
A similar impairment in CA3 mossy fibre LTP in the R6/2 mouse model of Huntington's disease and in...
12
A similar impairment in CA3 mossy fibre LTP in the R6 ⁄ 2 mouse model of Huntington’s disease and in the complexin II knockout mouse Helen E. Gibson, 2 Kerstin Reim, 3 Nils Brose, 3 A. Jennifer Morton 2 and Susan Jones 1 1 Department of Anatomy, University of Cambridge, Downing Street, Cambridge CB2 3DY, UK 2 Department of Pharmacology, University of Cambridge, Cambridge, UK 3 Max-Planck-Institut fu ¨ r Experimentelle Medizin, Go ¨ ttingen, Germany Keywords: associational ⁄ commissural LTP, hippocampus, spatial learning, synaptic vesicle release Abstract Complexin II is reduced in Huntington’s disease (HD) patients and in the R6 ⁄ 2 mouse model of HD. Mice lacking complexin II (Cplx2 – ⁄ – mice) show selective cognitive deficits that reflect those seen in R6 ⁄ 2 mice. To determine whether or not there is a common mechanism that might underlie the cognitive deficits, long-term potentiation (LTP) was examined in the CA3 region of hippocampal slices from R6 ⁄ 2 mice and Cplx2 – ⁄ – mice. While associational ⁄ commissural (A ⁄ C) LTP was not significantly different, mossy fibre (MF) LTP was significantly reduced in slices from R6 ⁄ 2 mice and Cplx2 – ⁄ – mice compared with wild-type (WT) and Cplx2 + ⁄ + control mice. MF field excitatory postsynaptic potentials (fEPSPs) in response to paired stimuli were not significantly different between control mice and R6 ⁄ 2 or Cplx2 – ⁄ – mice, suggesting that MF basal glutamate release is unaffected. Forskolin (30 lm) caused an increase in glutamate release at MF synapses in slices from R6 ⁄ 2 mice and from Cplx2 – ⁄ – mice that was not significantly different from that seen in control mice, indicating that the capacity for increased glutamate release is not diminished. Thus, R6 ⁄ 2 mice and Cplx2 – ⁄ – mice have a common selective impairment of MF LTP in the CA3 region. Together, these data suggest that complexin II is required for MF LTP, and that depletion of complexin II causes a selective impairment in MF LTP in the CA3 region. This impairment in MF LTP could contribute to spatial learning deficits observed in R6 ⁄ 2 and Cplx2 – ⁄ – mice. Introduction Huntington’s disease (HD) is a genetic disorder characterized by motor, cognitive and emotional deficits. R6 ⁄ 2 mice are transgenic for exon 1 of the human HD gene, carrying an expanded CAG repeat (Mangiarini et al., 1996), and show progressive motor and cognitive impairments (Carter et al., 1999; Lione et al., 1999). Increasing evidence suggests that a progressive decline in neuronal function precedes the overt symptoms and neuronal death in HD patients and in R6 ⁄ 2 mice (Lione et al., 1999; Li et al., 2003). R6 ⁄ 2 mice have spatial learning deficits in the Morris water maze (Lione et al., 1999). Spatial learning is hippocampal-dependent (Burgess et al., 2002) and synaptic plasticity, such as long-term potentiation (LTP), is a proposed neural substrate of learning and memory in the hippocampus (Malenka, 2003). Impaired synaptic plasticity is observed in the CA1 and dentate gyrus regions of the R6 ⁄ 2 mouse hippocampus (Murphy et al., 2000). Thus, a decline in hippocampal synaptic function might contribute to cognitive deficits in R6 ⁄ 2 mice. In the CA3 region of the hippocampus, two main forms of LTP are observed (Zalutsky & Nicoll, 1990; Nicoll & Malenka, 1995; Henze et al., 2000). Associational ⁄ commissural (A ⁄ C) inputs form synapses with CA3 pyramidal cells that exhibit N-methyl-d-aspartate (NMDA) receptor-dependent LTP (Zalutsky & Nicoll, 1990). Conversely, mossy fibre (MF) synapses with CA3 pyramidal cells, characterized by a large paired-pulse facilitation (Salin et al., 1996) and inhibition by Group II metabotropic glutamate receptor agonists (Manzoni et al., 1995), exhibit NMDA receptor-independent LTP (Zalutsky & Nicoll, 1990). MF LTP involves an increase in glutamate release (Weisskopf & Nicoll, 1995) and is impaired in mice lacking the synaptic vesicle protein Rab3A (Castillo et al., 1997), and in mice lacking the primary active zone constituent RIM1a (Castillo et al., 2002). Thus, MF LTP requires the interaction of proteins at the interface of synaptic vesicles and the active zone. Complexin II is a presynaptic protein that modulates neurotrans- mitter release (Reim et al., 2001; Rizo & Su ¨dhof, 2002). It is expressed in all regions of the hippocampus (Ishizuka et al., 1999; Freeman & Morton, 2004a). Complexin II is depleted in the brains of HD patients and R6 ⁄ 2 mice (Morton et al., 2001; Morton & Edwardson, 2001; Freeman & Morton, 2004b). Notably, although they do not show overt neurological symptoms, mice lacking the complexin II gene (Cplx2 – ⁄ – mice) show cognitive deficits in the Morris water maze that resemble those seen in R6 ⁄ 2 mice (Glynn et al., 2003). Cplx2 – ⁄ – mice have impaired potentiation of population spikes in the hippocampus, suggesting that synaptic plasticity might be dysfunctional (Takahashi et al., 1999; Huang et al., 2000). We hypothesized that complexin II is selectively required for presynaptic MF LTP, and that depletion of complexin II would lead to impaired MF LTP in R6 ⁄ 2 mice. To test this, we measured LTP in the CA3 region of hippocampal slices from R6 ⁄ 2 mice and Cplx2 – ⁄ – mice, and found that MF LTP is selectively impaired. Correspondence: Dr Susan Jones, as above. E-mail: [email protected] Received 20 May 2005, revised 3 August 2005, accepted 4 August 2005 European Journal of Neuroscience, Vol. 22, pp. 1701–1712, 2005 ª Federation of European Neuroscience Societies doi:10.1111/j.1460-9568.2005.04349.x
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of A similar impairment in CA3 mossy fibre LTP in the R6/2 mouse model of Huntington's disease and in...

A similar impairment in CA3 mossy fibre LTP in the R6 ⁄2mouse model of Huntington’s disease and in thecomplexin II knockout mouse
Helen E. Gibson,2 Kerstin Reim,3 Nils Brose,3 A. Jennifer Morton2 and Susan Jones11Department of Anatomy, University of Cambridge, Downing Street, Cambridge CB2 3DY, UK2Department of Pharmacology, University of Cambridge, Cambridge, UK3Max-Planck-Institut fur Experimentelle Medizin, Gottingen, Germany
Keywords: associational ⁄ commissural LTP, hippocampus, spatial learning, synaptic vesicle release
Abstract
Complexin II is reduced in Huntington’s disease (HD) patients and in the R6 ⁄ 2 mouse model of HD. Mice lacking complexin II(Cplx2– ⁄ – mice) show selective cognitive deficits that reflect those seen in R6 ⁄ 2 mice. To determine whether or not there is acommon mechanism that might underlie the cognitive deficits, long-term potentiation (LTP) was examined in the CA3 region ofhippocampal slices from R6 ⁄ 2 mice and Cplx2– ⁄ – mice. While associational ⁄ commissural (A ⁄C) LTP was not significantly different,mossy fibre (MF) LTP was significantly reduced in slices from R6 ⁄ 2 mice and Cplx2– ⁄ – mice compared with wild-type (WT) andCplx2+ ⁄ + control mice. MF field excitatory postsynaptic potentials (fEPSPs) in response to paired stimuli were not significantlydifferent between control mice and R6 ⁄ 2 or Cplx2– ⁄ – mice, suggesting that MF basal glutamate release is unaffected. Forskolin(30 lm) caused an increase in glutamate release at MF synapses in slices from R6 ⁄ 2 mice and from Cplx2– ⁄ – mice that was notsignificantly different from that seen in control mice, indicating that the capacity for increased glutamate release is not diminished.Thus, R6 ⁄ 2 mice and Cplx2– ⁄ – mice have a common selective impairment of MF LTP in the CA3 region. Together, these datasuggest that complexin II is required for MF LTP, and that depletion of complexin II causes a selective impairment in MF LTP in theCA3 region. This impairment in MF LTP could contribute to spatial learning deficits observed in R6 ⁄ 2 and Cplx2– ⁄ – mice.
Introduction
Huntington’s disease (HD) is a genetic disorder characterized bymotor, cognitive and emotional deficits. R6 ⁄ 2 mice are transgenic forexon 1 of the human HD gene, carrying an expanded CAG repeat(Mangiarini et al., 1996), and show progressive motor and cognitiveimpairments (Carter et al., 1999; Lione et al., 1999). Increasingevidence suggests that a progressive decline in neuronal functionprecedes the overt symptoms and neuronal death in HD patients and inR6 ⁄ 2 mice (Lione et al., 1999; Li et al., 2003).
R6 ⁄ 2 mice have spatial learning deficits in the Morris water maze(Lione et al., 1999). Spatial learning is hippocampal-dependent(Burgess et al., 2002) and synaptic plasticity, such as long-termpotentiation (LTP), is a proposed neural substrate of learning andmemory in the hippocampus (Malenka, 2003). Impaired synapticplasticity is observed in the CA1 and dentate gyrus regions of theR6 ⁄ 2 mouse hippocampus (Murphy et al., 2000). Thus, a decline inhippocampal synaptic function might contribute to cognitive deficits inR6 ⁄ 2 mice.
In the CA3 region of the hippocampus, two main forms of LTP areobserved (Zalutsky & Nicoll, 1990; Nicoll & Malenka, 1995; Henzeet al., 2000). Associational ⁄ commissural (A ⁄C) inputs form synapseswith CA3 pyramidal cells that exhibit N-methyl-d-aspartate (NMDA)receptor-dependent LTP (Zalutsky & Nicoll, 1990). Conversely, mossy
fibre (MF) synapses with CA3 pyramidal cells, characterized by a largepaired-pulse facilitation (Salin et al., 1996) and inhibition by Group IImetabotropic glutamate receptor agonists (Manzoni et al., 1995), exhibitNMDA receptor-independent LTP (Zalutsky & Nicoll, 1990). MF LTPinvolves an increase in glutamate release (Weisskopf & Nicoll, 1995)and is impaired in mice lacking the synaptic vesicle protein Rab3A(Castillo et al., 1997), and in mice lacking the primary active zoneconstituent RIM1a (Castillo et al., 2002). Thus, MF LTP requires theinteraction of proteins at the interface of synaptic vesicles and the activezone.Complexin II is a presynaptic protein that modulates neurotrans-
mitter release (Reim et al., 2001; Rizo & Sudhof, 2002). It isexpressed in all regions of the hippocampus (Ishizuka et al., 1999;Freeman & Morton, 2004a). Complexin II is depleted in the brains ofHD patients and R6 ⁄ 2 mice (Morton et al., 2001; Morton &Edwardson, 2001; Freeman & Morton, 2004b). Notably, although theydo not show overt neurological symptoms, mice lacking thecomplexin II gene (Cplx2– ⁄ – mice) show cognitive deficits in theMorris water maze that resemble those seen in R6 ⁄ 2 mice (Glynnet al., 2003). Cplx2– ⁄ – mice have impaired potentiation of populationspikes in the hippocampus, suggesting that synaptic plasticity mightbe dysfunctional (Takahashi et al., 1999; Huang et al., 2000). Wehypothesized that complexin II is selectively required for presynapticMF LTP, and that depletion of complexin II would lead to impairedMF LTP in R6 ⁄ 2 mice. To test this, we measured LTP in the CA3region of hippocampal slices from R6 ⁄ 2 mice and Cplx2– ⁄ – mice, andfound that MF LTP is selectively impaired.
Correspondence: Dr Susan Jones, as above.E-mail: [email protected]
Received 20 May 2005, revised 3 August 2005, accepted 4 August 2005
European Journal of Neuroscience, Vol. 22, pp. 1701–1712, 2005 ª Federation of European Neuroscience Societies
doi:10.1111/j.1460-9568.2005.04349.x

Materials and methods
Animals (R6 ⁄ 2 and Cplx2 mice)
R6 ⁄ 2 transgenic mice were taken from a colony established in theDepartment of Pharmacology,University of Cambridge, andmaintainedby backcrossing R6 ⁄ 2 males to CBA · C57BL ⁄ 6 F1 females. Geno-typing was confirmed by PCR (Mangiarini et al., 1996). Repeat lengthsof the mutation carried by R6 ⁄ 2 mice were 243 ± 3 (range 214–278;n ¼ 26 mice). This reflects a progressive expansion in the repeat length(from 145 CAGs in the founder mouse) which is typical in colonies thatbreed through the male line. We have seen neither deterioration in thebehavioural phenotype nor a change in neuropathology associated withthis expansion (A. J. Morton, unpublished data), although this clearlywarrants systematic investigation. Mice with deletion mutations in themurine Cplx2 gene (Cplx2– ⁄ –) and their respective wild-type (WT)controls (Cplx2+ ⁄ +) were generated as described by Reim et al. (2001).Cplx2mice used in this studywere taken fromcolonies established in theDepartment of Pharmacology, University of Cambridge. The founderbreeders (Cplx2+ ⁄ + andCplx2– ⁄ –) used to start the colonywere obtainedfrom the Max-Planck-Institute for Experimental Medicine, Gottingen.Experimentswere carried out using 48 slices from 42WTmice, 28 slicesfrom 26 R6 ⁄ 2mice, 35 slices from 24Cplx2– ⁄ –mice and 27 slices from21Cplx2+ ⁄ +mice. All experiments were carried out in accordance withthe UK Animals (Scientific Procedures) Act 1986 and with local ethicalapproval.
Western blotting
For Western blot analyses of hippocampi from littermate Cplx2+ ⁄ + andCplx2– ⁄ –mice (aged 8 weeks). Animals were killed by cervical disloca-tion, and pairs of hippocampi from individual mice were dissected andhomogenized in phosphate-buffered saline. Hippocampal homogenates(20 lg per lane) were separated by SDS-PAGE (Laemmli, 1970), andseparated proteins were transferred to nitrocellulose membranes(Towbin et al., 1979). Blots were probed with antibodies to Munc13-1(Rhee et al., 2002), RIM (BD Transduction Laboratories, Heidelberg,Germany), NMDAR1 (Brose et al., 1994), complexin I ⁄ II (SynapticSystems,Gottingen, Germany) andRab3 (Synaptic Systems,Gottingen,Germany). Immunoreactive proteins were visualized with enhancedchemiluminescence (Amersham Biosciences, Freiburg, Germany).
Hippocampal slice preparation
Slices were prepared from male WT, R6 ⁄ 2, Cplx2+ ⁄ + and Cplx2– ⁄ –
mice aged between 4 and 19 weeks. Mice were killed by cervicaldislocation and the brains were rapidly removed into ice-cold sucrose-based Ringer of the following composition (in mm): sucrose, 75;NaCl, 70; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; CaCl2, 0.5; MgCl2,14; and d-glucose, 10. Horizontal slices (400 lm) were prepared(DTK-1000 microslicer, Dosaka, Japan) and held after cutting in asubmersion chamber containing sucrose–Ringer at 30 �C and satur-ated with 95% O2 and 5% CO2.
Electrophysiological recordings
Slices were transferred to a recording chamber 1–6 h after preparation,suspended between two nylon nets and continuously perfused at 2–3 mL ⁄min with artificial cerebrospinal fluid (ACSF) of the followingcomposition (in mm): NaCl, 119; KCl, 2.5; NaH2PO4, 1.0; NaHCO3,26; CaCl2, 2.5; MgCl2, 1.3; d-glucose, 10; and the GABAA-receptorantagonist picrotoxin, 0.05; at 26–28 �C, and saturated with 95% O2
and 5% CO2. A bipolar stainless steel electrode (Frederick Haer andCo., Maine, USA) was placed in the granule cell–hilus border in theupper blade of the dentate gyrus to stimulate glutamatergic afferents toCA3 pyramidal cells. Paired test stimuli (100 ls duration, 50 msapart) were applied at 10 s intervals at a range of intensities to providea stimulus–response curve, and an intensity that evoked � 50% of theinitial maximum field excitatory postsynaptic potentials (fEPSPs)response was selected for experiments (Wang et al., 2003) to provideconsistency between slices. High frequency stimulation (HFS; three1 s trains at 100 Hz, intertrain interval 20 s) was used to induce LTP.Glass microelectrodes (5–15 MW when filled with ACSF) were placedin stratum radiatum of CA3 to record associational ⁄ commissural(A ⁄C)-evoked fEPSPs and in stratum lucidum of CA3 to recordMF-evoked fEPSPs. MF fEPSPs were defined as having a largepaired-pulse ratio (P2 ⁄ P1), where P1 is the response to the first of thepaired stimuli and P2 is the response to the second stimulus, and thepaired-pulse ratio was dependent on the interstimulus interval (Salinet al., 1996). MF fEPSPs showed frequency-dependent facilitation(Salin et al., 1996) and were inhibited by the Group II metabotropicreceptor agonist (2S,2¢R,3¢R)-2-(2¢,3¢Dicarboxycyclopropyl)glycine(DCG-IV), 1–3 lm (Kamiya et al., 1996; Li et al., 2001). LTP ofMF fEPSPs was not blocked by D(–)-2-Amino-5-phosphonopentanoicacid (D-AP5), 50 lm (Nicoll & Malenka, 1995). A ⁄C fEPSPs hadsmall paired-pulse ratios, showed little frequency-dependent facilita-tion and were not inhibited by DCG-IV (1–3 lm), and LTP of A ⁄CfEPSPs was blocked by D-AP5 (50 lm). The fEPSPs were amplifiedusing an Axoprobe 1A amplifier (Axon instruments, CA, USA), low-pass filtered at 1–2 kHz and digitally sampled to a PC at 10–20 kHzusing a Micro1401 interface (Cambridge Electronic Design, Cam-bridge, UK).The initial slope of the fEPSP was measured using the computer
program Spike 2 (Version 4; Cambridge Electronic Design). Todetermine the change in fEPSP slope following HFS or drug addition,fEPSPs were normalized to control (pre-HFS or pre-drug) fEPSP slopevalues. The fEPSP slope values were analyzed using anovawith a posthoc Dunnett’s test (GraphPad Prism, Version 4, San Diego, CA, USA),and a significant increase (P < 0.05) in fEPSP slope in 10 min periodsfollowing HFS that persisted > 30 min post-HFS indicated that LTP hadbeen induced. The number of slices from control vs. R6 ⁄ 2 or Cplx2– ⁄ –
mice showing LTP was compared using Fisher’s exact test. The fEPSPslope values 30–50 min post-HFS were compared between slices fromcontrol and R6 ⁄ 2 or Cplx2– ⁄ – mice using the t-test (with Welch’scorrection if the variances between the groups were unequal). In order tomeasure the paired-pulse ratio (P2 ⁄ P1) before HFS, P1 and P2 werecalculated from the average of 300 fEPSPs recorded 5 min immediatelyprior toHFS. In order tomeasure the paired-pulse ratio followingHFSordrug addition, P1 and P2 were calculated from the average of 1200fEPSPs 30–50 min post-HFS or 300 fEPSPs 15–20 min post-drug. Tocalculate the effects of drug application (DCG-IV, forskolin), normal-ized fEPSPs were averaged in the final 5 min of drug application andcompared with fEPSPs prior to drug application. The n-values reportedrefer to the number of slices. All combined data are shown asmean ± SEM. A critical P-value of P < 0.05 was considered significantfor the statistical tests used throughout this study.
Materials
All standard laboratory salts were obtained from BDH LaboratorySupplies (Poole, UK). Drugs were obtained from Sigma-Aldrich(Dorset, UK), except for DCG-IV and forskolin, which were obtainedfrom Tocris Cookson (Bristol, UK).
1702 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

Results
Determination of mossy fibre vs. A ⁄C fEPSPs
When paired stimuli are given at short intervals, MF fEPSPs show alarge response (P2) to the second stimulus relative to the response (P1)to the first stimulus; this is known as paired-pulse facilitation. In slicesfrom WT mice, MF fEPSPs showed paired-pulse facilitation that wasdependent on the stimulus interval. Typical MF fEPSPs illustratingpaired-pulse facilitation at stimulus intervals of 20 ms and 50 ms, butnot 200 ms, are shown in Fig. 1A, and the data are summarized inFig. 1B. With an interval of 50 ms, MF fEPSPs had a paired-pulseratio (P2 ⁄ P1) of 2.10 ± 0.05 (n ¼ 26). A ⁄C fEPSPs showed signi-ficantly less paired-pulse facilitation than MF fEPSPs at all stimulusintervals, as illustrated in Fig. 1A and B. With a stimulus interval of50 ms, A ⁄C fEPSPs had a P2 ⁄ P1 of 1.10 ± 0.03 (n ¼ 11 P < 0.001).
When the frequency of stimulation of mossy fibres was increasedfrom 0.1 to 0.5 Hz, MF fEPSPs exhibited frequency-dependent
facilitation (Fig. 1C and D). By contrast, A ⁄C fEPSPs show littlefrequency-dependent facilitation. The Group II metabotropic glutam-ate receptor agonist DCG-IV (1–3 lm) inhibited MF fEPSPs, causingan inhibition of 89.4 ± 4.6% (n ¼ 5; Fig. 1C and D) with 3 lm DCG-IV. This concentration had no effect on A ⁄C fEPSPs (0.26 ± 2.1% ofbaseline, n ¼ 5).
Selective impairment in MF LTP in the CA3 region of R6 ⁄ 2 mice
The effect of HFS on both MF and A ⁄C fEPSPs was compared in theCA3 region of WT and R6 ⁄ 2 mice. HFS induced LTP of MF fEPSPsin slices from WT mice that was not blocked by D-AP5 (50 lm). Atypical example of LTP of MF fEPSPs in the presence of D-AP5 isshown in Fig. 2A and B. LTP of MF fEPSPs in slices from WT micewas 197.7 ± 18.2% (n ¼ 21; 21 ⁄ 21 slices had LTP; Fig. 2C and D),and MF LTP was not significantly different in the presence of D-AP5
Fig. 1. MF and A ⁄ C fEPSPs in the CA3 region of hippocampal slices from WT mice. (A) Top trace shows typical MF fEPSPs, showing paired-pulse facilitation,with a larger response to the second of the paired stimuli (P2) given at 20- and 50-ms intervals after the first stimulus (P1). Bottom trace shows typical A ⁄ C fEPSPs,showing little paired-pulse facilitation at all stimulus intervals. (B) Bar graph summarizes the paired-pulse ratio (P2 ⁄ P1) in MF fEPSPs compared with A ⁄ C fEPSPsat stimulus intervals of 20, 50 and 200 ms (***P < 0.001). (C) Graph shows MF fEPSPs and A ⁄ C fEPSPs from five experiments (mean ± SEM). Increasing thefrequency of stimulation from 0.1 to 0.5 Hz (indicated by the bar) caused a large facilitation of MF fEPSPs (279 ± 4.4%) but had little effect on A ⁄ C fEPSPs(116 ± 0.8%). The Group II mGluR agonist DCG-IV (1–3 lm) inhibited MF fEPSPs (89.4 ± 4.6% with 3 lm) but not A ⁄ C fEPSPs (0.26 ± 2.1% with 3 lm).(D) Traces from a single experiment show MF fEPSPs (top) and A ⁄ C fEPSPs (bottom) in response to (1) 0.1 Hz stimulation, (2) during 0.5 Hz stimulation, (3)after returning to 0.1 Hz stimulation and (4) during perfusion with 3 lm DCG-IV.
Impaired mossy fibre LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice 1703
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

(196.4 ± 2.5%, n ¼ 10; Fig. 2D). By contrast, MF LTP was impairedin slices from R6 ⁄ 2 mice. None of the slices from R6 ⁄ 2 mice showedsignificant LTP (significantly different from WT mice; Fisher’s exacttest, P < 0.001), and the magnitude of fEPSPs 30–50 min post-HFSwas significantly smaller than in slices from WT mice (86.2 ± 5.2%,n ¼ 17; Fig. 2C and D). MF LTP was impaired in slices from R6 ⁄ 2mice in the presence of D-AP5 (92.0 ± 4.4%, n ¼ 6) compared withMF LTP in WT slices in D-AP5 (Fig. 2D). No differences wereobserved in input–output curves in slices from WT and R6 ⁄ 2 mice,
indicating that the same number of active synapses could be recruited(Fig. 2E).To determine when the onset of the impairment of LTP of MF
fEPSPs occurs in R6 ⁄ 2 mice, the magnitude of LTP was compared inslices taken from mice in three age groups. Mice aged between 4 and7 weeks represented ‘presymptomatic’ mice, mice aged between 8and 11 weeks represented mice in an ‘early symptomatic’ age group,and mice aged 12–19 weeks represented mice that were ‘latesymptomatic’. MF LTP was significantly impaired in slices from all
Fig. 2. MF LTP was impaired in hippocampal slices from R6 ⁄ 2 mice. (A) Graph shows a typical example of HFS-induced (arrow) LTP of MF fEPSPs in a slicefrom a WT mouse in the presence of 50 lm D-AP5. In this experiment, P2 ⁄ P1 was 1.94 ± 0.02 prior to HFS, and the fEPSP slope 30–50 min post-HFS was199.4 ± 0.9% of baseline (% LTP). (B) Traces show MF fEPSPs (1) before and (2) after HFS. (C) Graph shows normalized MF fEPSPs (mean ± SEM) duringLTP experiments using 21 slices from 17 WT mice aged 4–19 weeks (d) and 17 slices from 16 R6 ⁄ 2 mice aged 4–19 weeks (s). (D) Left-hand bar graphsummarizes LTP (mean ± SEM) measured at 30–50 min post-HFS in slices from WT and R6 ⁄ 2 mice, in the absence or presence of D-AP5, as indicated on thegraph. Right-hand bar graph summarizes the number of slices with a significant increase in fEPSP slope 30–50 min post-HFS in slices from WT and R6 ⁄ 2 mice(***P < 0.001). (E) Graph shows normalized MF fEPSPs (mean ± SEM) in response to different stimulation intensities in six slices from five WT and six slicesfrom five R6 ⁄ 2 mice. There was no significant difference in the stimulus–response curves (two-way anova, P ¼ 0.21). (F) Bar graph shows that LTP(mean ± SEM) of MF fEPSPs was significantly reduced in R6 ⁄ 2 mice (open bars) when compared with WT mice (solid bars) at all age groups examined(**P < 0.01, ***P < 0.001).
1704 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

R6 ⁄ 2 mouse age groups, including the presymptomatic 4- to 7-week-old R6 ⁄ 2 mice (Fig. 2F).
HFS induced LTP of A ⁄C fEPSPs that was blocked by D-AP5.Typical examples of LTP of A ⁄C fEPSPs in the absence and presenceof D-AP5 are shown in Fig. 3A and B. In contrast to MF LTP, A ⁄CLTP was not significantly reduced in slices from R6 ⁄ 2 mice (aged 4–19 weeks; 132.0 ± 5.0%, n ¼ 6) when compared with that seen inslices from WT mice (124.0 ± 1.7%, n ¼ 6; Fig. 3C and D). Thesedata indicate that MF LTP, but not A ⁄C LTP, is impaired in the CA3region of hippocampal slices from R6 ⁄ 2 mice.
Selective impairment in MF LTP in the CA3 regionof Cplx2– ⁄ – mice
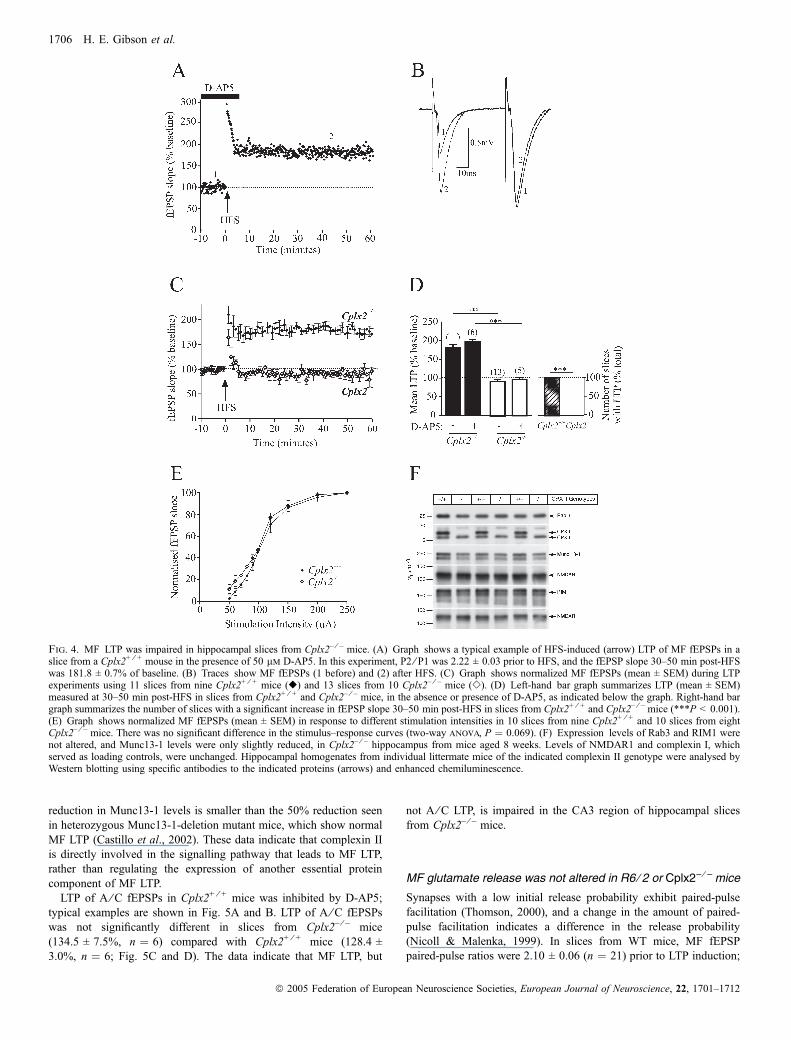
To determine whether or not the loss of complexin II might underliethe selective impairment of MF LTP, MF and A ⁄C LTP weremeasured in Cplx2+ ⁄ + mice and Cplx2– ⁄ – mice. A typical example ofLTP of MF fEPSPs in Cplx2+ ⁄ + mice is shown in Fig. 4A and B. Inslices from Cplx2+ ⁄ + mice, LTP of MF fEPSPs was 178.9 ± 6.9%(n ¼ 11, 11 ⁄ 11 slices had LTP; Fig. 4C and D), and MF LTP was not
significantly different in the presence of D-AP5 (193.3 ± 6.8%,n ¼ 6; Fig. 4D). By contrast, none of the slices from Cplx2– ⁄ – miceshowed LTP of MF fEPSPs (significantly different from Cplx2+ ⁄ +
mice; Fisher’s exact test, P < 0.001), and the magnitude of fEPSPs30–50 min post-HFS was significantly smaller than in Cplx2+ ⁄ + mice(91.3 ± 4.5%, n ¼ 13; Fig. 4C and D). MF LTP was impaired in slicesfrom Cplx2– ⁄ – mice in the presence of D-AP5 (96.1 ± 4.3%, n ¼ 5)compared with MF LTP in slices from Cplx2+ ⁄ + mice in D-AP5(Fig. 4D). No differences in input–output curves were observed,indicating that the same number of active synapses could be recruited(Fig. 4E).MF LTP is abolished not only in Cplx2– ⁄ – mice but also in mice
lacking Rab3A (Castillo et al., 1997) and RIM1a (Castillo et al.,2002). Thus, we next examined whether the impaired MF LTP inCplx2– ⁄ – mice is a secondary consequence of aberrant Rab3 or RIM1expression. For this purpose, we determined Rab3 and RIM1 proteinlevels in the hippocampi of Cplx2– ⁄ – mice and WT littermate controls(Fig. 4F). We found that neither Rab3 levels nor RIM1 levels werereduced in Cplx2– ⁄ – hippocampi. The levels of Munc13-1 tended to besomewhat lower in Cplx2– ⁄ – hippocampi. However, this apparent
Fig. 3. A ⁄ C LTP was not impaired in hippocampal slices from R6 ⁄ 2 mice. (A) The graph shows a typical example of HFS-induced (arrow) LTP of A ⁄ C fEPSPsin a slice from a WT mouse in the absence of 50 lm D-AP5 (n). In the control experiment, P2 ⁄ P1 was 1.14 ± 0.31 prior to HFS, and the fEPSP slope 30–50 minpost-HFS was 133.5 ± 0.5% of baseline.h, Typical example of HFS-induced (arrow) LTP of A ⁄ C fEPSPs in a slice from a WT mouse in the presence of 50 lm D-AP5. In this experiment, P2 ⁄ P1 was 1.11 ± 0.26 prior to HFS, and the fEPSP slope 30–50 min post-HFS was 100.7 ± 0.7% of baseline. (B) Top traces show A ⁄ CfEPSPs in control solutions (1) before and (2) after HFS. Bottom traces show A ⁄ C fEPSPs (1) before and (2) after HFS in the presence of D-AP5. (C) The graphshows HFS-induced (arrow) LTP of A ⁄ C fEPSPs in six slices from six WT mice (filled symbols) and six slices from five R6 ⁄ 2 mice. (D) Bar graph summarizesLTP (mean ± SEM) measured at 30–50 min post-HFS in WT mice in the absence of D-AP5, WT mice in the presence of D-AP5 (six slices from four WT mice), andR6 ⁄ 2 mice (***P < 0.001).
Impaired mossy fibre LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice 1705
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712
reduction in Munc13-1 levels is smaller than the 50% reduction seenin heterozygous Munc13-1-deletion mutant mice, which show normalMF LTP (Castillo et al., 2002). These data indicate that complexin IIis directly involved in the signalling pathway that leads to MF LTP,rather than regulating the expression of another essential proteincomponent of MF LTP.LTP of A ⁄C fEPSPs in Cplx2+ ⁄ + mice was inhibited by D-AP5;
typical examples are shown in Fig. 5A and B. LTP of A ⁄C fEPSPswas not significantly different in slices from Cplx2– ⁄ – mice(134.5 ± 7.5%, n ¼ 6) compared with Cplx2+ ⁄ + mice (128.4 ±3.0%, n ¼ 6; Fig. 5C and D). The data indicate that MF LTP, but
not A ⁄C LTP, is impaired in the CA3 region of hippocampal slicesfrom Cplx2– ⁄ – mice.
MF glutamate release was not altered in R6 ⁄ 2 or Cplx2– ⁄ – mice
Synapses with a low initial release probability exhibit paired-pulsefacilitation (Thomson, 2000), and a change in the amount of paired-pulse facilitation indicates a difference in the release probability(Nicoll & Malenka, 1999). In slices from WT mice, MF fEPSPpaired-pulse ratios were 2.10 ± 0.06 (n ¼ 21) prior to LTP induction;
Fig. 4. MF LTP was impaired in hippocampal slices from Cplx2– ⁄ – mice. (A) Graph shows a typical example of HFS-induced (arrow) LTP of MF fEPSPs in aslice from a Cplx2+ ⁄ + mouse in the presence of 50 lm D-AP5. In this experiment, P2 ⁄ P1 was 2.22 ± 0.03 prior to HFS, and the fEPSP slope 30–50 min post-HFSwas 181.8 ± 0.7% of baseline. (B) Traces show MF fEPSPs (1 before) and (2) after HFS. (C) Graph shows normalized MF fEPSPs (mean ± SEM) during LTPexperiments using 11 slices from nine Cplx2+ ⁄ + mice (r) and 13 slices from 10 Cplx2– ⁄ – mice (e). (D) Left-hand bar graph summarizes LTP (mean ± SEM)measured at 30–50 min post-HFS in slices from Cplx2+ ⁄ + and Cplx2– ⁄ – mice, in the absence or presence of D-AP5, as indicated below the graph. Right-hand bargraph summarizes the number of slices with a significant increase in fEPSP slope 30–50 min post-HFS in slices from Cplx2+ ⁄ + and Cplx2– ⁄ – mice (***P < 0.001).(E) Graph shows normalized MF fEPSPs (mean ± SEM) in response to different stimulation intensities in 10 slices from nine Cplx2+ ⁄ + and 10 slices from eightCplx2– ⁄ – mice. There was no significant difference in the stimulus–response curves (two-way anova, P ¼ 0.069). (F) Expression levels of Rab3 and RIM1 werenot altered, and Munc13-1 levels were only slightly reduced, in Cplx2– ⁄ – hippocampus from mice aged 8 weeks. Levels of NMDAR1 and complexin I, whichserved as loading controls, were unchanged. Hippocampal homogenates from individual littermate mice of the indicated complexin II genotype were analysed byWestern blotting using specific antibodies to the indicated proteins (arrows) and enhanced chemiluminescence.
1706 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

at 30–50 min following HFS, the paired-pulse ratio fell to 1.14 ± 0.04(n ¼ 21; Fig. 6A). This significant reduction in paired-pulse facilita-tion indicated an increase in the probability of release during LTP. Inslices from R6 ⁄ 2 mice, MF fEPSP paired-pulse ratios were1.94 ± 0.07 (n ¼ 17; Fig. 6A) prior to LTP induction, and were notsignificantly different from pre-HFS paired-pulse ratios measured inWT mice. However, at 30–50 min following HFS, the paired-pulseratio in slices from R6 ⁄ 2 mice was not reduced (2.05 ± 0.10, n ¼ 17),reflecting the absence of LTP and therefore no change in theprobability of release.
In slices from Cplx2+ ⁄ + mice, MF fEPSP paired-pulse ratios were2.16 ± 0.09 (n ¼ 11) prior to LTP induction; at 30–50 min followingHFS, the paired-pulse ratio fell to 1.07 ± 0.06 (n ¼ 11; Fig. 6B),a significant reduction in paired-pulse facilitation. In slices fromCplx2– ⁄ – mice, MF fEPSP paired-pulse ratios were 1.96 ± 0.05(n ¼ 13) prior to LTP induction, and were not significantly differentfrom pre-HFS paired-pulse ratios in Cplx2+ ⁄ + mice. As with slicesfrom R6 ⁄ 2 mice, the paired-pulse ratio in slices from Cplx2– ⁄ – mice at30–50 min following HFS was not reduced (2.06 ± 0.08, n ¼ 13;Fig. 6B), again reflecting the absence of LTP. These data indicate that
the probability of release at MF CA3 synapses is not altered either inR6 ⁄ 2 mice or in Cplx2– ⁄ – mice.Induction of LTP of MF fEPSPs involves activation of adenylyl
cyclase, and direct activation of adenylyl cyclase using forskolin causesan increase in glutamate release probability (Weisskopf et al., 1994). Theeffect of forskolin (30 lm) on MF fEPSPs was investigated in slicesfrom R6 ⁄ 2 and WT mice and Cplx2– ⁄ – and Cplx2+ ⁄ + mice. In theexample experiments illustrated in Fig. 6C and D,MF fEPSPs exhibitedfrequency-dependent facilitation and were sensitive to inhibition byDCG-IV. In these experiments, forskolin increased MF fEPSPs in allexperimental groups. Overall, forskolin increased MF fEPSPs to203.6 ± 7.0% (n ¼ 5) of baseline in WT mice and 180.7 ± 17.1%(n ¼ 5) in Cplx2+ ⁄ + mice. There was no difference in the effect offorskolin on MF fEPSPs in slices from R6 ⁄ 2 mice (212.6 ± 11.9%,n ¼ 5) compared with slices from WT mice (Fig. 6E) or in slices fromCplx2– ⁄ – mice (186.9 ± 18.8%, n ¼ 5) compared with slices fromCplx2+ ⁄ + mice (Fig. 6F). The percentage inhibition of MF fEPSPs byDCG-IV (3 lm) is given for each experimental group in Fig. 6E and F.These data indicate that MF synapses in R6 ⁄ 2 mice and Cplx2– ⁄ – miceretain the capacity to increase the probability of glutamate release.
Fig. 5. A ⁄ C LTP was not impaired in hippocampal slices from Cplx2– ⁄ – mice. (A) Graph shows a typical example of HFS-induced (arrow) LTP of A ⁄ C fEPSPsin a slice from a Cplx2+ ⁄ + mouse in the absence of 50 lm D-AP5 (m). In this experiment, P2 ⁄ P1 was 1.17 ± 0.02 prior to HFS, and the fEPSP slope 30–50 minpost-HFS was 130.0 ± 0.6% of baseline. n, typical example of HFS-induced (arrow) LTP of A ⁄ C fEPSPs in a slice from a Cplx2+ ⁄ + mouse in the presence of50 lm D-AP5. In this experiment, P2 ⁄ P1 was 1.16 ± 0.03 prior to HFS, and the fEPSP slope 30–50 min post-HFS was 98.7 ± 0.4% of baseline. (B) Top tracesshow A ⁄ C fEPSPs (1) before and (2) after HFS in control solutions. Bottom traces show A ⁄ C fEPSPs (1) before and (2) after HFS in the presence of D-AP5.(C) Graph shows HFS-induced (arrow) LTP of A ⁄ C fEPSPs in six slices from six Cplx2+ ⁄ + mice (m) and six slices from three Cplx2– ⁄ – mice (n). (D) Bar graphsummarizes LTP (mean ± SEM) measured at 30–50 min post-HFS in Cplx2+ ⁄ + mice and Cplx2– ⁄ – mice, in control solution or in the presence of D-AP5 (five slicesfrom four Cplx2+ ⁄ + mice), as indicated below the graph (***P < 0.001).
Impaired mossy fibre LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice 1707
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

DiscussionThis study revealed a selective impairment in MF LTP in R6 ⁄ 2 miceand in mice lacking the presynaptic protein complexin II in the CA3
region of the hippocampus. These data suggest that complexin II ispart of the molecular machinery necessary for MF LTP, and thatdepletion of this protein in R6 ⁄ 2 mice leads to the loss of MF LTP.
Fig. 6. Glutamate release from mossy fibres was not altered in R6 ⁄ 2 or Cplx2– ⁄ – mice. (A) Bar graph summarizes paired-pulse ratios (P2 ⁄ P1) pre-HFS and post-HFS, in slices from WT and R6 ⁄ 2 mice. In WT mice, HFS caused a significant reduction in the paired-pulse ratio (P < 0.001). In R6 ⁄ 2 mice, the paired-pulse ratioprior to HFS was not significantly different from that seen in WT mice, but HFS did not induce a significant reduction in paired-pulse ratio. (B) Bar graphsummarizes paired-pulse ratios (P2 ⁄ P1) pre-HFS and post-HFS, in slices from Cplx2+ ⁄ + and Cplx2– ⁄ – mice. In Cplx2+ ⁄ + mice, HFS caused a significant reductionin the paired-pulse ratio (P < 0.001). As with R6 ⁄ 2 mice, the paired-pulse ratio prior to HFS in slices from Cplx2– ⁄ – mice was not significantly different from thatseen in Cplx2+ ⁄ + mice, and HFS did not induce a significant reduction in paired-pulse ratio. (C) Graph shows a typical recording of MF fEPSPs (showingfrequency-dependent facilitation and sensitivity to DCG-IV) in which forskolin (30 lm) induced an overlapping increase in MF fEPSPs in a slice from a WT mouseand from an R6 ⁄ 2 mouse. (D) Graph shows a typical recording of MF fEPSPs (showing frequency-dependent facilitation and sensitivity to DCG-IV) in whichforskolin (30 lm) induced an overlapping increase in MF fEPSPs in a slice from a Cplx2+ ⁄ + mouse and from a Cplx2– ⁄ – mouse. (E) Left-hand bar graph showsthat forskolin (30 lm) induced an increase in MF fEPSPs that was not significantly different in slices from WT mice (n ¼ 5 slices from five mice) and R6 ⁄ 2 mice(n ¼ 5 slices from five mice). Right-hand bar graph summarizes the percentage inhibition observed with 3 lm DCG-IV in these experiments. (F) Left-hand bargraph shows that forskolin (30 lm) induced an increase in MF fEPSPs that was not significantly different in slices from Cplx2+ ⁄ + mice (n ¼ 5 slices from five mice)and Cplx2– ⁄ – mice (n ¼ 5 slices from five mice). Right-hand bar graph summarizes the percentage inhibition observed with 3 lm DCG-IV in these experiments.
1708 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

R6 ⁄ 2 mice have impaired MF LTP
Two forms of CA3 LTP have been described previously (Zalutsky &Nicoll, 1990). We have distinguished the two forms of LTP usingseveral criteria. Firstly, MF fEPSPs exhibited large paired-pulsefacilitation that was dependent on the stimulus interval, as describedby Salin et al. (1996). The paired-pulse ratio for our MF fEPSPsrecorded at 50 ms intervals from WT mice is slightly larger than thatreported by Salin et al. (1996), but accords well with the MF fEPSPpaired-pulse ratio reported by Wang et al. (2003). Secondly, MFfEPSPs, but not A ⁄C fEPSPs, were facilitated by 279% uponincreasing the frequency of stimulation from 0.1 to 0.5 Hz, similar tothe characteristic frequency-dependent facilitation of MF fEPSPsreported by Salin et al. (1996). Thirdly, MF fEPSPs were inhibited by89% by 3 lm DCG-IV whereas A ⁄C fEPSPs were unaffected (Liet al., 2001). Finally, the same amplitude of LTP of MF fEPSPs wasinduced in the presence of D-AP5 as in control conditions, consistentwith the fact that MF LTP is NMDA receptor-independent (Zalutsky& Nicoll, 1990). By contrast, A ⁄C LTP was completely blocked byD-AP5.
We show that MF LTP was impaired in slices from R6 ⁄ 2 micecompared with WT mice, whereas A ⁄C LTP was not affected. Thus,R6 ⁄ 2 mice have a selective deficit in MF LTP in the CA3 region.Impaired MF LTP was also observed in presymptomatic R6 ⁄ 2 mice.The fact that the deficit occurs before an overt behavioural phenotypeis observed (Mangiarini et al., 1996; Carter et al., 1999; Lione et al.,1999) supports the idea that neuronal dysfunction is an early hallmarkin HD mice.
The expression of MF LTP is believed to involve an increase inglutamate release (Weisskopf et al., 1994; Xiang et al., 1994;Weisskopf & Nicoll, 1995; Lopez-Garcıa et al., 1996; Tong et al.,1996), induced by either presynaptic (Castillo et al., 1994; Weisskopfet al., 1994; Weisskopf & Nicoll, 1995; Villacres et al., 1998) orpostsynaptic (Kapur et al., 1998; Yeckel et al., 1999) Ca2+-, adenylylcyclase- and protein kinase A (PKA)-dependent mechanisms. Targetsfor PKA are not resolved, but possibilities include the PKA substrateRIM1a (Castillo et al., 2002; Lonart et al., 2003). RIM1a interactswith Rab3A, which is also required for MF LTP (Castillo et al., 1997).
The HD gene encodes huntingtin, a cytosolic protein of unknownfunction (Huntington’s Disease Collaborative Research Group, 1993)that associates with synaptic vesicles and has binding partnersincluding synaptic proteins (DiFiglia et al., 1995; Li et al., 2003). Alarge number of proteins are dysregulated in HD and in R6 ⁄ 2 mice(Luthi-Carter et al., 2002; Morton et al., 2005). Complexin II is apresynaptic protein expressed throughout the brain, including thehippocampus (Ishizuka et al., 1999; Yamada et al., 1999; Eastwood &Harrison, 2000; Freeman & Morton, 2004a). Complexin II is dysreg-ulated in some neurons as early as 3 weeks of age in R6 ⁄ 2 mice(Freeman & Morton, 2004b), while other presynaptic proteinsmeasured, including a-SNAP and synaptophysin, are unaffected(Morton & Edwardson, 2001). Expression of mutant huntingtin inPC12 cells causes a selective reduction in complexin II expression anda concomitant decrease in neurotransmitter release (Edwardson et al.,2003). One explanation for the loss of MF LTP in R6 ⁄ 2 mice is thatearly changes in complexin II expression contribute to synapticdysfunction in R6 ⁄ 2 mice.
Cplx2– ⁄ – mice have impaired MF LTP
As in R6 ⁄ 2 mice, MF LTP was significantly impaired in slices fromCplx2– ⁄ – mice at all ages tested, whereas A ⁄C LTP was unaffected.Thus, in the CA3 hippocampus of Cplx2– ⁄ – mice, LTP is selectively
impaired at MF synapses. Complexin II is expressed throughout thehippocampus, and so we tested whether other presynaptic proteinsundergo compensatory changes in the hippocampus when complex-in II is removed. Given that the expression levels of Rab3 and RIM1are not altered and the Munc13-1 levels are only marginally reduced inCplx2– ⁄ – hippocampi, the impaired MF LTP in Cplx2– ⁄ – mice isunlikely to be due to changes in other components of the proteinmachinery that regulate MF LTP. Indeed, Munc13-1 levels can bereduced to 50% of WT levels without consequences for MF LTP(Castillo et al., 2002), and no changes in MF LTP were reported forheterozygous RIM1 and Rab3A deletion mutant hippocampi (Castilloet al., 1997 and 2002). Therefore, our data indicate that complexin IIis an integral part of the molecular machinery required for MF LTP.A previous study in Cplx2– ⁄ – mice reported a reduction in the
potentiation of population spikes in the CA3 region (Takahashi et al.,1999). Population spikes are generated by summation of all excitatorysynaptic inputs to CA3 pyramidal cell populations, including MFfEPSPs and A ⁄C fEPSPs. Our data are consistent with the findings ofTakahashi et al. (1999) because a reduction in the strength of MFsynaptic transmission (impaired MF LTP in the absence of complexinII), but not in A ⁄C synaptic transmission, might reduce the efficiencyof summation of EPSPs in generating the population spikes measuredin their recordings. Our novel demonstration of a role for complexin IIspecifically in LTP at CA3 mossy fibre synapses supports the idea thatMF LTP is expressed through presynaptic modifications dependentupon the synaptic vesicle machinery.
How is complexin II necessary for MF LTP?
Some evidence suggests that complexin II is a negative regulator ofneurotransmitter release (Ono et al., 1998; Itakura et al., 1999). In theabsence of complexin II, release probability might be increased to aceiling level that occludes MF LTP. However, in our experiments thedeficits in MF LTP are unlikely to be secondary to alterations in basalglutamate release for two reasons. Firstly, in slices from R6 ⁄ 2 andCplx2– ⁄ – mice, paired-pulse ratios were not significantly differentfrom control mice. Secondly, the effect of forskolin on MF fEPSPswas similar in R6 ⁄ 2 and Cplx2– ⁄ – mice and their respective controls.Thus, MF synapses in R6 ⁄ 2 and Cplx2– ⁄ – mice show an initial releaseprobability similar to control mice, and MF synapses in these mice cansupport an increase in glutamate release.Other studies suggest that complexin II is a positive regulator of
neurotransmitter release (Reim et al., 2001; Tokumaru et al., 2001;Edwardson et al., 2003). Synaptic vesicles docked at the active zonemust be primed for release by formation of ‘SNARE’ complexes(SNARE: soluble N-ethylmaleimide-sensitive factor attachment pro-tein receptor; Sudhof, 2004). Complexin II interacts with SNAREcomplexes and is thought to stabilize them for Ca2+-dependent fusion(McMahon et al., 1995; Chen et al., 2002; Rizo & Sudhof, 2002;Sudhof, 2004). In hippocampal cell cultures from mice lackingcomplexins I and II, the probability of fast synchronous neurotrans-mitter release and the Ca2+-sensitivity of release are both decreased(Reim et al., 2001). These data suggest that complexins regulate theefficiency of the release process at the level of the Ca2+-sensitivefusion event.We propose that, in MFs, complexin II is required to act as a
‘molecular switch’ to increase glutamate release during MF LTP. Themost likely site where complexin II could act would be at the level ofthe SNARE complex, stabilizing the complex in a tight conformationnecessary for fast synchronous neurotransmitter release, and regula-ting the action of the Ca2+ sensor so that the likelihood of Ca2+ entry
Impaired mossy fibre LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice 1709
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

being converted to a fusion event is increased (Marz & Hanson, 2002;Sudhof, 2004).One interesting question arising from this work is why synaptic
stimulation does not induce MF LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice butaddition of forskolin causes an increase in glutamate release probab-ility. Forskolin activates adenylyl cyclase, and is thought to initiate asignalling cascade identical to that initiated during synapticallyinduced MF LTP (Weisskopf et al., 1994). The observation thatforskolin can induce MF glutamate release in mice that have impairedMF LTP has also been reported in Rab3A knockout mice (Castilloet al., 1997) and in RIM1a knockout mice (Castillo et al., 2002). It ispossible that forskolin is a more powerful stimulus than synapticstimulation and can more effectively compensate for the lack ofimportant presynaptic proteins, including complexin II. Alternatively,forskolin may activate many isoforms of adenylyl cyclase, activating anumber of signalling pathways that converge on the release machin-ery, whereas synaptic stimulation may activate only a single pathwaythat requires complexin II. Indeed, two isoforms of adenylyl cyclase(Type 1 and Type 8) are known to be expressed in the CA3 region;both are activated by forskolin, but forskolin is still effective in theabsence of either isoform (Villacres et al., 1998; Wang et al., 2003).Finally, it is possible that the signalling mechanism normally activatedduring synaptic induction of MF LTP does not involve PKA but doesinvolve synaptic vesicle-associated proteins, including Rab3A, RIM1aand complexin II, and that forskolin activates a PKA signallingpathway that causes an increase in glutamate release that resembles,but is separate from, MF LTP. However, this seems unlikely becauseforskolin-induced glutamate release has been shown to occlude MFLTP (Weisskopf et al., 1994).A common, selective deficit in MF LTP was observed in R6 ⁄ 2 and
Cplx2– ⁄ – mice, suggesting that depletion of complexin II contributesto the loss of MF LTP in R6 ⁄ 2 mice. It is interesting that mossy fibreLTP is completely absent in R6 ⁄ 2 mice aged 4–7 weeks, the earliestage tested, despite the fact that R6 ⁄ 2 mice show a progressive declinein complexin II that mirrors the progression of behavioural symptoms(Morton & Edwardson, 2001). One explanation for this is that even a20% loss of complexin II, as seen in 3-week-old R6 ⁄ 2 mice (Morton& Edwardson, 2001), is sufficient to induce synaptic dysfunction butthat the behavioural consequences of this are masked by compensatorymechanisms. Furthermore, in R6 ⁄ 2 mice deficits in synaptic proteinsand synaptic plasticity are complex. By 8 weeks of age, a number ofproteins that might be important for LTP are dysregulated (Mortonet al., 2005). Thus, changes in synaptic function in R6 ⁄ 2 mice areunlikely to be explained by the loss of complexin II alone. However,given our Cplx2– ⁄ – mouse data, a plausible explanation for the earlyselective loss of CA3 MF LTP in R6 ⁄ 2 mice is the loss ofcomplexin II.
Significance of MF LTP in hippocampal function and dysfunction
Spatial learning depends on an intact hippocampus (Burgess et al.,2002; Lynch, 2004). R6 ⁄ 2 and Cplx2– ⁄ – mice show spatial learningdeficits from the earliest age tested (Lione et al., 1999; Glynn et al.,2003). Deficits in plasticity in the CA1 region have been reported forboth R6 ⁄ 2 mice (Murphy et al., 2000) and Cplx2– ⁄ – mice (Takahashiet al., 1999) that might contribute to their spatial learning deficits. Inthe CA3 region of the hippocampus, the sparse but powerful MF inputis thought to be an important ‘teaching signal’ in initiating learning(Rolls & Treves, 1994; Henze et al., 2000). Lesions of the CA3 regionin rats cause impairments in paired associative learning (Gilbert &Kesner, 2003) and disrupt spatial learning in the Morris water maze
(Handelmann & Olton, 1981; Stubley-Weatherly et al., 1996). Theselearning deficits, particularly in reversal learning, resemble those seenin Cplx2– ⁄ – and R6 ⁄ 2 mice.Mice lacking other presynaptic proteins and showing deficits in MF
LTP exhibit variable impairments in spatial learning, and so a role forMF LTP in learning is not clearly defined. Although genetic ablationof PKA impairs mossy fibre LTP it has no effect on spatial learning(Huang et al., 1995). However, RIM1a-knockout mice lack MF LTP(Castillo et al., 2002) and show impairments in spatial learning(Powell et al., 2004). Mice lacking Rab3A have been reported to showno difference (Hensbroek et al., 2003; Powell et al., 2004) or to showreversal deficits (D’Adamo et al., 2004) in spatial learning. Ourdemonstration of a common selective impairment in CA3 MF LTP intwo lines of mice that have similar cognitive deficits lends newsupport for a role for MF LTP in spatial learning. In rats, LTP ofpopulation spikes at MF synapses in CA3 coincides with learning inthe radial arm maze (Ishihara et al., 1997), a test of working spatialmemory. Together, these data support the idea that impaired MF LTP,as well as impaired CA1 plasticity, could contribute to spatial learningdeficits.In summary, we find that CA3 MF LTP is selectively impaired from
an early age in R6 ⁄ 2 mice and in Cplx2– ⁄ – mice. Loss of complexin IIalone does not affect basal neurotransmitter release, but complexin IIis necessary for the induction or expression of MF LTP. Furthermore,our data suggest that loss of complexin II in R6 ⁄ 2 mice couldcontribute to a decline in MF LTP even prior to the overt R6 ⁄ 2phenotype. This supports the idea that neuronal dysfunction precedesneuronal loss in HD.
Acknowledgements
We thank Dr J. A. Kauer and Dr J. R. Morgan for helpful comments on themanuscript. This work was supported by the Hereditary Disease FoundationUSA, High Q Foundation USA, the UK Medical Research Council (studentshipto H.E.G.), and the DFG Center for the Molecular Physiology of the Brain(N.B.).
Abbreviations
A ⁄ C, associational and ⁄ or commissural; Cplx2+ ⁄ + and Cplx2– ⁄ – mice, micewith and without complexin II; D-AP5, D(–)-2-amino-5-phosphonopentanoicacid; DCG-IV, (2S,2¢R,3¢R)-2-(2¢,3¢Dicarboxycyclopropyl)glycine; fEPSPs,field excitatory postsynaptic potentials; HD, Huntington’s disease; HFS, highfrequency stimulation; LTP, long-term potentiation; MF, mossy fibre; NMDA,N-methyl-d-aspartate; P1 and P2, responses to the first and second, respect-ively, of a pair of stimuli; P2 ⁄ P1, paired-pulse ratio; PKA, protein kinase A;WT, wild-type.
References
Brose, N., Huntley, G.W., Stern-Bach, Y., Sharma, G., Morrison, J.H. &Heinemann, S.F. (1994) Differential assembly of coexpressed glutamatereceptor subunits in neurons of rat cerebral cortex. J. Biol. Chem., 269,16780–16784.
Burgess, N., Maguire, E.A. & O’Keefe, J. (2002) The human hippocampus andspatial and episodic memory. Neuron, 35, 625–641.
Carter, R.J., Lione, L.A., Humby, T., Mangiarini, L., Mahal, A., Bates, G.P.,Dunnett, S.B. & Morton, A.J. (1999) Characterization of progressive motordeficits in mice transgenic for the human Huntington’s disease mutation.J. Neurosci., 19, 3248–3257.
Castillo, P.E., Janz, R., Sudhof, T.C., Tzounopoulos, T., Malenka, R.C. &Nicoll, R.A. (1997) Rab3A is essential for mossy fibre long-termpotentiation in the hippocampus. Nature, 388, 590–593.
Castillo, P.E., Schoch, S., Schmitz, F., Sudhof, T.C. & Malenka, R.C. (2002)RIM1a is required for presynaptic long-term potentiation. Nature, 415, 327–330.
1710 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

Castillo, P.E., Weisskopf, M.G. & Nicoll, R.A. (1994) The role of Ca2+
channels in hippocampal mossy fiber synaptic transmission and long-termpotentiation. Neuron, 12, 261–269.
Chen, X., Tomchick, D.R., Kovrigin, E., Arac, D., Machius, M., Sudhof, T.C.& Rizo, J. (2002) Three-dimensional structure of the complexin ⁄ SNAREcomplex. Neuron, 33, 397–409.
D’Adamo, P., Wolfer, D.P., Kopp, C., Tobler, I., Toniolo, D. & Lipp, H.-P.(2004) Mice deficient for the synaptic vesicle protein Rab3a show impairedspatial reversal learning and increased explorative activity but none of thebehavioral changes shown by mice deficient for the Rab3a regulator Gdi1.Eur. J. Neurosci., 19, 1895–1905.
DiFiglia, M., Sapp, E., Chase, K., Schwarz, C., Meloni, A., Young, C., Martin,E., Vonsattel, J.-P., Carraway, R., Reeves, S.A., Boyce, F.M. & Aronin, N.(1995) Huntingtin is a cytoplasmic protein associated with vesicles in humanand rat brain neurons. Neuron, 14, 1075–1081.
Eastwood, S.L. & Harrison, P.J. (2000) Hippocampal synaptic pathology inschizophrenia, bipolar disorder and major depression: a study of complexinmRNAs. Mol. Psychiatry, 5, 425–432.
Edwardson, J.M., Wang, C.-T., Gong, B., Wyttenbach, A., Bai, J., Jackson,M.B., Chapman, E.R. & Morton, A.J. (2003) Expression of mutanthuntingtin blocks exocytosis in PC12 cells by depletion of complexin II.J. Biol. Chem., 278, 30849–30853.
Freeman, W. & Morton, A.J. (2004a) Differential messenger RNA expressionof complexins in mouse brain. Brain Res. Bull., 63, 33–44.
Freeman, W. & Morton, A.J. (2004b) Regional and progressive changes inbrain expression of complexin II in a mouse transgenic for the Huntington’sDisease mutation. Brain Res. Bull., 63, 45–55.
Gilbert, P.E. & Kesner, R.P. (2003) Localization of function within the dorsalhippocampus: the role of the CA3 subregion in paired-associate learning.Behav. Neurosci., 117, 1385–1394.
Glynn, D., Bortnick, R.A. & Morton, A.J. (2003) Complexin II is essential fornormal neurological function in mice. Hum. Mol. Genet., 12, 1–18.
Handelmann, G.E. & Olton, D.S. (1981) Spatial memory following damage tohippocampal CA3 pyramidal cells with kainic acid: impairment and recoverywith preoperative training. Brain Res., 217, 41–58.
Hensbroek, R., Kamal, A., Baars, A., Verhage, M. & Spruijt, B. (2003)Spatial, contextual and working memory are not affected by the absence ofmossy fiber long-term potentiation and depression. Behav. Brain Res., 138,215–223.
Henze, D.A., Urban, N.N. & Barrionuevo, G. (2000) The multifarioushippocampal mossy fiber pathway: a review. Neuroscience, 98, 407–427.
Huang, Y.Y., Kandel, E.R., Varshavsky, L., Brandon, E.P., Qi, M., Idzerda,R.L., McKnight, G.S. & Bourtchouladze, R. (1995) A genetic test of theeffects of mutations in PKA on mossy fiber LTP and its relation to spatial andcontextual learning. Cell, 83, 1211–1222.
Huang, G.-Z., Ujihara, H., Takahashi, S.-i., Kaba, H., Yagi, T. & Inoue, S.(2000) Involvement of complexin II in synaptic plasticity in the CA1 regionof the hippocampus: the use of complexin II-lacking mice. Jpn.J. Pharmacol., 84, 179–187.
Huntington’s Disease Collaborative Research Group (1993) A novel genecontaining a trinucleotide repeat that is expanded and unstable onHuntington’s disease chromosomes. Cell, 72, 971–983.
Ishihara, K., Mitsuno, K., Ishikawa, M. & Sasa, M. (1997) Behavioral LTPduring learning in rat hippocampal CA3. Behav. Brain Res., 83, 235–238.
Ishizuka, T., Saisu, H., Odani, S., Kumanishi, T. & Abe, T. (1999) Distinctregional distribution in the brain of messenger RNAs for the two isoforms ofsynaphin associated with the docking ⁄ fusion complex. Neuroscience, 88,295–306.
Itakura, M., Misawa, H., Sekiguchi, M., Takahashi, S.-i. & Takahashi, M.(1999) Transfection analysis of functional roles of complexin I and II in theexocytosis of two different types of secretory vesicles. Biochem. Biophys.Res. Comm., 265, 691–696.
Kamiya, H., Shinozaki, H. & Yamamoto, C. (1996) Activation of metabotropicglutamate receptor type 2 ⁄ 3 suppresses transmission at rat hippocampalmossy fibre synapses. J. Physiol. (Lond.), 493, 447–455.
Kapur, A., Yeckel, M.F., Gray, R. & Johnston, D. (1998) L-type calciumchannels are required for one form of hippocampal mossy fiber LTP.J. Neurophysiol., 79, 2181–2190.
Laemmli U.K. (1970) Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature, 227, 680–685.
Li, Y., Hough, C.J., Frederickson, C.J. & Sarvey, J.M. (2001) Induction ofmossy fiber-CA3 long-term potentiation requires translocation of synapti-cally released Zn2+. J. Neurosci., 21, 8015–8025.
Li, J.-Y., Plomann, M. & Brundin, P. (2003) Huntington’s disease: asynaptopathy? Trends Mol. Med., 9, 414–420.
Lione, L.A., Carter, R.J., Hunt, M.J., Bates, G.P., Morton, A.J. & Dunnett, S.B.(1999) Selective discrimination learning impairments in mice expressing thehuman Huntington’s disease mutation. J. Neurosci., 19, 10428–10437.
Lonart, G., Schoch, S., Kaeser, P.S., Larkin, C.J., Sudhof, T.C. & Linden, D.J.(2003) Phosphorylation of RIM1a by PKA triggers presynaptic long-termpotentiation at cerebellar parallel fiber synapses. Cell, 115, 49–60.
Lopez-Garcıa, J.C., Arancio, O., Kandel, E.R. & Baranes, D. (1996) Apresynaptic locus for long-term potentiation of elementary synaptictransmission at mossy fiber synapses in culture. Proc. Natl Acad. Sci.USA, 93, 4712–4717.
Luthi-Carter, R., Hanson, S.A., Strand, A.D., Bergstroom, D.A., Chun, W.,Peters, N.L., Woods, A.M., Chan, E.Y., Kooperberg, C., Krainc, D., Young,A.B., Tapscott, S.J. & Olson, J.M. (2002) Dysregulation of gene expressionin the R6 ⁄ 2 model of polyglutamine disease: parallel changes in muscle andbrain. Hum. Mol. Genet., 11, 1911–1926.
Lynch, M.A. (2004) Long-term potentiation and memory. Physiol. Rev., 84,87–136.
Malenka, R.C. (2003) The long-term potential of LTP. Nat. Rev. Neurosci., 4,923–926.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hethering-ton, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W. & Bates, G.P.(1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient tocause a progressive neurological phenotype in transgenic mice. Cell, 87,493–506.
Manzoni, O.J., Castillo, P.E. & Nicoll, R.A. (1995) Pharmacology ofmetabotropic glutamate receptors at the mossy fiber synapses of the guineapig hippocampus. Neuropharmacol., 34, 965–971.
Marz, K.E. & Hanson, P.I. (2002) Sealed with a twist: complexin and thesynaptic SNARE complex. Trends Neurosci., 25, 381–383.
McMahon, H.T., Missler, M., Li, C. & Sudhof, T.C. (1995) Complexins:cytosolic proteins that regulate SNAP receptor function. Cell, 83, 111–119.
Morton, A.J. & Edwardson, J.M. (2001) Progressive depletion of complexin IIin a transgenic mouse model of Huntington’s disease. J. Neurochem., 76,166–172.
Morton, A.J., Faull, R.L.M. & Edwardson, J.M. (2001) Abnormalities in thesynaptic vesicle fusion machinery in Huntington’s disease. Brain Res. Bull.,56, 111–117.
Morton, A.J., Hunt, M.J., Hodges, A.K., Lewis, P.D., Redfern, A.J., Dunnett,S.B. & Jones, L. (2005) A combination drug therapy improves cognition andreverses gene expression changes in a mouse model of Huntington’s disease.Eur. J. Neurosci., 21, 855–870.
Murphy, K.P.S.J., Carter, R.J., Lione, L.A., Mangiarini, L., Mahal, A., Bates,G.P., Dunnett, S.B. & Morton, A.J. (2000) Abnormal synaptic plasticity andimpaired spatial cognition in mice transgenic for exon 1 of the humanHuntington’s disease mutation. J. Neurosci., 20, 5115–5123.
Nicoll, R.A. & Malenka, R.C. (1995) Contrasting properties of two forms oflong-term potentiation in the hippocampus. Nature, 377, 115–118.
Nicoll, R.A. & Malenka, R.C. (1999) Expression mechanisms underlyingNMDA receptor-dependent long-term potentiation. Ann. NY Acad. Sci., 868,515–525.
Ono, S., Baux, G., Sekiguchi, M., Fossier, P., Morel, N.F., Nihonmatsu, I.,Hirata, K., Awaji, T., Takahashi, S.-i. & Takahashi, M. (1998) Regulatoryroles of complexins in neurotransmitter release from mature presynapticnerve terminals. Eur. J. Neurosci., 10, 2143–2152.
Powell, C.M., Schoch, S., Monteggia, L., Barrot, M., Matos, M.F., Feldmann,N., Sudhof, T.C. & Nestler, E.J. (2004) The presynaptic active zoneprotein RIM1a is critical for normal learning and memory. Neuron, 42,143–153.
Reim, K., Mansour, M., Varoqueaux, F., McMahon, H.T., Sudhof, T.C., Brose,N. & Rosenmund, C. (2001) Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell, 104, 71–81.
Rhee, J.-S., Betz, A., Pyott, S., Reim, K., Varoqueaux, F., Augustin, I., Hesse,D., Sudhof, T.C., Takahashi, M., Rosenmund, C. & Brose, N. (2002) bPhorbol ester- and diacylglycerol-induced augmentation of transmitterrelease is mediated by Munc13s and not by PKCs. Cell, 108, 121–133.
Rizo, J. & Sudhof, T.C. (2002) SNAREs and Munc-18 in synaptic vesiclefusion. Nat. Rev. Neurosci., 3, 641–653.
Rolls, E.T. & Treves, A. (1994) Neural networks in the brain involved inmemory and recall. Prog. Brain Res., 102, 335–341.
Salin, P.A., Scanziani, M., Malenka, R.C. & Nicoll, R.A. (1996) Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc. NatlAcad. Sci. USA, 93, 13304–13309.
Stubley-Weatherly, L., Harding, J.W. & Wright, J.W. (1996) Effects of discretekainic acid-induced hippocampal lesions on spatial and contextual learningand memory in rats. Brain Res., 716, 29–38.
Impaired mossy fibre LTP in R6 ⁄ 2 and Cplx2– ⁄ – mice 1711
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712

Sudhof, T.C. (2004) The synaptic vesicle cycle. Ann. Rev. Neurosci., 27, 509–547.
Takahashi, S.-i., Ujihara, H., Huang, G.-Z., Yagyu, K.-i., Sanbo, M., Kaba, H.& Yagi, T. (1999) Reduced hippocampal LTP in mice lacking a presynapticprotein: complexin II. Eur. J. Neurosci., 11, 2359–2366.
Thomson, A.M. (2000) Facilitation, augmentation and potentiation at centralsynapses. Trends Neurosci., 23, 305–312.
Tokumaru, H., Umayahara, K., Pellegrini, L.L., Ishizuka, T., Saisu, H., Betz,H., Augustine, G.J. & Abe, T. (2001) SNARE complex oligomerization bysynaphin ⁄ complexin is essential for synaptic vesicle exocytosis. Cell, 104,421–432.
Tong, G., Malenka, R.C. & Nicoll, R.A. (1996) Long-term potentiation incultures of single hippocampal granule cells: a presynaptic form of plasticity.Neuron, 16, 1147–1157.
Towbin, H., Staehelin, T. & Gordon, J. (1979) Electrophoretic transfer ofproteins from polyacrylamide gels to nitrocellulose sheets: procedure andsome applications. Proc. Natl Acad. Sci. USA, 76, 4350–4354.
Villacres, E.C., Wong, S.T., Chavkin, C. & Storm, D.R. (1998) Type I adenylylcyclase mutant mice have impaired mossy fiber long-term potentiation.J. Neurosci., 18, 3186–3194.
Wang, H., Pineda, V.V., Chan, G.C.K., Wong, S.T., Muglia, L.J. & Storm,D.R. (2003) Type 8 adenylyl cyclase is targeted to excitatory synapses
and required for mossy fiber long-term potentiation. J. Neurosci., 23,9710–9718.
Weisskopf, M.G., Castillo, P.E., Zalutsky, R.A. & Nicoll, R.A. (1994)Mediation of hippocampal mossy fiber long-term potentiation by cyclicAMP. Science, 265, 1878–1882.
Weisskopf, M.G. & Nicoll, R.A. (1995) Presynaptic changes during mossyfibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature,376, 256–259.
Xiang, Z., Greenwood, A.C., Kairiss, E.W. & Brown, T.H. (1994) Quantalmechanism of long-term potentiation in hippocampal mossy-fibre synapses.J. Neurophys., 71, 2552–2556.
Yamada, M., Saisu, H., Ishizuka, T., Takahashi, H. & Abe, T. (1999)Immunohistochemical distribution of the two isoforms of synaphin ⁄ com-plexin involved in neurotransmitter release: localization at the distinctcentral nervous system regions and synaptic types. Neuroscience, 93, 7–18.
Yeckel, M.F., Kapur, A. & Johnston, D. (1999) Multiple forms of LTP inhippocampal CA3 neurons use a common postsynaptic mechanism. Nat.Neurosci., 2, 625–633.
Zalutsky, R.A. & Nicoll, R.A. (1990) Comparison of two forms of long-term potentiation in single hippocampal neurons. Science, 248, 1619–1624.
1712 H. E. Gibson et al.
ª 2005 Federation of European Neuroscience Societies, European Journal of Neuroscience, 22, 1701–1712




















