
Ab initio study of electronic structure and magnetic properties in ferromagnetic Be 1−x Mn x Se...
10
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/authorsrights
Transcript of Ab initio study of electronic structure and magnetic properties in ferromagnetic Be 1−x Mn x Se...

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Ab initio study of electronic structure and magnetic propertiesin ferromagnetic Be1−xMnxSe and Be1−xMnxTe alloys
N.A. Noor a, S.M. Alay-e-Abbas b,c, Y. Saeed b, S.M. Ghulam Abbas d, A. Shaukat c,⁎
a Department of Physics, University of the Punjab, Quaid-e-Azam Campus, 54590 Lahore, Pakistanb Department of Physics, GC University Faisalabad, Allama Iqbal Road, Faisalabad 38000, Pakistanc Department of Physics, University of Sargodha, Sargodha 40100, Pakistand Department of Chemistry, University of Agriculture, Faisalabad 38040, Pakistan
a r t i c l e i n f o
Article history:Received 11 January 2012Received in revised form21 February 2013Available online 14 March 2013
Keywords:First principles calculationDensity functional theoryElectronic structureMagnetic property
a b s t r a c t
Structural, electronic and magnetic properties of Mn-doped BeSe and BeTe have been studied byemploying the full-potential linear augmented plane waves plus local orbitals (FP-LAPWþ lo) methodwithin the spin-polarized density functional theory (DFT). The investigations are carried out by varyingthe Mn concentration, “x”, in BeSe and BeTe host matrices for x¼0.25, 0.5 and 0.75. The results of spin-polarized calculations manifest the presence of ferromagnetic band structures with both spin-up andspin-down alignments. Our calculated results of band structures reveal that for x¼0.25, 0.5 and 0.75, Be1−xMnxSe has a half-metallic (HM) band structure profile showing 100% spin polarization at the Fermilevel. On the other hand, the Be1−xMnxTe band structure shows complete 100% spin polarization at theFermi level only for x¼0.25 and 0.5. Spin-dependent charge densities have been calculated to study thebonding nature, and the values of the exchange constants, N0α and N0β, are also determined which areconsistent with the values from the typical magneto-optical experiment. In addition, the calculations ofspin exchange splitting, ΔX ðdÞ, exhibit important indication regarding the attractive effective potential forminority spin rather than majority spin. For each concentration x, the value of total magnetic momenthas been estimated to be 5μB, which reveals that addition of Mn impurity does not affect the holeconcentration of a perfect BeSe (Te) crystal.
& 2013 Elsevier B.V. All rights reserved.
1. Introduction
Wide band-gap semiconductors, with spin degree of freedomintroduced by substituting very small concentration of magneticions such as Cr or Mn, lead to the formation of important materialscalled diluted magnetic semiconductors (DMS). These magneticmaterials have attracted considerable research interest due to theirpotential applications in innovative spin-electronic devices, whichare regarded as an alternative to conventional microelectronics.Among various applications, “spin field effect transistors”, “spinvalves” and “spin qubits” are interesting applications of electronicspin as well as charge coordinate dependent materials [1–5]. Onthe other hand, another group of materials showing half-metallicferromagnetism consists of transition metal (TM)-doped semicon-ductors in which the concentration of the magnetic ion can be ashigh as that of the host component that has been investigated inthe recent past [6–8]. Due to the introduction of magnetic ion as adopant, these compounds are distinct from the conventional octet
isovalent semiconductor alloys and exhibit interesting propertiesresulting out of a combination of magnetism and semiconductivityas well as band structure characteristics. Contrary to the DMSs, inwhich the impurity atom interacts with the host material throughconduction electrons, in TM-doped semiconductor alloys, the TMatom is ordered in a periodical way in the lattice thereby inter-acting directly with the host atoms. Initial experimental effortshave shown that in the presence of an external magnetic field, thespin polarized electrons aligned by the paramagnetic II–VI semi-conductor layer could be injected into an AlGaAs light emittingdiode [9]. In further efforts, it was desired to remove the externalmagnetic field and to evolve a mechanism so that a combination offerromagnetic (FM) material with a semiconductor can be used forthe orientation of electron or hole spins. The obvious difficulty ininjecting spin alignment carriers from a ferromagnetic metal into asemiconductor can be removed by using a ferromagnetic semi-conductor with smaller conductivity than metals for complete spinpolarization [10–11].
To the best of our knowledge, no experimental investigation hasbeen carried out for magnetic properties of the Be1−xMnxSe alloyssystem in the zinc blende phase so far; however, substantial experi-mental as well as theoretical investigations have been performed on
Contents lists available at SciVerse ScienceDirect
journal homepage: www.elsevier.com/locate/jmmm
Journal of Magnetism and Magnetic Materials
0304-8853/$ - see front matter & 2013 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.jmmm.2013.02.040
⁎ Corresponding author. Tel.: þ92 48 9230618; fax: þ92 48 9230671.E-mail address: [email protected] (A. Shaukat).
Journal of Magnetism and Magnetic Materials 339 (2013) 11–19

Author's personal copy
Mn doped IIB–Se alloys. One such example in the zinc blende phase isZn1−xMnxSe, which has been extensively studied both in FM andantiferromagnetic (AFM) phases [12–14]. Contrary to the case of Mndoped BeSe alloys, molecular-beam epitaxy (MBE) grown Be1−xMnxTe [15] show that these alloys possess perfect lattice matchingto GaAs and ZnSe, which is essential for the growth of heterostruc-tures with reduced concentration of defects and exclusively high p-type dupability. Importantly, the MBE grown Be1−xMnxTe have astable FM phase at low temperature having a positive Curie–Weisstemperature (Tcw) and open hysteresis loop below Tcw [15]. On theother hand, surprisingly low value of the exchange constant N0βobtained from magneto-optical measurements of Be1−xMnxTe/ZnTeheterostructures [16] has led to a number of theoretical investiga-tions on the magnetic properties of Mn-doped IIA–VI semiconductors[17–18]. The theoretical study carried out by Picozzi et al. on the Be1−xMnxTe ordered alloys for selected low concentration of Mn in thezinc blende phase shows that these systems are semiconductors andfavor an AFM spin configuration that can be attributed to the FMcoupling between Mn d states and anion p states [18]. In spite of thefact that first-principles calculations like the ones reported in Ref.[18] show that Mn-doped Be chalcogenide prefers AFMas the stable magnetic phase at ambient conditions, the magneticproperties of these novel materials are still under thorough investi-gation [19].
From the abovementioned experimental and theoretical studiesit is clear that alloying of Mnwith BeSe and BeTe can give ascent tointeresting magnetic materials. No systematic study is available forboth low as well as high concentration of Mn in FM Be1−xMnxSeand Be1−xMnxTe so far. In the present study, our interest ispertinent to perform accurate electronic structure calculationsand to investigate magnetic properties of the FM Be1−xMnxSeand Be1−xMnxTe alloys by utilizing the state-of-the-art full poten-tial linear augmented plane wave plus local orbital (FP-LAPWþ lo)approach to get self-consistent solutions for the Kohn–Sham typeequations. As DFT calculations are temperature independent, inthis paper we hope to provide a detailed account of the hypothe-tical FM ordering in Mn-doped BeSe and BeTe in terms ofelectronic structure and magnetic properties.
2. Method of calculations
As stated earlier, the calculations for Be1−xMnxSe andBe1−xMnxTe alloys in the FM zinc blende phase have been
performed using the FP-LAPWþ lo method to solve the Kohn–Sham equations as implemented in the WIEN2K code [20].A muffin-tin (MT) model for crystal potential is assumed and theelectrons of constituent atoms are partitioned into core andvalence states. The core states have been treated fully relativisti-cally relaxed in the spherical approximation, while the valencestates have been treated scalar-relativistically. For these calcula-tions no spin–orbit effects have been included. The binary com-pounds (BeSe and BeTe) have been modelled in the zinc blendedstructure with space group F-43m (# 216), whereas the alloys unitcells are obtained for the compositions x¼0.25, 0.50 and 0.75 byreplacing one, two and three atoms, respectively, in the end binarycompound's supercell. In case of x¼0.25 and 0.75 the obtainedstructure is an eight-atom cubic lattice (space group P-43m, #215), while for x¼0.50 the smallest ordered structure is a four-atom tetragonal cell (space group P-4m2, # 115) [21].
Within the semi-relativistic approximation, the valence states(3d state of transition metal Mn, and the s and p states of Se andTe) are treated self-consistently, while the core states ([He] forBe and [Ar] for Mn and Se, and [Kr] for Te) are treated self-consistently as well as fully relativistically relaxed in the sphericalapproximation. The expansion of basis functions, charge densityand potential is carried out within the radius of muffin-tin spheresalong with spherical harmonics up to a cut-off value lmax¼10 andthe plane wave Fourier series in the interstitial region. Theparameter RMT×Kmax¼8 is used to find the matrix size, whereKmax is the plane wave cut-off and RMT is the smallest of all atomicradii. For Be1−xMnxSe and Be1−xMnxTe alloys, the values of muffin-tin radii are chosen as 2.5, 2.4, 2.26 and 2.0 a.u. for Mn, Te, Se andBe, respectively. We used 3000 k-points in these calculations thatresulted in 70, 129 and 70 k-points in the irreducible wedge of theBrillouin zone for x¼0.25, 0.50 and 0.75, respectively. The con-vergence of crystal's total energy to a value of less than 0.00001 Ryis achieved by the method of iteration.
3. Results and discussion
3.1. Structural properties
At ambient temperature and pressure, BeSe and BeTe possesszinc blende structure with lattice constants a¼5.137 Å anda¼5.617 Å, respectively [22]. At increased pressure, the hostbinary semiconductors undergo a structural phase transition from
Table 1Calculated value of lattice parameters and bulk modulus for Be1−xMnxSe and Be1−xMnxTe alloys, and their binary compounds compared to available experimental and othertheoretical results.
x Lattice constant a0 (A) Bulk Modulus B (GPa)
This work Expt. Other calculations This work Expt. Other calculations
0 5.13 5.137a 5.183b, 5.037c 75.86 92.2a 74.569b, 98.0c
0.25 5.41 – – 68.52 – –
Be1−xMnxSe 0.50 5.70 – – 52.30 – –
0.75 5.79 – – 48.79 – –
1.0 5.84 5.90d 5.28e 42.79 – 130.88e
Be1−xMnxTe 0 5.61 5.617a 5.671b, 5.531c 60.86 68.8a 56.128b, 70.0c
0.25 6.01 – – 45.57 – –
Be1−xMnxTe 0.50 6.14 – – 41.52 – –
0.75 6.24 – – 39.78 – –
1.0 6.27 6.33d 6.25f, 5.65e 35.36 37e 45.01f, 91.47e
a Ref. [38].b Ref. [39].c Ref. [40].d Ref. [41].e Ref. [42].f Ref. [43].
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–1912

Author's personal copy
zinc blende structure to NiAs type structure, which is the groundstate structure for AFM MnTe. On the contrary, under ambientconditions, MnSe (AFM phase) shows NaCl type structure. Thecompounds BeSe/BeTe, MnSe and MnTe, which are the end binarycompounds in this study, possess different stable crystal structuresunder the same conditions. It had been shown experimentally thatartificially stable zinc blende Mn-chalcogenides can be epitaxially-grown successfully or by alloying them with II–VI semiconductors;for example, the zinc blende MnTe and ZnMnTe alloys wereprepared experimentally with MBE [23].
As structural optimization plays a key role in the computationalinvestigation of ground state properties of materials [24], thestructural properties of the Be1−xMnxSe and Be1−xMnxTe alloyshave been computed by using the structural optimization tool ofthe WIEN2K code. We have calculated the total energies for severalvolumes of the unit cell of these alloys in FM ordering using thegeneralized gradient approximation functional proposed by Wuand Cohen (WC GGA) [25] and the ground state structural para-meters have been computed by fitting these data to the empiricalequation of states [26]. Our calculated values of equilibrium latticeconstants a0, and bulk moduli, B0, are presented in Table 1 and thecalculated total energies plotted as a function of unit cell volumesfor Be1−xMnxSe and Be1−xMnxTe at x¼0.25, 0.5 and 0.75 aredepicted in Fig. 1. Structural optimization curves were alsoobtained for the MnSe and MnTe compounds in the zinc blendeferromagnetic phase and the optimized lattice constants and bulkmoduli are presented in Table 1. Theoretical evidences showa large deviation of MnTe lattice constants [18,27] calculated byusing the local spin density approximation (LSDA) as compared tothat calculated by GGA results [28]. Table 1 reveals a closeagreement of our calculated MnTe lattice constant to that of earlierGGA results. The improvement of results can be attributed to theGGA functional which validates the accuracy by removing the socalled “overbinding” introduced by the LSDA parameterizationscheme. For the rest of the concentration range, Table 1 clearlyshows that the variation of concentration value from 0.25 to1.0 results in a corresponding increase of lattice constant anddecrease of bulk modulus which means that the alloys becomemore and more softer with the replacement of Be cations by Mn.Fig. 2 depicts the non-linear variation in the calculated equilibriumlattice constants of Be1−xMnxSe and Be1−xMnxTe alloys versus Mnconcentration. A deviation from Vegard's law, with large bowing inthe lattice constant, is clearly visible.
In order to exploit the solubility of Mn in BeSe and BeTe, wehave computed the heat of formation (ΔH) for all the Mn-doped
binary compounds using the method reported in Ref. [21]. Ourcomputed ΔH values for Be0.75Mn0.25Se, Be0.50Mn0.50Se andBe0.25Mn0.75Se are −1.10 eV, −0.90 eV and −0.68 eV, and forBe0.75Mn0.25Te, Be0.50Mn0.50Te and Be0.25Mn0.75Te are −1.43 eV,−1.22 eV and −0.50 eV, respectively. It is clear that on increasingthe concentration of Mn-dopant the heat of formation is decreasedsuggesting that with the increasing Mn content the stability of thecompound decreases which is consistent with the decreasing bulkmodulus values listed in Table 1. In spite of the fact that it isdifficult to draw a quantitative conclusion regarding the stability ofthese materials based only on the heat of formation for the wholeconcentration range without taking into account the entropy ofthe system under study, it is worth pointing out here that ourcalculated heat of formation values are nonetheless very reason-able as they, for the whole concentration range of the Mn-dopedBeSe and BeTe, have negative ΔH values which are always thelowest for binary FM beryllium chalcogenide [21].
3.2. Electronic band structures and density of states
By using the calculated equilibrium lattice parameters theFP-LAPWþ lo method was employed to calculate the FM phasespin polarized band structures of the Be1−xMnxSe and Be1−xMnxTe(x¼0.25, 0.50 and 0.75) alloys, which are presented in Figs. 3 and4, respectively. As one can see, for all compounds at any givenconcentration, the band structures correspond to spin-up and
Fig. 1. Structural optimization plots for the ferromagnetic (FM) phase of (a) BexMn1−xSe and (b) BexMn1−xTe at x¼0.25, 0.5, and 0.75.
Fig. 2. Non-linear variation in the lattice constants of BexMn1−xSe and BexMn1−xTealloys as a function of Mn in x.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–19 13

Author's personal copy
spin-down alignments. In the ferromagnetic phase of materialsunder study, the valence band maxima (VBM) of the minority-spincarriers are situated below the VBM in the majority-spin channel.This large (and negative) spin splitting of the VBM is common toall Mn doped II–VI DMS materials [18,27,28] and can be attributed
to the negative (antiferromagnetic) p–d exchange interaction.Furthermore, from the band structure presented in Figs. 3 and 4one can easily see that the abovementioned p–d exchange inter-action splitting increases with the increase in Mn concentration.The important FM energy gaps (i.e. the spin-down gap Gg-min and
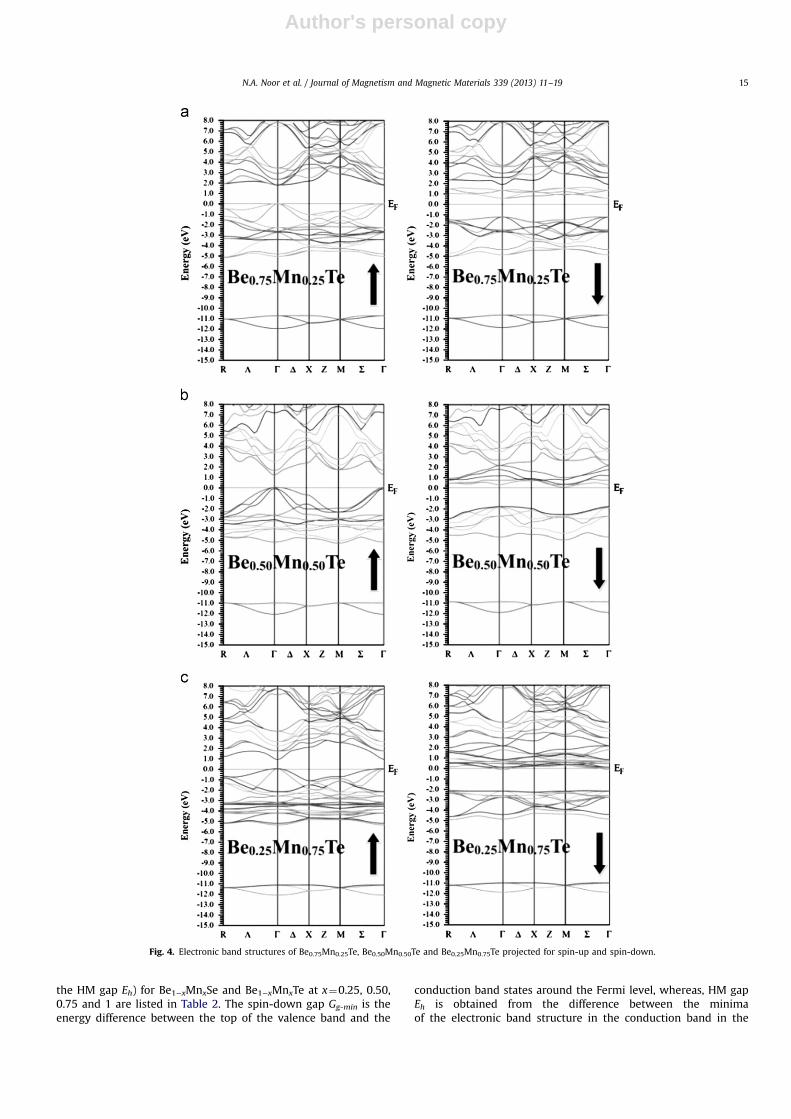
Fig. 3. Electronic band structures of Be0.75Mn0.25Se, Be0.50Mn0.50Se and Be0.25Mn0.75Se projected for spin up and spin down.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–1914
Author's personal copy
the HM gap Eh) for Be1−xMnxSe and Be1−xMnxTe at x¼0.25, 0.50,0.75 and 1 are listed in Table 2. The spin-down gap Gg-min is theenergy difference between the top of the valence band and the
conduction band states around the Fermi level, whereas, HM gapEh is obtained from the difference between the minimaof the electronic band structure in the conduction band in the
Fig. 4. Electronic band structures of Be0.75Mn0.25Te, Be0.50Mn0.50Te and Be0.25Mn0.75Te projected for spin-up and spin-down.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–19 15
Author's personal copy
spin-down channel and the Fermi level. One can see an increase inGg-min and decrease in Eh with increasing Mn concentration. Ourcomputed values of the band gaps for BeSe and BeTe and spin-down and HM gaps for FM Mn-doped binaries are in conformitywith earlier theoretical results [17,18,21]. It is evident that forBe1−xMnxSe at 0>x>1.0 and Be1−xMnxTe at 0>x>0.75 the spin-polar-ized band structures have an HM nature. This can be clearly seenin the density of states calculations discussed later in this section.We have also obtained band structures (not presented here) for bothFM MnSe and MnTe in the zinc blende phase that show half-metalliccharacter in consistent with earlier studies [29–30].
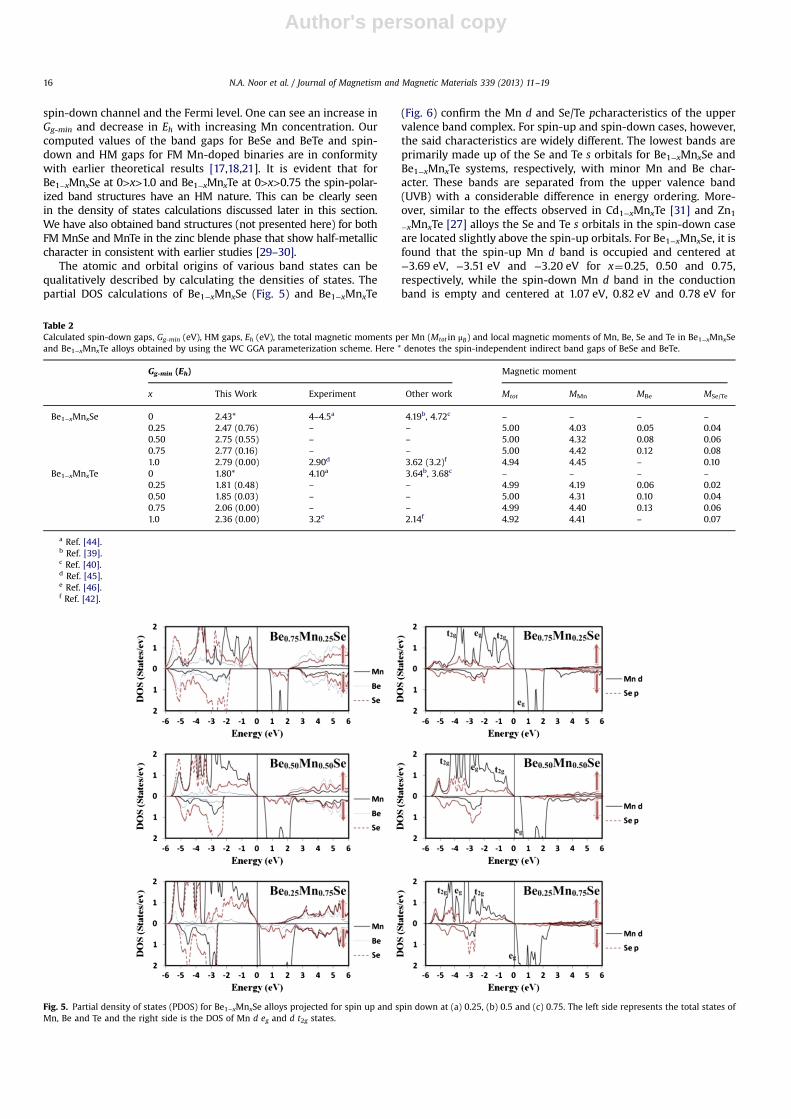
The atomic and orbital origins of various band states can bequalitatively described by calculating the densities of states. Thepartial DOS calculations of Be1−xMnxSe (Fig. 5) and Be1−xMnxTe
(Fig. 6) confirm the Mn d and Se/Te pcharacteristics of the uppervalence band complex. For spin-up and spin-down cases, however,the said characteristics are widely different. The lowest bands areprimarily made up of the Se and Te s orbitals for Be1−xMnxSe andBe1−xMnxTe systems, respectively, with minor Mn and Be char-acter. These bands are separated from the upper valence band(UVB) with a considerable difference in energy ordering. More-over, similar to the effects observed in Cd1−xMnxTe [31] and Zn1
−xMnxTe [27] alloys the Se and Te s orbitals in the spin-down caseare located slightly above the spin-up orbitals. For Be1−xMnxSe, it isfound that the spin-up Mn d band is occupied and centered at−3.69 eV, −3.51 eV and −3.20 eV for x¼0.25, 0.50 and 0.75,respectively, while the spin-down Mn d band in the conductionband is empty and centered at 1.07 eV, 0.82 eV and 0.78 eV for
Table 2Calculated spin-down gaps, Gg-min (eV), HM gaps, Eh (eV), the total magnetic moments per Mn (Mtot in μB) and local magnetic moments of Mn, Be, Se and Te in Be1−xMnxSeand Be1−xMnxTe alloys obtained by using the WC GGA parameterization scheme. Here * denotes the spin-independent indirect band gaps of BeSe and BeTe.
Gg-min (Eh) Magnetic moment
x This Work Experiment Other work Mtot MMn MBe MSe/Te
Be1−xMnxSe 0 2.43* 4–4.5a 4.19b, 4.72c – – – –
0.25 2.47 (0.76) – – 5.00 4.03 0.05 0.040.50 2.75 (0.55) – – 5.00 4.32 0.08 0.060.75 2.77 (0.16) – – 5.00 4.42 0.12 0.081.0 2.79 (0.00) 2.90d 3.62 (3.2)f 4.94 4.45 – 0.10
Be1−xMnxTe 0 1.80* 4.10a 3.64b, 3.68c – – – –
0.25 1.81 (0.48) – – 4.99 4.19 0.06 0.020.50 1.85 (0.03) – – 5.00 4.31 0.10 0.040.75 2.06 (0.00) – – 4.99 4.40 0.13 0.061.0 2.36 (0.00) 3.2e 2.14f 4.92 4.41 – 0.07
a Ref. [44].b Ref. [39].c Ref. [40].d Ref. [45].e Ref. [46].f Ref. [42].
Fig. 5. Partial density of states (PDOS) for Be1−xMnxSe alloys projected for spin up and spin down at (a) 0.25, (b) 0.5 and (c) 0.75. The left side represents the total states ofMn, Be and Te and the right side is the DOS of Mn d eg and d t2g states.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–1916
Author's personal copy
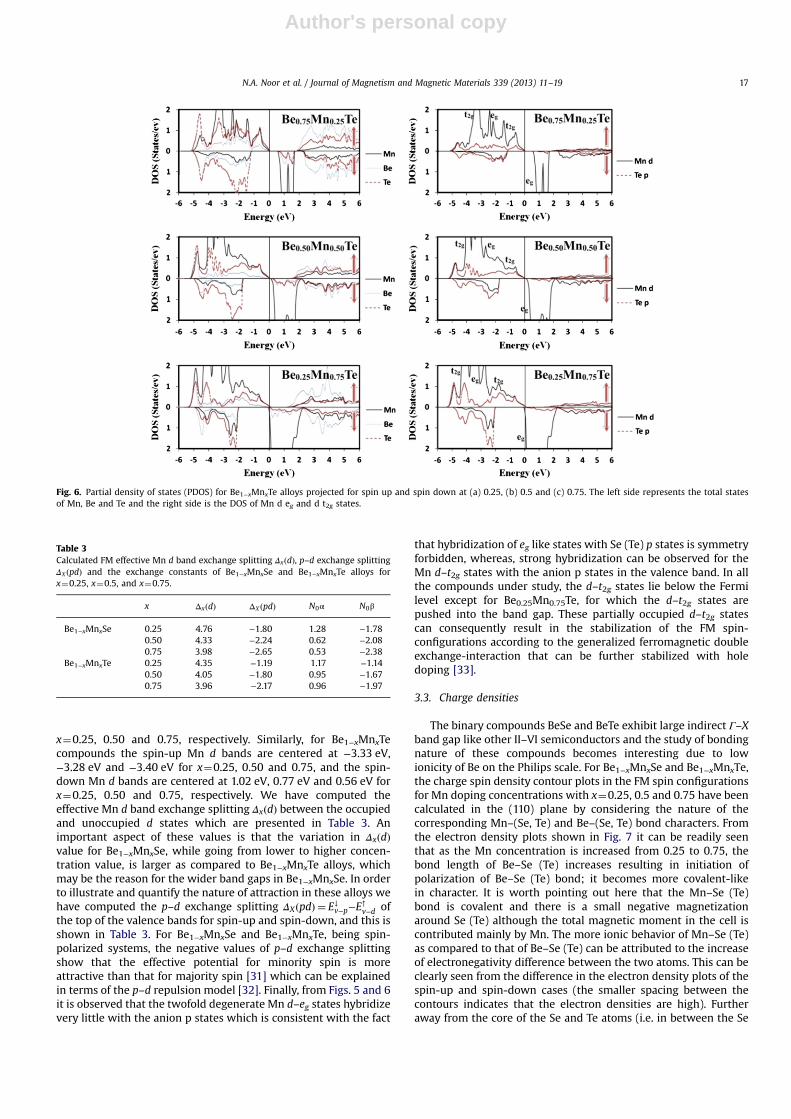
x¼0.25, 0.50 and 0.75, respectively. Similarly, for Be1−xMnxTecompounds the spin-up Mn d bands are centered at −3.33 eV,−3.28 eV and −3.40 eV for x¼0.25, 0.50 and 0.75, and the spin-down Mn d bands are centered at 1.02 eV, 0.77 eV and 0.56 eV forx¼0.25, 0.50 and 0.75, respectively. We have computed theeffective Mn d band exchange splitting ΔxðdÞ between the occupiedand unoccupied d states which are presented in Table 3. Animportant aspect of these values is that the variation in ΔxðdÞvalue for Be1−xMnxSe, while going from lower to higher concen-tration value, is larger as compared to Be1−xMnxTe alloys, whichmay be the reason for the wider band gaps in Be1−xMnxSe. In orderto illustrate and quantify the nature of attraction in these alloys wehave computed the p–d exchange splitting ΔXðpdÞ ¼ E↓v−p−E
↑v−d of
the top of the valence bands for spin-up and spin-down, and this isshown in Table 3. For Be1−xMnxSe and Be1−xMnxTe, being spin-polarized systems, the negative values of p–d exchange splittingshow that the effective potential for minority spin is moreattractive than that for majority spin [31] which can be explainedin terms of the p–d repulsion model [32]. Finally, from Figs. 5 and 6it is observed that the twofold degenerate Mn d–eg states hybridizevery little with the anion p states which is consistent with the fact
that hybridization of eg like states with Se (Te) p states is symmetryforbidden, whereas, strong hybridization can be observed for theMn d–t2g states with the anion p states in the valence band. In allthe compounds under study, the d–t2g states lie below the Fermilevel except for Be0.25Mn0.75Te, for which the d–t2g states arepushed into the band gap. These partially occupied d–t2g statescan consequently result in the stabilization of the FM spin-configurations according to the generalized ferromagnetic doubleexchange-interaction that can be further stabilized with holedoping [33].
3.3. Charge densities
The binary compounds BeSe and BeTe exhibit large indirect Γ–Xband gap like other II–VI semiconductors and the study of bondingnature of these compounds becomes interesting due to lowionicity of Be on the Philips scale. For Be1−xMnxSe and Be1−xMnxTe,the charge spin density contour plots in the FM spin configurationsfor Mn doping concentrations with x¼0.25, 0.5 and 0.75 have beencalculated in the (110) plane by considering the nature of thecorresponding Mn–(Se, Te) and Be–(Se, Te) bond characters. Fromthe electron density plots shown in Fig. 7 it can be readily seenthat as the Mn concentration is increased from 0.25 to 0.75, thebond length of Be–Se (Te) increases resulting in initiation ofpolarization of Be–Se (Te) bond; it becomes more covalent-likein character. It is worth pointing out here that the Mn–Se (Te)bond is covalent and there is a small negative magnetizationaround Se (Te) although the total magnetic moment in the cell iscontributed mainly by Mn. The more ionic behavior of Mn–Se (Te)as compared to that of Be–Se (Te) can be attributed to the increaseof electronegativity difference between the two atoms. This can beclearly seen from the difference in the electron density plots of thespin-up and spin-down cases (the smaller spacing between thecontours indicates that the electron densities are high). Furtheraway from the core of the Se and Te atoms (i.e. in between the Se
Fig. 6. Partial density of states (PDOS) for Be1−xMnxTe alloys projected for spin up and spin down at (a) 0.25, (b) 0.5 and (c) 0.75. The left side represents the total statesof Mn, Be and Te and the right side is the DOS of Mn d eg and d t2g states.
Table 3Calculated FM effective Mn d band exchange splitting ΔxðdÞ, p–d exchange splittingΔΧ ðpdÞ and the exchange constants of Be1−xMnxSe and Be1−xMnxTe alloys forx¼0.25, x¼0.5, and x¼0.75.
x ΔxðdÞ ΔΧ ðpdÞ N0α N0β
Be1−xMnxSe 0.25 4.76 −1.80 1.28 −1.780.50 4.33 −2.24 0.62 −2.080.75 3.98 −2.65 0.53 −2.38
Be1−xMnxTe 0.25 4.35 −1.19 1.17 −1.140.50 4.05 −1.80 0.95 −1.670.75 3.96 −2.17 0.96 −1.97
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–19 17

Author's personal copy
(Te) atoms and the Mn atom), there are some differences in theelectron density. This is due to the electrons in the valence p statesfrom the Se and Te atoms since other states from both the Se andTe atoms are confined to the core. A contour plot for concentrationvalue at x¼0.5, reveals more covalent bonding for both Mn and Beselenide and telluride bonds but the latter has a comparativelystronger bond. Another important result corresponding to semi-core Be 2s charge density is its considerable values surroundingthe anion positions. The valence 2s electrons from the Be atomsinteract with the d states electrons of the Mn atoms which leads tostrong s–d hybridization and also explains the prominent role ofthese electrons in bonding properties in IIA–VIB based semicon-ductors. It also reveals that the structural as well as electronicstructure properties of the compounds under study depend uponsemicore Be 2s electrons. Spin density is mainly produced due toenhanced participation of Mn d states in bonding by preserving itslocalization on Mn atom. It is further added that only Mn 3d↑ isimportant in this regard, while Mn 3d↓ is absent in the valenceband.
3.4. Exchange coupling and magnetic properties
Estimation of exchange constants N0α and N0β is crucial fordescribing the contribution of valence and conduction bands inthe exchange and splitting process of the band structure atΓ symmetry point. This section provides some discussion of theeffect of s, p, and d exchange on band structure of a hostsemiconductor in the mean field approximation [34,35]. In orderto estimate the contributions of these bands in the process ofexchange and splitting, the s–p exchange constant N0α (conduc-tion band) and the p–d exchange constant N0β (valence band) arecalculated for Be1−xMnxSe and Be1−xMnxTe alloys for x¼0.25, 0.50,0.75 and 1.0 with the help of the following formulae [34]:
N0α¼ΔEcx⟨S⟩
, N0β¼ΔEvx⟨S⟩
Here N0 denotes the concentration of the cations, ΔEc and ΔEvrepresent the conduction band and valence band edge splittingsrespectively and ⟨S⟩x is the average magnetization of Mn atom. Thevalues of estimated exchange constants are presented in Table 2and one can see that exchange coupling between the conductionbands of Be, Mn and Se (Te) impurities confirms the magneticcharacter of these materials.
The total and local magnetic moments as well as that ofinterstitial sites are displayed in Table 2. In the present class ofMn-doped semiconductors [36], an appealing possibility of Mndoping is the addition of five majority spin levels, per Mn impurity,to the valence band without affecting the valence minority spinelectron concentration. Importantly, the above scenario is corro-borated by our results for Be1−xMnxSe and Be1−xMnxTe due to thefact that Mn (Be) has 7 (2) valence electrons and it turns out thatthose 5 net electrons are contributed by each Mn impurity to fillthe majority spin band. These findings imply that hole concentra-tion of BeSe (Te) host crystal remains unaffected by Mn doping and5μB (μB is the Bohr magneton) is the calculated value of totalmagnetic moments. It is worth adding here that the unfilled Mn 3dshell gives rise to permanent magnetic moments in these semi-conductor alloys. These exchange interactions between themagnetic ions and the electrons or holes near the band edgesignificantly control the properties of these alloys [37]. The aboveinterpretation implies that quite large p–d hybridization is foundbetween Mn and Se (Te) states at the upper valence band. This p–dhybridization is very effective not only in reducing the localmagnetic moment of Mn, but also in generating small localmagnetic moment on the non-magnetic Be and Te sites.
4. Conclusions
In this study, we have applied the FP-LAPWþ lo method, whichenables the direct evaluation of the spin-effect electronic andmagnetic properties of the Be1−xMnxSe and Be1−xMnxTe at x¼0.25,0.50, 0.75 alloys in the ferromagnetic phase. The analysis of thetotal and partial densities of states makes it possible to evaluateboth the exchange splitting values ΔΧðdÞ and ΔΧðpdÞ. Our resultsprovide unambiguous evidence for effective potential, clarifying itsattractiveness for minority-spin than that for majority-spin case.Another important result regarding the total and local magneticmoments is that the addition of Mn ions introduces no hole carrierto the host BeSe and BeTe matrices. The value of total magneticmoments is estimated to be 5μB. Furthermore, it is concluded thatthe unfilled Mn 3d shell together with p–d hybridization results ina significant decrease in the local magnetic moment at the Mn sitefrom its free space value, leading to the generation of a small localmagnetic moment at Be and Te sites.
Fig. 7. Calculated spin-dependent charge density contours for Be1−xMnxSe (left panel) and Be1−xMnxTe (right panel) at (a) x¼0.25, (b) x¼0.5 and (c) x¼0.75.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–1918

Author's personal copy
References
[1] J.C. Zheng, J.W. Davenport, Physical Review B 69 (2004) 144415.[2] S.A. Wolf, D.D. Awschalom, R.A. Buhrman, J.M. Daughton, S. Molnar, M.
L. Roukes, A.Y. Chtehelkanova, D.M. Treger, Science 294 (2001) 1488.[3] S. Datta, B. Das, Applied Physics Letters 56 (1990) 665.[4] G. Prinz, Science 282 (1998) 1660.[5] G. Burkard, D. Loss, D.P. DiVincenzo, Physical Review B 59 (1999) 2070.[6] S.H. Wei, A. Zunger, Physical Review B 35 (1987) 2340.[7] Y.H. Zhao, P. Mahadevan, A. Zunger, Journal of Applied Physics 98 (2005)
113901.[8] Y. Liu, B.G. Liu, Journal of Magnetism and Magnetic Materials 307 (2006) 245.[9] R. Fiederling, M. Keim, G. Reuscher, W. Ossau, G. Schmidt, A. Waag, L.
W. Molenkamp, Nature (London) 402 (1999) 787.[10] G. Schmidt, D. Ferrand, L.W. Molenkamp, A.T. Fillip, B.J. van Wees, Physical
Review B 62 (2000) R4790.[11] L. Hansen, D. Ferrand, G. Richter, M. Thierley, V. Hock, N. Schwarz, G. Reuscher,
G. Schmidt, L.W. Molenkamp, A. Waag, Applied Physics Letters 79 (2001) 3125.[12] P. Klosowski, T.M. Giebultowicz, J.J. Rhyne, N. Samarth, H. Luo, J.K. Furdyna,
Journal of Applied Physics 69 (1991) 6109.[13] J.K. Furdyna, Journal of Applied Physics 64 (1988) R29.[14] N. Benkhettou, D. Bensaid, Open Condensed Matter Physics Journal 1 (2008) 29.[15] M. Sawicki, L.V. Khoi, L. Hansen, D. Ferrand, L.W. Molenkamp, A. Waag, T. Dietl,
Physica Status Solidi B 229 (2002) 717.[16] D.R. Yakovlev, C. Sas, B. Köning, L. Hansen, W. Ossau, G. Landwehr, A. Waag,
Applied Physics Letters 78 (2001) 1870.[17] D. Segev, S.-H. Wei, Physical Review B 70 (2004) 184401.[18] S. Picozzi, T. Shishidou, A.J. Freeman, B. Delley, Physical Review B 67 (2003) 165203.[19] A. Nakamura, Y. Takeda, N. Imaoka, AIP Conference Proceedings 1199 (2010) 399.[20] P. Blaha, K. Schwarz, G.K.H. Madsen, D. Kvasnicka, J. Luitz, WIEN2K, An
augmented plane waveþ local orbitals program for calculating crystal proper-ties, Karlheinz Schwarz, Technischen Universitat, Vienna, Austria, 2001.
[21] S.M. Alay-e-Abbas, K.M. Wong, N.A. Noor, A. Shaukat, Y. Lei, Solid StateSciences 14 (2012) 1525.
[22] H. Luo, K. Ghandehari, R.G. Greene, A.L. Ruoff, S.S. Trail, F.J. DiSalvo, PhysicalReview B 52 (1995) 7058.
[23] A.E. Merad, M.B. Kanoun, S. Goumri-Said, Journal of Magnetism and MagneticMaterials 302 (2006) 536.
[24] K.M. Wong, S.M. Alay-e-Abbas, A. Shaukat, Y. Fang, Y. Lei, Journal of AppliedPhysics 113 (2013) 014304.
[25] Z. Wu, R.E. Cohen, Physical Review B 73 (2006) 235116.[26] S.M. Alay-e-Abbas, A. Shaukat, Solid State Sciences 13 (2011) 1052.[27] U.P. Verma, Sonu Sharma, Nisha Devi, P.S. Bisht, P. Rajaramand, Journal of
Magnetism and Magnetic Materials 323 (2011) 394.[28] S.A. Touat, F. Litimein, A. Tadjer, B. Bouhafs, Physica B 405 (2010) 625.[29] A. Fleszar, M. Potthoff, W. Hanke, Physica Status Solidi C 4 (2007) 3270.[30] I. Galanakis, P. Mavropoulos, Physical Review B 67 (2003) 104417.[31] V.L. Morozzi, J.F. Janak, A.R. Williams, Calculated Electronic Properties of
Metals, Pergamon, New York, 1978.[32] J.E. Jaffe, A. Zunger, Physical Review B 29 (1984) 1882.[33] H. Akai, Physical Review Letters 81 (1998) 3002.[34] S. Sanvito, P. Ordejon, N.A. Hill, Physical Review B 63 (2001) 165206.[35] C. Echeverria-Arrondo, J. Perez-Conde, A. Ayuela, Physical Review B 82 (2010)
205419.[36] T.C. Schulthess, W.H. Butler, Journal of Applied Physics 89 (2001) 7021.[37] J.A. Gaj, R. Planel, G. Fishman, Solid State Communications 29 (1984) 435.[38] H. Luo, K. Ghandehair, R.G. Geene, A.L. Ruoff, S.S. Trail, F.J. DiSalvo, Physical
Review B 32 (1995) 7058.[39] H. Baaziz, Z. Charifi, F. El Haj Hassan, S.J. Hashemifar, H. Akbarzadeh, Physica
Status Solidi B 243 (2006) 1296.[40] M. González-Diáz, P. Rodríguez-Hermández, A. Munoz, Physical Review B 55
(1997) 14043.[41] J.K.J. Furdyna, Applied Physics 64 (1988) R29.[42] N. Benkhettou, D. Bensaid, Open Condensed Matter Physics Journal 1 (2008) 29.[43] N.A. Noor, S. Ali, W. Tahir, A. Shaukat, A.H. Reshak, Journal of Alloys and
Compounds 509 (2011) 8137.[44] W.M. Yim, J.P. Dismukes, E.J. Stofko, R.J. Paff, Journal of Physics and Chemistry
of Solids 33 (1972) 501.[45] M. Imamura, A. Okada, IEEE Transactions on Magnetics 42 (10) (2006) 3078.[46] R.L. Gunshor, A.V. Nurmikko, L.A. Kodziejski, M. Kobayashi, N. Otsuka, Journal
of Crystal Growth 101 (1990) 14.
N.A. Noor et al. / Journal of Magnetism and Magnetic Materials 339 (2013) 11–19 19




















