
Gold Nanoparticles Tethered to Gold Surfaces Using Nitroxyl Radicals
8
Published: March 25, 2011 r2011 American Chemical Society 7347 dx.doi.org/10.1021/jp200842u | J. Phys. Chem. C 2011, 115, 7347–7354 ARTICLE pubs.acs.org/JPCC Gold Nanoparticles Tethered to Gold Surfaces Using Nitroxyl Radicals Olga Swiech, Natalia Hrynkiewicz-Sudnik, Barbara Palys, Andrzej Kaim, and Renata Bilewicz* Department of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland ABSTRACT: We describe the interaction of Au with >NO radicals—the basis of a new method of binding gold nanoparticles to gold substrates. In the sandwich-like system, the gold electrode is separated from the gold nanoparticle layer by thiolated TEMPO radicals. The formation of the NOAu bond was confirmed by FAR IR spectroscopy. The properties of the Au substrateAu nanoparticle assembly were studied by cyclic voltammetry and scanning tunneling microscopy. Binding of nanoparticles by means of nitroxyl radicals instead of linking them using an alkanedithiol leads to a higher population of nanoparticles at the electrode surface, as shown by scanning tunneling microscopy. ’ INTRODUCTION TEMPO (2,2,6,6-tetramethypiperidine-1-oxyl radical) and its derivatives have been widely used as spin labels, 1 spin traps, 2 and antioxidants 3 in biomedical applications; mediators in “living/ controlled” free radical polymerization; 4 catalysts in aerobic oxidation processes; 5 and key components for electrode-active organic coatings. 6 They were used for the studies of the kinetics of surface partitioning and lateral diffusion of redox active amphiphiles in the aqueous/vapor interfacial region. 7 Thiolated TEMPO molecules have been anchored on gold electrodes as self-assembled monolayers and studied as versatile electroactive centers in aqueous and organic media. 810 Zhang and co-work- ers used stable paramagnetic probes to study interactions of gold nanoparticles and nitroxyl radicals. 1113 Interaction between nitroxyl radicals, such as TEMPO and TEMPAMINE, and gold nanoparticles (NPs) resulted in the loss of the electron para- magnetic resonance (EPR) signal. Following Freed and co- workers, 14 these authors suggested that the exchange interaction of unpaired electrons with conduction-band electrons of the metallic particle is responsible for eliminating the EPR signal. More recently, Krukowski and co-workers used scanning tunnel- ing microscopy (STM), cyclic voltammetry (CV), and EPR spectroscopy to investigate the behavior of 2,2,6,6-tetramethyl- piperidine, known as TMP, on Au(111). The authors suggest that TMP could be oxidized to the nitroxyl TEMPO radical, which adsorbs on Au in the form of an oxoammonium cation. Such a oxoammonium cation has been postulated to form a complex with gold at 0.5 V that is easily desorbed during STM imaging. 15 Our aim was to prepare gold nanoparticles (NPs) modified with a TEMPO derivativealkanethiol mixed monolayer and anchor them to gold electrodes in order to produce sandwich-like devices consisting of two conductive planes separated by an organic linker. Such nanostructured electrodes with a TEMPO organocatalyst can be employed for the chemoselective oxida- tions, for example, of primary and secondary alcohols. 15 In the present study, bis[2-(4-oxy-2,2,6,6-tetramethylpiperidine-1-oxyl) ethyl] disulfide (TEMPO-DiSS) (Figure 1a) and alkanethiols with 3 (C4SH) (Figure 1c) or 11 (C12SH) (Figure 1d) methylene units in the chain were used for the formation of monolayer- protected nanoparticles building one of these planes. Bare gold, 1,9-nonanedithiol, or TEMPO thiol-modified gold electrodes formed the other side of the sandwich. CV, STM, and infrared spectroscopy (FAR IR) revealed the interaction of the gold nanoparticles with the gold electrode by means of the TEMPO radical. This new way of binding AuNPs to gold Received: January 26, 2011 Revised: March 13, 2011
Transcript of Gold Nanoparticles Tethered to Gold Surfaces Using Nitroxyl Radicals

Published: March 25, 2011
r 2011 American Chemical Society 7347 dx.doi.org/10.1021/jp200842u | J. Phys. Chem. C 2011, 115, 7347–7354
ARTICLE
pubs.acs.org/JPCC
Gold Nanoparticles Tethered to Gold Surfaces Using Nitroxyl RadicalsOlga Swiech, Natalia Hrynkiewicz-Sudnik, Barbara Palys, Andrzej Kaim, and Renata Bilewicz*
Department of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
ABSTRACT:
We describe the interaction of Au with >NO radicals—the basis of a newmethod of binding gold nanoparticles to gold substrates. Inthe sandwich-like system, the gold electrode is separated from the gold nanoparticle layer by thiolated TEMPO radicals. Theformation of the NO�Au bond was confirmed by FAR IR spectroscopy. The properties of the Au substrate�Au nanoparticleassembly were studied by cyclic voltammetry and scanning tunneling microscopy. Binding of nanoparticles by means of nitroxylradicals instead of linking them using an alkanedithiol leads to a higher population of nanoparticles at the electrode surface, as shownby scanning tunneling microscopy.
’ INTRODUCTION
TEMPO (2,2,6,6-tetramethypiperidine-1-oxyl radical) and itsderivatives have been widely used as spin labels,1 spin traps,2 andantioxidants3 in biomedical applications; mediators in “living/controlled” free radical polymerization;4 catalysts in aerobicoxidation processes;5 and key components for electrode-activeorganic coatings.6 They were used for the studies of the kineticsof surface partitioning and lateral diffusion of redox activeamphiphiles in the aqueous/vapor interfacial region.7 ThiolatedTEMPO molecules have been anchored on gold electrodes asself-assembled monolayers and studied as versatile electroactivecenters in aqueous and organic media.8�10 Zhang and co-work-ers used stable paramagnetic probes to study interactions of goldnanoparticles and nitroxyl radicals.11�13 Interaction betweennitroxyl radicals, such as TEMPO and TEMPAMINE, and goldnanoparticles (NPs) resulted in the loss of the electron para-magnetic resonance (EPR) signal. Following Freed and co-workers,14 these authors suggested that the exchange interactionof unpaired electrons with conduction-band electrons of themetallic particle is responsible for eliminating the EPR signal.More recently, Krukowski and co-workers used scanning tunnel-ing microscopy (STM), cyclic voltammetry (CV), and EPRspectroscopy to investigate the behavior of 2,2,6,6-tetramethyl-piperidine, known as TMP, on Au(111). The authors suggestthat TMP could be oxidized to the nitroxyl TEMPO radical,
which adsorbs on Au in the form of an oxoammonium cation.Such a oxoammonium cation has been postulated to form acomplex with gold at 0.5 V that is easily desorbed during STMimaging.15
Our aim was to prepare gold nanoparticles (NPs) modifiedwith a TEMPO derivative�alkanethiol mixed monolayer andanchor them to gold electrodes in order to produce sandwich-likedevices consisting of two conductive planes separated by anorganic linker. Such nanostructured electrodes with a TEMPOorganocatalyst can be employed for the chemoselective oxida-tions, for example, of primary and secondary alcohols.15 In thepresent study, bis[2-(4-oxy-2,2,6,6-tetramethylpiperidine-1-oxyl)ethyl] disulfide (TEMPO-DiSS) (Figure 1a) and alkanethiols with3 (C4SH) (Figure 1c) or 11 (C12SH) (Figure 1d) methyleneunits in the chain were used for the formation of monolayer-protected nanoparticles building one of these planes.
Bare gold, 1,9-nonanedithiol, or TEMPO thiol-modified goldelectrodes formed the other side of the sandwich. CV, STM, andinfrared spectroscopy (FAR IR) revealed the interaction of thegold nanoparticles with the gold electrode by means of theTEMPO radical. This new way of binding AuNPs to gold
Received: January 26, 2011Revised: March 13, 2011

7348 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
surfaces was compared with AuNP attachment by alkanedithiollinkers.
Both the TEMPO-modified nanoparticles and the surfaces ofelectrodes covered by TEMPO-modified nanoparticles can findseveral applications. The use of the nanostructures in controlled“living” radical polymerization will lead to well-defined polymerchains on the gold surface. These hybrid structures with plannedarchitectures containing polymeric materials and metallic coresshould have different properties, for example, higher strengthand modified conductivity, compared with the conventionalpolymers.
Blends of polymers, for example, polystyrene, with goldnanoparticles capped with the appropriate thiol can serve aselectrode-sensitive bipolar resistive switchings.16 Thus, instead ofblends, TEMPO-coated gold nanoparticles grafted with poly-styrene chains synthesized according to nitroxide mediatedradical polymerization (NMRP) will probably enhance theprocessing and chemical stability of the devices.
TEMPO thiol attached with the thiol terminal group to thegold NP and with the other terminal moiety, NO, to another NPor the gold electrode can provide new types of networkedasymmetric NP junctions.17�19 Gold nanoparticles coated withTEMPO radicals can be employed as a new class of spin markers,for the studies of dynamics of biological systems, for example,membranes and proteins, and new materials for nanoparticle-based spintronics.20
’MATERIALS AND METHODS
Synthesis of Bis[2-(4-oxy-2,2,6,6-tetramethylpiperidine-1-oxyl)ethyl] Disulfide (TEMPO-DiSS). TEMPO-DiSS wassynthesized according to the procedure given by Matyjaszewskiet al.21 A round-bottom flask was filled with 3,3-dithiodi-propionic acid (6.3084 g), 4-hydroxy-TEMPO (15.5032 g),
dicyclohexylcarbodiimide (18.5826 g), and 4-(dimethylamino)pyridine (2.209 g) and purged for 15 min with dry oxygen-freeargon. Anhydrous tetrahydrofuran (50 mL) was then added. Themixture became turbid and orange-red. Subsequently, the con-tent of the flask was stirred at room temperature. After stirring for12 h, the reaction mixture was filtered and the solvent vaporized,which created a red oil. On the TLC plate (cyclohexane/ethylacetate, 8:2), apart from obtaining the required product(TEMPO-DiSS), a trace of an unreacted substrate of 4-hydro-xy-TEMPO was observed. Flash chromatography was used topurify this biradical: the column was filled with Al2O3 (activity I),and cyclohexane/ethyl acetate 8:2 was used as a developingsystem. The product was recrystallized from n-hexane. Orangecrystals (4.0066 g, yield = 26%) with the melting point of 94.4 �Cwere then obtained. MS (Mass Spectrometer Quattro LC), 1HNMR, and 13C NMR (Varian Unity Plus 200 MHz, CDCl3, inthe presence of phenyl hydrazine) proved the TEMPO-DiSSidentity.Preparation of Nanoparticles Covered with C4SH, C12SH,
C4SH/TEMPO Thiol, and C12SH/TEMPO Thiol.GoldNPs wereprepared as previously described22 using a modified Brust�Schiffrin method.23 An aqueous solution of hydrogen tetrachloro-aurate (90 mL, 30 mmol dm�3) was extracted (three times with200 mL) with a toluene solution of methyltrioctylammoniumbromide (5.38 g, 1.38 mmol). Most of the tetrachloroaurate ionswere transferred to the organic layer as the yellow aqueous layerfaded until colorless. Alkanethiol was added to the toluenesolution (2:1mol ratio of alkanethiol to AuCl4�), and themixturewas reduced at 15 �Cwith a freshly prepared aqueous solution ofsodium borohydride (1.40 g, 30 mmol in 10 mL of deionizedH2O). A solution of borohydride was quickly added undervigorous stirring. After further stirring for 3 h, the organic phasewas separated, washed with pure water (2� 50 mL), evaporatedto 5 mL in a rotary evaporator, and mixed with 200 mL of
Figure 1. Structures of (a) bis[2-(4-oxy-2,2,6,6-tetramethylpiperidine-1-oxyl)ethyl] disulfide (TEMPO-DiSS) and Au nanoparticles protected with(b) C4SH, (c) C4SH/TEMPO thiol, and (d) C12SH/TEMPO thiol monolayers.

7349 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
absolute ethanol to precipitate NPs. The mixture was kept for12 h at�4 �C. The dark brown precipitate was sonicated for 60 sand centrifuged (5 min, 13 000 rpm). Again, precipitate wasdissolved in a small amount of toluene (5 mL), precipitated withethanol (100 mL), and centrifuged. Finally, all samples weredissolved in 20 mL of hexane and centrifuged for 5 min. Theprocedure was repeated until no trace of excess thiol was found,as determined by 1H NMR spectra and TLC. Obtained nano-particles were characterized by X-ray measurements. The goldcore size was ca. 2 nm.Ligand-Exchange Reaction. An organic ligand (20 mg, bis-
[2-(4-oxy-2,2,6,6-tetramethylpiperidine-1-oxyl)ethyl] disulfide,DiSS) was added to the toluene solution (20 mL) of startinggold NPs (20 mg), and the reaction proceeded upon mixing atroom temperature for ca. 72 h. As no precipitation and/or colorchange occurred, we inferred that this ligand-exchange reactionwas not accompanied by side effects, such as irreversible aggrega-tion or significant gold core size modifications. The clearburgundy solution was rotary evaporated, and the blackish solidwas dissolved in a minimum volume of toluene (max. 2 mL). Inthe next step, the NPs were precipitated using acetone, cooled to4 �C, and centrifuged 2 h later (13 000 rpm, 5min). This washingprocess was repeated four times until no trace of excess DiSS wasfound, as determined by TLC.Preparing Electrode Surfaces Coated by NPs Modified
with C4SH, C4SH/TEMPO Thiol, C12SH/TEMPO Thiol, and1,9-Nonanedithiol. For electrochemical measurements, a goldelectrode (BAS) was polished mechanically with 1.0, 0.3, and0.05 μm alumina powder on a Buehler polishing cloth to obtain agold mirror. After several washings with water, the electrodes wereplaced in an ultrasound bath for 10 min. Next, electrochemicalcleaning was carried out in 1 M NaOH by holding the electrodeat�1.5�1V for 60 s and scanning five times in the potential rangeof 1.5�1V. The last step of the electrode pretreatment waselectrochemical cycling in 0.5 M H2SO4 from �0.2 to 1.6 V untilthe characteristic shape of the voltammogram for a gold electrodewas observed.24 For the SPM experiments, gold evaporated onglass slides with a chromium underlayer was used (Arrandee) andannealed several times over a flame and then in an oven at 700 �C for30 min. For the IR experiments, the gold electrodes were cleaned ina mixture of 25% ammonia, 30% hydrogen peroxide, and water in a1:1:5 ratio at 80 �C for 30�60 min. Pretreated electrodes wereimmediately used for monolayer deposition.Self-assembly of the thiolated TEMPO derivative (TEMPO
thiol) on the gold electrode was carried out in a 5 mM solutionof TEMPO-DiSS in acetonitrile for 18 h at room temperature.During self-assembly, the solution was deoxygenated usingargon.For further comparison experiments, the monolayer of non-
anedithiol was also self-assembled from 1 mM 1,9-nonanedithiolsolution in ethanol for 24 h.The electrodes were immersed into the solution of nanopar-
ticles in toluene for 18 h; after vigorous washing with toluene andethanol, they were used in the experiments.Electrochemistry. Electrochemical measurements were per-
formed using the PGSTAT Autolab (Eco Chemie BV, Utrecht,The Netherlands). All electrochemical experiments were done ina three-electrode arrangement with a silver/silver chloride (Ag/AgCl) electrode as the reference, the platinum foil as the counter,and the Au electrode (BAS, 2 mm diameter) as the workingelectrode. The supporting electrolyte solution was 0.1 MTBAHFP in acetonitrile.
Scanning Tunneling Microscopy. STMmeasurements wereperformed using a Nanoscope IIIa (Digital Instruments, SantaBarbara, CA) and commercially available Pt�Ir tips. The imageswere taken under ambient conditions.Infrared Reflection Adsorption Spectroscopy.The infrared
(IR) experiments were carried out using the Nicolet 8700spectrometer (Thermo Scientific). The reflectance spectra oflayers adsorbed on plates were recorded using the variable-angleaccessory by Thermo Electron. For the far IR range (200�600 cm�1), the DTGS detector with a polyethylene window wasused. The spectra in the mid-IR range (400�4000 cm�1) wererecorded using the DTGS detector with a KBr window.
’RESULTS AND DISCUSSION
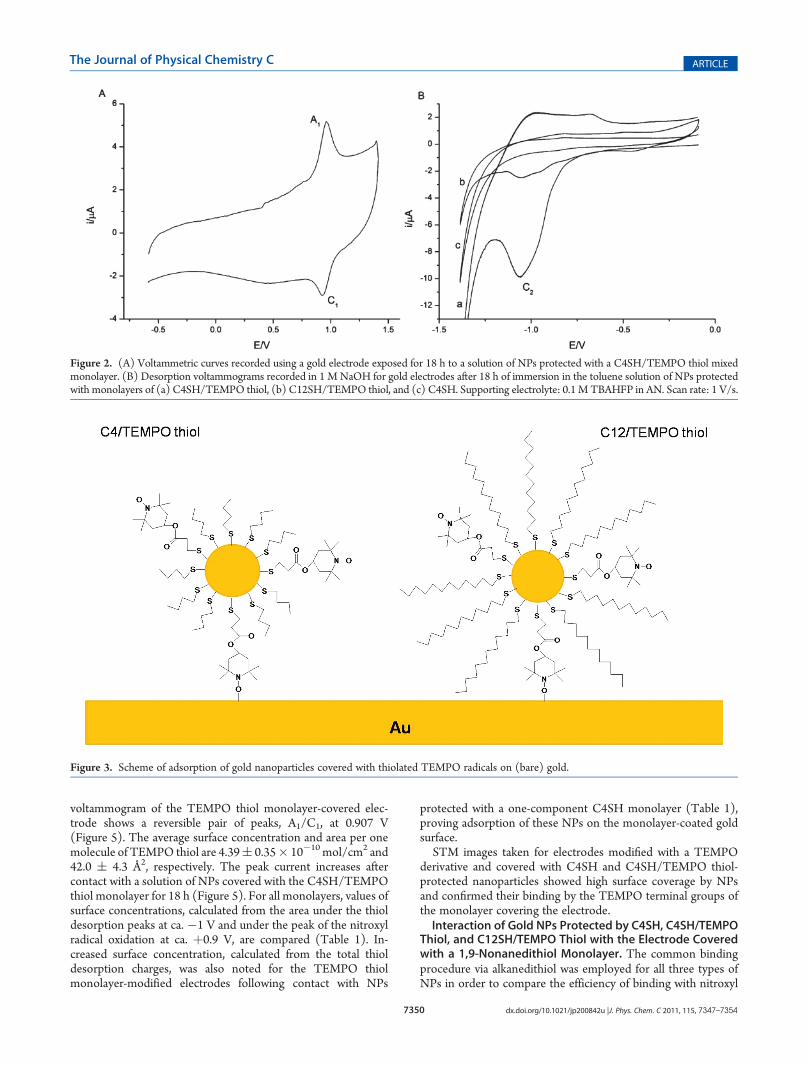
Interaction of Gold Nanoparticles Covered with C4SH,C4SH/TEMPO Thiol, and C12SH/TEMPO Thiol with a BareGold Electrode Surface. Figure 2A shows the voltammetriccurve recorded for a bare gold electrode immersed for 18 h in asolution of NP C4SH/TEMPO thiol. A pair of peaks, A1/C1 at0.950 V, characteristic of the TEMPO/TEMPOþ redox couple,proves the adsorption of TEMPO-decorated NPs on the surfaceof the bare gold electrode. When C12SH was used as the diluentin the monolayer protecting the NPs, the A1/C1 peaks could notbe resolved from the high background current (not shown).However, the desorption peak at �1 V was observed for bothC4SH/TEMPO thiol- and C12SH/TEMPO-protectedNPs self-assembled on gold electrodes. (Figure 2B, curve b) It confirmedthe presence of some adsorbed nanoparticles on the bare goldsurface also for C12SH/TEMPO-protected NPs, hence coveredwith a long-chain diluent thiol. No desorption peak was observedfor gold electrodes after 18 h of contact with a solution of C4SHNP devoid of TEMPO radicals (Figure 2B, curve c). This resultexcludes any physical adsorption of monolayer-protected nano-particles on the bare gold electrode surface. It also confirms thatthe presence of TEMPO groups in the coating area of the NP isindispensable to adsorb NPs on the gold surface. Therefore, anitroxyl radical�gold electrode interaction was considered(Figure 3).This suggested that C4SH/TEMPO thiol-protectedNPs, with
the short alkanethiol as the diluent in the monolayer, could beuseful for the construction of a sandwich-like assembly consistingof a gold NP layer and a gold electrode. The gold electrodescovered with these NPs were, therefore, imaged by STM. After18 h of contact with a solution of C4SH/TEMPO thiol NPs, thenanoparticles were tightly packed on the bare gold electrodesurface (Figure 4A) and could not be removed by thoroughrinsing of the gold surface with toluene and ethanol. However, inthe case of C12SH/TEMPO thiol-modifiedNPs, the STM imageshowed the presence of only a few NPs (Figure 4B), which wasattributed to the too long alkyl chains of the diluent C12SH. TheTEMPO groups were buried in the C12SH diluent, which madetheir access to the gold electrode surface much more difficult(Figure 3).Interaction of Gold NPs Coveredwith C4SH, C4SH/TEMPO
Thiol, and C12SH/TEMPO Thiol with a TEMPO Thiol-Mod-ified Electrode. To confirm binding through nitroxyl radicals,we conducted a series of experiments with electrodes covered byTEMPO thiol monolayers. When thiol and TEMPO terminalgroups are competing in binding to the gold substrate, theTEMPO groups of the monolayer are directed towardthe solution while thiol binds to the gold electrode. The
7350 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
voltammogram of the TEMPO thiol monolayer-covered elec-trode shows a reversible pair of peaks, A1/C1, at 0.907 V(Figure 5). The average surface concentration and area per onemolecule of TEMPO thiol are 4.39( 0.35� 10�10 mol/cm2 and42.0 ( 4.3 Å2, respectively. The peak current increases aftercontact with a solution of NPs covered with the C4SH/TEMPOthiol monolayer for 18 h (Figure 5). For all monolayers, values ofsurface concentrations, calculated from the area under the thioldesorption peaks at ca. �1 V and under the peak of the nitroxylradical oxidation at ca. þ0.9 V, are compared (Table 1). In-creased surface concentration, calculated from the total thioldesorption charges, was also noted for the TEMPO thiolmonolayer-modified electrodes following contact with NPs
protected with a one-component C4SH monolayer (Table 1),proving adsorption of these NPs on the monolayer-coated goldsurface.STM images taken for electrodes modified with a TEMPO
derivative and covered with C4SH and C4SH/TEMPO thiol-protected nanoparticles showed high surface coverage by NPsand confirmed their binding by the TEMPO terminal groups ofthe monolayer covering the electrode.Interaction of Gold NPs Protected by C4SH, C4SH/TEMPO
Thiol, and C12SH/TEMPO Thiol with the Electrode Coveredwith a 1,9-Nonanedithiol Monolayer. The common bindingprocedure via alkanedithiol was employed for all three types ofNPs in order to compare the efficiency of binding with nitroxyl
Figure 2. (A) Voltammetric curves recorded using a gold electrode exposed for 18 h to a solution of NPs protected with a C4SH/TEMPO thiol mixedmonolayer. (B) Desorption voltammograms recorded in 1 M NaOH for gold electrodes after 18 h of immersion in the toluene solution of NPs protectedwith monolayers of (a) C4SH/TEMPO thiol, (b) C12SH/TEMPO thiol, and (c) C4SH. Supporting electrolyte: 0.1M TBAHFP in AN. Scan rate: 1 V/s.
Figure 3. Scheme of adsorption of gold nanoparticles covered with thiolated TEMPO radicals on (bare) gold.

7351 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
radical bonding. Figure 6 shows the voltammetric curves re-corded for a 1,9-nonanedithiol-modified gold electrode im-mersed for 18 h in a solution of NP: C4SH/TEMPO thiol(Figure 6, curve a) and C12SH/TEMPO thiol (Figure 6, curve b).Gold NPs covered with a C4SH/TEMPO thiol monolayer gave apair of peaks at a formal potential equal to 0.950 V, identicalwith the formal potential for the same NPs immobilized on baregold surfaces. Gold NPs coated with mixed monolayers witha longer alkanethiol—C12SH/TEMPO thiol—gave a pair ofpeaks with a formal potential equal to 0.993 V. The more positivevalue potential of the A1/C1 couple reflects the more hydrophobicenvironment of the nitroxyl radical groups. The values of surfaceconcentrations calculated from the voltammetric curves of theTEMPO moiety oxidation (A1 peak) and desorption curves (C2
peak) are compared in Table 1. The alkanedithiol approach allowsthe binding of NPs protected with both long and short alkanethiols
as diluents, but the population of NPs is always larger in the case ofnitroxyl radicals as the binding unit.STM pictures recorded for the 1,9-nonanedithiol-modified
electrode after 18 h of contact with NPs protected with a C4SH/TEMPO thiol mixed monolayer confirm that the electrodesurface is less densely covered with the NPs than in cases ofthe bare gold substrate or modified with TEMPO thiol. Inaddition, NPs are shown to agglomerate and occupy mainlythe edges of the terraces and cracks of the electrode (Figure 7).IR Studies of NP-Covered Electrodes.To prove that binding
of NO�Au is responsible for the immobilization of the NPs atthe gold electrode surface, the FAR IR spectra were recorded.The curve a in Figure 8 shows the spectrum of the gold electrodeafter 18 h of contact with a toluene solution of TEMPOmolecules not containing thiol groups. For these molecules,the bond of NO�Au (electrode) is the onlymeans to immobilize
Figure 5. Voltammograms of TEMPO/TEMPOþ electrode processes in a TEMPO thiol monolayer-covered Au electrode. Self-assembly solution:5mMTEMPO-DiSS in AN. Voltammograms recorded before (a) and after 18 h of contact with a toluene solution of C4SH/TEMPO thiol (b) or C4SH(c) protected NPs. Supporting electrolyte: 0.1 M TBAHFP in AN. Scan rate: 1 V/s.
Figure 4. STM images of gold electrodes coated with gold nanoparticles protected with C4SH/TEMPO thiol (A) and C12SH/TEMPO thiol(B) mixed monolayers. NP deposition time: 18 h.

7352 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
them on the surface of gold. The spectrum is characterized by agroup of three peaks at frequencies of 530, 470, and 435 cm�1
and a single peak at the frequency of 280 cm�1, which corre-sponds to the binding of NO�Au (electrode). The observedvibration at frequencies higher than 430 cm�1 can be assigned toONC andNCCdeformationmodes. In the spectrum of TEMPOregistered in the KBr pellet (not shown), wide overlapping bandsbetween 450 and 600 cm�1 were also observed. The appearanceof that group of bands in the TEMPO spectrum in the absence ofa gold surface proves that the peaks at frequencies of 530, 470,and 435 cm�1 cannot correspond to the molecule�gold surfaceinteraction.The ONC and NCC deformation modes shift toward lower
frequencies in the spectra of the TEMPO monolayer-coveredelectrode immersed for 18 h in the C4SH/TEMPO thiol-protected NP solution (Figure 8, spectrum b). The frequencyshift is probably caused by the interaction with gold NPs and theC4SH neighboring the TEMPO molecules. Indeed, this type ofinteraction may lead to the high population of NPs observed inthe STM images (Figure 4).In addition, the spectra of a pure gold electrode after placing
on it C4SH NPs in toluene solution and evaporating the solvent(not shown) do not show the peaks at frequencies about 400 and280 cm�1, confirming the assignment of these vibration fre-quency peaks to binding by means of the NO�Au interaction ofthe NPs modified with TEMPO thiol.Spectrum c in Figure 8 was recorded for the gold electrode
coated with a monolayer of TEMPO thiol. In this case, weobserved the intense peak at a frequency of 360 cm�1, whichcorresponds to the S�Au bond. This confirms that thiols have alarger affinity to the gold substrates than the nitroxyl moiety.After 18 h of contact with a toluene solution of NP C4SH/TEMPO, we observed the appearance of peaks at a frequency of420 cm�1, hence corresponding to that of the ONC and NCCdeformation modes, and peaks at a frequency of 280 cm�1,corresponding to the NO�Au (NP) bond (Figure 8, spectrumd). The low-intensity peaks compared to the peak of the S�Aubond can be explained by the small population of NO in themixed monolayer covering the NPs, resulting in a small numberof NO�Au electrode bonds and by the good organization of theTEMPO thiol monolayer, providing vertical orientation of theS�Au bound molecules. Selection rules for the reflectancespectra enhance vibration perpendicular to the metal surface.25
Because the fraction of TEMPO molecules bonded to gold NPsoriented parallel to the gold electrode is small, their contributionis not visible in the spectrum.
’CONCLUSION
In summary, we propose building sandwich assemblies con-sisting of a gold electrode and a layer of gold nanoparticlesseparated by a TEMPO derivative with the nitroxyl radicalplaying the role of a binding unit. It is a new binding schemecompared to the common approach based on alkanedithiols. Theinteractions of Au with the >NO• radicals were noted earlier.14,15
Decreased EPR signals were observed in the gold�TEMPOsystem and ascribed to the exchange interaction of unpairedelectrons with conduction-band electrons of the metallic particle.In the present study, gold�gold NP assemblies separated bythiolated TEMPO radicals have been successfully developedusing the to and from approach. In the to approach, Au�NPassemblies are formed due to NO�Au bond via the NO• radicalof the TEMPO thiol-modified gold NP pointed toward the bare
Table 1. Values of TEMPOThiol Surface Concentrations Calculated from the Area under the TEMPOOxidation Peak (ΓTEMPO)and the Thiol Desorption Peak (Γthiol)
covered with NPs
without NPs C4SH/TEMPO thiola C12SH/TEMPO thiolb C4SHc
electrode
Γthiol [mol/cm2]
from desorption
(10�10)
ΓTEMPO [mol/
cm2] TEMPO
groups (10�10)
Γthiol [mol/cm2]
from desorption
(10�10)
ΓTEMPO [mol/
cm2] TEMPO
groups (10�10)
Γthiol [mol/cm2]
from desorption
(10�10)
ΓTEMPO [mol/
cm2] TEMPO
groups (10�10)
Γthiol [mol/cm2]
from desorption
(10�10)
ΓTEMPO [mol/
cm2] TEMPO
groups (10�10)
Au 57.8( 5.7 1.48( 0.11 11.2( 2.5
alkanedithiol-
modified Au
8.23( 0.31 17.8 ( 1.5 0.78( 0.07 10.9( 1.1 0.34( 0.05 11.8( 1.2
TEMPO thiol-
modified Au
4.84( 0.86 4.39( 0.35 18.1 ( 4.0 4.66( 0.3 4.18( 0.6 4.12( 0.36 12.9( 3.6 3.45( 0.2
aC4SH/TEMPO thiol: NPs modified with a mixed monolayer of butanethiol and TEMPO thiol. bC12SH/TEMPO thiol: NPs modified with a mixedmonolayer of dodecanethiol and TEMPO thiol. cC4SH: nanoparticles modified with a monolayer of butanethiol.
Figure 6. Voltammograms recorded in 0.1 M TBAHFP/AN solutionusing a 1,9-nonanedithiol modified-gold electrode after 18 h of immer-sion in a toluene solution of NPs protected with (a) C12SH/TEMPOthiol and (b) C4SH/TEMPO thiol. Scan rate: 1 V/s.

7353 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
gold surface. The complementary from approach generatesAu�Au NPs sandwich assemblies in which the NO• radical-forming NO�Au bond resides on the gold electrode surfaces.Thiolated TEMPO adsorbs on the NP via the thiol because of itsstronger affinity toward gold. Indeed, the S�Au bond is strongerthan NO�Au, and the latter cannot compete in binding to gold.In addition, the TEMPO group is much larger than the thiolgroup and has bulky substituents: four methyl groups in the ring.Therefore, in the FAR IR spectrum of the TEMPO monolayer,the band corresponding to the NO�Au bond is absent. When allthiol moieties formed from the disulfide groups are anchored on
gold (either on NPs or on the Au electrode), then bonding bymeans of the NO radical can take place, and it is easily recognizedin the spectra.
The monolayer of TEMPO thiol self-assembled on the goldelectrode is able to bind all kinds of AuNPs bymeans of the sameNO�Au binding. We also show that linking through nitroxylradicals leads to gold surfaces more densely covered by NPs thanin the case of binding by an alkanedithiol. The smaller populationof NPs in the case of alkanedithiol binding is due to the overlytight packing of 1,9-nonanedithiol molecules on the gold surfaceand restricted access of the terminal thiol group to the surface ofthe NPs. Nitroxyl groups present in a small population in thebutanethiol monolayer protecting the NPs (ca. 10%) protrudeabove it and, therefore, provide easily accessible sites for tether-ing the NPs to the electrode surface.
’AUTHOR INFORMATION
Corresponding Author*Tel:þ48 22 8220211. Fax:þ48 22 8225996. E-mail: [email protected].
’ACKNOWLEDGMENT
The authors would like to thank Michal Wojcik and WiktorLewandowski from the Laboratory of Natural Products Chem-istry for their assistance in preparing C4SH and C12SH mono-layer-protected nanoparticles.
’REFERENCES
(1) Borbat, P. P.; Costa-Filho, J.; Earle, K. A.; Moscicki, J. K.; Freed,J. H. Science 2001, 291, 266–269.
(2) Mason, R. P. Free Radical Biol. Med. 2004, 36, 1214–1223.(3) Mitchell, J. B.; Krishna, M. C.; Kuppusamy, P.; Cook, J. A.;
Russo, A. Exp. Biol. Med. (Maywood, NJ, U.S.) 2001, 226, 620–621.
Figure 7. STM images of a gold electrode coated with a monolayer of 1,9-nonanedithiol and AuNPs protected with a C4SH/TEMPO thiol monolayer.NP deposition time: 18 h.
Figure 8. IR spectra of a Au electrode after 18 h of contact with(a) TEMPO and (b) C4SH/TEMPO thiol-protected NP solution andspectra of TEMPO thiol SAMs on a Au electrode before (c) and after(d) 18 h of contact with C4SH/TEMPO thiol-protected NP solution intoluene.

7354 dx.doi.org/10.1021/jp200842u |J. Phys. Chem. C 2011, 115, 7347–7354
The Journal of Physical Chemistry C ARTICLE
(4) Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101,3661–3688.(5) Minisci, F.; Recupero, F.; Pedulli, G. F.; Lucarini, M. J. Mol.
Catal. A: Chem. 2003, 204�205, 63–90.(6) Takeshi, I.; Rainer, B.; Frings, B.; Lachowicz, A.; Soichi, K.;
Hiroyuki, N. Chem. Commun. 2010, 46, 3475–3477.(7) Glandut, N.; Monson, C. F.; Majda, M. Langmuir 2006, 22,
10697–10704.(8) Finklea, H. O.; Madhiri, N. J. Electroanal. Chem. 2008, 621,
129–133.(9) Al�eveque, O.; Seladji, F.; Gautier, Ch.; Dias, M.; Breton, T.;
Levillain, E. ChemPhysChem 2009, 10, 2401–2404.(10) Gautier, Ch.; Al�eveque, O.; Seladji, F.; Dias, M.; Breton, T.;
Levillain, E. Electrochem. Commun. 2010, 12, 79–82.(11) Zhang, Z.; Berg, A.; Levanon, H.; Fessenden, R. W.; Meisel, D.
J. Am. Chem. Soc. 2003, 125, 7959–7963.(12) Daniel, M. C.; Astruc, D. Chem. Rev. 2004, 104, 293–346.(13) Sch€atz, A.; Reiser, O.; Stark, W. J. Chem.—Eur. J. 2010,
16, 8950–8967.(14) Barkley, P. G.; Hornak, J. P.; Freed, J. H. J. Chem. Phys. 1986,
84, 1886–1900.(15) Krukowski, P.; Kowalczyk, P. J.; Krzyczmonik, P.; Olejniczak,
W.; Klusek, Z.; Puchalski, M.; Gwozdzinski, K. Appl. Surf. Sci. 2009,255, 3946–3952.(16) Ouyang, J. Y.; Yang, Y Appl. Phys. Lett. 2010, 96, 063506.(17) Sek, S.; Bilewicz, R.; Slowinski, K. Chem. Commun. 2004,
404–405.(18) Stolarczyk, K.; Bilewicz, R. Electrochim. Acta 2004, 51,
2358–2365.(19) Sek, S.; Tolak, A.; Misicka, A.; Bilewicz, R. J. Phys. Chem. B
2005, 109, 18433–18438.(20) Sugawara, T.; Matsushita, M. M. J. Mater. Chem. 2009, 19,
1738–1753.(21) Nicolay, R.; Marx, L.; H�emery, P.; Matyjaszewski, K. Macro-
molecules 2007, 40, 9217–9223.(22) Wojcik, M.; Lewandowski, W.; Matraszek, J.; Mieczkowski, J.;
Borysiuk, J.; Pociecha, D.; Gorecka, E. Angew. Chem., Int. Ed. 2009,48, 5167–5169.(23) Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D. J.; Whyman, R.
J. Chem. Soc., Chem. Commun. 1994, 801–802.(24) Hoare, J. P. J. Electrochem. Soc. 1984, 131, 1808–1815.(25) Greenler, R. G. J. Chem. Phys. 1966, 44, 310–315.




















