 0.061 (5) 0.011 (4) -0.001 (4) 0.000 (3)
Geometric parameters (Å, º)
Au1—Br1 2.3967 (7) C15—C14 1.371 (10)
Au1—P1 2.2399 (16) C4—C5 1.399 (8)
P1—C1 1.816 (6) C6—H6 0.9300
P1—C13 1.809 (7) C6—C5 1.374 (9)
P1—C7 1.815 (6) C13—C14 1.394 (9)
O1—C4 1.363 (8) C7—C8 1.388 (9)
O1—C19 1.434 (8) C17—H17 0.9300
C2—H2 0.9300 C17—C16 1.373 (10)
C2—C3 1.400 (9) C16—H16 0.9300
C2—C1 1.378 (8) C14—H14 0.9300
C18—H18 0.9300 C8—H8 0.9300

183
C18—C13 1.389 (8) C8—C9 1.406 (9)
C18—C17 1.379 (9) C11—H11 0.9300
C3—H3 0.9300 C11—C10 1.373 (10)
C3—C4 1.379 (9) C5—H5 0.9300
C12—H12 0.9300 C9—H9 0.9300
C12—C7 1.361 (9) C9—C10 1.386 (10)
C12—C11 1.363 (8) C10—H10 0.9300
C1—C6 1.390 (8) C19—H19A 0.9600
C15—H15 0.9300 C19—H19B 0.9600
C15—C16 1.387 (9) C19—H19C 0.9600
P1—Au1—Br1 176.43 (5) C14—C13—P1 117.5 (5)
C1—P1—Au1 114.4 (2) C12—C7—P1 122.2 (5)
C13—P1—Au1 112.20 (19) C12—C7—C8 118.0 (6)
C13—P1—C1 107.1 (3) C8—C7—P1 119.8 (5)
C13—P1—C7 104.2 (3) C18—C17—H17 119.9
C7—P1—Au1 111.7 (2) C16—C17—C18 120.1 (6)
C7—P1—C1 106.6 (3) C16—C17—H17 119.9
C4—O1—C19 116.8 (5) C15—C16—H16 120.1
C3—C2—H2 119.4 C17—C16—C15 119.8 (7)
C1—C2—H2 119.4 C17—C16—H16 120.1
C1—C2—C3 121.2 (6) C15—C14—C13 120.9 (6)
C13—C18—H18 119.6 C15—C14—H14 119.5
C17—C18—H18 119.6 C13—C14—H14 119.5
C17—C18—C13 120.9 (6) C7—C8—H8 120.2
C2—C3—H3 120.5 C7—C8—C9 119.6 (7)
C4—C3—C2 119.0 (6) C9—C8—H8 120.2
C4—C3—H3 120.5 C12—C11—H11 120.5
C7—C12—H12 118.2 C12—C11—C10 118.9 (7)
C7—C12—C11 123.7 (7) C10—C11—H11 120.5
C11—C12—H12 118.2 C4—C5—H5 120.0
C2—C1—P1 118.6 (5) C6—C5—C4 120.1 (6)
C2—C1—C6 119.1 (6) C6—C5—H5 120.0
C6—C1—P1 122.3 (5) C8—C9—H9 120.1
C16—C15—H15 120.0 C10—C9—C8 119.8 (7)
C14—C15—H15 120.0 C10—C9—H9 120.1
C14—C15—C16 120.0 (7) C11—C10—C9 119.9 (6)
O1—C4—C3 124.8 (5) C11—C10—H10 120.0
O1—C4—C5 115.2 (6) C9—C10—H10 120.0
C3—C4—C5 120.0 (6) O1—C19—H19A 109.5

184
C1—C6—H6 119.7 O1—C19—H19B 109.5
C5—C6—C1 120.6 (6) O1—C19—H19C 109.5
C5—C6—H6 119.7 H19A—C19—H19B 109.5
C18—C13—P1 124.3 (5) H19A—C19—H19C 109.5
C18—C13—C14 118.2 (6) H19B—C19—H19C 109.5
Au1—P1—C1—C2 -46.8 (6) C1—P1—C7—C8 97.0 (5)
Au1—P1—C1—C6 132.4 (5) C1—C2—C3—C4 -0.8 (10)
Au1—P1—C13—C18 130.5 (5) C1—C6—C5—C4 0.9 (11)
Au1—P1—C13—C14 -52.1 (5) C13—P1—C1—C2 78.2 (6)
Au1—P1—C7—C12 151.1 (5) C13—P1—C1—C6 -102.6 (6)
Au1—P1—C7—C8 -28.6 (6) C13—P1—C7—C12 29.8 (6)
P1—C1—C6—C5 -178.0 (6) C13—P1—C7—C8 -150.0 (5)
P1—C13—C14—C15 -176.2 (5) C13—C18—C17—C16 -1.4 (9)
P1—C7—C8—C9 -179.9 (5) C7—P1—C1—C2 -170.7 (5)
O1—C4—C5—C6 178.4 (6) C7—P1—C1—C6 8.5 (7)
C2—C3—C4—O1 -178.6 (6) C7—P1—C13—C18 -108.5 (5)
C2—C3—C4—C5 2.9 (10) C7—P1—C13—C14 68.9 (5)
C2—C1—C6—C5 1.2 (10) C7—C12—C11—C10 -0.1 (10)
C18—C13—C14—
C15
1.3 (9) C7—C8—C9—C10 -2.1 (11)
C18—C17—C16—
C15
1.0 (10) C17—C18—C13—P1 177.6 (4)
C3—C2—C1—P1 177.9 (5) C17—C18—C13—C14 0.2 (9)
C3—C2—C1—C6 -1.3 (10) C16—C15—C14—C13 -1.7 (10)
C3—C4—C5—C6 -3.0 (11) C14—C15—C16—C17 0.6 (10)
C12—C7—C8—C9 0.3 (10) C8—C9—C10—C11 2.8 (11)
C12—C11—C10—C9 -1.7 (10) C11—C12—C7—P1 -179.0 (5)
C1—P1—C13—C18 4.2 (6) C11—C12—C7—C8 0.8 (10)
C1—P1—C13—C14 -178.4 (5) C19—O1—C4—C3 5.3 (10)
C1—P1—C7—C12 -83.3 (6) C19—O1—C4—C5 -176.1 (6)

185
Chapter 7
The single crystal structures of some gold(I) halide complexes
containing the PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and
PPh(4-C6H4NMe2)3 ligands.

186
7. The single crystal structures of some gold(I) halide complexes
containing the PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and PPh(4-
C6H4NMe2)3 ligands.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
School of Chemistry, The University of Manchester, Manchester, M13 9PL, UK.
7.1 Abstract
The structure of the two-coordinate gold(I) phosphine halide complexes [AuX(P)] (P =
PPh2(4-C6H4NMe2), X=Cl and I, PPh(4-C6H4NMe2)2, X = Br and P(4-C6H4NMe2)3, X =
Cl, Br) have been determined by single crystal X-ray diffraction in order to investigate
the existence of short Au···Au (aurophilic) interactions. The molecules have an average
Au–P bond length of 2.241(2) Å and Au–X of 2.2809(9) Å (X = Cl), 2.402(1) Å (X =
Br) and 2.5553(6) Å (X = I). Aurophilic interactions were not evident in the complexes,
where all Au···Au distances were longer than twice the Van der Walls radii of gold. The
molecules of all complexes pack with C–H···X interactions.
7.2 Introduction
Au(I) phosphine compounds find several practical applications, in medicine, molecular
biology (Fackler Jr et al., 2009; Ott, 2009) and catalysis (Hashmi, 2007). Notably,
gold(I) phosphine derivatives have presented greater activity like anticancer agents
when compared with other gold compounds that have been studied (Caruso, 2008;
Horvath et al., 2012). Moreover, many gold(I) phosphines have found applications in
synthetic organic chemistry (Hashmi and Asiri, 2016) and so many gold coordination
compounds have been described, containing different phosphine ligands, for example
amides (Horvath et al., 2012), pyridyl (Brandys et al., 2000) and dithiolate (Hunks et
al., 2006).
Theoretical research on Au(I) phosphine complexes have shown significant
development over the last decade (Hutchings et al., 2008; Pyykkö , 2008; Pyykkö,
2004; Schmidbaur and Schier, 2008), because some of the complexes show the
interesting behaviour of characteristic short range Au···Au distances (2.8–3.5 Å),

187
widely called ‘‘aurophilic’’ interactions. These interactions are in the range of a weak
bonds (Schmidbaur et al., 1988; Scherbaum et al., 1988; Schmidbaur, 1990;
Schmidbaur, 1995) and arises from relativistic effects that are maximized in gold
compared with other heavy metals (Pyykkö, 2004; Schmidbaur and Schier, 2008;
Sculfort and Braunstein, 2011; Pyykkö, 2012). The relevant energies have been found to
be in the same order as the energies of standard hydrogen bonds (ca. 24-50 kJmol-1
)
when Au(I)---Au(I) distances lie between 2.8-3.2 Å. The formation of Au···Au
interactions in the crystal structures of gold complexes is a topic of much interest to
chemists; in part because these compounds of gold display exciting luminescence
properties, in the solid-state (Larson et al., 1995; Forward et al., 1999; Tiekink, 2009;
Lima and Rodriguez, 2011)
Typically, in all cases the aurophilic interactions are identified by means of X-ray
diffraction studies when the Au(I)···Au(I) distances are less than twice the van der
Waals radius of gold. For example, in the gold complex possessing a para-methyl
substituent on the aromatic ring, ie [AuCl(4-MeC6H4)3P] the Au···Au distance is 3.375
Å (Bott et al., 2004). For complexes containing ligands that are p-methoxy substituted
hydrogen bonding dominates, and no aurophilic interactions are observed. However, by
replacing the MeO- group with Me2N- it is hoped that the lower number and delocalised
nature of the nitrogen lone pair might reduce the degree of hydrogen bonding, and so
favour more aurophilic interactions. But of all the possible Au(I) complexes of Cl, Br
and I with P(Me2NC6H4)3, or PhP(Me2NC6H4)2 and Ph2P(Me2NC6H4), ie 9 possibilities
only the three chloride complexes have had their structure determined (Schmidbaur et
al., 1992).
7.3 Experimental
7.3.1 Synthesis and crystallization
7.3.1.1 Synthesis of [AuCl{PPh2(4-C6H4NMe2)}] (1a), [AuCl{PPh(4-C6H4NMe2)2}]
(2a) and [AuCl{P(4-C6H4NMe2)3}] (3a).
The synthesis of compounds 1a, 2a and 3a were carried out by adjusting the previously
reported two-step method (Brisdon et al., 2010). Initially, AuCl(tht) was formed by
dissolved K[AuCl4] (0.2 g, 0.5 mmol) in EtOH (5 cm3) and water (2 cm
3).
Tetrahydrothiophene (0.1 cm3, 1.1 mmol) was added dropwise, and the mixture was
allowed to stir for 15 min, during which time the solution changed from yellow to white

188
in colour. The resulting solution was filtered and the white solid allowed to dry in
vacuo. AuCl(tht) (0.1 g, 0.3 mmol) was placed in a round-bottom flask containing
CH2Cl2 (5 cm3) and PPh2(4-C6H4NMe2) (0.1 g, 0.32 mmol), PPh(4-C6H4NMe2)2 (0.1 g,
0.31 mmol) or P(4-C6H4 NMe2)3 (0.1 g, 0.25 mmol). The resulting mixture was stirred
overnight at room temperature, after which time the volatiles were removed in vacuo.
The resulting gold compounds were isolated as white solid products. 1a: 31
P{1H} NMR
(CDCl3): 31.47 ppm (s); 2a: 31
P{1H} NMR (CDCl3): 29.17 ppm (s); 3a:
31P{
1H} NMR
(CDCl3): δ 27.54 ppm (s). Crystals suitable for X-ray diffraction of complexes 1a and
3a were grown by slow evaporation from a saturated CH2Cl2 solution. Unfortunately we
were unable to obtain good quality crystals of complex 2a.
7.3.1.2 Synthesis of [AuI{PPh2(4-C6H4NMe2)}] (1c)
Compound 1c was synthesized via halide exchange (McAuliffe et al., 1979), by
dissolving 1a (0.1 g, 0.19 mmol) in ethanol (10 mL) and adding KI (0.03 g, 0.18 mmol),
then the mixture was allowed to stir for 15 minutes at 80 ºC, after this time the solvent
was removed under vacuo. 31
P{1H} NMR (CDCl3): δ 37.52 ppm (s).
The gold complex was dissolved in CH2Cl2, from which crystals were grown by slow
evaporation.
7.3.1.3 Synthesis of [AuBr{PPh(4-C6H4NMe2)2}] (2b) and [AuBr{P(4-C6H4NMe2)3}]
(3b).
The compounds 2b and 3b were synthesized through halide exchange (McAuliffe et al.,
1979) by dissolving 2a (0.1 g, 0.17 mmol) or 3a (0.1 g, 0.16 mmol) in ethanol (10 mL)
and adding LiBr, and stirring the mixture for 15 minutes at 80 ºC, after that time the
solvent was removed under vacuo. The gold complexes were dissolved in toluene, from
which crystals were grown by slow evaporation. 2b: 31
P{1H} NMR (CDCl3): δ 31.64
ppm (s); 3b: 31
P{1H} NMR (CDCl3): δ 30.15 ppm (s).
7.4 Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1.
Table 1
Experimental details

189
Crystal data 1a 1c 2b 3a 3b
Chemical
formula C20H20AuClNP C20H20AuINP C22H25AuBrN2P C24H30AuClN3P C24H30AuBrN3P·C7H8
Mr 537.76 629.21 625.29 623.89 760.49
Crystal system,
space group Monoclinic, I2/a Triclinic, P-1 Triclinic, P-1 Monoclinic,P21/n Triclinic, P-1
Temperature (K) 150 150 150 293 150
a, b, c (Å)
20.9350(7),
8.9304(3),
20.7905(15)
8.9310(3),
10.4647(4),
12.9546(5)
10.6462(5),
10.9053(5),
11.2894(4)
14.0725(6),
11.3108(4),
15.4429(6)
7.3065(4), 12.7271(9),
16.7388(10)
(°) 102.364(4)
75.422(4),
72.086(4),
68.510(4)
101.733(4),
98.778(4),
114.311(5)
96.637(4)
90.462(5),
90.550(4),
102.143(5)
V (Å3) 3796.8(3) 1058.65(8) 1127.75(10) 2441.60(16) 1521.56(16)
Z 8 2 2 4 2
Radiation type Mo K Mo K Mo K Mo K Mo K
(mm-1) 7.98 8.48 8.37 6.22 6.22
Crystal size
(mm) 0.16 × 0.12 × 0.08 0.08 × 0.04 × 0.04 0.1 × 0.08 × 0.04 0.12 × 0.06 × 0.04 0.2 × 0.18 × 0.16
Data collection
Diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
SuperNova, Single
source at offset,
Eos
diffractometer
Xcalibur, Sapphire2,
large Be window
diffractometer
SuperNova, Single
source at offset, Eos
diffractometer
Absorption
correction
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan
(CrysAlis PRO;
Agilent, 2014)
Multi-scan (CrysAlis
PRO; Agilent, 2014)
Tmin, Tmax 0.844, 1.000 0.648, 1.000 0.737, 1.000 0.622, 1.000 0.765, 1.000
No. of measured,
independent and
observed [I >
2(I)] reflections
4267, 2511, 2127 7164, 4138, 3603 7638, 4420, 3767 8189, 4954, 4024 5016, 3619, 3269
Rint 0.038 0.036 0.048 0.037 0.027
(sin /)max (Å-1) 0.594 0.617 0.617 0.628 0.595
Refinement
R[F2 > 2(F2)],
wR(F2), S 0.032, 0.058, 0.98 0.046, 0.103, 1.06
0.045, 0.084,
1.02 0.039, 0.081, 1.07 0.037, 0.114, 0.58
No. of reflections 2511 4138 4420 4954 3619
No. of
parameters 219 219 248 277 335
No. of restraints - - - - 86
H-atom
treatment
H-atom parameters
constrained
H-atom parameters
constrained
H-atom parameters
constrained
H-atom parameters
constrained
H-atom parameters
constrained
max, min (e Å-3) 1.15, -1.11 3.06, -1.44 1.43, -1.49 2.48, -1.89 0.88, -0.59

190
7.5 Results and discussion
The formation of a series of gold(I) phosphine halide complexes, [AuCl{PPh2(4-
C6H4NMe2)}] 1a, [AuI{PPh2(4-C6H4NMe2)}] 1c, [AuBr{PPh(4-C6H4NMe2)2}] 2b,
[AuCl{P(4-C6H4NMe2)3}] 3a and [AuBr{P(4-C6H4NMe2)3}] 3b were confirmed by
31P{
1H} NMR spectroscopy, where a shift in the observed phosphorus resonance is
detected, and the structure of each complex was obtained by single crystal X-ray
diffraction. Selected bond lengths and angles for all complexes are listed in Table 2.
Table 2
Selected bond lengths (Å) and bond angles (o) of complexes.
Complex 1a 1c 2b 3a 3b
Au–P /Å 2.226 (2) 2.261(2) 2.236(2) 2.237(2) 2.246(1)
Au–X /Å 2.287(2) 2.555(1) 2.394(1) 2.293(2) 2.409(1)
P–Au–X /o 178.92 (8) 177.79(9) 175.90(6) 178.33(6) 175.347(17)
Au···Au /Å 4.2079(6) 5.9812(5) 6.5946(5) 6.8886(4) 7.3065(5)
The complex [AuCl{PPh2(4-C6H4NMe2)}] 1a (Fig. 1) crystallises in the monoclinic
space group I2/a and contains 8 molecules in the unit cell. The analogous iodide
complex [AuI{PPh2(4-C6H4NMe2)}] 1c (Fig. 2) crystallises in the triclinic space group
P-1, with two molecules in the unit cell. These analogues are not isomorphous and
display different intermolecular packing. A structure for complex 1a has been reported
previously (Schmidbaur et al., 1992). However, our complex crystallises in a different
space group with different unit cell dimensions {cell data for that previously reported
(Schmidbaur et al., 1992): a = 9.198(1), b = 9.406(1), c = 11.304(1) Å, α = 86.45(1), β =
80.96(1), γ = 84.12(1)°, Z = 2; space group Triclinic, P-1}.

191
Figure 1 A view of the molecular structure of 1a, showing the atom-labelling scheme and with
displacement ellipsoids drawn at the 50% probability level.
Figure 2 A view of the molecular structure of 1b, showing the atom-labelling scheme and with
displacement ellipsoids drawn at the 50% probability level.
The bond lengths between gold and phosphorus for 1a and 1b are significantly
different, indicating the influence of the different halide (2.226(2) and 2.261(2) Å for 1a
and 1b, respectively). These values are also comparable to those found in a previous

192
determination of 1a d(Au‒P) = 2.232(1) Å (Schmidbaur et al., 1992) and [AuCl(PPh3)]
d(Au‒P) = 2.235(3) Å (Baenziger et al., 1976). In the chloro derivative 1a, the Au–Cl
bond length is 2.287(2) Å similar to those found in the previous reported complexes
[d(Au‒Cl)= 2.282(1) Å; Schmidbaur et al., 1992] and [AuCl(PPh3)] 2.279(3) Å
(Baenziger et al., 1976).
The Au–I bond length in the iodo analog 1b, 2.555(1) Å is similar to that found in
reported complex [AuI(PPh3)] d(Au‒I) = 2.553(1) Å (Ahrland et al., 1987).
The shortest Au···Au distance in complexes 1a and 1b are 4.2079(6) and 5.9812(5) Å,
respectively, which are longer than twice the van der Waals radius of Au, 3.60 Å, thus
suggesting that no significant Au···Au interactions occur in either complex. However,
the Au···Au distances are shorter than those found in the 4-MeOC6H4 derivatives.
The distance in 1a are somewhat different with that found in the previous determination
(Schmidbaur et al., 1992) d(Au···Au) = 5.3953(6) Å.
In the crystal structure of 1a, molecules are connected via weak intermolecular
hydrogen bonding between the hydrogen of the phenyl group and the chloride
(Cl(1)···H(10): 2.863 Å, Cl(1)···H(11): 2.893 Å and Cl(1)···H(17): 2.788 Å) (Fig. 3
and Table 3), all these distances are less than the sum of van der Waals radii of
hydrogen and chlorine of 2.95 Å. Whereas, the previous reported complex (Schmidbaur
et al., 1992) identifies weak intermolecular hydrogen bonding between the hydrogen of
the methyl group and the chloride.
By contrast, in the crystal packing of 1b there are no intermolecular interactions
between hydrogen and iodide.
Table 3
Hydrogen-bond geometry (Å , °) for (1a).
D—H···A D—H H···A D···A D—H···A
C10—H10···Cl1i 0.929 2.863 3.745(7) 159.0
C11—H11···Cl1ii 0.931 2.893 3.795(7) 163.6
C17—H17···Cl1iii
0.928 2.788 3.693(9) 165.1
Symmetry codes: (i) -x,-y,-z; (ii) x,1+y,z; (iii) 1/2-x,y,-z.

193
Figure 3 A view of the packing diagram of 1a showing the C—H···Cl interactions, viewed
along the b-axis.
The crystal structure of complex 2b (Fig. 4) crystallises in the triclinic system; space
group P-1 with two molecules in the unit cell. This complex is isomophous with the
analogous chloro complex reported previously (Schmidbaur et al., 1992) {comparative
cell data: a = 10.513(1), b = 11.130(1), c = 11.273(1) Å, α = 102.12(1)º, β = 98.76(1)º, γ
= 115.07(1)º, Z = 2; space group, P-1}.
Figure 4 A view of the molecular structure of 2b, showing the atom-labelling scheme and with
displacement ellipsoids drawn at the 50% probability level.

194
The Au‒P bond length in 2b (2.236(2) Å) is similar to that found in the complex
[AuBr(PPh3)] (Barron et al., 1987) d(Au‒P) = 2.252(6) Å, Whereas it is significantly
different to that reported in the isostructural chloro complex (Schmidbaur et al., 1992)
d(Au‒P) = 2.299(1) Å . The Au‒Br distance (2.4084(8) Å) is also similar to that found
in [AuBr(PPh3)] (Barron et al., 1987) d(Au‒P) = 2.406(3) Å. The shortest Au···Au
distance at 6.5946(5) Å is considerably larger than twice the van der Waals radius of
gold (3.60 Å), so no Au···Au interaction is found for this complex.
The crystal packing of compound 2b show that molecules are connected to each other
through intermolecular C–H···Br interactions, which are less than sum of the van der
Waals radii of hydrogen and bromine at 3.05 Å (Fig. 5 and Table 4).
Table 4
Hydrogen-bond geometry (Å , °) for (2b).
D—H···A D—H H···A D---A D—H···A
C13—H13···Bri 0.930 2.8974 3.723(8) 148.7
C21—H21···Brii 0.930 2.906 3.63(1) 135.9
Symmetry codes: (i) -x,-y,1-z; (ii) 1+x,y,z.
Figure 5 A view of the packing of adjacent chains in 2b, showing C–H···Br contacts viewed
down b-axis.

195
Complex 3a (Fig. 6) crystallises in the monoclinic space group P21/n with 4 molecules
in the unit cell. Whereas the bromo analog 3b (Fig. 7) crystallises as a toluene solvate in
the triclinic space group P-1 with two molecules in the unit cell. The structure of 3a has
been reported previously (Schmidbaur et al., 1992) which exhibited a similar space
group but different unit cell {comparative cell data: a = 9.940(1), b = 20.895(2), c =
12.237(2) Å, β= 106.33 (1)°, Z = 4; space group, P21/n,T = 223}. The different unit
cells may arise from the fact that in this work toluene was the solvent used, while the
previous structure was crystallised from THF. The structure of 3b was solved with the
presence of disordered toluene molecules.
Figure 6 A view of the molecular structure of 3a, showing the atom-labelling scheme and with
displacement ellipsoids drawn at the 50% probability level.

196
Figure 7 A view of the molecular structure of 3b, showing the atom-labelling scheme and with
displacement ellipsoids drawn at the 50% probability level, disorder solvent molecules are
omitted for clarity.
By comparing both the Au–P and Au–Cl distances of 3a, with the previously reported
complex [AuCl{P(4-C6H4NMe2)3}] (Schmidbaur et al., 1992) d(Au‒P) = 2.241(1) and
d(Au‒Cl) = 2.299 (1) Å, its shows that there is no significant difference in the distances.
The Au–P and Au–Br bond length of 3b are similar to that found in the reported
complex [AuBr(PPh3)] (Barron et al., 1987) d(Au‒P) = 2.252(6) and d(Au‒Br) =
2.406(3) Å.
The shortest of Au···Au distances in 3a and 3b are 6.8886(4) and 7.3065(5) Å,
respectively; these values are similar to that found in the previous reported complex
d(Au···Au) = 7.276(1) Å [ Schmidbaur et al., 1992], and longer than twice the Van der
Walls radii of gold. This suggest that no Au···Au interaction is observed in these
complexes.
In the extended structure of 3a, molecules are linked through C–H···Cl hydrogen bonds
to give pairs that run parallel to the b axis (Fig. 8 and Table 5). Whereas, the previous
reported complex (Schmidbaur et al., 1992) detects weak intermolecular hydrogen
bonding between the hydrogen and nitrogen (d(N3···H5) = 2.573 Å), and
intermolecular hydrogen bonding between the hydrogen and chloride (d(Cl1···H10) =
2.944 Å and d(Cl1···H21) = 2.854 Å) as shown in Fig. 9.

197
In 3b, molecules are connected via one C–H···Br hydrogen bond that is parallel to the
b axis (Fig. 10).
Table 5
Hydrogen-bond geometry (Å , °) for (3a and 3b).
Symmetry codes: (i)-x,-y,-z+1; (ii) 1-x,1-y,1-z.
Figure 8 A view of the packing of adjacent chains in 3a, showing C–H···Cl contacts viewed
down b-axis.
D—H···A D—H H···A D---A D—H···A
C21—H21···Cl1i 0.93 2.92 3.736(6) 147
C24A—H24A···Cl1i 0.96 2.94 3.841(7) 156
C23—H23C···Br1iii
0.96 2.9989 3.796(7) 141.3

198
Figure 9 A view of the packing of adjacent chains in [AuCl{P(4-C6H4NMe2)3}] (Schmidbaur et
al., 1992), showing C–H···Cl and C–H···N contacts.
Figure 10 A view of the packing of adjacent chains in 3b, showing C–H···Br contacts viewed
down b-axis, the disorder solvent molecules are omitted for clarity.
7.6 Summary
In summary, we have obtained and described the crystal structure of five gold(I)
phosphine halide complexes for the PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and P(4-

199
C6H4NMe2)3 ligand systems. As expected, the Au–X bond distance increases in the
order X = Cl, Br, I. For the Au–P distances a much smaller difference is observed on
changing the halide. In these systems a significant number of secondary C–H···X
interactions are observed in all of the complexes. However, unlike for the analogous –
OMe substituted systems, the –NMe2 analogues do not display hydrogen bonds
involving the nitrogen atom. However, no short Au···Au interactions are observed.

200
References
Abdou, H. E., Mohamed, A. A., Fackler Jr, J. P., Burini, A., Galassi, R., López-de
Luzuriaga, J. M. & Olmos, M. E. (2009). Coord. Chem. Rev. 253, 1661-1669.
Ahrland, S., Dreisch, K., Noren, B. & Oskarsson, A. (1987). Acta Chem. Scand.
A41, 173-177.
Asiria, A. M. & Hashmi, A. S.K. (2016). Chem. Soc. Rev. 45, 4471-4503.
Baenziger, N. C., Bennett, W. E. & Soboroff, D. M. (1976). Acta Cryst. B32, 962-
963.
Barnes, N. A., Brisdon, A. K., Brown, F. R. W., Cross, W. I, Crossley, I. R., Fish,
C. Herbert, C. J., Pritchard, R. G. & Warren, J. E. (2011). Dalton Trans. 40, 1743-
1750.
Bondi, A. (1964). J. Phys. Chem. 68, 441-451.
Barron, P. F., Engelhardt, L. H., Healy, P. C., Oddy, J. & White, A.H. (1987). Aust.
J. Chem. 40, 1545-1555.
Bott, R. C., Healy, P. C. & Smith, G. (2004). Aust. J. Chem. 57, 213.
Brandys, M. C., Jennings, M. C. & Puddephatt, R. J. (2000). J. Chem. Soc., Dalton
Trans. 4601-4606.
Carus, F., Pettinari, C., Paduano, F., Villa, R., Marchetti, F., Monti, E. & Rossi, M.
(2008). J. Med. Chem. 51 1584-1591.
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H.
(2009). J. Appl. Cryst. 42, 339-341.
Dziwok, K., Lachmann, J.,Wilkinson, D. L., Müller, G. & Schmidbaur, H. (1990).
Chem. Ber. 123, 423-431.
Forward, J. M., Fackler, J. P. Jr., Assefa, Z., in: D.M. Roundhill, J.P. Fackler Jr.
(Eds.), Plenum Press, New York,1999, pp. 195-229, Chapter 6.
Hashmi, A. S. K. (2007). Chem. Rev. 107, 180-3211.
Horvath, U. E. I., Dobrzańska, L., Strasser, C. E., Bouwer, W., Joone, G., van
Rensburg, C. E. J., Cronje, S. & Raubenheimer, H. G. (2012). Inorg. Biochem. 111,

201
80-90.
Hunks, W. J., Jennings, M. C. & Puddephatt, R. J. (2006). Inorg. Chim. Acta. 359,
3605-3616.
Hutchings, G. J., Brust, M. & Schmidbaur, H. (2008). Chem. Soc. Rev. 37, 1759-
1765.
Larson, L. J., McCauley, E. M., Weissbart, B. & Tinti, D. S. (1995). J. Phys. Chem.
99, 7218-7226.
Lima, J. C. & Rodriguez, L. (2011). Chem. Soc. Rev. 40, 5442-5456.
McAuliffe, C. A., Parish, R. V. & Randall, D. (1979). J.C.S.Dalton. 1730-1735.
Mirabelli, C. K., Hill, D. T., Faucette, L. F., McCabe, F. L., Girard, G. R., Bryan, D.
B., Sutton, B. M., Leary Barus, J. O., Crooke, S. T. & Johnson, R. K. (1987). J.
Med. Chem. 30, 2181-2190.
Ott, I. (2009). Coord. Chem. Rev. 253, 1670-1681.
Pyykkö, P. (2004). Angew. Chem., Int. Ed. 43, 4412-4456.
Pyykkö, P. (2008). Chem. Soc. Rev. 37, 1967-1997.
Pyykkö, P., Johnson, M. A. & Martinez, T. J. (2012). R ev. Phys. Chem. 63, 45-64.
Schmidbaur, H., Brachthäuser, B., Gampe, Sr., Schier, A. & Steigeimann, O.
(1992). Z. Naturforsch. 47b, 1725-1735.
Schmidbaur, H. (1995). Chem. Soc. Rev. 24, 391-400.
Schmidbaur, H., Graf, W. & Müller, G. (1988). Angew. Chem., Int. Ed. Engl. 27,
417-419.
Schmidbaur, H. & Schier, A. (2008). Chem. Soc. Rev. 37, 1931-1951.
Sculfort, S. & Braunstein, P. (2011). Chem. Soc. Rev. 40, 2741-27-60.
Sheldrick, G. M. (2008). Acta Cryst. A64, 112-122.
Sheldrick, G. M. (2015). Acta Cryst. C71, 3-8.
Tiekink, E. R. T. & Kang, J. -G. (2009). Coord. Chem. Rev. 253, 1627-1648.

202
Supporting information
The single crystal structures of some gold(I) halide complexes containing the
PPh2(4-C6H4NMe2), PPh(4-C6H4NMe2)2 and PPh(4-C6H4NMe2)3 ligands.
Arij T. Addaraidi, Alan K. Brisdon, Robin G. Pritchard
Computing details
Data collection: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33 (release 27-03-2014
CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); cell refinement: CrysAlis PRO, Agilent
Technologies, Version 1.171.37.33 (release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27
2014,17:12:48); data reduction: CrysAlis PRO, Agilent Technologies, Version 1.171.37.33
(release 27-03-2014 CrysAlis171 .NET) (compiled Mar 27 2014,17:12:48); program(s) used to
solve structure: SHELXS (Sheldrick, 2008); program(s) used to refine structure: SHELXL
(Sheldrick, 2015); molecular graphics: Olex2 (Dolomanov et al., 2009); software used to
prepare material for publication: Olex2 (Dolomanov et al., 2009).
[AuCl{PPh2(4-C6H4NMe2)}] 1a
Crystal data,
C20H20AuClNP F(000) = 2064
Mr = 537.76 Dx = 1.882 Mg m-3
Monoclinic, I2/a Mo K radiation, = 0.71073 Å
a = 20.9350 (7) Å Cell parameters from 2231 reflections
b = 8.9304 (3) Å = 3.8–27.9°
c = 20.7905 (15) Å = 7.98 mm-1
= 102.364 (4)° T = 150 K
V = 3796.8 (3) Å3 clear colourless
Z = 8 0.16 × 0.12 × 0.08 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
2511 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source 2127 reflections with I > 2(I)
Mirror monochromator Rint = 0.038
Detector resolution: 8.0714 pixels mm-1
max = 25.0°, min = 3.1°

203
scans h = -1224
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1010
Tmin = 0.844, Tmax = 1.000 l = -2318
4267 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.032 H-atom parameters constrained
wR(F2) = 0.058 w = 1/[
2(Fo
2)]
where P = (Fo2 + 2Fc
2)/3
S = 0.98 (/)max < 0.001
2511 reflections max = 1.15 e Å-3
219 parameters min = -1.11 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.09850 (2) 0.05614 (3) 0.03776 (2) 0.02073 (11)
P1 0.15951 (8) -0.0762 (2) 0.11892 (12) 0.0200 (6)
Cl1 0.03529 (8) 0.19511 (19) -0.04419 (11) 0.0261 (5)
C1 0.1193 (3) -0.1098 (7) 0.1856 (4) 0.018 (2)
C19 0.3143 (3) 0.1204 (8) 0.2456 (5) 0.037 (3)
H19 0.3267 0.1402 0.2904 0.044*
C5 0.1009 (3) -0.2642 (8) 0.2747 (4) 0.020 (2)
H5 0.1090 -0.3533 0.2981 0.025*
C15 0.2355 (3) 0.0166 (7) 0.1529 (5) 0.020 (2)

204
C20 0.2555 (3) 0.0464 (7) 0.2199 (5) 0.029 (2)
H20 0.2291 0.0165 0.2483 0.035*
C12 0.2065 (4) -0.5539 (9) 0.0640 (5) 0.041 (3)
H12 0.2150 -0.6528 0.0546 0.049*
C2 0.0765 (3) -0.0043 (7) 0.2028 (5) 0.021 (2)
H2 0.0676 0.0832 0.1783 0.025*
C18 0.3539 (3) 0.1637 (8) 0.2032 (6) 0.034 (3)
H18 0.3932 0.2123 0.2198 0.041*
C6 0.1302 (3) -0.2392 (7) 0.2233 (4) 0.023 (2)
H6 0.1586 -0.3113 0.2131 0.028*
C10 0.1330 (3) -0.3511 (8) 0.0568 (4) 0.024 (2)
H10 0.0915 -0.3116 0.0414 0.029*
C4 0.0585 (3) -0.1581 (8) 0.2930 (5) 0.022 (2)
C9 0.1812 (3) -0.2618 (7) 0.0938 (4) 0.021 (2)
C14 0.2440 (3) -0.3217 (8) 0.1160 (5) 0.028 (2)
H14 0.2772 -0.2645 0.1415 0.034*
C11 0.1453 (3) -0.4956 (8) 0.0425 (5) 0.031 (2)
H11 0.1121 -0.5545 0.0181 0.037*
N1 0.0288 (3) -0.1858 (6) 0.3445 (4) 0.0272 (19)
C3 0.0472 (3) -0.0273 (8) 0.2552 (5) 0.026 (2)
H3 0.0194 0.0455 0.2657 0.031*
C16 0.2765 (3) 0.0638 (8) 0.1122 (5) 0.030 (2)
H16 0.2642 0.0470 0.0671 0.036*
C17 0.3353 (3) 0.1354 (8) 0.1375 (5) 0.035 (3)
H17 0.3622 0.1641 0.1095 0.042*
C8 0.0560 (3) -0.3009 (9) 0.3927 (5) 0.035 (3)
H8A 0.0529 -0.3968 0.3715 0.053*
H8B 0.0320 -0.3027 0.4272 0.053*
H8C 0.1011 -0.2786 0.4111 0.053*
C13 0.2555 (4) -0.4663 (8) 0.0994 (5) 0.036 (3)
H13 0.2973 -0.5058 0.1125 0.043*
C7 -0.0067 (4) -0.0643 (9) 0.3693 (5) 0.049 (3)
H7A 0.0233 0.0144 0.3866 0.074*
H7B -0.0267 -0.1020 0.4035 0.074*
H7C -0.0399 -0.0258 0.3340 0.074*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.01995 (15) 0.02205 (17) 0.0208 (3) 0.00195 (11) 0.00583 (12) 0.00213 (16)
P1 0.0189 (9) 0.0212 (10) 0.0206 (19) 0.0021 (7) 0.0056 (8) 0.0014 (11)

205
Cl1 0.0250 (9) 0.0299 (10) 0.0230 (17) 0.0055 (7) 0.0041 (8) 0.0063 (10)
C1 0.017 (4) 0.019 (4) 0.018 (7) -0.001 (3) 0.004 (3) -0.009 (4)
C19 0.025 (4) 0.024 (4) 0.059 (10) 0.007 (3) 0.004 (4) -0.005 (5)
C5 0.020 (4) 0.019 (4) 0.025 (8) 0.000 (3) 0.008 (3) 0.004 (4)
C15 0.016 (4) 0.018 (4) 0.025 (8) 0.004 (3) 0.004 (3) 0.001 (4)
C20 0.020 (4) 0.019 (4) 0.048 (9) 0.005 (3) 0.006 (4) 0.002 (5)
C12 0.047 (5) 0.030 (5) 0.052 (10) 0.011 (4) 0.025 (5) -0.009 (5)
C2 0.019 (4) 0.019 (4) 0.024 (8) 0.003 (3) 0.004 (3) 0.004 (4)
C18 0.022 (4) 0.021 (4) 0.061 (10) -0.008 (3) 0.010 (4) -0.019 (5)
C6 0.020 (4) 0.020 (4) 0.030 (8) 0.004 (3) 0.010 (3) -0.003 (4)
C10 0.026 (4) 0.029 (4) 0.020 (7) 0.000 (3) 0.012 (3) -0.002 (4)
C4 0.014 (3) 0.022 (4) 0.031 (8) -0.005 (3) 0.009 (3) 0.002 (4)
C9 0.020 (4) 0.020 (4) 0.023 (7) 0.000 (3) 0.005 (3) 0.003 (4)
C14 0.024 (4) 0.029 (4) 0.031 (8) 0.000 (3) 0.006 (4) -0.006 (4)
C11 0.035 (4) 0.032 (5) 0.028 (8) -0.007 (3) 0.013 (4) -0.016 (5)
N1 0.038 (4) 0.022 (4) 0.026 (7) 0.002 (3) 0.016 (3) 0.003 (4)
C3 0.023 (4) 0.021 (4) 0.037 (8) 0.012 (3) 0.015 (4) 0.002 (4)
C16 0.034 (4) 0.032 (5) 0.026 (8) 0.001 (3) 0.008 (4) 0.011 (5)
C17 0.027 (4) 0.028 (5) 0.057 (10) -0.002 (3) 0.021 (4) 0.002 (5)
C8 0.033 (4) 0.049 (5) 0.028 (8) -0.001 (4) 0.014 (4) 0.010 (5)
C13 0.037 (5) 0.035 (5) 0.038 (9) 0.013 (4) 0.013 (4) -0.002 (5)
C7 0.057 (6) 0.041 (5) 0.062 (11) 0.010 (4) 0.041 (5) -0.003 (6)
Geometric parameters (Å, º)
Au1—P1 2.226 (2) C12—C11 1.367 (9)
Au1—Cl1 2.2866 (19) C12—C13 1.372 (11)
P1—C1 1.795 (8) C2—C3 1.375 (11)
P1—C15 1.800 (7) C18—C17 1.362 (12)
P1—C9 1.824 (7) C10—C9 1.384 (9)
C1—C2 1.398 (9) C10—C11 1.361 (9)
C1—C6 1.388 (9) C4—N1 1.373 (11)
C19—C20 1.399 (9) C4—C3 1.399 (10)
C19—C18 1.386 (13) C9—C14 1.402 (8)
C5—C6 1.359 (11) C14—C13 1.371 (9)
C5—C4 1.405 (9) N1—C8 1.463 (10)
C15—C20 1.390 (12) N1—C7 1.469 (9)
C15—C16 1.393 (11) C16—C17 1.387 (10)
P1—Au1—Cl1 178.92 (8) C17—C18—C19 120.3 (7)
C1—P1—Au1 112.7 (2) C5—C6—C1 122.2 (7)

206
C1—P1—C15 107.1 (4) C11—C10—C9 121.2 (7)
C1—P1—C9 104.9 (3) N1—C4—C5 120.6 (7)
C15—P1—Au1 111.7 (3) N1—C4—C3 122.6 (6)
C15—P1—C9 106.1 (3) C3—C4—C5 116.8 (7)
C9—P1—Au1 113.8 (3) C10—C9—P1 119.1 (5)
C2—C1—P1 121.1 (6) C10—C9—C14 119.0 (7)
C6—C1—P1 121.9 (5) C14—C9—P1 121.7 (5)
C6—C1—C2 117.0 (7) C13—C14—C9 118.6 (7)
C18—C19—C20 119.1 (10) C10—C11—C12 119.8 (7)
C6—C5—C4 121.3 (7) C4—N1—C8 119.1 (6)
C20—C15—P1 122.2 (6) C4—N1—C7 119.2 (7)
C20—C15—C16 117.3 (7) C8—N1—C7 115.6 (8)
C16—C15—P1 120.5 (8) C2—C3—C4 121.3 (6)
C15—C20—C19 121.6 (8) C17—C16—C15 121.4 (9)
C11—C12—C13 119.9 (7) C18—C17—C16 120.4 (8)
C3—C2—C1 121.4 (7) C14—C13—C12 121.4 (7)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].
[AuI{PPh2(4-C6H4NMe2)}] 1b
Crystal data
C20H20AuINP Z = 2
Mr = 629.21 F(000) = 588
Triclinic, P¯1 Dx = 1.974 Mg m-3
a = 8.9310 (3) Å Mo K radiation, = 0.71073 Å
b = 10.4647 (4) Å Cell parameters from 2812 reflections
c = 12.9546 (5) Å = 3.7–27.5°
= 75.422 (4)° = 8.48 mm-1
= 72.086 (4)° T = 150 K
= 68.510 (4)° colourless
V = 1058.65 (8) Å3 0.08 × 0.04 × 0.04 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
4138 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source, Incoatec Is
3603 reflections with I > 2(I)
Mirror monochromator Rint = 0.036
Detector resolution: 8.0714 pixels mm-1
max = 26.0°, min = 3.4°
scans h = -118

207
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1212
Tmin = 0.648, Tmax = 1.000 l = -1515
7164 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.046 H-atom parameters constrained
wR(F2) = 0.103 w = 1/[
2(Fo
2) + (0.037P)
2 + 3.6937P]
where P = (Fo2 + 2Fc
2)/3
S = 1.06 (/)max < 0.001
4138 reflections max = 3.06 e Å-3
219 parameters min = -1.44 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.14277 (4) 0.13011 (3) 0.27535 (3) 0.02082 (12)
I1 0.16822 (8) 0.36718 (6) 0.27007 (6) 0.0397 (2)
P1 0.1251 (3) -0.0829 (2) 0.28547 (17) 0.0164 (4)
C1 0.1975 (9) -0.2020 (8) 0.4004 (6) 0.0178 (17)
C9 0.2424 (10) -0.1645 (8) 0.1656 (7) 0.0218 (18)
C15 -0.0859 (10) -0.0761 (8) 0.3011 (6) 0.0176 (17)
C5 0.2668 (9) -0.2420 (8) 0.5735 (7) 0.0185 (17)
H5 0.2821 -0.2058 0.6267 0.022*
C6 0.2189 (10) -0.1531 (9) 0.4813 (6) 0.0209 (18)
H6 0.2012 -0.0584 0.4747 0.025*

208
C3 0.2676 (10) -0.4345 (8) 0.5038 (7) 0.0210 (18)
H3 0.2817 -0.5286 0.5108 0.025*
C2 0.2233 (10) -0.3458 (8) 0.4126 (6) 0.0200 (18)
H2 0.2102 -0.3809 0.3579 0.024*
C16 -0.1728 (10) -0.1366 (8) 0.3979 (7) 0.0222 (18)
H16 -0.1176 -0.1901 0.4523 0.027*
C4 0.2922 (9) -0.3848 (8) 0.5876 (7) 0.0198 (18)
C20 -0.1707 (12) 0.0057 (9) 0.2205 (8) 0.029 (2)
H20 -0.1160 0.0498 0.1563 0.034*
C19 -0.3348 (12) 0.0206 (10) 0.2367 (9) 0.039 (3)
H19 -0.3898 0.0720 0.1818 0.046*
C17 -0.3399 (11) -0.1189 (9) 0.4150 (7) 0.026 (2)
H17 -0.3965 -0.1596 0.4802 0.031*
C18 -0.4207 (12) -0.0402 (10) 0.3342 (9) 0.034 (2)
H18 -0.5326 -0.0275 0.3446 0.041*
C14 0.3935 (11) -0.1443 (9) 0.1091 (7) 0.026 (2)
H14 0.4292 -0.0839 0.1304 0.031*
C10 0.1948 (12) -0.2561 (9) 0.1319 (7) 0.028 (2)
H10 0.0946 -0.2718 0.1694 0.034*
C11 0.2917 (13) -0.3247 (9) 0.0443 (7) 0.036 (2)
H11 0.2568 -0.3848 0.0221 0.043*
C12 0.4406 (13) -0.3025 (9) -0.0096 (7) 0.037 (3)
H12 0.5074 -0.3493 -0.0682 0.045*
C13 0.4925 (12) -0.2137 (10) 0.0207 (8) 0.039 (3)
H13 0.5933 -0.1993 -0.0172 0.046*
N1 0.3331 (9) -0.4709 (7) 0.6791 (6) 0.0276 (17)
C8 0.3426 (12) -0.6166 (9) 0.6963 (8) 0.037 (2)
H8A 0.2369 -0.6225 0.6984 0.055*
H8B 0.3722 -0.6627 0.7647 0.055*
H8C 0.4250 -0.6606 0.6373 0.055*
C7 0.3560 (15) -0.4196 (11) 0.7654 (8) 0.044 (3)
H7A 0.4392 -0.3739 0.7350 0.066*
H7B 0.3906 -0.4961 0.8210 0.066*
H7C 0.2534 -0.3549 0.7972 0.066*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.02157 (19) 0.01461 (17) 0.02488 (19) -0.00630
(13)
-0.00182
(12)
-0.00441
(13)
I1 0.0413 (4) 0.0217 (3) 0.0517 (4) -0.0184 (3) 0.0117 (3) -0.0136 (3)

209
P1 0.0180 (11) 0.0143 (10) 0.0163 (11) -0.0049 (9) -0.0023 (8) -0.0038 (9)
C1 0.016 (4) 0.023 (4) 0.015 (4) -0.008 (3) 0.001 (3) -0.007 (3)
C9 0.021 (4) 0.018 (4) 0.017 (4) -0.001 (3) 0.001 (3) -0.003 (4)
C15 0.019 (4) 0.017 (4) 0.017 (4) -0.005 (3) -0.003 (3) -0.006 (3)
C5 0.018 (4) 0.021 (4) 0.021 (4) -0.010 (4) -0.002 (3) -0.008 (4)
C6 0.024 (5) 0.020 (4) 0.017 (4) -0.009 (4) 0.003 (3) -0.006 (4)
C3 0.026 (5) 0.010 (4) 0.021 (4) -0.001 (3) -0.002 (3) -0.004 (3)
C2 0.024 (5) 0.024 (4) 0.018 (4) -0.007 (4) -0.005 (3) -0.015 (4)
C16 0.024 (5) 0.016 (4) 0.027 (5) -0.006 (4) -0.006 (4) -0.006 (4)
C4 0.011 (4) 0.021 (4) 0.025 (5) 0.000 (3) -0.003 (3) -0.009 (4)
C20 0.037 (5) 0.022 (5) 0.032 (5) -0.015 (4) -0.015 (4) 0.004 (4)
C19 0.039 (6) 0.035 (6) 0.053 (7) -0.005 (5) -0.031 (5) -0.011 (5)
C17 0.023 (5) 0.023 (5) 0.033 (5) -0.010 (4) -0.001 (4) -0.011 (4)
C18 0.023 (5) 0.029 (5) 0.059 (7) -0.007 (4) -0.019 (5) -0.009 (5)
C14 0.030 (5) 0.024 (5) 0.022 (5) -0.009 (4) -0.003 (4) -0.002 (4)
C10 0.039 (6) 0.021 (5) 0.024 (5) -0.011 (4) -0.002 (4) -0.007 (4)
C11 0.061 (7) 0.016 (5) 0.025 (5) -0.008 (5) -0.002 (5) -0.009 (4)
C12 0.062 (7) 0.019 (5) 0.010 (4) 0.000 (5) 0.006 (4) -0.004 (4)
C13 0.038 (6) 0.030 (5) 0.025 (5) -0.003 (5) 0.008 (4) 0.004 (4)
N1 0.035 (4) 0.021 (4) 0.021 (4) -0.001 (3) -0.009 (3) -0.003 (3)
C8 0.045 (6) 0.024 (5) 0.040 (6) -0.011 (5) -0.016 (5) 0.004 (5)
C7 0.076 (8) 0.034 (6) 0.029 (6) -0.022 (6) -0.022 (5) 0.002 (5)
Geometric parameters (Å, º)
Au1—I1 2.5553 (6) C20—C19 1.370 (13)
Au1—P1 2.261 (2) C19—H19 0.9300
P1—C1 1.805 (8) C19—C18 1.395 (14)
P1—C9 1.802 (8) C17—H17 0.9300
P1—C15 1.809 (8) C17—C18 1.374 (13)
C1—C6 1.364 (11) C18—H18 0.9300
C1—C2 1.410 (11) C14—H14 0.9300
C9—C14 1.387 (11) C14—C13 1.395 (13)
C9—C10 1.384 (12) C10—H10 0.9300
C15—C16 1.392 (11) C10—C11 1.380 (12)
C15—C20 1.400 (11) C11—H11 0.9300
C5—H5 0.9300 C11—C12 1.372 (14)
C5—C6 1.393 (11) C12—H12 0.9300
C5—C4 1.399 (11) C12—C13 1.358 (14)
C6—H6 0.9300 C13—H13 0.9300
C3—H3 0.9300 N1—C8 1.458 (11)

210
C3—C2 1.371 (11) N1—C7 1.450 (12)
C3—C4 1.418 (11) C8—H8A 0.9600
C2—H2 0.9300 C8—H8B 0.9600
C16—H16 0.9300 C8—H8C 0.9600
C16—C17 1.388 (11) C7—H7A 0.9600
C4—N1 1.355 (10) C7—H7B 0.9600
C20—H20 0.9300 C7—H7C 0.9600
P1—Au1—I1 177.79 (6) C20—C19—C18 121.0 (9)
C1—P1—Au1 111.5 (3) C18—C19—H19 119.5
C1—P1—C15 107.6 (4) C16—C17—H17 120.5
C9—P1—Au1 114.5 (3) C18—C17—C16 119.1 (9)
C9—P1—C1 106.3 (4) C18—C17—H17 120.5
C9—P1—C15 104.9 (4) C19—C18—H18 120.0
C15—P1—Au1 111.6 (3) C17—C18—C19 120.1 (9)
C6—C1—P1 120.2 (6) C17—C18—H18 120.0
C6—C1—C2 118.4 (7) C9—C14—H14 119.7
C2—C1—P1 121.3 (6) C9—C14—C13 120.6 (9)
C14—C9—P1 119.3 (7) C13—C14—H14 119.7
C10—C9—P1 122.7 (6) C9—C10—H10 119.1
C10—C9—C14 117.8 (8) C11—C10—C9 121.9 (9)
C16—C15—P1 121.2 (6) C11—C10—H10 119.1
C16—C15—C20 118.8 (8) C10—C11—H11 120.6
C20—C15—P1 119.6 (6) C12—C11—C10 118.8 (9)
C6—C5—H5 119.4 C12—C11—H11 120.6
C6—C5—C4 121.2 (8) C11—C12—H12 119.3
C4—C5—H5 119.4 C13—C12—C11 121.4 (9)
C1—C6—C5 121.4 (8) C13—C12—H12 119.3
C1—C6—H6 119.3 C14—C13—H13 120.2
C5—C6—H6 119.3 C12—C13—C14 119.6 (9)
C2—C3—H3 119.5 C12—C13—H13 120.2
C2—C3—C4 121.0 (7) C4—N1—C8 120.0 (8)
C4—C3—H3 119.5 C4—N1—C7 121.5 (8)
C1—C2—H2 119.5 C7—N1—C8 118.4 (7)
C3—C2—C1 121.0 (7) N1—C8—H8A 109.5
C3—C2—H2 119.5 N1—C8—H8B 109.5
C15—C16—H16 119.3 N1—C8—H8C 109.5
C17—C16—C15 121.4 (8) H8A—C8—H8B 109.5
C17—C16—H16 119.3 H8A—C8—H8C 109.5
C5—C4—C3 117.0 (7) H8B—C8—H8C 109.5
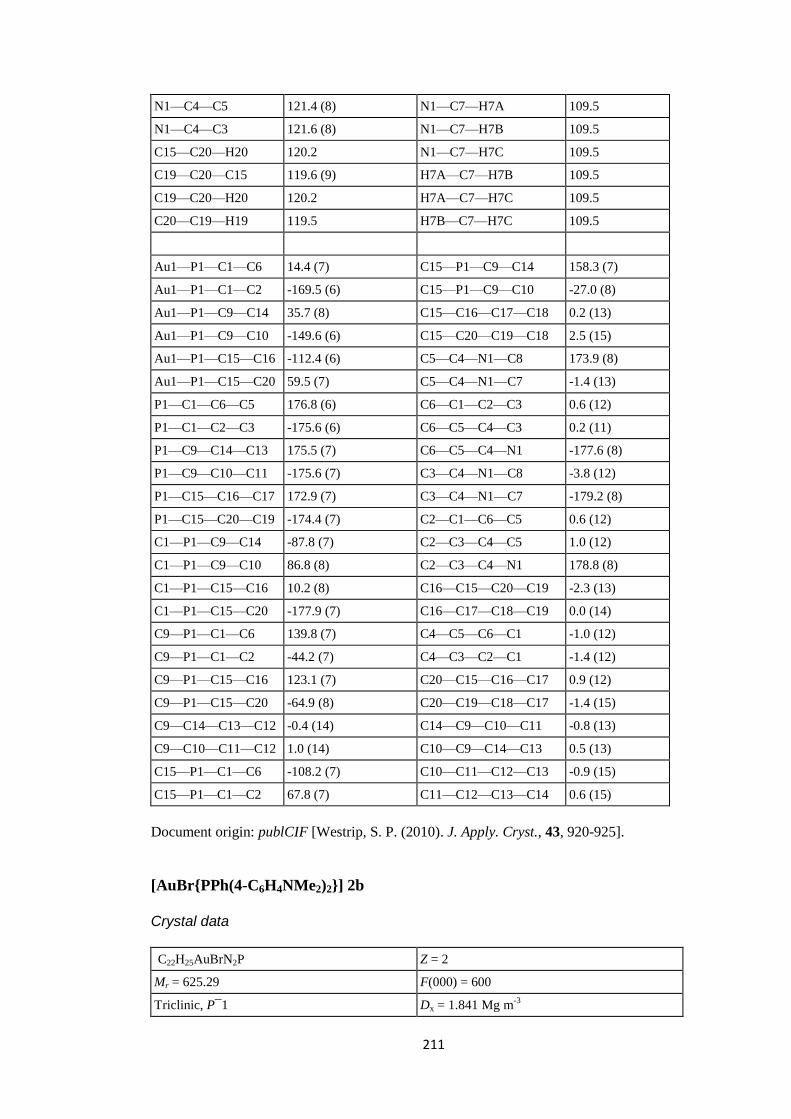
211
N1—C4—C5 121.4 (8) N1—C7—H7A 109.5
N1—C4—C3 121.6 (8) N1—C7—H7B 109.5
C15—C20—H20 120.2 N1—C7—H7C 109.5
C19—C20—C15 119.6 (9) H7A—C7—H7B 109.5
C19—C20—H20 120.2 H7A—C7—H7C 109.5
C20—C19—H19 119.5 H7B—C7—H7C 109.5
Au1—P1—C1—C6 14.4 (7) C15—P1—C9—C14 158.3 (7)
Au1—P1—C1—C2 -169.5 (6) C15—P1—C9—C10 -27.0 (8)
Au1—P1—C9—C14 35.7 (8) C15—C16—C17—C18 0.2 (13)
Au1—P1—C9—C10 -149.6 (6) C15—C20—C19—C18 2.5 (15)
Au1—P1—C15—C16 -112.4 (6) C5—C4—N1—C8 173.9 (8)
Au1—P1—C15—C20 59.5 (7) C5—C4—N1—C7 -1.4 (13)
P1—C1—C6—C5 176.8 (6) C6—C1—C2—C3 0.6 (12)
P1—C1—C2—C3 -175.6 (6) C6—C5—C4—C3 0.2 (11)
P1—C9—C14—C13 175.5 (7) C6—C5—C4—N1 -177.6 (8)
P1—C9—C10—C11 -175.6 (7) C3—C4—N1—C8 -3.8 (12)
P1—C15—C16—C17 172.9 (7) C3—C4—N1—C7 -179.2 (8)
P1—C15—C20—C19 -174.4 (7) C2—C1—C6—C5 0.6 (12)
C1—P1—C9—C14 -87.8 (7) C2—C3—C4—C5 1.0 (12)
C1—P1—C9—C10 86.8 (8) C2—C3—C4—N1 178.8 (8)
C1—P1—C15—C16 10.2 (8) C16—C15—C20—C19 -2.3 (13)
C1—P1—C15—C20 -177.9 (7) C16—C17—C18—C19 0.0 (14)
C9—P1—C1—C6 139.8 (7) C4—C5—C6—C1 -1.0 (12)
C9—P1—C1—C2 -44.2 (7) C4—C3—C2—C1 -1.4 (12)
C9—P1—C15—C16 123.1 (7) C20—C15—C16—C17 0.9 (12)
C9—P1—C15—C20 -64.9 (8) C20—C19—C18—C17 -1.4 (15)
C9—C14—C13—C12 -0.4 (14) C14—C9—C10—C11 -0.8 (13)
C9—C10—C11—C12 1.0 (14) C10—C9—C14—C13 0.5 (13)
C15—P1—C1—C6 -108.2 (7) C10—C11—C12—C13 -0.9 (15)
C15—P1—C1—C2 67.8 (7) C11—C12—C13—C14 0.6 (15)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].
[AuBr{PPh(4-C6H4NMe2)2}] 2b
Crystal data
C22H25AuBrN2P Z = 2
Mr = 625.29 F(000) = 600
Triclinic, P¯1 Dx = 1.841 Mg m-3

212
a = 10.6462 (5) Å Mo K radiation, = 0.71073 Å
b = 10.9053 (5) Å Cell parameters from 2963 reflections
c = 11.2894 (4) Å = 3.8–27.8°
= 101.733 (4)° = 8.37 mm-1
= 98.778 (4)° T = 150 K
= 114.311 (5)° colourless
V = 1127.75 (10) Å3 0.1 × 0.08 × 0.04 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
4420 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source, Incoatec Is
3767 reflections with I > 2(I)
Mirror monochromator Rint = 0.048
Detector resolution: 8.0714 pixels mm-1
max = 26.0°, min = 3.3°
scans h = -1312
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1313
Tmin = 0.737, Tmax = 1.000 l = -1313
7638 measured reflections
Refinement
Refinement on F2 Primary atom site location: iterative
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.045 H-atom parameters constrained
wR(F2) = 0.084 w = 1/[
2(Fo
2) + (0.0088P)
2]
where P = (Fo2 + 2Fc
2)/3
S = 1.02 (/)max = 0.001
4420 reflections max = 1.43 e Å-3
248 parameters min = -1.49 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)

213
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.07214 (3) 0.18179 (3) 0.29750 (2) 0.02367 (10)
Br1 0.29507 (8) 0.25378 (11) 0.44432 (7) 0.0419 (2)
P1 -0.14134 (19) 0.1203 (2) 0.17194 (16) 0.0198 (4)
C22 -0.3686 (8) 0.1607 (9) 0.2398 (6) 0.0256 (18)
H22 -0.4268 0.0663 0.1956 0.031*
C10 -0.3631 (7) -0.1439 (8) 0.0254 (6) 0.0226 (16)
H10 -0.3506 -0.1113 -0.0440 0.027*
N2 -0.6131 (7) -0.4539 (8) 0.0966 (6) 0.0420 (19)
C21 -0.4264 (9) 0.2409 (10) 0.3001 (6) 0.037 (2)
H21 -0.5236 0.1998 0.2949 0.045*
C11 -0.4734 (7) -0.2740 (8) 0.0099 (6) 0.0240 (17)
H11 -0.5325 -0.3279 -0.0701 0.029*
C5 -0.2533 (8) 0.1736 (9) -0.1677 (6) 0.032 (2)
H5 -0.3284 0.1834 -0.2113 0.039*
C19 -0.1996 (11) 0.4412 (11) 0.3713 (8) 0.050 (2)
H19 -0.1433 0.5367 0.4129 0.060*
N1 -0.1472 (7) 0.1809 (7) -0.3442 (5) 0.0335 (17)
C13 -0.4007 (8) -0.2449 (9) 0.2288 (7) 0.0314 (19)
H13 -0.4112 -0.2796 0.2973 0.038*
C9 -0.2697 (7) -0.0597 (8) 0.1429 (6) 0.0226 (16)
C12 -0.4992 (8) -0.3272 (8) 0.1101 (7) 0.0303 (19)
C17 -0.2227 (7) 0.2225 (8) 0.2456 (6) 0.0221 (17)
C6 -0.2492 (7) 0.1641 (8) -0.0468 (6) 0.0280 (18)
H6 -0.3208 0.1678 -0.0105 0.034*
C18 -0.1395 (9) 0.3622 (9) 0.3139 (7) 0.038 (2)
H18 -0.0417 0.4040 0.3217 0.045*
C3 -0.0375 (7) 0.1518 (8) -0.1567 (6) 0.0244 (17)
H3 0.0338 0.1461 -0.1924 0.029*
C4 -0.1476 (8) 0.1689 (8) -0.2262 (6) 0.0251 (17)
C14 -0.2914 (7) -0.1165 (8) 0.2445 (6) 0.0231 (17)
H14 -0.2293 -0.0646 0.3239 0.028*
C2 -0.0346 (7) 0.1436 (7) -0.0358 (6) 0.0225 (16)
H2 0.0401 0.1340 0.0089 0.027*
C20 -0.3429 (10) 0.3777 (10) 0.3663 (7) 0.041 (2)
H20 -0.3823 0.4292 0.4086 0.049*

214
C1 -0.1387 (7) 0.1491 (8) 0.0203 (6) 0.0213 (16)
C7 -0.0423 (9) 0.1636 (9) -0.4043 (7) 0.042 (2)
H7A 0.0519 0.2315 -0.3539 0.063*
H7B -0.0570 0.1776 -0.4856 0.063*
H7C -0.0524 0.0704 -0.4134 0.063*
C16 -0.6410 (9) -0.5086 (10) 0.2018 (8) 0.057 (3)
H16A -0.5556 -0.5060 0.2473 0.085*
H16B -0.7154 -0.6040 0.1713 0.085*
H16C -0.6705 -0.4520 0.2563 0.085*
C15 -0.7283 (9) -0.5231 (10) -0.0187 (8) 0.059 (3)
H15A -0.7617 -0.4578 -0.0371 0.089*
H15B -0.8055 -0.6025 -0.0086 0.089*
H15C -0.6937 -0.5545 -0.0865 0.089*
C8 -0.2773 (10) 0.1672 (14) -0.4241 (8) 0.073 (4)
H8A -0.3579 0.0833 -0.4249 0.110*
H8B -0.2678 0.1617 -0.5080 0.110*
H8C -0.2915 0.2477 -0.3919 0.110*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.01933 (16) 0.03245 (19) 0.01819 (16) 0.00993 (14) 0.00467 (12) 0.00979 (13)
Br1 0.0274 (4) 0.0634 (7) 0.0307 (5) 0.0206 (4) 0.0020 (4) 0.0101 (4)
P1 0.0179 (9) 0.0240 (11) 0.0170 (9) 0.0078 (8) 0.0048 (8) 0.0088 (8)
C22 0.024 (4) 0.039 (5) 0.016 (4) 0.015 (4) 0.004 (3) 0.011 (3)
C10 0.023 (4) 0.022 (4) 0.020 (4) 0.010 (3) 0.003 (3) 0.004 (3)
N2 0.040 (4) 0.035 (5) 0.039 (4) 0.004 (4) 0.003 (4) 0.020 (4)
C21 0.040 (5) 0.066 (7) 0.026 (4) 0.041 (5) 0.012 (4) 0.015 (4)
C11 0.022 (4) 0.022 (4) 0.022 (4) 0.009 (3) -0.003 (3) 0.005 (3)
C5 0.028 (4) 0.051 (6) 0.024 (4) 0.020 (4) 0.007 (4) 0.019 (4)
C19 0.069 (7) 0.046 (6) 0.050 (6) 0.038 (6) 0.018 (5) 0.017 (5)
N1 0.039 (4) 0.045 (5) 0.019 (3) 0.018 (4) 0.009 (3) 0.018 (3)
C13 0.039 (5) 0.032 (5) 0.025 (4) 0.018 (4) 0.003 (4) 0.014 (4)
C9 0.021 (4) 0.027 (5) 0.021 (4) 0.013 (3) 0.002 (3) 0.010 (3)
C12 0.034 (4) 0.025 (5) 0.041 (5) 0.017 (4) 0.012 (4) 0.019 (4)
C17 0.022 (4) 0.032 (5) 0.010 (3) 0.011 (4) 0.002 (3) 0.007 (3)
C6 0.019 (4) 0.037 (5) 0.020 (4) 0.005 (4) 0.005 (3) 0.011 (4)
C18 0.031 (5) 0.036 (6) 0.043 (5) 0.015 (4) 0.004 (4) 0.009 (4)
C3 0.018 (4) 0.024 (4) 0.024 (4) 0.000 (3) 0.007 (3) 0.010 (3)
C4 0.034 (4) 0.018 (4) 0.020 (4) 0.010 (4) 0.004 (3) 0.006 (3)
C14 0.025 (4) 0.029 (5) 0.013 (3) 0.013 (4) 0.001 (3) 0.003 (3)

215
C2 0.016 (4) 0.022 (4) 0.025 (4) 0.004 (3) 0.004 (3) 0.008 (3)
C20 0.059 (6) 0.058 (7) 0.029 (5) 0.046 (5) 0.021 (4) 0.015 (5)
C1 0.016 (4) 0.024 (4) 0.021 (4) 0.007 (3) 0.005 (3) 0.006 (3)
C7 0.055 (6) 0.040 (6) 0.028 (4) 0.014 (5) 0.016 (4) 0.017 (4)
C16 0.049 (6) 0.035 (6) 0.071 (7) 0.003 (5) 0.023 (5) 0.015 (5)
C15 0.032 (5) 0.038 (6) 0.067 (7) -0.006 (5) -0.016 (5) 0.004 (5)
C8 0.069 (7) 0.146 (12) 0.031 (5) 0.061 (8) 0.021 (5) 0.047 (6)
Geometric parameters (Å, º)
Au1—Br1 2.3943 (8) C13—H13 0.9300
Au1—P1 2.2361 (17) C13—C12 1.417 (9)
P1—C9 1.795 (8) C13—C14 1.357 (10)
P1—C17 1.822 (7) C9—C14 1.416 (9)
P1—C1 1.805 (7) C17—C18 1.379 (10)
C22—H22 0.9300 C6—H6 0.9300
C22—C21 1.391 (10) C6—C1 1.385 (9)
C22—C17 1.400 (9) C18—H18 0.9300
C10—H10 0.9300 C3—H3 0.9300
C10—C11 1.373 (10) C3—C4 1.409 (9)
C10—C9 1.393 (8) C3—C2 1.382 (9)
N2—C12 1.371 (10) C14—H14 0.9300
N2—C16 1.451 (10) C2—H2 0.9300
N2—C15 1.456 (9) C2—C1 1.374 (9)
C21—H21 0.9300 C20—H20 0.9300
C21—C20 1.352 (11) C7—H7A 0.9600
C11—H11 0.9300 C7—H7B 0.9600
C11—C12 1.386 (10) C7—H7C 0.9600
C5—H5 0.9300 C16—H16A 0.9600
C5—C6 1.384 (9) C16—H16B 0.9600
C5—C4 1.403 (10) C16—H16C 0.9600
C19—H19 0.9300 C15—H15A 0.9600
C19—C18 1.387 (11) C15—H15B 0.9600
C19—C20 1.378 (12) C15—H15C 0.9600
N1—C4 1.365 (8) C8—H8A 0.9600
N1—C7 1.448 (9) C8—H8B 0.9600
N1—C8 1.466 (9) C8—H8C 0.9600
P1—Au1—Br1 175.87 (5) C19—C18—H18 119.5
C9—P1—Au1 114.3 (2) C17—C18—C19 121.0 (8)
C9—P1—C17 104.4 (3) C17—C18—H18 119.5
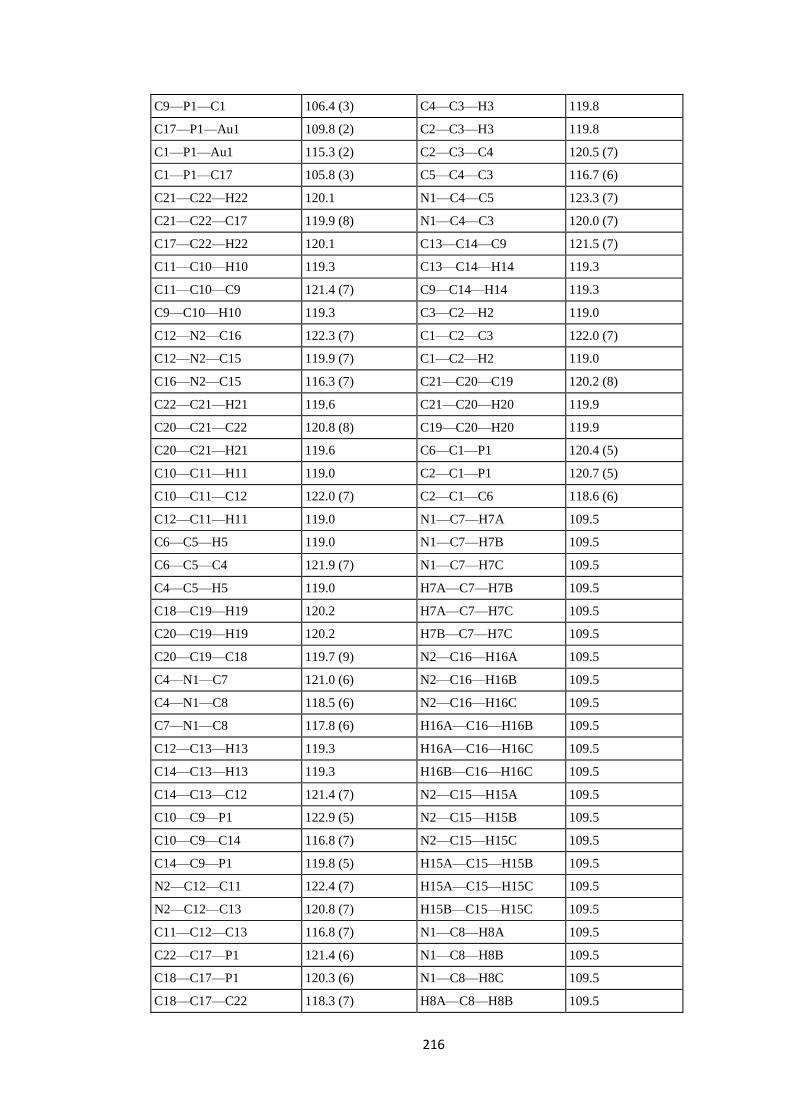
216
C9—P1—C1 106.4 (3) C4—C3—H3 119.8
C17—P1—Au1 109.8 (2) C2—C3—H3 119.8
C1—P1—Au1 115.3 (2) C2—C3—C4 120.5 (7)
C1—P1—C17 105.8 (3) C5—C4—C3 116.7 (6)
C21—C22—H22 120.1 N1—C4—C5 123.3 (7)
C21—C22—C17 119.9 (8) N1—C4—C3 120.0 (7)
C17—C22—H22 120.1 C13—C14—C9 121.5 (7)
C11—C10—H10 119.3 C13—C14—H14 119.3
C11—C10—C9 121.4 (7) C9—C14—H14 119.3
C9—C10—H10 119.3 C3—C2—H2 119.0
C12—N2—C16 122.3 (7) C1—C2—C3 122.0 (7)
C12—N2—C15 119.9 (7) C1—C2—H2 119.0
C16—N2—C15 116.3 (7) C21—C20—C19 120.2 (8)
C22—C21—H21 119.6 C21—C20—H20 119.9
C20—C21—C22 120.8 (8) C19—C20—H20 119.9
C20—C21—H21 119.6 C6—C1—P1 120.4 (5)
C10—C11—H11 119.0 C2—C1—P1 120.7 (5)
C10—C11—C12 122.0 (7) C2—C1—C6 118.6 (6)
C12—C11—H11 119.0 N1—C7—H7A 109.5
C6—C5—H5 119.0 N1—C7—H7B 109.5
C6—C5—C4 121.9 (7) N1—C7—H7C 109.5
C4—C5—H5 119.0 H7A—C7—H7B 109.5
C18—C19—H19 120.2 H7A—C7—H7C 109.5
C20—C19—H19 120.2 H7B—C7—H7C 109.5
C20—C19—C18 119.7 (9) N2—C16—H16A 109.5
C4—N1—C7 121.0 (6) N2—C16—H16B 109.5
C4—N1—C8 118.5 (6) N2—C16—H16C 109.5
C7—N1—C8 117.8 (6) H16A—C16—H16B 109.5
C12—C13—H13 119.3 H16A—C16—H16C 109.5
C14—C13—H13 119.3 H16B—C16—H16C 109.5
C14—C13—C12 121.4 (7) N2—C15—H15A 109.5
C10—C9—P1 122.9 (5) N2—C15—H15B 109.5
C10—C9—C14 116.8 (7) N2—C15—H15C 109.5
C14—C9—P1 119.8 (5) H15A—C15—H15B 109.5
N2—C12—C11 122.4 (7) H15A—C15—H15C 109.5
N2—C12—C13 120.8 (7) H15B—C15—H15C 109.5
C11—C12—C13 116.8 (7) N1—C8—H8A 109.5
C22—C17—P1 121.4 (6) N1—C8—H8B 109.5
C18—C17—P1 120.3 (6) N1—C8—H8C 109.5
C18—C17—C22 118.3 (7) H8A—C8—H8B 109.5

217
C5—C6—H6 119.9 H8A—C8—H8C 109.5
C5—C6—C1 120.3 (7) H8B—C8—H8C 109.5
C1—C6—H6 119.9
Au1—P1—C9—C10 -139.2 (5) C17—P1—C1—C6 -35.3 (7)
Au1—P1—C9—C14 48.7 (6) C17—P1—C1—C2 150.7 (6)
Au1—P1—C17—C22 -139.2 (5) C17—C22—C21—C20 -1.0 (11)
Au1—P1—C17—C18 37.8 (6) C6—C5—C4—N1 -178.6 (8)
Au1—P1—C1—C6 -156.9 (5) C6—C5—C4—C3 0.9 (11)
Au1—P1—C1—C2 29.2 (7) C18—C19—C20—C21 -3.6 (13)
P1—C9—C14—C13 170.5 (6) C3—C2—C1—P1 173.7 (6)
P1—C17—C18—C19 -179.6 (6) C3—C2—C1—C6 -0.3 (11)
C22—C21—C20—C19 2.2 (12) C4—C5—C6—C1 -0.2 (12)
C22—C17—C18—C19 -2.5 (11) C4—C3—C2—C1 1.1 (11)
C10—C11—C12—N2 176.7 (7) C14—C13—C12—N2 -176.9 (7)
C10—C11—C12—C13 -3.9 (11) C14—C13—C12—C11 3.7 (11)
C10—C9—C14—C13 -2.0 (10) C2—C3—C4—C5 -1.4 (11)
C21—C22—C17—P1 178.2 (5) C2—C3—C4—N1 178.2 (7)
C21—C22—C17—C18 1.1 (10) C20—C19—C18—C17 3.8 (13)
C11—C10—C9—P1 -170.5 (6) C1—P1—C9—C10 -10.8 (7)
C11—C10—C9—C14 1.8 (10) C1—P1—C9—C14 177.1 (5)
C5—C6—C1—P1 -174.2 (6) C1—P1—C17—C22 95.8 (6)
C5—C6—C1—C2 -0.2 (11) C1—P1—C17—C18 -87.2 (6)
C9—P1—C17—C22 -16.2 (6) C7—N1—C4—C5 -174.8 (7)
C9—P1—C17—C18 160.8 (6) C7—N1—C4—C3 5.7 (11)
C9—P1—C1—C6 75.3 (7) C16—N2—C12—C11 -179.2 (8)
C9—P1—C1—C2 -98.6 (6) C16—N2—C12—C13 1.5 (12)
C9—C10—C11—C12 1.2 (11) C15—N2—C12—C11 -13.2 (12)
C12—C13—C14—C9 -0.8 (11) C15—N2—C12—C13 167.5 (8)
C17—P1—C9—C10 100.8 (6) C8—N1—C4—C5 -13.8 (12)
C17—P1—C9—C14 -71.3 (6) C8—N1—C4—C3 166.7 (8)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].
[AuCl{P(4-C6H4NMe2)3}] 3a
Crystal data
C24H30AuClN3P F(000) = 1224
Mr = 623.89 Dx = 1.697 Mg m-3
Monoclinic, P21/n Mo K radiation, = 0.71073 Å

218
a = 14.0725 (6) Å Cell parameters from 3094 reflections
b = 11.3108 (4) Å = 3.2–27.7°
c = 15.4429 (6) Å = 6.22 mm-1
= 96.637 (4)° T = 293 K
V = 2441.60 (16) Å3 0.12 × 0.06 × 0.04 mm
Z = 4
Data collection
Xcalibur, Sapphire2, large Be window
diffractometer
4954 independent reflections
Radiation source: Enhance (Mo) X-ray Source 4024 reflections with I > 2(I)
Graphite monochromator Rint = 0.037
Detector resolution: 8.3367 pixels mm-1
max = 26.5°, min = 3.2°
scans h = -1517
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.36.28a (release 18-03-2013 CrysAlis171
.NET) (compiled Mar 18 2013,11:47:30)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -714
Tmin = 0.622, Tmax = 1.000 l = -1219
8189 measured reflections
Refinement
Refinement on F2 Primary atom site location: structure-invariant
direct methods
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.039 H-atom parameters constrained
wR(F2) = 0.081 w = 1/[
2(Fo
2) + (0.0208P)
2 + 1.8456P]
where P = (Fo2 + 2Fc
2)/3
S = 1.07 (/)max = 0.004
4954 reflections max = 2.48 e Å-3
277 parameters min = -1.89 e Å-3
0 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.

219
Fractional atomic coordinates and isotropic or equivalent isotropic displacement
parameters (Å2)
x y z Uiso*/Ueq
Au1 0.08922 (2) 0.23068 (2) 0.63054 (2) 0.01599 (8)
P1 0.20858 (11) 0.29862 (13) 0.56048 (10) 0.0157 (3)
Cl1 -0.03027 (12) 0.16147 (13) 0.70612 (11) 0.0270 (4)
N1 0.2266 (4) 0.8271 (4) 0.5998 (3) 0.0213 (12)
C17 0.1930 (4) 0.2666 (5) 0.4466 (4) 0.0131 (12)
C9 0.3208 (4) 0.2328 (5) 0.6056 (4) 0.0149 (12)
N3 0.1621 (4) 0.1746 (5) 0.1810 (3) 0.0249 (12)
C10 0.3395 (4) 0.2170 (5) 0.6949 (4) 0.0166 (13)
H10 0.2977 0.2491 0.7310 0.020*
C4 0.2246 (4) 0.7058 (5) 0.5897 (4) 0.0179 (14)
C1 0.2198 (4) 0.4571 (5) 0.5704 (4) 0.0144 (13)
N2 0.5553 (4) 0.0311 (4) 0.7154 (3) 0.0213 (12)
C19 0.2356 (4) 0.2949 (5) 0.3002 (4) 0.0179 (13)
H19 0.2721 0.3343 0.2627 0.021*
C22 0.1289 (4) 0.1793 (5) 0.4136 (4) 0.0155 (13)
H22 0.0931 0.1399 0.4516 0.019*
C11 0.4180 (4) 0.1554 (5) 0.7315 (4) 0.0173 (13)
H11 0.4294 0.1492 0.7918 0.021*
C3 0.1441 (4) 0.6481 (5) 0.5458 (4) 0.0168 (13)
H3 0.0916 0.6922 0.5221 0.020*
C13 0.4644 (4) 0.1208 (5) 0.5890 (4) 0.0166 (13)
H13 0.5065 0.0899 0.5526 0.020*
C12 0.4815 (4) 0.1014 (5) 0.6798 (4) 0.0161 (13)
C18 0.2456 (4) 0.3239 (5) 0.3876 (4) 0.0185 (13)
H18 0.2885 0.3830 0.4078 0.022*
C8 0.3118 (5) 0.8859 (6) 0.6368 (4) 0.0302 (17)
H8A 0.3295 0.8573 0.6950 0.045*
H8B 0.3004 0.9695 0.6384 0.045*
H8C 0.3626 0.8702 0.6020 0.045*
C21 0.1169 (5) 0.1494 (5) 0.3263 (4) 0.0211 (14)
H21 0.0729 0.0913 0.3065 0.025*
C6 0.2996 (4) 0.5139 (5) 0.6126 (4) 0.0180 (13)
H6 0.3524 0.4693 0.6347 0.022*
C5 0.3021 (4) 0.6360 (5) 0.6223 (4) 0.0204 (14)
H5 0.3565 0.6717 0.6510 0.024*
C2 0.1429 (4) 0.5266 (5) 0.5378 (4) 0.0169 (13)

220
H2 0.0886 0.4902 0.5097 0.020*
C14 0.3865 (4) 0.1846 (5) 0.5539 (4) 0.0172 (13)
H14 0.3770 0.1962 0.4939 0.021*
C20 0.1709 (4) 0.2065 (5) 0.2669 (4) 0.0190 (14)
C24 0.0857 (5) 0.0961 (6) 0.1456 (4) 0.0304 (17)
H24A 0.0905 0.0226 0.1769 0.046*
H24B 0.0911 0.0816 0.0851 0.046*
H24C 0.0250 0.1322 0.1513 0.046*
C23 0.2120 (5) 0.2403 (6) 0.1189 (4) 0.0338 (18)
H23A 0.1882 0.3198 0.1146 0.051*
H23B 0.2013 0.2029 0.0628 0.051*
H23C 0.2793 0.2413 0.1383 0.051*
C15 0.5596 (5) 0.0009 (6) 0.8071 (4) 0.0263 (15)
H15A 0.5610 0.0721 0.8412 0.040*
H15B 0.6163 -0.0445 0.8242 0.040*
H15C 0.5042 -0.0448 0.8167 0.040*
C16 0.6014 (5) -0.0502 (6) 0.6615 (4) 0.0303 (17)
H16A 0.5555 -0.1067 0.6361 0.045*
H16B 0.6521 -0.0907 0.6965 0.045*
H16C 0.6273 -0.0072 0.6160 0.045*
C7 0.1450 (5) 0.8976 (5) 0.5648 (5) 0.0338 (18)
H7A 0.1359 0.8901 0.5025 0.051*
H7B 0.1563 0.9790 0.5803 0.051*
H7C 0.0888 0.8703 0.5885 0.051*
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.01525 (13) 0.01493 (13) 0.01843 (13) 0.00000 (10) 0.00466 (9) 0.00000 (10)
P1 0.0149 (8) 0.0152 (8) 0.0173 (8) 0.0008 (6) 0.0035 (6) -0.0014 (7)
Cl1 0.0219 (8) 0.0272 (9) 0.0341 (9) -0.0026 (7) 0.0121 (7) 0.0052 (7)
N1 0.024 (3) 0.010 (3) 0.029 (3) 0.002 (2) 0.000 (2) 0.002 (2)
C17 0.011 (3) 0.018 (3) 0.010 (3) 0.003 (2) 0.000 (2) 0.002 (2)
C9 0.015 (3) 0.013 (3) 0.015 (3) -0.004 (2) -0.004 (2) -0.003 (2)
N3 0.025 (3) 0.036 (3) 0.014 (3) -0.001 (3) 0.004 (2) -0.005 (3)
C10 0.022 (3) 0.014 (3) 0.014 (3) 0.001 (3) 0.001 (3) 0.000 (2)
C4 0.022 (3) 0.018 (3) 0.015 (3) 0.001 (3) 0.008 (3) 0.000 (3)
C1 0.016 (3) 0.010 (3) 0.018 (3) 0.001 (2) 0.003 (3) 0.003 (2)
N2 0.026 (3) 0.014 (3) 0.024 (3) 0.006 (2) 0.002 (2) -0.002 (2)
C19 0.018 (3) 0.019 (3) 0.018 (3) -0.001 (3) 0.004 (3) 0.001 (3)
C22 0.015 (3) 0.014 (3) 0.018 (3) 0.000 (3) 0.003 (2) 0.001 (3)

221
C11 0.024 (4) 0.017 (3) 0.011 (3) -0.001 (3) 0.003 (3) 0.001 (2)
C3 0.014 (3) 0.016 (3) 0.019 (3) 0.003 (3) -0.002 (3) 0.002 (3)
C13 0.014 (3) 0.013 (3) 0.024 (3) -0.001 (2) 0.008 (3) -0.006 (3)
C12 0.015 (3) 0.011 (3) 0.023 (3) -0.006 (3) 0.004 (3) -0.002 (3)
C18 0.014 (3) 0.020 (3) 0.021 (3) 0.006 (3) 0.000 (3) 0.003 (3)
C8 0.032 (4) 0.022 (4) 0.035 (4) -0.005 (3) 0.003 (3) 0.001 (3)
C21 0.021 (4) 0.017 (3) 0.025 (4) 0.000 (3) 0.002 (3) -0.002 (3)
C6 0.018 (3) 0.017 (3) 0.019 (3) 0.003 (3) 0.002 (3) 0.001 (3)
C5 0.017 (3) 0.022 (3) 0.022 (3) -0.005 (3) 0.002 (3) -0.001 (3)
C2 0.018 (3) 0.015 (3) 0.017 (3) -0.004 (3) -0.003 (3) 0.000 (3)
C14 0.016 (3) 0.020 (3) 0.017 (3) 0.000 (3) 0.004 (3) 0.002 (3)
C20 0.017 (3) 0.022 (3) 0.018 (3) 0.012 (3) 0.000 (3) -0.002 (3)
C24 0.043 (5) 0.023 (4) 0.024 (4) 0.002 (3) -0.001 (3) -0.010 (3)
C23 0.030 (4) 0.053 (5) 0.020 (4) 0.008 (4) 0.011 (3) 0.000 (3)
C15 0.030 (4) 0.021 (3) 0.028 (4) 0.006 (3) 0.001 (3) 0.007 (3)
C16 0.036 (4) 0.026 (4) 0.028 (4) 0.016 (3) 0.000 (3) 0.003 (3)
C7 0.033 (4) 0.016 (3) 0.051 (5) 0.002 (3) -0.004 (4) 0.000 (3)
Geometric parameters (Å, º)
Au1—P1 2.2367 (16) C3—H3 0.9300
Au1—Cl1 2.2925 (15) C3—C2 1.380 (8)
P1—C17 1.785 (6) C13—H13 0.9300
P1—C9 1.811 (6) C13—C12 1.412 (8)
P1—C1 1.805 (5) C13—C14 1.371 (8)
N1—C4 1.380 (7) C18—H18 0.9300
N1—C8 1.431 (8) C8—H8A 0.9600
N1—C7 1.450 (8) C8—H8B 0.9600
C17—C22 1.395 (8) C8—H8C 0.9600
C17—C18 1.396 (8) C21—H21 0.9300
C9—C10 1.384 (8) C21—C20 1.414 (8)
C9—C14 1.401 (8) C6—H6 0.9300
N3—C20 1.366 (7) C6—C5 1.389 (8)
N3—C24 1.452 (8) C5—H5 0.9300
N3—C23 1.456 (8) C2—H2 0.9300
C10—H10 0.9300 C14—H14 0.9300
C10—C11 1.372 (8) C24—H24A 0.9600
C4—C3 1.411 (8) C24—H24B 0.9600
C4—C5 1.394 (8) C24—H24C 0.9600
C1—C6 1.388 (8) C23—H23A 0.9600
C1—C2 1.385 (8) C23—H23B 0.9600

222
N2—C12 1.371 (7) C23—H23C 0.9600
N2—C15 1.452 (7) C15—H15A 0.9600
N2—C16 1.444 (7) C15—H15B 0.9600
C19—H19 0.9300 C15—H15C 0.9600
C19—C18 1.381 (8) C16—H16A 0.9600
C19—C20 1.409 (8) C16—H16B 0.9600
C22—H22 0.9300 C16—H16C 0.9600
C22—C21 1.380 (8) C7—H7A 0.9600
C11—H11 0.9300 C7—H7B 0.9600
C11—C12 1.404 (8) C7—H7C 0.9600
P1—Au1—Cl1 178.33 (6) N1—C8—H8B 109.5
C17—P1—Au1 113.2 (2) N1—C8—H8C 109.5
C17—P1—C9 107.4 (3) H8A—C8—H8B 109.5
C17—P1—C1 106.6 (3) H8A—C8—H8C 109.5
C9—P1—Au1 110.03 (19) H8B—C8—H8C 109.5
C1—P1—Au1 111.32 (19) C22—C21—H21 119.7
C1—P1—C9 108.1 (3) C22—C21—C20 120.5 (6)
C4—N1—C8 120.8 (5) C20—C21—H21 119.7
C4—N1—C7 119.9 (5) C1—C6—H6 119.3
C8—N1—C7 118.9 (5) C1—C6—C5 121.3 (6)
C22—C17—P1 120.3 (4) C5—C6—H6 119.3
C22—C17—C18 117.4 (5) C4—C5—H5 119.5
C18—C17—P1 122.3 (4) C6—C5—C4 121.0 (6)
C10—C9—P1 119.6 (5) C6—C5—H5 119.5
C10—C9—C14 117.0 (5) C1—C2—H2 118.9
C14—C9—P1 123.0 (4) C3—C2—C1 122.1 (5)
C20—N3—C24 120.5 (5) C3—C2—H2 118.9
C20—N3—C23 120.6 (5) C9—C14—H14 119.0
C24—N3—C23 117.1 (5) C13—C14—C9 122.0 (5)
C9—C10—H10 119.1 C13—C14—H14 119.0
C11—C10—C9 121.9 (6) N3—C20—C19 121.6 (6)
C11—C10—H10 119.1 N3—C20—C21 121.0 (6)
N1—C4—C3 121.3 (5) C19—C20—C21 117.4 (5)
N1—C4—C5 121.1 (6) N3—C24—H24A 109.5
C5—C4—C3 117.6 (5) N3—C24—H24B 109.5
C6—C1—P1 123.9 (4) N3—C24—H24C 109.5
C2—C1—P1 118.4 (4) H24A—C24—H24B 109.5
C2—C1—C6 117.6 (5) H24A—C24—H24C 109.5
C12—N2—C15 118.1 (5) H24B—C24—H24C 109.5

223
C12—N2—C16 120.6 (5) N3—C23—H23A 109.5
C16—N2—C15 116.2 (5) N3—C23—H23B 109.5
C18—C19—H19 119.6 N3—C23—H23C 109.5
C18—C19—C20 120.9 (5) H23A—C23—H23B 109.5
C20—C19—H19 119.6 H23A—C23—H23C 109.5
C17—C22—H22 119.0 H23B—C23—H23C 109.5
C21—C22—C17 122.0 (5) N2—C15—H15A 109.5
C21—C22—H22 119.0 N2—C15—H15B 109.5
C10—C11—H11 119.3 N2—C15—H15C 109.5
C10—C11—C12 121.4 (5) H15A—C15—H15B 109.5
C12—C11—H11 119.3 H15A—C15—H15C 109.5
C4—C3—H3 119.9 H15B—C15—H15C 109.5
C2—C3—C4 120.3 (5) N2—C16—H16A 109.5
C2—C3—H3 119.9 N2—C16—H16B 109.5
C12—C13—H13 119.6 N2—C16—H16C 109.5
C14—C13—H13 119.6 H16A—C16—H16B 109.5
C14—C13—C12 120.7 (5) H16A—C16—H16C 109.5
N2—C12—C11 121.8 (5) H16B—C16—H16C 109.5
N2—C12—C13 121.4 (5) N1—C7—H7A 109.5
C11—C12—C13 116.8 (5) N1—C7—H7B 109.5
C17—C18—H18 119.1 N1—C7—H7C 109.5
C19—C18—C17 121.7 (6) H7A—C7—H7B 109.5
C19—C18—H18 119.1 H7A—C7—H7C 109.5
N1—C8—H8A 109.5 H7B—C7—H7C 109.5
Au1—P1—C17—C22 -17.7 (5) C1—P1—C9—C10 82.0 (5)
Au1—P1—C17—C18 164.3 (4) C1—P1—C9—C14 -105.2 (5)
Au1—P1—C9—C10 -39.8 (5) C1—C6—C5—C4 -0.2 (9)
Au1—P1—C9—C14 133.0 (5) C22—C17—C18—C19 -0.6 (8)
Au1—P1—C1—C6 116.2 (5) C22—C21—C20—N3 177.5 (5)
Au1—P1—C1—C2 -60.7 (5) C22—C21—C20—C19 -0.8 (9)
P1—C17—C22—C21 -178.0 (5) C3—C4—C5—C6 -0.7 (8)
P1—C17—C18—C19 177.4 (4) C12—C13—C14—C9 -0.2 (9)
P1—C9—C10—C11 172.3 (4) C18—C17—C22—C21 0.0 (9)
P1—C9—C14—C13 -170.7 (4) C18—C19—C20—N3 -178.1 (5)
P1—C1—C6—C5 -176.5 (4) C18—C19—C20—C21 0.2 (9)
P1—C1—C2—C3 177.4 (5) C8—N1—C4—C3 -173.9 (6)
N1—C4—C3—C2 -178.8 (5) C8—N1—C4—C5 5.8 (8)
N1—C4—C5—C6 179.6 (5) C6—C1—C2—C3 0.4 (9)
C17—P1—C9—C10 -163.4 (4) C5—C4—C3—C2 1.4 (8)

224
C17—P1—C9—C14 9.5 (6) C2—C1—C6—C5 0.4 (9)
C17—P1—C1—C6 -119.9 (5) C14—C9—C10—C11 -1.0 (8)
C17—P1—C1—C2 63.2 (5) C14—C13—C12—N2 175.5 (5)
C17—C22—C21—
C20
0.7 (9) C14—C13—C12—C11 -3.1 (8)
C9—P1—C17—C22 103.9 (5) C20—C19—C18—C17 0.5 (9)
C9—P1—C17—C18 -74.0 (5) C24—N3—C20—C19 -171.0 (6)
C9—P1—C1—C6 -4.8 (6) C24—N3—C20—C21 10.8 (9)
C9—P1—C1—C2 178.3 (5) C23—N3—C20—C19 -6.9 (9)
C9—C10—C11—C12 -2.4 (9) C23—N3—C20—C21 174.9 (6)
C10—C9—C14—C13 2.3 (8) C15—N2—C12—C11 7.7 (8)
C10—C11—C12—N2 -174.2 (5) C15—N2—C12—C13 -170.8 (5)
C10—C11—C12—
C13
4.4 (8) C16—N2—C12—C11 161.6 (6)
C4—C3—C2—C1 -1.3 (9) C16—N2—C12—C13 -16.9 (8)
C1—P1—C17—C22 -140.4 (5) C7—N1—C4—C3 0.0 (9)
C1—P1—C17—C18 41.7 (6) C7—N1—C4—C5 179.7 (6)
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].
[AuBr{P(4-C6H4NMe2)3}] 3b
Crystal data
C24H30AuBrN3P·C7H8 Z = 2
Mr = 760.49 F(000) = 748
Triclinic, P¯1 Dx = 1.660 Mg m-3
a = 7.3065 (4) Å Mo K radiation, = 0.71073 Å
b = 12.7271 (9) Å Cell parameters from 2643 reflections
c = 16.7388 (10) Å = 4.0–28.1°
= 90.462 (5)° = 6.22 mm-1
= 90.550 (4)° T = 150 K
= 102.143 (5)° colourless
V = 1521.56 (16) Å3 0.2 × 0.18 × 0.16 mm
Data collection
SuperNova, Single source at offset, Eos
diffractometer
3619 independent reflections
Radiation source: SuperNova (Mo) X-ray
Source, Incoatec Is
3269 reflections with I > 2(I)
Mirror monochromator Rint = 0.027
Detector resolution: 8.0714 pixels mm-1
max = 25.0°, min = 3.2°

225
scans h = -88
Absorption correction: multi-scan
CrysAlis PRO, Agilent Technologies, Version
1.171.37.33 (release 27-03-2014 CrysAlis171
.NET) (compiled Mar 27 2014,17:12:48)
Empirical absorption correction using spherical
harmonics, implemented in SCALE3
ABSPACK scaling algorithm.
k = -1515
Tmin = 0.765, Tmax = 1.000 l = -1019
5016 measured reflections
Refinement
Refinement on F2 Primary atom site location: dual
Least-squares matrix: full Hydrogen site location: inferred from
neighbouring sites
R[F2 > 2(F
2)] = 0.032 H-atom parameters constrained
wR(F2) = 0.074 w = 1/[
2(Fo
2) + (0.030P)
2 + 0.425P]
where P = (Fo2 + 2Fc
2)/3
S = 1.07 (/)max = 0.001
3619 reflections max = 0.88 e Å-3
334 parameters min = -0.59 e Å-3
86 restraints
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated
using the full covariance matrix. The cell esds are taken into account individually in the
estimation of esds in distances, angles and torsion angles; correlations between esds in cell
parameters are only used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
Au1 0.47180 (3) 0.46111 (2) 0.25213 (2) 0.03045 (12)
Br1 0.55129 (10) 0.28828 (6) 0.23336 (6) 0.0464 (3)
P1 0.3794 (2) 0.61809 (14) 0.26179 (11) 0.0283 (5)
C1 0.5690 (8) 0.7337 (5) 0.2617 (4) 0.0264 (19)
C17 0.2469 (8) 0.6310 (5) 0.3507 (4) 0.0271 (19)
C6 0.5618 (9) 0.8273 (6) 0.3019 (5) 0.042 (2)
H6 0.4576 0.8305 0.3325 0.050*
N3 -0.0519 (7) 0.6675 (5) 0.5576 (3) 0.0333 (18)
C2 0.7257 (8) 0.7317 (5) 0.2146 (4) 0.031 (2)
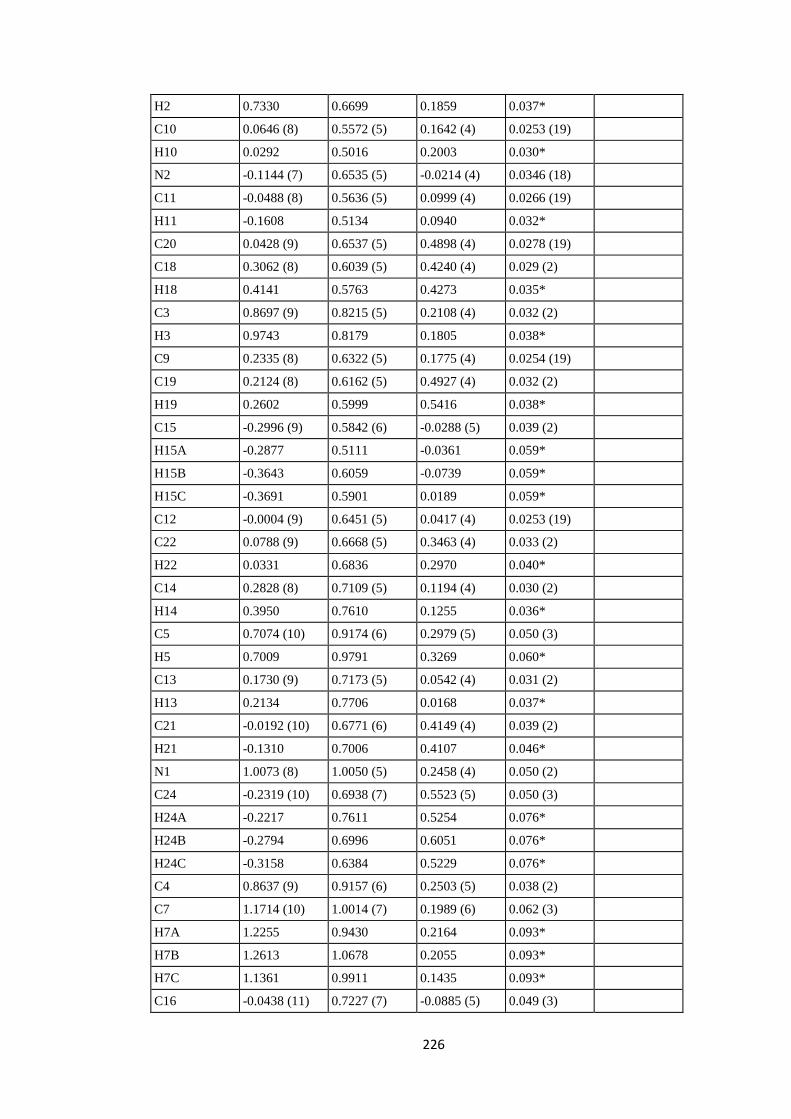
226
H2 0.7330 0.6699 0.1859 0.037*
C10 0.0646 (8) 0.5572 (5) 0.1642 (4) 0.0253 (19)
H10 0.0292 0.5016 0.2003 0.030*
N2 -0.1144 (7) 0.6535 (5) -0.0214 (4) 0.0346 (18)
C11 -0.0488 (8) 0.5636 (5) 0.0999 (4) 0.0266 (19)
H11 -0.1608 0.5134 0.0940 0.032*
C20 0.0428 (9) 0.6537 (5) 0.4898 (4) 0.0278 (19)
C18 0.3062 (8) 0.6039 (5) 0.4240 (4) 0.029 (2)
H18 0.4141 0.5763 0.4273 0.035*
C3 0.8697 (9) 0.8215 (5) 0.2108 (4) 0.032 (2)
H3 0.9743 0.8179 0.1805 0.038*
C9 0.2335 (8) 0.6322 (5) 0.1775 (4) 0.0254 (19)
C19 0.2124 (8) 0.6162 (5) 0.4927 (4) 0.032 (2)
H19 0.2602 0.5999 0.5416 0.038*
C15 -0.2996 (9) 0.5842 (6) -0.0288 (5) 0.039 (2)
H15A -0.2877 0.5111 -0.0361 0.059*
H15B -0.3643 0.6059 -0.0739 0.059*
H15C -0.3691 0.5901 0.0189 0.059*
C12 -0.0004 (9) 0.6451 (5) 0.0417 (4) 0.0253 (19)
C22 0.0788 (9) 0.6668 (5) 0.3463 (4) 0.033 (2)
H22 0.0331 0.6836 0.2970 0.040*
C14 0.2828 (8) 0.7109 (5) 0.1194 (4) 0.030 (2)
H14 0.3950 0.7610 0.1255 0.036*
C5 0.7074 (10) 0.9174 (6) 0.2979 (5) 0.050 (3)
H5 0.7009 0.9791 0.3269 0.060*
C13 0.1730 (9) 0.7173 (5) 0.0542 (4) 0.031 (2)
H13 0.2134 0.7706 0.0168 0.037*
C21 -0.0192 (10) 0.6771 (6) 0.4149 (4) 0.039 (2)
H21 -0.1310 0.7006 0.4107 0.046*
N1 1.0073 (8) 1.0050 (5) 0.2458 (4) 0.050 (2)
C24 -0.2319 (10) 0.6938 (7) 0.5523 (5) 0.050 (3)
H24A -0.2217 0.7611 0.5254 0.076*
H24B -0.2794 0.6996 0.6051 0.076*
H24C -0.3158 0.6384 0.5229 0.076*
C4 0.8637 (9) 0.9157 (6) 0.2503 (5) 0.038 (2)
C7 1.1714 (10) 1.0014 (7) 0.1989 (6) 0.062 (3)
H7A 1.2255 0.9430 0.2164 0.093*
H7B 1.2613 1.0678 0.2055 0.093*
H7C 1.1361 0.9911 0.1435 0.093*
C16 -0.0438 (11) 0.7227 (7) -0.0885 (5) 0.049 (3)

227
H16A 0.0676 0.7035 -0.1085 0.074*
H16B -0.0154 0.7964 -0.0708 0.074*
H16C -0.1372 0.7136 -0.1301 0.074*
C8 0.9858 (11) 1.1058 (6) 0.2776 (6) 0.061 (3)
H8A 0.8767 1.1247 0.2544 0.092*
H8B 1.0943 1.1601 0.2655 0.092*
H8C 0.9718 1.1006 0.3345 0.092*
C23 0.0142 (10) 0.6388 (6) 0.6347 (4) 0.035 (2)
H23A 0.0243 0.5648 0.6335 0.053*
H23B -0.0725 0.6492 0.6752 0.053*
H23C 0.1348 0.6835 0.6464 0.053*
C1_1 0.190 (3) 0.964 (2) 0.4327 (18) 0.205 (18)* 0.5
H1A_1 0.2117 0.9777 0.3769 0.308* 0.5
H1B_1 0.1138 1.0104 0.4534 0.308* 0.5
H1C_1 0.1272 0.8903 0.4395 0.308* 0.5
C2_1 0.372 (2) 0.9842 (14) 0.4766 (11) 0.080* 0.5
C3_1 0.516 (2) 1.0692 (15) 0.4591 (12) 0.071 (6)* 0.5
H3_1 0.5023 1.1168 0.4187 0.086* 0.5
C4_1 0.679 (2) 1.0824 (14) 0.5024 (11) 0.066 (5)* 0.5
H4_1 0.7755 1.1400 0.4905 0.079* 0.5
C5_1 0.706 (3) 1.0151 (17) 0.5621 (14) 0.102 (7)* 0.5
H5_1 0.8173 1.0272 0.5912 0.123* 0.5
C6_1 0.567 (2) 0.9308 (13) 0.5775 (10) 0.063 (5)* 0.5
H6_1 0.5825 0.8828 0.6173 0.075* 0.5
C7_1 0.403 (2) 0.9152 (13) 0.5351 (10) 0.043 (4)* 0.5
H7_1 0.3088 0.8558 0.5463 0.051* 0.5
C1_3 0.301 (3) 1.1149 (17) -0.0165 (14) 0.086 (7)* 0.5
H1A_3 0.1871 1.0754 -0.0409 0.129* 0.5
H1B_3 0.3577 1.1719 -0.0512 0.129* 0.5
H1C_3 0.2725 1.1448 0.0336 0.129* 0.5
C2_3 0.4301 (17) 1.0425 (11) -0.0030 (8) 0.047 (4)* 0.5
C3_3 0.5433 (19) 1.0634 (12) 0.0631 (8) 0.030 (4)* 0.5
H3_3 0.5401 1.1212 0.0970 0.036* 0.5
C4_3 0.663 (2) 0.9945 (13) 0.0771 (10) 0.060 (4)* 0.5
H4_3 0.7376 1.0033 0.1230 0.072* 0.5
C5_3 0.673 (3) 0.9151 (15) 0.0252 (11) 0.077 (7)* 0.5
H5_3 0.7604 0.8726 0.0343 0.092* 0.5
C6_3 0.559 (2) 0.8954 (12) -0.0405 (10) 0.058 (4)* 0.5
H6_3 0.5642 0.8377 -0.0743 0.070* 0.5
C7_3 0.435 (2) 0.9617 (13) -0.0567 (11) 0.058 (6)* 0.5
228
H7_3 0.3585 0.9515 -0.1021 0.069* 0.5
Atomic displacement parameters (Å2)
U11
U22
U33
U12
U13
U23
Au1 0.02898 (16) 0.03131 (16) 0.03211 (19) 0.00907 (12) -0.00434
(15)
-0.00303
(15)
Br1 0.0456 (4) 0.0369 (4) 0.0603 (6) 0.0182 (4) -0.0153 (5) -0.0107 (5)
P1 0.0275 (9) 0.0319 (10) 0.0263 (11) 0.0080 (8) -0.0014 (10) -0.0019 (10)
C1 0.025 (3) 0.031 (4) 0.023 (4) 0.006 (3) -0.007 (4) -0.007 (4)
C17 0.029 (4) 0.028 (4) 0.024 (4) 0.004 (3) 0.000 (4) -0.003 (4)
C6 0.028 (4) 0.047 (5) 0.049 (5) 0.004 (3) 0.010 (5) -0.009 (5)
N3 0.029 (3) 0.046 (4) 0.025 (3) 0.006 (3) -0.002 (3) -0.001 (4)
C2 0.033 (4) 0.037 (4) 0.027 (4) 0.017 (3) 0.002 (4) 0.002 (4)
C10 0.025 (3) 0.027 (4) 0.025 (4) 0.007 (3) -0.004 (4) -0.004 (4)
N2 0.026 (3) 0.045 (4) 0.029 (4) -0.001 (3) -0.007 (3) 0.005 (4)
C11 0.017 (3) 0.030 (4) 0.030 (4) -0.001 (3) 0.002 (4) -0.006 (4)
C20 0.028 (4) 0.032 (4) 0.020 (4) -0.001 (3) -0.010 (4) -0.006 (4)
C18 0.021 (3) 0.031 (4) 0.035 (4) 0.008 (3) -0.009 (4) -0.001 (4)
C3 0.028 (3) 0.038 (4) 0.034 (5) 0.015 (3) 0.006 (4) 0.001 (4)
C9 0.027 (3) 0.026 (4) 0.023 (4) 0.006 (3) -0.002 (4) -0.005 (4)
C19 0.028 (4) 0.041 (4) 0.025 (4) 0.007 (3) -0.008 (4) 0.005 (4)
C15 0.028 (4) 0.056 (5) 0.032 (4) 0.007 (3) -0.005 (4) -0.003 (5)
C12 0.022 (3) 0.030 (4) 0.021 (4) 0.001 (3) -0.004 (4) -0.004 (4)
C22 0.037 (4) 0.035 (4) 0.028 (4) 0.011 (3) -0.009 (4) -0.002 (4)
C14 0.023 (3) 0.034 (4) 0.032 (4) 0.002 (3) -0.003 (4) -0.005 (4)
C5 0.048 (5) 0.039 (4) 0.062 (7) 0.008 (4) 0.014 (5) -0.017 (5)
C13 0.028 (4) 0.038 (4) 0.023 (4) 0.002 (3) -0.002 (4) 0.004 (4)
C21 0.036 (4) 0.051 (5) 0.036 (5) 0.024 (4) 0.006 (4) 0.007 (5)
N1 0.046 (4) 0.033 (3) 0.067 (6) -0.002 (3) 0.013 (5) -0.012 (4)
C24 0.045 (4) 0.076 (6) 0.035 (5) 0.025 (4) 0.006 (5) 0.001 (5)
C4 0.039 (4) 0.032 (4) 0.042 (5) 0.009 (3) 0.004 (5) 0.000 (5)
C7 0.045 (5) 0.059 (5) 0.078 (7) -0.002 (4) 0.021 (6) 0.004 (6)
C16 0.048 (5) 0.068 (6) 0.030 (5) 0.009 (4) -0.007 (5) 0.012 (5)
C8 0.062 (5) 0.038 (5) 0.078 (8) -0.004 (4) 0.009 (6) -0.009 (6)
C23 0.038 (4) 0.044 (5) 0.022 (4) 0.004 (4) -0.007 (4) -0.005 (4)
Geometric parameters (Å, º)
Au1—Br1 2.4092 (7) N1—C8 1.425 (10)
Au1—P1 2.2445 (16) C24—H24A 0.9600
P1—C1 1.796 (7) C24—H24B 0.9600

229
P1—C17 1.808 (6) C24—H24C 0.9600
P1—C9 1.793 (7) C7—H7A 0.9600
C1—C6 1.375 (10) C7—H7B 0.9600
C1—C2 1.401 (7) C7—H7C 0.9600
C17—C18 1.369 (9) C16—H16A 0.9600
C17—C22 1.398 (8) C16—H16B 0.9600
C6—H6 0.9300 C16—H16C 0.9600
C6—C5 1.393 (10) C8—H8A 0.9600
N3—C20 1.365 (8) C8—H8B 0.9600
N3—C24 1.425 (8) C8—H8C 0.9600
N3—C23 1.450 (9) C23—H23A 0.9600
C2—H2 0.9300 C23—H23B 0.9600
C2—C3 1.383 (10) C23—H23C 0.9600
C10—H10 0.9300 C1_1—H1A_1 0.9600
C10—C11 1.366 (10) C1_1—H1B_1 0.9600
C10—C9 1.407 (10) C1_1—H1C_1 0.9600
N2—C15 1.455 (9) C1_1—C2_1 1.488 (19)
N2—C12 1.357 (9) C2_1—C3_1 1.374 (14)
N2—C16 1.462 (7) C2_1—C7_1 1.369 (13)
C11—H11 0.9300 C3_1—H3_1 0.9300
C11—C12 1.421 (8) C3_1—C4_1 1.366 (14)
C20—C19 1.418 (8) C4_1—H4_1 0.9300
C20—C21 1.385 (10) C4_1—C5_1 1.361 (14)
C18—H18 0.9300 C5_1—H5_1 0.9300
C18—C19 1.369 (9) C5_1—C6_1 1.341 (14)
C3—H3 0.9300 C6_1—H6_1 0.9300
C3—C4 1.374 (10) C6_1—C7_1 1.364 (14)
C9—C14 1.396 (8) C7_1—H7_1 0.9300
C19—H19 0.9300 C1_3—H1A_3 0.9600
C15—H15A 0.9600 C1_3—H1B_3 0.9600
C15—H15B 0.9600 C1_3—H1C_3 0.9600
C15—H15C 0.9600 C1_3—C2_3 1.470 (16)
C12—C13 1.412 (10) C2_3—C3_3 1.364 (12)
C22—H22 0.9300 C2_3—C7_3 1.368 (13)
C22—C21 1.379 (8) C3_3—H3_3 0.9300
C14—H14 0.9300 C3_3—C4_3 1.383 (13)
C14—C13 1.361 (10) C4_3—H4_3 0.9300
C5—H5 0.9300 C4_3—C5_3 1.341 (14)
C5—C4 1.402 (8) C5_3—H5_3 0.9300
C13—H13 0.9300 C5_3—C6_3 1.364 (14)

230
C21—H21 0.9300 C6_3—H6_3 0.9300
N1—C4 1.378 (10) C6_3—C7_3 1.389 (13)
N1—C7 1.448 (8) C7_3—H7_3 0.9300
P1—Au1—Br1 175.27 (6) H24A—C24—H24C 109.5
C1—P1—Au1 113.81 (19) H24B—C24—H24C 109.5
C1—P1—C17 105.9 (3) C3—C4—C5 117.5 (7)
C17—P1—Au1 113.84 (19) C3—C4—N1 121.9 (5)
C9—P1—Au1 109.4 (2) N1—C4—C5 120.5 (8)
C9—P1—C1 106.2 (3) N1—C7—H7A 109.5
C9—P1—C17 107.3 (3) N1—C7—H7B 109.5
C6—C1—P1 122.6 (4) N1—C7—H7C 109.5
C6—C1—C2 118.1 (6) H7A—C7—H7B 109.5
C2—C1—P1 119.1 (6) H7A—C7—H7C 109.5
C18—C17—P1 120.9 (4) H7B—C7—H7C 109.5
C18—C17—C22 118.1 (5) N2—C16—H16A 109.5
C22—C17—P1 121.0 (5) N2—C16—H16B 109.5
C1—C6—H6 119.3 N2—C16—H16C 109.5
C1—C6—C5 121.4 (5) H16A—C16—H16B 109.5
C5—C6—H6 119.3 H16A—C16—H16C 109.5
C20—N3—C24 120.1 (6) H16B—C16—H16C 109.5
C20—N3—C23 120.4 (5) N1—C8—H8A 109.5
C24—N3—C23 118.8 (5) N1—C8—H8B 109.5
C1—C2—H2 119.9 N1—C8—H8C 109.5
C3—C2—C1 120.2 (7) H8A—C8—H8B 109.5
C3—C2—H2 119.9 H8A—C8—H8C 109.5
C11—C10—H10 119.0 H8B—C8—H8C 109.5
C11—C10—C9 121.9 (5) N3—C23—H23A 109.5
C9—C10—H10 119.0 N3—C23—H23B 109.5
C15—N2—C16 118.4 (7) N3—C23—H23C 109.5
C12—N2—C15 121.0 (5) H23A—C23—H23B 109.5
C12—N2—C16 120.2 (5) H23A—C23—H23C 109.5
C10—C11—H11 119.1 H23B—C23—H23C 109.5
C10—C11—C12 121.7 (6) H1A_1—C1_1—H1B_1 109.5
C12—C11—H11 119.1 H1A_1—C1_1—H1C_1 109.5
N3—C20—C19 121.5 (6) H1B_1—C1_1—H1C_1 109.5
N3—C20—C21 122.0 (5) C2_1—C1_1—H1A_1 109.5
C21—C20—C19 116.5 (5) C2_1—C1_1—H1B_1 109.5
C17—C18—H18 118.9 C2_1—C1_1—H1C_1 109.5
C17—C18—C19 122.3 (5) C3_1—C2_1—C1_1 122.3 (16)
231
C19—C18—H18 118.9 C7_1—C2_1—C1_1 119.8 (15)
C2—C3—H3 118.9 C7_1—C2_1—C3_1 117.9 (13)
C4—C3—C2 122.3 (5) C2_1—C3_1—H3_1 120.7
C4—C3—H3 118.9 C4_1—C3_1—C2_1 118.6 (13)
C10—C9—P1 120.0 (4) C4_1—C3_1—H3_1 120.7
C14—C9—P1 123.6 (5) C3_1—C4_1—H4_1 118.5
C14—C9—C10 116.3 (7) C5_1—C4_1—C3_1 123.0 (14)
C20—C19—H19 119.7 C5_1—C4_1—H4_1 118.5
C18—C19—C20 120.6 (6) C4_1—C5_1—H5_1 121.0
C18—C19—H19 119.7 C6_1—C5_1—C4_1 118.0 (15)
N2—C15—H15A 109.5 C6_1—C5_1—H5_1 121.0
N2—C15—H15B 109.5 C5_1—C6_1—H6_1 119.8
N2—C15—H15C 109.5 C5_1—C6_1—C7_1 120.3 (13)
H15A—C15—H15B 109.5 C7_1—C6_1—H6_1 119.8
H15A—C15—H15C 109.5 C2_1—C7_1—H7_1 119.0
H15B—C15—H15C 109.5 C6_1—C7_1—C2_1 122.0 (12)
N2—C12—C11 122.1 (6) C6_1—C7_1—H7_1 119.0
N2—C12—C13 122.3 (5) H1A_3—C1_3—H1B_3 109.5
C13—C12—C11 115.7 (7) H1A_3—C1_3—H1C_3 109.5
C17—C22—H22 120.0 H1B_3—C1_3—H1C_3 109.5
C21—C22—C17 120.1 (6) C2_3—C1_3—H1A_3 109.5
C21—C22—H22 120.0 C2_3—C1_3—H1B_3 109.5
C9—C14—H14 118.7 C2_3—C1_3—H1C_3 109.5
C13—C14—C9 122.5 (6) C3_3—C2_3—C1_3 116.4 (14)
C13—C14—H14 118.7 C3_3—C2_3—C7_3 124.0 (12)
C6—C5—H5 119.8 C7_3—C2_3—C1_3 119.6 (14)
C6—C5—C4 120.4 (8) C2_3—C3_3—H3_3 121.6
C4—C5—H5 119.8 C2_3—C3_3—C4_3 116.8 (13)
C12—C13—H13 119.1 C4_3—C3_3—H3_3 121.6
C14—C13—C12 121.9 (6) C3_3—C4_3—H4_3 119.6
C14—C13—H13 119.1 C5_3—C4_3—C3_3 120.8 (14)
C20—C21—H21 118.8 C5_3—C4_3—H4_3 119.6
C22—C21—C20 122.4 (5) C4_3—C5_3—H5_3 119.3
C22—C21—H21 118.8 C4_3—C5_3—C6_3 121.5 (15)
C4—N1—C7 120.2 (7) C6_3—C5_3—H5_3 119.3
C4—N1—C8 120.5 (5) C5_3—C6_3—H6_3 120.1
C8—N1—C7 118.7 (6) C5_3—C6_3—C7_3 119.7 (14)
N3—C24—H24A 109.5 C7_3—C6_3—H6_3 120.1
N3—C24—H24B 109.5 C2_3—C7_3—C6_3 117.0 (13)
N3—C24—H24C 109.5 C2_3—C7_3—H7_3 121.5

232
H24A—C24—H24B 109.5 C6_3—C7_3—H7_3 121.5
Document origin: publCIF [Westrip, S. P. (2010). J. Apply. Cryst., 43, 920-925].

233
Chapter 8
Conclusions

234
8.1 Conclusions
This thesis is concerned with the preparation and characterisation of a series of gold(I)
halide complexes possessing a tertiary aryl phosphine ligand containing one or more
electron withdrawing substituent positioned, generally, so as to have little steric impact
on the size of the phosphine. These complexes were targeted in order to investigate the
presence, or otherwise, of aurophilic interactions, characterised by Au–Au distances less
than twice the van der Waals radius of gold (3.60 Å). Aurophilic interactions have been
extensively studied before and the findings of these studies have been summarised in
some of the reviews published.1 However, there is still no clear understanding of when
an aurophilic interaction will, or will not, be observed. For example, it is know that the
complex [AuCl(P(C6H5)3)]2 does not exhibit an aurophilic interaction, while
[AuCl(P(C6F5)3)]3 does, but it is not obvious whether this is a steric, electronic or
packing-related effect. Therefore to complement the previously reported data a
systematic study of a series of gold(I) halide phosphine complexes were prepared in
which phosphines containing either a mixture of phenyl and pentafluorophenyl
substituents or partially fluorinated rings were investigated, as were others with similar
substitution patterns containing CF3, OMe and NMe2 substituents.
The complexes were prepared using a stepwise procedure; firstly K[AuCl4] was reduced
with tetrahydrothiophene (tht) to form AuCl(tht) which was reacted with the appropriate
phosphine to form the phosphine gold(I) chloride complex. Subsequently the bromide
and iodide analogues were prepared by halide exchange reactions using LiBr or KI. The
formation of each complex was confirmed by a change of ca. 30 – 41 ppm in 31
P NMR
chemical shift on coordination. For a total of 29 complexes, the structure of the
compounds was determined by single-crystal X-ray crystallography. As expected in
each case the gold centre adopts a near-linear geometry with the bond angles ranging
from 170.50(12) to 179.19(6) for Cl–Au–P, 169.32(6) to 179.10(6)° for Br–Au–P and
165.35(7)° to 178.62(7)° for I–Au–P. For the Au–P distances a much smaller difference
is observed on changing the halide, while, as expected, the Au–X bond distance
increases in the order X = Cl, Br, I. For all of the complexes the Au–Au distance was
determined, and these are listed in Table 8.1. From this data it is clear that there is a
wide variation in the Au–Au distances observed. Most of the structures also showed a
significant number of other interactions less than the sum of the relevant van der Waal’s

235
radii, such as C–H···X, C–H···F and F···F interactions, the shortest of these for each
structure is also listed in Table 8.1.
Table 8.1 A summary of the shortest Au···Au distances (Å) and average intermolecular
distances (Å) for all complexes reported in this thesis.
Complex d(Au···Au) d(C–H···X)av d(C–H···F)av d(F···F)av
[AuCl{P(4-C6H4F)3}] 4.3592(5) 2.884 2.5404 -
[AuBr{P(4-C6H4F)3}] 4.3260(4) 2.923 2.659(1) -
[AuCl{P(3-C6H4F)3}] 6.982 (2) 2.846 2.415 -
[AuBr{P(3-C6H4F)3}] 7.121(1) 2.963 2.43 -
[AuI{P(3-C6H4F)3}] 7.179(1) 3.155 2.405 2.90(9)
[AuCl{P(3,5-C6H3F2)3}] 7.1873(6) 2.807 2.571 2.86(1)
[AuBr{P(3,5-C6H3F2)3}] 7.2895(6) 2.827 2.343 2.86(1)
[AuI{P(3,5-C6H3F2)3}] 8.3507(7) 3.18 2.575 2.76(1)
AuCl{P(3,4,5-C6H2F3)3}] 3.1273(7) 2.624 2.442 2.833(1)
[AuBr{P(3,4,5-C6H2F3)3}] 3.2322(5) 2.826 2.547 2.835(8)
[AuI{P(3,4,5-C6H2F3)3}] 3.2341(7) 3.043 2.485 2.821(1)
[AuCl{PPh2(C6F5)}] 4.9795(4) 2.8853 2.653 2.838(6)
[AuBr{PPh2(C6F5)}] 5.0178(6) 3.003 - 2.8745(7)
[AuI{PPh2(i-C3F7)}] 4.6975(8) - 2.636 2.855(1)
[AuCl{PPh2(3,5-(CF3)2C6H3)}] 3.7519(8) 2.8975 2.61 -
[AuBr{PPh2(3,5-(CF3)2C6H3)}] 3.8067(6) 3.006 2.602 2.883
[AuI{PPh2(3,5-(CF3)2C6H3)}] 4.3393(6) - 2.554 -
[AuCl{P(3,5-(CF3)2C6H3)3}] 9.0059(6) 2.763 2.545 2.833(8)
[AuBr{P(3,5-(CF3)2C6H3)3}] 3.3196(1) 2.843 2.578 2.8393
[AuI{P(3,5-(CF3)2C6H3)3}] 3.262(1) 3.102 2.565 2.852
[AuCl{P(4-MeOC6H4)3}] 6.2246(4) 2.839 - -
[AuCl{PPh(4-MeOC6H4)2}] 7.133(1) 2.865 - -
[AuCl{PPh2(4-MeOC6H4)}] 6.5239(6) 2.896 - -
[AuBr{PPh2(4-MeOC6H4)}] 6.5643(6) 2.988 - -
[AuCl{PPh2(4-C6H4NMe2)}] 4.2079(6) 2.848 - -
[AuI{PPh2(4-C6H4NMe2)}] 5.9812(5) - - -
[AuBr{PPh(4-C6H4NMe2)2}] 6.5946(5) 2.902 - -
[AuCl{P(4-C6H4NMe2)3}] 6.8886(4) 2.93 - -

236
[AuBr{P(4-C6H4NMe2)3}] 7.3065(5) 2.998 - -
As mentioned above, the gold chloride complex of triphenylphosphine does not give
rise to a short Au···Au distance, whereas that of tris(pentafluorophenyl)phosphine does.
A search of the Cambridge Structural Database shows that there were no structure
reports for complexes for the mixed systems of the type [AuX{P(C6H5)n(C6F5)3-n}]
(X=Cl, Br, I). In chapter 3 the structure of the corresponding complex containing
PPh2(C6F5) is reported and shown to possess an Au···Au distance which is longer than
twice the van der Waals radius of gold and its most significant intermolecular
interaction appears to be π-stacking. It would be interesting in the future to investigate
the structure of the missing complex in the series, ie that of PPh(C6F5)2.
For systems containing partially fluorinated aromatic substituents (chapter 2) the
complexes containing phosphines that are fluorinated only at the meta-position, ie (3-
FC6H4) or (3,5-F2C6H3) give rise to Au···Au distances that are longer than ca. 7 Å, and
this is in agreement with previously published work in the case of the chloride,4
however, for systems containing either (4-FC6H4) or (3,4,5-F3C6H2) very much shorter
distances are observed in the range ca. 3.2 – 4.5 Å. These complexes also exhibit C–
H···X interactions between the gold-bound halide and an ortho proton of the aromatic
rings. This motif is also a feature in a number of other systems.
On the other hand, for the complexes containing the P(3,5-(CF3)2C6H3)3 ligand (chapter
4) while the chloride-containing complex shows a long Au···Au distance (9.0059(6) Å),
the corresponding bromide and iodide produce much shorter distances of 3.3196(1) and
3.262(1) Å respectively. There is therefore a distinct difference between the systems
containing the P(3,5-F2C6H3)3 and P(3,5-(CF3)2C6H3)3 ligands. This system shows,
although in an extreme way, the reverse of the general trend for observing shorter
Au···Au distances in the chloride-containing complex compared with the analogous
bromide and iodide. The usual trend can be seen in all the other data presented in table
8.1. For example, the shortest of the Au···Au distances observed for the complexes
containing PPh2(3,5-(CF3)2C6H3), although all longer than twice the van der Waals
radius of gold, show the usual trend, that is the Au···Au distance increases as the halide
changes from Cl, Br, I (3.7519(8), 3.8067(6) and 4.3393(6) Å, respectively). During
this study the gold complex, [AuCl{P(3,5-(CF3)2C6H3)3}] was found to crystallise in the

237
monoclinic space group I2/a. However, in previous work on the same complex three
different crystals were identified, two were monoclinic, while the other was triclinic.
Structural data were only reported for one of these crystals,5
which resulted in the
determination of a very different gold-gold separation; it would be worth this complex
being revisited in future to verify the original data by repeating the literature
preparation.
Because the gold complexes with phosphines containing the (4-FC6H4) group were
shown to exhibit Au–Au bond lengths in the range 3.2 – 4.5 Å the complexes containing
phosphines with different electronegative groups in the para-position were investigated.
For the ligands P(4-MeOC6H4)3, PPh(4-MeOC6H4)2, PPh2(4-MeOC6H4), PPh2(4-
C6H4NMe2), PPh(4-C6H4NMe2)2 and P(4-C6H4NMe2)3, none of the complexes were
found to exhibit an aurophilic interaction. For the methoxy-containing systems the
shortest Au–Au distances ranged from ca. 6.2 – 7.1 Å. Significant intermolecular
interactions included C–H···X and C–H···O hydrogen bonding. For the complexes of
the NMe2-containing ligands the shortest Au···Au distance although longer than twice
the van der Waals radius of gold, are a little shorter, ranging from ca. 4.2 – 7.3 Å.
Instead the extended structures of these complexes are dominated by C–H···X
interactions; there are no C–H···N interactions equivalent to that found in the methoxy
system due to the lone pair on the nitrogen being delocalised into the π-system.
Considering all these results it appears that in these cases changing the phosphine has a
greater significance on whether a family of AuX(PR3)complexes exhibit an aurophilic
interaction than changing the halide in the complex. For example none of the complexes
containing P(4-C6H4NMe2)3 or P(4-MeOC6H4)3 result in short aurophilic interactions,
whereas all of the halide complexes of phosphines containing either (4-FC6H4) or
(3,4,5-F3C6H2) give rise to shorter Au···Au distances.
It is also clear that all of the complexes exhibit other non-bonded interactions, those
identified include extensive C–H···X hydrogen bonding interactions, C–H···F and F···F
intermolecular contacts. The process of crystallisation is likely to result in a packing
arrangement that maximises the number of all of these interactions, and given that the
energies associated with aurophilic interactions and hydrogen bonds are considered to
be similar, it is likely that there is a fine balance between all the different possible weak
interactions rather than aurophilic interactions being a dominant driving force.

238
While the work in this thesis has been principally structural in nature, having set out to
obtain structures in a systematic way of a series of gold(I) phosphine halide complexes
to compliment the data that are already known for related systems, the next step in using
the newly acquired data is to attempt to rationalise the large amount of data that is now
available. As should be clear from the data presented in this thesis there appears to be
no simple predictive ability of whether a gold-gold interaction will be observed based
on the size or electronic properties of either the halide or phosphine. Indeed if there was
then this link would probably have been made before given the amount of pre-existing
data. Instead, as the amount of data accumulates it would seem that this is a system that
might be usefully probed by multivariate analysis to try and ascertain what features of
the ligand set have a significant impact on the observation, or otherwise of gold-gold
interactions. It is hoped that the work in this thesis might usefully help in such a future
endeavour.

239
8.2 References
1. H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931-1951; P. Pyykkö,
Angew. Chem., Int. Ed., 2004, 43, 4412-4456; P. Pyykkö, W. Schneider, A. Bauer, A.
Bayler and H. Schmidbaur, Chem. Commun., 1997, 1111-1112; R. Pretorius, M. R.
Fructos, H. Müller-Bunz, R. A. Gossage, P. J. Pérez and M. Albrecht, Dalton Trans.,
2016, 45, 14591-14602.
2. N. C. Baenziger, W. E. Bennett and D. M. Soboroff, Acta Cryst., 1976, B32, 962-
963.
3. H. W. Chen and E. R. T. Tiekink, Acta Cryst., 2003, E59, m50-m52.
4. O. B. Shawkataly, A. Tariq, S. S. Ghani, C. S. Yeap and H. K. Fun, Acta Cryst.,
2010, E66, m1217-m1218.
5. A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organomet., 2012, 31, 644-
661.
240
Appendix:
Code Chapter Complex
argp214 2 1a
argp213 2 1b
orgp123 2 2a
orgp229 2 2b
orgp148 2 2c
argp480 2 3a
argp489 2 3b
argp501 2 3c
orgp150 2 4a
argp154 2 4b
argp67 2 4c
argp176 3 I
argp225 3 II
argp65 4 1
argp481 5 1a
argp490 5 1b
argp500 5 1c
orgp132 5 2a
argp444 5 2b
argp64 5 2c
argp59 6 1
argp159 6 2
argp120 6 3a
argp177 6 3b
argp60 7 1a
argp121 7 1c
argp122 7 2b
orgp122 7 3a
argp123 7 3b




















