
Tropane alkaloids as medicinally useful natural products and ...

lable at ScienceDirect
European Journal of Medicinal Chemistry 45 (2010) 3885e3894
Contents lists avai
European Journal of Medicinal Chemistry
journal homepage: http: / /www.elsevier .com/locate/ejmech
Original article
Aconitum and Delphinium alkaloids of curare-like activity. QSAR analysis andmolecular docking of alkaloids into AChBP
M.A. Turabekova a,b, B.F. Rasulev a,b, F.N. Dzhakhangirov b, D. Leszczynska a,c,*, J. Leszczynski a,d
a Interdisciplinary Center for Nanotoxicity, Jackson State University, 1400 J.R. Lynch Street, P.O. Box 17910, Jackson, MS 39217, USAb Institute of Chemistry of Plant Substances AS RUz, Kh.Abdullaev Street, Tashkent 100170, UzbekistancDepartment of Civil and Environmental Engineering, Jackson State University, 1400 J.R. Lynch Street, P.O. Box 17068, Jackson, MS 39217, USAdDepartment of Chemistry and Biochemistry, Jackson State University, 1400 J.R. Lynch Street, P.O. Box 17068, Jackson, MS 39217, USA
a r t i c l e i n f o
Article history:Received 20 February 2010Received in revised form18 May 2010Accepted 19 May 2010Available online 26 May 2010
Keywords:AlkaloidsQSARMolecular dockingToxicityCurare-like activitynAChR-bindersAChBP
* Corresponding author. Department of Civil andJackson State University, 1400 J.R. Lynch St, PO Box 17Tel.: þ1 601 979 1091.
E-mail address: [email protected] (D
0223-5234/$ e see front matter � 2010 Elsevier Masdoi:10.1016/j.ejmech.2010.05.042
a b s t r a c t
Early studies have shown that some of diterpenoid alkaloids, found in highly toxic plants of the generaAconitum and Delphinium, act at neuronal nicotinic acetylcholine receptors (nAChRs) and exhibit potentN-cholinolytic activity. In the current study, GA-MLRA and GA-PLS approaches have been used to buildQSAR models to predict N-cholinolytic activity measured in vivo (blockade of neuromuscular conduc-tivity, BNMC and third eyelid relaxing activity, TYRA) and in vitro (suppression of frog’s abdominalstraight muscles on acetylcholine, SAM) for a series of diterpenoid alkaloids. Random splitting of a dataset (five trials in total) produced QSAR models of a good level of correlation between experimental invitro/in vivo and calculated N-cholinolytic activity expressed as log(1/ED50) with following averagestatistical parameters: log BNMC (r2 ¼ 0.87, s ¼ 0.14, q2 ¼ 0.82), log TYRA (r2 ¼ 0.80, s ¼ 0.29, q2 ¼ 0.67),log SAM (r2 ¼ 0.84, s ¼ 29, q2 ¼ 0.64). QSAR results suggest descriptors accounting for H-bond capabilityof molecules influence all three type of N-cholinolytic activity with additional contribution of steric andreactivity features as identified for TYRA and SAM data, respectively.
The alkaloid-receptor complexes were further analyzed by means of AutoDock Vina docking programusing the binding site of MLA complexed with AChBP (homolog of the ligand binding domain of nAChRs)as template. All compounds were shown to be well fitted in the binding pocket of native MLA with goodcorrelation exhibited between their ED50 and AutoDock Vina binding free energy. An analysis of thepossible factors significant for the ligand recognition has been enhanced by comparative docking studiesperformed for structurally related lycoctonine-type alkaloids (lappaconitine and aconitine) that areknown to bind to voltage-gated Naþ channel, but not to nAChRs.
� 2010 Elsevier Masson SAS. All rights reserved.
1. Introduction
Early studies have shown that some of diterpenoid alkaloidsoccurring naturally in Aconitum and Delphinium sp. are of curare-like activity and, therefore, act at neuronal nicotinic acetylcholinereceptors (nAChRs) and exhibit potent N-cholinolytic activity[1e3]. The nAChRs are prototypes for Cys-loop receptor family ofpentameric ligand-gated ion channels. They are considered as animportant drug targets since neuronal nAChRs are involved in highbrain function and neurodegenerative pathologies [4e7]. Inparticular, dysfunction of human heteromeric a4b2 and homomeric
Environmental Engineering,068, Jackson, MS 39217, USA.
. Leszczynska).
son SAS. All rights reserved.
a7 nAChRs (most abundant subtypes in the central nervous system,CNS) have been associated with a number of human diseases suchas schizophrenia, Alzheimer’s and Parkinson’s diseases, epilepsy,anxiety and depression. Also, these receptors are known to beinvolved in nicotine addiction and pain. At present, several nico-tinic receptor ligands are being clinically investigated [6,7].Nevertheless, discovery of novel and efficient drugs selectivelytargeting a distinct nAChR subtype remains a challenge due toseveral reasons [9]. In-depth structural investigations of thedifferent ligand binding sites followed by structure-based drugdesign cannot be performed directly due to lack of high-resolutionX-ray structure of nAChRs. Having taken into account thecomplexity in structure and function of nAChRs this become defi-nitely an issue challenging scientists to seek alternative approachesin gaining better knowledge of binding sites and their relativity toreceptor function [4,5,8]. Thus, an important breakthrough camefrom the resolved crystal structure of molluscan acetylcholine

M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e38943886
binding protein (AChBP), a water-soluble homolog of the extracel-lular ligand-binding domain of nicotinic receptors. To date, crystalstructure of several AChBPs from different species have beenreported and all the AChBPs were found to display a pharmaco-logical profile close to that of the homopentameric a7 nAChRs [8].Overall, starting from the last decade, the X-ray structural charac-terization of AChBP in complex with various ligands has beenproviding good alternative in search for subtype-specific nicotinicreceptor ligands [9e11]. Additionally, these complexes served asa good template for building models subsequently used for in silicoscreening procedures including docking simulations of the ligand-receptor interactions [10,12,13].
For the last twenty years subtype-selective agonist ligands,either full or partial, are being actively pursued as promising noveltherapeutics, while little attention has been focused on subtype-selective nicotinic receptor antagonists [6,14,15]. Thus, the majorityof nAChR-selective compounds, including those approved formarketing or still undergoing clinical trials, are nicotinic agonists.However, over the next decade, emerging considerable interest isanticipated in nAChR antagonists as potential drug candidates [6].Expanding research to antagonists will aid in more clear under-standing of nAChRs function related to certain diseases and,subsequently, result in discovery of new medications withimproved efficiency. The development of selective antagonists isrelatively new area that requires more complex and sophisticatedresearch approach for characterization of a drug candidate asa nAChR antagonist [14]. Significant limitation in drug discoveryand pharmacophore development comes from the fact that most ofthe known antagonists are not nAChR subtype-selective [2]. In fact,very little work published on structure activity relationship (SAR)studies performed for nicotinic antagonist molecules and structuralsimilarities between well known nAChR antagonists are yet to beestablished [14].
Fig. 1. Molecular structures of studied Aconitu
Methyllycaconitine (MLA, Fig. 1: 12), a tertiary diterpenoidalkaloid isolated from Delphinium and Aconitum sp., is one of themost potent, selective, competitive nonpeptide antagonists withapproximately 100-, 1000-, and 10 000-fold higher affinity for thea7 subtype, compared to a3b2, a4b2, and muscle nAChR [16].Furthermore, recent studies suggested MLA can interact withpresynaptic a3 and/or a6 subunits together with b2 and b3 [16].Structurally similar to MLA molecules, including its derivatives andsynthetic analogs, have also been assessed and the importance ofmethylsuccinimidobenzoyl group for nAChR inhibition activity wasclearly established. Hence, several SAR studies revealed thatremoval of the methyl group, methylsuccino, or entire methyl-succinimidobenzoate moiety results in concomitant 20-, 100-, or2000-fold decrease in affinity [14]. Additional SAR investigationsfor MLA analogs showed that different substituents of the 2-methylsuccinimido group alter a7 subtype binding affinity [17].Somewhat interesting results have been observed for the ring Eanalogs of MLA. Thus, while one group of ring E analogs of MLAexhibited similar affinity to the a7 subtype indicating the crucialrole of methylsuccinimidobenzoyl portion of a molecule in binding[18], the other group of ring E analogs showed high affinity for a3b4albeit with little or no inhibitory effects on binding to a7 [19,20].
Recent advances in the modeling of ligand interactions withnAChR binding sites enhanced by extensive SAR studies arepromising technique of improved successful predictive power innew nicotinic drugs design. This approach can be extremely usefulwhen applied to nAChR agonists for which strong limitations intranslation from screening models to clinical applications areobserved [7,21]. Very often, many of them would fail as medicinaldrug candidates because of their poor pharmacological profile. Thisis also true for nAChR antagonists [14,22] particularly in the light ofstrong evidence of lack of this type combined investigations asrevealed by literature survey. In line with our previous molecular
m and Delphinium diterpenoid alkaloids.

M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e3894 3887
modeling and QSAR studies performed for the series of Aconitumand Delphinium diterpenoid alkaloids [23e25], herein we reportresults of our current computational investigations on N-chol-inolytic activity. It was believed that QSAR analysis complementedby molecular docking of alkaloids into nAChBP binding site mayserve as a good template for revealing useful rules for enhancingpotency and selectivity of studied antagonists.
2. Materials and methods
2.1. Model preparation of Ac-AChBP and antagonist alkaloids
An in-house compounds collection of total 19 curare-like alka-loids has been used (Fig. 1). AM1 semiempirical method [26] wasapplied to optimize the structures. For each inhibitor, the confor-mation with the lowest energy was chosen as the docking ligandstructure. The lowest energy structures were identified by theprocedure applied and described earlier [23].
Crystal structures of AChBP from Aplysia californica in the apoform cocrystallized with MLA (PDB ID code: 2BYR [11]) with reso-lution 2.45 Å was downloaded from Brookhaven Protein Data Bankto serve as the docking template. The crystallographic water andligand molecules were removed from the AChBP model.
2.2. Automated docking setup
All the docking calculations were performed applying recentlyintroduced by Scripps Research Institute Autodock Vina program[27]. AChBP was firstly modified by adding polar hydrogens andthen kept rigid in docking process. The docking areawas defined bya box, centered on the MLA. Grid points of 36 � 30 � 38 with 1.0 Åspacing were calculated around the docking area for all the ligandatom types using Autodock Vina default optimization parameters.The torsional bonds of ligands were set free by Ligand module inAutoDock Tools-ADT [28]. The docking results from each calcula-tion were clustered on the basis of root-mean square deviation(RMSD) between the Cartesian coordinates of the ligand atoms andwere ranked according to the binding free energy. The structurewith relative lower binding free energy and the most clustermembers was chosen for the optimum docking conformation.
2.3. Biological data
Toxicity and N-cholinolytic activity data used in this study wereutilized from the early report [3]. All original in vivo LD50 toxicitydata (mg/kg) and ED50 miorelaxant data (mg/kg) such as blockadeof neuromuscular conductivity (BNMC), third eyelid relaxingactivity (TYRA) and obtained in vitro suppression of frog’s abdom-inal straight muscles on acetylcholine (SAM) were expressed as thelogarithm of the inverse molar concentration and (log 1/ED50)response variables.
2.4. QSAR and statistics
Models with good statistical qualities were obtained by geneticalgorithm-based multiple linear regression analysis (GA-MLRA) forboth BNMC and TYRA data and partial least squares (GA-PLS)regression for the in vitro suppression of frog’s abdominal straightmuscles on acetylcholine data.
Preliminary models selection was performed by means of GA-MLRA [29] technique as implemented in the BuildQSAR [30]program. This approach allows selection of the models with thefollowing characteristics for the better performance: high correla-tion coefficient R, low standard deviation S and the least number ofdescriptors involved. Next, the MiniTab [31] professional software
package was applied for detailed statistical analysis of the modelsobtained. A number of QSAR models have been built followed bythe statistical significance of each model being evaluated by thesquared correlation coefficient r2, standard error s, Fisher coeffi-cient F and non-collinearity of descriptors. A final set of QSARs wasidentified by applying the “leave-one-out” technique with its pre-dicting ability being evaluated and confirmed by cross validationcoefficient q2 based on predictive error sum of squares (PRESS).Additionally, all QSAR models have been subjected to externalvalidation. For this, the data set was divided into training [n ¼ 14(z75%)] and test [n ¼ 4(z25%)] sets in a random manner (fivetrials). In this strategy, we randomly split experimental data of allthree endpoints according to the structure (#1e4) and the activity(#5) ranking. The QSAR models for these five training sets weregenerated by using the same descriptors and validated on the basison their statistics. The resulting models of the training sets wereused to predict each type of N-cholinolytic activities of thecompounds present in their respective test sets.
Physicochemical and quantum-chemical descriptors used inthis study have been calculated applying the DRAGON program[32] and semiempirical AM1 [26] quantum-chemical calculationsrespectively.
3. Results and discussions
3.1. QSAR analysis
Keeping in mind that successful QSARs are developed for theligands of uniform mode of action and congeneric chemicalframeworks, for our QSAR studies we selected N-cholinolytic datadetermined in vivo and in vitro that had been reported by Dzha-khangirov [3]. Chemical structures and bioactivity of studiedcompounds are shown in Fig. 1 and collected in Table 1, respec-tively. In total 84 descriptors have been applied in our studyincluding so called physicochemical, constitutional, geometrical,charge, functional groups, properties and quantum-chemicaldescriptors [33].
It must be stressed that despite the fact of nAChR ligands beingthe subject of great deal of experimental work, extensive QSAR and3D-QSAR studies are still lacking for both agonists and antagonists[34,35]. This problem is mainly attributed to their vast structuraldiversity and the absence of experimental data of high homoge-neity. A survey of early research reports on SAR studies showedsome useful pharmacophores were identified though mostly foragonist molecules [34]. However, the main drawback of thosephrarmacophore models is their limited significance accounted toweak reliability when applied for the newer nAChR ligands.Oftentimes, the features defining the pharmacophore are derivedfor the series of molecules interacting with specific nAChR subtype.Subsequently, this would result in model’s failure to explainreasonably correct activity for various nAChR ligands of differentbindingmodes. In this respect it is clear that more QSARmodels arerequired to be developed for the each set of compounds followed byanalysis and establishment of their common pharmacophorefragments.
The best QSAR models obtained for three types of miorelaxantactivity were selected by GA. Hence, a one-parameter QSAR anal-ysis on blockade of neuromuscular conductivity data of 17 diter-penoid alkaloids afforded a linear equation with TPSA (TopologicalPolar Surface Area [33] determined as a van der Waals area ofelectron donor and electron acceptor atoms). TPSA is described asa polar part of the molecule associated with the oxygen, nitrogen,sulfur atoms, and also hydrogen atoms connected to theseheteroatoms. In general, this descriptor is attributed to H-bondingcapability of a molecule and the positive coefficient with this term

Table 1Biological data and descriptor values used in obtained QSAR equations.
Cmpd lgTox lgTYRA lgBNMC lgSAM HOMO TPSA RBF SPP nOHs DGb,Lcal/mol
1 3.80 5.47 4.37 4.80 �9.117 30.93 0.16 0.645 2 �8.32 4.00 5.13 4.17 4.71 �9.221 21.70 0.14 0.636 2 �8.13 3.86 5.07 4.07 4.62 �9.158 12.47 0.12 0.642 2 �8.44 3.43 5.05 4.23 4.97 �9.010 40.16 0.18 0.649 1 �7.95 4.39 5.48 4.72 5.74 �9.201 48.00 0.16 0.778 1 �8.06 4.41 5.46 4.79 5.82 �9.041 48.00 0.17 0.780 1 �7.77 3.71 5.28 4.32 4.96 �9.006 30.93 0.17 0.649 1 �7.88 3.44 4.59 a 3.82 �8.591 40.16 0.18 0.651 0 �7.89 3.54 4.39 a 3.05 �8.688 30.93 0.17 0.656 1 �7.810 5.41 6.43 5.52 4.83 �8.562 103.84 0.16 0.840 0 �10.511 5.60 6.51 5.75 5.01 �8.689 120.91 0.17 0.840 0 �10.312 5.24 6.36 5.48 4.53 �8.583 103.84 0.16 0.843 0 �10.213 4.46 5.47 4.99 4.89 �8.583 66.46 0.18 0.846 0 �9.214 4.48 a 4.91 a �8.683 100.60 0.21 0.851 0 �9.515 4.20 4.85 4.66 4.21 �8.689 57.23 0.18 0.855 1 �9.016 4.60 5.24 5.09 4.91 �8.643 83.53 0.19 0.855 0 �9.117 4.39 4.94 4.69 4.01 �8.742 74.30 0.19 0.845 1 �9.818 4.71 5.70 5.04 4.91 �8.751 100.60 0.20 0.847 0 �9.719 4.84 5.54 5.02 4.84 �8.655 83.53 0.19 0.850 0 �9.7
a Data not available.
M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e38943888
suggests that larger size of polar surface area promotes the BNMCwith the corresponding derivatives. Two-parameter regressionmodel developed for the third eyelid relaxing activity (TYRA) ofditerpenoid alkaloids, allow Eq. (2) pointing out quite similarH-bonding capability effects well accounted for by TPSA parameter.Furthermore, a notable aspect concerns the second selectedparameter RBF (Rotatable Bond Fraction), which is associated withthe flexibility of a molecule. Hence, increase of this descriptorresults in reduced N-cholinolytic activity for which greater flexi-bility of a structure is unfavorable. This can be well explained byboth the “drug-likeness” concept [36] and the ligand-receptorbinding dynamics. According to the first one, “non-drug”moleculeswith unreasonably many rotatable bonds (general case of RBF) areof weak membrane permeability that would hamper them fromreaching target protein for inducing therapeutic action. On theother hand, rotatable bonds can be considered as additional sterichindrance once molecule is at the receptor site. Thus, flexibility ofa compound might move the equilibrium towards increase rate ofreverse reaction (i.e. increase of dissociation constant) resulted inits lower affinity. In case of the SAM data one- and two-parametermodels did not result in good correlation and at least threedescriptors were required to produce meaningful equation.However, at this point we had to replace MLRA technique by GA-PLS due to the high descriptor collinearity. Among the modelsproposed, three-descriptor equation containing HOMO energyvalues, subpolarity parameter SPP and number of secondaryaliphatic alcohols nOHs was selected as the best one with accept-able quality. As can be seen from the Equation 3A-E, the variablesused in this equation can explain in average 85% of the variance inthe N-cholinolytic activity. Unlike two other types of N-cholinolyticactivities, suppression of frog’s abdominal straight muscles activityis modulated by electronic effects as accounted for by HOMOenergies. The negative term of HOMO and nOHs descriptors suggestthat higher HOMO energy values and lower number of secondaryOH groups favor activity of alkaloids Table 2.
To the best of our knowledge, with the regards to nAChRantagonist there are very few examples on classical QSAR appli-cation to quaternary ammonium salts [37,38] and 3D QSAR studiesof MLA-analogues [39]. In the first case MLRA and ANN QSARmodels indicated the importance of several descriptors includingthe length of the N-alkyl chain attached to quaternary ammoniumhead group, Moriguchi octanolewater partitioning coefficient,molecular surface area, WHIM type descriptors. Ghose-Grippen
molar refractivity, and the energy of lowest unoccupied molecularenergy also contributed to the model though of relatively lowervalues [36,37]. Authors of the 3D-QSAR report constructed phar-macophore (three hydrophobes, two hydrogen bond acceptorsHBAs, and one positively charged nitrogen atom) that further wasexploited for the screening two commercial molecule databasesfor novel inhibitors [39]. Successfully retrieved eight moleculesdemonstrated high inhibition potency on a3b4* AChRs. Our resultsare in a good consistency with 3D-QSAR findings indicatingH-bond properties contribute most to the ligand-receptor complexstability. In particular, as reported [39], COMFA model identifiedthe positively charged amino group and negatively charged estercarbonyl group to be essential for binding, while COMSIAemphasized the important role of amid carbonyl group from thesuccinimide ring for docking. Thus, important for binding activityelectrostatic and H-bond interactions and attributed to the above-mentioned groups are reflected by generalizing descriptor TPSA inEq. (1), and Eq. (2). It is interesting to note, that HOMO energyterm appeared to be related to in vitro N-cholinolytic activity in Eq.(3) only. This indicates the stronger nucleophilic properties ofcompounds improve binding affinity; i.e ligands become morepotent as electron removal energy decreases. According to theisosurface plot analysis the HOMOs of each compound werelocalized predominantly on lycoctonine nitrogen atom and thecarbons in its close neighborhood. These observations emphasizethe role of nitrogen atom on the N-cholinolytic activity and are ina good consistency with our previous findings [23e25]. Thus, thedetailed AM1/DFT calculations performed for a series of Aconitumand Delphinium alkaloids confirmed lycoctonine carcass nitrogenmostly contributes to the HOMOs and in this manner defines theactivity of molecules.
Further, the correlation between toxicity data and each partic-ular type of N-cholinolytic data has been examined. It is note-worthy to mention, that both measured in vivo BNMC and TYRAdata appeared to be highly correlated with toxicity unlike in vitrosuppression of frog’s abdominal straight muscles for which nosubstantial correlation was observed with LD50 (lgTox).
lgTox ¼ þ0:973ð�0:291Þ lgTYRA � 0:906ð�1:577Þ
�n ¼ 18; r ¼ 0:87; s ¼ 0:33; F ¼ 49:71; q2 ¼ 0:71;SPRESS ¼ 0:36�
ð4Þ
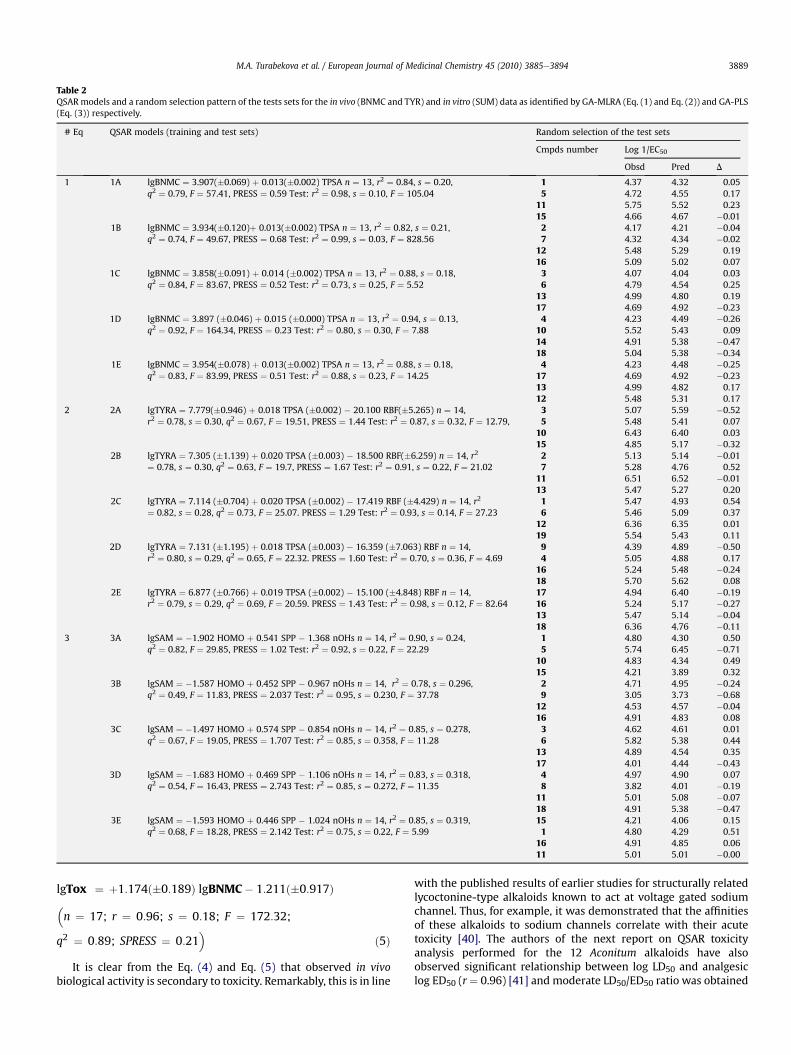
Table 2QSARmodels and a random selection pattern of the tests sets for the in vivo (BNMC and TYR) and in vitro (SUM) data as identified by GA-MLRA (Eq. (1) and Eq. (2)) and GA-PLS(Eq. (3)) respectively.
# Eq QSAR models (training and test sets) Random selection of the test sets
Cmpds number Log 1/EC50
Obsd Pred D
1 1A lgBNMC ¼ 3.907(�0.069) þ 0.013(�0.002) TPSA n ¼ 13, r2 ¼ 0.84, s ¼ 0.20,q2 ¼ 0.79, F ¼ 57.41, PRESS ¼ 0.59 Test: r2 ¼ 0.98, s ¼ 0.10, F ¼ 105.04
1 4.37 4.32 0.055 4.72 4.55 0.17
11 5.75 5.52 0.2315 4.66 4.67 �0.01
1B lgBNMC ¼ 3.934(�0.120)þ 0.013(�0.002) TPSA n ¼ 13, r2 ¼ 0.82, s ¼ 0.21,q2 ¼ 0.74, F ¼ 49.67, PRESS ¼ 0.68 Test: r2 ¼ 0.99, s ¼ 0.03, F ¼ 828.56
2 4.17 4.21 �0.047 4.32 4.34 �0.02
12 5.48 5.29 0.1916 5.09 5.02 0.07
1C lgBNMC ¼ 3.858(�0.091) þ 0.014 (�0.002) TPSA n ¼ 13, r2 ¼ 0.88, s ¼ 0.18,q2 ¼ 0.84, F ¼ 83.67, PRESS ¼ 0.52 Test: r2 ¼ 0.73, s ¼ 0.25, F ¼ 5.52
3 4.07 4.04 0.036 4.79 4.54 0.25
13 4.99 4.80 0.1917 4.69 4.92 �0.23
1D lgBNMC ¼ 3.897 (�0.046) þ 0.015 (�0.000) TPSA n ¼ 13, r2 ¼ 0.94, s ¼ 0.13,q2 ¼ 0.92, F ¼ 164.34, PRESS ¼ 0.23 Test: r2 ¼ 0.80, s ¼ 0.30, F ¼ 7.88
4 4.23 4.49 �0.2610 5.52 5.43 0.0914 4.91 5.38 �0.4718 5.04 5.38 �0.34
1E lgBNMC ¼ 3.954(�0.078) þ 0.013(�0.002) TPSA n ¼ 13, r2 ¼ 0.88, s ¼ 0.18,q2 ¼ 0.83, F ¼ 83.99, PRESS ¼ 0.51 Test: r2 ¼ 0.88, s ¼ 0.23, F ¼ 14.25
4 4.23 4.48 �0.2517 4.69 4.92 �0.2313 4.99 4.82 0.1712 5.48 5.31 0.17
2 2A lgTYRA ¼ 7.779(�0.946) þ 0.018 TPSA (�0.002) � 20.100 RBF(�5.265) n ¼ 14,r2 ¼ 0.78, s ¼ 0.30, q2 ¼ 0.67, F ¼ 19.51, PRESS ¼ 1.44 Test: r2 ¼ 0.87, s ¼ 0.32, F ¼ 12.79,
3 5.07 5.59 �0.525 5.48 5.41 0.07
10 6.43 6.40 0.0315 4.85 5.17 �0.32
2B lgTYRA ¼ 7.305 (�1.139) þ 0.020 TPSA (�0.003) � 18.500 RBF(�6.259) n ¼ 14, r2
¼ 0.78, s ¼ 0.30, q2 ¼ 0.63, F ¼ 19.7, PRESS ¼ 1.67 Test: r2 ¼ 0.91, s ¼ 0.22, F ¼ 21.022 5.13 5.14 �0.017 5.28 4.76 0.52
11 6.51 6.52 �0.0113 5.47 5.27 0.20
2C lgTYRA ¼ 7.114 (�0.704) þ 0.020 TPSA (�0.002) � 17.419 RBF (�4.429) n ¼ 14, r2
¼ 0.82, s ¼ 0.28, q2 ¼ 0.73, F ¼ 25.07. PRESS ¼ 1.29 Test: r2 ¼ 0.93, s ¼ 0.14, F ¼ 27.231 5.47 4.93 0.546 5.46 5.09 0.37
12 6.36 6.35 0.0119 5.54 5.43 0.11
2D lgTYRA ¼ 7.131 (�1.195) þ 0.018 TPSA (�0.003) e 16.359 (�7.063) RBF n ¼ 14,r2 ¼ 0.80, s ¼ 0.29, q2 ¼ 0.65, F ¼ 22.32. PRESS ¼ 1.60 Test: r2 ¼ 0.70, s ¼ 0.36, F ¼ 4.69
9 4.39 4.89 �0.504 5.05 4.88 0.17
16 5.24 5.48 �0.2418 5.70 5.62 0.08
2E lgTYRA ¼ 6.877 (�0.766) þ 0.019 TPSA (�0.002) � 15.100 (�4.848) RBF n ¼ 14,r2 ¼ 0.79, s ¼ 0.29, q2 ¼ 0.69, F ¼ 20.59. PRESS ¼ 1.43 Test: r2 ¼ 0.98, s ¼ 0.12, F ¼ 82.64
17 4.94 6.40 �0.1916 5.24 5.17 �0.2713 5.47 5.14 �0.0418 6.36 4.76 �0.11
3 3A lgSAM ¼ �1.902 HOMO þ 0.541 SPP � 1.368 nOHs n ¼ 14, r2 ¼ 0.90, s ¼ 0.24,q2 ¼ 0.82, F ¼ 29.85, PRESS ¼ 1.02 Test: r2 ¼ 0.92, s ¼ 0.22, F ¼ 22.29
1 4.80 4.30 0.505 5.74 6.45 �0.71
10 4.83 4.34 0.4915 4.21 3.89 0.32
3B lgSAM ¼ �1.587 HOMO þ 0.452 SPP � 0.967 nOHs n ¼ 14, r2 ¼ 0.78, s ¼ 0.296,q2 ¼ 0.49, F ¼ 11.83, PRESS ¼ 2.037 Test: r2 ¼ 0.95, s ¼ 0.230, F ¼ 37.78
2 4.71 4.95 �0.249 3.05 3.73 �0.68
12 4.53 4.57 �0.0416 4.91 4.83 0.08
3C lgSAM ¼ �1.497 HOMO þ 0.574 SPP � 0.854 nOHs n ¼ 14, r2 ¼ 0.85, s ¼ 0.278,q2 ¼ 0.67, F ¼ 19.05, PRESS ¼ 1.707 Test: r2 ¼ 0.85, s ¼ 0.358, F ¼ 11.28
3 4.62 4.61 0.016 5.82 5.38 0.44
13 4.89 4.54 0.3517 4.01 4.44 �0.43
3D lgSAM ¼ �1.683 HOMO þ 0.469 SPP � 1.106 nOHs n ¼ 14, r2 ¼ 0.83, s ¼ 0.318,q2 ¼ 0.54, F ¼ 16.43, PRESS ¼ 2.743 Test: r2 ¼ 0.85, s ¼ 0.272, F ¼ 11.35
4 4.97 4.90 0.078 3.82 4.01 �0.19
11 5.01 5.08 �0.0718 4.91 5.38 �0.47
3E lgSAM ¼ �1.593 HOMO þ 0.446 SPP � 1.024 nOHs n ¼ 14, r2 ¼ 0.85, s ¼ 0.319,q2 ¼ 0.68, F ¼ 18.28, PRESS ¼ 2.142 Test: r2 ¼ 0.75, s ¼ 0.22, F ¼ 5.99
15 4.21 4.06 0.151 4.80 4.29 0.51
16 4.91 4.85 0.0611 5.01 5.01 �0.00
M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e3894 3889
lgTox ¼ þ1:174ð�0:189Þ lgBNMC� 1:211ð�0:917Þ
�n ¼ 17; r ¼ 0:96; s ¼ 0:18; F ¼ 172:32;q2 ¼ 0:89; SPRESS ¼ 0:21�
ð5Þ
It is clear from the Eq. (4) and Eq. (5) that observed in vivobiological activity is secondary to toxicity. Remarkably, this is in line
with the published results of earlier studies for structurally relatedlycoctonine-type alkaloids known to act at voltage gated sodiumchannel. Thus, for example, it was demonstrated that the affinitiesof these alkaloids to sodium channels correlate with their acutetoxicity [40]. The authors of the next report on QSAR toxicityanalysis performed for the 12 Aconitum alkaloids have alsoobserved significant relationship between log LD50 and analgesiclog ED50 (r ¼ 0.96) [41] and moderate LD50/ED50 ratio was obtained

M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e38943890
for local anesthetic activity (r ¼ 0.71) [42]. Also, the regressionanalysis performed by our group resulted in high correlationcoefficient (r ¼ 0.88) between antiarrhythmic and toxicity dataavailable for 63 alkaloids [26].
Consequently, while developing new medicines on the basis ofstudied compounds one has to bear in mind, that their in vivotherapeutic action is strongly accompanied by the toxicity effect.Indeed, this is quite commonly observed situationwhen increase intherapeutic activity inevitably leads to stronger toxicity of thera-peutics, especially in case with neuronal active compounds. Addi-tionally, this is supported by the fact, that descriptor TPSA wasidentified as the best related to toxicity in our earlier QSAR toxicitystudies performed for the same set of compounds [23e25]. Onceagain, our findings confirm that the hydrogen bonding is largelyresponsible factor for the toxicity as well as N-cholinolytic activityinduced by the studied compounds.
Fig. 2. (a) Superimposition of native crystal structure MLA (green) and the best docked connetwork of MLA (12) within the binding pocket of Ac-AChBP. (c) The superimposed lowest eSuperimposition of the best docking conformations of the ligands 1e19 inside the bindingreferred to the web version of this article).
3.2. Docking calculations
As discussed above, the crystal structure at atomic resolution ofAChBP provided first molecular insights concerning nAChRsstructure. Unlike the emerged situation on strong need for QSARstudies, the literature dealing with AChBP used for generatingnAChR homology models and docking simulations is rapidlygrowing. In fact, current extensive application of this alternativeapproach provides high rate success in the rational discovery ofnew lead molecules for both agonists and antagonists withimproved subtype selectivity for human nAChR [10]. Nevertheless,as evidenced by recent publications, more attention has beendevoted by theoreticians to model interaction for nAChR antago-nists of peptide nature [13,43,44], while somewhat scares dockingstudies were reported for the low molecular antagonists. At thesame time, according to the “drug-likeness” concept, which is alsoknown as Lipinski’s rule of five, one of the characteristics respon-sible for the potential of a molecule to become a drug is molecularweight �500 Da [36,45]. Also, about 50% of worldwide marketeddrugs are based on natural substances [46] of mostly lowmolecular
formation obtained by AutoDock Vina (red). (b) Hydrogen-bonding and polar contactsnergy conformations of the ligands 1e19 obtained by docking at MLA’s binding site. (d)site (For interpretation of the references to colour in this figure legend, the reader is

Table 3Selected H-bond and Van der Waals contacts corresponding to docking poses of thelowest binding free energy (DGb) and of the molecule’s orientation similar to thenative MLA’s pose within the binding pocket.
Compound DGb(kcal/mol)
Hydrogen bonds and polar contactsbetween atoms of compounds and amino acids
Atom ofcompound
Aminoacid
Distance(Å)
12 (X-ray) NEt O of Trp147 2.93C1-OMe O of Trp147 3.05Succim-Oxo OH Ser167 (H-bond) 2.45Succim-Oxo OH Ser167 2.99Est-Oxo OH-Tyr55 3.02
12 �10.2 NEt O of Trp147 3.36C1-OMe O of Trp147 3.29Succim-Oxo OH Ser167 (H-bond) 2.64Succim-Oxo OH Ser167 2.89Est-Oxo OH-Tyr55 2.90
20 �9.3 C16-OMe NH2 of Gln57 2.31O-Benzoyl-NH OH of Tyr188 2.13Est-Oxo OH of Tyr93 2.67
20a �8.4 O-Benzoyl-NHHC(O)CH3-Oxo
OH of Ser167 2.15
21 �8.1 C3-OH O of Ser146 1.92C8-OAcetyl OH of Thr36 2.49
21a �7.3 C3-OH O of Trp147 2.32C8-OAcetyl-Oxo NH2 Gln57 2.86
22 �9.7 C13-OH OH of Tyr195 2.02Est-Oxo OH of Thr36 3.07
22a �9.5 Succim-Oxo OH of Ser167 2.75C-14-O-Benzoyl-Oxo
SeS of 190Cys-191Cys
3.12
NEt Trp147 (centre) 4.6723 �9.6 Est-OC NH2 of Gln57 2.47
C13-OH OH of Tyr195 2.2C2-NEt Trp147 8.8O-benzoyl-NH2 O of Asp159 3.11
23a �8.9 O-Benzoyl-NH2 OH of Tyr55 2.39C8-OCOMe OH of Thr36 2.20NEt Trp147 (centre) 5.10
a Docked pose similar to the position of native MLA in the binding pocket.
Fig. 3. Plot of the AutoDock Vina binding free energy (DGb) vs lgToxicity (1/logLD50)(a) and lgBNMC (1/logED50) (b).
Fig. 4. MLA’s structurally related compounds 20e23 submitted to additional dockingcalculations.
M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e3894 3891
weights suggesting this type of compounds, including presentlystudied secondary plant metabolites, still remain as highly prom-ising and attractive research targets in drug discovery and devel-opment. In line with this statement, docking of low molecular 20antagonists and 7 agonists into the open- and closed-channel statesof modeled a4b2 receptor was performed recently [47]. Authorsproposed computational strategy elegantly distinguishing whetherreceptor ligand is an agonist or antagonist based on calculation ofthe relative binding free energies only [47]. Next, relaxed-complexmethod combiningmolecular dynamics simulations with Autodockdocking was applied for virtual screening study of a large databaseagainst three species of AChBP [48]. Autodock is shown to predictfairly accurate poses for the majority of well-known AChR binderswith available crystal structures in the complex with AChBP.However, the trend was observed indicating the reported RMSDvalue between docked and crystal structures increases as theligands size and the number of rotatable bonds increase (<1 ÅRMSD for nicotine and epibatidine and >5 Å RMSD for MLA) [48].Also, water molecules behavior analysis in and around nicotine andcarbamylcholine binding pocket revealed five complex stabilizingzones occupied by waters [49]. Positions of some antagonistsrelatively to these water occupancy zones were examined, indi-cating little or no importance of water molecules with respect toantagonist binding [49]. With regards to competitive antagonists,binding of d-tubocurarine and metocurine to AChBP has beenstudied using computational methods together with mutagenesisand ligand binding measurements [50]. Surprisingly, these twoligands are shown to bind in distinctly different orientations
suggesting versality of the binding site in AChBP to dock curariformligands.
As can be seen, the previous computational studies were mostlyof comparative nature considering lowweight antagonists togetherwith agonists molecules and no further pharmacophore model hadbeen sketched out for the particular class of nicotinic antagonists.Therefore, for the present study we have selected a homologuesseries of curare-like diterpenoid alkaloids in order to providecomprehensive data on investigated phenomena by examination oftheir interactions with the receptor site followed by identificationof pharmacophore groups crucial for binding affinity.
With the aim to a better understanding of the influence of thedifferent substitutions, we have examined the lower dockingenergy pose of MLA and its related compounds with the lycocto-nine skeleton in the binding pocket of AChBP. The validation of thefunction implemented in AutoDock Vina was done by docking thenative ligand (12, MLA) into its binding site. A grid spacing of 1 Å

Fig. 5. Differential binding modes of 20e23 in the Ac-AChBP binding site. Panels (a),(c), (e) and (g) show the best docked pose and DGb (Kcal/mol) value of 20, 21, 22 and23 respectively corresponding to the lowest binding free energy calculated by
M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e38943892
was used as no significant differences in docking accuracy wereobserved using a grid spacing ranging from 0.3 to 1.0 Å. The dockedresults were compared to the crystal structure of the bound MLA-nAChBP complex. Fig. 3 illustrates that the obtained success rate isexcellent as the docked MLA appeared to be superimposed almostexactly on the native molecule with obtained binding free energy(DGb) of �10.2 kcal/mol (Table 1, Fig. 2a). The docked ligandexhibited hydrogen bonds and van der Waals interactions with thesame atoms of amino acids involved with the native ligand (Fig. 2b,Table 3). As evidenced by distances depicted in Table 3, pipiredinemoiety of dockedmolecule 12 slightly shifted from its experimentallocation. The minor difference of 0.5 Å observed for the tertiaryamine (NEt), while eOCH3 at C1 (C1-OMe), oxygen of succinimide(Succi-Oxo) and carbonyl oxygen of flexible ester linkage (Est-Oxo)were locatedwithin 1.0O 1.2 Å of X-ray structure. Next, the bindingaffinity of the rest 18 diterpenoid alkaloids was evaluated and freebinding energy values are collected in Table 1. A superposition ofthe examined compounds at the binding pocket is shown in Fig. 2cand d. All molecules in the series were located in very similarposition to the MLA with lycoctonine tertiary amine being at thedistance of z5 Å to the Trp147 enabling cation-p interactionbetween Trp147 and N-ethylpiperidine moiety. For those structureswith large anthranilate or succinimidoylanthranilate groups at C-18(10e19) the binding energy valueswere relatively lower due to theirability to form additional H-bond network with the aminoresidues.Thus, they exhibited additional interactions involving a carbonyl ofthree fragments (flexible ester linkage, anthranoyl and methyl-succino) stabilized by H-bond with Tyr55, Tyr188 and Ser167respectively. According to the results displayed in Table 1, threecompounds 10e12 have the lowest binding energy values which isin good consistency with previous experimental evidence [14,16].
The overall good correlation between the toxicity and BNMC ofstudied diterpenoid alkaloids and the binding affinities predictedby AutoDock Vinawasmade clear as indicated in Fig. 3. It was foundthat the stronger binding affinity, the more potent BNMC ortoxicity. Furthermore, binding free energy values (DGb) showedfairly good correlationwith TYRA data (z50%) while no parallelismbetween DGb and in vitro SAM activity data was observed.
Additional docking simulations were performed for the MLA’sstructurally related alkaloids: lappaconitine (20) and aconitine (21).These alkaloids share the same basic lycoctonine skeleton with -O-Benzoyl substitution at C-14 and C-18 for 21 and 20, respectively.Remarkably, the observed minor alteration in structural patternresults in diminished cuare-like activity. Instead, 20 and 21 are wellknown as modulators of voltage-gated Naþ channels. As reportedelsewhere, MLA-A-AChBP complex structure revealed that all aco-nitine substituents can easily be accommodated in A-AChBP [8].Although two structures reasonably fitted into binding pocket, ourdocking protocol indicated that 20 and 21 adopt a distinct orien-tation with different interactions (Fig. 5(a,c) and Table 3). Thedocking conformation of the lowest binding free energy(DGb ¼ �9.3 kcal/mol) for lappaconitine is depicted in Fig. 5a forwhich three H-bonds are observed between eOCH3 at C16, eNHgroup of anthranoyl and carbonyl of ester linkage with Gln57,Tyr188 and Tyr93 correspondingly. For aconitine molecule thelowest energy conformation (DGb ¼ �8.1 kcal/mol) depicted inFig. 5c is accompanied by formation of two H-bonds: between OHat C3 and O of Ser146, and also between carbonyl of eOAcetyl at C8and OH of Thr36. Further, among the list of low energy posesidentified by docking calculations for 20 and 21 we have examined
AutoDock Vina. Panels (b), (d), (f) and (h) show the docked conformation with cor-responding DGb (Kcal/mol) value for 20e23 identified by AutoDock Vina as maximallysimilar to the orientation of native MLA in the crystal structure of MLA-Ac-AChBPcomplex.

M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e3894 3893
those with lycoctonine core structure located similarly to theexperimental orientation of MLA’s lycoctonine ring. These dockingposes are illustrated in Fig. 5b and d fromwhich one cannot expectthese compounds to be reasonable candidates for nAChR inhibition.Most striking difference is observed for aconitine with O-Benzoylgroup pointing out from the binding pocket and, therefore, unfa-vouring a proper fixation of lycoctonine ring and resulting ininstability of the whole receptor-ligand system. Despite of thegreater structural similarity to MLA, lappaconitine did notdemonstrate highly enhanced binding affinity (DGb ¼ �8.4 kcal/mol). In fact, only one H-bond between carbonyl of eC6H5NHC(O)CH3 and OH of Ser167 and sudden drop of binding energy wereobserved. Apparently, �C6H5NHC(O)CH3 group attached to C4 viaester linkage in 20 is shorter then succinimidoylanthranilate groupof MLA and cannot form H-bond network required for the tighterbinding followed by induction of curare-like activity (Fig. 4)
As earlier reported in the literature, interesting results wereobtained on synthesis of two anthranilate esters of aconitine inorder to mimic the structure of MLA [51]. Remarkably, addition of2-aminobenzoyl and 2-(methylsuccinimido)benzoyl groups toaconitine produced compounds with nicotinic potency comparableto that of MLA, but with abolished ability to activate voltage-gatedsodium channel. These two synthesized compounds 22 (3-deoxy-18-O-desmethyl[(methylsuccinimido)benzoyl]aconitine) and 23(3-deoxy-18-O-desmethyl(2-aminobenzoyl)aconitine) were thesubject of our further docking investigations. According to ourdocking protocols, the lowest energy conformations of 22 and 23are tend to occupy the binding pocket in somewhat differentmanner toMLA (Fig. 5e and g). As evidenced by the data collected inTable 3, two compounds in their lowest energy poses exhibit verysimilar binding affinity (�9.7 kcal/mol for 22 and �9.6 kcal/mol for23). Having also located in very similar orientation within thereceptor, 22 and 23 still form different hydrogen bonds and van derWaals interactions (Table 3) determined by their distinct substit-uents at C-4. An exception is H-bond between OH at C13 and OH ofTyr195 observed for both structures. An examination of the bestdocking poses obtained for 22 and 23 revealed for the compound22 relatively stable positionwhich is very similar to the native MLAorientation (Fig. 5f) with DGb ¼ �9.5 kcal/mol. Compound 23 alsoappears to be partially superimposed on the native MLA (Fig. 5h),though with notable distortion and of lower binding energy(DGb ¼ �8.9 kcal/mol). Likewise to aconitine molecule, its twoanthranilate ester derivatives have -O-aromatic substituent at C-14perpendicularly pointing out from and lycoctonine core beingburied deeply in the binding site.
These theoretical findings are in consistency with experimentalresults indicating compound 22 was at least 2 orders of magnitudemore potent then compound 23 at Brain [125I]aBgt binding site [51].Also, our theoretical results confirm the crucial role of anthranoyland succinimidoylanthranilate groups for AChBP binding, while thepresence of large substituents at lycoctonine ring can result inworsened binding affinity of a compound to the receptor. Thisclearly can be seenwhen free binding energy of 22 and 23 is turnedout to be relatively higher when compared to the one ofcompounds 10e19 with no large side chain substituents at C14. Onthe whole, there is a reasonable correspondence between experi-mental data and theoretical predictions for studied hereincompounds. As no strict comparison can be presently attempted,the current molecular docking results still provide good template tobe further considered from a qualitative standpoint.
4. Conclusion
QSAR and molecular docking techniques had been successfullyapplied for 19 Aconitum and Delpinium diterpenoid alkaloids from
our in-house library. The H-bond interactions are revealed to be theprevalent factors modulating N-cholinolytic activity by both QSARanalysis and docking studies.
Our QSAR study demonstrated that polar part of the moleculeassociated with the oxygen, nitrogen, sulfur atoms and alsohydrogen atoms connected to these heteroatoms and modeled byTPSA descriptor is the most important parameter for in vivodetermined N-cholinolytic activity (both BNMC and TYRA). TheQSAR models included additionally descriptors encoding elec-tronic/reactivity properties (energy of HOMO), presence of H-bonddonors (OHs), steric hindrance (RBF) and charge-related (SPP)descriptors, which indicates that in vivo and in vitro N-cholinolitycactivity of the studied compounds is also controlled by these majorfactors. Determined in vivo data is highly correlated with toxicityindicating that the miorelaxant activity of compounds is secondaryto their toxicity, while no such parallelism was observed betweentoxicity and the data of in vitro studies.
The AutoDock Vina investigations carried out for the presentseries of compounds showed that they bind to AChBP with orien-tation and position very close to that resulting from the crystallo-graphic analysis in the AChBP complex with MLA. The stability ofthe ligandeprotein complexes is controlled by electrostatic, stericand hydrogen bonds network. The anthranoyl and succinimidoy-lanthranilate moieties are shown to be important for compounds tobe able to exert potent N-cholinolytic activity.
Additional docking studies were performed for lappaconitine(20) and aconitine (21) alkaloids that are structurally related toMLA, but of distinct properties. Docking results suggested thesecompounds occupy different orientation in the binding pocketindicating once more the importance of anthranoyl and succini-midoylanthranilate groups. In general, the docking calculations of21 evidence that the large substituents at C14 accounts for a worseMLA-type interactions between the lycoctonine ring and bindingmoiety, lowering the consensus score. This effect is howeveroverwhelmed by insertion of succinimidoylanthranilate ester at C4which forms required network of van der Waals and H-bondinteractions residing in binding pocket. This is clearly demon-strated by the docking results of compound 22 that tend to adoptorientation similar to native MLA molecule.
To conclude, our findings showed that coordinated applicationof classical QSAR and molecular docking methods yields significantand complementary insights into the main physicochemical inter-actions underlying important chemical and biological processes.
Acknowledgments
This work was supported in part by the ICPS (Uzbekistan). Wethank the National Science Foundation (NSF/RISE HRD-07346445and NSF/CREST HRD-0833178), EPSCoR Award #: 362492-190200-01\NSFEPS-0903787 and HPCDNM: W912HZ-09-C-0108 forgenerous financial support.
References
[1] M.N. Benn, J.M. Jacyno, in: S.W. Pelletier (Ed.), The Alkaloids: Chemical andBiological Perspectives, John Willey, New York, 1984, pp. 153e210.
[2] J.W. Daly, Cell. Mol. Neurobiol. 25 (2005) 513e552.[3] F.N. Dzhakhangirov, B.T. Salimov, I.A. Bessonova, M.N. Sultankhodzhaev,
Chem. Nat. Compd. 31 (1995) 708e712.[4] R.C. Hogg, M. Raggenbass, D. Bertrand, Rev. Physiol. Biochem. Pharmacol. 147
(2003) 1e46.[5] C. Gotti, F. Clementi, Prog. Neurobiol. 74 (2004) 363e396.[6] S.P. Arneric, M. Holladay, M. Williams, Biochem. Pharmacol. 74 (2007)
1092e1101.[7] B.K. Cassels, I. Bermudez, F. Dajas, J.A.A. -Carriquiry, S. Wonnacot, Drug
Discovery Today 10 (2005) 1657e1665.[8] P. Rucktooa, A.B. Smit, T.K. Sixma, Biochem. Pharmacol. 78 (2009) 777e787.

M.A. Turabekova et al. / European Journal of Medicinal Chemistry 45 (2010) 3885e38943894
[9] P. Taylor, T.T. Talley, Z. Radic, S.B. Hansen, R.E. Hibbs, J. Shi, Biochem. Phar-macol. 74 (2007) 1164e1171.
[10] V. Tsetlin, F. Hucho, Curr. Opin. Pharmacol. 9 (2009) 306e310.[11] S.B. Hansen, G. Sulzenbacher, T. Huxford, P. Marchot, P. Taylor, Y. Bourne, Eur.
Mol. Biol. Org. 24 (2005) 3635e3646.[12] C. Ulens, A. Akdemir, A. Jongejan, R. van Elk, S. Bertrand, A. Perrakis, R. Leurs, A.
B. Smit, T.K. Sixma,D. Bertrand, I.J.P. deEsch, J.Med. Chem.52 (2009) 2372e2383.[13] I.E. Kasheverov, Y.N. Utkin, V.I. Tsetlin, Curr. Pharm. Des. 15 (2009) 2430e2452.[14] L.P. Dwoskin, P.A. Crooks, J. Pharmacol. Exp. Ther. 298 (2001) 395e402.[15] M.N. Romanelli, P. Gratteri, L. Guandalini, E. Martini, C. Bonaccini, F. Gualtieri,
ChemMedChem. 2 (2007) 746e767.[16] A.J. Mogg, P. Whiteaker, J.M. Mcintosh, M. Marks, A.C. Collins, S. Wonnacott, J.
Pharmacol. Exp. Ther. 302 (2002) 197e204.[17] F.I. Carroll, W. Ma, H.A. Navarro, P. Abraham, S.A. Wolckenhauer, M.I. Damaj,
B.R. Martin, Bioorg. Med. Chem. 15 (2007) 678e685.[18] S.C. Bergmeier, D.J. Lapinsky, R.B. Free, D.B. McKay, Bioorg. Med. Chem. Lett. 9
(1999) 2263e2266.[19] D.L. Bryant, R.B. Free, S.M. Thomasy, D.J. Lapinsky, K.A. Ismail, S.B. McKay, S.
C. Bergmeier, D.B. McKay, Neurosci. Res. 42 (2002) 57e63.[20] S.C. Bergmeier, K.A. Ismail, K.A. Arason, S.B. McKay, D.L. Bryant, D.B. McKay,
Bioorg. Med. Chem. Lett. 14 (2004) 3739e3742.[21] B. Tasso, C.C. Boido, E. Terranova, C. Gotti, L. Riganti, F. Clementi, R. Artali,
G. Bombieri, F. Meneghetti, F. Sparatore, J. Med. Chem. 52 (2009) 4345e4357.[22] L.P. Dwoskin, R. Xu, J.T. Ayers, P.A. Crooks, Expert. Opin. Ther. Pat. 10 (2000)
1561e1581.[23] M.A. Turabekova, B.F. Rasulev, M.G. Levkovich, J. Leszczynski, N.D. Abdullaev,
Comput. Biol. Chem. 32 (2008) 88e101.[24] M.A. Turabekova, B.F. Rasulev, Chem. Nat. Compd. 41 (2005) 213e217.[25] M.A. Turabekova, B.F. Rasulev, F.N. Dzhakhangirov, Sh.I. Salikhov, Envir. Toxic.
Pharmacol. 25 (2008) 310e320.[26] D. Young, Computational Chemistry: a Practical Guide for Applying Tech-
niques to Real World Problems. John-Wiley & Sons, New York, 2001.[27] O. Trott, A.J. Olson, J. Comput. Chem. 31 (2009) 455e461.[28] M.F. Sanner, J. Mol. Graph. Model. 17 (1999) 57e61.[29] J. Devillers, Genetic Algorithms in Molecular Modeling. Academic Press Ltd.,
London, 1996.[30] D.B. de Oliveira, A.C. Gaudio, Quant. Struct.-Act. Relat. 19 (2000) 599e604.
[31] Statistical Analysis Software MiniTab (2009).[32] DRAGON, Web Version 3.0 for Windows (2003).[33] R. Todeschini, V. Consonni, Handbook of molecular descriptors. in:
R. Mannhold, H. Kubinyi, H. Timmerman (Eds.), Methods and Principles inMedicinal Chemistry. WileyeVCH Verlag GmbH, Weinheim, 2000, p. 668.
[34] O. Nicolotti, M. Pellegrini-Calace, C. Altomare, A. Carrierri, A. Carotti, F. Sanz,Curr. Med. Chem. 9 (2002) 1e29.
[35] H. Zhang, H. Li, Q. Ma, J. Mol. Graph. Model. 26 (2007) 226e235.[36] I. Muegge, Med. Res. Rev. 23 (2003) 302e321.[37] F. Zheng, E. Bayram, S.P. Sumithran, J.T. Ayers, C.-G. Zhan, J.D. Schmitt, L.
P. Dwoskin, P.A. Crooks, Bioorg. Med. Chem. 14 (2006) 3017e3037.[38] F. Zheng, M.J. McConnell, C.-G. Zhan, L.P. Dwoskin, P.A. Crooks, Bioorg. Med.
Chem. 17 (2009) 4477e4485.[39] D.B. McKay, C. Chang, T.F. Gonzalez-Cestari, S.B. McKay, R.A. El-Hajj, D.
L. Bryant, M.X. Zhu, P.W. Swaan, K.M. Arason, A.B. Pulipaka, C.M. Orac, S.C. Bergmeier, Mol. Pharmacol. 71 (2007) 1288e1297.
[40] J. Friese, J. Gleitz, U.T. Guster, J.F. Heubach, T. Matthiesen, B. Wilffert, N. Selve,Eur. J. Pharmacol. 337 (1997) 165e174.
[41] A.M. Bello-Ramirez, J. Buendia-Orozco, A.A. Nava-Ocampo, Fundam. Clin.Pharmacol. 17 (2003) 575e580.
[42] A.M. Bello-Ramirez, A.A. Nava-Ocampo, Fundam. Clin. Pharmacol. 18 (2004)157e161.
[43] D.Yu. Mordvitsev, Ya.L. Polyak, D.A. Kuzmin, O.V. Levtsova, Ye.V. Tourleigh, Yu.N. Utkin, K.V. Shaitan, V.I. Tsetlin, Comput. Biol. Chem. 31 (2007) 72e81.
[44] S. Dutertre, R.J. Lewis, Eur. J. Biochem. 271 (2004) 2327e2334.[45] C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Adv. Drug Delivery Rev.
23 (1997) 3e25.[46] G.A. Cordell, M.L. Quinn-Beattie, N.R. Farnsworth, Phytother. Res. 15 (2001)
183e205.[47] X. Huang, F. Zheng, C.-G. Zhan, J. Am. Chem. Soc. 130 (2008) 16691e16696.[48] A. Babakhani, T.T. Talley, P. Taylor, J.A. McCammon, Comput. Biol. Chem. 33
(2009) 160e170.[49] S. Amiri, M.S.P. Sansom, P.C. Biggin, Prot. Eng. Des. Sel 20 (2007) 353e359.[50] F. Gao, N. Bren, A. Little, H.-L. Wang, S.B. Hansen, T.T. Talley, P. Taylor, S.M. Sin,
J. Biol. Chem. 278 (2003) 23020e23026.[51] D.J. Hardick, G. Cooper, T. Scott-Ward, I.S. Blagbrough, B.V.L. Potter,
S. Wonnacott, FEBS Lett. 365 (1995) 79e82.
Copyright © 2022 FDOKUMEN




















