
a Mouse Model of Allergic Asthma in Cytokines, and Airway Eosinophilia Sensitization, Serum IgE, Th2...
11
of March 17, 2014. This information is current as Mouse Model of Allergic Asthma Cytokines, and Airway Eosinophilia in a Allergic Sensitization, Serum IgE, Th2 -Inducing Factor (IL-18) Increases γ IFN- and Sanjiv Sur Mohammed S. Siddiqui, Rafeul Alam, Masashi Kurimoto James S. Wild, Anastasia Sigounas, Nilanjana Sur, http://www.jimmunol.org/content/164/5/2701 2000; 164:2701-2710; ; J Immunol References http://www.jimmunol.org/content/164/5/2701.full#ref-list-1 , 32 of which you can access for free at: cites 55 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2000 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on March 17, 2014 http://www.jimmunol.org/ Downloaded from by guest on March 17, 2014 http://www.jimmunol.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of a Mouse Model of Allergic Asthma in Cytokines, and Airway Eosinophilia Sensitization, Serum IgE, Th2...

of March 17, 2014.This information is current as
Mouse Model of Allergic AsthmaCytokines, and Airway Eosinophilia in aAllergic Sensitization, Serum IgE, Th2
-Inducing Factor (IL-18) IncreasesγIFN-
and Sanjiv SurMohammed S. Siddiqui, Rafeul Alam, Masashi Kurimoto James S. Wild, Anastasia Sigounas, Nilanjana Sur,
http://www.jimmunol.org/content/164/5/27012000; 164:2701-2710; ;J Immunol
Referenceshttp://www.jimmunol.org/content/164/5/2701.full#ref-list-1
, 32 of which you can access for free at: cites 55 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

IFN-g-Inducing Factor (IL-18) Increases Allergic Sensitization,Serum IgE, Th2 Cytokines, and Airway Eosinophilia in aMouse Model of Allergic Asthma1
James S. Wild,* Anastasia Sigounas,† Nilanjana Sur,* Mohammed S. Siddiqui,† Rafeul Alam,*Masashi Kurimoto,‡ and Sanjiv Sur2*
We investigated the effects of IFN-g-inducing factor (IL-18) in a ragweed (RW) mouse model of allergic asthma. Administrationof IL-18 in conjunction with allergic sensitization and challenge in wild-type, but not IFN-g 2/2 mice, inhibited the bronchoal-veolar lavage (BAL) eosinophilia induced by RW challenge, and increased serum levels of RW-specific IgG2a and production ofIFN-g from splenocytes cultured with RW, indicating a critical role for IFN- g in mediating these effects. Paradoxically, the sametreatment schedule in WT mice increased serum levels of RW-specific IgE and IgG1,and production of IL-4 and IL-5 fromsplenocytes cultured with RW. When the effects of the same IL-18 treatment schedule were allowed to mature for 3 wk, theinhibition of lung eosinophil recruitment was replaced by augmentation of lung eosinophil recruitment. In another experiment,IL-18 administered only with allergic sensitization increased BAL eosinophilia and lung expression of IL-5 and IFN-g, while IL-18administered only with RW challenge decreased BAL eosinophilia and increased lung IFN-g expression, while lung expression ofIL-5 remained unchanged. IL-18 administered without RW or adjuvant to naive mice increased total serum IgE levels. Finally,intrapulmonary administrations of IL-18 plus RW in naive mice dramatically increased Th2 cytokine production, IgE levels,eosinophil recruitment, and airway mucus, demonstrating induction of allergic sensitization. This is the first report demonstratingthat IL-18 promotes a Th2 phenotype in vivo, and potently induces allergic sensitization. These results suggest that IL-18 maycontribute to the pathogenesis of allergic asthma. The Journal of Immunology,2000, 164: 2701–2710.
L ate phase pulmonary inflammation in allergic asthma ismodulated by Th cells that have been classified into Th1and Th2 types in accordance with their cytokine patterns.
Substantial evidence has been presented implicating Th2 cytokinesIL-4 and IL-5 in the pathology of allergic asthma and demonstrat-ing the protective effect of Th1 cells. The number of cells produc-ing Th2 cytokine mRNA is increased during allergic inflammation(1, 2). IL-4 knockout (KO)3 mice and animals treated with anti-IL-4 demonstrate inhibited eosinophil recruitment (3, 4). Also, anti-IL-5 Ab treatment inhibits allergen-induced eosinophil recruitmentand bronchial hyperresponsiveness in animals (5, 6). In contrast,the Th1 cytokine IFN-g has been shown to attenuate eosinophilrecruitment (7, 8). These data suggest that agents that shift the Tcell population from a Th2 profile to a Th1 profile are likely toprotect against eosinophilic inflammation in asthma. This hypoth-esis is supported by the observation that IL-12, a cytokine thatenhances production of the Th1 cytokine IFN-g, inhibits Th2 cy-
tokine synthesis and IgE synthesis in vitro and in vivo (9–11). Weand others have shown that IL-12 inhibits allergen-induced eosin-ophil recruitment, airway responsiveness, and allergen-specificIgE synthesis in murine models of asthma (12–14).
Originally called IFN-g-inducing factor, IL-18 has been re-ported to have properties similar to IL-12, including its ability tostimulate production of IFN-g in T cells, NK cells, and B cells(15). IL-18 and IL-12 act synergistically to promote IFN-g pro-duction (16), but IL-18 has also been reported to produce host-defense functions independent of IL-12 and IFN-g (17). Amongthe immunoregulatory effects of IL-18 that appear to be distinctfrom costimulation of IFN-g production are induction of Fas li-gand (18, 19), TNF-a, IL-1b, and CC and CXC chemokines (20).More recently, IL-18 was reported to induce the production of theTh2 cytokine, IL-13, from T cells and NK cells in vitro (21).
We have recently shown that CpG DNA, motifs that are over-represented in bacterial DNA and induce IFN-g, IL-12, and IL-18(22), protect against eosinophilic lung inflammation in a mousemodel of asthma (23). Because both IFN-g and IL-12 inhibit eo-sinophilic lung inflammation, we sought to determine whetherIL-18 would contribute similar or opposing effects in a mousemodel of asthma. The results of our study indicate that IL-18 pro-duced an early inhibition of allergen-specific lung eosinophilia thatwas mediated by IFN-g, but also increased allergen-specific serumIgE levels, IL-4 and IL-5 production from splenocytes culturedwith the RW, and increased eosinophilic lung inflammation.
Materials and MethodsExperimental animals
Six- to 8-wk-old female BALB/c mice were purchased from Harlan Lab-oratories (Indianapolis, IN) to perform all experiments except those requir-ing IFN-g2/2 and IFN-g1/1 mice. The latter (IFN-g2/2 and IFN-g1/1
mice) were 5-wk-old female BALB/c mice purchased from The Jackson
*Department of Internal Medicine, Division of Allergy and Immunology, Universityof Texas Medical Branch, Galveston, TX 77555;†Department of Medicine, EastCarolina University School of Medicine, Greenville, NC; and‡Fujisaki Institute, Ha-yashibara Biochemical Labs, Okayama, Japan
Received for publication September 22, 1999. Accepted for publication December22, 1999.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported in part by National Institutes of Health Grants K08AI01539 and P01 AI46004, and the James W. McLaughlin Fellowship Fund.2 Address correspondence and reprint requests to Dr. Sanjiv Sur, Department of In-ternal Medicine, Division of Allergy and Immunology, University of Texas MedicalBranch, Galveston, TX 77555-0762. E-mail address: [email protected] Abbreviations used in this paper: KO, knockout;b2m, b2-microglobulin; BAL,bronchoalveolar lavage; i.n., intranasal; i.t., intratracheal; RW, ragweed allergen; WT,wild-type.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

Laboratory (Bar Harbor, ME). All mice were maintained in a specificpathogen-free environment throughout the experiment at University ofTexas Medical Branch (Galveston, TX).
Two-week model of allergic sensitization and challenge
Using a protocol described previously (14), mice were sensitized by twoi.p. injections of RW (150mg) and alum on days 0 and 4. On day 11, anintratracheal (i.t.) or intranasal (i.n.) instillation of RW (200mg) was per-formed in anesthetized mice. Three days later on day 14, the mice weresacrificed and bronchoalveolar lavage (BAL) fluid, blood, and lung spec-imens were collected.
Murine rIL-18
Murine rIL-18 (MuIGIF) was generously provided by the Fujisaki Institute,Hayashibara Biochemical Labs (Okayama, Japan). The 18.1-kDa cytokinewas expressed inEscherichia coli, and the purity was determined to be.99% by SDS-PAGE. The cytokine preparation was sterilized by filtra-tion, and the endotoxin content was 1 ng/ml, as determined by Limulusamebocyte lysate assay.
Experimental protocols
IL-18 was diluted in PBS containing 1% autologous BALB/c mouse serumas a carrier protein. All PBS injections for the control groups also contained1% autologous serum. Several protocols were used to administer IL-18 tomice (Fig. 1). We have previously shown that seven 1mg IL-12 i.p. in-jections on days 0, 4, 5, 6 (to interfere with allergic sensitization) and days11 and 12 (to interfere with the late phase inflammation induced by RWchallenge) inhibited RW-specific IgE and eosinophil recruitment in amouse model of asthma (14). We wished to determine whether an identicaldosage and schedule of IL-18 administration would interfere with IgE pro-duction and eosinophil recruitment. To accomplish this, in the first exper-iment, BALB/c mice were sensitized and challenged with RW, and thetreatment group was injected with seven 1mg doses of i.p. IL-18 (RW1IL-18/RW1IL-18), whereas the control group was injected with PBS (RW/RW) (Fig. 1,protocol I,II).
Because IL-18 is a potent inducer of IFN-g, in the second experimentwe sought to define IFN-g-dependent and independent effects by repeatingthe same protocol of IL-18 administration in WT and IFN-g2/2 mice (Fig.1, protocol I,II).
In the third experiment, IL-18 or PBS was administered to mice sensi-tized and challenged with RW in conjunction with allergen sensitization orallergen challenge. IL-18 was administered i.p. in 1mg doses in conjunc-tion with allergic sensitization on days 0, 4, 5, and 6 in the RW1IL-18/RWgroup (Fig. 1,protocol IIIa), or in conjunction with allergen challenge on
days 11 and 12 in the RW/RW1IL-18 group (Fig. 1,protocol IIIb). Micein the control group (RW/RW) were injected with seven i.p. injections ofPBS on days 0, 4, 5, 6, 11 (two injections), and 12, and mice in all groupswere sacrificed on day 14.
The fourth experiment was designed to examine the effects of IL-18administered without RW (Fig. 1,protocol IV). One group of naive micewas injected with seven 1mg doses of IL-18 administered i.p. on days 0,4, 5, 6, 11 (two injections), and 12 (IL-18 naive group). The correspondingcontrol group of naive mice (PBS naive group) received seven i.p. admin-istrations of PBS, and mice in both groups were sacrificed on day 14.
The fifth experiment was designed to examine the early and late effectsof IL-18. Mice were sensitized with RW and treated with seven injectionsof i.p. PBS or 1mg IL-18, as was done for the RW/RW and RW1IL-18/RW1IL-18 groups. However, in this experiment, the RW challenge andanimal sacrifice were staggered to occur at early and later intervals. One setof PBS control- and IL-18-treated mice was challenged with RW on day 11and sacrificed on day 14 (RW/RW, Day 14, and RW1IL-18/RW1IL-18,Day 14, respectively; Fig. 1,protocol Va). Another set of control andtreatment mice was challenged with RW on day 18 and sacrificed on day21 (RW/RW, day 21, and RW1IL-18/RW1IL-18, day 21; Fig. 1,protocolVb). A final set of control and treatment animals was challenged with RWon day 32, and sacrificed on day 35 (RW/RW, day 35, and RW1IL-18/RW1IL-18, day 35; Fig. 1,protocol Vc).
The sixth experiment was designed to examine the ability of IL-18 toaugment allergic sensitization. Mice were administered three i.n. doses ofRW (200mg) on days 0, 2, and 4, with concomitant administration of 500ng IL-18 or PBS. Mice were then challenged with i.n. RW or PBS on day11 and sacrificed on day 14 (Fig. 1,protocol VI).
Sample preparation
Mice were euthanized with an i.p. injection of ketamine and xylazine toperform BAL, as previously described (14). Briefly, the BAL fluids wereobtained by cannulating the trachea, and lavaging the lungs with two0.7-ml aliquots of ice-cold Dulbecco’s PBS (Sigma, St. Louis, MO). TheBAL cells were pelleted, washed, and Wright Giemsa stained. The numberof eosinophils, neutrophils, lymphocytes, and macrophages was deter-mined by performing a differential count on at least 200 cells/slide of acytocentrifuge preparation. Whole mouse lungs from different treatmentgroups were stored at280°C for later mRNA extraction. Animals werebled by cardiac puncture and serum was stored at280°C until use.
Th1 and Th2 cytokine production by splenocytes culturedwith RW
Spleens were removed aseptically from experimental mice, minced, andpassed through a wire mesh to generate single cell suspensions. Erythro-cytes were lysed and splenocytes were incubated at 53 106 cells/ml witheither diluent or 100mg/ml RW for 4 days. The cells were incubated incomplete medium (RPMI 1640 supplemented with 10% FCS) at 37°C in ahumidified incubator with 5% CO2 atmosphere. The cell supernatants wereexamined for IL-4, IL-5, and IFN-g levels by ELISA.
Lung histology
Treatment and control mouse lungs were perfusion fixed with 1 ml of i.t.instilled Formalin solution (10% neutral buffered formaldehyde; Sigma).Subsequently, the lungs were embedded in paraffin, sectioned at a thicknessof 4 um, and stained with hemoxylin and eosin or hemoxylin and periodicacid-Schiff for mucus staining. Lung inflammation was assessed by a pa-thologist blinded to the treatment groups. The degree of peribronchial andperivascular inflammation was evaluated on a subjective scale of 0, 1, 2, 3,and 4 corresponding to none, mild, moderate, marked, or severe inflam-mation, respectively, with an increment of 0.5 if the inflammation fellbetween two integers, as described previously (14, 23, 24). The total lunginflammation was defined as the sum of peribronchial and perivascularinflammation scores.
IL-4, IL-5, IFN-g, and total IgE ELISA
Two-site immunoenzymetric assays were used for measuring IL-4, IL-5,IFN-g, and total IgE, as previously described (14). Briefly, 96-well Immu-lon 4 microtiter plates (Dynex Technologies, Chantilly, VA) were coatedand incubated overnight at 4°C with rat anti-mouse IL-4 mAb (clone 24G2;Endogen, Woburn, MA), rat anti-mouse/human IL-5 mAb (clone TRFK5;PharMingen, San Diego, CA), rat anti-mouse IFN-g mAb (clone R4-6A2;PharMingen), or rat anti-mouse IgE (clone R35-72; PharMingen). Afterwashing, the plates were blocked with 10% Seablock (Pierce, Rockford,
FIGURE 1. Experimental protocols for allergic sensitization and chal-lenge and IL-18 administration. The roman numerals correspond to thenumbered experiments described in detail inMaterials and Methods. Fol-lowing termination, BAL was performed and serum and other sampleswere collected.
2702 IL-18 INDUCES ALLERGIC SENSITIZATION IN A MURINE MODEL OF ASTHMA
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

IL), then washed and incubated for 2 h at room temperature with the ap-propriate detection Abs: biotinylated rat anti-mouse IL-4 (clone 24G2; En-dogen), biotinylated rat anti-mouse IL-5 (clone TRFK4; PharMingen), bi-otinylated rat anti-mouse IFN-g (clone XMG1.2; PharMingen), orbiotinylated rat anti-mouse IgE (clone R35-118; PharMingen). Followingwashing, the plates were incubated with avidin-conjugated alkaline phos-phatase (Sigma) for 1 h, then washed and developed withp-nitrophenolphosphate (Sigma). The lower limits of the assay systems were 1 pg/ml forIL-4, 5 pg/ml for IL-5, 9 pg/ml for IFN-g, and 96 pg/ml for total IgE.
RW-specific serum IgE, IgG1, and IgG2a ELISA
Ninety-six-well Immulon 4 microtiter plates were coated with 10mg/ml ofRW protein overnight at 4°C, washed, and blocked. After washing, plateswere overlaid with diluted sera from control and treatment animals, incu-bated for 12 h at 4°C, washed, then incubated with biotin-conjugated ratanti-mouse IgG1 (clone G1-6.5; PharMingen), IgG2a (clone R19-5;PharMingen), or IgE (clone R35-72; PharMingen). After 2 h of incubation,the plates were washed and incubated with avidin-conjugated alkalinephosphatase, followed by washing and development withp-nitrophenolphosphate. Serum Ab concentrations were determined by comparison witha serially diluted high-titered positive control serum.
Semiquantitative RT-PCR of lung IL-5 and IFN-g mRNA
Total RNA was extracted from lungs using Triazol (Life Technologies,Grand Island, NY), according to the manufacturer’s specifications. ThemRNA was reverse transcribed to first strandcDNA using oligo(dT12–15)primer (Life Technologies). Amplification ofb2-microglobulin (b2m), IL-5,and IFN-g was performed by PCR. The primers forb2m (forward, ATGGCTCGCTCGGTGACCCTAG, and reverse, TCATGATGCTTGATCACATGTCTCG), IL-5 (forward, GACAAGCAATGAGACGATG, and reverse,GTCACCATGGAGCAGCTCAGCC), and IFN-g (forward, TACTGCCACGGCACAGTCATTGAA,andreverse,TGGACCTGTGGGTTGTTGACCTCAAACTTGGC) were synthesized by Biosynthesis (Lewisville, TX). A 20-mlPCR was performed using PCR buffer, deoxynucleotide triphosphates, MgCl2,
and Taq polymerase (Amplitaq Gold; Perkin-Elmer, Foster City, CA) in aDNA thermal cycler (Perkin-Elmer 9700). The PCRincluded a hot startmodification at an initial denaturing step of 94°C for 10 min, followed bycycles of annealing at 55°C for 30 s, extension at 72°C for 60 s, anddenaturation at 94°C for 30 s. Cycle numbers corresponding to the expo-nential phase were individually determined for each primer set. The cyclenumbers were 19, 27, and 29 forb2m, IFN-g, and IL-5, respectively. ThePCR products were electrophoresed in a 3% agarose gel (Seakem LE Aga-rose; FMC Bioproducts, Rockland, ME), stained with ethidium bromide,and photographed using type 57 polaroid film (Sigma) and a UV camera.The intensity of the bands on photographs of the agarose gels was quan-tified by scanning the photographs with a contrast scanner (Sharp JX-330;Sharp Electronics, Mahwah, NJ) using optical software (ImageQuant, ver-sion 3.3; Molecular Dynamics, Sunnyvale, CA). The values obtained fromindividual cytokines were expressed as a ratio of cytokine band intensity tothe intensity of the housekeeping gene,b2m.
Data analysis
The difference in BAL cell counts, serum Ig levels, cytokine levels, andcytokine/b2m expression levels between treatment groups was analyzed byStudent’st test for two groups and ANOVA for more than two groups.Significant ANOVAs were further analyzed by the Bonferroni posthoc test.The comparison of histology scores between the two treatment groups wasanalyzed using the Mann-WhitneyU test.
ResultsIL-18 administered in conjunction with allergic sensitization andchallenge inhibits BAL eosinophilia via production of IFN-g
Compared with the RW/RW group, IL-18 administered in con-junction with RW sensitization and challenge (RW1IL-18/RW1IL-18) reduced BAL eosinophils by 71% (p # 0.0001; Fig.2A). The same protocol also decreased BAL total cells, lympho-
FIGURE 2. Effect of IL-18 administered in conjunction with allergic sensitization and challenge on BAL eosinophilia and peribronchial and perivascularinflammation.A, BAL eosinophil cell numbers. BALB/c mice (Harlan Laboratories) were sensitized with 150mg of RW and alum administered i.p. on days0 and 4, followed by allergen challenge with 200mg of RW administered i.t. on day 11 and sacrifice on day 14. The RW1IL-18/RW1IL-18 group received1 mg of i.p. IL-18 on days 0, 4, 5, 6, 11 (twice), and 12. The RW/RW group received i.p. PBS by the same dosage schedule.B, Effect of the same IL-18dosage schedule on peribronchial and perivascular lung inflammation. A pathologist blinded to the treatment groups evaluated the degree of peribronchialand perivascular inflammation on a scale of 0–4, with an increment of 0.5 if the inflammation fell between two integers. The total lung inflammation wasdefined as the sum of peribronchial and perivascular inflammation scores. Values are expressed as mean6 SEM for 10 (RW/RW), or 13 (RW1IL-18/RW1IL-18) mice.p, p # 0.05; pp, p # 0.01; pppp, p # 0.0001 compared with the corresponding RW/RW group.
2703The Journal of Immunology
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from
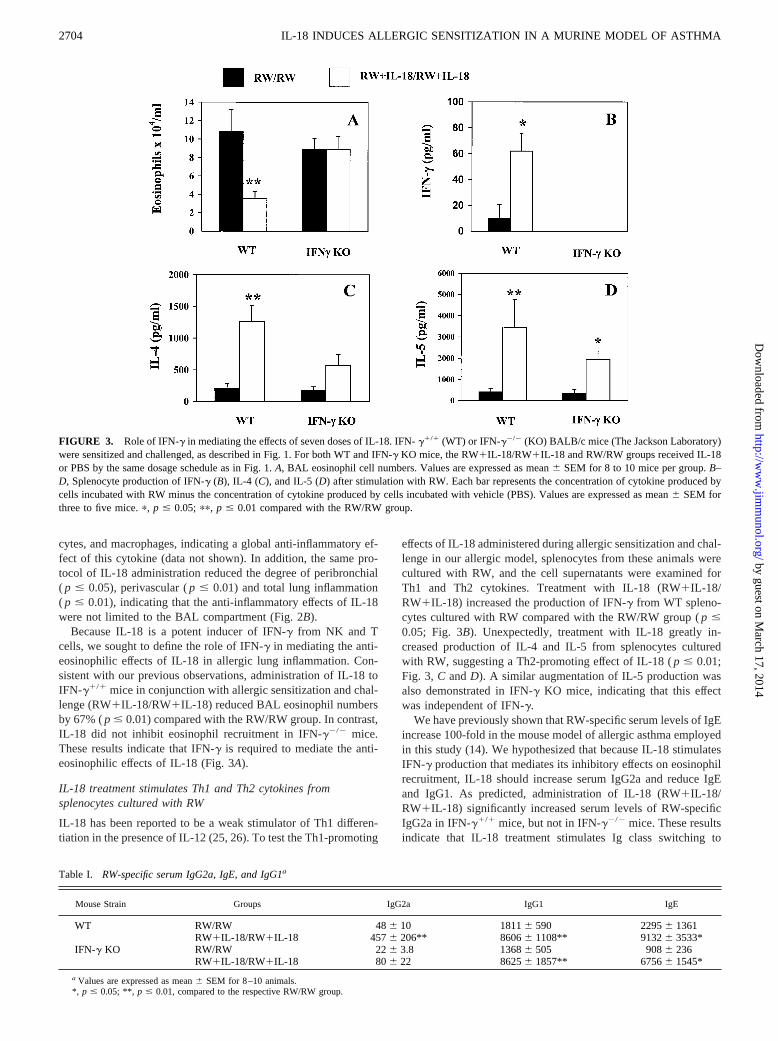
cytes, and macrophages, indicating a global anti-inflammatory ef-fect of this cytokine (data not shown). In addition, the same pro-tocol of IL-18 administration reduced the degree of peribronchial( p # 0.05), perivascular (p # 0.01) and total lung inflammation( p # 0.01), indicating that the anti-inflammatory effects of IL-18were not limited to the BAL compartment (Fig. 2B).
Because IL-18 is a potent inducer of IFN-g from NK and Tcells, we sought to define the role of IFN-g in mediating the anti-eosinophilic effects of IL-18 in allergic lung inflammation. Con-sistent with our previous observations, administration of IL-18 toIFN-g1/1 mice in conjunction with allergic sensitization and chal-lenge (RW1IL-18/RW1IL-18) reduced BAL eosinophil numbersby 67% (p # 0.01) compared with the RW/RW group. In contrast,IL-18 did not inhibit eosinophil recruitment in IFN-g2/2 mice.These results indicate that IFN-g is required to mediate the anti-eosinophilic effects of IL-18 (Fig. 3A).
IL-18 treatment stimulates Th1 and Th2 cytokines fromsplenocytes cultured with RW
IL-18 has been reported to be a weak stimulator of Th1 differen-tiation in the presence of IL-12 (25, 26). To test the Th1-promoting
effects of IL-18 administered during allergic sensitization and chal-lenge in our allergic model, splenocytes from these animals werecultured with RW, and the cell supernatants were examined forTh1 and Th2 cytokines. Treatment with IL-18 (RW1IL-18/RW1IL-18) increased the production of IFN-g from WT spleno-cytes cultured with RW compared with the RW/RW group (p #0.05; Fig. 3B). Unexpectedly, treatment with IL-18 greatly in-creased production of IL-4 and IL-5 from splenocytes culturedwith RW, suggesting a Th2-promoting effect of IL-18 (p # 0.01;Fig. 3, C andD). A similar augmentation of IL-5 production wasalso demonstrated in IFN-g KO mice, indicating that this effectwas independent of IFN-g.
We have previously shown that RW-specific serum levels of IgEincrease 100-fold in the mouse model of allergic asthma employedin this study (14). We hypothesized that because IL-18 stimulatesIFN-g production that mediates its inhibitory effects on eosinophilrecruitment, IL-18 should increase serum IgG2a and reduce IgEand IgG1. As predicted, administration of IL-18 (RW1IL-18/RW1IL-18) significantly increased serum levels of RW-specificIgG2a in IFN-g1/1 mice, but not in IFN-g2/2 mice. These resultsindicate that IL-18 treatment stimulates Ig class switching to
FIGURE 3. Role of IFN-g in mediating the effects of seven doses of IL-18. IFN-g1/1 (WT) or IFN-g2/2 (KO) BALB/c mice (The Jackson Laboratory)were sensitized and challenged, as described in Fig. 1. For both WT and IFN-g KO mice, the RW1IL-18/RW1IL-18 and RW/RW groups received IL-18or PBS by the same dosage schedule as in Fig. 1.A, BAL eosinophil cell numbers. Values are expressed as mean6 SEM for 8 to 10 mice per group.B–D, Splenocyte production of IFN-g (B), IL-4 (C), and IL-5 (D) after stimulation with RW. Each bar represents the concentration of cytokine produced bycells incubated with RW minus the concentration of cytokine produced by cells incubated with vehicle (PBS). Values are expressed as mean6 SEM forthree to five mice.p, p # 0.05; pp, p # 0.01 compared with the RW/RW group.
Table I. RW-specific serum IgG2a, IgE, and IgG1a
Mouse Strain Groups IgG2a IgG1 IgE
WT RW/RW 486 10 18116 590 22956 1361RW1IL-18/RW1IL-18 4576 206** 86066 1108** 91326 3533*
IFN-g KO RW/RW 226 3.8 13686 505 9086 236RW1IL-18/RW1IL-18 806 22 86256 1857** 67566 1545*
a Values are expressed as mean6 SEM for 8–10 animals.*, p # 0.05; **, p # 0.01, compared to the respective RW/RW group.
2704 IL-18 INDUCES ALLERGIC SENSITIZATION IN A MURINE MODEL OF ASTHMA
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

IgG2a and that the effect is mediated by IFN-g (Table I). Unex-pectedly, IL-18 administration increased production of RW-spe-cific IgE and IgG1 in serum, effects that are consistent with aug-mentation of a Th2 phenotype (Table I).
To assess the ability of IL-18 to promote IgE production inde-pendent of allergic sensitization and challenge, naive mice wereadministered seven 1mg doses of IL-18 by the same dosage sched-ule used in allergic animals. IL-18 treatment of naive mice (IL-18naive) increased total serum IgE concentration to 287.76 71.4ng/ml, a significant 5-fold increase (p # 0.01) compared with the54.46 6 ng/ml total serum IgE concentration of the PBS-treatednaive mice (PBS naive). These results indicate that IL-18 increasesIgE independent of the effects of allergen or alum. However, IL-18treatment of naive mice did not alter BAL eosinophils, total cells,neutrophils, lymphocytes, and macrophages (data not shown).
Differential effects of IL-18 administered during sensitizationand challenge
We wished to test whether the anti-eosinophilic effects of IL-18were dependent on timing of IL-18 administration with respect toallergic sensitization or allergen challenge. Administration ofIL-18 at the time of allergen challenge (RW/RW1IL-18) de-creased BAL eosinophil numbers by 89% (p # 0.01) comparedwith the corresponding RW/RW group (Fig. 4A). In contrast, ad-ministration of IL-18 in conjunction with allergic sensitization(RW1IL-18/RW) increased BAL eosinophil numbers by 137%( p # 0.05) compared with the RW/RW group. These results in-dicate that the time of IL-18 administration in relation to allergicsensitization and challenge influences the anti-eosinophilic effects
of IL-18, and that IL-18 administered in conjunction with allergicsensitization can exacerbate allergic lung eosinophilia. The admin-istration of IL-18 in conjunction with allergic sensitization(RW1IL-18/RW) significantly augmented RW-specific serumIgG1 levels relative to the corresponding RW/RW group (TableII). Serum IgE and IgG2a levels were not altered in the RW1IL-18/RW or RW/RW1IL-18 groups. IL-18 administered in conjunc-tion with allergic sensitization, but not allergen challenge, alsoincreased IL-4 production from splenocytes cultured with RW(Fig. 4C). These results are consistent with a Th2-promoting effectof IL-18 administered at the time of allergic sensitization.
Administration of IL-18 in conjunction with allergic sensitiza-tion or allergen challenge induced differential patterns of cytokinemRNA expression in the lung determined by semiquantitative RT-PCR. IL-18 administered with allergen challenge (RW/RW1IL-18) increased lung expression of IFN-g compared with theRW/RW control group without altering IL-5 mRNA expression(Fig. 5). In contrast, IL-18 administered with allergic sensitization
FIGURE 4. Differential effects of IL-18 administered in conjunction with allergic sensitization or challenge. BALB/c mice (Harlan Laboratories) weresensitized and challenged, as described in Fig. 1. Mice in the RW1IL-18/RW group received 1mg of IL-18 administered i.p. on days 0, 4, 5, and 6 inconjunction with allergic sensitization. The RW/RW1IL-18 group received the same dose of IL-18 on day 11 (twice) and day 12 in conjunction withallergic challenge. The RW/RW group received i.p. PBS on days 0, 4, 5, 6, 11 (twice), and 12.A, BAL eosinophil numbers for the three treatment groups.Values are expressed as mean6 SEM for five to seven animals per group.B–D, RW-stimulated production of IFN-g (B), IL-4 (C), and IL-5 (D), fromsplenocytes derived from individual mice. Each bar represents the concentration of cytokine produced by cells incubated with RW minus the concentrationof cytokine produced by cells incubated with vehicle (PBS). Values are expressed as mean6 SEM for five or six mice.p, p # 0.05;pp, p # 0.01 comparedwith the RW/RW group;1, p # 0.05 compared with the RW1IL-18/RW group.
Table II. The effects of IL-18 dosage timing on RW-specific serum Iga
Groups IgG2a IgG1 IgE
RW/RW 56.06 21.0 1202.56 585.4 5319.36 4384.0RW1IL-18/RW 87.06 35.3 5958.86 339.7* 3555.66 1253.2RW/RW1IL-18 31.16 10.4 967.36 384.1 817.86 270.3
Values are expressed as mean6 SEM for five to seven animals.*, p # 0.001 compared to the RW/RW group.
2705The Journal of Immunology
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

(RW1IL-18/RW) up-regulated lung IL-5 and IFN-g mRNA ex-pression. These mRNA expression patterns suggest that increasedlung IL-5 production may have stimulated the allergen-inducedBAL eosinophilia demonstrated by the RW1IL-18/RW group,whereas increased lung IFN-g production without alteration ofIL-5 production may have reduced BAL eosinophilia in the RW/RW1IL-18 group.
Augmentation of eosinophil recruitment with maturation of IL-18 effects
Because our findings indicated that IL-18 administration in con-junction with allergic sensitization and challenge inhibited eosin-ophilic lung inflammation while simultaneously promoting theproduction of IgE and Th2 cytokines, we sought to determinewhether the latter effects would eventually override the IFN-g-mediated suppression of eosinophil recruitment. Experiments wereconducted in which sensitized mice received seven injections ofPBS or IL-18, as described previously, followed by allergen chal-lenge and animal sacrifice at three different time points. As dem-onstrated before, when mice were challenged with RW on day 11,IL-18 administration inhibited BAL eosinophil numbers on day 14( p # 0.05; Fig. 6). In contrast, the allergen-induced BAL eosin-ophil recruitment increased with the time elapsed after IL-18 ad-ministration, and by day 35, 23 days following the last adminis-tration of IL-18, BAL eosinophil numbers were increased by 111%in the IL-18-treated group compared with the RW/RW controlgroup (p # 0.05). These results indicate that IL-18 has biphasiceffects in the mouse model of asthma. Initially, IL-18 treatment
inhibits allergen-induced BAL eosinophil recruitment via IFN-gproduction. Subsequently, as its effect on the animal matures andthe initially dominant IFN-g-dependent effects dissipate, IL-18augments eosinophil recruitment.
IL-18 treatment augments allergic sensitization
To determine whether IL-18 could promote allergic sensitizationin conjunction with airway exposure to RW, mice were adminis-tered three i.n. doses of RW mixed with either PBS or IL-18, thenchallenged 1 wk later with PBS or RW. As expected, PBS chal-lenge of mice 7 days after the last dose of i.n. RW1PBS in thecontrol group (i.n. RW/PBS) did not induce BAL eosinophilia.Unexpectedly, PBS challenge of mice 7 days after the last dose ofi.n. RW1IL-18 (i.n. RW1IL-18/PBS) induced 900% greater BALeosinophil numbers (p # 0.0001; Fig. 7A) compared with the con-trol group. These results suggest that the substitution of PBS withIL-18 during the three initial i.n. doses of RW rapidly increasedrecruitment of eosinophils to the lung, and some of the recruitedeosinophils were retained in the lung 10 days later. Similarly, thei.n. RW/PBS group did not demonstrate appreciable mucus secre-tion by airway epithelium, but mucus-containing goblet cells weresubstantially increased in the i.n. RW1IL-18/PBS group (Fig. 8).To examine the mechanism of these unexpected findings, we quan-tified RW-specific serum IgE and RW-induced Th2 cytokine pro-duction by splenocytes from the mice challenged with PBS. Thei.n. RW1IL-18/PBS group demonstrated 66-fold greater RW-spe-cific serum IgE (p # 0.01) than the control group, and 63-fold( p # 0.01) and 44-fold (p # 0.001) greater IL-4 and IL-5 pro-duction, respectively, from splenocytes cultured with RW (Fig. 7,B, C, andD). These results strongly suggest that IL-18 adminis-tered i.n. with RW increased allergic sensitization to RW. Furtherevidence of an induction of allergic sensitization by i.n. adminis-tration of IL-18 with RW comes from the observed effects of RWchallenge performed in the two groups of mice 7 days after thethree i.n. doses of RW1PBS or RW1IL-18. Administration ofIL-18 in conjunction with RW in the i.n. RW1IL-18/RW groupincreased BAL eosinophil recruitment 456% (p # 0.0001) com-pared with the control group (i.n. RW/RW) (Fig. 7A). Peribron-chial and perivascular inflammation was also increased in the i.n.RW1IL-18/RW group, indicating that the proinflammatory effectsof IL-18 were not confined to the BAL compartment (Fig. 9).
FIGURE 5. Lung expression of IL-5 and IFN-g, as measured by semi-quantitative RT-PCR. Total RNA was isolated from the lungs of mice inthe RW1IL-18/RW, RW/RW1IL-18, and RW/RW groups. RT-PCR wasperformed using primers specific for murine IL-5, IFN-g, andb2m. A, Theratio of lung IL-5 expression to lungb2m expression.B, The ratio of lungIFN-g expression to lungb2m expression. Values are expressed as themean6 SEM for three to seven mice per group.pp, p # 0.01;ppp, p #
0.001 compared with the RW/RW group.
FIGURE 6. The early and late occurring effects of IL-18 administrationon allergen-induced BAL eosinophilia. Mice were sensitized with i.p. RWand alum on days 0 and 4, and administered PBS or IL-18 on days 0, 4, 5,6, 11 (twice), and 12. One set of control (RW/RW)- and IL-18-treated(RW1IL-18/RW1IL-18) mice were challenged with i.n. RW (200mg) onday 11 and sacrificed on day 14. Another set of control and treatment micewas challenged with i.n. RW on day 18 and sacrificed on day 21, and thefinal set of control and treatment animals was challenged with RW on day32, and sacrificed on day 35. The sacrifice/BAL day for each set of controland IL-18 treatment animals is shown on the abscissa. Values are expressedas the mean6 SEM for three to six mice per group.p, p # 0.05 comparedwith the respective RW/RW group.
2706 IL-18 INDUCES ALLERGIC SENSITIZATION IN A MURINE MODEL OF ASTHMA
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

Taken together, these results provide compelling evidence thatIL-18 administered i.n. with RW dramatically increased allergicsensitization to RW.
DiscussionThe results of this study indicate that administration of IL-18 inconjunction with allergic sensitization and allergen challenge in-hibited recruitment of eosinophils to the lung. This effect, likemany previously described in vivo effects of IL-18, was criticallymediated by IFN-g. Similarly, IL-18 administered in conjunctionwith allergen challenge reduced BAL eosinophilia. Paradoxically,administration of IL-18 in conjunction with allergic sensitizationand allergen challenge increased serum IgE and IgG1 levels, andincreased IL-4 and IL-5 production from splenocytes cultured withRW. When the effects of the same IL-18 treatment schedule wereallowed to mature for 3 wk, the inhibition of eosinophil recruit-ment was replaced by augmentation of eosinophil recruitment.Also, IL-18 administered in naive mice induced IgE production.Finally, intrapulmonary administration of IL-18 with RW dramat-ically increased BAL eosinophilia, airway mucus, RW-specific se-rum IgE production, and RW-stimulated Th2 cytokine productionfrom splenocytes, indicating that IL-18 augments allergicsensitization.
The effects of IL-18 have been previously assessed in murinemodels of allergic asthma by Hofstra et al. (27), and Kumano et al.(28), with differing outcomes. In the study by Hofsta et al., eventhough the combination of IL-18 and IL-12 inhibited Ag-inducedairway hyperresponsiveness, lung eosinophilia, and serum IgE, ad-ministration of either IL-12 or IL-18 alone failed to modulate anyof the allergic responses. Although the present study and the studyby Hofstra et al. employed similar IL-18 dosages, the two studiesdiffer strikingly in the allergic models that were used. The presentstudy employed a 2-wk model involving two sensitizing doses ofRW plus alum, and a single i.t. or i.n. RW challenge, and the studyby Hofstra et al. utilized an immunization protocol involving seveni.p. OVA injections without adjuvant for sensitization, and eightaerosolized OVA challenges. In a recent study by Kumano et al.,two 10 or 20mg doses of IL-18 administered i.p. before OVAchallenge enhanced eosinophil recruitment into the lungs of sen-sitized mice 48 h after challenge (28). These results are consistent
with some of the findings in the present study, but elevated Th2cytokines were not demonstrated in the Kumano et al. study. Al-though it is difficult to pinpoint the reasons IL-18 stimulated vari-able effects in the three studies, it is possible that the differences inthe models may explain differences in the findings. As increasingknowledge of the effects of IL-18 in allergy emerge, elucidation ofthe specific immunological environments presented by differentstudy models may prove valuable in understanding the scope ofIL-18 activity.
Although IFN-g was a critical mediator of the anti-eosinophi-lic effects of IL-18 administered with allergic sensitization andchallenge in this study, the cellular source of IFN-g was not de-termined. Because IL-18 treatment increased IFN-g from spleno-cytes cultured with RW, it is possible that IL-18 stimulated thedevelopment of Th1 cells. IL-18 has been reported to induce Th1differentiation independent of IL-12, although the effect was morepronounced in the presence of IL-12 (25, 26). However, in anotherstudy, Robinson et al. (29) suggested that IL-18 was unable toindependently drive Th1 development in BALB/c mice, the exper-imental animals that were used in the present study. Indeed, theamount of IFN-g produced by RW-stimulated splenocytes in thisstudy is much smaller than what we have demonstrated in a pre-vious study following the same protocol of administration of IL-12in the same murine model of asthma (14). Also, in the mice thatreceived IL-18 only in conjunction with allergic challenge (RW/RW1IL-18 group), BAL eosinophilia was significantly reducedand lung IFN-g expression was significantly augmented, but pro-duction of IFN-g from spleen cells cultured with RW was notincreased. These findings suggest that IL-18 administration in thisstudy may have stimulated IFN-g production from a variety ofcells, such as B cells and/or NK cells, rather than primarily induc-ing development of Ag-specific Th1 cells.
In our study, IL-18 dramatically stimulated allergic sensitiza-tion. The administration of i.n. IL-18 with RW increased produc-tion of BAL eosinophilia, RW-specific IgE, airway mucus, andTh2 cytokines from splenocytes cultured with RW. In this exper-iment, IL-18 acted as mucosal sensitization adjuvant when admin-istered with a protein immunogen such as RW. However, IL-18administration to naive animals increased production of total se-rum IgE, suggesting that IL-18 can stimulate IgE class switching
FIGURE 7. Administration of IL-18 plus RW i.n.augments allergic sensitization to RW. Mice were ad-ministered 200mg of i.n. RW on days 0, 2, and 4, thenchallenged i.n. with PBS or 200mg RW on day 11,followed by sacrifice on day 14. In the IL-18 treatmentgroups (i.n. RW1IL-18/RW and i.n. RW1IL-18/PBS), mice received 500 ng of i.n. IL-18 in conjunc-tion with the RW doses on days 0, 2, and 4. In thecontrol groups (i.n. RW/RW and RW/PBS), mice re-ceived i.n. PBS in the place of IL-18.A, BAL eosin-ophils/ml for control- and IL-18-treated mice follow-ing i.n. challenge with PBS or RW. Values areexpressed as mean6 SEM for 4–5 animals (PBS chal-lenge), or 10–11 mice (RW challenge).B, Serum lev-els of RW-specific IgE for PBS-challenged mice. Val-ues are expressed as mean6 SEM for four to fivemice.C andD, RW-stimulated production of IL-4 andIL-5 from splenocytes obtained from PBS-challengedmice. Values are expressed as mean6 SEM for four tofive mice. pp, p , 0.01; ppp, p , .001; pppp, p ,0.0001.
2707The Journal of Immunology
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

in the absence of a protein immunogen. These effects could in-volve a variety of cellular interactions, including direct or indirectstimulation of B cell IgE class switching or Th2 cell differentiationand/or Th2 cell proliferation, and elucidation of the precise mech-anisms will require further study.
Hypothetically, the IL-18-induced production of IgE and IgG1may have resulted from direct effects of IL-18 on B cell classswitching, or secondary to increased Th2 cytokines resulting fromIL-18 administration. IL-18 administration in conjunction with al-lergic sensitization and allergen challenge stimulated IL-4 produc-tion from splenocytes cultured with RW. A similar increase inAg-induced IL-4 production in vivo may account for the increasedserum IgE, as IL-4 is a known inducer of IgE class switching (30).IL-18 in combination with IL-12 has been shown to suppress theproduction of IgE in activated B cells (31), and IL-18 alone wasreported to have no effect on IgE production in purified splenic Bcells (32). Thus, in the present study, it seems probable that theincreased serum IgE was mediated by increased IL-4 productionfrom Th2 cells, and not direct B cell activation by IL-18.
A plausible scenario is that IL-18 induces the production ofother cytokines that promote Th2 differentiation or proliferation.
IL-4 strongly promotes Th2 differentiation and IgE class switch-ing; however, it has been reported that IL-18 did not induce IL-4production from splenocytes derived from animals pretreated withIL-2 (21) or from a Th2 cell clone (33). Given IL-18 induction ofIL-4 has only been examined in these specifically conditioned cellpopulations, more study in this area seems warranted, particularlyin cell populations known to express the IL-18R. Although Th2cells readily synthesize IL-4, Th2 cells reportedly do not express
FIGURE 8. Intranasally administered IL-18 augments the number ofmucus-secreting goblet cells in the airway epithelium. The lungs of micefrom the i.n. RW/PBS and i.n. RW1IL-18/PBS groups were fixed in For-malin, embedded in paraffin, sectioned, and stained with hematoxylin andperiodic acid-Schiff. Representative sections are shown at3100 magnifi-cation.A, A lung section from the i.n. RW/PBS group showing few mucus-containing cells.B, A lung section from the i.n. RW1IL-18/PBS groupshowing hypertrophy and hyperplasia of bronchial goblet cells containingmucus. Epithelial cells, mucus-staining goblet cells, bronchi, and vesselsare denoted by e, m, b, and v, respectively.
FIGURE 9. Intranasally administered IL-18 augments peribronchialand perivascular inflammation. The photographs are of lung sections fromrepresentative animals in each group at340 magnification. Vessels andbronchi are denoted by v and b, respectively.A, A lung from the i.n.RW/RW group with grade 1.0 peribronchial inflammation, grade 0 perivas-cular inflammation, and grade 1.0 total lung inflammation.B, A lung fromthe i.n. RW1IL-18/RW group with grade 4.0 peribronchial inflammation,grade 2.5 perivascular inflammation, and grade 6.5 total lung inflammation.C, The peribronchial, perivascular, and total lung inflammation scores formice in the i.n. RW/RW and i.n. RW1IL-18/RW groups. Values are ex-pressed as mean6 SEM for five or six mice.p, p # 0.05;pp, p # 0.01.
2708 IL-18 INDUCES ALLERGIC SENSITIZATION IN A MURINE MODEL OF ASTHMA
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

the IL-18R (34). IL-13 is also an important regulator of Th2 dif-ferentiation and contributes to IgE class switching in mice (35, 36).Recently, Hoshino et al. (21) reported that in vitro treatment withIL-18 plus IL-2 was able to stimulate production of IL-13 from NKand T cells obtained from C57BL/6 mice. IL-13 was only induced,however, in splenocyte populations with expanded T cell and NKcell components produced by pretreating the mice with IL-2, andwas not induced in splenocytes from untreated mice. Although it isnot known what role IL-13 may have played in the present study,the mice were not pretreated with IL-2, and thus IL-18 may nothave stimulated IL-13. IL-18 has also been reported to stimulateIL-10 production in mouse spleen cell cultures (37). IL-10 is not astrong candidate for mediating the effects of IL-18, however, be-cause it has not been reported to stimulate Th2 differentiation, andprior studies disagree as to whether IL-10 stimulates IgE produc-tion (38) or inhibits IgE production (39, 40). Finally, the inductionof IL-1b by IL-18 could represent a proinflammatory pathwaycontributing to Th2 cell proliferation. IL-1a and IL-1b have beenreported to stimulate Th2 cell proliferation via the IL-1R afterTCR costimulation (41). Also, in a manner similar to the effects ofIL-18 in the present study, IL-1a and IL-1b were recently reportedto be effective adjuvants for mucosal and systemic immune re-sponses when coadministered with protein immunogens (42).
As indicated above, the Th2-promoting effects of IL-18 can benarrowed down to the most plausible sites of action (Fig. 10). IL-4and IL-13 stimulate Th2 differentiation, and IL-1b promotes Th2cell proliferation, so it seems plausible that IL-18 may produce anexpanded Th2 cell population via the actions of one or more ofthese cytokines. It is unlikely that IL-18 directly stimulates Th2cell differentiation, because IL-18 reportedly does not directlystimulate Th2 cells (29, 33), and Th2 cells reportedly do not ex-press the IL-18R (34). Mixed T cell populations, when culturedwith IL-12, but before Th1 differentiation, have been shown toexpress IL-18R, however, indicating that non-Th1 T cells at spe-cific stages of differentiation may be directly responsive to IL-18(43). Similarly, CD41, CD81, and CD42CD82 T cells have been
shown to express the IL-18R, but only upon stimulation in thepresence of IL-12 (44). Although it is uncertain what effects IL-18could produce in these cell populations in variable in vivo envi-ronments, because of the obligatory preexposure to IL-12, IFN-gsynthesis would be the expected result. Still, IL-18 has been shownto stimulate IL-13 protein synthesis from purified T cells (21),suggesting that under some conditions, IL-18 can stimulate T cellsexpressing the IL-18R to produce a Th2 cytokine. NK cells mayalso play a role in mediating the effects of IL-18 as they express theIL-18R (45), and produce IL-13 and IL-5 (21, 46). Consistent withthis premise, NK cells have been reported to contribute to thedevelopment of Ag-specific IgE, increased BAL levels of IL-4 andIL-5, and eosinophilic airway inflammation in a murine model ofallergic asthma (47). Similarly, NK1 T cells have been reported torapidly produce large quantities of IL-4 upon ligation of surfaceCD1 (48). IL-18 has been shown to stimulate the lytic activity ofisolated NK1 T cells (49), suggesting the IL-18R is present in thiscell population, and that IL-18 potentially stimulates other NK1 Tcell activities, including the rapid production of IL-4.
In view of reports that bacterial sinusitis can exacerbate asthmain children and adults (50, 51), it is tempting to speculate thatIL-18 may mediate these effects particularly in view of its abilityto augment allergic sensitization. Microbial products such as LPS(52, 53), and specific bacterial DNA sequences (22) have beenreported to stimulate macrophage production of IL-18, suggestingthat bacterial infections can stimulate the production of IL-18. Inconformity with human asthma, in the present study, IL-18 stim-ulated prolonged airway eosinophilia and increased mucus produc-tion by airway epithelium. Recently, IL-18 was reported to beconstitutively expressed in normal human lung, and down-regu-lated in the lungs of six mild asthmatics (54). Although it is pre-mature to draw major conclusions from a study with a small sam-ple size, this evidence may suggest that endogenous IL-18 does notaugment airway eosinophilia in ongoing mild asthma, but it doesnot exclude the possibility that IL-18 is up-regulated in asthmaexacerbations initiated by bacterial sinusitis. Also, in the presentstudy, intrapulmonary administration of IL-18 with RW potentlyinduced allergic sensitization to RW, suggesting that IL-18 may bea critical factor initiating allergic sensitization in asthmatic patients.Indeed, large scale epidemiological studies strongly suggest that al-lergic sensitization manifested by atopy and allergic rhinitis precedesthe onset of asthma symptoms by a period of years (55–57).
In summary, this is the first report demonstrating that IL-18differentially modulates allergen-induced eosinophilic inflamma-tion, promotes a Th2 phenotype, and potently induces allergic sen-sitization. These results suggest that IL-18 may contribute to thepathogenesis of allergic asthma.
References1. Robinson, D., Q. Hamid, A. Bentley, S. Ying, A. B. Kay, and S. R. Durham.
1993. Activation of CD41 T cells, increased TH2-type cytokine mRNA expres-sion, and eosinophil recruitment in bronchoalveolar lavage after allergen inhala-tion challenge in patients with atopic asthma.J. Allergy Clin. Immunol. 92:313.
2. Robinson, D. S., Q. Hamid, S. Ying, A. Tsicopoulos, J. Barkans, A. M. Bentley,C. Corrigan, S. R. Durham, and A. B. Kay. 1992. Predominant TH2-like bron-choalveolar T-lymphocyte population in atopic asthma.N. Engl. J. Med. 326:298.
3. Coyle, A. J., G. Le Gros, C. Bertrand, S. Tsuyuki, C. H. Heusser, M. Kopf, andG. P. Anderson. 1995. Interleukin-4 is required for the induction of lung Th2mucosal immunity.Am. J. Respir. Cell Mol. Biol. 13:54.
4. Brusselle, G., J. Kips, G. Joos, H. Bluethmann, and R. Pauwels. 1995. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and in-terleukin-4-deficient mice.Am. J. Respir. Cell Mol. Biol. 12:254.
5. Nakajima, H., I. Iwamoto, S. Tomoe, R. Matsumura, H. Tomioka, K. Takatsu,and S. Yoshida. 1992. CD41 T-lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea.Am. Rev. Respir. Dis.146:374.
6. Mauser, P. J., A. M. Pitman, X. Fernandez, S. K. Foran, G. K. Adams,W. Kreutner, R. W. Egan, and R.W. Chapman. 1995. Effects of an antibody to
FIGURE 10. Possible IL-18-induced pathways for Th2 differentiation andproliferation. Immune cells that may contribute to the production of IL-4 andIL-13 may include NK or NK1 T cells. Alternately, IL-18 may directly stim-ulate undifferented T cells that express the IL-18R in some conditions. Theimmune cells that produce IL-18 are predominantly mononuclear cells.
2709The Journal of Immunology
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

interleukin-5 in a monkey model of asthma.Am. J. Respir. Crit. Care Med.152:467.
7. Nakajima, H., I. Iwamoto, and S. Yoshida. 1993. Aerosolized recombinant in-terferon-g prevents antigen-induced eosinophil recruitment in mouse trachea.Am.Rev. Respir. Dis. 148:1102.
8. Iwamoto, I., H. Nakajima, H. Endo, and S. Yoshida. 1993. Interferong regulatesantigen-induced eosinophil recruitment into the mouse airways by inhibiting theinfiltration of CD41 T cells.J. Exp. Med. 177:573.
9. Afonso, L. C., T. M. Scharton, L. Q. Vieira, M. Wysocka, Trinchieri, andP. Scott. 1994. The adjuvant effect of interleukin-12 in a vaccine againstLeish-mania major.Science 263:235.
10. Hsieh, C. S., S. E. Macatonia, C. S. Tripp, S. F. Wolf, A. O’Garra, andK. M. Murphy. 1993. Development of TH1 CD41 T cells through IL-12 pro-duced byListeria-induced macrophages.Science 260:547.
11. Seder, R. A., R. Gazzinelli, A. Sher, and W. E. Paul. 1993. Interleukin 12 acts directlyon CD41 T cells to enhance priming for interferong production and diminishesinterleukin 4 inhibition of such priming.Proc. Natl. Acad. Sci. USA 90:10188.
12. Gavett, S. H., D. J. O’Hearn, X. Li, S. K. Huang, F. D. Finkelman, andM. Wills-Karp. 1995. Interleukin 12 inhibits antigen-induced airway hyperrespon-siveness, inflammation, and Th2 cytokine expression in mice.J. Exp. Med. 182:1527.
13. Kips, J. C., G. J. Brusselle, G. F. Joos, R. A. Peleman, J. H. Tavernier,R. R. Devos, and R. A. Pauwels. 1996. Interleukin-12 inhibits antigen-inducedairway hyperresponsiveness in mice.Am. J. Respir. Crit. Care Med. 153:535.
14. Sur, S., J. Lam, P. Bouchard, A. Sigounas, D. Holbert, and W. J. Metzger. 1996.Immunomodulatory effects of IL-12 on allergic lung inflammation depend ontiming of doses.J. Immunol. 157:4173.
15. Yoshimoto, T., H. Okamura, Y. I. Tagawa, Y. Iwakura, and K. Nakanishi. 1997.Interleukin 18 together with interleukin 12 inhibits IgE production by inductionof interferon-g production from activated B cells.Proc. Natl. Acad. Sci. USA94:3948.
16. Munder, M., M. Mallo, K. Eichmann, and M. Modolell. 1998. Murine macrophagessecrete interferong upon combined stimulation with interleukin (IL)-12 and IL-18: anovel pathway of autocrine macrophage activation.J. Exp. Med. 187:2103.
17. Osaki, T., J. M. Peron, Q. Cai, H. Okamura, P. D. Robbins, M. Kurimoto,M. T. Lotze, and H. Tahara. 1998. IFN-g-inducing factor/IL-18 administrationmediates IFN-g- and IL-12-independent antitumor effects.J. Immunol. 160:1742.
18. Ohtsuki, T., M. J. Micallef, K. Kohno, T. Tanimoto, M. Ikeda, and M. Kurimoto.1997. Interleukin 18 enhances Fas ligand expression and induces apoptosis inFas-expressing human myelomonocytic KG-1 cells.Anticancer Res. 17:3253.
19. Tsutsui, H., K. Matsui, N. Kawada, Y. Hyodo, N. Hayashi, H. Okamura, K.Higashino, K. Nakanishi. 1997. IL-18 accounts for both TNF-a- and Fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice.J. Im-munol. 159:3961.
20. Puren, A. J., G. Fantuzzi, Y. Gu, M. S. Su, and C. A. Dinarello. 1998. Interleu-kin-18 (IFNg-inducing factor) induces IL-8 and IL-1b via TNFa production fromnon-CD141 human blood mononuclear cells.J. Clin. Invest. 101:711.
21. Hoshino, T. W. R. 1999. IL-18 is a potent coinducer of IL-13 in NK and T cells:a new potential role for IL-18 in modulating the immune response.J. Immunol.162:5070.
22. Roman, M., E. Martin-Orozco, J. S. Goodman, M. D. Nguyen, Y. Sato,A. Ronaghy, R. S. Kornbluth, D. D. Richman, D. A. Carson, and E. Raz. 1997.Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants.Nat. Med. 3:849.
23. Sur, S., J. S. Wild, B. K. Choudhury, N. Sur, R. Alam, and D. M. Klinman. 1999.Long term prevention of allergic lung inflammation in a mouse model of asthmaby CpG oligodeoxynucleotides.J. Immunol. 162:6284.
24. Ashcroft, T., J. M. Simpson, and V. Timbrell. 1988. Simple method of estimatingseverity of pulmonary fibrosis on a numerical scale.J. Clin. Pathol. 41:467.
25. Micallef, M. J., T. Ohtsuki, K. Kohno, F. Tanabe, S. Ushio, M. Namba, T. Tanimoto,K. Torigoe, M. Fujii, M. Ikeda, et al. 1996. Interferon-g-inducing factor enhances Thelper 1 cytokine production by stimulated human T cells: synergism with interleu-kin-12 for interferon-g production.Eur. J. Immunol. 26:1647.
26. Takeda, K., H. Tsutsui, T. Yoshimoto, O. Adachi, N. Yoshida, T. Kishimoto,H. Okamura, K. Nakanishi, and S. Akira. 1998. Defective NK cell activity andTh1 response in IL-18-deficient mice.Immunity 8:383.
27. Hofstra, C. L., I. Van Ark, G. Hofman, M. Kool, F. P. Nijkamp, andA. J. Van Oosterhout. 1998. Prevention of Th2-like cell responses by coadmin-istration of IL-12 and IL-18 is associated with inhibition of antigen-induced air-way hyperresponsiveness, eosinophilia, and serum IgE levels.J. Immunol. 161:5054.
28. Kumano, K., A. Nakao, H. Nakajima, F. Hayashi, M. Kurimoto, H. Okamura,Y. Saito, and I. Iwamoto. 1999. Interleukin-18 enhances antigen-induced eosinophilrecruitment into the mouse airways.Am. J. Respir. Crit. Care Med. 160:873.
29. Robinson, D., K. Shibuya, A. Mui, F. Zonin, E. Murphy, Sana, S. B. Hartley,S. Menon, R. Kastelein, F. Bazan, and A. O’Garra. 1997. IGIF does not drive Th1development but synergizes with IL-12 for interferon-g production and activatesIRAK and NFkB. Immunity 7:571.
30. Coffman, R. L., J. Ohara, M. W. Bond, J. Carty, A. Zlotnik, and W. E. Paul. 1986.B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells.J. Immunol. 136:4538.
31. Yoshimoto, T., N. Nagai, K. Ohkusu, H. Ueda, H. Okamura, and K. Nakanishi.1998. LPS-stimulated SJL macrophages produce IL-12 and IL-18 that inhibit IgEproduction in vitro by induction of IFN-g production from CD3intIL-2Rb1 Tcells.J. Immunol. 161:1483.
32. Lauwerys, B. R., J. C. Renauld, and F. A. Houssiau. 1998. Inhibition of in vitroimmunoglobulin production by IL-12 in murine chronic graft-vs.-host disease:synergism with IL-18.Eur. J. Immunol. 28:2017.
33. Kohno, K., J. Kataoka, T. Ohtsuki, Y. Suemoto, I. Okamoto, M. Usui, M. Ikeda,and M. Kurimoto. 1997. IFN-g-inducing factor (IGIF) is a costimulatory factoron the activation of Th1 but not Th2 cells and exerts its effect independently ofIL-12. J. Immunol. 158:1541.
34. Xu, D., W. L. Chan, B. P. Leung, D. Hunter, K. Schulz, R. W. Carter,I. B. McInnes, J. H. Robinson, and F. Y. Liew. 1998. Selective expression andfunctions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells.J. Exp. Med. 188:1485.
35. McKenzie, G. J., C. L. Emson, S. E. Bell, S. Anderson, P. Fallon, G. Zurawski,R. Murray, R. Grencis, and A. N. McKenzie. 1998. Impaired development of Th2cells in IL-13-deficient mice.Immunity 9:423.
36. Emson, C. L., S. E. Bell, A. Jones, W. Wisden, and A. N. McKenzie. 1998.Interleukin (IL)-4-independent induction of immunoglobulin (Ig)E, and pertur-bation of T cell development in transgenic mice expressing IL-13.J. Exp. Med.188:399.
37. Micallef, M. J., T. Tanimoto, K. Kohno, M. Ikeda, H. Ikegami, and M. Kurimoto.1998. Augmentation of in vitro interleukin 10 production after in vivo adminis-tration of interleukin 18 is activated macrophage-dependent and is probably notinvolved in the antitumor effects of interleukin 18.Anticancer Res. 18:4267.
38. Ohmori, H., T. Kanda, T. Takai, and M. Hikida. 1995. Induction of antigen-specific IgE response in murine lymphocytes by IL-10.Immunol. Lett. 47:127.
39. Punnonen, J., M. de Waal, P. van Vlasselaer, J. F. Gauchat, and J. E. de Vries.1993. IL-10 and viral IL-10 prevent IL-4-induced IgE synthesis by inhibiting theaccessory cell function of monocytes.J. Immunol. 151:1280.
40. Dobber, R., M. Tielemans, and L. Nagelkerken. 1995. The in vivo effects ofneutralizing antibodies against IFN-g, IL-4, or IL-10 on the humoral immuneresponse in young and aged mice.Cell. Immunol. 160:185.
41. Huber, M., A. Rutherford, W. Meister, A. Weiss,M. Rollinghoff, and M. Lohoff.1996. TCR- and IL-1-mediated co-stimulation reveals an IL-4-independent wayof Th2 cell proliferation.Int. Immunol. 8:1257.
42. Staats, H. F., and F. A. Ennis Jr. 1999. IL-1 is an effective adjuvant for mucosaland systemic immune responses when coadministered with protein immunogens.J. Immunol. 162:6141.
43. Yoshimoto, T., K. Takeda, T. Tanaka, K. Ohkusu, S. Kashiwamura, H. Okamura,S. Akira, and K. Nakanishi. 1998. IL-12 up-regulates IL-18 receptor expressionon T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-g production.J. Immunol. 161:3400.
44. Tomura, M., S. Maruo, J. Mu, X. Y. Zhou, H. J. Ahn, T. Hamaoka, H. Okamura,K. Nakanishi, S. Clark, M. Kurimoto, and H. Fujiwara. 1998. Differential capac-ities of CD41, CD81, and CD42CD82 T cell subsets to express IL-18 receptorand produce IFN-g in response to IL-18.J. Immunol. 160:3759.
45. Kunikata, T., K. Torigoe, S. Ushio, T. Okura, C. Ushio, H. Yamauchi, M. Ikeda,H. Ikegami, and M. Kurimoto. 1998. Constitutive and induced IL-18 receptorexpression by various peripheral blood cell subsets as determined by anti-hIL-18R monoclonal antibody.Cell. Immunol. 189:135.
46. Walker, C., J. Checkel, S. Cammisuli, P. J. Leibson, and G. J. Gleich. 1998. IL-5production by NK cells contributes to eosinophil infiltration in a mouse model ofallergic inflammation.J. Immunol. 161:1962.
47. Korsgren, M., C. G. Persson, F. Sundler, T. Bjerke, T. Hansson, B. J. Chambers,S. Hong, L. Van Kaer, H. G. Ljunggren, and O. Korsgren. 1999. Natural killercells determine development of allergen-induced eosinophilic airway inflamma-tion in mice.J. Exp. Med. 189:553.
48. Bendelac, A., R. D. Hunziker, and O. Lantz. 1996. Increased interleukin 4 andimmunoglobulin E production in transgenic mice overexpressing NK1 T cells.J. Exp. Med. 184:1285.
49. Dao, T., W. Z. Mehal, and I. N. Crispe. 1998. IL-18 augments perforin-dependentcytotoxicity of liver NK-T cells.J. Immunol. 161:2217.
50. Rachelefsky, G. S., R. M. Katz, and S. C. Siegel. 1984. Chronic sinus diseasewith associated reactive airway disease in children.Pediatrics 73:526.
51. Nishioka, G. J., P. R. Cook, W. E. Davis, and J. P. McKinsey. 1994. Functionalendoscopic sinus surgery in patients with chronic sinusitis and asthma.Otolar-yngol. Head Neck Surg. 110:494.
52. Nakamura, K., H. Okamura, M. Wada, K. Nagata, and T. Tamura. 1989. Endo-toxin-induced serum factor that stimulatesg interferon production.Infect. Immun.57:590.
53. Okamura, H., K. Nagata, T. Komatsu, T. Tanimoto, Y. Nukata, F. Tanabe,K. Akita, K. Torigoe, T. Okura, and S. Fukuda. 1995. A novel costimulatoryfactor for g interferon induction found in the livers of mice causes endotoxicshock.Infect. Immun. 63:3966.
54. Cameron, L. A. 1999. Airway epithelium expresses interleukin-18.Eur. Respir.J. 14:553.
55. Sporik, R., S. T. Holgate, and J. J. Cogswell. 1991. Natural history of asthma inchildhood: a birth cohort study.Arch. Dis. Child. 66:1050.
56. Van Asperen, P. P., and A. Mukhi. 1994. Role of atopy in the natural history ofwheeze and bronchial hyper-responsiveness in childhood.Pediat. Allergy Immu-nol. 5:178.
57. Settipane, R. J., G. W. Hagy, and G. A. Settipane. 1994. Long-term risk factorsfor developing asthma and allergic rhinitis: a 23-year follow-up study of collegestudents.Allergy Proc. 15:21.
2710 IL-18 INDUCES ALLERGIC SENSITIZATION IN A MURINE MODEL OF ASTHMA
by guest on March 17, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from




















