
Structural Changes in Li2 MnO 3 Cathode Material for Li- Ion Batteries

PHYSICAL REVIEW B VOLUME 50, NUMBER 8
Ab initio study of Mno and Nio
15 AUGUST 1994-II
M. D. Towler and N. L. AllanSchool of Chemistry, Bristol University, Cantock's Close, Bristol B$8 ITS, United Kingdom
N. M. Harrison and V. R. Saunders
W. C. MackrodtDepartment of Theoretical Chemistry, Oxford University, 5, South Parks Road, Oxford OX1 3UB, United Kingdom
E. ApraDepartment ofInorganic, Physical and Materials Chemistry, University of Torino, via Giuria 5, I 10125-Torino, Italy
(Received 7 February 1994)
The ground-state electronic structure of NiO and MnO has been calculated within the Hartree-Fockapproximation using local Gaussian basis sets. For both, a qualitatively correct ground-state electronicstructure is obtained in which the wide-band-gap insulating character of these materials is seen to be aresult of large on-site Coulomb interactions. The materials are correctly predicted to be antiferromag-netic with the AF& structure. The relative energy differences between various magnetic structures areconsistent with the ratio of the Neel temperatures. The structural, elastic, and vibrational properties arein reasonable agreement with available experimental data.
I. INTRODUCTION
Since their unusual insulating and magnetic propertieswere first recognized in the 1930s, the first-rowtransition-metal monoxides have provided considerablechallenges to commonly accepted theories of electronicstructure and bonding. In this paper we consider MnOand NiO, which crystallize in the rock-salt structure andwhich at low temperatures are found to be high-spin anti-ferromagnetic insulators with conductivities' in therange 10 ' -10 ' 0 'cm ' and Neel temperatures(Ttt) of 116 and 525 K, respectively. The insulatingcharacter and local magnetic moments of both are main-tained above Ttt, with optical band gaps of 3.6 and 3.8 eVwhich are essentially independent of T. This is in markedcontrast to many materials which undergo metal-insulator transitions such as VO2, NiS, and V203, wherethe optical gaps are all less than 0.6 eV and decreasemarkedly with increasing temperature. The persistenceof the insulating gap and local moments above the spin-disordering temperature appears to be the best phenome-nological way of defining the Mott-insulator state, al-though a recent development has been the differentiationbetween Mott-Hubbard and charge-transfer insulators,based on whether the observed gap is of 1~d or p —+dcharacter. '
The majority of previous theoretical treatments of thetransition meta1 oxides have been based on the local-spin-density approximation (LSDA), which encountersserious problems in describing such cases. ' The work ofBrandow ' and others has suggested that a useful mod-el for Mott insulators is provided by unrestricted
Hartree-Fock (UHF) theory. Indeed, the most recentdensity-functional calculations have tended to includemodifications such as self-interaction-corrected (SIC)LSDA (Ref. 11) and LSDA+U (Ref. 12) in an apparentattempt to emulate features of the Hartree-Fock Hamil-tonian. These studies have offered improved descriptionsof the Mott-insulating state.
In the present work, therefore, we apply the ab initiolinear-combination of atomic orbitals (LCAO) periodicHartree-Fock approach, embodied in the cRYsTAL92code, ' ' to MnO and NiO using extended basis sets andthe UHF scheme for describing open shell states. ' Thisscheme has been used previously to examine a range ofother oxides, including Li20, ' ' MgO is and A1203 i9,20
Preliminary results from a study of the ground-stateproperties of a selection of the first-row monoxides, in-cluding CaO and the metallic oxide VO, have beenpresented in a previous paper. ' In this work we providea much more detailed study of MnO and NiO and extendthe treatment previously given to include elastic proper-ties and phonon frequencies. We are aware of only twoprevious ab initio Hartree-Fock studies of these materi-als, one due to Kunz, the other to Janssen andNieuwpoort, who consider a [Ni06]' cluster in apoint ion field.
Recent experimental and theoretical evidence has ledto the speculation that the upper valence band in thesematerials is primarily 0 2p in character. " This hasimportant consequences regarding both the nature ofhole states and the valency of the higher oxides, and hasled to the suggested classification of NiO as a charge-transfer insulator. '
0163-1829/94/50(8)/5041(14)/$06. 00 50 5041 1994 The American Physical Society

5042 M. D. TOWLER et al. 50
The structure of the present paper is as follows. InSec. II some general points will be made regardingmethodology and the local basis sets used. In Sec. III awide range of results for MnO and NiO are presented in-cluding the calculated lattice structure, magnetostriction,magnetic moments, Neel temperatures, band structure,density of states, charge density, elastic constants, andphonon frequencies. The electronic structure is analyzedin terms of a single site model.
II. COMPUTATIONAL METHOD
The implementation of the all-electron ab initio self-consistent field (SCF}Hartree-Fock LCAO computation-al scheme for periodic systems within the computer codecRYSTAL92 has been described previously. ' ' There area number of limitations to the accuracy of calculationsusing this scheme. The main source of error (electroncorrelation) stems directly from the Hartree-Fock ap-proximation. In previous work, this has generally led toan underestimate of the binding energies by about 30%,and to overestimates of the lattice parameter by approxi-
xnately 2%, although successful attempts have been madeto include correlation corrections using functionals of theHartree-Fock density.
The second inaccuracy stems from numerical approxi-mations introduced in the implementation of the SCFequations. These approximations appear in the recipro-cal space integration to reconstruct the electronic chargedistribution and in the evaluation of the Coulomb and ex-change series. In these series, the Gaussian integrals areclassified according to overlap or penetration criteria. In-tegrals of sufficiently low overlap or penetration may beneglected or approximated, the cutofrs being controlledby five parameters. Our calculations were performedwith these tolerances set to 10, 10, 10, 10, and10 ', which provide high numerical accuracy. The de-tailed e8'ect of these parameters is discussed elsewhere.The reciprocal space integration was performed by sam-pling the Brillouin zone at a regular set of points definedby a shrinking factor of 8. Errors are greatest for numer-ically evaluated second derivatives; we estimate that thenumerical error is less than 2%%uo for elastic constants andphonon frequencies and negligible for the lattice struc-ture.
TABLE I. Core and valence basis sets for MnO and NiO.
Core basesShell
No. type ExponentMn
Coefficients Exponent Coefficients Exponent0
Coefficients
SP
SP
292 601.042 265.0
8 947.292 330.32
702.047242.90794.95539.5777
732.14175.55158.509323.1299.75363.4545
38.38915.43676.17812.8235
0.000 2270.001 90.011 1
0.050 1
0.170 5
0.369 1
0.403 5
0.143 7—0.0053—0.0673—0.1293
0.25350.63450.27140.0157
—0.2535—0.8648
0.9337
0.00860.06120.21350.40180.40120.2222
—0.0311—0.0969
0.25631.6552
367 916.052 493.911 175.82 925.4
882.875305.538119.55149.9247
924.525223.044
74.421129.621112.47214.2461
56.658121.2063
8.49143.6152
0.000 2270.001 9290.011 1
0.050 00.170 30.369 00.403 5
0.142 6—0.0052—0.0679—0.1319
0.25760.63570.28380.0124
—0.2218—0.8713
1.0285
0.00860.06090.21350.39440.39730.2586
—0.0180—0.0800
0.20891.2550
8020.01338.0255.469.2223.909.2643.8511.212
49.4310.473.2351.217
0.001 080.008 040.053 240.168 1
0.358 1
0.385 5
0.146 8
0.072 8—0.008 83 0.009 58—0.091 5 0.069 6—0.040 2 0.206 5
0.379 0 0.347 0
ShellNo. type Exponent
MnCoefficients
Valence bases
Exponent Coefficients Exponent0
CoefBcients
SP
SPd
1.20860.4986
22.59296.16742.06380.74010.2490
1.01.0
0.07080.30440.54690.5102
1.0
1.01.0
1.51450.6144
41.080011.41303.85611.33020.4110
1.01.0
0.04050.20220.43380.4897
1.0
1.01.0
0.4980'0.1690'
1.01.0
1.01.0
These are from an original CaO oxygen basis (Ref. 21). Optimized values for MnO are 0.4763 and 0.1760 bohr and for NiO 0.4764and 0.1802 bohr .Given incorrectly in our previous paper (Ref. 21).

50 AS INITIO STUDY OF MnO AND NiO 5043
The final source of error relates to the choice of basisset. Extended Gaussian basis sets were generated forboth oxides composed of 27 atomic orbitals for Mn andNi and 13 for oxygen, where each orbital is a linear com-bination (contraction) of Gaussian-type functions. Thebasis sets were derived in the following manner. Com-plete sets of contraction coefficients and exponents for thecation bases were obtained by energy minimization of thefree-ion +2 state. Together with a previously derivedbasis set for oxygen in CaO, ' the free-ion bases were thenused to derive a basis set appropriate for a description ofthe solid state by minimizing the ground-state energy ofthe solid in a single unit-cell description at the experi-mental lattice constants. ' The exponents and contrac-tion coefficients for the cation d shells and for the twooutermost sp shells of the cation and oxygen bases wereoptimized in this way. The final part of the procedure in-volved optimizing the geometry and the same basis setparameters in turn until self-consistency was achieved.The basis sets adopted are given in Table I.
III. RESULTS
A. Geometry and magnetic properties
In the observed low-temperature antiferromagneticstate the individual atomic moments of MnO and NiOare aligned in ferromagnetic (111) sheets, with adjacentsheets having antiparallel spin. This is generally referredto as the AF2 spin arrangement. In Table II we list thecalculated equilibrium lattice constants for this state andcompare these with the available x-ray data. For com-parison, predicted geometries using the same basis setsare given for the hypothetical ferromagnetic and AF,magnetic states, where the latter consists of (100) fer-romagnetic sheets with adjacent sheets antiparallel.These calculations were all performed by minimization ofthe total energy of cells containing two formula units.For both systems the calculated lattice constants arearound 2/o greater than the experimental values, whichis ty ical of previous studies of closed-shell ox-ides. ' ' ' Table II also contains a comparison ofHartree-Fock binding energies, derived from thedifference between the crystal and atomic solutions, andthe corresponding experimental values. The underesti-mate of approximately 30% due to the neglect of the
TABLE II. Calculated lattice constants and binding energiescompared with experiment.
electron correlation is again compatible with earlierwork.
Calculated and experimental magnetic moments arecompared in Table III. The calculated values are de-duced from Mulliken population analyses of the UHFwave function. Our values for MnO and NiO, 4.92p~and 1.92p~, may be compared with SIC-LSD values of4.49p~ and 1.53p~, respectively, calculated by Svane andGunnarsson. "The inclusion of correlation effects wouldtend to decrease our predicted moments, whereas spin-orbit coupling (which is neglected) will tend to increasethe moments. However, perhaps a more useful compar-ison would be between experimental and theoreticalneutron-scattering factors. '
The relative energies of the closed-shell and variousferromagnetic states (differing in their magnetic mo-ments) at the calculated ground-state lattice constants areshown in Table IV. From these it is clear that Hartree-Fock calculations correctly predict both MnO and NiOto be high-spin insulators. The relative stability of theantiferromagnetic and ferromagnetic states are reportedin Table V at the calculated ground-state lattice con-stants. The AF2 state is correctly predicted to be themost stable spin configuration followed by the ferromag-netic and then the AF, . Furthermore this order does notchange when calculations are performed at the optimalgeometry of either of the latter two configurations. Wecan write the energy difference between any two magneticstates in the form hnE~+b, n'Ez„where E„andEz,refer to contributions from second-neighbor superex-change and first-neighbor direct exchange, respectively.An and hn' are the difFerences in the number of such in-teractions between the two states. Given the energydifferences listed in Table V it is straightforward to esti-mate values of E and Ed, for MnO and NiO. For MnO,E and Ed, are —2.0X10 and —5.0X10 eV, re-spectively, ' the corresponding values for NiO are—7.0X 10 and —1.5 X 10 eV. As expected,~E ~
& ~Ed, ~for both compounds.
In preliminary work, using a pseudopotential modelto represent the core electron distribution in thetransition-metal ions, the variation of the energydifference b,E„,b between the ferromagnetic and antifer-romagnetic ( AF2) solutions as a function of lattice pa-rameter was investigated. On increasing the cation-anionseparation, EEst,b decreases markedly due to the reducedM-0-M superexchange interaction. At very high latticeparameters, the effects of ordinary exchange increasinglyfavor the pairing of electron spins, and the ferromagneticstate becomes more stable.
Using our values for AE„,b together with the localmagnetic moments LM, it is possible to produce an estimate
Oxide
Mno (AF, )
NiO (AF )MnO (ferro)NiO (ferro)MnO (AF&)NiO (AF, )
4.52604.26384.52394.26464.52724.2699
4 AAA8
4.16846.26.2
9.59.5
0Lattice constant (A) Binding energy (eV)Calc. Expt. (Ref. 31) Calc. Expt. (Ref. 63)
Oxide Calc.m, (pq)
Expt.
MnONiO
4.9231.924
4.58 (Ref. 64), 4.79 (Ref. 65)1.64 (Ref. 66), 1.77 (Ref. 65), 1.90 (Ref. 64)
TABLE III. Calculated spin magnetic moments m, (pz) forthe AF2 state compared with experimental values.

M. D. TOILER et al.
TABLE IV. Comparison of ferromagnetic state energies(eV/cell) at the calculated AF2 lattice constant (o.=2S, / cell).
TABLE VI. Deviations from ideal cubic (90') angles in theAF& state due to exchange magnetostriction.
MnO NiO Oxide Calc. Expt.State State
t 2geg(o. =5)
closed shell
0.0+ 10.69+ 11.43+ 12.79
t~geg(cr =2)closed shellt zg eg (o.=0)
0.0+ 11.31+ 11.86
for the ratio of the Neel temperatures of the two systems.The UHF wave function is an eigenfunction of S, but notof S, and thus most closely corresponds to an Ising mod-el of the spin. Within mean-field theory the transitiontemperature of the Ising model is
2zIJIS(S+1)
3kii
where S is the total spin moment, kz is the Boltzmannconstant, and z is the number of second-nearest-neighbormetal ions which are involved in the superexchange in-teraction. In the Ising model AE„,b is 2S
~J iz. Hence
~ES~abT~ cc (1+S ') .
B
TABLE V. Calculated antiferromagnetic stabilization ener-
gies relative to the ferromagnetic state (eV/double cell) at thecalculated AF& lattice constant. Calculated and experimentalNeel temperatures (K).
TN TlV
Oxide EEstab( AF2 ) AEstab( AF, ) (Estimated) (Expt. .)
MnONiO
—0.0115—0.0331
+0.0004+0.0119
94392ratio:4.17
116525ratio:4.53
The experimental ratio Tz(NiO)/TN(MnO) of 4.52(Table V} compared favorably with our calculated ratio of4.17. It is interesting to note that, assuming a propor-tionality constant of —„directestimates of Tz are in fairlyclose agreement with experiment.
The observed ground-state spin arrangement of succes-sively antiparallel ferromagnetic (111}planes gives rise toa magnetostriction effect within the crystal. This takesthe form of a rhombohedral contraction normal to theferromagnetic planes; at low temperature the crystal axesare inclined at 0.62' (MnO) and 0. 1' (NiO) from the idealcubic directions. ' The distortion increases with in-
creasing temperature.Figures 1(a) and 1(b} show the calculated variation of
the total energy of the AF2 state as a function of avolume-conserving rhombohedral distortion. Analogouscurves for the ferromagnetic state are also shown. Forboth materials, the shift in the minimum for the AF2state corresponds to a contraction along the [11 lj axis in
MnONiO
90.47'90.075
90.62 (Ref. 34)90.1' (Ref. 35)
accordance with experiment. As one would expect, thecalculation produced no distortion from cubic symmetryin the ferromagnetic state. Furthermore, the calculatedequilibrium deviations from the ideal cubic angles in theAI'2 case are in good agreement with experiment (TableVI).
B. Electronic structure
We first consider the Mulliken population analysespresented in Table VII. The charge transfer of around1.9e indicates that both compounds can be qualitativelydescribed as classical ionic oxides. This is supported fur-ther by the 3d populations and atomic spin moments,given in Table VIII, which are characteristic of tz e
(Mn) and tz es (Ni) configurations of the free +2 cationsin an octahedral environment. Electron-densitydifference maps [Figs. 2(a) and 2(b)] were obtained bysubtracting the spherical atomic charge distributions (de-rived from the free ion solutions) away from the bulkdensity. Both charge densities refer to the basis set opti-mized in the solid. The figures, which show the basal(100) plane of the unit cell, demonstrate significant con-tractions of the atomic orbitals of both anions and cat-ions in the solid relative to the free ions. This contrac-tion is presumably due to short-range exchange repul-sions and, for the anion, to the confining effect of theMadelung potential. This qualitative picture may explainthe success of ionic model calculations for the lattice anddefect properties of the transition-metal monoxides.There is clear evidence for the preferential occupation ofthe tz levels in NiO in the nonspherical symmetry ofboth the electron-density difference and spin-densitymaps shown in Figs. 2(b) and 2(d}, respectively, as ob-served experimentally. The corresponding maps forMnO, Figs. 2(a) and 2(c), show equally clearly the spheri-cal d (t 2 e ) electron configuration of Mn +.
The calculated band structures for the ferromagneticand AF2 states are given in Fig. 3. Selected directionsand special points in the Brillouin zones, which are illus-
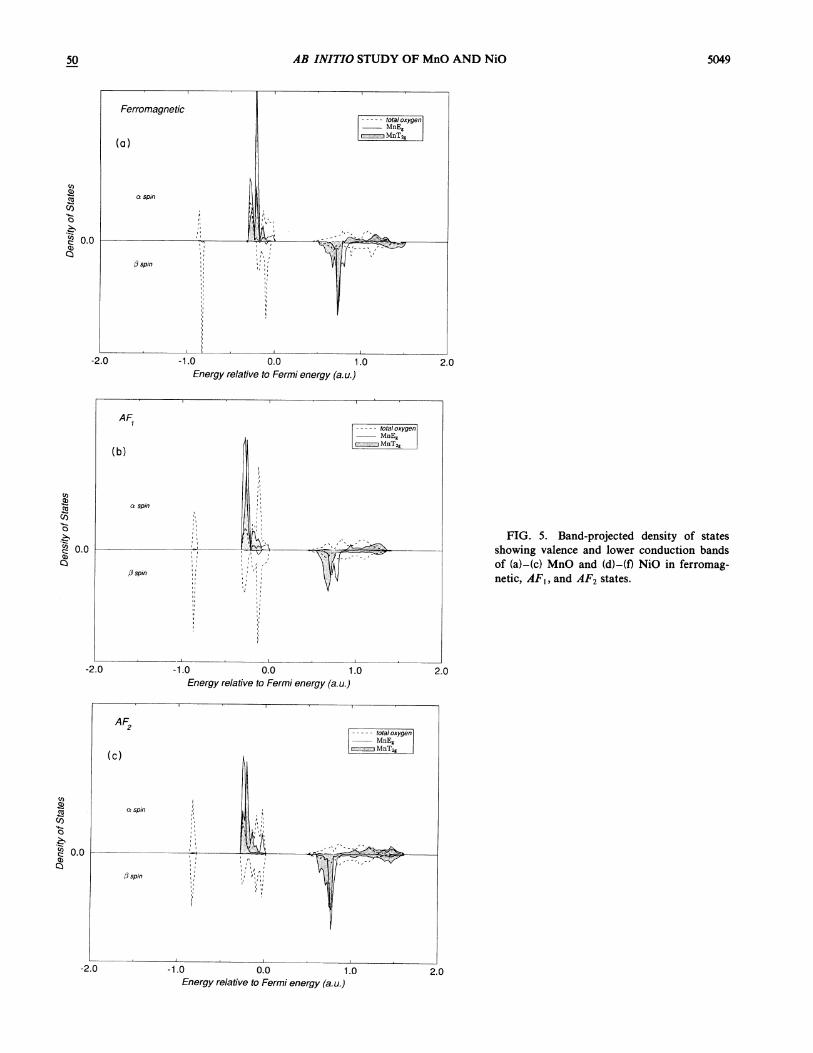
trated in Fig. 4, follow the notation given by Bradley andCracknell and Slater ' for the ferromagnetic (fcc) andthe AF~ (rhombohedral} cases, respectively. The calcu-lated band structures are broadly similar for each mag-netic state. In NiO the valence bands are centered at—0. 18 hartree with a bandwidth of 0.36 hartree and aband gap of 0.5 hartree. The projected density of states(DOS) are shown in Fig. 5. The lower and upper valencebands have predominantly Ni d and 0 p character, re-spectively and hybridize strongly in the center of theband. The conduction band includes contributions fromboth Ni and 0 orbitals, although the lower edge is largely

50 AB INITIO STUDY OF MnO AND NiO
ferromagnetic caseAF2 case
-1224.8260q
- 1 224.8262
Q
-1224.8264
-1224.8266
-1224.8268-0.020
(b)
-0.010 0.0005
ferromagnetic case-AF2 case
0.010 0.020
FIG. 1. Energetic response of (a) MnO and(b) NiO to an elastic C44 distortion. Thenonzero components of the symmetric La-grangian elastic tensor are 5/2.
-1581.8085 $
(g
-1581.8090—p
-1581.8095 I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
-1581.8100-0.020 -0.010 0.000 0.010 0.020
TABLE VII. Distribution of charge among atomic orbitals in the AF2 state following Mulliken pop-ulation analysis.
Oxide Atom 1s 3$p Ssp
Mulliken populationorbital
4sp 3d
MnO
NiO
Mn
0Ni
2.000
1.9972.000
1.997
8.082
2.6408.147
2.647
2.164
2.6282.403
2.664
4.293
2.6024.262
2.566
1.487
1.222
4.502t2R 2.652eR1.85
6.659t2R4. 911eR1.748
0.611t2RO. 375eR0.236
1.430t2R 1 08eRO. 35
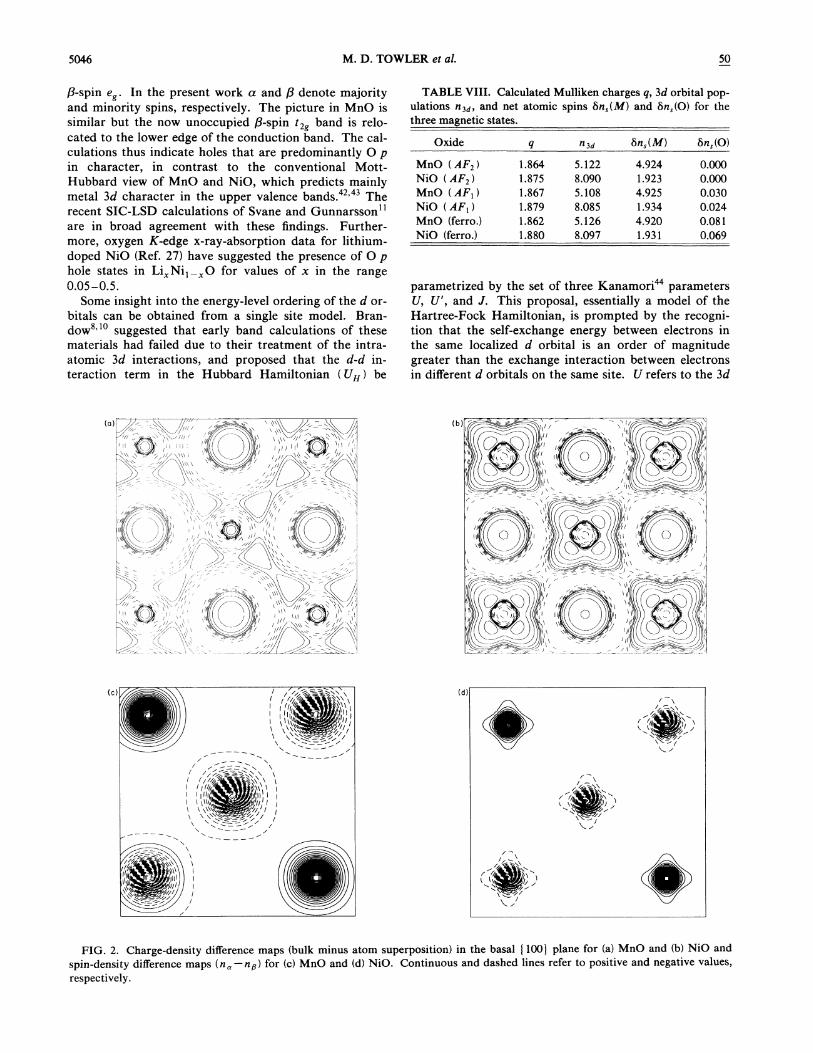
5046 M. D. TOWLER et al. 50
P-spin es. In the present work a and P denote majorityand minority spins, respectively. The picture in MnO issimilar but the now unoccupied P-spin t2 band is relo-cated to the lower edge of the conduction band. The cal-culations thus indicate holes that are predominantly 0 pin character, in contrast to the conventional Mott-Hubbard view of MnO and NiO, which predicts mainlymetal 3d character in the upper valence bands. " ' Therecent SIC-LSD calculations of Svane and Gunnarsson"are in broad agreement with these findings. Further-more, oxygen K-edge x-ray-absorption data for lithium-doped NiO (Ref. 27) have suggested the presence of 0 phole states in Li„Ni& 0 for values of x in the range0.05—0.5.
Some insight into the energy-level ordering of the d or-bitals can be obtained from a single site model. Bran-dows' suggested that early band calculations of thesematerials had failed due to their treatment of the intra-atomic 3d interactions, and proposed that the d-d in-teraction term in the Hubbard Hamiltonian (UH) be
Oxide
MnO ( AF, )
NiO (AF )
MnO (AF))NiO ( AFq )
MnO (ferro. )
NiO (ferro. )
1.8641.8751.8671.8791.8621.880
5.1228.0905.1088.0855.1268.097
5n, {M)
4.9241.9234.9251.9344.9201.931
6n, (O)
0.0000.0000.0300.0240.0810.069
parametrized by the set of three Kanamori parametersU, U', and J. This proposal, essentially a model of theHartree-Fock Hamiltonian, is prompted by the recogni-tion that the self-exchange energy between electrons inthe same localized d orbital is an order of magnitudegreater than the exchange interaction between electronsin different d orbitals on the same site. U refers to the 3d
TABLE VIII. Calculated Mulliken charges q, 3d orbital pop-ulations n3d, and net atomic spins 5n, (M) and 5n, (O) for thethree magnetic states.
(b)
/ //
/,Ig(
/
]///
Jl)/
/I(I
//~'(
/'(( /
I
I//
I
i') &
)
S )I
(c)
/
IJI r I' /it'll ~
//// /
//
/
/'
/r /~
4 IIVI))gh
'
iA f/~ j&
// /j/
//
FIG. 2. Charge-density difference maps (bulk minus atom superposition) in the basal t 100) plane for (a) MnO and (b) NiO and
spin-density difference maps (n —n&) for (c) MnO and (d} NiO. Continuous and dashed lines refer to positive and negative values,
respectively.

50 AS INITIO STUDY OF MnO AND NiO 5047
self-interaction term, which is equivalent to the directCoulomb repulsion between electrons in the same d orbit-al, U' to the on-site Coulomb repulsion between electronsin different d orbitals, and J to the on-site exchange in-teraction between difFerent d orbitals (which onlyoperates between electrons of the same spin). Note thatU' and J are averaged over all possible pairs of orbitalsinvolved in the interactions, and that after such averaging
Consider first a high-spin d s Ni + ion in the octahedralfield of the crystalline environment. The tz~ and es sub-
bands are split through electrostatic crystal-field effects,with e higher in energy by hcF. Intra-atomic interac-gtions produce further shifts, the magnitudes of which canbe deduced from a consideration of the ground-state elec-tronic configuration (a t2-g) (P t2-~) (a eg-) . Such con-siderations lead to the following splittings:
hE, =E(a-ti )—E(a e)=-2J hc„—,EE&=E(P t& )-E(—a t& )-=2J,
bEi=E(P-es) E(P—-t,g)= U 3J+—hcF .
.80 .80
.60.
.40. 4Q
pg .20.
.00 EF
~~)~.00
-.20.~~
EF
-.40 !
r Z K A UBK
MnO AF2
-.40Y r Z KAUBK
NiO AF2
.80 .80
.60.(c)
.60.(d)
~ 40. 4Q
c5
bO
h1~ 00 EF
~ 20.
.00
-.20 -.20.
—.40W L I X W K
NiO ferromagnetic
40W L r X W K
MnO ferromagnetic
FIG. 3. E(k) curves along high-symmetry directions for the AF2 and ferromagne'
orna netic states of MnO and NiO. Minority-spin elec-tron states are indicated with a dashed line for the ferromagnetic case.

5048 M. D. TOILER et al. 50
{a}
FIG. 4. First Brillouin zones of (a) AF2 and (b) ferromagnet-ic unit cells.
The corresponding energy-level diagram is given inFig. 6(a). We have evaluated the necessary integrals atthe experimental lattice parameter using Ni d radial func-tions optimized in the bulk, yielding values of 1.026,0.037, and 0.012 hartree for U, J, and hc„,respectively.The predicted splittings are therefore 0.062, 0.074, and0.927 hartree for EE„EE2,and b,E3, respectively, to becompared with 0.051, 0.078, and 0.867 hartree foundfrom the calculated band structure at the I' point. Thediscrepancies between these two sets of results are wellwithin the calculated bandwidths, the latter being due tointersite interactions (covalency). The a-spin es statesare at a lower energy than the a-spin tz states, indisagreement with simple crystal-field rules, because ofintra-atomic interactions. This splitting persists even ifmeasured from the centroid of the bands [Fig. 5(d)].These interactions introduce an energy band between thevalence and conduction bands of order U, and the insu-lating nature of NiO is thus primarily the result of on-siteCoulombic repulsion, which is sufficient to open a largergap. Furthermore, this band-splitting pattern should ap-ply to all possible spin configurations, since U is verymuch larger than J, so that we expect the insulatingproperties will persist above Tz as experimentally ob-served. It is worth noting that Brandow had deducedthe values 0.213, 0.026, and 0.040 hartree for U, J, andECF, respectively, from spectroscopic data, giving rise toa much smaller band gap than that found in the presentwork. However, it should be remembered thatdifferences in Hartree-Pock eigenvalues do not strictly
correspond to excitation energies even within a frozen or-bital picture; the experimental data will also be affectedby correlation, and probably more importantly, relaxa-tion sects which will considerably lower the observedexcitation energies. It can therefore safely be concludedthat the Hartree-Fock band gap will be a considerableoverestimate of the observed optical gap, and that is itnot appropriate to interpret spectroscopic data in termsof Hartree-Fock eigenvalues. Some success has beenachieved through applying hSCF and configuration-interaction techniques to compute excitation energies.Janssen and Nieuwpoort estimated the correlationcorrection in charge-transfer excitation energies to be1 —2 eV in [Ni06]' clusters, and Massidda, Posternak,and Baldereschi used a screened Coulomb interaction toestimate correlation effects on the band gap in CaCuOi tobe —5 eV. The reason for the failure of the LSDA topredict a sufficiently large gap ' is because the valueof U obtained from a local homogeneous electron-gas ap-proximation is very much smaller than that evaluateddirectly. '
A similar analysis of the band structure of MnO yields
hE, =E(a e~ ) —E-(a-t2g ) = b,c„,DER =E(p
t2s ) E-(a e—s ) = U-+4J b,cF, —
DER =E(p e) E(p-t2 —)=b,cF-,
and the resulting energy-level diagram is shown in Fig.6(b). We have again evaluated the necessary integrals atthe experimental lattice parameter using Mn d-orbital ra-dial functions optimized in the bulk, resulting in 0.861,0.031, and 0.017 hartree for U, J, and hc„,respectively.The predicted splittings are therefore 0.017, 0.968, and0.017 hartree for hE, , EEz, and EE3, respectively, to becompared with —0.001, 0.861, and 0.054 hartree foundfrom the calculated band structure at the I point. Thediscrepancies between these two sets of results are againwithin the calculated bandwidths. A surprising feature ofthe MnO density of states is the amount of a-e densitywhich lies below the a-tz band. This is primari1y due tometal-oxygen covalent 0 bonding which stabilizes the oc-cupied e orbitals relative to the tzg. Even at I, wherethere is no such mixing, the interatomic terms are suckthat e lies at slightly lower energy. Overall, the splittingis small compared to the subband widths, and it is notclear that it would persist if measured from the band cen-troids [Fig. 5(a)].
C. Elastic properties
The elastic constants and phonon dispersion curves ofionic and partially ionic compounds have usually beencalculated using classical approaches based on semi-empirical formulas of the Born type. ' The central-forcepotentials used to represent the ground-state energy ofthe solid as a function of the nuclear positions in thesemethods require a set of empirical parameters to specifytheir functional form. These are usually determined byfitting to available experimental data such as binding en-
50 AB INITIO STUDY OF MnO AND NiO 5049
Ferromagnetic————— total oxygen
MnE~i::::::::-::.:::::::::::::::1MnTg~
0)Q)
0)0
0.0Q
n spin
P spin
I
I
I
I
I
I
I
I
I
I
I
I
I
III
I
I
I111:
~I
I
VI
hI 1
II I
I
I II I
II
II
IL r I
-2.0 -1.01
0.0I
1.0 2.0Energy relative to Fermi energy (a.u.)
AF,
(b)
———- - total oxygenMnEg
1MnTg~
9)
0)O
0.0Cj
a spin
P spin
\
I II II
I
I
I
I
I
I I
I
I I
I I
II
I I
I I
II11IIIIII
II
II
1
I
11
III
I
II
I
I
I
I
I
I%I I
I II
I LI II I
I I
I II
II I
I
II I
I
I1
II
I
I
I
FIG. 5. Band-projected density of statesshowing valence and lower conduction bandsof (a)-(c) MnO and (d)-(f) NiO in ferromag-netic, AF&, and AF2 states.
-2.0 -1.0 0.0 1.0Energy relative to Fermi energy (a.u.)
2.0
AF
(c)
————— total oxygenMnEg
: -1MnTg~
0)
0
0.0Q
a spin
P spin
11
II
III I
I
II
I
I
I
I
I
II
I
II
II
II
II
II
II
II
I
I11 I
I II I I
II, 111I I 111
I
I III
I
i II
-2.0I I I
-1.0 0.0 1.0Energy relati ve to Fermi energy (a.u.)
2.0

5050 M. D. TO%'LER et al. 50
Ferromagnetictotal oxygenNiE~
-I Ni Tg~
a spin
I0
g o.o Pq) I
Q9 spin
-2.0 -1.0 0.0 1.0Energy relative to Fermi energy (a.u. )
2.0
AF,
(e)
—- —- - total oxygenNiEg
I::::::::::INiTg~
V)Q)
(f)
0
0.0O
a spin
g spin
FIG. S. (Continued j.
-2.0I
-1.0 0.0 1.0Energy relative to Fermi energy (a.u.)
2.0
AFz— — — — total oxygen
NiE~::::::::.INi Tgq
V)
CQ
O
~ 00
e spin
spin
I
II
I
II
II
II
II
I
-1.0 0.0 1.0Energy relative to Fermi energy (a.u. )
2.0

50 AB INITIO STUDY OF MnO AND NiO 5051
e,
2J I~ ~ W W 2g
t2g
kg
U+4J~
FIG. 6. Splitting of the 3d subbands in (a) NiO and (b) MnOfollowing the scheme presented in the text.
ergy, lattice parameter, and dielectric constants. TheHartree-Fock approach, as implemented in CRYSTAL, al-lows a parameter-free ab initio determination of suchproperties even for materials where experimental data isunavailable, e.g., Refs. 17 and 52. Here calculations ofthe elastic constants and phonon frequencies of MnO andNiO are reported.
1. Elastic constants
The static elastic constants C;J are second derivativesof the energy density with respect to strain components,
where V is the volume of the cell and E is the energy percell. The total energies of NiO and MnO were calculatedas a function of elastic strains of up to 2%, then fourth-order polynomial fits of the resulting data mere employedto obtain the appropriate second derivatives at the energyminimum.
By simple variation of the lattice constant of the cubicunit cell, an isotropic deformation of the crystal structurewas obtained, yielding both the equilibrium geometry andthe bulk elastic modulus 8. The three independent com-ponents of the elastic tensor were then derived as follows.C» was given directly by deforming one of the threeedges of the cubic unit cell. C,2 was calculated indirect-ly, via the linear combination C»-C, 2, from a volume-conserving strain involving the contraction of two edgesand the stretching of the other by twice as much. Theshear constant C44 resulted from the application of avolume-conserving rhombohedral strain along the (111)direction. As an internal consistency cheek one ean com-pute the bulk modulus from the formulaB=(C»+2C&z )I3. Strains of 2% correspond to tetrago-
nal distortions of the cell sides by 0.08—0.09 A in the C»and C»-C, z cases, and deviations of the cell angles from60' of about 1.3' in the rhombohedral case (C44). Sinceall the atoms are at centrosymmetric sites no internal po-sitional relaxations are possible.
The calculated constants for both ferromagnetic andAF2 magnetic states are shown in Table IX, togetherwith experimental data from a number of sources. Ourcalculations ignore any lattice-vibrational contributionsto the free energy and thus should be compared to mea-surements extrapolated to the athermal limit. Unfor-tunately the wide variation in the published experimentaldata for NiO (Refs. 53—55) makes detailed comparisondiIcult. For MnO room-temperature measurements us-ing ultrasonic techniques are broadly in agreement witheach other, ' although attempts to measure the con-stants below the antiferromagnetic ordering temperaturehave met with mixed success. In particular the C44 shearconstant softens dramatically on approaching Tz, andbelow T& acoustic attenuation appears to be too strong toobtain an accurate measurement. In many instances,sharp discontinuities in elastic constants have been ob-served on passing through the Neel temperature of anti-ferromagnetic materials, so any attempt to extrapolatefrom linear curves in the paramagnetic region to absolutezero is likely to produce large errors. Hence the value ofC44 in the athermal limit is unclear. The theoretical pre-dictions for the other constants, Table IX(a), are seen tobe in reasonable agreement with experiment, errors rang-ing from +3 to 28%. The value of C44 for the AF2 stateis around 5% less than that calculated for the ferromag-netic state, whereas the other constants are insensitive tothe magnetic order.
For comparative purposes the elastic constants of CaO,MnO, and NiO were also calculated using a semiempiri-cal classical technique. A two-body interatomic potentialshell model was employed within an ionic model frame-work. The consistent set of potentials and shell modelparameters used is due to Stoneham and Sangster. As iswell known, two-body forces cannot account for the ex-perimentally observed Cauchy violation. Neverthelessthere are clear trends in the calculated values along theseries Ca to Ni and, bearing in mind the limitations of thetwo-body model, the values support the ab initio calcula-tions.
2. Phonon frequencies
The phonon spectra of both materials have been deter-mined experimentally. ' ' %e have calculated the pho-non frequencies at the I and L points in the first Bril-louin zone using the frozen-phonon approximation; meassume that the acoustic and optical modes do not cou-ple. Ionic displacements of 0.025 A were used, and ener-gy curves fitted with fourth-order polynomials. Coulombterms were summed using the Ewald convention and thusno macroscopic fields are generated; therefore the I-point distortion corresponds to the transverse-optic (TO)mode. The calculated values, reported in Table X, tendto be somewhat higher than experiment. This is con-

M. D. TOWLER et al. 505052
(a)MnO elastic constants (GPa)
Calc.Expt.Ref. 57 Ref. 58 Ref. 54 Ref. 56 Ferror (K) AFq
-231—119—112
28116611596
170
C11-C12C
B
123.6
90.6169—152
230.5119.5111
C11-C12Ci~
B
50123.4
150.8
231.4120.9110.5
~48149.3
116(TN) C11-C12
Ci~
B
124.6
~40
22310312079
154.3
222109.9112.178.3
148.6
C11-C12Ci~
B
298116
7877
(b)NiO elastic constants (GPa)
Expt.Ref. 67 Ref. 55 Ref. 68
Calc.Ref. 53 Ref. 54 Ref. 69 Ferro. AF&T (K) C;,
21190
121109145
C11-C12Ci~C44B
399272127121215
115214
344.6203.3141.340
205
22413297
110137
Cl 1 C12
Ci~C44B
271145125105173
298
190 199189
(c)Elastic constants (GPa)
MnO NiOCao
450163163258
272128128175
Ci~C44B
2179494
134
TABLE IX. (a) Calculated elastic constant and bulk modulus data for MnO compared with experi-mental data at various temperatures. (b) Calculated elastic constant and bulk modulus data for NiOcompared with experimental data at various temperatures. (c) Elastic constants calculated using asemiempirical two-body potential model for CaO, MnO, and NiO. An ab initio Hartree-Fock study ofCaO gave a value of 128 GPa for the bulk modulus (Ref. 28).

50 AB INITIO STUDY OF MnO AND NiO
TABLE X. I -point and L-point phonon frequencies (THz). [Expt. (Refs. 60 aud 61)].
I pointTO TA
L pointLO TO
Oxide Expt. Calc. Expt. Calc. Expt. Calc. Expt. Calc. Expt. Calc.
MnONiO
7.911.6
9.511.8
9.010.0
10.9111.28
4.86.2
5.706.24
15.616.3
17.8817.81
8.010.4
9.4611.32
sistent with previous experience of molecular and period-ic Hartree-Pock calculations which tend to overestimatevibrational frequencies. A posteriori density-functionalcorrelation corrections soften all of the lattice vibrationsat the I, X, and l. points in MgO. The neglect of cou-pling between the L-point modes may also be a source oferror.
Coulomb interactions. The materials are correctly pre-dicted to be antiferromagnetic with AF2 structure. Therelative energy differences between various magneticstructures are consistent with the ratio of the Neel tem-peratures. The structural, elastic, and vibrational proper-ties are in reasonable agreement with available experi-mental data.
IV. CONCLUSIONS ACKNOWLEDGMENTS
The Hartree-Fock method generates a qualitativelycorrect ground-state electronic structure in NiO andMnO in which the wide-band-gap insulating character ofthese materials is seen to be a result of the large on-site
M.D.T. wishes to thank SERC and ICI Chemicals kPolymers Ltd. for 6nancial support. The work was sup-ported, in part, by SERC Grant No. GR/HO7160.W.C.M. thanks Wolfson College, Oxford for support.
D. Adler, in Solid State Physics, edited by H. Ehrenreich andD. Turnbull (Academic, New York, 1968), Vol. 21, p. 1.
A. B.Kunz, J. Phys. C 14, L455 (1981).C. Kittel, Introduction to Solid-State Physics (Wiley, New
York, 1986).4J. Zaanen, G. A. Sawatzky, and J. W. Allen, Phys. Rev. Lett.
55, 418 (1984).5J. B. Torrance, P. Lacorre, C. Asavaroengchai, and R. M.
Metzger, Physica C 182, 351 (1991).K. Schwartz, in Basic Properties ofBinary Oxides, edited by A.
Dominguez Rodriguez, J. Castaing, and R. Marquez (Serviciode Publicaciones de la Universidad de Sevilla, Seville, 1984),p. 43.
K. Terakura, A. R. Williams, T. Oguchi, and J. Kubler, Phys.Rev. B 30, 4734 (1984).
B.H. Brandow, Adv. Phys. 24, 651 (1977).9B. Brandow, in Narrow Band Pheno-mena Influen—ce of Elec
trons with Both Band and Localised Character, edited by J. C.Fuggle, G. A. Sawatzky, and J. W. Allen (Plenum, NewYork, 1988),p. 97.B.Brandow, J. Alloys Compounds 181, 377 (1992).A. Svane and O. Gunnarsson, Phys. Rev. Lett. 65, 1148 (1990).V. I. Anisimov, J. Zaanen, and O. K. Andersen, Phys. Rev. B44, 943 {1991).C. Pisani, R. Dovesi, and C. Roetti, Hartree-Fock Ab InitioTreatment of Crystalline Systems (Springer-Verlag, Berlin,1988).
R. Dovesi, V. R. Saunders, and C. Roetti, cRYsTAL92, UserManual (Universita di Torino and SERC Daresbury Labora-tory, 1992)
' J. A. Pople and R. K. Nesbet, J. Chem. Phys. 22, 571 (1954).R. Dovesi, Solid State Commun. 54, 183 {1985).
~R. Dovesi, C. Roetti, C. Freyria-Fava, M. Prencipe, and V. R.Saunders, Chem. Phys. I56, 11 (1991).M. Causa, R. Dovesi, C. Pisani, and C. Roetti, Phys. Rev. B33, 1388 (1986);34, 2939 (1986).M. Causa, R. Dovesi, C. Roetti, and V. R. Saunders, Chem.
Phys. Lett. 140, 20 (1987).L. Salasco, R. Dovesi, R. Orlando, M. Causa, and V. R.Saunders, Mol. Phys. 72, 267 (1991).W. C. Mackrodt, N. M. Harrison, V. R. Saunders, N. L. Al-
lan, M. D. Towler, E. Apra, and R. Dovesi, Philos. Mag. A68, 653 (1993).
2 G. J. M. Janssen and W. C. Nieuwpoort, Phys. Rev. B 38,3449 (1988).A. Fujimori, F. Minami, and S. Sugano, Phys. Rev. B 29, 5225(1984).
24A. Fujimori and F. Minami, Phys. Rev. B 30, 957 (1984).G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett. 53, 2239(1984).
6S. Hiifner and T. Riesterer, Phys. Rev. B 33, 7267 (1986).P. Kuiper, G. Kruizinga, J. Ghijsen, G. A. Sawatzky, and H.Verweij, Phys. Rev. Lett. 62, 221 (1989).R. Dovesi, C. Roetti, C. Freyria-Fava, E. Apra, V. R.Saunders, and N. M. Harrison, Philos. Trans. R. Soc. Lon-don Ser. A 341, 203 (1992).
29R. Dovesi, M. Causa, R. Orlando, C. Roetti, and V. R.Saunders, J. Chem. Phys. 92, 7402 (1990).H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188(1976).R. W. G. WyckoB; Crystal Structures (Interscience, NewYork, 1964).
2E. Apra, Ph.D. thesis, Universita di Torino, 1993.J. H. van Vleck, J. Chem. Phys. 9, 85 (1941).H. Shaked, J. Faber, and R. L. Hittermann, Phys. Rev. B 38,11 901 (1988).
G. A. Slack, J. Appl. Phys. 3j., 1571 (1960).C. R. A. Catlow, B. E. F. Fender, and D. G. Muxworthy, J.Phys. (Paris) C 7, 67 (1975).M. J. L. Sangster and A. M. Stoneham, Philos. Mag. B 43, 609(1980).W. C. Mackrodt, Solid State Ionics 12, 175 (1984).S. Sasaki, K. Fujino, and Y. Takeuchi, Proc. Jpn. Acad. B 55,43 (1979).

5054 M. D. TOWLER et al. 50
~C. J. Bradley and A. P. Cracknell, The Mathematical Theory
ofSymmetry in Solids: Representation Theory for Point Groupsand Space Groups (Clarendon, Oxford, 1970).
~tJ. C. Slater, Quantum Theory of Molecules and Solids, Vol I.I:Symmetry and Energy Bands in Crystals (McGraw-Hill, NewYork, 1965).
4~P. A. Cox, The Transition Metal Oxides (Oxford UniversityPress, Oxford, 1992).
43K. Terakura, A. R. Williams, T. Oguchi, and J. Kiibler, Phys.Rev. Lett. 52, 1830 (1984).
~J. Kanamori, Prog. Theor. Phys. 30, 275 (1963).45F. A. Cotton and G. Wilkinson, Aduanced Inorganic Chemis-
try, 2nd ed. (Wiley, New York, 1966), p. 660.S. Massidda, M. Posternak, and A. Baldereschi, Phys. Rev. 846, 11 705 (1992).
470. K. Andersen, H. L. Skriver, H. Nohl, and B. Johannsson,Pure Appl. Chem. 52, 93 (1979)~
T. Oguchi, K. Terakura, and A. R. Williams, Phys. Rev. B 28,6443 (1983).J. Kubler and A. R. Williams, J. Magn. Magn. Mater. 54-57,603 (1986).L. Severin, M. S. S. Brooks, and B. Johannsson, Phys. Rev.Lett. 71, 3214 (1993).
5'C. R. A. Catlow, M. Dixon, and W. C. Mackrodt, in Comput-er Simulation of Solids, edited by C. R. A. Catlow and W. C.Mackrodt (Springer-Verlag, Berlin, 1982), p. 130.M. Catti, A. Pavese, R. Dovesi, C. Roetti, and M. Causa,Phys. Rev. B 44, 3509 (1991).P. d. V. DuPlessis, S. J. Van Tonder, and L. Alberts, J. Phys.C 4, 1983 (1971).
54N. Uchida and A. Saito, J. Acoust. Soc. Am. 51, 1602 (1972).J. Wang, E. S. Fisher, and M. H. Manghnzmi, Chin. Phys.Lett. 8, 153 (1991).
56D. W. Oliver, J. Appl. Phys. 40, 893 (1969).S. B. Palmer and A. Waintal, Solid State Commun. 34, 663(1980).D. Seino, J. Magn. Magn. Mater. 28, 55 (1982).
59A. M. Stoneham and M. J. L. Sangster, Philos. Mag. B 52,717 (1985).
soV. Wagner, W. Reichhardt, and W. Kress, in Proceedings ofthe Conference on Neutron Scattering, Gatlinburg, edited by
R. M. Moon (NTIS, Springfield, VA, 1976), Vol. 1, p. 175.W. Reichardt, V. %'agner, and W. Kress, J. Phys. C 8, 3955(1975).M. I. McCarthy and N. M. Harrison, Phys. Rev. B 49, 8574(1994).
s3I. Barin, Thermochemical Data of Pure Substances (VCH,Cambridge, 1989).
~A. K. Cheetham and D. A. O. Hope, Phys. Rev. B 27, 6964(1983).B. E. F. Fender, A. J. Jacobson, and F. A. Wedgewood, J.Chem. Phys. 48, 990 (1968).H. A. Alperin, J. Phys. Soc. Jpn. Suppl. B 17, 12 (1962).I. Wakabayashi, H. Kobayashi, H. Nagasaki, and S. Minomu-
ra, J. Phys. Soc. Jpn. 25, 227 (1968).8M. R. Notis, R. M. Spriggs, and W. C. Hahn, J. Geophys.
Rev. 76, 7052 (1971).R. L. Clenenden and H. G. Drickamer, J. Chem. Phys. 44,4223 (1966).


Copyright © 2022 FDOKUMEN



















