
A Monte Carlo Model of DNA Double-Strand Break Clustering and Rejoining Kinetics for the Analysis of...
11
453 RADIATION RESEARCH 162, 453–463 (2004) 0033-7587/ 04 $15.00 q 2004 by Radiation Research Society. All rights of reproduction in any form reserved. A Monte Carlo Model of DNA Double-Strand Break Clustering and Rejoining Kinetics for the Analysis of Pulsed-Field Gel Electrophoresis Data M. Pinto, 1 K. M. Prise and B. D. Michael Gray Cancer Institute, Mount Vernon Hospital, Northwood, HA6 2JR Middlesex, United Kingdom Pinto, M., Prise, K. M. and Michael, B. D. A Monte Carlo Model of DNA Double-Strand Break Clustering and Rejoin- ing Kinetics for the Analysis of Pulsed-Field Gel Electropho- resis Data. Radiat. Res. 162, 453–463 (2004). In studies of radiation-induced DNA fragmentation and re- pair, analytical models may provide rapid and easy-to-use methods to test simple hypotheses regarding the breakage and rejoining mechanisms involved. The random breakage model, according to which lesions are distributed uniformly and in- dependently of each other along the DNA, has been the model most used to describe spatial distribution of radiation-induced DNA damage. Recently several mechanistic approaches have been proposed that model clustered damage to DNA. In gen- eral, such approaches focus on the study of initial radiation- induced DNA damage and repair, without considering the ef- fects of additional (unwanted and unavoidable) fragmentation that may take place during the experimental procedures. While most approaches, including measurement of total DNA mass below a specified value, allow for the occurrence of back- ground experimental damage by means of simple subtractive procedures, a more detailed analysis of DNA fragmentation necessitates a more accurate treatment. We have developed a new, relatively simple model of DNA breakage and the re- sulting rejoining kinetics of broken fragments. Initial radia- tion-induced DNA damage is simulated using a clustered breakage approach, with three free parameters: the number of independently located clusters, each containing several DNA double-strand breaks (DSBs), the average number of DSBs within a cluster (multiplicity of the cluster), and the maximum allowed radius within which DSBs belonging to the same cluster are distributed. Random breakage is simulated as a special case of the DSB clustering procedure. When the model is applied to the analysis of DNA fragmentation as mea- sured with pulsed-field gel electrophoresis (PFGE), the hy- pothesis that DSBs in proximity rejoin at a different rate from that of sparse isolated breaks can be tested, since the kinetics of rejoining of fragments of varying size may be followed by means of computer simulations. The problem of how to ac- count for background damage from experimental handling is also carefully considered. We have shown that the conven- tional procedure of subtracting the background damage from the experimental data may lead to erroneous conclusions dur- ing the analysis of both initial fragmentation and DSB rejoin- ing. Despite its relative simplicity, the method presented al- lows both the quantitative and qualitative description of ra- diation-induced DNA fragmentation and subsequent rejoining of double-stranded DNA fragments. q 2004 by Radiation Research Society INTRODUCTION Radiation-induced DNA damage (1, 2) and subsequent repair may be described both qualitatively and quantita- tively by means of analytical or numerical models. Focus- ing mainly on the DNA double-strand break as a relevant biological lesion, the most simple and perhaps most popular approach is the random breakage model. This was initially proposed to describe breakage of long-chain polymers (3– 5) and was reassessed more recently to provide analytical functions for the analysis of DNA fragmentation using more advanced techniques, including gene probing (6, 7 ). Random breakage models are consistent with experimental data on DNA fragmentation induced by sparsely ionizing radiations. These include soft X or g rays and also fast electrons and high-energy protons. Ionizing radiation caus- es damage along the intersection between charged-particle tracks and biological targets, such as the chromatin fiber. The combination of charged-particle track structure and the organization of chromatin at the cell cycle interphase is instrumental in the formation of clustered damage to DNA at both the nanometer and micrometer levels. At the scale of DNA associated with nucleosomes, lesions may be of several orders of complexity, classified as locally multiply damaged sites (LMDS) by Ward (8). The complexity of individual LMDS is a function of radiation quality (9), and the higher the level of complexity, the more biologically relevant the lesion is believed to be. Due to the three-di- mensional organization of the DNA double helix in the form of the 30-nm chromatin fiber, supercoiled structures 1 Address for correspondence: Department of Radiology, Division of Radiation Research, University of Medicine and Dentistry of New Jersey, New Jersey Medical School, MSB F-451 185 South Orange Avenue, University Heights, Newark, NJ 07103-2714; e-mail: pintoma@umdnj. edu.
Transcript of A Monte Carlo Model of DNA Double-Strand Break Clustering and Rejoining Kinetics for the Analysis of...

453
RADIATION RESEARCH 162, 453–463 (2004)0033-7587/04 $15.00q 2004 by Radiation Research Society.All rights of reproduction in any form reserved.
A Monte Carlo Model of DNA Double-Strand Break Clustering andRejoining Kinetics for the Analysis of Pulsed-Field Gel
Electrophoresis Data
M. Pinto,1 K. M. Prise and B. D. Michael
Gray Cancer Institute, Mount Vernon Hospital, Northwood, HA6 2JR Middlesex, United Kingdom
Pinto, M., Prise, K. M. and Michael, B. D. A Monte CarloModel of DNA Double-Strand Break Clustering and Rejoin-ing Kinetics for the Analysis of Pulsed-Field Gel Electropho-resis Data. Radiat. Res. 162, 453–463 (2004).
In studies of radiation-induced DNA fragmentation and re-pair, analytical models may provide rapid and easy-to-usemethods to test simple hypotheses regarding the breakage andrejoining mechanisms involved. The random breakage model,according to which lesions are distributed uniformly and in-dependently of each other along the DNA, has been the modelmost used to describe spatial distribution of radiation-inducedDNA damage. Recently several mechanistic approaches havebeen proposed that model clustered damage to DNA. In gen-eral, such approaches focus on the study of initial radiation-induced DNA damage and repair, without considering the ef-fects of additional (unwanted and unavoidable) fragmentationthat may take place during the experimental procedures.While most approaches, including measurement of total DNAmass below a specified value, allow for the occurrence of back-ground experimental damage by means of simple subtractiveprocedures, a more detailed analysis of DNA fragmentationnecessitates a more accurate treatment. We have developed anew, relatively simple model of DNA breakage and the re-sulting rejoining kinetics of broken fragments. Initial radia-tion-induced DNA damage is simulated using a clusteredbreakage approach, with three free parameters: the numberof independently located clusters, each containing severalDNA double-strand breaks (DSBs), the average number ofDSBs within a cluster (multiplicity of the cluster), and themaximum allowed radius within which DSBs belonging to thesame cluster are distributed. Random breakage is simulatedas a special case of the DSB clustering procedure. When themodel is applied to the analysis of DNA fragmentation as mea-sured with pulsed-field gel electrophoresis (PFGE), the hy-pothesis that DSBs in proximity rejoin at a different rate fromthat of sparse isolated breaks can be tested, since the kineticsof rejoining of fragments of varying size may be followed bymeans of computer simulations. The problem of how to ac-count for background damage from experimental handling isalso carefully considered. We have shown that the conven-tional procedure of subtracting the background damage fromthe experimental data may lead to erroneous conclusions dur-ing the analysis of both initial fragmentation and DSB rejoin-
ing. Despite its relative simplicity, the method presented al-lows both the quantitative and qualitative description of ra-diation-induced DNA fragmentation and subsequent rejoiningof double-stranded DNA fragments.q 2004 by Radiation Research Society
INTRODUCTION
Radiation-induced DNA damage (1, 2) and subsequentrepair may be described both qualitatively and quantita-tively by means of analytical or numerical models. Focus-ing mainly on the DNA double-strand break as a relevantbiological lesion, the most simple and perhaps most popularapproach is the random breakage model. This was initiallyproposed to describe breakage of long-chain polymers (3–5) and was reassessed more recently to provide analyticalfunctions for the analysis of DNA fragmentation usingmore advanced techniques, including gene probing (6, 7).Random breakage models are consistent with experimentaldata on DNA fragmentation induced by sparsely ionizingradiations. These include soft X or g rays and also fastelectrons and high-energy protons. Ionizing radiation caus-es damage along the intersection between charged-particletracks and biological targets, such as the chromatin fiber.The combination of charged-particle track structure and theorganization of chromatin at the cell cycle interphase isinstrumental in the formation of clustered damage to DNAat both the nanometer and micrometer levels. At the scaleof DNA associated with nucleosomes, lesions may be ofseveral orders of complexity, classified as locally multiplydamaged sites (LMDS) by Ward (8). The complexity ofindividual LMDS is a function of radiation quality (9), andthe higher the level of complexity, the more biologicallyrelevant the lesion is believed to be. Due to the three-di-mensional organization of the DNA double helix in theform of the 30-nm chromatin fiber, supercoiled structures
1 Address for correspondence: Department of Radiology, Division ofRadiation Research, University of Medicine and Dentistry of New Jersey,New Jersey Medical School, MSB F-451 185 South Orange Avenue,University Heights, Newark, NJ 07103-2714; e-mail: [email protected].

454 PINTO, PRISE AND MICHAEL
known as loops/rosettes/coils and chromosome territories[see for example refs. (10–12)], damaged sites that are cor-related in Euclidean space along the direction of thecharged-particle track will also be spatially correlated alongthe genomic distance of the DNA double helix. Lesionsproduced in spatial proximity may thus be at a genomicdistance that can range from a few tens to several millionbase pairs. For each order of chromatin structure that is hit,there is a corresponding level of regionally multiply dam-aged site (RMDS), defined by Rydberg (13) as the damageclustered on nucleosomes and the 30-nm chromatin fiber.
A number of numerical models have been developed toallow for deviations from predictions based on the randombreakage model as well as to simulate DNA lesion com-plexity at the nanometer level. Some models use an atom-istic description of the target, the DNA double helix, andits three-dimensional association with histones as well as adetailed description of the chain of physical and chemicalevents that follow the passage of a charged particle (or aphoton) in an aqueous environment. One example is thePARTRAC approach [see ref. (14) and references therein],which has also been used for the quantification of DNAfragmentation measured with PFGE techniques, with anemphasis on the formation of fragments that arise fromRMDS at higher-order chromatin structures (15–18). An-other example of the Monte Carlo approach for modelinginitial radiation-induced DNA damage is RADACK [(19)and references therein], which focuses on the radiolytic at-tack to DNA, or the charged-particle track structure-basedmodels described by Nikjoo (20, 21). Some investigatorshave developed approaches that focus on the radiation-in-duced formation of very small DNA segments originatingfrom multiple SSBs and DSBs on the 30-nm chromatinfiber (13, 22–24), while other recent studies have focusedon the importance of extrapolating data on DNA fragmen-tation to low doses of ionizing radiation. In particular, thelatter works have concentrated on genomic distance asso-ciation of DSBs (which will be referred to as DSB cluster-ing below) at the Mbp scale using a combination of ana-lytical and numerical approaches (25–29).
Essentially, all the above-mentioned computer modelsagree well with experimental data on initial DNA damageyield and distribution, while the random breakage modeloffers a much more limited tool, particularly for high-LETradiation. Recent experiments on radiation-induced DNAfragmentation, measured by gel electrophoresis techniques,suggest that in addition to damage caused by the ionizingradiation, manipulation of the sample itself may cause ex-tensive unwanted DNA damage (30–33). The unwantedDNA damage is difficult to account for experimentally, andconventional ways to correct for it may lead to a significantbias in the results. Briefly, when correcting data for theexperimental background damage measured in the unirra-diated controls by simply subtracting the fragmentation pat-terns, distortions arise in the fragmentation patterns them-selves. For fragments that would be generated by radiation
in the absence of background damage, such distortions mayeventually lead to an overestimation of relatively smallDNA fragments (hundreds of kbp and below, although theactual value may depend on both radiation dose and theextent of background damage, which varies from experi-ment to experiment) accompanied by an underestimation oflarger, Mbp-sized DNA fragments. As a result, this maylead to incorrect interpretations of both the mechanism ofinitial DNA fragmentation (random or non-random) and theproperties of its rejoining kinetics [see for example ref.(34)].
None of the numerical approaches reported above fo-cused on the issue of the influence of background damageto DNA on the experimental outcome. Recently, however,Friedland et al. have shown that if the background damageto DNA is incorporated in a Monte Carlo analysis of DNAfragmentation produced by protons of varying energy aswell as 60Co g rays, the radiation-induced DSB yields arenot affected (18). On the other hand, analytical methodshave been developed recently to make quantitative assess-ments that are less biased by the presence of experimentalbackground DNA damage (31, 35), although these are oflimited applicability since they rely on the random breakagemodel.
The rejoining kinetics of DSBs induced by ionizing ra-diation has commonly been used as an indicator of the bi-ological relevance of the lesion inflicted. After incubationof cells in conditions that favor repair of damage, or afterincubation of damaged DNA in the presence of cell extractsthat contain repair enzymes, data on the rate of disappear-ance of DSBs as a function of repair time are normallyfound to suggest the presence of two kinetic phases, andpossibly a residual, unrepairable damage component (36–41). Underlying single or multi-exponential decay compo-nents is the assumption that repair is a first-order process;i.e., the repair rate is proportional to the number of repair-able species (N), the individual DSBs, with a repair rateconstant, l.
dN5 2lN. (1)
dt
Several other models of rejoining kinetics exist, some ofwhich also account for DSB misrejoining events and theirrole in rejoining kinetics (42–44) as well as in the formationof chromosome aberrations (45). A comprehensive reviewis beyond the scope of this work [see also (46–48)].
To describe radiation-induced DNA fragmentation thatcan be measured using the techniques available, as well asDSB rejoining kinetics, we have developed a novel quali-tative and quantitative descriptive approach. The approachpresented here uses the clustered breakage formalism, in anumerical implementation, and does not use either an at-omistic description of DNA organization or charged-parti-cle transport in water. Throughout the paper, DSB cluster-ing refers to the spatial association of individual DSBs,

455MONTE CARLO MODEL OF RADIATION-INDUCED DSB CLUSTERING AND REJOINING
FIG. 1. The fragmentation that is produced from localization of four randomly distributed breaks (panel a) and four spatially associated breaks(panel b), and the superimposition of all eight breaks (panel c). Below each representation of the broken DNA fragments is the corresponding frequencydistribution of fragments of varying size.
regardless of their level of local complexity. Despite itssimplicity, however, the model is directly applicable for thedescription of DNA fragmentation data. The approach de-scribed below has been applied successfully to quantifyDNA fragmentation after exposure to both sparsely anddensely ionizing radiation as well as changes in the frag-ment size histograms due to DNA DSB rejoining (manu-script in preparation). While it was being developed, themethod was used in a pilot study in which the role of spatialproximity of lesions containing DSBs in determining DSBrejoining kinetics was investigated (34).
MATERIALS AND METHODS
Background Damage to DNA
A key feature of the newly developed numerical approach for the anal-ysis of DNA fragmentation is the consideration of the influence that back-ground DNA damage has on the experimental inference of radiation-induced DNA damage. Background damage to DNA caused during ex-perimental handling may be evaluated from an investigation of the un-irradiated control samples, but its influence on the complete fragmentationpattern, due to the combined action with radiation, is so complex that thefragmentation patterns may not be subtracted from each other. Also, back-ground damage appears to vary with preparation. Figure 1 shows thefragmentation of a single stretched segment of double-stranded DNA, ina stick-like representation. In panel a, four breaks are distributed random-ly on the DNA segment. In panel b, the same number of breaks arelocated in spatial proximity. The third representation in panel c showsthe situation in which all of the eight breaks (located on the DNA segmentas shown in panels a and b) are distributed on the same piece of DNA.Below each representation of DNA with locations of the breakage sites,a diagram shows the frequency size distribution using six histogram bins.The distribution after clustering of four breaks (panel b) shows a higherfrequency of small DNA fragments and only one very large fragment(whose size approaches that of the original intact fragment) compared tothe size distribution produced after randomly locating four breaks (dia-gram in panel a, histogram at the bottom). We assume that the randomly
induced breaks are caused by radiation (which is deemed to be an ex-cellent approximation for low-LET radiation), and the clustered breaksare introduced during the sample manipulation procedures (33, 35, 36).Experimentally, one can only measure the fragmentation profile as shownin panel b (unirradiated controls) and that produced by the superimpo-sition of both types of DSBs, as shown in panel c. This is due to somelevel of experimental background damage being unavoidable, and a directmeasurement of radiation-induced DNA fragmentation alone is not pos-sible. Our aim is to extract the frequency histogram shown in the lowerpanel a, starting from the direct measurement of the other two. If onewere to subtract histogram b from histogram c, the resulting histogram(b 2 c) would be different from that shown in panel a. On the subtractedhistogram, there is a relative depletion of frequency of large fragments,with even a negative value in bin 6. Similarly, adding histograms a 1 bdoes not produce the histogram shown in panel c. While the total numberof breaks adds up, the fragmentation patterns should not be combinedarithmetically. The example presented above is a simplification of whatcan be measured in an experiment, but negative frequencies of Mbp-sizeddouble stranded fragments, at the higher molecular weight end of themolecular weight spectrum that may be resolved using PFGE, can indeedbe observed when subtractions of the kind shown are carried out (datanot shown). This practice, which was adopted extensively in the 1990s,is therefore discouraged.
Ideally one should have a model that does not require manipulation ofexperimental data to allow for the experimental background damage. Theapproach proposed here is to model the action of both fragmentationmechanisms to produce a simulated fragmentation profile that may becompared to the experimental data. Distributing ‘‘background breaks’’ onDNA requires knowledge of the underlying damaging mechanism thatproduces them. In PFGE experiments, DNA must be extracted from intactcells using a lysis protocol that employs lauryl-sarcosyl and proteinaseK in EDTA buffer, generally carried out overnight at 508C (49), althoughrecent experiments recommend lower temperatures to avoid conversionof heat-labile sites into breaks (50, 51). The fragmentation patterns in theunirradiated controls appear to be produced by clustered breakage (30–33), although the underlying mechanism is unknown. It is unlikely thatthe background damage to DNA observed in an experiment is indicativeof damage really present in untreated cells. Recently, an attempt has beenmade to give a mechanistic interpretation to the background damage inPFGE (52). While lacking a mechanistic model of background DNA dam-
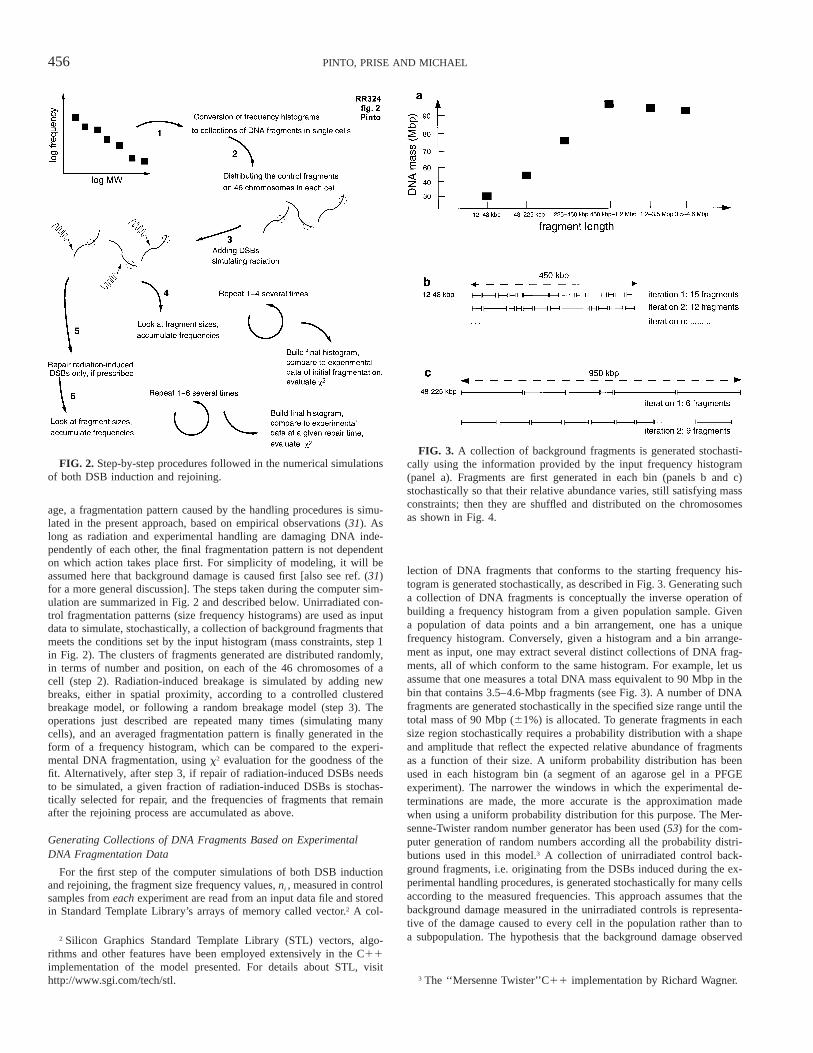
456 PINTO, PRISE AND MICHAEL
FIG. 2. Step-by-step procedures followed in the numerical simulationsof both DSB induction and rejoining.
FIG. 3. A collection of background fragments is generated stochasti-cally using the information provided by the input frequency histogram(panel a). Fragments are first generated in each bin (panels b and c)stochastically so that their relative abundance varies, still satisfying massconstraints; then they are shuffled and distributed on the chromosomesas shown in Fig. 4.
age, a fragmentation pattern caused by the handling procedures is simu-lated in the present approach, based on empirical observations (31). Aslong as radiation and experimental handling are damaging DNA inde-pendently of each other, the final fragmentation pattern is not dependenton which action takes place first. For simplicity of modeling, it will beassumed here that background damage is caused first [also see ref. (31)for a more general discussion]. The steps taken during the computer sim-ulation are summarized in Fig. 2 and described below. Unirradiated con-trol fragmentation patterns (size frequency histograms) are used as inputdata to simulate, stochastically, a collection of background fragments thatmeets the conditions set by the input histogram (mass constraints, step 1in Fig. 2). The clusters of fragments generated are distributed randomly,in terms of number and position, on each of the 46 chromosomes of acell (step 2). Radiation-induced breakage is simulated by adding newbreaks, either in spatial proximity, according to a controlled clusteredbreakage model, or following a random breakage model (step 3). Theoperations just described are repeated many times (simulating manycells), and an averaged fragmentation pattern is finally generated in theform of a frequency histogram, which can be compared to the experi-mental DNA fragmentation, using x2 evaluation for the goodness of thefit. Alternatively, after step 3, if repair of radiation-induced DSBs needsto be simulated, a given fraction of radiation-induced DSBs is stochas-tically selected for repair, and the frequencies of fragments that remainafter the rejoining process are accumulated as above.
Generating Collections of DNA Fragments Based on ExperimentalDNA Fragmentation Data
For the first step of the computer simulations of both DSB inductionand rejoining, the fragment size frequency values, ni , measured in controlsamples from each experiment are read from an input data file and storedin Standard Template Library’s arrays of memory called vector.2 A col-
2 Silicon Graphics Standard Template Library (STL) vectors, algo-rithms and other features have been employed extensively in the C11implementation of the model presented. For details about STL, visithttp://www.sgi.com/tech/stl.
lection of DNA fragments that conforms to the starting frequency his-togram is generated stochastically, as described in Fig. 3. Generating sucha collection of DNA fragments is conceptually the inverse operation ofbuilding a frequency histogram from a given population sample. Givena population of data points and a bin arrangement, one has a uniquefrequency histogram. Conversely, given a histogram and a bin arrange-ment as input, one may extract several distinct collections of DNA frag-ments, all of which conform to the same histogram. For example, let usassume that one measures a total DNA mass equivalent to 90 Mbp in thebin that contains 3.5–4.6-Mbp fragments (see Fig. 3). A number of DNAfragments are generated stochastically in the specified size range until thetotal mass of 90 Mbp (61%) is allocated. To generate fragments in eachsize region stochastically requires a probability distribution with a shapeand amplitude that reflect the expected relative abundance of fragmentsas a function of their size. A uniform probability distribution has beenused in each histogram bin (a segment of an agarose gel in a PFGEexperiment). The narrower the windows in which the experimental de-terminations are made, the more accurate is the approximation madewhen using a uniform probability distribution for this purpose. The Mer-senne-Twister random number generator has been used (53) for the com-puter generation of random numbers according all the probability distri-butions used in this model.3 A collection of unirradiated control back-ground fragments, i.e. originating from the DSBs induced during the ex-perimental handling procedures, is generated stochastically for many cellsaccording to the measured frequencies. This approach assumes that thebackground damage measured in the unirradiated controls is representa-tive of the damage caused to every cell in the population rather than toa subpopulation. The hypothesis that the background damage observed
3 The ‘‘Mersenne Twister’’C11 implementation by Richard Wagner.

457MONTE CARLO MODEL OF RADIATION-INDUCED DSB CLUSTERING AND REJOINING
FIG. 4. The collection of control DNA fragments is distributed equallyon each of the 46 chromosomes on a cell-by-cell basis. Backgroundbreaks are assumed to be concentrated in one region, which is also ac-cessible to radiation-induced DSBs. In the example shown, each of theseregions covers 7% of the chromosome size, with 7% being the experi-mentally determined FAR value. Solid circles on each chromosome areschematic representations of the centromeres.
is the result of DNA fragmentation in a cell subpopulation could be made,although it can be shown that this is unlikely to occur.4
Distributing the Control Fragments on 46 Chromosomes throughoutthe Genome
Once a collection of background damage-induced DSBs has been gen-erated in a single cell, the breaks need to be distributed throughout thegenome. No information is currently available regarding the distributionof the background damage on different chromosomes, such as whetherthey are preferentially located on a subset of chromosomes. For simplic-ity, it is assumed that each chromosome is equally likely to be damagedduring the experimental procedures.
For the purpose of distributing background breaks on chromosomes, itis important to note that PFGE allows the experimenter to study DNAfragmentation only below a critical fragment threshold size, currently notgreater than 10 Mbp. For studies of human DNA, this is only a fractionof the size of even the smallest human chromosome. As a consequence,only a portion of the human genome fragmentation may be studied. Thefraction of DNA mass that becomes available for the analysis is thatwhich gets extracted in the gel, consisting of fragments below a giventhreshold size, and is quantified as the fraction of (radio)activity released5
(FAR 5 mass of DNA measured below the threshold size/total DNAmass). In particular, the computer-generated background fragments are allsmaller than 10 Mbp. Hence, as shown in Fig. 4, in the present model,it is assumed that every chromosome is occupied by background damage-generated breaks, not more than 10 Mbp apart, such that they span aregion that is only a fraction of a chromosome length, such fraction beingthe experimentally measured FAR value. Forty-six chromosomes of equalsize (the average human chromosome size, 139 Mbp) are considered percell. The simplification of chromosomes having equal size may not holdat very low doses of ionizing radiation, where the initial size of the intactfragments has a role in determining the shape of the fragmentation patternafter breakage [see also refs. (4, 54)]. However in the case of PFGEexperiments, it may be taken as a good approximation. In principle, theremay be several regions within a single chromosome, separated by severaltens of Mbp, that are damaged by the experimental procedures. Never-theless, radiation-induced DSBs located between those regions occupiedby background fragments would result in a relatively high yield of Mbp-sized double-stranded fragments at low doses, which does not seem tobe supported by measurements in our laboratory (data not shown). Foreach chromosome, the location of the region populated by backgroundDSBs is chosen randomly, using a uniform probability distribution (seeFig. 4). A different approach was used by Friedland et al., where thebackground damage was located near the telomere regions (18). Sincebackground and radiation-induced DSBs are assumed to be caused byindependent actions, the region populated by background breaks is alsomade accessible to radiation. After background breaks have been distrib-uted on all the chromosomes, they are selectively labeled to distinguishthem from the chromosome ends and from the radiation-induced DSBsthat are about to be allocated with the subsequent computer programmodule.
Radiation-Induced Initial DNA Fragmentation and x2 Fitting
By means of x2 minimization, the purpose of the computer simulationsof radiation-induced DNA fragmentation is to estimate the best value ofthree model parameters:
1. The number of independently located clusters of DSBs, Ncl .
4 M. Pinto, Induction and rejoining of DNA double strand breaks inhuman cells after exposure to ionising radiation: An experimental andmodelling approach. Ph.D. Thesis, Department of Oncology, UniversityCollege London, 2002.
5 The name FAR was proposed when quantification of DNA mass wasachieved by counting disintegrations of radionuclide-labeled DNA.
2. The average multiplicity of each cluster, m.3. The maximum cluster extent, 2r, a genomic distance of r in either
direction from the cluster center.
Minimization of the x2 value is sought through discrete sampling ofNcl , m and r values. Values for these three model parameters are readfrom an input data file along with the number of cells in which DNAfragmentation is going to be simulated (set to a constant value of 100;see below). Since the experimental data sets of DNA fragmentation spanseveral orders of magnitude, the x2 for the goodness of the fit is evaluatedas shown in Eq. (2):
i i 2[log( f ) 2 log( f )]exp sim2x 5 . (2)Oizlog( f )zi exp
For each molecular weight region (agarose gel section), i, andi if fsim exp
are the computer-simulated and the experimentally determined fragmentfrequency values, respectively. When the x2 was evaluated on raw data(without the logarithmic transformation as in Eq. 2), x2 minimization wasfound to be heavily dependent on the quality of fit at the low-molecular-weight end of the experimental data range, where frequency values arelargest. Due to the high precision of the approach used in the computersimulations, which handles individual DNA fragments in single cells,simulation of DNA fragmentation in 100 cells was found to be sufficientto match the accuracy of PFGE experimental determinations, where ittakes several tens of thousands cells before one can measure an averageDNA fragmentation profile. A x2 value for the goodness of fit is evaluatedusing Eq. (2) for each set of model parameters read from the input datafile. When random breakage is simulated, DSBs are located independentlyof each other; that is, the cluster multiplicity is null. For random breakage,

458 PINTO, PRISE AND MICHAEL
FIG. 5. Panel a: The intersection of a charged-particle track with sev-eral chromatin loops produces the fragmentation pattern shown in panelb. In panel c, the particle intersects chromatin loops with a different angle,producing the fragmentation pattern shown in panel d. If one considersevery order of chromatin organization, for a given radiation quality thereshould be a theoretical, expected fragmentation pattern, shown schemat-ically in panel e as a solid line. The pattern expected from random break-age is also shown for comparison with a dashed line. The peaks in thetheoretical distribution correspond to the many orders of chromatin or-ganization that are known.
FIG. 6. The maximum cluster extent 2r used in the computer simu-lation is related to radiation quality and chromatin structure. For ultrasoftX rays, for example, clustering of DNA damage due to a single photonmay extend up to only a few hundreds of base pairs due to the limitedrange of photoelectrons in water. For a relatively slow electron, whoserange is sufficient to travel across the 30-nm chromatin fiber, clusteringof DNA damage is expected to extend further, up to about 1 kbp. For ahigh-LET particle, clustered damage may extend up to several Mbp, anda single particle may cross a whole cell nucleus, intersecting one or morechromosome territories, depending on cell thickness. For each radiationquality hitting the orders of chromatin organization shown, there is acorresponding r value.each chromosome is selected randomly [see also ref. (47)]. Also, the
assumption is made that no hot spots exist in individual chromosomes;hence each DSB is located randomly along a chromosome according toa uniform probability distribution.
The DSB Clustering Procedure
As an extension of random breakage, the DSB clustering procedurehas been designed to simulate the clustering of DSBs that is expected tooriginate from the intersection of a single charged-particle track withchromatin structures, using the simplest approach possible. DSBs havebeen distributed using a simplistic abstraction of both track structure andinterphase chromosome organization. Clustering of DSBs is describedquantitatively by a set of model parameters that serve to summarize theentire physical and physical-chemical events caused by the passage of acharged particle through the nucleus of a mammalian cell. Other typesof DNA damage have not been accounted for in the present approach.Although the computer simulation does not explicitly distinguish betweenbreaks of different orders of complexity, the integration of an analyticalmodel in the design of the approach presented here addresses this prob-lem.
Computer simulation of DSBs of different orders of complexity is notnecessary for the scope of this work, since all DSBs are detected in thesame way with conventional PFGE. The presence of complex DSBs may
be inferred indirectly from results of computer-modeled rejoining kinet-ics, as described in below.
The origin of a cluster is a randomly located DSB. The extent thatdefines the region the cluster spans is then chosen randomly, using auniform deviate with 2r defining the upper limit of the cluster extent.Interchromosome clustering is not considered in the present stage of de-velopment of the model. The choice of a uniform probability distributionis an approximation that cannot account for fine structures in the expectedfragment length distributions, as shown schematically in Fig. 5 (diagrame). As a result of this choice, production of small fragments due to DSBclustering is favored compared to random breakage; this agrees with ex-perimental evidence of DNA fragmentation after exposure to high-LETradiation. The maximum cluster extent 2r may be viewed as a parameterrelated to both radiation quality and chromatin structure (Fig. 6). For avery short-ranged charged particle, one would expect that correlated DNAdamage would not extend beyond a certain number of base pairs fromthe primary damaged site. With low-energy photons, for example, whichlead to the production of photoelectrons that can travel up to tens ofnanometers away from the site at which they are produced, one expectscorrelated breakage at the level of the repeated units that define the 30-nm chromatin structure, so the maximum cluster extent may be in the

459MONTE CARLO MODEL OF RADIATION-INDUCED DSB CLUSTERING AND REJOINING
FIG. 7. How DSBs are clustered along chromosomes to simulate theeffect of a traversal of a single charged particle. In the example shown,the chromosome contains three clusters of DSBs, one with four breaksall together, one containing three breaks, and one an isolated break.
FIG. 8. DNA fragment length distributions may be measured for frag-ments that originate from background breaks as well as from fragmentswhose ends are radiation-induced breaks, in each case ignoring the pres-ence of other types of breaks. Frequencies of fragments whose ends areof any type can also be counted (not shown in figure).
range of a few kbp. Ultrasoft X rays are known to produce highly locallyclustered DNA damage, typical of high-LET radiation, which includesstrand breaks associated with base damage and base loss but not spatiallycorrelated damage at levels of 100 kbp or above. For charged particlesof longer range, the maximum cluster extent 2r may be of the order ofa few tens or hundreds of kbp if a single track intersects more than onechromatin stretch within a single looped chromatin domain. For particlesthat can cross the whole cell nucleus, the maximum cluster extent maybe more a property of chromatin organization than of radiation quality.For very heavy ions, particle velocity and radial dose distribution mayhave to be modeled, but this level of accuracy was beyond the scope ofthis work [see also ref. (55)].
The track structure of energy deposition events, and consequently DNAdamage, is modeled using another parameter, the expected multiplicity ofone DSB cluster, m. This is the number of expected additional breaksdistributed around the cluster origin, and it is related to the density ofDSB lesions along the part of chromatin being hit by a single charged-particle track. The multiplicity of a single cluster is considered here tobe a Poisson-distributed variable, to be chosen randomly (Fig. 7). Insummary, for each cluster, the combination of a variable cluster extentand a variable cluster multiplicity aims to simulate the effect of the in-tersection of a charged-particle track of varying radiation quality with themany orders of chromatin structures that can be probed for with PFGEtechniques. Additional DSBs (up to the value drawn for m) are distributedin each cluster within the limits set by the current randomly chosen clusterextent. A uniform probability distribution acts as the proximity functionfor the localization of the breaks within the same cluster. The choice ofa uniform distribution may be regarded as a simplification since it couldbe expected that, depending on the chromatin structure that is being hit,fragments should be produced of size resonant to the size of a repeatedunit, characteristic of the specific chromatin structure [as shown in Fig.
5 and by Rydberg (22) and Friedland et al. (18)]. Nevertheless, the agree-ment with PFGE experimental data is remarkable (Fig. 9 and manuscriptin preparation), suggesting that a finely structured probability distributionfor the localization of clustered breaks is not necessary when fitting PFGEdata. This may be due to the limited sensitivity of the PFGE technique,which makes detection of fine structures in the fragment size distributiondifficult. However, fine structure seems to be an important issue whenfocusing on formation of fragments of 1 kbp or less (13, 18, 22, 24).Once the prescribed number of DSBs has been distributed in a clusterwithin limits of the cluster extent, a new cluster is generated. The newcluster will fall on a chromosome that is chosen randomly.
Accumulation of DNA Fragments Size Frequencies
When the simulation of fragmentation in one cell is completed, frag-ment size frequencies are accumulated in preparation for x2 evaluationfor the goodness of fit to a PFGE fragmentation profile. When the sim-ulation for 100 cells is complete, a final frequency histogram is built,reflecting the fragmentation pattern averaged over all the 100 cells sim-ulated. Since the design of the computer codes written for the simulationsallows telomeres, radiation-induced breaks and background breaks to bedistinguished from each other, one can measure the size distribution offragments that are delimited by any type of DSB, particularly radiation-induced breaks, omitting the background breaks produced by PFGE. Thismay be desirable if one wants to focus on radiation-induced fragmentationalone (see Fig. 8) and not be influenced by the constraints set by theexperimental technique.
DSB Rejoining Kinetics
Background PFGE DSBs presumably are not present as frank breaksin viable cells, so only rejoining of radiation-induced DSBs is consideredin the computer simulations. Although the PFGE experimental data oninitial DNA fragmentation do not allow us to distinguish between un-wanted background DSBs and radiation-induced DSBs, the computersimulation can distinguish between the two. The radiation-induced frag-mentation pattern that has been computer-simulated using the randombreakage/clustering module described previously may then be employedas the substrate for the simulation of rejoining kinetics. For this purpose,a good agreement between the computer-generated best fit and the mea-sured initial DNA fragmentation profile is an essential condition for thecomputer-simulated rejoining process to produce good-quality fits to thedata on DSB rejoining kinetics.
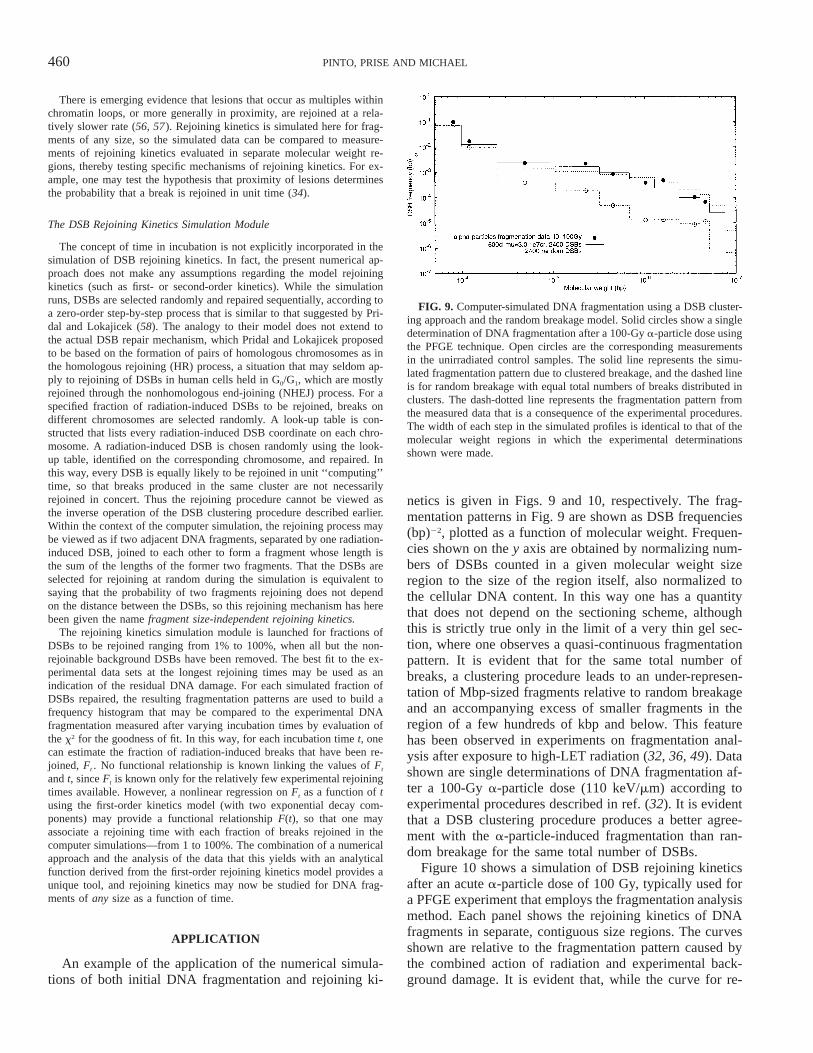
460 PINTO, PRISE AND MICHAEL
FIG. 9. Computer-simulated DNA fragmentation using a DSB cluster-ing approach and the random breakage model. Solid circles show a singledetermination of DNA fragmentation after a 100-Gy a-particle dose usingthe PFGE technique. Open circles are the corresponding measurementsin the unirradiated control samples. The solid line represents the simu-lated fragmentation pattern due to clustered breakage, and the dashed lineis for random breakage with equal total numbers of breaks distributed inclusters. The dash-dotted line represents the fragmentation pattern fromthe measured data that is a consequence of the experimental procedures.The width of each step in the simulated profiles is identical to that of themolecular weight regions in which the experimental determinationsshown were made.
There is emerging evidence that lesions that occur as multiples withinchromatin loops, or more generally in proximity, are rejoined at a rela-tively slower rate (56, 57). Rejoining kinetics is simulated here for frag-ments of any size, so the simulated data can be compared to measure-ments of rejoining kinetics evaluated in separate molecular weight re-gions, thereby testing specific mechanisms of rejoining kinetics. For ex-ample, one may test the hypothesis that proximity of lesions determinesthe probability that a break is rejoined in unit time (34).
The DSB Rejoining Kinetics Simulation Module
The concept of time in incubation is not explicitly incorporated in thesimulation of DSB rejoining kinetics. In fact, the present numerical ap-proach does not make any assumptions regarding the model rejoiningkinetics (such as first- or second-order kinetics). While the simulationruns, DSBs are selected randomly and repaired sequentially, according toa zero-order step-by-step process that is similar to that suggested by Pri-dal and Lokajicek (58). The analogy to their model does not extend tothe actual DSB repair mechanism, which Pridal and Lokajicek proposedto be based on the formation of pairs of homologous chromosomes as inthe homologous rejoining (HR) process, a situation that may seldom ap-ply to rejoining of DSBs in human cells held in G0/G1, which are mostlyrejoined through the nonhomologous end-joining (NHEJ) process. For aspecified fraction of radiation-induced DSBs to be rejoined, breaks ondifferent chromosomes are selected randomly. A look-up table is con-structed that lists every radiation-induced DSB coordinate on each chro-mosome. A radiation-induced DSB is chosen randomly using the look-up table, identified on the corresponding chromosome, and repaired. Inthis way, every DSB is equally likely to be rejoined in unit ‘‘computing’’time, so that breaks produced in the same cluster are not necessarilyrejoined in concert. Thus the rejoining procedure cannot be viewed asthe inverse operation of the DSB clustering procedure described earlier.Within the context of the computer simulation, the rejoining process maybe viewed as if two adjacent DNA fragments, separated by one radiation-induced DSB, joined to each other to form a fragment whose length isthe sum of the lengths of the former two fragments. That the DSBs areselected for rejoining at random during the simulation is equivalent tosaying that the probability of two fragments rejoining does not dependon the distance between the DSBs, so this rejoining mechanism has herebeen given the name fragment size-independent rejoining kinetics.
The rejoining kinetics simulation module is launched for fractions ofDSBs to be rejoined ranging from 1% to 100%, when all but the non-rejoinable background DSBs have been removed. The best fit to the ex-perimental data sets at the longest rejoining times may be used as anindication of the residual DNA damage. For each simulated fraction ofDSBs repaired, the resulting fragmentation patterns are used to build afrequency histogram that may be compared to the experimental DNAfragmentation measured after varying incubation times by evaluation ofthe x2 for the goodness of fit. In this way, for each incubation time t, onecan estimate the fraction of radiation-induced breaks that have been re-joined, Ft . No functional relationship is known linking the values of Ft
and t, since Ft is known only for the relatively few experimental rejoiningtimes available. However, a nonlinear regression on Ft as a function of tusing the first-order kinetics model (with two exponential decay com-ponents) may provide a functional relationship F(t), so that one mayassociate a rejoining time with each fraction of breaks rejoined in thecomputer simulations—from 1 to 100%. The combination of a numericalapproach and the analysis of the data that this yields with an analyticalfunction derived from the first-order rejoining kinetics model provides aunique tool, and rejoining kinetics may now be studied for DNA frag-ments of any size as a function of time.
APPLICATION
An example of the application of the numerical simula-tions of both initial DNA fragmentation and rejoining ki-
netics is given in Figs. 9 and 10, respectively. The frag-mentation patterns in Fig. 9 are shown as DSB frequencies(bp)22, plotted as a function of molecular weight. Frequen-cies shown on the y axis are obtained by normalizing num-bers of DSBs counted in a given molecular weight sizeregion to the size of the region itself, also normalized tothe cellular DNA content. In this way one has a quantitythat does not depend on the sectioning scheme, althoughthis is strictly true only in the limit of a very thin gel sec-tion, where one observes a quasi-continuous fragmentationpattern. It is evident that for the same total number ofbreaks, a clustering procedure leads to an under-represen-tation of Mbp-sized fragments relative to random breakageand an accompanying excess of smaller fragments in theregion of a few hundreds of kbp and below. This featurehas been observed in experiments on fragmentation anal-ysis after exposure to high-LET radiation (32, 36, 49). Datashown are single determinations of DNA fragmentation af-ter a 100-Gy a-particle dose (110 keV/mm) according toexperimental procedures described in ref. (32). It is evidentthat a DSB clustering procedure produces a better agree-ment with the a-particle-induced fragmentation than ran-dom breakage for the same total number of DSBs.
Figure 10 shows a simulation of DSB rejoining kineticsafter an acute a-particle dose of 100 Gy, typically used fora PFGE experiment that employs the fragmentation analysismethod. Each panel shows the rejoining kinetics of DNAfragments in separate, contiguous size regions. The curvesshown are relative to the fragmentation pattern caused bythe combined action of radiation and experimental back-ground damage. It is evident that, while the curve for re-
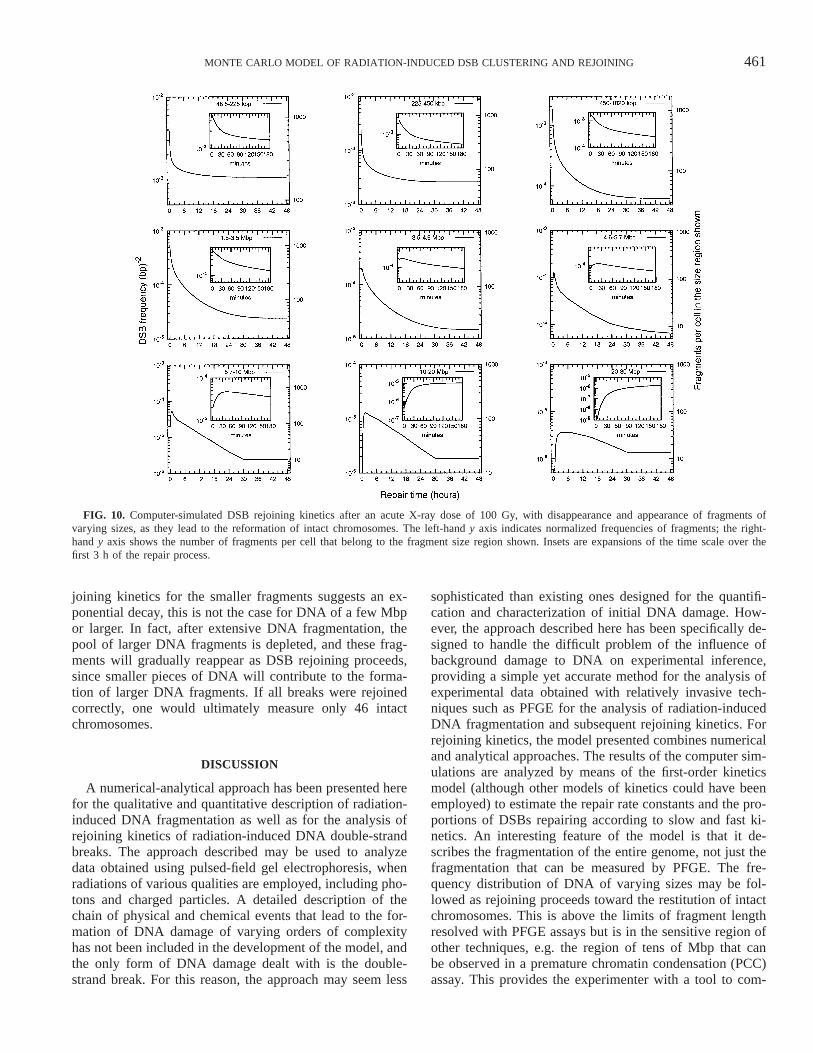
461MONTE CARLO MODEL OF RADIATION-INDUCED DSB CLUSTERING AND REJOINING
FIG. 10. Computer-simulated DSB rejoining kinetics after an acute X-ray dose of 100 Gy, with disappearance and appearance of fragments ofvarying sizes, as they lead to the reformation of intact chromosomes. The left-hand y axis indicates normalized frequencies of fragments; the right-hand y axis shows the number of fragments per cell that belong to the fragment size region shown. Insets are expansions of the time scale over thefirst 3 h of the repair process.
joining kinetics for the smaller fragments suggests an ex-ponential decay, this is not the case for DNA of a few Mbpor larger. In fact, after extensive DNA fragmentation, thepool of larger DNA fragments is depleted, and these frag-ments will gradually reappear as DSB rejoining proceeds,since smaller pieces of DNA will contribute to the forma-tion of larger DNA fragments. If all breaks were rejoinedcorrectly, one would ultimately measure only 46 intactchromosomes.
DISCUSSION
A numerical-analytical approach has been presented herefor the qualitative and quantitative description of radiation-induced DNA fragmentation as well as for the analysis ofrejoining kinetics of radiation-induced DNA double-strandbreaks. The approach described may be used to analyzedata obtained using pulsed-field gel electrophoresis, whenradiations of various qualities are employed, including pho-tons and charged particles. A detailed description of thechain of physical and chemical events that lead to the for-mation of DNA damage of varying orders of complexityhas not been included in the development of the model, andthe only form of DNA damage dealt with is the double-strand break. For this reason, the approach may seem less
sophisticated than existing ones designed for the quantifi-cation and characterization of initial DNA damage. How-ever, the approach described here has been specifically de-signed to handle the difficult problem of the influence ofbackground damage to DNA on experimental inference,providing a simple yet accurate method for the analysis ofexperimental data obtained with relatively invasive tech-niques such as PFGE for the analysis of radiation-inducedDNA fragmentation and subsequent rejoining kinetics. Forrejoining kinetics, the model presented combines numericaland analytical approaches. The results of the computer sim-ulations are analyzed by means of the first-order kineticsmodel (although other models of kinetics could have beenemployed) to estimate the repair rate constants and the pro-portions of DSBs repairing according to slow and fast ki-netics. An interesting feature of the model is that it de-scribes the fragmentation of the entire genome, not just thefragmentation that can be measured by PFGE. The fre-quency distribution of DNA of varying sizes may be fol-lowed as rejoining proceeds toward the restitution of intactchromosomes. This is above the limits of fragment lengthresolved with PFGE assays but is in the sensitive region ofother techniques, e.g. the region of tens of Mbp that canbe observed in a premature chromatin condensation (PCC)assay. This provides the experimenter with a tool to com-

462 PINTO, PRISE AND MICHAEL
pare the yield of DSBs measured with PFGE to the yieldof excess prematurely condensed chromosomes in condi-tions in which repair of DNA damage is minimized. To dateit has been difficult to compare DSB yields obtained bythese two techniques. For extensions of the DSB rejoiningkinetics model to the analysis of chromosome aberrations,algorithms of misrejoining of incorrect ends should be ex-plicitly considered. The model described here has been usedto analyze an extensive data set of initial DNA fragmen-tation and subsequent rejoining of DSBs induced inAG01522 primary human fibroblasts by 240 kVp X raysor 238Pu a particles. Since a complete discussion of theresults obtained from best-fitting PFGE data on the induc-tion of DSBs and rejoining kinetics would have overshad-owed the relevant aspects of the model, the application ofthe model will be presented separately. However, it is an-ticipated that, for DSB rejoining kinetics, when asking thequestion of whether proximity of lesions determines therejoining kinetics, one would obtain different conclusionsdepending on whether the background distribution of DSBsis subtracted or treated according to the method describedhere. This was also shown in a pilot study when the modelwas under development (34). In conclusion, although theeffect of the subtraction procedure to correct for the back-ground damage does not appear to change the estimates ofthe yields of radiation-induced DSBs (18, 32), it may leadto erroneous conclusions when investigating the shapes ofthe fragmentation patterns and ultimately the mechanismsof radiation-induced fragmentation and rejoining.
ACKNOWLEDGMENTS
The authors wish to thank Dr. James Heald for stimulating discussionson the design of part of the codes written for the development of thenumerical approach. The Cancer Research Campaign, the Sir SamuelScott of Yews Trust, and the Nuclear Fission Safety Program of the Eu-ropean Community generously supported this work with a Ph.D. fellow-ship to MP.
Received: December 9, 2003; accepted: May 21, 2004
REFERENCES
1. W. Burkart, T. Jung and G. Frasch, Damage pattern as a function ofradiation quality and other factors. CR Acad. Sci. III 322, 89–101(1999).
2. D. T. Goodhead, The initial physical damage produced by ionizingradiations. Int. J. Radiat. Biol. 56, 623–634 (1989).
3. E. W. Montroll and R. Simha, Theory of depolymerization of longchain molecules. J. Chem. Phys. 8, 721–727 (1940).
4. A. Charlesby, Molecular-weight changes in the degradation of longchain polymers. J. R. Stat. Soc. 120–128 (1953).
5. S. Litwin, The distribution of radioactive recovery in randomly cutand segmented DNA. J. Appl. Probab. 6, 275–284 (1969).
6. V. E. Cook and R. K. Mortimer, A quantitative model of DNA frag-ments generated by ionizing radiation and possible experimental ap-plications. Radiat. Res. 125, 102–106 (1991).
7. C. R. Contopoulou, V. E. Cook and R. K. Mortimer, Analysis ofDNA double strand breakage and repair using orthogonal field alter-nation gel electrophoresis. Yeast 3, 71–76 (1987).
8. J. F. Ward, Some biochemical consequences of the spatial distributionof ionizing radiation-produced free radicals. Radiat. Res. 86, 185–195 (1981).
9. K. M. Prise, Use of radiation quality as a probe for DNA lesioncomplexity. Int. J. Radiat. Biol. 65, 43–48 (1994).
10. J. Widom, Structure, dynamics, and function of chromatin in vitro.Annu. Rev. Biophys. Biomol. Struct. 27, 285–327 (1998).
11. J. Mirkovitch, S. M. Gasser and U. K. Laemmli, Relation of chro-mosome structure and gene expression. Phil. Trans. R. Soc. Lond. BBiol. Sci. 317, 563–574 (1987).
12. J. Filipski, J. Leblanc, T. Youdale, M. Sikorska and P. R. Walker,Periodicity of DNA folding in higher order chromatin structures.EMBO J. 9, 1319–1327 (1990).
13. B. Rydberg, Clusters of DNA damage induced by ionizing radiation:Formation of short DNA fragments. II. Experimental detection. Ra-diat. Res. 145, 200–209 (1996).
14. F. Ballarini, M. Biaggi, M. Merzagora, A. Ottolenghi, M. Dingfelder,W. Friedland, P. Jacob and H. G. Paretzke, Stochastic aspects anduncertainties in the prechemical and chemical stages of electrontracks in liquid water: A quantitative analysis based on Monte Carlosimulations. Radiat. Environ. Biophys. 39, 179–188 (2000).
15. W. Friedland, P. Jacob, H. G. Paretzke, M. Merzagora and A. Otto-lenghi, Simulation of DNA fragment distributions after irradiationwith photons. Radiat. Environ. Biophys. 38, 39–47 (1999).
16. W. Friedland, P. Jacob, H. G. Paretzke and T. Stork, Monte Carlosimulation of the production of short DNA fragments by low-linearenergy transfer radiation using higher-order DNA models. Radiat.Res. 150, 170–182 (1998).
17. W. Friedland, P. Jakob, H. G. Paretzke and T. Stork, Simulation ofstrand breaks and short DNA fragments in the biophysical modelPARTRAC. In Microdosimetry: An Interdisciplinary Approach (P.O’Neill, H. G. Menzel and D. T. Goodhead, Eds.), pp. 43–46. TheRoyal Society of Chemistry, Cambridge, 1997.
18. W. Friedland, P. Jacob, P. Bernhardt, H. G. Paretzke and M. Ding-felder, Simulation of DNA damage after proton irradiation. Radiat.Res. 159, 401–410 (2003).
19. M. Begusova, M. Spotheim-Maurizot, D. Sy, V. Michalik and M.Charlier, RADACK, a stochastic simulation of hydroxyl radical at-tack to DNA. J. Biomol. Struct. Dyn. 19, 141–158 (2001).
20. H. Nikjoo, P. O’Neill, M. Terrissol and D. T. Goodhead, Quantitativemodelling of DNA damage using Monte Carlo track structure meth-od. Radiat. Environ. Biophys. 38, 31–38 (1999).
21. H. Nikjoo, C. E. Bolton, R. Watanabe, M. Terrissol, P. O’Neill andD. T. Goodhead, Modelling of DNA damage induced by energeticelectrons (100 eV to 100 keV). Radiat. Prot. Dosim. 99, 77–80(2002).
22. B. Rydberg, W. R. Holley, I. S. Mian and A. Chatterjee, Chromatinconformation in living cells: Support for a zig-zag model of the 30nm chromatin fiber. J. Mol. Biol. 284, 71–84 (1998).
23. B. Rydberg, L. Heilbronn, W. R. Holley, M. Lobrich, C. Zeitlin, A.Chatterjee and P. K. Cooper, Spatial distribution and yield of DNAdouble-strand breaks induced by 3–7 MeV helium ions in humanfibroblasts. Radiat. Res. 158, 32–42 (2002).
24. W. R. Holley and A. Chatterjee, Clusters of DNA induced by ionizingradiation: Formation of short DNA fragments. I. Theoretical model-ing. Radiat. Res. 145, 188–199 (1996).
25. A. L. Ponomarev, F. A. Cucinotta, R. K. Sachs and D. J. Brenner,Monte Carlo predictions of DNA fragment-size distributions for largesizes after HZE particle irradiation. Phys. Med. 17 (Suppl.), 153–156(2001).
26. A. L. Ponomarev, D. Brenner, L. R. Hlatky and R. K. Sachs, Apolymer, random walk model for the size-distribution of large DNAfragments after high linear energy transfer radiation. Radiat. Environ.Biophys. 39, 111–120 (2000).
27. A. L. Ponomarev, F. A. Cucinotta, R. K. Sachs, D. J. Brenner andL. E. Peterson, Extrapolation of the DNA fragment-size distributionafter high-dose irradiation to predict effects at low doses. Radiat. Res.156, 594–597 (2001).

463MONTE CARLO MODEL OF RADIATION-INDUCED DSB CLUSTERING AND REJOINING
28. R. K. Sachs, A. L. Ponomarev, P. Hahnfeldt and L. R. Hlatky, Lo-cations of radiation-produced DNA double strand breaks along chro-mosomes: A stochastic cluster process formalism. Math. Biosci. 159,165–187 (1999).
29. R. K. Sachs, D. J. Brenner, P. J. Hahnfeldt and L. R. Hlatky, Aformalism for analysing large-scale clustering of radiation-inducedbreaks along chromosomes. Int. J. Radiat. Biol. 74, 185–206 (1998).
30. E. Hoglund, E. Blomquist, J. Carlsson and B. Stenerlow, DNA dam-age induced by radiation of different linear energy transfer: Initialfragmentation. Int. J. Radiat. Biol. 76, 539–547 (2000).
31. M. Pinto, H. C. Newman, K. M. Prise and B. D. Michael, Quantifi-cation of DNA damage by PFGE: Development of an analytical ap-proach to correct for the background distribution. Int. J. Radiat. Biol.76, 741–748 (2000).
32. M. Pinto, K. M. Prise and B. D. Michael, Quantification of radiationinduced DNA double-strand breaks in human fibroblasts by PFGE:Testing the applicability of random breakage models. Int. J. Radiat.Biol. 78, 375–388 (2002).
33. M. Belli, R. Cherubini, M. Dalla Vecchia, V. Dini, G. Esposito, G.Moschini, O. Sapora, G. Simone and M. A. Tabocchini, DNA frag-mentation in V79 cells irradiated with light ions as measured bypulsed-field gel electrophoresis. I. Experimental results. Int. J. Ra-diat. Biol. 78, 475–482 (2002).
34. M. Pinto, K. M. Prise and B. D. Michael, Double strand break re-joining after irradiation of human fibroblasts with X rays or alphaparticles: PFGE studies and numerical models. Radiat. Prot. Dosim.99, 133–136 (2002).
35. M. Belli, R. Cherubini, M. Dalla Vecchia, V. Dini, G. Esposito, G.Moschini, O. Sapora, C. Signoretti, G. Simone and M. A. Tabocchini,DNA fragmentation in mammalian cells exposed to various lightions. Adv. Space. Res. 27, 393–399 (2001).
36. H. Hoglund and B. Stenerlow, Induction and rejoining of DNA dou-ble-strand breaks in normal human skin fibroblasts after exposure toradiation of different linear energy transfer: Possible roles of trackstructure and chromatin organization. Radiat. Res. 155, 818–825(2001).
37. B. Stenerlow, E. Hoglund and J. Carlsson, Induction and rejoiningof large DNA fragments after ion irradiation. Radiat. Res. 151, 642–648 (1999).
38. B. Stenerlow, E. Hoglund, J. Carlsson and E. Blomquist, Rejoiningof DNA fragments produced by radiations of different linear energytransfer. Int. J. Radiat. Biol. 76, 549–557 (2000).
39. B. Stenerlow and E. Hoglund, Rejoining of double-stranded DNA-fragments studied in different size-intervals. Int. J. Radiat. Biol. 78,1–7 (2002).
40. B. P. Kysela, J. E. Arrand and B. D. Michael, Relative contributionsof levels of initial damage and repair of double-strand breaks to theionizing radiation-sensitive phenotype of the Chinese hamster cellmutant, XR-V15B. Part II. Neutrons. Int. J. Radiat. Biol. 64, 531–538 (1993).
41. B. P. Kysela, B. D. Michael and J. E. Arrand, Relative contributionsof levels of initial DNA damage and repair of double strand breaksto the ionizing radiation-sensitive phenotype of the Chinese hamstercell mutant, XR-V15B. Part I. X-rays. Int. J. Radiat. Biol. 63, 609–616 (1993).
42. T. Radivoyevitch, D. G. Hoel, P. Hahnfeldt and R. K. Sachs, Sizedistributions of misrejoining DNA fragments in irradiated cells.Math. Biosci. 149, 107–136 (1998).
43. T. Radivoyevitch, D. G. Hoel, A. M. Chen and R. K. Sachs, Misre-joining of double-strand breaks after X irradiation: Relating moderateto very high doses by a Markov model. Radiat. Res. 149, 59–67(1998).
44. T. Radivoyevitch, D. G. Hoel, P. J. Hahnfeldt, B. Rydberg and R. K.Sachs, Recent data obtained by pulsed-field gel electrophoresis sug-gest two types of double-strand breaks. Radiat. Res. 149, 52–58(1998).
45. W. R. Holley, I. S. Mian, S. J. Park, B. Rydberg and A. Chatterjee,A model for interphase chromosomes and evaluation of radiation-induced aberrations. Radiat. Res. 158, 568–580 (2002).
46. N. Foray, C. Badie, G. Alsbeih, B. Fertil and E. P. Malaise, A newmodel describing the curves for repair of both DNA double-strandbreaks and chromosome damage. Radiat. Res. 146, 53–60 (1996).
47. R. K. Sachs, A. M. Chen and D. J. Brenner, Proximity effects in theproduction of chromosome aberrations by ionizing radiation. Int. J.Radiat. Biol. 71, 1–19 (1997).
48. J. F. Fowler, Is repair of DNA strand break damage from ionizingradiation second-order rather than first-order? A simpler explanationof apparently multiexponential repair. Radiat. Res. 152, 124–136(1999).
49. H. C. Newman, K. M. Prise, M. Folkard and B. D. Michael, DNAdouble-strand break distributions in X-ray and alpha-particle irradi-ated V79 cells: Evidence for non-random breakage. Int. J. Radiat.Biol. 71, 347–363 (1997).
50. B. Rydberg, Radiation-induced heat-labile sites that convert intoDNA double-strand breaks. Radiat. Res. 153, 805–812 (2000).
51. B. Stenerlow, K. H. Karlsson, B. Cooper and B. Rydberg, Measure-ment of prompt DNA double-strand breaks in mammalian cells with-out including heat-labile sites: Results for cells deficient in nonho-mologous end joining. Radiat. Res. 159, 502–510 (2003).
52. I. K. Khvostunov, S. G. Andreev and Y. Eidelman, Biophysical anal-ysis of radiation induced initial DNA fragmentation. Radiat. Prot.Dosim. 99, 151–152 (2002).
53. M. Matsumoto and T. Nishimura, Mersenne Twister: A 623-dimen-sionally equidistributed uniform pseudo-random number generator.ACM Trans. Model. Comput. Simul. 8, 3–30 (1998).
54. B. E. Cedervall and T. J. McMillan, The fraction of DNA releasedon pulsed-field gel electrophoresis gels may differ significantly be-tween genomes at low levels of double-strand breaks. Radiat. Res.158, 247–249 (2002).
55. F. Kraxenberger, K. J. Weber, A. A. Friedl, F. Eckardt-Schupp, M.Flentje, P. Quicken and A. M. Kellerer, DNA double-strand breaksin mammalian cells exposed to gamma-rays and very heavy ions.Fragment-size distributions determined by pulsed-field gel electro-phoresis. Radiat. Environ. Biophys. 37, 107–115 (1998).
56. P. Johnston, S. MacPhail, T. Stamato, C. Kirchgessner and P. Olive,Higher-order chromatin structure-dependent repair of DNA double-strand breaks: Involvement of the V(D)J recombination double-strandbreak repair pathway. Radiat. Res. 149, 455–462 (1998).
57. B. Gauter, O. Zlobinskaya and K. J. Weber, Rejoining of radiation-induced DNA double-strand breaks: Pulsed-field gel electrophoresisanalysis of fragment size distributions after incubation for repair. Ra-diat. Res. 157, 721–733 (2002).
58. I. Pridal and M. V. Lokajicek, A model of DSB-repair kinetics. J.Theor. Biol. 111, 81–90 (1984).




















