
Advanced oxidation of the pharmaceutical drug diclofenac with UV/H 2O 2 and ozone
9
Water Research 38 (2004) 414–422 Advanced oxidation of the pharmaceutical drug diclofenac with UV/H 2 O 2 and ozone Davide Vogna a , Raffaele Marotta b , Alessandra Napolitano a , Roberto Andreozzi b, *, Marco d’Ischia a a Dip. di Chimica Organica e Biochimica, Federico II, Facolta di Scienze, Universita degli Studi di Napoli Complesso Universitario M. S. Angelo, via Cinthia, 4, 80126, Napoli, Italy b Dip. di Ingegneria Chimica, Facolta di Ingegneria, Universita degli Studi di Napoli FedericoII Prazzale Tecchio, 80, 80125 Napoli, Italy Received 24 October 2002; received in revised form 2 September 2003; accepted 19 September 2003 Abstract Diclofenac, a widely used anti-inflammatory drug, has been found in many Sewage Treatment Plant effluents, rivers and lake waters, and has been reported to exhibit adverse effects on fish. Advanced oxidation processes, ozonation and H 2 O 2 /UV were investigated for its degradation in water. The kinetic of the degradation reaction and the nature of the intermediate products were still poorly defined. Under the conditions adopted in the present study, both ozonation and H 2 O 2 /UV systems proved to be effective in inducing diclofenac degradation, ensuring a complete conversion of the chlorine into chloride ions and degrees of mineralization of 32% for ozonation and 39% for H 2 O 2 /UV after a 90 min treatment. The reactions were found to follow similar, but not identical, reaction pathways leading to hydroxylated intermediates (e.g. 2-[(2,6-dichlorophenyl)amino]-5-hydroxyphenylacetic acid) and C–N cleavage products (notably 2,5- dihydroxyphenylacetic acid) through competitive routes. Subsequent oxidative ring cleavage leads to carboxylic acid fragments via classic degradation pathways. In the pH range 5.0–6.0 kinetic constants (1.76 10 4 –1.84 10 4 M 1 s 1 ) were estimated for diclofenac ozonation. r 2003 Elsevier Ltd. All rights reserved. Keywords: Diclofenac; Advanced oxidation processes; Ozone; UV/H 2 O 2 ; Kinetics; Drugs 1. Introduction In recent years pharmaceutical drugs have emerged as a novel class of water contaminants for which public and scientific concern is increasing steadily because of the potential impact on human health and the environment even at trace levels [1–5]. Huge amounts of these chemicals in terms of thousands of tons [6–8] are annually used for therapeutic purposes or in animal farming in each European country, and may be excreted both unmetabolized and as active metabolites. Improper disposal or industrial waste may also contribute to their occurrence in aquatic environments, due in part to ineffective degradation in sewage treatment plants [9–11]. A typical case is (2-[2 0 ,6 0 -dichlorophenyl)amino]phe- nylacetic acid) (diclofenac, 1), a popular non-steroidal anti-inflammatory drug widely used to treat inflamma- tory and painful diseases of rheumatic and non- rheumatic origin which has been detected in many municipal sewage treatment plant (STP) effluents [12,13]. Although it is susceptible to photodegradation by complex mechanisms depending on environmental conditions [14], its presence has been documented in river and lake waters [14–16]. Preliminary investigations concerning diclofenac impact on aquatic life indicated some adverse effects on rainbow trouts exposed to water ARTICLE IN PRESS *Corresponding author. Tel.: +390817682251; fax: +390815936936. E-mail address: [email protected] (R. Andreozzi). 0043-1354/$ - see front matter r 2003 Elsevier Ltd. All rights reserved. doi:10.1016/j.watres.2003.09.028
Transcript of Advanced oxidation of the pharmaceutical drug diclofenac with UV/H 2O 2 and ozone

Water Research 38 (2004) 414–422
ARTICLE IN PRESS
*Correspond
+39081593693
E-mail addr
0043-1354/$ - se
doi:10.1016/j.w
Advanced oxidation of the pharmaceutical drug diclofenacwith UV/H2O2 and ozone
Davide Vognaa, Raffaele Marottab, Alessandra Napolitanoa,Roberto Andreozzib,*, Marco d’Ischiaa
aDip. di Chimica Organica e Biochimica, Federico II, Facolta di Scienze, Universita degli Studi di Napoli Complesso Universitario M. S.
Angelo, via Cinthia, 4, 80126, Napoli, ItalybDip. di Ingegneria Chimica, Facolta di Ingegneria, Universita degli Studi di Napoli FedericoII Prazzale Tecchio, 80, 80125 Napoli, Italy
Received 24 October 2002; received in revised form 2 September 2003; accepted 19 September 2003
Abstract
Diclofenac, a widely used anti-inflammatory drug, has been found in many Sewage Treatment Plant effluents, rivers
and lake waters, and has been reported to exhibit adverse effects on fish. Advanced oxidation processes, ozonation and
H2O2/UV were investigated for its degradation in water. The kinetic of the degradation reaction and the nature of the
intermediate products were still poorly defined. Under the conditions adopted in the present study, both ozonation and
H2O2/UV systems proved to be effective in inducing diclofenac degradation, ensuring a complete conversion of the
chlorine into chloride ions and degrees of mineralization of 32% for ozonation and 39% for H2O2/UV after a 90min
treatment.
The reactions were found to follow similar, but not identical, reaction pathways leading to hydroxylated
intermediates (e.g. 2-[(2,6-dichlorophenyl)amino]-5-hydroxyphenylacetic acid) and C–N cleavage products (notably 2,5-
dihydroxyphenylacetic acid) through competitive routes. Subsequent oxidative ring cleavage leads to carboxylic acid
fragments via classic degradation pathways. In the pH range 5.0–6.0 kinetic constants (1.76� 104–1.84� 104M�1 s�1)
were estimated for diclofenac ozonation.
r 2003 Elsevier Ltd. All rights reserved.
Keywords: Diclofenac; Advanced oxidation processes; Ozone; UV/H2O2; Kinetics; Drugs
1. Introduction
In recent years pharmaceutical drugs have emerged as
a novel class of water contaminants for which public and
scientific concern is increasing steadily because of the
potential impact on human health and the environment
even at trace levels [1–5]. Huge amounts of these
chemicals in terms of thousands of tons [6–8] are
annually used for therapeutic purposes or in animal
farming in each European country, and may be excreted
both unmetabolized and as active metabolites. Improper
ing author. Tel.: +390817682251; fax:
6.
ess: [email protected] (R. Andreozzi).
e front matter r 2003 Elsevier Ltd. All rights reserve
atres.2003.09.028
disposal or industrial waste may also contribute to their
occurrence in aquatic environments, due in part to
ineffective degradation in sewage treatment plants [9–11].
A typical case is (2-[20,60-dichlorophenyl)amino]phe-
nylacetic acid) (diclofenac, 1), a popular non-steroidal
anti-inflammatory drug widely used to treat inflamma-
tory and painful diseases of rheumatic and non-
rheumatic origin which has been detected in many
municipal sewage treatment plant (STP) effluents
[12,13]. Although it is susceptible to photodegradation
by complex mechanisms depending on environmental
conditions [14], its presence has been documented in
river and lake waters [14–16]. Preliminary investigations
concerning diclofenac impact on aquatic life indicated
some adverse effects on rainbow trouts exposed to water
d.

ARTICLE IN PRESS
Nomenclature
Do3 diffusivity of ozone in water,
1.77� 10�7 dm2 s�1
E enhancement factor, dimensionless
kLo gas–liquid phase mass transfer coefficient
without chemical reaction,
4.26� 10�4 dm s�1
kLoa gas–liquid phase volumetric of mass transfer
coefficient without chemical reaction,
0.045 s�1
ko3 rate constant of diclofenac ozonation,
M�1 s�1
I ionic strength, 0.1M
[O3]B ozone bubbles concentration, M
[O3]F ozone freeboard concentration, M
[O3]in initial ozone concentration, in the gas phase,
1.0� 10�3MQ gas flow rate, 0.01L s�1
rdiclofenac reaction rate, M�1 s�1
VB bubbles volume, 0.034L
VF freeboard volume, 0.2 L
VL volume of solution for ozonation experi-
ments, 0.80L
z stoichiometric coefficient, dimensionless
Greek symbols
a Ostwald coefficient, 0.186 dimensionless
b selectivity degree, dimensionless
s standard deviation, %
D. Vogna et al. / Water Research 38 (2004) 414–422 415
concentrations of 1.0 mgL�1 for 28 days [17]. Although
no conclusive data is available about the possible
environmental effect of diclofenac in surface waters, an
evaluation of the suitability of currently available
processes for removal of organic pollutants from STP
effluents seemed appropriate.
Recently, some investigations on the removal of
diclofenac from contaminated and drinking waters by
means of advanced oxidation processes (O3, O3/H2O2 and
photo–Fenton), have appeared [18–20]; however, neither
detailed kinetic assessments nor intermediate/product
analysis during the degradation pathways were provided.
In the present study, we report the oxidation behavior
of diclofenac to ozone and UV/H2O2, two widely used
oxidizing systems with established effectiveness and a
high level of technical development for industrial
application. Specific aims of the study were to investi-
gate the kinetics of the processes and to assess the nature
of the main intermediates along the route to mineraliza-
tion. This latter aspect is of particular relevance, since
potentially toxic species may be produced by incomplete
degradation of organic pollutants and their assessment
is an absolute requirement should the method be applied
for water remediation.
2. Materials and methods
2.1. Chemicals
Hydrogen peroxide (30% w/w) was purchased from
Fluka. Diclofenac (2-[20,60-dichlorophenyl)amino]phe-
nylacetic acid) (1) was purchased as sodium salt from
Sigma. Glyoxal and oxalic, formic, glyoxalic, ketoma-
lonic, malonic, succinic, fumaric, tartronic, hydroxysuc-
cinic (malic), oxaloacetic, D,L tartaric, 2,5-
dihydroxyphenylacetic (homogentisic acid), 2-hydroxy-
phenylacetic acids and 2,6-dichloroaniline were pur-
chased from Aldrich. All other chemicals were of the
highest grade commercially available and were used as
received. Doubly glass-distilled water was used for the
preparation of standard aqueous solutions.
2.2. Ozonation
A Fischer Model 502 apparatus was used for the
production of ozone. The ozone stream (36Lh�1) was
continuously introduced through a porous sparger in the
semi-batch glass reactor [21] contanining the aqueous
solution (1.090L), thermostated at 25�C. The ozone in
the freeboard phase was evaluated continuously by
means of a Varian UV spectrophotometer at 253 nm
(e03=3200M�1 cm�1) equipped with a quartz cell
(optical length=2.0mm).
For kinetic experiments, ozonation runs were carried
out at three different pH values (5.0, 5.5 and 6.0) in the
presence of a radical scavenger (tert-butyl alcohol,
4.0� 10�3L) to prevent radical oxidation. Phosphate
buffers were used to fix ionic strength at 0.1M, and the
pH of the ozonation mixtures was monitored throughout
the reaction course. Aliquots of the mixtures were with-
drawn periodically and immediately analyzed by HPLC.
Batch ozonation experiments were performed to
check the reliability of kinetic constants. In the case of
ozonation, the experiments were conducted by rapidly
mixing 5.0mL of an aqueous solution, buffered at pH
5.0 and containing tert-butyl alcohol (0.2mL), diclofe-
nac (1.0� 10�4M) and phenol (1.0� 10�4M) with
5.0mL of an ozone-saturated aqueous solution
(1.0� 10�4M) at the same pH. The experiments wererepeated with varying starting ozone concentrations.
2.3. UV/H2O2 oxidation
Irradiation experiments were carried out in an
annular reactor (0.420L) thermostated at 25�C and

ARTICLE IN PRESSD. Vogna et al. / Water Research 38 (2004) 414–422416
equipped with a 17W low-pressure mercury monochro-
matic lamp emitting at 254 nm (Helios Italquartz).
The irradiating power of the lamp (I0=2.7� 10�6 E s�1)
was measured by an actinometric procedure [22].
Hydrogen peroxide concentration during actinometric
runs was determined by a modified iodometric
method [23].
The pH of 1 solutions was adjusted to the desired
values with perchloric acid and/or sodium hydroxide.
The reaction course was monitored by periodical HPLC
analysis.
2.4. Analytical methods
GC-MS analyses were carried out on a Saturn
2000 apparatus (Varian) equipped with an Ion Trap
detector. The flow rate of the carrier gas (Helium)
was set at 1mLmin�1. A DB5-MS fused silica
column (Zorbax, 30m� 0.25mm ID, 0.25 mm film
thickness) was used. The temperature program was
as follows: 80�C for 1min, 7�Cmin�1 up to 150�C,
hold time 5min, 7�Cmin�1 up to 200�C, hold time
5min. The injector and transferline temperatures were
250�C and 170�C respectively. The MS detector
was operated in the EI mode, scanning in the range
40–640 amu. Benzopyrene was used as internal stan-
dard in all runs. Formation yields of reaction pro-
ducts were determined by comparing integrated
peak areas with external calibration curves obtained
for 1.
HPLC measurements were performed on a Hewlett
Packard 1100 liquid chromatograph using a UV diode
array detector. For analysis of 1 a Sinergy RP-max
column was used, eluant CH3CN 0.07M phosphate
buffer pH 2 containing 5% CH3OH, 60:40. The flow
rate was 1.0mLmin�1, and the detection wavelength
was set at 274 nm.
To monitor free chloride formation during the
ozonation and H2O2/UV processes an Orion 96-17B
combinated electrode was used.1H and 13C NMR spectra were recorded at 400.1
and 100.6MHz, respectively. An instrument fitted
with a 5mm 1H/broadband gradient probe with
inverse geometry was used. Experiments used were
standard Bruker implementations of gradient-selected
versions of inverse (1H detected) heteronuclear multi-
ple quantum coherence (HMQC) and heteronuclear
multiple bond correlation (HMBC) experiments. The
HMBC experiments used a 100ms long-range coupling
delay.
Total organic carbon (TOC) was monitored by a TOC
analyzer (Shimadzu 5000 A). The molar extinction
coefficient of diclofenac at 254 nm was determined
under the reaction conditions. The absorbance of the
UV radiation mixtures was determined at different
reaction times.
2.5. Product analysis
Aliquots (1.0mL) of the ozonation mixture, buffered
at pH 7.0, with 1 at 5.0� 10�3 M, were withdrawn atdifferent reaction times, lyophilized (Labconco) and
derivatized according to a reported procedure [24]. After
centrifugation, the mixtures were analyzed by GC-MS.
National Institute of Standards and Technology Library
searching was used for compound identification. Identi-
fication of the most abundant components was secured
by comparison of the chromatographic behavior and
fragmentation patterns with those of authentic samples.
A similar procedure was used for analysis of the UV/
H2O2 oxidation mixture of 1 (1.0� 10�3 M), with
hydrogen peroxide concentration at 0.1 or 1.0M.
2.6. Isolation and characterization of 2-[20,60-
dichlorophenyl)amino]-5-hydroxyphenylacetic acid (2)
A solution of 1 (1.0� 10�3 M) in water (200mL)
adjusted to pH 7.0 was bubbled with ozone for 5min
and then nitrogen to purge ozone. The mixture was
extracted with ethyl acetate (3� 100mL), and the
combined organic phases dried with anhydrous Na2SO4and evaporated to dryness. Fractionation by preparative
thin layer chromatography on precoated silica gel F-254
plates from Merck (0.50mm) allowed isolation of pure 2
as colorless oil (Rf 0.73, CHCl3: CH3OH 9:1, 5mg, 10%
yield); UV lmax (MeOH) 278 nm;1H-NMR and 13C-
NMR (see Table 2).
2.7. Preparation of 2-aminophenylacetic acid (9)
A solution of 2-nitrophenylacetic acid (0.5 g) in 0.2M
carbonate buffer pH 10 (40mL) was treated with solid
Na2S2O4 (4.6 g) at room temperature under stirring.
After 15min, the mixture was extracted with ethyl
acetate (3� 30mL). The combined organic layers wererepeatedly washed with brine and water, dried over
sodium sulfate and taken to dryness to yield compound
2 as a colourless solid (390mg, 93% yield). GC-MS
analysis (O-TMS derivative) Rt=11.45min, m/z 295
(M+).
3. Results and discussion
3.1. Ozonation and H2O2/UV processes
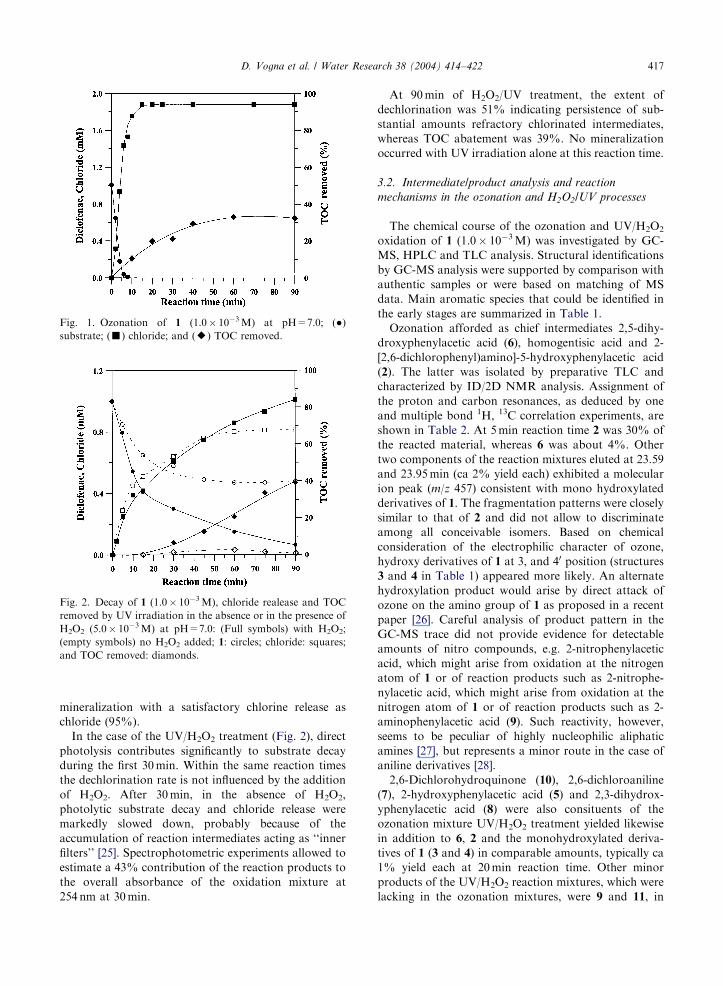
Figs. 1 and 2 show the removal of 1 (1.0� 10�3M) inaqueous solutions buffered at pH=7.0 by ozonation
and H2O2/UV oxidation. Data show complete abate-
ment of 1 within 10min by the ozonation process and
after more than 90min by the UV/H2O2 system. After
90min, the ozonation process induced about 32% of
ARTICLE IN PRESS
Fig. 2. Decay of 1 (1.0� 10�3M), chloride realease and TOCremoved by UV irradiation in the absence or in the presence of
H2O2 (5.0� 10�3M) at pH=7.0: (Full symbols) with H2O2;
(empty symbols) no H2O2 added; 1: circles; chloride: squares;
and TOC removed: diamonds.
Fig. 1. Ozonation of 1 (1.0� 10�3M) at pH=7.0; (�)substrate; (’) chloride; and (E) TOC removed.
D. Vogna et al. / Water Research 38 (2004) 414–422 417
mineralization with a satisfactory chlorine release as
chloride (95%).
In the case of the UV/H2O2 treatment (Fig. 2), direct
photolysis contributes significantly to substrate decay
during the first 30min. Within the same reaction times
the dechlorination rate is not influenced by the addition
of H2O2. After 30min, in the absence of H2O2,
photolytic substrate decay and chloride release were
markedly slowed down, probably because of the
accumulation of reaction intermediates acting as ‘‘inner
filters’’ [25]. Spectrophotometric experiments allowed to
estimate a 43% contribution of the reaction products to
the overall absorbance of the oxidation mixture at
254 nm at 30min.
At 90min of H2O2/UV treatment, the extent of
dechlorination was 51% indicating persistence of sub-
stantial amounts refractory chlorinated intermediates,
whereas TOC abatement was 39%. No mineralization
occurred with UV irradiation alone at this reaction time.
3.2. Intermediate/product analysis and reaction
mechanisms in the ozonation and H2O2/UV processes
The chemical course of the ozonation and UV/H2O2oxidation of 1 (1.0� 10�3M) was investigated by GC-MS, HPLC and TLC analysis. Structural identifications
by GC-MS analysis were supported by comparison with
authentic samples or were based on matching of MS
data. Main aromatic species that could be identified in
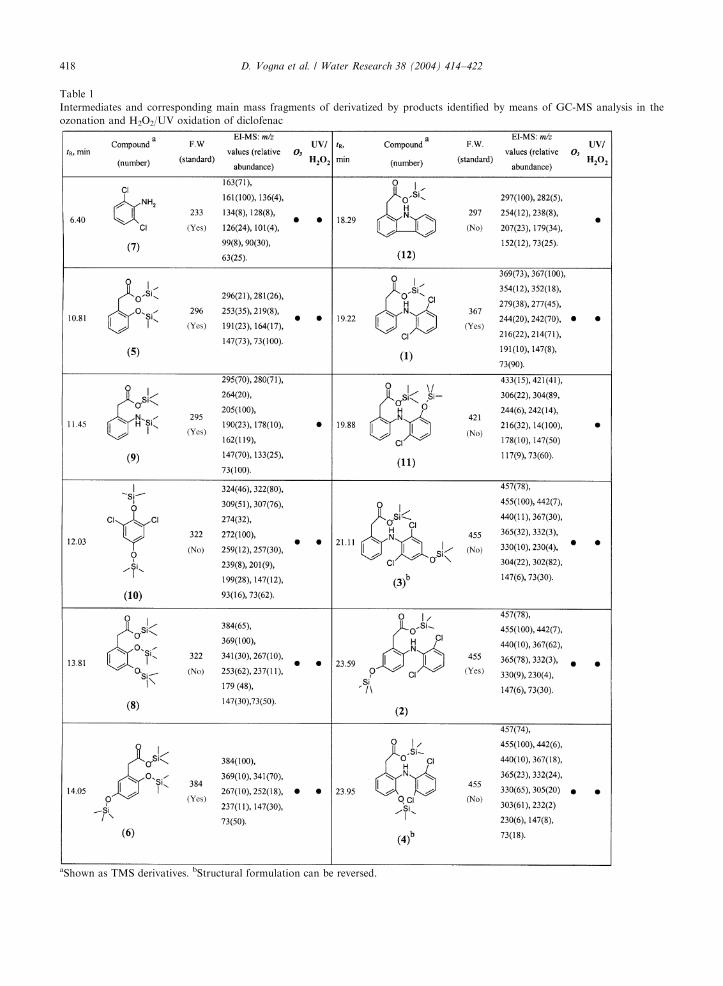
the early stages are summarized in Table 1.
Ozonation afforded as chief intermediates 2,5-dihy-
droxyphenylacetic acid (6), homogentisic acid and 2-
[2,6-dichlorophenyl)amino]-5-hydroxyphenylacetic acid
(2). The latter was isolated by preparative TLC and
characterized by ID/2D NMR analysis. Assignment of
the proton and carbon resonances, as deduced by one
and multiple bond 1H, 13C correlation experiments, are
shown in Table 2. At 5min reaction time 2 was 30% of
the reacted material, whereas 6 was about 4%. Other
two components of the reaction mixtures eluted at 23.59
and 23.95min (ca 2% yield each) exhibited a molecular
ion peak (m/z 457) consistent with mono hydroxylated
derivatives of 1. The fragmentation patterns were closely
similar to that of 2 and did not allow to discriminate
among all conceivable isomers. Based on chemical
consideration of the electrophilic character of ozone,
hydroxy derivatives of 1 at 3, and 40 position (structures
3 and 4 in Table 1) appeared more likely. An alternate
hydroxylation product would arise by direct attack of
ozone on the amino group of 1 as proposed in a recent
paper [26]. Careful analysis of product pattern in the
GC-MS trace did not provide evidence for detectable
amounts of nitro compounds, e.g. 2-nitrophenylacetic
acid, which might arise from oxidation at the nitrogen
atom of 1 or of reaction products such as 2-nitrophe-
nylacetic acid, which might arise from oxidation at the
nitrogen atom of 1 or of reaction products such as 2-
aminophenylacetic acid (9). Such reactivity, however,
seems to be peculiar of highly nucleophilic aliphatic
amines [27], but represents a minor route in the case of
aniline derivatives [28].
2,6-Dichlorohydroquinone (10), 2,6-dichloroaniline
(7), 2-hydroxyphenylacetic acid (5) and 2,3-dihydrox-
yphenylacetic acid (8) were also consituents of the
ozonation mixture UV/H2O2 treatment yielded likewise
in addition to 6, 2 and the monohydroxylated deriva-
tives of 1 (3 and 4) in comparable amounts, typically ca
1% yield each at 20min reaction time. Other minor
products of the UV/H2O2 reaction mixtures, which were
lacking in the ozonation mixtures, were 9 and 11, in
ARTICLE IN PRESS
Table 1
Intermediates and corresponding main mass fragments of derivatized by products identified by means of GC-MS analysis in the
ozonation and H2O2/UV oxidation of diclofenac
aShown as TMS derivatives. bStructural formulation can be reversed.
D. Vogna et al. / Water Research 38 (2004) 414–422418
ARTICLE IN PRESS
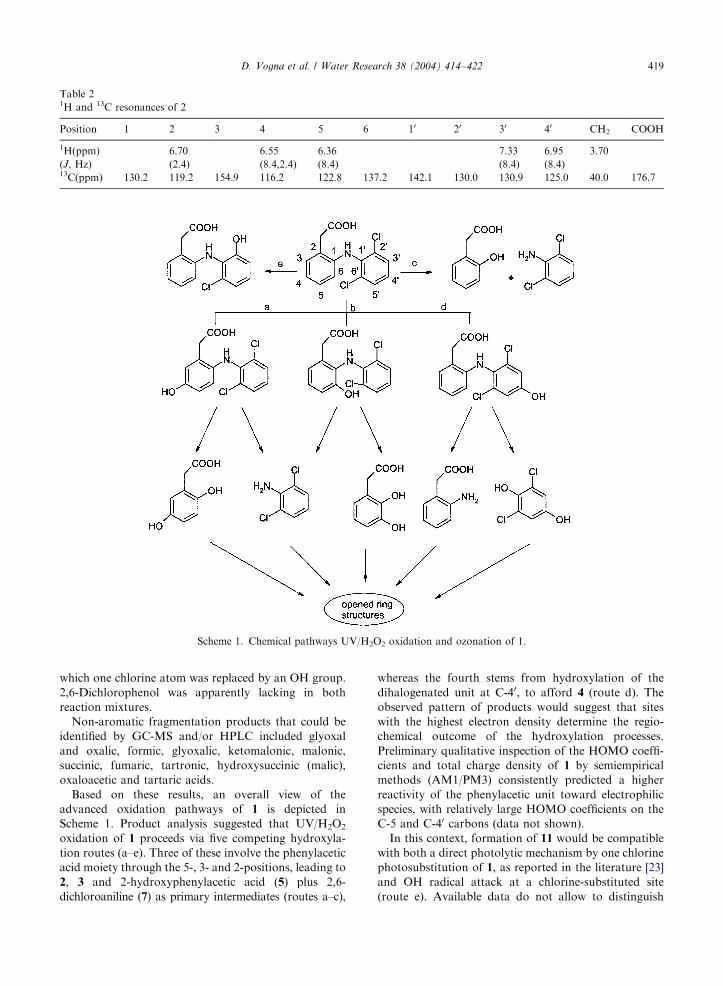
Table 21H and 13C resonances of 2
Position 1 2 3 4 5 6 10 20 30 40 CH2 COOH
1H(ppm) 6.70 6.55 6.36 7.33 6.95 3.70
(J, Hz) (2.4) (8.4,2.4) (8.4) (8.4) (8.4)13C(ppm) 130.2 119.2 154.9 116.2 122.8 137.2 142.1 130.0 130.9 125.0 40.0 176.7
Scheme 1. Chemical pathways UV/H2O2 oxidation and ozonation of 1.
D. Vogna et al. / Water Research 38 (2004) 414–422 419
which one chlorine atom was replaced by an OH group.
2,6-Dichlorophenol was apparently lacking in both
reaction mixtures.
Non-aromatic fragmentation products that could be
identified by GC-MS and/or HPLC included glyoxal
and oxalic, formic, glyoxalic, ketomalonic, malonic,
succinic, fumaric, tartronic, hydroxysuccinic (malic),
oxaloacetic and tartaric acids.
Based on these results, an overall view of the
advanced oxidation pathways of 1 is depicted in
Scheme 1. Product analysis suggested that UV/H2O2oxidation of 1 proceeds via five competing hydroxyla-
tion routes (a–e). Three of these involve the phenylacetic
acid moiety through the 5-, 3- and 2-positions, leading to
2, 3 and 2-hydroxyphenylacetic acid (5) plus 2,6-
dichloroaniline (7) as primary intermediates (routes a–c),
whereas the fourth stems from hydroxylation of the
dihalogenated unit at C-40, to afford 4 (route d). The
observed pattern of products would suggest that sites
with the highest electron density determine the regio-
chemical outcome of the hydroxylation processes.
Preliminary qualitative inspection of the HOMO coeffi-
cients and total charge density of 1 by semiempirical
methods (AM1/PM3) consistently predicted a higher
reactivity of the phenylacetic unit toward electrophilic
species, with relatively large HOMO coefficients on the
C-5 and C-40 carbons (data not shown).
In this context, formation of 11 would be compatible
with both a direct photolytic mechanism by one chlorine
photosubstitution of 1, as reported in the literature [23]
and OH radical attack at a chlorine-substituted site
(route e). Available data do not allow to distinguish

ARTICLE IN PRESS
Scheme 2. Proposed mechanisms of formation of 2 in the ozonation or UV/H2O2 oxidation of 1.
D. Vogna et al. / Water Research 38 (2004) 414–422420
between these options. It should be noted that direct UV
photolysis of 1, in the absence of H2O2, resulted in about
50% substrate consumption after 90min irradiation
without a defined pattern of products (Fig. 2).
Whereas in the UV/H2O2 oxidation treatment all of
the possible hydroxylation routes seemed to be opera-
tive, the ozonation reaction generated 2 and homo-
gentisic acid (6) as the principal intermediates (routes
a,c), suggesting that hydroxylation at C-5 is the chief
decomposition route of 1 with ozone. The apparent
selectivity associated with ozonation would indicate
substantial mechanistic differences with respect to the
UV/H2O2 oxidation process, in which most of the
degradative paths are mediated by the highly reactive,
relatively unselective OH radicals. Representative hy-
droxylation mechanisms at C-5 leading to 2 are depicted
in Scheme 2 for both processes.
Further hydroxylation of 2 at the activated 2-position
would then induce fission of the C–N bond leading to
2,5-dihydroxyphenylacetic (6) acid and 2,6 dichloroani-
line (7). Similarly, hydroxylation of 3 would trigger an
alternate C–N cleavage route leading to 2,3-dihydrox-
yphenylacetic acid along with 2,6-dichloroaniline (7).
The presence of the OH group on the C–40 in 4 would
direct oxidation chemistry toward the otherwise poorly
reactive dichloroaniline moiety favoring OH radical
attack at the 10-position. Subsequent cleavage of the C–
N bond would then yield 2,6-dichlorohydroquinone (10)
and 2-aminophenylacetic acid (9). This latter product
was demonstrated in small amounts only in the UV/
H2O2 process, suggesting that the relevant route is of
minor importance in the case of the ozonation process.
Failure to detect 2,6-dichlorophenol among main aro-
matic intermediates indicates that of the two NH-
bearing positions, the 10-position on the dichlorinated
ring is not sufficiently activated toward OH radical
attack compared to the 2-position on the phenylacetic
moiety.
Photochemical decomposition of 1 in water was
reported to afford as principal products carbazole-1-
acetic acid (in the absence of H-atom donors), 8-
chlorocarbazole-1-acetic acid and 8-hydroxycarbazole-
1-acetic acid, via dechlorination paths that have been
implicated to account for the behavior and fate of 1 in
aquatic environments [29,30]. It appears, on this basis,
that carbazole derivatives can be taken as specific
indicators of purely photolytic processes. Careful
analysis of the UV/H2O2 oxidation mixtures of 1
revealed only trace amounts of carbazole derivatives
(Table 1, 12), thus suggesting that most of the UV
radiation was absorbed by H2O2 and that direct
photolysis of 1 was not a main contributing reaction
pathway under the specific conditions of this study.
3.3. Kinetic studies
A suitable kinetic model was developed to describe the
consumption of diclofenac in early ozonation stages. It
is assumed that the process develops under a ‘‘fast’’
kinetic regime of absorption with reaction. This model
combines a fluid-dynamic submodel [31] with a simpli-
fied kinetic one in which the ozone attack on diclofenac
is described by one single reaction:
where b parameter indicates the selectivity of the
oxidation for pathway 1.
The complete model includes the differential mass
balance equations for ozone in the bubble ([O3]B),
freeboard ([O3]F) and liquid ([O3]L) phases, each one
considered as a well-mixed stirred reactor:
d½O3�Bdt
¼Q
VBð½O3�in � ½O3�BÞ �
k0La E ð½O3�B a� ½O3�LÞVB
VL;
ð1Þ

ARTICLE IN PRESS
Table 3
Kinetic parameters obtained from experimental runs at
different pH values. [Diclofenac]o=3.0� 10�4M
pH 5.0 5.5 6.0
104 ko3 (M�1 s�1) 1.7670.14 1.6970.10 1.8470.15
% s Diclofenac 8.5 6.3 6.2
% sCl 8.3 6.3 10.5
% so3 freeb 3.4 2.3 4.1
D. Vogna et al. / Water Research 38 (2004) 414–422 421
d½O3�Fdt
¼Q
VFð½O3�B � ½O3�FÞ; ð2Þ
d½O3�Ldt
¼ k0La E ð½O3�B a� ½O3�LÞ � z rDiclofenac ð3Þ
where the enhancement factor E was calculated through
the following formulae:
E ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi1þ
Do3ko3 ½S�z
k02
L
s: ð4Þ
The overall diclofenac consumption and chloride ion
production rates are given by
d½S�dt
¼ �1
zko3 ½S�½O3�L ¼ rDiclofenac; ð5Þ
d½Cl�dt
¼ �bko3 ½S�½O3�L: ð6Þ
For kL0a, kL
0 , a, Do3 ; VB VL and VF the values reported in
the legend (p. 19) were adopted whereas from the data
collected during the runs a selectivity (b) equal to 0.70and a stoichiometric coefficient (z) equal to 2.0 were
calculated.
The model allowed thus to estimate, by means
of an optimizing procedure through which the minimum
of the objective function is determined [32], the best
values of kinetic constant ko3 (Table 3). Statistical
indices such as the percentage standard deviations (s)were used to evaluate the model adequacy. They are
defined as
1
%Y�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiPðYi � CiÞ
2
N � 1
s
in which Yi and Ci are calculated and experimental
concentrations at the time ti, N and %Y the number of
experimental data and the mean concentration in a
single run, respectively. Data analysis indicated satisfac-
tory accuracy in the estimation of the kinetic constant as
well as low values of percentage standard deviations for
the measured species (substrate, chloride and ozone in
the liquid bulk).
Huber and co-authors [26] reported a kinetic constant
for diclofenac ozonation of B1.0� 106M�1 s�1 deter-
mined by a comparison method with phenol in the pH
range 6.7–7.0. Since this value does not agree with the
kinetic constants estimated above, a new set of batch
experiments were carried out at pH 5.0, by submitting
diclofenac and phenol simultaneously to ozonation. A
kphenol/kdiclofenac ratio of kinetic constants equal to 1.24
was thus calculated from the results of these experi-
ments. By taking into account the values reported by
Hoign"e and Bader [33] for phenol and phenolate ion, a
value for the kinetic constant of ozonation of phenol
(kphenol) equal to 1.89� 104M�1 s�1 was derived at pH
5.0. On the basis of this result and the previously
calculated kinetic constant ratio, a value for kdiclofenacequal to 1.52� 104M�1 s�1 was obtained which well
agrees with those reported in Table 3 and estimated in
the semibatch ozonation runs on diclofenac.
For the H2O2/UV system the determination of the
kinetic constant for OH attack on the substrate by
means of the competition kinetics method [34] was
prevented by the occurrence of the direct photolytic
degradation of diclofenac (Fig. 2).
4. Conclusions
In aqueous solutions under the experimental condi-
tions adopted, both ozonation and H2O2/UV systems
are effective in inducing diclofenac degradation. After
90min of treatment, the ozonation process ensures a
complete conversion of chlorine into chloride ions with a
degree of mineralization of 32% whereas only 52% of
chlorine was converted into chloride ions with H2O2/UV
system with a mineralization of 39%.
The reactions were found to follow similar, but not
identical, reaction pathways leading to hydroxylated
intermediates (e.g. 2-[(2,6-dichlorophenyl)amino]-5-hy-
droxyphenylacetic acid (2)) and C–N cleavage products
(notably 2,5-dihydroxyphenylacetic acid (6, homogenti-
sic acid) through competitive routes. Subsequent oxida-
tive ring cleavage leads to carboxylic acid fragments via
classic degradation pathways.
In the pH range 5.0–6.0 kinetic constants (1.76� 104–1.84� 104M�1 s�1) were estimated for diclofenac ozo-
nation. Limited solubility at lower pH and foaming of
diclofenac aqueous solutions at pH>6.0 prevented the
investigation in a broader range.
Acknowledgements
We wish to thank the Commission of the European
Communities for the financial support of this work
under Grant No. EVK1-CT-2000-00048. We also thank
the ‘‘Centro Interdipartimentale di Metodologie Chimi-
co Fisiche’’ of Naples University for NMR facilities,
Mrs. Silvana Corsani and Dr. Italo Giudicianni for
assistance.

ARTICLE IN PRESSD. Vogna et al. / Water Research 38 (2004) 414–422422
References
[1] Halling-Sorensen B, Nielsen SN, Lanzky PF, Ingerslev F,
Lutzhorft HC, Jorgensen SE. Occurrence, fate and effects
of pharmaceutical substances in the environment—a
review. Chemosphere 1998;36:357–93.
[2] Daughton CG, Ternes TA. Pharmaceuticals and personal
care products in the environment: agents of subtle change?
Environ Health Perspect 1999;107:907–38.
[3] Buser HR, Poiger T, Muller MD. Occurrence and
environmental behavior of the chiral pharmaceutical drug
Ibuprofen in surface waters and in wastewater. Environ Sci
Technol 1999;33:2529–35.
[4] Heberer T. Occurrence, fate and removal of pharmaceu-
tical residues in the aquatic environment: a review of recent
research data. Toxicol Lett 2002;131:5–17.
[5] Bort R, Ponsoda X, Jover R, Gomez-Lechon J, Castell JV.
Diclofenac toxicity to hepatocytes: a role for drug
metabolism in cell toxicity. J Pharmacol Exp Ther 1999;
288:65–72.
[6] Tolls J. Sorption of veterinary pharmaceuticals in soils: a
review. Environ Sci Technol 2001;35(17):3397–406.
[7] Jones QAH, Voulvoulis N, Lester JN. Aquatic environ-
mental assessmet of the top 25 English prescription
pharmaceuticals. Water Res 2002;36:5013–22.
[8] Kummerer K, editor. Pharmaceuticals in the environment:
sources, fate, effects and risks, (1st ed.) Berlin: Springer,
2001.
[9] Holm JH, Rugge K, Bjerg PL, Christensen TH. Occur-
rence and distribution of pharmaceutical organic com-
pounds in the groundwater downgradient of a landfill
(Griendsted, Denmark). Environ Sci Technol 1995;29:
1415–20.
[10] Hirsch R, Ternes T, Haberer K, Kratz KL. Occurrence of
antibiotics in the aquatic environment. Sci Tot Environ
1999;225:109–18.
[11] Zuccato E, Calamari D, Natangelo M, Fanelli R. Presence
of therapeutic drugs in the environment. Lancet 2000;
355:1789–90.
[12] Ternes TA. Occurrence of drugs in German sewage
treatment plants and rivers. Water Res 1998;32:3245–60.
[13] Ternes TA, Bonerz M, Schmidt T. Determination of
neutral pharmaceutical in wastewater and rivers by liquid
chromatography–electrospray tandem mass spectrometry.
J. Chromatogr A 2001;938:175–85.
[14] Buser H, Poiger T, Muller MD. Occurrence and fate of the
pharmaceutical drug diclofenac in surface waters. Environ
Sci Technol 1998;32:3449–56.
[15] Buser HR, Muller MD, Norbert T. Occurrence of the
pharmaceutical drug clofibric acid and the herbicide
mecoprop in various Swiss lakes and in the North Sea.
Environ Sci Technol 1998;32:188–92.
[16] Ahrer W, Scherwenik E, Buchberger W. Determination of
drug residues in water by the combination of liquid
chromatography or capillary electrophoresis with electro-
spray mass spectrometry. J Chromatogr A 2001;919:69–78.
[17] Triebskorn R, Heyd A, Casper H, Kohler Heinz-R,
Ferling H, Negele R, Schwaiger J. Nephrotoxicity of
anti-inflammatory drug diclofenac in rainbow trout
(Oncorhynchus mykiss). Book of Abstracts of SETAC
Europe 12th Annual Meeting: Challenges in Environmen-
tal Risk Assessment and Modeling, Linking basic and
applied research, Vienna, Austria; 2002.
[18] Zwiener C, Frimmel FH. Oxidative treatment of pharma-
ceuticals in water. Water Res 2000;34:1881–5.
[19] Ravina M, Campanella L, Kiwi J. Accelerated mineraliza-
tion of the drug Diclofenac via Fenton reactions in a
concentric Photo-reactor. Water Res 2002;36:3553–60.
[20] Ternes TA, Meisenheimer M, Mcdowell D, Sacher F,
Brauch HJ, Haist-Glude B, Press G, Wilme U, Zulei-
Seibert N. Removal of pharmaceuticals during dringing
water treatment. Environ Sci Technol 2002;36:3855–63.
[21] Caprio V, Insola A. Aniline and anilinium ion ozonation
in aqueous solution. Ozone Sci Eng 1985;7:169–73.
[22] Nicole I, De Laat J, Dor"e M, Duguet JP, Bonnel C. Use of
UV radiation in water treatment: measurement of photo-
nic flux by hydrogen peroxide actinometry. Water Res
1990;24:157–68.
[23] Treadwell FP, Hall J. Analytical chemistry, vol. II, 8th ed.
New York: Wiley; 1935.
[24] Li X, Cubbage JW, Jenks WS. Photocatalytic degradation
of 4-chlorophenol. 2. The 4-chlorocatechol pathway. J Org
Chem 1999;64:8525–36.
[25] Beltran FJ, Gonzales M, Gonzales F. Industrial waste-
water advanced oxidation. Part 1. UV radiation in the
presence and absence of hydrogen peroxide. Water Res
1997;31:2405–14.
[26] Huber MM, Canonica S, Park GY, Von Gunten U.
Oxidation of pharmaceuticals during ozonation and
advanced oxidation processes. Environ Sci Technol 2003;
37:1016–24.
[27] Munoz F, von Sonntag C. The reactions of ozone with
tertiary amines including the complexing agents nitrilo-
triacetic acid (NTA) and ethylenediaminetetraacetic acid
(EDTA) in aqueous solution. J Chem Soc Perkin Trans
2000;2:2029–33.
[28] Sarasa J, Cort"es S, Ormad P, Gracia R, Ovelleiro JL.
Study of the aromatic by-products formed from ozonation
of anilines in aqueous solution. Water Res 2002;17:185–94.
[29] Poiger T, Buser HR, Muller DM. Photodegradation of the
pharmaceutical drug diclofenac in a lake: pathway, field
measurements and mathematical modeling. Environ Sci
Technol 2001;20:256–63.
[30] Moore BE, Thomson SR, Zhen D, Duke CC. Photo-
chemical studies of anti-inflammatory drug diclofenac.
Photochem Photobiol 1990;52:685–90.
[31] Andreozzi R, Caprio V, Insola A, Tufano V. Measuring
ozonation rate constants in gas–liquid reaction under the
kinetic-diffusional transition regime. Chem Eng Commun
1996;143:195–7.
[32] Reklaitis GV, Ravindran A, Regsdell KM. Engineering
optimization. New York: Wiley; 1983.
[33] Hoign!e J, Bader H. Rate constants of reactions of ozone
with organic and inorganic compounds in water-II.
Dissociating organic compounds. Water Res 1983;17:
185–94.
[34] Onstein P, Stefan MI, Bolton JR. Competition kinetics
method for the determination of rate constants for the
reaction of hydroxyl radicals with organic pollutants using
UV/H2O2 advanced oxidation technology: the rate con-
stants for the tert-butyl formate esters and 2,4-dinitrophe-
nol. J Adv Oxidation Technol 1999;4(2):231–6.




















