
Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to...
12
The FASEB Journal • Research Communication Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells Ce ´sar Ca ´rdenas,* ,‡,1 Nevenka Juretic ´,* ,1 Jorge A. Bevilacqua,* ,†,§ Isaac E. García,* Reinaldo Figueroa,* Ricardo Hartley,* Ana L. Taratuto, Roger Gejman, ¶ Nora Riveros,* Jordi Molgo ´, ‡ and Enrique Jaimovich* ,2 *Centro de Estudios Moleculares de la Ce ´lula and † Programa de Anatomía y Biología del Desarrollo, Instituto de Ciencias Biome ´dicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile; ‡ Department of Physiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA; § Departamento de Neurología y Neurocirugía, Hospital Clínico Universidad de Chile, Independencia, Chile; Departamento de Neuropatología, Instituto de Investigaciones Neurolo ´ gicas, FLENI, Buenos Aires, Argentina; ¶ Departamento de Anatomía Patolo ´ gica, Facultad de Medicina, Pontificia Universidad Cato ´lica de Chile, Santiago, Chile; and # Centre National de la Recherche Scientifique, Institut de Neurobiologie Alfred Fessard, FRC2118, Laboratoire de Neurobiologie Cellulaire et Mole ´culaire UPR9040, Gif sur Yvette, France ABSTRACT Inositol 1,4,5-trisphosphate (IP 3 ) recep- tors (IP 3 Rs) drive calcium signals involved in skeletal muscle excitation-transcription coupling and plasticity; IP 3 R subtype distribution and downstream events evoked by their activation have not been studied in human muscle nor has their possible alteration in Duchenne muscular dystrophy (DMD). We studied the expression and localization of IP 3 R subtypes in normal and DMD human muscle and in normal (RCMH) and dystrophic (RCDMD) human muscle cell lines. In nor- mal muscle, both type 1 IP 3 Rs (IP 3 R1) and type 2 IP 3 Rs (IP 3 R2) show a higher expression in type II fibers, whereas type 3 IP 3 Rs (IP 3 R3) show uniform distribu- tion. In DMD biopsies, all fibers display a homoge- neous IP 3 R2 label, whereas 24 7% of type II fibers have lost the IP 3 R1 label. RCDMD cells show 5-fold overexpression of IP 3 R2 and down-regulation of IP 3 R3 compared with RCMH cells. A tetanic stimulus induces IP 3 -dependent slow Ca 2 transients significantly larger and faster in RCDMD cells than in RCMH cells as well as significant ERK1/2 phosphorylation in normal but not in dystrophic cells. Excitation-driven gene expres- sion was different among cell lines; 44 common genes were repressed in RCMH cells and expressed in RCDMD cells or vice versa. IP 3 -dependent Ca 2 release may play a significant role in DMD pathophysiology.—Ca ´rdenas, C., Juretic ´, N., Bevilacqua, J. A., García, I. E., Figueroa, R., Hartley, R., Taratuto, A. L., Gejman, R., Riveros, N., Molgo ´ , J., Jaimovich, E. Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 24, 3210 –3221 (2010). www.fasebj.org Key Words: skeletal muscle excitation-transcription coupling dystrophin muscle plasticity myofiber types Duchenne muscular dystrophy (DMD) is an X- linked recessive disease that affects 1 in 3500 live male births. It is well established that it is caused by the absence of functional dystrophin, due to deletions or point mutations in the dystrophin gene (1). Several lines of evidence support the idea of an alteration of Ca 2 homeostasis in dystrophic cells (re- viewed in ref. 2); however, the extent of the sarcoplas- mic reticulum (SR) involvement in the abnormal Ca 2 handling is not clear. Apparently the proteins involved in excitation-contraction coupling are normally ex- pressed in dystrophic muscle. The expression of 1-, 2-, and -subunits of the dihydropyridine receptor (DHPR) were similar in control and mdx mice, a murine Duchenne model (3). Type 1 ryanodine recep- tors (RyR) and the fast isoform of the Ca 2 -ATPase (SERCA1) were found to exist in comparable amounts in control and dystrophin-deficient muscles (3). How- ever, we have found that the amount of inositol 1,4,5- trisphosphate (IP 3 ) receptors (IP 3 Rs), as well as the total mass of IP 3 , are largely increased in an mdx mouse-derived cell line compared with normal cells (4). IP 3 -dependent pathways regulate intracellular Ca 2 1 These authors contributed equally to this work. 2 Correspondence: Centro de Estudios Moleculares de la Ce ´lula, Instituto de Ciencias Biome ´dicas, Facultad de Medi- cina, Universidad de Chile, Independencia 1027, Santiago 7, Chile. E-mail: [email protected] doi: 10.1096/fj.09-152017 3210 0892-6638/10/0024-3210 © FASEB
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to...

The FASEB Journal • Research Communication
Abnormal distribution of inositol 1,4,5-trisphosphatereceptors in human muscle can be related to alteredcalcium signals and gene expression in Duchennedystrophy-derived cells
Cesar Cardenas,*,‡,1 Nevenka Juretic,*,1 Jorge A. Bevilacqua,*,†,§ Isaac E. García,*Reinaldo Figueroa,* Ricardo Hartley,* Ana L. Taratuto,� Roger Gejman,¶
Nora Riveros,* Jordi Molgo,‡ and Enrique Jaimovich*,2
*Centro de Estudios Moleculares de la Celula and †Programa de Anatomía y Biología del Desarrollo,Instituto de Ciencias Biomedicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile;‡Department of Physiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA;§Departamento de Neurología y Neurocirugía, Hospital Clínico Universidad de Chile,Independencia, Chile; �Departamento de Neuropatología, Instituto de Investigaciones Neurologicas,FLENI, Buenos Aires, Argentina; ¶Departamento de Anatomía Patologica, Facultad de Medicina,Pontificia Universidad Catolica de Chile, Santiago, Chile; and #Centre National de la RechercheScientifique, Institut de Neurobiologie Alfred Fessard, FRC2118, Laboratoire de NeurobiologieCellulaire et Moleculaire UPR9040, Gif sur Yvette, France
ABSTRACT Inositol 1,4,5-trisphosphate (IP3) recep-tors (IP3Rs) drive calcium signals involved in skeletalmuscle excitation-transcription coupling and plasticity;IP3R subtype distribution and downstream eventsevoked by their activation have not been studied inhuman muscle nor has their possible alteration inDuchenne muscular dystrophy (DMD). We studied theexpression and localization of IP3R subtypes in normaland DMD human muscle and in normal (RCMH) anddystrophic (RCDMD) human muscle cell lines. In nor-mal muscle, both type 1 IP3Rs (IP3R1) and type 2 IP3Rs(IP3R2) show a higher expression in type II fibers,whereas type 3 IP3Rs (IP3R3) show uniform distribu-tion. In DMD biopsies, all fibers display a homoge-neous IP3R2 label, whereas 24 � 7% of type II fibershave lost the IP3R1 label. RCDMD cells show 5-foldoverexpression of IP3R2 and down-regulation of IP3R3compared with RCMH cells. A tetanic stimulus inducesIP3-dependent slow Ca2� transients significantly largerand faster in RCDMD cells than in RCMH cells as wellas significant ERK1/2 phosphorylation in normal butnot in dystrophic cells. Excitation-driven gene expres-sion was different among cell lines; 44 common genes wererepressed in RCMH cells and expressed in RCDMD cells orvice versa. IP3-dependent Ca2� release may play asignificant role in DMD pathophysiology.—Cardenas,C., Juretic, N., Bevilacqua, J. A., García, I. E., Figueroa,R., Hartley, R., Taratuto, A. L., Gejman, R., Riveros,N., Molgo, J., Jaimovich, E. Abnormal distribution ofinositol 1,4,5-trisphosphate receptors in human musclecan be related to altered calcium signals and geneexpression in Duchenne dystrophy-derived cells. FASEBJ. 24, 3210–3221 (2010). www.fasebj.org
Key Words: skeletal muscle � excitation-transcription coupling� dystrophin � muscle plasticity � myofiber types
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease that affects 1 in 3500 live malebirths. It is well established that it is caused by theabsence of functional dystrophin, due to deletions orpoint mutations in the dystrophin gene (1).
Several lines of evidence support the idea of analteration of Ca2� homeostasis in dystrophic cells (re-viewed in ref. 2); however, the extent of the sarcoplas-mic reticulum (SR) involvement in the abnormal Ca2�
handling is not clear. Apparently the proteins involvedin excitation-contraction coupling are normally ex-pressed in dystrophic muscle. The expression of �1-,�2-, and �-subunits of the dihydropyridine receptor(DHPR) were similar in control and mdx mice, amurine Duchenne model (3). Type 1 ryanodine recep-tors (RyR) and the fast isoform of the Ca2�-ATPase(SERCA1) were found to exist in comparable amountsin control and dystrophin-deficient muscles (3). How-ever, we have found that the amount of inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs), as well as the totalmass of IP3, are largely increased in an mdx mouse-derivedcell line compared with normal cells (4).
IP3-dependent pathways regulate intracellular Ca2�
1 These authors contributed equally to this work.2 Correspondence: Centro de Estudios Moleculares de la
Celula, Instituto de Ciencias Biomedicas, Facultad de Medi-cina, Universidad de Chile, Independencia 1027, Santiago 7,Chile. E-mail: [email protected]
doi: 10.1096/fj.09-152017
3210 0892-6638/10/0024-3210 © FASEB

signaling in many cell types (5), including skeletalmuscle cells (6, 7). Elevated K� or electrical stimulationtrigger a slow Ca2�-release transient, dependent on IP3production and not related to the excitation-contrac-tion coupling process (6, 8, 9) but associated withtransient phosphorylation in both the ERK1/2 and thecAMP response element-binding protein (CREB) (7,10). In particular, ERK1/2 phosphorylation has beenshown to depend on calcium signals in muscle primarycultures and cell lines (7, 10, 24). Moreover, this slowCa2� transient regulates the expression of early andlate genes (10–12). In fact, a number of genes becomeexpressed or repressed after a short membrane depo-larization of cultured muscle cells (12).
The possible role of IP3Rs in the pathophysiology ofdystrophinopathy has never been explored. IP3Rs couldbe a putative candidate to explain Ca2� dynamicsderegulation in these patients, and, owing to their rolein the regulation of gene expression, altered IP3Rscould be the cause of the severe impairment observedin dystrophic muscle cells.
Although an abnormal pattern of gene expressionhas been well documented in dystrophy (13, 14), themechanisms involved are unclear. Recent studies pro-vided evidence that ERK1/2 activation in dystrophicmuscle is related to mechanical stress (15) and tread-mill exercise (16).
In this work, we provide evidence indicating thatboth expression and localization of IP3Rs are differentin normal and dystrophic human skeletal muscle and innormal (RCMH) and dystrophic (RCDMD) human celllines. In DMD biopsies, we determined that part of fast(type II) fibers have lost type 1 IP3R (IP3R1) labelingcompared with age-matched controls. Furthermore,using the human-derived cell lines, we show that IP3-dependent Ca2� transients evoked by electrical stimu-lation are both larger and faster in RCDMD cells thanin normal cells and that ERK1/2 phosphorylation isdiminished compared with that in RCMH cells. Inaddition, microarray analysis after electrical stimulationrevealed a different pattern of gene expression betweenthe cell lines.
MATERIALS AND METHODS
Reagents
Antibodies directed against dystrophin, type 2 IP3Rs (IP3R2),PLC�, and phosphorylated forms of ERK1/2 were obtainedfrom Santa Cruz Biotechnology (Santa Cruz, CA, USA), andantibodies against fast and slow myosin heavy chains werefrom Sigma-Aldrich Corp. (St. Louis, MO, USA). Antibodyraised against the epitope-purified IP3R1 was obtained fromAffinity Bioreagents (Golden, CO, USA), and antibodyagainst lamina-associated polypeptide 2 (LAP2) was from BDTransduction Laboratories (Franklin Lakes, NJ, USA). Anti-body directed against specific epitope-regions in the C termi-nus of type 3 IP3R (IP3R3) (17) was kindly provided by Dr.G. A. Mignery (Loyola College, Chicago, IL, USA). PAN-IP3Rantibody, that recognizes a conserved region on the C termi-nus, was from Calbiochem (San Diego, CA, USA). Secondary
horseradish peroxidase-conjugated anti-rabbit, anti-mouse, oranti-goat IgG was from Pierce Biotechnology (Rockford, IL,USA). Alexa 488- and Alexa 633-conjugated anti-rabbit oranti-mouse IgG was from Molecular Probes (Eugene, OR,USA).
ECL reagents were from Pierce Biotechnology or Amer-sham Biosciences (Piscataway, NJ, USA). TPA and U73122were from Sigma-Aldrich Corp.
Human samples
Adult normal skeletal muscle tissue was obtained from volun-tary patients of the Hospital Clínico Universidad de Chileundergoing cardiac or orthopedic surgery not related tomyopathic diseases. Informed consent of the patients wasobtained in advance, according to a questionnaire adminis-tered to the patient or relatives with the approval of the ethicscommittee of the institution. Tissue samples of sartorius andquadriceps muscles were used. Biopsy samples from pediatricpatients with a clinical diagnosis of DMD were obtained fromthe Instituto de Investigaciones Neurologicas, FLENI (Bue-nos Aires, Argentina) and from the Departamento de Anat-omía Patologica, Facultad de Medicina, Pontificia Univer-sidad Catolica de Chile (Santiago, Chile). Control samples(i.e., from biopsies performed for diagnostic purposes forwhich results were normal) were from the same source.Samples were immediately frozen in isopentane precooled inliquid nitrogen and stored at �70°C until their utilization asdescribed elsewhere (18). Serial sections of normal or dystro-phic muscle from at least 3 unrelated patients were assayed.Histopathological diagnosis of DMD was made by an experi-enced pathologist (A.L.T. or R.G.); complete absence ofdystrophin was demonstrated by immunohistochemical anal-ysis according to standardized diagnostic protocols.
Cell cultures
Dystrophic human myoblasts of the RCDMD cell line (19)and normal RCMH human myoblasts (20) were cultured inDMEM supplemented with 10% FBS and 10% horse serum at37°C and 5% CO2. Myogenic differentiation was accom-plished by horse serum reduction to 1%.
Immunostaining
Serial cryosections of skeletal muscle (7–10 �m thick) wereplaced on slides and air dried at room temperature. Sectionswere fixed in methanol for 10 min at 4°C and rinsed with 0.1M PBS, pH 7.4, and Triton X-100 (0.03%). Blockade ofunspecific binding sites was performed with PBS-3% BSA.Sections were incubated with primary antibodies (1:100)overnight at 4°C, washed with PBS-3% BSA and incubatedwith fluorescent secondary antibodies (1:500) during 60 minat room temperature. Negative controls were obtained aftersections were incubated with secondary antibodies in theabsence of primary antibodies. Specificity of anti-IP3R anti-bodies was established previously using preimmune serum,antigenic peptide, or Western blots (6, 7, 21–23). Slides weremounted in Vectashield (Vector Laboratories, Burlingame,CA, USA). Representative confocal images were acquired witha Carl Zeiss Axiovert 135 microscope (LSM Microsystems,Jena, Germany). The images reproduced herein were manip-ulated in Adobe Photoshop (Adobe Systems, San Jose, CA,USA) to improve clarity; no information was added or deletedby those adjustments.
Immunofluorescence for cultured cells was performed asreported previously (24). Dilution of primary antibodies was1:100, and dilution of secondary antibodies was 1:500. Con-
3211IP3Rs IN NORMAL AND DYSTROPHIC CELLS

focal image sections of 1 �m were acquired with a Carl ZeissAxiovert 200, LSM 5 Pascal system.
Western blot analysis
Cell lysis, determination of protein concentration, separationof lysate proteins, transfer to PDVF membranes, and blockingwere performed as described previously (21). Membraneswere incubated overnight with the appropriate primary anti-body. Dilution of IP3R antibodies was 1:1000. PLC� andphosphorylated forms of ERKs and ERK2 were used at 1:2000.Immunoreactive proteins were detected using ECL reagentsaccording to the manufacturer’s instructions. To correct forloading, membranes were stripped in buffer containing 62.5mM Tris-HCl (pH 6.8), 2% SDS, and 50 mM �-mercaptoetha-nol at 50°C for 30 min and reprobed with the correspondingantibodies. Films were scanned, and densitometric analysis ofthe bands was performed with the Scion Image program(http://www.scioncorp.com).
Electrical stimulation
Electrical stimulation for Ca2� signal acquisition was per-formed as described previously (9). In brief, a 1-ml capacitychamber with the cells previously loaded with the Ca2�
indicator Fluo-3-acetoximetyl ester (Molecular Probes) wasplaced in the stage of an inverted microscope equipped withepifluorescence illumination (Olympus T041; Olympus, NewHyde Park, NY, USA) and a charge-coupled device cooledcamera (MCD600; Spectra Source Instrument, Westlake Vil-lage, CA, USA). The electrical pulse train stimulation con-sisted of 400 pulses of 1-ms duration at 45 Hz. To determineERK1/2 phosphorylation, all cultured myotubes in 60-mmdishes were homogeneously stimulated using a device consist-ing of a row of six platinum wires intercalated 1 cm apart withalternated polarity across a circular plastic holder that fits inthe dish. The two terminals were connected to a stimulator.Square pulses were monitored by displaying signals in anoscilloscope.
Microarray analysis
Total RNAs from RCMH and RCDMD cells were obtainedusing TRIzol reagent (Invitrogen, Carlsbad, CA, USA) accord-ing to the manufacturer’s protocol. cDNA from control anddepolarized samples was labeled with Alexa 555 or 647 dyes,respectively. In brief, cDNA was obtained by priming 20 �g oftotal RNA with 2 �M oligo(dT) in the presence of either 0.02mM Alexa 555 or Alexa 647-AHA-dUTP (Invitrogen), 0.5 mMconcentrations of each dATP, dGTP, and dCTP, 0.08 mMdTTP, 2 U of RNAsin, and 1� buffer RT in a total volume of50 �l. Reaction was started by addition of 1.5 �l of Super-Script II reverse transcriptase. To suppress gene-specific dyeeffects, reciprocal labeling was performed. After 3 h ofincubation at 37°C, the reaction was stopped with 5 �l of 0.5M EDTA. Template RNA was hydrolyzed with 1 �l of 10 MNaOH during 15 min at 70°C, and then 10 �l of 2 M Tris-HCl(pH 7.5) was added. Cleanup of labeled cDNA was performedusing Mini Bio-Spin columns (Bio-Rad Laboratories, Her-cules, CA, USA). An Illumina 22K human oligonucleotidemicroarray was kindly provided by Dr. D. J. Munroe [NationalCancer Institute (NCI), Frederick, MD, USA]. Hybridizationand scanning were performed as described previously (12).
Microarrays data processing
Images were analyzed with GenePix Pro III software (AxonInstruments, Foster City, CA, USA). GenePix Pro files (gpr)
and JEPG images were deposited at the NCI’s MicroarrayDatabase (mAdb; http://nciarray.nci.nih.gov). Annotatedfiles were retrieved from mAdb and print-tip loess normal-ized; scale was corrected by means of DNMAD (ref. 25;http://dnmad.bioinfo.cnio.es/). After the elimination ofgenes found in �70% of arrays, missing values were estimatedby KNN-Impute in the Preprocessor of Gene ExpressionProfile Analysis Suite (GEPAS) (http://gepas.bioinfo.cipf.es/). ANOVA was performed with Pomelo (ref. 26; http://pomelo.bioinfo.cnio.es/). Differentially expressed geneswere selected using an unadjusted P � 0.01 as cutoff. Then,filtered data were averaged and expressed as mean log2 ratios.Clustering was done with the clustering hierarchical tool ofGEPAS. The relationship between genes (and keywords) wasestablished using the Chilibot program (http://www.chilibot.net/).
Statistical analysis
Statistical analysis of data was performed using GraphPadSoftware (GraphPad, San Diego, CA, USA). All experimentswere performed in different preparations and in triplicate.Results are expressed as means se. The significance ofdifferences among treatments was evaluated using Student’s ttest for paired data.
RESULTS
Distribution of IP3R subtypes in human skeletalmuscle
The presence and distribution of IP3Rs in normalhuman skeletal muscle sections was revealed by immu-nostaining with an antibody (PAN-IP3R) recognizing allthree IP3R subtypes (Fig. 1A, left panel). Anti-slowmyosin heavy chain I (MHCI) antibody, which specifi-cally labeled slow myofibers, was used to distinguishslow (I) and fast (II) fiber types (Fig. 1A, middle panel).As shown in a transverse section of sartorius muscle inFig. 1A, both type I and type II fibers showed a stronglabeling for PAN-IP3R antibody, with a “dotted” patternsuggesting a SR-associated distribution of the receptor(Fig. 1A, left panel). A longitudinal section showingstriations with a high degree of colocalization betweenthe IP3R1 label and SR marker calreticulin is shown inSupplemental Fig. 1. MHCI also showed an intracellu-lar punctuate pattern (Fig. 1A, middle panel) somehowcolocalizing with the IP3R (Fig. 1A, right panel). Know-ing that IP3Rs are expressed in all normal humanskeletal muscle fibers, we explored the location of eachIP3R subtype individually. Both IP3R1 and IP3R2showed a specific “chessboard” pattern of immuno-staining (Fig. 1B, C, left panel). By double-labeling thesections with anti-slow MHCI antibody, we revealed thatIP3R1 and IP3R2 subtypes selectively labeled type IIfibers (Fig. 1B, C, right panel). On the other hand, thestaining for IP3R3 showed homogeneous labeling inboth fiber types (Fig. 1D). The intracellular dottedpattern of staining observed with the PAN-IP3R anti-body was seen equally with all three receptors subtype-specific antibodies.
Similarly to what was described in skeletal muscle
3212 Vol. 24 September 2010 CARDENAS ET AL.The FASEB Journal � www.fasebj.org
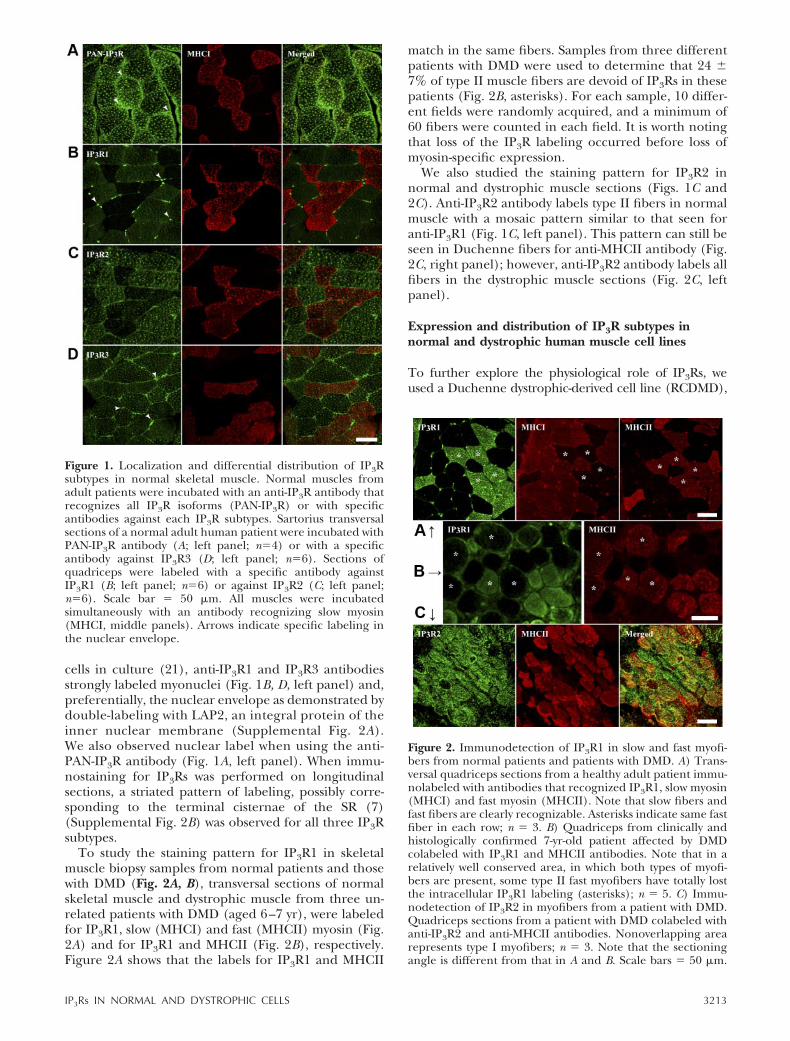
cells in culture (21), anti-IP3R1 and IP3R3 antibodiesstrongly labeled myonuclei (Fig. 1B, D, left panel) and,preferentially, the nuclear envelope as demonstrated bydouble-labeling with LAP2, an integral protein of theinner nuclear membrane (Supplemental Fig. 2A).We also observed nuclear label when using the anti-PAN-IP3R antibody (Fig. 1A, left panel). When immu-nostaining for IP3Rs was performed on longitudinalsections, a striated pattern of labeling, possibly corre-sponding to the terminal cisternae of the SR (7)(Supplemental Fig. 2B) was observed for all three IP3Rsubtypes.
To study the staining pattern for IP3R1 in skeletalmuscle biopsy samples from normal patients and thosewith DMD (Fig. 2A, B), transversal sections of normalskeletal muscle and dystrophic muscle from three un-related patients with DMD (aged 6–7 yr), were labeledfor IP3R1, slow (MHCI) and fast (MHCII) myosin (Fig.2A) and for IP3R1 and MHCII (Fig. 2B), respectively.Figure 2A shows that the labels for IP3R1 and MHCII
match in the same fibers. Samples from three differentpatients with DMD were used to determine that 24 7% of type II muscle fibers are devoid of IP3Rs in thesepatients (Fig. 2B, asterisks). For each sample, 10 differ-ent fields were randomly acquired, and a minimum of60 fibers were counted in each field. It is worth notingthat loss of the IP3R labeling occurred before loss ofmyosin-specific expression.
We also studied the staining pattern for IP3R2 innormal and dystrophic muscle sections (Figs. 1C and2C). Anti-IP3R2 antibody labels type II fibers in normalmuscle with a mosaic pattern similar to that seen foranti-IP3R1 (Fig. 1C, left panel). This pattern can still beseen in Duchenne fibers for anti-MHCII antibody (Fig.2C, right panel); however, anti-IP3R2 antibody labels allfibers in the dystrophic muscle sections (Fig. 2C, leftpanel).
Expression and distribution of IP3R subtypes innormal and dystrophic human muscle cell lines
To further explore the physiological role of IP3Rs, weused a Duchenne dystrophic-derived cell line (RCDMD),
Figure 2. Immunodetection of IP3R1 in slow and fast myofi-bers from normal patients and patients with DMD. A) Trans-versal quadriceps sections from a healthy adult patient immu-nolabeled with antibodies that recognized IP3R1, slow myosin(MHCI) and fast myosin (MHCII). Note that slow fibers andfast fibers are clearly recognizable. Asterisks indicate same fastfiber in each row; n 3. B) Quadriceps from clinically andhistologically confirmed 7-yr-old patient affected by DMDcolabeled with IP3R1 and MHCII antibodies. Note that in arelatively well conserved area, in which both types of myofi-bers are present, some type II fast myofibers have totally lostthe intracellular IP3R1 labeling (asterisks); n 5. C) Immu-nodetection of IP3R2 in myofibers from a patient with DMD.Quadriceps sections from a patient with DMD colabeled withanti-IP3R2 and anti-MHCII antibodies. Nonoverlapping arearepresents type I myofibers; n 3. Note that the sectioningangle is different from that in A and B. Scale bars 50 �m.
Figure 1. Localization and differential distribution of IP3Rsubtypes in normal skeletal muscle. Normal muscles fromadult patients were incubated with an anti-IP3R antibody thatrecognizes all IP3R isoforms (PAN-IP3R) or with specificantibodies against each IP3R subtypes. Sartorius transversalsections of a normal adult human patient were incubated withPAN-IP3R antibody (A; left panel; n4) or with a specificantibody against IP3R3 (D; left panel; n6). Sections ofquadriceps were labeled with a specific antibody againstIP3R1 (B; left panel; n6) or against IP3R2 (C; left panel;n6). Scale bar 50 �m. All muscles were incubatedsimultaneously with an antibody recognizing slow myosin(MHCI, middle panels). Arrows indicate specific labeling inthe nuclear envelope.
3213IP3Rs IN NORMAL AND DYSTROPHIC CELLS
and its normal counterpart (RCMH). Despite the factthat both cell lines were well characterized years ago (4,19, 20), immunofluorescence studies to detect dystro-phin expression in RCMH and RCDMD cells wereperformed to test possible reversions in either the phe-notype or the genotype that could have occurred duringseveral culture passages. We found that RCMH cellsretained full expression of dystrophin on the cell mem-brane region (Supplemental Fig. 3A), whereas RCDMDcells did not show any staining (Supplemental Fig. 3B).
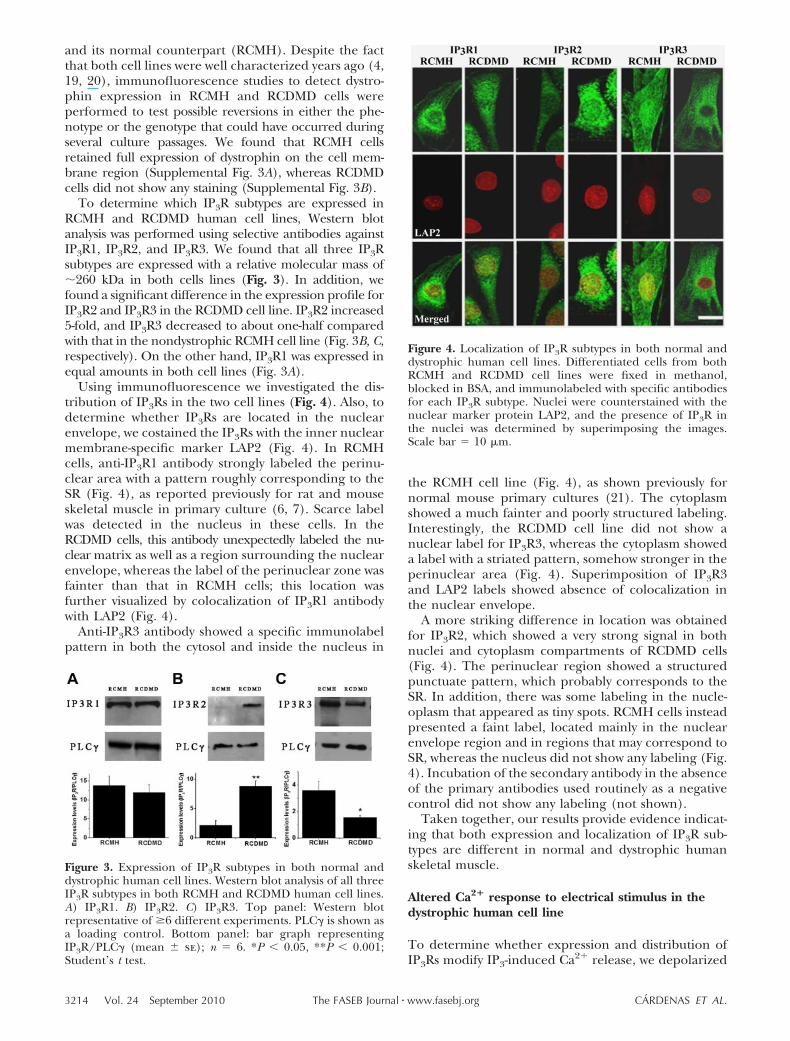
To determine which IP3R subtypes are expressed inRCMH and RCDMD human cell lines, Western blotanalysis was performed using selective antibodies againstIP3R1, IP3R2, and IP3R3. We found that all three IP3Rsubtypes are expressed with a relative molecular mass of�260 kDa in both cells lines (Fig. 3). In addition, wefound a significant difference in the expression profile forIP3R2 and IP3R3 in the RCDMD cell line. IP3R2 increased5-fold, and IP3R3 decreased to about one-half comparedwith that in the nondystrophic RCMH cell line (Fig. 3B, C,respectively). On the other hand, IP3R1 was expressed inequal amounts in both cell lines (Fig. 3A).
Using immunofluorescence we investigated the dis-tribution of IP3Rs in the two cell lines (Fig. 4). Also, todetermine whether IP3Rs are located in the nuclearenvelope, we costained the IP3Rs with the inner nuclearmembrane-specific marker LAP2 (Fig. 4). In RCMHcells, anti-IP3R1 antibody strongly labeled the perinu-clear area with a pattern roughly corresponding to theSR (Fig. 4), as reported previously for rat and mouseskeletal muscle in primary culture (6, 7). Scarce labelwas detected in the nucleus in these cells. In theRCDMD cells, this antibody unexpectedly labeled the nu-clear matrix as well as a region surrounding the nuclearenvelope, whereas the label of the perinuclear zone wasfainter than that in RCMH cells; this location wasfurther visualized by colocalization of IP3R1 antibodywith LAP2 (Fig. 4).
Anti-IP3R3 antibody showed a specific immunolabelpattern in both the cytosol and inside the nucleus in
the RCMH cell line (Fig. 4), as shown previously fornormal mouse primary cultures (21). The cytoplasmshowed a much fainter and poorly structured labeling.Interestingly, the RCDMD cell line did not show anuclear label for IP3R3, whereas the cytoplasm showeda label with a striated pattern, somehow stronger in theperinuclear area (Fig. 4). Superimposition of IP3R3and LAP2 labels showed absence of colocalization inthe nuclear envelope.
A more striking difference in location was obtainedfor IP3R2, which showed a very strong signal in bothnuclei and cytoplasm compartments of RCDMD cells(Fig. 4). The perinuclear region showed a structuredpunctuate pattern, which probably corresponds to theSR. In addition, there was some labeling in the nucle-oplasm that appeared as tiny spots. RCMH cells insteadpresented a faint label, located mainly in the nuclearenvelope region and in regions that may correspond toSR, whereas the nucleus did not show any labeling (Fig.4). Incubation of the secondary antibody in the absenceof the primary antibodies used routinely as a negativecontrol did not show any labeling (not shown).
Taken together, our results provide evidence indicat-ing that both expression and localization of IP3R sub-types are different in normal and dystrophic humanskeletal muscle.
Altered Ca2� response to electrical stimulus in thedystrophic human cell line
To determine whether expression and distribution ofIP3Rs modify IP3-induced Ca2� release, we depolarized
Figure 3. Expression of IP3R subtypes in both normal anddystrophic human cell lines. Western blot analysis of all threeIP3R subtypes in both RCMH and RCDMD human cell lines.A) IP3R1. B) IP3R2. C) IP3R3. Top panel: Western blotrepresentative of �6 different experiments. PLC� is shown asa loading control. Bottom panel: bar graph representingIP3R/PLC� (mean se); n 6. *P � 0.05, **P � 0.001;Student’s t test.
Figure 4. Localization of IP3R subtypes in both normal anddystrophic human cell lines. Differentiated cells from bothRCMH and RCDMD cell lines were fixed in methanol,blocked in BSA, and immunolabeled with specific antibodiesfor each IP3R subtype. Nuclei were counterstained with thenuclear marker protein LAP2, and the presence of IP3R inthe nuclei was determined by superimposing the images.Scale bar 10 �m.
3214 Vol. 24 September 2010 CARDENAS ET AL.The FASEB Journal � www.fasebj.org
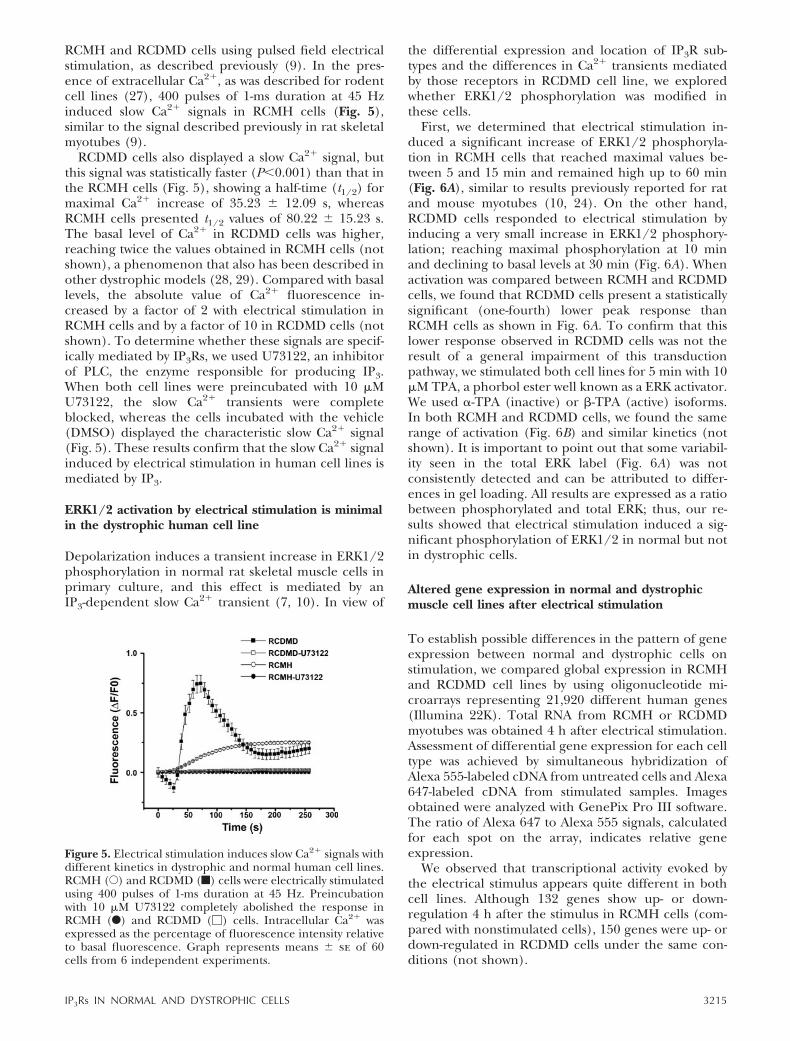
RCMH and RCDMD cells using pulsed field electricalstimulation, as described previously (9). In the pres-ence of extracellular Ca2�, as was described for rodentcell lines (27), 400 pulses of 1-ms duration at 45 Hzinduced slow Ca2� signals in RCMH cells (Fig. 5),similar to the signal described previously in rat skeletalmyotubes (9).
RCDMD cells also displayed a slow Ca2� signal, butthis signal was statistically faster (P�0.001) than that inthe RCMH cells (Fig. 5), showing a half-time (t1/2) formaximal Ca2� increase of 35.23 12.09 s, whereasRCMH cells presented t1/2 values of 80.22 15.23 s.The basal level of Ca2� in RCDMD cells was higher,reaching twice the values obtained in RCMH cells (notshown), a phenomenon that also has been described inother dystrophic models (28, 29). Compared with basallevels, the absolute value of Ca2� fluorescence in-creased by a factor of 2 with electrical stimulation inRCMH cells and by a factor of 10 in RCDMD cells (notshown). To determine whether these signals are specif-ically mediated by IP3Rs, we used U73122, an inhibitorof PLC, the enzyme responsible for producing IP3.When both cell lines were preincubated with 10 �MU73122, the slow Ca2� transients were completeblocked, whereas the cells incubated with the vehicle(DMSO) displayed the characteristic slow Ca2� signal(Fig. 5). These results confirm that the slow Ca2� signalinduced by electrical stimulation in human cell lines ismediated by IP3.
ERK1/2 activation by electrical stimulation is minimalin the dystrophic human cell line
Depolarization induces a transient increase in ERK1/2phosphorylation in normal rat skeletal muscle cells inprimary culture, and this effect is mediated by anIP3-dependent slow Ca2� transient (7, 10). In view of
the differential expression and location of IP3R sub-types and the differences in Ca2� transients mediatedby those receptors in RCDMD cell line, we exploredwhether ERK1/2 phosphorylation was modified inthese cells.
First, we determined that electrical stimulation in-duced a significant increase of ERK1/2 phosphoryla-tion in RCMH cells that reached maximal values be-tween 5 and 15 min and remained high up to 60 min(Fig. 6A), similar to results previously reported for ratand mouse myotubes (10, 24). On the other hand,RCDMD cells responded to electrical stimulation byinducing a very small increase in ERK1/2 phosphory-lation; reaching maximal phosphorylation at 10 minand declining to basal levels at 30 min (Fig. 6A). Whenactivation was compared between RCMH and RCDMDcells, we found that RCDMD cells present a statisticallysignificant (one-fourth) lower peak response thanRCMH cells as shown in Fig. 6A. To confirm that thislower response observed in RCDMD cells was not theresult of a general impairment of this transductionpathway, we stimulated both cell lines for 5 min with 10�M TPA, a phorbol ester well known as a ERK activator.We used �-TPA (inactive) or �-TPA (active) isoforms.In both RCMH and RCDMD cells, we found the samerange of activation (Fig. 6B) and similar kinetics (notshown). It is important to point out that some variabil-ity seen in the total ERK label (Fig. 6A) was notconsistently detected and can be attributed to differ-ences in gel loading. All results are expressed as a ratiobetween phosphorylated and total ERK; thus, our re-sults showed that electrical stimulation induced a sig-nificant phosphorylation of ERK1/2 in normal but notin dystrophic cells.
Altered gene expression in normal and dystrophicmuscle cell lines after electrical stimulation
To establish possible differences in the pattern of geneexpression between normal and dystrophic cells onstimulation, we compared global expression in RCMHand RCDMD cell lines by using oligonucleotide mi-croarrays representing 21,920 different human genes(Illumina 22K). Total RNA from RCMH or RCDMDmyotubes was obtained 4 h after electrical stimulation.Assessment of differential gene expression for each celltype was achieved by simultaneous hybridization ofAlexa 555-labeled cDNA from untreated cells and Alexa647-labeled cDNA from stimulated samples. Imagesobtained were analyzed with GenePix Pro III software.The ratio of Alexa 647 to Alexa 555 signals, calculatedfor each spot on the array, indicates relative geneexpression.
We observed that transcriptional activity evoked bythe electrical stimulus appears quite different in bothcell lines. Although 132 genes show up- or down-regulation 4 h after the stimulus in RCMH cells (com-pared with nonstimulated cells), 150 genes were up- ordown-regulated in RCDMD cells under the same con-ditions (not shown).
Figure 5. Electrical stimulation induces slow Ca2� signals withdifferent kinetics in dystrophic and normal human cell lines.RCMH (E) and RCDMD (f) cells were electrically stimulatedusing 400 pulses of 1-ms duration at 45 Hz. Preincubationwith 10 �M U73122 completely abolished the response inRCMH (F) and RCDMD (�) cells. Intracellular Ca2� wasexpressed as the percentage of fluorescence intensity relativeto basal fluorescence. Graph represents means se of 60cells from 6 independent experiments.
3215IP3Rs IN NORMAL AND DYSTROPHIC CELLS
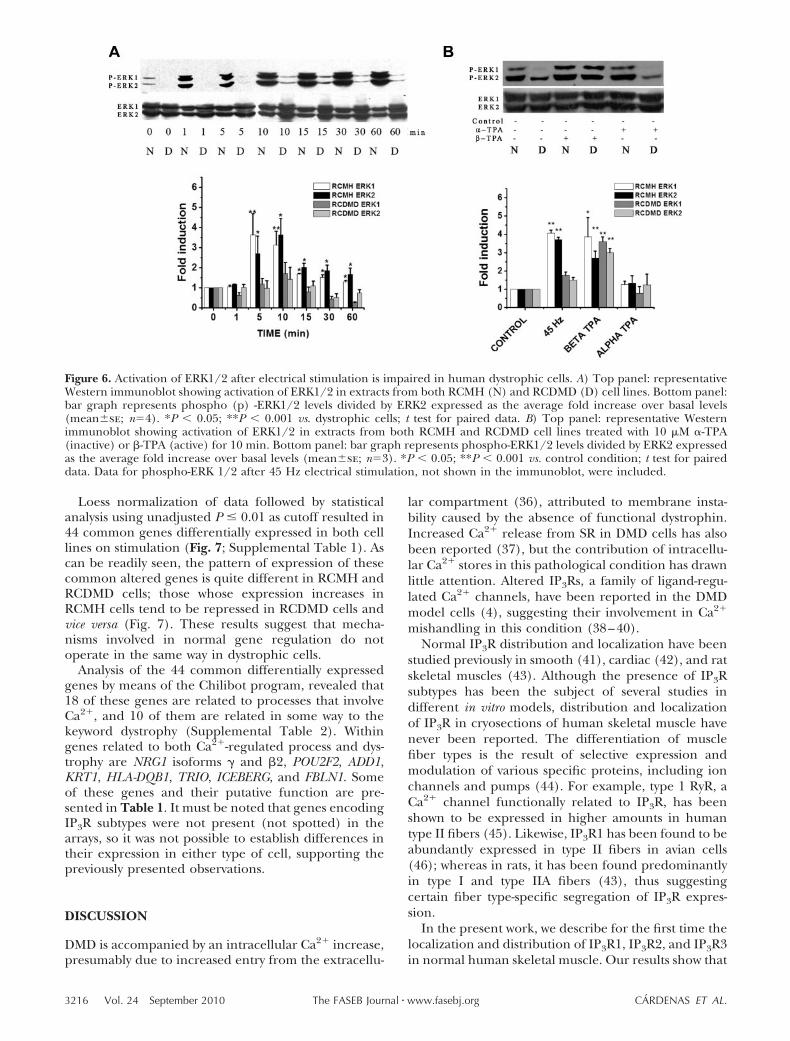
Loess normalization of data followed by statisticalanalysis using unadjusted P � 0.01 as cutoff resulted in44 common genes differentially expressed in both celllines on stimulation (Fig. 7; Supplemental Table 1). Ascan be readily seen, the pattern of expression of thesecommon altered genes is quite different in RCMH andRCDMD cells; those whose expression increases inRCMH cells tend to be repressed in RCDMD cells andvice versa (Fig. 7). These results suggest that mecha-nisms involved in normal gene regulation do notoperate in the same way in dystrophic cells.
Analysis of the 44 common differentially expressedgenes by means of the Chilibot program, revealed that18 of these genes are related to processes that involveCa2�, and 10 of them are related in some way to thekeyword dystrophy (Supplemental Table 2). Withingenes related to both Ca2�-regulated process and dys-trophy are NRG1 isoforms � and �2, POU2F2, ADD1,KRT1, HLA-DQB1, TRIO, ICEBERG, and FBLN1. Someof these genes and their putative function are pre-sented in Table 1. It must be noted that genes encodingIP3R subtypes were not present (not spotted) in thearrays, so it was not possible to establish differences intheir expression in either type of cell, supporting thepreviously presented observations.
DISCUSSION
DMD is accompanied by an intracellular Ca2� increase,presumably due to increased entry from the extracellu-
lar compartment (36), attributed to membrane insta-bility caused by the absence of functional dystrophin.Increased Ca2� release from SR in DMD cells has alsobeen reported (37), but the contribution of intracellu-lar Ca2� stores in this pathological condition has drawnlittle attention. Altered IP3Rs, a family of ligand-regu-lated Ca2� channels, have been reported in the DMDmodel cells (4), suggesting their involvement in Ca2�
mishandling in this condition (38–40).Normal IP3R distribution and localization have been
studied previously in smooth (41), cardiac (42), and ratskeletal muscles (43). Although the presence of IP3Rsubtypes has been the subject of several studies indifferent in vitro models, distribution and localizationof IP3R in cryosections of human skeletal muscle havenever been reported. The differentiation of musclefiber types is the result of selective expression andmodulation of various specific proteins, including ionchannels and pumps (44). For example, type 1 RyR, aCa2� channel functionally related to IP3R, has beenshown to be expressed in higher amounts in humantype II fibers (45). Likewise, IP3R1 has been found to beabundantly expressed in type II fibers in avian cells(46); whereas in rats, it has been found predominantlyin type I and type IIA fibers (43), thus suggestingcertain fiber type-specific segregation of IP3R expres-sion.
In the present work, we describe for the first time thelocalization and distribution of IP3R1, IP3R2, and IP3R3in normal human skeletal muscle. Our results show that
Figure 6. Activation of ERK1/2 after electrical stimulation is impaired in human dystrophic cells. A) Top panel: representativeWestern immunoblot showing activation of ERK1/2 in extracts from both RCMH (N) and RCDMD (D) cell lines. Bottom panel:bar graph represents phospho (p) -ERK1/2 levels divided by ERK2 expressed as the average fold increase over basal levels(meanse; n4). *P � 0.05; **P � 0.001 vs. dystrophic cells; t test for paired data. B) Top panel: representative Westernimmunoblot showing activation of ERK1/2 in extracts from both RCMH and RCDMD cell lines treated with 10 �M �-TPA(inactive) or �-TPA (active) for 10 min. Bottom panel: bar graph represents phospho-ERK1/2 levels divided by ERK2 expressedas the average fold increase over basal levels (meanse; n3). *P � 0.05; **P � 0.001 vs. control condition; t test for paireddata. Data for phospho-ERK 1/2 after 45 Hz electrical stimulation, not shown in the immunoblot, were included.
3216 Vol. 24 September 2010 CARDENAS ET AL.The FASEB Journal � www.fasebj.org

IP3Rs are distributed in an organized fashion at histo-logical and cellular levels. Expression of receptor sub-types is segregated according to the fiber subtype, asshown by double immunostaining with slow myosin(Fig. 1). At the cellular level, we observed that labelingfollows distinctive patters that coincide with SR (IP3R1,IP3R2, and IP3R3), sarcolemmal (IP3R1 and IP3R3), ornuclear localization (IP3R1 and IP3R3). Although in alow proportion, receptor subtypes are also present inslow (type I) fibers compared with negative controls;fast (type II) muscle fibers show much more intenselabeling for both IP3R1 and IP3R2 subtypes than type Ifibers (Fig. 1). We know that muscle satellite cellsexpress IP3Rs (22), but we were unable to discriminatewhether at least some of the nuclei we have seenlabeled could correspond to muscle satellite cells. Thispossibility should be further investigated.
In biopsy samples from patients with DMD, we foundthat 24 7% of the type II fibers lost IP3R1 labeling(Fig. 2B, asterisks) and type I fibers gained IP3R2,before they lost myosin-specific expression. BecauseIP3R has been shown to regulate myosin-type expres-sion in chicken cultured myotubes (46), it is possiblethat the expression of IP3Rs is necessary to maintaincell type differentiation, which is actually very poor indystrophic muscle, in accordance with a low capacity ofmyofibers to maintain their function in the absence ofdystrophin. In contrast to what we found in tissuederived from patients with DMD, RCDMD cells present
the same levels of IP3R1 subtype as do normal RCMHcells (Fig. 3A). However, IP3R2 levels are largely in-creased in RCDMD cells (Fig. 3B), a phenomenonsimilar to that observed in DMD tissue, in which IP3R2is widespread in both type I and type II fibers (Fig. 2C),whereas in normal tissue it is restricted to type II fibers(Fig. 1C).
The cellular distribution of IP3Rs in the normalRCMH cell line (Fig. 4) is compatible with what wefound in adult human muscle (Fig. 1): IP3R1 and IP3R2in the cytoplasm, possibly in the SR region, and IP3R3located mainly in the nuclei (Fig. 4). However, wefound clear differences in all three IP3R locations inRCDMD dystrophic cells with respect to the normal cellline: IP3R1 localization changed from perinuclear/SRto nuclear (nuclear envelope and nucleoplasm), IP3R2is overexpressed and located de novo in the nucleo-plasm, a location that has been reported in nonskeletalmuscle cells (47), whereas IP3R3 changed from thenucleus to the SR (Fig. 4). These changes in IP3Rlocalization in RCDMD cells could possibly be attrib-uted to lack of dystrophin, because the absence ofdystrophin causes the misallocation of several otherproteins (1). Although a direct relationship betweenIP3Rs and dystrophin has not been described, it hasbeen reported that transfection of mouse-derived dys-trophic cells with mini-dystrophin can modulate theIP3-dependent Ca2� signals (40). The different IP3Rsubtypes display differences in affinity and sensitivity to
Figure 7. Differentially expressed genes inRCMH and RCDMD cell lines after electricalstimulation. ANOVA and t tests were conductedfor 9 arrays at 4 h after electrical stimulation (5arrays for RCMH and 4 arrays for RCDMD).Unadjusted P � 0.01 was obtained for 44clones. Clusters were made using clusteringhierarchical tool of GEPAS. Results are ex-pressed as mean log2 ratios; differences in geneexpression are shown in color according to theright bottom scale. For further details, see Ma-terials and Methods.
3217IP3Rs IN NORMAL AND DYSTROPHIC CELLS

IP3 (48). IP3R2 is the subtype most sensitive to IP3,mobilizes more Ca2� than the other IP3R isoforms, andis not inhibited by Ca2� as IP3R1 is (17, 49); thus,overexpression of IP3R2 observed in RCDMD cells (Fig.3B) could be playing an important role in the Ca2�
mishandling reported for DMD. In contrast to ourresults, a slight down-regulation of IP3R2 was recentlydescribed in a mouse-derived dystrophic cell line (40).This difference, however, could be explained as speciesdifferences and could possibly be related to the differ-ent phenotype and characteristics of the disease ob-served between mice and humans.
IP3-dependent Ca2� release can be activated in skel-etal muscle cells by membrane depolarization (6, 9)and has been characterized as the mechanism forexcitation-transcription coupling (10–12). Activationof DHPR as a voltage sensor triggers ATP release, whichactivates a G-protein-coupled purinergic receptor(P2Y) and PI3 kinase, which in turn activates PLC toproduce IP3 and diacyl glycerol (27, 50, 51). A slowCa2� transient produced by this mechanism can beseen in cultured muscle cells but has not been studiedin human skeletal muscle. Therefore, it was importantto determine whether electrical stimulation in humancell lines induced IP3R-dependent Ca2� responses andwhether this signal has kinetics similar to those re-ported previously in mouse and rat muscle cell cultures(6, 9). Effectively, normal RCMH human cells (stimu-lated with 400 1-ms pulses at 45 Hz) displayed a slowCa2� signal with a half-time (t1/2) for a maximal Ca2�
increase of 80.22 15.23 s (Fig. 5), very similar to thatpreviously reported in rat myotubes (t1/2 98.407.92 s)(9). In addition, this signal was completely blocked byU73122, an inhibitor of PLC, the enzyme responsiblefor IP3 production, confirming the source of this signal(Fig. 5). On the other hand, dystrophic RCDMD cellsdisplayed a transient, IP3-dependent, slow Ca2� releasethat is much larger and faster compared with that ofRCMH cells (Fig. 5). A previous report showed a higherincrease of IP3 mass in dystrophic cells after depolar-ization and also a larger number of IP3Rs (4). Thelarger amount of IP3R2s revealed by Western blot (Fig.3B), together with immunofluorescence labeling ofnuclei in dystrophic cells (Fig. 4), suggests that RCDMDcells have a much higher content of IP3R2 located inthe nuclei. These results may explain the larger (andfaster) IP3-dependent Ca2� signal observed in RCDMDcells. In dystrophic cells, IP3 pathway deregulation hasalso been implicated in abnormal subsarcolemmalCa2� increases induced by carbachol in mdx-derivedmyotubes (38). In addition, high K� depolarization ofSolC1 cells, dystrophic mouse-derived myotubes, in-duces an IP3-dependent Ca2� increase higher than thatin normal counterparts, which was restored by mini-dystrophin transfection, suggesting that the IP3 path-way is regulated by dystrophin (39).
It has been noted that the Ca2� increase mediated byIP3 in normal mice skeletal muscle cells induces anincrease in ERK1/2 and CREB phosphorylation (10).Kolodziejczyk et al. in 2001 (52) claimed that no
TABLE 1. Examples of genes differentially regulated in normal and dystrophic muscle cells
Gene symbol, description Condition Suggested physiological role
HLA-DQB1, major histocompatibilitycomplex class II DQ � 1
Unchanged in RCMH;up-regulated in RCDMD
Role in antigen processing and presentation;HLA-DR genes are up-regulated in humanDMD skeletal muscle (30)
NRG1, neuregulin 1 transcript variantHRG
Down-regulated in RCMH;up-regulated in RCDMD
NRG-3 is involved in proliferation anddifferentiation of satellite cells in mdx mice(31) as well as in up-regulation ofutrophin in mdx mouse (32)
POU2F2, POU domain class 2 transcriptionfactor 2
Down-regulated in RCMH;up-regulated in RCDMD
Controls muscle-specific gene expressionduring mammalian myogenesis (33)
ICEBERG, caspase-1 inhibitor Down-regulated in RCMH;up-regulated in RCDMD
Inhibits apoptosis induced by perturbationof cell-ECM associations (34)
FBLN1, fibulin 1 transcript variant C Down-regulated in RCMH;up-regulated in RCDMD
Glycoprotein component of fibrillar ECM;involved in developmental processes andthe organization of ECM architecture;binds to laminin and nidogen withcalcium requirement (Source Searchsoftware tool; http://smd-www.stanford.edu/cgi-bin/source/sourceSearch)
ADD1, adducin 1 � transcript variant 1 Down-regulated in RCMH;unchanged in RCDMD
Membrane cytoskeleton-associated protein;promotes the assembly of spectrin-actinnetwork; binds with high affinity to Ca2�/calmodulin and is a substrate for PKA andPKC (Source Search tool)
TRIO, triple functional domain PTPRinteracting
Down-regulated in RCMH;up-regulated in RCDMD
Regulates actin cytoskeleton reorganization,cell adhesion, and gene transcription viaactivation of Rho GTPases; essential forlate embryonic development, fetal skeletalmuscle organization and neural tissueformation (35)
3218 Vol. 24 September 2010 CARDENAS ET AL.The FASEB Journal � www.fasebj.org

differences in ERK activity existed in either skeletal orcardiac dystrophic cells. However, higher levels ofERK1/2 activation to mechanical stretch (15) or tread-mill exercise (16) were described. In RCDMD cells theERK1/2 pathway was clearly impaired, showing a dis-crete activation after electrical stimulation comparedwith that in RCMH cells (Fig. 6). The fact that ERKactivation could be reduced on a much larger andfaster Ca2� signal points to tight regulation of thisactivity by either the kinetics or the location of the Ca2�
source. This finding also suggests that either dystrophinor dystrophin-associated proteins have a central role inERK activation. Suppression of this pathway could possi-bly be linked to impairment of the developmental pro-gram and death of Duchenne dystrophy cells. Similarinactivation was found in the mdx mouse, in which disrup-tion between laminin and dystroglycan inhibits AKT acti-vation, leading to a loss of muscle cell survival (34).
It is also possible that both the magnitude andkinetics of IP3-dependent Ca2� signals could be criticalin the regulation of gene expression. We have recentlyreported that K� depolarization of C2C12 skeletal mus-cle cell lines induces differential transcriptional activityof a limited number of genes, mainly associated withmetabolism, cell communication, and response tostress (12). A number of genes related to Ca2� signalingpathways are induced at 4 h, reinforcing the potential roleof Ca2� in early steps of signal transduction that leads togene expression (12). Moreover, IP3-dependent slowCa�2 transients are involved in the expression of mostanalyzed genes, such as c-fos, c-jun (10), interleukin-6(11), troponin I (12), hsp70, and hmox-1 (53).
To establish eventual differences in the gene expres-sion pattern on electrical stimulation between normaland dystrophic cells, we compared global expression inRCMH and RCDMD cells by using oligonucleotidemicroarrays. Several genes show differential expressionin RCMH and RCDMD cells after stimulation; never-theless, the expression of only 44 of them is modified inboth cell lines (Fig. 7; Supplemental Table 1). Thepattern of expression (up- or down-regulation) of thesecommon genes is strikingly different between cell lines,and they appear to be regulated in opposite ways (Fig. 7).
Gene expression comparison of human biopsy sam-ples from DMD and normal skeletal muscle has shownthat many of the differentially expressed genes areconsistent with reported histopathological changes(54). For example, immune response signals and extra-cellular matrix (ECM) genes are overexpressed in DMDmuscle, an indication of the infiltration of inflamma-tory cells and connective tissue (54). cDNA analyses ofindividual patients with DMD have shown that genesrelated to the immune response, sarcomere, ECM, andsignaling/cell growth were increased. Up-regulation ofthese genes accompanies dystrophic changes in DMDmuscles such as myofiber necrosis, inflammation, andmuscle regeneration (30). In agreement with this find-ing, within these 44 genes we identified genes related tothe immune response (HLA-DQB1), cytoskeleton/ECMproteins (ADD1, KRT1, and FBLN1), and signaling
(NRG and POU2F2), among others. We found that 18 ofthese 44 genes are related to processes associated withcalcium, and 10 of them have been related in some wayto dystrophy (Supplemental Table 2).
Within the genes whose expression increases inRCDMD cells, particularly interesting in relation tomuscle function and development (Table 1) are thosecoding for the two isoforms of neuregulin (NGR1-�2and NRG1-�) and the POU2F2 gene. NRG1 is a growthfactor that potentiates myogenesis and may play animportant role in differentiation of satellite cells inmuscle regeneration (55). Moreover, NRG stimulatesCa2�-induced glucose transport during contraction(56) and is implicated in the metabolic and prolifera-tive response of muscle to exercise (57). POU2F2 hasbeen described as a transcription factor expressed indeveloping mouse skeletal muscle (33). Interestingly,we recently demonstrated that IP3-dependent slowCa2� transients stimulate NRG1-� expression in bothRCMH and RCDMD cells (unpublished results).
In addition, we found variations in the expression ofICEBERG, HLA-DQB1, ADD1, FBLN1, and TRIO genes thatalso have been associated with Ca2� and dystrophy (Sup-plemental Table 2). Considering that changes observed inDMD muscle biopsies have been related to elevation ofintracellular Ca2� concentration, which could activateCa2�-dependent degradation pathways, resulting in myo-fibril disruption and muscle necrosis (58), it will beinteresting to analyze the roles described for the above-mentioned genes (Table 1). To our knowledge, there areno studies describing the role of membrane depolariza-tion on the expression of these genes, and further studiesare needed to explore the involvement of IP3-mediatedslow Ca2� signals in the expression of some of theseparticular genes in skeletal muscle cells.
In conclusion, we have demonstrated that the spe-cific distribution of IP3Rs is partially lost in dystrophichuman muscle, even while fibers still retain their mo-lecular identity (as judged by the expression of myo-sin). It is, thus, theoretically possible that structuralalterations provoked by the absence of dystrophininterfere with appropriate location and functioning ofIP3Rs within the cell-modifying gene expression. Pro-tein interactions within the dystroglycan complex are ofsuch a nature that the absence of one component ofthe complex modifies the expression of several others(59). In this sense, because depolarization induces IP3production and consequent IP3R activation via a com-plex that includes G protein-associated receptors, aninteraction of dystroglycan complex with this pathwayat the membrane level is quite possible (39, 40, 50).This proposed mechanism has yet to be explored andmay account for some of the unexplained physiopatho-logical processes that occur in dystrophin-related mus-cular dystrophy.
The authors are grateful to Monica Silva for culturepreparations and to Dr. Ulises Urzua for microarray labora-tory facilities. The authors thank Dr. Marcos Ganga (Depart-ment of Traumatology, Hospital Clínico Universidad deChile, Independencia, Chile) and Drs. Christian Bermudez
3219IP3Rs IN NORMAL AND DYSTROPHIC CELLS

and Ernesto Larraín (Department of Cardiology, HospitalClínico Universidad de Chile) for their collaboration inobtaining normal tissue samples. This work was supported byFondo de Investigacion Avanzada en Areas Prioritarias(FONDAP; grant 15010006) and Fondo Nacional de Investi-gacion Científica y Tecnologica (grant 1080120 to E.J.),ECOS Sud-CONICYT exchange program (C03S02), ProyectoOAIC (OAIC 106-05), Hospital Clínico Universidad de Chile(J.A.B. and C.C.), and the Association Francaise Contre lesMyopathies (AFM) (J.M.). C.C. was supported by a postdoc-toral fellowship from FONDAP and by a short-term fellowshipfrom AFM at Laboratoire de Neurobiologie Cellulaire etMoleculaire (France). N.J. was supported by a postdoctoralfellowship from project PSD24, Programa Bicentenario deCiencia y Tecnología.
REFERENCES
1. Batchelor, C. L., and Winder, S. J. (2006) Sparks, signals andshock absorbers: how dystrophin loss causes muscular dystro-phy. Trends Cell Biol. 16, 198–205
2. Gailly, P. (2002) New aspects of calcium signaling in skeletalmuscle cells: implications in Duchenne muscular dystrophy.Biochim. Biophys. Acta 1600, 38–44
3. Culligan, K., Banville, N., Dowling, P., and Ohlendieck, K.(2002) Drastic reduction of calsequestrin-like proteins andimpaired calcium binding in dystrophic mdx muscle. J. Appl.Physiol. 92, 435–445
4. Liberona, J. L., Powell, J. A., Shenoi, S., Petherbridge, L.,Caviedes, R., and Jaimovich, E. (1998) Differences in bothinositol 1,4,5-trisphosphate mass and inositol 1,4,5-trisphos-phate receptors between normal and dystrophic skeletal musclecell lines. Muscle Nerve 21, 902–909
5. Patterson, R. L., Boehning, D., and Snyder, S. H. (2004) Inositol1,4,5-trisphosphate receptors as signal integrators. Annu. Rev.Biochem. 73, 437–465
6. Jaimovich, E., Reyes, R., Liberona, J. L., and Powell, J. A. (2000)IP3 receptors, IP3 transients, and nucleus-associated Ca2� signalsin cultured skeletal muscle. Am. J. Physiol. Cell Physiol. 278,C998–C1010
7. Powell, J. A., Carrasco, M. A., Adams, D. S., Drouet, B., Rios, J.,Muller, M., Estrada, M., and Jaimovich, E. (2001) IP3 receptorfunction and localization in myotubes: an unexplored Ca2�
signaling pathway in skeletal muscle. J. Cell Sci. 114(Pt. 20),3673–3683
8. Jaimovich, E., and Rojas, E. (1994) Intracellular Ca2� transientsinduced by high external K� and tetracaine in cultured ratmyotubes. Cell Calcium 15, 356–568
9. Eltit, J. M., Hidalgo, J., Liberona, J. L., and Jaimovich, E. (2004)Slow calcium signals after tetanic electrical stimulation in skel-etal myotubes. Biophys. J. 86, 3042–3051
10. Carrasco, M. A., Riveros, N., Rios, J., Muller, M., Torres, F.,Pineda, J., Lantadilla, S., and Jaimovich, E. (2003) Depolariza-tion-induced slow calcium transients activate early genes inskeletal muscle cells. Am. J. Physiol. Cell Physiol. 284, C1438–C1447
11. Juretic, N., Garcia-Huidobro, P., Iturrieta, J. A., Jaimovich, E.,and Riveros, N. (2006) Depolarization-induced slow Ca2� tran-sients stimulate transcription of IL-6 gene in skeletal musclecells. Am. J. Physiol. Cell Physiol. 290, C1428–C1436
12. Juretic, N., Urzua, U., Munroe, D. J., Jaimovich, E., and Riveros,N. (2007) Differential gene expression in skeletal muscle cellsafter membrane depolarization. J. Cell. Physiol. 210, 819–830
13. Chen, Y. W., Zhao, P., Borup, R., and Hoffman, E. P. (2000)Expression profiling in the muscular dystrophies: identificationof novel aspects of molecular pathophysiology. J. Cell Biol. 151,1321–1336
14. Haslett, J. N., Sanoudou, D., Kho, A. T., Han, M., Bennett, R. R.,Kohane, I. S., Beggs, A. H., and Kunkel, L. M. (2003) Geneexpression profiling of Duchenne muscular dystrophy skeletalmuscle. Neurogenetics 4, 163–171
15. Kumar, A., Khandelwal, N., Malya, R., Reid, M. B., and Boriek,A. M. (2004) Loss of dystrophin causes aberrant mechanotrans-duction in skeletal muscle fibers. FASEB J. 18, 102–113
16. Nakamura, A., Yoshida, K., Ueda, H., Takeda, S., and Ikeda, S.(2005) Up-regulation of mitogen activated protein kinases inmdx skeletal muscle following chronic treadmill exercise. Bio-chim. Biophys. Acta 1740, 326–331
17. Ramos-Franco, J., Caenepeel, S., Fill, M., and Mignery, G.(1998) Single channel function of recombinant type-1 inositol1,4,5-trisphosphate receptor ligand binding domain splice vari-ants. Biophys. J. 75, 2783–2793
18. Dubowitz, V. (1985) Muscle Biopsy: A Practical Approach, BaillereTindal, London
19. Caviedes, R., Caviedes, P., Liberona, J. L., and Jaimovich, E.(1994) Ion channels in a skeletal muscle cell line from aDuchenne muscular dystrophy patient. Muscle Nerve 17, 1021–1028
20. Caviedes, R., Liberona, J. L., Hidalgo, J., Tascon, S., Salas, K.,and Jaimovich, E. (1992) A human skeletal muscle cell lineobtained from an adult donor. Biochim. Biophys. Acta 1134,247–255
21. Cardenas, C., Liberona, J. L., Molgo, J., Colasante, C., Mignery,G. A., and Jaimovich, E. (2005) Nuclear inositol 1,4,5-trisphos-phate receptors regulate local Ca2� transients and modulatecAMP response element binding protein phosphorylation. J. CellSci. 118(Pt. 14), 3131–3140
22. Powell, J. A., Molgo, J., Adams, D. S., Colasante, C., Williams, A.,Bohlen, M., and Jaimovich, E. (2003) IP3 receptors and associ-ated Ca2� signals localize to satellite cells and to components ofthe neuromuscular junction in skeletal muscle. J. Neurosci. 23,8185–8192
23. Ibarra, C., Estrada, M., Carrasco, L., Chiong, M., Liberona, J. L.,Cardenas, C., Díaz-Araya, G., Jaimovich, E., and Lavandero, S.(2004) Insulin-like growth factor-1 induces an inositol 1,4,5-trisphosphate-dependent increase in nuclear and cytosolic calciumin cultured rat cardiac myocytes. J. Biol. Chem. 279, 7554–7565
24. Cardenas, C., Muller, M., Jaimovich, E., Perez, F., Buchuk, D.,Quest, A. F., and Carrasco, M. A. (2004) Depolarization ofskeletal muscle cells induces phosphorylation of cAMP responseelement binding protein via calcium and protein kinase C�.J. Biol. Chem. 279, 39122–39131
25. Vaquerizas, J. M., Dopazo, J., and Diaz-Uriarte, R. (2004)DNMAD: web-based diagnosis and normalization for microarraydata. Bioinformatics 20, 3656–3658
26. Herrero, J., Vaquerizas, J. M., Al-Shahrour, F., Conde, L.,Mateos, A., Díaz-Uriarte, J. S., and Dopazo, J. (2004) Newchallenges in gene expression data analysis and the extendedGEPAS. Nucleic Acids Res. 32(Web Server issue), W485–W491
27. Eltit, J. M., García, A. A., Hidalgo, J., Liberona, J. L., Chiong, M.,Lavandero, S., Maldonado, E., and Jaimovich, E. (2006) Mem-brane electrical activity elicits inositol 1,4,5-trisphosphate-de-pendent slow Ca2� signals through a G��/phosphatidylinositol3-kinase � pathway in skeletal myotubes. J. Biol. Chem. 281,12143–12154
28. Imbert, N., Cognard, C., Duport, G., Guillou, C., and Raymond,G. (1995) Abnormal calcium homeostasis in Duchenne muscu-lar dystrophy myotubes contracting in vitro. Cell Calcium 18,177–186
29. Robert, V., Massimino, M. L., Tosello, V., Marsault, R., Cantini,M., Sorrentino, V., and Pozzan, T. (2001) Alteration in calciumhandling at the subcellular level in mdx myotubes. J. Biol. Chem.276, 4647–4651
30. Noguchi, S., Tsukahara, T., Fujita, M., Kurokawa, R., Tachikawa,M., Toda, T., Tsujimoto, A., Arahata, K., and Nishino, I. (2003)cDNA microarray analysis of individual Duchenne musculardystrophy patients. Hum. Mol. Genet. 12, 595–600
31. Turk, R., Sterrenburg, E., de Meijer, E. J., van Ommen, G. J.,den Dunnen, J. T., and ’t Hoen, P. A. (2005) Muscle regener-ation in dystrophin-deficient mdx studied by gene expressionprofiling. BMC Genomics 6, 98
32. Krag, T. O., Bogdanovich, S., Jensen, C. J., Fischer, M. D.,Hansen-Schwartz, J., Javazon, E. H., Flake, A. W., Edvinsson, L.,and Khurana, T. S. (2004) Heregulin ameliorates the dystrophicphenotype in mdx mice. Proc. Natl. Acad. Sci. U. S. A. 101,13856–13860
33. Dominov, J. A., and Miller, J. B. (1996) POU homeodomaingenes and myogenesis. Dev. Genet. 19, 108–118
34. Langenbach, K. J., and Rando, T. A. (2002) Inhibition ofdystroglycan binding to laminin disrupts the PI3K/AKT pathwayand survival signaling in muscle cells. Muscle Nerve 26, 644–653
3220 Vol. 24 September 2010 CARDENAS ET AL.The FASEB Journal � www.fasebj.org

35. O’Brien, S. P., Seipel, K., Medley, Q. G., Bronson, R., Segal, R.,and Streuli, M. (2000) Skeletal muscle deformity and neuronaldisorder in Trio exchange factor-deficient mouse embryos. Proc.Natl. Acad. Sci. U. S. A. 97, 12074–12078
36. Alderton, J. M., and Steinhardt, R. A. (2000) How calcium influxthrough calcium leak channels is responsible for the elevatedlevels of calcium-dependent proteolysis in dystrophic myotubes.Trends Cardiovasc. Med. 10, 268–272
37. Deval, E., Levitsky, D. O., Marchand, E., Cantereau, A.,Raymond, G., and Cognard, C. (2002) Na�/Ca2� exchange inhuman myotubes: intracellular calcium rises in response toexternal sodium depletion are enhanced in DMD. Neuromuscul.Disord. 12, 665–673
38. Basset, O., Boittin, F. X., Dorchies, O. M., Chatton, J. Y., vanBreemen, C., and Ruegg, U. T. (2004) Involvement of inositol1,4,5-trisphosphate in nicotinic calcium responses in dystrophicmyotubes assessed by near-plasma membrane calcium measure-ment. J. Biol. Chem. 279, 47092–47100
39. Balghi, H., Sebille, S., Constantin, B., Patri, S., Thoreau, V.,Mondin, L., Mok, E., Kitzis, A., Raymond, G., and Cognard, C.(2006) Mini-dystrophin expression down-regulates overactiva-tion of G protein-mediated IP3 signaling pathway in dystrophin-deficient muscle cells. J. Gen. Physiol. 127, 171–182
40. Balghi, H., Sebille, S., Mondin, L., Cantereau, A., Constantin, B.,Raymond, G., and Cognard, C. (2006) Mini-dystrophin expres-sion down-regulates IP3-mediated calcium release events inresting dystrophin-deficient muscle cells. J. Gen. Physiol. 128,219–230
41. Tasker, P. N., Michelangeli, F., and Nixon, G. F. (1999) Expres-sion and distribution of the type 1 and type 3 inositol 1,4,5-trisphosphate receptor in developing vascular smooth muscle.Circ. Res. 84, 536–542
42. Moschella, M. C., and Marks, A. R. (1993) Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes. J. CellBiol. 120, 1137–1146
43. Moschella, M. C., Watras, J., Jayaraman, T., and Marks, A. R.(1995) Inositol 1,4,5-trisphosphate receptor in skeletal muscle:differential expression in myofibres. J. Muscle Res. Cell Motil. 16,390–400
44. Froemming, G. R., Murray, B. E., Harmon, S., Pette, D., andOhlendieck, K. (2000) Comparative analysis of the isoform expres-sion pattern of Ca2�-regulatory membrane proteins in fast-twitch,slow-twitch, cardiac, neonatal and chronic low-frequency stimu-lated muscle fibers. Biochim. Biophys. Acta 1466, 151–168
45. Salanova, M., Schiffl, G., Rittweger, J., Felsenberg, D., andBlottner, D. (2008) Ryanodine receptor type-1 (RyR1) expres-sion and protein S-nitrosylation pattern in human soleus myo-fibres following bed rest and exercise countermeasure. Histo-chem. Cell Biol. 130, 105–118
46. Jordan, T., Jiang, H., Li, H., and DiMario, J. X. (2005) Regula-tion of skeletal muscle fiber type and slow myosin heavy chain 2gene expression by inositol trisphosphate receptor 1. J. Cell Sci.118, 2295–2302
47. Laflamme, K., Domingue, O., Guillemette, B. I., andGuillemette, G. (2002) Immunohistochemical localization of
type 2 inositol 1,4,5-trisphosphate receptor to the nucleus ofdifferent mammalian cells. J. Cell. Biochem. 85, 219–228
48. Newton, C. L., Mignery, G. A., and Sudhof, T. C. (1994)Co-expression in vertebrate tissues and cell lines of multipleinositol 1,4,5-trisphosphate (InsP3) receptors with distinct affin-ities for InsP3. J. Biol. Chem. 269, 28613–28619
49. Kasri, N. N., Kocks, S. L., Verbert, L., Hebert, S. S., Callewaert,G., Parys, J. B., Missiaen, L., and De Smedt, H. (2006) Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 isresponsible for a decreased endoplasmic-reticulum Ca2� con-tent in presenilin double knock-out cells. Cell Calcium 40, 41–51
50. Araya, R., Liberona, J. L., Cardenas, J. C., Riveros, N., Estrada,M., Powell, J. A., Carrasco, M. A., and Jaimovich, E. (2003)Dihydropyridine receptors as voltage sensors for a depolariza-tion-evoked, IP3R-mediated, slow calcium signal in skeletalmuscle cells. J. Gen. Physiol. 121, 3–16
51. Buvinic, S., Almarza, G., Bustamante, M., Casas, M., Lopez, J.,Riquelme, M., Saez, J. C., Huidobro-Toro, J. P., and Jaimovich,E. (2009) ATP released by electrical stimuli elicits calciumtransients in skeletal muscle. J. Biol. Chem. 284, 34490–34505
52. Kolodziejczyk, S. M., Walsh, G. S., Balazsi, K., Seale, P., Sandoz,J., Hierlihy, A. M., Rudnicki, M. A., Chamberlain, J. S., Miller,F. D., and Megeney, L. A. (2001) Activation of JNK1 contributesto dystrophic muscle pathogenesis. Curr. Biol. 11, 1278–1282
53. Jorquera, G., Juretic, N., Jaimovich, E., and Riveros, N. (2009)Membrane depolarization induces calcium-dependent upregu-lation of Hsp70 and Hmox-1 in skeletal muscle cells. Am. J.Physiol. Cell Physiol. 297, C581–C590
54. Haslett, J. N., Sanoudou, D., Kho, A. T., Bennett, R. R.,Greenberg, S. A., Kohane, I. S., Beggs, A. H., and Kunkel, L. M.(2002) Gene expression comparison of biopsies from Duch-enne muscular dystrophy (DMD) and normal skeletal muscle.Proc. Natl. Acad. Sci. U. S. A. 99, 15000–15005
55. Hirata, M., Sakuma, K., Okajima, S., Fujiwara, H., Inashima, S.,Yasuhara, M., and Kubo, T. (2007) Increased expression ofneuregulin-1 in differentiating muscle satellite cells and inmotoneurons during muscle regeneration. Acta Neuropathol.113, 451–459
56. Canto, C., Chibalin, A. V., Barnes, B. R., Glund, S., Suarez, E.,Ryder, J. W., Palacín, M., Zierath, J. R., Zorzano, A., and Guma,A. (2006) Neuregulins mediate calcium-induced glucose trans-port during muscle contraction. J. Biol. Chem. 281, 21690–21697
57. Lebrasseur, N. K., Cote, G. M., Miller, T. A., Fielding, R. A., andSawyer, D. B. (2003) Regulation of neuregulin/ErbB signalingby contractile activity in skeletal muscle. Am. J. Physiol. CellPhysiol. 284, C1149–C1155
58. Turner, P. R., Schultz, R., Ganguly, B., and Steinhardt, R. A.(1993) Proteolysis results in altered leak channel kinetics andelevated free calcium in mdx muscle. J. Membr. Biol. 133, 243–251
59. Gillis, J. M. (1999) Understanding dystrophinopathies: an in-ventory of the structural and functional consequences of theabsence of dystrophin in muscles of the mdx mouse. J. MuscleRes. Cell Motil. 20, 605–625
Received for publication December 28, 2009.Accepted for publication March 28, 2010.
3221IP3Rs IN NORMAL AND DYSTROPHIC CELLS




















