
Ab-initio study of electronic and elastic properties of B 2-type ductile YM (M=Cu, Zn and Ag)...
11
Ab-initio study of the electronic and elastic properties of beryllium chalcogenides BeX (X=S, Se and Te) This article has been downloaded from IOPscience. Please scroll down to see the full text article. 2011 Phys. Scr. 84 035704 (http://iopscience.iop.org/1402-4896/84/3/035704) Download details: IP Address: 137.99.20.175 The article was downloaded on 05/11/2011 at 19:49 Please note that terms and conditions apply. View the table of contents for this issue, or go to the journal homepage for more Home Search Collections Journals About Contact us My IOPscience
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Ab-initio study of electronic and elastic properties of B 2-type ductile YM (M=Cu, Zn and Ag)...

Ab-initio study of the electronic and elastic properties of beryllium chalcogenides BeX (X=S,
Se and Te)
This article has been downloaded from IOPscience. Please scroll down to see the full text article.
2011 Phys. Scr. 84 035704
(http://iopscience.iop.org/1402-4896/84/3/035704)
Download details:
IP Address: 137.99.20.175
The article was downloaded on 05/11/2011 at 19:49
Please note that terms and conditions apply.
View the table of contents for this issue, or go to the journal homepage for more
Home Search Collections Journals About Contact us My IOPscience

IOP PUBLISHING PHYSICA SCRIPTA
Phys. Scr. 84 (2011) 035704 (10pp) doi:10.1088/0031-8949/84/03/035704
Ab-initio study of the electronic andelastic properties of berylliumchalcogenides BeX (X = S, Se and Te)N Munjal1, V Sharma2, G Sharma1, V Vyas1, B K Sharma3
and J E Lowther2
1 Department of Physics, Banasthali University, Banasthali-304022, India2 School of Physics and DST/NRF Centre of Excellence in Strong Materials,University of the Witwatersrand, Johannesburg-2050, South Africa3 Department of Physics, University of Rajasthan, Jaipur-302004, India
E-mail: [email protected] (G Sharma)
Received 5 July 2011Accepted for publication 26 July 2011Published 31 August 2011Online at stacks.iop.org/PhysScr/84/035704
AbstractAb-initio methods have been employed to investigate the electronic and elastic properties ofberyllium chalcogenides (namely BeS, BeSe and BeTe). The electron momentum density,autocorrelation function and energy band gap have been computed using the linearcombination of atomic orbitals method. Using the full potential linearized augmentedplane-wave and projector-augmented wave methods, the energy bands and density of states(DOS) along with elastic properties are also calculated. The electronic band structure, totaland partial DOS and elastic moduli obtained from the present calculations are found to be ingood agreement with available earlier data. The calculated valence band width, equal valenceelectron density curve and bulk modulus confirm the trend of ionicity BeS> BeSe> BeTe.
PACS numbers: 71.15.Ap, 71.15.Dx, 71.15.Mb, 71.15.Nc, 71.20.Nr, 78.70.−g
(Some figures in this article are in colour only in the electronic version.)
1. Introduction
II–VI semiconductors are technologically important becausethey are used in the fabrication of light-emitting devicesthat are employed in optical processing, detection systemsfor environmental pollution and color-displaying modules.In particular, the beryllium chalcogenides BeS, BeSe andBeTe are the II–VI compounds that crystallize in the fourfoldcoordinated zinc-blende (B3) structure at low pressure. TheseBeX (X = S, Se and Te) compounds have structure andbinding similar to those of the III-P group of semiconductors.They have large band gaps (2.7–5.5 eV) and a high value ofthe bulk modulus (B0) that results in an increased hardnessand stability [1–2].
A number of theoretical studies on electronic, optical,elastic and structural properties of BeX compounds have beenreported [3–23]. In these studies, different methods have beenused to study the electronic properties of these compounds.For instance, Stukle [3] used non-relativistic self-consistent
calculations based on Slater’s local exchange potentialapproach, while Sarkar and Chatterjee [4] used the compositewave variational version of the augmented plane wave(APW) method in conjunction with the linear combinationof atomic orbitals (LCAO) method to calculate the energyband structure of BeX (X = S, Se and Te). Some of theearlier calculations are non-relativistic [5–7], first-principlesself-consistent [5], pseudopotential (PP) [8] and tight-bindinglinear muffin-tin orbital (TB-LMTO) [9]. Besides these,Fleszar and Hanke [10] used the ab-initio GW method tocalculate the electronic band structure of these compounds.Moreover, the full potential linearized augmented plane-wave(FP-LAPW) method within the local density approximation(LDA) and generalized gradient approximation (GGA) pluslocal orbitals (APW + lo) has been used by a number ofworkers [11–15]. Likewise, Hassan and Akbarzadeh [13]found indirect band gaps for all BeX compounds except BeTe,and Heciri et al [15] observed a non-negligible differencein the chemical bonding of BeS, BeSe and BeTe. The
0031-8949/11/035704+10$33.00 Printed in the UK & the USA 1 © 2011 The Royal Swedish Academy of Sciences

Phys. Scr. 84 (2011) 035704 N Munjal et al
plane wave PP method [16] has also been used to studythese compounds. Rached et al [17] used the FP-LMTOmethod to calculate the ground state properties along withstructural phase transformation, and the results of thestructural phase transition and pressure dependence of bandgap confirmed the existence of reversible pressure-inducedfirst-order phase transition between the zinc-blende (B3)and NiAs (B8) phases in these compounds. Using the DeLaunay model that is based on the atomic model and angularcentral forces, the elastic properties of BeSe and BeTehave been reported by Doyen-Lang et al [18]. Nagelstraßeret al [19] performed angle-resolved synchrotron-radiationphotoemission spectroscopy measurements to study theelectronic structure of BeTe. These authors also performedfirst-principles calculation to compare the experimentalresults. Yadav et al [20] performed ab-initio calculations ofthe structural, electronic and optical absorption properties ofberyllium chalcogenides (BeS, BeSe and BeTe) using the PPmethod within GGA. The elastic and structural properties ofthe B3 and B8 structures of BeS have been studied by Saiband Bouarissa [21] employing the ab-initio PP method and alinear response approach within LDA. Kong and Jiang [22]have reported the first-principles calculations of structuralphase transition, elastic and thermodynamics properties ofBeSe in B3 phase within GGA. Using the FP-LMTO method,the structural and electronic properties of ZnSe, BeSe andtheir alloy have been studied by Ameri et al [23], and theyfound that the bulk modulus for the alloy increases with Beconcentration.
However, due to the high degree of toxicity of thesecompounds, only a few experimental studies [19], [24–28]have been carried out so far. Yim et al [24] have made opticalabsorption measurements on BeX (X = S, Se and Te) todetermine the energy band gaps. The existence of a reversiblepressure-induced first-order phase transition between B3 andB8 structures in the beryllium chalcogenides has also beenreported by Luo et al [25]. Among other experimental studies,molecular-beam epitaxy of beryllium-chalcogenide-basedthin films [26], reversible first-order phase transition of BeSfrom the B3 to B8 phase [27] and the dielectric function ofBeTe and Bex Zn1−x Se by spectroscopic ellipsometry in theultraviolet and vacuum ultraviolet ranges [28] have also beenreported.
As the above review suggests, a number of studies havebeen reported for beryllium chalcogenides, but studies ofother ground state properties such as the electron momentumdensity (EMD), autocorrelation function (AF) and Comptonprofile have been rarely attempted. Therefore, in this paper,the electron momentum density, autocorrelation function,density of states (DOS) and energy band gap for BeS, BeSeand BeTe compounds using the LCAO method embodiedin the CRYSTAL code are studied. The aim of this paperis also to investigate the electronic and elastic propertiesin these chalcogenides by means of the FP-LAPW andprojector-augmented wave (PAW) methods as embodied inWIEN2k and Vienna ab-initio simulation package (VASP)codes, respectively.
The paper is organized as follows. The computationalmethods used in this study are described in section 2. Insection 3, our main calculated results on various electronic
and elastic properties along with Compton profiles and theiranalysis are discussed. Finally, the summary is presented insection 4.
2. Computational details
2.1. LCAO method
The ab-initio periodic LCAO calculations are performedwherein one solves the Kohn–Sham equationsself-consistently under the density functional theory(DFT) [29, 30]. There are a few fundamental schemesfor constructing the Hamiltonian for the periodic solids.Hartree–Fock (HF) approximation and DFT [29–31]are well-known approaches among these schemes. TheCRYSTAL06 code [31] provides a platform to calculate theelectronic structure of periodic systems with Gaussian basisemploying HF, DFT and hybrid schemes. In this method,each crystalline orbital ψi(r, k) is a linear combination of theBloch functions ϕµ(r, k) defined in terms of local functionsϕµ(r), normally referred to as atomic orbitals. The localfunctions are expressed as a linear combination of a certainnumber of individually normalized Gaussian-type functions.For Be, S, Se and Te, the local functions were constructedfrom the Gaussian-type basis sets (www.tcm.phy.cam.ac.uk/).The Kohn–Sham Hamiltonian was constructed whileconsidering the exchange scheme of Becke and thePerdew–Burke–Ernzerhof (PBE) correlation scheme [32].Calculations were performed for zinc-blende structure (B3)(space group F43m) of all compounds. The self-consistentcalculations were performed considering 145 k points inthe irreducible Brillouin zone with sufficient tolerances. Toachieve self-consistency, mixing of successive cycles wasconsidered and self-consistency was achieved within sevencycles. The respective values of lattice parameters used inthe present computations are 9.204, 9.708 and 10.616 a.u. forBeS, BeSe and BeTe. Using this method, along with DOSand energy band gaps, we have also calculated the electronmomentum density and AF for all BeX compounds.
Regarding Compton profiles, it has been well establishedthat valence EMD is an important ground state property. Inthe independent particle model, the momentum density n(p)is given by [33, 34]
n(p)∝
∑i
∣∣∣∣∫ ψi (r)exp (ip · r)dr
∣∣∣∣2
(1)
where n(p) is the three-dimensional momentum density andψ(r) represents the electron wave function. The summation isover the occupied states. The Compton profile can be derivedfrom the ground state momentum density n(p) as
J (pz)=
∫ +∞
−∞
∫ +∞
−∞
n(p) dpx dpy . (2)
The spherically averaged Compton profile is determinedby the averaged momentum density as
J (pz)= 2π∫
∞
pz
〈n(p)〉 pdp. (3)
2

Phys. Scr. 84 (2011) 035704 N Munjal et al
Table 1. Unconvoluted theoretical directional and average valence Compton profile calculated using the PBE correlation functional withinthe DFT–LCAO method for BeS, BeSe and BeTe.
pz J(pz) (e a.u.−1)
(a.u.) BeS BeSe BeTe
J100 J110 J111 Jave J100 J110 J111 Jave J100 J110 J111 Jave
0.0 4.7414 4.7671 4.6046 4.7436 5.0379 5.0067 4.9146 4.9936 5.5219 5.5215 5.2644 5.50240.1 4.7060 4.7276 4.5992 4.7002 4.9965 4.9658 4.8716 4.9509 5.4755 5.4664 5.2680 5.43940.2 4.6107 4.6058 4.5410 4.5784 4.8721 4.8173 4.7476 4.8176 5.3388 5.2875 5.2241 5.25660.3 4.4532 4.3693 4.3845 4.3862 4.6568 4.5385 4.5502 4.5917 5.0596 4.9002 5.0145 4.95650.4 4.1921 4.0161 4.1259 4.1178 4.3318 4.1762 4.2763 4.2825 4.5920 4.3946 4.6028 4.55480.5 3.8066 3.6445 3.7819 3.7777 3.8846 3.8006 3.9094 3.8962 3.9816 3.9464 4.0565 4.04750.6 3.3285 3.3089 3.3745 3.3726 3.3311 3.4099 3.4379 3.4248 3.2526 3.4195 3.4089 3.38640.7 2.7898 2.9301 2.8986 2.8918 2.7179 2.9344 2.8771 2.8642 2.4420 2.6828 2.6398 2.58830.8 2.2197 2.4329 2.3623 2.3439 2.1056 2.3407 2.2712 2.2448 1.6848 1.8711 1.8337 1.79061.0 1.2150 1.2978 1.3193 1.2729 1.0912 1.1217 1.1636 1.1142 0.7054 0.6728 0.7106 0.69321.2 0.6111 0.5598 0.6160 0.5896 0.5065 0.4311 0.4832 0.4703 0.3240 0.2594 0.2959 0.29541.4 0.3239 0.2787 0.2892 0.2967 0.2553 0.2078 0.2138 0.2294 0.2051 0.2023 0.1882 0.19791.6 0.1936 0.1794 0.1588 0.1790 0.1577 0.1523 0.1322 0.1486 0.1638 0.1705 0.1553 0.16071.8 0.1307 0.1301 0.1077 0.1239 0.1176 0.1268 0.1068 0.1157 0.1432 0.1375 0.1305 0.13752.0 0.0982 0.0993 0.0878 0.0953 0.0993 0.1028 0.0943 0.0979 0.1261 0.1207 0.1120 0.12113.0 0.0478 0.0481 0.0503 0.0472 0.0523 0.0527 0.0550 0.0516 0.0468 0.0469 0.0485 0.04614.0 0.0265 0.0265 0.0261 0.0252 0.0224 0.0223 0.0220 0.0209 0.0161 0.0161 0.0164 0.01495.0 0.0143 0.0143 0.0144 0.0130 0.0097 0.0097 0.0097 0.0082 0.0092 0.0092 0.0092 0.00806.0 0.0081 0.0081 0.0081 0.0068 0.0051 0.0051 0.0051 0.0036 0.0066 0.0066 0.0066 0.00537.0 0.0048 0.0048 0.0048 0.0035 0.0033 0.0033 0.0033 0.0019 0.0045 0.0045 0.0045 0.00328.0 0.0030 0.0030 0.0030 0.0017 0.0024 0.0024 0.0024 0.0010 0.0030 0.0030 0.0030 0.00179.0 0.0020 0.0020 0.0020 0.0006 0.0019 0.0019 0.0019 0.0004 0.0019 0.0019 0.0019 0.000610.0 0.0014 0.0014 0.0014 0.0001 0.0015 0.0015 0.0015 0.0000 0.0013 0.0013 0.0013 0.0000
The quantity J (pz) can also be determinedexperimentally through the measurement of spectraldistribution of inelastically scattered radiation as
d2σ
d� dω2= C(ω1, ω2, pz, φ)J (pz). (4)
This way the Compton profile study enables a directtest of the theoretical methods used in the determination ofthe electronic structure and related properties [33, 34]. TheCompton profiles can also be analyzed in real space by takingtheir Fourier transforms. The real space transform of themomentum density, and thereby the Compton profile data, isnormally known as AF or the reciprocal form factor B(z). Thisfunction is directly associated with the formation of bonds andfacilitates the identification of bonding in solids [34–36]. Itcan be written as
B(z)=
∫ +∞
−∞
J (pz) exp(i pzz) dpz . (5)
2.2. FP-LAPW method
To determine the energy band structure along with total andpartial DOS, and elastic properties, the FP-LAPW method,as employed in the WIEN2k code, is used [37]. The recentnon-empirical GGA approach of Wu and Cohen [38] has beenconsidered. To ensure sufficient accuracy in convergence, thetotal energy of the crystal was converged to 0.01 mRyd. Theconvergence was achieved by considering basis functions upto RMT = 8/Kmax, where RMT is the radius of muffin-tinsphere and Kmax is the magnitude of the largest k vectorin the combined basis set of LAPWs. The maximum radialexpansion lmax was set to 10. The Brillouin zone integration
was performed for 330 k points. The independent elasticconstants of beryllium chalcogenides are derived from thederivative of total energy as a function of lattice strain in fulldetail. The bulk modulus (B0) and its first pressure derivativeare obtained by fitting total energy versus volume data to aMurnaghan equation of state (EOS). The choice of atomicradii RMT in a.u. was the following: Be (1.6), S (1.82), Se(1.98) and Te (2.29).
2.3. PAW method
In PAW calculations, VASP [39–41] has been employed.The exchange–correlation interaction was treated within bothLDA [42] and GGA using the PBE functional [32]. The totalenergy was minimized by calculating the Hellmann–Feynmanforces and the stress tensor. The relaxation of atomic positionsand optimization of lattice parameters have been doneby the conjugate gradient method. The atomic relaxationwas stopped when forces acting on atoms were less than0.01 eV Å−1. The total energy was converged to less than0.001 eV per atom and an 8 × 8 × 8 grid of Monkhorst–Packpoints was used. The total and projected DOS have beencalculated by the linear tetrahedron method with Blöchlcorrections [43]. The theoretical equilibrium volume V0 andbulk modulus (B0) are obtained by fitting the total energiesversus volume according to the Murnaghan EOS [44]. Theatomic basis vectors perpendicular to the applied strain aresimultaneously relaxed until the stress components vanish.
Elastics properties are important as they provide valuableinformation on the bonding characteristics between adjacentatomic planes, the anisotropic character of bonding and thestructural stability of the compound. To calculate the elasticconstants, one requires knowledge of the curvature of the
3
Phys. Scr. 84 (2011) 035704 N Munjal et al
0 1 2 3 4-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
(a)
BeS
ΔJ (
e/a.
u.)
pz (a.u.)
[100]-[110] [100]-[111] [110]-[111]
0 1 2 3 4-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
(b)
BeSe
ΔJ (
e/a.
u.)
pz (a.u.)
[100]-[110] [100]-[111] [110]-[111]
0 1 2 3 4-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
(c)
BeTe
Δ J (
e/a.
u.)
pz (a.u.)
[100]-[110] [100]-[111] [110]-[111]
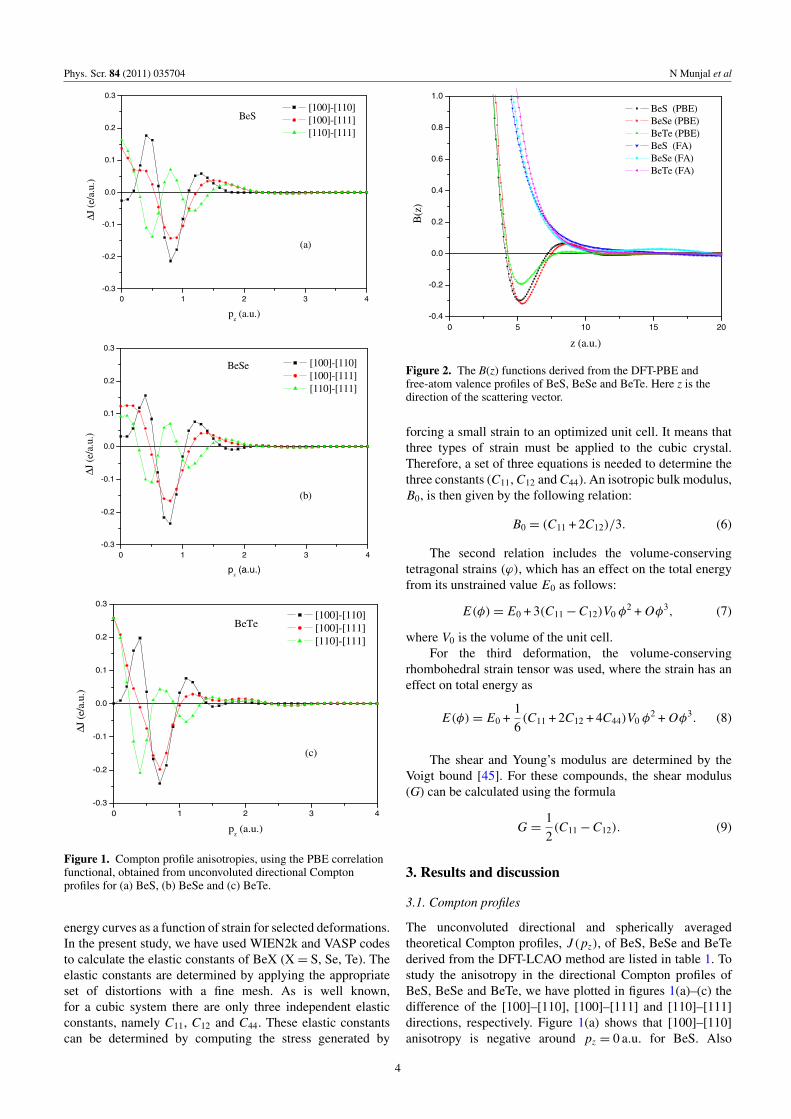
Figure 1. Compton profile anisotropies, using the PBE correlationfunctional, obtained from unconvoluted directional Comptonprofiles for (a) BeS, (b) BeSe and (c) BeTe.
energy curves as a function of strain for selected deformations.In the present study, we have used WIEN2k and VASP codesto calculate the elastic constants of BeX (X = S, Se, Te). Theelastic constants are determined by applying the appropriateset of distortions with a fine mesh. As is well known,for a cubic system there are only three independent elasticconstants, namely C11, C12 and C44. These elastic constantscan be determined by computing the stress generated by
0 5 10 15 20-0.4
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
B(z
)
z (a.u.)
BeS (PBE) BeSe (PBE) BeTe (PBE) BeS (FA) BeSe (FA) BeTe (FA)
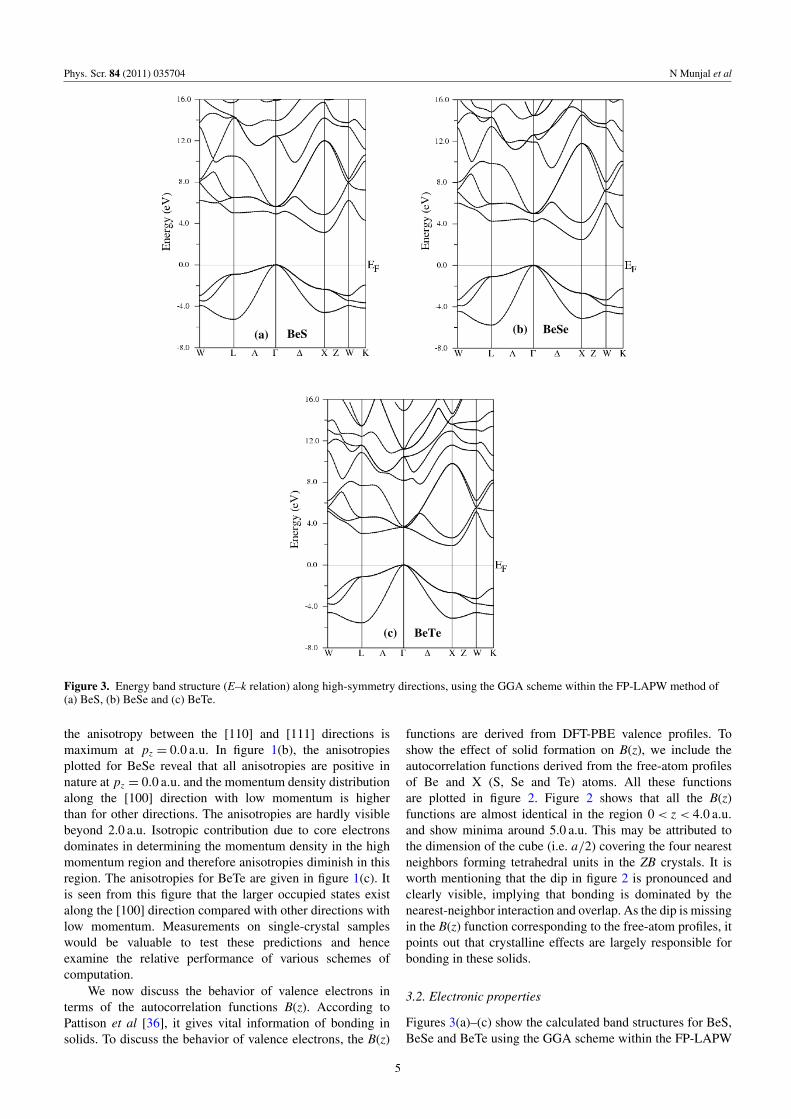
Figure 2. The B(z) functions derived from the DFT-PBE andfree-atom valence profiles of BeS, BeSe and BeTe. Here z is thedirection of the scattering vector.
forcing a small strain to an optimized unit cell. It means thatthree types of strain must be applied to the cubic crystal.Therefore, a set of three equations is needed to determine thethree constants (C11, C12 and C44). An isotropic bulk modulus,B0, is then given by the following relation:
B0 = (C11 + 2C12)/3. (6)
The second relation includes the volume-conservingtetragonal strains (ϕ), which has an effect on the total energyfrom its unstrained value E0 as follows:
E(φ)= E0 + 3(C11 − C12)V0 φ2 + Oφ3, (7)
where V0 is the volume of the unit cell.For the third deformation, the volume-conserving
rhombohedral strain tensor was used, where the strain has aneffect on total energy as
E(φ)= E0 +1
6(C11 + 2C12 + 4C44)V0 φ
2 + Oφ3. (8)
The shear and Young’s modulus are determined by theVoigt bound [45]. For these compounds, the shear modulus(G) can be calculated using the formula
G =1
2(C11 − C12). (9)
3. Results and discussion
3.1. Compton profiles
The unconvoluted directional and spherically averagedtheoretical Compton profiles, J (pz), of BeS, BeSe and BeTederived from the DFT-LCAO method are listed in table 1. Tostudy the anisotropy in the directional Compton profiles ofBeS, BeSe and BeTe, we have plotted in figures 1(a)–(c) thedifference of the [100]–[110], [100]–[111] and [110]–[111]directions, respectively. Figure 1(a) shows that [100]–[110]anisotropy is negative around pz = 0 a.u. for BeS. Also
4
Phys. Scr. 84 (2011) 035704 N Munjal et al
BeSe(b)
BeTe (c)
BeS (a)
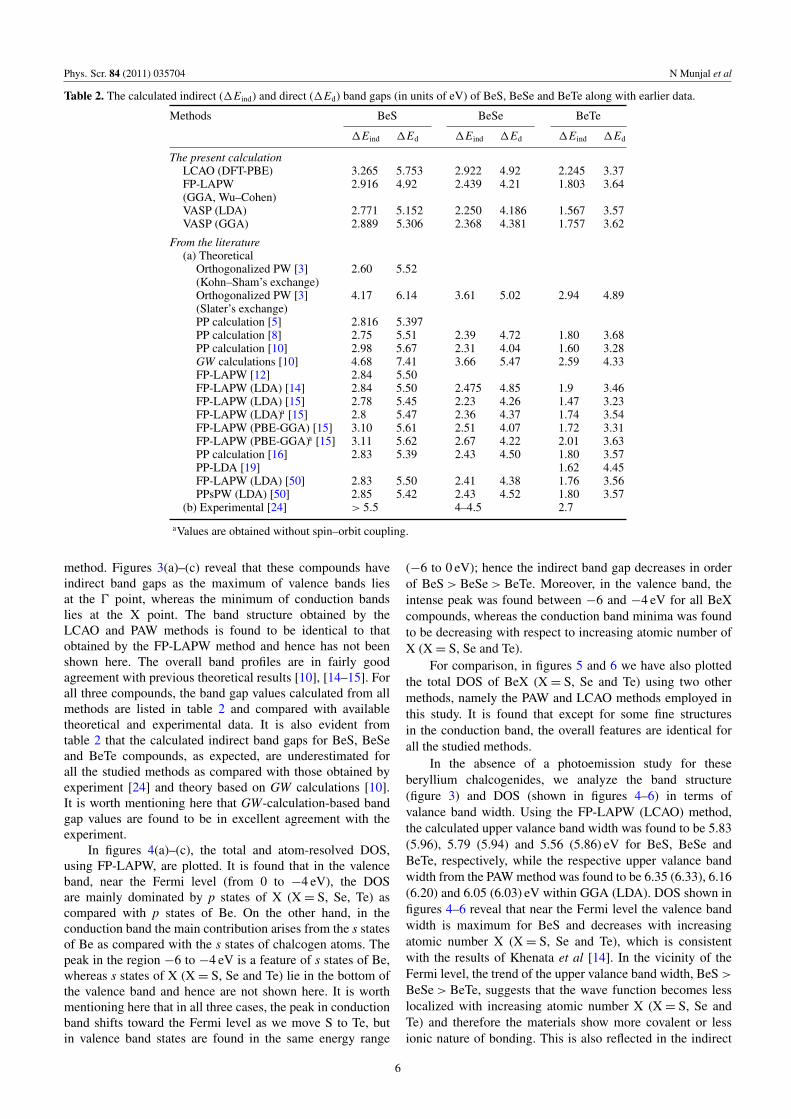
Figure 3. Energy band structure (E–k relation) along high-symmetry directions, using the GGA scheme within the FP-LAPW method of(a) BeS, (b) BeSe and (c) BeTe.
the anisotropy between the [110] and [111] directions ismaximum at pz = 0.0 a.u. In figure 1(b), the anisotropiesplotted for BeSe reveal that all anisotropies are positive innature at pz = 0.0 a.u. and the momentum density distributionalong the [100] direction with low momentum is higherthan for other directions. The anisotropies are hardly visiblebeyond 2.0 a.u. Isotropic contribution due to core electronsdominates in determining the momentum density in the highmomentum region and therefore anisotropies diminish in thisregion. The anisotropies for BeTe are given in figure 1(c). Itis seen from this figure that the larger occupied states existalong the [100] direction compared with other directions withlow momentum. Measurements on single-crystal sampleswould be valuable to test these predictions and henceexamine the relative performance of various schemes ofcomputation.
We now discuss the behavior of valence electrons interms of the autocorrelation functions B(z). According toPattison et al [36], it gives vital information of bonding insolids. To discuss the behavior of valence electrons, the B(z)
functions are derived from DFT-PBE valence profiles. Toshow the effect of solid formation on B(z), we include theautocorrelation functions derived from the free-atom profilesof Be and X (S, Se and Te) atoms. All these functionsare plotted in figure 2. Figure 2 shows that all the B(z)functions are almost identical in the region 0< z < 4.0 a.u.and show minima around 5.0 a.u. This may be attributed tothe dimension of the cube (i.e. a/2) covering the four nearestneighbors forming tetrahedral units in the ZB crystals. It isworth mentioning that the dip in figure 2 is pronounced andclearly visible, implying that bonding is dominated by thenearest-neighbor interaction and overlap. As the dip is missingin the B(z) function corresponding to the free-atom profiles, itpoints out that crystalline effects are largely responsible forbonding in these solids.
3.2. Electronic properties
Figures 3(a)–(c) show the calculated band structures for BeS,BeSe and BeTe using the GGA scheme within the FP-LAPW
5
Phys. Scr. 84 (2011) 035704 N Munjal et al
Table 2. The calculated indirect (1Eind) and direct (1Ed) band gaps (in units of eV) of BeS, BeSe and BeTe along with earlier data.
Methods BeS BeSe BeTe
1Eind 1Ed 1Eind 1Ed 1Eind 1Ed
The present calculationLCAO (DFT-PBE) 3.265 5.753 2.922 4.92 2.245 3.37FP-LAPW 2.916 4.92 2.439 4.21 1.803 3.64(GGA, Wu–Cohen)VASP (LDA) 2.771 5.152 2.250 4.186 1.567 3.57VASP (GGA) 2.889 5.306 2.368 4.381 1.757 3.62
From the literature(a) Theoretical
Orthogonalized PW [3] 2.60 5.52(Kohn–Sham’s exchange)Orthogonalized PW [3] 4.17 6.14 3.61 5.02 2.94 4.89(Slater’s exchange)PP calculation [5] 2.816 5.397PP calculation [8] 2.75 5.51 2.39 4.72 1.80 3.68PP calculation [10] 2.98 5.67 2.31 4.04 1.60 3.28GW calculations [10] 4.68 7.41 3.66 5.47 2.59 4.33FP-LAPW [12] 2.84 5.50FP-LAPW (LDA) [14] 2.84 5.50 2.475 4.85 1.9 3.46FP-LAPW (LDA) [15] 2.78 5.45 2.23 4.26 1.47 3.23FP-LAPW (LDA)a [15] 2.8 5.47 2.36 4.37 1.74 3.54FP-LAPW (PBE-GGA) [15] 3.10 5.61 2.51 4.07 1.72 3.31FP-LAPW (PBE-GGA)a [15] 3.11 5.62 2.67 4.22 2.01 3.63PP calculation [16] 2.83 5.39 2.43 4.50 1.80 3.57PP-LDA [19] 1.62 4.45FP-LAPW (LDA) [50] 2.83 5.50 2.41 4.38 1.76 3.56PPsPW (LDA) [50] 2.85 5.42 2.43 4.52 1.80 3.57
(b) Experimental [24] > 5.5 4–4.5 2.7
aValues are obtained without spin–orbit coupling.
method. Figures 3(a)–(c) reveal that these compounds haveindirect band gaps as the maximum of valence bands liesat the 0 point, whereas the minimum of conduction bandslies at the X point. The band structure obtained by theLCAO and PAW methods is found to be identical to thatobtained by the FP-LAPW method and hence has not beenshown here. The overall band profiles are in fairly goodagreement with previous theoretical results [10], [14–15]. Forall three compounds, the band gap values calculated from allmethods are listed in table 2 and compared with availabletheoretical and experimental data. It is also evident fromtable 2 that the calculated indirect band gaps for BeS, BeSeand BeTe compounds, as expected, are underestimated forall the studied methods as compared with those obtained byexperiment [24] and theory based on GW calculations [10].It is worth mentioning here that GW-calculation-based bandgap values are found to be in excellent agreement with theexperiment.
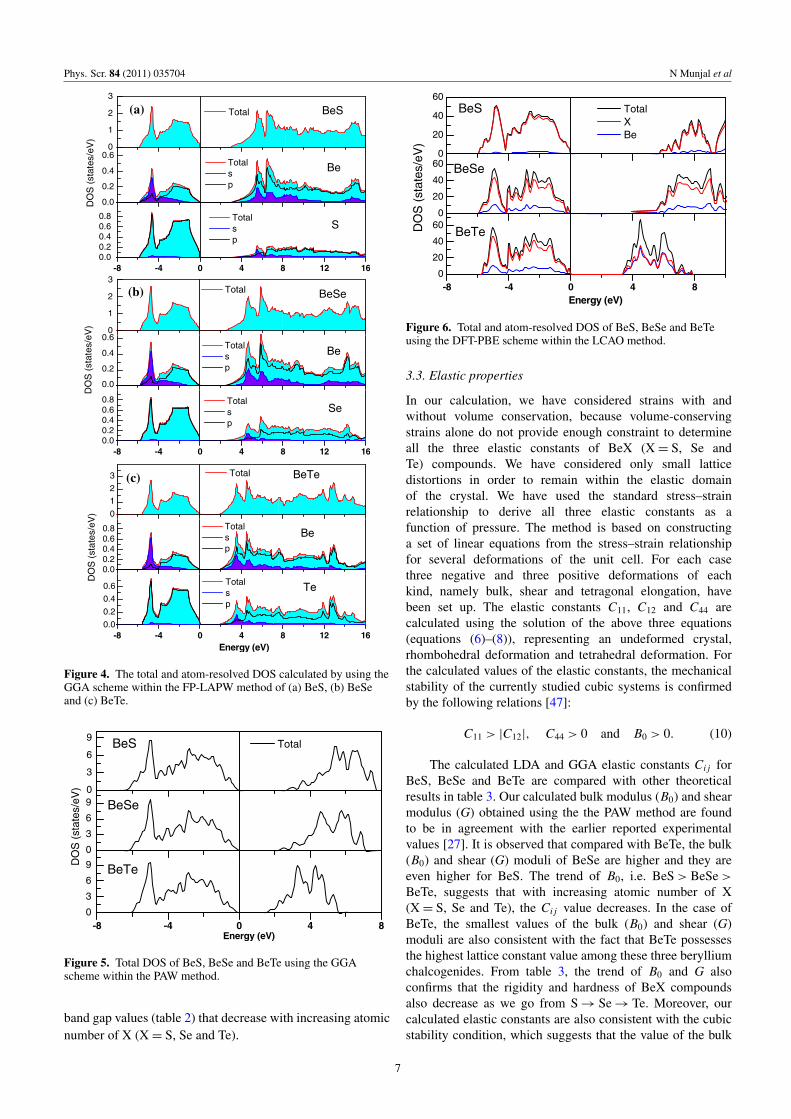
In figures 4(a)–(c), the total and atom-resolved DOS,using FP-LAPW, are plotted. It is found that in the valenceband, near the Fermi level (from 0 to −4 eV), the DOSare mainly dominated by p states of X (X = S, Se, Te) ascompared with p states of Be. On the other hand, in theconduction band the main contribution arises from the s statesof Be as compared with the s states of chalcogen atoms. Thepeak in the region −6 to −4 eV is a feature of s states of Be,whereas s states of X (X = S, Se and Te) lie in the bottom ofthe valence band and hence are not shown here. It is worthmentioning here that in all three cases, the peak in conductionband shifts toward the Fermi level as we move S to Te, butin valence band states are found in the same energy range
(−6 to 0 eV); hence the indirect band gap decreases in orderof BeS> BeSe> BeTe. Moreover, in the valence band, theintense peak was found between −6 and −4 eV for all BeXcompounds, whereas the conduction band minima was foundto be decreasing with respect to increasing atomic number ofX (X = S, Se and Te).
For comparison, in figures 5 and 6 we have also plottedthe total DOS of BeX (X = S, Se and Te) using two othermethods, namely the PAW and LCAO methods employed inthis study. It is found that except for some fine structuresin the conduction band, the overall features are identical forall the studied methods.
In the absence of a photoemission study for theseberyllium chalcogenides, we analyze the band structure(figure 3) and DOS (shown in figures 4–6) in terms ofvalance band width. Using the FP-LAPW (LCAO) method,the calculated upper valance band width was found to be 5.83(5.96), 5.79 (5.94) and 5.56 (5.86) eV for BeS, BeSe andBeTe, respectively, while the respective upper valance bandwidth from the PAW method was found to be 6.35 (6.33), 6.16(6.20) and 6.05 (6.03) eV within GGA (LDA). DOS shown infigures 4–6 reveal that near the Fermi level the valence bandwidth is maximum for BeS and decreases with increasingatomic number X (X = S, Se and Te), which is consistentwith the results of Khenata et al [14]. In the vicinity of theFermi level, the trend of the upper valance band width, BeS>BeSe> BeTe, suggests that the wave function becomes lesslocalized with increasing atomic number X (X = S, Se andTe) and therefore the materials show more covalent or lessionic nature of bonding. This is also reflected in the indirect
6
Phys. Scr. 84 (2011) 035704 N Munjal et al
(a)
(b)
-8 -4 0 4 8 12 160.00.20.40.60.8
Se Total s p
0.0
0.2
0.4
0.6
Be
BeSe
DO
S (
stat
es/e
V)
Total s p
0
1
2
3
Total
-8 -4 0 4 8 12 160.00.20.40.60.8
S Total s p
0.0
0.2
0.4
0.6
Be
BeS
DO
S (
stat
es/e
V)
Total s p
0
1
2
3
Total
-8 -4 0 4 8 12 160.0
0.2
0.4
0.6
Te Total s p
Energy (eV)
0.00.20.40.60.8 Be
BeTe
DO
S (
stat
es/e
V)
Total s p
0
1
2
3
Total(c)
Figure 4. The total and atom-resolved DOS calculated by using theGGA scheme within the FP-LAPW method of (a) BeS, (b) BeSeand (c) BeTe.
-8 -4 0 4 80
3
6
9 BeTe
Energy (eV)
DO
S (
stat
es/e
V)
0
3
6
9 BeSe0
3
6
9 TotalBeS
Figure 5. Total DOS of BeS, BeSe and BeTe using the GGAscheme within the PAW method.
band gap values (table 2) that decrease with increasing atomicnumber of X (X = S, Se and Te).
-8 -4 0 4 80
20
40
60BeTe
Energy (eV)
DO
S (
stat
es/e
V)
0
20
40
60 BeSe0
20
40
60 Total X Be
BeS
Figure 6. Total and atom-resolved DOS of BeS, BeSe and BeTeusing the DFT-PBE scheme within the LCAO method.
3.3. Elastic properties
In our calculation, we have considered strains with andwithout volume conservation, because volume-conservingstrains alone do not provide enough constraint to determineall the three elastic constants of BeX (X = S, Se andTe) compounds. We have considered only small latticedistortions in order to remain within the elastic domainof the crystal. We have used the standard stress–strainrelationship to derive all three elastic constants as afunction of pressure. The method is based on constructinga set of linear equations from the stress–strain relationshipfor several deformations of the unit cell. For each casethree negative and three positive deformations of eachkind, namely bulk, shear and tetragonal elongation, havebeen set up. The elastic constants C11, C12 and C44 arecalculated using the solution of the above three equations(equations (6)–(8)), representing an undeformed crystal,rhombohedral deformation and tetrahedral deformation. Forthe calculated values of the elastic constants, the mechanicalstability of the currently studied cubic systems is confirmedby the following relations [47]:
C11 > |C12|, C44 > 0 and B0 > 0. (10)
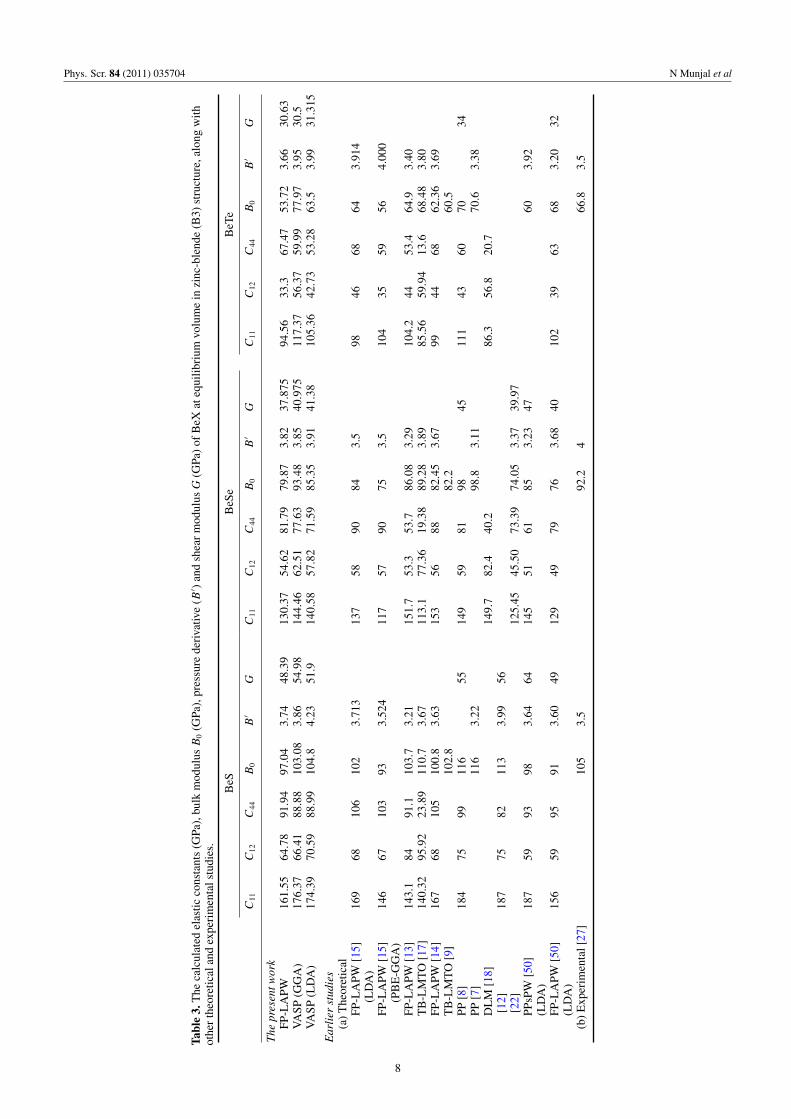
The calculated LDA and GGA elastic constants Ci j forBeS, BeSe and BeTe are compared with other theoreticalresults in table 3. Our calculated bulk modulus (B0) and shearmodulus (G) obtained using the the PAW method are foundto be in agreement with the earlier reported experimentalvalues [27]. It is observed that compared with BeTe, the bulk(B0) and shear (G) moduli of BeSe are higher and they areeven higher for BeS. The trend of B0, i.e. BeS> BeSe>BeTe, suggests that with increasing atomic number of X(X = S, Se and Te), the Ci j value decreases. In the case ofBeTe, the smallest values of the bulk (B0) and shear (G)moduli are also consistent with the fact that BeTe possessesthe highest lattice constant value among these three berylliumchalcogenides. From table 3, the trend of B0 and G alsoconfirms that the rigidity and hardness of BeX compoundsalso decrease as we go from S → Se → Te. Moreover, ourcalculated elastic constants are also consistent with the cubicstability condition, which suggests that the value of the bulk
7
Phys. Scr. 84 (2011) 035704 N Munjal et al
Tabl
e3.
The
calc
ulat
edel
astic
cons
tant
s(G
Pa),
bulk
mod
ulus
B0
(GPa
),pr
essu
rede
riva
tive
(B′ )
and
shea
rm
odul
usG
(GPa
)of
BeX
ateq
uilib
rium
volu
me
inzi
nc-b
lend
e(B
3)st
ruct
ure,
alon
gw
ithot
her
theo
retic
alan
dex
peri
men
tals
tudi
es.
BeS
BeS
eB
eTe
C11
C12
C44
B0
B′
GC
11C
12C
44B
0B
′G
C11
C12
C44
B0
B′
G
The
pres
entw
ork
FP-L
APW
161.
5564
.78
91.9
497
.04
3.74
48.3
913
0.37
54.6
281
.79
79.8
73.
8237
.875
94.5
633
.367
.47
53.7
23.
6630
.63
VA
SP(G
GA
)17
6.37
66.4
188
.88
103.
083.
8654
.98
144.
4662
.51
77.6
393
.48
3.85
40.9
7511
7.37
56.3
759
.99
77.9
73.
9530
.5V
ASP
(LD
A)
174.
3970
.59
88.9
910
4.8
4.23
51.9
140.
5857
.82
71.5
985
.35
3.91
41.3
810
5.36
42.7
353
.28
63.5
3.99
31.3
15
Ear
lier
stud
ies
(a)
The
oret
ical
FP-L
APW
[15]
169
6810
610
23.
713
137
5890
843.
598
4668
643.
914
(LD
A)
FP-L
APW
[15]
146
6710
393
3.52
411
757
9075
3.5
104
3559
564.
000
(PB
E-G
GA
)FP
-LA
PW[1
3]14
3.1
8491
.110
3.7
3.21
151.
753
.353
.786
.08
3.29
104.
244
53.4
64.9
3.40
TB
-LM
TO
[17]
140.
3295
.92
23.8
911
0.7
3.67
113.
177
.36
19.3
889
.28
3.89
85.5
659
.94
13.6
68.4
83.
80FP
-LA
PW[1
4]16
768
105
100.
83.
6315
356
8882
.45
3.67
9944
6862
.36
3.69
TB
-LM
TO
[9]
102.
882
.260
.5PP
[8]
184
7599
116
5514
959
8198
4511
143
6070
34PP
[7]
116
3.22
98.8
3.11
70.6
3.38
DL
M[1
8]14
9.7
82.4
40.2
86.3
56.8
20.7
[12]
187
7582
113
3.99
56[2
2]12
5.45
45.5
073
.39
74.0
53.
3739
.97
PPsP
W[5
0]18
759
9398
3.64
6414
551
6185
3.23
4760
3.92
(LD
A)
FP-L
APW
[50]
156
5995
913.
6049
129
4979
763.
6840
102
3963
683.
2032
(LD
A)
(b)
Exp
erim
enta
l[27
]10
53.
592
.24
66.8
3.5
8
Phys. Scr. 84 (2011) 035704 N Munjal et al
0 1 2 3 40
1
2
3
4
5
6
0 1 2 3 4-0.15
-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
0.25
Δ [J
(pz)*
p f]
pz/p
f
BeS-BeSe BeS-BeTe
J(p z)*
p f
pz/p
f
BeS BeSe BeTe
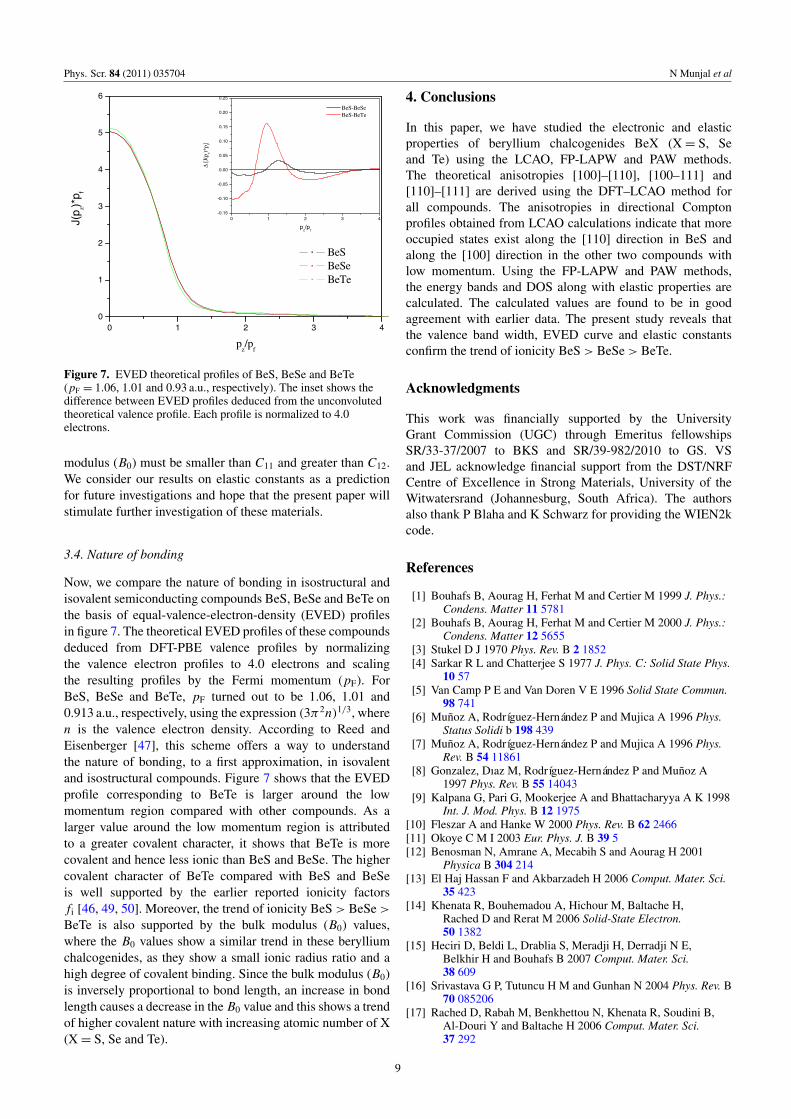
Figure 7. EVED theoretical profiles of BeS, BeSe and BeTe(pF = 1.06, 1.01 and 0.93 a.u., respectively). The inset shows thedifference between EVED profiles deduced from the unconvolutedtheoretical valence profile. Each profile is normalized to 4.0electrons.
modulus (B0) must be smaller than C11 and greater than C12.We consider our results on elastic constants as a predictionfor future investigations and hope that the present paper willstimulate further investigation of these materials.
3.4. Nature of bonding
Now, we compare the nature of bonding in isostructural andisovalent semiconducting compounds BeS, BeSe and BeTe onthe basis of equal-valence-electron-density (EVED) profilesin figure 7. The theoretical EVED profiles of these compoundsdeduced from DFT-PBE valence profiles by normalizingthe valence electron profiles to 4.0 electrons and scalingthe resulting profiles by the Fermi momentum (pF). ForBeS, BeSe and BeTe, pF turned out to be 1.06, 1.01 and0.913 a.u., respectively, using the expression (3π2n)1/3, wheren is the valence electron density. According to Reed andEisenberger [47], this scheme offers a way to understandthe nature of bonding, to a first approximation, in isovalentand isostructural compounds. Figure 7 shows that the EVEDprofile corresponding to BeTe is larger around the lowmomentum region compared with other compounds. As alarger value around the low momentum region is attributedto a greater covalent character, it shows that BeTe is morecovalent and hence less ionic than BeS and BeSe. The highercovalent character of BeTe compared with BeS and BeSeis well supported by the earlier reported ionicity factorsfi [46, 49, 50]. Moreover, the trend of ionicity BeS> BeSe>BeTe is also supported by the bulk modulus (B0) values,where the B0 values show a similar trend in these berylliumchalcogenides, as they show a small ionic radius ratio and ahigh degree of covalent binding. Since the bulk modulus (B0)is inversely proportional to bond length, an increase in bondlength causes a decrease in the B0 value and this shows a trendof higher covalent nature with increasing atomic number of X(X = S, Se and Te).
4. Conclusions
In this paper, we have studied the electronic and elasticproperties of beryllium chalcogenides BeX (X = S, Seand Te) using the LCAO, FP-LAPW and PAW methods.The theoretical anisotropies [100]–[110], [100–111] and[110]–[111] are derived using the DFT–LCAO method forall compounds. The anisotropies in directional Comptonprofiles obtained from LCAO calculations indicate that moreoccupied states exist along the [110] direction in BeS andalong the [100] direction in the other two compounds withlow momentum. Using the FP-LAPW and PAW methods,the energy bands and DOS along with elastic properties arecalculated. The calculated values are found to be in goodagreement with earlier data. The present study reveals thatthe valence band width, EVED curve and elastic constantsconfirm the trend of ionicity BeS> BeSe> BeTe.
Acknowledgments
This work was financially supported by the UniversityGrant Commission (UGC) through Emeritus fellowshipsSR/33-37/2007 to BKS and SR/39-982/2010 to GS. VSand JEL acknowledge financial support from the DST/NRFCentre of Excellence in Strong Materials, University of theWitwatersrand (Johannesburg, South Africa). The authorsalso thank P Blaha and K Schwarz for providing the WIEN2kcode.
References
[1] Bouhafs B, Aourag H, Ferhat M and Certier M 1999 J. Phys.:Condens. Matter 11 5781
[2] Bouhafs B, Aourag H, Ferhat M and Certier M 2000 J. Phys.:Condens. Matter 12 5655
[3] Stukel D J 1970 Phys. Rev. B 2 1852[4] Sarkar R L and Chatterjee S 1977 J. Phys. C: Solid State Phys.
10 57[5] Van Camp P E and Van Doren V E 1996 Solid State Commun.
98 741[6] Muñoz A, Rodríguez-Hernández P and Mujica A 1996 Phys.
Status Solidi b 198 439[7] Muñoz A, Rodríguez-Hernández P and Mujica A 1996 Phys.
Rev. B 54 11861[8] Gonzalez, Dıaz M, Rodríguez-Hernández P and Muñoz A
1997 Phys. Rev. B 55 14043[9] Kalpana G, Pari G, Mookerjee A and Bhattacharyya A K 1998
Int. J. Mod. Phys. B 12 1975[10] Fleszar A and Hanke W 2000 Phys. Rev. B 62 2466[11] Okoye C M I 2003 Eur. Phys. J. B 39 5[12] Benosman N, Amrane A, Mecabih S and Aourag H 2001
Physica B 304 214[13] El Haj Hassan F and Akbarzadeh H 2006 Comput. Mater. Sci.
35 423[14] Khenata R, Bouhemadou A, Hichour M, Baltache H,
Rached D and Rerat M 2006 Solid-State Electron.50 1382
[15] Heciri D, Beldi L, Drablia S, Meradji H, Derradji N E,Belkhir H and Bouhafs B 2007 Comput. Mater. Sci.38 609
[16] Srivastava G P, Tutuncu H M and Gunhan N 2004 Phys. Rev. B70 085206
[17] Rached D, Rabah M, Benkhettou N, Khenata R, Soudini B,Al-Douri Y and Baltache H 2006 Comput. Mater. Sci.37 292
9

Phys. Scr. 84 (2011) 035704 N Munjal et al
[18] Doyen-Lang S, Pages O, Lang L and Hugel J 2002 Phys.Status Solidi b 229 563
[19] Nagelstraßer M et al 1998 Phys. Rev. B 58 10394[20] Yadav P S, Yadav R K, Agrawal S and Agrawal B K 2007
Physica E 36 79[21] Saib S and Bouarissa N 2010 Solid State Sci. 12 563[22] Kong F and Jiang G 2009 Physica B 404 3935[23] Ameri M, Rached D, Rabaha M, Khenata R, Benkhettou N,
Bouhafs B and Maachou M 2007 Mater. Sci. Semicond.Process. 10 6
[24] Yim W M, Dismakes J B, Stofko E J and Paff R J 1972J. Phys. Chem. Solids 33 501
[25] Luo H, Ghandehari K, Greene R G, Ruoff A L, Trail S S andDiSalvo F J 1995 Phys. Rev. B 52 7058
[26] Waag A et al 1996 J. Appl. Phys. 80 792[27] Narayana C, Nesamony V J and Ruoff A L 1997 Phys. Rev. B
56 14338[28] Wilmers K et al 1999 J. Electron. Mater. 28 670[29] Dovesi R, Orlando R, Roetti C, Pisani C and Saunders V R
2000 Phys. Status Solidi b 217 63[30] Pisani C, Dovesi R and Roetti C 1988 Hartree–Fock Ab Initio
Treatment of Crystalline Solids (Lecture Notes in Chemistryvol 48) (Heidelberg: Springer)
[31] Dovesi R et al 2006 CRYSTAL06 User’s Manual (Torino:University of Torino)
[32] Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett.77 3865
[33] Cooper M J 1985 Rep. Prog. Phys. 48 415Williams B 1977 Compton Scattering (London: McGraw-Hill)
[34] Cooper M J, Mijnarends P E, Shiotani N, Sakai N and BansilA 2004 X-Ray Compton Scattering (Oxford: OxfordPublishing Press)
[35] Weyrich W, Pattison P and Williams B 1979 Chem. Phys.41 271
[36] Pattison P, Hansen N K and Schneider J R 1981 Chem. Phys.59 231
[37] Blaha P, Schwarz K, Madsen G K H, Kvasnicka D and Luitz J2001 WIEN2K, an Augmented Plane Wave + Local OrbitalsProgram for Calculating Crystal Properties (Wien: Techn.Universität Wien)
[38] Wu Z and Cohen R 2006 Phys. Rev. B 73 235116[39] Kresse G and Hafner J 1994 Phys. Rev. B 49 14251[40] Kresse G and Furthmüller J 1996 Comput. Mater. Sci. 6 15
Kresse G and Furthmüller J 1996 Phys. Rev. B 54 11169[41] Kresse G and Joubert J 1999 Phys. Rev. B 59 1758[42] Perdew J P and Zunger A 1981 Phys. Rev. B 23 5048[43] Blöchl P, Jepsen O and Andersen O K 1994 Phys. Rev. B
49 16223[44] Murnaghan F D 1944 Proc. Natl Acad. Sci. USA 30 244[45] Hill R 1952 Proc. Phys. Soc. A 65 349[46] Berghout A, Zaoui A and Hugel J 2006 J. Phys.: Condens.
Matter 18 10365[47] Sinko G V and Smirnow N A 2002 J. Phys.: Condens. Matter
14 6989[48] Reed W A and Eisenberger P 1972 Phys. Rev. B 6 4596[49] Christensen N E, Satpathy S and Pawlowska Z 1987 Phys. Rev.
B 36 1032[50] Phillips J C 1973 Bonds and Bands in Semiconductor (New
York: Academic)
10




















