
An Alternative Root for the Eukaryote Tree of Life
6
Current Biology 24, 465–470, February 17, 2014 ª2014 Elsevier Ltd All rights reserved http://dx.doi.org/10.1016/j.cub.2014.01.036 Report An Alternative Root for the Eukaryote Tree of Life Ding He, 1,2 Omar Fiz-Palacios, 1,2 Cheng-Jie Fu, 1 Johanna Fehling, 1,3 Chun-Chieh Tsai, 1 and Sandra L. Baldauf 1, * 1 Program in Systematic Biology, Department of Organismal Biology, Uppsala University, Norbyva ¨ gen 18D, 75236 Uppsala, Sweden Summary The root of the eukaryote tree of life defines some of the most fundamental relationships among species. It is also critical for defining the last eukaryote common ancestor (LECA), the shared heritage of all extant species. The unikont-bikont root has been the reigning paradigm for eukaryotes for more than 10 years [1] but is becoming increasingly controversial [2–4]. We developed a carefully vetted data set, consisting of 37 nuclear-encoded proteins of close bacterial ancestry (euBacs) and their closest bacterial relatives, augmented by deep sequencing of the Acrasis kona (Heterolobosea, Discoba) transcriptome. Phylogenetic analysis of these data produces a highly robust, fully resolved global phy- logeny of eukaryotes. The tree sorts all examined eukaryotes into three megagroups and identifies the Discoba, and potentially its parent taxon Excavata [5], as the sister group to the bulk of known eukaryote diversity, the proposed Neozoa (Amorphea + Stramenopila+Alveolata+Rhizaria+ Plantae [SARP] [6]). All major alternative hypotheses are rejected with as little as w50% of the data, and this resolu- tion is unaffected by the presence of fast-evolving alignment positions or distant outgroup sequences. This ‘‘neozoan- excavate’’ root revises hypotheses of early eukaryote evolution and highlights the importance of the poorly stud- ied Discoba for understanding the evolution of eukaryotic diversity and basic cellular processes. Results Thirty-Seven Universal euBac Proteins Multigene phylogenies now assign most known eukaryotes to a few major groups, and these to three proposed megagroups—Amorphea, Stramenopila+Alveolata+Rhizaria+ Plantae (SARP or Diaphoretickes), and Excavata [5]. Howev- er, little effort has been made to explore the eukaryote root with multigene phylogeny, and attempts to root the tree with macromolecular characters have given widely different results [1, 3, 4, 6]. We sought to develop a multigene data set tailored to the study of deep eukaryote phylogeny, including a close outgroup to root the tree. Since mitochon- dria or their remnants are unique to eukaryotes and universal among them [7], universal eukaryotic genes of bacterial origin (euBacs) should provide one of the closest possible outgroups to root the tree. Two parallel protocols employing a combination of homolo- gous clustering and phylogenetic screening were used to identify proteins suitable for deep eukaryote phylogeny (see Supplemental Experimental Procedures and Figure S1 available online). Screening identified genes that appear to be (1) of bacterial origin, (2) present in the last eukaryote com- mon ancestor (LECA) (universal or nearly universal among eukaryotes), and (3) with strong phylogenetic signal (out- paralog free and consistent with well-supported eukaryote phylogeny [5]). Of the 281 universal euBac proteins identified, most failed the latter criteria, primarily due to early gene dupli- cation and lineage-specific losses. Thirty-seven euBacs survived all screening protocols, 33 of which are known or predicted to function in the mitochondrion (Table S1). To increase sampling for Excavata, we sequenced the Acrasis kona (Heterolobosea, Discoba) transcriptome, yielding a full set of 37 euBac proteins. Outgroup taxa included the closest bacterial relatives of the 37 euBacs (Table S2). All euBacs in the final data set reproduce eight or more of the ten major eukaryote groups represented, and 36 euBacs show >60% maximum-likelihood bootstrap (mlBP) support for six or more of these major groups (Table S4). A Rooted Phylogeny of Eukaryotes Phylogenetic analysis of a concatenation of the 37 euBac protein sequences produces a highly resolved phylogeny of eukaryotes (Figure 1). All major nodes are strongly supported based on mlBP [8] and Bayesian inference posterior proba- bilities (biPP) [9]. All ten major eukaryote clades represented are reproduced as monophyletic (1.0/100% biPP/mlBP; Figure 1). These major clades are further organized into three megagroups (1.0/98%–100% biPP/mlBP; Figure 1): Discoba (Excavata), Amorphea (formerly Unikonta), and SA[R]P (Stramenopila+Alveolata+[Rhizaria not represented]+Plantae). Among these, Amorphea and SA[R]P form a clade to the exclusion of Discoba (1.0/90% biPP/mlBP; Figure 1, matrix M). This identifies Discoba, and potentially its parent taxon Excavata, as the sister lineage to the bulk of known eukaryote diversity (‘‘Neozoa’’ [6]), thus yielding a ‘‘neozoan- excavate’’ root. A major concern in a rooted phylogeny is possible artifactual attraction of long ingroup branches toward the potentially long branches of a distant outgroup (long-branch attraction, or LBA) [10]. We tested the euBac phylogeny for possible sources of LBA, particularly signal dependence on fast-evolving align- ment positions and distant outgroup sequences. Removal of the fastest-evolving alignment positions (3,017 sites, or 21% of the data set), defined in a tree-independent manner [11], yielded an identical maximum-likelihood tree with strong support for the neozoan-excavate root (0.99/92% biPP/ mlBP; Figure 1, matrix M 0 ). Sequences with distant bacterial homologs were identified based on the maximum likelihood branch length connecting ingroup and outgroup, corrected for the number of informative sites (‘‘IOScore,’’ Table S1; also see Experimental Procedures). Masking bacterial sequences for the ten euBacs with IOScores > 1.0 increases rather than decreases support for the neozoan-excavate root (0.99/100% biPP/mlBP; Figure 1 matrix M 00 ). 2 These authors contributed equally to this work 3 Deceased *Correspondence: [email protected]
Transcript of An Alternative Root for the Eukaryote Tree of Life

An Alternative Root
Current Biology 24, 465–470, February 17, 2014 ª2014 Elsevier Ltd All rights reserved http://dx.doi.org/10.1016/j.cub.2014.01.036
Report
for the Eukaryote Tree of Life
Ding He,1,2 Omar Fiz-Palacios,1,2 Cheng-Jie Fu,1
Johanna Fehling,1,3 Chun-Chieh Tsai,1
and Sandra L. Baldauf1,*1Program in Systematic Biology, Department of OrganismalBiology, Uppsala University, Norbyvagen 18D, 75236 Uppsala,Sweden
Summary
The root of the eukaryote tree of life defines someof themost
fundamental relationships among species. It is also criticalfor defining the last eukaryote common ancestor (LECA),
the shared heritage of all extant species. The unikont-bikontroot has been the reigning paradigm for eukaryotes for more
than 10 years [1] but is becoming increasingly controversial[2–4]. We developed a carefully vetted data set, consisting of
37 nuclear-encoded proteins of close bacterial ancestry(euBacs) and their closest bacterial relatives, augmented
by deep sequencing of the Acrasis kona (Heterolobosea,Discoba) transcriptome. Phylogenetic analysis of these
data produces a highly robust, fully resolved global phy-logeny of eukaryotes. The tree sorts all examined eukaryotes
into three megagroups and identifies the Discoba, andpotentially its parent taxon Excavata [5], as the sister group
to the bulk of known eukaryote diversity, the proposedNeozoa (Amorphea + Stramenopila+Alveolata+Rhizaria+
Plantae [SARP] [6]). All major alternative hypotheses are
rejected with as little as w50% of the data, and this resolu-tion is unaffected by the presence of fast-evolving alignment
positions or distant outgroup sequences. This ‘‘neozoan-excavate’’ root revises hypotheses of early eukaryote
evolution and highlights the importance of the poorly stud-ied Discoba for understanding the evolution of eukaryotic
diversity and basic cellular processes.
Results
Thirty-Seven Universal euBac ProteinsMultigene phylogenies now assign most known eukaryotesto a few major groups, and these to three proposedmegagroups—Amorphea, Stramenopila+Alveolata+Rhizaria+Plantae (SARP or Diaphoretickes), and Excavata [5]. Howev-er, little effort has been made to explore the eukaryote rootwith multigene phylogeny, and attempts to root the treewith macromolecular characters have given widely differentresults [1, 3, 4, 6]. We sought to develop a multigene dataset tailored to the study of deep eukaryote phylogeny,including a close outgroup to root the tree. Since mitochon-dria or their remnants are unique to eukaryotes and universalamong them [7], universal eukaryotic genes of bacterialorigin (euBacs) should provide one of the closest possibleoutgroups to root the tree.
2These authors contributed equally to this work3Deceased
*Correspondence: [email protected]
Two parallel protocols employing a combination of homolo-gous clustering and phylogenetic screening were used toidentify proteins suitable for deep eukaryote phylogeny (seeSupplemental Experimental Procedures and Figure S1available online). Screening identified genes that appear tobe (1) of bacterial origin, (2) present in the last eukaryote com-mon ancestor (LECA) (universal or nearly universal amongeukaryotes), and (3) with strong phylogenetic signal (out-paralog free and consistent with well-supported eukaryotephylogeny [5]). Of the 281 universal euBac proteins identified,most failed the latter criteria, primarily due to early gene dupli-cation and lineage-specific losses.Thirty-seven euBacs survived all screening protocols, 33 of
which are known or predicted to function in the mitochondrion(Table S1). To increase sampling for Excavata, we sequencedthe Acrasis kona (Heterolobosea, Discoba) transcriptome,yielding a full set of 37 euBac proteins. Outgroup taxa includedthe closest bacterial relatives of the 37 euBacs (Table S2). AlleuBacs in the final data set reproduce eight or more of theten major eukaryote groups represented, and 36 euBacsshow >60% maximum-likelihood bootstrap (mlBP) supportfor six or more of these major groups (Table S4).
A Rooted Phylogeny of EukaryotesPhylogenetic analysis of a concatenation of the 37 euBacprotein sequences produces a highly resolved phylogeny ofeukaryotes (Figure 1). All major nodes are strongly supportedbased on mlBP [8] and Bayesian inference posterior proba-bilities (biPP) [9]. All ten major eukaryote clades representedare reproduced as monophyletic (1.0/100% biPP/mlBP;Figure 1). These major clades are further organized into threemegagroups (1.0/98%–100% biPP/mlBP; Figure 1): Discoba(Excavata), Amorphea (formerly Unikonta), and SA[R]P(Stramenopila+Alveolata+[Rhizaria not represented]+Plantae).Among these, Amorphea and SA[R]P form a clade to theexclusion of Discoba (1.0/90% biPP/mlBP; Figure 1, matrixM). This identifies Discoba, and potentially its parent taxonExcavata, as the sister lineage to the bulk of knowneukaryote diversity (‘‘Neozoa’’ [6]), thus yielding a ‘‘neozoan-excavate’’ root.Amajor concern in a rooted phylogeny is possible artifactual
attraction of long ingroup branches toward the potentially longbranches of a distant outgroup (long-branch attraction, orLBA) [10].We tested the euBac phylogeny for possible sourcesof LBA, particularly signal dependence on fast-evolving align-ment positions and distant outgroup sequences. Removal ofthe fastest-evolving alignment positions (3,017 sites, or 21%of the data set), defined in a tree-independent manner [11],yielded an identical maximum-likelihood tree with strongsupport for the neozoan-excavate root (0.99/92% biPP/mlBP; Figure 1, matrix M0). Sequences with distant bacterialhomologs were identified based on the maximum likelihoodbranch length connecting ingroup and outgroup, correctedfor the number of informative sites (‘‘IOScore,’’ Table S1;also see Experimental Procedures). Masking bacterialsequences for the ten euBacs with IOScores > 1.0 increasesrather than decreases support for the neozoan-excavate root(0.99/100% biPP/mlBP; Figure 1 matrix M00).
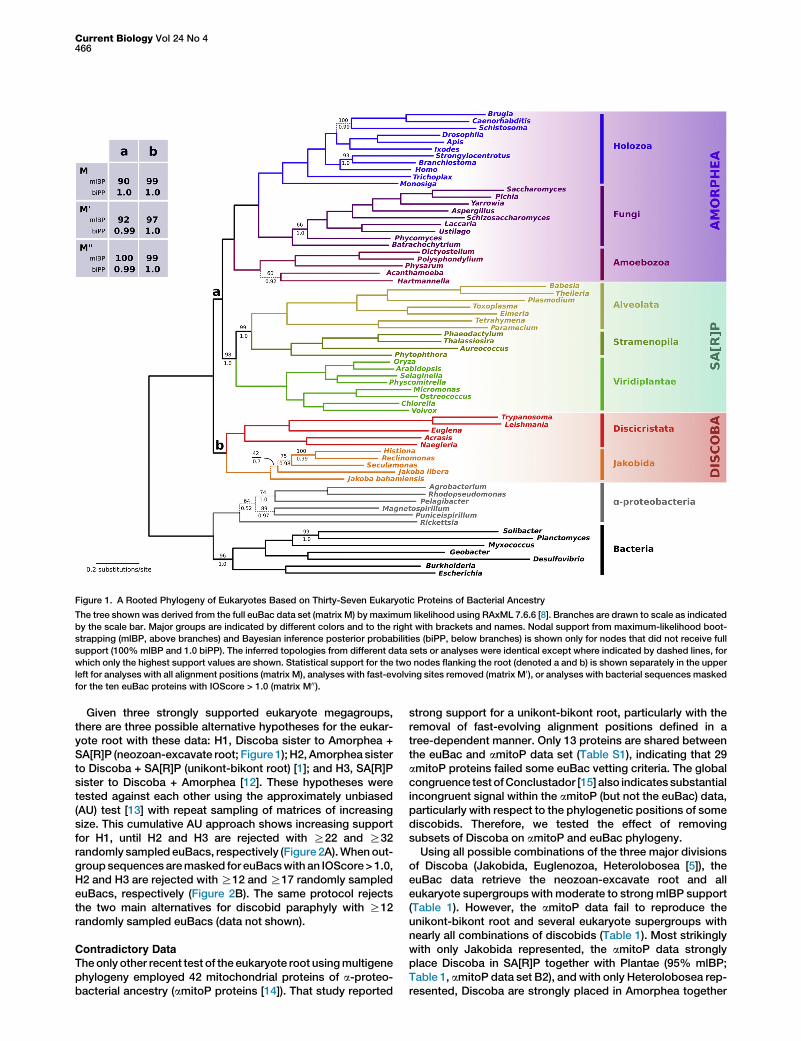
Figure 1. A Rooted Phylogeny of Eukaryotes Based on Thirty-Seven Eukaryotic Proteins of Bacterial Ancestry
The tree shown was derived from the full euBac data set (matrix M) by maximum likelihood using RAxML 7.6.6 [8]. Branches are drawn to scale as indicated
by the scale bar. Major groups are indicated by different colors and to the right with brackets and names. Nodal support from maximum-likelihood boot-
strapping (mlBP, above branches) and Bayesian inference posterior probabilities (biPP, below branches) is shown only for nodes that did not receive full
support (100% mlBP and 1.0 biPP). The inferred topologies from different data sets or analyses were identical except where indicated by dashed lines, for
which only the highest support values are shown. Statistical support for the two nodes flanking the root (denoted a and b) is shown separately in the upper
left for analyses with all alignment positions (matrix M), analyses with fast-evolving sites removed (matrix M0), or analyses with bacterial sequences masked
for the ten euBac proteins with IOScore > 1.0 (matrix M00).
Current Biology Vol 24 No 4466
Given three strongly supported eukaryote megagroups,there are three possible alternative hypotheses for the eukar-yote root with these data: H1, Discoba sister to Amorphea +SA[R]P (neozoan-excavate root; Figure 1); H2, Amorphea sisterto Discoba + SA[R]P (unikont-bikont root) [1]; and H3, SA[R]Psister to Discoba + Amorphea [12]. These hypotheses weretested against each other using the approximately unbiased(AU) test [13] with repeat sampling of matrices of increasingsize. This cumulative AU approach shows increasing supportfor H1, until H2 and H3 are rejected with R22 and R32randomly sampled euBacs, respectively (Figure 2A).Whenout-group sequencesaremasked for euBacswith an IOScore>1.0,H2 and H3 are rejected with R12 and R17 randomly sampledeuBacs, respectively (Figure 2B). The same protocol rejectsthe two main alternatives for discobid paraphyly with R12randomly sampled euBacs (data not shown).
Contradictory DataTheonly other recent test of the eukaryote root usingmultigenephylogeny employed 42 mitochondrial proteins of a-proteo-bacterial ancestry (amitoP proteins [14]). That study reported
strong support for a unikont-bikont root, particularly with theremoval of fast-evolving alignment positions defined in atree-dependent manner. Only 13 proteins are shared betweenthe euBac and amitoP data set (Table S1), indicating that 29amitoP proteins failed some euBac vetting criteria. The globalcongruence test ofConclustador [15] also indicates substantialincongruent signal within the amitoP (but not the euBac) data,particularly with respect to the phylogenetic positions of somediscobids. Therefore, we tested the effect of removingsubsets of Discoba on amitoP and euBac phylogeny.Using all possible combinations of the three major divisions
of Discoba (Jakobida, Euglenozoa, Heterolobosea [5]), theeuBac data retrieve the neozoan-excavate root and alleukaryote supergroups with moderate to strongmlBP support(Table 1). However, the amitoP data fail to reproduce theunikont-bikont root and several eukaryote supergroups withnearly all combinations of discobids (Table 1). Most strikinglywith only Jakobida represented, the amitoP data stronglyplace Discoba in SA[R]P together with Plantae (95% mlBP;Table 1, amitoP data set B2), and with only Heterolobosea rep-resented, Discoba are strongly placed in Amorphea together
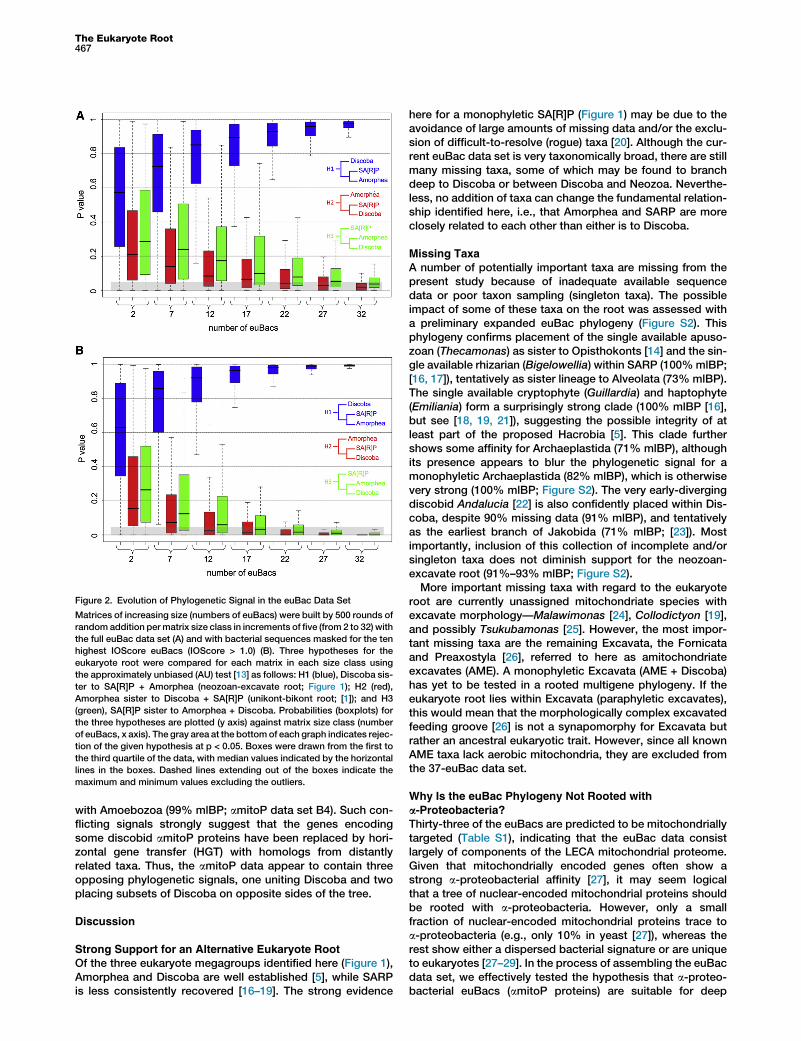
Figure 2. Evolution of Phylogenetic Signal in the euBac Data Set
Matrices of increasing size (numbers of euBacs) were built by 500 rounds of
random addition permatrix size class in increments of five (from 2 to 32) with
the full euBac data set (A) and with bacterial sequences masked for the ten
highest IOScore euBacs (IOScore > 1.0) (B). Three hypotheses for the
eukaryote root were compared for each matrix in each size class using
the approximately unbiased (AU) test [13] as follows: H1 (blue), Discoba sis-
ter to SA[R]P + Amorphea (neozoan-excavate root; Figure 1); H2 (red),
Amorphea sister to Discoba + SA[R]P (unikont-bikont root; [1]); and H3
(green), SA[R]P sister to Amorphea + Discoba. Probabilities (boxplots) for
the three hypotheses are plotted (y axis) against matrix size class (number
of euBacs, x axis). The gray area at the bottomof each graph indicates rejec-
tion of the given hypothesis at p < 0.05. Boxes were drawn from the first to
the third quartile of the data, with median values indicated by the horizontal
lines in the boxes. Dashed lines extending out of the boxes indicate the
maximum and minimum values excluding the outliers.
The Eukaryote Root467
with Amoebozoa (99% mlBP; amitoP data set B4). Such con-flicting signals strongly suggest that the genes encodingsome discobid amitoP proteins have been replaced by hori-zontal gene transfer (HGT) with homologs from distantlyrelated taxa. Thus, the amitoP data appear to contain threeopposing phylogenetic signals, one uniting Discoba and twoplacing subsets of Discoba on opposite sides of the tree.
Discussion
Strong Support for an Alternative Eukaryote RootOf the three eukaryote megagroups identified here (Figure 1),Amorphea and Discoba are well established [5], while SARPis less consistently recovered [16–19]. The strong evidence
here for a monophyletic SA[R]P (Figure 1) may be due to theavoidance of large amounts of missing data and/or the exclu-sion of difficult-to-resolve (rogue) taxa [20]. Although the cur-rent euBac data set is very taxonomically broad, there are stillmany missing taxa, some of which may be found to branchdeep to Discoba or between Discoba and Neozoa. Neverthe-less, no addition of taxa can change the fundamental relation-ship identified here, i.e., that Amorphea and SARP are moreclosely related to each other than either is to Discoba.
Missing TaxaA number of potentially important taxa are missing from thepresent study because of inadequate available sequencedata or poor taxon sampling (singleton taxa). The possibleimpact of some of these taxa on the root was assessed witha preliminary expanded euBac phylogeny (Figure S2). Thisphylogeny confirms placement of the single available apuso-zoan (Thecamonas) as sister to Opisthokonts [14] and the sin-gle available rhizarian (Bigelowellia) within SARP (100%mlBP;[16, 17]), tentatively as sister lineage to Alveolata (73% mlBP).The single available cryptophyte (Guillardia) and haptophyte(Emiliania) form a surprisingly strong clade (100% mlBP [16],but see [18, 19, 21]), suggesting the possible integrity of atleast part of the proposed Hacrobia [5]. This clade furthershows some affinity for Archaeplastida (71% mlBP), althoughits presence appears to blur the phylogenetic signal for amonophyletic Archaeplastida (82% mlBP), which is otherwisevery strong (100% mlBP; Figure S2). The very early-divergingdiscobid Andalucia [22] is also confidently placed within Dis-coba, despite 90% missing data (91% mlBP), and tentativelyas the earliest branch of Jakobida (71% mlBP; [23]). Mostimportantly, inclusion of this collection of incomplete and/orsingleton taxa does not diminish support for the neozoan-excavate root (91%–93% mlBP; Figure S2).More important missing taxa with regard to the eukaryote
root are currently unassigned mitochondriate species withexcavate morphology—Malawimonas [24], Collodictyon [19],and possibly Tsukubamonas [25]. However, the most impor-tant missing taxa are the remaining Excavata, the Fornicataand Preaxostyla [26], referred to here as amitochondriateexcavates (AME). A monophyletic Excavata (AME + Discoba)has yet to be tested in a rooted multigene phylogeny. If theeukaryote root lies within Excavata (paraphyletic excavates),this would mean that the morphologically complex excavatedfeeding groove [26] is not a synapomorphy for Excavata butrather an ancestral eukaryotic trait. However, since all knownAME taxa lack aerobic mitochondria, they are excluded fromthe 37-euBac data set.
Why Is the euBac Phylogeny Not Rooted with
a-Proteobacteria?Thirty-three of the euBacs are predicted to be mitochondriallytargeted (Table S1), indicating that the euBac data consistlargely of components of the LECA mitochondrial proteome.Given that mitochondrially encoded genes often show astrong a-proteobacterial affinity [27], it may seem logicalthat a tree of nuclear-encoded mitochondrial proteins shouldbe rooted with a-proteobacteria. However, only a smallfraction of nuclear-encoded mitochondrial proteins trace toa-proteobacteria (e.g., only 10% in yeast [27]), whereas therest show either a dispersed bacterial signature or are uniqueto eukaryotes [27–29]. In the process of assembling the euBacdata set, we effectively tested the hypothesis that a-proteo-bacterial euBacs (amitoP proteins) are suitable for deep
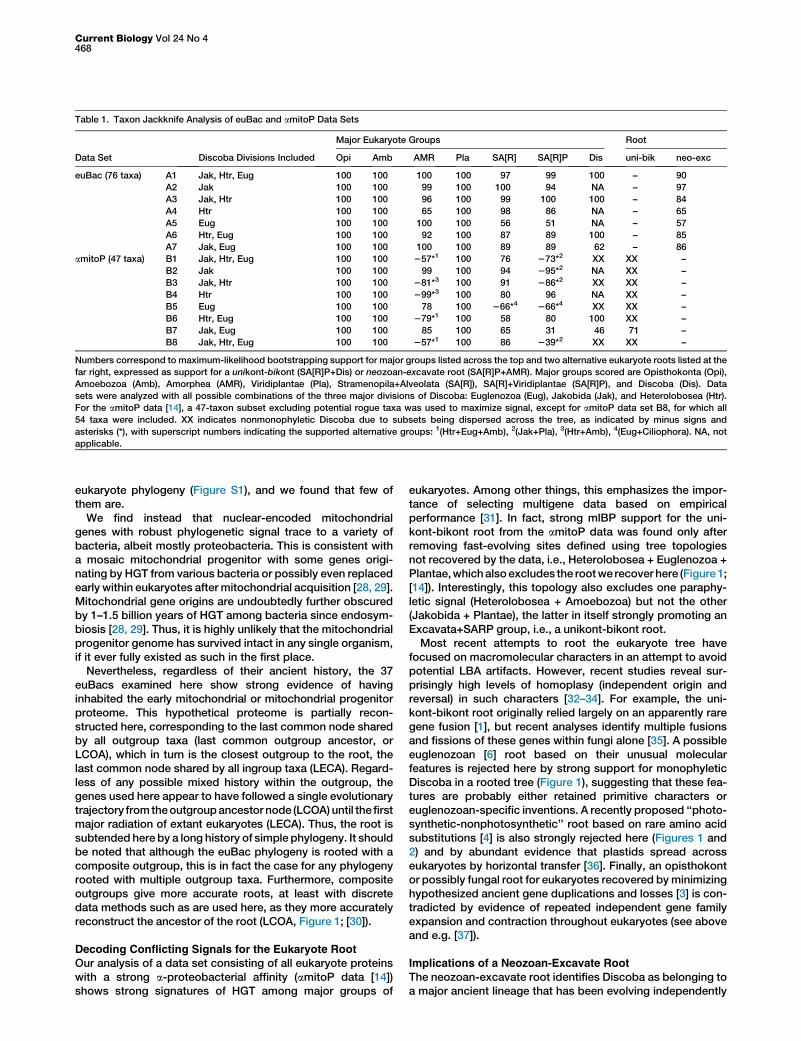
Table 1. Taxon Jackknife Analysis of euBac and amitoP Data Sets
Data Set Discoba Divisions Included
Major Eukaryote Groups Root
Opi Amb AMR Pla SA[R] SA[R]P Dis uni-bik neo-exc
euBac (76 taxa) A1 Jak, Htr, Eug 100 100 100 100 97 99 100 – 90
A2 Jak 100 100 99 100 100 94 NA – 97
A3 Jak, Htr 100 100 96 100 99 100 100 – 84
A4 Htr 100 100 65 100 98 86 NA – 65
A5 Eug 100 100 100 100 56 51 NA – 57
A6 Htr, Eug 100 100 92 100 87 89 100 – 85
A7 Jak, Eug 100 100 100 100 89 89 62 – 86
amitoP (47 taxa) B1 Jak, Htr, Eug 100 100 257*1 100 76 273*2 XX XX –
B2 Jak 100 100 99 100 94 295*2 NA XX –
B3 Jak, Htr 100 100 281*3 100 91 286*2 XX XX –
B4 Htr 100 100 299*3 100 80 96 NA XX –
B5 Eug 100 100 78 100 266*4 266*4 XX XX –
B6 Htr, Eug 100 100 279*1 100 58 80 100 XX –
B7 Jak, Eug 100 100 85 100 65 31 46 71 –
B8 Jak, Htr, Eug 100 100 257*1 100 86 239*2 XX XX –
Numbers correspond to maximum-likelihood bootstrapping support for major groups listed across the top and two alternative eukaryote roots listed at the
far right, expressed as support for a unikont-bikont (SA[R]P+Dis) or neozoan-excavate root (SA[R]P+AMR). Major groups scored are Opisthokonta (Opi),
Amoebozoa (Amb), Amorphea (AMR), Viridiplantae (Pla), Stramenopila+Alveolata (SA[R]), SA[R]+Viridiplantae (SA[R]P), and Discoba (Dis). Data
sets were analyzed with all possible combinations of the three major divisions of Discoba: Euglenozoa (Eug), Jakobida (Jak), and Heterolobosea (Htr).
For the amitoP data [14], a 47-taxon subset excluding potential rogue taxa was used to maximize signal, except for amitoP data set B8, for which all
54 taxa were included. XX indicates nonmonophyletic Discoba due to subsets being dispersed across the tree, as indicated by minus signs and
asterisks (*), with superscript numbers indicating the supported alternative groups: 1(Htr+Eug+Amb), 2(Jak+Pla), 3(Htr+Amb), 4(Eug+Ciliophora). NA, not
applicable.
Current Biology Vol 24 No 4468
eukaryote phylogeny (Figure S1), and we found that few ofthem are.
We find instead that nuclear-encoded mitochondrialgenes with robust phylogenetic signal trace to a variety ofbacteria, albeit mostly proteobacteria. This is consistent witha mosaic mitochondrial progenitor with some genes origi-nating by HGT from various bacteria or possibly even replacedearly within eukaryotes after mitochondrial acquisition [28, 29].Mitochondrial gene origins are undoubtedly further obscuredby 1–1.5 billion years of HGT among bacteria since endosym-biosis [28, 29]. Thus, it is highly unlikely that the mitochondrialprogenitor genome has survived intact in any single organism,if it ever fully existed as such in the first place.
Nevertheless, regardless of their ancient history, the 37euBacs examined here show strong evidence of havinginhabited the early mitochondrial or mitochondrial progenitorproteome. This hypothetical proteome is partially recon-structed here, corresponding to the last common node sharedby all outgroup taxa (last common outgroup ancestor, orLCOA), which in turn is the closest outgroup to the root, thelast common node shared by all ingroup taxa (LECA). Regard-less of any possible mixed history within the outgroup, thegenes used here appear to have followed a single evolutionarytrajectory fromtheoutgroupancestor node (LCOA) until thefirstmajor radiation of extant eukaryotes (LECA). Thus, the root issubtended here by a long history of simple phylogeny. It shouldbe noted that although the euBac phylogeny is rooted with acomposite outgroup, this is in fact the case for any phylogenyrooted with multiple outgroup taxa. Furthermore, compositeoutgroups give more accurate roots, at least with discretedata methods such as are used here, as they more accuratelyreconstruct the ancestor of the root (LCOA, Figure 1; [30]).
Decoding Conflicting Signals for the Eukaryote RootOur analysis of a data set consisting of all eukaryote proteinswith a strong a-proteobacterial affinity (amitoP data [14])shows strong signatures of HGT among major groups of
eukaryotes. Among other things, this emphasizes the impor-tance of selecting multigene data based on empiricalperformance [31]. In fact, strong mlBP support for the uni-kont-bikont root from the amitoP data was found only afterremoving fast-evolving sites defined using tree topologiesnot recovered by the data, i.e., Heterolobosea + Euglenozoa +Plantae,whichalsoexcludes the rootwe recover here (Figure1;[14]). Interestingly, this topology also excludes one paraphy-letic signal (Heterolobosea + Amoebozoa) but not the other(Jakobida + Plantae), the latter in itself strongly promoting anExcavata+SARP group, i.e., a unikont-bikont root.Most recent attempts to root the eukaryote tree have
focused on macromolecular characters in an attempt to avoidpotential LBA artifacts. However, recent studies reveal sur-prisingly high levels of homoplasy (independent origin andreversal) in such characters [32–34]. For example, the uni-kont-bikont root originally relied largely on an apparently raregene fusion [1], but recent analyses identify multiple fusionsand fissions of these genes within fungi alone [35]. A possibleeuglenozoan [6] root based on their unusual molecularfeatures is rejected here by strong support for monophyleticDiscoba in a rooted tree (Figure 1), suggesting that these fea-tures are probably either retained primitive characters oreuglenozoan-specific inventions. A recently proposed ‘‘photo-synthetic-nonphotosynthetic’’ root based on rare amino acidsubstitutions [4] is also strongly rejected here (Figures 1 and2) and by abundant evidence that plastids spread acrosseukaryotes by horizontal transfer [36]. Finally, an opisthokontor possibly fungal root for eukaryotes recovered byminimizinghypothesized ancient gene duplications and losses [3] is con-tradicted by evidence of repeated independent gene familyexpansion and contraction throughout eukaryotes (see aboveand e.g. [37]).
Implications of a Neozoan-Excavate Root
The neozoan-excavate root identifies Discoba as belonging toa major ancient lineage that has been evolving independently

The Eukaryote Root469
since the first speciation events of extant eukaryotes.Expressed in terms of more familiar taxa, the neozoan-exca-vate root means that Discoba diverged from the main line ofeukaryote descent well before the origin of the separate line-ages that eventually gave rise to plants, animals, and fungi.Nonetheless, other than the well-studied parasites Trypano-soma and Leishmania, most molecular information on disco-bids concerns their mitochondrial genomes (mtDNA). Theserange from the large, gene-rich bacterial-like mtDNAs of jako-bids [23] to the highly fragmented and/or massively editedmtDNAs of Euglenozoa [38]. This limited molecular samplingsuggests that Discoba could harbor a further wealth of biolog-ical novelty.
The neozoan-excavate root also emphasizes the impor-tance of Discoba, and probably also AME, for understandingearly eukaryote evolution. Extensive similarities have beenidentified between Neozoa and the only fully sequencedfree-living discobid, Naegleria gruberi [39]. However,Naegleria is still only a single data point in the vast stretch ofevolutionary time separating Discoba and the bulk of knowneukaryotes. Further data are clearly needed from a broadersampling of Excavata in order to distinguish possibly uniquefeatures of Naegleria from the common heritage of extanteukaryotes.
Experimental Procedures
Acrasis kona Protein Sequences
A cDNA library was constructed from total RNA extracted from a
laboratory-grown culture of Acrasis kona ATCC strain MYA-3509 (formerly
Acrasis rosea [40]) and sequenced on a 454 GS FLX Titanium
platform. For details of cell culture, RNA extraction, sequencing, assembly,
and transcript screening, please see the Supplemental Experimental
Procedures.
Identification of Universal euBacs
Two separate pipelines were used to identify euBacs and determine their
suitability for deep eukaryote phylogeny (Figure S1). Both protocols used
the predicted proteomes from 12 completely sequenced eukaryote
genomes as a starting point (Supplemental Experimental Procedures; Fig-
ure S1). The 12 included at least one representative each from five of the
six currently recognized supergroups of eukaryotes (Opisthokonta,
Amoebozoa, Archaeplastida, Chromalveolata, and Excavata; [5]), as sub-
stantial genomic data were unavailable from Rhizaria during these analyses
(Table S2).
Final euBac Data Set Assembly
Eukaryotic sequenceswere taken primarily from the predicted proteomes of
completely sequenced genomes (Table S2). Missing entries and data from
taxa without complete genome sequences were obtained by BLASTp
searches of the NCBI nr or Expressed Sequence Tags (EST) databases or
other publically available genome data, as needed. Some sequences were
also assembled by hand from EST data, particularly for jakobids. Whenever
possible, sequences were taken from a single target genome (Table S2), but
for many taxa one or two genes could be found only in related taxa. In these
cases, sequences were taken from the most closely related taxon possible,
which was always in the same genus. Bacterial sequences were retrieved
separately using a taxonomic hierarchy protocol (Supplemental Experi-
mental Procedures; Table S3). During assembly, the data set was checked
regularly and, especially when complete, by single-gene control trees to
assure that all sequences were orthologs. The final 37 trimmed alignments
were assembled by hand into a single interleaved full data matrix M. Two
modified versions of this matrix were generated by (1) removing the
fastest-evolving alignment positions (matrix M0; see below) and (2) removing
bacterial sequences for the ten proteins with IOScores > 1.0 (matrix M00; seebelow and Table S1).
Phylogenetic Analyses
Maximum-likelihood bootstrap analyses consisted of 100 rapid bootstrap
replicates (mlBP) performed using theWAG amino acid substitution models
corrected for observed amino acid frequencies and among-site rate varia-
tion using a four-discrete-category gamma distribution as implemented in
RAxML 7.6.6 [8]. Bayesian inference analyses allowing for site-heteroge-
neous amino acid replacement processes and rate variation among sites
and among lineages utilized the CAT+G4+covarion model with constant
sites removed. These analyses were run on two independent chains using
PhyloBayes 3.3 [9]. The convergence of two root-defining nodes (a and b;
Figure 1) among chains was diagnosed based on a comparison of the
frequency of all bipartitions after discarding w50% of the cycles as burn-
in (maxdiff < 0.1). All analyses were run on a local cluster, the Uppsala
Multidisciplinary Center for Advanced Computational Science (UPPMAX,
http://www.uppmax.uu.se), BioPortal (http://www.mn.uio.no/ibv/bioportal/
index.html), or the Cyberinfrastructure for Phylogenetic Research (CIPRES,
http://www.phylo.org; [41]), depending on availability.
Vetting Sources of Artifacts and Incongruence
The corrected distances between ingroup and outgroup (IOScore) were
calculated from individual maximum-likelihood control trees [8]. The value
corresponds to the length of the branch connecting ingroup and outgroup
(dIO), divided by the number of informative sites (InfSites), multiplied by
1,000 for ease of interpretation [(dIO/InfSites) 3 1,000)]. The value for
InfSites was calculated with PAUP* [42] under the heuristic search function
and corresponds to all sites with at least two different states shared by a
minimum of two taxa each. Fast-evolving sites were determined in a tree-
independent manner using the program TIGER [11] with default settings.
Sites were assigned into ten rate bins. Topology-based congruent tests
were run using Conclustador with maximum-likelihood bootstrap method
as described in [15].
Cumulative AU Test
Alternative hypotheses were tested using the approximately unbiased (AU)
test [13] on matrices of increasing size from 2 to 32 in increments of 5 due to
computational constraints. Matrices were built by random sampling without
replacement of the 37-euBac protein data set. Sampling consisted of 500
independent replicates for each matrix size class, yielding a total of 3,500
unique matrices, with any identical replicates automatically deleted. AU
tests for alternative phylogenetic hypotheses were performed on each
matrix under the WAG substitution model with 100,000 bootstrap replicates
using the program TREEFINDER [43]. The median p values for each
hypothesis were then calculated from the p values for all samples in each
matrix size class.
Accession Numbers
Deduced amino acids sequences for the 37 euBacs from Acrasis kona have
been deposited in GenBank with accession numbers JL968957–JL968959,
JL968961–JL968982, JL968984, and JL968986–JL968996. The 37-euBac
protein data matrices have been deposited in TreeBASE (http://treebase.
org/; Study ID S14693).
Supplemental Information
Supplemental Information includes two figures, four tables, and Supple-
mental Experimental Procedures and can be found with this article online
at http://dx.doi.org/10.1016/j.cub.2014.01.036.
Acknowledgments
We thank P. Ajawatanawong, E. Lundin, and L. Ponnuswamy for assistance
in writing scripts; J.W. Leigh for assistance in running Conclustador; and M.
Brown and F. Spiegel for kindly providing Acrasis kona cultures. We also
thank the Joint Genome Institute, Protist Database Project, andWashington
University Genome Institute for the use of unpublished sequences and
UPPMAX, BioPortal, and CIPRES for the use of computational resources.
This work was supported by grants from the Swedish Research Council
(Vetenskapradet). This paper is dedicated to the memory of Dr. Johanna
Fehling (1979–2011).
Received: November 4, 2013
Revised: December 12, 2013
Accepted: January 16, 2014
Published: February 6, 2014

Current Biology Vol 24 No 4470
References
1. Stechmann, A., and Cavalier-Smith, T. (2002). Rooting the eukaryote
tree by using a derived gene fusion. Science 297, 89–91.
2. Roger, A.J., and Simpson, A.G.B. (2009). Evolution: revisiting the root of
the eukaryote tree. Curr. Biol. 19, R165–R167.
3. Katz, L.A., Grant, J.R., Parfrey, L.W., and Burleigh, J.G. (2012). Turning
the crown upside down: gene tree parsimony roots the eukaryotic tree
of life. Syst. Biol. 61, 653–660.
4. Rogozin, I.B., Basu, M.K., Csuros, M., and Koonin, E.V. (2009). Analysis
of rare genomic changes does not support the unikont-bikont phylog-
eny and suggests cyanobacterial symbiosis as the point of primary
radiation of eukaryotes. Genome Biol. Evol. 1, 99–113.
5. Adl, S.M., Simpson, A.G., Lane, C.E., Luke�s, J., Bass, D., Bowser, S.S.,
Brown, M.W., Burki, F., Dunthorn, M., Hampl, V., et al. (2012). The
revised classification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–493.
6. Cavalier-Smith, T. (2010). Kingdoms Protozoa and Chromista and the
eozoan root of the eukaryotic tree. Biol. Lett. 6, 342–345.
7. Hjort, K., Goldberg, A.V., Tsaousis, A.D., Hirt, R.P., and Embley, T.M.
(2010). Diversity and reductive evolution of mitochondria among micro-
bial eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 713–727.
8. Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap
algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771.
9. Lartillot, N., Lepage, T., and Blanquart, S. (2009). PhyloBayes 3: a
Bayesian software package for phylogenetic reconstruction andmolec-
ular dating. Bioinformatics 25, 2286–2288.
10. Wagele, J.W., and Mayer, C. (2007). Visualizing differences in phyloge-
netic information content of alignments and distinction of three classes
of long-branch effects. BMC Evol. Biol. 7, 147.
11. Cummins, C.A., and McInerney, J.O. (2011). A method for inferring the
rate of evolution of homologous characters that can potentially improve
phylogenetic inference, resolve deep divergence and correct system-
atic biases. Syst. Biol. 60, 833–844.
12. Wideman, J.G., Gawryluk, R.M.R., Gray, M.W., and Dacks, J.B. (2013).
The ancient and widespread nature of the ER-mitochondria encounter
structure. Mol. Biol. Evol. 30, 2044–2049.
13. Shimodaira, H. (2002). An approximately unbiased test of phylogenetic
tree selection. Syst. Biol. 51, 492–508.
14. Derelle, R., and Lang, B.F. (2012). Rooting the eukaryotic tree with mito-
chondrial and bacterial proteins. Mol. Biol. Evol. 29, 1277–1289.
15. Leigh, J.W., Schliep, K., Lopez, P., and Bapteste, E. (2011). Let them fall
where they may: congruence analysis in massive phylogenetically
messy data sets. Mol. Biol. Evol. 28, 2773–2785.
16. Hackett, J.D., Yoon, H.S., Li, S., Reyes-Prieto, A., Rummele, S.E., and
Bhattacharya, D. (2007). Phylogenomic analysis supports the mono-
phyly of cryptophytes and haptophytes and the association of rhizaria
with chromalveolates. Mol. Biol. Evol. 24, 1702–1713.
17. Burki, F., Shalchian-Tabrizi, K., and Pawlowski, J. (2008).
Phylogenomics reveals a new ‘megagroup’ including most photosyn-
thetic eukaryotes. Biol. Lett. 4, 366–369.
18. Parfrey, L.W., Grant, J., Tekle, Y.I., Lasek-Nesselquist, E., Morrison,
H.G., Sogin, M.L., Patterson, D.J., and Katz, L.A. (2010). Broadly
sampled multigene analyses yield a well-resolved eukaryotic tree of
life. Syst. Biol. 59, 518–533.
19. Zhao, S., Burki, F., Brate, J., Keeling, P.J., Klaveness, D., and Shalchian-
Tabrizi, K. (2012). Collodictyon—an ancient lineage in the tree of eukary-
otes. Mol. Biol. Evol. 29, 1557–1568.
20. Aberer, A.J., Krompass, D., and Stamatakis, A. (2013). Pruning rogue
taxa improves phylogenetic accuracy: an efficient algorithm and
webservice. Syst. Biol. 62, 162–166.
21. Burki, F., Okamoto, N., Pombert, J.F., and Keeling, P.J. (2012). The
evolutionary history of haptophytes and cryptophytes: phylogenomic
evidence for separate origins. Proc. Biol. Sci. 279, 2246–2254.
22. Lara, E., Chatzinotas, A., andSimpson, A.G. (2006). Andalucia (n. gen.)—
the deepest branch within jakobids (Jakobida; Excavata), based on
morphological and molecular study of a new flagellate from soil.
J. Eukaryot. Microbiol. 53, 112–120.
23. Burger, G., Gray, M.W., Forget, L., and Lang, B.F. (2013). Strikingly
bacteria-like and gene-rich mitochondrial genomes throughout jakobid
protists. Genome Biol. Evol. 5, 418–438.
24. Hampl, V., Hug, L., Leigh, J.W., Dacks, J.B., Lang, B.F., Simpson,
A.G.B., and Roger, A.J. (2009). Phylogenomic analyses support the
monophyly of Excavata and resolve relationships among eukaryotic
‘‘supergroups’’. Proc. Natl. Acad. Sci. USA 106, 3859–3864.
25. Yabuki, A., Nakayama, T., Yubuki, N., Hashimoto, T., Ishida, K.-I., and
Inagaki, Y. (2011). Tsukubamonas globosa n. gen., n. sp., a novel exca-
vate flagellate possibly holding a key for the early evolution in
‘‘Discoba’’. J. Eukaryot. Microbiol. 58, 319–331.
26. Simpson, A.G.B. (2003). Cytoskeletal organization, phylogenetic affin-
ities and systematics in the contentious taxon Excavata (Eukaryota).
Int. J. Syst. Evol. Microbiol. 53, 1759–1777.
27. Karlberg, O., Canback, B., Kurland, C.G., and Andersson, S.G. (2000).
The dual origin of the yeast mitochondrial proteome. Yeast 17, 170–187.
28. Huynen, M.A., Duarte, I., and Szklarczyk, R. (2013). Loss, replacement
and gain of proteins at the origin of the mitochondria. Biochim.
Biophys. Acta 1827, 224–231.
29. Thiergart, T., Landan, G., Schenk,M., Dagan, T., andMartin,W.F. (2012).
An evolutionary network of genes present in the eukaryote common
ancestor polls genomes on eukaryotic and mitochondrial origin.
Genome Biol. Evol. 4, 466–485.
30. Graham, S.W., Olmstead, R.G., and Barrett, S.C.H. (2002). Rooting
phylogenetic trees with distant outgroups: a case study from the
commelinoid monocots. Mol. Biol. Evol. 19, 1769–1781.
31. Salichos, L., and Rokas, A. (2013). Inferring ancient divergences re-
quires genes with strong phylogenetic signals. Nature 497, 327–331.
32. Bapteste, E., and Philippe, H. (2002). The potential value of indels as
phylogenetic markers: position of trichomonads as a case study. Mol.
Biol. Evol. 19, 972–977.
33. Han, K.-L., Braun, E.L., Kimball, R.T., Reddy, S., Bowie, R.C., Braun,
M.J., Chojnowski, J.L., Hackett, S.J., Harshman, J., Huddleston, C.J.,
et al. (2011). Are transposable element insertions homoplasy free?: an
examination using the avian tree of life. Syst. Biol. 60, 375–386.
34. Ajawatanawong, P., and Baldauf, S.L. (2013). Evolution of protein indels
in plants, animals and fungi. BMC Evol. Biol. 13, 140.
35. Leonard, G., and Richards, T.A. (2012). Genome-scale comparative
analysis of gene fusions, gene fissions, and the fungal tree of life.
Proc. Natl. Acad. Sci. USA 109, 21402–21407.
36. Archibald, J.M. (2009). The puzzle of plastid evolution. Curr. Biol. 19,
R81–R88.
37. Breviario, D., Gianı, S., and Morello, L. (2013). Multiple tubulins: evolu-
tionary aspects and biological implications. Plant J. 75, 202–218.
38. Flegontov, P., Gray, M.W., Burger, G., and Luke�s, J. (2011). Gene frag-
mentation: a key to mitochondrial genome evolution in Euglenozoa?
Curr. Genet. 57, 225–232.
39. Fritz-Laylin, L.K., Prochnik, S.E., Ginger, M.L., Dacks, J.B., Carpenter,
M.L., Field, M.C., Kuo, A., Paredez, A., Chapman, J., Pham, J., et al.
(2010). The genome of Naegleria gruberi illuminates early eukaryotic
versatility. Cell 140, 631–642.
40. Brown,M.W., Silberman, J.D., and Spiegel, F.W. (2012). A contemporary
evaluation of the acrasids (Acrasidae, Heterolobosea, Excavata). Eur. J.
Protistol. 48, 103–123.
41. Miller, M.A., Pfeiffer, W., and Schwartz, T. (2010). Creating the CIPRES
Science Gateway for inference of large phylogenetic trees. In
Proceedings of the Gateway Computing Environments Workshop
(GCE) 2010 (New York: Institute of Electrical and Electronics
Engineers), pp. 1–8.
42. Swofford, D.L. (1998). PAUP*: Phylogenetic Analysis Using Parsimony
(*and Other Methods), v4.0 (Sunderland: Sinauer Associates).
43. Jobb, G., von Haeseler, A., and Strimmer, K. (2004). TREEFINDER: a
powerful graphical analysis environment for molecular phylogenetics.
BMC Evol. Biol. 4, 18.




















