
Ph.D Thesis Diversity and Antagonistic Activity of Endophytic ...
Upload
independentCategory
view
0download
0
,
*Departamento de Bioquımica y Biologıa Molecular y Fisiologıa, Facultad de Medicina, Universidad de Valladolid, Instituto de
Biologıa y Genetica Molecular, CSIC, Ciber de Enfermedades Respiratorias, CIBERES, Instituto de Salud Carlos III, Valladolid,
Spain
�Department of Pharmacology, Faculty of Medical Sciences, New University of Lisbon, Lisbon, Portugal
Carotid bodies (CB) are peripheral chemoreceptors that sensechanges in arterial PO2, PCO2 and pH levels. Hypoxia,acidosis and hypercapnia activate CB which responds byincreasing the frequency of sensory fibers of the carotid sinusnerve (CSN). Central projections of the CSN terminate in thebrainstem, where the firing frequency is integrated by centralrespiratory control system inducing compensatory ventilatoryresponses. The structures that sense PO2, PCO2 and pHlevels are chemoreceptor cells, which are synapticallyconnected with the sensory nerve endings of the CSN (fora review see Gonzalez et al. 1994). Several neurotransmittersystems participate in this sensory synapse. Among them,dopamine (DA) and adenosine have received great attention(Gonzalez et al. 1994; Gauda 2002), but there are aspects ofthe physiology of these neurotransmitter systems (e.g.,interaction between them) that have received little attention.
Adenosine has been defined as an excitatory neurotrans-mitter in the CB because adenosine administration increased
CSN chemosensory activity and ventilation in several species(McQueen and Ribeiro 1981, 1983; Monteiro and Ribeiro1987, 1989; Runold et al. 1990; Ribeiro and Monteiro 1991)including humans (Watt and Routledge 1985; Uematsu et al.,2000). The physiological significance of these findingsemerge from the observation that mild hypoxia increasesadenosine release in an in vitro preparation of rat CB in aspecific manner (Conde and Monteiro 2004). Recentexperiments, also in rat CB in vitro, have established that
Received June 3, 2008; revised manuscript received July 31, 2008;accepted September 14, 2008.Address correspondence and reprint requests to Silvia V. Conde,
Department of Pharmacology, Faculty of Medical Sciences, New Uni-versity of Lisbon, Campo Martires da Patria, 130, 1169-056 Lisbon,Portugal. E-mail: [email protected] used: ADA, adenosine deaminase; CA, catecholamines;
CB, carotid body; CSN, carotid sinus nerve; DA, dopamine; NECA, 5¢-(N-ethylcarboxamido)adenosine.
Abstract
We have previously demonstrated that adenosine controls
the release of catecholamines (CA) from carotid body (CB)
acting on A2B receptors. Here, we have tested the hypothesis
that the control is exerted via an interaction between aden-
osine A2B and dopamine D2 receptors present in chemore-
ceptor cells. Experiments were performed in vitro in CB from
3 months rats. The effect of A2B adenosine and D2 dopamine
agonists and antagonists applied alone or in combination
were studied on basal (20%O2) and hypoxia (10%O2)-
evoked release of CA and cAMP content of CB. We have
found that adenosine A2 agonists and D2 antagonists dose-
dependently increased basal and evoked release CA from
the CB while A2 antagonists and D2 agonists had an inhibi-
tory action. The existence of A2B-D2 receptor interaction was
established because the inhibitory action of A2 antagonists
was abolished by D2 antagonists, and the stimulatory action
of A2 agonists was abolished by D2 agonists. Further, A2
agonists increased and D2 agonist decreased cAMP content
in the CB; their co-application eliminated the response. The
present results provide direct pharmacological evidence that
an antagonistic interaction between A2B adenosine and D2
dopamine receptors exist in rat CB and would explain the
dopamine-adenosine interactions on ventilation previously
observed.
Keywords: adenosine, adenosine A2B receptors, carotid
body, D2 dopamine receptors, interactions.
J. Neurochem. (2008) 107, 1369–1381.
JOURNAL OF NEUROCHEMISTRY | 2008 | 107 | 1369–1381 doi: 10.1111/j.1471-4159.2008.05704.x
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381 1369

adenosine mediates up to 60% of the low PO2-induced CSNactivity (Conde et al. 2006). About half of this effect waspost-synaptic and mediated by A2A receptors and the rest waspre-synaptic mediated by A2B receptors and associated to anaugmentation of the release of catecholamine (CA) fromchemoreceptor cells (Conde et al. 2006).
Chemoreceptor cells have a high DA content in all speciesincluding the rat, and an ample series of physiological andpharmacological experiments indicate that it is an inhibitoryneurotransmitter in all species (except might be in the rabbit;Fidone et al. 1982; Gonzalez et al. 1994; Iturriaga andAlcayaga 2004). CB expresses dopamine D1 and D2
receptors differentially located. D1 receptors seem to belocated in the vasculature of the organ (Almaraz et al., 1991)while D2 receptors are located pre-synaptically on chemore-ceptor cells and post-synaptically on CSN sensory nerveendings (Dinger et al. 1981; Gauda 2002). Early studies havesuggested dopamine-adenosine interactions in the CB. Forexample, Ribeiro and McQueen (1983) found that adenosinepotentiated the inhibitory effect of DA on cat CSN activity.Similarly, Monteiro and Ribeiro (2000) in in vivo experi-ments in the rat observed that the inhibitory effect of D2
dopamine agonists on ventilation was attenuated or evenreversed by the blockage of A2 receptors.
Interactions between adenosine and dopamine receptorsare well known to occur in the central nervous system. Theinteractions imply that the occupancy of the DA receptorsmodify the effectiveness of activation (or inhbition) of Adoreceptors and vice versa (Ferre et al. 1997; Fuxe et al. 2007;Fredholm 2007). Molecular mechanisms of these interactionsare complex and include formation of heteroreceptors andchange in the rate of receptor trafficking between the cellinterior and the plasma membrane (Fuxe et al. 2007).Additionally, A2 and D2 receptors are G-protein coupledreceptors linked to adenylyl cyclases in opposite manners: A2
receptors are coupled to Gs protein and their activationincreases cAMP levels (Fredholm, 2007) while D2 receptorsare coupled inhibitory Gi/Go proteins and their activationdecreases cAMP levels (Zeng et al. 2007). Therefore,interaction between both neurotransmitter systems can alsooccur at the second messenger level and its signallingcascade (Ferre et al. 1997; Xu et al. 2005; Fuxe et al. 2007).
Our hypothesis is that previously observed activation ofCA release induced by adenosine in chemoreceptors cells ismediated by an interaction between D2 and A2B receptors.Our findings have confirmed the hypothesis on demonstrat-ing that A2 agonists and D2 antagonists dose-dependentlyincreased basal and evoked release CA from the CB while A2
antagonists and D2 agonists had an inhibitory action; it wasalso observed that inhibition produced by A2 antagonists wasabolished by D2 antagonists and stimulation produced by A2
agonists was abolished by D2 agonists and by an A2B
antagonist. The effects of the A2B and D2 agonists andantagonists tested on CA release are reproducible at the level
of cAMP, implying that this second messenger mediates A2B
and D2 interactions. All these findings reveal for the first timethe existence of an antagonistic receptor-receptor interactionbetween A2B receptors and D2 receptors in carotid bodychemoreceptor cells.
Material and methods
Animals and surgical proceduresExperiments were performed in Wistar adult rats of both sexes
(250–350 g) obtained from vivarium of the Faculty of Medicine of
the University of Valladolid and from the vivarium of the Faculty of
Medical Sciences of the New University of Lisbon. The Institutional
Committee of the University of Valladolid and New University of
Lisbon for Animal Care and Use approved the protocols. Rats were
anaesthetized with sodium pentobarbital (60 mg/kg i.p.), tracheos-
tomized and the carotid arteries were dissected past the carotid
bifurcation. The carotid bifurcation was placed in a Lucite chamber
in ice-cold 95% O2-equilibrated Tyrode (in mM: NaCl 140; KCl 5;
CaCl2 2; MgCl2 1.1; HEPES 10; glucose 5.5, pH 7.40 and the CB
was cleaned free of CSN and nearby connective tissue (Vicario et al.2000). For CA release experiments 8–12 CBs/experiment were used
and for cAMP CB content experiments 4–9 CB/experiment were
used. In all instances animals were killed by an intracardiac
overdose of sodium pentobarbital.
Labelling of catecholamines stores: release of 3H-CATo monitor the release of CAwe have used a radioisotopic method as
described by Fidone et al. (1982) and Vicario et al. (2000). In brief,
CA stores of the chemoreceptor cells were labelled by incubating the
CBs during 2 h in Tyrode solution containing the natural precursor3H-tyrosine (30 lM) (specific activity of 45 Ci/mmol) and 6-methyl-
tetrahydropterine (100 lM) and ascorbic acid (1 mM), cofactors of
tyrosine hydroxylase and dopamine-b-hydroxylase, respectively
(Fidone and Gonzalez 1982). After the labelling period, individual
CBs were transferred to vials containing 4 mL of precursor-free
Tyrode-bicarbonate solution (composition as above except for the
substitution of 24 mM of NaCl by 24 mM of NaHCO3). Solutions
were continuously bubbled with 20%O2/5% CO2/75% N2 saturated
with water vapour, except when applied hypoxic stimuli. The
solutions of the initial incubation periods (3 · 20 min) were
discarded to washout the precursor and the readily releasable pool
of labelled CA (Almaraz et al. 1986) and thereafter renewed at fixed
times (every 10 min; see Results) and collected for subsequent
analysis of their 3H-CA content. Specific protocols for stimulus and
drug applications are provided in the Results. Stimulus included
hypoxia 10% O2-equilibrated solutions (PO2 � 66 mmHg) and high
external K+ (30 mM) The agonist and antagonists used were:
caffeine (a non-selective adenosine antagonist, 1 mM), domperidone
(a D2 dopamine antagonist, 0.5 nM–5 lM) haloperidol (a D2
dopamine antagonist, 0.01–10 lM), 8-[4-[((4-cyanophenyl)car-
bamoylmethyl)-oxy]phenyl]-1,3-di(n-propyl)xanthine (MRS 1754,
a selective A2B adenosine antagonist, 200 nM) 5¢-(N-ethylcarbox-amido)adenosine (NECA, an adenosine A2 agonist; 0.1–100 lM),
propylnorapomorphine (a D2 dopamine agonist, 0.02–200 nM) and
sulpiride (a D2 dopamine antagonist,1 lM). Incubating solutions
collected were acidified with glacial acetic acid and 1 mM ascorbic
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1370 | S. V. Conde et al.

acid to pH = 3 and maintained at 4�C until analysis to prevent 3H-
CA degradation. 3H-CA present in the incubating solutions were
analyzed by adsorption onto alumina at alkaline pH (pH = 8.6) and
bulk elution with hydrochloric acid (1 N) and quantified by
scintillation counting. CB were homogenized (glass to glass; 4�C)in 200 lL of perchloric acid 0.6 M and centrifuged for 10 min (4�C;12 000 g), in a microfuge (Beckmann, Madrid, Spain). Supernatants
were analyzed for their 3H-catechol content as described above for
the incubating solutions. Previous HPLC analyses have shown that
over 90% of the 3H-catechols present in the alumina eluates
correspond to 3H-DA and its catabolite 3H-DOPAC (Vicario et al.2000).
Release data are presented either as dpm of 3H-CA released or
expressed as percentage of 3H-CA of the tissue content; this last
presentation corrects for the potential variations in CB size and/or
rate of 3H-CA synthesis.
Quantification of cAMP carotid body contentImmediately after surgical removal, the CBs were pre-incubated for
15 min at 37�C in a shaker bath in 95% O2/5% CO2-equilibrated
Tyrode (composition as above except for the substitution of 24 mM
of NaCl by 24 mM of NaHCO3) to allow the recovery of the
preparation. After the pre-incubation period, the CBs were placed in
2 mL Eppendorf tubes containing 1 mL of fresh incubating solution
of identical composition and containing 500 lM isobutylmethyl-
xanthine (IBMX, Sigma-Aldrich, Madrid, Spain), a phosphodies-
terase inhibitor. The incubating solutions were equilibrated 20%O2/
5%CO2/75%N2 (normoxia), lasted 30 min and was also carried out
in the metabolic bath at 37�C. Specific protocols for drug
applications are provided in the Results. The agonist and antagonists
used were: adenosine deaminase (ADA, enzyme that catabolise
adenosine, 2 U/L), 2-p-(2-carboxyethyl)phenethyl-amino-5¢-N-ethylcaboxamido-adenosine hydrochloride (CGS 21680, A2A selec-
tive adenosine receptor antagonist, 0.03 and 1 lM), dopamine
(300 lM), forskolin (FSK, an activator of adenylyl cyclase, 3 lM),
8-[4-[((4-cyanophenyl)carbamoylmethyl)-oxy]phenyl]-1,3-di(n-pro-
pyl) xanthine (MRS 1754, a selective A2B adenosine receptor
antagonist, 200 nM) NECA (an adenosine A2 receptor agonist; 1
and 10 lM), propylnorapomorphine (a D2 dopamine receptor
agonist, 200 nM). Extraction of cyclic nucleotides and cAMP
quantification were performed as previously described by Batuca
et al. (2003). In brief, to extract cyclic nucleotides from the CBs
they were immersed in cold 6% (w/v) trichloroacetic acid (600 lL)for 10 min, homogenised using a Potter glass to glass homogeniser
and centrifuged at 12 000 g for 10 min (4�C). The supernatants
were washed four times in 3 mL of water-saturated diethyl ether
solution, the remaining aqueous phase was lyophilised, and the
sealed samples were stored at )20�C until cAMP was assayed using
an EIA commercial kit (GE Healthcare Bio-Sciences AB, Uppsala,
Sweden). Data for cAMP levels are expressed pmole/CB.
Drugs and chemicals6-Methyl-tetrahydropterine, adenosine deaminase, ascorbic acid,
caffeine, CGS 21860, dopamine, domperidone, forskolin, haloper-
idol, MRS 1754, NECA, propylnorapomorphine and sulpiride were
all obtained from Sigma-Aldrich. 3H-tyrosine was obtained from
Amersham (Madrid, Spain). Domperidone, haloperidol, NECA and
sulpiride were prepared as 5 mM stock solutions in dimethylsulf-
oxide; the final concentration of dimethylsulfoxide was always
below 1/500 which by itself lacks effects on our preparations.
Propylnorapomorphine was prepared in a 5 mM stock solution in
methanol.
Data analysisData were evaluated using a Graph Pad Prism Software, version 4
(GraphPad Software Inc., San Diego, CA, USA) and were presented
as mean ± SEM. Dose-response curves were fitted to Sigmoidal
dose-response curves and the IC50, EC50 and the Emax values were
obtained directly from the Sigmoidal dose-response equation. The
significance of the differences between the means was calculated by
unpaired Student’s t-test and by One and Two-Way Analysis of
Variance (ANOVA) with Dunnett’s and Bonferroni multiple compa-
rison tests, respectively. p values of 0.05 or less were considered to
represent significant differences.
Results
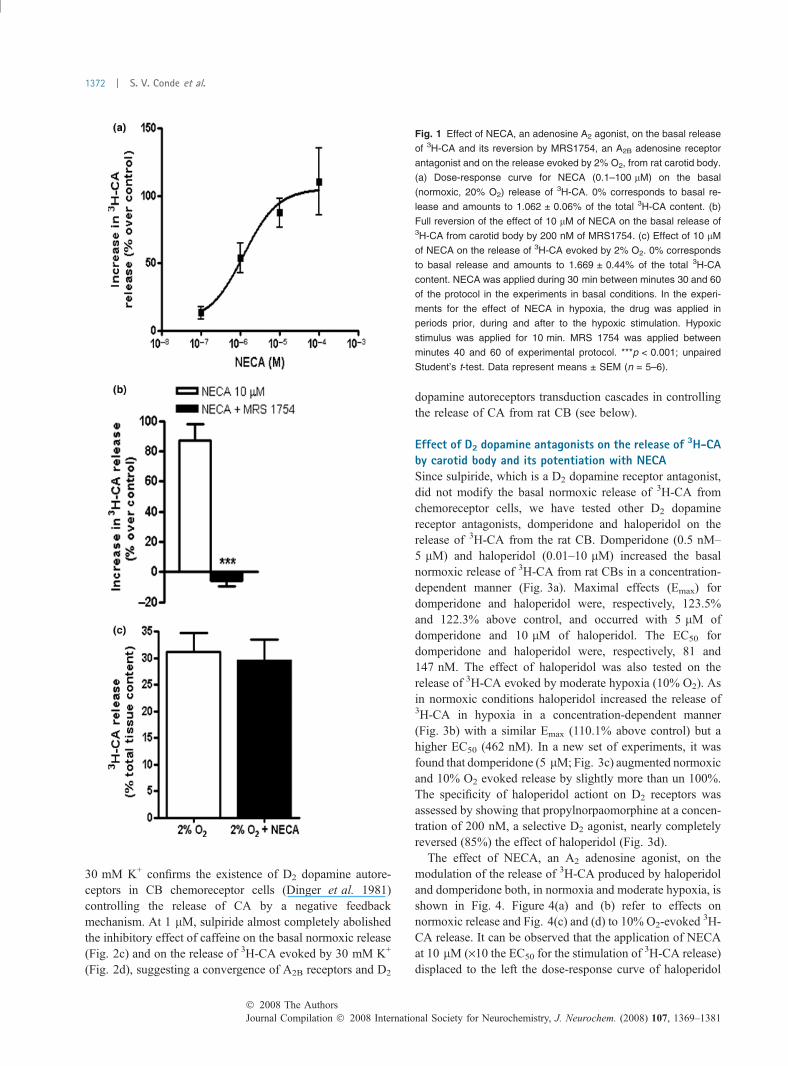
Effect of NECA, an A2 adenosine agonist on the release of3H-CA from rat carotid bodyCaffeine at low concentrations is a non-selective antagonistof adenosine receptors that inhibits the basal and low, but nothigh intensity, stimuli-evoked release of 3H-CA from CBchemoreceptor cells (Conde et al. 2006). Figure 1(a) showsthat NECA increases the release of 3H-CA in basal normoxicconditions (PO2 �133 mmHg) in a concentration-dependent;NECA maximal effect (Emax) was 105.3% (i.e., NECA morethan doubled basal release) and had an EC50 of 1.22 lM(Fig. 1a). This figure also shows that 200 nM of MRS1754, aspecific A2B specific antagonist reversed in full the activationof normoxic release of 3H-CA induced by NECA (Fig. 1b),indicating, as previously concluded (Conde et al. 2006), thatadenosine actions on the release of 3H-CA result fromspecific actions on A2B adenosine receptors located inchemoreceptor cells.
Previously, we have also shown (Conde et al. 2006) thatNECA stimulates the release of 3H-CA evoked by moderatehypoxia. Now we show that NECA, as it was the case withcaffeine and other adenosine agonists and antagonists (Condeet al. 2006), was not able to modify the release of 3H-CAevoked by intense hypoxic stimulation (incubation with 2%O2-equilibrated solution; PO2 � 22 mmHg; Fig. 1c).
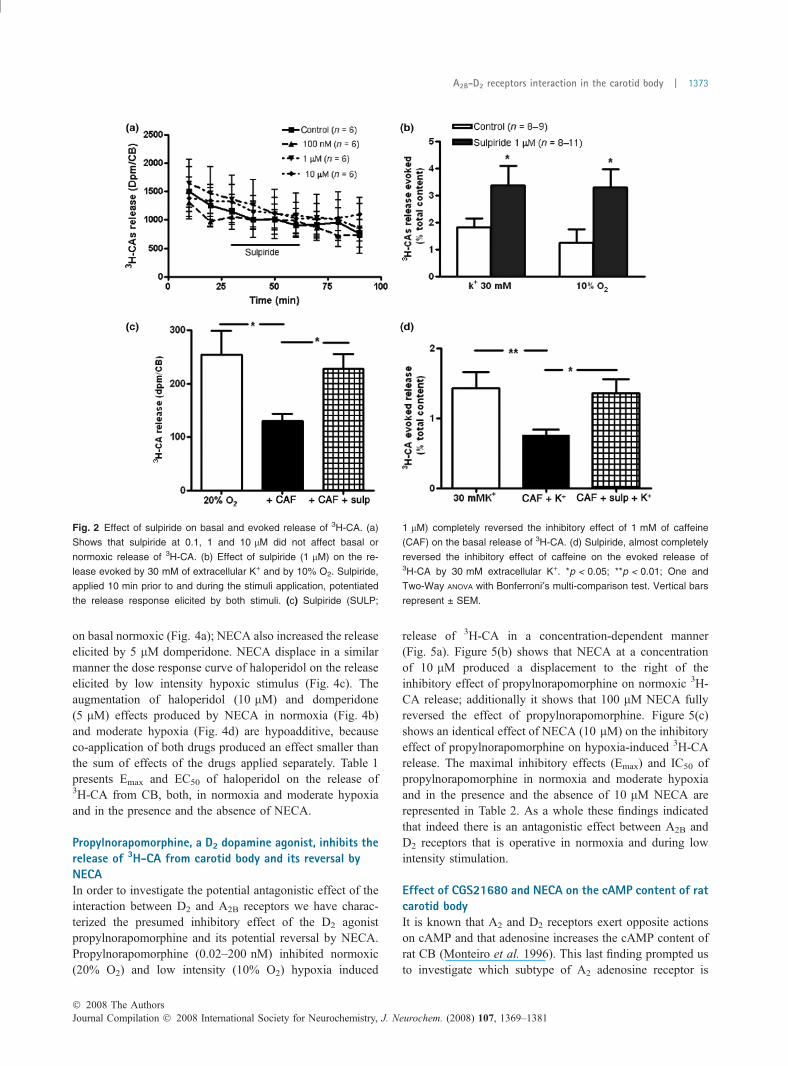
Reversion of the inhibitory effect of caffeine on the 3H-CAby a D2 dopamine antagonist, sulpirideSulpiride, which is a D2 dopamine receptor antagonist, didnot modify the basal release of 3H-CA from rat CB in therange of concentrations of 0.1–10 lm (Fig. 2a). However, at1 lM it produced an increase of 106.1% and 163.5% in therelease of 3H-CA evoked, respectively, by 30 mM K+ and10% O2 (Fig. 2b). The fact that sulpiride induced an increasein the release of 3H-CA evoked by moderate hypoxia and
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1371
30 mM K+ confirms the existence of D2 dopamine autore-ceptors in CB chemoreceptor cells (Dinger et al. 1981)controlling the release of CA by a negative feedbackmechanism. At 1 lM, sulpiride almost completely abolishedthe inhibitory effect of caffeine on the basal normoxic release(Fig. 2c) and on the release of 3H-CA evoked by 30 mM K+
(Fig. 2d), suggesting a convergence of A2B receptors and D2
dopamine autoreceptors transduction cascades in controllingthe release of CA from rat CB (see below).
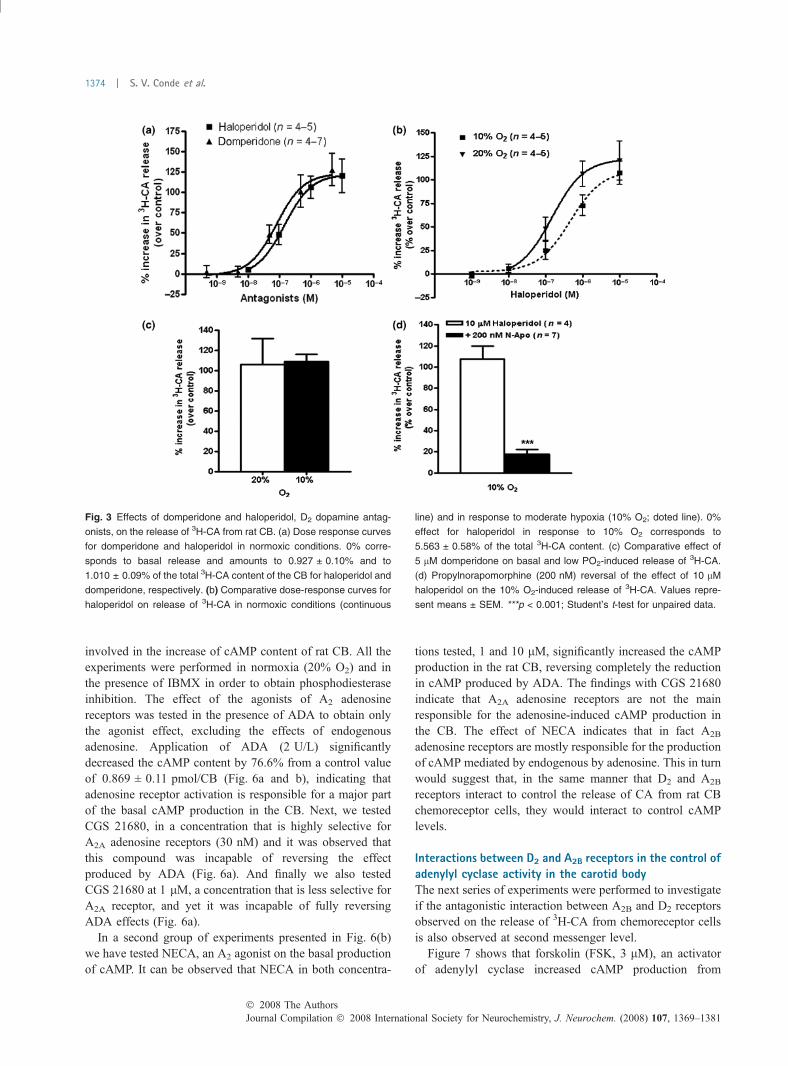
Effect of D2 dopamine antagonists on the release of 3H-CAby carotid body and its potentiation with NECASince sulpiride, which is a D2 dopamine receptor antagonist,did not modify the basal normoxic release of 3H-CA fromchemoreceptor cells, we have tested other D2 dopaminereceptor antagonists, domperidone and haloperidol on therelease of 3H-CA from the rat CB. Domperidone (0.5 nM–5 lM) and haloperidol (0.01–10 lM) increased the basalnormoxic release of 3H-CA from rat CBs in a concentration-dependent manner (Fig. 3a). Maximal effects (Emax) fordomperidone and haloperidol were, respectively, 123.5%and 122.3% above control, and occurred with 5 lM ofdomperidone and 10 lM of haloperidol. The EC50 fordomperidone and haloperidol were, respectively, 81 and147 nM. The effect of haloperidol was also tested on therelease of 3H-CA evoked by moderate hypoxia (10% O2). Asin normoxic conditions haloperidol increased the release of3H-CA in hypoxia in a concentration-dependent manner(Fig. 3b) with a similar Emax (110.1% above control) but ahigher EC50 (462 nM). In a new set of experiments, it wasfound that domperidone (5 lM; Fig. 3c) augmented normoxicand 10% O2 evoked release by slightly more than un 100%.The specificity of haloperidol actiont on D2 receptors wasassessed by showing that propylnorpaomorphine at a concen-tration of 200 nM, a selective D2 agonist, nearly completelyreversed (85%) the effect of haloperidol (Fig. 3d).
The effect of NECA, an A2 adenosine agonist, on themodulation of the release of 3H-CA produced by haloperidoland domperidone both, in normoxia and moderate hypoxia, isshown in Fig. 4. Figure 4(a) and (b) refer to effects onnormoxic release and Fig. 4(c) and (d) to 10% O2-evoked
3H-CA release. It can be observed that the application of NECAat 10 lM (·10 the EC50 for the stimulation of 3H-CA release)displaced to the left the dose-response curve of haloperidol
Fig. 1 Effect of NECA, an adenosine A2 agonist, on the basal release
of 3H-CA and its reversion by MRS1754, an A2B adenosine receptor
antagonist and on the release evoked by 2% O2, from rat carotid body.
(a) Dose-response curve for NECA (0.1–100 lM) on the basal
(normoxic, 20% O2) release of 3H-CA. 0% corresponds to basal re-
lease and amounts to 1.062 ± 0.06% of the total 3H-CA content. (b)
Full reversion of the effect of 10 lM of NECA on the basal release of3H-CA from carotid body by 200 nM of MRS1754. (c) Effect of 10 lM
of NECA on the release of 3H-CA evoked by 2% O2. 0% corresponds
to basal release and amounts to 1.669 ± 0.44% of the total 3H-CA
content. NECA was applied during 30 min between minutes 30 and 60
of the protocol in the experiments in basal conditions. In the experi-
ments for the effect of NECA in hypoxia, the drug was applied in
periods prior, during and after to the hypoxic stimulation. Hypoxic
stimulus was applied for 10 min. MRS 1754 was applied between
minutes 40 and 60 of experimental protocol. ***p < 0.001; unpaired
Student’s t-test. Data represent means ± SEM (n = 5–6).
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1372 | S. V. Conde et al.
on basal normoxic (Fig. 4a); NECA also increased the releaseelicited by 5 lM domperidone. NECA displace in a similarmanner the dose response curve of haloperidol on the releaseelicited by low intensity hypoxic stimulus (Fig. 4c). Theaugmentation of haloperidol (10 lM) and domperidone(5 lM) effects produced by NECA in normoxia (Fig. 4b)and moderate hypoxia (Fig. 4d) are hypoadditive, becauseco-application of both drugs produced an effect smaller thanthe sum of effects of the drugs applied separately. Table 1presents Emax and EC50 of haloperidol on the release of3H-CA from CB, both, in normoxia and moderate hypoxiaand in the presence and the absence of NECA.
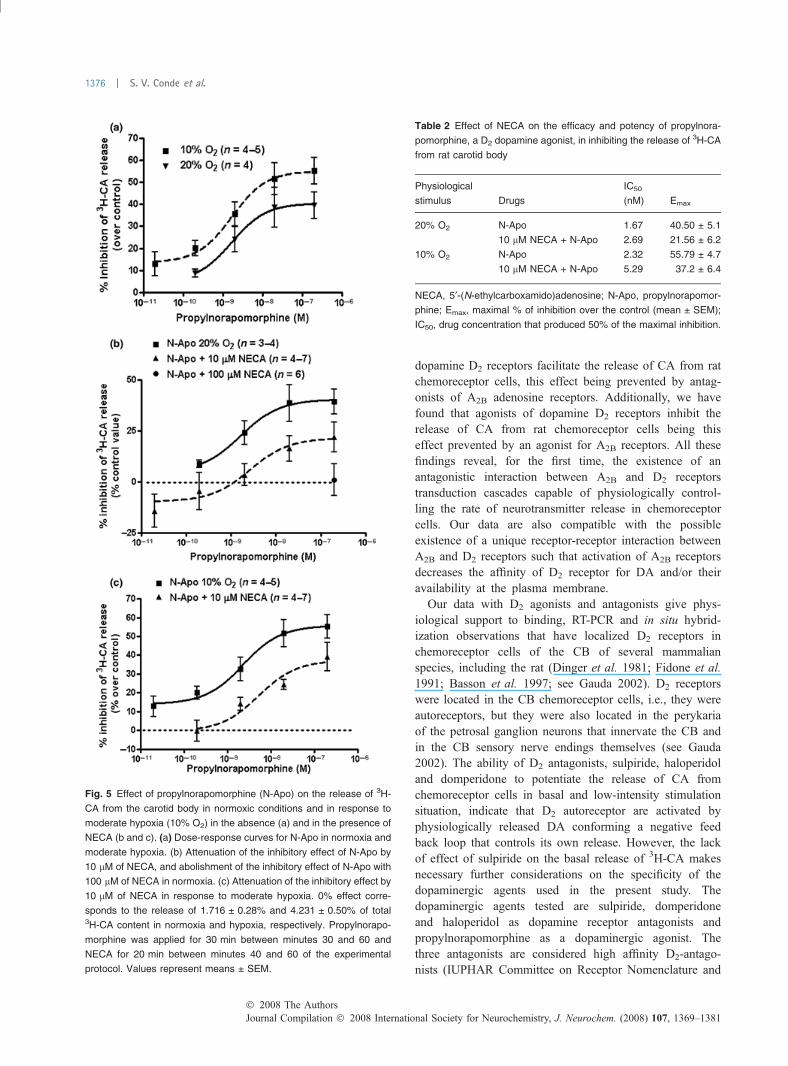
Propylnorapomorphine, a D2 dopamine agonist, inhibits therelease of 3H-CA from carotid body and its reversal byNECAIn order to investigate the potential antagonistic effect of theinteraction between D2 and A2B receptors we have charac-terized the presumed inhibitory effect of the D2 agonistpropylnorapomorphine and its potential reversal by NECA.Propylnorapomorphine (0.02–200 nM) inhibited normoxic(20% O2) and low intensity (10% O2) hypoxia induced
release of 3H-CA in a concentration-dependent manner(Fig. 5a). Figure 5(b) shows that NECA at a concentrationof 10 lM produced a displacement to the right of theinhibitory effect of propylnorapomorphine on normoxic 3H-CA release; additionally it shows that 100 lM NECA fullyreversed the effect of propylnorapomorphine. Figure 5(c)shows an identical effect of NECA (10 lM) on the inhibitoryeffect of propylnorapomorphine on hypoxia-induced 3H-CArelease. The maximal inhibitory effects (Emax) and IC50 ofpropylnorapomorphine in normoxia and moderate hypoxiaand in the presence and the absence of 10 lM NECA arerepresented in Table 2. As a whole these findings indicatedthat indeed there is an antagonistic effect between A2B andD2 receptors that is operative in normoxia and during lowintensity stimulation.
Effect of CGS21680 and NECA on the cAMP content of ratcarotid bodyIt is known that A2 and D2 receptors exert opposite actionson cAMP and that adenosine increases the cAMP content ofrat CB (Monteiro et al. 1996). This last finding prompted usto investigate which subtype of A2 adenosine receptor is
Fig. 2 Effect of sulpiride on basal and evoked release of 3H-CA. (a)
Shows that sulpiride at 0.1, 1 and 10 lM did not affect basal or
normoxic release of 3H-CA. (b) Effect of sulpiride (1 lM) on the re-
lease evoked by 30 mM of extracellular K+ and by 10% O2. Sulpiride,
applied 10 min prior to and during the stimuli application, potentiated
the release response elicited by both stimuli. (c) Sulpiride (SULP;
1 lM) completely reversed the inhibitory effect of 1 mM of caffeine
(CAF) on the basal release of 3H-CA. (d) Sulpiride, almost completely
reversed the inhibitory effect of caffeine on the evoked release of3H-CA by 30 mM extracellular K+. *p < 0.05; **p < 0.01; One and
Two-Way ANOVA with Bonferroni¢s multi-comparison test. Vertical bars
represent ± SEM.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1373
involved in the increase of cAMP content of rat CB. All theexperiments were performed in normoxia (20% O2) and inthe presence of IBMX in order to obtain phosphodiesteraseinhibition. The effect of the agonists of A2 adenosinereceptors was tested in the presence of ADA to obtain onlythe agonist effect, excluding the effects of endogenousadenosine. Application of ADA (2 U/L) significantlydecreased the cAMP content by 76.6% from a control valueof 0.869 ± 0.11 pmol/CB (Fig. 6a and b), indicating thatadenosine receptor activation is responsible for a major partof the basal cAMP production in the CB. Next, we testedCGS 21680, in a concentration that is highly selective forA2A adenosine receptors (30 nM) and it was observed thatthis compound was incapable of reversing the effectproduced by ADA (Fig. 6a). And finally we also testedCGS 21680 at 1 lM, a concentration that is less selective forA2A receptor, and yet it was incapable of fully reversingADA effects (Fig. 6a).
In a second group of experiments presented in Fig. 6(b)we have tested NECA, an A2 agonist on the basal productionof cAMP. It can be observed that NECA in both concentra-
tions tested, 1 and 10 lM, significantly increased the cAMPproduction in the rat CB, reversing completely the reductionin cAMP produced by ADA. The findings with CGS 21680indicate that A2A adenosine receptors are not the mainresponsible for the adenosine-induced cAMP production inthe CB. The effect of NECA indicates that in fact A2B
adenosine receptors are mostly responsible for the productionof cAMP mediated by endogenous by adenosine. This in turnwould suggest that, in the same manner that D2 and A2B
receptors interact to control the release of CA from rat CBchemoreceptor cells, they would interact to control cAMPlevels.
Interactions between D2 and A2B receptors in the control ofadenylyl cyclase activity in the carotid bodyThe next series of experiments were performed to investigateif the antagonistic interaction between A2B and D2 receptorsobserved on the release of 3H-CA from chemoreceptor cellsis also observed at second messenger level.
Figure 7 shows that forskolin (FSK, 3 lM), an activatorof adenylyl cyclase increased cAMP production from
Fig. 3 Effects of domperidone and haloperidol, D2 dopamine antag-
onists, on the release of 3H-CA from rat CB. (a) Dose response curves
for domperidone and haloperidol in normoxic conditions. 0% corre-
sponds to basal release and amounts to 0.927 ± 0.10% and to
1.010 ± 0.09% of the total 3H-CA content of the CB for haloperidol and
domperidone, respectively. (b) Comparative dose-response curves for
haloperidol on release of 3H-CA in normoxic conditions (continuous
line) and in response to moderate hypoxia (10% O2; doted line). 0%
effect for haloperidol in response to 10% O2 corresponds to
5.563 ± 0.58% of the total 3H-CA content. (c) Comparative effect of
5 lM domperidone on basal and low PO2-induced release of 3H-CA.
(d) Propylnorapomorphine (200 nM) reversal of the effect of 10 lM
haloperidol on the 10% O2-induced release of 3H-CA. Values repre-
sent means ± SEM. ***p < 0.001; Student’s t-test for unpaired data.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1374 | S. V. Conde et al.

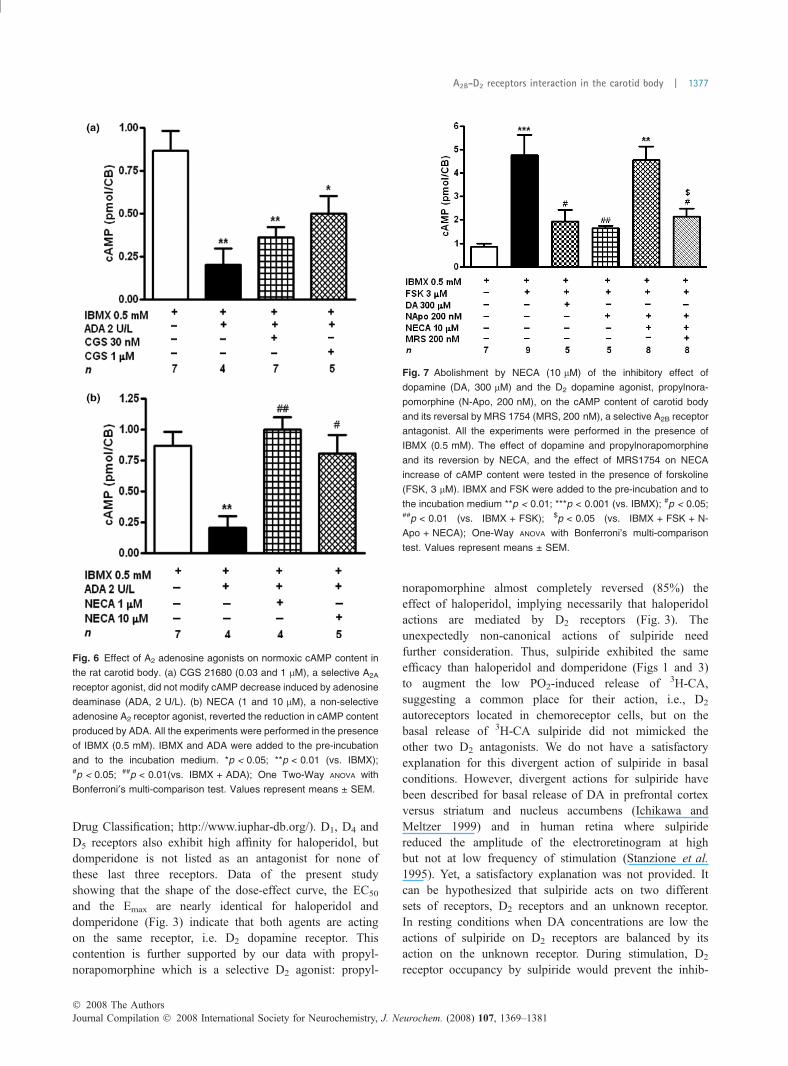
0.869 ± 0.11 pmol/CB (CBs incubated only with IBMX)to 4.772 ± 0.87 pmol/CB (449% effect). Figure 7 alsoshows that exogenous dopamine (300 lM) and the D2
dopamine agonist propylnorapomorphine at 200 nM (aconcentration maximally effective inhibiting 3H-CA re-lease) reduced by about two thirds the stimulating actionof forskolin. NECA (10 lM) fully reversed the inhibitoryaction of propylnorapomorphine and MRS 1754 decreasedthis effect of NECA by 86%, indicating the existence ofan antagonistic interaction between D2 and A2B receptorsat the cAMP level.
Discussion
Present results provide direct pharmacological evidencethat adenosine A2B receptors and dopamine D2 transduc-tion cascades in chemoreceptor cells converge to controlcAMP levels in rat CB O2-sensing chemoreceptor cellsand their rate of CA release. We have found that agonistsof dopamine D2 receptors inhibit cAMP production in theCB this effect being prevented by an agonist for A2B
adenosine receptors. We also found that antagonists of
Fig. 4 NECA, an A2 adenosine receptor agonist, augments the effect
of haloperidol and domperidone, D2 dopamine receptor antagonists,
on 3H-CA release from CB. (a) and (b), normoxic conditions (20% O2).
(c) and (d), in response to moderate hypoxia (10% O2). In (b) and (d)
the dotted lines represent the absolute values of the sum of the both
drugs (NECA and haloperidol/domperidone) applied separately in
normoxia and hypoxia, respectively. Note that the effect of both drugs
is hypoadditive. 0% effect corresponds to the release of
0.927 ± 0.10% and to 5.563 ± 0.58% of total CA content in normoxia
and hypoxia, respectively. NECA was applied for 30 min between
minutes 30 and 60 and haloperidol and domperidone for 20 min
between minutes 40 and 60 of the experimental protocol. Values
represent means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. One
way ANOVA with Bonferroni multicomparison test.
Table 1 Effect of NECA (10 lM) on the efficacy and potency of
haloperidol, a D2 dopamine antagonist, in stimulating the release
of 3H-CA from rat carotid body
Physiological
stimulus Drugs
EC50
(nM) Emax
20% O2 Haloperidol 147 122.3 ± 13.0
NECA + Haloperidol 78 161.5 ± 9.2
10% O2 Haloperidol 462 110.1 ± 10.8
NECA + Haloperidol 79 152.7 ± 12.7
NECA by itself in this concentration caused 87.35 ± 10.73% increase
in 3H-CA (over control).
NECA, 5¢-(N-ethylcarboxamido)adenosine; Emax, maximal increase
(%) in the release of 3H-CA (mean ± SEM); EC50, drug concentration
that produced 50% of maximal effect.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1375
dopamine D2 receptors facilitate the release of CA from ratchemoreceptor cells, this effect being prevented by antag-onists of A2B adenosine receptors. Additionally, we havefound that agonists of dopamine D2 receptors inhibit therelease of CA from rat chemoreceptor cells being thiseffect prevented by an agonist for A2B receptors. All thesefindings reveal, for the first time, the existence of anantagonistic interaction between A2B and D2 receptorstransduction cascades capable of physiologically control-ling the rate of neurotransmitter release in chemoreceptorcells. Our data are also compatible with the possibleexistence of a unique receptor-receptor interaction betweenA2B and D2 receptors such that activation of A2B receptorsdecreases the affinity of D2 receptor for DA and/or theiravailability at the plasma membrane.
Our data with D2 agonists and antagonists give phys-iological support to binding, RT-PCR and in situ hybrid-ization observations that have localized D2 receptors inchemoreceptor cells of the CB of several mammalianspecies, including the rat (Dinger et al. 1981; Fidone et al.1991; Basson et al. 1997; see Gauda 2002). D2 receptorswere located in the CB chemoreceptor cells, i.e., they wereautoreceptors, but they were also located in the perykariaof the petrosal ganglion neurons that innervate the CB andin the CB sensory nerve endings themselves (see Gauda2002). The ability of D2 antagonists, sulpiride, haloperidoland domperidone to potentiate the release of CA fromchemoreceptor cells in basal and low-intensity stimulationsituation, indicate that D2 autoreceptor are activated byphysiologically released DA conforming a negative feedback loop that controls its own release. However, the lackof effect of sulpiride on the basal release of 3H-CA makesnecessary further considerations on the specificity of thedopaminergic agents used in the present study. Thedopaminergic agents tested are sulpiride, domperidoneand haloperidol as dopamine receptor antagonists andpropylnorapomorphine as a dopaminergic agonist. Thethree antagonists are considered high affinity D2-antago-nists (IUPHAR Committee on Receptor Nomenclature and
Fig. 5 Effect of propylnorapomorphine (N-Apo) on the release of 3H-
CA from the carotid body in normoxic conditions and in response to
moderate hypoxia (10% O2) in the absence (a) and in the presence of
NECA (b and c). (a) Dose-response curves for N-Apo in normoxia and
moderate hypoxia. (b) Attenuation of the inhibitory effect of N-Apo by
10 lM of NECA, and abolishment of the inhibitory effect of N-Apo with
100 lM of NECA in normoxia. (c) Attenuation of the inhibitory effect by
10 lM of NECA in response to moderate hypoxia. 0% effect corre-
sponds to the release of 1.716 ± 0.28% and 4.231 ± 0.50% of total3H-CA content in normoxia and hypoxia, respectively. Propylnorapo-
morphine was applied for 30 min between minutes 30 and 60 and
NECA for 20 min between minutes 40 and 60 of the experimental
protocol. Values represent means ± SEM.
Table 2 Effect of NECA on the efficacy and potency of propylnora-
pomorphine, a D2 dopamine agonist, in inhibiting the release of 3H-CA
from rat carotid body
Physiological
stimulus Drugs
IC50
(nM) Emax
20% O2 N-Apo 1.67 40.50 ± 5.1
10 lM NECA + N-Apo 2.69 21.56 ± 6.2
10% O2 N-Apo 2.32 55.79 ± 4.7
10 lM NECA + N-Apo 5.29 37.2 ± 6.4
NECA, 5¢-(N-ethylcarboxamido)adenosine; N-Apo, propylnorapomor-
phine; Emax, maximal % of inhibition over the control (mean ± SEM);
IC50, drug concentration that produced 50% of the maximal inhibition.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1376 | S. V. Conde et al.
Drug Classification; http://www.iuphar-db.org/). D1, D4 andD5 receptors also exhibit high affinity for haloperidol, butdomperidone is not listed as an antagonist for none ofthese last three receptors. Data of the present studyshowing that the shape of the dose-effect curve, the EC50
and the Emax are nearly identical for haloperidol anddomperidone (Fig. 3) indicate that both agents are actingon the same receptor, i.e. D2 dopamine receptor. Thiscontention is further supported by our data with propyl-norapomorphine which is a selective D2 agonist: propyl-
norapomorphine almost completely reversed (85%) theeffect of haloperidol, implying necessarily that haloperidolactions are mediated by D2 receptors (Fig. 3). Theunexpectedly non-canonical actions of sulpiride needfurther consideration. Thus, sulpiride exhibited the sameefficacy than haloperidol and domperidone (Figs 1 and 3)to augment the low PO2-induced release of 3H-CA,suggesting a common place for their action, i.e., D2
autoreceptors located in chemoreceptor cells, but on thebasal release of 3H-CA sulpiride did not mimicked theother two D2 antagonists. We do not have a satisfactoryexplanation for this divergent action of sulpiride in basalconditions. However, divergent actions for sulpiride havebeen described for basal release of DA in prefrontal cortexversus striatum and nucleus accumbens (Ichikawa andMeltzer 1999) and in human retina where sulpiridereduced the amplitude of the electroretinogram at highbut not at low frequency of stimulation (Stanzione et al.1995). Yet, a satisfactory explanation was not provided. Itcan be hypothesized that sulpiride acts on two differentsets of receptors, D2 receptors and an unknown receptor.In resting conditions when DA concentrations are low theactions of sulpiride on D2 receptors are balanced by itsaction on the unknown receptor. During stimulation, D2
receptor occupancy by sulpiride would prevent the inhib-
Fig. 6 Effect of A2 adenosine agonists on normoxic cAMP content in
the rat carotid body. (a) CGS 21680 (0.03 and 1 lM), a selective A2A
receptor agonist, did not modify cAMP decrease induced by adenosine
deaminase (ADA, 2 U/L). (b) NECA (1 and 10 lM), a non-selective
adenosine A2 receptor agonist, reverted the reduction in cAMP content
produced by ADA. All the experiments were performed in the presence
of IBMX (0.5 mM). IBMX and ADA were added to the pre-incubation
and to the incubation medium. *p < 0.05; **p < 0.01 (vs. IBMX);#p < 0.05; ##p < 0.01(vs. IBMX + ADA); One Two-Way ANOVA with
Bonferroni¢s multi-comparison test. Values represent means ± SEM.
Fig. 7 Abolishment by NECA (10 lM) of the inhibitory effect of
dopamine (DA, 300 lM) and the D2 dopamine agonist, propylnora-
pomorphine (N-Apo, 200 nM), on the cAMP content of carotid body
and its reversal by MRS 1754 (MRS, 200 nM), a selective A2B receptor
antagonist. All the experiments were performed in the presence of
IBMX (0.5 mM). The effect of dopamine and propylnorapomorphine
and its reversion by NECA, and the effect of MRS1754 on NECA
increase of cAMP content were tested in the presence of forskoline
(FSK, 3 lM). IBMX and FSK were added to the pre-incubation and to
the incubation medium **p < 0.01; ***p < 0.001 (vs. IBMX); #p < 0.05;##p < 0.01 (vs. IBMX + FSK); $p < 0.05 (vs. IBMX + FSK + N-
Apo + NECA); One-Way ANOVA with Bonferroni’s multi-comparison
test. Values represent means ± SEM.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1377

itory action of DA and the potentiating effect of sulpirideon DA release emerges, in other words the action ofsulpiride on the unknown receptor is overridden. Thishypothesis would imply that the unknown receptor isexcitatory and that it plays a major role in setting basalactivity in chemoreceptor cells. Receptors meeting thesecriteria are 5HT2a receptors (Zhang et al. 2003), andsulpiride does indeed interact with 5HT2a receptors withhigher affinity than with D2 receptors (Kuroki et al. 1999).It might be argued that haloperidol can also interact with5HT2a receptors and therefore it also should havedivergent effects on basal versus stimulus-induced releaseof 3H-CA, but the lower affinity of 5HT2a receptors incomparison to D2 receptors (Ma et al. 2006) wouldminimize its effects on 5HT2a receptors. We exclude D3
receptors present in the CB (Wang et al. 2001) ascandidates for the unknown receptor, because as pre-synaptic receptors they feedback control the release of DAand its inhibition would produce an augmentation of therelease of DA (Zapata et al. 2001; Joseph et al. 2002) inthe same manner that inhibition of D2 receptors does. Thesame arguments are valid to exclude a-adrenergic receptorsknown to be present in chemoreceptor cells (Almaraz et al.1997).
At any given concentration haloperidol was more effectiveto augment the release of CA from chemoreceptor cells innormoxia than in response to moderate hypoxia (Fig. 3b).This would reflect that the extra DA present in the milieusurrounding the cells due cell stimulation is competing withhaloperidol and displacing it from the receptors with theresulting decreased haloperidol effect. Conversely, theinhibitory effect of the D2 agonist propylnorapomorphineon the release of CAwas greater in moderate hypoxia than innormoxia (Fig. 5a). This would reflect that at any givenconcentration of propylnorapomorphine the amount of DAcooperating to inhibit its own release is higher undermoderate hypoxic stimulation than in normoxia. In thiscontext we should also comment on our findings withNECA. We have previously observed with the non-specificadenosine antagonist caffeine inhibits the release of CA inbasal and low intensity of stimulation, being ineffective athigh level of stimulation (Conde et al. 2006), NECApotentiated the release of 3H-CA in normoxic and lowintensity stimulation (Fig. 4) but it was ineffective tomodulate the release induced by high intensity stimulation(Fig. 1). We consider that in this last situation the high levelsof DA in the surrounding milieu (released by a high intensitystimulus) would saturate D2 receptors masking the effects ofthe interactions between adenosine on dopamine receptors(see below under Fig. 8).
Although NECA is a non-selective A2 adenosine receptoragonist, we are going to assign all its effects on CBchemoreceptor cells to the specific activation of A2B
adenosine receptors. This attribution is based on a prior
comprehensive pharmacological characterization of caffeineand NECA effects on the release of 3H-CA from CBchemoreceptor cells which allowed to conclude that theiractions were mediated by A2B receptors; additionally, A2B
receptors were immunocytochemically localized in chemo-receptor cells (Conde et al. 2006; see also Fig. 1). In thecurrent study, our measurements of cAMP would conformthat conclusion. From Fig. 6 it is evident that NECA iscapable of reversing the effect of the enzymatic destructionof the physiologically released adenosine. However,CGS21680 at concentrations specific for A2A receptors,and even increasing CGS21680 to a concentration where itmight have some effects on A2B receptors, reversed onlypartially the effect of adenosine destruction. We wouldpropose that A2A receptors mediated effect is produced innon-chemoreceptor cells, while A2B receptors mediatedeffect (difference between NECA and CGS21680 effects) ismostly produced on chemoreceptor cells. The proposal issubstantiated from our findings of Fig. 7. From this figure itis evident that around 65–75% of the increase in cAMP
Fig. 8 Mechanisms of interaction between A2B and D2 receptors in
chemoreceptor cells that modulate the release of CA from rat carotid
body. The antagonist interaction between A2B and D2 receptors is
evident at the adenylyl cyclase level. Both receptors exert opposite
actions on adenylyl cyclase and therefore on cAMP levels, and,
application of NECA reverted completely the inhibitory effect of D2
dopamine agonist on cAMP production. Additionally, it can be
hypothesized a receptor-receptor coupling at the plasma membrane
level between A2B and D2 receptors. This hypothetical coupling would
decrease the affinity of D2 receptors for DA when the A2B are occupied
by adenosine, and thereby endogenously released adenosine would
be tonically activating in normoxia the release of CA from chemore-
ceptor cells as it hinders the inhibitory action of DA. On inhibition of
A2B receptors with caffeine, full inhibitory power of D2 receptors ap-
pears, and thereby the decrease in CA release induced by caffeine
and other A2B antagonists. Indeed, the activation of A2B adenosine
receptors with NECA decreases the affinity of the D2 receptor agonist,
propylnorapomorphine in CB chemoreceptor cells. The same behav-
iour will hold true for moderate hypoxic stimulation, but under intense
stimulation, adenosine would lose its effectiveness to modulate CA
release because of the saturation of D2 receptors in spite of the de-
creased affinity caused by the A2B agonist. AC, adenylyl cyclase;
cAMP, cyclic 3¢,5¢-adenosine monophosphate.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1378 | S. V. Conde et al.

produced by forskolin activation of adenylyl cyclase mustoccur in CB structures containing D2 receptors, because DAand the D2 agonist propylnorapomorphine prevented thatpercentage activation; the remaining adenylyl cyclase activ-ity should be located in D2 receptors-free structures.Although D2 are present in chemoreceptor cells and in nerveendings of the CSN, the additional findings that NECAreversed the action of propylnorapomorphine and that theA2B receptor antagonist MRS1754 inhibited the effect ofNECA clearly evidence that D2-A2B interactions is occurringin chemoreceptor cells. Or stating it in a different manner,the potential interaction D2 and A2A receptors present in thesensory nerve endings do not contribute significantly to thefindings presented in Fig. 7. However, this conclusion doesnot exclude the existence of interactions at the adenylylcyclase level between D2 and A2A receptors present in thesensory nerve endings; yet, the small percentage of the CBmass represented by the sensory nerve endings would renderimpossible to solve it analytically.
A final consideration from the data of Fig. 6 refers to thefact that destruction of endogenously released adenosine bythe addition of adenosine deaminase lowers cAMP levels andthat 1 and 10 lM NECA reverses the effect of adenosinedeaminase. Taking into account that NECA and adenosine areequipotent on activating adenylyl cyclase in cloned humanA2B receptors (Fredholm et al. 2001), data would imply thatthe concentration of adenosine in the milieu bathing the cellsshould be in the low micromolar range. Indeed this figure isnot dislike to that obtained in real time adenosine measure-ments in brain slices (Pearson et al. 2003).
The nature of D2-A2B receptor effects on chemoreceptorcells should be defined as an antagonistic, because the effectsof activation of D2 receptors with propylnorapomorphinediminished with increasing concentrations of NECA, whichactivates A2B receptors. Although in the present series ofexperiments, we have not specifically examined the intimatemechanisms of this antagonistic interaction (see Ferre et al.2007; Fuxe et al. 2007), we consider that the modelpresented in Fig. 8 reasonably explains present and previousfindings form different laboratories. Chemoreceptor cells D2
autoreceptors form a negative feedback controlling therelease of CA from chemoreceptor cells in the rabbit (Fidoneet al. 1991; Basson et al. 1997) and in the rat (currentresults). The antagonist interaction between A2B and D2
receptors is evident at the adenylyl cyclase level. First, bothreceptors exert opposite actions on adenylyl cyclase andtherefore on cAMP levels (Figs 6 and 7); and second,application of NECA reverted completely the inhibitoryeffect of D2 dopamine agonist on cAMP production (Fig. 7).Additionally, our data do not exclude an antagonisticinteraction at the A2B-D2 receptor level, similar to thatdescribed in the CNS for A2A-D2 receptors (Fuxe et al.2007), but further experiments are required to affirm or denyits existence. If this receptor-receptor interaction does indeed
occur it should operate in such a manner that activation ofA2B receptors would decrease the affinity of D2 receptor forDA or would trigger internalization of D2 receptors (Fuxeet al. 2007).
In present discussion we have not yet referred to the factthat two laboratories have identified A2A receptors in ratchemoreceptor cells immunocytochemically and by in situhybridization techniques (Gauda et al. 2000; Kobayashiet al., 2000). The obvious question in the context of presentwork is to ask which are the effects of those putative A2A
located in chemoreceptor cells. Neither in present nor in ourprevious work (Conde et al. 2006) we have detected effectson the control of the release of 3H-CA or in the regulationof adenylyl cyclase in chemoreceptor cells that might beattributed to A2A receptors. In this context, it should bementioned that the two existing studies addressing thefunctional significance of A2A receptors on rat chemore-ceptor cells have provided opposite results. Thus Kobayashiet al. (2000) found that adenosine did not modify basalnormoxic intracellular Ca2+ levels, but via A2A receptors itinhibited hypoxia-induced increase in Ca2+ rise as well asCa2+ currents. Contrary to that Xu et al. (2006) found thatadenosine augmented normoxic Ca2+ levels by promotingchemoreceptor cell depolarization and did not modifyhypoxia-induced increase in intracellular Ca2+. We cannotprovide a satisfactory explanation to these groups offindings.
A final consideration relates to the fact that D2
interactions, particularly in the central nervous system,are most commonly described with A2A receptors. How-ever, the interaction between catecholamine autoreceptorsand A2B receptors is not unique to chemoreceptor cells.For example, Talaia et al. (2005) found that adenosineacting via A2B receptors augments the release of nor-adrenaline from the sympathetic endings innervating thevas deferens, this effect being abolished by yohimbine,a blocker of pre-synaptic a2-adrenoreceptors (see alsoQueiroz et al. 2004).
To summarise, we can state chemoreceptor cells func-tion, exemplified by their levels of cAMP and their releaseof CA, is reciprocally modulated by adenosine acting onA2B receptors and dopamine acting on D2 receptors. Thisdual control operates in basal conditions as well as inhypoxia since both, adenosine and dopamine are releasedtonically in normoxia and their release augments inhypoxia.
Acknowledgements
We want to thank Mª de los Llanos Bravo for technical assistance.
The work was supported by BFU2007-61848 (DGICYT), CIBER
CB06/06/0050 (FISS-ICiii) and CEPR/FCT. SV Conde was funded
by a PhD grant by Portuguese Foundation for Science and
Technology (SFRH/BD/14178/2003).
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1379

References
Almaraz L., Gonzalez C. and Obeso A. (1986) Effects of high potassiumon the release of [3H]dopamine from the cat carotid body in vitro.J. Physiol. 379, 293–307.
Almaraz L., Perez-Garcia M. T. and Gonzalez C. (1991) Presence of D1receptors in the rabbit carotid body. Neurosci. Lett. 132, 259–262.
Almaraz L., Perez-Garcıa M. T., Gomez-Nino A. and Gonzalez C.(1997) Mechanisms of alpha2-adrenoceptor-mediated inhibition inrabbit carotid body. Am. J. Physiol. 272, C628–C637.
Basson H., Bairam A., Cottet-Emard J. M., Pequignot J. M. and MarchalF. (1997) Carotid body dopamine content and release by short-termhypoxia: effect of haloperidol and alpha methyl paratyrosine. Arch.Physiol. Biochem. 105, 3–9.
Batuca J. R., Monteiro T. C. and Monteiro E. C. (2003) Contribution ofdopamine D2 receptors for the cAMP levels at the carotid body.Adv. Exp. Med. Biol. 536, 367–373.
Conde S. V. and Monteiro E. C. (2004) Hypoxia induces adenosinerelease from the rat carotid body. J. Neurochem. 89, 1148–1156.
Conde S. V., Obeso A., Vicario I., Rigual R., Rocher R. and Gonzalez C.(2006) Caffeine inhibition of rat carotid body chemoreceptors ismediated by A2A and A2B adenosine receptors. J. Neurochem. 98,616–628.
Dinger B., Gonzalez C., Yoshizaki K. and Fidone S. (1981) [3H]spir-operidol binding in normal and denervated carotid bodies.Neurosci. Lett. 21, 51–55.
Ferre S., Fredholm B. B., Morelli M., Popoli P. and Fuxe K. (1997)Adenosine-dopamine receptor-receptor interactions as an integra-tive mechanism in the basal ganglia. Trends Neurosci. 20, 482–487.
Ferre S., Ciruela F., Woods A. S., Lluis C. and Franco R. (2007)Functional relevance of neurotransmitter receptor heteromers in thecentral nervous system. Trends Neurosci. 30, 440–446.
Fidone S. J. and Gonzalez C. (1982) Catecholamine synthesis in rabbitcarotid body in vitro. J. Physiol. 333, 69–79.
Fidone S. J., Gonzalez C. and Yoshizaki K. (1982) Effects of low oxygenon the release of dopamine from the rabbit carotid body in vitro.J. Physiol. 333, 93–110.
Fidone S. J., Gonzalez C., Dinger B., Gomez-Nino A., Obeso A. andYoshizaki K. (1991) Cellular aspects of peripheral chemorecep-tor function, in The Lung: Scientific Foundations (CristalR. G., West J. B., Barnes P. J., Cherniack S. and Weibel E. R.,eds), pp. 1319–1332. Raven Press, Ltd., NY.
Fredholm B. B. (2007) Adenosine, an endogenous distress signal,modulates tissue damage and repair. Cell Death Differ. 14, 1315–1323.
Fredholm B. B., Irenius E., Kull B. and Schulte G. (2001) Comparisonof the potency of adenosine as an agonist at human adenosinereceptors expressed in Chinese hamster ovary cells. Biochem.Pharmacol. 61, 443–448.
Fuxe K., Canals M., Torvinen M. et al. (2007) Intramembrane receptor–receptor interactions: a novel principle in molecular medicine.J. Neural. Transm. 114, 49–75.
Gauda E. B. (2002) Gene expression in peripheral arterial chemore-ceptors. Microsc. Res. Tech. 59, 153–167.
Gauda E. B., Northington F. J., Linden J. and Rosin D. L. (2000) Dif-ferential expression of A2a, A1-adenosine and D2-dopaminereceptor genes in rat peripheral arterial chemoreceptors duringpostnatal development. Brain Res. 872, 1–10.
Gonzalez C., Almaraz L., Obeso A. and Rigual R. (1994) Carotid bodychemoreceptors: from natural stimuli to sensory discharges.Physiol. Rev. 74, 829–898.
Ichikawa J. and Meltzer H. Y. (1999) R(+)-8-OH-DPAT, a serotonin(1A)receptor agonist, potentiated S(-)-sulpiride-induced dopamine
release in rat medial prefrontal cortex and nucleus accumbens butnot striatum. J. Pharmacol. Exp. Ther. 291, 1227–1232.
Iturriaga R. and Alcayaga J. (2004) Neurotransmission in the carotidbody: transmitters and modulators betweeen glomus cells andpetrosal ganglion nerve terminals. Brain Res. Brain Res. Rev. 47,46–53.
Joseph J. D., Wang Y. M., Miles P. R., Budygin E. A., Picetti R.,Gainetdinov R. R., Caron M. G. and Wightman R. M. (2002)Dopamine autoreceptor regulation of release and uptake in mousebrain slices in the absence of D3 receptors. Neuroscience 112, 39–49.
Kobayashi S., Conforti L. and Millhorn D. E. (2000) Gene expressionand function of A2A receptor in the rat carotid body. Am. J. Physiol.Lung Cell Mol. Physiol. 279, L273–L282.
Kuroki T., Meltzer H. Y. and Ichikawa J. (1999) Effects of antipsychoticdrugs on extracellular dopamine levels in rat medial prefrontalcortex and nucleus accumbens. J. Pharmacol. Exp. Ther. 288, 774–781.
Ma J., Ye N. and Cohen B. M. (2006) Expression of noradrenergic a1,serotoninergic 5HT2a and dopaminergic D2 receptors on neuronsactivated by typical and atypical antipsychotic drugs. Prog.Neuropsychopharmacol. Biol. Psychiatry 30, 647–657.
McQueen D. S. and Ribeiro J. A. (1981) Effect of adenosine oncarotid chemoreceptor activity in the cat. Br. J. Pharmacol. 74,129–136.
McQueen D. S. and Ribeiro J. A. (1983) On the specificity and type ofreceptor involved in carotid body chemoreceptor activation byadenosine in the cat. Br. J. Pharmacol. 80, 347–354.
Monteiro E. C. and Ribeiro J. A. (1987) Ventilatory effects of adenosinemediated by carotid body chemoreceptors in the rat. Naunyn- Schmiedeberg’s Arch. Pharmacol. 335, 143–148.
Monteiro E. C. and Ribeiro J. A. (1989) Adenosine deaminase andadenosine uptake inhibitors facilitate ventilation in rats. Naunyn-Schmiedeberg¢s Arch. Pharmacol. 340, 230–238.
Monteiro E. C. and Ribeiro J. A. (2000) Adenosine-dopamine inter-actions and ventilation mediated through carotid body chemore-ceptors. Adv. Exp. Med. Biol. 475, 671–684.
Monteiro E. C., Vera-Cruz P., Monteiro T. C. and Silva e Sousa M. A.(1996) Adenosine increases the cAMP content of the rat carotidbody in vitro. Adv. Exp. Med. Biol. 410, 299–303.
Pearson T., Currie A. J., Etherington L. A., Gadalla A. E., Damian K.,Llaudet E., Dale N. and Frenguelli B. G. (2003) Plasticity of purinerelease during cerebral ischemia: clinical implications? J. Cell.Mol. Med. 7, 362–375.
Queiroz G., Quintas C., Talaia C. and Goncalves J. (2004) Coupling toprotein kinases A and C of adenosine A2B receptors involved in thefacilitation of noradrenaline release in the prostatic portion of ratvas deferens. Neuropharmacology 47, 216–224.
Ribeiro J. A. and McQueen D. S. (1983) On the muscular depression andcarotid chemoreceptor activation caused by adenosine, in Physi-ology and Pharmacology of Adenosine Derivatives (Daly J. W.,Kuroda Y., Phillis J. W., Shimizu H. and Ui M., eds), pp. 179–188.Raven Press, New York.
Ribeiro J. A. and Monteiro E. C. (1991) On the adenosine receptorinvolved in the excitatory action of adenosine on respiration:antagonist profile. Nucleosides Nucleotides 10, 945–953.
Runold M., Cherniak N. S. and Prabhakar N. R. (1990) Effect ofadenosine on isolated and superfused cat carotid body activity.Neurosci. Lett. 113, 111–114.
Stanzione P., Pierantozzi M., Semprini R., Tagliati M., Traversa R.,Peppe A., Pierelli F. and Bernardi G. (1995) Increasing doses ofl-sulpiride reveal dose- and spatial frequency-dependent effects ofD2 selective blockade in the human electroretinogram. Vision Res.35, 2659–2664.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381� 2008 The Authors
1380 | S. V. Conde et al.

Talaia C., Queiroz G., Quintas C. and Goncalves J. (2005) Interactionbetween A2B-receptors and alpha2-adrenoreceptors on the modu-lation of noradrenaline release in the rat vas deferens: possibleinvolvement of a group 2 adenylyl cyclase isoform. Neurochem.Int. 47, 418–429.
Uematsu T., Kozawa O., Matsuno H., Niwa M., Yoshikoshi H., Oh-uchiM., Cono K., Nagashima S. and Kanamaru M. (2000) Pharmaco-kinetics and tolerability of intravenous infusion of adenosine(SUNY4001) in healthy volunteers. Br. J. Clin. Pharmacol. 50,177–181.
Vicario I., Rigual R., Obeso A. and Gonzalez C. (2000) Characterizationof the synthesis and release of catecholamine in the rat carotid bodyin vitro. Am. J. Physiol. Cell Physiol., 278, C490–C499.
Wang Z. Y., Herman J. K., O’Halloran K. D., Keith I. M. and BisgardG. E. (2001) Pharmacological and immunochemical evidence ofthe dopamine D3 receptor in the goat carotid body. Adv. Exp. Med.Biol. 499, 49–53.
Watt A. H. and Routledge P. A. (1985) Adenosine stimulates respirationin man. Br. J. Pharmacol. 20, 503–506.
Xu K., Bastia E. and Schwarzachild M. (2005) Therapeutic potential ofadenosine A2A receptor antagonists in Parkinson’s disease.Pharmacol. Ther. 105, 267–310.
Xu F., Xu J., Tse F. W. and Tse A. (2006) Adenosine stimulates depo-larization and rise in cytoplasmic Ca2+ concentration in type I cellsof rat carotid bodies. Am. J. Physiol. Cell Physiol. 290, 1592–1598.
Zapata A., Witkin J. M. and Shippenberg T. S. (2001) Selective D3
receptor agonist effects of ±PD 128907 on dialysate dopamine atlow doses. Neuropharmacology 41, 351–359.
Zeng C., Zhang M., Asico L. D., Eisner G. M. and Jose P. A. (2007) Thedopaminergic system in hypertension. Clin. Sci. 112, 583–597.
Zhang M., Fearon I. M., Zhong H. and Nurse C. A. (2003) Presynapticmodulation of rat arterial chemoreceptor function by 5-HT: role ofK+ channel inhibition via protein kinase C. J. Physiol. 551, 825–842.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1369–1381
A2B-D2 receptors interaction in the carotid body | 1381
Copyright © 2022 FDOKUMEN







![FINANCIAL MANAGEMENT 2B [BSR2B01, FNM02B2] LAST ...](https://static.fdokumen.com/doc/165x107/633c134708bfd70fe20afb65/financial-management-2b-bsr2b01-fnm02b2-last-.jpg)




![financial management 2b [bsr2b01, fnm02b2] last assessment ...](https://static.fdokumen.com/doc/165x107/631aa5b5c51d6b41aa04e9e3/financial-management-2b-bsr2b01-fnm02b2-last-assessment-.jpg)







