
Adenylate Cyclase Toxin Subverts Phagocyte Function by RhoA Inhibition and Unproductive Ruffling
12
of January 14, 2016. This information is current as Unproductive Ruffling Function by RhoA Inhibition and Adenylate Cyclase Toxin Subverts Phagocyte and Peter Sebo Jana Vojtova, Irena Linhartova, Oldrich Benada, Ingo Just Jana Kamanova, Olga Kofronova, Jiri Masin, Harald Genth, http://www.jimmunol.org/content/181/8/5587 doi: 10.4049/jimmunol.181.8.5587 2008; 181:5587-5597; ; J Immunol References http://www.jimmunol.org/content/181/8/5587.full#ref-list-1 , 42 of which you can access for free at: cites 55 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2008 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on January 14, 2016 http://www.jimmunol.org/ Downloaded from by guest on January 14, 2016 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Adenylate Cyclase Toxin Subverts Phagocyte Function by RhoA Inhibition and Unproductive Ruffling

of January 14, 2016.This information is current as
Unproductive RufflingFunction by RhoA Inhibition and Adenylate Cyclase Toxin Subverts Phagocyte
and Peter SeboJana Vojtova, Irena Linhartova, Oldrich Benada, Ingo Just Jana Kamanova, Olga Kofronova, Jiri Masin, Harald Genth,
http://www.jimmunol.org/content/181/8/5587doi: 10.4049/jimmunol.181.8.5587
2008; 181:5587-5597; ;J Immunol
Referenceshttp://www.jimmunol.org/content/181/8/5587.full#ref-list-1
, 42 of which you can access for free at: cites 55 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on January 14, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Adenylate Cyclase Toxin Subverts Phagocyte Function byRhoA Inhibition and Unproductive Ruffling1
Jana Kamanova,* Olga Kofronova,* Jiri Masin,* Harald Genth,† Jana Vojtova,*Irena Linhartova,* Oldrich Benada,* Ingo Just,† and Peter Sebo2*
Adenylate cyclase toxin (CyaA or ACT) is a key virulence factor of pathogenic Bordetellae. It penetrates phagocytes expressing the�M�2 integrin (CD11b/CD18, Mac-1 or CR3) and paralyzes their bactericidal capacities by uncontrolled conversion of ATP intoa key signaling molecule, cAMP. Using pull-down activity assays and transfections with mutant Rho family GTPases, we show thatcAMP signaling of CyaA causes transient and selective inactivation of RhoA in mouse macrophages in the absence of detectableactivation of Rac1, Rac2, or RhoG. This CyaA/cAMP-induced drop of RhoA activity yielded dephosphorylation of the actinfilament severing protein cofilin and massive actin cytoskeleton rearrangements, which were paralleled by rapidly manifestedmacrophage ruffling and a rapid and unexpected loss of macropinocytic fluid phase uptake. As shown in this study for the firsttime, CyaA/cAMP signaling further caused a rapid and near-complete block of complement-mediated phagocytosis. Induction ofunproductive membrane ruffling, hence, represents a novel sophisticated mechanism of down-modulation of bactericidal activitiesof macrophages and a new paradigm for action of bacterial toxins that hijack host cell signaling by manipulating cellular cAMPlevels. The Journal of Immunology, 2008, 181: 5587–5597.
B ordetella species pathogenic to mammals secrete an ade-nylate cyclase toxin (CyaA, ACT or AC-Hly) that targetshost myeloid phagocytic cells expressing the �M�2 inte-
grin receptor (CD11b/CD18, CR3 or Mac-1), such as macro-phages, neutrophils, and dendritic cells (DC).3 Following CD11b/CD18 binding, the toxin penetrates the cytoplasmic membrane ofphagocytes and delivers into their cytosol its adenylate cyclase(AC) enzyme domain. There the AC is activated by binding ofintracellular calmodulin and subverts cellular signaling by unreg-ulated conversion of ATP to cAMP, a key second messenger mol-ecule (see Ref. 1 for review). Rapid elevation of intracellularcAMP concentration by CyaA then yields suppression of bacteri-cidal functions of phagocytes, such as chemotaxis, FcR-mediatedphagocytosis, or superoxide production (2–5) and eventually pro-vokes apoptosis (6). This capacity of CyaA appears to account forthe key role of CyaA in virulence of Bordetella species in mam-
mals, enabling the bacteria to escape destruction by the first-linedefense of innate immune system (7, 8). Recent work further sug-gests that cAMP signaling of CyaA may play an even more ver-satile role in fooling the host defense by promoting incomplete oraberrant maturation of DCs into a so-called semimature state (9–11). This may shape the induction of adaptive immune responsetoward tolerance of the pathogen.
Although suppressive effects of CyaA action on bactericidalfunctions of myeloid phagocytes were repeatedly reported, littleattention was paid to the underlying mechanisms of toxin-medi-ated cAMP signaling and to the corresponding downstream effec-tors, which remain to be identified. In general, the effects of cAMPsignaling are rather complex, cell-type dependent, and quite pleio-tropic, being mediated through activation of protein kinase A(PKA), the guanine exchange proteins directly activated by cAMP(Epac-1 and Epac-2) and the cAMP-activated channels, respec-tively. In particular, the spatio-temporal control of cAMP signalingappears to be crucial for differential regulation of cellular targetsinvolved in various cAMP signaling cascades (see Ref. 12 for re-view). For example, an asymmetric distribution of active PKA isrequired for establishment of polarity and migration of neutrophils(13). PKA was further shown to regulate functional plasticity ofDCs, via inhibition of Src-like kinases (14) and to control thesynthesis and release of cytokines in macrophages (15). cAMPsignaling was also demonstrated to suppress oxidative burst andFcR- and CR3-mediated phagocytosis by monocytes and macro-phages (15–17). Among other cAMP effects, the actin cytoskeletonhomeostasis was also found to be perturbed by elevated cAMPlevels. cAMP was, indeed, shown to inhibit actin assembly onphagosomes of macrophages (18) and microfilament assembly inneutrophils (19). cAMP-induced activation of PKA caused disso-lution of stress fibers and yielded a cell rounding phenotype offibroblasts (20), or a stellate morphology of human neuroblastomacells (21).
The key regulators of actin cytoskeletal dynamics are the Rhofamily GTPases. These signaling proteins act as molecularswitches, being inactive when bound to GDP and becoming
*Cellular and Molecular Microbiology Division, Institute of Microbiology of theAcademy of Sciences of the Czech Republic, Prague 4, Czech Republic and †Instituteof Toxicology, Hannover Medical School, Hannover, Germany
Received for publication January 25, 2008. Accepted for publication August 18, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the European Union 6th FP Contract LSHB-CT-2003-503582 THERAVAC (to J.V.), Deutsche Forschungs-gemein-schaft Grant SPP1150GE1247/1-3 (to I.J. and H.G.), the Institutional Research Concept MSM0021620858(to J.M.), and Grants 1M0506 (to J.K.) and 2B06161 (to I.L.) from the Ministry ofEducation, Youth, and Sports and concept AV0Z50200510 and the Grant No. GA310/08/0447 (to P.S.) of the National Science Foundation of the Czech Republic.2 Address correspondence and reprint requests to Dr. Peter Sebo, Institute of Micro-biology AS CR, v.v.i., Videnska 1083, Prague 4, Czech Republic. E-mail address:[email protected] Abbreviations used in this paper: DC, dendritic cell; AC, adenylate cyclase enzymedomain; PKA, protein kinase A; GEF, guanine nucleotide exchange factor; CR3,complement receptor 3; db-cAMP, N6,2�-O-dibutyryladenosine 3�,5�-cyclic mono-phosphate; LY, lucifer yellow; BMM, bone marrow macrophage-like cells; CyaA, B.pertussis adenylate cyclase; CyaA-AC�, enzymatically inactive adenylate cyclasetoxoid; ROK, Rho kinase.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

activated upon a GDP/GTP exchange mediated by guanine nucle-otide exchange factors (GEFs). RhoA, Rac1, and Cdc42 are themost extensively characterized members of this protein family,where RhoA regulates formation of contractile actin/myosin stressfibers in fibroblasts and actin cables in macrophages, Rac is in-volved in formation of lamellipodia and membrane ruffles, andCdc42 governs formation of filopodia and microspikes in both fi-broblasts, as well as macrophages (22). The Rho GTPases exertcrucial mechanistic functions in innate and adaptive immunity andhave a pivotal role in the biology of infection (see Ref. 23 forreview). For example, FcR-mediated phagocytosis is initiated byengagement of the Ig receptor FcR and depends on the function ofCdc42 and Rac. In turn, the other phagocytic mechanism mediatedby complement receptor 3 (CR3) engagement depends on RhoA(24). Not surprisingly then, targeting of Rho GTPases representsan important strategy used by bacterial pathogens in subversion ofhost cell functions (23).
In this study, we show for the first time that cAMP signalingeffects of low doses of CyaA toxin cause rapid and transient in-activation of RhoA, massive actin cytoskeleton rearrangements,nonproductive membrane ruffling, inhibition of macropinocyticuptake and most importantly, an instantaneous and near-completeinhibition of CR3-mediated phagocytosis in macrophages.
Materials and MethodsReagents
In brief, 3-isobutyl-1-methylxanthin, N6,2�-O-dibutyryladenosine 3�,5�-cy-clic monophosphate (db-cAMP), PMA, wortmannin, and C5-deficient se-rum were obtained from Sigma-Aldrich and Y-27632 was obtained fromCalbiochem. The Abs used in this study were anti-Rac1 (610650, BDTransduction Laboratories), anti-Rac2 (07-604, Upstate), anti-RhoG (sc-1007, Santa Cruz Biotechnology), anti-RhoA (2117, Cell Signaling Tech-nology), anti-NTAL (NAP-07, gift of P. Angelisova, Institute of MolecularGenetics, Prague, Czech Republic), anti-cofilin (3318, Cell SignalingTechnology), anti-phospho-cofilin (ser3, 3311, Cell Signaling Technol-ogy), and anti-sheep RBC IgM Abs (CL9000-M, Cedarlane Laboratories).
Cell cultures, growth conditions, and handling of cells
J774A.1 mouse macrophages (ATCC TIB 67), RAW 264.7 mouse mac-rophages (ATCC TIB 71) and bone marrow macrophage-like cells wereused. The J774A.1 and RAW 264.7 cells were maintained in RPMI 1640medium containing 10% FCS (FCS, Life Technologies) and antibiotic an-timycotic solution (0.1 mg/ml streptomycin, 1000 U/ml penicillin and 0.25�g/ml amphotericin, Sigma-Aldrich). Bone marrow macrophage-like cellswere obtained from femoral and tibial bones of 6- to 8-wk-old femaleC57BL/6 mice by cultivating bone marrow-derived cells in RPMI 1640medium supplemented with 10% FCS, 20 ng/ml GM-CSF and antibioticantimycotic solution for 7 days, as previously described by (25). At day 7,the nonadherent cells were removed, whereas adherent cells were physi-cally scrapped and washed before use. The expression of surface Ags(% positive of adherent cell population) was 47% for F4/80 (FITC-anti-mouse-F4/80 Ab, clone BM8, eBioscience), 20% for Gr-1 (FITC-anti-mouse-Gr-1 Ab, clone RB6-8C5, eBioscience), 92% for CD11c (APC-anti-mouse-CD11c Ab, clone N418, eBioscience), and 99% for CD11b(PE-anti-mouse-CD11b Ab, clone M1/70, BD Pharmingen) as deter-mined using a FACS LSR II instrument (BD Biosciences) and gating forlive cells. During all experiments the RPMI 1640 medium used forcultivating of cells was replaced by DMEM (1.9 mM Ca2�) withoutFCS to avoid uncontrollable chelation of calcium ions by the phosphateions contained in RPMI 1640 medium, as calcium is required for CyaAactivity. 0.1 mM 3-isobutyl-1-methylxanthin was systematically addedto incubation medium to inhibit cellular phosphodiesterases when ex-posing cells to db-cAMP.
Production, purification, and handling of CyaA
The wild-type CyaA and CyaA-AC� (CyaA mutant devoid of adenylatecyclase activity due to insertion of a GlyPhe dipeptide into the ATP bind-ing site of the AC domain; Ref. 26) were produced upon IPTG induction(1 mM) in liquid cultures in the presence of the activating protein CyaC byusing the Escherichia coli strain XL-1 Blue (Stratagene) transformed withthe appropriate constructs derived from pT7CACT1 (27), and purified as
described (28). In the final step, the proteins were eluted with 8 M urea, 2mM EDTA, 50 mM Tris-HCl (pH 8.0), and stored at �20°C. The endo-toxin content in the samples was determined by the LAL assay (QCL-1000;Cambrex), according to the manufacturer’s instructions and was below 50EU/mg of purified protein. The CyaA proteins were prediluted to 100 or1000 times the final indicated concentration using 50 mM Tris-HCl (pH8.0), 8 M urea, and 0.2 mM CaCl2 buffer. Before addition ofCyaA to cells,it was diluted in DMEM without FCS and mixed with cell suspension toreach the final indicated toxin concentration. This resulted in a final ureaconcentration of 80 mM, or less, which had no effect on cell physiology ormorphology whatsoever, as verified by appropriate controls with cells in-cubated at identical urea concentrations.
Determination of intracellular cAMP, phagocytic, andmacropinocytic activities
For determination of intracellular cAMP, J774A.1 cells (2 � 105 per well)were incubated with 10 or 100 ng/ml CyaA in DMEM without FCS forindicated time intervals before the reaction was stopped by addition of0.2% Tween 20 in 50 mM HCl and the cAMP concentration was deter-mined by immunoassay as described elsewhere (29).
FcR-mediated phagocytosis was assessed using paraformaldehyde-inac-tivated, FITC-labeled E. coli BioParticles (E-2861, Molecular Probes) op-sonized with E. coli-specific rabbit polyclonal IgG (E-2870, MolecularProbes) according to the manufacturer’s instructions. Opsonized IgG-FITC-E. coli were added to cells at a ratio of �10 particles per macrophagecell. Upon incubation in the dark at 37°C for 30 min, the uningested par-ticles were washed-out with PBS and fluorescence of extracellularly at-tached IgG-FITC-E. coli particles was quenched by treatment with trypanblue (300 �g/ml, Molecular Probes) for 1 min. Macrophage cells wererinsed in PBS and the fluorescence of phagocytosed E. coli was determinedupon J774A.1 cell lysis in 50 mM Tris-HCl (pH 9.0), 150 mM NaCl, 5 mMEDTA and 0.5% Nonidet P-40 using a microplate reader (483ex/525em,Safire2, Schoeller Instruments). Data were corrected for the fluorescence ofunquenched, extracellular bacteria, determined as fluorescence of cells in-cubated with IgG-FITC-E. coli at 4°C.
CR3-mediated phagocytosis was assessed as internalization of C3bi-opsonized sheep RBCs by J774A.1 cells as described (24). In brief, 5 �108 of RBCs/ml were incubated for 30 min at 37°C in the presence ofanti-sheep RBCs IgM Abs (1/250). RBCs were washed and IgM-coatedRBCs were opsonized by incubation in 10% (v/v) C5-deficient humanserum for 30 min at 37°C. Under these conditions, C3b is rapidly fixed toIgM-coated RBCs and is completely converted into C3bi (30). Opsoniza-tion with C3bi was checked by flow cytometry following incubation withgoat anti-C3 (Sigma-Aldrich) and staining by FITC-conjugated anti-goatIgG (Sigma-Aldrich). Before phagocytic challenge, the J774.A1 cells wereinduced for efficient CR3-mediated phagocytosis by 150 ng/ml PMA for 15min at 37°C, as previously described (31). iC3b-RBCs were added to cellsat a ratio of �25 RBCs per J774A.1 cell and incubated for 30 min at 37°Cto allow phagocytosis. Unbound RBCs were washed away with ice-coldPBS, whereas adherent but nonphagocytosed RBCs were hypotonicallylysed by a 30 s incubation pulse with distilled H2O, followed by wash inPBS. This lysis-wash procedure was repeated two times for complete re-moval of noninternalized RBCs. The quantity of phagocytosed iC3b-RBCswas next determined by measuring the amount of fluorene blue formedfrom 2,7-diaminofluorene (Sigma-Aldrich) by the pseudoperoxidase activ-ity of hemoglobin released from RBCs upon lysis of J774A.1 cells (32).Absorbance was read at 610 nm using a microplate reader (Safire2, Schoe-ller Instruments) and corrected against a blank of lysed J774A.1 cells in-cubated with 2,7-diaminofluorene.
To determine the level of fluid-phase uptake (macropinocytosis),J774A.1 cells were incubated with either lucifer yellow (LY, 500 �g/ml,Molecular Probes) or FITC-dextran (1 mg/ml, Mw 4 kDa, Sigma-Aldrich)for 30 min at 37°C. LY or FITC-dextran uptake was stopped by threewashes with ice-cold PBS and fluorescence of cell surface-bound LY orFITC-dextran was quenched in trypan blue as above. Cells were rinsed inPBS and lysed as above. The fluorescence of internalized LY or FITC-dextran was then read in a microplate reader (405ex/580em for LY or 483ex/525em for FITC-dextran, Safire2, Schoeller Instruments) and data were cor-rected for the fluorescence of unquenched, extracellular LY, or FITC-dextran (fluorescence of cells incubated with LY or FITC-dextran at 4°C).
Transfection experiments
Transfections of pEGFP-C1 constructs bearing cDNA of Rac1, Cdc42,RhoA, and their mutants Rac1-T17N, Cdc42-T17N and RhoA-G14V, ormock pEGFP-C1 construct, were performed at 50% confluence of RAW264.7 macrophages using the FuGENE-6 transfection reagent (Roche) ac-cording to the manufacturer’s instructions. Imaging of actin cytoskeleton
5588 cAMP SIGNALING OF CyaA SUBVERTS PHAGOCYTE FUNCTION
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from
and determination of cell surface level of CD11b/CD18 were performed12 h after transfection. Expression of CD11b/CD18 on transfected cellswas analyzed by flow cytometry after incubation with PE-conjugated anti-CD11b Ab (clone M1/70, BD Pharmingen) using a FACS LSR II instru-ment (BD Biosciences) and gating for live GFP-positive cells. Level ofCD11b/CD18 was deduced from the mean fluorescence intensity and ex-pressed as the percentage of the CD11b level of control (nontransfected)cells.
Microscopy of J774A.1, RAW 264.7, and bone marrowmacrophage-like cells
For scanning electron microscopy, the cells were fixed with 3% glutaral-dehyde and washed with a cacodylate buffer. The samples were dehydratedin an alcohol series followed by absolute acetone, and dried in a critical-point device (Balzers 010). Finally, the samples were sputter-coated withgold in a Polaron sputter-coater (Series 11HD) and examined in anAquasem scanning electron microscope (Tescan) at 15 kV.
For fluorescence microscopy of the actin cytoskeleton, cells werewashed with PBS and fixed with 4% paraformaldehyde in PBS (20 min,room temperature). After three washes with PBS (3 � 5 min), the cellswere permeabilized with 0.1% Triton X-100 in PBS (5 min), and washedwith PBS (3 � 5 min). F-actin was stained with TRITC-conjugated phal-loidin (0.5 �g/ml, Sigma-Aldrich) in PBS supplemented with 2% BSAfor 30 min. Images were taken using an Olympus BX60 fluorescencemicroscope.
For microscopy analysis of macropinosome formation, J774A.1 cellswere treated as indicated in the presence of lysine-fixable FITC-dextran (1mg/ml, Mw 3 kDa, Molecular Probes). After two washes with PBS, cellswere fixed with 4% paraformaldehyde in PBS (20 min, RT) and examinedby Olympus BX60 fluorescence microscope.
Quantification of ruffling
Quantification of ruffling was performed according to Cox et al. (33). Ruf-fling was defined by the presence of F-actin-rich submembranous foldsusing fluorescence microscopy. The extent of ruffling of each cell wasscored using a scale of 0–2, where 0 indicates that no ruffles were present,1 indicates that ruffling was confined to one area of the cell only (�25% ofthe cell’s circumference), and 2 indicates that two or more discrete areas ofthe cell contained ruffles. The ruffling index was recorded as the sum of theruffling scores of 100 cells.
Affinity precipitation of cellular GTP-Rac1/Rac2, GTP-RhoG,and GTP-RhoA
GTP-bound active forms of Rac1/2, RhoG, and RhoA were detected usinga pull-down assay using Cdc42/Rac interactive-binding region of PAK(GST-CRIB), RhoG docking protein (GST-ELMO1), and RhoA interac-tive-binding region of rhotekin (GST-RBD), respectively. In total, 2 � 106
J774A.1 cells were treated with the indicated agent (CyaA, CyaA-AC�,db-cAMP) at 37°C for the indicated time, washed with ice-cold PBS andlysed for 5 min in ice-cold buffer containing 1% Nonidet P-40, 40 mMTris-HCl (pH 7.4), 120 mM NaCl, 4 mM MgCl2, 5 mM DTT, 1 mM PMSF,and Complete Mini protease inhibitors (EDTA free, Roche). Lysates wereclarified by centrifugation at 10 000 � g for 3 min at 4°C and incubated at4°C for 45 min with glutathione-Sepharose 4B beads (Amersham Bio-sciences) preloaded with the binding proteins fused to GST (GST-CRIB,GST-ELMO1 or GST-RBD, respectively). The beads were washed threetimes in lysis buffer. The bound GTP-Rac1/Rac2, GTP-RhoG, and GTP-RhoA proteins were eluted by boiling in Laemmli buffer for 5 min andamounts of pulled-down proteins were quantified by Western blotting us-ing specific Abs and chemilumiscent detection (see below). For positivecontrols, pull-downs were performed with lysates in which cellular Rac1/2,RhoG, and RhoA were preloaded with GTP�S, as a nonhydrolysable GTPanalog, as follows. Lysates were treated with 100 �M GTP�S in the pres-ence of 10 mM EDTA for 15 min at 30°C. The reaction was terminated andGTP�S was locked into the proteins by addition of 60 mM MgCl2 on ice.
Preparations of cell membranes for determination of RhoAcontent
For preparation of the fraction enriched in plasma membrane, 2 � 106
J774A.1 cells were washed with PBS and incubated in ice-cold hypotonicbuffer containing 20 mM HEPES (pH 7.5), 2 mM EDTA, and CompleteMini protease inhibitors. After 30 min of incubation on ice the cells werelysed by three cycles of freezing and thawing followed by three passagesthrough a 25-gauge needle. After pelleting of unlysed cells (10 min, 800 �g, 4°C), the membranes were collected by ultra-centrifugation (30 min, 100000 � g). Membrane pellet was resuspended in 1% Nonidet P-40, the
samples were mixed with Laemmli buffer, heated for 5 min at 95°C andanalyzed by Western blots.
Preparation of cell lysates for determination of phospho-cofilin
Two � 106 J774A.1 cells were washed with PBS and lysed in 150 �l ofice-cold lysis buffer containing 1% Nonidet P-40, 20 mM Tris-HCl (pH8.0), 100 mM NaCl, 10 mM EDTA, 10 mM Na4P2O7, 1 mM Na3VO4, 50mM NaF, 10 nM CalyculinA, and Complete Mini protease inhibitors perwell of a 6-well culture dish. After scraping, lysates were incubated on icefor 15 min and briefly vortexed. Samples were clarified by centrifugation(3 min, 10 000 � g) and analyzed by Western blot.
Western blot analysis
The proteins were separated by 15% SDS-PAGE, transferred onto nitro-cellulose membrane, and probed overnight with primary Abs, as indicatedin the figure legends and revealed by HRP-conjugated secondary Ab (di-lution 1/4,000, GE Healthcare) using the West Femto Maximum Sensitiv-ity Substrate (Pierce). Western blot signals were quantified using LAS-1000 (Luminiscence Analyzing System, Fuji) and AIDA 1000/1D ImageAnalyzer software, version 3.28 (Raytest Isotopenmessgeraete GmbH).
Statistical analysis
Significance of differences in values was assessed by Student’s t test.
ResultscAMP signaling of CyaA toxin rapidly haltscomplement-mediated phagocytosis
We first analyzed the effects of toxin action on phagocytosis inmodel mouse J774A.1 monocyte/macrophage-like cells (29). As
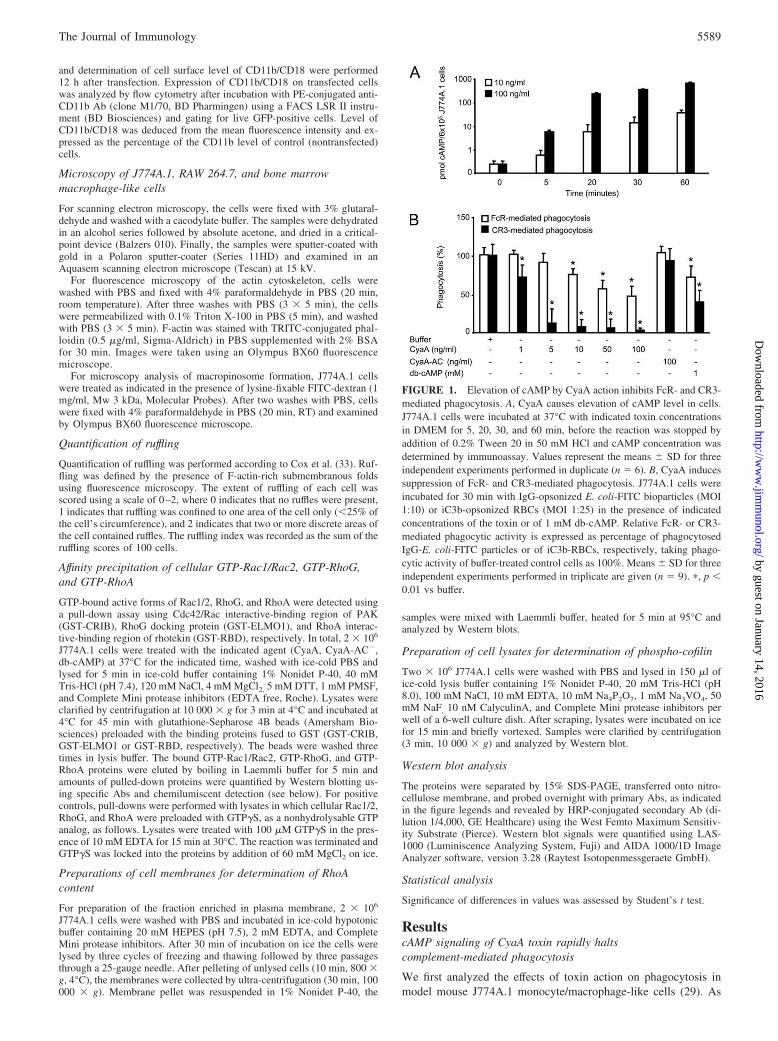
FIGURE 1. Elevation of cAMP by CyaA action inhibits FcR- and CR3-mediated phagocytosis. A, CyaA causes elevation of cAMP level in cells.J774A.1 cells were incubated at 37°C with indicated toxin concentrationsin DMEM for 5, 20, 30, and 60 min, before the reaction was stopped byaddition of 0.2% Tween 20 in 50 mM HCl and cAMP concentration wasdetermined by immunoassay. Values represent the means � SD for threeindependent experiments performed in duplicate (n � 6). B, CyaA inducessuppression of FcR- and CR3-mediated phagocytosis. J774A.1 cells wereincubated for 30 min with IgG-opsonized E. coli-FITC bioparticles (MOI1:10) or iC3b-opsonized RBCs (MOI 1:25) in the presence of indicatedconcentrations of the toxin or of 1 mM db-cAMP. Relative FcR- or CR3-mediated phagocytic activity is expressed as percentage of phagocytosedIgG-E. coli-FITC particles or of iC3b-RBCs, respectively, taking phago-cytic activity of buffer-treated control cells as 100%. Means � SD for threeindependent experiments performed in triplicate are given (n � 9). �, p �0.01 vs buffer.
5589The Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

previously reported, already at as low toxin concentrations as 10ng/ml, a rapid accumulation of supraphysiological concentrationsof cAMP in J774A.1 cells was observed, as shown in Fig. 1A. Atthis toxin concentration, a moderate but statistically significant im-pairment of FcR-mediated phagocytosis of IgG-opsonised E. colibioparticles by J774A.1 cells was seen and the extent of phago-cytosis inhibition further increased at higher toxin doses (Fig. 1B).
Importantly, as further shown in Fig. 1B, the impact of CyaAaction on CR3-mediated phagocytic capacity of macrophages wasmuch more pronounced. This pathway depends on opsonization ofpathogens by the C3bi component of complement and is mediatedby the CR3 (known also as �M�2 integrin, CD11b/CD18, or Mac-1). Intriguingly, CD11b/CD18 serves also as the cellular receptorof the CyaA toxin itself. However, the observed inhibitory effect ofCyaA on CR3-mediated phagocytosis at the used toxin dose wasclearly not due to competition of CyaA for the receptor with theC3bi-opsonized particles. No effect on CR3-mediated phagocyto-sis was, indeed, seen at 100 ng/ml of the enzymatically inactiveCyaA-AC� toxoid (Fig. 1B), that lacks the capacity to convert
ATP into cAMP, while being fully competent in receptor binding(34). In turn, a statistically significant inhibition of CR3-mediatedphagocytosis was observed already at 1 ng/ml CyaA and the effectof CyaA could be mimicked by exposing J774A.1 cells to 1 mMdb-cAMP, a membrane-permeable cAMP analog. Collectively,hence, these results unambiguously show that it was the capacityof CyaA to elevate cAMP levels in J774A.1 cells that accountedfor the inhibition of CR3-mediated phagocytosis by the toxin.
Phagocytosis inhibition is accompanied by actin cytoskeletonrearrangements and membrane ruffling
Because phagocytic functions depend on the coordinated rear-rangements of actin cytoskeleton, we examined the morphologicalconsequences of toxin treatment using two different murine mac-rophage cell lines (J774A.1 and RAW 264.7) and primary bonemarrow-derived macrophage-like cells (BMM), respectively. Asrevealed by scanning electron micrographs (upper panels) andTRITC-phalloidin staining of F-actin (lower panels) shown in Fig.2, exposure of all three macrophage types to CyaA resulted in
FIGURE 2. Elevation of cytosolic cAMP by CyaAinduces pronounced alterations of macrophage cy-toskeleton and morphology. Murine J774A.1 macro-phage cells, RAW 264.7 macrophage cells or BMM-like cells were treated as indicated. For examination ofcell morphology (M), cells were fixed with 3% glutar-aldehyde and examined by scanning electron micros-copy. For examination of cell cytoskeleton (C), cellswere fixed with 4% paraformaldehyde, stained for F-actin with TRITC-phalloidin and examined using anOlympus BX60 fluorescence microscope. Scale bar,10 �m. The shown micrographs are representative ofthree independent experiments from which the ruf-fling index (see Materials and Methods) was deter-mined and represents the mean � SD value (n � 3).�, p � 0.01 vs buffer.
5590 cAMP SIGNALING OF CyaA SUBVERTS PHAGOCYTE FUNCTION
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

massive formation of sheet-like cell membrane extensions com-monly referred as lamellipodia, or membrane ruffles. Moreover, inprimary BMM also disappearance of actin cables and podosomeswas observed to result from CyaA action. In turn, no ruffling wasobserved with cells exposed to the enzymatically inactive CyaA-AC� toxoid purified in the same way as CyaA, thus excluding thepossibility that cell ruffling might have been promoted by traces ofsome contaminating bacterial components present in the CyaA andCyaA-AC� preparations. Moreover, comparable membrane ruf-fling was also observed upon exposure of macrophage cells to 2mM db-cAMP, demonstrating that elevation of cAMP in cells dueto CyaA action was itself accounting for the massive ruffling ofmurine macrophages.
As further shown in Fig. 2 and determined by calculating theruffling index using the counting of actin-rich submembrane foldsaccording to Cox et al. (33), all three examined macrophage typesresponded by similar morphological changes to CyaA or db-cAMPexposure, albeit at differing toxin doses and at a differing percent-age of responding cells. The J774A.1 cells and BMM were com-parably sensitive to CyaA action and ruffled vigorously already at10 ng/ml CyaA, with however only about half of the cell popula-tion responding by ruffling to CyaA or db-cAMP treatment. More-over, this proportion of ruffling cells was not enhanced even at a10-fold higher (100 ng/ml) concentration of CyaA (see Fig. 3 forJ774A.1 cells). In contrast, while the RAW 264.7 cells were lesssensitive to CyaA action and up to 100 ng/ml of the toxin had tobe used to observe ruffling in 10 min of exposure to toxin, theruffling response of RAW 264.7 cells was quite homogeneous withessentially all cells in the population responding to CyaA or db-cAMP treatment by ruffling. A likely explanation of these differ-ences in homogeneity of ruffling responses would be that it de-pends on the proportion of cells that was differentiated into
FIGURE 3. CyaA-induced membrane ruffling is transient and wanesfaster at higher toxin concentration. J774A.1 cells were treated with 10 or100 ng/ml CyaA for indicated time. Morphology alterations were assessedby scanning electron microscopy. Cytoskeleton rearrangements were fol-lowed by fluorescence microscopy of F-actin staining with TRITC-phal-loidin (data not shown) to determine the ruffling index (see Materials andMethods) that represents the mean � SD value (n � 3). �, p � 0.01 vsbuffer. The shown micrographs are representative of three independentexperiments. Scale bar, 10 �m.
FIGURE 4. CyaA suppresses fluid phase uptake by macrophages. A, CyaA suppresses the basal, as well as bacterial-lysate-induced fluid phase uptake.LY (500 �g/ml) or FITC-dextran (1 mg/ml) were added to J774A.1 either concomitantly with CyaA or db-cAMP, or following preincubation of J774A.1cells for 30 min with indicated concentrations of CyaA or db-cAMP. Where indicated, E. coli cell lysate (800 �g/ml) was added to boost the macropinocyticactivity of cells. Uptake of LY or FITC-dextran in 30 min of incubation was expressed as percentage of the amount of LY or FITC-dextran taken up bymock-treated cells. Results represent the means � SD of two independent experiments performed in triplicate (n � 6). �, p � 0.01 vs buffer. B, CyaA actioninhibits formation of macropinosomes by macrophages. J774A.1 cells were treated with CyaA (100 ng/ml) in the presence of E. coli cell lysate (800 �g/ml)and lysine-fixable FITC-dextran (1 mg/ml) for 30 min. Cells were fixed with 4% paraformaldehyde and examined using an Olympus BX60 fluorescencemicroscope. Inhibition of macropinosome formation by pretreatment of cells with 100 nM wortmannin for 30 min was used as control. Representativeresults of two independent experiments are shown.
5591The Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

macrophages. While the RAW 264.7 cell population is quite ho-mogeneous, consisting essentially of macrophage-like cells, theJ774A.1 cell population consists of a mixture of monocyte- andmacrophage-like cells, similarly to the primary BMM culture, thelatter comprising also bone marrow DCs and granulocytes (seeMaterials and Methods for phenotype characterization of BMMculture). Alternatively, also the cell-cycle dependence of the ruf-fling response of the RAW 264.7 and J774A.1 cell and BMMpopulation may differ.
CyaA-induced actin rearrangements and cell ruffling aretransient and wane faster at higher toxin concentration
To characterize the CyaA-induced macrophage ruffling phenotypein more detail, kinetics of morphological and actin cytoskeletonalterations of J774A.1 cells were examined in time at a high (100ng/ml) and a low (10 ng/ml) toxin concentration, respectively. Asdetermined by quantification of F-actin accumulation in cell pe-riphery and confirmed by scanning electron micrographs (Fig. 3),membrane ruffling of J774A.1 cells was transient and could beobserved already in 5 min of exposure to CyaA at both 10 ng/mland 100 ng/ml concentrations of CyaA. However, while cellstreated with 10 ng/ml CyaA still formed membrane ruffles at 30min and started to lose them only after 60 min of exposure to toxin,cells exposed to 100 ng/ml CyaA had already retracted the rufflesin 30 min of toxin treatment, showing that ruffling waned faster athigher CyaA concentration. The hypothesis would be that athigher toxin concentrations an inhibitory signaling threshold,such as activation of cAMP-dependent protein phosphatases in-terfering with cAMP signal transmission, and/or exhaustion ofactin rearrangement machinery is reached earlier than at lowertoxin concentrations.
CyaA-induced membrane ruffling subverts macropinocytic fluidphase uptake
Membrane ruffling is a typical feature of myeloid phagocytes ac-tivated by TLR signaling of pathogen components, which serves to
FIGURE 5. CyaA action does not yield detectable activation of Rac1,Rac2, or RhoG. Active cellular Rac1, Rac2, and RhoG were affinity pre-cipitated from lysates of toxin-treated J774A.1 cells using GST-CRIB orGST-ELMO1-coated agarose beads. Rac1-GTP, total Rac1, Rac2-GTP,total Rac2, RhoG-GTP, and total RhoG were detected by Western blotusing anti-Rac1, anti-Rac2 or anti-RhoG Abs. Relative activities of Rac1,Rac2, and RhoG were determined as the amounts of CRIB or ELMO1-bound Rac1, Rac2, or RhoG by normalization to total detected amounts ofRac1, Rac2, or RhoG, respectively. Results represent the means � SD forat least five independent experiments (n � 5). No statistically significantdifferences (p � 0.05) of amounts of active Rac1, Rac2, or RhoG wereobserved between toxin-treated and untreated cells.
FIGURE 6. Rac1 activity is required for induction of actin cytoskeleton rearrangements by CyaA. RAW 264.7 cells were transfected to express wild-typeor dominant negative (d.n.) variants of Rac1 or Cdc42 fused to GFP, or GFP alone (mock treatment). Production of the proteins was allowed to proceedfor 12 h before the cells were exposed to 100 ng/ml CyaA for 10 min. Cell morphology was assessed by F-actin staining with TRITC-phalloidin andtransfected cells were visualized by GFP expression. The morphology of transfected cells in the shown micrographs is representative of images from threeindependent experiments for which the mean � SD ruffling index was calculated (n � 3). Cell surface level of CD11b/CD18 on GPF-positive Rac1-T17N-transfected cells was determined by flow cytometry and expressed as percentage of CD11b level of control (nontransfected) cells. No statisticallysignificant difference (p � 0.05) of cell surface level of CD11b/CD18 was observed between control and Rac1-T17N-transfected cells.
5592 cAMP SIGNALING OF CyaA SUBVERTS PHAGOCYTE FUNCTION
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

enhance environment sampling through macropinocytic fluidphase uptake and allows enhanced Ag presentation and inductionof adaptive immune response against infection (35). Moreover,active enhancement of membrane ruffling and macropinocytosis isexploited by numerous bacterial pathogens for cell entry. There-fore, we examined whether CyaA-induced ruffling of J774A.1 cellswas also accompanied by enhancement of macropinocytic activity.Unexpectedly, however, as shown in Fig. 4A and measured asuptake of LY and of FITC-labeled dextran, the basal, as well asenhanced fluid phase uptake induced by addition of bacterial lysateto J774A.1 cells, were both importantly inhibited already in thepresence of 10 ng/ml CyaA. Moreover, this inhibition was occur-ring rapidly upon cell exposure to toxin, because essentially thesame extent of uptake inhibition was observed no matter whetherFITC-dextran and CyaA were added concomitantly to cells, orwhether cells were preincubated for 30 min with the toxin beforeFITC-dextran addition. Furthermore, no inhibition of fluid phaseuptake was observed with 100 ng/ml CyaA-AC�, while 1 mMdb-cAMP caused a comparably important decrease of dye uptake
as did treatment with 10 ng/ml CyaA, showing that it was theelevation of intracellular cAMP concentration by toxin actionthat accounted for the inhibition of macropinocytosis. As fur-ther confirmed by fluorescence microscopy, action of CyaA at100 ng/ml caused a comparable block of FITC-dextran uptakeas did 100 nM wortmannin, a potent inhibitor of macropino-some formation (Fig. 4B).
Steady-state Rac1 activity is required for CyaA/cAMP-inducedactin rearrangements
The CyaA-induced actin rearrangements suggested that cAMP-signaling modulated the activity of Rho family GTPases control-ling the homeostasis of actin cytoskeleton. As a first choice, weexamined the activation of Rac subfamily proteins in cells treatedwith CyaA, as activation of Rac-like GTPases is known to beinvolved in formation of membrane ruffles in macrophages (22).However, as determined by pull-down assays for active (GTP-loaded) Rac1, Rac2, and RhoG with the Cdc42/Rac interactive-binding region of PAK (GST-CRIB) and the RhoG effector protein
FIGURE 7. CyaA-induced membrane ruffling is due to transient inactivation of RhoA. A, CyaA transiently inactivates RhoA and provokes its relocal-ization from cell membranes. Isolation of cell membranes and affinity-precipitation of active cellular RhoA-GTP on GST-RBD-coated agarose beads wereperformed on J774A.1 cells treated with db-cAMP (2 mM, 10 min) or 10 or 100 ng/ml CyaA (for indicated times). RhoA-GTP pulled-down from wholecell lysates, or the membrane-associated RhoA, were detected by Western blotting and normalized to total RhoA detected in whole cell lysates, or toamounts of the integral membrane protein marker NTAL, respectively. The results represent the means � SD from at least five independent experiments(n � 5). �, p � 0.05 vs untreated cells. B, Expression of constitutively active RhoA prevents CyaA-induced membrane ruffling. RAW 264.7 cells weretransfected to express wild-type or the constitutively active (c.a.) variant of RhoA fused to GFP. Production of RhoA-GFP was allowed to proceed for 12 hbefore the cells were exposed to 100 ng/ml CyaA for 10 min. Cell morphology was assessed by F-actin staining with TRITC-phalloidin and transfectedcells were visualized by GFP expression. The morphology of transfected cells in the shown micrographs is representative of images from three independentexperiments for which the mean � SD ruffling index was calculated (n � 3). Cell surface level of CD11b/CD18 on GPF-positive RhoA and RhoA-G14V-transfected cells was determined by flow cytometry and expressed as percentage of CD11b level of control (nontransfected) cells. No statisticallysignificant difference (p � 0.05) of cell surface level of CD11b/CD18 was observed between control, RhoA, and RhoA-G14V-transfected cells.
5593The Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

ELMO1 (GST-ELMO1), respectively, no detectable activation ofRac1, Rac2, or RhoG was observed in CyaA-exposed cells atthe time points at which pronounced cell ruffling occurred inresponse to CyaA treatment, as shown in Fig. 5.
To further corroborate these observations, the capacity of CyaAto induce ruffling was examined on macrophages transiently trans-fected with constructs for expression of the wild-type and domi-nant negative variants of Rac1 fused to GFP. Because J774A.1cells are particularly resilient to transfection, this set of experi-ments was performed using RAW 264.7 murine macrophages thatresponded by identical morphological alterations to toxin treat-ment as did J774A.1 cells, although at increased CyaA concentra-tions (100 ng/ml). As documented in Fig. 6, the mock-transfectedcells, or cells transfected with wild type Rac1-GFP, both re-sponded to CyaA treatment by typical ruffling. In contrast, expres-sion of the dominant negative variant of Rac1-GFP (Rac1-T17N)efficiently protected cells from undergoing CyaA-induced ruffling.This requirement for Rac1 function for CyaA-induced ruffling tooccur appeared to be specific, as expression of the dominant neg-ative variant of Cdc42-GFP (Cdc42-T17N) used as control had amodest effect on toxin induced ruffling, as also shown in Fig. 6.Moreover, the loss of ruffling response to CyaA action upon Rac1-T17N expression was not a secondary effect of a decrease in toxinreceptor expression in transfected cells, as flow cytometric analysisconfirmed that the amounts of surface-exposed CyaA receptor,CD11b/CD18, on Rac1-T17N-transfected and nontransfected cellswere identical (Fig. 6). Hence, while no activation of Rac1 wasdetected in CyaA-exposed cells, a steady state level of Rac1 ac-tivity still appeared to be specifically required for CyaA-inducedcell ruffling to occur.
cAMP signaling of CyaA causes transient inactivation of RhoAin macrophages
We reasoned that in the absence of Rac activation, the observedruffling might have been due to inhibition of the signaling pathwayof the Rac antagonist, RhoA. Indeed, formation of membrane pro-trusions in human THP1 monocytes was previously seen upon in-hibition of the RhoA effector kinase ROK (36). Therefore, RhoAactivity was first examined in whole cell lysates of CyaA-treatedcells. As documented in Fig. 7A by results of pull-down assays foractive RhoA using RhoA interactive-binding region of rhotekin(GST-RBD), exposure of J774A.1 cells to 10 ng/ml CyaA yieldeda transient inactivation of RhoA. At this low toxin concentration asignificant decrease in the pulled-down amounts of active RhoAwas observed at 5 and 30 min of cell exposure to CyaA, with arecovery of active RhoA levels in the cells by 60 min of exposureto toxin. At the higher CyaA concentration (100 ng/ml), the de-crease in active RhoA-GTP level was observed only at 5 min aftertoxin addition and restoration of active RhoA levels occurred al-ready in 30 min. This observation was further corroborated byanalyzing the content of RhoA in the cell membrane fraction, asRhoA in its active GTP-bound conformation is associated withcellular membranes, while inactive GDP-bound RhoA can be se-questered in the cytosol by Rho-GDI proteins. As further shown inFig. 7A, the exposure of J774A.1 cells to CyaA resulted in a tran-sient decrease of RhoA amounts associated with the membranefraction of cells, supporting the conclusion that activity of RhoAwas transiently decreased upon cell exposure to CyaA. Hence, asimilarly transient and toxin concentration-dependent effect ofCyaA action on RhoA-GTP levels was observed as when cellularmorphology and ruffling in response to CyaA action was examined(cf. Fig. 3).
To test the hypothesis that inactivation of RhoA accounted foractin rearrangements and cell ruffling in response to elevation of
cAMP in cells by CyaA, we examined the ruffling of toxin-treatedRAW 264.7 macrophages that were transfected with wild-type orconstitutively active RhoA variants fused to GFP. Although againthe levels of CD11b/CD18 receptor on cells were not altered upontransfection with the used RhoA constructs and the expression ofwild-type RhoA-GFP in transfected cells had a modest impact ontoxin-induced cell ruffling, the expression of the constitutively ac-tive RhoA-GFP variant (RhoA-G14V) effectively protected cellsfrom undergoing CyaA-induced ruffling (Fig. 7B). Altogether,hence, these results show that inactivation of RhoA by a cAMP-dependent signaling mechanism was both a consequence of thetoxin action, as well as a prerequisite for the ruffling response tooccur.
To further corroborate this result, activity of cofilin, an effectordownstream of RhoA was assessed in CyaA-treated cells. Cofilinhas been identified as an important actin-filament severing and
FIGURE 8. CyaA induces activation of the actin filament severing pro-tein cofilin. A, J774A.1 cells were treated for 5 min with 10 or 100 ng/mlCyaA or with db-cAMP (2 mM, 10 min) and the inactive 3-Ser-phosphor-ylated cofilin or total cofilin amounts in whole cell lysates were detected byWestern blot. Relative dephosphorylation of cofilin is expressed as phos-phorylated cofilin/total cofilin with normalization to untreated cells. Re-sults represent the means � SD of four independent experiments (n � 4).�, p � 0.05 vs untreated cells. B, ROK kinase inhibitor Y-27632 enhancesmembrane protrusive activity of macrophages and activates the actin fila-ment severing protein cofilin. J774A.1 cells were treated with 10 �MY-27632 for indicated time intervals and ruffling index (see Materials andMethods) or the 3-Ser-phosphorylated cofilin or total cofilin amounts inwhole cell lysates were determined. Ruffling index represents the mean �SD from three independent experiments (n � 3). �, p � 0.01 vs untreatedcells. Relative activation (dephosphorylation) of cofilin with normalizationto untreated cells represent the mean � SD of three independent experi-ments (n � 3). �, p � 0.05 vs untreated cells.
5594 cAMP SIGNALING OF CyaA SUBVERTS PHAGOCYTE FUNCTION
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

depolymerizing protein that is regulated by inhibitory phosphory-lation at its Ser 3 residue and is involved in initiation of actin-driven membrane protrusions in regions of rapid actin assembly(see Ref. 37 for review). As shown in Fig. 8A, when lysates fromcells exposed to CyaA or to db-cAMP were probed with Abs se-lectively recognizing only the inactive phosphocofilin, or detectingboth phosphocofilin and active cofilin, a significant cofilin activa-tion (dephosphorylation) was observed in lysates of CyaA or db-cAMP treated cells. Moreover, as shown in Fig. 8B, cells treatedwith the inhibitor of the RhoA effector ROK kinase, Y-27632,exhibited a clear albeit somewhat delayed enhancement of mem-brane protrusive activity and cofilin dephosphorylation, thus mim-icking the outcome of CyaA action. These results go well with theconclusion that cAMP signaling provokes phagocyte ruffling by amechanism that involves RhoA inactivation and activation ofcofilin.
DiscussionThe present study unravels the capacity of CyaA to suppress thebactericidal activities of macrophages through unproductive mem-brane ruffling and shows for the first time that cAMP signaling oflow, likely physiologically relevant, concentrations of the CyaAtoxin causes a rapid and complete inhibition of CR3-mediatedphagocytosis. This may be of crucial importance in the early stagesof bacterial colonization of naive (unvaccinated) infants that lackspecific Abs to B. pertussis, because this bacterium resists lysis bycomplement itself (38) and would benefit from evading destructionensuing CR3-mediated phagocytosis, to which it is in turn highlysensitive (39, 40).
We report that the molecular mechanism of the repeatedly doc-umented capacity of CyaA to undermine bactericidal activities of�M�2 integrin-expressing myeloid phagocytic cells may well relyprimarily on RhoA inactivation as a result of cAMP signaling, assummarized in the model proposed in Fig. 9. An essential role ofRhoA GTPase in innate immunity functions of macrophages has,indeed, been previously established through the work of severalgroups. An active RhoA signaling pathway was found to be es-sential for CR3-mediated phagocytosis (24), where the RhoA-de-pendent activation of ROK allows recruitment of myosin-IIA andinitial assembly of actin and Arp2/3 complex in the phagocytic cup(31). Furthermore, inactivation of RhoA signaling was found toaffect chemotactic properties of primary monocytes, probably byderegulation of actin cytoskeleton coordination (36). Moreover,RhoA was also found to be involved in signaling leading to su-peroxide formation through CR3- and Fc�-receptor stimulation(41). Hence, the repeatedly observed interference of CyaA with allthree of the above mentioned activities would go well with a mech-anism of toxin action relying on inactivation of RhoA throughCyaA/cAMP signaling observed in this study.
RhoA is, indeed, a frequent target of virulence factors of severalpathogenic bacteria. For example, covalent modification of theRhoA switch 1 domain by large clostridial toxins (C. difficile Aand B, C. sordellii hemorrhagic toxins and C. novyi � toxins)results in inhibition of RhoA effector binding, whereas spatial reg-ulation of RhoA is targeted by the C3 transferase of C. botulinum,or YopT of Yersinia (see Ref. 42 for review). In this study, weshow that CyaA of Bordetella inactivates RhoA by yet another,cAMP-signaling dependent mechanism. This sets a new paradigm,because the same mechanism may be used also by other toxinspotentially targeting macrophages and manipulating the intracel-lular concentration of cAMP, such as the edema factor of Bacillusanthracis, ExoY of Pseudomonas aeruginosa, or the adenylate cy-clase of Yersinia pestis.
Membrane ruffling in macrophages is known to be mediated byactivation of Rac-like protein subfamily signaling (22), while ac-tivation of RhoA and of its downstream effector, ROK may sup-press this membrane protrusive activity of Rac-like GTPases (43).Indeed, Rac and RhoA appear to exhibit an antagonistic relation-ship, with the two proteins counterbalancing each other’s activity.For example, in migrating leukocytes a low RhoA activity wasdetected at the protruding, leading edge of the cell, while highlevels of RhoA activity were detected by RhoA biosensors at therear and at the sides of the cell, where formation of protrusions issuppressed (44). Furthermore, inhibition of the RhoA/ROK sig-naling pathway enhances the membrane protrusive activity ofmonocytes and results in competing membrane lamellipodia (36).Thus, the observed requirement for a functional steady-state Rac1signaling (Fig. 6) in the presence of cAMP signaling-mediatedinactivation of RhoA (Fig. 7A) is well compatible with the ob-served induction of membrane ruffling in macrophages in responseto toxin action of CyaA toxin (Fig. 2).
RhoA was, indeed, reported to be phosphorylated by cAMP-activated PKA at the Ser188 residue near its C terminus (45) andthis appears to inhibit RhoA function, possibly by impairing RhoAinteraction with ROK� (21) and/or by enhancing RhoA interactionwith RhoGDI (Rho guanine nucleotide dissociation inhibitor) (45,46). Effects of cAMP elevation on stress fiber dissolution and cellmorphology could, indeed, be prevented by overexpression ofboth, a phosphorylation-resistant S188A RhoA mutant (46) and ofROK� (21). In our hands, however, expression of the S188ARhoA mutant did not protect cells from undergoing CyaA-inducedmembrane ruffling (data not shown). This was only prevented byexpression of the constitutively active G14V RhoA mutant (Fig.7B). It remains, hence, to be clarified whether expression of S188ARhoA in the RAW 264.7 cells did not exert a sufficiently dominantphenotype on the background of the native RhoA produced in thecells, or whether other mechanisms than direct RhoA phosphory-lation at Ser 188 are involved in RhoA pathway inactivation andinduction of membrane ruffling in macrophages by CyaA/cAMPsignaling. Recent studies indicate that PKA can also regulate theactivity of upstream activators of RhoA, such as G�13 (47) and/orpromote also the inhibition of RhoA-GEF activity of the AKAP-Lbc signaling complex (48). Besides that, also the cAMP/Epac1/Rap1 pathway may potentially contribute to cAMP signaling-me-diated inhibition of RhoA. It has, indeed, been demonstrated that
FIGURE 9. Model of CyaA-mediated subversion of phagocyte func-tions. Following CD11b/CD18 binding, CyaA mediates rapid elevation ofcellular cAMP levels, the signaling of which results in inactivation ofRhoA signaling pathway. Toxin-induced drop of active RhoA level wouldresult in inhibition of CR3-mediated phagocytosis (24) and allow thesteady-state activity of Rac1 to prevail and provoke cell membrane ruffling.This is, however, unproductive (subversive), because accompanied by in-hibition of both macropinocytic fluid phase uptake and FcR-mediatedphagocytosis by an as yet not defined mechanism.
5595The Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

the RhoA-GAP activity of ARAP3 is activated upon binding ofRap (49). It should be also noted that the ROK inhibitor aloneinduced ruffles that were more localized and brush-like, whileCyaA induced sheet-like ruffles covering the complete cell surface(Fig. 2 vs Fig. 8B), suggesting that CyaA/cAMP signaling-inducedruffling of macrophage cells may involve mechanisms additional toRhoA-mediated decrease of ROK activity.
As a part of their colonization strategies, various pathogenicbacteria (e.g., Salmonella typhimurium and Shigella spp.) injecteffectors into cells to trigger membrane ruffling and exploit en-hanced formation of macropinosomes for cell invasion. Intrigu-ingly, the primarily extracellular pathogen B. pertussis appears todown-modulate its own invasion into the tracheal epithelial cellsby the action of CyaA (50). The results reported in this studyindicate that this may be due to the capacity of CyaA to promotesubversive membrane ruffling and to inhibit at the same time themacropinocytotic activity of cells. It will, hence, be of interest toelucidate the details of the mechanism by which CyaA/cAMP sig-naling leads to inhibition of macropinocytosis.
Membrane ruffling is intimately linked to the stimulation ofmacropinocytosis via formation of macropinosomes, which origi-nate primarily as actin-rich ruffles that close to form intracellularvesicles. Although ruffling is a prerequisite for macropinosomeformation, indeed, additional activities may be required to trans-form a ruffle into a closed intracellular vesicle. For example, Arakiet al. showed that PI3K activity is necessary for the completion ofmacropinocytosis but not for the initial ruffling phase in macro-phages (51). Further, Rah, a small GTPase of the Rab family, wasshown to be required for efficient macropinosome formation frommembrane ruffles (52). In the light of the results reported in thisstudy, it is plausible to hypothesize that cAMP/CyaA signalingmay target also the signaling pathway(s) accounting for closure ofmacropinosomes and phagosomes and their transformation intointracellular vesicles. This would go well with the here observedpersistence of inhibition of CR3-mediated phagocytosis also uponrestoration of the initial RhoA levels at 30 min of macrophageexposure to the higher CyaA dose of 100 ng/ml (Fig. 7 and data notshown). Alternatively, the latter phagocytic impotence of CyaA-treated cells may, however, be also caused by depleted ATP levelsdue to CyaA action (29), as well as due to mislocalization of com-ponents needed to be recruited for productive assembly of phago-cytic machinery by the preceding massive ruffling.
Macropinocytosis was also shown to account for an importantpart of Ag internalization and presentation on MHC molecules byprofessional APCs, such as DCs and macrophages (53–55). More-over, a burst of cell macropinocytic activity upon encounter ofTLR ligands was shown to direct an enhanced Ag presentation byDCs (35). It is, hence, plausible to propose that inhibiting mac-ropinocytosis through CyaA action might contribute to Bordetellasurvival on tracheal epithelia also by hampering induction of adap-tive immune response. Experiments are underway to test the hy-pothesis that CyaA activity inhibits Ag uptake and presentationby DCs.
AcknowledgmentsWe thank Johannes Huelsenbeck and Hana Kubinova for assistance. Thegift of bone marrow cells and analysis of their phenotype by Irena Adkinsis gratefully acknowledged.
DisclosuresThe authors have no financial conflict of interest.
References1. Vojtova, J., J. Kamanova, and P. Sebo. 2006. Bordetella adenylate cyclase toxin:
a swift saboteur of host defense. Curr. Opin. Microbiol. 9: 69–75.
2. Confer, D. L., and J. W. Eaton. 1982. Phagocyte impotence caused by an invasivebacterial adenylate cyclase. Science 217: 948–950.
3. Friedman, R. L., R. L. Fiederlein, L. Glasser, and J. N. Galgiani. 1987. Bordetellapertussis adenylate cyclase: effects of affinity-purified adenylate cyclase on hu-man polymorphonuclear leukocyte functions. Infect. Immun. 55: 135–140.
4. Njamkepo, E., F. Pinot, D. Francois, N. Guiso, B. S. Polla, and M. Bachelet.2000. Adaptive responses of human monocytes infected by Bordetella pertussis:the role of adenylate cyclase hemolysin. J. Cell. Physiol. 183: 91–99.
5. Weingart, C. L., P. S. Mobberley-Schuman, E. L. Hewlett, M. C. Gray, andA. A. Weiss. 2000. Neutralizing antibodies to adenylate cyclase toxin promotephagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 68:7152–7155.
6. Khelef, N., A. Zychlinsky, and N. Guiso. 1993. Bordetella pertussis inducesapoptosis in macrophages: role of adenylate cyclase-hemolysin. Infect. Immun.61: 4064–4071.
7. Weiss, A. A., and M. S. Goodwin. 1989. Lethal infection by Bordetella pertussismutants in the infant mouse model. Infect. Immun. 57: 3757–3764.
8. Goodwin, M. S., and A. A. Weiss. 1990. Adenylate cyclase toxin is critical forcolonization and pertussis toxin is critical for lethal infection by Bordetella per-tussis in infant mice. Infect. Immun. 58: 3445–3447.
9. Bagley, K. C., S. F. Abdelwahab, R. G. Tuskan, T. R. Fouts, and G. K. Lewis.2002. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussisactivate human monocyte-derived dendritic cells and dominantly inhibit cytokineproduction through a cAMP-dependent pathway. J. Leukocyte Biol. 72: 962–969.
10. Skinner, J. A., A. Reissinger, H. Shen, and M. H. Yuk. 2004. Bordetella type IIIsecretion and adenylate cyclase toxin synergize to drive dendritic cells into asemimature state. J. Immunol. 173: 1934–1940.
11. Boyd, A. P., P. J. Ross, H. Conroy, N. Mahon, E. C. Lavelle, and K. H. Mills.2005. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptiveimmune responses: distinct roles for acylation and enzymatic activity in immu-nomodulation and cell death. J. Immunol. 175: 730–738.
12. Griffioen, G., and J. M. Thevelein. 2002. Molecular mechanisms controlling thelocalisation of protein kinase A. Curr. Genet. 41: 199–207.
13. Jones, S. L., and Y. Sharief. 2005. Asymmetrical protein kinase A activity es-tablishes neutrophil cytoskeletal polarity and enables chemotaxis. J. LeukocyteBiol. 78: 248–258.
14. Galgani, M., V. De Rosa, S. De Simone, A. Leonardi, U. D’Oro, G. Napolitani,A. M. Masci, S. Zappacosta, and L. Racioppi. 2004. Cyclic AMP modulates thefunctional plasticity of immature dendritic cells by inhibiting Src-like kinasesthrough protein kinase A-mediated signaling. J. Biol. Chem. 279: 32507–32514.
15. Aronoff, D. M., C. Canetti, C. H. Serezani, M. Luo, and M. Peters-Golden. 2005.Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles ofprotein kinase A and exchange protein directly activated by cAMP-1. J. Immunol.174: 595–599.
16. Bryn, T., M. Mahic, J. M. Enserink, F. Schwede, E. M. Aandahl, and K. Tasken.2006. The cyclic AMP-Epac1-Rap1 pathway is dissociated from regulation ofeffector functions in monocytes but acquires immunoregulatory function in ma-ture macrophages. J. Immunol. 176: 7361–7370.
17. Newman, S. L., L. K. Mikus, and M. A. Tucci. 1991. Differential requirementsfor cellular cytoskeleton in human macrophage complement receptor- and Fcreceptor-mediated phagocytosis. J. Immunol. 146: 967–974.
18. Kalamidas, S. A., M. P. Kuehnel, P. Peyron, V. Rybin, S. Rauch, O. B. Kotoulas,M. Houslay, B. A. Hemmings, M. G. Gutierrez, E. Anes, and G. Griffiths. 2006.cAMP synthesis and degradation by phagosomes regulate actin assembly andfusion events: consequences for mycobacteria. J. Cell Sci. 119: 3686–3694.
19. Downey, G. P., E. L. Elson, B. Schwab 3rd, S. C. Erzurum, S. K. Young, andG. S. Worthen. 1991. Biophysical properties and microfilament assembly in neu-trophils: modulation by cyclic AMP. J. Cell Biol. 114: 1179–1190.
20. Lamb, N. J., A. Fernandez, M. A. Conti, R. Adelstein, D. B. Glass, W. J. Welch,and J. R. Feramisco. 1988. Regulation of actin microfilament integrity in livingnonmuscle cells by the cAMP-dependent protein kinase and the myosin lightchain kinase. J. Cell Biol. 106: 1955–1971.
21. Dong, J. M., T. Leung, E. Manser, and L. Lim. 1998. cAMP-induced morpho-logical changes are counteracted by the activated RhoA small GTPase and theRho kinase ROK�. J. Biol. Chem. 273: 22554–22562.
22. Allen, W. E., G. E. Jones, J. W. Pollard, and A. J. Ridley. 1997. Rho, Rac andCdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci.110: 707–720.
23. Aktories, K., and J. T. Barbieri. 2005. Bacterial cytotoxins: targeting eukaryoticswitches. Nat. Rev. Microbiol. 3: 397–410.
24. Caron, E., and A. Hall. 1998. Identification of two distinct mechanisms of phago-cytosis controlled by different Rho GTPases. Science 282: 1717–1721.
25. Lutz, M. B., N. Kukutsch, A. L. Ogilvie, S. Rossner, F. Koch, N. Romani, andG. Schuler. 1999. An advanced culture method for generating large quantities ofhighly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223:77–92.
26. Fayolle, C., A. Osickova, R. Osicka, T. Henry, M. J. Rojas, M. F. Saron, P. Sebo,and C. Leclerc. 2001. Delivery of multiple epitopes by recombinant detoxifiedadenylate cyclase of Bordetella pertussis induces protective antiviral immunity.J. Virol. 75: 7330–7338.
27. Osicka, R., A. Osickova, T. Basar, P. Guermonprez, M. Rojas, C. Leclerc, andP. Sebo. 2000. Delivery of CD8� T-cell epitopes into major histocompatibilitycomplex class I antigen presentation pathway by Bordetella pertussis adenylatecyclase: delineation of cell invasive structures and permissive insertion sites.Infect. Immun. 68: 247–256.
28. Tartz, S., J. Kamanova, M. Simsova, P. Sebo, S. Bolte, V. Heussler, B. Fleischer,and T. Jacobs. 2006. Immunization with a circumsporozoite epitope fused to
5596 cAMP SIGNALING OF CyaA SUBVERTS PHAGOCYTE FUNCTION
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from

Bordetella pertussis adenylate cyclase in conjunction with cytotoxic T-lympho-cyte-associated antigen 4 blockade confers protection against Plasmodiumberghei liver-stage malaria. Infect. Immun. 74: 2277–2285.
29. Basler, M., J. Masin, R. Osicka, and P. Sebo. 2006. Pore-forming and enzymaticactivities of Bordetella pertussis adenylate cyclase toxin synergize in promotinglysis of monocytes. Infect. Immun. 74: 2207–2214.
30. Newman, S. L., and L. K. Mikus. 1985. Deposition of C3b and iC3b onto par-ticulate activators of the human complement system: quantitation with monoclo-nal antibodies to human C3. J. Exp. Med. 161: 1414–1431.
31. Olazabal, I. M., E. Caron, R. C. May, K. Schilling, D. A. Knecht, andL. M. Machesky. 2002. Rho-kinase and myosin-II control phagocytic cup for-mation during CR, but not Fc�R, phagocytosis. Curr. Biol. 12: 1413–1418.
32. Gebran, S. J., E. L. Romano, H. A. Pons, L. Cariani, and A. N. Soyano. 1992. Amodified colorimetric method for the measurement of phagocytosis and antibody-dependent cell cytotoxicity using 2,7-diaminofluorene. J. Immunol. Methods 151:255–260.
33. Cox, D., P. Chang, Q. Zhang, P. G. Reddy, G. M. Bokoch, and S. Greenberg.1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phago-cytosis in leukocytes. J. Exp. Med. 186: 1487–1494.
34. El-Azami-El-Idrissi, M., C. Bauche, J. Loucka, R. Osicka, P. Sebo, D. Ladant,and C. Leclerc. 2003. Interaction of Bordetella pertussis adenylate cyclase withCD11b/CD18: role of toxin acylation and identification of the main integrin in-teraction domain. J. Biol. Chem. 278: 38514–38521.
35. West, M. A., R. P. Wallin, S. P. Matthews, H. G. Svensson, R. Zaru,H. G. Ljunggren, A. R. Prescott, and C. Watts. 2004. Enhanced dendritic cellantigen capture via toll-like receptor-induced actin remodeling. Science 305:1153–1157.
36. Worthylake, R. A., and K. Burridge. 2003. RhoA and ROCK promote migrationby limiting membrane protrusions. J. Biol. Chem. 278: 13578–13584.
37. DesMarais, V., M. Ghosh, R. Eddy, and J. Condeelis. 2005. Cofilin takes the lead.J. Cell Sci. 118: 19–26.
38. Pishko, E. J., D. J. Betting, C. S. Hutter, and E. T. Harvill. 2003. Bordetellapertussis acquires resistance to complement-mediated killing in vivo. Infect. Im-mun. 71: 4936–4942.
39. Schneider, B., R. Gross, and A. Haas. 2000. Phagosome acidification has oppositeeffects on intracellular survival of Bordetella pertussis and B. bronchiseptica.Infect. Immun. 68: 7039–7048.
40. Lenz, D. H., C. L. Weingart, and A. A. Weiss. 2000. Phagocytosed Bordetellapertussis fails to survive in human neutrophils. Infect. Immun. 68: 956–959.
41. Kim, J. S., B. A. Diebold, J. I. Kim, J. Kim, J. Y. Lee, and J. B. Park. 2004. Rhois involved in superoxide formation during phagocytosis of opsonized zymosans.J. Biol. Chem. 279: 21589–21597.
42. Boquet, P., and E. Lemichez. 2003. Bacterial virulence factors targeting RhoGTPases: parasitism or symbiosis? Trends Cell Biol. 13: 238–246.
43. Burridge, K., and R. Doughman. 2006. Front and back by Rho and Rac. Nat. CellBiol. 8: 781–782.
44. Wong, K., O. Pertz, K. Hahn, and H. Bourne. 2006. Neutrophil polarization:spatiotemporal dynamics of RhoA activity support a self-organizing mechanism.Proc. Natl. Acad. Sci. USA 103: 3639–3644.
45. Lang, P., F. Gesbert, M. Delespine-Carmagnat, R. Stancou, M. Pouchelet, andJ. Bertoglio. 1996. Protein kinase A phosphorylation of RhoA mediates the mor-phological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBOJ. 15: 510–519.
46. Ellerbroek, S. M., K. Wennerberg, and K. Burridge. 2003. Serine phosphoryla-tion negatively regulates RhoA in vivo. J. Biol. Chem. 278: 19023–19031.
47. Manganello, J. M., J. S. Huang, T. Kozasa, T. A. Voyno-Yasenetskaya, andG. C. Le Breton. 2003. Protein kinase A-mediated phosphorylation of the Gal-pha13 switch I region alters the G���13-G protein-coupled receptor complexand inhibits Rho activation. J. Biol. Chem. 278: 124–130.
48. Diviani, D., L. Abuin, S. Cotecchia, and L. Pansier. 2004. Anchoring of bothPKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling com-plex. EMBO J. 23: 2811–2820.
49. Krugmann, S., R. Williams, L. Stephens, and P. T. Hawkins. 2004. ARAP3 is aPI3K- and rap-regulated GAP for RhoA. Curr. Biol. 14: 1380–1384.
50. Bassinet, L., P. Gueirard, B. Maitre, B. Housset, P. Gounon, and N. Guiso. 2000.Role of adhesins and toxins in invasion of human tracheal epithelial cells byBordetella pertussis. Infect. Immun. 68: 1934–1941.
51. Araki, N., M. T. Johnson, and J. A. Swanson. 1996. A role for phosphoinositide3-kinase in the completion of macropinocytosis and phagocytosis by macro-phages. J. Cell Biol. 135: 1249–1260.
52. Sun, P., H. Yamamoto, S. Suetsugu, H. Miki, T. Takenawa, and T. Endo. 2003.Small GTPase Rah/Rab34 is associated with membrane ruffles and macropino-somes and promotes macropinosome formation. J. Biol. Chem. 278: 4063–4071.
53. Norbury, C. C., L. J. Hewlett, A. R. Prescott, N. Shastri, and C. Watts. 1995.Class I MHC presentation of exogenous soluble antigen via macropinocytosis inbone marrow macrophages. Immunity 3: 783–791.
54. Norbury, C. C., B. J. Chambers, A. R. Prescott, H. G. Ljunggren, and C. Watts.1997. Constitutive macropinocytosis allows TAP-dependent major histocompat-ibility complex class I presentation of exogenous soluble antigen by bone mar-row-derived dendritic cells. Eur. J. Immunol. 27: 280–288.
55. Sallusto, F., M. Cella, C. Danieli, and A. Lanzavecchia. 1995. Dendritic cells usemacropinocytosis and the mannose receptor to concentrate macromolecules in themajor histocompatibility complex class II compartment: downregulation by cy-tokines and bacterial products. J. Exp. Med. 182: 389–400.
5597The Journal of Immunology
by guest on January 14, 2016http://w
ww
.jimm
unol.org/D
ownloaded from




















