
Synthesis and optical characterization of nanocrystalline CdTe thin films
Upload
independentCategory
view
4download
0
ARTICLE IN PRESS
0043-1354/$ - se
doi:10.1016/j.w
�Correspondfax: +1201 216
E-mail addr1New Jersey
P.O. Box 409, T
Water Research 39 (2005) 2327–2337
www.elsevier.com/locate/watres
Adsorption of As(V) and As(III) by nanocrystallinetitanium dioxide
Maria E. Pena, George P. Korfiatis, Manish Patel1, Lee Lippincott1,Xiaoguang Meng�
Center for Environmental Systems, Stevens Institute of Technology, Hoboken, NJ 07030, USA
Received 15 May 2004; received in revised form 11 March 2005
Available online 17 May 2005
Abstract
This study evaluated the effectiveness of nanocrystalline titanium dioxide (TiO2) in removing arsenate [As(V)] and
arsenite [As(III)] and in photocatalytic oxidation of As(III). Batch adsorption and oxidation experiments were
conducted with TiO2 suspensions prepared in a 0.04M NaCl solution and in a challenge water containing the
competing anions phosphate, silicate, and carbonate. The removal of As(V) and As(III) reached equilibrium within 4 h
and the adsorption kinetics were described by a pseudo-second-order equation. The TiO2 was effective for As(V)
removal at pHo8 and showed a maximum removal for As(III) at pH of about 7.5 in the challenge water. The
adsorption capacity of the TiO2 for As(V) and As(III) was much higher than fumed TiO2 (Degussa P25) and granular
ferric oxide. More than 0.5mmol/g of As(V) and As(III) was adsorbed by the TiO2 at an equilibrium arsenic
concentration of 0.6mM. The presence of the competing anions had a moderate effect on the adsorption capacities of
the TiO2 for As(III) and As(V) in a neutral pH range. In the presence of sunlight and dissolved oxygen, As(III) (26.7 mMor 2mg/L) was completely converted to As(V) in a 0.2 g/L TiO2 suspension through photocatalytic oxidation within
25min. The nanocrystalline TiO2 is an effective adsorbent for As(V) and As(III) and an efficient photocatalyst.
r 2005 Elsevier Ltd. All rights reserved.
Keywords: Adsorption; Challenge water; TiO2; Photocatalytic oxidation; Arsenic speciation
1. Introduction
Arsenic is one of the most toxic contaminants found
in the environment and has been recognized as a toxic
element for centuries. Arsenic contamination has
become a worldwide epidemic, especially in developing
countries where a significant percentage of the popula-
e front matter r 2005 Elsevier Ltd. All rights reserve
atres.2005.04.006
ing author. Tel.: +1201 216 8014;
8303.
ess: [email protected] (X. Meng).
Department of Environmental Protection,
renton, NJ 08625, USA.
tion depends on groundwater for drinking. Elevated
concentrations of As in groundwater are found in many
countries such as India, Bangladesh, Vietnam and Chile
(Berg et al., 2001). Chronic exposure to arsenic-
contaminated drinking water can result in serious health
problems, such as skin lesions and cancers (Davis et al.,
1996). To minimize the health impact of arsenic, the
United States Environmental Protection Agency
adopted a new maximum contaminant level of 10 mg/Lin drinking water on January 22, 2001.
In natural waters, inorganic arsenic occurs primarily
in two oxidation states, As(V) and As(III). At circum-
neutral pH levels, the predominant As(V) species are the
d.

ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–23372328
monovalent (H2AsO4�) and divalent (HAsO4
2�), while
the predominant arsenite species is neutral H3AsO3(Cullen and Reimer, 1989; Meng et al., 2000). As(V)
generally exhibits a low mobility in aquifer and sediment
systems due to its retention on mineral surfaces
controlled primarily by adsorption reactions with metal
hydroxide (Picheler et al., 1999). As(III) is more mobile
and more toxic (25–60 times) than As(V). This elevated
toxicity is due to its preferential reaction with sulfhydryl
groups in mammalian enzymes (Korte and Quintus,
1991).
Several treatment techniques are available for remov-
ing As in water, such as: coagulation/precipitation, ion
exchange, lime softening, reverse osmosis and electro-
dialysis. Coagulation/precipitation with ferric and alu-
minum salts is one of the conventional methods for
arsenic removal from aqueous systems (Gregor, 2001;
McNeill and Edwards, 1997; Gulledge and O’connor,
1973). However, this technique is usually encumbered by
problems associated with the treatment and disposal of
the resulting waste sludge. To overcome this problem,
adsorption processes using granular media have been
explored for their ability to remove arsenic from water.
Numerous laboratory and field filtration tests have been
conducted to assess the removal capacity of adsorbents
such as iron oxide-coated sand, granular ferric hydro-
xide, sulfur-modified iron and activated alumina (Hatch,
2002; Driehaus et al., 1998; Vaishya and Gupta, 2003).
Most technologies require the pre-oxidation of As(III)
to As(V) to enhance removal of As(III), since As(V)
adsorbs more strongly onto the solid phase than As(III).
Table 1
Photocatalytic reactions in TiO2 and As(III) system
Formation of radicals
1TiO2�!
hvhþ þ e�
2 hþ þ e� ! heat
3 e� þO2 ! Od�2
4 Od�2 þHþ ! HOd2
5 2HOd2 ! H2O2 þO26 HOd2 þOd�
2 ! HO�2 þO2
7 HO�2 þ hþ
! HOd28 hþþ2H2Oad ! OHdadþH
þ
9 hþþOH�ad ! OHdad
Possible As(III) oxidation reactions
10 AsðIIIÞ þ hþ ! AsðIVÞ
11 As(III)+OHd-As(IV)
12 As(III)+O2d�-As(IV)+H2O2
13 As(III)+H2O2-As(IV)+2OH�
14 As(IV)+O2-As(V)+O2d�
15 As(IV)+OHd-As(V)
16 As(IV)+O2d� -As(V)
17 As(IV)+O2+H+-As(V)+HO
As(III) oxidation is carried out by oxidants such as
chlorine compounds, ozone, H2O2, permanganate or
manganese oxide and Fenton’s reagent (Pettine et al.,
1999; Kim and Nriagu, 2000; Pettine and Millero, 2000;
Hug and Leupin, 2003). Photochemical oxidation of
As(III) by iron complexes such as Fe(III)OH2+,
Fe(III)Cl2+ and solid iron phases was also investigated
(Hug et al., 2001; Emett and Khoe, 2001; Kocar and
Inskeep, 2003). Photocatalytic oxidation with TiO2, as a
semiconductor, is effective in destroying a wide range of
contaminants in gaseous and aqueous phases. Experi-
mental results have demonstrated that As(III) can be
oxidized to As(V) in UV-illuminated TiO2 (Degussa
P25) suspension (Yang et al., 1999; Lee and Choi, 2002;
Bissen et al., 2001).
The photocatalytic oxidation involves absorption of
light with wavelength shorter than 387.5 nm by TiO2,
which results in the excitation of an electron from the
valence band (vb) to the conduction band (cb). This
excitation creates a positively charged hole, h+, in the
valence band (Table 1, reaction 1). An electron–hole
pair forms at the surface of TiO2, which may react with
adsorbed species such as oxygen to generate free radicals
such as Od�2 ;HOd2 ;OH
d (Table 1). Although the vastmajority of previous TiO2 photocatalytic oxidation
studies have focused on organic compounds, inorganic
species have also been investigated (Gruebel et al., 1995).
For the oxidation reactions to take place, it is necessary
that the valence band or conduction band is more
oxidizing than the oxidation potential of the species in
question.
Low et al. (1991)
Low et al. (1991)
Wong and Chu (2003)
Low et al. (1991)
Kocar and Inskeep (2003)
Low et al. (1991)
Low et al. (1991)
Chen and Ray (1998)
Chen and Ray (1998)
Lee and Choi (2002)
Hug and Leupin (2003)
Hug et al. (2001)
Lee and Choi (2002)
Klaning et al. (1989)
Lee and Choi (2002)
Lee and Choi (2002)
2d Emett and Khoe (2001)

ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–2337 2329
As(III) may be oxidized to As(IV) by h+, hydroxyl
radical, superoxide ion, and hydrogen peroxide in a
TiO2/UV system (reactions 10–13). As(IV) is expected to
be oxidized rapidly to As(V) through reactions 14–17.
There is dispute over which reaction controls the As(III)
oxidation process. Lee and Choi (2002) and Ryu and
Choi (2004) showed that superoxide is the dominant
oxidant for As(III) oxidation, which was supported by
Ferguson et al. (2005). On other hand, Dutta et al.
(2005) demonstrated that hydroxyl radical was mainly
responsible for As(III) oxidation.
Based on the literature results, TiO2 is a promising
material for treatment of arsenic, especially As(III).
However, the TiO2 product (Degussa P25) used in most
of the research has very low adsorptive capacity for
arsenic. In the present study, the effectiveness of a
nanocrystalline TiO2 for treatment of arsenic was
examined. The main objectives were to: (a) determine
its adsorption capacity for arsenic; (b) establish the
kinetics of removal of As(V) and As(III); (c) investigate
the photocatalytic oxidation of As(III) to As(V); and (d)
evaluate the effects of common anions in natural water
on As(V) and As(III) adsorption.
2. Experimental methods
2.1. Materials and chemicals
All solutions were prepared using Fisher Scientific
ACS grade chemicals and deionized water (DI). Stock
solutions of 1000mg/L of arsenic were prepared using
sodium salt heptahydrate (Na2HAsO4 � 7H2O) and
sodium arsenite (Na3AsO3). Challenge water containing:
Mg2+ ¼ 12 ppm, SO42�
¼ 50 ppm, NO3�¼ 6 ppm, F� ¼
1ppm, SiO2 ¼ 20ppm, PO43�
¼ 40ppb, Ca2+ ¼ 40ppm,
CO32�
¼ 179ppm, and total chlorine ¼ 0.25–0.75ppm
(chlorine was added only to As(V)-contaminated water)
was prepared daily before the experiments were run
(USEPA/600/R-01/021).
The nanocrystalline TiO2 was produced by hydrolysis
of titanium sulfate solution (Meng et al., 2003). X-ray
powder diffraction analysis indicated that the oxide had
a primary crystallite size of about 6 nm. The material in
powder form costs less than commercial granular ferric
hydroxide (GFH) and granular ferric oxide (GFO). The
nanocrystalline TiO2 slurry was washed with DI water
to remove sulfate ions which could interfere with As
adsorption, and was dried at 105 1C. This metal oxide
had a specific surface area of 330m2/g and a total pore
volume of 0.42 cm3/g. The point of zero charge pH
(pHpzc) of the TiO2 was 5.8 which was determined in a
suspension containing 0.01 g of TiO2 in 0.04M NaCl
using ZetaSizer 3000 (Malvern Instrument). Scanning
electron microscopic images indicated that the nano-
crystalline TiO2 agglomerated to form particles with
sizes of 0.5–2.0 mm.
2.2. Adsorption kinetics
The rate of arsenic uptake is an important factor for
arsenic removal, and is influenced by the surface and
pore properties of the adsorbent. Batch experiments
were conducted to determine the reaction time required
to reach adsorption equilibrium. All suspensions were
prepared in a 0.04M NaCl solution in 1-L glass beakers.
Aliquots of As(V) and As(III) stock solutions were
added to make 2.0mg/L (i.e. 26.7 mmol/L) of As(V) orAs(III) concentration. After the solution pH was
adjusted to 7.0 by adding hydrochloric acid and sodium
hydroxide at room temperature (21–25 1C), TiO2 was
added to attain a 0.2 g/L suspension. The suspension
was mixed with a magnetic stirrer, and the pH was
maintained at 7.070.1 throughout the experiment byaddition of the acid and base solutions. Approximately
25ml aliquots were taken from the suspension at the
following intervals: 0.1, 0.15, 0.25, 0.3, 0.6, 0.9, 2, 4, 6,
10, 20, 40 and 72 h of reaction. The samples were filtered
through a 0.45mm membrane filter. Total As in the
filtered solution was determined using a Furnace Atomic
Absorption Spectrometer (FAAS) (Varian Spectra AA-
400). Speciation of As(III) and As(V) in the solution
samples was performed by passing a solution sample
through an arsenic speciation cartridge (Meng et al.,
2001). The cartridge removed all of the As(V), but did
not remove As(III). The soluble As(V) concentration
was calculated from the difference between the total
soluble arsenic and the soluble As(III) concentrations.
For quality control purposes, the batch experiments
were performed at least in duplicate.
In order to evaluate the effect of dissolved oxygen and
light irradiation on the photocatalytic oxidation of
As(III) in TiO2 suspensions, As(III) removal tests were
conducted in five experimental systems: (1) air and light
system (air–light), where the suspension was open to the
air and under room light (natural and florescent light);
(2) air–dark system, where the reactor was wrapped with
aluminum foil and oxygen in the air was dissolved in the
suspension; (3) N2–dark system, which was obtained by
purging the suspension with high purity nitrogen gas in
the dark; (4) air–sunlight system, where the suspension
in a 1-L glass beaker was mixed directly under the
sunlight outside of the laboratory (this experiment was
performed between 9 a.m. and 5 p.m. on a sunny
summer day (July 8, 2003)), and (5) N2–light system.
2.3. Effect of pH on As adsorption
A suspension containing 0.04M NaCl, 1.0mg/L of
As(V) or As(III) and 0.2 g/L of TiO2 was prepared in a
1-L beaker. Then, 50ml of the uniform suspension was

ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–23372330
transferred into 10 50-ml polyethylene terephthalate
(PETE) tubes. The pH values of the suspension in each
tube were adjusted to between 3 and 13 using dilute
NaOH and HCl solutions. The tubes were placed in a
tumbler and mixed for 22 h. After mixing, the final pH
of the suspension was measured as the equilibrium pH.
The solids were then separated from the solution by
centrifugation for 35min at 13,000 rpm in a Model IEC/
Micromax centrifuge. The solutions were acidified using
HNO3 and the total soluble arsenic concentrations were
determined by FAAS. Similar batch tests were also
carried out using challenge water to determine the effects
of common anions on arsenic adsorption.
2.4. Adsorption isotherms
Adsorption isotherms were obtained by adding
different amounts of stock solutions of As(V) and
As(III) in a suspension containing 1.0 g/L TiO2.
Separate batch tests were performed using 0.04M NaCl
and challenge water. The pH value was adjusted to
7.070.1 by adding hydrochloric acid and sodium
hydroxide at room temperature. After 22 h of mixing,
suspension samples were withdrawn and centrifuged for
35min. Arsenic concentrations in the supernatant
solution were analyzed by FAAS.
3. Results and discussion
3.1. Adsorption kinetics
Arsenic removal by TiO2 occurred rapidly in all
systems except the air–light system (Fig. 1). The uptake
of As(V) and As(III) in the air–dark and N2–dark
systems reached equilibrium in approximately 4 h.
Adsorption kinetics of As(III) in the sunlight system
was similar to that of As(V). The amount of As(III)
adsorbed in the air–light system increased at a slow pace
and reached the same level as that observed in the As(V)
system after 14 h of mixing. The adsorption of As(III) by
commercial TiO2 (Degussa P25) reached equilibrium in
about 1 h, which was similar to the reported rate of
As(V) removal by P25 (Dutta et al., 2004). P25 was
made of nonporous TiO2 particles and the nanocrystal-
line TiO2 was composed of aggregated particles with
high porosity. Therefore, it was expected that longer
time was needed to reach adsorption equilibrium in
porous adsorbent.
The total arsenic to TiO2 ratio in all the suspensions
was 133mmol-As/g–TiO2. At equilibrium approximately120 mmol/g of arsenic or 90% of the total arsenic was
adsorbed in the As(V), As(III)-air–light and As(III)-
air–sunlight systems. The removal of As(III) in the
N2–dark system was lower than that of As(V), reaching
an adsorption density of 95 mmol/g. It should be noted
that only about 50 mmol/g of As(III) in the air–lightsystem was adsorbed by P25 TiO2. The results indicated
that the nanocrystalline TiO2 used in this study had a
much higher adsorption capacity than P25. This could
be attributed to much higher specific surface area of the
nanocrystalline TiO2 than that of P25 (Table 2). The
lower pHpzc value of the TiO2 than that of P25 indicated
that the titanyl group, Ti-OH, on the TiO2 was more
acidic than the functional groups on P25.
Several kinetic models including pseudo-first-order of
Lagergren, pseudo-second-order, Elovich, parabolic
diffusion and power function were tested for simulation
of the experimental data in Fig. 1 (Ho and McKay,
1998; Sparks, 1989). The experimental results were best
described by pseudo-second-order and Elovich kinetic
equations.
A pseudo-second-order rate expression is defined by
in Eq. (1) (Sag and Aktay, 2002) and the integrated form
is given in Eq. (2).
dqt=dt ¼ Kadðqe � qtÞ2, (1)
qe � qt ¼ 1=ðð1=qeÞ þ ðKad � tÞÞ, (2)
where qe and qt are the amount (mmol g�1) of arsenic
adsorbed at equilibrium and at time t, respectively. Kadis the rate constant of sorption (mmol g�1 h�1).The Elovich kinetic equation is defined by Eq (3) and
the integrated form is expressed in Eq. (4) (Sparks,
1989).
dqt=dt ¼ a expð�bqtÞ, (3)
qt ¼ ð1=bÞ ln ðt þ toÞ � ð1=bÞ ln to, (4)
where a is an experimental fitting constant and 1=b ¼
kad is the reaction rate constant (Liang and Tsai, 1995).
The Elovich equation was previously used to study the
sorption kinetics of arsenic (Elkhabit et al., 1984) and
phosphate (Chien and Clayton, 1980) on soils, and
cadmium adsorption by bone char (Cheung et al., 2001).
The lines in Fig. 1 were calculated using the kinetic
equations. The pseudo-second-order kinetic equation
described the experimental data well for all systems
except the air–light condition. The adsorption kinetic
data in the air–light system was simulated well by the
Elovich equation. The modeling results suggested that
the mechanism controlling the rate of arsenic adsorption
in the air–light system was different from that in the
other systems. The best fit constants were summarized in
Table 3. The goodness of the model was quantified by
the mean weighted square error (MWSE). The small
MWSE values (o0.005) indicated that the model fittedthe data accurately.
The rate constants (Kad) of the pseudo-second-order
equation were 0.118–0.199 and 0.073 mmol g�1 h�1 forAs(III) and As(V) systems, respectively (Table 3). The
rate values indicated that the removal of As(III) was
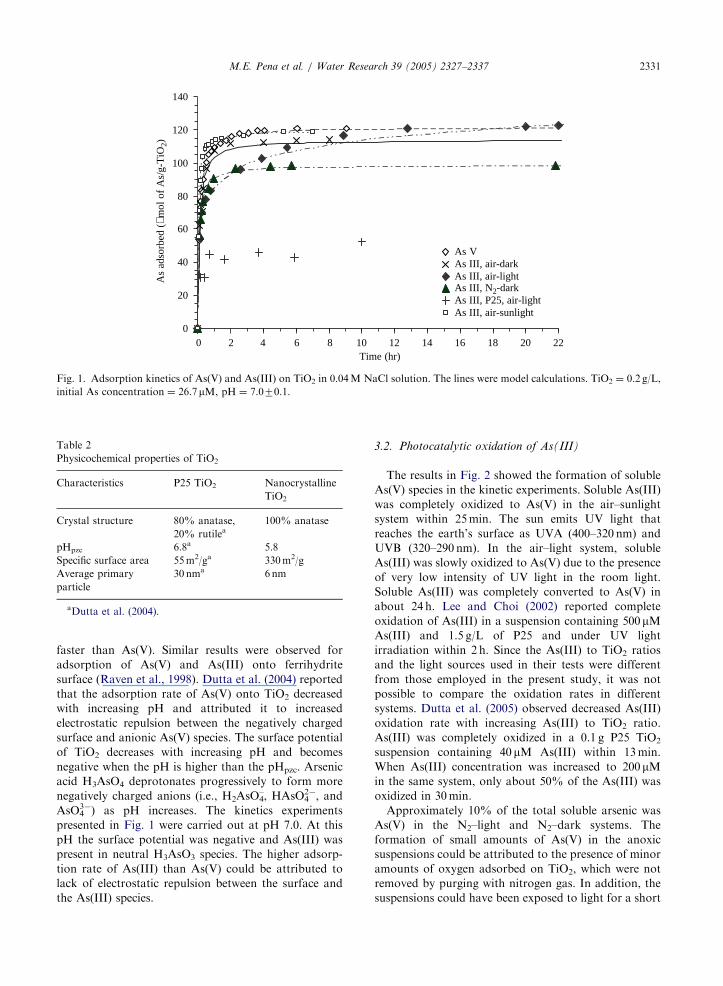
ARTICLE IN PRESS
Table 2
Physicochemical properties of TiO2
Characteristics P25 TiO2 Nanocrystalline
TiO2
Crystal structure 80% anatase,
20% rutilea100% anatase
pHpzc 6.8a 5.8
Specific surface area 55m2/ga 330m2/g
Average primary
particle
30 nma 6 nm
aDutta et al. (2004).
0
20
40
60
80
100
120
140
0 10 12 14 16 18 20 22Time (hr)
As
adso
rbed
(µm
ol o
f A
s/g-
TiO
2)
As VAs III, air-darkAs III, air-lightAs III, N2-darkAs III, P25, air-lightAs III, air-sunlight
8642
Fig. 1. Adsorption kinetics of As(V) and As(III) on TiO2 in 0.04M NaCl solution. The lines were model calculations. TiO2 ¼ 0.2 g/L,
initial As concentration ¼ 26.7mM, pH ¼ 7.070.1.
M.E. Pena et al. / Water Research 39 (2005) 2327–2337 2331
faster than As(V). Similar results were observed for
adsorption of As(V) and As(III) onto ferrihydrite
surface (Raven et al., 1998). Dutta et al. (2004) reported
that the adsorption rate of As(V) onto TiO2 decreased
with increasing pH and attributed it to increased
electrostatic repulsion between the negatively charged
surface and anionic As(V) species. The surface potential
of TiO2 decreases with increasing pH and becomes
negative when the pH is higher than the pHpzc. Arsenic
acid H3AsO4 deprotonates progressively to form more
negatively charged anions (i.e., H2AsO4–, HAsO4
2�, and
AsO43�) as pH increases. The kinetics experiments
presented in Fig. 1 were carried out at pH 7.0. At this
pH the surface potential was negative and As(III) was
present in neutral H3AsO3 species. The higher adsorp-
tion rate of As(III) than As(V) could be attributed to
lack of electrostatic repulsion between the surface and
the As(III) species.
3.2. Photocatalytic oxidation of As(III)
The results in Fig. 2 showed the formation of soluble
As(V) species in the kinetic experiments. Soluble As(III)
was completely oxidized to As(V) in the air–sunlight
system within 25min. The sun emits UV light that
reaches the earth’s surface as UVA (400–320 nm) and
UVB (320–290 nm). In the air–light system, soluble
As(III) was slowly oxidized to As(V) due to the presence
of very low intensity of UV light in the room light.
Soluble As(III) was completely converted to As(V) in
about 24 h. Lee and Choi (2002) reported complete
oxidation of As(III) in a suspension containing 500mMAs(III) and 1.5 g/L of P25 and under UV light
irradiation within 2 h. Since the As(III) to TiO2 ratios
and the light sources used in their tests were different
from those employed in the present study, it was not
possible to compare the oxidation rates in different
systems. Dutta et al. (2005) observed decreased As(III)
oxidation rate with increasing As(III) to TiO2 ratio.
As(III) was completely oxidized in a 0.1 g P25 TiO2suspension containing 40 mM As(III) within 13min.
When As(III) concentration was increased to 200mMin the same system, only about 50% of the As(III) was
oxidized in 30min.
Approximately 10% of the total soluble arsenic was
As(V) in the N2–light and N2–dark systems. The
formation of small amounts of As(V) in the anoxic
suspensions could be attributed to the presence of minor
amounts of oxygen adsorbed on TiO2, which were not
removed by purging with nitrogen gas. In addition, the
suspensions could have been exposed to light for a short
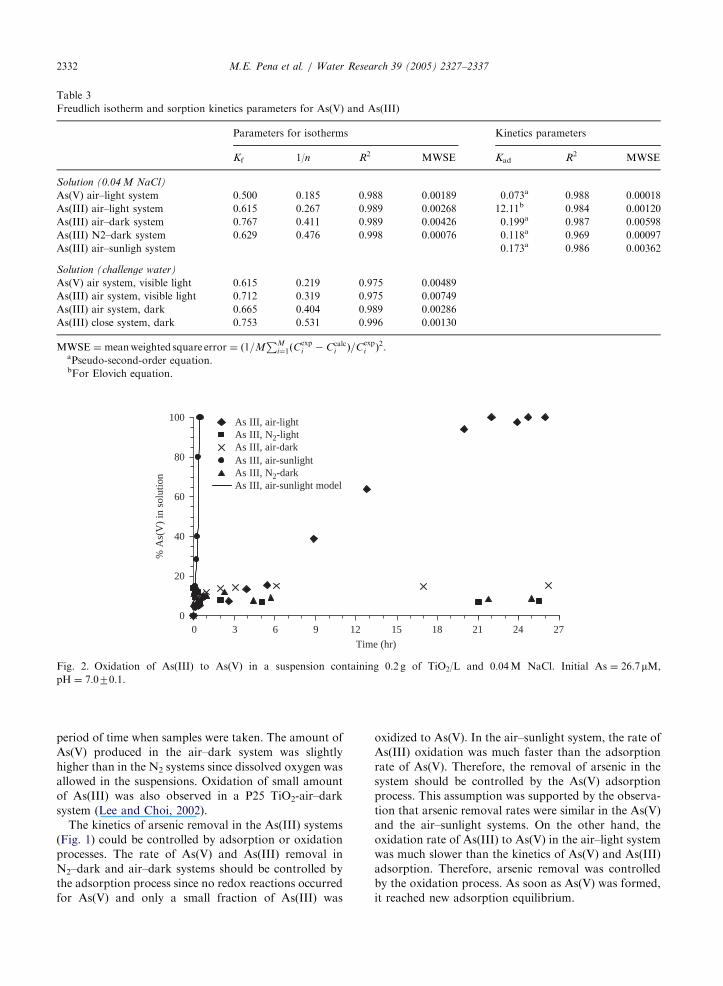
ARTICLE IN PRESS
Table 3
Freudlich isotherm and sorption kinetics parameters for As(V) and As(III)
Parameters for isotherms Kinetics parameters
Kf 1/n R2 MWSE Kad R2 MWSE
Solution (0.04 M NaCl)
As(V) air–light system 0.500 0.185 0.988 0.00189 0.073a 0.988 0.00018
As(III) air–light system 0.615 0.267 0.989 0.00268 12.11b 0.984 0.00120
As(III) air–dark system 0.767 0.411 0.989 0.00426 0.199a 0.987 0.00598
As(III) N2–dark system 0.629 0.476 0.998 0.00076 0.118a 0.969 0.00097
As(III) air–sunligh system 0.173a 0.986 0.00362
Solution (challenge water)
As(V) air system, visible light 0.615 0.219 0.975 0.00489
As(III) air system, visible light 0.712 0.319 0.975 0.00749
As(III) air system, dark 0.665 0.404 0.989 0.00286
As(III) close system, dark 0.753 0.531 0.996 0.00130
MWSE ¼ meanweighted square error ¼ ð1=MPM
i¼1ðCexpi � Ccalci Þ=C
expi Þ
2.aPseudo-second-order equation.bFor Elovich equation.
0
20
40
60
80
100
0 3 6 9 12 15 18 21 24 27
Time (hr)
% A
s(V
) in
sol
utio
n
As III, air-lightAs III, N2-lightAs III, air-darkAs III, air-sunlightAs III, N2-darkAs III, air-sunlight model
Fig. 2. Oxidation of As(III) to As(V) in a suspension containing 0.2 g of TiO2/L and 0.04M NaCl. Initial As ¼ 26.7mM,pH ¼ 7.070.1.
M.E. Pena et al. / Water Research 39 (2005) 2327–23372332
period of time when samples were taken. The amount of
As(V) produced in the air–dark system was slightly
higher than in the N2 systems since dissolved oxygen was
allowed in the suspensions. Oxidation of small amount
of As(III) was also observed in a P25 TiO2-air–dark
system (Lee and Choi, 2002).
The kinetics of arsenic removal in the As(III) systems
(Fig. 1) could be controlled by adsorption or oxidation
processes. The rate of As(V) and As(III) removal in
N2–dark and air–dark systems should be controlled by
the adsorption process since no redox reactions occurred
for As(V) and only a small fraction of As(III) was
oxidized to As(V). In the air–sunlight system, the rate of
As(III) oxidation was much faster than the adsorption
rate of As(V). Therefore, the removal of arsenic in the
system should be controlled by the As(V) adsorption
process. This assumption was supported by the observa-
tion that arsenic removal rates were similar in the As(V)
and the air–sunlight systems. On the other hand, the
oxidation rate of As(III) to As(V) in the air–light system
was much slower than the kinetics of As(V) and As(III)
adsorption. Therefore, arsenic removal was controlled
by the oxidation process. As soon as As(V) was formed,
it reached new adsorption equilibrium.
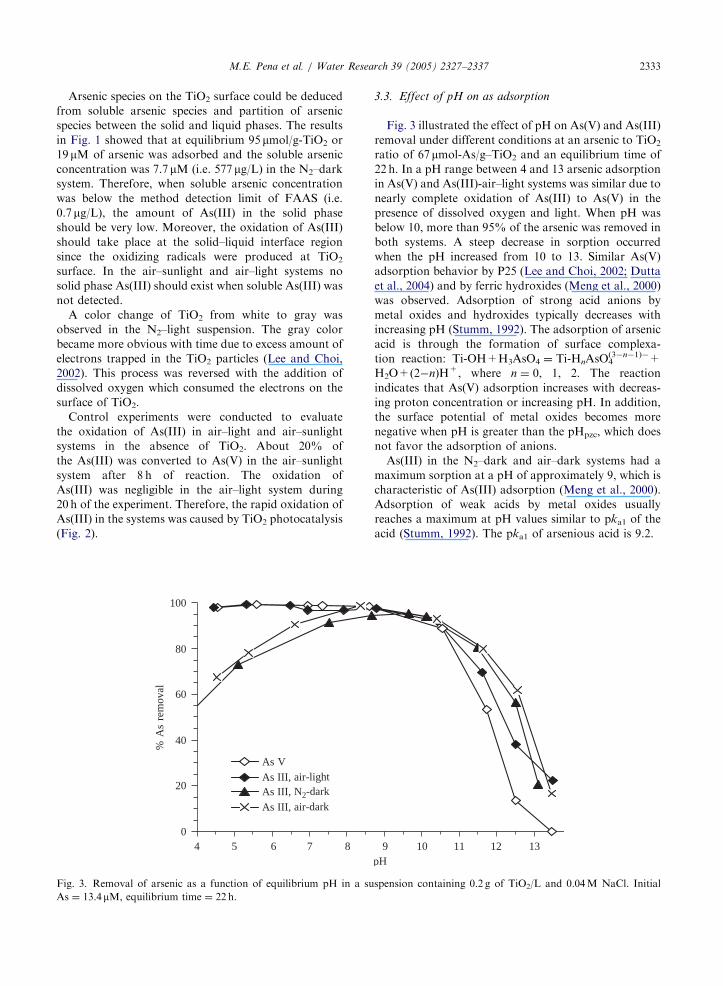
ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–2337 2333
Arsenic species on the TiO2 surface could be deduced
from soluble arsenic species and partition of arsenic
species between the solid and liquid phases. The results
in Fig. 1 showed that at equilibrium 95 mmol/g-TiO2 or19mM of arsenic was adsorbed and the soluble arsenic
concentration was 7.7 mM (i.e. 577 mg/L) in the N2–darksystem. Therefore, when soluble arsenic concentration
was below the method detection limit of FAAS (i.e.
0.7mg/L), the amount of As(III) in the solid phaseshould be very low. Moreover, the oxidation of As(III)
should take place at the solid–liquid interface region
since the oxidizing radicals were produced at TiO2surface. In the air–sunlight and air–light systems no
solid phase As(III) should exist when soluble As(III) was
not detected.
A color change of TiO2 from white to gray was
observed in the N2–light suspension. The gray color
became more obvious with time due to excess amount of
electrons trapped in the TiO2 particles (Lee and Choi,
2002). This process was reversed with the addition of
dissolved oxygen which consumed the electrons on the
surface of TiO2.
Control experiments were conducted to evaluate
the oxidation of As(III) in air–light and air–sunlight
systems in the absence of TiO2. About 20% of
the As(III) was converted to As(V) in the air–sunlight
system after 8 h of reaction. The oxidation of
As(III) was negligible in the air–light system during
20 h of the experiment. Therefore, the rapid oxidation of
As(III) in the systems was caused by TiO2 photocatalysis
(Fig. 2).
0
20
40
60
80
100
4 6
% A
s re
mov
al
As V
As III, air-lightAs III, N2-dark
As III, air-dark
875
Fig. 3. Removal of arsenic as a function of equilibrium pH in a su
As ¼ 13.4mM, equilibrium time ¼ 22 h.
3.3. Effect of pH on as adsorption
Fig. 3 illustrated the effect of pH on As(V) and As(III)
removal under different conditions at an arsenic to TiO2ratio of 67mmol-As/g–TiO2 and an equilibrium time of22 h. In a pH range between 4 and 13 arsenic adsorption
in As(V) and As(III)-air–light systems was similar due to
nearly complete oxidation of As(III) to As(V) in the
presence of dissolved oxygen and light. When pH was
below 10, more than 95% of the arsenic was removed in
both systems. A steep decrease in sorption occurred
when the pH increased from 10 to 13. Similar As(V)
adsorption behavior by P25 (Lee and Choi, 2002; Dutta
et al., 2004) and by ferric hydroxides (Meng et al., 2000)
was observed. Adsorption of strong acid anions by
metal oxides and hydroxides typically decreases with
increasing pH (Stumm, 1992). The adsorption of arsenic
acid is through the formation of surface complexa-
tion reaction: Ti-OH+H3AsO4 ¼ Ti-HnAsO4(3�n�1)�+
H2O+(2�n)H+, where n ¼ 0, 1, 2. The reaction
indicates that As(V) adsorption increases with decreas-
ing proton concentration or increasing pH. In addition,
the surface potential of metal oxides becomes more
negative when pH is greater than the pHpzc, which does
not favor the adsorption of anions.
As(III) in the N2–dark and air–dark systems had a
maximum sorption at a pH of approximately 9, which is
characteristic of As(III) adsorption (Meng et al., 2000).
Adsorption of weak acids by metal oxides usually
reaches a maximum at pH values similar to pka1 of the
acid (Stumm, 1992). The pka1 of arsenious acid is 9.2.
9 10 11 12 13pH
spension containing 0.2 g of TiO2/L and 0.04M NaCl. Initial
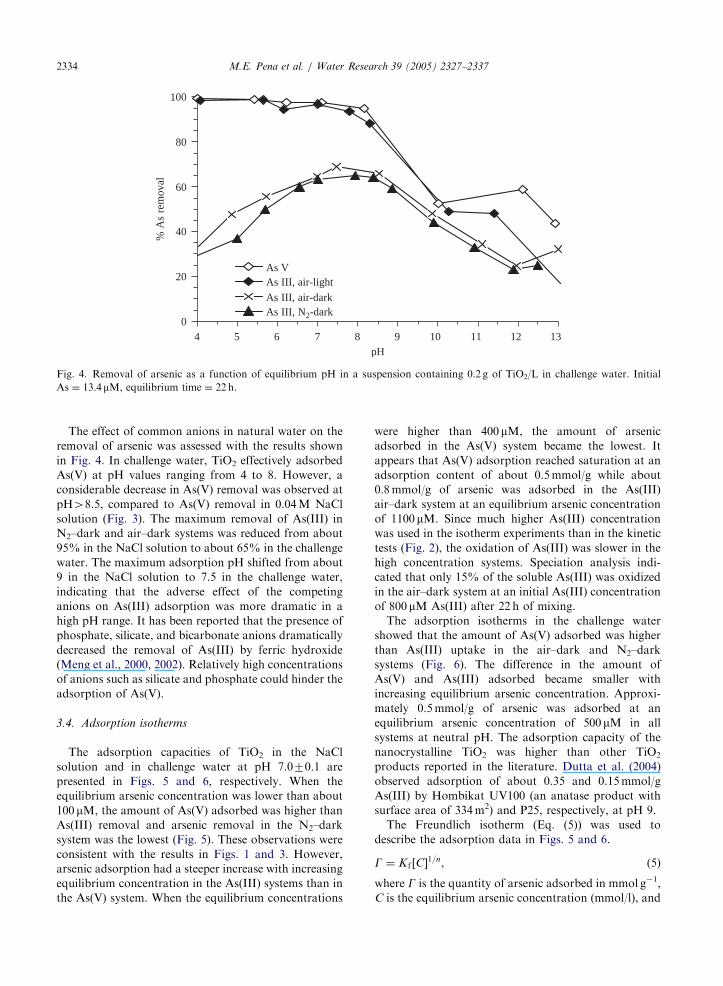
ARTICLE IN PRESS
0
20
40
60
80
100
4 5 6 7 8 9 10 11 12 13pH
% A
s re
mov
al
As VAs III, air-light
As III, air-darkAs III, N2-dark
Fig. 4. Removal of arsenic as a function of equilibrium pH in a suspension containing 0.2 g of TiO2/L in challenge water. Initial
As ¼ 13.4mM, equilibrium time ¼ 22 h.
M.E. Pena et al. / Water Research 39 (2005) 2327–23372334
The effect of common anions in natural water on the
removal of arsenic was assessed with the results shown
in Fig. 4. In challenge water, TiO2 effectively adsorbed
As(V) at pH values ranging from 4 to 8. However, a
considerable decrease in As(V) removal was observed at
pH48.5, compared to As(V) removal in 0.04M NaCl
solution (Fig. 3). The maximum removal of As(III) in
N2–dark and air–dark systems was reduced from about
95% in the NaCl solution to about 65% in the challenge
water. The maximum adsorption pH shifted from about
9 in the NaCl solution to 7.5 in the challenge water,
indicating that the adverse effect of the competing
anions on As(III) adsorption was more dramatic in a
high pH range. It has been reported that the presence of
phosphate, silicate, and bicarbonate anions dramatically
decreased the removal of As(III) by ferric hydroxide
(Meng et al., 2000, 2002). Relatively high concentrations
of anions such as silicate and phosphate could hinder the
adsorption of As(V).
3.4. Adsorption isotherms
The adsorption capacities of TiO2 in the NaCl
solution and in challenge water at pH 7.070.1 arepresented in Figs. 5 and 6, respectively. When the
equilibrium arsenic concentration was lower than about
100 mM, the amount of As(V) adsorbed was higher thanAs(III) removal and arsenic removal in the N2–dark
system was the lowest (Fig. 5). These observations were
consistent with the results in Figs. 1 and 3. However,
arsenic adsorption had a steeper increase with increasing
equilibrium concentration in the As(III) systems than in
the As(V) system. When the equilibrium concentrations
were higher than 400mM, the amount of arsenicadsorbed in the As(V) system became the lowest. It
appears that As(V) adsorption reached saturation at an
adsorption content of about 0.5mmol/g while about
0.8mmol/g of arsenic was adsorbed in the As(III)
air–dark system at an equilibrium arsenic concentration
of 1100 mM. Since much higher As(III) concentrationwas used in the isotherm experiments than in the kinetic
tests (Fig. 2), the oxidation of As(III) was slower in the
high concentration systems. Speciation analysis indi-
cated that only 15% of the soluble As(III) was oxidized
in the air–dark system at an initial As(III) concentration
of 800 mM As(III) after 22 h of mixing.
The adsorption isotherms in the challenge water
showed that the amount of As(V) adsorbed was higher
than As(III) uptake in the air–dark and N2–dark
systems (Fig. 6). The difference in the amount of
As(V) and As(III) adsorbed became smaller with
increasing equilibrium arsenic concentration. Approxi-
mately 0.5mmol/g of arsenic was adsorbed at an
equilibrium arsenic concentration of 500 mM in all
systems at neutral pH. The adsorption capacity of the
nanocrystalline TiO2 was higher than other TiO2products reported in the literature. Dutta et al. (2004)
observed adsorption of about 0.35 and 0.15mmol/g
As(III) by Hombikat UV100 (an anatase product with
surface area of 334m2) and P25, respectively, at pH 9.
The Freundlich isotherm (Eq. (5)) was used to
describe the adsorption data in Figs. 5 and 6.
G ¼ K f ½C1=n, (5)
where G is the quantity of arsenic adsorbed in mmol g�1,C is the equilibrium arsenic concentration (mmol/l), and

ARTICLE IN PRESS
Equilibrium As (µmol/L)0 100 200 300 400 500 600 700
As
adso
rbed
(m
mol
/g)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8As V
As III, air-light
As III, air-dark
As III, N2-dark
As V, GFO
As III, air-dark, GFO
Fig. 6. Arsenic adsorption isotherms in a 1.0 TiO2 and challenge water at equilibrium pH 7.070.1, equilibrium time ¼ 22 h. The lineswere model calculations.
Equilibrium As (µmol/L)0 200 400 600 800 1000 1200
As
adso
rbed
(m
mol
/g)
0.0
0.2
0.4
0.6
0.8
1.0As VAs III, air-lightAs III, air-dark
As III, N2-dark
0 20 40 60 80 1000.0
0.1
0.2
0.3
0.4
Fig. 5. Arsenic adsorption isotherms in a 1.0 TiO2 and 0.04M NaCl suspension at equilibrium pH 7.070.1, equilibrium time ¼ 22 h.The lines were model calculations.
M.E. Pena et al. / Water Research 39 (2005) 2327–2337 2335
Kf and n are constants. The optimal constants in Table 3
were determined by fitting model-calculated values to
the experimental data. As(III) and As(V) adsorption on
TiO2 were found to obey the Freundlich isotherm in
NaCl solution and challenge water (Figs. 5 and 6).
Arsenic removal by a common iron-based adsorbent,
GFO (Apyron Technologies Inc, Georgia) was also
tested to compare with the adsorption capacity of the
TiO2 in Fig. 6. The GFO product was in granular form
with particle size between 0.5 and 10mm. In order to

ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–23372336
compare with the TiO2 powder, the adsorbent was
pulverized to pass 60 mesh standard sieve (0.25mm
opening) and used in the adsorption tests. The BET
surface area of the GFO powder was 138m2/g. The
isotherms in Fig. 6 indicated that the TiO2 had a higher
adsorption capacity for As(III) and As(V) than the GFO
powder. At an equilibrium arsenic concentration of
600mmol/L, approximately 0.3mmol/g of arsenic was
adsorbed by the GFO and more than 0.5mmol/g of
arsenic was adsorbed by TiO2.
4. Conclusions
Adsorption of As(V) and As(III) by nanocrystalline
TiO2 reached equilibrium within 4 h and the adsorption
followed pseudo-second-order kinetics. Gradual oxida-
tion of As(III) to As(V) in the air–light system resulted
in a slow removal rate which could be described by the
Elovich equation. The TiO2 caused rapid photocatalytic
oxidation of As(III) to As(V) in the presence of oxygen
in sunlight. The adsorbent effectively removed As(V) in
a pH range of less than 8 and had a maximum
adsorption for As(III) at pH of approximately 7.5 in
the challenge water. The presence of silicate, carbonate,
and phosphate had a moderate effect on the adsorption
of As(V) and As(III) at pH 7. The nanocrystalline TiO2was an effective adsorbent for As(V) and As(III).
References
Berg, M., Tran, H.C., Nguyen, T.C., Pham, H.V., Schertenleib,
R., Giger, W., 2001. Arsenic contamination of groundwater
and drinking water in Vietnam: A human health threat.
Environ. Sci. Technol. 35 (13), 2621–2626.
Bissen, M., Vieillard-Baron, M.M., Schindelin, A.J., Frimmel,
F.H., 2001. TiO2-catalyzed photooxidation of arsenite(III)
to arsenate(V) in aqueous samples. Chemosphere 44,
751–757.
Chen, D.W., Ray, A.K., 1998. Photodegradation kinetics of 4-
nitrophenol in TiO2 suspension. Water Res. 32 (11),
3223–3234.
Cheung, C.W., Porter, J.F., Mckay, G., 2001. Sorption kinetics
analysis for the removal of cadmium ions from effluents
using bone char. Water Res. 35, 605–612.
Chien, S.H., Clayton, W.R., 1980. Application of Elovich
equation to the kinetics of phosphate release and sorption in
soils. Soil Sci. Soc. Am. J. 44, 265–268.
Cullen, W.R., Reimer, K.J., 1989. Arsenic speciation in the
environment. Chem. Rev. 89, 713–764.
Davis, A., Ruby, M.V., Bloom, M., Schoof, R., Freeman, G.,
Bergstrom, P.D., 1996. Mineralogic constraints on the
biovailabililty of arsenic in smelter-impacted soils. Environ.
Sci. Technol. 30, 392–399.
Driehaus, W., Jekel, M., Hildebrandt, U., 1998. Granular ferric
hydroxide—a new adsorbent for the removal of arsenic
from natural water. J. Water SRT–Aqua 47, 30–35.
Dutta, P.K., Ray, A.K., Sharma, V.K., Millero, F.J., 2004.
Adsorption of arsenate and arsenite on titanium dioxide
suspensions. J. Colloid Interface Sci. 278, 270–275.
Dutta, P.K., Pehkonen, S.O., Sharma, V.K., Ray, A.K., 2005.
Photocatalytic oxidation of Arsenic (III): evidence of
hydroxyl radicals. Environ. Sci. Technol. 39 (6), 1827–1834.
Emett, M.T., Khoe, G.H., 2001. Photochemical oxidation of
arsenic by oxygen and iron in acidic solutions. Water Res.
35 (3), 649–656.
Elkhabit, E.A., Bennett, O.L., Wright, R.J., 1984. Kinetics of
arsenate sorption in soils. Soil Sci. Soc. Am. J. 48, 758.
Ferguson, M.A., Hoffmann, M.R., Hering, J.G., 2005. TiO2-
photocatalyzed As(III) oxidation in aqueous suspensions:
reaction kinetics and effects of adsorption. Environ. Sci.
Technol. ASAP Article.
Gregor, J., 2001. Arsenic removal during conventional alumi-
num-base drinking water treatment. Water Res. 35 (7),
1659–1664.
Gruebel, K.A., Davis, J.A., Leckie, J.O., 1995. kinetics of
oxidation of selenite to selenate in the presence of oxygen,
titania, and light. Environ. Sci. Technol. 29, 586–594.
Gulledge, J.H., O’connor, J.T., 1973. Removal of arsenic(V)
from water by adsorption on aluminum and ferric hydro-
xides. J. Am. Water Works Assoc. 65 (8), 548–552.
Hatch, G.L., 2002. Meeting the new arsenic standard with a
new iron-based adsorbent media: POU applications. Water
Conditioning and Purification, pp. 52–56.
Ho, Y.S., McKay, G.A., 1998. Comparison of chemisorption
kinetics models applied to pollutant removal on various
sorbents. Inst. Chem. Eng. 76b, 332–340.
Hug, S.J., Leupin, O., 2003. Iron-catalyzed oxidation of arsenic
(III) by oxygen and by hydrogen peroxide: pH-dependent
formation of oxidants in the Fenton reaction. Environ. Sci.
Technol. 37 (12), 2734–2742.
Hug, S.J., Canonica, L., Wegelin, M., Gechter, D., Von
Gunten, U.V., 2001. Solar oxidation and removal of arsenic
at circumneutral pH in iron containing waters. Environ. Sci.
Technol. 35 (10), 2114–2121.
Kim, M.J., Nriagu, J., 2000. Oxidation of arsenite in ground-
water using ozone and oxygen. Sci. Total Environ. 247,
71–79.
Klaning, U.K., Bielski, B.H.J., Sehested, K., 1989. Arsenic
(IV). A pulse-radiolysis study. Inorg. Chem. 28 (14),
2717–2724.
Kocar, B.D., Inskeep, W.P., 2003. Photochemical oxidation of
As(III) in ferrioxalate solutions. Environ. Sci. Technol. 37,
1581–1588.
Korte, N.E., Quintus, F., 1991. A review of arsenic (III) in
groundwater. Crit. Rev. Environ. Control 21 (1), 1–39.
Lee, H., Choi, W., 2002. Photocatalytic oxidation of arsenite in
TiO2 suspension: kinetics and mechanisms. Environ. Sci.
Technol. 36, 3872–3878.
Liang, T., Tsai, J.Y., 1995. Sorption kinetics of cesium on
natural mordenite. Appl. Radiat. Isot. 46, 7–12.
Low, G.K.C., McEvoy, S.R., Matthews, R., 1991. Formation
of nitrate and ammonium ions in titanium dioxide mediated
photocatalytic degradation of organic compounds contain-
ing nitrogen atoms. Environ. Sci. Technol. 25, 460–467.
McNeill, L.S., Edwards, M., 1997. Predicting As removal
during metal hydroxide precipitation. J. Am. Water Works
Assoc. 89 (1), 75–86.

ARTICLE IN PRESSM.E. Pena et al. / Water Research 39 (2005) 2327–2337 2337
Meng, X.G., Bang, S.B., Korfiatis, G.P., 2000. Effect of silicate,
sulfate and carbonate on arsenic removal by ferric hydro-
xide. Water Res. 34, 1255–1261.
Meng, X.G., Korfiatis, G.P., Chuanyong, J., Christodoulatos,
C., 2001. Redox transformations of arsenic and iron in
water treatment sludge during aging and TCLP extraction.
Environ. Sci. Technol. 35, 3476–3481.
Meng, X.G., Korfiatis, G.P., Bang, S.B., Bang, K.W., 2002.
Combined effects of anions on arsenic removal by iron
hydroxides. Toxicol. Lett. 133, 103–111.
Meng, X.G., Dadachov, M., Korfiatis, G.P., Christodoulatos, C.,
2003. Method of preparing a surface-activated titanium oxide
product and of using the same in water treatment processes.
Patent pending, application number 20030155302.
Pettine, M., Millero, F.J., 2000. Effect of metals on the
oxidation of As(III) with H2O2. Mar. Chem. 70, 223–234.
Pettine, M., Campanella, L., Millero, F.J., 1999. Arsenite
oxidation by H2O2 in aqueous solutions. Geochim. Cosmo-
chim. 63 (18), 2727–2733.
Picheler, T., Veizer, J., Hall, G.E.M., 1999. Natural impute of
arsenic into a coral-reef ecosystem by hydrothermal fluids
and its removal by Fe(III) oxyhydroxides. Environ. Sci.
Technol. 33, 1373–1378.
Raven, K.P., Jain, A., Loeppert, R.H., 1998. Arsenite and
arsenate adsorption on ferrihydrite: kinetics, equilibrium
and adsorption envelopes. Environ. Sci. Technol. 32,
344–349.
Ryu, J., Choi, W., 2004. Effects of TiO2 surface modi-
fications on photocatalytic oxidation of arsenite: the
role of superoxides. Environ. Sci. Technol. 38 (10),
2928–2933.
Sag, Y., Aktay, Y., 2002. Kinetic studies on sorption of Cr(VI)
and Cu(II) ions by chitin, chitosan and rhizopus arrhizus.
Biochem. Eng. J. 12, 143–153.
Sparks, D.L., 1989. Kinetics of Soil Chemical Processes.
Academic Press, New York.
Stumm, W., 1992. Chemistry of the Solid–Water Interface.
Wiley-Interscience, New York, pp. 23–26.
USEPA, 2001. Laboratory study on the oxidation of arsenic III
to arsenic V, EPA/600/R-01/021, March 2001.
Vaishya, R.C., Gupta, S.K., 2003. Arsenic removal from
groundwater by iron impregnated sand. J. Environ. Eng.
129 (1), 89–92.
Wong, C.C., Chu, W., 2003. The direct photolysis and
photocatalytic degradation of alachlor at different TiO2and UV sources. Chemosphere 50 (8), 981–987.
Yang, H., Lin, W.Y., Rajeshwar, K., 1999. Homogeneous and
heterogeneous photocatalytic reactions involving As(III)
and As(V) species in aqueous media. J. Photochem.
Photobiol. 123, 137–143.
Copyright © 2022 FDOKUMEN




















