



AIRE's CARD Revealed, a New Structure for Central Tolerance Provokes Transcriptional Plasticity
19
1 AIRE’S CARD REVEALED; A NEW STRUCTURE FOR CENTRAL TOLERANCE PROVOKES TRANSCRIPTIONAL PLASTICITY* Brian J. Ferguson #2 , Clare Alexander #2 , Simona W. Rossi ‡ , Ingrid Liiv † , Ana Rebane † , Catherine L. Worth + , Joyce Wong + , Martti Laan † , Pärt Peterson † , Eric J. Jenkinson ‡ , Graham Anderson ‡ , Hamish S. Scott § , Anne Cooke # & Tina Rich #1, # Department of Pathology, Divisions of Immunology and Cellular Pathology, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QP, UK. ‡ MRC Centre for Immune Regulation, Institute for Biomedical Research, University of Birmingham, B15 2TT, UK. † Molecular Pathology, University of Tartu, Biomedicum, 50411, Estonia. + Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QP, UK § Walter and Eliza Hall Institute of Medical Research, 3050 Melbourne, Australia. Running Title: AIRE CARD domain drives nuclear CBP accumulation Address correspondence to: T. Rich, Institute of Comparative Medicine, University of Glasgow, G61 1QH, UK, Scotland, UK. Tel: (44)-(0)141-3308213; Fax: (44)-(0)141-3305602; E-mail: [email protected] Developing T cells encounter peripheral self antigens in the thymus in order to delete autoreactive clones. It is now known that the Autoimmune Regulator protein (AIRE) which is expressed in thymic medullary epithelial cells plays a key role in regulating the thymic transcription of these peripheral tissue specific antigens. Mutations in the AIRE gene are associated with a severe multi-organ autoimmune syndrome (APECED) and autoimmune reactivities are manifest in AIRE deficient mice. Functional AIRE protein is expressed as distinct nuclear puncta though no structural basis existed to explain their relevance to disease. In addressing the cell biologic basis for APECED we made the unexpected discovery that an AIRE mutation hotspot lies in a caspase recruitment domain (CARD). Combined homology modeling and in- vitro data now show how APECED mutations influence the activity of this transcriptional regulator. We also provide novel in-vivo evidence for AIRE’s association with a global transcription cofactor, which may underlie AIRE’s focal, genome wide, alteration of the transcriptome. Central tolerance remains a conundrum for Immunologists (1). At the cellular level, we know that depletion of self-reactive thymocytes is preceded by their thymic preview of tissue- specific antigens. This antigenic display, and the transcriptional alterations required to generate it, are attributed to the activity of a single protein, autoimmune regulator (AIRE), whose molecular mode of action remains largely unknown. The necessity for AIRE during the generation of tolerance is illustrated by the multi organ autoimmune disease that results upon its mutation. This autoimmune syndrome, termed autoimmune- polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) or autoimmune- polyglandular syndrome type 1 (APS-1), is diagnosed when two of the following three symptoms are present; hypoparathyroidism, adrenal insufficiency or chronic mucocutaneous candidal infections. Microarray analyses have shown that the AIRE gene (2,3) encodes an activity that imposes large scale alterations to the transcriptome of medullary thymic epithelial cells (mTECs) to favor transcripts of tissue specific antigens (TSAs) (4,5). Whilst mTEC expression of TSAs incurs protection from autoimmunity (6-8) the molecular mechanisms of AIRE function have been difficult to deduce. AIRE mutations in APECED patients http://www.jbc.org/cgi/doi/10.1074/jbc.M707211200 The latest version is at JBC Papers in Press. Published on November 1, 2007 as Manuscript M707211200 Copyright 2007 by The American Society for Biochemistry and Molecular Biology, Inc. by on November 22, 2007 www.jbc.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of AIRE's CARD Revealed, a New Structure for Central Tolerance Provokes Transcriptional Plasticity

1
AIRE’S CARD REVEALED; A NEW STRUCTURE FOR CENTRAL TOLERANCE PROVOKES
TRANSCRIPTIONAL PLASTICITY*
Brian J. Ferguson#2, Clare Alexander#2, Simona W. Rossi‡, Ingrid Liiv†, Ana Rebane†, Catherine L. Worth+, Joyce Wong+, Martti Laan†, Pärt Peterson†, Eric J. Jenkinson‡, Graham Anderson‡, Hamish S.
Scott§, Anne Cooke# & Tina Rich#1, #Department of Pathology, Divisions of Immunology and Cellular Pathology, University of Cambridge, Tennis
Court Road, Cambridge, CB2 1QP, UK. ‡MRC Centre for Immune Regulation, Institute for Biomedical Research, University of Birmingham, B15 2TT,
UK. †Molecular Pathology, University of Tartu, Biomedicum, 50411, Estonia.
+Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QP, UK §Walter and Eliza Hall Institute of Medical Research, 3050 Melbourne, Australia.
Running Title: AIRE CARD domain drives nuclear CBP accumulation
Address correspondence to: T. Rich, Institute of Comparative Medicine, University of Glasgow, G61 1QH, UK, Scotland, UK. Tel: (44)-(0)141-3308213; Fax: (44)-(0)141-3305602; E-mail:
[email protected] Developing T cells encounter peripheral self antigens in the thymus in order to delete autoreactive clones. It is now known that the Autoimmune Regulator protein (AIRE) which is expressed in thymic medullary epithelial cells plays a key role in regulating the thymic transcription of these peripheral tissue specific antigens. Mutations in the AIRE gene are associated with a severe multi-organ autoimmune syndrome (APECED) and autoimmune reactivities are manifest in AIRE deficient mice. Functional AIRE protein is expressed as distinct nuclear puncta though no structural basis existed to explain their relevance to disease. In addressing the cell biologic basis for APECED we made the unexpected discovery that an AIRE mutation hotspot lies in a caspase recruitment domain (CARD). Combined homology modeling and in-vitro data now show how APECED mutations influence the activity of this transcriptional regulator. We also provide novel in-vivo evidence for AIRE’s association with a global transcription cofactor, which may underlie AIRE’s focal, genome wide, alteration of the transcriptome.
Central tolerance remains a conundrum for Immunologists (1). At the cellular level, we know that depletion of self-reactive thymocytes is preceded by their thymic preview of tissue-specific antigens. This antigenic display, and the transcriptional alterations required to generate it, are attributed to the activity of a single protein, autoimmune regulator (AIRE), whose molecular mode of action remains largely unknown. The necessity for AIRE during the generation of tolerance is illustrated by the multi organ autoimmune disease that results upon its mutation. This autoimmune syndrome, termed autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) or autoimmune-polyglandular syndrome type 1 (APS-1), is diagnosed when two of the following three symptoms are present; hypoparathyroidism, adrenal insufficiency or chronic mucocutaneous candidal infections. Microarray analyses have shown that the AIRE gene (2,3) encodes an activity that imposes large scale alterations to the transcriptome of medullary thymic epithelial cells (mTECs) to favor transcripts of tissue specific antigens (TSAs) (4,5). Whilst mTEC expression of TSAs incurs protection from autoimmunity (6-8) the molecular mechanisms of AIRE function have been difficult to deduce. AIRE mutations in APECED patients
http://www.jbc.org/cgi/doi/10.1074/jbc.M707211200The latest version is at JBC Papers in Press. Published on November 1, 2007 as Manuscript M707211200
Copyright 2007 by The American Society for Biochemistry and Molecular Biology, Inc.
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

2
are often of limited value in analyzing AIRE function as many generate truncates or no protein at all. However a series of missense mutations in one mutation hotspot has been informative (9); the amino terminal sequence labeled the homogeneously staining region, (HSR) which drives AIRE oligomerisation (9). However, ‘HSR’ is a descriptive moniker and implies no structural information which has hampered efforts to discover the mechanism of AIRE self-assembly into nuclear foci. At the time of writing, no supra-molecular structure has been described for any sub-nuclear domain though, in many cases, the determination of the structural and cellular criteria for puncta assembly have been critical milestones in determining function. This has proven to be the case for nuclear domains of promyelocytic leukemia protein (PML-NDs) which modulate transcriptional responses to stress (10). Unlike AIRE, the protein assembly motifs that drive nucleation of PML puncta are evident if one examines the PML primary amino acid sequence. However, protein homologues that share identical folds can evade detection by standard sequence based search algorithms if sequence identity is low (structural conservation may require few amino acid matches) (11). With this in mind, we took advantage of a powerful structure based sequence alignment program to reveal previously hidden homologies in AIRE’s HSR (12). We now show that the amino terminus of AIRE expresses a previously un-described caspase recruitment (CARD) domain whose integrity is needed for AIRE trans-activation of TSAs. This finding presents fresh insights into how missense AIRE mutations found in APECED patients disrupt AIRE function to cause disease. AIRE’s function in altering the transcriptome is likely to involve interactions with histone modifying activities. This possibility has been raised by in-vitro experiments showing that AIRE interacts with a global transcription cofactor, the histone acetyltransferase, CREB binding protein (CBP) to enhance AIRE’s transactivating potential (13,14). However the biologic relevance of these findings are questionable given other studies showing CBP sequestration by PML-NDs (15,14). We used an experimental system that recapitulates the in vivo environment; fetal thymic organ culture (FTOC), to study the spatio-temporal regulation of CBP and AIRE following stimulation of the receptor activator of NF-kappaB (RANK)
pathway that induces AIRE protein (16). This treatment provokes the focal nuclear accumulation of CBP with AIRE puncta, irrespective of the PML-ND fraction. However, RANK signaling in AIRE null thymi failed to provoke any nuclear concentration of CBP. These data provide novel mechanistic insights into the molecular and cellular regulation of a key effector of central tolerance, the AIRE protein.
EXPERIMENTAL PROCEDURES
Sequence alignments and homology modeling- Disorder prediction was with the VL-XT predictor http://www.pondr.com/. The first 100 amino-acid residues of AIRE were subjected to a sequence-structure homology recognition analysis using the FUGUE software (12). FUGUE takes a given query sequence and scans its database of structure profiles for potential matches. The summation of environment-specific substitution tables and structure-dependent gap penalties for each amino acid alignment can be used to generate a compatibility score, indicating the accuracy of each prediction. Manual sequence alignment and editing was carried out using the BioEdit software. The homology modeling software MODELLER (17) was used to create 30 potential models of this domain which were analyzed using the PROCHECK (18), WHAT IF (19) and Verify3D (20) algorithms to ensure the satisfaction of stereochemical restraints. The model with the lowest energy and fewest spatial violations was selected as the most accurate representation of this domain. Site Directed Mutator (21) was used to calculate a stability score for specific point mutations in the AIRE CARD model. 3D structure visualization and image generation was carried out using Pymol (22). Cell culture- HeLa and HEK293 human cell lines were cultured in RPMI and DMEM, respectively, supplemented with 10% FCS (v/v) and 100µg penicillin and streptomycin. Plasmids- To generate the luciferase reporter plasmid pBL-INV, the promoter (3737 nt) of the human involucrin gene was excised from pTZhINV-nlbgal (a gift from A.Männik, FitBiotech, Estonia) and sub-cloned into pBL-KS (a gift from K.Saksela, University of Tampere, Finland). pBL-IFNβ was cloned by PCR amplification of 711 nt of the IFNβ promoter
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

3
(using genomic DNA from HEK293 cells as a template) and inserted into pBL-KS. The pC-AIRE-L28P, pC-AIRE L29P and pC-AIRE K83E clones were generated by PCR using pGST-AIRE L28P (13), pGST-AIRE-L29P and pGST-AIRE- K83E as templates. pC-AIRE-L28P and L29P were sub-cloned into pCDNA 3.1- B myc/his (Invitrogen, Carlsbad, CA) and pC-AIRE K83E into pCDNA 3.1- A myc/his (Invitrogen). All clones were verified by sequencing, and expression of each mutant AIRE protein was verified by Western blot. pC-AIRE has been described (23), p-EGFP-AIRE was a kind gift of Professor Matsumoto, University of Tokushima. AIRE∆CARD (aa 101-545) was generated by PCR and subcloned into pAcGFP1-C3 (Clontech, Mountain View, CA). Transient transfections were with FuGENE 6 (Roche Applied Science, Switzerland), Lipofectamine 2000 (Invitrogen), or Exgen in vitro reagent (Fermentas, Burlington, Ontario). Transactivation assays- Luciferase reporter Assays, RNA purification, quantitative RT-PCR and immunofluorescence were performed as in (14). For luciferase assays, transfections were with 0.1 µg of reporter constructs, 0.3µg of AIRE constructs or the control (pCDNA 3.1 B myc/his) and 0.5 µg of CBP. For endogenous promoter activation assays, 0.5µg of AIRE construct or control DNA (pCDNA 3.1- B myc/his) with 0.9 µg of CBP (pRc/RSV-mCBP-HA-RK, a gift from R. Goodman, Oregon Health and Science Institute, USA) were transfected. cDNA samples were analyzed in triplicate by quantitative PCR (qPCR) using qPCRTM SYBR® Green Core Kit (Eurogentec) and ABI Prism 7900HT. The relative gene expression levels were calculated using comparative Ct (∆∆Ct) method (Applied Biosystems). The following primers were used for quantitative RT-PCR: for HPRT as an internal reference, HPRTexon6 and GACTTTGCTTTCCTTGGTCAGGHPRTexon7 AGTCTGGCTTATATCCAACACTTCG. Primer sequences to amplify the involucrin and S100A8 cDNAs were as follows: hINV fwd: GCCTTACTGTGAGTCTGGTTGACA, hINV rev: GGAGGAACA GTCTTGAGGAGCT, S100A8 fwd: CTCAGTATATCAGGAAAAGGGTGCAGAC, S100A8 rev: CACGCCCATCTTTATCACCAGAATGAG.
Mice- AIRE null and wild type C57BL/6 mice were bred at the University of Tartu and the University of Birmingham respectively, and all experiments were performed in accordance with UK Home Office regulations. Aire-deficient mice (C57BL/6J) were generated at The Walter and Eliza Hall Institute (Melbourne, Australia). NOD and C57BL/6 mice were maintained in the Biological Services facility of the Department of Pathology at the University of Cambridge. Thymi were removed and snap frozen. 10 micron sections were cut using a cryostat and adhered to polylysine coated slides (BDH). Sections were immediately fixed with acetone prior to storage at -80ºC. Thymic Organ Culture and AIRE induction Thymic lobes were dissected from E15 BALB/c embryos. Each lobe was placed at the air- medium interface on top of a 0.8µm isopore membrane filter (Millipore, Watford,UK), supported by an Artiwrap sponge (Medipost Ltd., Weymouth, Dorset, UK). The filter and sponge was submerged in DMEM-10 (DMEM supplemented with 10% FCS, 1x nonessential amino acids, 10mM HEPES, 5x10-5M 2-Mercaptoethanol, 4 mM L-Glutamine, 100 IU/ml penicillin and 100 µg/ml streptomycin) in 10%CO2, 37°C. 1.35 mM 2-Deoxyguanosine was added to the media for 6 days to deplete T cells and DCs. Lobes were briefly washed in fresh DMEM and 1 lobe per well of a Terasaki-plate was incubated in 20µl DMEM-10 with 10 µg/ml anti-Rank antibody (R&D) alone or with 10 µg/ml anti LTβR (4H8 WH2, Alexis) for 2 or 4 days. For the 4 day stimulation, the medium containing fresh antibodies was replaced after 2 days. A control group was cultured in DMEM-10 only. The lobes were then snap frozen and cut at 6 µm thickness for histological analysis. Immunofluorescence- Immunostaining of transfectants was as described previously (24). Immunostaining reagents were: anti-PML (clone N19, Santa Cruz), anti-murine AIRE (rabbit polyclonal, a kind gift from Professor Peltonen, Biomedicum, Helsinki, Finland) and anti-human AIRE (mouse monoclonal AIRE6.1 as described previously (23). Acetone-fixed thymus sections were rehydrated in PBS, permeabilized and stained as above, with the following: anti-PML (clone 36-1-104); anti-keratin 5 (Covance) anti-Aire (clone B1/02-5H12-2) and anti-CBP (clone
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

4
C-20, Santa Cruz). Secondary antibodies used for transfectants and thymus sections were Alexa-fluor conjugated secondary antibodies (Molecular Probes, Oregon, USA). Cells and sections were mounted and counterstained with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA). Microscopy- Micrographs of transfectants were obtained using a Zeiss Axiophot fluorescence microscope (100x oil emersion objective), DM IRBE Leica Confocal microscope (Leica, Vienna, Austria) with a 63x 1.32 oil immersion objective or with or with Nikon Eclipse TE2000-U (60 x water-immersion objective). Confocal microscopy of FTOC sections was with a LSM10 Zeiss confocal microscope.
RESULTS AIRE contains an N-terminal CARD domain which is required for nuclear puncta formation. The AIRE1 gene encodes a 545 amino-acid residue protein with a molecular mass of 58 kDa. Previous sequence based analyses had identified four distinct protein domains; an amino terminal Homogeneously Staining Region (HSR) formed by the first 100 amino acids of AIRE, two plant homeodomain (PHD) fingers as well as an Sp100, AIRE, NucP41/P75, DEAF-1 (SAND) domain (25). All but the first of these domains are implicated in interactions with chromatin and nucleic acids (9,25). Key observations that led to our study of the HSR domain were that two thirds of all of APECED-associated missense mutations are clustered in this domain (25) which is also the most highly conserved sequence in the AIRE protein (Supplementary Fig. 1). These observations suggest that the HSR is vital for AIRE’s function. Primary sequence-based database searches identify AIRE’s homology to the Sp100 protein family (Supplementary Fig. 2), (26) which contain the HSR-SAND domain structure, suggesting a common evolutionary ancestor (27). As for AIRE, the HSR domain of Sp100 proteins can drive their oligomerisation to form nuclear puncta (28,29). Despite this homology, the HSR nomenclature is entirely non-functional (30) and this domain remains structurally uncharacterized. Disorder prediction (31) of full length AIRE suggested that the HSR is ordered with a high propensity to assemble as a globular folded
domain, whilst the remaining protein showed regions of disorder and flexibility; a feature commonly found in proteins that interact with nucleic acids (Fig. 1A). To further analyze the amino terminal HSR domain three-dimensional structure-based sequence analysis was utilized (12) allowing the identification of homologous proteins from their conserved three-dimensional structures, even in the absence of high primary sequence identity. In this case, a sequence-structure homology recognition analysis indicated that the HSR region may correspond to a previously uncharacterized CARD domain. The target AIRE sequence was found to align well with the CARD domain from apoptosis activating factor 1 (Apaf-1) (protein data bank code 1ygs_p). This alignment was subsequently extended to include the sequences of three further CARD domains with fully characterized three-dimensional structures (Fig. 1B). A key feature of the primary sequences of the CARD family is a series of highly conserved hydrophobic residues that form the core of the folded domain. The six amphipathic alpha-helices that constitute the secondary structure of the CARD domain are presumed to fold around this cluster of hydrophobic residues, all of which are conserved in solved CARD domain structures (Fig. 1B). This pattern of residues, due to both their importance in defining the CARD fold and their high level of conservation can be identified as the signature of this protein family. Combining the AIRE/Apaf-1 alignment with the CARD family alignment indicated that this CARD signature is present in the N-terminal region of AIRE giving a very strong indication as to the accuracy of the FUGUE assignment (Fig. 1B). This detailed multiple sequence alignment was used to create a hypothetical model of the AIRE CARD domain structure using homology modeling techniques. Importantly, the CARD model was found to fully satisfy spatial and stereochemical restraints presenting a PROCHECK G-factor of -0.07 and with 94.5% of residues found in the favored regions (and 0% in the disallowed regions) of a Ramachandran plot. The model generated exhibits all of the requisite features of a CARD domain with 6 alpha-helices presented with Greek key topology along with spatial conservation of the key hydrophobic core residues (Fig. 2A). Further, the domain shows the polarized distribution of electrostatic surface
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

5
charges characteristic of CARD domains (Fig. 2B) that drives homotypic protein/protein interactions (32). Superimposition of the AIRE CARD model with the structures of the Apaf-1 and ICEBERG CARDs reveals the striking structural conservation (Fig. 2C). The N-terminal domain of AIRE has been previously described as vital for the protein’s transactivation function and specifically for its oligomerisation (29). As confirmation, an AIRE construct devoid of the CARD domain but still expressing a nuclear localization sequence, pGFP-AIRE∆CARD, was transiently transfected into HeLa cells. Ablation of the CARD domain led to a diffuse staining phenotype with both cytoplasmic and nuclear expression evident (Fig. 2D). Our assignment of a CARD domain to the amino terminus of AIRE was also supported by AIRE expression data gathered from transient transfectants (Supplementary Fig. 3A). HeLa cells transfected with GFP-AIRE showed nuclear punctate, cytoplasmic filament and aggregate fluorescence patterns; all consistent with previous reports (33), distributions that contrast with in vivo staining patterns that are exclusively nuclear punctate (Supplementary Fig. 3B). Over expression of death-fold superfamily-containing proteins, which includes the CARD domain, commonly results in the nucleation of artifactual death effector filaments (34) at intermediate filaments and microtubules (15). The cytoplasmic filaments of AIRE protein that we detect in cell culture are therefore likely to be death effector filaments; an assertion strengthened by their known association with microtubules and intermediate filaments (15). Correlation of CARD domain disrupting mutants with transactivation. The availability of a structural model of the AIRE CARD domain permits us to analyze disease-associated APECED missense mutations. The positions of the 12 AIRE CARD residues that are mutated in APECED were mapped onto the CARD model and were found to lie either on the surface of the domain, where their substitution may disrupt protein/protein interactions, or at the hydrophobic core, which may lead to incorrect folding and inactivation (Fig. 3A). Three APECED-associated substitutions, Leucine 28 to Proline (L28P), Leucine 29 to Proline (L29P) and Lysine 83 to Glutamate (K83E), were further analyzed for their effects on
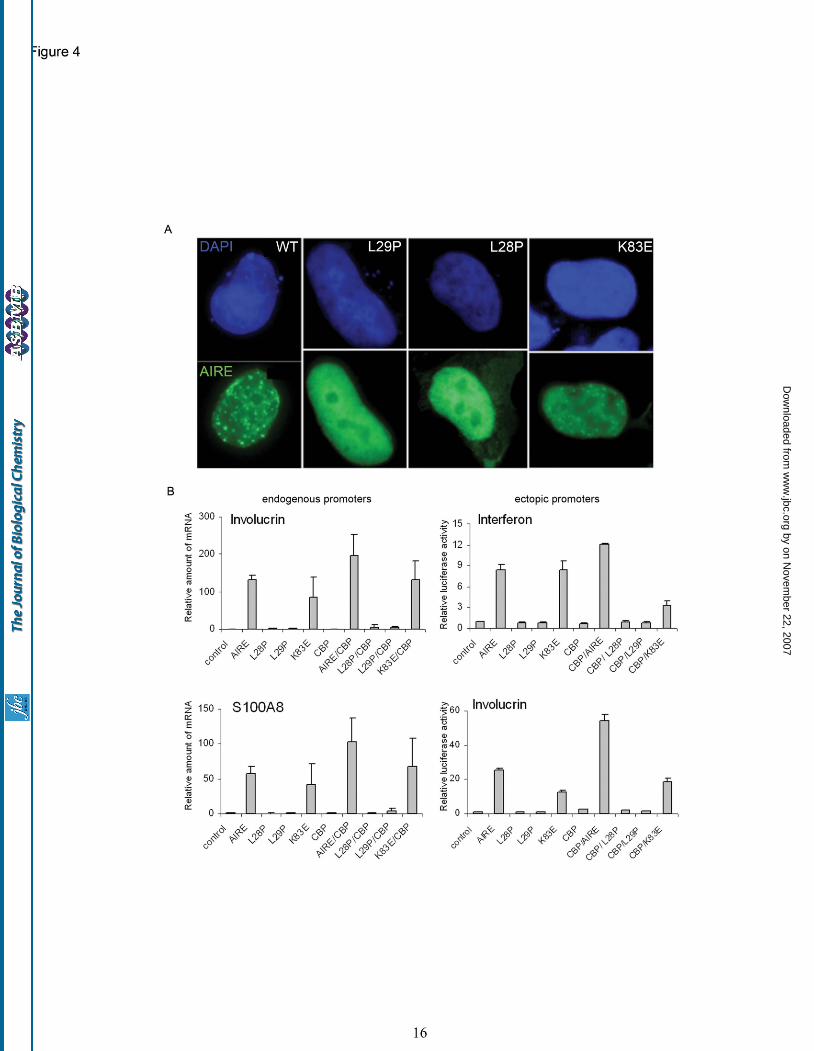
AIRE distribution and transactivation potential. These mutations were chosen as they lie either at the core (L28, L29) or surface (K83) of the CARD domain (Fig. 3B). Expression of the L28P or L29P mutants leads to a diffuse nuclear pattern of AIRE, contrasting with the punctate distribution characteristic of the wild-type AIRE protein (Fig. 4A). The K83E mutant exhibits an intermediate distribution with a speckled nuclear pattern. In keeping with our model showing that homotypic CARD domain interactions drive AIRE puncta formation, mutations at L28 and L29 have been shown to prevent AIRE self association (9) whereas K83E has an intermediate phenotype which is more severe when analyzing ectopic versus endogenous promoters. The effects of these mutations on AIRE’s ability to initiate the expression of endogenous and ectopic gene promoters were then studied. As ectopic gene targets we used the luciferase gene under the control of the full-length interferon beta and involucrin promoters. From analyses of AIRE deficient mice it was known that the endogenous genes involucrin and S100A8 are down-regulated in an AIRE null background (GEO DataSets, record GDS2274) and hence these genes were used to assay endogenous gene transcription. We also transfected each AIRE construct with or without co-transfection of CBP followed by measurements of luciferase activity or real-time quantitative PCR (qRT-PCR) analysis of transcript levels. In each case, we found that the L28 and L29 mutations prevented transactivation, regardless of CBP expression (Fig. 4B), whilst the effects of the K83E mutation were minimal. Notably, wild type AIRE induced transactivation was always augmented by CBP. From our CARD model the two Leu-Pro mutations would be predicted to partially or entirely unfold the CARD domain, abrogating its function as a protein/protein interaction module. In contrast, the K83E mutation is predicted only to alter the charge distribution on the domain surface and not to disrupt the overall fold of the domain. These predictions were tested using software capable of estimating the level of structural destabilization encoded by point mutations. Site Directed Mutator (SDM) (21,35) uses a statistical potential energy function that predicts the effect that a specific amino-acid substitution may have on the stability of that protein and produces a score analogous to the free energy difference
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

6
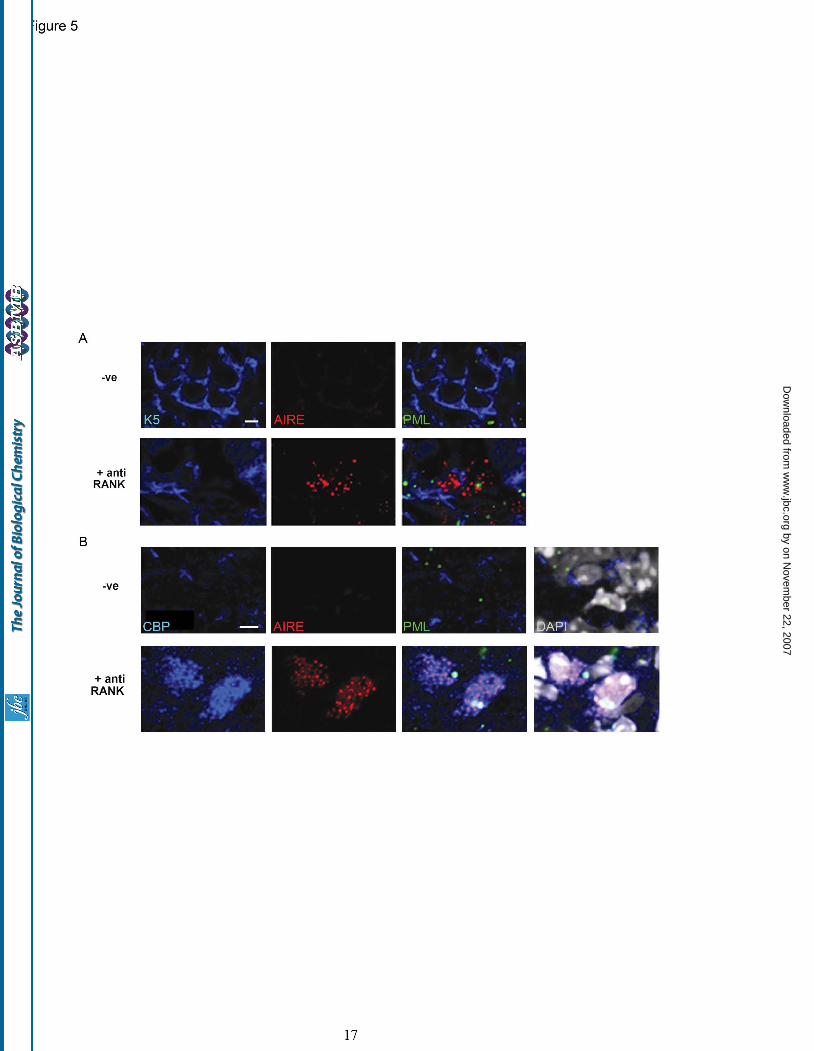
between a wild-type and mutant protein. The L28P and L29P substitutions were shown to induce structural alterations with corresponding pseudo-∆∆G values of -3.75 kcal/mol, implying overall gross destabilization of the CARD domain (a value of less than -2 from this analysis indicates a large and general fold destabilization). The K83E mutation was, on the other hand, predicted to be partially stabilizing, with a pseudo-∆∆G value of 0.29 kcal/mol. These results emphasize the excellent overall correlation between the modeling and experimental analyses of these APECED-associated mutants. RANK stimulation promotes the nuclear co-localization of CBP and AIRE. Given that AIRE’s CARD drives puncta formation, which indicates transactivation competent protein, we next addressed the question of whether AIRE protein co-localizes with the global transcriptional activator, CBP, in-vivo. Synchronous AIRE induction required our use of an agent naturally involved in AIRE induction. Anti-lymphotoxin beta receptor (LTβR) antibody, a previously described inducer of AIRE-positive mTEC development (36), as well as anti-RANK (receptor activator of nuclear factor-kappaB antibody), a newly described AIRE agonist (16), were used to induce endogenous AIRE expression in cell lines. However, no AIRE induction was achieved following incubation of mTEC derived cell lines with either the anti-LTβR or anti-RANK antibodies. Additionally, in these lines endogenous AIRE was dispersed, and not punctate (data not shown), which would suggest that cell architecture is important for AIRE induction. In consequence we used ex-vivo fetal thymic organ culture (FTOC) in which to test AIRE induction as this experimental system more closely resembles the intimate cell / cell contacts achieved in thymic architecture. Dual immunostains for AIRE and keratin 5 (a marker specific for the thymic medulla) revealed that AIRE induction was only achieved by anti-RANK stimulation (Fig. 5A and Supplementary Fig. 4). Prior to induction of AIRE, the distribution of CBP was predominantly cytoplasmic and punctate, a distribution that has been described in oocyte development (37). After induction, focal CBP and AIRE were observed in nuclei, with cytoplasmic CBP puncta clustered around AIRE negative nuclei (Fig. 5B). Nuclear
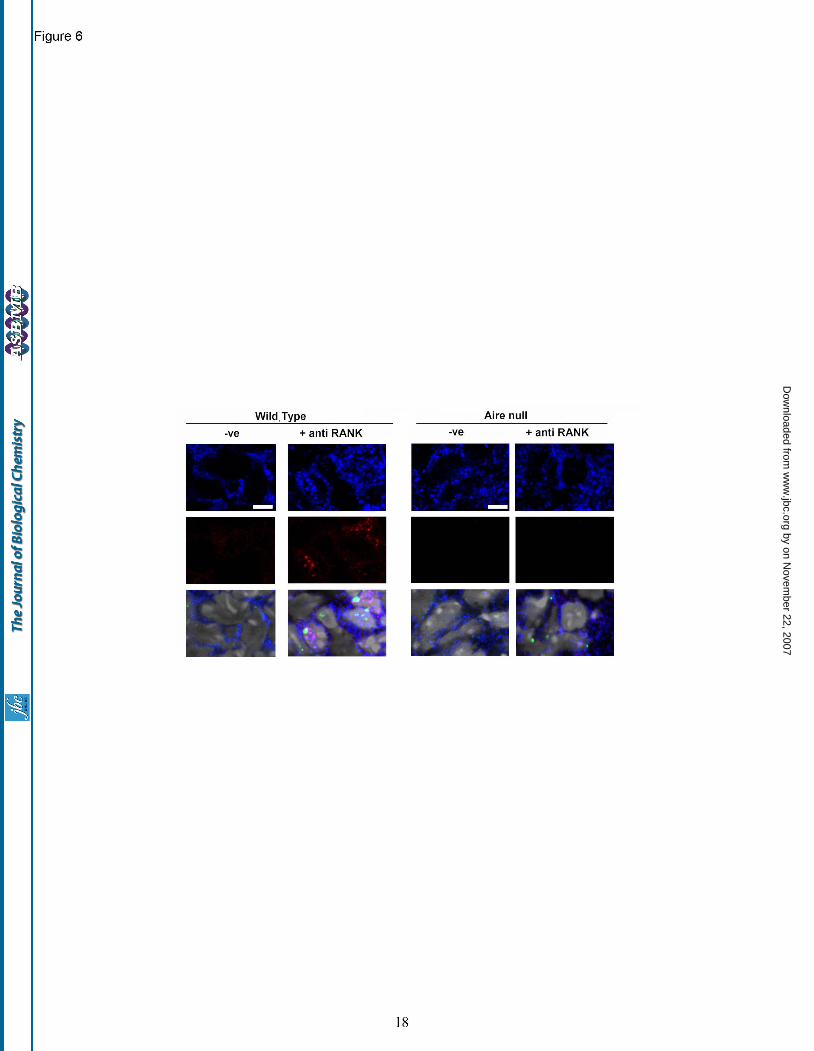
puncta of CBP were excluded from the DAPI bright portions of nuclei that typify heterochromatin territories. These data suggest that RANK stimulation drives the focal nuclear accumulation of both CBP and AIRE. Interestingly, immunostains for PML protein in FTOC revealed unusually low numbers of PML-NDs both before and after RANK treatment, with no overt sequestration of CBP (Fig. 5A and B). Nuclear accumulation of CBP following RANK stimulation requires AIRE expression. To examine whether AIRE expression was requisite to achieve a nuclear concentration of CBP, FTOCs derived from AIRE knockout versus wild type mice were generated. The fetal thymi were treated with anti-RANK antibody, anti-RANK and anti-LTβR antibodies or anti-LTβR antibody alone. In thymi taken from wild type mice, AIRE induction was achieved solely with anti-RANK treatment. Further, following this treatment, there was prominent nuclear staining of CBP and AIRE. Conversely, in thymi derived from AIRE knock-out animals, no nuclear accumulation of CBP (or AIRE) was detected following anti-RANK stimulation (Fig. 6) indicating the necessity for AIRE expression in the translocation of CBP into the nucleus.
DISCUSSION These data address two critical aspects of AIRE function; oligomerisation into transactivation competent complexes, and association with the histone modifier CBP. Replacement of the non-functional designation, HSR, with CARD, immediately suggests that AIRE is part of a novel tolerance-inducing molecular complex, almost certainly involving other CARD expressing proteins. There is a growing acceptance of the importance of CARD domains in the formation of signaling machines that trigger apoptosis, inflammation and innate immune recognition (32,38-40). CARD domain proteins neither act solely as caspase recruitment and activation machines, nor do they reside exclusively in the cytoplasm to regulate the activity of CARD-containing complexes (40-42). Future work will establish the protein partners of AIRE’s CARD domain. The presence of acidic and basic charge clusters on opposing exposed surfaces of AIRE’s CARD moiety is predicted to drive self-
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

7
association and interaction with other CARD domains, as exemplified by the caspase9:Apaf-1 CARD complex structure (32,43). Homotypic interaction of CARD domains is typically used to orientate protein scaffolds (32) and to activate effector proteins, although homo-oligomers of CARD domains are less common than death or death-effector domain dimers (32). Our structural data were collected using human AIRE sequences and alignment of the murine and human proteins show that sequence conservation is greatest at the amino terminus. Critically, all the residues identified in our alignment as being critical to maintain the CARD fold are conserved in the mouse sequence, as well as in other metazoans that express AIRE. These conserved residues are also the targets for mutation in APECED which underlines their importance for AIRE function. When three such APECED-associated mutants were analyzed biochemically an excellent correlation was noted between the predicted structural severity of the substitution and their effects on subcellular localization and transactivation. The L28P and L29P substitutions were expected to be highly destabilizing and to potentially unfold the CARD domain, thereby abrogating the function of AIRE protein, and, indeed, the transactivation activity of these mutants was found to be null. The K83E mutant, however, was predicted to be stable and to only affect the charge distribution on the CARD domain surface. When analyzed experimentally, these predictions were borne out by this mutant having residual transactivation activity and retaining the ability to form nuclear puncta. Given the functional role of CARD domains, it is probable that this mutation causes APECED by disrupting electrostatic protein/protein interactions between AIRE and unidentified interaction partners. It is unfortunate that genotype/phenotype associations have not been reported for APECED patients other than the mild phenotype (hypothyroidism alone) found in patients expressing the founder mutation in Iranian Jews with APECED, Y85C. Y85C is, however, somewhat analogous to K83E in that it lies at the surface of the CARD domain, where it would not alter the CARD fold but potentially disrupt specific protein/protein interactions. Our modeling data allow us to reassign CARD domains to both AIRE and its structural relatives, the Sp100 family proteins26. Sp100
proteins are the alternatively spliced forms of a single gene and appear to regulate chromatin structure and transcriptional activity. The prototypic member of this family was the first PML-ND cargo protein to be identified (Sp100) and its ability to oligomerise has been attributed to the N-terminal sequence that we now know contains a CARD domain. The identification of another CARD domain expressing protein within the PML-ND, caspase-2 (41), could implicate PML-NDs as scaffolds for caspase-2 mediated cell death, perhaps using Sp100 as an intermediary. If this is the case, then this effect must be restricted to particular cell types as we have surveyed caspase-2 expression in several transformed and untransformed cell types and find it to be largely constrained to the golgi apparatus (data not shown). Given that PML-NDs generally repress the function of their cargo proteins it would appear more likely that PML-ND resident caspase-2 is held in an inactive form. The identification of a CARD domain in Sp100 proteins may be of more relevance in regulating their anti-viral functions. Sp100-B, -C and -HMG repress the herpes simplex virus type-1 protein ICP0, but only when dispersed from PML-NDs (44). On the other hand, Sp100-A is permissive for ICP0 expression, which may encourage viral replication at the site of PML-NDs. All these Sp100 proteins express N-terminal CARD domains and their scaffold partners within and around PML-NDs may direct their immune regulatory activities. The molecular details of AIRE induction via RANK signaling are not yet known though the spectrum of defects seen in RANKL and RANK null mice (osteoporosis, defective T and B cell maturation and failed lactation) suggest the lack of a differentiation stimulus (45,46). In this case, RANK signaling may drive mTEC differentiation, which is in agreement with the description of multiple mTECs lineages (47). A differentiated cell status is associated with reduced PML-ND numbers, which may also be cytoprotective given the role of these structures in detecting injury and engaging apoptosis (48). Neither the cytoplasmic distribution nor the large size of CBP puncta in mTECs were expected as CBP has been shown to be nuclear, forming puncta that approach the resolution limits of the microscope (~200nm diameter) (49). Previous experiments had questioned epitope availability for CBP sequestered to puncta versus the
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

8
nucleoplasmic fraction (50). As we were interested in any flux between nucleoplasmic CBP and that fraction localized to AIRE puncta, we first confirmed that our CBP antibody could stain both these fractions in AIRE transfectants (data not shown). These experiments showed that AIRE puncta and CBP could co-localize, which we subsequently confirmed in our organ culture experiments. The identification of cytoplasmic CBP puncta was uniquely made in organ culture experiments and suggests that cytoplasmic CBP oligomerizes or is sequestered to unknown cytoplasmic structures. CBP entry to the nucleus is likely to be tightly regulated and may be coupled to the developmental plan of embryogenesis. The rarity of PML-NDs in mTECs also illustrates how monolayer cell culture experiments, in which numerous PML-NDs out-compete AIRE puncta for CBP, are inappropriate models with which to study mTEC function. The nuclear concentration of CBP that occurs following RANK stimulation both provides an elegant mechanism for AIRE function and obviates any need to recruit CBP from other nuclear compartments as has been proposed (14). The finding that AIRE expression is requisite for the translocation of CBP from the cytosol to
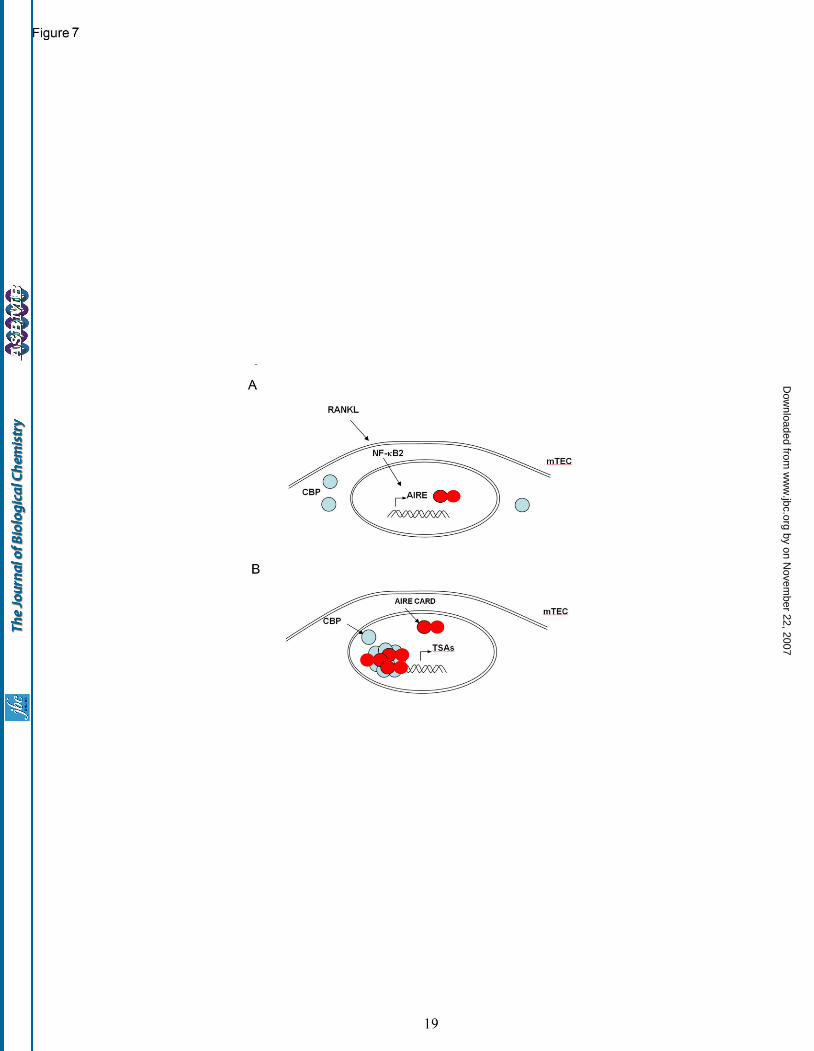
nucleus is intriguing. One could speculate that AIRE deficient mTECs lack the cellular architecture and signaling hardware needed to trigger the nuclear accumulation of CBP, resulting in a key transcriptional activator failing to reach its nuclear substrates in AIRE null mTECs. From the transactivation data, this study has shown convincingly that co-expression of wild type AIRE and CBP accelerates AIRE’s transactivation potential. Consequently, the nuclear accumulation of CBP and AIRE in anti-RANK-stimulated nuclei may drive focal induction of tissue specific transcripts. The regulation of AIRE expression by RANKL is consistent with reports that this member of the tumor necrosis factor superfamily can activate nuclear factor kappa B2 (NF-κB2, also named p100) (51) and that NF-κB2 is essential for the expression of AIRE in mTECs (52). In conclusion, our model for thymic expression of TSAs as regulated by AIRE protein is postulated to require RANKL activation of NF-κB2 leading to AIRE and CBP co-accumulation in the nucleus, an activity driven by CARD dependent interactions and resulting in large-scale alterations in the mTEC transcriptome (Fig. 7).
REFERENCES
1. Venanzi, E. S., Benoist, C., and Mathis, D. (2004) Curr Opin Immunol 16(2), 197-202 2. Nagamine, K., Peterson, P., Scott, H. S., Kudoh, J., Minoshima, S., Heino, M., Krohn, K. J.,
Lalioti, M. D., Mullis, P. E., Antonarakis, S. E., Kawasaki, K., Asakawa, S., Ito, F., and Shimizu, N. (1997) Nat Genet 17(4), 393-398
3. Consortium, T. F.-G. A. (1997) Nat Genet 17(4), 399-403 4. Anderson, M. S., Venanzi, E. S., Klein, L., Chen, Z., Berzins, S. P., Turley, S. J., von Boehmer,
H., Bronson, R., Dierich, A., Benoist, C., and Mathis, D. (2002) Science 298(5597), 1395-1401 5. Johnnidis, J. B., Venanzi, E. S., Taxman, D. J., Ting, J. P., Benoist, C. O., and Mathis, D. J.
(2005) Proc Natl Acad Sci U S A 102(20), 7233-7238 6. Gavanescu, I., Kessler, B., Ploegh, H., Benoist, C., and Mathis, D. (2007) Proc Natl Acad Sci U S
A 104(11), 4583-4587 7. DeVoss, J., Hou, Y., Johannes, K., Lu, W., Liou, G. I., Rinn, J., Chang, H., Caspi, R. R., Fong, L.,
and Anderson, M. S. (2006) J Exp Med 203(12), 2727-2735 8. Taubert, R., Schwendemann, J., and Kyewski, B. (2007) Eur J Immunol 37(3), 838-848 9. Halonen, M., Kangas, H., Ruppell, T., Ilmarinen, T., Ollila, J., Kolmer, M., Vihinen, M., Palvimo,
J., Saarela, J., Ulmanen, I., and Eskelin, P. (2004) Hum Mutat 23(3), 245-257 10. Maul, G. G., Negorev, D., Bell, P., and Ishov, A. M. (2000) J Struct Biol 129(2-3), 278-287 11. Pearson, W. (1997) Comput Appl Biosci, 325-332 12. Shi, J., Blundell, T. L., and Mizuguchi, K. (2001) J Mol Biol 310(1), 243-257
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

9
13. Pitkanen, J., Doucas, V., Sternsdorf, T., Nakajima, T., Aratani, S., Jensen, K., Will, H., Vahamurto, P., Ollila, J., Vihinen, M., Scott, H. S., Antonarakis, S. E., Kudoh, J., Shimizu, N., Krohn, K., and Peterson, P. (2000) J Biol Chem 275(22), 16802-16809
14. Pitkanen, J., Rebane, A., Rowell, J., Murumagi, A., Strobel, P., Moll, K., Saare, M., Heikkila, J., Doucas, V., Marx, A., and Peterson, P. (2005) Biochem Biophys Res Commun 333(3), 944-953
15. Akiyoshi, H., Hatakeyama, S., Pitkanen, J., Mouri, Y., Doucas, V., Kudoh, J., Tsurugaya, K., Uchida, D., Matsushima, A., Oshikawa, K., Nakayama, K. I., Shimizu, N., Peterson, P., and Matsumoto, M. (2004) J Biol Chem 279(32), 33984-33991
16. Rossi, S., Kim, M.-Y., Leibbrandt, A., Parnell, S. M., Jenkinson, W. E., Glanville, S. H., McConnell, F. M., Penninger, J. M., Jenkinson, E. J., Lane, P. L., and Anderson, G. (2007) J Exp Med 204(6):1267-72.
17. Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. (2000) Annu Rev Biophys Biomol Struct 29, 291-325
18. Laskowski, R. A., Moss, D. S., and Thornton, J. M. (1993) J Mol Biol 231(4), 1049-1067 19. Vriend, G. (1990) J Mol Graph 8(1), 52-56, 29 20. Luthy, R., Bowie, J. U., and Eisenberg, D. (1992) Nature 356(6364), 83-85 21. Topham, C. M., Srinivasan, N., and Blundell, T. L. (1997) Protein Eng 10(1), 7-21 22. DeLano, W. (2002) DeLano Scientific, Palo Alto, CA, USA. 23. Heino, M., Peterson, P., Kudoh, J., Nagamine, K., Lagerstedt, A., Ovod, V., Ranki, A., Rantala, I.,
Nieminen, M., Tuukkanen, J., Scott, H. S., Antonarakis, S. E., Shimizu, N., and Krohn, K. (1999) Biochem Biophys Res Commun 257(3), 821-825
24. Varadaraj, A., Dovey, C., Laredj, L., Ferguson, B., Alexander, C., Lubben, N., Wyllie, A., and Rich, T. (2007) J Path 211(4), 471-80.
25. Su, M. A., and Anderson, M. S. (2004) Curr Opin Immunol 16(6), 746-752 26. Sternsdorf, T., Grotzinger, T., Jensen, K., and Will, H. (1997) Immunobiology 198(1-3), 307-331 27. Blechschmidt, K., Schweiger, M., Wertz, K., Poulson, R., Christensen, H. M., Rosenthal, A.,
Lehrach, H., and Yaspo, M. L. (1999) Genome Res 9(2), 158-166 28. Sternsdorf, T., Jensen, K., Reich, B., and Will, H. (1999) J Biol Chem 274(18), 12555-12566 29. Ramsey, C., Bukrinsky, A., and Peltonen, L. (2002) Hum Mol Genet 11(26), 3299-3308 30. Grotzinger, T., Jensen, K., Guldner, H. H., Sternsdorf, T., Szostecki, C., Schwab, M., Savelyeva,
L., Reich, B., and Will, H. (1996) Mol Cell Biol 16(3), 1150-1156 31. Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005) Febs
Journal 272, 5129-5148 32. Park, H. H., Lo, Y. C., Lin, S. C., Wang, L., Yang, J. K., and Wu, H. (2007) Annu Rev Immunol
25, 561-586 33. Pitkanen, J., Vahamurto, P., Krohn, K., and Peterson, P. (2001) J Biol Chem 276(22), 19597-
19602 34. Siegel, R. M., Martin, D. A., Zheng, L., Ng, S. Y., Bertin, J., Cohen, J., and Lenardo, M. J. (1998)
J Cell Biol 141(5), 1243-1253 35. Worth, C. L., Bickerton, G. R. J., Cheng, T. M. K., Lee, S., Schreyer, A., Forman, J. R., Gong, S.,
Burke, D. F., and Blundell, T. L. (2007) Journal of Bioinformatics and Computational Biology In press
36. Chin, R. K., Lo, J. C., Kim, O., Blink, S. E., Christiansen, P. A., Peterson, P., Wang, Y., Ware, C., and Fu, Y. X. (2003) Nat Immunol 4(11), 1121-1127
37. Kwok, R. P., Liu, X. T., and Smith, G. D. (2006) Mol Reprod Dev 73(7), 885-894 38. Ponting, C. P., and Russell, R. R. (2002) Annu Rev Biophys Biomol Struct 31, 45-71 39. Mariathasan, S., and Monack, D. M. (2007) Nat Rev Immunol 7(1), 31-40 40. Park, H. H., Logette, E., Raunser, S., Cuenin, S., Walz, T., Tschopp, J., and Wu, H. (2007) Cell
128(3), 533-546 41. Tang, J., Xie, W., and Yang, X. (2005) Cancer Biol Ther 4(6), 645-649 42. Milovic-Holm, K., Krieghoff, E., Jensen, K., Will, H., and Hofmann, T. G. (2007) EMBO 26,
391-401
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

10
43. Qin, H., Srinivasula, S. M., Wu, G., Fernandes-Alnemri, T., Alnemri, E. S., and Shi, Y. (1999) Nature 399(6736), 549-557
44. Negorev, D.G., Vladimirova, O.V., Ivanov, A., Rauscher, F. 3rd., Maul, G.G. (2006) J Virol 80, 8019-8029
45. Li, J., Sarosi, I., Yan, X. Q., Morony, S., Capparelli, C., Tan, H. L., McCabe, S., Elliott, R., Scully, S., Van, G., Kaufman, S., Juan, S. C., Sun, Y., Tarpley, J., Martin, L., Christensen, K., McCabe, J., Kostenuik, P., Hsu, H., Fletcher, F., Dunstan, C. R., Lacey, D. L., and Boyle, W. J. (2000) Proc Natl Acad Sci U S A 97(4), 1566-1571
46. Fata, J. E., Kong, Y. Y., Li, J., Sasaki, T., Irie-Sasaki, J., Moorehead, R. A., Elliott, R., Scully, S., Voura, E. B., Lacey, D. L., Boyle, W. J., Khokha, R., and Penninger, J. M. (2000) Cell 103(1), 41-50
47. Hamazaki, Y., Fujita, H., Kobayashi, T., Choi, Y., Scott, H. S., Matsumoto, M., and Minato, N. (2007) Nat Immunol 8(3), 304-311
48. Wang, Z. G., Ruggero, D., Ronchetti, S., Zhong, S., Gaboli, M., Rivi, R., and Pandolfi, P. P. (1998) Nat Genet 20, 266-272
49. McManus, K. J., Stephens, D. A., Adams, N. M., Islam, S. A., Freemont, P. S., and Hendzel, M. J. (2006) PLoS Comput Biol 2(10), e139
50. Doucas, V., Tini, M., Egan, D. A., and Evans, R. M. (1999 ) Proc Natl Acad Sci U S A. 16, 2627-2632 51. Novack, D. V., Yin, L., Hagen-Stapleton, A., Schreiber, R. D., Goeddel, D. V., Ross, F. P., and
Teitelbaum, S. L. (2003) J Exp Med 198(5), 771-781 52. Zhu, M., Chin, R. K., Christiansen, P. A., Lo, J. C., Liu, X., Ware, C., Siebenlist, U., and Fu, Y.
X. (2006) J Clin Invest 116(11), 2964-2971 53. Anderson, M. S., Venanzi, E. S., Chen, Z., Berzins, S. P., Benoist, C., and Mathis, D. (2005)
Immunity 23(2), 227-239
FOOTNOTES *This work was supported by grants from the BBSRC; T.R. and B.J.F: BBC5098231; T.R., A.C. and C.E.A.: BBSB0370X. A.R., I.L., ML and P.P. were supported by the grants from Wellcome Trust and EU Framework program 6 (Thymaide and Euraps) and by Estonian Science Foundation (6663 and 6490). CW, JW are supported by BBSRC studentships; GA, EJ and SR are supported by an MRC Programme Grant and the EU Thymaide project. H.S.S is supported by NHMRC fellowships (171601 and 461204), NHMRC program grants (257501 and 264573), Eurothymaide, 6th FP of the EU, and the Nossal Leadership Award from the Walter & Eliza Hall Institute of Medical Research. The abbreviations used are: AIRE, Autoimmune Regulator; mTEC, Medullary Thymic Epithelial Cell; TSA, Tissue Specific Antigen; APECED, Autoimmune-polyendocrinopathy-candidiasis-ectodermal Dystrophy; APS-1, Autoimmune Polyglandular Syndrome Type; HSR, Homogeneously Staining Region; CARD, Caspase Recruitment Domain; CREB, Cyclic AMP-Response Element Binding Protein; CBP, CREB Binding Protein; RANKL, Receptor Activator of Nuclear Factor-kappaB Ligand; SAND, Sp100, AIRE, NucP41/P75, DEAF-1; APAF-1, Apoptosis Protease Activating Factor 1; FTOC, Fetal Thymic Organ Culture; LTβR, Lymphotoxin Beta Receptor; PML-ND, Promyelocytic Leukemia Nuclear Domain. 1Present address; Institute of Comparative Medicine, University of Glasgow, G61 1QH, UK, Scotland, UK 2These authors contributed equally to this study and should be considered joint first authors
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

11
ACKNOWLEDGEMENTS We thank Gabrielle Reeves, Marc Cortese and Nick Gay for advice and discussions.
FIGURE LEGENDS
Fig. 1: AIRE contains an N-terminal CARD domain. (A) PONDR VL-XT prediction for AIRE protein showing a structured amino terminus. PONDR scores below the boundary (0.5) signify a propensity for order, with scores above 0.5 indicating a high likelihood of disorder. (B) Structure-based sequence alignment of AIRE (residues 8-96) with the sequences of four other CARD domains that have known three-dimensional structures. APAF1: Apaf-1 (PDB code: 3YGS_P), CASP9: caspase-9 (PDB code: 3YGS_C), ICBR: Iceberg (PDB code: 1DGN) and RAIDD: RIP-associated ICH1/CED3 homologous protein with death domain (PDB code: 3CRD). Amino-acid residue numbers are indicated for each sequence. The key hydrophobic residues that constitute the core of these domains when correctly folded are shown in red. Yellow boxes indicate the positions of the six alpha-helices of the Apaf-1 CARD characteristic of this domain’s topology. The position of the putative CARD domain in the AIRE polypeptide is indicated in the schematic shown beneath the alignment which also indicates positions of the SAND domain, two PHD fingers, LxxLL motifs and nuclear localisation signal (NLS). Fig. 2. Three dimensional model of the putative AIRE CARD domain created using MODELLER software. (A) The AIRE CARD model (red) compared to the crystal structure of the Apaf-1 CARD domain (blue) in two equivalent orientations. The AIRE CARD model exhibits the expected secondary structure and topology (alpha-helices denoted α1-α6) as well as spatial conservation of the key hydrophobic residues (shown in ball-and-stick representation). (B) Polarized distribution of electrostatic surface charges exhibited by the AIRE CARD model. The orientation in the left panel shows the domain surface of helices 1, 4 and 6 which contains a high proportion of negative charge (red). This contrasts with the domain surface consisting of helices 2, 3 and 5, shown in the right panel, which is predominantly positively charged (blue). (C) Spatial conservation of hydrophobic core residues of AIRE CARD model. Superposition of alpha helices 5 and 6 of AIRE (red), Apaf-1 (blue) and ICEBERG (yellow). The positions of residues F77, L81 and L93 (AIRE) indicate their importance in maintaining the structural integrity of the fold. These amino acids are highly conserved in all three CARD domains. (D) Expression of pGFP-∆CARD results in a loss of nuclear puncta. HeLa cells transfected with pGFP-∆CARD exhibit diffuse nuclear and cytoplasmic AIRE protein distribution (centre and right panels). Nuclei are counterstained with DAPI (left panel). Scale bar: 20 microns. Fig. 3. Location of AEPCED-associated mutants in the AIRE CARD domain. (A) Sites of all missense mutations reported in APECED patients are mapped onto the AIRE CARD model. (B) Residues L28, L29 and K83, corresponding to mutation sites chosen for further study, are labeled in blue. Fig. 4. Functional analyses of APECED-associated mutations located in the AIRE CARD domain. (A) Fluorescent micrographs of HEK-293 cells transfected with AIRE and mutant AIRE constructs. (B) Loss of transactivation function of the AIRE protein. Transactivation assays were carried out using empty vector (control), wild type AIRE or the mutants L28P, L29P and K83E. Where indicated, co-transfection of CBP was used; otherwise, quantity of total transfected DNA was kept constant by adding comparable amounts of control vector. Relative expression levels of endogenous involucrin and S100A8 genes are shown compared to the involucrin or S100A8 mRNA levels in control transfected cells (=1) (left panels). Relative luciferase activity from ectopic involucrin and interferon beta promoters (right panels) is shown compared to background activation provided by co-transfection of the control (=1). The values (+/- standard error) are means from at least two independent transfections. Scale bar: 5 microns.
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from

12
Fig. 5. Rank stimulation induces the nuclear accumulation of AIRE and CBP. (A) Lymphocyte depleted thymic lobes from BALB/c embryos (-ve) were treated with anti-RANK antibody to induce AIRE expression (red) in mTECs stained with keratin 5 (blue). PML protein is indicated by green fluorescence. (B) Incubation with anti-RANK antibody results in the nuclear accumulation of CBP (false colored blue); DAPI (false colored grey); PML (stained green). Scale bar: 5 microns. Fig. 6. AIRE expression is required for focal nuclear accumulation of CBP. Anti-RANK stimulation of wild type (left panels) results in thymic AIRE expression (red) nuclear accumulation of CBP (blue). In thymi from AIRE null mice (right panels) CBP remains cytoplasmic following anti-RANK treatment. Bottom panels also show PML (green staining) and DAPI (grayscale). Scale bar : 5microns. Fig. 7. Schematic model of AIRE function in response to RANK stimulation in mTECs. (A) RANK stimulation provokes the transcription of AIRE potentially via the activity of NF-κB2. Prior to stimulation CBP expression is predominantly cytoplasmic and punctate. (B) AIRE expression is requisite for the nuclear accumulation of CBP. CARD dependent oligomerisation of AIRE is postulated to drive the transactivation of tissue specific antigens.
by on Novem
ber 22, 2007 w
ww
.jbc.orgD
ownloaded from




















