
Potential cosmeceutical lamellar liquid crystals containing ...
Upload
independentCategory
view
0download
0
Ab Initio Calculation of Elastic Constants of Ceramic Crystals
Hongzhi Yao,z Lizhi Ouyang,y and Wai-Yim Chingw,z
zDepartment of Physics, University of Missouri-Kansas City, Kansas City, Missouri 64110yDepartment of Mathematics and Physics, Tennessee State University, Nashville, Tennessee 37209
An effective computational scheme to calculate the complete setof independent elastic constants as well as other structuralparameters including bulk modulus, shear modulus, Young’smodulus, and Poisson’s ratio for crystals is reported. Thescheme is based on the stress–strain analysis approach withthe appropriate selection of strain governed by symmetry con-sideration. The first principles Vienna ab initio simulationpackage (VASP) is used in stress calculations. Comprehensivetests were performed for a-SiO2 and spinel MgAl2O4 withdifferent exchange-correlation potentials, and different sets ofcomputational parameters to investigate the relative accuraciesof the calculations. A wide range of oxides, nitrides, and car-bonate crystals with different crystal symmetries were chosen totest the scheme under both LDA and GGA approximations atzero temperature and pressure. Some of these calculations forlarge complex crystals are believed to be attempted for the firsttime. The calculated elastic constants show quite good agree-ment with the existing experimental data for almost all the ex-amined systems with the exception of the relatively soft materialsuch as a-SiO2 and the C14 parameter of some trigonal crystalsexpressed in the hexagonal form such as in a-Al2O3. Otherstructural properties derived from the elastic constants alsoshow good agreements with the measured values.
I. Introduction
WITH the increasing interest in the advanced applications ofceramic materials especially at the nano-scale, the me-
chanical properties of these materials become an important partin the overall evaluation of the quality and suitability of a par-ticular application.1 In ceramics, most of the materials are poly-crystalline with the presence of many different types ofmicrostructures and defects; the mechanical properties arevery difficult to evaluate and characterize because they are in-variably sample dependent. Single-crystal data of ceramic ma-terials are rare and, for many of them, non-existent. Theincreased complexity for multi-component ceramic materialsand the difficulty in obtaining pure single crystals of sufficientsize make accurate experimental determination of the mechan-ical properties an extremely challenging task.2,3
In recent years, owing to the increased computational powerand much-improved methodology, ab initio calculations of me-chanical properties of crystalline solids have reached a level ofaccuracy such that the calculated parameters are sufficientlyclose to the measured values.4 Among these studies, calculationof elastic constants is the most essential because many otherparameters related to mechanical properties can be derived from
elastic constants. They are the fundamental parameters inunderstanding the interatomic interaction, mechanical stability,phase transitions, materials strength, and the internal structureof the materials, and much more. As a result, ab initio calcula-tion of elastic constants becomes a useful tool to understand andeven predict the mechanical properties of many materials, notonly for materials with promising applications but also for ma-terials under extreme conditions such as those found in the geo-logical formations and settings.5–8
There are two closely related approaches in calculating theelastic constants from first principles. One is based on the anal-ysis of the calculated total energy of a crystal as a function of itsvolume or pressure. Elastic constants are related to the second-order expansions of the total energy with respect to the latticestrain components:
EðV ; eÞ ¼ EðV0Þ þ VX6
i¼1siei þ
V
2
X6
i;j¼1Cijeiej þ � � � (1)
The elastic constants are extracted by fitting the total energiesderived under different amounts of strain to a parabola near theenergy minimum.9
The second approach originated from the 1983 paper byNielsen and Martin10 on ab initio calculation of stress–strainrelationship and its application to obtain the second-, third-, andfourth-order elastic constants. The method is based on the anal-ysis of changes in the calculated stress values resulting fromchanges in the strain. In this approach, stress tensors calculatedfrom each deformation are used to construct a set of linearequations. Using orthogonal matrix factorizations, maximumlikelihood estimation of the solutions of linear system (equiva-lent to the best-least-square-fit) could be easily found. Addition-al developments and many applications of this approach havesince appeared and been reported in a wide range of physics,materials, geophysics, and mineralogy journals.11–17
In the present work, we report the results obtained using anautomated computational scheme based on the stress–strain ap-proach to evaluate elastic constants and the related mechanicalproperties of a large number of complex ceramic crystals withdifferent crystal symmetries, some of them for the first time. Webelieve that the best way to assess a particular methodology andits efficiency is to apply it to a large number of systems ratherthan relying on a meticulous calculation of a single material. Theuniform application of ab initio density functional theory(DFT)-based method can fulfill this goal. Furthermore, for crys-tals where experimental data either are unavailable or are inconflict with each other, our calculated results with the con-comitant accuracy analysis will certainly help the experimenta-lists to better assess the reliability of their data. In the followingsection, we outline our method and approach. The main resultsfor crystals with different symmetries are presented and dis-cussed in Section III.
II. Method of Calculation
To increase the efficiency and accuracy of first principles elasticconstants calculation, a set of programs and scripts were
I. Tanaka—contributing editor
Paper presented at the 107th Annual Meeting of the American Ceramic Society,Cincinnati, OH.
This work was financially supported by the U.S. Department of Energy under the GrantNo. DE-FG02-84DR45170. This research used the resources of NERSC supported by theOffice of Science of DOE under the contract No. DE-AC03-76SF00098.
wAuthor to whom correspondence should be addressed. e-mail: [email protected]
Manuscript No. 22793. Received February 9, 2007; approved June 15, 2007.
Journal
J. Am. Ceram. Soc., 90 [10] 3194–3204 (2007)
DOI: 10.1111/j.1551-2916.2007.01931.x
r 2007 The American Ceramic Society
3194

developed to optimize and automate the symmetry analysis,atomic position determination, strain selection andmodification,and procedures for elastic tensor extraction. As mentioned inSection I, the basic idea of extracting elastic constants of a crys-tal using the stress–strain approach is very straightforward. It isbased on the well-known tensorial form of the Hooks law, whichdescribes the linear dependency of stress component si (i5 1–6)and the applied strain ej (j5 1–6) under a small deformation:
si ¼X6
j¼1Cijej (2)
Here, Cij are the elastic constants of the crystal whose struc-ture has been fully relaxed under a given set of exchange-corre-lation potential functions (see next paragraph) and attained anequilibrium structure with a minimum total energy. Equation (2)constitutes a set of six linear equations with 27 variables, name-ly, the six components of stress and 21 elastic constants (withCij 5Cji). In our scheme, solving this set of linear equations re-quires seven separate ab initio calculations with seven differentlevels of applied strain (�0.03,�0.02, �0.01, 0.0, 0.01, 0.02, and0.03). This is a computationally very intensive task especially forcrystals with complex structures and large unit cells. In order toincrease the efficiency, symmetry constraints must be fully takeninto consideration such that the solution of the above linearequations is reduced to solving only the independent elasticconstants in accordance with the symmetry of the system. Inother words, if a strain ek can be transformed into a previouslygenerated strain ej under a symmetry operation, we should beable to transform the previously computed stress using tensortransformation rather than carrying out additional ab initio cal-culations. Based on the Institute of Radio Engineers (IRE), wehave the appropriate strains required for the 14 crystal types.18
For crystal systems with triclinic, mononclinic, and orthorhom-bic symmetries, all six strains (e1–e6) need to be calculated. Fortetragonal crystals, the list is reduced to only four (e1, e3, e4, e6),and for trigonal and hexagonal crystals, it is further reduced tothree (e1, e3, e4). For the highly symmetric cubic crystals, onlye1 and e4 are needed. In the actual implementation, we select astrain magnitude e (typicallyB1%) and then generate a series oftasks for ab initio computation with a single strain. We theneliminate the tasks whose strains are symmetry related to thepreviously generated tasks with the same strains. For instance,in cubic systems, only the tasks for e1 and e4 are executed, but inorthorhombic systems, we have to consider all six types ofstrains. In some cases, all six deformed structures are comput-ed. The resulting elastic tensor elements are then symmetrized(averaged over the symmetrically equivalent elements). Wefound that the differences between symmetry equivalent off-di-agonal elements are very small, of the order of 1 kbar, which isthe level of accuracy attained in the stress calculation. The di-agonal elements are supposedly to be fully symmetric. In thisway we minimize the errors related to numerical uncertainties.
To obtain sufficient precision for the elastic constants, accurateab initio stress calculation is mandatory. We used the VASP (Vi-enna Ab-initio Simulation Package)19,20 for the ab initio stresscalculation. VASP is a density functional theory (DFT)21–23-based method using plane waves. We adopted both ultra-softVanderbilt pseudo-potentials (US-PP)24 and the projector-aug-mented wave (PAW) potential25,26 with sufficient k-point sam-pling using the Monkhorst-Pack27 scheme and a high kineticenergy cut-off. Using VASP, fully relaxed atomic positions withnear-zero residue forces can be obtained as well as the full-stresstensor. The magnitude of applied strain should be chosen withextreme care. There is a trade-off between the desired accuracyand the overall computational resources needed. A relativelysmall strain value is necessary for the deformation to be withinthe elastic region, but this will require a concomitant higher levelof accuracy in the total energy and the restoring forces, hence thecomputional time. In practice, a 1% strain was used in ourscheme for most of the systems. For relatively soft crystals such
as a-quartz, a 2% strain was used instead. Moreover, in additionto positive strain (stretching the cell), one can also apply a neg-ative strain (compressing the cell) and take the average of the twoto be the final stress value. With this procedure, uncertainty in theerror estimation of the final stress values during the deformationcan be reduced to a minimum and improve the overall accuracy.
From the calculated elastic constants, other structural prop-erties such as bulk modulus (K), shear modulus (G), Young’smodulus (E), and Poisson’s ratio (Z) can be derived using Voigt–Reuss–Hill (VRH) approximation.28–30 Voigt’s and Reuss’sschemes represent the upper and lower bounds for the structur-al parameters, respectively, and the final values can be taken asthe averages of the two:
K ¼ ðKV þ KRÞ=2; G ¼ ðGV þ GRÞ=2;E ¼ 9KG=ð3K þ GÞ; and Z ¼ ð3K � 2GÞ=2ð3K þ GÞ
(3)
The relationships between the upper and lower bound of bulkand shear modulus and elastic constants defined in VRHapproximation are given as
KV ¼19ðC11 þ C22 þ C33Þ þ 2
9ðC12 þ C13 þ C23Þ
KR ¼1
ðS11 þ S22 þ S33Þ þ 2ðS12 þ S13 þ S23Þ(4)
and
GV ¼ 115ðC11 þ C22 þ C33 � C12 � C13 � C23Þþ 1
5ðC44 þ C55 þ C66Þ
GR ¼15
4ðS11 þ S22 þ S33Þ � 4ðS12 þ S13 þ S23Þ þ 3ðS44 þ S55 þ S66Þ(5)
where Cij and Sij are elastic constants and elastic compliances,respectively.30
III. Results and Discussion
In any accurate first principles simulations, extensive tests mustbe conducted to ensure that the convergence in k-point samplingis adequate. It is also necessary to test the relative accuracy of theexchange-correlation potential forms used and the number ofplane waves used (energy cut-off) is adequate. However, compu-tational cost increases exponentially when the system under studybecomes large and complex. Hence one must balance the requiredaccuracy for the problem at hand and the resources it demands toreach that accuracy. Furthermore, there are conflicting reports inthe published literature as to which form of the exchange-corre-lation potential gives the more accurate results in comparisonwith experiments. The fact is that it depends on the type of thematerials system and on the nature of the properties being stud-ied. For more conclusive evidences and a fair assessment, it isnecessary to carry out calculations on a large number of crystalsusing the same computational scheme and input parameters.
Based on the above considerations, we performed compre-hensive test calculations under different energy cutoff, numberof k-points in the Brillouin zone (BZ), and exchange-correlationpotentials on two systems: a-quartz and spinel MgAl2O4. Wethen carry out the elastic constant calculations on a large num-ber of ceramic crystals, including those with very complex struc-tures. In many of these crystals, experimental data are scatteredor unreliable because of the difficulty in obtaining large single-phase, homogeneous, defect-free pure crystals for accurate mea-surements. We have studied the elastic constants of a total of 44oxide, nitride, and carbide crystals with different structures us-ing our highly integrated scheme. They are listed in Table I to-gether with basic information for each crystal type. In somecases, it is more expedient to use a larger cell with orthogonal
October 2007 Ab initio Calculation of Elastic Constants of Ceramic Crystals 3195

axes rather than the smaller primitive cell for elastic constantcalculations using our scheme.
(1) Comprehensive Tests on a-Quartz and Spinel MgAl2O4
Our comparative studies of the effects of energy cut-off, numberof k-points in the BZ, and exchange-correlation potentials on a-quartz and spinel MgAl2O4 were summarized in Figs. 1–3. Inthese tests, a range of 300–1000 eV in energy cut-off, and 17 to398 k-points in BZ sampling were used for a-quartz. InMgAl2O4, two types of pseudopotentials (Ultrasoft and PAWpseudopotential) under two different exchange-correlation ap-proximations (LDA and GGA) were used.
The top panel of Fig. 1 shows the elastic constants of a-quartzof our calculated results under different energy cut-offs for afixed set of k-point mesh and exchange-correlation potential.The absolute percentage differences of the lattice constants andcell volume between experiment data and calculation are shownin the lower panel. Clearly, the discrepancies in lattice constantsand crystal volume decrease as a function of the cut-off energy.
Both the lattice and elastic constants converge reasonably well atthe energy cut-off of 500 eV. Similarly, we display our test re-sults for a-quartz with different numbers of k-points at a fixedenergy cut-off (600 eV) in Fig. 2. The sensitivity on the numberof k-points is much less than that of the energy cut-off. A rea-sonable number of k-points (88 k-points with a 7� 7� 6 mesh)is more than adequate for a-quartz. However, it should bepointed out that the adequacy of k-point mesh also dependson the crystal volume. A larger crystal with a correspondingsmaller BZ may not require as many k-points as a simple crystalwith a small unit cell. More importantly, all the crystals inves-tigated in this paper are insulators with a band gap. In metallicsystems where a Fermi level exists, a much larger number of k-points is needed for accurate results. In Fig. 3, we show the de-pendence of the elastic constants and the lattice parameters ofspinel MgAl2O4 under different exchange-correlation potentials.It shows that the ultrasoft PP generally gives a smaller lattice con-stant and larger elastic constant than the PAW type of potentials.It also shows that LDA approximation generally overestimates theelastic constants while GGA approximation underestimates them
Table I. Size and Symmetry of Crystals Used in Our Elastic Constants Calculations
Crystals Name Symmetry Space group number Number of atoms/cell
Oxides a-Al2O3 Hexagonal 167 30a-SiO2 Hexagonal 154 9SiO2 (stishovite) Tetragonal 136 6MgO Cubic 225 8CaO Cubic 225 8ZnO Hexagonal 186 4SnO2 Tetragonal 136 6WO2 Tetragonal 136 6Y2O3 Cubic 206 80In2O3 Cubic 206 80TiO2 (rutile) Tetragonal 136 6TiO2 (anatase) Tetragonal 141 12TiO2 (brookite) Orthorhombic 136 6TiO2 (II) Orthorhombic 136 6MgAl2O4 Cubic 227 56MgSiO3 (perovskite) Orthorhombic 125 20MgSiO3 (post-perovskite) Orthorhombic 63 20Mg2SiO4 Orthorhombic 125 28b-Mg2SiO4 Orthorhombic 74 56g-Mg2SiO4 Cubic 227 56ZrSiO4 Tetragonal 141 24SrTiO3 Cubic 221 5BSO (Bi12SiO20) Cubic 197 66YAG Cubic 230 160
Nitrides a-Si3N4 Hexagonal 159 28b-Si3N4 Hexagonal 176 14g-Si3N4 Cubic 227 56Si2N2O Orthorhombic 36 20c-BN Cubic 227 4h-BN Hexagonal 194 4TiN Cubic 225 4HfN Cubic 225 8g-Sn3N4 Cubic 227 56
Carbonides Diamond Cubic 227 8SiC-2H Hexagonal 186 12SiC-4H Hexagonal 186 16SiC-6H Hexagonal 186 28SiC-3C Cubic 216 8
Others c-ZnS Cubic 216 8Andalusite Orthorhombic 58 32Kyanite Triclinic 2 32Sillimanite Orthorhombic 62 32FAP (fluorapatite) Hexagonal 176 42HAP (hydroxyapatite) Hexagonal 176 44
3196 Journal of the American Ceramic Society—Yao et al. Vol. 90, No. 10

when compared with the measured values. Another observation inour tests is that using PAW types of potentials always consumesabout 30% more computational time. This is understandable be-cause the PAW type of potentials with smaller radial cutoffs andthe reconstruction of the exact valence wave functions with allnodes in the core region are considered to be more accurate.However, our test calculations show no clear evidence that theelastic constants results were improved using the PAW potential.
In summary, our test calculations on a-quartz and spinelMgAl2O4 indicate that to increase both the accuracy and com-putational efficiency, careful selection of energy cut-off andnumber of K-points is very important. Up to a certain level, ahigher parameter setting will not improve the result much butwill require a lot more computational time. Generally speaking,a 600 eV energy cut-off and a relatively small number of k-points are sufficient for insulating crystals. Also, for most sys-tems we investigated, ultrasoft types of pseudo-potentials wereused in the calculations because of the relatively higher compu-tational speed. However, the appropriate choice of exchangecorrelation potential appears to be highly system dependentwithout a clear preference. We will return to this point after theanalysis of results on many crystal systems to be presented be-
low. (We have actually performed additional but less-extensivetests on other systems such as the trigonal a-Al2O3 expressed ashaving a hexagonal structure.)
(2) Elastic Constants and Structural Properties of Ceramics
Using a predetermined k-point mesh and energy cut-off underboth LDA and GGA approximations using the ultrasoft pseu-dopotential, we have calculated the complete set of independentelastic constants of a large number of selected ceramic crystals(see Table I). Our calculations were performed with the follow-ing common parameters: (1) the strain increment was set to 0.01and �0.01, using RMM-DIIS algorithm31; (2) a high accuracyfor electronic ground-state convergence (10�8 eV); (3) a smalltolerance for ionic relaxation convergence (�10�5); (4) maxi-mum ionic steps of 50 for all cases (It is unusual that the ionicconvergence cannot be achieved in 50 steps for fixed volume re-laxation. This may happen in certain hexagonal crystals. Inthose cases, the stress convergences are carefully monitored. Ifthe stress fluctuations are less than 0.05 kbar, the results aretaken as is. If not, more ionic steps are used to ensure the desiredconveregence is reached); and (5) an adequate number of k-points in the reciprocal space integration. The calculated latticeparameters, complete elastic constants, and other structural pa-rameters are presented in Table II for cubic crystals, Table IIIfor hexagonal and tetragonal crystals, and Table IV for ortho-rhombic crystals. Table V lists the results for kyanite, a crystalwith triclinic symmetry. Also listed are the available experimen-tal data for comparison. Overall, our scheme was very efficientand has been successfully applied to all 44 crystals. The generalagreements with the existing experimental data are quite good.In a few cases, such as the C13, C33, C44 in hexagonal boronnitride (h-BN) and C12, C14, C13 in a-quartz, large discrepancieswere found. The large differences found in C44 are possibly dueto the very small values of the coefficients. These types of dis-crepancies always exist both in calculations and in measure-ments because these constants have relatively small magnitudesand their accuracies are more difficult to attain. For a-quartz,which is a relatively soft material, accurate elastic constants cal-culation is much more challenging because the stress responseis quite large even for a small applied strain of the order of1% incremental increase. In the present case, the incrementalincrease is increased to 1.5% to cover a large range of stress re-sponse. Of the 44 crystals investigated, about 55% have betteragreements with experimental data with LDA approximationthan with the GGA approximation. Thus there is no clear
200 300 400 500 600 700 800 900 1000 11000
5
10
15
20
25
020406080
100120140160180200220
Ela
stic
Co
nst
ants
(G
Pa)
Lat
tice
Par
amet
ers
Ch
ang
ing
(%
)
Energy Cut-off (eV)
(a)
(b)
Fig. 1. Calculated elastic constants (a) and lattice parameters and cellvolume (b) for a-SiO2 as a function of energy cut-off. A 7� 7� 6 k-pointmesh in the Brillouin zone and the LDA/ultrasoft pseudo-potentials isused.
(a)
(b)
Fig. 2. Calculated elastic constants (a) and lattice parameters and cellvolume (b) for a-SiO2 as a function of k-point mesh in the Brillouin zonewith the energy cutoff fixed at 600 eV and the LDA/ultrasoft pseudo-potentials is used.
LDA/USPP GGA/USPP LDA/PAW GGA/PAW
Exchange-Correlation Potential
(a)
(b)
Fig. 3. Calculated elastic constants (a) and lattice parameters and cellvolume (b) for spinel MgAl2O4 for four different types of exchange-cor-relation potentials with a 4� 4� 4 k-point mesh in the Brillouin zoneand an energy cutoff of 600 eV.
October 2007 Ab initio Calculation of Elastic Constants of Ceramic Crystals 3197

evidence to indicate the preference for LDA or GGA in theelastic constants calculations.
Elastic constants were also calculated for some large crystalssuch as yttria (Y2O3), indium oxide (In2O3), yttrium aluminumoxide (YAG or Y3Al5O12), and bismuth silicate (BSO orBi12SiO20). We found that in these crystals, it is more conve-nient to use the cubic cell rather than the primitive cells so thenumber of atoms per cell in the Y2O3, In2O3, YAG, and BSOcrystals are, respectively, 80, 80, 160, and 66 in our calculations.To our knowledge, the independent elastic constants for systemslike In2O3, g-Sn3N4, TiO2 (brookite, and II), WO2, and Si2N2Ohave not been previously reported and our first-time results can
be used for future comparisons when experimental data or othertheoretical calculations become available. We discuss the specificresults below in a more detailed fashion.
(A) Cubic Systems: In Table II, we list the results of cu-bic crystals with three independent elastic constants under bothLDA and GGA approximations. It is very clear that the elasticconstant obtained with LDA approximation are always largerthan those with GGA approximation. This has been observed inmany other calculations independent of the methods used.87–90
The reason is that GGA approximation leads to a larger ex-change-correlation energy, thus favoring longer bond lengthsor larger lattice constants, and softer vibration frequencies or
Table II. Elastic Constants, Bulk Modulus, Young’s Modulus, Shear Modulus, and Poisson’s Ratio for Cubic Ceramic Crystals withboth LDA/USPP and GGA/USPP Exchange-Correlation Potentials
System Exchange correlation
# of
k-Points a (A) V0 (A3)
C11 C12 C44 K G E Z
(GPa)
MgO LDA/USPP 4.2309 75.74 291 92 156 158.3 130.3 306.7 0.1772GGA/USPP 180 4.2317 75.78 276 86 149 149.3 124.4 292.1 0.1740Experiment 32,33 4.2130 74.78 294 93 155 160.0 130.3 — 0.18
CaO LDA/USPP 4.5677 95.30 240.5 48.2 93.2 112.3 94.4 221.2 0.1718GGA/USPP 150 4.7668 108.30 207.8 49.9 79.5 102.5 79.3 189.1 0.1926Experiment33–35 4.8071 111.08 224 60 80.6 114.7 81.2 — —
Y2O3 LDA/USPP 10.4942 1155.64 241.9 128.0 85.1 166.0 72.4 189.7 0.3095GGA/USPP 2 10.6822 1218.86 213.6 112.9 72.6 155.0 66.6 174.8 0.3121Experiment 36,37 10.6073 1193.48 224 112 74.6 150. 66.3 — —
In2O3 LDA/USPP 9.9839 995.10 269.9 138.8 70.5 181.8 68.1 181.5 0.3336GGA/USPP 2 10.2374 1072.91 234.3 107.2 62.7 149.6 63 165.8 0.3152Experiment38 10.1170 1035.51 — — — — — — —
MgAl2O4 LDA/USPP 8.0200 515.82 273.6 149.6 150.7 191.0 105.6 267.4 0.2666GGA/USPP 4 8.1610 543.50 256.5 133.2 142.4 174.3 101.8 255.6 0.2556Experiment 32,39 8.0860 528.69 282.9 155.4 154.8 197.9 108.5 — —
g-Mg2SiO4 LDA/USPP 7.9557 503.53 338.3 111.8 130.0 187.3 128.6 314.0 0.2206GGA/USPP 4 8.1139 534.17 299.6 103.3 128.2 168.7 115.2 281.5 0.2219Experiment 40,39 8.1700 545.34 327 112 126 184 119 — —
SrTiO3 LDA/USPP 3.8572 57.38 397.2 100.8 114.1 199.6 126.7 313.7 0.2380GGA/USPP 80 3.9403 61.17 324.7 89.8 106.3 168.1 110.6 272.2 0.2301Experiment 32,39 3.9040 59.50 335.0 105 127
YAG LDA/USPP 11.8465 1662.37 356.7 122.6 114.3 200.6 115.4 290.5 0.2587Experiment32,41 1 12.0000 1728.00 333 113 115 — — — —
BSO LDA/USPP 9.9152 974.74 150.1 36.5 31.4 74.4 39.9 101.5 0.2724GGA/USPP 2 10.2227 1068.27 116.5 30.1 23.9 58.9 30.4 77.7 0.2801Experiment 42,43 10.1043 1031.62 129.8 30.2 24.7 — — — —
c-BN LDA/USPP 3.5725 45.6 825 193 475 404 403 908 0.125GGA/USPP 260 3.6653 49.2 783 172 444 376 382 856 0.120Experiment.32,44 3.6150 47.2 820 190 480 400 405 — —
g-Si3N4 LDA/USPP 7.7861 472.0 529 169 334 289 261 601 0.153GGA/USPP 4 7.7868 472.1 504 177 327 286 248 576 0.164Experiment 45,46 7.8369 481.3 — — — 300 — — —
g-Sn3N4 LDA/USPP 8.9521 717.38 282.1 139.5 140.4 187 107 269.5 0.2598GGA/USPP 12 9.1171 757.82 246.1 116.9 128.0 160.0 97.3 242.6 0.2472Expr.47 9.037 738.03 — — — — — — —
TiN LDA/USPP 4.1865 73.4 688 124 171 312 209 513 0.226GGA/USPP 180 4.2606 77.3 680 130 171 313 207 509 0.229Experiment 32,48 4.2387 76.2 625 165 163 325 192 — —
HfN LDA/USPP 4.4446 87.80 734.8 105.8 129.4 315.5 186.3 467.1 0.2532GGA/USPP 150 4.5219 92.46 628.9 103.6 119.5 278.7 164.8 413 0.2530Experiment 32,49 4.5400 93.58 679 119 150 306 202 387 0.15
Diamond LDA/USPP 3.5268 43.86 1107.2 144.7 598.1 465.5 548.3 1181.1 0.0771GGA/USPP 80 3.5674 45.40 1055.0 120.4 559.0 431.9 520.3 1113.7 0.0702Expr.32,50 3.5670 45.38 1079 124 578 443.0 535.7 — —
SiC-3C LDA/USPP 4.3194 80.58 401.9 136.4 255.7 224.9 196.5 456.6 0.1616GGA/USPP 80 4.3694 83.42 384.5 121.5 243.3 209.2 190.1 437.6 0.1513Experiment.51,52 4.3597 82.86 390 142 256 225 — — —
c-ZnS LDA/USPP 5.3087 149.60 116.6 71.8 50.9 86.7 36.6 96.3 0.3150GGA/USPP 80 5.4512 161.98 96.5 56.5 44.9 69.8 32.5 84.3 0.2988Experiment53,54 5.4102 158.36 102 64.6 44.6 77.1 31.5 — —
3198 Journal of the American Ceramic Society—Yao et al. Vol. 90, No. 10

Table
III.
Elastic
Constants,Bulk
Modulus,Young’sModulus,ShearModulus,andPoisson’sRatioforHexagonalandTetragonalCeramic
Crystalswithboth
LDA/U
SPPandGGA/
USPPExchange-CorrelationPotentials
System
Exchange-correlation
#of
k-Points
a(A
)c/a
V0(A
3)
C11
C12
C13
C14
C33
C44
C66
KG
E@
(GPa)
Hexagonal
a-Al 2O
3LDA/U
SPP
4.7457
2.7251
252.24
476.8
157.6
119.4
19.4
476.6
145.5
159.6
246.9
158.5
391.6
0.2356
GGA/U
SPP
4.8195
2.7284
264.51
437.2
144.3
101.5
21
443.3
125.5
146.4
223.4
143.0
353.6
0.2362
Experim
ent32,33
4.7620
2.7270
255.03
495
160
115
-23
497
146
—251.7
162.5
——
a-SiO
2LDA/U
SPP
51
4.8761
1.1010
110.55
76.1
6.0
6.1
20
94.2
52.8
35
31.3
43.2
88.7
0.0270
GGA/U
SPP
4.8861
1.1022
111.35
83.4
13.3
15.6
17.1
110.3
57.2
35.1
40.3
45.1
98.5
0.0926
Experim
ent33,55
4.9160
1.0996
113.13
86.8
7.1
14.4
�18
107.5
58.2
—36.4
31.1
97.2
0.16
ZnO
LDA/U
SPP
208
3.1929
1.6159
45.55
220.8
139.7
124.4
236.7
37.5
40.6
161.7
42.0
116.0
0.3805
GGA/U
SPP
3.2779
1.6138
49.22
187.0
111.7
95.0
203.1
36.7
37.6
131.2
40.1
109.2
0.3612
Experim
ent33,56
3.2490
1.6026
47.60
209
120
104
218
44.1
—143.5
46.8
——
h-BN
LDA/U
SPP
255
2.4806
2.5965
34.32
925.9
209.6
2.5
31.2
5.8
358.1
143.4
99.0
241.4
0.2194
GGA/U
SPP
2.5040
3.2093
43.78
720.9
154.1
0.2
3.8
0.7
283.4
99.3
72.3
174.4
0.2072
Experim
ent51,57
2.5040
2.6602
36.17
811
169
0.0
27.0
7.7
——
——
—a-Si 3N
4LDA/U
SPP
88
7.6988
0.7247
286.41
415.0
142.0
122.9
15.0
454.4
129.2
136.5
228.8
138.3
345.2
0.2486
GGA/U
SPP
7.6659
0.7251
282.90
417.7
146.8
127.5
15.7
458.4
128
135.4
233.0
137.1
343.8
0.2541
Experim
ent32
7.6405
0.7263
280.56
——
——
——
—229
——
—b-Si 3N
4LDA/U
SPP
25
7.5522
0.3818
142.42
418.1
189.4
106.4
548.6
101.8
114.4
243.0
124.4
318.8
0.2814
GGA/U
SPP
7.6366
0.3819
147.29
400.9
171.9
95.7
519.8
103
114.5
227.4
124.6
316.0
0.2684
Experim
ent32,58
7.6220
0.3818
146.42
433
195
127
574
108
119.0
259
——
—SiC-2H
LDA/U
SPP
172
3.0496
1.6413
40.31
521.7
107.8
52.0
564.0
157.5
207.0
225.7
194.0
452.4
0.1659
GGA/U
SPP
3.0845
1.6416
41.72
496.5
94.7
43.4
533.8
154.5
200.9
210.0
188.6
435.5
0.1543
Experim
ent32
3.0760
1.6573
41.36
——
——
——
——
——
SiC-4H
LDA/U
SPP
86
3.0516
3.2739
80.57
514.5
111.4
53.0
564.6
164.9
201.6
225.4
195.4
454.8
0.1637
GGA/U
SPP
3.0868
3.2742
83.40
488.7
98.0
44.8
533.8
161.2
195.4
209.6
189.5
436.8
0.1526
Experim
ent51,59
3.0817
3.2713
82.90
507
108
52
547
159
—221
——
—SiC-6H
LDA/U
SPP
62
3.0525
4.9068
120.86
511.7
112.6
53.3
565.2
167.6
199.5
225.2
195.8
455.5
0.1629
GGA/U
SPP
3.0877
4.9072
125.10
486.2
98.9
45.3
534.0
163.5
193.6
209.5
189.8
437.3
0.1521
Experim
ent51,59
3.0817
4.9055
124.33
502
95
565
169
203
FAP
PBE/PAW
10
9.4818
0.7283
537.69
132.4
38.5
62.8
170.2
43.3
47.0
83.2
44.6
113.4
0.2726
Experim
ent60,61
9.4615
0.7239
530.99
152.0
50.0
63.1
185.7
42.75
51.2
92.8
48.0
122.7
HAP
PBE/PAW
10
9.5339
0.7240
543.36
126.4
35.6
65.1
167.4
44.3
45.4
81.2
43.4
110.5
0.2731
Experim
ent60,62
9.4300
0.7308
530.69
137
42.5
54.9
172
39.6
47.3
82.6
44.6
113.3
0.2715
Tetragonal
SiO
2(Stishovite)
LDA/U
PP
36
4.1653
0.6413
46.34
439.7
213.1
186.8
716.2
244.5
313.0
302.5
244
576.8
0.1822
GGA/U
PP
4.2411
0.6364
48.55
381.1
166.3
172.2
666.4
220.2
278.6
264.3
218.2
513.3
0.1763
Experim
ent32,63
4.1790
0.6377
46.54
453
211
203
776
252
302
316
220
——
TiO
2(R
utile)
LDA/U
PP
252
4.5669
0.6416
61.11
293.8
225.5
171.0
516.7
122.7
246.2
245.2
146.3
366.1
0.2511
GGA/U
PP
4.6545
0.6394
64.47
270.1
172.0
147.0
467.6
115.9
216.3
211.3
135.5
335.0
0.2359
Experim
ent54,64
4.5936
0.6441
62.43
269
177
146
480
124
192
215.5
112.4
——
October 2007 Ab initio Calculation of Elastic Constants of Ceramic Crystals 3199
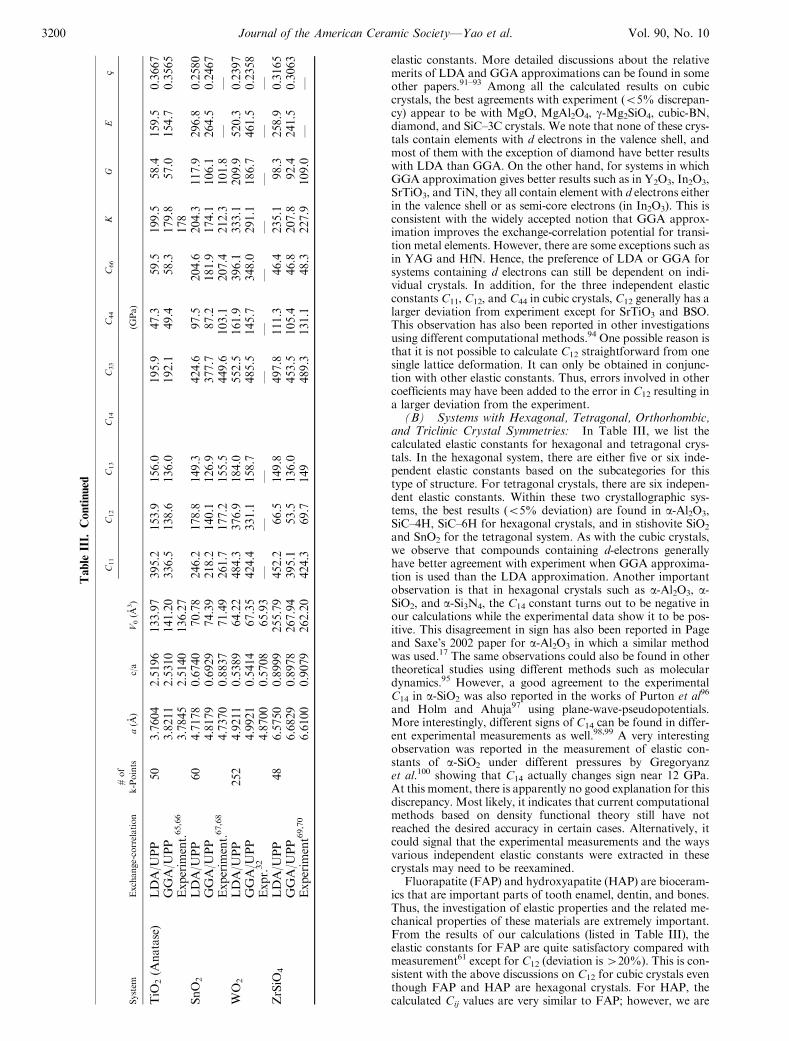
elastic constants. More detailed discussions about the relativemerits of LDA and GGA approximations can be found in someother papers.91–93 Among all the calculated results on cubiccrystals, the best agreements with experiment (o5% discrepan-cy) appear to be with MgO, MgAl2O4, g-Mg2SiO4, cubic-BN,diamond, and SiC–3C crystals. We note that none of these crys-tals contain elements with d electrons in the valence shell, andmost of them with the exception of diamond have better resultswith LDA than GGA. On the other hand, for systems in whichGGA approximation gives better results such as in Y2O3, In2O3,SrTiO3, and TiN, they all contain element with d electrons eitherin the valence shell or as semi-core electrons (in In2O3). This isconsistent with the widely accepted notion that GGA approx-imation improves the exchange-correlation potential for transi-tion metal elements. However, there are some exceptions such asin YAG and HfN. Hence, the preference of LDA or GGA forsystems containing d electrons can still be dependent on indi-vidual crystals. In addition, for the three independent elasticconstants C11, C12, and C44 in cubic crystals, C12 generally has alarger deviation from experiment except for SrTiO3 and BSO.This observation has also been reported in other investigationsusing different computational methods.94 One possible reason isthat it is not possible to calculate C12 straightforward from onesingle lattice deformation. It can only be obtained in conjunc-tion with other elastic constants. Thus, errors involved in othercoefficients may have been added to the error in C12 resulting ina larger deviation from the experiment.
(B) Systems with Hexagonal, Tetragonal, Orthorhombic,and Triclinic Crystal Symmetries: In Table III, we list thecalculated elastic constants for hexagonal and tetragonal crys-tals. In the hexagonal system, there are either five or six inde-pendent elastic constants based on the subcategories for thistype of structure. For tetragonal crystals, there are six indepen-dent elastic constants. Within these two crystallographic sys-tems, the best results (o5% deviation) are found in a-Al2O3,SiC–4H, SiC–6H for hexagonal crystals, and in stishovite SiO2
and SnO2 for the tetragonal system. As with the cubic crystals,we observe that compounds containing d-electrons generallyhave better agreement with experiment when GGA approxima-tion is used than the LDA approximation. Another importantobservation is that in hexagonal crystals such as a-Al2O3, a-SiO2, and a-Si3N4, the C14 constant turns out to be negative inour calculations while the experimental data show it to be pos-itive. This disagreement in sign has also been reported in Pageand Saxe’s 2002 paper for a-Al2O3 in which a similar methodwas used.17 The same observations could also be found in othertheoretical studies using different methods such as moleculardynamics.95 However, a good agreement to the experimentalC14 in a-SiO2 was also reported in the works of Purton et al96
and Holm and Ahuja97 using plane-wave-pseudopotentials.More interestingly, different signs of C14 can be found in differ-ent experimental measurements as well.98,99 A very interestingobservation was reported in the measurement of elastic con-stants of a-SiO2 under different pressures by Gregoryanzet al.100 showing that C14 actually changes sign near 12 GPa.At this moment, there is apparently no good explanation for thisdiscrepancy. Most likely, it indicates that current computationalmethods based on density functional theory still have notreached the desired accuracy in certain cases. Alternatively, itcould signal that the experimental measurements and the waysvarious independent elastic constants were extracted in thesecrystals may need to be reexamined.
Fluorapatite (FAP) and hydroxyapatite (HAP) are bioceram-ics that are important parts of tooth enamel, dentin, and bones.Thus, the investigation of elastic properties and the related me-chanical properties of these materials are extremely important.From the results of our calculations (listed in Table III), theelastic constants for FAP are quite satisfactory compared withmeasurement61 except for C12 (deviation is420%). This is con-sistent with the above discussions on C12 for cubic crystals eventhough FAP and HAP are hexagonal crystals. For HAP, thecalculated Cij values are very similar to FAP; however, we are
Table
III.
Continued
System
Exchange-correlation
#of
k-Points
a(A
)c/a
V0(A
3)
C11
C12
C13
C14
C33
C44
C66
KG
E@
(GPa)
TiO
2(A
natase)
LDA/U
PP
50
3.7604
2.5196
133.97
395.2
153.9
156.0
195.9
47.3
59.5
199.5
58.4
159.5
0.3667
GGA/U
PP
3.8211
2.5310
141.20
336.5
138.6
136.0
192.1
49.4
58.3
179.8
57.0
154.7
0.3565
Experim
ent.65,66
3.7845
2.5140
136.27
178
SnO
2LDA/U
PP
60
4.7178
0.6740
70.78
246.2
178.8
149.3
424.6
97.5
204.6
204.3
117.9
296.8
0.2580
GGA/U
PP
4.8179
0.6929
74.39
218.2
140.1
126.9
377.7
87.2
181.9
174.1
106.1
264.5
0.2467
Experim
ent.67,68
4.7370
0.8837
71.49
261.7
177.2
155.5
449.6
103.1
207.4
212.3
101.8
——
WO
2LDA/U
PP
252
4.9211
0.5389
64.22
484.3
376.9
184.0
552.5
161.9
396.1
333.1
209.9
520.3
0.2397
GGA/U
PP
4.9921
0.5414
67.35
424.4
331.1
158.7
485.5
145.7
348.0
291.1
186.7
461.5
0.2358
Expr.32
4.8700
0.5708
65.93
——
——
——
——
——
ZrSiO
4LDA/U
PP
48
6.5750
0.8999
255.79
452.2
66.5
149.8
497.8
111.3
46.4
235.1
98.3
258.9
0.3165
GGA/U
PP
6.6829
0.8978
267.94
395.1
53.5
136.0
453.5
105.4
46.8
207.8
92.4
241.5
0.3063
Experim
ent69,70
6.6100
0.9079
262.20
424.3
69.7
149
489.3
131.1
48.3
227.9
109.0
——
3200 Journal of the American Ceramic Society—Yao et al. Vol. 90, No. 10

Table
IV.
Elastic
Constants
forOrthorhombicCeramic
Crystalswithboth
LDA/U
PPandGGA/U
PPExchange-CorrelationPotentials
System
Exchange-correlation
#ofk-Points
a(A
)b(A
)c(A
)
C11
C12
C13
C22
C23
C33
C44
C55
C66
KG
EC
(GPa)
MgSiO
3LDA/U
PP
75
4.7357
4.8982
6.8603
464.5
122.8
127.3
530.8
141.1
457.6
191.3
170.4
149.0
248.0
172.1
419.4
0.2181
GGA/U
PP
4.8253
4.9756
6.9871
425.7
107.8
120.1
476.3
130.4
402.4
174.3
156.1
127.9
224.1
153.6
375.1
0.2210
Experim
ent71,72
4.7754
4.9292
6.8969
482
144
147
537
146
485
204
186
147
MgSiO
3(Post
Perovskite)
LDA/U
PP
22
2.6718
9.0046
6.6146
605.6
39.0
80.3
423.5
109.3
500.2
79.3
99.1
77.0
218.8
124.0
313.0
0.2616
GGA/U
PP
2.7189
9.1969
6.7363
543.7
26.0
73.8
381.7
90.0
432.0
60.0
76.3
33.2
191.4
91.3
236.2
0.2943
Experim
ent73,74w
2.4650
8.0420
6.0930
624
54
84
433
122
524
89
110
98
231
136
Mg2SiO
4LDA/U
PP
18
4.6938
10.0159
5.8791
335.6
69.4
69.4
207.3
72.9
242.1
70.4
83.3
85.5
132.2
84.6
209.2
0.2363
GGA/U
PP
4.7796
10.2824
6.0143
299.9
56.6
59.7
182.0
64.7
216.7
59.3
74.1
72.6
116.0
74.1
183.3
0.2365
Experim
ent75,76
4.7560
10.1943
5.9807
337
205.6
241.1
69.5
81
83.4
b-Mg2SiO
4LDA/U
PP
65.7446
11.2548
8.0430
372.0
56.4
98.3
360.4
97.0
277.5
102.8
124.4
95.1
167.6
113.0
276.9
0.2246
GGA/U
PP
5.8501
11.4804
8.2302
331.8
45.1
92.9
322.3
87.5
244.9
93.4
119.3
76.2
149.5
100.0
245.4
0.2265
Experim
ent77,78
5.6983
11.4380
8.2566
360
75
110
383
105
273
112
118
98
TiO
2(Brookite)
LDA/U
PP
80
9.1291
5.4113
5.0949
315.1
191.0
184.2
350.8
163.6
310.4
112.6
105.8
106.9
228.3
91.9
243.0
0.3225
GGA/U
PP
9.2859
5.5080
5.1910
285.1
156.3
158.5
302.4
140.9
289.1
101.9
95.2
87.6
198.7
83.8
220.3
0.3152
Experim
ent79
9.1740
5.4490
5.1380
TiO
2(II)
LDA/U
PP
96
4.5147
5.4545
4.8645
340.9
196.3
187.7
350.8
153.9
383.1
92.9
126.1
95.2
238.9
96.9
256.1
0.3214
GGA/U
PP
4.5922
5.5650
4.9482
309.5
163.1
161.1
309.9
139.5
353.7
87.2
119.0
72.8
210.9
88.1
232.1
0.3166
Experim
ent80,81
4.5318
5.5019
4.9063
253
Si 2N
2O
LDA/U
PP
16
8.8338
5.4692
4.8209
316.0
81.3
51.5
241.6
31.0
320.7
139.1
59.7
76.2
132.7
97.1
234.2
0.2059
GGA/U
PP
8.9314
5.5315
4.8810
313.9
84.0
53.6
252.3
36.5
320.3
136.7
61.6
76.0
136.4
97.3
235.8
0.2118
Experim
ent82,83
8.8717
5.4990
4.8504
93.1
221.6
Andalusite
LDA/U
PP
10
7.7199
7.8554
5.5344
221.4
91.7
102.3
293.9
91.6
339.4
92.0
79.9
115.2
156.1
93.7
234.3
0.2499
GGA/U
PP
7.8753
7.9881
5.6104
195.1
75.2
87.3
251.8
79.9
320.5
88.2
76.0
102.5
136.3
86.9
215.0
0.2371
Experim
ent84
7.7382
7.8571
5.5338
233.4
97.7
116.2
289.0
81.4
380.1
99.5
87.8
112.3
Sillimanite
LDA/U
PP
10
7.4404
7.6060
5.7420
283.4
103.6
92.3
223.9
122.5
392.7
115.2
74.5
81.6
167.0
90.1
229.2
0.2713
GGA/U
PP
7.5601
7.7759
5.8138
273.8
89.5
74.2
200.2
108.5
372.8
109.0
69.9
74.6
150.7
85.7
216.2
0.2609
Expreriment85,86
7.4146
7.5739
5.7450
287.3
158.6
83.4
231.9
94.7
388.4
122.4
80.7
89.3
w Thecalculationresultsfrom
Tsuchiya’spaper.
October 2007 Ab initio Calculation of Elastic Constants of Ceramic Crystals 3201

unable to locate any measured data for comparison. It is notedthat the calculations for HAP take more time to reach necessaryconvergence during relaxation than FAP. This is most likely dueto the lower crystal symmetry of HAP when the OH group re-places F in FAP.
Our calculated results for crystals with orthorhombic sym-metries were summarized in Table IV. There are nine indepen-dent elastic constants in orthorhombic crystals. Very goodagreements were found for the diagonal elements of elastic ten-sor when compared with the existing experimental values. How-ever, larger deviations were noted in C12 and C13. The previousarguments for C14 in hexagonal crystals could also be appliedhere. We cannot find any experimentally determined elastic con-stants for some of these crystals like TiO2(II) and Si2N2O. Butthe measured bulk modulus for TiO2(II)
81 and Young’s modu-lus and shear modulus for Si2N2O
83 match very well to our cal-culated values. For MgSiO3 in the post-perovskite phase, whichcan only exist at a high-temperature and high-pressure environ-ment, there is no experimental data so far. Our results werecompared with Tsuchiya and Tsuchiya’s calculation under zeropressure.74 The agreements are very satisfactory. This also con-firms the reliability of our calculations.
Another set of important materials are the three aluminosil-icate (Al2SiO5) polymorphs: kyanite, sillimanite, and andalusite.They are good thermal and electrical insulators that can with-stand high temperatures, corrosive environments, or other ad-verse conditions. Andalusite and sillimanite have orthorhombicsymmetry whereas kyanite is triclinic. Our calculated results arein reasonably good agreement with experimental data for and-alusite84 and sillimanite.86 For kyanite, which is of the lowestsymmetry type among all the crystals studied in this paper, it hasall 21 independent elastic constants. We list the entire elastictensor in Table V along with the results by Page and Saxe’scalculation17 using the similar method and same computationalparameters, except that we used a relatively higher energy cut-off. The results match very well except for those coefficients withrelatively small values such as C15, C16, C25, C26, C35, C36, C45,C46, and C56, some of them even have different signs. We believeour results are more reliable because of the higher convergency.
IV. Summary
In the present work, we report the results of the complete set ofelastic constants and other structural parameters for a largenumber of ceramic crystals using an improved scheme based onthe stress–strain approach. The scheme is applied to a series ofoxides, nitrides, carbides, and other complex materials underzero temperature and pressure. Our calculations show highcomputational efficiency with results generally in good agree-ment with experimental data. It shows that the GGA exchange-correlation potential gives better agreement for crystals contain-ing d electrons, otherwise there is no clear indication of GGAbeing a better approximation than the LDA potential. We havealso calculated the elastic constants for some complex crystalsthat have not been reported in the literature or have not beenmeasured. These results can be confidently viewed as reasonabletheoretical predictions. However, there are still some intriguingproblems that require further investigations such as the compu-
tational accuracy required when dealing with relatively soft ma-terials such as a-quartz and the disagreement in sign with theexperimental value on the elastic constant C14 in some hexag-onal crystals. It is also necessary to further investigate the pre-cise nature of the influence of the exchange-correlation potentialon crystals containing d electrons. The availability of a highlyefficient computational scheme for lattice deformation calcula-tion is the first step toward the ab initio phonon calculations thatwould be required in the evaluation of temperature-and-pressuredependent Gibbs free energy. Such comprehensive studies of thethermodynamics of ceramic materials will be attempted and re-ported in future publications.
References
1W. F. Brinkman, Physics through the 1990s. National Academy Press,Washington, DC, 1986.
2Y. Kawazoe, ‘‘Can ab initio Simulation Really Predict Properties of MaterialsPrior to Experimental,’’ Bull. Mater. Sci., 22, 901–4 (1999).
3B. D. Dunietz, M. D. Beachy, Y. Cao, D. A. Whittington, S. J. Lippard, andR. A. Friesner, ‘‘Large Scale Ab-initio Quantum Chemical Calculation of theIntermediates in the Soluble Methane Monooxygenase Catalytic Cycle,’’ J. Am.Chem. Soc., 122, 2828–39 (2000).
4B. Winkler, ‘‘An Introduction to Computational Crystallography,’’ Z. Kristal-logr., 212, 506–11 (1999).
5A. B. Alchagirov, J. P. Perdew, J. C. Boettger, R. C. Albers, and C. Fiolhais,‘‘Energy and Pressure Versus Volume: Equations of State Motivated by theStabilized Jellium Model,’’ Phys. Rev. B, 63, 224115–30 (2001).
6P. Kackell, B. Wenzien, and F. Bechstedt, ‘‘Influence of Atomic Relaxations onthe Structural Properties of SiC Polytypes from ab initio Calculations,’’ Phys. Rev.B, 50, 17037–46 (1994).
7E. Wachowicz and A. Kiejna, ‘‘Bulk and Surface Properties of Hexagonal-Close-Packed Be and Mg,’’ J. Phys.: Condens. Matter, 13, 10767–76 (2001).
8Y. Yourdshahyan, C. Ruberto, M. Halvarsson, L. Bengtsson, V. Langer, B. I.Lundqvist, S. RUSPPi, and U. Rolander, ‘‘Theoretical Structure Determination ofa Complex Material: k-Al2O3,’’ J. Am. Ceram. Soc., 82, 1365–80 (1999).
9R. Stadler, W. Wolf, R. Podloucky, G. Kresse, J. Furthmuller, and J. Hafner,‘‘Ab initio Calculations of the Cohesive, Elastic, and Dynamical Properties ofCoSi2 by Pseudopotential and All-Electron Techniques,’’ Phys. Rev. B, 54, 1729–34 (1996).
10H. Nielsen and R. M. Martin, ‘‘First-Principles Calculation of Stress,’’ Phys.Rev. Lett., 50, 697–700 (1983).
11S. Wei, D. C. Allan, and J. W. Wilkins, ‘‘Elastic Constants of a Si/Ge Super-lattice and of Bulk Si and Ge,’’ Phys. Rev. B, 46, 12411–20 (1992).
12S. G. Shen, ‘‘Calculation of the Elastic Properties of Semiconductors,’’J. Phys.: Condens. Matter, 6, 8733–44 (1994).
13G. De Sandre, L. Colombo, and C. Bottani, ‘‘Calculation of Elastic Constantsin Defected and Amorphous Silicon by Quantum Simulations,’’ Phys. Rev. B, 54,11857–60 (1996).
14B. B. Karki, L. Stixrude, S. J. Clark, M. C. Warren, G. J. Ackland, andJ. Crain, ‘‘Structure and Elasticity of MgO at High Pressure,’’ Am. Miner., 82,51–60 (1997).
15B. Kiefer, L. Stixrude, and R. M. Wentzcovitch, ‘‘Calculated Elastic Con-stants and Anisotropy of Mg2SiO4 Spinel at High Pressure,’’ Geophys. Res. Lett.,24 (22) 2841–4 (1997).
16W. Duan, R. M. Wentzcovitch, and K. T. Thomson, ‘‘First-Principles Studyof High-Pressure Alumina Polymorphs,’’ Phys. Rev. B, 57, 10363–9 (1998).
17Page Y. L. and P. Saxe, ‘‘Symmetry-General Least-Squares Extraction ofElastic Data for Strained Materials from ab initio Calculations of Stress,’’ Phys.Rev. B, 65, 104104–17 (2002).
18J. G. Brainerd, A. G. Jensen, L. G. Cumming, R. R. Batcher, S. G. Begun,H. S. Black, G. M. Grown, C. R. Burrows, H. Busignies, W. G. Cady, P. S. Carter,A. B. Chamberlain, D. Chambers, E. J. Content, J. W. Forrester, R. A. Hack-busch, L. R. Hafstad, J. V. L. Hofan, J. E. Keister, E. A. Laport, W. Mason, J. F.McDonald, L. S. Nergaard, A. F. Pomeroy, G. Rappaport, E. S. Seeley, R. F.Shea, J. R. Steen, W. N. Tuttle, L. C. Van Atta, K. S. Van Dyke, D. E. Watts,L. E. Whittemore, R. A. Sykes, C. F. Baldwin, W. L. Bond, J. K. Clapp,C. Frondel, H. Jaffe, W. P. Mason, P. L. Smith, and J. M. Wolfskill, ‘‘Standardson Piezoelectric Crystals,’’ Proc. IRE, 37, 1378 (1949).
Table V. Full Elastic Tensor with Cij for Kyanite with Triclinic Symmtry under LDA/UPP Exchange-Correlation Potentials
Elastic constants 1 2 3 4 5 6
1 363.3 (376) 102.3 (108) 76.0 (70) 10.1 (0) 23.6 (3) 7.0 (�3)2 102.3 (108) 363.4 (357) 107.8 (112) �21.8 (�20) �0.1 (�1) 2.7 (3)3 76.3 (70) 107.8 (112) 363.3 (370) �27 (�22) �13.7 (3) �6.1 (2)4 10.1 (0) �21.8 (�20) �27 (�22) 164 (169) �15.7 (0) �2.4 (2)5 23.6 (3) 1 (�1) �13.7 (3) �15.7 (0) 103.7 (90) 1.9 (�7)6 7.0 (�3) 2.7 (3) �6.1 (2) �2.4 (2) 1.9 (�7) 120.9 (121)
wThe values in the parentheses are those from Page and Saxe.17
3202 Journal of the American Ceramic Society—Yao et al. Vol. 90, No. 10

19C. da Silva, L. Stixrude, and R. M. Wentzcovitch, ‘‘Elastic Constants andAnistropy of Forsterite at High Pressure,’’ Geophys. Res. Lett., 24 [15] 1963–6(1997).
20G. Kresse and J. Hafner, ‘‘Ab initio Molecular Dynamics for Open-ShellTransition Metals,’’ Phys. Rev. B, 48, 13115–8 (1993).
21G. Kresse and J. Hafner, ‘‘Ab initio Molecular-Dynamics Simulation of theLiquid-Metal–Amorphous-Semiconductor Transition in Germanium,’’ Phys. Rev.B, 49, 14251–69 (1994).
22P. Hohenberg and W. Kohn, ‘‘Inhomogeneous Electron Gas,’’ Phys. Rev.,136, B864–71 (1964).
23W. Kohn and L. J. Sham, ‘‘Self-Consistent Equations Including Exchange andCorrelation Effects,’’ Phys. Rev., 140, A1133–8 (1965).
24D. Vanderbilt, ‘‘Soft Self-Consistent Pseudopotentials in a Generalized Eigen-value Formalism,’’ Phys. Rev. B, 41, 7892–5 (1990).
25P. E. Blochl, ‘‘Projector Augmented-Wave Method,’’ Phys. Rev. B, 50, 17953–79 (1994).
26G. Kresse and J. Joubert, ‘‘From Ultrasoft Pseudopotentials to the ProjectorAugmented Wave Method,’’ Phys. Rev. B, 59, 1758–75 (1999).
27H. J. Monkhorst and J. D. Pack, ‘‘Special Points for Brillouin-Zone Integra-tions,’’ Phys. Rev. B, 13, 5188–92 (1976).
28W. Voigt, Lehrbuch der Kristallphysik. Taubner, Leipzig, 1928.29A. Reuss and Z. Angew, ‘‘Berchung der Fiessgrenze von Mischkristallen
auf Grund der Plastiziatsbedingung fur Einkristalle,’’ Math. Mech., 9, 55(1929).
30R. Hill, ‘‘The Elastic Behaviour of a Crystalline Aggregate,’’ Proc. Phys. Soc.Lond., 65, 350 (1952).
31P. Pulay, ‘‘Convergence Acceleration of Iterative Sequences: The Case of ScfIteration,’’ Chem. Phys. Lett., 73, 393–8 (1980).
32The crystal structures for these crystals are the same as used by the UMKCElectronic Structure Group (ESG) [http://cas.umkc.edu/physics/ching] and in theirprevious publications.
33R. F. S. Hearmon, ‘‘The Elastic Constants of Crystals and Other AnisotropicMaterials’’; p. 854 in Lansolt-Bornstein Tables, III/11, Edited by K. H. Hellwegeand A. M. Hellwege. Springer-Verlag, Berlin, 1979.
34R. Ganguly, V. Siruguri, and I. Gopalakrishnan, ‘‘Stability of the LayeredSr3Ti2O7 Structure in La1.2(Sr1-xCax)1.8Mn2O7,’’ J. Phys.: Condens. Matter, 12,1683–9 (2000).
35N. Soga, ‘‘Elastic Constants of Garnet Under Pressure and Temperature,’’J. Geophys. Res., 72, 4227 (1967).
36M. Facucher and J. Pannetire, ‘‘Refinement of the Structure at 77 K,’’ Acta.Cryst., Sect. B: Struct. Crystallogr. Cryst. Chem., 36, 3209–11 (1980).
37J. W. Palko, W.M. Kriven, S. V. Sinogeikin, J. D. Bass, and A. Sayir, ‘‘ElasticConstants of Yttria (Y2O3) Monocrystals to High Temperatures,’’ J. Appl. Phys.,89 [12] 7791–6 (2001).
38S. Kohiki, M. Sasaki, Y. Murakawa, K. Hori, K. Okoda, H. Shimooka,T. Tajiri, H. Deguchi, S. Matsuch, M. Oku, ‘‘Doping of Fe to In2O3,’’ Thin SolidFilms, 505, 122–5 (2005).
39A. Yoneda, ‘‘Pressure Derivatives of Elastic Constants of Single Crystal MgOand MgAl2O4,’’ J. Phys. Earth, 38, 19–55 (1990).
40W. H. Baur, ‘‘Computer-Simulated Crystal Structures of Observed and Hy-pothetical Mg2SiO4 Polymorphs of Low and High Density,’’Am. Mineralogist, 57,709–31 (1972).
41V. F. Kitaeva, N. N. Sobolev, I. L. Chistyj, E. Zharikov, V. V. Osiko, M. I.Timoshechkin, and A. C. Zolot’ko, ‘‘Molecular Light Scattering in Garnets,’’ Sov.Solid State Phys., 22 [5] 1379 (1980).
42S. C. Arahams, J. L. Berstein, and C. Sbensson, ‘‘Crystal Structure and Ab-solute Piezoelectric d14 Coefficient in Laevorotatory Bi12SiO20,’’ J. Chem. Phys.,71, 788–92 (1979).
43S. Lee, C. Lee, H. S. Lee, and J. H. Kim, ‘‘The Study on the Elastic and theElasto-Optic Properties of the Bismuth Germanium Oxide Single Crystals,’’ Sae-Mulli, 33, 283–8 (1993).
44M. Grimsditch, E. S. Zouboulis, and A. Polian, ‘‘Elastic Constants of BoronNitride,’’ J. Appl. Phys., 76, 832–4 (1994).
45A. Zerr, G. Miehe, and G. Serghiou et al., ‘‘Synthesis of Cubic SiliconNitride,’’ Nature, 400, 340–2 (1999).
46E. Soignard, Mi Somayazulu, and J. Dong, ‘‘High Pressure-High TemperatureSynthesis and Elasticity of the Cubic Nitride Spinel g-Si3N4,’’ J. Phys. Condens.Matter, 13, 557–63 (2001).
47N. Scotti, H. Jacobs, and W. Kockelmann, ‘‘Sn3N4, ein Zinn(IV)-Nitrid -Synthese und erste Strukturbestimmung einer binaren Zinn-Stickstoff-Verbin-dung,’’ Z. Anorg. Allg. Chem., 625 [9] 1435–9 (1999).
48J. O. Kim, J. D. Achenbach, and P. B. Mirkarimi, ‘‘Elastic Constants of Sin-gle-Crystal Transition-Metal Nitride Films Measured by Line-Focus AcousticMicroscopy,’’ J. Appl. Phys., 72, 1805–11 (1992).
49X.-J. Chen, V. V. Struzhkin, Z. Wu, M. Somayazulu, and J. Qian, ‘‘HardSuperconducting Nitrides,’’ Appl. Phys. Sci., PNAS, 102 [9] 3199 (2005).
50H. J. McShimin and W. L. Bond, ‘‘Elastic Moduli of Diamond as a Functionof Pressure and Temperature,’’ J. Appl. Phys., 43, 2944–8 (1972).
51A. Taylor and B. J. Kagle, ‘‘Crystallographic Data on Metal and Alloy Struc-tures,’’ Dover Publications, Inc., New York, 1962.
52W. R. L. Lambrecht, B. Segall, M. Methfessel, andM.van Schilfgaarde, ‘‘Cal-culated Elastic Constants and Deformation Potentials of Cubic SiC,’’ Phys. Rev.B, 44, 3685–94 (1991).
53J. C. Jamieson and H. H. Demarest, ‘‘A Note on the Compression of CubicZnS,’’ J. Phys. Chem. Solids, 41 [9] 963–4 (1980).
54R. F. S. Hearmon, ‘‘The Elastic Constants of Crystals and Other AnisotropicMaterials’’; pp. 1154 in Lansolt-Bornstein Tables, III/11, Edited by K. H. Hellwegeand A. M. Hellwege. Springer-Verlag, Berlin, 1984.
55L. Levien, T. Charles, Prewitt, and J. Donald, ‘‘Structure and Elastic Prop-erties of Quartz at Pressure,’’ Am. Miner., 65, 920–3 (1980).
56T. B. Rymer and G. D. Archard, ‘‘Lattice Constants of Zinc Oxide,’’Research,5, 292 (1952).
57A. Bosak, J. Serrano, M. Krisch, K. Watanabe, T. Taniguchi, and H. Kanda,‘‘Elasticity of Hexagonal Boron Nitride: Inelastic X-Ray Scattering Measure-ments,’’ Phys. Rev. B, 73 (4) 41402 (2006).
58R. Vogelgesang, M. Grimsditch, and J. S. Wallace, ‘‘The Elastic Constants ofSingle Crystal b-Si3N4,’’ Appl. Phys. Lett., 76 [8] 982–4 (2000).
59K. Kkanitani, M. Grimsditch, J. C. Nipko, C. K. Loong, M. Okada, andI. Kimura, ‘‘The Elastic Constants of Silicon Carbide: A Brillouin-ScatteringStudy of 4H and 6H SiC Single Crystal,’’ J. Appl. Phys., 82, 3152–4 (1997).
60J. Y. Kim, R. R. Fenton, B. A. Hunter, and B. J. Kennedy, ‘‘Powder Diffrac-tion Studies of Synthetic Calcium and Lead Apatites,’’ Aust. J. Chem., 53, 679–86(2000).
61M. C. Sha, Z. Li, and R. C. Bradt, ‘‘Single-Crystal Elastic Constants of Fluo-rapatite,’’ J. Appl. Phys., 75 [12] 7784–7 (1994).
62J. L. Katz and K. Ukraincik, ‘‘On the Anisotropic Elastic Properties ofHydroxyapatite,’’ J. Biomech., 4, 221–7 (1971).
63D. J. Weidner, J. D. Bass, A. E. Ringwood, and W. Sinclair, ‘‘The SingleCrystal Elastic Moduli of Stishovite,’’ J. Geophys. Res., 87, 4740 (1982).
64S. C. Abrahams and J. L. Bernstein, ‘‘Rutile: Normal Probability Plot Anlysisand Accurate Measurement of Crystal Structure,’’ J. Chem. Phys., 55, 3206–11(1971).
65C. J. Howard, T. M. Sabine, and F. Dickson, ‘‘Structural and Thermal Pa-rameters for Rutile and Anatase,’’ Acta Crystallogr. B, 47, 462–8 (1991).
66V. Swamy, J. D. Gale, and L. S. Dubrovinsky, ‘‘Atomistic Simulation of theCrystal Structures and Bulk Moduli of TiO2 Polymorphs,’’ J. Phys. Chem. Solids,62, 887 (2001).
67R. M. Hazen and L. W. Finger, ‘‘Bulk Moduli and High-Pressure CrystalStructures of Rutile-Type Compounds,’’ J. Phys. Chem. Solids, 42, 143–51 (1981).
68E. Chang and E. K. Graham, ‘‘The Elastic Constants of Cassiterite SnO2 andtheir Pressure and Temperature Dependence,’’ J. Geophys. Res., 80, 2595 (1975).
69Z. Mursic, T. Vogt, H. Boysen, and F. Frey, ‘‘Single-Crystal Neutron Diffrac-tion study of Metamict Zircon up to 2000 K,’’ J. Appl. Crystallogr., 25, 519–23(1992).
70H. Ozkan and J. C. Jamieson, ‘‘Pressure Dependence of the Elastic Constantsof Nonmetamict Zircon,’’ Phys. Chem. Miner., 2, 215 (1978).
71Y. Kudoh, E. Ito, and H. Takeda, ‘‘Effect of Pressure on the Crystal Structureof Perovskite-Type MgSiO3,’’ Phys. Chem. Miner., 14, 350–4 (1987).
72A. Yeganeh-Haeri, ‘‘Synthesis and Re-Investigation of the Elastic Propertiesof Single-Crystal Magnesium Silicate Perovskite,’’ Phys. Earth Planet. Inter., 87,111–2 (1994).
73M. Murakami and K. Hirose, ‘‘Post-Perovskite Phase Transition in MgSiO3,’’Science, 304, 855–8 (2004).
74T. Tsuchiya, J. Tsuchiya, K. Umemoto, and R. M. Wentzcovitch, ‘‘Elasticityof Post-Perovskite MgSiO3,’’ Geophys. Res. Lett., 31 [1–4] L14603 (2004).
75K. Fujino, S. Sasaki, Y. Takeuchi, and R. Sadanaga, ‘‘X-ray Determination ofElectron Distributions in Forsterite, Fayalite and Tephroite,’’ Acta Crystallogr. B,37, 513–8 (1981).
76I. Suzuki, O. L. Ander, and Y. Sumino, ‘‘Elastic Properties of a Single-CrystalForsterite Mg2SiO4 up to 1200 K,’’ Phys. Chem. Miner., 10 [1] 38–46 (1983).
77H. Horiuchi and H. Sawamoto, ‘‘p-Mg2SiO4: Single-Crystal X-Ray Diffrac-tion Study,’’ Am. Miner., 66, 568–75 (1981).
78H. Sawamoto, D. J. Weidner, S. Sasaki, and M. Kumazawa, ‘‘Single CrystalElastic Properties of the Modified Spinel (Beta) Phase of Magnesium Orthosili-cate,’’ Science, 224, 749–51 (1984).
79E. P. Meagaher and G. A. Lager, ‘‘Polyhedral Thermal Expansion in the TiO2
Polymorphs: Refinement of the Crystal Structures of Rutile,’’ Can. Mineral., 17,77–85 (1979).
80I. E. Grey, C. Li, I. C. Madsen, and G. Braunshausen, ‘‘TiO sub 2 –II. Am-bient Pressure Preparation and Structure Refinement,’’ Mater. Res. Bull., 23 [5]743–5 (1988).
81M. Akaogi, K. Kusaba, J. Susaki, and T. Yagi, ‘‘High-Pressure High-Temperature Stability of a-PbO, 2-Type TiO2 and MgSiO3 Majorite: Calorimetricand In Situ X-Ray Diffraction Studies’’; pp. 447–455 in High-Pressure Research:Application to Earth and Planetary Sciences (Geophysical Monograph), Edited byY. Syono and M. H. Manghnani. Amer Geophysical Union, Washington, DC,1992.
82J. Sjoberg, G. Helgesson, and I. Idrestedt, ‘‘Refinement of the Structure ofSi2N2O,’’ Acta Cryst. C, 47, 2438–41 (1991).
83P. Boch and J. C. Glandus, ‘‘Elastic Properties of Silicon Oxynitride,’’J. Mater. Sci., 14, 379–85 (1979).
84R. L. Ralph, L. W. Finger, and R. M. Hazen, ‘‘Compressibility and CrystalStructure of Andalusite at High Pressures 25 kbar Step-Scan,’’ Am. Mineral., 69,513–9 (1984).
85H. Yang, R. M. Hazen, L. W. Finger, C. T. Prewitt, and R. T. Downs,‘‘Compressibility and Crystal Structure of Sillimanite Al2SiO5, at High Pressure,’’Phys. Chem. Miner., 25, 39–47 (1997).
86R. K. Verma, ‘‘Elasticity of Some High Density Crystals,’’ J. Geophys. Res.,65, 757–66 (1960).
87H. Bross, ‘‘LDA and GGA Investigations of Some Ground State Properties ofAluminium With The All Electron MAPW Method,’’ Eur. Phys. J. B–Condens.Matter Complex Syst., 37 (2) 405–11 (2003).
88C. Kocer, N. Hirosake, and S. Ogata, ‘‘Ab Initio Calculation of the IdealTensile and Shear Strength of Cubic Silicon Nitride,’’ Phys. Rev. B, 67, 35210(2003).
89V. Kanchana, G. Vaitheeswaran, A. Svane, and A. Delin, ‘‘First-PrinciplesStudy of Elastic Properties of CeO2, ThO2 and PoO2,’’ J. Phys.: Condens. Matter,18, 9615–24 (2006).
90J. S. Tse, D. D. Klug, K. Uehara, and Z. Q. Li, ‘‘Elastic Properties of PotentialSuperhard Phases of RuO2,’’ Phys. Rev. B, 61, 10029–34 (2000).
October 2007 Ab initio Calculation of Elastic Constants of Ceramic Crystals 3203

91J. P. Perdew and M. Ernzerhof, Electronic Density Functional Theory: RecentProgress and New Directions. Plenum Press, New York, 1998.
92A. Zupan, P. Blaha, K. Schwarz, and J. P. Perdew, ‘‘Pressure-Induced PhaseTransitions in Solid Si, SiO2, and Fe: Performance of Local-Spin-Density andGeneralized-Gradient-Approximation Density Functionals,’’ Phys. Rev. B, 58,11266 (1998).
93V. N. Staroverov, G. E. Scuseria, J. Tao, and J. P. Perdew, ‘‘Tests of a Ladder ofDensity Functionals for Bulk Solids and Surfaces,’’ Phys. Rev. B, 69, 075102 (2004).
94P. Ravindran, L. Fast, P. A. Korzhavyi, B. Johansson, J. Wills, and O. Eriks-son, ‘‘Density Functional Theory for Calculation of Elastic Properties of Ortho-rhombic Crystals: Application to TiSi2,’’ J. Appl. Phys., 84, 4891 (1998).
95H. Kimizuka, H. Kaburaki, and Y. Kogure, ‘‘Molecular-Dynamics Study ofthe High-Temperature Elasticity of Quartz above the a-b Phase Transition,’’ Phys.Rev. B, 67, 024105 (2003).
96J. Purton, R. Jones, C. R. A. Catlow, and M. Lesie, ‘‘Ab Initio Potentials forthe Calculation of the Dynamical and Elastic Properties of a-Quartz,’’ Phys.Chem. Miner., 19, 392–400 (1993).
97B. Holm and R. Ahuja, ‘‘Ab Initio Calculation of Elastic Constants of SiO2
Stishovite and a-Quartz,’’ J. Chem. Phys., 111 [5] 271 (1999).98R. Bechmann, ‘‘Elastic and Piezoelectric Constants of Alpha-Quartz,’’ Phys.
Rev., 110, 1060–1 (1958).99W. P. Mason, ‘‘Piezoelectric Crystals and their Applications to ul-trasonics,’’
Van Nostrand, New York, 84, 508 (1950).100E. Gregoryanz, R. J. Hemley, H.-K. Mao, and P. Gillet, ‘‘High-Pressure
Elasticity of a-Quartz: Instability and Ferroelastic Transition,’’ Phys. Rev. Lett.,84, 3117–20 (2000). &
3204 Journal of the American Ceramic Society—Yao et al. Vol. 90, No. 10
Copyright © 2022 FDOKUMEN




















