
15424.pdf - IRUA - Institutional Repository van de Universiteit ...
201
TACKLING ETIOLOGICALLY UNSOLVED PROGRESSIVE MUSCLE DISORDERS: FROM RARE INHERITED MYOPATHIES TO SPORADIC INCLUSION BODY MYOSITIS HET ONTRAFELEN VAN ETIOLOGISCH ONOPGEHELDERDE, PROGRESSIEVE SPIERZIEKTEN: VAN ZELDZAME ERFELIJKE SPIERZIEKTEN TOT SPORADISCHE ‘INCLUSION BODY’-MYOSITIS Thesis submitted for the degree of Doctor of Medical Sciences at the University of Antwerp, Faculty of Medicine and Health Sciences to be defended by Willem DE RIDDER Promotors: Prof. Dr. Jonathan Baets Em. Prof. Dr. Peter De Jonghe Antwerp, 2020
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of 15424.pdf - IRUA - Institutional Repository van de Universiteit ...

TACKLING ETIOLOGICALLY UNSOLVED PROGRESSIVE MUSCLE DISORDERS:
FROM RARE INHERITED MYOPATHIES TO SPORADIC INCLUSION BODY
MYOSITIS
HET ONTRAFELEN VAN ETIOLOGISCH ONOPGEHELDERDE, PROGRESSIEVE
SPIERZIEKTEN: VAN ZELDZAME ERFELIJKE SPIERZIEKTEN TOT SPORADISCHE
‘INCLUSION BODY’-MYOSITIS
Thesis submitted for the degree of Doctor of Medical Sciences at the University of Antwerp,
Faculty of Medicine and Health Sciences
to be defended by
Willem DE RIDDER
Promotors:
Prof. Dr. Jonathan Baets
Em. Prof. Dr. Peter De Jonghe Antwerp, 2020


“Once you eliminate the impossible, whatever remains, no matter how improbable, must be
the truth.”
Arthur Conan Doyle
– one extra quote, for the initiated –
“If you throw a banana at a wall, there’s a small possibility that it will pass through the
wall.”
Garth Risk Hallberg


MEMBERS OF THE JURY
Promotors
Prof. Dr. Jonathan Baets (MD, PhD), University of Antwerp, Belgium
Em. Prof. Dr. Peter De Jonghe (MD, PhD), University of Antwerp, Belgium
Chair
Prof. Dr. Patrick Cras (MD, PhD), University of Antwerp, Belgium
Internal jury member
Prof. Dr. Bart Loeys (MD, PhD), University of Antwerp, Belgium
External jury members
Prof. Dr. Werner Stenzel (MD, PhD), Charité – Universitätsmedizin Berlin, Germany
Dr. Umesh A. Badrising (MD, PhD), Leiden University Medical Center, the Netherlands


7
CONTENT
GENERAL INTRODUCTION: ………………...……………………………………………………………………………….9
AIMS AND OUTLINE: ………………………………………………………………………………………………………….25
RESULTS: .…………………………………………………………………………………………………………………………..31
PART 1: Genetics of patients with suspected IMD and the study of molecular and/or
biological pathomechanisms of rare or novel causes of IMD………………………………………….33
CHAPTER 1: Extending the clinical and mutational spectrum of TRIM32-related
myopathies in a non-Hutterite population………………………………………………………………35
CHAPTER 2: Muscular dystrophy with arrhythmia caused by loss-of-function
mutations in BVES…………………………………………………………………………………………………..61
CHAPTER 3: High prevalence of sporadic late-onset nemaline myopathy in a cohort of
whole-exome sequencing negative myopathy patients..…………………………………………83
PART 2: A proteomic approach to study disease signatures in muscle tissue of myopathy
patients in an unbiased way…..…………………………………………………………………………………….103
CHAPTER 4: A tale of the unexpected: multisystem proteinopathy due to a
homozygous p.Arg159His VCP mutation……………………………………………………………….105
CHAPTER 5: Ageing signatures and disturbed muscle regeneration and differentiation
in sporadic inclusion body myositis……………………………………………………………………….133
GENERAL DISCUSSION: …………………………………………………………………………………………………….159
SUMMARY – SAMENVATTING: …………………………………………………………………………………………179
LIST OF COMMONLY USED ABBREVIATIONS: ……………………………………………………………………183
CURRICULUM VITAE: ……………………………………………………………………………………………………….187
ACKNOWLEDGEMENTS – DANKWOORD…………………………………………………………………………..195

8

9
GENERAL INTRODUCTION

10
11
DISEASES OF THE SKELETAL MUSCLE
Primary muscle disorders constitute a large group of inherited and acquired diseases that
affect muscle structure, metabolism, or the function of muscle ion channels.1 The diagnosis
of a muscle disorder (or myopathy) is initially suspected based on history, clinical neurologic
examination and routine investigations such as measurement of creatine kinase (CK) levels
in blood and electrodiagnostic studies.1, 2 Muscle disorders most typically present with
progressive muscle weakness, yet associated clinical symptoms such as muscle atrophy or
rather hypertrophy, cardiac or respiratory involvement and contractures can point towards
a more specific diagnosis. Detailed evaluation of these clinical features, combined with the
age at onset, pace of progression and the pattern of muscle involvement based on clinical
examination and whole-body muscle MRI can help to delineate a specific diagnosis.3 As
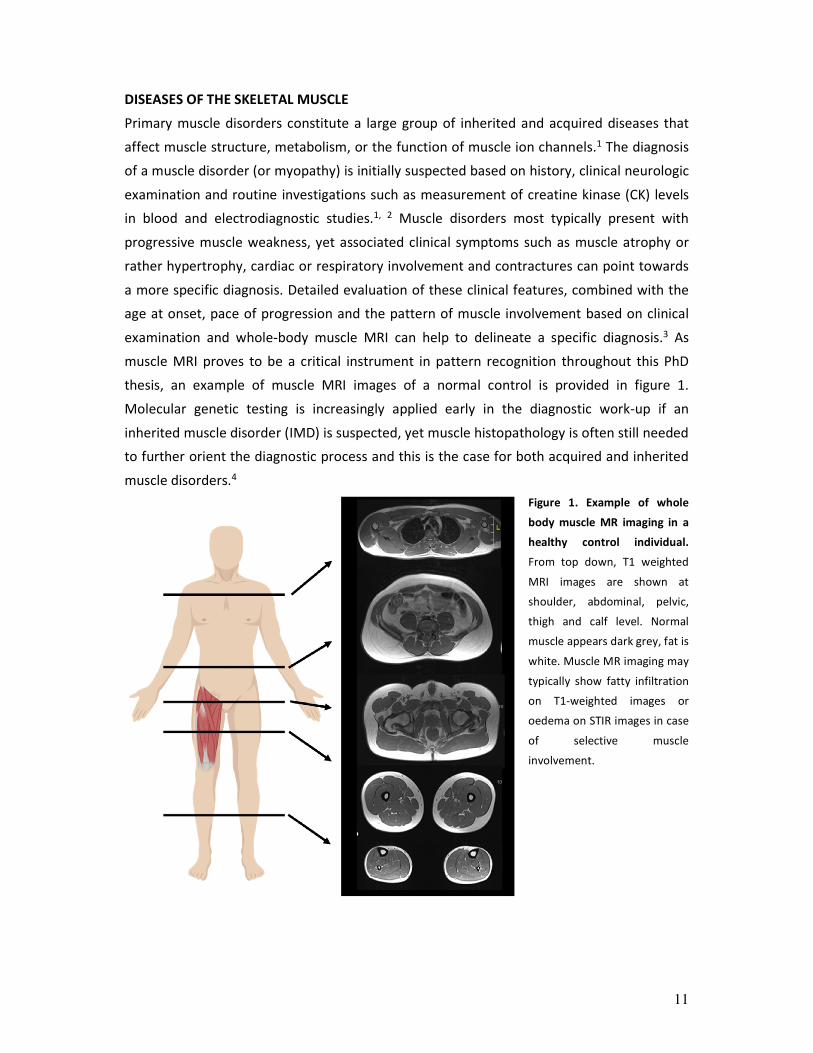
muscle MRI proves to be a critical instrument in pattern recognition throughout this PhD
thesis, an example of muscle MRI images of a normal control is provided in figure 1.
Molecular genetic testing is increasingly applied early in the diagnostic work-up if an
inherited muscle disorder (IMD) is suspected, yet muscle histopathology is often still needed
to further orient the diagnostic process and this is the case for both acquired and inherited
muscle disorders.4
Figure 1. Example of whole
body muscle MR imaging in a
healthy control individual.
From top down, T1 weighted
MRI images are shown at
shoulder, abdominal, pelvic,
thigh and calf level. Normal
muscle appears dark grey, fat is
white. Muscle MR imaging may
typically show fatty infiltration
on T1-weighted images or
oedema on STIR images in case
of selective muscle
involvement.
12
MUSCLE BIOPSY
A muscle biopsy is a relatively non-invasive and useful procedure to directly study diseased
muscle tissue. Different techniques are performed on a muscle specimen to evaluate fibre
atrophy, structural changes, inflammatory features and signs suggestive of a metabolic
disorder. These include histological, histochemical and histoenzymatic stainings as well as
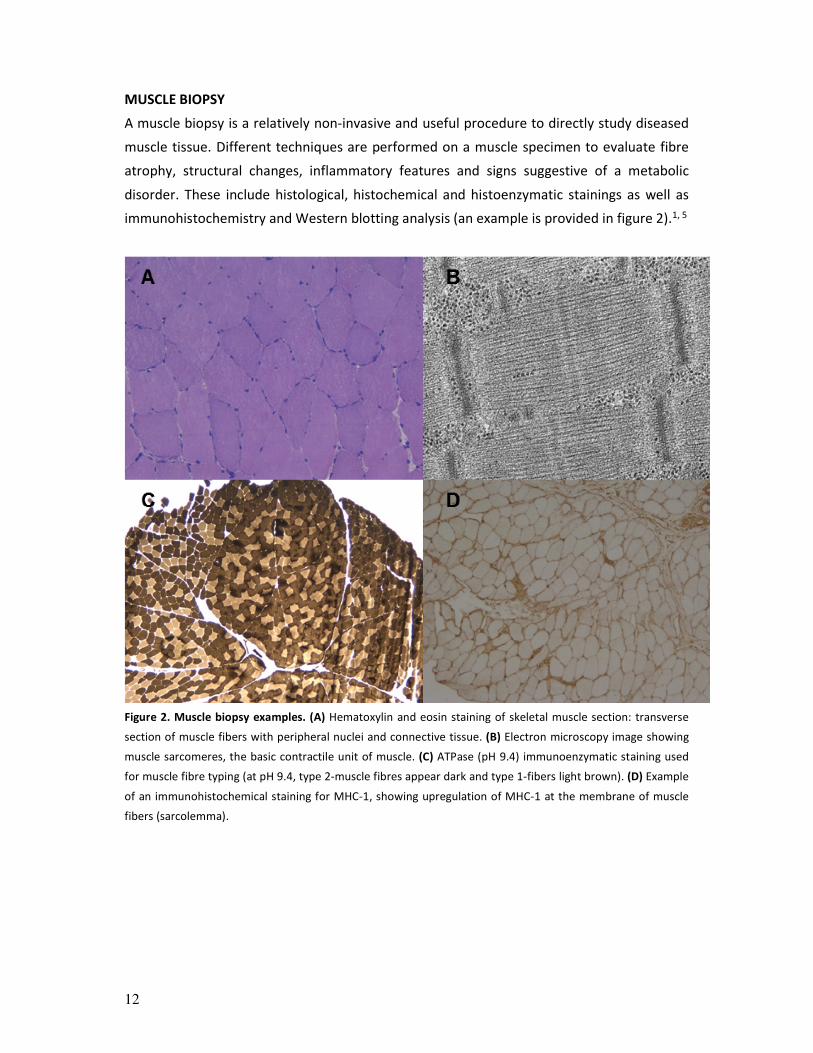
immunohistochemistry and Western blotting analysis (an example is provided in figure 2).1, 5
Figure 2. Muscle biopsy examples. (A) Hematoxylin and eosin staining of skeletal muscle section: transverse
section of muscle fibers with peripheral nuclei and connective tissue. (B) Electron microscopy image showing
muscle sarcomeres, the basic contractile unit of muscle. (C) ATPase (pH 9.4) immunoenzymatic staining used
for muscle fibre typing (at pH 9.4, type 2-muscle fibres appear dark and type 1-fibers light brown). (D) Example
of an immunohistochemical staining for MHC-1, showing upregulation of MHC-1 at the membrane of muscle
fibers (sarcolemma).

13
MUSCLE DISORDERS CHARACTERISED BY SLOWLY PROGRESSIVE MUSCULAR WEAKNESS
Throughout this PhD thesis, I focus on muscle disorders presenting with slowly progressive
muscular weakness. Given that only very few acquired muscle disorders present with slowly
progressive muscular weakness, an IMD is typically suspected in case of this clinical
presentation. IMD constitute a clinically, genetically and histopathologically very
heterogeneous group of rare diseases with more than 160 genetically distinct entities
identified, rendering the diagnostic process complex.2, 6 Occasionally, basic diagnostic work-
up suffices to make a correct clinical diagnosis that is subsequently confirmed by focused
molecular genetic testing. Notable examples are myotonic dystrophy type 1,
facioscapulohumeral dystrophy (FSHD) and dystrophinopathies. In many cases the initial
evaluation nonetheless precludes a correct diagnosis due to the extensive overlap in clinical
presentation.4, 7
Besides IMD, idiopathic inflammatory myopathies (IIM) constitute another important
subgroup among primary muscle disorders, currently classified in five subtypes based on
distinct clinical and pathological features in conjunction with specific autoantibodies:
dermatomyositis, immune-mediated necrotising myopathy, overlap myositis, polymyositis
and sporadic inclusion body myositis (sIBM).8 Dermatomyositis, immune-mediated
necrotising myopathy, overlap myositis and polymyositis typically present with subacute
muscle weakness and respond well to immune-suppression.8 In many ways sIBM however
differs from the other IIM, due to its slow progression, strict late-onset nature and the
striking, selective and often asymmetric involvement of distal muscle groups (particularly
long finger flexors).8 Moreover and most importantly, trials using immune-modulating drugs
in sIBM have failed to show effect.9 sIBM constitutes the most important differential
diagnosis of IMD in case of slowly progressive muscle weakness in patients over 50 years of
age.8
A few other atypically presenting acquired muscle disorders might however also constitute
relevant differential diagnoses. Recent literature suggested that IIM with anti-HMGCR
antibodies might present with slowly progressive muscle weakness.10 The same has been
suggested for an enigmatic, supposedly very rare, putatively immune-mediated acquired
myopathy, called sporadic late-onset nemaline myopathy (SLONM).11

14
GENETICS OF INHERITED MUSCLE DISORDERS
Subgroups of IMD
IMD have historically been classified based on clinical and histopathological features, or a
combination of both. Clinically, limb-girdle muscular weakness (LGMW), with predominant
proximal weakness, is the most frequent pattern of muscle weakness. In case of distal
muscle weakness, the disease is classified as a ‘distal myopathy’, yet other patterns of
muscle weakness are considered, such as scapuloperoneal of facioscapulohumeral
patterns.1 Based on histopathological features on the other hand, different categories have
been defined as well: muscular dystrophies, myofibrillar myopathies, metabolic myopathies,
mitochondrial myopathies and congenital myopathies marked by structural abnormalities of
muscle fibres.12, 13
With more than 160 genetically distinct entities identified, increasing clinical and
histopathological overlap and often complex genotype-phenotype correlations, this
classification system has become more and more confusing.2, 6 An improved classification
system and nomenclature are clearly needed and a diagnosis should probably be focused on
the molecular genetic cause and inheritance pattern, rather than on the varying clinical and
histopathological features of specific genetic entities. Such an effort has recently been done
for limb-girdle muscular dystrophies (LGMDs),14 clinically characterized by predominant
proximal weakness and histopathologically by muscle fibre necrosis and replacement by fat
and connective tissue.6, 15
Reliable prevalence estimates for IMD are generally lacking. For recessive LGMDs
(encompassing approximately 90% of all LGMD patients), collective prevalence was
estimated at 1:15.000 in 2003.16 There is considerable regional variation in relative
prevalence of LGMD subtypes, but CAPN3-related myopathy, FKRP-related myopathy and
DYSF-related myopathy are generally among the most frequently encountered subtypes.14
On the other hand, for many rare subtypes, the low incidence and the limited amount of
epidemiology data lead to difficulties in getting robust prevalence estimates.17 Currently,
efforts are being done to estimate the prevalence for LGMD based on public sequencing
databases such as GnomAD.17 Considering the debilitating nature of IMD and the reduced
life expectancy for many IMD subtypes due to cardiac, respiratory or bulbar involvement,
IMD result in a considerable personal, societal and economic burden.
Inheritance patterns and genetic variation (basics and beyond)
The human genetic code (genome), formed by deoxyribonucleic acid (DNA), contains 23
pairs of chromosomes, encompassing over 3 billion base pairs (bp). The code is formed by a

15
sequence of four chemical bases (nucleotides), adenine (A), cytosine (C), guanine (G), and
thymine (T). DNA consists of a double helix with A and T and C and G forming base pairs. The
protein coding part of the genome (1-2% of the genome) comprises approximately 20.000
protein-coding sequences (genes).
Each individual has two copies of a gene, one inherited from each parent. ‘Normal’ genetic
variation, consisting of small differences in the genetic code (different ‘alleles’ of a specific
gene), contributes to differences between individuals. Genetic variation (genotypes) can
however also be linked to diseases (phenotypes) such as IMD. In the introduction of this
thesis I focus on monogenic disorders (Mendelian disorders), caused by rare variants in a
single gene having a high impact on disease risk.18 This contrasts with multifactorial
diseases, in which common genetic variants make small contributions to disease risk, in
combination with multiple environmental factors.18
Mendelian disorders show a few major modes of inheritance: (1) in autosomal dominant
inheritance, one disease allele is needed to develop a phenotype; (2) autosomal recessive
inheritance, with two copies of a disease allele being required to express a phenotype; (3) X-
linked (dominant or recessive) inheritance, with males being hemizygous for X-linked genes
as they have only one X chromosome. Different factors, such as a varying or late age at
onset, incomplete penetrance, variable expressivity, and phenocopy may complicate
pedigrees and putative inheritance patterns.18 In IMD, variable age at onset constitutes the
most frequently encountered problem.
IMD are linked to different types of disease-causing genetic variants, each requiring
different molecular genetic techniques to be detected. Most frequent types of variation in
IMD range from ‘large variants’ (affecting more than 50 bp, e.g. copy number variants
(CNVs) such as deletions or duplications) to single nucleotide variants (SNVs) or small
deletions or insertions. SNVs can have different consequences, such as a single amino acid
alteration (missense variants), a shift of the reading frame leading to insertion of a
premature stop codon (frameshift variants) or a direct insertion of a premature stop codon
(nonsense variants) and splicing alteration (splice variants). Different molecular mechanisms
may underlie pathogenicity of these variants: these variants may exert a loss-of-function
(LOF), gain-of-function (GOF) or dominant-negative effect. In case of a loss-of-function
mechanism, the normal function of a protein is disrupted, through loss of expression or
alteration of the structure of the encoded protein. Gain-of-function mutations generally
(with few exceptions) show a dominant inheritance pattern and typically result in a new
toxic function of the protein or increased activity. Dominant-negative mutations typically

16
result in a structural change of the protein encoded by this allele, interfering with the
function of the normal protein encoded by the other.
Alterations to repetitive regions in the DNA, exonic or intronic, constitute another well-
known mutational mechanism. Trinucleotide repeat expansion disorders are the most
frequent repeat disorders.19 In contrast to e.g. cerebellar ataxias,20 only few muscle
disorders are known repeat disorders and all four of them show a distinct clinical
phenotype: FSHD is caused by a reduction of (D4Z4) repeat units and oculopharyngeal
muscular dystrophy (OPMD) and myotonic dystrophy type 1 and 2 are repeat expansion
disorders.
Molecular genetic testing
Recently, considerable progress has been made in molecular genetic testing. Sanger
sequencing, a technique with a limited throughput and high cost, due to the need of a single
DNA fragment for each sequencing reaction, remained the golden standard method for a
long period, but next generation sequencing (NGS) strategies are more and more available
and rapidly decrease in costs: targeted panel sequencing, whole-exome sequencing (WES)
and whole-genome sequencing (WGS).7 Targeted panel sequencing of genes focuses on a
predefined set of genes of interest. In WES, the entire set of exons (‘exome’, comprising
approximately 1-2% of the genome) is targeted, in WGS the complete genome. Studying
only the exome however still is a relevant approach as most pathogenic variants underlying
Mendelian disorders disrupt protein-coding sequences.21 The exome is a highly enriched
subset of the genome in which variants with large effect sizes are found.21
The strategic use of these new techniques, in combination with systematic analysis of
clinical, radiological and histopathological data, can lead to the identification of novel genes
or novel genotype-phenotype correlations. These new techniques are being implemented in
new guidelines on diagnostic testing in muscle disorders.6 A caveat of WES is that it is
commonly considered to be intractable to structural variant (i.e. genomic rearrangements
larger than 50 bp) detection. However, specialised analytical software is now starting to
permit this facet of analysis.22
Recent data of larger cohorts of patients with a suspected IMD in which targeted panel
sequencing was applied, showed success rates that varied between 20% and 45%.7, 23, 24
Interpreting genetic variation
The results of NGS techniques, interrogating a large part of the exome or genome, have to
be rigorously interpreted with regard to the pathogenicity of variants. Current WES-
pipelines yield a dataset of approximately 70,000 to 80,000 SNVs.25, 26 Identifying (a)

17
disease-causing variant(s) within this dataset is a challenging task. Guidelines of the
American College of Medical Genetics and Genomics, capitalizing on different types of
evidence, aiding in filtering and interpretation of variants, are at hand.27 Standard
terminology is advised to describe variants identified in Mendelian disorders: ‘pathogenic’,
‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’, or ‘benign’.27
The following elements are crucial in gathering genetic evidence with regard to potential
pathogenicity of variants:18, 27 (1) variant frequency in unaffected control individuals (with
ExAC28 and gnomAD databases currently being the largest available datasets); (2)
cosegregation data (with filtering based on the – suspected – inheritance pattern); (3)
computational and predictive data of in silico prediction algorithms evaluating pathogenicity
or conservation: concordance of different algorithms has the strongest predictive power.29
Algorithms question the impact of variants based on knowledge about the protein’s
function, structure, and evolutionary conservation, which has to be consistent with the
known disease mechanism.18
Additional functional evidence may be needed to assess the impact of variants at the mRNA
or protein level.27 In IMD, patient’s diseased tissue is usually at hand to study molecular and
functional consequences of variants. Alternatively, additional in vitro or in vivo modelling
may be needed, particularly in case of potential ‘novel’ genes.
One category of evidence classified as ‘other’, besides these categories of genetic and
functional evidence, is very relevant with regard to IMD. ‘Using phenotype to support
variant claims’ is very pertinent in IMD, definitely in comparison with some other inherited
disorders such as intellectual disability, as deep phenotyping (clinical, detailed muscle
biopsy analysis, muscle MRI) may indeed yield features characteristic of a specific genetic
entity.
Relevance of studying rare IMD
Although clearly challenging, it is very important to establish an exact genetic diagnosis.
Firstly, this aids in estimating long-term prognosis. Furthermore it differentiates without
doubt inherited from acquired myopathies, thereby for example avoiding unnecessary
immunosuppressive treatment in hereditary myopathies histopathologically resembling IIM.
Last but not least, a definite diagnosis assists in directing genetic counselling and if desired
pre-symptomatic and pre-implantation diagnostics.6 Scientifically, the identification of
patients with very rare or novel LGMDs is relevant too: (1) functionally studying
pathomechanisms in specific entities yields valuable information with regard to pathways
that are crucially involved in muscle homeostasis, their disturbance leading to muscle
degeneration.30 (2) Last but not least, for rapid translation of basic research to the clinic,

18
proper genotyping and phenotyping of patients with rare muscle disorders is crucial.30
Clinicians need to be prepared to treat patients with sometimes extremely rare IMD: a
genetic diagnosis is currently essential with regard to trial readiness and will result in
therapies in the nearby future.
SPORADIC INCLUSION BODY MYOSITIS: PUTATIVE PATHOMECHANISMS
sIBM is considered to be the most common IIM among patients over 50 years of age, with a
prevalence of approximately 2.48 to 4.56/100,000.31 Patients typically develop progressive
muscle weakness of the long finger flexors and quadriceps muscles. Muscle biopsy reveals
two cardinal features, (1) inflammatory changes including endomysial inflammatory
infiltrates and major histocompatibility complex I (MHC-I) upregulation, and (2) marked
degenerative changes, including rimmed vacuoles and protein aggregates.32 In absence of a
sIBM diagnostic gold-standard test,32, 33 different diagnostic categories exist with high
specificity (≥97%), but variable sensitivity.34 The European Neuromuscular Centre (ENMC)
criteria are most widely used.32 sIBM is historically classified as one of the IIM but is
refractory to immunosuppression.35 Due to lack of effective therapy, steady decline of
muscle strength results in loss of ambulation and ultimately reduced life expectancy due to
swallowing difficulties and respiratory complications. Diagnosis is often delayed for several
years, leading to unnecessary and potentially harmful immunosuppressive treatments.36
The interplay of diverse potential mechanisms underlying sIBM pathogenesis remains
unclear as is illustrated by the dual inflammatory-degenerative pathology. Of particular
interest is the role of protein dyshomeostasis, evident by the aggregation of β-amyloid (Aβ),
phosphorylated tau, ubiquitin, α-synuclein and prion protein.33 Other mechanisms have
been extensively studied: (1) mitochondrial dysfunction; (2) ER stress and the Unfolded
Protein Response; (3) oxidative stress; (4) dysregulation of the Myostatin pathway; (5)
inflammation.33, 37 Concerning the inflammatory features, key roles have been attributed to
cytotoxic T cells, immune-regulatory secondary signals, regulatory T cells and possibly also
humoral responses.33, 38 The central question remains if sIBM is in origin inflammatory or
degenerative;33, 37, 38 lack of response to immune-modulating therapies certainly is in favour
of the latter. sIBM is unmistakably an age-related disorder and multiple pathological
commonalities with neurodegenerative disorders have been described.39 However, the
central hallmarks of ageing (genomic instability, telomere attrition, epigenetic alterations,
loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular
senescence, stem cell exhaustion and altered intercellular communication) have not been
systematically studied in sIBM.40 Endo-lysosomal dysfunction is the assumed converging
mechanism driving loss of proteostasis in protein-aggregation disorders.41, 42 Similarities

19
between neurodegenerative disorders and sIBM are embodied by a dominantly-inherited
multisystem proteinopathy (MSP1), caused by in the valosin containing protein gene (VCP),
presenting with a high diversity of combinations of phenotypes, including inclusion body
myopathy (IBM), early-onset Paget disease of bone (PDB), frontotemporal dementia (FTD),
amyotrophic lateral sclerosis (ALS) and parkinsonism.43
PROTEOMICS IN MUSCLE DISORDERS
Proteomics, or the systematic study of the complete inventory of proteins (‘proteome’), is a
powerful approach to dissect disease signatures in an unbiased way.44 Mass spectrometry
(MS)-based techniques have evolved dramatically over time and are capable of quantifying
thousands of proteins across collections of large numbers of samples with a high degree of
reproducibility.44 Studying expression alterations with MS methods in muscle disorders is
highly relevant, as this approach interrogates alterations closest to the biology and
pathomechanisms of the disease. Although transcriptomics is the most common technology
for functional genomics, MS-based proteomics has clear complementary advantages:
altered mRNA levels are not always reflected in the proteome and many changes arise from
protein modifications rather than changes in gene expression. Other mechanisms such as
post-transcriptional and translational regulation contribute at least as much as transcription
itself in the determination of protein concentrations.45 Therefore proteomic analyses
generate high-dimensionality data, likely the most proximal to the generation of the disease
phenotype. Proteomics studies have indeed increasingly been applied in acquired and
inherited muscle disorders as patient diseased tissue is readily available.46, 47
Many proteomic studies on diseased muscle tissue focused on dysregulation of single
proteins rather than on describing general disease patterns. This is particularly the case for
muscle disorders with different types of protein aggregates in muscle fibers. Researchers
performing an ‘unbiased’ proteomic study on patients’ muscle tissue often focus on the
proteins present in protein aggregates by performing laser capture microdissection and
subsequent MS analysis of these aggregates. Examples can be found in myofibrillar
myopathies.48 This methodology is also applied by some of the few studies using proteomics
to investigate sIBM, which also show methodological weaknesses such as small samples
sizes, insufficient sample matching leading to important variability of protein abundances
and the use of low sensitivity MS methods without fractionation techniques.49-54 A recent
study combined quantitative proteomics on laser dissected aggregates in sIBM muscle and
WES to focus on top upregulated proteins in the search for genetic risk factors.53
The uncertainty concerning the key pathomechanisms of sIBM complicates the design of
reliable experimental cell- or animal-models emphasizes the relevance of well-designed

20
experiments using human disease tissue, applying an unbiased proteomic approach. The
approach is however also highly relevant in other acquired myopathies or IMD for which
disease mechanisms downstream of the molecular genetic cause still need to be further
unravelled.
REFERENCES
1. Jackson CE. A clinical approach to muscle diseases. Seminars in neurology 2008;28:228-240.
2. Mercuri E, Muntoni F. Muscular dystrophies. Lancet 2013;381:845-860.
3. Straub V, Carlier PG, Mercuri E. TREAT-NMD workshop: pattern recognition in genetic muscle diseases
using muscle MRI: 25-26 February 2011, Rome, Italy. Neuromuscular disorders : NMD 2012;22 Suppl
2:S42-53.
4. Malfatti E, Romero NB. Diseases of the skeletal muscle. Handbook of clinical neurology 2017;145:429-
451.
5. Dubowitz V, Sewry CA, Oldfors A. Muscle Biopsy: A Practical Approach 2013.
6. Narayanaswami P, Weiss M, Selcen D, et al. Evidence-based guideline summary: diagnosis and
treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of
the American Academy of Neurology and the practice issues review panel of the American Association
of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-1463.
7. Nigro V, Savarese M. Next-generation sequencing approaches for the diagnosis of skeletal muscle
disorders. Current opinion in neurology 2016;29:621-627.
8. Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen
AL. Classification and management of adult inflammatory myopathies. The Lancet Neurology
2018;17:816-828.
9. Naddaf E, Barohn RJ, Dimachkie MM. Inclusion Body Myositis: Update on Pathogenesis and
Treatment. Neurotherapeutics : the journal of the American Society for Experimental
NeuroTherapeutics 2018;15:995-1005.
10. Mohassel P, Landon-Cardinal O, Foley AR, et al. Anti-HMGCR myopathy may resemble limb-girdle
muscular dystrophy. Neurology(R) neuroimmunology & neuroinflammation 2019;6:e523.
11. Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, et al. Sporadic late-onset nemaline myopathy:
clinico-pathological characteristics and review of 76 cases. Orphanet journal of rare diseases
2017;12:86.
12. Dubowitz V. SC, Oldfors A. Muscle Biopsy: A Practical Approach, 4th ed. Philadelphia, PA:
Saunders/Elsevier, 2014.
13. Meola G, Bugiardini E, Cardani R. Muscle biopsy. Journal of neurology 2012;259:601-610.
14. Straub V, Murphy A, Udd B. 229th ENMC international workshop: Limb girdle muscular dystrophies -
Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017.
Neuromuscular disorders : NMD 2018;28:702-710.
15. Emery AE. The muscular dystrophies. Lancet 2002;359:687-695.
16. Nigro V. Molecular bases of autosomal recessive limb-girdle muscular dystrophies. Acta myologica :
myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology / edited
by the Gaetano Conte Academy for the study of striated muscle diseases 2003;22:35-42.

21
17. Liu W, Pajusalu S, Lake NJ, et al. Estimating prevalence for limb-girdle muscular dystrophy based on
public sequencing databases. Genetics in medicine : official journal of the American College of
Medical Genetics 2019.
18. Strande NT, Brnich SE, Roman TS, Berg JS. Navigating the nuances of clinical sequence variant
interpretation in Mendelian disease. Genetics in medicine : official journal of the American College of
Medical Genetics 2018;20:918-926.
19. Paulson H. Repeat expansion diseases. Handbook of clinical neurology 2018;147:105-123.
20. Storey E. Genetic cerebellar ataxias. Seminars in neurology 2014;34:280-292.
21. Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene
discovery. Nature reviews Genetics 2011;12:745-755.
22. Tattini L, D'Aurizio R, Magi A. Detection of Genomic Structural Variants from Next-Generation
Sequencing Data. Frontiers in bioengineering and biotechnology 2015;3:92.
23. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a
large LGMD cohort of 4656 patients. Annals of clinical and translational neurology 2018;5:1574-1587.
24. Reddy HM, Cho KA, Lek M, et al. The sensitivity of exome sequencing in identifying pathogenic
mutations for LGMD in the United States. Journal of human genetics 2017;62:243-252.
25. Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome
sequencing for detecting exome variants. Proceedings of the National Academy of Sciences of the
United States of America 2015;112:5473-5478.
26. Lindor NM, Schahl KA, Johnson KJ, et al. Whole-Exome Sequencing of 10 Scientists: Evaluation of the
Process and Outcomes. Mayo Clinic proceedings 2015;90:1327-1337.
27. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a
joint consensus recommendation of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Genetics in medicine : official journal of the American College of
Medical Genetics 2015;17:405-424.
28. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans.
Nature 2016;536:285-291.
29. Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant
interpretation guidelines. Genome biology 2017;18:225.
30. Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for
translational research. Nature reviews Neurology 2016;12:294-309.
31. Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;372:1734-1747.
32. Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011,
Naarden, The Netherlands. Neuromuscular disorders : NMD 2013;23:1044-1055.
33. Benveniste O, Stenzel W, Hilton-Jones D, Sandri M, Boyer O, van Engelen BG. Amyloid deposits and
inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the
degenerative chicken. Acta Neuropathol 2015;129:611-624.
34. Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and
construction of diagnostic criteria for inclusion body myositis. Neurology 2014;83:426-433.
35. Rose MR, Jones K, Leong K, et al. Treatment for inclusion body myositis. Cochrane Database Syst Rev
2015;6:Cd001555.
36. Molberg O, Dobloug C. Epidemiology of sporadic inclusion body myositis. Curr Opin Rheumatol
2016;28:657-660.

22
37. Askanas V, Engel WK, Nogalska A. Sporadic inclusion-body myositis: A degenerative muscle disease
associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochimica et
biophysica acta 2015;1852:633-643.
38. Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nature reviews
Rheumatology 2019;15:257-272.
39. Askanas V, Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible
pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta
Neuropathol 2008;116:583-595.
40. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell
2013;153:1194-1217.
41. Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-
related diseases. Nature communications 2014;5:5659.
42. Wang C, Telpoukhovskaia MA, Bahr BA, Chen X, Gan L. Endo-lysosomal dysfunction: a converging
mechanism in neurodegenerative diseases. Curr Opin Neurobiol 2017;48:52-58.
43. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease
pathogenesis. Journal of cell science 2014;127:3877-3883.
44. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature
2016;537:347-355.
45. Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and
transcriptomic analyses. Nature reviews Genetics 2012;13:227-232.
46. Dowling P, Murphy S, Ohlendieck K. Proteomic profiling of muscle fibre type shifting in neuromuscular
diseases. Expert review of proteomics 2016;13:783-799.
47. Gelfi C, Vasso M, Cerretelli P. Diversity of human skeletal muscle in health and disease: contribution of
proteomics. Journal of proteomics 2011;74:774-795.
48. Maerkens A, Olive M, Schreiner A, et al. New insights into the protein aggregation pathology in
myotilinopathy by combined proteomic and immunolocalization analyses. Acta neuropathologica
communications 2016;4:8.
49. Li J, Yin C, Okamoto H, et al. Proteomic analysis of inclusion body myositis. Journal of neuropathology
and experimental neurology 2006;65:826-833.
50. Hutchinson DO, Jongbloed B. Two-dimensional gel electrophoresis in inclusion body myositis. Journal
of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2008;15:440-444.
51. Parker KC, Kong SW, Walsh RJ, et al. Fast-twitch sarcomeric and glycolytic enzyme protein loss in
inclusion body myositis. Muscle & nerve 2009;39:739-753.
52. Doppler K, Lindner A, Schutz W, Schutz M, Bornemann A. Gain and loss of extracellular molecules in
sporadic inclusion body myositis and polymyositis--a proteomics-based study. Brain pathology (Zurich,
Switzerland) 2012;22:32-40.
53. Guttsches AK, Brady S, Krause K, et al. Proteomics of rimmed vacuoles define new risk allele in
inclusion body myositis. Annals of neurology 2017;81:227-239.
54. Roos A, Preusse C, Hathazi D, Goebel HH, Stenzel W. Proteomic Profiling Unravels a Key Role of
Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Frontiers in immunology
2019;10:1040.

23

24

25
AIMS AND OUTLINE

26
AIMS THESIS
The ultimate aim of this PhD thesis is to gain insights in pathomechanisms of muscle
disorders characterized by slowly progressive muscle weakness, inherited or acquired,
which are unmistakably marked by progressive muscle degeneration. Dissecting disease
signatures of different disorders will reveal commonalities (‘final common pathways’) as
well as differences, which can yield valuable information with regard to muscle proteostasis,
degeneration and regeneration in particular. Muscle disorders presenting with slowly
progressive muscle weakness after all constitute a group of disorders eminently suited for a
multi-level pattern recognition approach, clinically as well as biologically. Therefore, we
aimed to:
(1) Identify rare or novel molecular genetic causes of inherited muscle disorders (IMD) in
patients presenting with slowly progressive muscle weakness through whole-exome
sequencing (WES). We participated in a large multicentre study, MYO-SEQ, in which the
exomes of 2.000 suspected IMD patients have been sequenced.
(2) Study genotype-phenotype correlations and molecular and/or biological
pathomechanisms of these IMD to contribute to our knowledge of these disorders of which
we know the upstream (genetic) mechanism, though often not precise downstream
pathomechanisms ultimately leading to muscle degeneration and disease (left panel of
figure 1).
(3) Perform an unbiased quantitative proteomic approach on diseased tissue of sporadic
(sIBM) muscle disorder, the most frequent age-related acquired muscle disorder
characterized by relentless muscle degeneration, with the aim to identify upstream
regulators of disease mechanisms (right panel of figure 1).
(4) The overarching aim of this PhD thesis was the identification of potential ‘common
pathways’ underlying muscle degeneration in both acquired and inherited muscle disorders
(middle panel of figure 1).
In the long term, better understanding of the pathomechanisms underlying muscle
degeneration in muscle disorders presenting with slowly progressive muscle weakness, will
aid in the search for disease biomarkers and therapeutic strategies.

27
Figure 1. Studying disease mechanisms in inherited muscle disorders (IMD, left panel) characterized by
progressive muscle degeneration and the (at least partially) degenerative acquired sporadic inclusion body
myositis (sIBM, right panel). Representation of: 1) the principal levels that can be studied, with important
examples of alterations in case of muscle disease (middle panel); 2) different core methodological approaches
in the study of both IMD and sIBM (left and right panel respectively). These studies directly in (diseased)
skeletal muscle allow a search for common pathways involved in muscle degeneration. FSHD,
facioscapulohumeral dystrophy; NGS, next generation sequencing; WES, whole exome sequencing; WGS,
whole genome sequencing; WB, western blotting; IHC, immunohistochemistry; NMD, nonsense mediated
mRNA-decay.
OUTLINE
Part 1. Genetics of patients with suspected IMD and the study of molecular and/or
biological pathomechanisms of rare or novel causes of IMD
Part 1 is focused on the identification of patients with rare IMD, deep phenotyping of these
patients and the study of molecular and/or biological pathomechanisms in diseased muscle
tissue. In these studies, we contribute to our knowledge of genotype-phenotype
correlations of these specific disorders, allowing a prompt diagnosis, as well as to our
understanding of pathomechanisms of the specific genetic entity and by extension of
muscle degeneration or failure of muscle regeneration.
In chapter 1, we describe deep phenotyping data of TRIM32-related myopathy patients, a
recessive IMD that appears to be extremely rare in non-Hutterite patients. In particular, we
highlight characteristics of the pattern of muscle involvement on muscle MRI studies.
Findings on these MR images, as well as other clinical and histopathological findings,
contrast with what is observed in patients carrying a homozygous missense variant of

28
unknown significance (VUS), not residing in the protein domain in which pathogenic
missense mutations had previously been described. By studying the functional effect at the
transcript level of one of the identified frameshift mutations, we contribute to insights in
molecular mechanisms of the disorder.
In chapter 2, we study genetic and phenotypic details of four individuals from three families
harbouring recessive mutations in BVES. Previously a single family had been identified with
multiple individuals showing variable skeletal muscle and cardiac involvement harbouring a
homozygous p.Ser201Phe missense variant in BVES. This disorder appears to have a low
prevalence although it is probably underdiagnosed due to its striking phenotypic variability
and often subtle yet clinically relevant manifestations. Again, we contribute to insights in
molecular mechanisms of the disorder, by studying the functional effect at the transcript
level of the different mutations and at the protein level by immunohistochemical
techniques.
Chapter 3: this third chapter elegantly bridges part 1 and part 2 of this thesis. We
performed a systematic study of the 18 isolated yet suspected inherited myopathy (IMD)
patients included in the Antwerp subcohort of MYO-SEQ, showing late-onset, slowly
progressive limb-girdle muscular weakness (LGMW), which remained unsolved after
thorough WES data analysis. The success rate of genetically solving patients in this subgroup
was markedly lower than in the complete group, potentially suggesting an
overrepresentation of a previously unrecognized acquired myopathy. After detailed re-
phenotyping, we identified marked nemaline rods on muscle biopsy for 10 out of 18
patients, with a monoclonal gammopathy of unknown significance (MGUS) in four, highly
suggesting an enrichment of patients with slowly progressing sporadic late-onset nemaline
myopathy (SLONM) in WES-unsolved suspected IMD patient cohorts. We describe the
clinical details of patients of this cohort and advocate an active and prospective search for
slowly progressing SLONM cases in homogeneous cohorts, such as WES-unsolved suspected
IMD patient cohorts.
Part 2. A proteomic approach to study disease signatures in muscle tissue of myopathy
patients in an unbiased way
Chapter 4 focuses on VCP, a gene associated with a rare, dominantly inherited multisystem
proteinopathy (MSP1), which presents with a high diversity of combinations of phenotypes,
including inclusion body myopathy (IBM), early-onset Paget disease of bone (PDB) and
different neurodegenerative phenotypes. In this study, we describe the first patient
manifesting a multisystem proteinopathy due to a homozygous VCP mutation, previously
reported to be pathogenic in heterozygous state. Capitalizing on the identification of this

29
unique patient and the availability of diseased muscle tissue, we performed in depth
phenotypic studies, as well as functional studies by means of proteomic experiments on
muscle tissue of the index patient, his father, three additional VCP-related myopathy
patients and three control individuals. This study: 1) yields valuable insights on VCP-related
disease mechanisms; 2) uncovers additional phenotypic and possibly also pathomechanistic
parallels between VCP-related IBM and sIBM and therefore elegantly bridges part 1 and
part 2 of this PhD thesis, too.
Chapter 5: in this final chapter, we capitalize on a unique proteomic dataset on muscle
tissue of 28 sIBM patients and 28 control individuals. This unbiased proteomic study
provided unique insights in the proteomic landscape of sIBM and allowed us to prioritize a
promising potential upstream regulator predicted to be activated, KDM5A, a histone
demethylase involved in the DNA damage response (DDR) and (myogenic) differentiation.
Further studies in human myoblasts and sIBM muscle tissue allowed us to raise the
hypothesis that KDM5A might play a central role in sIBM pathomechanisms or at least
represents a very relevant mediator with regard to the age-related nature of the disease.
General discussion
In the general discussion of this PhD thesis, I will summarize the findings in this PhD thesis in
light of its original aims and the broader context of the fast-moving field of myology. In my
discussion of part 1, I will highlight some other ongoing studies on rare or novel IMD, to
illustrate the unstoppable progress in the field. Furthermore, I will particularly highlight the
relevance of studying both inherited and acquired muscle disorders in search for common
pathways and therapeutic strategies.

30

31
RESULTS

32

33
PART 1
Genetics of patients with suspected IMD and the study
of molecular and/or biological pathomechanisms of
rare or novel causes of IMD

34

35
CHAPTER 1
Extending the clinical and mutational spectrum of
TRIM32-related myopathies in a non-Hutterite
population
Katherine Johnson*, Willem De Ridder*, Ana Töpf, Marta Bertoli, Lauren Phillips, Peter De
Jonghe, Jonathan Baets, Tine Deconinck, Vidosava Rakocevic Stojanovic, Stojan Perić, Hacer
Durmus, Shirin Jamal-Omidi, Shahriar Nafissi, Tiziana Mongini, Anna Łusakowska, Mark
Busby, James Miller, Fiona Norwood, Judith Hudson, Rita Barresi, Monkol Lek, Daniel G
MacArthur, Volker Straub
*Equal contribution
J Neurol Neurosurg Psychiatry. 2019;90(4):490-493 [Adapted version]

36
ABSTRACT
TRIM32-related myopathies represent a phenotypic spectrum of rare autosomal recessive
neuromuscular disorders that are associated with progressive wasting and weakness of
proximal skeletal muscles. The myopathies do not have any distinguishing clinical features
from others with limb-girdle weakness and so a diagnosis can be difficult to achieve. We
aimed to determine the frequency and phenotypic spectrum of TRIM32-related myopathies.
Whole exome sequencing was performed for 1 000 patients with unexplained limb-girdle
weakness. Genes known to be associated with limb-girdle weakness, including TRIM32,
were analysed for pathogenic variants. Variants were classified by current ACMG guidelines.
Deleteriousness according to in silico prediction tools, population frequencies, ClinVar
reports of pathogenicity and published literature were also considered. Thirty-six patients
with rare TRIM32 variants were identified; we characterised the clinical features of the
seven patients with pathogenic variants plus two patients who were diagnosed through
sequencing of TRIM32 in our diagnostic service. Disease onset varied from 10 to 45 years.
The presenting symptoms were related to proximal lower limb weakness. Serum creatine
kinase levels were moderately increased; only one normal value was reported. There were
no patients with a cardiomyopathy. Muscle MRI scans revealed a preferential affection of
the posterior compartment muscles at the thigh level. Muscle biopsies showed nonspecific
myopathic or dystrophic changes with occasional vacuoles. Overall, there are no
pathognomonic findings that enable a reliable clinical diagnosis of TRIM32-related
myopathies. Instead, a complementary approach of targeted sequencing and detailed
phenotyping will help diagnose these patients.

37
INTRODUCTION
Limb-girdle muscular dystrophies (LGMDs) are a heterogeneous group of rare genetic
conditions characterised by progressive wasting and weakness of the proximal musculature.
The two major sub-classifications are LGMD1 (autosomal dominant) and LGMD2 (autosomal
recessive),1 with over 30 subdivisions thereof.2 The presentations and phenotypes of the
different diseases are highly variable. Distinguishing clinical and histopathological features
may orient towards a specific diagnosis, but a large proportion of LGMD patients remain
without genetic diagnoses even after state-of-the-art diagnostic work-ups.3, 4 TRIM32-
related myopathies represent a phenotypic spectrum of muscle disorders including
LGMD2H (OMIM #254110). LGMD2H was first identified in the ethnoreligious Hutterite
population and the homozygous TRIM32 founder mutation, p.Asp487Asn, was later
identified as the cause of this disease.5 Only seven definite non-Hutterite TRIM32-related
myopathy patients have been reported in the literature.6-10 The disease is generally
described as a mild and progressive recessive myopathy without characteristic clinical
features. TRIM32 has also been associated with Bardet-Biedl syndrome-11 (BBS11; OMIM
#615988).11
The 14 kb TRIM32 gene on chromosome 9q31-q33, comprising one coding exon, encodes
the 653 amino acid tripartite motif containing 32 (TRIM32) protein. This widely-expressed
E3 ubiquitin ligase comprises a RING, B-box and coiled-coil domain in addition to six NHL
repeats. Two reported missense mutations associated with TRIM32-related myopathy reside
in the NHL repeats (figure 1A), possibly disrupting its protein structure, self-dimerization
and/or muscle-specific interactions.12, 13 TRIM32 deletions, frameshift and nonsense
mutations have also been reported.8-10 In skeletal muscle, TRIM32 localises to Z-lines at
sarcomeric boundaries14 and of the many interactors and substrates, actin, tropomyosin,
troponins and α-actinin are evident and crucial targets.15 TRIM32 has also been implicated
in skeletal muscle regeneration and consequently the function of satellite cells in
particular.16, 17
Here, we present seven patients with TRIM32-related myopathy caused by recessive
TRIM32 mutations detected by whole exome sequencing (WES). An additional two patients
were diagnosed through TRIM32 sequencing in the Northern Molecular Genetics Service
(NMGS) diagnostic laboratory (Newcastle, UK). Eight of the ten distinct pathogenic variants
were novel in their association to the disease, four of which reside outside the NHL repeats.
Furthermore, we report on three patients harbouring homozygous variants of unknown
significance (VUS) in the coiled-coil and intervening regions. We show that the clinical and

38
genetic spectrum of TRIM32-related myopathies is considerably more variable than
previously known, and suggest that targeted sequencing is invaluable in determining the
genetic cause of rare muscle diseases.
METHODS
Patient recruitment
Ethical approval was granted by the Newcastle and North Tyneside research ethics
committee (REC #09/H0906/28) and by the local ethics committees of the participating
centres. Anonymised phenotypic information for each patient was uploaded by the referring
clinician onto PhenoTips.18 Informed written consent was given by the patients, all of whom
presented with unexplained limb-girdle muscle weakness and/or elevated serum creatine
kinase (CK) activity.
Targeted sequencing
Patient DNA samples were submitted to the MRC Centre for Neuromuscular Diseases
Biobank (Newcastle University, UK). WES and data processing were performed by the
Genomics Platform at the Broad Institute of Harvard and MIT (Boston, MA, USA) as
described previously.19 Data were archived in the European Genome-phenome Archive
(EGAS00001002069). Additional TRIM32-related myopathy patients were diagnosed by the
NMGS diagnostic laboratory through routine panel sequencing of 32 LGMD genes, using a
HaloPlex Targeted Enrichment (Agilent) system followed by sequencing on an Illumina
MiSeq instrument.
Analysis of whole exome sequencing data
The variant call set was uploaded onto the Broad Institute of Harvard and MIT’s seqr
platform (https://seqr.broadinstitute.org). TRIM32 was analysed for biologically relevant
variants as described previously.20 Variants were classified according to current ACMG
guidelines.21, 22
Magnetic resonance imaging
Muscle magnetic resonance imaging (MRI) was performed for five of the nine patients on a
1.5T MRI platform at the respective referring centres and for each of the patients
harbouring a homozygous VUS. Cross-sections at the level of the thigh and calf were
assessed on T1-weighted images to evaluate patterns of muscle involvement. Fatty
replacement of muscle was graded according to the Mercuri scale.23

39
Muscle histopathology
Muscle biopsies were obtained for all patients from quadriceps, tibialis anterior, deltoid or
biceps brachii muscles and analysed following standard histologic techniques for light
microscopy.
RNA isolation and quantitative PCR
Lymphoblasts from patient 14, the son of patient 15 (ng222.3) and a healthy control were
cultured in RPMI medium and harvested when a stable and healthy culture was obtained.
Total RNA was obtained using QIAGEN RNeasy Mini Kit (QIAGEN) according to the
manufacturer’s instructions and treated with TURBO DNA-free Kit (Thermo Fisher Scientific).
cDNA was synthesised using the SuperScript III First-Strand Synthesis System (Thermo Fisher
Scientific). Quantitative PCR (qPCR) was performed on a ViiA7 Real-Time PCR System
(Applied Biosystems, Thermo Fisher Scientific) using a SYBR green master mix (Applied
Biosystems, Thermo Fisher Scientific). The signal for TRIM32 was normalised to those of the
housekeeping genes GAPDH and HPRT1, then fold-changes in mRNA levels were calculated
relative to the control sample. Experiments were performed in triplicate. Data analysis was
performed with qbase+ (Version 3.1; Biogazelle, Zwijnaarde, Belgium) and GraphPad Prism 6
(GraphPad Software Inc., USA). Additional information according to MIQE guidelines24 is
provided in the supplement.
Cycloheximide treatment
For the patient and control lymphoblasts, subcultures were stimulated at 37°C for 4 h with
(i) 150 µg/ml cycloheximide (CHX) to block nonsense-mediated mRNA decay (NMD) and 30
µl of dimethyl sulfoxide (DMSO) per 20 ml of cell culture volume, (ii) the same volume of
DMSO as negative solvent control or (iii) no extra compound.
RESULTS
Genetic findings
Of the 1000 MYO-SEQ patients, we identified 36 with rare coding variants in TRIM32 (minor
allele frequency < 1%; numbered 1-36 in supplementary table 1). Twenty-six patients had
single heterozygous variants; these were discarded from our analysis as the variants were
unlikely to be pathogenic in this autosomal recessive disease. Two further patients were
excluded from our analysis: patient 18 was heterozygous for a pathogenic DES mutation and
patient 10 was homozygous for the known pathogenic CAPN3 mutation (p.Arg572Trp)25.
This resulted in seven patients with suspected pathogenic TRIM32 variants and one patient
with a homozygous VUS. For patient 5, only the frameshift variant was considered

40
pathogenic since the missense occurred downstream and had to be classified as a VUS
regardless of the frameshift variant. We included four additional patients harbouring
homozygous rare TRIM32 variants (supplementary table 2): three patients for whom
diagnostic panel sequencing was performed by NMGS and one for whom WES was
performed in Tehran. Two patients harboured a pathogenic variant and two had a VUS.
Pathogenic variants outside the NHL repeats were frameshift mutations (figure 1B);
missense mutations in the coiled-coil region and intervening region were classified as a
VUS.26
Clinical phenotypes
A detailed overview of the clinical findings of patients with pathogenic TRIM32 variants is
described in the upper panel of table 1. Disease onset varied from 10 to 45 years. The main
presenting symptoms were related to proximal lower limb weakness. Over 77% (7/9) of
these patients also had proximal upper limb weakness and 67% (6/9) had distal lower limb
weakness. Only two of the nine patients had (mild) distal upper limb weakness. Axial, facial
and periscapular muscles were variably involved. Serum CK levels were moderately
increased (≤2 000 U/l); only one normal value was reported. Forced vital capacity (FVC) was
normal in the eight patients for whom we had reliable spirometry. An electrocardiogram
(ECG) and echocardiography was performed for every patient and no abnormalities were
noted. There were no patients with a cardiomyopathy. A few striking clinical features were
noted for the patients carrying a homozygous TRIM32 VUS (lower panel of table 1),
including marked distal upper limb weakness for patient 16 and childhood onset and high CK
for patient 38.
Muscle imaging
MRI scans of the patients with pathogenic TRIM32 variants revealed a preferential affection
of the posterior thigh compartment, evolving to a diffuse involvement of the anterior thigh
in later stages (figure 2). A consistent involvement of the posterior lower leg compartment
and the tibialis anterior muscle was observed, as well as the peronei muscles in later stages.
There was a relative sparing of the flexor hallucis longus, flexor digitorum longus and tibialis
posterior muscles. MRI images for the patients carrying a homozygous VUS did not reveal a
consistent pattern (supplementary figure 1).
Histological features
Muscle biopsies showed nonspecific myopathic or dystrophic changes. Vacuoles containing
basophilic material were noted in scattered muscle fibers of patients 14, 15 and 39

41
(supplementary figure 2). No specific histopathological features were noted on the biopsies
of patients carrying a homozygous VUS in TRIM32.
mRNA analysis of the mutant transcript
The functional effect of the p.Glu192GlyfsTer7 frameshift variant was studied in
lymphoblast cultures of patient 14 (compound heterozygous for the p.Ala388Val) and the
son of patient 15 (ng222.3) who is a healthy heterozygous carrier of the frameshift variant.
qPCR revealed a >20% decrease in the abundance of TRIM32 mRNA relative to the control in
both cases, which indicated partial NMD of the truncated transcript (supplementary figure
3A). Treatment of the lymphoblast cultures with the potent NMD inhibitor CHX revealed an
upregulation of total TRIM32 transcript in each cell line when quantified by qPCR
(supplementary figure 3B). However, cDNA sequencing qualitatively indicated a selective
increase of mutant TRIM32 transcript in the cell lines of patient 14 and ng222.3 (figure 3),
further implying that the p.Glu192GlyfsTer7 transcript is targeted but not completely
eliminated by NMD.
DISCUSSION
To our knowledge, other studies have never yielded such a large cohort of non-Hutterite
TRIM32-related myopathy patients and thus we present an extended understanding of this
rare neuromuscular disorder.
The highly conserved NHL domain was the focal region of the TRIM32-related myopathy-
associated missense variants in this study, which likely cause conformational changes that
lead to decreased TRIM32 stability.13 Since indisputable genetic, phenotypic and/or
functional evidence is lacking, two homozygous missense variants were reported as a VUS
according to current ACMG guidelines:21, 22 the variants were in the intervening region
(patient 16) and the coiled-coil domain (patients 38 and 40). The remaining variants outside
the NHL repeats were frameshift changes that were similarly in the C terminal of the
protein.
Despite WES allowing the genetic spectrum of diseases to be broadened, a caveat is that it is
commonly considered to be intractable to copy number variation (CNV) detection. However,
specialised analytical software is now starting to permit this facet of analysis.28 Indeed,
patient 36 appeared to be homozygous for p.Arg613Ter, but after a further evaluation of
her exome using a modified version of PennCNV on a custom Illumina Infinium Array,27 we
detected an overlapping heterozygous 63.5 kb deletion. Only a few large homozygous or
compound heterozygous deletions encompassing the whole gene have previously been
reported.9, 10 Repeat expansion disorders – including facioscapulohumeral dystrophy,

42
myotonic dystrophy type 1 and 2, and oculopharyngeal muscular dystrophy – were excluded
in case of clinical suspicion.
The phenotypes of the patients harbouring novel variants were not strikingly different from
those of the patients with known pathogenic variants. Detailed phenotyping revealed
findings that may orient towards a diagnosis and aided the interpretation of candidate
sequence variants obtained by WES. Only clearly described in one genetically confirmed
TRIM32-related myopathy patient so far,10 marked distal weakness in the upper limbs
seems to be rather exceptional, even in later stages of the disease. Consistent with the
current literature, axial, facial and periscapular muscles were variably involved.6, 29 Despite
TRIM32-related myopathy typically being described as a “mild” myopathy, four patients in
our cohort were largely wheelchair-dependent.5 Current guidelines advise systematic
cardiac evaluation,3 however, our data do not provide evidence in favour of marked cardiac
or respiratory involvement. Similarly prognostically relevant, no clear signs in favour of
bulbar involvement were noted.
Systematic evaluation of muscle MRI images of our TRIM32-related myopathy patient
cohort revealed a consistent pattern of muscle involvement affecting the posterior
compartment muscles at the thigh and calf level in early stages, and with relative sparing of
flexor digitorum longus, tibialis posterior and flexor hallucis longus muscles. In many
LGMDs, the sartorius and gracilis muscles are spared during disease progression,30 which
does not seem to be the case for TRIM32-related myopathy. A predominant involvement of
the posterior thigh muscles on MRI imaging was described in one patient so far.9 MRI
images were acquired around mid-thigh and mid-calf level, but the level slightly differed
from image to image.
The observed histological features in our patients encompassed the spectrum that has been
described in the literature: nonspecific myopathic changes or dystrophic features
sometimes accompanied by vacuoles.8, 29, 31 Schoser and colleagues first described a
vacuolar myopathy related to mutations in TRIM32: ultrastructural evaluation showed that
the small (empty) vacuoles consisted of focal dilations of the sarcoplasmatic reticulum,
hence the name ‘sarcotubular myopathy’ (STM).29 EM was not performed for many of our
patients and when it was, no such vacuoles were observed. The vacuoles observed on the
biopsies of patients 14, 15 and 38 appeared to contain basophilic material, as did the
vacuoles on muscle biopsies of later reported TRIM32 vacuolar myopathy patients.8, 31 The
appearance of vacuoles in scattered muscle fibers on the biopsies of patients with a

43
TRIM32-related myopathy seems to be a relatively common but not obligatory finding,
suggesting that these features are part of a histopathological spectrum, rather than
constituting a separate phenotype.
Additionally, we describe in detail the phenotype of three patients harbouring a
homozygous missense VUS in TRIM32; future phenotypic or functional data will provide
insight into their potential pathogenicity. For these patients, no other candidate variants in
known myopathy genes were identified. Phenotypically, some features contrast with those
of the patients harbouring definite pathogenic variants, such as marked distal weakness in
the upper limbs of patient 16 and the rather atypical disease progression. However, we
cannot exclude that these variants are causal in the disease.
Further investigation of the pathogenicity of these variants is needed, as is the case for the
exact genetic mechanisms in TRIM32-related myopathy. TRIM32 has only one coding exon,
so theoretically it is unlikely that the mRNA-transcript of a nonsense allele is destabilised by
NMD: a stop codon is only recognised as a premature termination codon if it is located more
than 50-55 nucleotides upstream of the last exon-exon junction.32 However, by studying the
functional effect of the p.Glu192GlyfsTer7 variant on the mRNA level in lymphoblasts, we
showed that the mutant transcript is at least partially targeted by NMD. Our documentation
of NMD of a TRIM32 nonsense transcript further reinforces the hypothesis that the
downstream consequences of different TRIM32 mutations seem to be similar, namely a
relative loss of TRIM32 protein abundance and/or function. However, we do not observe
the multi-systemic involvement observed in BBS as noted for the BBS11 variant located in
the B-box domain of TRIM32.11 Vice versa, no skeletal muscle involvement is noted in the
patient with this BBS11 variant.
Overall, we report nine TRIM32-related myopathy patients harbouring pathogenic TRIM32
variants and introduce three patients with a homozygous TRIM32 VUS. Complemented with
deep phenotyping, our application of WES enabled patients with TRIM32-related myopathy
to be identified. We propose that similar approaches of targeted sequencing and thorough
curation of phenotypic information will expedite future TRIM32-related myopathy
diagnoses.
ACKNOWLEDGEMENTS
We thank the patients for donating their tissue samples. Dr Tuomo Polvikoski provided the
pathology slides for patient 39.

44
FUNDING
This work was supported by Sanofi Genzyme, Ultragenyx, LGMD2I Research Fund, Samantha J Brazzo
Foundation, LGMD2D Foundation, Kurt+Peter Foundation, Muscular Dystrophy UK and Coalition to
Cure Calpain 3. This work was also supported by the Association Belge contre les Maladies
Neuromusculaire (ABMM) - Aide à la Recherche ASBL. JB is supported by a Senior Clinical
Researcher mandate of the Research Fund - Flanders (FWO).
45
FIGURES AND TABLES
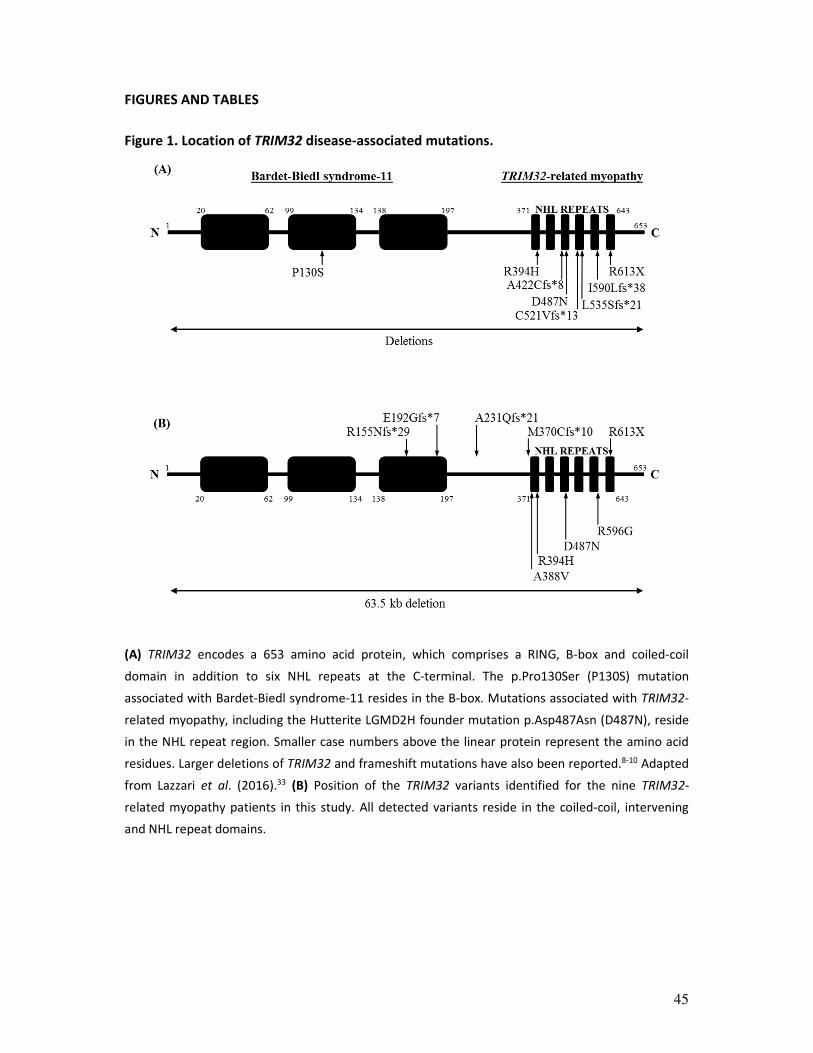
Figure 1. Location of TRIM32 disease-associated mutations.
(A) TRIM32 encodes a 653 amino acid protein, which comprises a RING, B-box and coiled-coil
domain in addition to six NHL repeats at the C-terminal. The p.Pro130Ser (P130S) mutation
associated with Bardet-Biedl syndrome-11 resides in the B-box. Mutations associated with TRIM32-
related myopathy, including the Hutterite LGMD2H founder mutation p.Asp487Asn (D487N), reside
in the NHL repeat region. Smaller case numbers above the linear protein represent the amino acid
residues. Larger deletions of TRIM32 and frameshift mutations have also been reported.8-10 Adapted
from Lazzari et al. (2016).33 (B) Position of the TRIM32 variants identified for the nine TRIM32-
related myopathy patients in this study. All detected variants reside in the coiled-coil, intervening
and NHL repeat domains.

46
Figure 2. T1-weighted axial magnetic resonance imaging (MRI) of the lower limbs of five
patients identified with pathogenic TRIM32 variants.
MRI images at mid-thigh level on the left and mid-calf level on the right for: (A) patient 14 at
approximately 14 years of disease duration; (B) patient 15 at 18 years of disease duration; (C)
patient 27 at 8 years of disease duration; (D) patient 28 at 28 years of disease duration; (E) patient
37 at 5 years disease duration. (A-C and E) MRI images at thigh level, with slight differences in the
exact level of acquisition of the image, revealed a preferential involvement of posterior
compartment muscles. (D) End-stage involvement of all thigh muscles was observed for patient 28.
(A-E) MRI images at calf level revealed a similar pattern for all patients, with predominant
involvement of the posterior compartment. White arrows indicate relatively spared muscles. FDL,
flexor digitorum longus; TP, tibialis posterior; FHL, flexor hallucis longus.
47
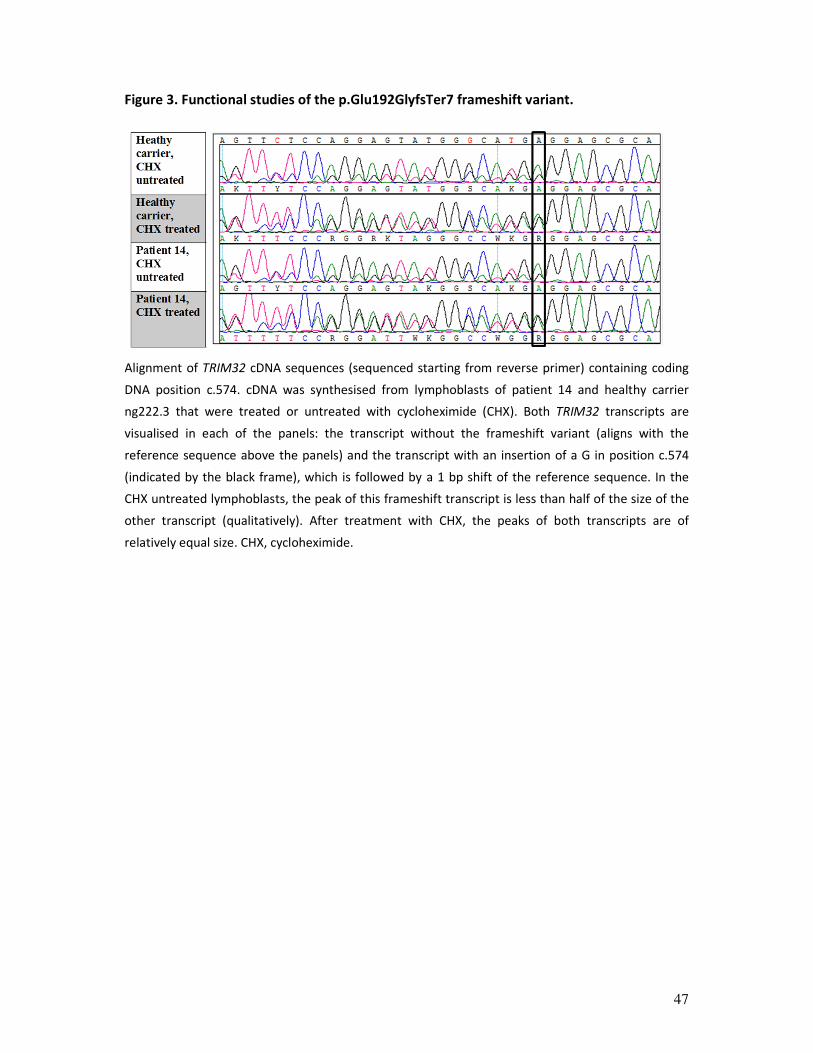
Figure 3. Functional studies of the p.Glu192GlyfsTer7 frameshift variant.
Alignment of TRIM32 cDNA sequences (sequenced starting from reverse primer) containing coding
DNA position c.574. cDNA was synthesised from lymphoblasts of patient 14 and healthy carrier
ng222.3 that were treated or untreated with cycloheximide (CHX). Both TRIM32 transcripts are
visualised in each of the panels: the transcript without the frameshift variant (aligns with the
reference sequence above the panels) and the transcript with an insertion of a G in position c.574
(indicated by the black frame), which is followed by a 1 bp shift of the reference sequence. In the
CHX untreated lymphoblasts, the peak of this frameshift transcript is less than half of the size of the
other transcript (qualitatively). After treatment with CHX, the peaks of both transcripts are of
relatively equal size. CHX, cycloheximide.
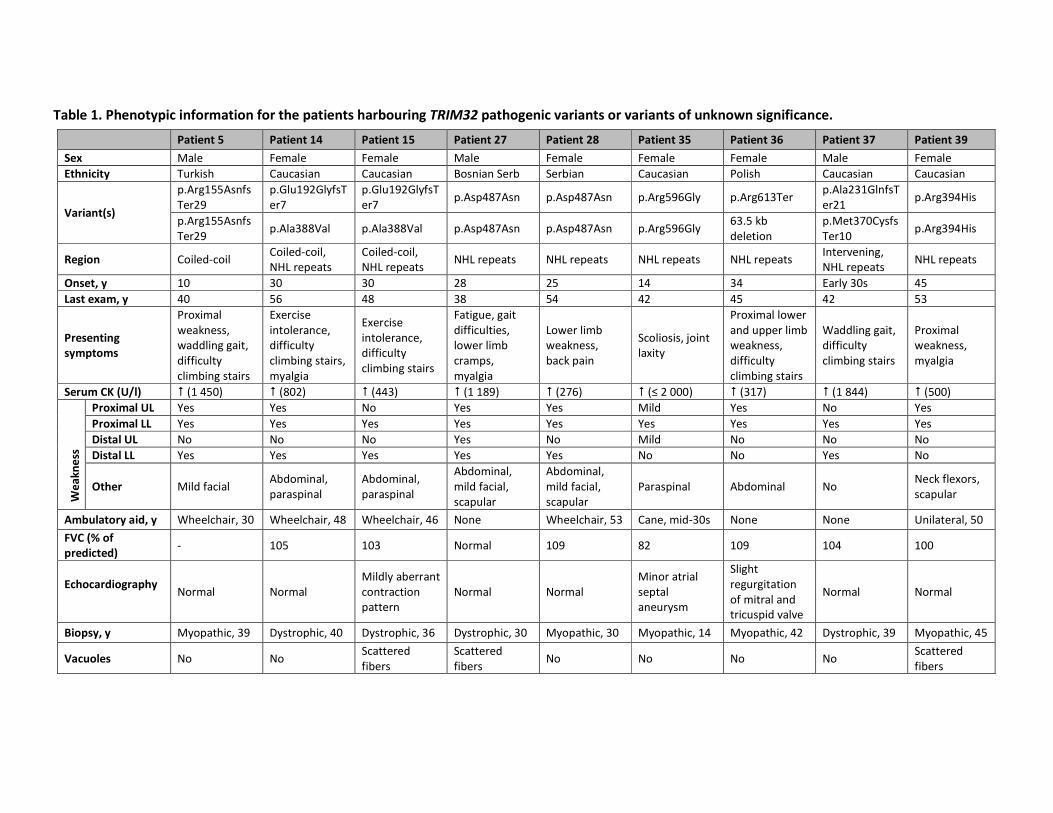
Table 1. Phenotypic information for the patients harbouring TRIM32 pathogenic variants or variants of unknown significance.
Patient 5 Patient 14 Patient 15 Patient 27 Patient 28 Patient 35 Patient 36 Patient 37 Patient 39
Sex Male Female Female Male Female Female Female Male Female
Ethnicity Turkish Caucasian Caucasian Bosnian Serb Serbian Caucasian Polish Caucasian Caucasian
Variant(s)
p.Arg155Asnfs
Ter29
p.Glu192GlyfsT
er7
p.Glu192GlyfsT
er7 p.Asp487Asn p.Asp487Asn p.Arg596Gly p.Arg613Ter
p.Ala231GlnfsT
er21 p.Arg394His
p.Arg155Asnfs
Ter29 p.Ala388Val p.Ala388Val p.Asp487Asn p.Asp487Asn p.Arg596Gly
63.5 kb
deletion
p.Met370Cysfs
Ter10 p.Arg394His
Region Coiled-coil Coiled-coil,
NHL repeats
Coiled-coil,
NHL repeats NHL repeats NHL repeats NHL repeats NHL repeats
Intervening,
NHL repeats NHL repeats
Onset, y 10 30 30 28 25 14 34 Early 30s 45
Last exam, y 40 56 48 38 54 42 45 42 53
Presenting
symptoms
Proximal
weakness,
waddling gait,
difficulty
climbing stairs
Exercise
intolerance,
difficulty
climbing stairs,
myalgia
Exercise
intolerance,
difficulty
climbing stairs
Fatigue, gait
difficulties,
lower limb
cramps,
myalgia
Lower limb
weakness,
back pain
Scoliosis, joint
laxity
Proximal lower
and upper limb
weakness,
difficulty
climbing stairs
Waddling gait,
difficulty
climbing stairs
Proximal
weakness,
myalgia
Serum CK (U/l) � (1 450) � (802) � (443) � (1 189) � (276) � (≤ 2 000) � (317) � (1 844) � (500)
We
ak
ne
ss
Proximal UL Yes Yes No Yes Yes Mild Yes No Yes
Proximal LL Yes Yes Yes Yes Yes Yes Yes Yes Yes
Distal UL No No No Yes No Mild No No No
Distal LL Yes Yes Yes Yes Yes No No Yes No
Other Mild facial Abdominal,
paraspinal
Abdominal,
paraspinal
Abdominal,
mild facial,
scapular
Abdominal,
mild facial,
scapular
Paraspinal Abdominal No Neck flexors,
scapular
Ambulatory aid, y Wheelchair, 30 Wheelchair, 48 Wheelchair, 46 None Wheelchair, 53 Cane, mid-30s None None Unilateral, 50
FVC (% of
predicted) - 105 103 Normal 109 82 109 104 100
Echocardiography
Normal Normal
Mildly aberrant
contraction
pattern
Normal Normal
Minor atrial
septal
aneurysm
Slight
regurgitation
of mitral and
tricuspid valve
Normal Normal
Biopsy, y Myopathic, 39 Dystrophic, 40 Dystrophic, 36 Dystrophic, 30 Myopathic, 30 Myopathic, 14 Myopathic, 42 Dystrophic, 39 Myopathic, 45
Vacuoles No No Scattered
fibers
Scattered
fibers No No No No
Scattered
fibers
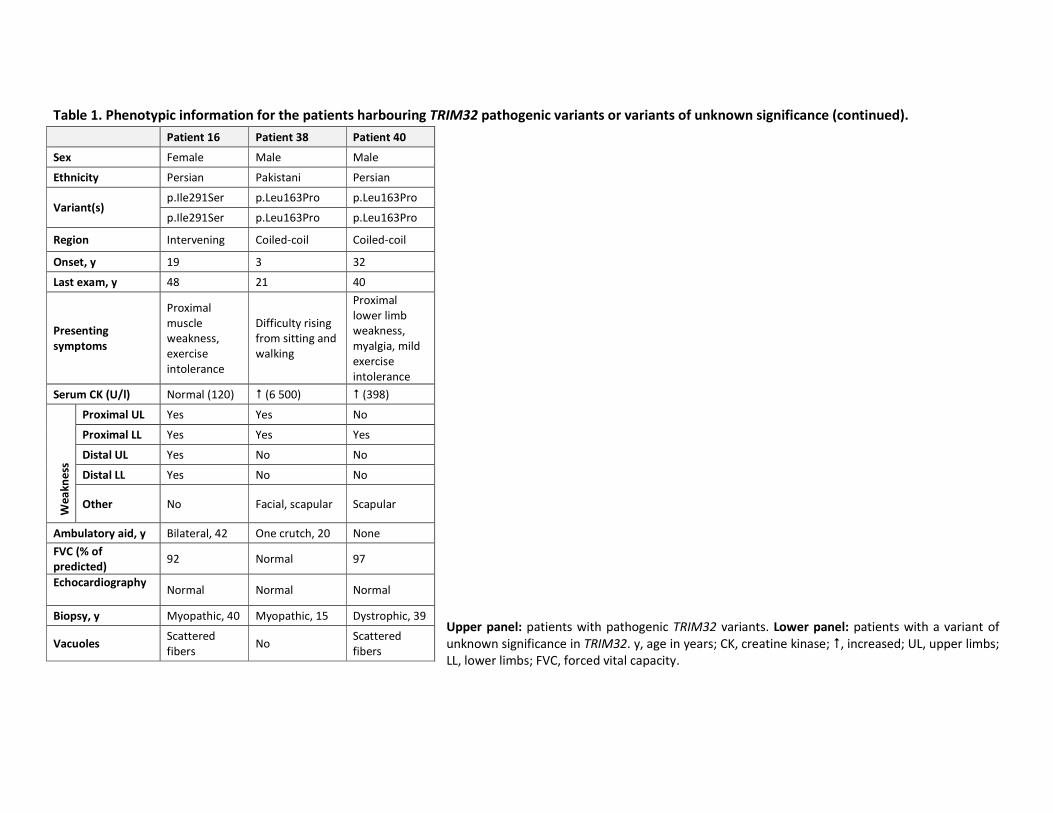
Table 1. Phenotypic information for the patients harbouring TRIM32 pathogenic variants or variants of unknown significance (continued).
Upper panel: patients with pathogenic TRIM32 variants. Lower panel: patients with a variant of
unknown significance in TRIM32. y, age in years; CK, creatine kinase; �, increased; UL, upper limbs;
LL, lower limbs; FVC, forced vital capacity.
Patient 16 Patient 38 Patient 40
Sex Female Male Male
Ethnicity Persian Pakistani Persian
Variant(s) p.Ile291Ser p.Leu163Pro p.Leu163Pro
p.Ile291Ser p.Leu163Pro p.Leu163Pro
Region Intervening Coiled-coil Coiled-coil
Onset, y 19 3 32
Last exam, y 48 21 40
Presenting
symptoms
Proximal
muscle
weakness,
exercise
intolerance
Difficulty rising
from sitting and
walking
Proximal
lower limb
weakness,
myalgia, mild
exercise
intolerance
Serum CK (U/l) Normal (120) � (6 500) � (398)
We
ak
ne
ss
Proximal UL Yes Yes No
Proximal LL Yes Yes Yes
Distal UL Yes No No
Distal LL Yes No No
Other No Facial, scapular Scapular
Ambulatory aid, y Bilateral, 42 One crutch, 20 None
FVC (% of
predicted) 92 Normal 97
Echocardiography
Normal Normal Normal
Biopsy, y Myopathic, 40 Myopathic, 15 Dystrophic, 39
Vacuoles Scattered
fibers No
Scattered
fibers

50
REFERENCES
1. Guglieri M, Straub V, Bushby K, Lochmuller H. Limb-girdle muscular dystrophies. Current opinion in
neurology 2008;21:576-584.
2. Vissing J. Limb girdle muscular dystrophies: classification, clinical spectrum and emerging therapies.
Current opinion in neurology 2016;29:635-641.
3. Narayanaswami P, Weiss M, Selcen D, et al. Evidence-based guideline summary: diagnosis and
treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of
the American Academy of Neurology and the practice issues review panel of the American Association
of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-1463.
4. Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for
translational research. Nature reviews Neurology 2016;12:294-309.
5. Frosk P, Weiler T, Nylen E, et al. Limb-girdle muscular dystrophy type 2H associated with mutation in
TRIM32, a putative E3-ubiquitin-ligase gene. American journal of human genetics 2002;70:663-672.
6. Saccone V, Palmieri M, Passamano L, et al. Mutations that impair interaction properties of TRIM32
associated with limb-girdle muscular dystrophy 2H. Human mutation 2008;29:240-247.
7. Cossee M, Lagier-Tourenne C, Seguela C, et al. Use of SNP array analysis to identify a novel TRIM32
mutation in limb-girdle muscular dystrophy type 2H. Neuromuscular disorders : NMD 2009;19:255-
260.
8. Borg K, Stucka R, Locke M, et al. Intragenic deletion of TRIM32 in compound heterozygotes with
sarcotubular myopathy/LGMD2H. Human mutation 2009;30:E831-844.
9. Neri M, Selvatici R, Scotton C, et al. A patient with limb girdle muscular dystrophy carries a TRIM32
deletion, detected by a novel CGH array, in compound heterozygosis with a nonsense mutation.
Neuromuscular disorders : NMD 2013;23:478-482.
10. Nectoux J, de Cid R, Baulande S, et al. Detection of TRIM32 deletions in LGMD patients analyzed by a
combined strategy of CGH array and massively parallel sequencing. European journal of human
genetics : EJHG 2015;23:929-934.
11. Chiang AP, Beck JS, Yen HJ, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3
ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proceedings of the National Academy of
Sciences of the United States of America 2006;103:6287-6292.
12. Koliopoulos MG, Esposito D, Christodoulou E, Taylor IA, Rittinger K. Functional role of TRIM E3 ligase
oligomerization and regulation of catalytic activity. 2016;35:1204-1218.
13. Kudryashova E, Struyk A, Mokhonova E, Cannon SC, Spencer MJ. The common missense mutation
D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in
knock-in mice resulting in a Trim32-null phenotype. Human molecular genetics 2011;20:3925-3932.
14. Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Human
molecular genetics 2009;18:2344-2358.
15. Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-
bands, and thin filaments in muscle atrophy. The Journal of cell biology 2012;198:575-589.
16. Kudryashova E, Kramerova I, Spencer MJ. Satellite cell senescence underlies myopathy in a mouse
model of limb-girdle muscular dystrophy 2H. The Journal of clinical investigation 2012;122:1764-1776.
17. Nicklas S, Otto A, Wu X, et al. TRIM32 regulates skeletal muscle stem cell differentiation and is
necessary for normal adult muscle regeneration. PloS one 2012;7:e30445.
18. Girdea M, Dumitriu S, Fiume M, et al. PhenoTips: patient phenotyping software for clinical and
research use. Human mutation 2013;34:1057-1065.
19. Peric S, Glumac JN, Topf A, et al. A novel recessive TTN founder variant is a common cause of distal
myopathy in the Serbian population. European journal of human genetics : EJHG 2017;25:572-581.
20. Johnson K, Topf A, Bertoli M, et al. Identification of GAA variants through whole exome sequencing
targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness. Orphanet journal
of rare diseases 2017;12:173.

51
21. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a
joint consensus recommendation of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Genetics in medicine : official journal of the American College of
Medical Genetics 2015;17:405-424.
22. Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the
ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genetics in
medicine : official journal of the American College of Medical Genetics 2016;18:1165.
23. Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited
neuromuscular disorders: past, present, and future. Journal of magnetic resonance imaging : JMRI
2007;25:433-440.
24. Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of
quantitative real-time PCR experiments. Clinical chemistry 2009;55:611-622.
25. Richard I, Brenguier L, Dincer P, et al. Multiple independent molecular etiology for limb-girdle
muscular dystrophy type 2A patients from various geographical origins. American journal of human
genetics 1997;60:1128-1138.
26. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a
joint consensus recommendation of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Genet Med 2015;17:405-424.
27. Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-
resolution copy number variation detection in whole-genome SNP genotyping data. Genome research
2007;17:1665-1674.
28. Tattini L, D'Aurizio R, Magi A. Detection of Genomic Structural Variants from Next-Generation
Sequencing Data. Frontiers in bioengineering and biotechnology 2015;3:92.
29. Schoser BG, Frosk P, Engel AG, Klutzny U, Lochmuller H, Wrogemann K. Commonality of TRIM32
mutation in causing sarcotubular myopathy and LGMD2H. Annals of neurology 2005;57:591-595.
30. Diaz-Manera J, Llauger J, Gallardo E, Illa I. Muscle MRI in muscular dystrophies. Acta myologica :
myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology / edited
by the Gaetano Conte Academy for the study of striated muscle diseases 2015;34:95-108.
31. Liewluck T, Tracy JA, Sorenson EJ, Engel AG. Scapuloperoneal muscular dystrophy phenotype due to
TRIM32-sarcotubular myopathy in South Dakota Hutterite. Neuromuscular disorders : NMD
2013;23:133-138.
32. Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB
reports 2017;50:175-185.
33. Lazzari E, Meroni G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: From muscular
dystrophy to tumours. The international journal of biochemistry & cell biology 2016;79:469-477.
52
SUPPLEMENTARY METHODS, FIGURES AND TABLES
Supplementary methods. Detailed methodology of quantitative PCR.
Quantitative PCR (qPCR) target information and qPCR oligonucleotides
• Target: TRIM32
o Primers (manufactured by Integrated DNA Technologies (IDT))
� CAGAGGGAGGCAGGCGG: located in exon 1 (non-coding)
� GGGCATCCAGGTTCAGG: located in exon 2 (coding)
o Location of amplicon (hg19): chr9:119,449,581-119,463,579 (forward strand)
o 2 splice variants are targeted:
� >uc004bjx.2__TRIM32 (ENST00000373983):21+198: 178 bp
� >uc004bjw.2__TRIM32 (ENST00000450136):21+201: 181 bp
qPCR validation
PCR efficiency for the TRIM32 primers calculated from slope (E), standard error (SE) for E, calibration
curves with slope and y intercept and R2 of calibration curve are presented in the table below.
Analysis was performed with qbase+ (Version 3.1; Biogazelle, Zwijnaarde, Belgium).
E
E (SE) R2 Slope Slope error Intercept
Intercept
error
Computed
Efficiency
(%) Computed Computed Computed Computed Computed Computed
GAPDH 1.983 98.3 0.01 0.999 -3.364 0.024 20.166 0.034
HPRT1 2.015 101.5 0.022 0.998 -3.606 0.095 24.88 0.135
TRIM32 1.994 99.4 0.062 0.982 -3.336 0.151 30.822 0.188
Data analysis
Data analysis was performed with qbase+ (Version 3.1; Biogazelle, Zwijnaarde, Belgium), data were
presented using Excel. The experiment was performed in triplicate. Outliers were identified and
discarded (CT values >0.5 difference). The signal for TRIM32 was normalised to those of the
housekeeping genes GAPDH and HPRT1.

53
Supplementary figure 1. T1-weighted axial magnetic resonance imaging (MRI) of the lower
limbs of patient 38 identified with a homozygous TRIM32 variant of unknown significance.
MRI images at mid-thigh level on the left and mid-calf level on the right for: (A) patient 16 acquired
at approximately 29 years of disease duration; (B) patient 38 at 11 years of disease duration; (C)
patient 40 at 8 years of disease duration. (A) MRI images of patient 16 showed end-stage
involvement of all muscles at the level of the thigh and the calf, with the exception of marked
sparing of the gracilis muscle and the lateral gastrocnemius. Arrows indicate relatively spared
muscles. (B) MRI images of patient 38 revealed a diffuse mild fatty infiltration of muscles of the
anterior and posterior compartment of the thigh. Arrows indicate predominantly involved muscles
at the calf level, i.e. muscles of the anterior and lateral compartment, accompanied by asymmetric
involvement of the medial gastrocnemius muscle (atrophy more prominent than fatty replacement).
(C) MRI images of patient 40 showed end-stage involvement of all thigh muscles. At the calf level,
end-stage involvement of muscles of the lateral compartment and of the gastrocnemius and soleus
muscles was noted, with the remaining muscle groups being moderately involved. GR, gracilis
muscle; LG, lateral gastrocnemius; MG, medial gastrocnemius.
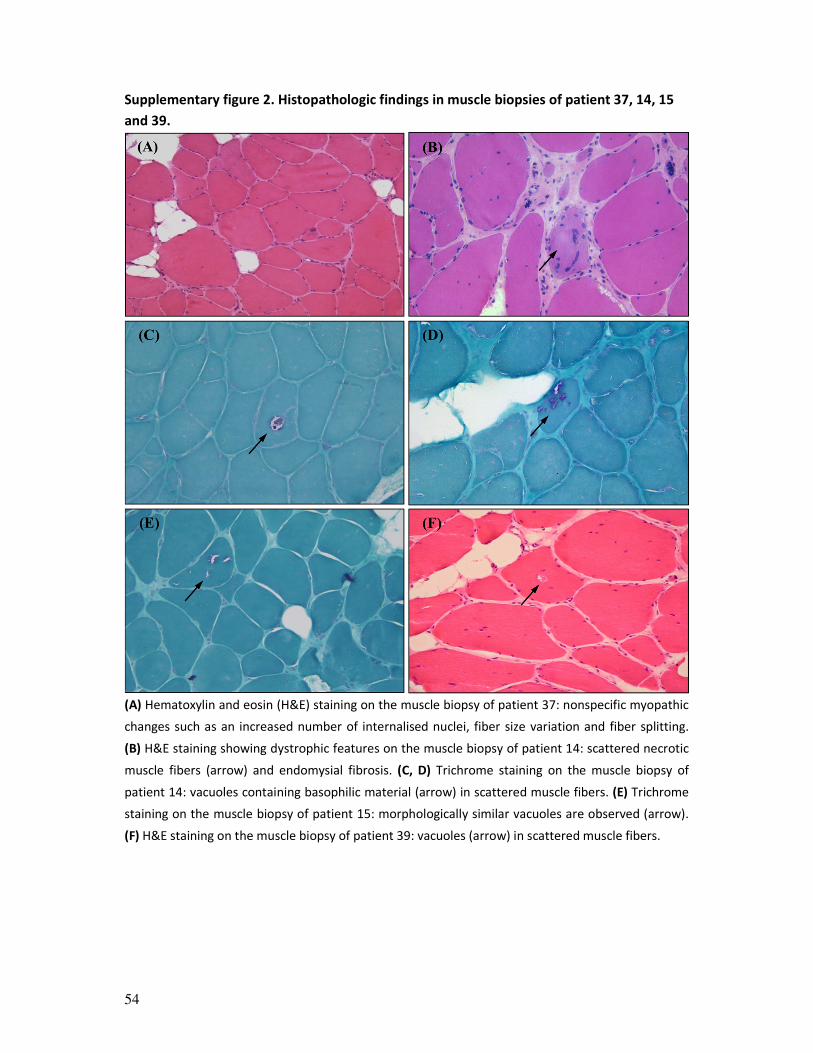
54
Supplementary figure 2. Histopathologic findings in muscle biopsies of patient 37, 14, 15
and 39.
(A) Hematoxylin and eosin (H&E) staining on the muscle biopsy of patient 37: nonspecific myopathic
changes such as an increased number of internalised nuclei, fiber size variation and fiber splitting.
(B) H&E staining showing dystrophic features on the muscle biopsy of patient 14: scattered necrotic
muscle fibers (arrow) and endomysial fibrosis. (C, D) Trichrome staining on the muscle biopsy of
patient 14: vacuoles containing basophilic material (arrow) in scattered muscle fibers. (E) Trichrome
staining on the muscle biopsy of patient 15: morphologically similar vacuoles are observed (arrow).
(F) H&E staining on the muscle biopsy of patient 39: vacuoles (arrow) in scattered muscle fibers.

55
Supplementary figure 3. Functional studies of the p.Glu192GlyfsTer7 frameshift variant.
(A) TRIM32 mRNA analysis in lymphoblasts. TRIM32 mRNA levels were first normalised internally to
GAPDH and HPRT1 levels; subsequently, TRIM32 mRNA levels of the cell lines of patient 14 and the
healthy carrier (ng222.3) were normalised (as a percentage) to the control. Error bars: standard
error of the mean (SEM). (B) TRIM32 mRNA analysis in lymphoblasts: cycloheximide (CHX) untreated
vs. treated. For each respective cell line, TRIM32 mRNA levels of the CHX-treated cells were
normalised (as a percentage) to those of the untreated cells, after internal normalisation to GAPDH
and HPRT1 levels. There is an increase in TRIM32 mRNA in the cells of patient 14 and the healthy
carrier (ng222.3), but for the control as well. Error bars: SEM of the replicates. CHX, cycloheximide.
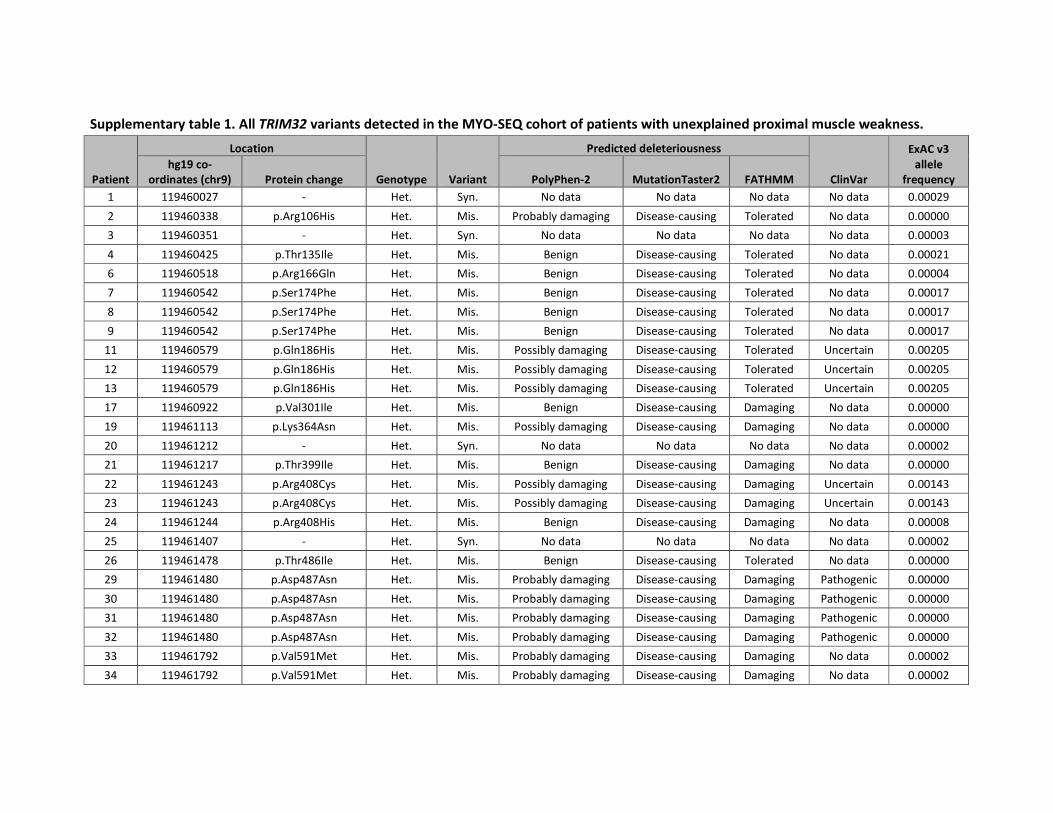
Supplementary table 1. All TRIM32 variants detected in the MYO-SEQ cohort of patients with unexplained proximal muscle weakness.
Patient
Location
Genotype Variant
Predicted deleteriousness
ClinVar
ExAC v3
allele
frequency
hg19 co-
ordinates (chr9) Protein change PolyPhen-2 MutationTaster2 FATHMM
1 119460027 - Het. Syn. No data No data No data No data 0.00029
2 119460338 p.Arg106His Het. Mis. Probably damaging Disease-causing Tolerated No data 0.00000
3 119460351 - Het. Syn. No data No data No data No data 0.00003
4 119460425 p.Thr135Ile Het. Mis. Benign Disease-causing Tolerated No data 0.00021
6 119460518 p.Arg166Gln Het. Mis. Benign Disease-causing Tolerated No data 0.00004
7 119460542 p.Ser174Phe Het. Mis. Benign Disease-causing Tolerated No data 0.00017
8 119460542 p.Ser174Phe Het. Mis. Benign Disease-causing Tolerated No data 0.00017
9 119460542 p.Ser174Phe Het. Mis. Benign Disease-causing Tolerated No data 0.00017
11 119460579 p.Gln186His Het. Mis. Possibly damaging Disease-causing Tolerated Uncertain 0.00205
12 119460579 p.Gln186His Het. Mis. Possibly damaging Disease-causing Tolerated Uncertain 0.00205
13 119460579 p.Gln186His Het. Mis. Possibly damaging Disease-causing Tolerated Uncertain 0.00205
17 119460922 p.Val301Ile Het. Mis. Benign Disease-causing Damaging No data 0.00000
19 119461113 p.Lys364Asn Het. Mis. Possibly damaging Disease-causing Damaging No data 0.00000
20 119461212 - Het. Syn. No data No data No data No data 0.00002
21 119461217 p.Thr399Ile Het. Mis. Benign Disease-causing Damaging No data 0.00000
22 119461243 p.Arg408Cys Het. Mis. Possibly damaging Disease-causing Damaging Uncertain 0.00143
23 119461243 p.Arg408Cys Het. Mis. Possibly damaging Disease-causing Damaging Uncertain 0.00143
24 119461244 p.Arg408His Het. Mis. Benign Disease-causing Damaging No data 0.00008
25 119461407 - Het. Syn. No data No data No data No data 0.00002
26 119461478 p.Thr486Ile Het. Mis. Benign Disease-causing Tolerated No data 0.00000
29 119461480 p.Asp487Asn Het. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000
30 119461480 p.Asp487Asn Het. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000
31 119461480 p.Asp487Asn Het. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000
32 119461480 p.Asp487Asn Het. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000
33 119461792 p.Val591Met Het. Mis. Probably damaging Disease-causing Damaging No data 0.00002
34 119461792 p.Val591Met Het. Mis. Probably damaging Disease-causing Damaging No data 0.00002
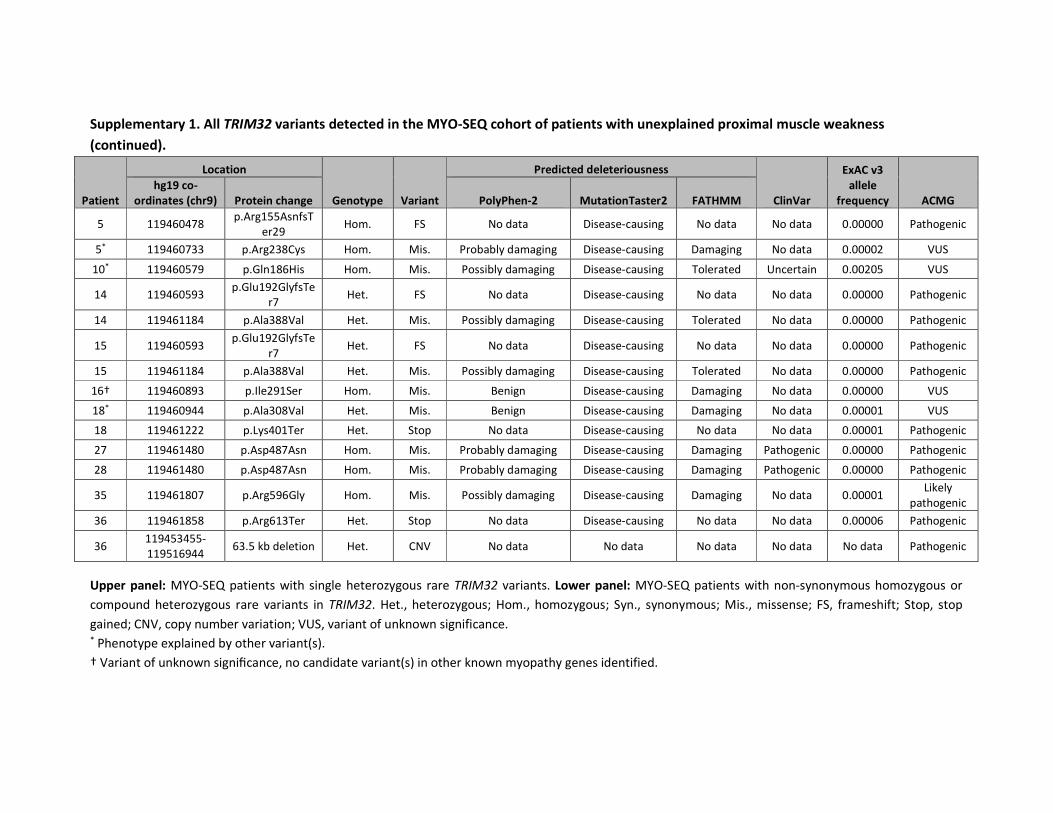
Supplementary 1. All TRIM32 variants detected in the MYO-SEQ cohort of patients with unexplained proximal muscle weakness
(continued).
Patient
Location
Genotype Variant
Predicted deleteriousness
ClinVar
ExAC v3
allele
frequency ACMG
hg19 co-
ordinates (chr9) Protein change PolyPhen-2 MutationTaster2 FATHMM
5 119460478 p.Arg155AsnfsT
er29 Hom. FS No data Disease-causing No data No data 0.00000 Pathogenic
5* 119460733 p.Arg238Cys Hom. Mis. Probably damaging Disease-causing Damaging No data 0.00002 VUS
10* 119460579 p.Gln186His Hom. Mis. Possibly damaging Disease-causing Tolerated Uncertain 0.00205 VUS
14 119460593 p.Glu192GlyfsTe
r7 Het. FS No data Disease-causing No data No data 0.00000 Pathogenic
14 119461184 p.Ala388Val Het. Mis. Possibly damaging Disease-causing Tolerated No data 0.00000 Pathogenic
15 119460593 p.Glu192GlyfsTe
r7 Het. FS No data Disease-causing No data No data 0.00000 Pathogenic
15 119461184 p.Ala388Val Het. Mis. Possibly damaging Disease-causing Tolerated No data 0.00000 Pathogenic
16† 119460893 p.Ile291Ser Hom. Mis. Benign Disease-causing Damaging No data 0.00000 VUS
18* 119460944 p.Ala308Val Het. Mis. Benign Disease-causing Damaging No data 0.00001 VUS
18 119461222 p.Lys401Ter Het. Stop No data Disease-causing No data No data 0.00001 Pathogenic
27 119461480 p.Asp487Asn Hom. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000 Pathogenic
28 119461480 p.Asp487Asn Hom. Mis. Probably damaging Disease-causing Damaging Pathogenic 0.00000 Pathogenic
35 119461807 p.Arg596Gly Hom. Mis. Possibly damaging Disease-causing Damaging No data 0.00001 Likely
pathogenic
36 119461858 p.Arg613Ter Het. Stop No data Disease-causing No data No data 0.00006 Pathogenic
36 119453455-
119516944 63.5 kb deletion Het. CNV No data No data No data No data No data Pathogenic
Upper panel: MYO-SEQ patients with single heterozygous rare TRIM32 variants. Lower panel: MYO-SEQ patients with non-synonymous homozygous or
compound heterozygous rare variants in TRIM32. Het., heterozygous; Hom., homozygous; Syn., synonymous; Mis., missense; FS, frameshift; Stop, stop
gained; CNV, copy number variation; VUS, variant of unknown significance. * Phenotype explained by other variant(s).
† Variant of unknown significance, no candidate variant(s) in other known myopathy genes identified.
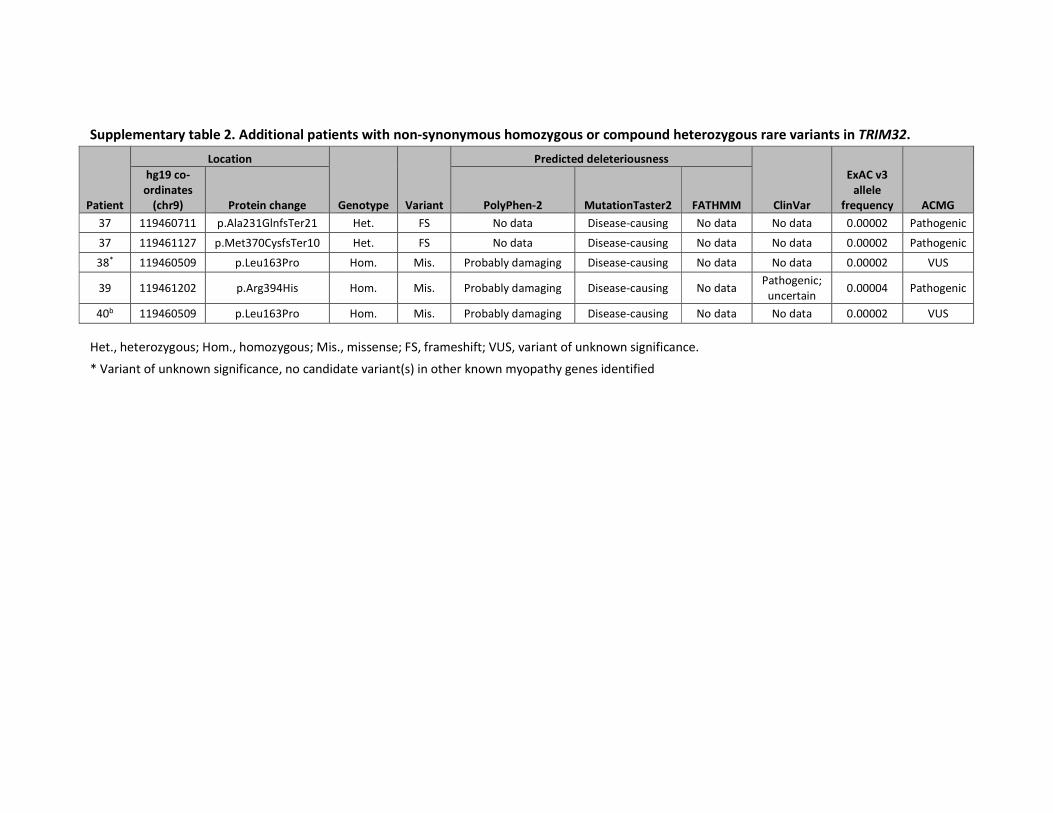
Supplementary table 2. Additional patients with non-synonymous homozygous or compound heterozygous rare variants in TRIM32.
Patient
Location
Genotype Variant
Predicted deleteriousness
ClinVar
ExAC v3
allele
frequency ACMG
hg19 co-
ordinates
(chr9) Protein change PolyPhen-2 MutationTaster2 FATHMM
37 119460711 p.Ala231GlnfsTer21 Het. FS No data Disease-causing No data No data 0.00002 Pathogenic
37 119461127 p.Met370CysfsTer10 Het. FS No data Disease-causing No data No data 0.00002 Pathogenic
38* 119460509 p.Leu163Pro Hom. Mis. Probably damaging Disease-causing No data No data 0.00002 VUS
39 119461202 p.Arg394His Hom. Mis. Probably damaging Disease-causing No data Pathogenic;
uncertain 0.00004 Pathogenic
40b 119460509 p.Leu163Pro Hom. Mis. Probably damaging Disease-causing No data No data 0.00002 VUS
Het., heterozygous; Hom., homozygous; Mis., missense; FS, frameshift; VUS, variant of unknown significance.
* Variant of unknown significance, no candidate variant(s) in other known myopathy genes identified

59

60

61
CHAPTER 2
Muscular dystrophy with arrhythmia caused by loss-of-
function mutations in BVES
Willem De Ridder*, Isabelle Nelson*, Bob Asselbergh, Boel De Paepe, Maud Beuvin, Rabah
Ben Yaou, Cécile Masson, Anne Boland, Jean-François Deleuze, Thierry Maisonobe, Bruno
Eymard, Sofie Symoens, Roland Schindler, Thomas Brand, Katherine Johnson, Ana Töpf,
Volker Straub, Peter De Jonghe, Jan L. De Bleecker, Gisèle Bonne†, Jonathan Baets†
* Equal contribution
† Equal contribujon
Neurol Genet. 2019;5(2):e321

62
ABSTRACT
Objective: To study the genetic and phenotypic spectrum of patients harboring recessive
mutations in BVES.
Methods: We performed whole exome sequencing in a multicenter cohort of 1929 patients
with a suspected hereditary myopathy, showing unexplained limb-girdle muscular weakness
and/or elevated creatine kinase level. Immunohistochemistry and mRNA experiments on
patients’ skeletal muscle tissue were performed to study the pathogenicity of identified
loss-of-function (LOF) variants in BVES.
Results: We identified four individuals from three families harboring homozygous LOF
variants in BVES, the gene which encodes for the Popeye domain containing protein 1
(POPDC1). Patients showed skeletal muscle involvement and cardiac conduction
abnormalities of varying nature and severity, but all exhibited at least subclinical signs of
both skeletal muscle and cardiac disease. All identified mutations lead to a partial or
complete loss of function of BVES through nonsense-mediated decay or through functional
changes to the POPDC1 protein.
Conclusions: We report the identification of homozygous LOF mutations in BVES, causal in a
young adult onset myopathy with concomitant cardiac conduction disorders in the absence
of structural heart disease. These findings underline the role of POPDC1, and by extension
other members of this protein family, in striated muscle physiology and disease. This
disorder appears to have a low prevalence although it is probably underdiagnosed due to its
striking phenotypic variability and often subtle yet clinically relevant manifestations,
particularly concerning the cardiac conduction abnormalities.

63
INTRODUCTION
Limb-girdle muscular dystrophies (LGMDs) comprise a phenotypically and genetically
heterogeneous group of autosomally inherited myopathies characterized by progressive
proximal muscle weakness.1 Cardiac involvement is common in LGMDs2 and practice
guidelines recommend referring patients for cardiac assessment.3 Hereditary cardiac
conduction disorders without structural cardiac disease are rare but an increasing number
of culprit genes are being identified.4
Previously, a homozygous missense mutation (p.Ser201Phe) in the blood vessel epicardial
substance (BVES) gene has been identified in three individuals from a single family, the
eldest presenting with an overt LGMD phenotype and all three showing elevated creatine
kinase (CK) levels and a second-degree atrioventricular (AV) block.5 The disease was
originally classified as LGMD2X (OMIM #616812). BVES encodes for a 360 amino acid
protein also known as POPDC1, part of the Popeye domain containing (POPDC) family of
proteins, which are cAMP-binding transmembrane proteins that are abundantly expressed
in striated muscle.6 In patients’ muscle, a marked reduction was observed in the membrane
localization of POPDC1 and POPDC2.5 In zebrafish popdc1 morphants and popdc1p.Ser191Phe
knock-in (KI) mutants, skeletal muscle abnormalities and AV conduction blocks have been
noted.5 In addition, previously reported homozygous Popdc1 null mutant mice showed
delayed skeletal muscle regeneration and an age-dependent stress-induced sinus node
dysfunction (SND).7, 8
Here, we present four individuals from three families harboring homozygous loss-of-
function (LOF) mutations in BVES. We show that the skeletal muscle and cardiac
involvement resulting from these BVES mutations is highly variable and emphasize the
relevance of BVES mutations with regard to hereditary cardiac conduction disorders.
MATERIAL AND METHODS
Standard protocol approvals, registrations, and patient consents
Ethical approval was granted by the relevant local Ethical Committees of the participating
centers. All patients gave their written consent for participation in the study.
Patients and clinical evaluation
We studied four patients harboring rare variants in BVES, identified by whole exome
sequencing (WES) of a cohort of 1929 unsolved cases with limb-girdle muscular weakness
(LGMW) and/or an elevated CK level, established through an international collaboration
between different clinical and genetic centers: the MYO-SEQ project, the Myocapture
project and the Center for Medical Genetics of the Ghent University Hospital. Patient 1 and

64
2 (family A) are siblings of consanguineous parents (figure 1). Patient 3 (family B) and
patient 4 (family C) are isolated cases, with the maternal grandfather and paternal
grandmother of patient 4 being first cousins.
Medical history taking and physical examination were focused on neuromuscular and
cardiac symptoms and signs. Muscle strength was evaluated by manual muscle testing (MRC
scale). An EMG was performed for all patients. Cardiac function was assessed by ECG, Holter
monitoring and echocardiography in all patients. In addition, arm ergometer stress testing
was performed in patient 3 and bicycle ergometer stress testing and a cardiac
electrophysiology study (EPS) in patient 2.
Muscle MRI
Muscle MRI studies were performed for patient 1, 2 and 3 on 1.5T MRI platforms at the
respective centers. Cross-sections at pelvic, thigh and calf levels were assessed on axial T1-
weighted images to evaluate patterns of muscle involvement. Fatty replacement of muscle
was graded according to the Mercuri scale.9
Analysis of WES data
For family A, WES was performed by the CNRGH on DNA samples from patient 1 and 2 and
their mother. Variants were annotated and filtered using an in-house-developed software
system (PolyWeb).10 WES data of patient 3 were processed by the Genomics Platform at the
Broad Institute MIT and Harvard (Boston, MA, USA) and analyzed by the team at the John
Walton Muscular Dystrophy Research Centre, Newcastle University as described
previously.11 For patient 4, WES was performed using the SureSelect XT Human All Exon V6
enrichment kit (Agilent Technologies) followed by paired-end sequencing (2x150bp) on the
HiSeq3000 sequencer (Illumina). Reads were mapped and variants were called and
annotated with the BCBio pipeline.
BVES (reference sequence: NM_001199563) was analyzed for biologically relevant variants.
Population frequencies were estimated using Exome Aggregation Consortium (ExAC)12, last
accessed in August 2018. MutationTaster213 and the Combined Annotation Dependent
Depletion (CADD)14 tool (version v1.3) were used as in silico prediction algorithms to predict
the pathogenicity of the identified variants. Variants with CADD scores >20 represent the 1%
highest ranked variants genome-wide with regard to potential deleteriousness. In silico
splice site predictions were obtained using the Alamut Batch Software v.2.8 (interactive
Biosoftware). Variants were validated by Sanger sequencing and segregation studies were
performed with available DNA samples.

65
Muscle biopsies
Muscle biopsies of quadriceps or deltoid muscle were obtained for patients 2, 3 and 4 and
analyzed following standard histologic and immunohistochemical (IHC) procedures.
Standard IHC stainings, including those for dystrophin, α-, β- and γ-sarcoglycans, α- and β-
dystroglycan, caveolin 3 and telethonin, were evaluated. Additionally, IHC stainings were
performed for POPDC1 and POPDC2. Frozen 10-μm sections of skeletal muscle biopsies of
patients and two controls were mounted on Superfrost Plus glass slides (Thermo Fisher
Scientific) and used for IHC using the following primary antibodies as described:5 anti-
POPDC1 (HPA014788, Sigma-Aldrich) and anti-POPDC2 (HPA024255, Sigma-Aldrich),
combined with anti-α-sarcoglycan (SGCA) (NCL-a-sarc, Leica) for membrane counterstaining.
The following secondary antibodies were employed for detection of the signal: Alexa Fluor
488-conjugated goat anti-rabbit (A11034, Life Technologies) and CY3-labeled goat anti-
mouse (115-166-071, Jackson ImmunoResearch).
Microscopy and image analysis
Images were acquired with a Zeiss LSM700 laser scanning confocal microscope using a
20x/0.8 Plan Apochromat objective. To avoid cross-talk between fluorescence channels,
line-by-line sequential scanning was employed. All images (16-bit, 512 x 512 pixels, 417 nm
x 417 nm per pixel) were taken with identical excitation and detection settings to allow
comparison of fluorescence intensities. For each sample, five images were acquired at
random positions. Intensities at the sarcolemma were quantified using the Fiji distribution
of ImageJ.15, 16 Raw image files (LSM5) were loaded in Fiji and background intensity levels
were corrected by subtracting the mean intensity of the Gaussian blurred image (sigma = 20
pixels) from the original image. On each image, six random membrane segments were
delineated (average line length per segment > 50µm) using the segmented-line-tool on the
sarcolemma channel and were stored as ImageJ ROI-files. Intensities in both channels
(POPDC1/POPDC2 and SGCA) were quantified as mean intensities of these lines, set at a line
width of 5 pixels (2µm). The analysis procedure was employed as an ImageJ macro,
measuring all images in batch.
mRNA studies
Total RNA was extracted from muscle biopsies (controls and patients) using standard
methods (TRIzol). First-strand cDNA was synthesized using the SuperScript III First-Strand
Synthesis System (Invitrogen-ThermoFisher Scientific) with Oligo-dTs. The obtained cDNA
was used for Sanger sequencing as well as quantitative PCR (qPCR) experiments with SYBR
green I dye incorporation (Light Cycler 480 system-Roche). The average Ct value obtained

66
with multiple BVES primers (figure 2A) was normalized against the housekeeping gene
RPLP0, then fold-changes in mRNA levels were calculated relative to the control sample.
Experiments were performed in triplicate. All primer sequences are available upon request.
Data analysis was performed with qBase + (Version 3.1; Biogazelle, Zwijnaarde, Belgium).
Data are presented using GraphPad Prism (GraphPad Software, La Jolla, CA) as mean values
with standard error of the mean (SEM). Data were analyzed using the unpaired t test. A p
value <0.05 was considered statistically significant.
Data availability statement
Anonymized data will be shared by request from any qualified investigator, only for
purposes of replicating procedures and results.
RESULTS
Clinical findings and case descriptions
A summarized overview of the highly variable clinical symptoms is provided in table 1.
Patients 1 and 2 are affected siblings, among five, of consanguineous parents (first cousins).
Patient 1, a 41-year-old female, manifested complaints of exercise-induced myalgia and
fatigue of the lower limb muscles as well as breathlessness from the age of 27 years. No
further skeletal muscle signs or symptoms were noted and clinical neurological examination
was normal. CK levels, repeatedly measured at that time, were elevated in the range of
1300-3661 IU/L. EMG was normal and cardiac assessment revealed a first-degree AV block
in the absence of structural cardiac disease on echocardiography. Muscle symptoms and
cardiac function remained stable during follow-up.
Patient 2, a currently 37-year-old male, was first investigated at 19 years of age, presenting
with presyncopal episodes and palpitations. During cardiac workup, a first-degree AV block
and a transient second-degree Mobitz type 1 AV block were noted on ECG and Holter
monitoring, with an echocardiography showing no structural abnormalities. There were no
symptoms suggestive of neuromuscular disease and clinical examination at the age of 20
years was normal. However, high CK levels (1074-3600 IU/L) were measured repeatedly,
EMG disclosed myopathic abnormalities and muscle biopsy showed mild dystrophic changes
with normal routine immunohistochemical and western blot studies. Intercurrently, the
patient manifested a symptomatic S1 radiculopathy on the right side at the age of 22 years,
with residual weakness and atrophy of the right gastrocnemius and soleus muscles. An EPS
at the age of 27 years confirmed an AV nodal block, with a normal HV interval and without
SND or ventricular hyperexcitability. During follow-up, this patient manifested multiple
presyncopal episodes and the cardiac conduction abnormalities progressed towards a

67
second-degree Mobitz type 2 AV block combined with an incomplete right bundle branch
block. A new EPS at the age of 29 years revealed a combined AV nodal and infrahissian block
(HV interval ranging 55 to 80 ms), again without SND or hyperexcitability. Pacemaker
implantation was advised but refused by the patient. Bicycle ergometer testing performed
at the age of 35 years showed a second-degree Mobitz type 2 AV block during recovery.
Apart from right S1 radiculopathy sequelae, no additional muscle weakness or wasting was
noted during follow-up but CK levels stayed consistently elevated (maximal 5500 IU/L).
Patient 3, a 65-year-old female, developed slowly progressive proximal weakness in the
lower limbs in her mid-thirties. Being very athletic up to that point, she noticed difficulties in
walking uphill and climbing stairs. A few years later she manifested slowly progressive
proximal weakness in the upper limbs too. When she was referred for neuromuscular work-
up for the first time at the age of 43 years, she climbed stairs on her hands and feet. Clinical
neurological examination at that time confirmed proximal weakness in the lower limbs, as
well as weakness of the anterior tibial muscles. Scapular winging was found. CK level was
1161 IU/L, EMG revealed myopathic changes. Over the following years, the muscle
weakness was slowly progressive, leading to loss of ambulation. Thorough cardiac work-up
revealed a first-degree AV block at night as well as a borderline first-degree AV block during
exercise. Furthermore, no rise in heart rate (85/min during the whole test) was observed
during arm ergometer testing, halted due to exhaustion at 50W. Both parents were
deceased, no neuromuscular or cardiac problems were reported. Of five siblings, one was
deceased and none reported muscle or cardiac symptoms. One sister agreed to be formally
examined, clinical examination was unremarkable.
Patient 4, a currently 44-year-old male, reported mild myalgia of the calves at age 39 years,
6 months after starting fibrate therapy for hypertriglyceridemia. A CK value of 8000 IU/L was
measured and he was referred to a cardiologist. The fibrate therapy was interrupted,
muscle complaints diminished and control CK-values averaged around 3500-4000 U/l. Initial
cardiac work-up, including ECG, echocardiography and Holter monitoring, yielded normal
results. The cardiologist referred the patient for neuromuscular work-up. By that time,
muscle complaints had resolved. A myopathic recruitment pattern was noted on EMG
though, as well as non-specific myopathic features on muscle biopsy. Clinical examination of
two brothers, a 7-year-old son and a 5-year-old daughter was unremarkable. For the
parents and the two brothers, a normal CK value was measured. Clinically, the patient
remained stable during five years of follow-up. Cardiac work-up was however repeated and
revealed a first-degree AV block and a nocturnal second-degree Mobitz type 2 AV block.
None of the patients presented with contractures, rigid spine, clinical myotonia or myotonic
discharges on EMG.

68
Muscle imaging
Muscle MRI studies of patient 1, 2 and 3 are shown in figure 3. For patient 1, muscle MRI
performed at the age of 28 years and repeated at the age of 39 years, did not reveal any
selective muscle involvement. For patient 2, initial CT imaging of muscle at the age of 20
years was normal. At the age of 30 years, muscle MRI showed moderate fatty replacement
and atrophy of the right gastrocnemius, soleus and biceps femoris muscles, visualizing the
clinically evident residual atrophy and weakness due to the S1 radiculopathy. Additional
muscle MRI studies at the age of 35 years confirmed these findings.
Muscle MRI of patient 3, showing the most severe skeletal muscle phenotype, revealed a
pattern of selective muscle involvement with preferential affection of the posterior thigh
compartment. In addition, an asymmetric moderate involvement of the left lateral vastus
and relative sparing of distal leg muscles were observed.
Genetic findings
We identified three different homozygous variants in BVES in three families (table 2).
Variants were absent from ExAC control database and in silico prediction algorithms were in
favor of pathogenicity. Segregation analyses were performed with available DNA samples
(figure 1). Only affected individuals harbored the variant in homozygous state. The
c.816+2T>C variant in intron 6 of BVES affects the highly conserved canonical T of the splice
donor site and is predicted to cause skipping of exon 6 and the loss of 56 amino acids
(p.Val217_Lys272del) within the Popeye domain of BVES. Two potential cryptic splice sites
could be activated alternatively: in intron 6 (c.816+46, p.Lys272fs4*) or within exon 6
(c.749*, p.Arg250Argfs20*). The c.262C>T variant (rs796206315) introduces a premature
stop codon at amino acid position 88, located in the second transmembrane domain of this
short protein. According to the MutationTaster2 algorithm,13 the c.1A>G variant is predicted
to result in a loss of the initiating methionine (cDNA position 218) and potential activation of
a downstream translation initiation site at cDNA position 354, resulting in a new reading
frame with insertion of a premature stop codon at amino acid position 2. As this potential
alternative initiation site is not embedded in a strong Kozak sequence, other initiation sites
might be activated at cDNA position 423 or 431. In both cases this would lead to an in-frame
deletion, of 66 or 72 amino acids respectively. Activation of different alternative start
codons located more downstream could however also result in a shift of the reading frame
with insertion of a premature stop codon.

69
Molecular consequences at mRNA level
To provide direct evidence for the predicted alterations at the transcript level, Sanger
sequencing as well as qPCR experiments were performed with mRNA extracted from muscle
biopsies of patients 2, 3 and 4. PCR products of BVES cDNA amplicons A, B and C (figure 2A)
could all be sequenced (data not shown) and all variants were confirmed at the cDNA level.
Gel electrophoresis of the amplicon B encompassing exon 4 up to and including 8 revealed a
shorter product for patient 2, sized approximately 350 base pairs (bp) instead of the
expected size of 507 bp (figure 2B). The sequencing of this fragment confirmed that exon 6
(168 bp long) was indeed spliced out. Nevertheless, fragment C could be amplified for
patient 2, although the forward primer is located in exon 6. qPCR data however showed that
these mRNA levels for amplicon C are close to 0 when compared to the controls, indicating
that the absolute level of mutant mRNA in which exon 6 is not skipped is extremely low.
Furthermore, mRNA levels for amplicon A are markedly decreased as well, indicating
nonsense-mediated mRNA decay (NMD) of the mutant transcript in any case. For patient 3,
qPCR for amplicons A and C revealed a marked decrease in BVES mRNA relative to the
controls (figure 2C). For patient 4, BVES mRNA levels are similar to these in the controls.
Reduction in membrane localization of POPDC1 and POPDC2
Non-specific myopathic features such as increased fiber size variation were noted on muscle
biopsies of patient 2, 3 and 4. Additionally, for patient 2 a few necrotic fibers were
observed. Standard IHC stainings were normal.
For the previously described patients harboring the p.Ser201Phe in BVES in homozygosity,
defective POPDC1 membrane trafficking was reported to result in strongly reduced
membrane localization of POPDC1 and POPDC2.5 To examine POPDC1 and POPDC2 at the
plasma membrane, we performed IHC stainings in patient and control muscle samples. Both
POPDC1 and POPDC2 were abundantly present at the plasma membrane of control skeletal
muscle. In all patient samples however, both POPDC1 and POPDC2 were drastically
diminished at the sarcolemma (figure 4). Levels of α-sarcoglycan, used as a control marker
for sarcolemmal proteins, remained normal in the patient samples with similar staining
patterns and intensities as for control samples. These results confirm that the predicted LOF
mutations in BVES indeed lead to a loss of membrane localization and consequently the
main function of POPDC1 and POPDC2 in patient muscle.

70
DISCUSSION
In the present study, we identified three different new homozygous LOF mutations in BVES
in four individuals from three families, showing early adult onset skeletal muscle and cardiac
conduction abnormalities of varying nature and severity.
The presenting symptoms vary between individuals, with patient 2 presenting with cardiac
symptoms due to an AV conduction defect within the second decade, and patients 1, 3 and
4 with skeletal muscle symptoms or signs. Only patient 3 showed progressive proximal
muscle weakness, while patient 1 had symptoms of exercise intolerance and patient 4 had
permanently high CK levels (>3500 IU/L after interruption of fibrate therapy) with transient
complaints of myalgia. During follow-up, all patients appeared to have at least subclinical
signs of both skeletal muscle and cardiac involvement. In patient 2 with a predominant
cardiac phenotype, a chronically elevated CK (>1000 U/l) was noted repeatedly, as well as
myopathic features on EMG and muscle biopsy. In patient 1, 3 and 4, initially showing a
predominant skeletal muscle phenotype of variable severity, a progressive cardiac
conduction disorder became apparent during follow-up.
Intrafamilial variability of the phenotype linked to mutations in BVES has already been
shown in the originally reported consanguineous family in which a pseudo-dominant
inheritance pattern of the p.Ser201Phe variant was described:5 the grandfather presented
with a LGMD phenotype at age 40 years, complicated by a symptomatic second-degree AV
block at age 59 years requiring pacemaker implantation, the grandsons with symptomatic
second-degree AV block respectively at age 17 years and 12 years. Cardiac conduction
disorders seemed to be confined to the AV node.5 However, for patient 2 definite His-
bundle involvement was shown on an EPS at the age of 29 years. Although not fulfilling the
exact criteria of chronotropic incompetence,17 the results of the arm ergometer testing in
patient 3 are strongly indicative of SND. Due to lower limb weakness, only less standardized
arm ergometer testing could be performed for this wheelchair bound patient. During an
incremental dynamic exercise test, workload was limited to 50 W, mainly due to weakness
of the biceps muscles.
Similarly, heterogeneity was observed on muscle imaging studies. For patient 3, a pattern of
selective muscle involvement was shown, with muscles of the posterior compartment of the
thighs being most severely involved. Muscle MRI studies of patient 1 yielded normal results.
The selective unilateral atrophy and fatty replacement of soleus, gastrocnemius and biceps
femoris muscles in patient 2 may be due to S1 radiculopathy.
As we identified additional unrelated families with a BVES-related myopathy, we propose
that this disorder should be classified as ‘LGMD R25 BVES-related’ according to the recently
published novel ENMC classification of LGMDs. ‘LGMD2X’ was excluded from the LGMD

71
nomenclature, based on the criterion that the condition must be described in at least two
unrelated families with affected individuals.18 All other criteria of the novel definition of an
‘LGMD’ are fulfilled too. Each patient achieved independent walking and has an elevated CK
activity. Degenerative changes on muscle imaging are clearly shown on muscle MRI of
patient 3 of the current study and dystrophic changes on histology, noted for the same
patient, had already been described for the eldest patient of the originally reported family
too.5, 18
IHC experiments revealed a pattern of consistent reduction of POPDC1 and POPDC2 at the
sarcolemma, similar to the pattern described in muscle of patients harboring the
homozygous p.Ser201Phe missense mutation and the popdc1p.Ser191Phe KI zebrafish.5 Of note,
for patient 3, harboring the homozygous p.Arg88Ter variant, no antibody targeting an
epitope at the N-terminal side of the premature stop codon was available. Additionally,
mRNA studies are supportive of a LOF mechanism at the mRNA level for the BVES variants in
patient 2 and 3, most likely due to NMD. For patient 2, qualitative and quantitative PCR data
illustrate that different splice variants are transcribed, though ultimately leading to
significantly decreased BVES mRNA levels. The low level of remaining mRNA probably
mainly consists of mRNA with in-frame skipping of exon 6, encoding for 56 amino acids
(p.Val217_Lys272del) which are part of the Popeye domain, the crucial functional domain of
the protein. For patient 4, BVES mRNA levels were similar to these in the controls.
Apparently, the mutant mRNA is not targeted by NMD, yet a non-functional protein is
translated, which is not recognized by the anti-POPDC1 antibody, raised against the C-
terminal part of the POPDC1 protein. In case of an in-frame deletion of 66 or 72 amino acids
at the N-terminal end of the protein, the antibody should still recognize this truncated
POPDC1 protein. This observation most likely implies that there is a shift of the reading
frame, with a premature stop codon less than 55 nucleotides upstream of the last exon-
exon border. In that case NMD will be skipped and a protein with a different amino acid
sequence is formed, not recognized by the anti-POPDC1 antibody. All mutations identified
lead to a partial or complete loss of function of BVES through NMD or through functional
changes to the POPDC1 protein.
By identifying and elaborating on LOF mutations in BVES, we expand the genetic spectrum
of the disorder and provide pathomechanistic insights in the disorder. Only the p.Ser201Phe
missense mutation had been previously identified in a single family. Identification of
patients harboring LOF mutations in BVES could however be anticipated based on functional
studies in homozygous zebrafish popdc1 morphants and Popdc1 null mutant mice, showing
a similar phenotype compared to the popdc1p.Ser191Phe KI zebrafish.5, 7, 8 Interestingly, the
Popdc1 null mutant mice revealed an age-dependent stress-induced SND with chronotropic

72
incompetence,8 a condition we also strongly suspect in patient 3. Further studies are
needed to unveil the exact functional consequences of the complete loss of function of the
POPDC1 protein in skeletal muscle and the heart. Crucially, POPDC2 appears to be a direct
interactor of POPDC1 and aberrant trafficking of POPDC1 also impairs membrane transport
of POPDC2. Heteromeric complexes might be formed, potentially explaining the secondary
loss of membrane localization of POPDC2, similarly shown for patients harboring the
p.Ser201Phe missense mutation.5 This appears to be pathomechanistically relevant, as
popdc2 zebrafish morphants show a severe muscular dystrophy phenotype and cardiac
abnormalities too.19
Our findings stress the importance of a thorough cardiac work-up in (unsolved) LGMD
patients. Cardiac work-up is often focused on structural cardiac evaluation,3 but as in
myotonic dystrophy,2 extensive and repeated screening for arrhythmias in the absence of
structural heart disease is definitely relevant in case of a BVES-related myopathy.
Furthermore this study highlights the diagnostic difficulties that can be faced in case of
pauci- or asymptomatic hyper-CKemia.20 We note chronically elevated CK values (>3 ULM) in
all patients. In the absence of marked dystrophic features on muscle biopsy, this may be an
indication of membrane instability linked to the interaction of POPDC1 with dystrophin,
dysferlin and caveolin-3.6, 21 Additionally, ultrastructural analysis of muscle of the
grandfather of the p.Ser201Phe family revealed membrane discontinuities.5 When further
neuromuscular work-up is advised in this clinical setting according to guidelines,20, 22 cardiac
screening could be relevant too, regardless of the results of the neuromuscular work-up.
The other way around, our findings suggest that BVES should be included in a candidate
gene list for progressive cardiac conduction disorders (PCCD) presenting as primary
electrical disease. The search for culprit genes has long been complicated by the fact that
the majority of cardiac conduction disorders are sporadic as they are highly prevalent and
usually associated with diverse (acquired) structural heart disease. Channelopathies are
evidently the predominant hereditary entities associated with PCCD.4, 23 Furthermore, a
short clinical neuromuscular evaluation, including CK measurement, could be of value when
a hereditary PCCD is suspected.
Identification of LOF mutations in BVES underlines by extension the role of the POPDC
protein family in striated muscle physiology and disease.6
We present four individuals from three families harboring homozygous LOF variants in BVES,
showing early adult onset skeletal muscle and cardiac conduction abnormalities of varying
nature and severity. Overall, this recessive disorder linked to mutations in BVES appears to

73
have a low prevalence, but is probably underdiagnosed due to its striking phenotypic
variability and often subtle yet clinically relevant manifestations.
ACKNOWLEDGEMENTS
The authors thank the patients and families for their cooperation and contributions; Sophie D’hose,
technician, Laboratory for Neuropathology, Division of Neurology, Ghent University Hospital, for
laboratory assistance; Ursula Herbort-Brand, technician, Imperial College London, for laboratory
assistance.

74
FIGURES AND TABLES
Figure 1. Segregation analysis of the three identified BVES mutations
Pedigrees of family A, B and C are shown. Unaffected family members for whom DNA was available
for segregation analysis of the BVES variants in the respective families, I-1, II-1, II-2, III-1, III-2, III-3
and III-4, were heterozygous for the respective variants.

75
Figure 2. BVES gene structure and mRNA analysis
(A) BVES mRNA transcript variant C (NM_001199563) contains eight exons, of which seven coding.
Transcript length is 5567 base pairs. Untranslated regions are filled in blue, translated in black. The
location of the three identified variants is marked with an arrow, position of the amplified fragments
for quantitative and quantitative PCR experiments with dashed lines. (B) Qualitative PCR: agarose gel
of PCR products (amplicon A, B and C) amplified from cDNA from control (C1) and patient 2 (P2). For
every amplicon, the two lanes on the right contain mRNA without reverse transcriptase and H2O
respectively. (C) BVES quantitative mRNA analysis for amplicon A and C, respectively. BVES mRNA
levels were first normalized internally to RPLP0 mRNA levels; subsequently, BVES mRNA levels of the
patients were normalized (as a percentage) to the mean of the two controls. For both amplicon A
and C there is a significant difference in mRNA levels between both controls and patient 2 and 3
respectively, which is not the case for patient 4. Error bars: standard error of the mean (SEM). *p <
0.05, **p < 0.01, using unpaired t test. Bp = base pairs; Ex = exon.
76
Figure 3. Muscle MRI findings in three patients harboring homozygous mutations in BVES
Axial T1-weighted images are shown for patient 1, 2 and 3, performed at the age of 39, 35 and 56
years respectively.
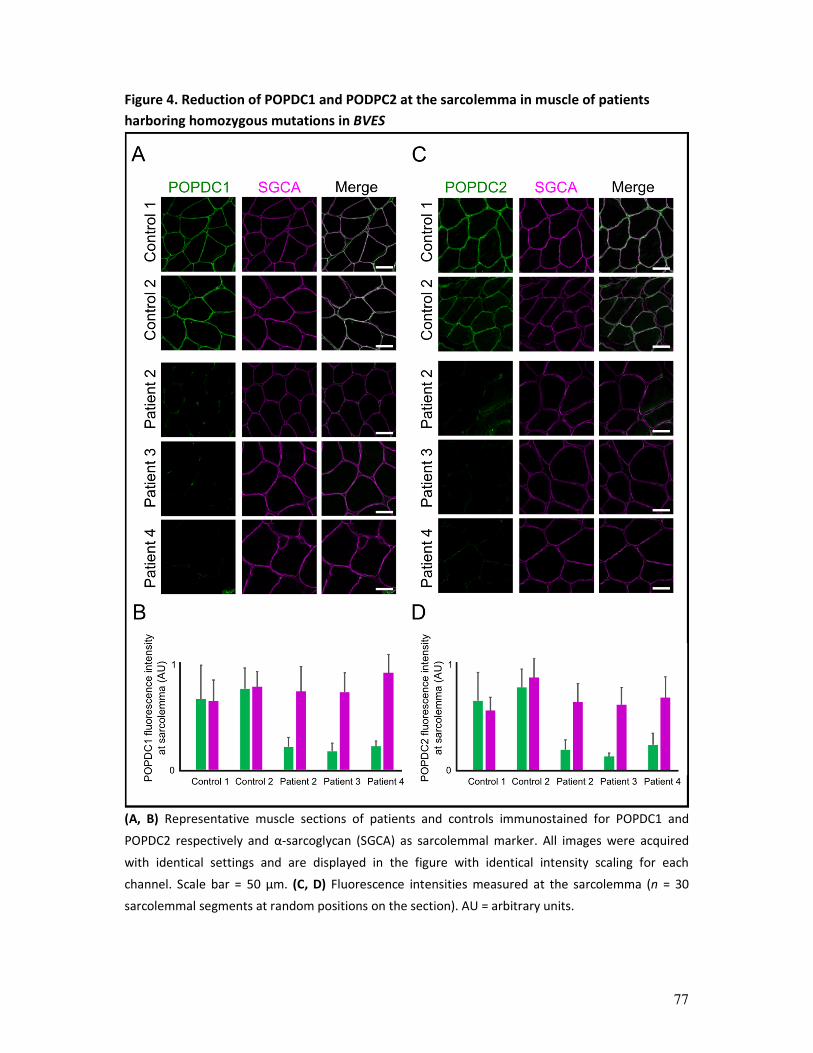
77
Figure 4. Reduction of POPDC1 and PODPC2 at the sarcolemma in muscle of patients
harboring homozygous mutations in BVES
(A, B) Representative muscle sections of patients and controls immunostained for POPDC1 and
POPDC2 respectively and α-sarcoglycan (SGCA) as sarcolemmal marker. All images were acquired
with identical settings and are displayed in the figure with identical intensity scaling for each
channel. Scale bar = 50 µm. (C, D) Fluorescence intensities measured at the sarcolemma (n = 30
sarcolemmal segments at random positions on the section). AU = arbitrary units.
Table 1. Phenotypic information for the patients harboring homozygous mutations in BVES
Patient 1 Patient 2 Patient 3 Patient 4
Sex Female Male Female Male
Ethnicity North African North African Caucasian (Belgium) Caucasian (Belgium)
BVES variant, cDNA position c.816+2T>C c.816+2T>C c.262C>T c.1A>G
Age at onset, y 27 19 35 39
Presenting symptoms
Exercise intolerance Palpitations, faintness,
elevated CK
Proximal weakness LL Myalgia, high CK
Age at last examination, y 41 37 65 44
We
ak
ne
ss
Proximal UL No No Yes No
Proximal LL No No Yes No
Distal UL No No Yes No
Distal LL No No Yes No
Other No Right calf muscles Periscapular No
Ambulation status Ambulatory Ambulatory Wheelchair bound Ambulatory
Serum CK (IU/L) 1300-3661 1074-5500 1918 3500-4000
EMG (age, y) Normal (28) Myopathic (30) Myopathic (45) Myopathic (43)
Resting ECG First-degree AV block First-degree AV block Normal Normal
Echocardiography Normal Normal Aortic valve stenosis Normal
Holter monitoring First-degree AV block Second-degree AV block
(Mobitz type 2), iRBBB
Nocturnal first-degree AV block Nocturnal second-degree AV
block (Mobitz type 2)
Bicycle/arm ergometer stress
testing
NA Bicycle ergometer: Mobitz type
2 second-degree AV block
during recovery
Arm ergometer: no rise in heart
rate during the test; borderline
first-degree AV block
NA
Biopsy (age, y) NA Myopathic (21) Myopathic (45) Myopathic (43)
Biopsied muscle NA Left deltoid Quadriceps Quadriceps
Abbreviations: y = years; CK = creatine kinase; UL = upper limbs; LL = lower limbs; AV = atrioventricular; iRBBB = incomplete right bundle branch block; NA =
not assessed
Table 2. BVES mutations identified in the present study
Patient
Variant location (hg19)
Genotype Variant
Predicted deleteriousness
ExAC allele
frequency Genomic (chr. 6) Coding
Protein
change MutationTaster2 CADD-score
1 105564574A>G c.816+2T>C p.? Hom. Splice donor No data No data 0
2 105564574A>G c.816+2T>C p.? Hom. Splice donor No data No data 0
3 105577343G>A c.262C>T p.Arg88Ter Hom. Stop gained Disease causing 39 0
4 105581452T>C c.1A>G p.? Hom. Start lost Disease causing 24.1 0
Abbreviations: Hom. = homozygous; CADD: Combined Annotation Dependent Depletion; Start lost = variant in start codon; ExAC = Exome Aggregation
Consortium.

80
REFERENCES
1. Vissing J. Limb girdle muscular dystrophies: classification, clinical spectrum and emerging therapies.
Current opinion in neurology 2016;29:635-641.
2. Silvestri NJ, Ismail H, Zimetbaum P, Raynor EM. Cardiac involvement in the muscular dystrophies.
Muscle & nerve 2018;57:707-715.
3. Narayanaswami P, Weiss M, Selcen D, et al. Evidence-based guideline summary: diagnosis and
treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of
the American Academy of Neurology and the practice issues review panel of the American Association
of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-1463.
4. Rezazadeh S, Duff HJ. Genetic Determinants of Hereditary Bradyarrhythmias: A Contemporary Review
of a Diverse Group of Disorders. The Canadian journal of cardiology 2017;33:758-767.
5. Schindler RF, Scotton C, Zhang J, et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by
affecting protein trafficking. The Journal of clinical investigation 2016;126:239-253.
6. Brand T, Schindler R. New kids on the block: The Popeye domain containing (POPDC) protein family
acting as a novel class of cAMP effector proteins in striated muscle. Cellular signalling 2017;40:156-
165.
7. Andree B, Fleige A, Arnold HH, Brand T. Mouse Pop1 is required for muscle regeneration in adult
skeletal muscle. Molecular and cellular biology 2002;22:1504-1512.
8. Froese A, Breher SS, Waldeyer C, et al. Popeye domain containing proteins are essential for stress-
mediated modulation of cardiac pacemaking in mice. The Journal of clinical investigation
2012;122:1119-1130.
9. Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited
neuromuscular disorders: past, present, and future. Journal of magnetic resonance imaging : JMRI
2007;25:433-440.
10. Gordon CT, Petit F, Kroisel PM, et al. Mutations in endothelin 1 cause recessive auriculocondylar
syndrome and dominant isolated question-mark ears. American journal of human genetics
2013;93:1118-1125.
11. Peric S, Glumac JN, Topf A, et al. A novel recessive TTN founder variant is a common cause of distal
myopathy in the Serbian population. European journal of human genetics : EJHG 2017;25:572-581.
12. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans.
Nature 2016;536:285-291.
13. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-
sequencing age. Nature methods 2014;11:361-362.
14. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating
the relative pathogenicity of human genetic variants. Nature genetics 2014;46:310-315.
15. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image
analysis. Nature methods 2012;9:676-682.
16. Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: ImageJ for the next generation of scientific image
data. BMC bioinformatics 2017;18:529.
17. Brubaker PH, Kitzman DW. Chronotropic incompetence: causes, consequences, and management.
Circulation 2011;123:1010-1020.
18. Straub V, Murphy A, Udd B. 229th ENMC international workshop: Limb girdle muscular dystrophies -
Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017.
Neuromuscular disorders : NMD 2018;28:702-710.
19. Kirchmaier BC, Poon KL, Schwerte T, et al. The Popeye domain containing 2 (popdc2) gene in zebrafish
is required for heart and skeletal muscle development. Developmental biology 2012;363:438-450.
20. Kyriakides T, Angelini C, Schaefer J, et al. EFNS guidelines on the diagnostic approach to pauci- or
asymptomatic hyperCKemia. European journal of neurology 2010;17:767-773.

81
21. Alcalay Y, Hochhauser E, Kliminski V, et al. Popeye domain containing 1 (Popdc1/Bves) is a caveolae-
associated protein involved in ischemia tolerance. PloS one 2013;8:e71100.
22. Silvestri NJ, Wolfe GI. Asymptomatic/pauci-symptomatic creatine kinase elevations (hyperckemia).
Muscle & nerve 2013;47:805-815.
23. Baruteau AE, Probst V, Abriel H. Inherited progressive cardiac conduction disorders. Current opinion
in cardiology 2015;30:33-39.

82

83
CHAPTER 3
High prevalence of sporadic late-onset nemaline
myopathy in a cohort of whole-exome sequencing
negative myopathy patients
Willem De Ridder, Peter De Jonghe, Magdalena Mroczek, Volker Straub, Martin Lammens,
Jonathan Baets
[Manuscript in preparation]

84
ABSTRACT
Objective: To systematically study (para)clinical and histopathological findings in a cohort of
18 isolated yet suspected inherited myopathy patients, showing late-onset, slowly
progressive limb-girdle muscular weakness (LGMW), that remained unsolved after whole-
exome sequencing (WES).
Methods: We studied 18 WES-unsolved patients, showing a (biopsy proven) myopathy with
late adult onset (> 40 years) LGMW. The presence of a monoclonal gammopathy of
unknown significance (MGUS) and anti-HMGCR antibodies was determined in blood.
Biopsies were systematically re-evaluated and systematic immunohistochemical and
electron microscopy studies were performed to particularly evaluate the presence of rods
and/or inflammatory features.
Results: Ten patients showed rods as core feature on muscle biopsy on re-evaluation, four
of these had an IgG κ MGUS in blood. As such, these ten patients represented suspected
slowly progressing sporadic late-onset myopathy cases (SLONM), with auxiliary data
supporting this diagnosis: 1) additional muscle biopsy features pointing towards Z-disk and
myofibrillar pathology; 2) a common selective pattern of muscle involvement on MRI; 3)
inflammatory features consisting of the presence of small inflammatory infiltrates and
sarcolemmal MHC-I upregulation.
Conclusions: The findings in this proof-of-concept study highlight the difficulties in reliably
diagnosing slowly progressing SLONM and the probably underestimated prevalence of this
sporadic entity in cohorts of WES negative myopathy patients, initially considered to have
an inherited myopathy.

85
INTRODUCTION
Muscle disorders, most typically presenting with progressive proximal muscle weakness,
comprise a heterogeneous group of acquired and inherited diseases affecting skeletal
muscle.1 With the exception of sporadic inclusion-body myositis (sIBM), only few acquired
muscle disorders present with slowly progressive muscle weakness and as such, an inherited
muscle disorder (IMD) is typically suspected in case of this clinical presentation. A few other
atypically presenting acquired muscle disorders might however also constitute relevant
differential diagnoses. Recent literature suggested that myopathies with anti-HMGCR
antibodies may present with slowly progressive muscle weakness.2 The same has been
shown for an enigmatic, supposedly very rare, putatively immune-mediated late-onset
myopathy, called sporadic late-onset nemaline myopathy (SLONM).3 Contrary to the highly
recognizable SLONM cases presenting with sub-acutely progressing severe muscle
weakness, the cases on the less fulminant side of the apparent spectrum are difficult to
detect in light of the rather non-specific key features, nemaline rods on muscle biopsy, with
or without presence of a monoclonal gammopathy of unknown significance (MGUS) in
blood.3
In this study, we present a case series of (para)clinically and histopathologically
systematically characterized sporadic yet suspected IMD patients showing late-onset, slowly
progressive limb-girdle muscular weakness (LGMW) that remained unsolved after whole-
exome sequencing (WES). Our findings in this proof-of-concept study highlight the
difficulties in reliably diagnosing slowly progressing SLONM and the probably
underestimated prevalence of this sporadic entity in WES-unsolved myopathy patient
cohorts.
SUBJECTS AND METHODS
Patient selection and (para)clinical evaluation
We studied 18 suspected IMD patients from the Antwerp University Hospital (UZA),
included in the MYO-SEQ project, showing a (biopsy proven) myopathy with late adult onset
(> 40 years) LGMW with or without a high creatine kinase (CK) level, that remained
genetically unsolved after: 1) directed molecular genetic testing prior to inclusion in MYO-
SEQ to exclude a dystrophinopathy, facioscapulohumeral dystrophy (FSHD),
oculopharyngeal muscular dystrophy (OPMD) or myotonic dystrophy type 1 or 2 in case of
clinical suspicion; 2) WES data analysis as described previously.4 An overview of the
complete cohort is provided in supplementary table 1: 37 out of all 65 UZA cases (56.9%)
included in MYO-SEQ were genetically solved after WES. Of 43 patients without family
history however, 26 (60.5%) remained without a genetic diagnosis. 18 of these showed late

86
adult onset LGMW. For these patients, we decided to systematically re-evaluate or
complete all (para)clinical, radiological, histopathological and lab features that could lead to
the alternative diagnosis of a previously unrecognized acquired myopathy. Muscle strength
was evaluated by manual muscle testing (MRC scale). The presence of an MGUS was
evaluated based on serum protein electrophoresis and immunofixation and a free light
chain (FLC) assay. Serum samples were screened for anti-HMGCR autoantibodies.
Muscle biopsies
Muscle biopsies of quadriceps, anterior tibial or deltoid muscles were obtained for all
patients and analysed following standard histological and immunohistochemical (IHC) light
microscopy and electron microscopy (EM) protocols. Biopsies were systematically re-
evaluated and systematic IHC studies were performed for: 1) myotilin, alpha-actinin and
desmin to evaluate the presence of rods and/or desmin aggregates; 2) MHC-1, MHC-II, CD8,
CD68 and C5b9 (MAC) to evaluate inflammatory muscle biopsy features.
Muscle MRI studies
Muscle MRI was performed on a 1.5T MRI platform at the UZA. Cross-sections at shoulder,
abdominal, pelvic, thigh and calf levels were assessed on T1-weighted images to evaluate
patterns of muscle involvement. Fatty replacement of muscle was graded according to the
Mercuri scale.5 For multiple patients, longitudinal follow-up MRI studies were performed
spanning multiple years of disease progression.
RESULTS
After systematic documentation of all relevant (para)clinical, radiological, histopathological
and lab features, the cohort of 18 patients showing late adult onset LGMW was divided in
two subgroups based on the presence (table 1) or absence (supplementary table 2) of rod-
like aggregates on muscle biopsy (on Gomori, IHC for Myotilin or α-actinin and/or on EM), as
this is the core feature of SLONM.
Ten patients showing rods on muscle biopsy (patient 1-10), all presented a rather non-
specific pattern of muscle weakness, with predominantly proximal muscle weakness, more
pronounced in lower than in upper limbs, though with marked weakness of gluteus
maximus in six patients, marked paraspinal weakness in five and periscapular weakness in
three. All patients showed slowly progressive weakness, though for patient 1-7 and patient
10 leading to important walking difficulties and the need of ambulatory aids (table 1). CK
levels ranged from normal to moderately increased levels. Four of these patients show an
IgG κ MGUS, with polyclonal increased κ and λ chains in two and an increased κ/λ ratio 2.41

87
in one patient. One patient (patient 3) showed a normal protein electrophoresis, yet had
markedly increased κ and λ chains on the FLC assay. Rods represented the key muscle
biopsy feature, yet the following additional features were also frequently documented: 1)
cytoplasmic bodies for five patients; 2) rimmed vacuoles for four patients; 3) the presence
of numerous lobulated fibres for six patients; 4) hyaline inclusions on haematoxylin and
eosin (H&E) stainings resembling spheroid bodies for three patients (representative images
are shown in figure 1A-F; additional illustrative muscle biopsy images are provided in
supplementary figure 1). Patient 7 showed atypical inclusions on Gomori staining, which
were partly immunoreactive to α-actinin, myotilin, desmin, nebulin and SERCA-2-ATPase
(supplementary figure 1).
For two out of eight patients (patient 11 and 18) for whom no rods were detected on
muscle biopsy, a specific lab or muscle biopsy feature oriented towards an alternative
diagnosis (details see supplementary table 2): 1) patient 11 had positive anti-HMGCR
antibodies, fitting the diagnosis of a HMGCR-related immune-mediated necrotizing
myopathy with selective muscle fibre necrosis; 2) patient 12 showed striking tubular
aggregates on muscle biopsy, of currently unknown aetiology. None of the 18 patients
manifested significant signs or symptoms of bulbar, cardiac or respiratory involvement.
A set of IHC stainings was performed to evaluate the presence of inflammatory features and
inflammatory cells on muscle biopsies of all patients, except for patient 2, 17 and 18 for
whom not enough spare muscle material was available. For five out of nine patients, diffuse
or patchy sarcolemmal MHC-I upregulation was noted and for four patients inflammatory
infiltrates, mainly consisting of CD68-positive macrophages. These inflammatory muscle
biopsy features were not detected for any of the patients not showing rods on muscle
biopsy.
On MRI, the suspected SLONM patients showed a very similar pattern of selective muscle
involvement, with early involvement of vastus intermedius, adductor magnus, biceps
femoris (caput longus) and soleus muscles (representative images see figure 1G-L; an
extensive overview of muscle MRI studies is provided in supplementary figure 2). Over years
of disease progression, MRI studies mainly showed further selective involvement of
posterior thigh muscles and lateral gastrocnemius muscles as well as progressive gluteal and
paraspinal muscle involvement. Patient 8 however showed a slightly different MRI pattern
with predominant anterior lower leg involvement, patient 9 showed only mild, yet also
selective involvement of adductor magnus, biceps femoris (caput longus) and soleus
muscles, for patient 7 this similar pattern of involvement was markedly asymmetric and for
patient 6 more patchy.

88
An overview of MRI images of patients showing no rods on muscle biopsy (patient 11-18) is
provided in supplementary figure 3.
DISCUSSION
We report on a cohort of 18 WES unsolved, (para)clinically and histopathologically
systematically characterized suspected IMD patients, though without any family history of
muscle disease, presenting with late-onset, slowly progressive LGMW. Our findings in this
cohort strongly suggested enrichment of isolated cases of LGMW with rods as core muscle
biopsy feature (10 out of 18 patients), with four of these patients having an IgG κ MGUS and
one markedly increased (polyclonal) κ and λ chains, which is also indicative of an
inflammatory disorder.6 As such, ten patients represented likely (slowly progressing) SLONM
cases, as based upon an exhaustive interpretation of the available diagnostic criteria. We
capitalized on findings in this homogeneous cohort in search for additional features
supporting this putative diagnosis.
In literature, SLONM is considered to constitute a clinical spectrum encompassing subacute
presenting SLONM patients, representing a highly recognizable clinical and histopathological
entity, as well as patients presenting with slowly progressive LGMW.3 For the latter,
diagnosis also still relies on the two core features that are not necessarily completely
specific: 1) nemaline rods are evidently typically observed in inherited nemaline rod
myopathies, yet can also be observed in other (inherited) myopathies such as in advanced
stage of muscular dystrophies;7 8 2) an MGUS is present in serum of approximately 3-5% of
healthy subjects over 70 years.9 This means that slowly progressing SLONM is to an
important extent a diagnosis of exclusion. Rigorous WES data analysis did not yield any
candidate variant in a known myopathy gene, which implies that this cohort underwent
systematic and rigorous testing of all known myopathy genes, contrasting with most
reported cases that have not been investigated this thoroughly from a genetic point of view.
3
Unlike in HMGCR-myopathy, where the diagnosis can be made based on a single lab test (as
in patient 11 of the current study), our current understanding of the enigmatic SLONM
entity does not allow us to easily diagnose earlier unidentified cases retrospectively. Clearly,
there is a pressing need for additional criteria supporting a probable diagnosis of slowly
progressing SLONM, which should be determined in patient cohorts, which are as
homogeneous as possible. Clinically, the pattern of muscle weakness appears to be rather
non-specific at first sight.3 All suspected SLONM patients in this cohort show LGMW, though
for most a marked gluteal and paraspinal muscle weakness is observed. This pattern is
corroborated by muscle MRI studies showing a selective pattern, which strikingly resembles

89
a pattern described in a cohort of subacute SLONM cases.10 Myofibrillar disintegration and
rimmed vacuoles constitute frequently reported additional histopathological findings,
features typically also observed in myofibrillar myopathies, for which pathology is primarily
located at the Z-disk; cores and lobulated fibres suggest myofibrillar disorganisation too.3 11
On muscle biopsies of the suspected SLONM patients of this cohort, numerous cytoplasmic
bodies and hyaline inclusions containing abnormal myofibrillar material resembling spheroid
bodies or atypical caps (patient 7) are also frequently observed, which might further suggest
a histopathological spectrum of (slowly progressing) SLONM, relevant with regard to the
(putatively immune-mediated) pathomechanisms appearing to primarily target sarcomeres,
the contractile apparatus of muscle.3
This enigmatic entity is thought to represent an atypical immune-mediated myopathy. This
is mainly implied by the marked enrichment of the prevalence of a MGUS in SLONM cohorts
(+-53%), the fragmentary documentation of inflammatory features on muscle biopsy and
the favourable outcome after stem cell therapy in a few cases.3 Most of the suspected
SLONM cases show some inflammatory features on muscle biopsy, evident by sarcolemmal
MHC-I upregulation or the presence of small endomysial inflammatory infiltrates, which is
suggestive, though not a proof of immune-mediated pathomechanisms.
Based on the current study, we strongly advocate an active and prospective search for
slowly progressing SLONM cases in large genetically well-studied patient cohorts, such as
WES-unsolved suspected IMD patient cohorts, in search for better biomarkers,
pathomechanistic insights and therapeutic strategies. The studies in the current literature
are likely biased towards patients showing the most conspicuous rods, probably resulting in
an underestimation of the histopathologic spectrum of this allegedly treatable disease.3 12
Based on this proof-of-concept study, we propose that a probable SLONM diagnosis, based
on the identification of rods on muscle biopsy of a sporadic late onset myopathy patient,
with or without identification of an MGUS, could be substantiated by supportive criteria
based on: 1) negative results of WES analysis; 2) muscle MRI imaging (suggestive pattern); 3)
findings of an MGUS on protein electrophoresis or increased κ and λ chains on a FLC assay;
4) serological exclusion of HMGCR-myopathy; 5) additional biopsy features such as
cytoplasmic bodies and hyaline inclusions; 6) presence of endomysial inflammatory
infiltrates and/or sarcolemmal MHC-I upregulation.

90
ACKNOWLEDGEMENTS
The authors thank the patients and families for their cooperation and contributions;
Natacha Camacho and Safoura Jafary, Laboratory of Neuromuscular Pathology, Institute
Born-Bunge, University of Antwerp, for laboratory assistance.
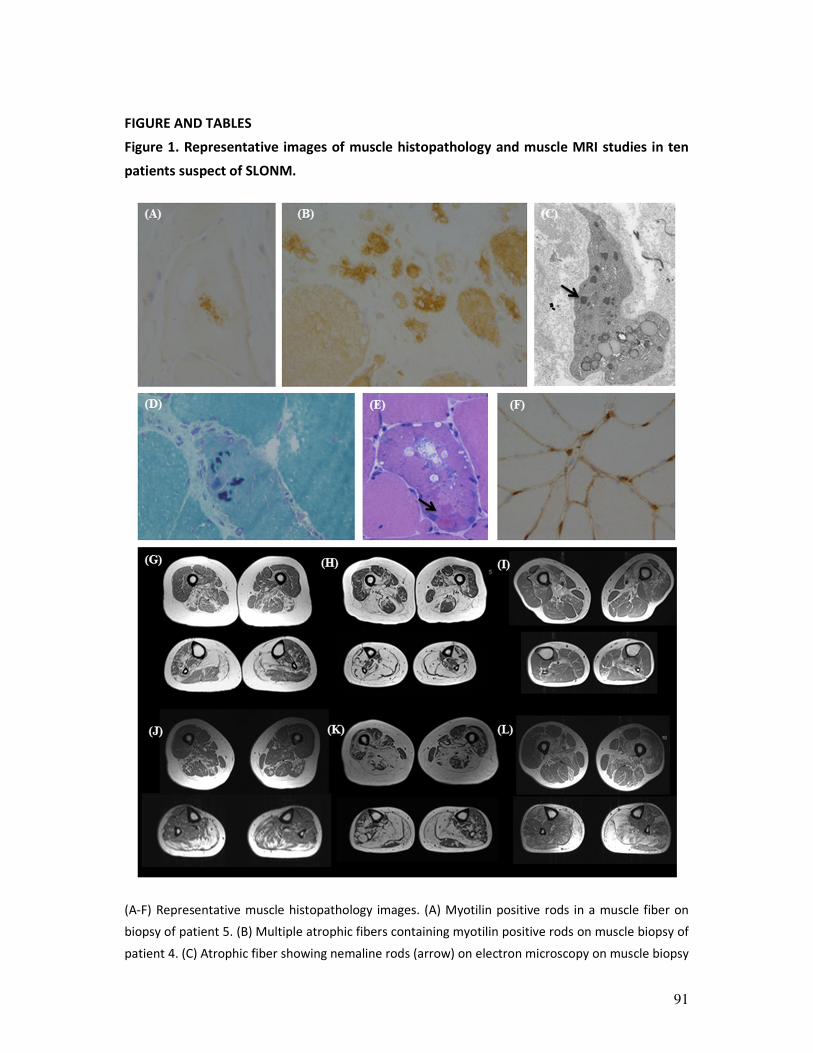
91
FIGURE AND TABLES
Figure 1. Representative images of muscle histopathology and muscle MRI studies in ten
patients suspect of SLONM.
(A-F) Representative muscle histopathology images. (A) Myotilin positive rods in a muscle fiber on
biopsy of patient 5. (B) Multiple atrophic fibers containing myotilin positive rods on muscle biopsy of
patient 4. (C) Atrophic fiber showing nemaline rods (arrow) on electron microscopy on muscle biopsy

92
of patient 6. (D) Cytoplasmic bodies on gomori trichrome staining for patient 3. (E) Spheroid bodies
(arrow) on H&E staining and (F) sarcolemmal upregulation of MHC-I on muscle biopsy of patient 1.
(G-L) Selection of muscle MR images at thigh and calf level, shown for: (G-H) patient 1, at age 64
years (G) and 75 years (H); (I) patient 3 at age 65 years; (J-K) patient 2, at age 63 years and 76 years;
(L) patient 5, at 76 years. SLONM, sporadic late-onset nemaline myopathy
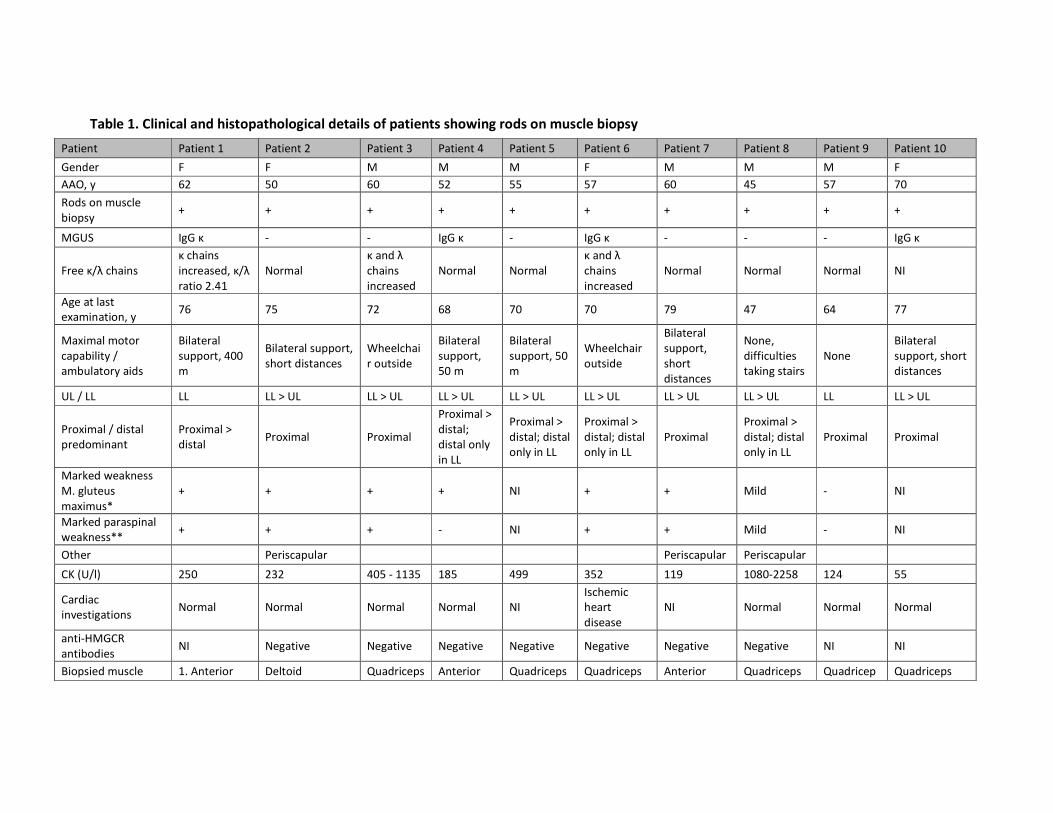
Table 1. Clinical and histopathological details of patients showing rods on muscle biopsy
Patient Patient 1 Patient 2 Patient 3 Patient 4 Patient 5 Patient 6 Patient 7 Patient 8 Patient 9 Patient 10
Gender F F M M M F M M M F
AAO, y 62 50 60 52 55 57 60 45 57 70
Rods on muscle
biopsy + + + + + + + + + +
MGUS IgG κ - - IgG κ - IgG κ - - - IgG κ
Free κ/λ chains
κ chains
increased, κ/λ
ratio 2.41
Normal
κ and λ
chains
increased
Normal Normal
κ and λ
chains
increased
Normal Normal Normal NI
Age at last
examination, y 76 75 72 68 70 70 79 47 64 77
Maximal motor
capability /
ambulatory aids
Bilateral
support, 400
m
Bilateral support,
short distances
Wheelchai
r outside
Bilateral
support,
50 m
Bilateral
support, 50
m
Wheelchair
outside
Bilateral
support,
short
distances
None,
difficulties
taking stairs
None
Bilateral
support, short
distances
UL / LL LL LL > UL LL > UL LL > UL LL > UL LL > UL LL > UL LL > UL LL LL > UL
Proximal / distal
predominant
Proximal >
distal Proximal Proximal
Proximal >
distal;
distal only
in LL
Proximal >
distal; distal
only in LL
Proximal >
distal; distal
only in LL
Proximal
Proximal >
distal; distal
only in LL
Proximal Proximal
Marked weakness
M. gluteus
maximus*
+ + + + NI + + Mild - NI
Marked paraspinal
weakness** + + + - NI + + Mild - NI
Other Periscapular Periscapular Periscapular
CK (U/l) 250 232 405 - 1135 185 499 352 119 1080-2258 124 55
Cardiac
investigations Normal Normal Normal Normal NI
Ischemic
heart
disease
NI Normal Normal Normal
anti-HMGCR
antibodies NI Negative Negative Negative Negative Negative Negative Negative NI NI
Biopsied muscle 1. Anterior Deltoid Quadriceps Anterior Quadriceps Quadriceps Anterior Quadriceps Quadricep Quadriceps
tibial; 2.
Quadriceps
tibial tibial s
Age at muscle
biopsy, y 1. 62; 2. 75 63 65 59 60 64 79 47 59 76
Cytoplasmic bodies + - + - + - - ++ + -
Rimmed vacuoles + + + - - - - + - -
Lobulated fibers
(numerous) + - + + + - + - + -
Cores + - - - - - - - + -
Desmin IR areas in
muscle fibers (on
IHC)
+ + - - - - + - - -
Spheroid bodies + + - - - - - + - -
Necrosis - - - - + - - + - -
Fatty infiltration Mild Mild - + + - - - - -
Endomysial fibrosis Mild Mild Mild + + Mild Mild - - -
Fiber type
predominance Type 1 Type 1 - Type 1 Type 1 Type 2 Type 1 Type 2 - -
Sarcolemmal MHC-I
upregulation Diffusely + - - Patchy + Patchy + - Patchy + - - Patchy +
Sarcolemmal MHC-
II upregulation - NI - - - - - - - -
Inflammatory
infiltrates
(endomysial)
- NI Small + + - - + - -
CD68-positive cells + NI + + + - + + - -
CD8-positive cells - NI - - - - - - - -
MAC (C5b9)-
labelling of non-
necrotic muscle
fibers
- NI - - - - - - - -
*MRC 3/5 or less; ** the xiphoid process cannot be lifted from the bed; F, female; M, male; AAO, age at onset; y, years; MGUS, monoclonal gammopathy of
unknown significance; UL, upper limbs; LL, lower limbs; NI, not investigated; CK, creatinine kinase; IR, immunoreactive; IHC, immunohistochemistry

95
REFERENCES 1. Jackson CE. A clinical approach to muscle diseases. Seminars in neurology 2008;28(2):228-40. doi:
10.1055/s-2008-1062266 [published Online First: 2008/03/21]
2. Mohassel P, Landon-Cardinal O, Foley AR, et al. Anti-HMGCR myopathy may resemble limb-girdle
muscular dystrophy. Neurology(R) neuroimmunology & neuroinflammation 2019;6(1):e523. doi:
10.1212/nxi.0000000000000523 [published Online First: 2018/12/28]
3. Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, et al. Sporadic late-onset nemaline myopathy: clinico-
pathological characteristics and review of 76 cases. Orphanet journal of rare diseases 2017;12(1):86.
doi: 10.1186/s13023-017-0640-2 [published Online First: 2017/05/12]
4. Peric S, Glumac JN, Topf A, et al. A novel recessive TTN founder variant is a common cause of distal
myopathy in the Serbian population. European journal of human genetics : EJHG 2017;25(5):572-81.
doi: 10.1038/ejhg.2017.16 [published Online First: 2017/03/16]
5. Mercuri E, Pichiecchio A, Allsop J, et al. Muscle MRI in inherited neuromuscular disorders: past, present,
and future. Journal of magnetic resonance imaging : JMRI 2007;25(2):433-40. doi: 10.1002/jmri.20804
[published Online First: 2007/01/30]
6. Brebner JA, Stockley RA. Polyclonal free light chains: a biomarker of inflammatory disease or treatment
target? F1000 medicine reports 2013;5:4. doi: 10.3410/m5-4 [published Online First: 2013/02/16]
7. Uruha A, Benveniste O. Sporadic late-onset nemaline myopathy with monoclonal gammopathy of
undetermined significance. Current opinion in neurology 2017;30(5):457-63. doi:
10.1097/wco.0000000000000477 [published Online First: 2017/07/06]
8. Jungbluth H, Treves S, Zorzato F, et al. Congenital myopathies: disorders of excitation-contraction
coupling and muscle contraction. Nature reviews Neurology 2018;14(3):151-67. doi:
10.1038/nrneurol.2017.191 [published Online First: 2018/02/03]
9. Mouhieddine TH, Weeks LD, Ghobrial IM. Monoclonal gammopathy of undetermined significance.
Blood 2019;133(23):2484-94. doi: 10.1182/blood.2019846782 [published Online First: 2019/04/24]
10. Monforte M, Primiano G, Silvestri G, et al. Sporadic late-onset nemaline myopathy: clinical, pathology
and imaging findings in a single center cohort. Journal of neurology 2018;265(3):542-51. doi:
10.1007/s00415-018-8741-y [published Online First: 2018/01/23]
11. Selcen D, Engel AG. Myofibrillar myopathies. Handbook of clinical neurology 2011;101:143-54. doi:
10.1016/b978-0-08-045031-5.00011-6 [published Online First: 2011/04/19]
12. Naddaf E, Milone M, Kansagra A, et al. Sporadic late-onset nemaline myopathy: Clinical spectrum,
survival, and treatment outcomes. Neurology 2019;93(3):e298-e305. doi:
10.1212/wnl.0000000000007777 [published Online First: 2019/06/07]
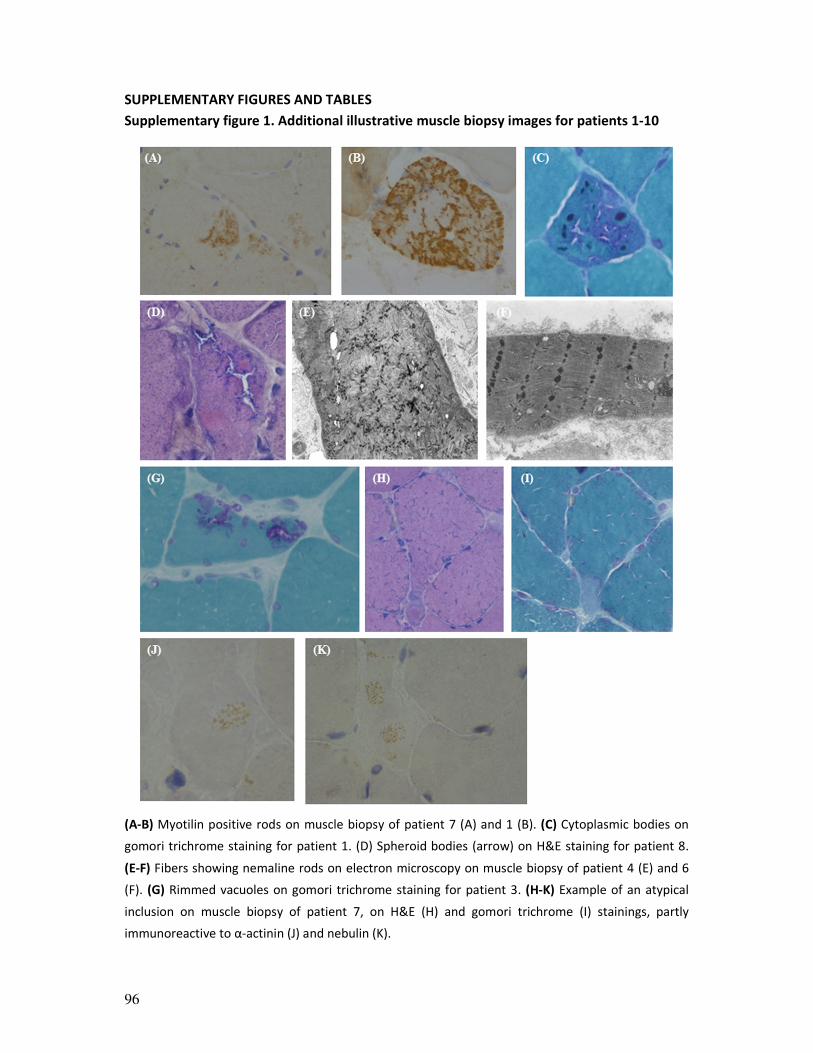
96
SUPPLEMENTARY FIGURES AND TABLES
Supplementary figure 1. Additional illustrative muscle biopsy images for patients 1-10
(A-B) Myotilin positive rods on muscle biopsy of patient 7 (A) and 1 (B). (C) Cytoplasmic bodies on
gomori trichrome staining for patient 1. (D) Spheroid bodies (arrow) on H&E staining for patient 8.
(E-F) Fibers showing nemaline rods on electron microscopy on muscle biopsy of patient 4 (E) and 6
(F). (G) Rimmed vacuoles on gomori trichrome staining for patient 3. (H-K) Example of an atypical
inclusion on muscle biopsy of patient 7, on H&E (H) and gomori trichrome (I) stainings, partly
immunoreactive to α-actinin (J) and nebulin (K).
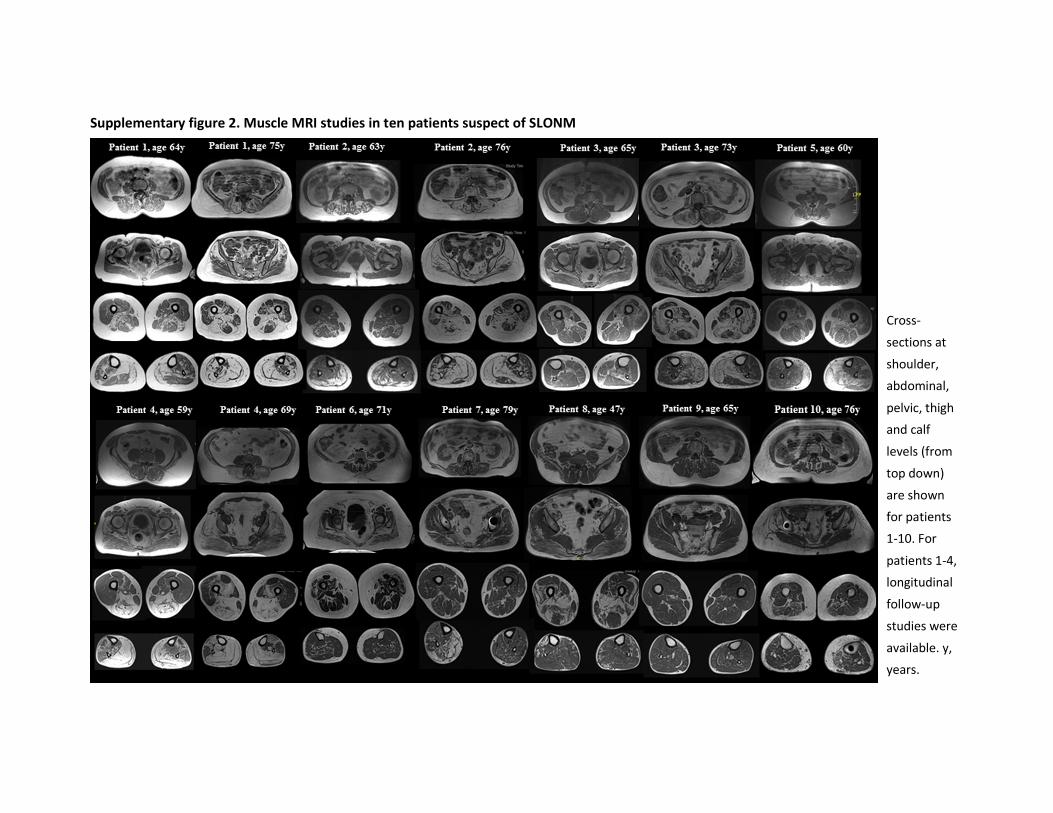
Supplementary figure 2. Muscle MRI studies in ten patients suspect of SLONM
Cross-
sections at
shoulder,
abdominal,
pelvic, thigh
and calf
levels (from
top down)
are shown
for patients
1-10. For
patients 1-4,
longitudinal
follow-up
studies were
available. y,
years.
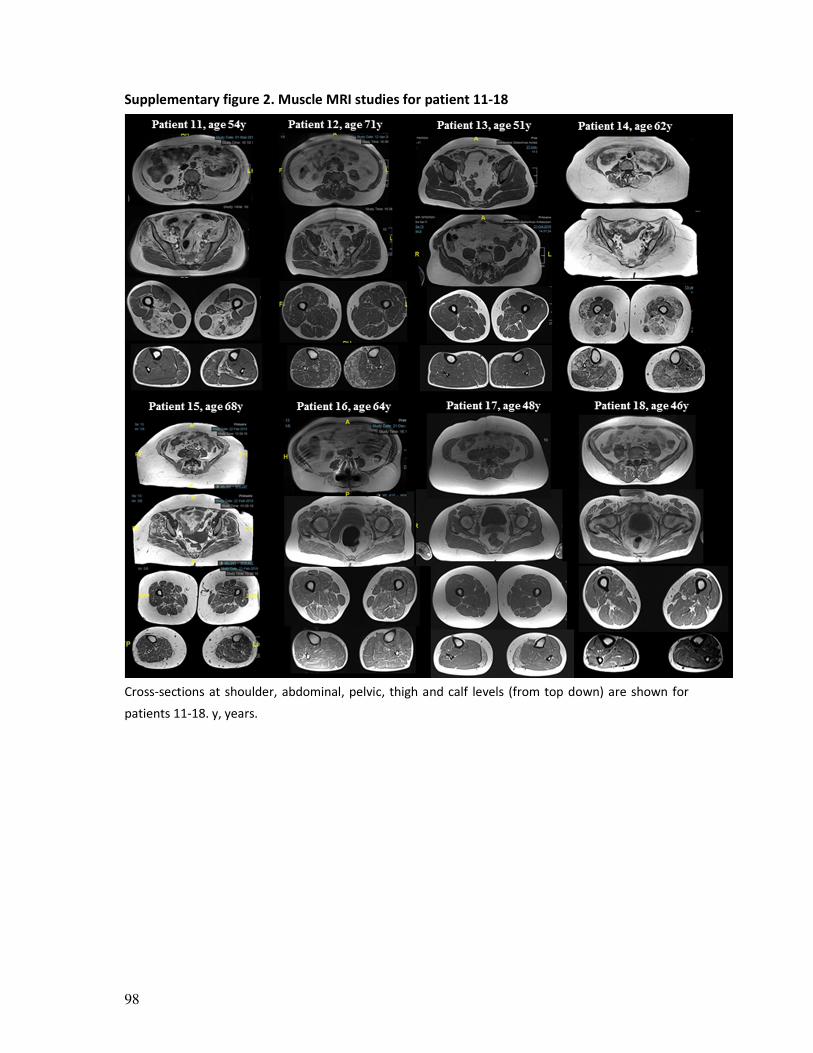
98
Supplementary figure 2. Muscle MRI studies for patient 11-18
Cross-sections at shoulder, abdominal, pelvic, thigh and calf levels (from top down) are shown for
patients 11-18. y, years.
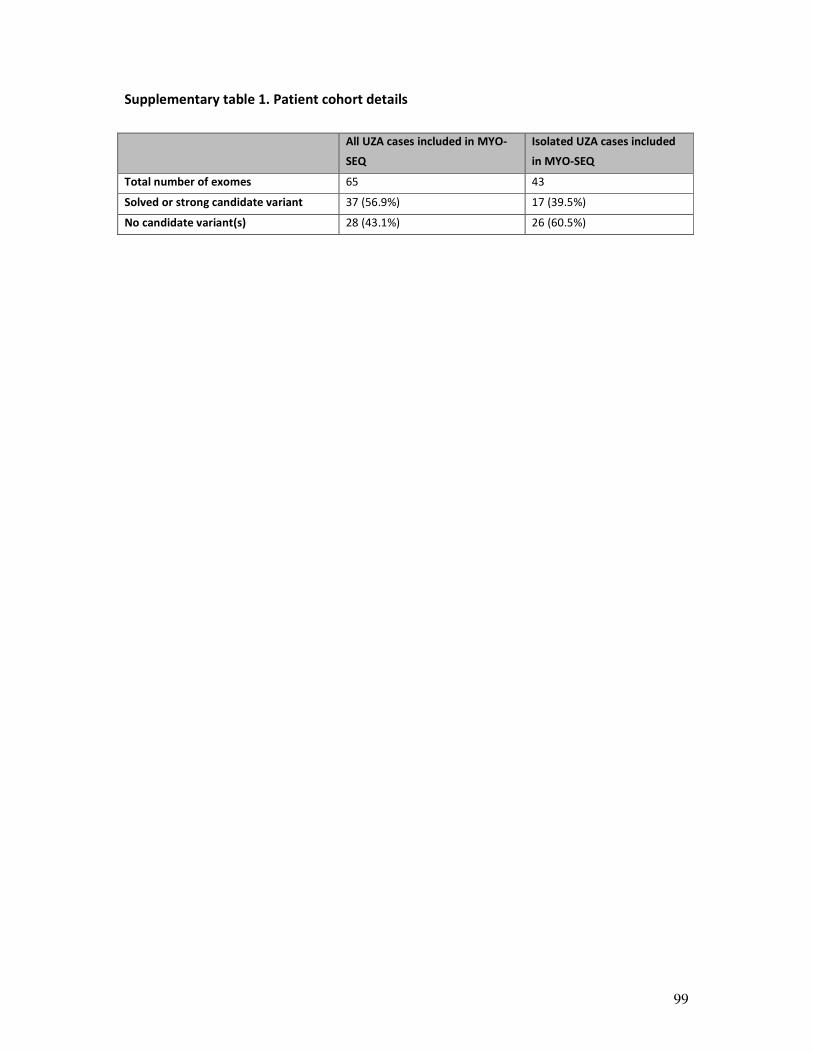
99
Supplementary table 1. Patient cohort details
All UZA cases included in MYO-
SEQ
Isolated UZA cases included
in MYO-SEQ
Total number of exomes 65 43
Solved or strong candidate variant 37 (56.9%) 17 (39.5%)
No candidate variant(s) 28 (43.1%) 26 (60.5%)
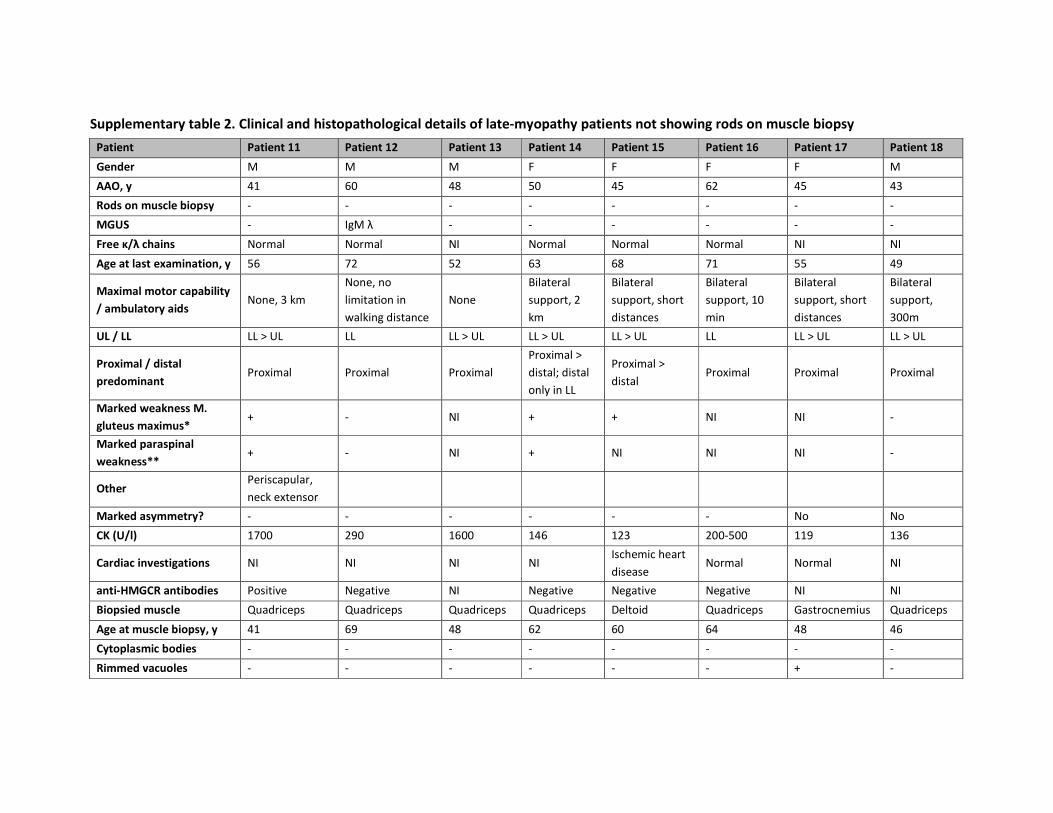
Supplementary table 2. Clinical and histopathological details of late-myopathy patients not showing rods on muscle biopsy
Patient Patient 11 Patient 12 Patient 13 Patient 14 Patient 15 Patient 16 Patient 17 Patient 18
Gender M M M F F F F M
AAO, y 41 60 48 50 45 62 45 43
Rods on muscle biopsy - - - - - - - -
MGUS - IgM λ - - - - - -
Free κ/λ chains Normal Normal NI Normal Normal Normal NI NI
Age at last examination, y 56 72 52 63 68 71 55 49
Maximal motor capability
/ ambulatory aids None, 3 km
None, no
limitation in
walking distance
None
Bilateral
support, 2
km
Bilateral
support, short
distances
Bilateral
support, 10
min
Bilateral
support, short
distances
Bilateral
support,
300m
UL / LL LL > UL LL LL > UL LL > UL LL > UL LL LL > UL LL > UL
Proximal / distal
predominant Proximal Proximal Proximal
Proximal >
distal; distal
only in LL
Proximal >
distal Proximal Proximal Proximal
Marked weakness M.
gluteus maximus* + - NI + + NI NI -
Marked paraspinal
weakness** + - NI + NI NI NI -
Other Periscapular,
neck extensor
Marked asymmetry? - - - - - - No No
CK (U/l) 1700 290 1600 146 123 200-500 119 136
Cardiac investigations NI NI NI NI Ischemic heart
disease Normal Normal NI
anti-HMGCR antibodies Positive Negative NI Negative Negative Negative NI NI
Biopsied muscle Quadriceps Quadriceps Quadriceps Quadriceps Deltoid Quadriceps Gastrocnemius Quadriceps
Age at muscle biopsy, y 41 69 48 62 60 64 48 46
Cytoplasmic bodies - - - - - - - -
Rimmed vacuoles - - - - - - + -

Lobulated fibers
(numerous) - - - - + + + -
Cores - - - - - - - +
Desmin IR areas in muscle
fibers (on IHC) - - NI - - - - -
Spheroid bodies - - - - - - - -
Necrosis + - - - - - - -
Fatty infiltration - - - + - - - -
Endomysial fibrosis - - - + - - + -
Fiber type predominance - - - - Type 1 Type 2 Type 1 Type 1
Other striking muscle
biopsy features /
Marked tubular
aggregates /
Mixed
myogenic
and
neurogenic
features
/ / /
Mixed
myogenic
and
neurogenic
features
Sarcolemmal MHC-I
upregulation - - - - - - NI NI
Sarcolemmal MHC-II
upregulation - - - - - - NI NI
Inflammatory infiltrates
(endomysial) - - - - - - NI NI
CD68-positive cells Isolated
necrotic fibers + - - - - NI NI
CD8-positive cells - - - - - - NI NI
MAC (C5b9)-labelling of
non-necrotic muscle
fibers
- - - - - NI NI
*MRC 3/5 or less; ** the xiphoid process cannot be lifted from the bed; F, female; M, male; AAO, age at onset; y, years; MGUS, monoclonal gammopathy of
unknown significance; UL, upper limbs; LL, lower limbs; NI, not investigated; CK, creatinine kinase; IR, immunoreactive; IHC, immunohistochemistry

102

103
Part 2
A proteomic approach to study disease signatures in
muscle tissue of myopathy patients in an unbiased way

104

105
CHAPTER 4
A tale of the unexpected: multisystem proteinopathy
due to a homozygous p.Arg159His VCP mutation
Willem De Ridder, Abdelkrim Azmi, Christoph S. Clemen, Ludwig Eichinger, Andreas
Hofmann, Rolf Schröder, Katherine Johnson, Ana Töpf, Volker Straub, Peter De Jonghe,
Stuart Maudsley, Jan L. De Bleecker, Jonathan Baets
Neurology. 2019 Dec 17 [Epub ahead of print]

106
ABSTRACT
Objective: To assess the clinical, radiological, myopathological, and proteomic findings in a
patient manifesting a multisystem proteinopathy due to a homozygous VCP mutation,
previously reported to be pathogenic in heterozygous state.
Methods: We studied a currently 36-year-old male index patient and his father both
presenting with progressive limb-girdle weakness. Muscle involvement was assessed by MRI
and muscle biopsies. We performed whole-exome sequencing and Sanger sequencing for
segregation analysis of the identified p.Arg159His VCP mutation. To dissect biological
disease signatures we applied state-of-the-art quantitative proteomics on muscle tissue of
the index case, his father, three additional VCP-related myopathy patients and three control
individuals.
Results: The index patient, homozygous for the known p.Arg159His mutation in VCP,
manifested a typical VCP-related myopathy phenotype, although with a markedly high
creatine kinase value and a relatively early disease onset, and Paget disease of bone. The
father exhibited a myopathy phenotype and discrete parkinsonism and multiple deceased
family members on the maternal side of the pedigree displayed either a dementia,
parkinsonism or myopathy phenotype. Bioinformatic analysis of quantitative proteomic
data revealed the degenerative nature of the disease, with evidence suggesting selective
failure of muscle regeneration and stress granule dyshomeostasis.
Conclusions: We report on a patient showing a multisystem proteinopathy due to a
homozygous VCP mutation. The patient manifests a severe phenotype, yet fundamental
disease characteristics are preserved. Proteomic findings provide further insights in VCP-
related pathomechanisms.

107
INTRODUCTION
Mutations in the valosin containing protein gene (VCP) are associated with a rare,
dominantly-inherited multisystem proteinopathy (MSP1), which presents with a high
diversity of combinations of phenotypes, including inclusion body myopathy (IBM), early-
onset Paget disease of bone (PDB), frontotemporal dementia (FTD), amyotrophic lateral
sclerosis (ALS) and parkinsonism.1 At present, more than 40 different heterozygous
missense mutations have been reported in VCP. Important intra- and interfamilial
phenotypic variability has been noted1, 2. Penetrance of the myopathy phenotype, PDB and
FTD are estimated at 90%, 50% and 30%, respectively.3 Onset of muscle weakness occurs
during adulthood, at a mean age of approximately 40-45 years.3, 4
Mutations cluster in the N and D1 domains of the VCP protein, which is involved in multiple
cellular processes and has a critical role in proteostasis, at the intersection of the ubiquitin-
proteasome system and autophagy.1, 5 VCP belongs to the AAA+ ATPase family (‘ATPases
associated with diverse cellular activities’), and uses energy from ATP hydrolysis to
segregate molecules from immobile cellular structures such as protein complexes or
aggregates, membranes and chromatin, in conjunction with a collection of cofactors and
adaptors.6, 7 Exact molecular mechanisms of VCP-related disease remain unknown,5
particularly with regard to the tissue specific nature of the disorder and genotype-
phenotype correlations.
Here, we present a MSP1 patient harboring in homozygosity the VCP p.Arg159His mutation,
previously only reported to be pathogenic in heterozygous state.8-13 Detailed study of this
patient demonstrates that the key MSP1 phenotypic characteristics are preserved in case of
homozygosity of this VCP mutation.
METHODS
Standard protocol approvals, registrations, and patient consents
Ethical approval was granted by the relevant local Ethical Committees of the participating
centers. All participants provided written informed consent prior to participation in the
study.
Patients and clinical evaluation
The index patient (patient A) and his father (patient B) presented with unexplained limb-
girdle muscular weakness and an elevated serum creatine kinase (CK) level. No other family
members agreed to be clinically evaluated, except for the mother of patient A. Nerve
conduction studies (NCS) and an electromyography (EMG) were performed. Muscle MRI
was performed on a 1.5T MRI platform at the Antwerp University Hospital (UZA). Cross-

108
sections at shoulder, abdominal, pelvic, thigh and calf levels were assessed on T1-weighted
images to evaluate patterns of muscle involvement. Fatty replacement of muscle was
graded according to the Mercuri scale.14 Pulmonary function was assessed by spirometry
testing (forced vital capacity, FVC) and cardiac function by ECG and echocardiography. Bone
scintigraphy was carried out and bone turnover markers in blood (alkaline phosphatase) and
urine (collagen crosslinks) were evaluated.
Analysis of exome sequencing data
A DNA sample of patient A was submitted to the MRC Centre for Neuromuscular Diseases
Biobank (Newcastle University, UK). Samples were processed and whole-exome sequencing
(WES) was performed and analyzed by a targeted approach as described previously.15 A
candidate variant in VCP (reference sequence: NM_007126) was validated by Sanger
sequencing and segregation analysis was performed with DNA samples of patient A, his
father patient B and his mother.
Muscle biopsies
Muscle biopsies of quadriceps muscle were obtained from patients A and B, three additional
patients with a VCP-related myopathy phenotype and three control individuals (patients C-E
and controls 1-3, clinical details see table 1) and analyzed following standard histological
and immunohistochemical light microscopy and electron microscopy (EM) protocols.
Patient D and E harboring the p.Gly125Asp mutation are siblings. Segregation studies
confirmed that the mutation was inherited from the affected father, showing a VCP-related
myopathy phenotype as well. For all of the patients, the choice to biopsy the quadriceps
muscle was similarly based on clinical and radiological selective but not yet end-stage
involvement of the muscle. Control individuals, biopsied for subjective myalgia, yet for
whom no clinical, morphological or electrodiagnostic abnormalities had been identified,
were selected based on biopsied muscle, sex and age.
Sample preparation for iTRAQ labeling and mass spectrometry analysis
Proteins were extracted from muscle biopsy specimens of patients A-E and three control
individuals, in a buffer containing 4% SDS, 100 mM TCEP, 50 mM Tris pH 7.8. Lysates were
heated at 95°C for 5 minutes. The extracted proteins were precipitated with trichloroacetic
acid (TCA) and re-solubilized in 8 M urea, 2 M thiourea, 0.1% SDS in 50 mM
triethylammonium bicarbonate (TEAB). After measuring protein concentrations using the
RCDC kit (Bio-Rad), equal amounts of proteins from each lysate were reduced and alkylated
with TCEP (tris-2-carboxyethyl phosphine) and MMTS (5-methyl-methanoethiosulphate),

109
respectively, followed by trypsin digestion. Peptides from each sample were labelled using
iTRAQ reagents 8plex (Sciex) according to the manufacturer's instructions. The mixed
peptides were separated on an offline 2D-liquid chromatography (LC) system (Dionex,
ULTIMATE 3000, ThermoScientific), consisting of a 15 cm strong cationic exchange column
and a 25 cm nano-RP C18 column. The nano-LC was coupled online to a QExactive™-Plus
Orbitrap (ThermoScientific) mass spectrometer (MS).
Bioinformatic analysis of mass spectrometry data
The generated raw data from the MS were processed with the Proteome Discoverer 2.1
(PD2.1) software (ThermoScientific). For protein identification, the search engine Sequest
HT was used against the human UniProt/SwissProt database with a false discovery rate
(FDR) of less than 1%. The quantitative proteomics data were then further statistically
analyzed with the Perseus software (version 1.6.1.1).16 Log2-transformed scaled abundance
values generated by the PD2.1 software were normalized by subtraction of the median
value of the respective column. The complete list of identified proteins with the original
scaled abundance values is available from Dryad (table e-1). Hierarchical clustering analyses
were performed using Euclidean algorithms. Volcano plot analyses, assessing statistical
significance (t-test) together with fold change (FDR = 0.05, S0 = 0.1), were performed to
identify significantly dysregulated proteins between patients and controls. The downstream
canonical pathways analysis (filtering based on p-value of overlap) and the upstream
regulator analysis (filtering based on |Z-score| ≥ 2) as a causal analysis approach were
applied on this set of dysregulated proteins, using the Ingenuity Pathway Analysis (IPA)
software (QIAGEN).17 To identify significant outlier values in the data of the index patient
compared to the other patients, a Significance A analysis in Perseus was performed,18
correcting for multiple hypothesis testing with the Benjamini-Hochberg FDR (FDR = 0.05).
Immunoblotting
Protein extractions of muscle tissue specimens from patients A-C and a pooled extract of
the control individuals used for MS analysis, were subjected to western blotting to validate
the MS dataset. Equal amounts of protein were loaded and separated on 4–12 % NuPAGE®
Bis-Tris gels (Life Technologies) and transferred on a polyvinylidene difluoride membrane
(PVDF, Hybond P, Amersham Biosciences). Membranes were probed with the following
selective primary antibodies: anti-PGAM2 (ab97800, Abcam); anti-LMNB1 (LS-B11184,
LSBio); anti-MFF (ab81127, Abcam); anti-GAPDH (GTX100118, GeneTex); anti-VCP (ab11433,
Abcam and #2648, Cell Signaling Technology). Immunodetection was performed using host-
specific secondary antibodies conjugated with horseradish peroxidase (HRP) and the ECL-

110
plus chemiluminescent detection system (Thermo Scientific). Western blot results were
visualized using the Amersham™ Imager 600 digital imaging system and quantified with
ImageQuant™ TL software (GE Healthcare Life Sciences). Quantitative data were normalized
to GAPDH expression levels. Expression data were visualized as ratios of the respective
sample relative to the pooled controls and compared to log2 iTRAQ expression ratios.
Data availability
Anonymized data not published within the article will be shared upon request by any
qualified investigator.
RESULTS
Genetic findings
A rare variant in VCP was identified in the exome of patient A. He appeared to harbor the
previously reported heterozygous pathogenic p.Arg159His (c.476G>A) mutation in
homozygosity.8-13 DNA of the parents was available for segregation analysis, both were
confirmed to be heterozygous carriers of the mutation (figure 1).
Clinical aspects
Patient A, a currently 36-year-old man of Belgian ancestry, presented with complaints
related to progressive proximal weakness in the lower limbs that started at the age of 29
years. Clinically, marked atrophy of lower limb muscles was noted. NCS yielded normal
results, an EMG showed a mixed pattern of neurogenic or myogenic discharges. Similarly, a
diagnostic muscle biopsy performed at that time appeared to show a mixed pattern of
myopathic and apparently neurogenic abnormalities. Weakness in the limbs progressed to
distal weakness in the lower limbs with a marked foot drop bilaterally and upper limb,
periscapular and paraspinal weakness. The patient was re-evaluated in our center at the age
of 31 years. DNA was sent for WES and the muscle biopsy was reassessed. MRI revealed an
asymmetric pattern of patchy muscle involvement with preferential involvement of
paraspinal and lower limb muscles (figure 2).
At the age of 63 years, patient B (the father) presented with complaints related to proximal
weakness in the lower limbs, with symptoms slowly progressing since the age of 58 years.
MRI studies revealed a similar patchy pattern of muscle involvement as observed for patient
A (figure 2). During follow-up, the patient noticed an impairment of his right hand function,
which could clinically be attributed to an asymmetric extrapyramidal syndrome.
The serum CK level for patient B was only mildly elevated (192 U/l), contrasting with the
high CK level for patient A (1138 U/l). Clinical details for patient A and B are summarized in

111
table 2 and a detailed description of the pattern of muscle weakness is available from Dryad
(table e-2).
Intriguingly, on the maternal side of the family tree (figure 1), the maternal grandfather had
exhibited a myopathy phenotype and many individuals had been diagnosed with a dementia
phenotype or parkinsonism in the past. Clinical examination of the mother at the age of 60
years showed no signs of a neuromuscular disorder, parkinsonism, dementia or Paget
disease of bone. She refused additional technical investigations. When studying the family
history in detail, there appeared to be distant consanguinity between the parents of the
index patient (figure 1).
After identification of the VCP p.Arg159His mutation, bone scintigraphy and baseline studies
of bone metabolism led to the diagnosis of an asymptomatic PDB lesion of vertebra L2 and
the ilium on the right side for patient A. For patient B, the urinary pyridoline/creatinine ratio
was borderline increased, a bone scintigraphy yielded normal results (table 2).
Muscle biopsies
Muscle biopsy of patient A (figure 3A-D) showed myopathic features, with increased fiber
size variation and multiple internalized nuclei, but also apparently neurogenic features,
demonstrated by the presence of angular atrophic fibers. There was no evident fiber type
grouping or grouped atrophy. Scattered necrotic and regenerating fibers as well as fibers
with rimmed vacuoles and endomysial inflammatory infiltrates were present.
Immunostainings demonstrated that inflammatory infiltrates mainly consisted of CD68+
macrophages (figure 3C). MHC-I was regionally upregulated at the sarcolemma (figure 3D).
EM analysis revealed 15 to 18 nm tubulofilamentous inclusions and rimmed vacuoles.
On the muscle biopsy of patient B, myopathic features consisting of an increased
percentage of internalized nuclei and an increased fiber size variation were also noted. A
few rimmed vacuoles were visualized, however no striking signs of necrosis or regeneration.
Ultrastructural analysis confirmed nonspecific myopathic features.
Muscle biopsies of patient C-E similarly revealed mainly myopathic features (table 1), with
some scattered angular atrophic fibers and endomysial inflammatory infiltrates with focal
invasion of non-necrotic muscle fibers (figure available from Dryad [figure e-1]). Muscle
biopsy studies of the control individuals yielded strictly normal results (table 1).
Proteomic investigation and bioinformatic interpretation of VCP-related disease
signatures in skeletal muscle
To investigate VCP-related myopathy pathomechanisms in more detail, an exploratory
comparative proteomic study was performed on skeletal muscle tissue lysates of patients A

112
and B, three additional patients (C-E) with a VCP-related myopathy phenotype, and three
control individuals. In total, 1656 iTRAQ labeled proteins were detected across the 8
samples. Visualizing the dataset by means of hierarchic clustering, patient A appeared to
cluster most closely with patient E (figure 4A). To create a general appreciation of VCP-
related disease signatures in VCP-related myopathy muscle first, a volcano plot was
generated to compare protein expression data of the four patients harboring a
heterozygous mutation in VCP and the three control individuals, followed by functional
annotation of significantly dysregulated proteins. Based on the volcano plot analysis, 390
proteins were significantly dysregulated, of which 207 downregulated, in patients as
compared to control individuals (figure 4B). A full list of these proteins is available from
Dryad (table e-3). Based on p-value of overlap, the top 10 of enriched pathways identified in
the canonical pathway analysis mainly reflected downstream consequences of disease
mechanisms, as evident by changes in metabolic pathways in muscle tissue of patients with
a VCP-related myopathy (upper panel of table 3). The pathway with the lowest (negative) Z-
score (predicted to be inhibited) was ‘oxidative phosphorylation’ and the one with the
highest Z-score (predicted to be activated) was ‘sirtuin signaling pathway. The set of
predicted potential ‘key regulators’, generated with the IPA upstream regulator analysis,
yielded additional pathomechanistic insights (full list available from Dryad, table e-4). Here,
the predicted activation or inhibition of multiple upstream regulators (such as TWIST1, mir-
1, MEF2C) reflected inhibition of myogenesis. Furthermore, inflammatory signatures were
evident from the analysis, with multiple cytokines being predicted to be activated, as well as
dysregulation of a key metabolic regulator, PPARGC1A. The occurrence of oxidative stress in
patient tissue was suggested by the predicted activation of NFE2L2, the key mediator the
Nrf2-mediated oxidative stress response, occurrence of ER stress and the unfolded protein
response (UPR) by the predicted activation of XBP1. The upstream regulator with the
highest positive Z-score was KDM5A, a histone demethylase involved in the DNA damage
response (DDR).19
Subsequently, a Significance A outlier analysis was performed in Perseus to search for
differences between the proteomes of patient A and the heterozygous VCP patients. This
yielded a shortlist of 19 proteins that appeared to show significant outlier values (lower
panel of table 3) for patient A. The top hit was ZFAND1, a protein involved in the regulation
of cytoplasmic stress granule turnover. Notably, RAD17 is a protein involved in the DDR and
WDR33 is an RNA processing protein linked to mRNA homeostasis. Most of the other
proteins, showing a less significant outlier value for the homozygous patient, represent
proteins of the contractile apparatus in skeletal muscle.

113
Finally, we validated MS data of a discrete set of proteins, PGAM2, LMNB and MMF, via
western blot analysis, with protein expression ratios between patients and pooled controls
corroborating the data (figure 4C-D). Based on the MS dataset, protein expression levels of
VCP were similar across patients and controls, which was also confirmed by western blotting
(figure available from Dryad [figure e-2]).
DISCUSSION
We report on a MSP1 patient harboring the p.Arg159His VCP mutation in homozygosity,
previously reported to be pathogenic in heterozygous state. The index patient presented
with young adult onset proximal weakness in the lower limbs resulting after 6 years of
evolution in walking distance restriction to 300 meters and inability to climb stairs. In
addition, distal weakness in upper and lower limbs with a marked bilateral foot drop and
scapular winging are typical clinical features of a VCP-related myopathy.2 Furthermore, an
asymptomatic PDB lesion was diagnosed. The father of the index patient, heterozygous for
the p.Arg159His mutation, exhibited a myopathy phenotype with onset at 58 years of age
and discrete asymmetric parkinsonism. The notion of a dementia or parkinsonism
phenotype in multiple deceased family members and the observation that multiple
obligatory carriers died presumably asymptomatic at an old age, further illustrates the
characteristic intra-familial phenotypic variability of MSP1, even for the otherwise rather
penetrant myopathy part of the disease spectrum.3
The p.Arg159His mutation has previously been reported to be pathogenic in heterozygous
state in multiple unrelated families showing intra- and interfamilial phenotypic variability,
encompassing IBM, PDB, FTD and ALS, which is typical for MSP1, without complete
penetrance.8-13
Muscle MRI revealed an asymmetric and patchy pattern of muscle involvement in patients A
and B, with paraspinal and lower limb muscles being preferentially involved. This patchy
involvement has been described previously and seems to contrast with the selective
patterns of muscle involvement in muscular dystrophies and sporadic IBM (sIBM).2, 20, 21
The muscle biopsy of patient A revealed myopathic features as well as the presence of
atrophic angulated fibers, which are generally considered as a neurogenic feature. However,
no diagnosis of a lower motor neuron disease (disease of the anterior horn cell) or motor
neuropathy (axonopathy) could be made based on clinical and electrophysiological
investigations. This mixture of myopathic and neurogenic features on muscle biopsy or on
EMG appears to be frequent in VCP-related myopathies. This observation might suggest the
occurrence of a subclinical mild motor axonopathy.13, 22 Rimmed vacuoles, as observed in
approximately 35-40% of patients exhibiting a VCP-related myopathy phenotype, have a

114
similar appearance as in other rimmed vacuolar myopathies such as sIBM.2 The 15 to 18 nm
tubulofilamentous inclusions, which were observed in muscle of patient A, have previously
been observed in muscle of VCP patients and are typically also observed in sIBM.13, 23 The
appearance of inflammatory infiltrates and MHC-I upregulation, as observed for patient A,
might further complicate the histopathological differential diagnosis with sIBM.23 On muscle
biopsies of patients C, D and E, some endomysial inflammatory infiltrates were also
observed, with focal invasion of non-necrotic fibers on muscle biopsies of patients D and E,
yet a systematic immunohistochemical study of inflammatory features could not be
performed within the scope of the manuscript. Based on literature, the appearance of
inflammatory changes on muscle biopsies of patients manifesting a VCP-related myopathy
appears to be rare or at least not conspicuous, although it has previously been mentioned
briefly that some biopsies may show MHC-I upregulation or small inflammatory infiltrates.2
Focal invasion of non-necrotic muscle fibers is typically observed in sIBM and appears to be
a very rare phenomenon in inherited muscle disorders.24
Homozygous mutations in hereditary autosomal-dominant disorders are uncommon events.
Generally, phenotypes are expected to be more pronounced, but systematic literature is
evidently scarce.25 In case of a direct loss-of-function mechanism of the mutation leading to
haploinsufficiency in heterozygous state, a much more severe phenotype is anticipated.
Dominant-negative mutations, however, are not expected to cause a much more severe
phenotype, and for mutations exhibiting a gain-of-function, consequences seem to be
variable.25 In the present case, the index patient exhibited a typical pattern of muscle
weakness. Yet the onset age of the myopathy phenotype is rather early for this age-related
disorder, compared to patients harboring the same p.Arg159His mutation and indeed the
complete group of MSP1 patients; the mean age at onset is approximately 40-45 years, with
only few patients being in their twenties.2, 3, 8-12, 26 Notably, CK levels for the index patient
are markedly elevated (1138 U/l), contrasting with CK levels generally reported to be normal
or mildly elevated in VCP-related myopathies.3, 4 Besides these anomalies, the two key
features of VCP-related disease are preserved, i.e. the slowly progressive, age-related
nature of the disorder and the tissue-specific phenotype.1 An example of another
autosomal-dominant age-related disorder for which homozygosity of a known variant
appeared to be viable and moderately accelerate the age at onset, is the familial Alzheimer
disease (FAD) linked to the E280A mutation in PSEN1.27 Other similar examples have been
described in other protein aggregate myopathies, i.e. myotilinopathies and
desminopathies.28, 29
Because of the singularity of the observation of a human patient with a homozygous VCP
mutation we decided to dissect biological disease signatures by means of quantitative

115
proteomic analyses. First of all, hierarchic clustering analyses did not yield arguments for
globally different patterns of protein dysregulation at the level of the proteome between
patient A and four other patients with a VCP-related myopathy, nor between patients
harboring the p.Arg159His mutation or the p.Gly125Asp mutation. Functional annotation of
proteins significantly dysregulated between heterozygous patients and control individuals
yielded a general appreciation of VCP-related disease signatures in muscle. Whereas the
downstream pathway analysis mainly pointed towards metabolic failure of skeletal muscle,
the upstream regulator analysis, reflected a broader landscape of potential
pathomechanisms, including failure of muscle regeneration, oxidative stress, ER stress and
the unfolded protein response. Furthermore, the appearance of KDM5A as a predicted
upstream regulator with the highest positive Z-score, also hinted towards involvement of
DDR signaling.19 Changes in cellular energy metabolism most likely represent a non-specific
downstream consequence of disease mechanisms in degenerative muscle disorders, as e.g.
also described in a proteomic study of muscle tissue of patients manifesting a GNE-related
myopathy.30 The focused outlier analysis conducted in the present study identified ZFAND1
as the top scoring differentially regulated protein when comparing the homozygous patient
with the heterozygous patients. This protein was recently identified as an evolutionarily
conserved regulator of stress granule turnover which recruits VCP and the 26S proteasome
to organize degradation of stress granules.31 The notion that ZFAND1 appears to be
upregulated in muscle tissue of the homozygous patient compared to the heterozygous
patients most likely indicates a compensatory mechanism, e.g. because of a more
pronounced loss of interaction with VCP or a marked aggregation of stress granules.
Intriguingly, the relevance of stress granules for pathomechanisms of VCP-related disease
had been suggested by the finding that C2C12 myoblast cell lines transfected with mutant
VCP showed delayed stress granule resolution on oxidative stress.32 Clinicopathologically,
there are important similarities between VCP-related disease and the multisystem
proteinopathies related to mutations in stress granule components such as HNRNPA1 and
HNRNPA2B1, further implicating a strong interconnection of proteostasis and stress granule
homeostasis in disease.31, 33
Structural appraisal of the p.Arg159His mutations using VCP three-dimensional models34
supports the hypothesis that this mutation impacts only subtly on the interactor binding
behavior of VCP. VCP is assembled as a homo-hexamer with each monomer comprising an
N-terminal domain (NTD) as well as two ATPase domains (D1 and D2). The D1 and D2
domains are arranged in coaxially stacked rings and the N-terminal domains are located at
the periphery of the ring formed by the D1 domains.1 Arg159 locates to the β-barrel moiety
of the N-terminal domain of VCP and its side chain is situated in the interface of the N-

116
terminal domain and the D1 domain (residues 155-159, 386, 387). Out of more than 40
reported disease mutations, 16 map to this interface.2 In general, amino acid residues
located at domain interfaces of complex multi-domain proteins are poised to engage in
inter-domain communications either through direct van der Waals or polar interactions or,
more subtly, through allosteric mechanisms. Indeed, recent NMR-based studies
investigating the conformational equilibrium of the N-terminal domain of VCP between the
‘up’ (VCP:ATP) and ‘down’ (VCP:ADP) states found that residues in this interface can cause
subtle changes to the ‘up’ or ‘down’ equilibrium which probably impacts on the binding
behavior of VCP to its partner proteins.35, 36 Visual inspection of the VCP hexamer from
Protein Data Bank (PDB) entry 1s3s34 indicates that the side chain of Arg159 possibly
engages in two interactions of importance, one with a residue in the same structural moiety
(Glu124, β-barrel moiety of the NTD) and one with a residue in the D1 domain (Ala232,
RecA-like moiety). Replacing the side chain of 159 with an imidazolyl (histidine side chain)
group would disrupt both interactions.
The clinical findings in this homozygous patient together with the results of the proteomic
analysis and a structural appraisal of the p.Arg159His mutation suggest subtle changes of
VCP dynamics, and, as a result, altered binding behavior of VCP with respect to specific
interactors, as a likely (dominant-negative) molecular mechanism for the observed disease
pathology. In particular, the present constellation allows the comparison of the proteomic
consequences of the unique in vivo situation of having VCP hexamers constituting only of
mutant VCP monomers rather than a situation of having variable combinations of wild type
or mutant proteins in different VCP hexamers. As such, the availability of homo- and
heterozygous patients presents a highly valuable resource as functional analysis of mutant
VCP protein in vitro is always biased towards full mutant hexamers.7 However, with our
current data we cannot experimentally disprove the gain-of-function hypothesis which has
recently been put forward,5 based on the observation that the regulation of ATPase activity
may be altered in VCP mutants.7 The ultimate set of upstream key regulators and key
pathways that are specifically affected by the VCP p.Arg159His (or indeed any pathogenic)
mutation remains to be unraveled.
Preservation of the age-related nature of the disorder in case of homozygosity of the VCP
p.Arg159His mutation contrasts with findings in the VCP p.Arg155His homozygous missense
mouse model, which exhibits growth retardation and early lethality (survival less than 21
days).37 Other animal models have been used for preclinical studies of compounds and to
study pathogenesis of VCP-related disease, of which the heterozygous VCP p.Arg155His+/-
knock-in mouse model seems to mimic the MSP1 phenotype.38 However, as different
missense mutations are being compared, these mouse models should be used with caution

117
in the study of the molecular mechanisms of VCP mutations especially considering the
striking early-onset phenotype in the homozygous mice. Directly studying pathomechanisms
in diseased tissue of patients is therefore highly relevant. This present study reports on a
proteomic dataset on muscle tissue of VCP patients. Albeit this study focused on a relatively
small yet relevant set of samples, it serves as a proof-of-concept for further investigations of
molecular mechanisms of VCP pathologies.
Clearly, homozygosity of the p.Arg159His mutation in VCP appears to be viable and key
features of VCP-related disease are preserved. Notably, the observed phenotype appears to
be slightly more severe than in patients harboring a heterozygous mutation in VCP. The
findings in this study hint at the complex molecular mechanisms of VCP-related disease and
illustrate the phenotypic variability and diagnostic pitfalls in MSP1.

118
FIGURES AND TABLES
Figure 1. Segregation analysis of the VCP p.Arg159His (c.476G>A) mutation
Only patient A and his parents were clinically examined. Segregation analysis confirmed that both
parents were heterozygous carriers of the mutation. The maternal grandfather of patient A would
have shown a myopathy phenotype and died at the age of 72 years. Arrow = index patient. Half-
filled symbols represent individuals diagnosed with a dementia phenotype in the past, the quarter
filled symbol an individual diagnosed with parkinsonism. † = for the presumably asymptomajc
obligatory carriers the age at death (y) is mentioned.
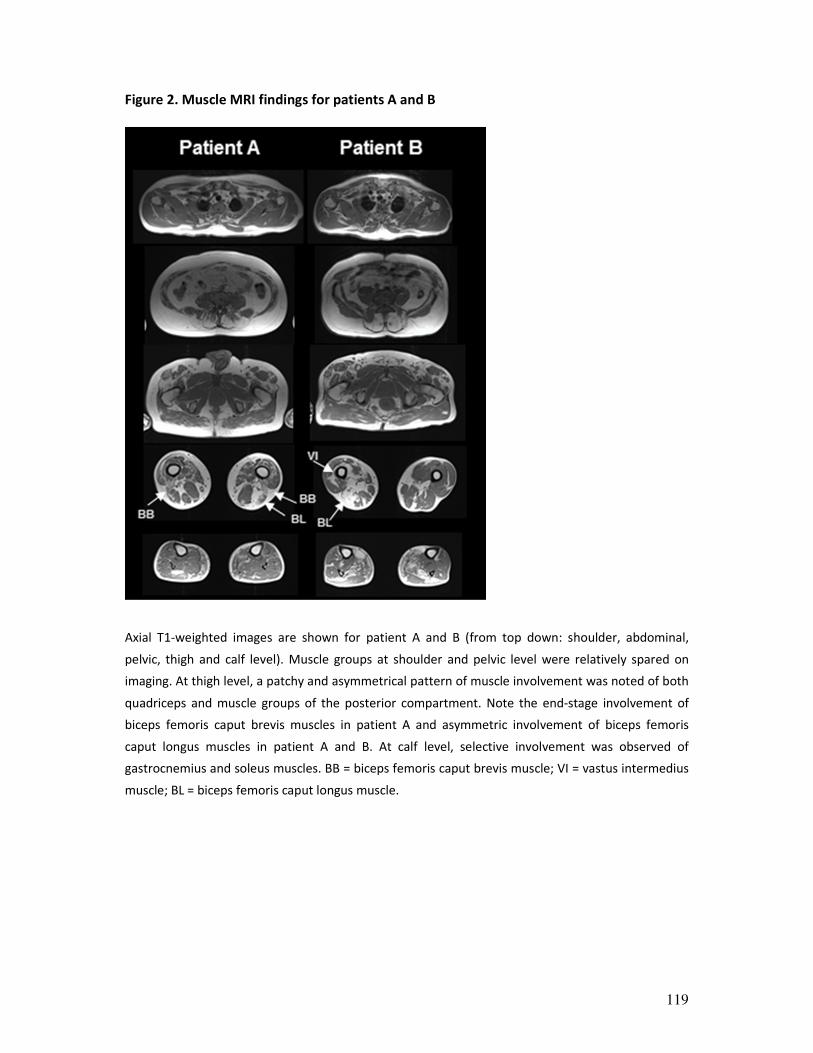
119
Figure 2. Muscle MRI findings for patients A and B
Axial T1-weighted images are shown for patient A and B (from top down: shoulder, abdominal,
pelvic, thigh and calf level). Muscle groups at shoulder and pelvic level were relatively spared on
imaging. At thigh level, a patchy and asymmetrical pattern of muscle involvement was noted of both
quadriceps and muscle groups of the posterior compartment. Note the end-stage involvement of
biceps femoris caput brevis muscles in patient A and asymmetric involvement of biceps femoris
caput longus muscles in patient A and B. At calf level, selective involvement was observed of
gastrocnemius and soleus muscles. BB = biceps femoris caput brevis muscle; VI = vastus intermedius
muscle; BL = biceps femoris caput longus muscle.
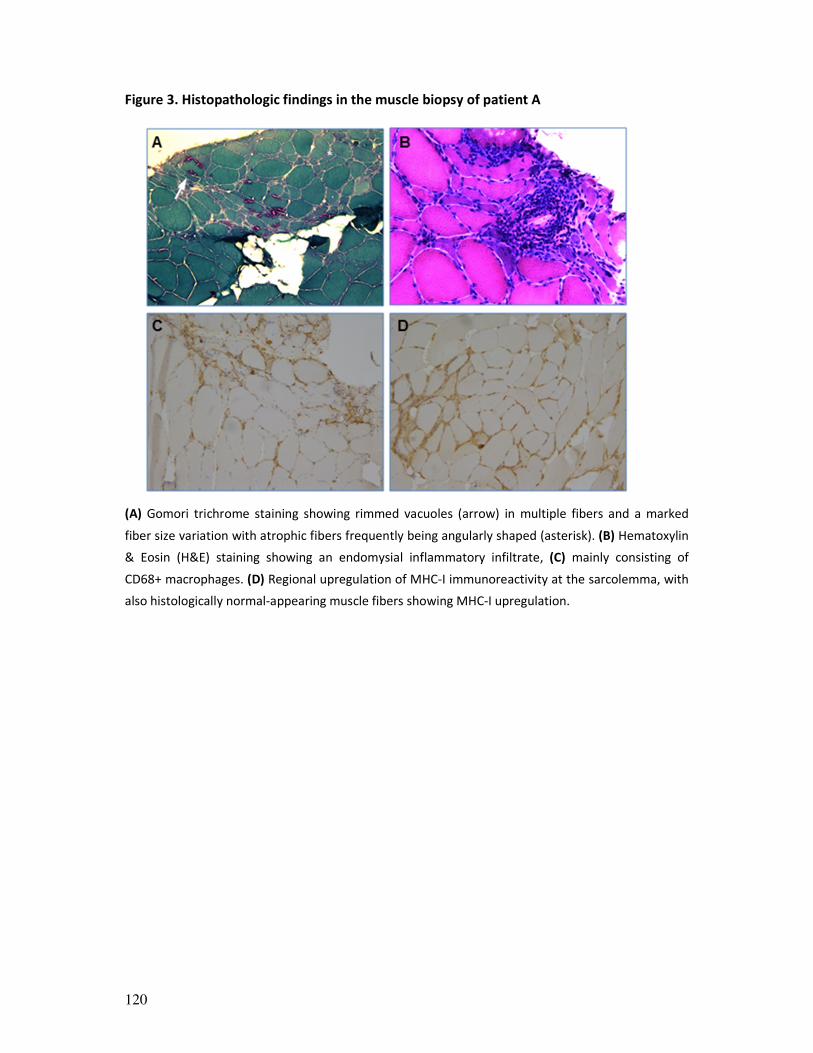
120
Figure 3. Histopathologic findings in the muscle biopsy of patient A
(A) Gomori trichrome staining showing rimmed vacuoles (arrow) in multiple fibers and a marked
fiber size variation with atrophic fibers frequently being angularly shaped (asterisk). (B) Hematoxylin
& Eosin (H&E) staining showing an endomysial inflammatory infiltrate, (C) mainly consisting of
CD68+ macrophages. (D) Regional upregulation of MHC-I immunoreactivity at the sarcolemma, with
also histologically normal-appearing muscle fibers showing MHC-I upregulation.

121
Figure 4. Visualization and validation of the quantitative proteomics data
(A) Hierarchic clustering analysis (Euclidean algorithms) based on the proteomic data of patient A-E
and the three control individuals. Color codes of the intensities corresponding to the values that
were normalized to the median across the complete dataset are shown below the heatmaps. (B)
Volcano plot analysis showing significantly dysregulated proteins (false discovery rate = 0.05, S0 =
0.1) between patient B-E and the three control individuals. The vertical axis corresponds to statistical
significance (−Log p), the horizontal axis shows the average fold change between patients and

122
control individuals (difference in Log2 values). (C) Among up- or downregulated proteins, PGAM2,
LMNB1 and MFF were validated using western blot analysis. (D) Figures showing iTRAQ ratios on the
left and protein expression data according to western blot analysis on the right for PGAM2, LMNB1
and MFF respectively. PC = pooled controls; C = patient C; B = patient B; A = patient A.
Table 1. Characteristics of five patients with a VCP-related myopathy phenotype and three control individuals
Patient Patient A Patient B Patient C Patient D Patient E
Sex Male Male Male Male Female
AAO, y 29 58 53 58 58
VCP mutation p.Arg159His
(homozygous)
p.Arg159His p.Arg159His p.Gly125Asp p.Gly125Asp
Presenting
symptoms
Proximal weakness
LL
Proximal weakness
LL
Proximal weakness
LL
Proximal weakness
LL
Proximal weakness
LL
Biopsy (age, y) Myopathic, rimmed
vacuoles,
endomysial
infiltrates (31)
Myopathic, rimmed
vacuoles (64)
Myopathic, rimmed
vacuoles,
endomysial
infiltrates (57)
Myopathic,
endomysial
infiltrates, with
invasion of non-
necrotic muscle
fibers (58)
Myopathic,
endomysial
infiltrates, with
invasion of non-
necrotic muscle
fibers (59)
Biopsied muscle Quadriceps Quadriceps Quadriceps Quadriceps Quadriceps
Control Control 1 Control 2 Control 3
Sex Male Male Male
Age at present, y 58 66 63
Age at biopsy, y 53 58 59
Biopsied muscle Quadriceps Quadriceps Quadriceps
AAO = age at onset; y = years; LL = lower limbs.
124
Table 2. Clinical characteristics of patient A and B
Patients Patient A Patient B
Sex Male Male
Age at present, y 36 66
AAO, y 29 58
Presenting symptoms Proximal weakness LL Proximal weakness LL
Maximal motor capability Walking 300 m Walking 25 m
Walking aids None None
Stairs Cannot climb stairs since age 33 y Cannot climb stairs since age 65 y
Marked muscle cramping Yes No
Cardiac symptoms No No
Age at last examination, y 35 64
Weakness Proximal UL Yes No
LL Yes Yes
Distal UL Yes No
LL Yes Yes
Other Periscapular, paraspinal, abdominal
muscles
Paraspinal
Skeletal muscle atrophy LL, distal and proximal muscle groups Quadriceps, forearms
Scapular winging Yes, marked No
Fasciculations No No
Reflexes Absent in UL, normal PTR, absent
ATR
Absent in LL and UL
Pyramidal tract signs No No
Extrapyramidal signs No Mild bradykinesia, rigidity and
tremor of the right arm
MoCA 28/30 27/30
Serum CK (U/l) 1138 192
EMG (age, y) Mixed myopathic / neurogenic
features; spontaneous activity with
positive sharp waves (30)
Myopathic features; no
spontaneous activity (63)
Resting ECG Normal Normal
Echocardiography Normal Mild left ventricular hypertrophy
Holter monitoring Normal Normal
FVC (% predicted) 81 74
Bone scintigraphy Paget lesion of L2 and right ileum Normal
Urinary Pyridoline/creatinine,
pmol/µmol (normal values: 5.5 -
69.4 pmol/µmol)
170 51
Urinary Deoxypyridoline/creatinine,
pmol/µmol (normal values: 1.0 -
16.9 pmol/µmol)
33.2 11.5
y = years; AAO = age at onset; LL = lower limbs; UL = upper limbs; PTR = patellar tendon reflex; ATR =
achilles tendon reflex; MoCA = Montreal Cognitive Assessment; CK = creatine kinase; FVC = forced
vital capacity.
125
Table 3. Bioinformatic interpretation of the proteomics data
Canonical pathways p-value of overlap Z-score
Oxidative Phosphorylation 3.981E-20 -4.491
Mitochondrial Dysfunction 1.259E-19 NaN
Sirtuin Signaling Pathway 6.310E-17 3
Glycolysis I 3.981E-15 -3.464
Gluconeogenesis I 1.995E-13 -3.317
Epithelial Adherens Junction
Signaling 7.586E-10 NaN
Calcium Signaling 1.778E-08 0
Actin Cytoskeleton Signaling 1.905E-08 0.243
Remodeling of Epithelial
Adherens Junctions
2.344E-08 NaN
TCA Cycle II (Eukaryotic) 1.072E-07 -2.646
Significance A
outlier analysis
p-value UniProt
identifier
Protein name
ZFAND1 1.520E-14 Q8TCF1 AN1-type zinc finger protein 1
AK7 3.220E-10 Q96M32 Adenylate kinase 7
RAD17 3.220E-10 O75943 DNA damage checkpoint control protein RAD17
MYH4 6.220E-07 Q9Y623 Myosin-4
WDR33 1.310E-06 Q9C0J8 pre-mRNA 3' end processing protein WDR33
IGHV3-72 5.240E-06 A0A087WW89 Immunoglobulin heavy variable 3-72
PLN 6.800E-06 P26678 Cardiac phospholamban
LRMP 1.350E-05 Q12912 Lymphoid-restricted membrane protein
MYLK 5.960E-05 Q15746 Myosin light chain kinase, smooth muscle
TNNC1 8.140E-05 P63316 Troponin C, slow skeletal and cardiac muscles
HBB 9.530E-05 P68871 Hemoglobin subunit beta
ILF3 1.274E-04 Q12906 Interleukin enhancer-binding factor 2
HBA1 1.997E-04 P69905 Hemoglobin subunit alpha
CRKL 2.411E-04 P46109 Crk-like protein
MYH1 2.769E-04 P12882 Myosin-1
STK10 2.899E-04 O94804 Serine/threonine-protein kinase 10
MYL2 3.432E-04 P10916 MYL2 protein
TNNT1 4.847E-04 P13805 Troponin T, slow skeletal muscle
NDST1 5.615E-04 P52848 Bifunctional heparan sulfate N-deacetylase/N-
sulfotransferase 1
Upper panel: results of the IPA canonical pathway analysis of proteins significantly dysregulated
between the four patients harboring a heterozygous mutation in VCP (patients B-E) and three
control individuals. Filtering based on p-value of overlap. Lower panel: results of the Significance A
outlier analysis in Perseus comparing protein expression data of patient A and patients harboring a
heterozygous mutation in VCP (patients B-E).

126
REFERENCES
1. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease
pathogenesis. Journal of cell science 2014;127:3877-3883.
2. Evangelista T, Weihl CC, Kimonis V, Lochmuller H. 215th ENMC International Workshop VCP-related
multi-system proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands.
Neuromuscular disorders : NMD 2016;26:535-547.
3. Mehta SG, Khare M, Ramani R, et al. Genotype-phenotype studies of VCP-associated inclusion body
myopathy with Paget disease of bone and/or frontotemporal dementia. Clinical genetics 2013;83:422-
431.
4. Figueroa-Bonaparte S, Hudson J, Barresi R, et al. Mutational spectrum and phenotypic variability of VCP-
related neurological disease in the UK. Journal of neurology, neurosurgery, and psychiatry 2016;87:680-
681.
5. van den Boom J, Meyer H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and
Signaling. Molecular cell 2018;69:182-194.
6. Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016;583:64-77.
7. Ye Y, Tang WK, Zhang T, Xia D. A Mighty "Protein Extractor" of the Cell: Structure and Function of the
p97/CDC48 ATPase. Frontiers in molecular biosciences 2017;4:39.
8. Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked
to a novel mutation in the VCP gene. Neurology 2005;65:1304-1305.
9. van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to
VCP p.Arg159His. Neurology 2009;73:626-632.
10. Koppers M, van Blitterswijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic
lateral sclerosis. Neurobiology of aging 2012;33:837.e837-813.
11. Papadimas GK, Paraskevas GP, Zambelis T, et al. The multifaceted clinical presentation of VCP-
proteinopathy in a Greek family. Acta myologica : myopathies and cardiomyopathies : official journal of
the Mediterranean Society of Myology / edited by the Gaetano Conte Academy for the study of striated
muscle diseases 2017;36:203-206.
12. Segers K, Glibert G, Callebaut J, Kevers L, Alcan I, Dachy B. Involvement of peripheral and central
nervous systems in a valosin-containing protein mutation. Journal of clinical neurology (Seoul, Korea)
2014;10:166-170.
13. Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with
valosin-containing protein myopathy associated with Paget's disease of bone and frontotemporal
dementia. Neuromuscular disorders : NMD 2009;19:316-323.
14. Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited
neuromuscular disorders: past, present, and future. Journal of magnetic resonance imaging : JMRI
2007;25:433-440.
15. Johnson K, Topf A, Bertoli M, et al. Identification of GAA variants through whole exome sequencing
targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness. Orphanet journal of
rare diseases 2017;12:173.
16. Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis of
(prote)omics data. Nature methods 2016;13:731-740.
17. Kramer A, Green J, Pollard J, Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway
Analysis. Bioinformatics (Oxford, England) 2014;30:523-530.
18. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass
accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367-1372.
19. Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. Histone demethylase KDM5A regulates the
ZMYND8-NuRD chromatin remodeler to promote DNA repair. The Journal of cell biology
2017;216:1959-1974.

127
20. Straub V, Carlier PG, Mercuri E. TREAT-NMD workshop: pattern recognition in genetic muscle diseases
using muscle MRI: 25-26 February 2011, Rome, Italy. Neuromuscular disorders : NMD 2012;22 Suppl
2:S42-53.
21. Tasca G, Monforte M, De Fino C, Kley RA, Ricci E, Mirabella M. Magnetic resonance imaging pattern
recognition in sporadic inclusion-body myositis. Muscle & nerve 2015;52:956-962.
22. Kazamel M, Sorenson EJ, McEvoy KM, et al. Clinical spectrum of valosin containing protein (VCP)-
opathy. Muscle & nerve 2016;54:94-99.
23. Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden,
The Netherlands. Neuromuscular disorders : NMD 2013;23:1044-1055.
24. Ikenaga C, Kubota A, Kadoya M, et al. Clinicopathologic features of myositis patients with CD8-MHC-1
complex pathology. Neurology 2017;89:1060-1068.
25. Zlotogora J. Dominance and homozygosity. American journal of medical genetics 1997;68:412-416.
26. Al-Obeidi E, Al-Tahan S, Surampalli A, et al. Genotype-phenotype study in patients with valosin-
containing protein mutations associated with multisystem proteinopathy. Clinical genetics 2018;93:119-
125.
27. Kosik KS, Munoz C, Lopez L, et al. Homozygosity of the autosomal dominant Alzheimer disease
presenilin 1 E280A mutation. Neurology 2015;84:206-208.
28. Rudolf G, Suominen T, Penttila S, et al. Homozygosity of the Dominant Myotilin c.179C>T (p.Ser60Phe)
Mutation Causes a More Severe and Proximal Muscular Dystrophy. Journal of neuromuscular diseases
2016;3:275-281.
29. Durmus H, Ayhan O, Cirak S, et al. Neuromuscular endplate pathology in recessive desminopathies:
Lessons from man and mice. Neurology 2016;87:799-805.
30. Sela I, Milman Krentsis I, Shlomai Z, et al. The proteomic profile of hereditary inclusion body myopathy.
PloS one 2011;6:e16334.
31. Turakhiya A, Meyer SR, Marincola G, et al. ZFAND1 Recruits p97 and the 26S Proteasome to Promote
the Clearance of Arsenite-Induced Stress Granules. Molecular cell 2018;70:906-919.e907.
32. Rodriguez-Ortiz CJ, Flores JC, Valenzuela JA, et al. The Myoblast C2C12 Transfected with Mutant
Valosin-Containing Protein Exhibits Delayed Stress Granule Resolution on Oxidative Stress. The
American journal of pathology 2016;186:1623-1634.
33. Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause
multisystem proteinopathy and ALS. Nature 2013;495:467-473.
34. Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS. Structural basis of the interaction
between the AAA ATPase p97/VCP and its adaptor protein p47. The EMBO journal 2004;23:1030-1039.
35. Schutz AK, Rennella E, Kay LE. Exploiting conformational plasticity in the AAA+ protein VCP/p97 to
modify function. Proceedings of the National Academy of Sciences of the United States of America
2017.
36. Schuetz AK, Kay LE. A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife
2016;5.
37. Nalbandian A, Llewellyn KJ, Kitazawa M, et al. The homozygote VCP(R(1)(5)(5)H/R(1)(5)(5)H) mouse
model exhibits accelerated human VCP-associated disease pathology. PloS one 2012;7:e46308.
38. Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated
diseases: inclusion body myopathy with Paget's disease of bone, frontotemporal dementia, and
amyotrophic lateral sclerosis. Journal of molecular neuroscience : MN 2011;45:522-531.

128
SUPPLEMENTARY TABLES AND FIGURES
Data available from Dryad: table e-1, table e-2, table e-3, table e-4, figure e-1, figure e-2
(https://datadryad.org/review?doi=doi:10.5061/dryad.60fn581).
Large tables (table e-1, table e-3 and table e-4) are not included in this PhD thesis, but can be
consulted online.
Table e-2. Pattern of muscle weakness in upper and lower limbs for patient A and B
Patient Patient A Patient B
UL Proximal m. sternocleidomastoïdeus 5 5
m. trapezius 5 5
m. deltoïdeus 4 5
m. biceps brachii 3 5
m. triceps 5- 5
Distal Wirst extensors 5 5
Wrist flexors 5- 5
m. extensor digitorum 4- 5
m. flexor digitorum 5- 5
m. opponens pollicis 5 5
m. interossei 5- 5
LL Proximal Hip flexion 3 4
Knee flexion 3 4
Knee extension 4+ 4+
Distal m. gastrocnemius 5- 5
m. tibialis anterior 3 5-
m. tibialis posterior 5 5
m. peroneus longus 5- 5
m. extensor hallucis 2 3
Muscle strength testing of upper and lower limbs according to the Medical Research Council (MRC)
Scale for patients A and B at age 35 years and 64 years respectively. Muscle groups showing mild
weakness are marked in light grey (5-/5 or 4+/5), muscle groups showing moderate to severe
weakness (4 or less) in dark grey.
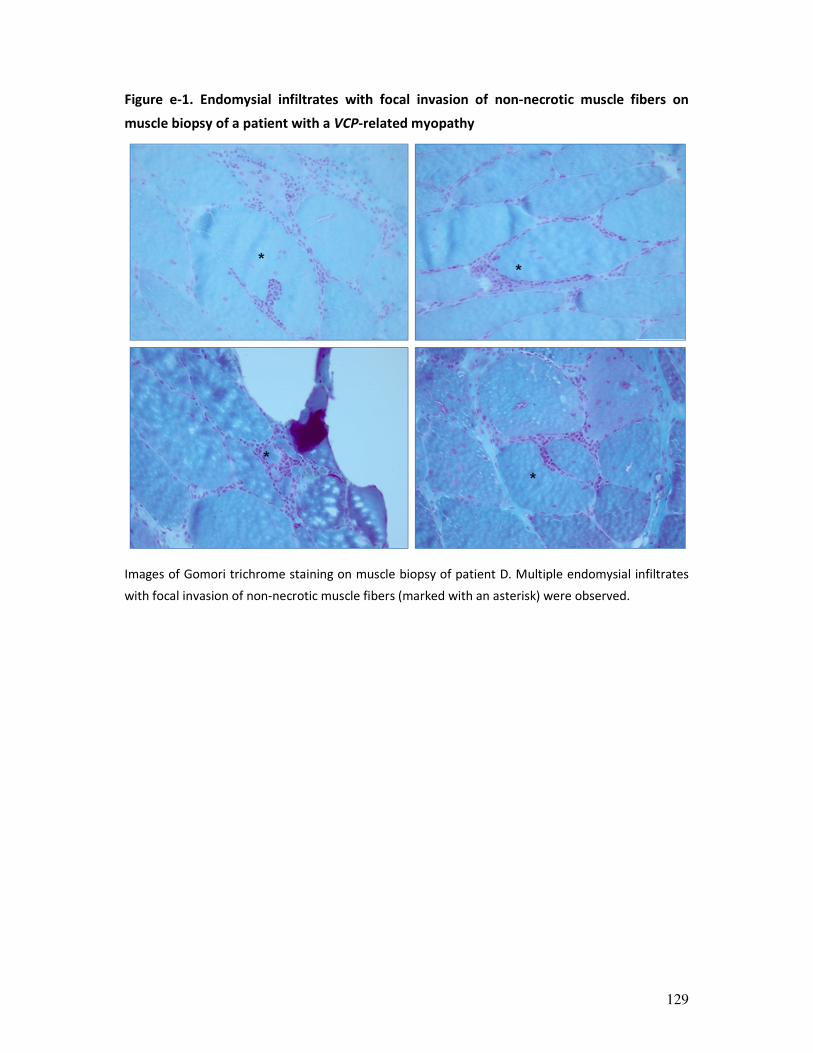
129
Figure e-1. Endomysial infiltrates with focal invasion of non-necrotic muscle fibers on
muscle biopsy of a patient with a VCP-related myopathy
Images of Gomori trichrome staining on muscle biopsy of patient D. Multiple endomysial infiltrates
with focal invasion of non-necrotic muscle fibers (marked with an asterisk) were observed.
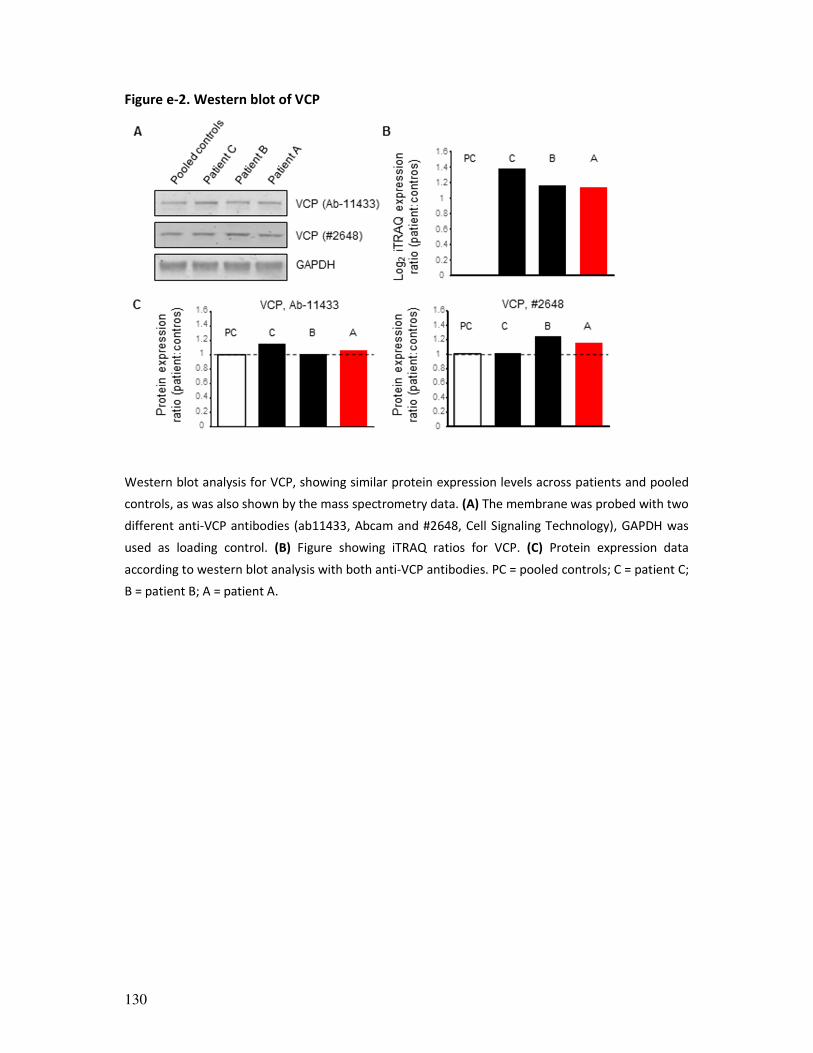
130
Figure e-2. Western blot of VCP
Western blot analysis for VCP, showing similar protein expression levels across patients and pooled
controls, as was also shown by the mass spectrometry data. (A) The membrane was probed with two
different anti-VCP antibodies (ab11433, Abcam and #2648, Cell Signaling Technology), GAPDH was
used as loading control. (B) Figure showing iTRAQ ratios for VCP. (C) Protein expression data
according to western blot analysis with both anti-VCP antibodies. PC = pooled controls; C = patient C;
B = patient B; A = patient A.

131

132

133
CHAPTER 5
Ageing signatures and disturbed muscle regeneration
and differentiation in sporadic inclusion body myositis
Willem De Ridder, Abdelkrim Azmi, Bob Asselbergh, Peter De Jonghe, Peter Van Den Bergh,
Vincent Mouly, Jan L. De Bleecker, Stuart Maudsley, Jonathan Baets
[Manuscript in preparation]

134
ABSTRACT
Objective: To study sporadic inclusion body myositis (sIBM) disease signatures by an
unbiased proteomic approach with the ultimate aim to identify potential novel ‘key
regulators’ of currently unknown disease mechanisms.
Methods: Proteins of muscle lysates of 28 sIBM patients and 28 control individuals were
analysed by a state-of-the-art mass spectrometry approach. To study the identified
potential novel key regulator KDM5A, immunohistochemical studies were performed on
patient and muscle and western blotting on a myoblast lysate of a healthy 53-year-old male
control individual.
Results: 617 proteins were significantly dysregulated between sIBM patients and control
individuals. The proteomic dataset most strongly reflected inflammatory signatures,
changes in cellular energy metabolism and altered myogenesis in sIBM muscle. The top hit
in the upstream regulator analysis predicted to be activated was KDM5A, a histone
demethylase involved in the DNA damage response (DDR) and (myogenic) differentiation
and interconnecting core sIBM disease signatures. We showed expression of KDM5A in a
myoblast lysate of a control individual and marked expression of KDM5A in myogenin-
positive regenerating fibres in sIBM muscle tissue.
Conclusions: This unbiased proteomic study provides unique insights in the proteomic
landscape of sIBM, captivating known core features of sIBM pathomechanisms as well as
highlighting strong signatures pointing towards selective failure of myogenesis. Failure of
muscle differentiation during regeneration appears to be linked to the DDR through KDM5A,
as well as to other interconnected ageing-associated signatures of sIBM.

135
INTRODUCTION
Sporadic inclusion-body myositis (sIBM) is the most common idiopathic inflammatory
myopathy (IIM) among patients over 50 years of age.1 In many of its clinical aspects
however sIBM differs from other IIM, particularly regarding its slowly progressive, strictly
late-onset nature and the striking, selective and often asymmetric involvement of distal
muscle groups (e.g. long finger flexors).1 Moreover, IIM typically respond well to immune-
suppression and a staged approach to immune-modulating therapy is the cornerstone of
treatment. In sharp contrast, trials using immune-modulating drugs in sIBM failed to show
effect.2
Different mechanisms underlying sIBM pathogenesis have been implicated but their
interplay remains unclear. The central question remains if sIBM is in origin an inflammatory
or degenerative disorder.3,4 Diagnosis of sIBM is often delayed and criteria rely on
characteristic clinical features and muscle biopsy findings typically showing inflammatory
infiltrates and MHC-I upregulation, as well as degenerative features under the form of
rimmed vacuoles and protein accumulation.5
Mass-spectrometry-based technologies have evolved dramatically, and currently available
platforms provide unprecedented insights into different facets of the proteome.6
Proteomics studies have increasingly been applied in muscle disorders as patient diseased
tissue is readily available.7
In this study we aimed to perform an unbiased dissection of sIBM disease signatures to
identify potential novel ‘key regulators’ of disease mechanisms. The proteomic dataset
sheds light on core features of sIBM pathomechanisms and inflammatory signatures,
changes in cellular energy metabolism and altered myogenesis in particular.
METHODS
Standard protocol approvals, registrations, and patient consents
Ethical approval was granted by the relevant local Ethical Committees of the participating
centres. All participants provided written informed consent prior to participation in the
study.
Patients
28 sIBM patients (12 women, 16 men, mean age 69.6±8.5 years at time of biopsy, range 55–
85 years) for whom freshly frozen muscle tissue was available were included in this study.
Clinical and myopathological details were rigorously documented, assuring homogeneity of
sIBM muscle specimens. Diagnosis of sIBM was based on the ENMC criteria.5 All patients
were ambulatory at the time of biopsy. Muscle biopsies were critically reassessed and for all

136
patients, endomysial inflammatory infiltrates and rimmed vacuoles were visualized; typical
15-18 nm filaments were documented for most patients, protein accumulation evident by
SMI-31-, TDP43- or p62-positive aggregates was shown for the remaining. 28 control
individuals, biopsied for subjective myalgia, yet for whom no clinical, morphological or
electrophysiological abnormalities had been identified, were matched based on biopsied
muscle, sex and age (12 women, 16 men, age 63.1±11.0 years at time of biopsy, range 49–
88 years). A brief overview of core clinical details of patients and controls is provided in
supplementary table 1.
Skeletal muscle specimens
Muscle biopsies of quadriceps muscle were obtained from quadriceps, tibialis anterior or
deltoid muscles and were initially analysed following standard histological and IHC light
microscopy and electron microscopy (EM) protocols. The complete set of muscle specimens
was used for unbiased proteomic analyses, further focused IHC experiments were
performed on a selection of muscle biopsies.
Sample preparation for iTRAQ labeling and mass spectrometry analysis
Proteins were extracted from muscle biopsy specimens in a buffer containing 4% SDS, 100
mM TCEP, 50 mM Tris pH 7.8. Lysates were heated at 95°C for 5 minutes. The extracted
proteins were precipitated with trichloroacetic acid (TCA) and re-solubilized in 8 M urea, 2
M thiourea, 0.1% SDS in 50 mM triethylammonium bicarbonate (TEAB). After measuring
protein concentrations using the RCDC kit (Bio-Rad), equal amounts of proteins from each
lysate were reduced and alkylated with TCEP (tris-2-carboxyethyl phosphine) and MMTS (5-
methyl-methanoethiosulphate), respectively, followed by trypsin digestion. Peptides from
each sample were labelled using iTRAQ reagents 8plex (Sciex) according to the
manufacturer's instructions. The mixed peptides were separated on an offline 2D-liquid
chromatography (LC) system (Dionex, ULTIMATE 3000, ThermoScientific), consisting of a 15
cm strong cationic exchange column and a 25 cm nano-RP C18 column. The nano-LC was
coupled online to a QExactive™-Plus Orbitrap (ThermoScientific) mass spectrometer (MS).
Bioinformatic analysis of mass spectrometry data
The generated raw data from the MS were processed with the Proteome Discoverer 2.1
(PD2.1) software (ThermoScientific). For protein identification, the search engine Sequest
HT was used against the human UniProt/SwissProt database with a false discovery rate
(FDR) of less than 1%. The quantitative proteomics data were then further statistically
analysed with the Perseus software (version 1.6.1.1).8 Log2-transformed scaled abundance

137
values generated by the PD2.1 software were normalized by subtraction of the median
value of the respective column. The complete list of identified proteins with the original
scaled abundance values is available on request. Principal component analysis (PCA) and
hierarchic clustering analyses using Euclidean algorithms were performed on 100% valid
values, for hierarchic clustering after filtering ANOVA-significantly dysregulated proteins
(FDR = 0.01). Further analyses were performed after selecting proteins that were detected
in at least 70% of the samples. A volcano plot analysis, assessing statistical significance (t-
test) together with fold change was performed to identify significantly dysregulated proteins
between patients and controls: a permutation-based FDR cut-off was determined with 250
randomisations and S0 = 0.1 (default). The downstream canonical pathways analysis
(filtering based on p-value of overlap) and the upstream regulator analysis (filtering based
on molecule type (genes, RNAs and proteins) and |Z-score| ≥ 2 and p-value of overlap ≤
0.05) as a causal analysis approach were applied on this set of dysregulated proteins, using
the Ingenuity Pathway Analysis (IPA) software (QIAGEN).9
Human myoblast culture
Human myoblasts isolated from muscle from a 53-year-old healthy man and immortalized
as previously described, were provided anonymously by MYOBANK, a tissue bank affiliated
to EUROBIOBANK.10 Human myoblasts were cultured in Skeletal Muscle Cell Media
(PromoCell, no. C-23060) supplemented with 20% FBS at 37 °C in a humidified atmosphere
containing 5% CO2.
Immunoblotting
Protein extracts of muscle tissue specimens used for MS analysis and myoblasts pellets were
subjected to western blotting. Proteins were loaded and separated on 4–12 % NuPAGE® Bis-
Tris gels (Life Technologies) and transferred on a polyvinylidene difluoride membrane (PVDF,
Hybond P, Amersham Biosciences). Membranes were probed with rabbit anti-KDM5A (1 in
1000, Cell Signaling Technology, #3876). Immunodetection was performed using host-
specific secondary antibodies conjugated with horseradish peroxidase (HRP) and the ECL-
plus chemiluminescent detection system (Thermo Scientific). Western blot results were
visualized using the Amersham™ Imager 600 digital imaging system and quantified with
ImageQuant™ TL software (GE Healthcare Life Sciences).
Immunohistochemistry
Frozen 7-μm sections of skeletal muscle biopsies of four patients and four controls were
mounted on Superfrost Plus glass slides (Thermo Fisher Scientific). Sections were air-dried

138
and encircled using the ImmEdge pen to create a hydrophobic barrier. Muscle sections were
fixed in ice cold aceton during 10 minutes. Muscle sections were initially used for enzymatic
(peroxidase-anti-peroxidase (PAP)) detection of rabbit anti-KDM5A (1 in 50, Cell signal,
#3876) following standard IHC protocols.
Next, immunofluorescence (IF) detection methods were applied for double stainings.
Sections were incubated with 100μl blocking solution containing 5% goat serum and 1% BSA
in TBS for 1 hour at room temperature. Subsequently, blocking solution was removed and
primary antibodies diluted in blocking solution were added and incubated overnight at 4°C.
Antibody solution was removed and sections were washed 3 times for 10 minutes, with Tris-
Buffered Saline (TBS). Secondary antibodies diluted in blocking solution were added to the
cover glasses and incubated for 1 hour at room temperature in the dark. The following
secondary antibodies were employed for detection of the signal: Alexa Fluor 594-conjugated
goat anti-rabbit (A11037, Life Technologies) and Alexa Fluor 488-conjugated goat anti-
mouse (A11001, Life Technologies). The antibody was removed and sections were washed
twice with TBS for 5 minutes. Slides were quenched in a solution of 0.1% Sudan Black/70%
ethanol for 10 minutes. Sections were washed twice with TBS for 5 minutes and nuclei were
counterstained with Hoechst solution (1:20.000 in TBS) for 5 minutes. The sections were
then mounted with DAKO onto microscope slides and stored at 4°C to allow the DAKO to
set. Primary antibodies used for IF stainings in this study were: rabbit anti-KDM5A (1 in 50,
Cell signal, #3876), mouse anti-myogenin (1 in 50, Santa Cruz, sc-12732), mouse anti-CD4 (1
in 50, Dako, MT310), mouse anti-CD8 (1 in 500, Dako, M707), mouse anti-CD68 (1 in 300,
Dako, M0718). Image stacks were acquired on a Zeiss LSM700 laser scanning confocal
microscope using a Plan-Apochromat 63x/1.40 or Plan-Neofluar 40x/1.3 objective and
maximum intensity projections were made in the Fiji distribution of ImageJ.
Data availability
Anonymized data not published within the article will be shared upon request by any
qualified investigator.
RESULTS
Proteomic investigation of sIBM-related disease signatures in skeletal muscle
To investigate sIBM pathomechanisms in an unbiased way, an exploratory proteomic study
was performed on skeletal muscle tissue lysates of 28 sIBM patients and 28 controls. In
total, 3057 iTRAQ labeled proteins were detected across the 56 samples, of which 1283 in at
least 70% of the samples and 832 in all.

139
The dataset was first visualized by means of hierarchic clustering analyses and PCA (figure
1A-B). The four outlier patients clustering in between controls, as shown by the heatmap
(figure 1A), are male patients; for three of them a protein extract of quadriceps muscle was
used and for the other of anterior tibial muscle. Except for these same four patients,
patients and controls clearly segregated based on component 1 and component 2 of the
PCA analysis, which accounted for 62.1% and 7.5% of proteins, respectively (figure 1B).
Based on these analyses, patients appeared not to selectively cluster based on gender,
muscle type or age at biopsy, suggesting strongly similar disease patterns across different
types of samples.
Subsequently, to create a general appreciation of sIBM disease signatures in patient’s
muscle, a volcano plot was generated to compare protein expression data of the 28 patients
and the 28 controls, followed by functional annotation of significantly dysregulated
proteins. Based on the volcano plot analysis (FDR = 0.01, S0 = 0.1), 617 proteins were
significantly dysregulated, of which 313 downregulated, in patients as compared to control
individuals (figure 2A). The alarmins S100A6 and S100A411 and the Proteasome activator
complex subunit 1 and 2 (PSME1 and PSME2), implicated in immunoproteasome assembly
and required for efficient antigen processing,12 were among the top 10 most consistently
dysregulated proteins (highest –Log p value). Based on p-value of overlap, the top 20 of
enriched pathways identified in the canonical pathway analysis mainly reflected changes in
metabolic pathways and cytoskeletal reorganization (figure 2B). Within the top 10 activated
and inhibited upstream regulators as predicted by IPA (table 1), multiple regulators
reflected: 1) inflammatory signatures of sIBM pathology (predicted activation of OSM, IL6
and IL4); 2) changes in cellular energy metabolism (predicted inhibition of PPARGC1A,
PPARGC1B, IGF1R, INSR, ESRRA, HBA1/HBA2 and TSC2 and activation of RICTOR); 3) altered
myogenesis (NRG1, MAP4K4, predicted to be activated and MEF2C and RB1 predicted to be
inhibited).
RICTOR and TSC2 are closely linked to with mTOR signalling, a central regulator of cellular
metabolism, growth and survival. MEF2C, predicted to be inhibited, is a cooperating factor
of myogenin, one of the key myogenic regulatory factors.13 NRG1, predicted to be activated
is a growth factor produced by both skeletal muscle and peripheral nerves, which has a role
in muscle homeostasis, satellite cell survival and neuromuscular junction gene expression.14
MAP4K4, also predicted to be activated, has been identified as a suppressor of skeletal
muscle differentiation.15 RB1 is a key regulator of cell division and RB1 inactivation, as
predicted based on our analysis of this dataset, was shown to expand satellite cells and
immature myoblast, at the cost of a deficit of muscle fibre formation and maturation in case
of sustained loss.16 The top hit in the analysis, showing the highest activation Z-score, was

140
however KDM5A, a histone demethylase involved in the DNA damage response (DDR)17,
with prominent roles in transcriptional regulation and cell differentiation.18
KDM5A is expressed in human skeletal muscle and myoblasts
In an unbiased way, KDM5A was prioritized as potential novel key regulator of sIBM
pathomechanisms for further studies in human skeletal muscle. Statistically, KDM5A was
the top predicted key regulator based on the upstream analysis, yet it was also biologically a
very relevant ‘candidate regulator’, interconnecting with many of the other top 20 predicted
key regulators. More specifically, KDM5A has been proposed to have an important role in
myogenic differentiation. In mouse embryonic fibroblasts differentiated to myoblasts
(through expression of MYOD), Kdm5a catalytic activity was shown to be involved in
inhibition of myogenic differentiation.19 Highly illustrative for the potential novelty of this
putative key regulator, we first had to prove that KDM5A is indeed expressed in human
skeletal muscle, as this had not yet been shown directly. When performing western blot
analyses for KDM5A in whole skeletal muscle lysates of controls and sIBM, used for MS
analysis, we failed to show a specific band, which however did succeed in human fibroblast
and lymphoblast cell lysates of a control individual (supplementary figure 1). As this protein
is mainly localized to the nucleus, we then tried to directly show KDM5A expression by
enzymatic IHC stainings in muscle of healthy control individuals, but again we did not
observe any specific signal. Based on literature,19 we subsequently hypothesized that
KDM5A may be localized specifically to (the nucleus of) immature, regenerating myoblasts.
For this purpose, we performed western blotting of KDM5A on a myoblast lysate of a 53-
year-old healthy control individual and indeed succeeded in showing a KDM5A specific band
(supplementary figure 1).
KDM5A expression in sIBM muscle is localized to myogenin positive regenerating fibres
We then reasoned that KMD5A expression in sIBM muscle would be specifically localized to
(activated) regenerating muscle fibres, which would explain the absence or at least very low
expression of KDM5A in healthy skeletal muscle. Given that myogenin, a marker of
myogenic cell differentiation, was shown to be strongly upregulated in s-IBM muscle,20 we
chose myogenin to show localization of KDM5A to immature muscle fibres and myogenic
nuclei in particular. We indeed showed that the highest signal of KDM5A was localized to
numerous small-diameter myogenin-positive regenerating muscle fibres in sIBM muscle
(figure 3A-B). The localization of the KDM5A-signal was strictly nuclear. All myogenin-
positive muscle fibres showed KDM5A-expression, yet we also noticed a less intense KDM5A
signal in myogenin-negative nuclei of cells localized to the endomysium, which were

141
immunohistochemically characterized as (CD4-, CD8- and CD68-positive) infiltrating
inflammatory cells (figure 3C-D). In control muscle samples, KDM5A immunoreactivity was
only detected in very scarce myogenin-positive nuclei.
DISCUSSION
In the present study, data from an unbiased, very sensitive proteomic approach shed unique
insights in the proteomic landscape and disease signatures of sIBM. This highly powered
dataset captivated known core features of sIBM pathomechanisms and allowed us to
prioritize KDM5A as a potential novel key disease regulator.
Previous studies in sIBM probably focused often on downstream phenomena of sIBM
pathology and the crucial pivotal mechanism early in the disease risks to remain hidden.
Clearly both inflammatory and degenerative features in sIBM do not fully explain the
disease and are perhaps often rather consequences than causes. The focus of this study was
to identify potential novel proximal regulators of disease, likely hidden in the deep
proteome, irrespective of any inflammatory or degenerative theory. Adding a layer of
complexity to the analysis of the sIBM proteomic dataset by means of conducting causal
(upstream) analyses was therefore crucial with regard to an unbiased search for potential
novel upstream regulators of disease, which are likely hidden in the deep proteome. For
further validation experiments, we focused on KDM5A as (biologically relevant) statistical
top hit, yet importantly, top predicted key regulators are highly interconnected.
Besides inflammatory changes and changes in cellular energy metabolism, the upstream
regulator analysis in IPA reflected strong signatures linked to altered myogenesis. The
predicted activation of NRG1 and MAP4K4 and inhibition of MEF2C and RB1 appears to
indicate a strong inhibition of differentiation of immature, regenerating muscle fibres.13-16
Skeletal muscle regeneration is facilitated by muscle stem cells (MuSCs), which reside in a
‘niche’ in muscle tissue, a membrane-enclosed anatomical space between the basal lamina
and plasma membrane of a mature myofiber.21 MuSCs become activated by stimuli such as
myofibre damage, cytokines and growth factors and undergo multiple rounds of self-
renewing divisions that are essential to keep their regenerative function.21 The remaining
myoblasts upregulate myogenic transcription factors and differentiate to form
multinucleated myofibers.22 Under physiological conditions, MuSCs do not need to migrate
away from their natural niche in order to achieve the task of adding nuclei to host
myofibres. Early cell-cycle withdrawal of the self-renewal cell prevents extensive shortening
of telomere length and associated accelerated senescence. Under situations of pronounced
muscle degeneration MuSCs are however recruited from neighbouring intact fibres too.22

142
Blocking of small-sized differentiating myoblasts in an immature stage could potentially
explain strong upregulation of myogenin in sIBM muscle.20 Pronounced and sustained RB1
inhibition could on the other hand at least partially explain this apparent aberrant cell cycle
re-entry of MuSC, leading to proliferation of immature myoblasts, though with a deficit of
muscle fibre formation and maturation.16,23
This elegantly brings us to KDM5A, the prioritized predicted upstream regulator for which
further experiments were performed in skeletal muscle tissue of sIBM patients. KDM5A is a
RB1-interacting protein, counteracting its role in promoting differentiation. The H3K4
(lysine) demethylase KDM5A was proposed to represent an epigenetic regulator controlling
the metabolic program that synchronizes energy homeostasis with a differentiation signal.19
As such, this protein would interconnect core sIBM disease signatures and would be a very
relevant mediator related to observed perturbations of muscle repair.19 We indeed showed
marked expression of KDM5A in all myogenin-positive nuclei in sIBM muscle. An extensive
evaluation of KDM5A expression in other IIM and other degenerative muscle disorders was
beyond the scope of this study.
Hitherto, we focused on the role of KDM5A in myogenesis, however its role in the DDR is
also highly relevant with regard to sIBM pathomechanisms. DNA damage with DNA double
strand breakes leading to myonuclear breakdown has been shown in sIBM.24 Transient and
controlled DNA damage in muscle fibres leads to activation of genes involved in myogenic
differentiation under physiological conditions; uncontrolled DNA damage however results in
pathological muscle differentiation and ageing.25 The role of KDM5A therefore particularly
reflects the close interplay of DNA damage, the DDR, cell cycle regulation, regulation of
differentiation signals and energy homeostasis in skeletal muscle.26
As of yet, ageing still is the only clinical factor that is strongly correlated with sIBM,3 yet core
features associated with ageing have not yet been systematically studied. DNA damage in
muscle fibres, induced by factors such as oxidative stress tends to accumulate with age.25
Genomic instability in general evidently is one of the central hallmarks of ageing, together
with epigenetic changes, telomere attrition and loss of proteostasis.27 Nuclear DNA damage
in sIBM has been described,24 yet has not been studied mechanistically. Mitochondrial DNA
damage and particularly protein dyshomeostasis have evidently been studied more
extensively.28 Early telomere shortening was shown in ‘young’ sIBM cultures, reflecting
increased ageing in these samples.29 Deregulated nutrient sensing, mitochondrial
dysfunction (both also reflected in our dataset) and cellular senescence represent
‘antagonistic’ hallmarks of ageing.27 Finally, ageing results in the ‘integrative’ hallmarks stem
cell exhaustion and altered intercellular communication.27 A typical characteristic of the

143
latter is ‘inflammaging’, with activation of NFkB, the NLRP3 inflammasome and other
proinflammatory pathways, finally leading to increased production of IL-1β, tumor necrosis
factor and interferons.27 Accumulation of intracellular damage in stem cells during ageing
particularly contributes to the functional decline, epigenome stability and deregulation of
developmental pathways in these cells crucial for tissue regeneration. This appears to be a
major driver of epigenetic drift, resulting in ageing-associated decline in the tissue function
and ageing-related disease.30
The focus of this study was to identify potential novel upstream regulators of disease,
irrespective of any inflammatory or degenerative theory, yet the proteomic dataset also
nicely captivates many known inflammatory and degenerative alterations in sIBM muscle.
Cytotoxic (CD8+) T cells appear to play a central role in inflammatory signatures, with
chemokines and cytokines, such as those identified in the analysis of the present dataset,
playing an important local role.31 The exact interplay of inflammatory and degenerative
signatures in sIBM remains unclear, yet might be related to strong upregulation of alarmins,
such as S100A6 and S100A4, or of the immunoproteasome, reflected by upregulation of
PSME1 and PSME2, proteins being directly identified in our proteomic dataset. Alarmins
constitute a group of proteins that can exert beneficial cellular housekeeping functions in
response to tissue damage, but their activation may result in deleterious uncontrolled
inflammation.11 It has already been suggested that HMGB1, another alarmin, might
constitute a link between inflammatory and degenerative signatures.32 Furthermore, the
immunoproteasome is required for efficient antigen processing by degrading intracellular
proteins to generate a source of peptides with MHC-I binding affinity.12 Moreover,
upregulation of the immunoproteasome was shown in muscle of different IIM, including
sIBM, and appears to be a key mediator of myokines and MHC-I expression.33
We propose that ageing of muscle fibres (MuSCs in particular) and their milieu, genetic risk
factors and selective, still to be identified epigenetic changes lead to ageing-associated
skeletal muscle dysfunction and especially primary failure of muscle regeneration in sIBM,
resulting in highly interconnected degenerative as well as inflammatory changes in skeletal
muscle. The DDR and KDM5A might play a central role in sIBM pathomechanisms or at least
represent very relevant factors with regard to the age-related nature of the disease. We
suggest that further studies in sIBM should consider the ageing aspect of sIBM, irrespective
of any inflammatory or degenerative theory.

144
The uncertainty concerning key sIBM pathomechanisms complicates the design of reliable
experimental cell- or animal-models, emphasizing the relevance of well-designed
experiments using human disease tissue. Prior proteomic studies had already shown the
strength of an unbiased proteomic approach in sIBM research, e.g. by identification of as a
new risk allele in sIBM.34 MS-based technologies have evolved dramatically over time and
our dataset is highly powered, containing the largest proteomic dataset in sIBM research so
far.6
This study shares its most critical weakness with most other current studies in sIBM
research, namely the fact that we can still only predict what pathomechanisms are really the
upstream drivers of the disease. Our observations remain indirect, yet this dataset is
eminently powered for an unbiased snapshot of such disease signatures very proximal to
the presumed etiology. Studies using induced pluripotent stem cells (iPSC) or even muscle
organoids have the potential to overcome this issue. Furthermore, studying the evolution of
proteomic signatures over time (and disease progression), could yield additional valuable
insights. We were unable to reliably stratify patients based on disease stage (there is e.g. no
validated scoring system for the severity of muscle biopsy changes) and we did not have
access to serial muscle biopsies. Strikingly however, sIBM disease signatures reflected in our
dataset appear to be very robust, as they were strongly similar across gender, muscle type,
disease duration and age at biopsy.
Due to lack of an effective therapy in sIBM, steady decline of muscle strength results in loss
of ambulation and ultimately reduced life expectancy. Targeting inflammatory pathways in
sIBM showed unsuccessful up to date. Similarly, targeting degenerative pathways such as
heat shock or myostatin pathways has not yet proven to be effective either.2 sIBM research
is in need of new therapeutic strategies based on novel and sufficiently strong hypotheses
and KDM5A as well as the DDR might represent relevant and druggable targets. Inhibitors of
KDM5 proteins are being developed in cancer research so there might also be an
opportunity for rapid trial readiness through drug repurposing.35,36
In summary, this unbiased proteomic study provides unique insights into the proteomic
landscape of sIBM, captivating known core features of sIBM pathomechanisms as well as
highlighting strong signatures pointing towards selective failure of myogenesis. Failure of
muscle differentiation during regeneration appears to be linked to the DDR through KDM5A,
as well as other interconnected ageing-associated signatures of sIBM.

145
Acknowledgements: The authors thank the patients and families for their cooperation and
contributions; Natacha Camacho and Safoura Jafary, Laboratory of Neuromuscular
Pathology, Institute Born-Bunge, University of Antwerp, for laboratory assistance.
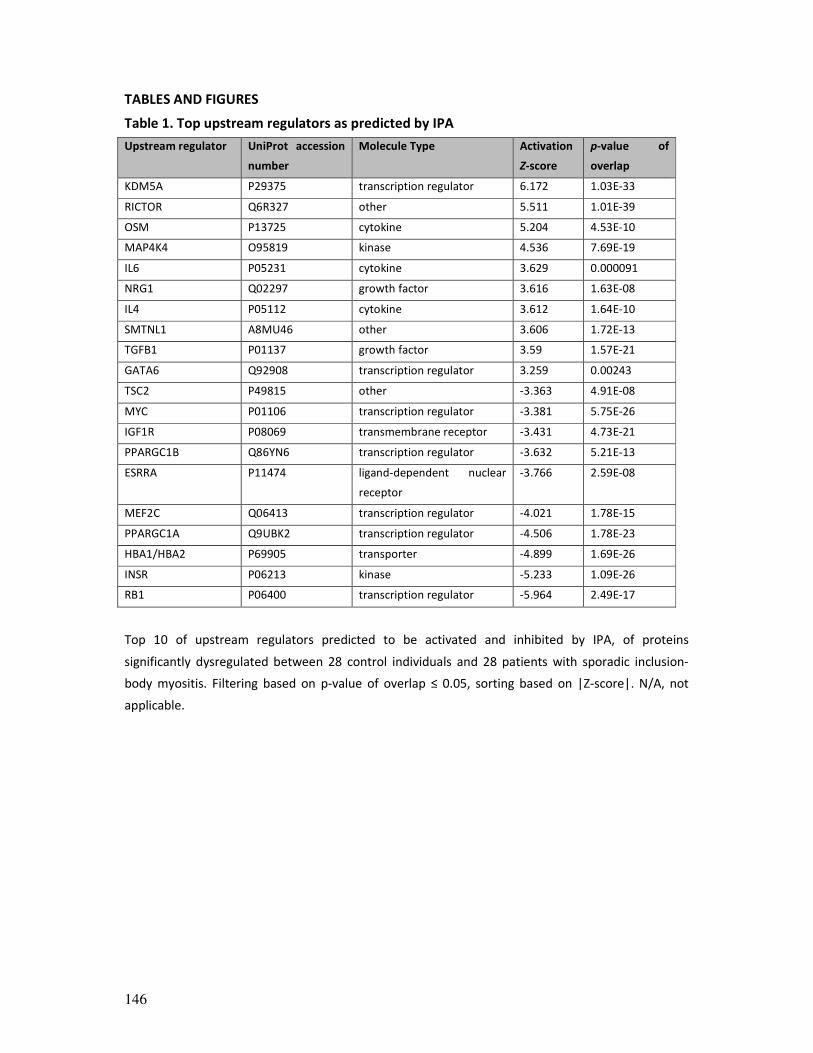
146
TABLES AND FIGURES
Table 1. Top upstream regulators as predicted by IPA
Upstream regulator UniProt accession
number
Molecule Type Activation
Z-score
p-value of
overlap
KDM5A P29375 transcription regulator 6.172 1.03E-33
RICTOR Q6R327 other 5.511 1.01E-39
OSM P13725 cytokine 5.204 4.53E-10
MAP4K4 O95819 kinase 4.536 7.69E-19
IL6 P05231 cytokine 3.629 0.000091
NRG1 Q02297 growth factor 3.616 1.63E-08
IL4 P05112 cytokine 3.612 1.64E-10
SMTNL1 A8MU46 other 3.606 1.72E-13
TGFB1 P01137 growth factor 3.59 1.57E-21
GATA6 Q92908 transcription regulator 3.259 0.00243
TSC2 P49815 other -3.363 4.91E-08
MYC P01106 transcription regulator -3.381 5.75E-26
IGF1R P08069 transmembrane receptor -3.431 4.73E-21
PPARGC1B Q86YN6 transcription regulator -3.632 5.21E-13
ESRRA P11474 ligand-dependent nuclear
receptor
-3.766 2.59E-08
MEF2C Q06413 transcription regulator -4.021 1.78E-15
PPARGC1A Q9UBK2 transcription regulator -4.506 1.78E-23
HBA1/HBA2 P69905 transporter -4.899 1.69E-26
INSR P06213 kinase -5.233 1.09E-26
RB1 P06400 transcription regulator -5.964 2.49E-17
Top 10 of upstream regulators predicted to be activated and inhibited by IPA, of proteins
significantly dysregulated between 28 control individuals and 28 patients with sporadic inclusion-
body myositis. Filtering based on p-value of overlap ≤ 0.05, sorting based on |Z-score|. N/A, not
applicable.

147
Figure 1. Visualization of the quantitative proteomic dataset
(A) Hierarchical clustering analysis (with Euclidean algorithms) of the proteomic data of muscle
tissue of 28 control individuals and 28 patients with sporadic inclusion-body myositis. Colour codes
of the intensities corresponding to the values that were normalized to the median across the
complete dataset are shown below the heatmap. On top of the heatmap, patients are marked in
pink, control individuals in blue. 12 patients and control individuals are female (sIBM1-12 and
control 1-12), 16 are male (sIBM13-28 and control 13-28). (B) Principle component analysis of
protein expression data of controls (black squares), female (green squares) and male patients (red
squares).
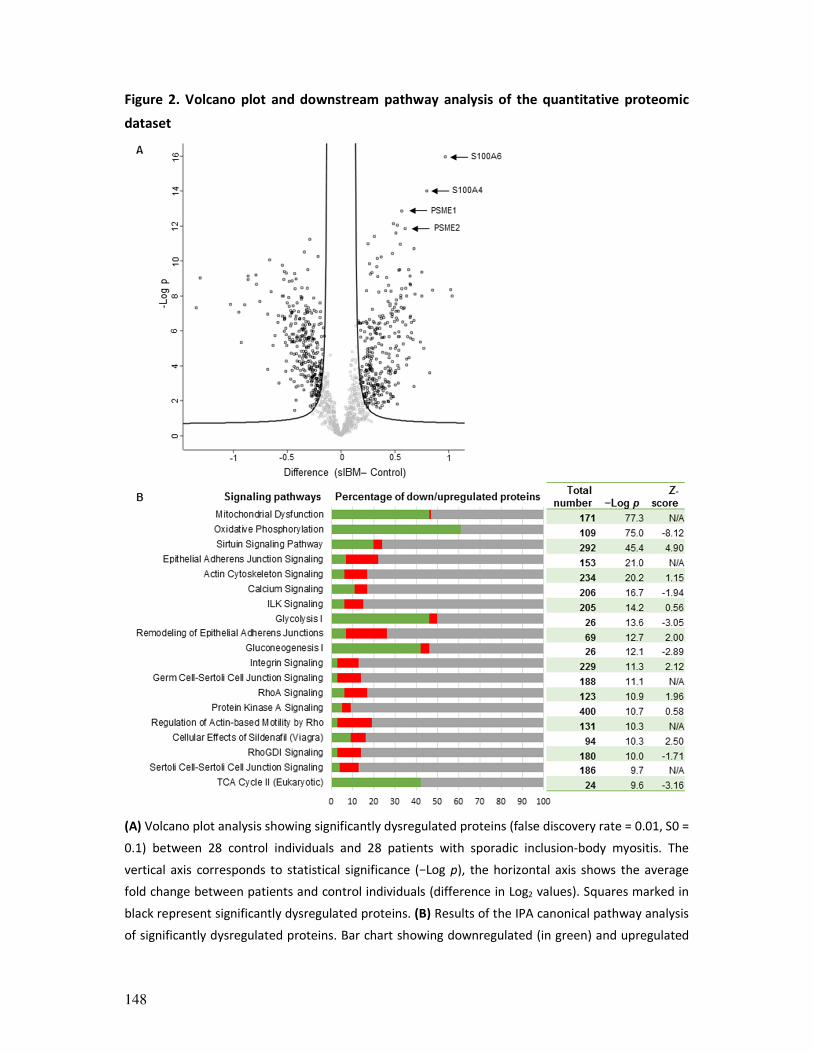
148
Figure 2. Volcano plot and downstream pathway analysis of the quantitative proteomic
dataset
(A) Volcano plot analysis showing significantly dysregulated proteins (false discovery rate = 0.01, S0 =
0.1) between 28 control individuals and 28 patients with sporadic inclusion-body myositis. The
vertical axis corresponds to statistical significance (−Log p), the horizontal axis shows the average
fold change between patients and control individuals (difference in Log2 values). Squares marked in
black represent significantly dysregulated proteins. (B) Results of the IPA canonical pathway analysis
of significantly dysregulated proteins. Bar chart showing downregulated (in green) and upregulated

149
(in red) proteins as a percentage of the total number of molecules of the pathway as annotated in
IPA. Filtering based on p-value of overlap. Pathways with a negative Z-score are predicted to be
inhibited, those with a positive Z-score predicted to be activated. N/A, no activity pattern available.

150
Figure 3. KDM5A expression in myogenin-positive muscle fibres in sIBM muscle
(A-B) Immunoreactivity for KDM5A is strictly nuclear (Hoechst staining for DNA) on
immunohistochemical stainings. The highest signal for KDM5A in sIBM muscle is localized to

151
myogenin-positive regenerating muscle fibres in sIBM muscle. Representative examples of nuclei
showing immunoreactivity for both KDM5A and myogenin (marked by arrowheads) on muscle
biopsy of patient sIBM2 (A) and patient sIBM20 (B). (C-D) A less intense KDM5A signal is localized to
CD4-, CD8 (C)- and CD68-positive (D) infiltrating inflammatory cells, representative examples on
muscle biopsy of patient sIBM20. Scale bar = 30 µm.

152
REFERENCES
1. Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen
AL. Classification and management of adult inflammatory myopathies. The Lancet Neurology.
2018;17(9):816-828.
2. Naddaf E, Barohn RJ, Dimachkie MM. Inclusion Body Myositis: Update on Pathogenesis and
Treatment. Neurotherapeutics : the journal of the American Society for Experimental
NeuroTherapeutics. 2018;15(4):995-1005.
3. Askanas V, Engel WK, Nogalska A. Sporadic inclusion-body myositis: A degenerative muscle disease
associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochimica et
biophysica acta. 2015;1852(4):633-643.
4. Benveniste O, Stenzel W, Hilton-Jones D, Sandri M, Boyer O, van Engelen BG. Amyloid deposits and
inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the
degenerative chicken. Acta Neuropathol. 2015;129(5):611-624.
5. Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011,
Naarden, The Netherlands. Neuromuscular disorders : NMD. 2013;23(12):1044-1055.
6. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature.
2016;537(7620):347-355.
7. Dowling P, Murphy S, Ohlendieck K. Proteomic profiling of muscle fibre type shifting in neuromuscular
diseases. Expert review of proteomics. 2016;13(8):783-799.
8. Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis
of (prote)omics data. Nature methods. 2016;13(9):731-740.
9. Kramer A, Green J, Pollard J, Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway
Analysis. Bioinformatics (Oxford, England). 2014;30(4):523-530.
10. Mamchaoui K, Trollet C, Bigot A, et al. Immortalized pathological human myoblasts: towards a
universal tool for the study of neuromuscular disorders. Skeletal muscle. 2011;1:34.
11. Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cellular &
molecular immunology. 2017;14(1):43-64.
12. Kaur G, Batra S. Emerging role of immunoproteasomes in pathophysiology. Immunology and cell
biology. 2016;94(9):812-820.
13. Ohtsubo H, Sato Y, Suzuki T, et al. APOBEC2 negatively regulates myoblast differentiation in muscle
regeneration. The international journal of biochemistry & cell biology. 2017;85:91-101.
14. Morano M, Ronchi G, Nicolo V, et al. Modulation of the Neuregulin 1/ErbB system after skeletal
muscle denervation and reinnervation. Scientific reports. 2018;8(1):5047.
15. Wang M, Amano SU, Flach RJ, Chawla A, Aouadi M, Czech MP. Identification of Map4k4 as a novel
suppressor of skeletal muscle differentiation. Molecular and cellular biology. 2013;33(4):678-687.
16. Hosoyama T, Nishijo K, Prajapati SI, Li G, Keller C. Rb1 gene inactivation expands satellite cell and
postnatal myoblast pools. The Journal of biological chemistry. 2011;286(22):19556-19564.
17. Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. Histone demethylase KDM5A regulates
the ZMYND8-NuRD chromatin remodeler to promote DNA repair. The Journal of cell biology.
2017;216(7):1959-1974.
18. Lopez-Bigas N, Kisiel TA, Dewaal DC, et al. Genome-wide analysis of the H3K4 histone demethylase
RBP2 reveals a transcriptional program controlling differentiation. Molecular cell. 2008;31(4):520-530.
19. Varaljai R, Islam AB, Beshiri ML, Rehman J, Lopez-Bigas N, Benevolenskaya EV. Increased
mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB-
deficient cells. Genes & development. 2015;29(17):1817-1834.
20. Wanschitz JV, Dubourg O, Lacene E, et al. Expression of myogenic regulatory factors and myo-
endothelial remodeling in sporadic inclusion body myositis. Neuromuscular disorders : NMD.
2013;23(1):75-83.

153
21. Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging.
Nat Med. 2015;21(8):854-862.
22. Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and
differentiation. Cell stem cell. 2008;2(1):22-31.
23. Kwon B, Kumar P, Lee HK, et al. Aberrant cell cycle reentry in human and experimental inclusion body
myositis and polymyositis. Human molecular genetics. 2014;23(14):3681-3694.
24. Nishii M, Nakano S, Nakamura S, et al. Myonuclear breakdown in sporadic inclusion body myositis is
accompanied by DNA double strand breaks. Neuromuscular disorders : NMD. 2011;21(5):345-352.
25. Bou Saada Y, Zakharova V, Chernyak B, et al. Control of DNA integrity in skeletal muscle under
physiological and pathological conditions. Cellular and molecular life sciences : CMLS.
2017;74(19):3439-3449.
26. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell.
2010;40(2):179-204.
27. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell.
2013;153(6):1194-1217.
28. De Paepe B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras.
Biomolecules. 2019;9(1).
29. Morosetti R, Broccolini A, Sancricca C, et al. Increased aging in primary muscle cultures of sporadic
inclusion-body myositis. Neurobiology of aging. 2010;31(7):1205-1214.
30. Ermolaeva M, Neri F, Ori A, Rudolph KL. Cellular and epigenetic drivers of stem cell ageing. Nature
reviews Molecular cell biology. 2018;19(9):594-610.
31. Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nature reviews
Rheumatology. 2019;15(5):257-272.
32. Muth IE, Zschuntzsch J, Kleinschnitz K, et al. HMGB1 and RAGE in skeletal muscle inflammation:
Implications for protein accumulation in inclusion body myositis. Experimental neurology.
2015;271:189-197.
33. Bhattarai S, Ghannam K, Krause S, et al. The immunoproteasomes are key to regulate myokines and
MHC class I expression in idiopathic inflammatory myopathies. Journal of autoimmunity. 2016;75:118-
129.
34. Guttsches AK, Brady S, Krause K, et al. Proteomics of rimmed vacuoles define new risk allele in
inclusion body myositis. Annals of neurology. 2017;81(2):227-239.
35. Mitsui E, Yoshida S, Shinoda Y, et al. Identification of ryuvidine as a KDM5A inhibitor. Scientific
reports. 2019;9(1):9952.
36. Vinogradova M, Gehling VS, Gustafson A, et al. An inhibitor of KDM5 demethylases reduces survival of
drug-tolerant cancer cells. Nature chemical biology. 2016;12(7):531-538.
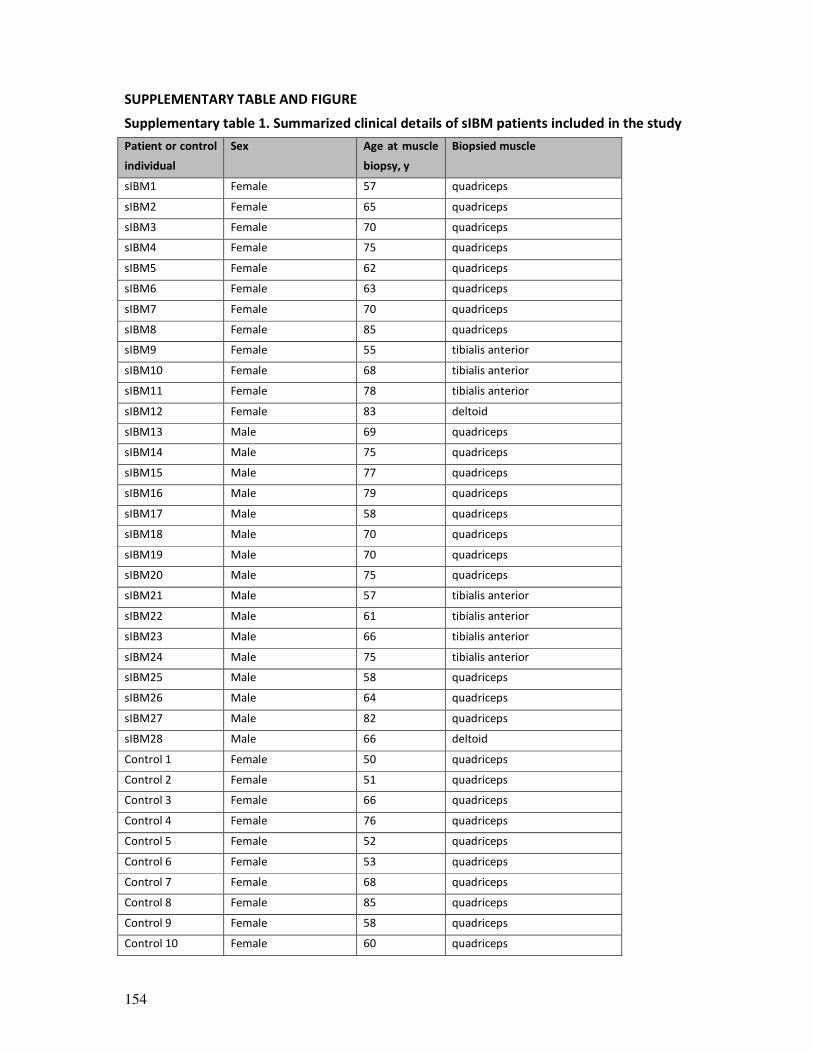
154
SUPPLEMENTARY TABLE AND FIGURE
Supplementary table 1. Summarized clinical details of sIBM patients included in the study
Patient or control
individual
Sex Age at muscle
biopsy, y
Biopsied muscle
sIBM1 Female 57 quadriceps
sIBM2 Female 65 quadriceps
sIBM3 Female 70 quadriceps
sIBM4 Female 75 quadriceps
sIBM5 Female 62 quadriceps
sIBM6 Female 63 quadriceps
sIBM7 Female 70 quadriceps
sIBM8 Female 85 quadriceps
sIBM9 Female 55 tibialis anterior
sIBM10 Female 68 tibialis anterior
sIBM11 Female 78 tibialis anterior
sIBM12 Female 83 deltoid
sIBM13 Male 69 quadriceps
sIBM14 Male 75 quadriceps
sIBM15 Male 77 quadriceps
sIBM16 Male 79 quadriceps
sIBM17 Male 58 quadriceps
sIBM18 Male 70 quadriceps
sIBM19 Male 70 quadriceps
sIBM20 Male 75 quadriceps
sIBM21 Male 57 tibialis anterior
sIBM22 Male 61 tibialis anterior
sIBM23 Male 66 tibialis anterior
sIBM24 Male 75 tibialis anterior
sIBM25 Male 58 quadriceps
sIBM26 Male 64 quadriceps
sIBM27 Male 82 quadriceps
sIBM28 Male 66 deltoid
Control 1 Female 50 quadriceps
Control 2 Female 51 quadriceps
Control 3 Female 66 quadriceps
Control 4 Female 76 quadriceps
Control 5 Female 52 quadriceps
Control 6 Female 53 quadriceps
Control 7 Female 68 quadriceps
Control 8 Female 85 quadriceps
Control 9 Female 58 quadriceps
Control 10 Female 60 quadriceps
155
Control 11 Female 64 deltoid
Control 12 Female 76 tibialis anterior
Control 13 Male 53 quadriceps
Control 14 Male 59 quadriceps
Control 15 Male 68 quadriceps
Control 16 Male 88 quadriceps
Control 17 Male 51 quadriceps
Control 18 Male 59 quadriceps
Control 19 Male 60 quadriceps
Control 20 Male 73 quadriceps
Control 21 Male 49 tibialis anterior
Control 22 Male 56 tibialis anterior
Control 23 Male 62 tibialis anterior
Control 24 Male 65 tibialis anterior
Control 25 Male 53 quadriceps
Control 26 Male 58 quadriceps
Control 27 Male 79 quadriceps
Control 28 Male 76 deltoid
sIBM, sporadic inclusion-body myositis; y, years.

156
Supplementary figure 1. KDM5A western blot
Western blot showing KDM5A expression in fibroblast and lymphoblast protein lysates of a healthy
control individual, as well as in a myoblast lysate of a 53 years old healthy control individual. 50 µg of
protein was loaded for every sample. Fib, fibroblasts; Lymph, lymphoblasts; Myob, myoblasts.

157

158

159
GENERAL DISCUSSION

160
Primary muscle disorders constitute a large group of inherited and acquired diseases that
affect muscle structure, metabolism, or the function of muscle ion channels.1 Given that
only very few acquired muscle disorders present with slowly progressive muscular
weakness, an inherited muscle disorder (IMD) is typically suspected in case of this clinical
presentation. IMD constitute a clinically, genetically and histopathologically very
heterogeneous group of rare diseases with more than 160 genetically distinct entities
identified, rendering the diagnostic process complex.2, 3 Recently, considerable progress has
been made in molecular genetic testing. Next generation sequencing (NGS) strategies are
more and more available and rapidly decrease in costs: targeted panel sequencing, whole-
exome sequencing (WES) and whole-genome sequencing (WGS).4 Sporadic inclusion body
myositis (sIBM) of course constitutes a very relevant, clinically recognizable differential
diagnosis in patients over 50 years of age.5 A few other atypically presenting acquired
muscle disorders might also present with slowly progressive muscle weakness. Recent
literature suggested that idiopathic inflammatory myopathies (IIM) with anti-HMGCR
antibodies might present with slowly progressive muscle weakness.6 The same has been
suggested for an enigmatic, supposedly very rare, putatively immune-mediated acquired
myopathy, called sporadic late-onset nemaline myopathy (SLONM).7
Muscle disorders presenting with slowly progressive muscle weakness constitute a group of
disorders eminently suited for a multi-level pattern recognition approach, clinically as well
as biologically. The ultimate aim of this PhD thesis was to gain insights in pathomechanisms
of this group of muscle disorders, inherited or acquired, which are unmistakably marked by
progressive muscle degeneration.
Taking part in MYO-SEQ, a large multi-centre WES study of unsolved suspected IMD
patients, we first aimed to identify patients with (very) rare IMD subtypes in order to be
able to further study pathomechanisms of these entities. I will first summarize the findings
of this part (part 1) of the Results section of my thesis and highlight a few other ongoing
studies on rare or novel IMD, to illustrate the unstoppable progress in the field.
Subsequently, I will discuss findings of the proteomic approach (part 2) to study sIBM
disease signatures and put this in perspective of the sIBM field. Here, I will also highlight
methodological clues for proteomic studies in IMD (VCP-related myopathy in particular) and
the relevance of studying both inherited and acquired muscle disorders in search for
common pathways and strategies.
Finally, I will put findings in this doctoral thesis in perspective of the field of myology
research, which is fast moving, forcing us to prepare rapid translation to the clinics.

161
GENOTYPE-PHENOTYPE CORRELATIONS IN RARE INHERITED MUSCLE DISORDERS AND
NOVEL GENE HUNTING: IMPORTANCE OF DEEP PHENOTYPING AND A MULTI-LEVEL
PATTERN RECOGNITION APPROACH
Importance of deep phenotyping and studying molecular mechanisms of rare IMD
In chapter 1, we describe deep phenotyping data of TRIM32-related myopathy patients, a
recessive IMD that appears to be extremely rare in non-Hutterite patients. The disease had
been generally described as a mild and progressive recessive myopathy without
characteristic clinical features, yet we were able to define a characteristic MRI pattern on
muscle MRI imaging. We also help to elucidate the histopathological spectrum of the
disease: the appearance of vacuoles in scattered muscle fibres on the biopsies of patients
with a TRIM32-related myopathy seems to be a relatively common but not obligatory
finding, suggesting that these features are part of a histopathological spectrum, rather than
constituting a separate phenotype. Furthermore, we showed that TRIM32 nonsense alleles
are at least partially targeted by nonsense mediated mRNA-decay, although theoretically
this would not be expected for a gene with only one coding exon.8 We clarify molecular
mechanisms, showing a straightforward TRIM32 protein loss of expression and/or function.
This renders putative molecular mechanisms of the reported homozygous missense variant
Bardet-Biedl syndrome-11 (BBS11) variant (located in the B-box domain of TRIM32) quite
puzzling. We do not observe the multi-systemic involvement as in BBS and vice versa, no
skeletal muscle involvement is noted in the patient with this BBS11 variant.9 By elucidating
molecular mechanisms of this rare disorder, we add to the biological knowledge of muscle
regeneration, as TRIM32 was shown to have selective a role in skeletal muscle
regeneration.10 Lastly, we describe the clinical details of patients carrying a missense
‘variant of unknown significance’ (VUS), not residing in the protein domain in which
pathogenic missense mutations had previously been described. Although some features
contrast with those of the patients harbouring definite pathogenic variants, pathogenicity of
the VUSs still is uncertain and deep phenotyping these patients will help in interpreting the
effect of such VUSs in future identified patients.
In chapter 2, we similarly add to our knowledge of an apparently extremely rare disorder
linked to recessive mutations in BVES and the role of the Popeye domain containing protein
1 (POPDC1) in striated muscle physiology and disease. We describe the very striking
phenotypic spectrum of the disease with varying cardiac and skeletal muscle involvement.
By showing loss of transcript or loss of protein expression or function of the different
homozygous variants, we further prove a straightforward LOF mechanism of the disease.
Previously only one family had been identified, with multiple individuals harbouring a
homozygous p.Ser201Phe missense variant in BVES.

162
In chapter 4, we capitalize on a combination of the clinical observation of a unique
homozygous VCP patient and the in-depth proteomic study of this unique in vivo situation of
having VCP hexamers constituting only of mutant VCP monomers, to add to our knowledge
of the complex pathomechanisms of this multi-faceted, normally dominantly inherited
disease. Our observations in this study originated from a clinical approach, however the
added layer of complexity resulting from the proteomic studies allowed to (conscientiously)
postulate a dominant-negative mechanism rather than a toxic gain-of-function (GOF)
mechanism.
Together, these chapters elegantly illustrate the importance of in-depth clinical and
functional studies of rare IMD, which not only allows us to better diagnose these patients,
yet also adds to our knowledge of skeletal muscle physiology and pathways involved in
muscle degeneration or regeneration. This diverse group of disorders characterized by
slowly progressive muscle weakness is eminently suited for a multi-level pattern recognition
approach, clinically as well as biologically, with a broad range of clinical and biological
studies (with direct access to the diseased tissue) being easily at hand (as illustrated in the
Aims section, figure 1).
Identifying and dissecting disease mechanisms of rare or novel IMD: a never-ending story
To illustrate the abiding progress in the field of IMD, I would like to briefly discuss a few
studies in progress, which did not yet have a place as a full-grown chapter in the Results
section of this PhD thesis but nicely illustrate the continuous evolution in this field.
Myosinopathies
We’re currently studying patients with very rare myosinopathies, in a collaborative effort,
and will again provide functional insights in disease mechanisms of these disorders. Myosins
constitute the thick filament of the sarcomeres, the basic contractile apparatus of muscle,
with myosin heavy chains (MyHC) being the motor of muscle.11 Mutations in different MyHC
isoforms have been linked to striated muscle disease. Mutations in MYH3 and MYH8 have
only been linked to a dominant congenital muscle disorder with distal arthrogryposis.11 For
mutations MYH7 and MYH2, molecular mechanisms appear to be more complex and will
likely encompass dominant negative as well as LOF effects: both dominant and recessive
myosinopathies have been described for both entities.11
Through WES, we identified the heterozygous p.Phe122Leu MYH2 variant, absent from
gnomAD12, with in silico prediction algorithms in favour of pathogenicity, segregating with

163
disease across three generations of a Belgian family. As of yet, only three different
autosomal dominantly inherited missense mutations have been reported in MYH2, exerting
a supposedly dominant negative effect on the function of this MyHC isoform. Patients
typically present with contractures in several joints at birth and show progressive muscle
weakness and wasting with juvenile to young adult onset,13-15 as was the case for affected
individuals in the current family. Recessive mutations in MYH2 exerting a loss-of-function
(LOF) effect appear to be more frequent. For dominantly inherited mutations, exact
pathomechanisms still remain to be unraveled.11 Genetically, this is a puzzling story as the
three different reported dominantly inherited missense mutations affect different domains
of the MYH2 protein. The originally reported p.Glu706Lys mutation is located in the MMD
domain,13 similarly as is the heterozygous p.Phe122Leu mutation. Recently, recessive
missense mutations affecting the myosin motor domain (MMD) have been reported as
well.16-20 Furthermore, VUSs are regularly encountered in this relatively large gene
(NM_017534.5 encompasses 40 exons, total annotated spliced exon length is 6086 base
pairs (bp)). Constraint scores in gnomAD reflect a relative intolerance to missense variation
in the gene with a positive Z-score (Z=2.32),12 yet still more than 1000 missense variants
have currently been noted, rendering the evaluation of VUSs in MYH2 in individual patients
difficult if no strong segregation data can be gathered. We’re currently performing
functional studies to evaluate the functional effect of different heterozygous missense
variants on the MYH2 protein, in collaboration with Prof. Julien Ochala (King's College
London). Studying myosin function through single fibre contractile recordings on muscle
fibres isolated from patients’ muscle and in vitro motility assays will add to our knowledge
of the probably residue specific effect of heterozygous missense variants in the gene. Deep
phenotyping of these patients is still ongoing as well. New insights in genotype-phenotype
correlations and pathomechanisms are needed to allow better diagnosing and counselling
for this rare disorder.
In collaboration, we’re currently also studying a particularly interesting novel candidate
gene, linked to a ‘secondary’ myosinopathy. UNC45B (unc45 myosin chaperone B) encodes a
myosin-specific co-chaperone essential for the folding, stability and maintenance of
sarcomeric myosins, thus facilitating proper assembly and function of myosins in skeletal
and cardiac muscle.21 We describe 3 unrelated patients with childhood-onset progressive
muscle weakness. Through WES, we identified rare homozygous variants in the C-terminal
UCS domain of UNC45B which is critical for myosin binding, with in silico prediction
algorithms being in favour of pathogenicity. The recurring p.Arg754Gln (c. 2261G>A)
UNC45B variant was identified in homozygosity in two patients, while the third patient was

164
found to harbour the rare missense p.Arg778Trp variant in compound heterozygosity with a
c.2261+5G>C splice site mutation resulting in aberrant splicing. Western blot revealed a
severe reduction of UNC-45B protein in patient muscle compared to control, and
immunofluorescence localization studies demonstrated abnormal relocalization of the
residual UNC-45B protein within the sarcomere as well as focal disruption of the myofibrillar
apparatus. Functional analyses of the effect of these UNC45B mutations on muscle fibre
mechanics, through force generation and myosin binding in patient’s muscle, in addition to
in vivo studies in C.elegans of the effect of these mutations on UNC45b function, will
provide further insights into this novel disease mechanism.22, 23 Our series establishes
recessive mutations in UNC45B as a cause of a novel form of progressive myopathy, which
we propose to be classified as a secondary myosinopathy.
Metabolic myopathies with polyglucosan accumulation
Muscle glycogen storage disorders (GSDs) are recessively inherited disorders of glycogen
metabolism that are histopathologically characterized by storage or depletion of glycogen in
muscle fibers.24, 25 Clinically, patients may present with exercise intolerance with muscle
pain and cramps, frequently followed by myoglobinuria, or they may present with
stationary, slowly progressive muscle weakness.26
The enzymes involved in muscle glycogen synthesis (glycogenesis) include glycogen synthase
(GYS1; GSD 0B), branching enzyme (GBE1; GSD IV), and glycogenin-1 (GYG1; GSD XV).
Glycogen storage disease XV (GSD XV) is caused by recessive mutations in the glycogenin-1
gene (GYG1) and presents as a predominant skeletal myopathy or cardiomyopathy. We
studied two patients, presenting with late onset slowly progressive muscle weakness,
carrying compound heterozygous GYG1 variants (manuscript currently in press: Hedberg-
Oldfors C, De Ridder W, et al. Functional characterization of GYG1 variants in two patients
with myopathy and glycogenin-1 deficiency. Neuromuscular disorders 2019). In patient 1,
the novel c.144-2A>G splice acceptor variant and the novel frameshift variant
p.Val211Cysfs*30 (c.631delG) were identified, and in patient 2, the previously described
p.Asp102His (c.304G>C)27 and p.Asp163Thrfs*5 (c.487delG) variants. Protein analysis
showed total absence of glycogenin-1 expression in patient 1 whereas in patient 2 there was
reduced expression of glycogenin-1, with the residual protein being non-functional. Both
patients showed glycogen and polyglucosan (amylopectin like material) storage in their
muscle fibres, as revealed by PAS staining and electron microscopy. Cardiac evaluation for
patient 1 did not show any specific abnormalities linked to the glycogenin-1 deficiency. In
patient 2, who was shown to express the p.Asp102His mutated glycogenin-1, cardiac
evaluation was still normal at age 77 years. This contrasts with the association of the

165
p.Asp102His variant in homozygosity with a severe cardiomyopathy in several cases with an
onset age between 30 and 50 years.27 This finding might indicate that the level of the
p.Asp102His missense mutated glycogenin-1 determines if a patient will develop a
cardiomyopathy. Slightly different mechanisms between complete LOF variants and
missense variants appear to confine tissue specificity.
We’re currently also adding to our knowledge of molecular mechanisms in another GSD
with abnormal glycogen synthesis, linked to recessive mutations in GBE1, which appear to
parallel this effect. Here, molecular mechanisms and genotype-phenotype are even more
puzzling. Recessive mutations in GBE1, encoding the glycogen branching enzyme (GBE) are
linked to glycogen storage disease type IV (GSD IV), a disorder with varying hepatic and
skeletal muscle involvement with infantile or childhood onset, as well as a presumed
separate entity, adult polyglucosan body disease (APBD), typically presenting with a late
adult-onset myelopathy, leukodystrophy, neurogenic bladder dysfunction and a peripheral
neuropathy. Deep phenotyping of a patient with an apparently typical APBD-like phenotype
however revealed an unmistakable myopathy, too. The perinatal and congenital forms of
GBE1-related disease at the most severe end of the clinical spectrum are caused by LOF
variants (nonsense, frameshift and splice site variants), in homozygosity or in some cases in
compound heterozygosity with a missense variant targeting the catalytic domain. All these
alleles result in very low levels of residual GBE enzymatic activity.28 On the other end of the
spectrum, APBD has never been linked to straightforward LOF variants. Currently reported
patients were compound heterozygous for missense variants or frameshift variants that are
predicted to skip nonsense mediated mRNA-decay.29, 30 At first sight, this phenotypic
spectrum appears to be at least partially explained by the difference in residual GBE activity
in case of the missense mutations. It is however puzzling that some missense variants
associated with APBD, such as the p.Thr254Ala variant identified in this study, have also
been associated with classic progressive hepatopathy in childhood when seen in a
homozygous or compound heterozygous state. This might suggest that an additional
molecular mechanism on top of the loss of enzymatic function of GBE confines tissue
specificity.
The missing ‘heritability’ in IMD
Findings in the IMD part of this PhD thesis, as summarized above, illustrate the strength of
WES in defining new genotype-phenotype correlations and hunting ‘novel’ genes. WES, as
one of the three main NGS methodologies, overcame the two major barriers of earlier
sequencing techniques, limited throughput and high sequencing costs.31 NGS techniques

166
have been continuously evolving over the last decade and are more and more available and
rapidly decrease in costs. In research, implementation of NGS led to an explosion in the
discovery of myopathy genes.32 Targeted gene panels were increasingly implemented in
clinical genetics (diagnostic setting) first, followed by WES. Recent data of larger cohorts of
patients with a suspected IMD in which targeted panel sequencing was applied, showed
success rates that varied between 20% and 45%.4, 33, 34 Although WGS not only interrogates
the complete genome, but also is more powerful than WES for detecting exome variants, it
has been less widely used than WES as of yet because of the higher costs and the complex
(time consuming) nature of data analysis of WGS datasets.35 In literature, cost estimates
regarding WES and WGS vary widely between platforms, yet generally WES appears to be
approximately four times as expensive as WES.36 WES as such still remains a cost-effective
sequencing strategy in medical genetics and research, yet a new revolution in NGS
technologies is coming up.31
Despite the countless advancements of NGS, these techniques are currently hindered by a
few important limitations. NGS techniques are typically characterized by short-read lengths
(∼150–300 bp) in order to preserve high read quality. The highly repetitive and complex
nature of the human genome however constitutes an important obstacle: errors in calling
genetic variants are introduced, certain genomic regions are not captured well and specific
types of pathogenic variants are considered to be intractable.31, 37 Apart from single
nucleotide variants (SNVs) and small indels, two important types of genetic variation
potentially leading to Mendelian disorders such as IMD are the following: 1) structural
variants (genomic rearrangements larger than 50 bp); 2) tandem repeat (i.e. copies of short,
1–6 bp, sequence units) expansions or contractions.37 Important examples in IMD are: 1)
approximately 70% of dystrophinopathies are caused by deletions or duplications targeting
DMD;38 2) repeat disorders facioscapulohumeral dystrophy (FSHD), oculopharyngeal
muscular dystrophy (OPMD) or myotonic dystrophy type 1 or 2 for which directed molecular
genetic testing is performed in case of clinical suspicion.
Specialised bioinformatics tools are now starting to permit the detection of copy number
variants (CNVs) in NGS data.39 These types of analysis have increasingly been applied in IMD
too.40 We also identified a 63.5 kb deletion targeting TRIM32 in a patient with a TRIM32-
related myopathy.41 Of current NGS technologies, low-coverage and paired-end WGS
strategies appear to be the most efficient in detecting CNVs, even outperforming array-
based CNV analyses.42 However, short read NGS approaches still often lack sensitivity, show
excess of false positive CNVs and misinterpret complex structural variants.37 Long read
sequencing (LRS) techniques (‘third generation sequencing’) hold promise to overcome
these difficulties in assessing structural variants, repetitive elements and complex genomic

167
regions in Mendelian disease, but their sequencing and bioinformatic methodologies still
need to be improved.37, 43
The rate of pathogenic CNVs in IMD is most probably underestimated, but in contrast to e.g.
cerebellar ataxias, only few muscle disorders are currently known repeat disorders.44 Other
more complex molecular mechanisms could explain an additional part of the ‘missing
heritability’ in IMD: deep intronic variants affecting splicing, for which performing
transcriptome sequencing has shown to be useful in IMD,45 mutations in regulatory
elements such as promotors, digenic or oligogenic inheritance.32
With regard to more complex genetic variation, but also to apparently ‘simple’ single
nucleotide polymorphisms (SNPs) or small indels, studying molecular mechanisms of rare
IMD is definitely crucial with respect to interpretation of identified variants.46 Elucidating
LOF, GOF or dominant negative mechanisms of genetic variants is crucial in the
interpretation of pathogenicity of e.g. missense mutations residing in specific protein
domains or nonsense variants. Collaborative efforts are crucial in studying these rare IMD.32
Chapter 3 of this PhD thesis nevertheless highlights another potential explanation of current
success rates of NGS generally staying below 50%. With the exception of sporadic sIBM, very
few acquired muscle disorders present with slowly progressive muscle weakness and as
such, an inherited muscle disorder (IMD) is typically suspected in case of this clinical
presentation. A few other atypically presenting acquired muscle disorders might however
also be relevant differential diagnoses and risk to remain undiagnosed.
The success rate of WES in the Antwerp University Hospital (UZA) subcohort of MYO-SEQ
dropped from 56.9% in all 65 suspected IMD cases included in MYO-SEQ to 39.5% in 43
myopathy cases without family history of muscle disease. Eighteen of these showed late
adult onset limb-girdle muscular weakness (LGMW). These suspected IMD patients
remained genetically unsolved after: 1) directed molecular genetic testing prior to inclusion
in MYO-SEQ to exclude a dystrophinopathy, FSHD, OPMD or myotonic dystrophy type 1 or 2
in case of clinical suspicion; 2) rigorous WES data analysis. Systematic (para)clinical and
histopathological characterization of these suspected IMD patients strongly suggested an
enrichment of isolated cases of LGMW with rods as core feature (10 out of 18 patients),
with four of these patients having an (IgG κ) monoclonal gammopathy of unknown
significance (MGUS) and one markedly increased (polyclonal) κ and λ chains.47 As such, ten
patients represented suspected slowly progressing SLONM cases. Based on this proof-of-
concept study in this genetically and clinically heterogeneous group of patients, we propose
that a probable SLONM diagnosis based on the identification of rods with or without an

168
MGUS could be substantiated by supportive criteria based on: 1) negative results of WES
analysis; 2) muscle MRI imaging (suggestive pattern); 3) findings of protein electrophoresis
as well as free light chain (FLC) assay; 4) serological exclusion of HMGCR-myopathy; 5)
additional biopsy features such as cytoplasmic bodies, hyaline inclusions, rimmed vacuoles;
6) immunohistochemical (IHC) analysis for MHC-I.
PROTEOMICS AND AN UNBIASED DISSECTION OF DISEASE SIGNATURES IN MUSCLE
DISORDERS
Proteomics is a powerful approach to dissect disease signatures in an unbiased way.48 Mass
spectrometry (MS)-based techniques have evolved dramatically over time and are capable
of quantifying thousands of proteins across collections of large numbers of samples with a
high degree of reproducibility.48 Proteomics studies on diseased muscle tissue have
increasingly been applied in muscle disorders as patient diseased tissue is readily
available.49, 50
Findings in this doctoral thesis highlight the strength of an ‘unbiased’ analysis of ‘unbiased’
proteome data in both acquired and inherited muscle disorders. We studied the proteome
of sIBM muscle and VCP-inclusion body myopathy (IBM) muscle, a rare IMD, part of an
intriguing multisystem proteinopathy. sIBM and VCP-IBM not only show important clinical
and histopathological similarities, disease signatures on muscle proteome level also appear
highly similar. The sIBM dataset is highly powered to detect proteins significantly
dysregulated between patients and control individuals. The VCP-IBM dataset on itself is also
sufficiently powered to detect dysregulated proteins, correcting for multiple hypothesis
testing.51 The expected percentage of differentially expressed proteins however strongly
influences power and sample size calculation of proteomic datasets. Regarding the strong
histopathological – and probably pathomechanistic – resemblances, larger numbers of
muscle biopsy specimens are clearly needed for direct comparison of VCP-IBM and sIBM
proteomes. We did not have access to an adequate number of VCP-IBM samples to establish
such a sufficiently powered proteomics dataset.
Our proteomic approach in VCP-IBM illustrates the importance of directly studying
pathomechanisms in diseased patients’ tissue and even the usefulness of performing
focused outlier analyses on proteome data of unique patients such as the reported
homozygous VCP patient. Based on our analysis of the proteomic dataset, the upstream
pathomechanistic role of selective failure of muscle regeneration and stress granule
dyshomeostasis in VCP-IBM appears to be underestimated.
The considerably more extensive proteomic dataset on sIBM very similarly highlights strong
signatures pointing towards selective failure of muscle differentiation during regeneration,

169
which appears to be linked to the DNA damage response (DDR) through KDM5A. sIBM might
represent a muscle disorder with selective ‘premature ageing’ of muscle, but this ageing of
muscle stem cells (MuSC) might be a common downstream pathway in other degenerative
muscle disorders too.
An ‘unbiased’ analysis of an ‘unbiased’ proteome dataset implies a largely hypothesis free
analysis of the dataset. In such an analysis of a dataset on muscle tissue, researchers have
the difficult task to find the middle ground between what is statistically significant and at
the same time biologically meaningful. Focussing on the first might lead to describing
biologically meaningless alterations, yet focussing on the latter might imply a biased analysis
of a supposedly unbiased dataset. Volcano plot analyses excellently illustrate the search for
this balance, assessing statistical significance (t-test, with correction for multiple hypothesis
testing) together with fold change to identify significantly dysregulated proteins between
patients and controls (an example can be found in figure 1A). Furthermore, the set of
identified proteins and consequently also of dysregulated proteins, is biased towards more
abundant proteins. Current high-sensitive MS pipelines allow identification of more than
10.000 different proteins in cell lysates,48 but it still is challenging to reach a few thousand
proteins in whole muscle lysates, as exemplified in a comprehensive deep proteomic study
of mouse skeletal muscle.52
Highlighting top dysregulated proteins directly identified in the dataset might yield some
pathomechanistic hints or lead to identification of potential biomarkers, yet looking at
patterns rather than single protein minimizes the risk of studying spurious clues, even in
case of statistical correction for multiple hypothesis testing, which was already mentioned
above; rigorous correction for multiple testing problem certainly is essential to correct for
the occurrence of false positives in the analysis of large datasets.
Moreover, upstream regulators of disease are likely hidden in the deep proteome.
Dysregulation of these proteins is reflected in dysregulation of more abundant proteins and
pathways, the so-called ‘functional echo’.53 Different bioinformatics strategies can be
applied to predict dysregulation of ‘upstream regulators’ based on a set of dysregulated
proteins (reflecting these patterns), yet particularly the causal analysis approaches of the
Ingenuity Pathway Analysis (IPA) software (QIAGEN), which constitute state-of-the-art
analyses, also widely used in the interpretation of high-throughput gene-expression data
(example in figure 1B).54 These IPA causal analyses, using manually curated data, allowed us
to prioritize KDM5A as top dysregulated proximal regulator based on our sIBM proteomic
dataset. KDM5A, predicted to be activated, was also biologically a very interesting player
with regard to putative sIBM pathomechanisms.
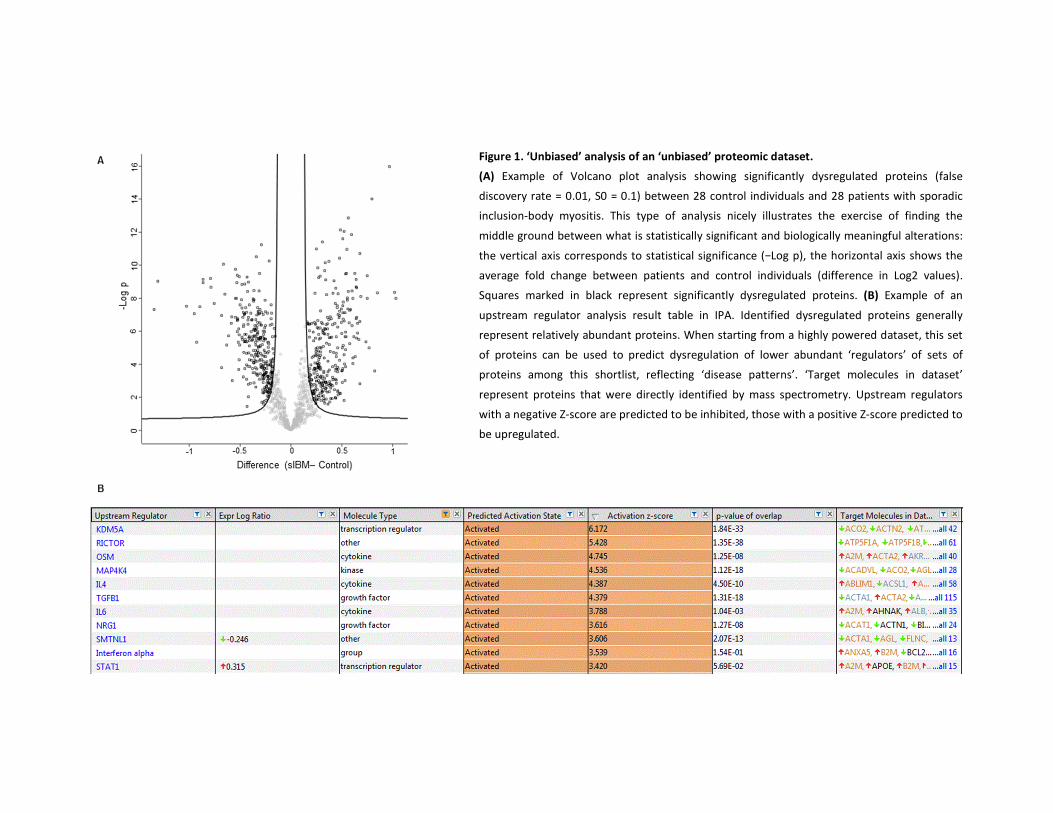
Figure 1. ‘Unbiased’ analysis of an ‘unbiased’ proteomic dataset.
(A) Example of Volcano plot analysis showing significantly dysregulated proteins (false
discovery rate = 0.01, S0 = 0.1) between 28 control individuals and 28 patients with sporadic
inclusion-body myositis. This type of analysis nicely illustrates the exercise of finding the
middle ground between what is statistically significant and biologically meaningful alterations:
the vertical axis corresponds to statistical significance (−Log p), the horizontal axis shows the
average fold change between patients and control individuals (difference in Log2 values).
Squares marked in black represent significantly dysregulated proteins. (B) Example of an
upstream regulator analysis result table in IPA. Identified dysregulated proteins generally
represent relatively abundant proteins. When starting from a highly powered dataset, this set
of proteins can be used to predict dysregulation of lower abundant ‘regulators’ of sets of
proteins among this shortlist, reflecting ‘disease patterns’. ‘Target molecules in dataset’
represent proteins that were directly identified by mass spectrometry. Upstream regulators
with a negative Z-score are predicted to be inhibited, those with a positive Z-score predicted to
be upregulated.

171
Although the methodology of proteomic studies and the bioinformatic interpretation of
these datasets are rapidly evolving, we have to stay aware of potential bias confounding
enrichment analyses of such ‘omics’ datasets, particularly sampling and detection bias as
discussed above (number of proteins that is detected across a number of samples), as well
as biological bias.55 For the latter, background gene lists, as also applied in IPA software, are
very useful; in skeletal muscle specifically, proteins of the contractile apparatus and proteins
involved in cellular metabolism and oxidative phosphorylation, constitute a large part of the
proteins that are highly expressed.
Contrasting with the set-up of our proteomic studies, focussing on a specific facet of the
proteome or a specific hypothesis, e.g. by laser capture microdissection of protein
aggregates, might be relevant and might yield valuable insights, but these protein
aggregates might just represent the downstream effect of protein dyshomeostasis too. With
this set-up the complete proteomic landscape of a disease in not interrogated in an
unbiased way.56 The few published proteomics studies in sIBM mainly applied this ‘targeted’
analysis methodology, ‘biased’ towards a specific hypothesis or part of the proteome. Small
samples sizes, insufficient sample matching leading to important variability of protein
abundances or the use of low sensitivity MS methods without fractionation techniques
represent other methodological issues of these studies.56-61 In our proteomic study of sIBM
we overcame these issues by establishing a highly powered state-of-the-art proteomics
dataset on a large set of whole muscle tissue lysates.
In summary, the design of our highly powered proteomics study in sIBM is exceptionally
suited for the study of disease patterns, rather than just single proteins, which reflect
dysregulation of key upstream regulators, the ‘drivers’ of the disease.
FROM MOLECULAR DIAGNOSIS OR DISEASE SIGNATURES IN MUSCLE TO THERAPEUTIC
STRATEGIES AND TRANSLATION TO THE CLINIC
Good general standards-of-care are available for different IMD, especially the muscular
dystrophies: medication, rehabilitation, early detection of complications, surgery, etc.3, 62, 63
A striking example is the improvement of life expectancy in Duchenne muscular dystrophy
due to optimal cardiac management, ventilator support, scoliosis surgery and corticosteroid
therapy3: epidemiological research shows an increase in survival rate at the age of 25 from
about 13.5% in patients born between 1961 and 1970 to about 50% in patients born
between 1981 and 1990.64 Furthermore, enzyme replacement therapy has proven its
positive effect on muscle strength, pulmonary function, and daily life activities in Pompe

172
disease.65 Despite these progresses, IMD generally still result in severe disability in the
absence of a curative treatment.
Translation of potentially curative genetic therapies to the clinics is a rapidly evolving field in
neuromuscular disorders and IMD in particular. Different therapeutic strategies are being
developed, depending of the molecular mechanism of the disorder. Gene therapies (sensu
stricto) are aimed at achieving durable expression of a therapeutic gene or ‘transgene’, to
‘replace’ a LOF allele. At least six gene therapy products have already been approved by the
European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA),
including for spinal muscular atrophy (SMA). Hundreds of programs are in clinical
development, including e.g. for dystrophinopathies.66 For IMD caused by GOF, dominant
negative, or also certain LOF mutations, alternative genetic therapeutic approaches are
available. Gene regulation strategies at the level of RNA through RNA therapeutics
(antisense oligonucleotides (ASO) oand short interfering RNA (siRNA)) are highly emerging.
A few compounds are already FDA approved and many others are tested in (pre-) clinical
studies.67 A notable example is the ASO nusinersen which modulates the splicing of SMN2 as
a treatment for SMA. I’m currently also taking part as a co-investigator in a phase 1/2 trial
studying an ASO in patients with a centronuclear myopathy. Genome engineering or editing,
using systems such as CRISPR-Cas9, represents a last emerging strategy as a genetic therapy
in neuromuscular disorders.68
As highlighted above, finding these patients with rare IMD and deciphering molecular
mechanisms (as illustrated in this PhD thesis), is crucial for rapid translation of therapeutic
strategies to the clinic. For these rare disorders, we need to rely on collaborative efforts
such as the MYO-SEQ project.32
The design of therapeutic strategies in pathomechanistically unsolved acquired myopathies
may be hampered by a lack of strong, unbiased hypotheses. Targeting inflammatory
pathways in sIBM showed unsuccessful up to date. Similarly, targeting degenerative
pathways such as heat shock or myostatin pathways has not yet sufficiently proven to be
promising either.69 sIBM research is in high need of new therapeutic strategies with a strong
hypothesis and as such, KDM5A might represent a relevant and druggable target. Inhibitors
of KDM5 proteins are already being developed in cancer research.70, 71 Furthermore, KDM5A
and the DDR might be involved in the regenerative decline in degenerative disorders and
might as such represent a common therapeutic target. Ultimately, therapeutic strategies
might comprise the combined targeting of different key mediators of disease.

173
CONCLUDING REMARKS AND FUTURE DIRECTIONS
In this thesis we particularly capitalized on ongoing revolutions in: 1) genetics of IMD to
identify patients with very rare IMD and to study disease mechanisms of these disorders; 2)
proteomic studies of muscle, allowing an unbiased dissection of disease signatures in both
acquired and inherited muscle disorders.
This thesis elegantly illustrates the relevance of studying both inherited and acquired muscle
disorders characterized by slowly progressive muscular weakness, or at least grasping
clinical and biological patterns underlying disease.
We are on the verge of fascinating and exciting times in myology research and clinical
myology. Diagnosing patients with rare IMD and dissecting disease mechanisms of
degenerative muscle disorders will remain highly relevant with regard to rapid translation of
therapies to the clinics. By taking advantage of new revolutions in molecular genetics,
keeping up with new insights in muscle MR imaging and directed muscle biopsy studies, 50-
60% of patients with very rare IMD will be diagnosed within 3 months’ time. In this, we will
have to keep up with new methodological revolutions in genomics, proteomics and other
‘omics’ studies not represented in this doctoral thesis.
Translation of these therapies to the clinics is not a futuristic fantasy pipe dream anymore,
multiple therapies for inherited neuromuscular disorders are already being tested in clinical
trials or have even reached the status of “standard treatment”. These evolutions will
transform our largely contemplative diagnostic and supportive neuromuscular subspecialty
into one of a multitude of highly advanced curative therapies. We need to be prepared to
deal with this rapidly approaching new reality and will be in high need of clinicians with
extensive expertise in the field.

174
REFERENCES
1. Jackson CE. A clinical approach to muscle diseases. Seminars in neurology 2008;28:228-240.
2. Narayanaswami P, Weiss M, Selcen D, et al. Evidence-based guideline summary: diagnosis and
treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of
the American Academy of Neurology and the practice issues review panel of the American Association
of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-1463.
3. Mercuri E, Muntoni F. Muscular dystrophies. Lancet 2013;381:845-860.
4. Nigro V, Savarese M. Next-generation sequencing approaches for the diagnosis of skeletal muscle
disorders. Current opinion in neurology 2016;29:621-627.
5. Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen
AL. Classification and management of adult inflammatory myopathies. The Lancet Neurology
2018;17:816-828.
6. Mohassel P, Landon-Cardinal O, Foley AR, et al. Anti-HMGCR myopathy may resemble limb-girdle
muscular dystrophy. Neurology(R) neuroimmunology & neuroinflammation 2019;6:e523.
7. Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, et al. Sporadic late-onset nemaline myopathy:
clinico-pathological characteristics and review of 76 cases. Orphanet journal of rare diseases
2017;12:86.
8. Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB
reports 2017;50:175-185.
9. Chiang AP, Beck JS, Yen HJ, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3
ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proceedings of the National Academy of
Sciences of the United States of America 2006;103:6287-6292.
10. Lazzari E, Meroni G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: From muscular
dystrophy to tumours. The international journal of biochemistry & cell biology 2016;79:469-477.
11. Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta neuropathologica
2013;125:3-18.
12. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans.
Nature 2016;536:285-291.
13. Tajsharghi H, Thornell LE, Darin N, et al. Myosin heavy chain IIa gene mutation E706K is pathogenic
and its expression increases with age. Neurology 2002;58:780-786.
14. D'Amico A, Fattori F, Bellacchio E, Catteruccia M, Servidei S, Bertini E. A new de novo missense
mutation in MYH2 expands clinical and genetic findings in hereditary myosin myopathies.
Neuromuscular disorders : NMD 2013;23:437-440.
15. Cabrera-Serrano M, Fabian VA, Boutilier J, et al. Adult onset distal and proximal myopathy with
complete ophthalmoplegia associated with a novel de novo p.(Leu1877Pro) mutation in MYH2.
Clinical genetics 2015;88:573-578.
16. Lossos A, Oldfors A, Fellig Y, Meiner V, Argov Z, Tajsharghi H. MYH2 mutation in recessive myopathy
with external ophthalmoplegia linked to chromosome 17p13.1-p12. Brain : a journal of neurology
2013;136:e238.
17. Willis T, Hedberg-Oldfors C, Alhaswani Z, Kulshrestha R, Sewry C, Oldfors A. A novel MYH2 mutation in
family members presenting with congenital myopathy, ophthalmoplegia and facial weakness. Journal
of neurology 2016;263:1427-1433.
18. Hernandez-Lain A, Esteban-Perez J, Montenegro DC, Dominguez-Gonzalez C. Myosin myopathy with
external ophthalmoplegia associated with a novel homozygous mutation in MYH2. Muscle & nerve
2017;55:E8-e10.
19. Findlay AR, Harms MB, Pestronk A, Weihl CC. Homozygous recessive MYH2 mutation mimicking
dominant MYH2 associated myopathy. Neuromuscular disorders : NMD 2018;28:675-679.

175
20. Tajsharghi H, Hammans S, Lindberg C, et al. Recessive myosin myopathy with external
ophthalmoplegia associated with MYH2 mutations. European journal of human genetics : EJHG
2014;22:801-808.
21. Hellerschmied D, Clausen T. Myosin chaperones. Current opinion in structural biology 2014;25:9-15.
22. Lee CF, Melkani GC, Bernstein SI. The UNC-45 myosin chaperone: from worms to flies to vertebrates.
International review of cell and molecular biology 2014;313:103-144.
23. Gazda L, Pokrzywa W, Hellerschmied D, et al. The myosin chaperone UNC-45 is organized in tandem
modules to support myofilament formation in C. elegans. Cell 2013;152:183-195.
24. Oldfors A, DiMauro S. New insights in the field of muscle glycogenoses. Current opinion in neurology
2013;26:544-553.
25. Hedberg-Oldfors C, Oldfors A. Polyglucosan storage myopathies. Mol Aspects Med 2015;46:85-100.
26. Kilimann MW, Oldfors A. Glycogen pathways in disease: new developments in a classical field of
medical genetics. Journal of inherited metabolic disease 2015;38:483-487.
27. Hedberg-Oldfors C, Glamuzina E, Ruygrok P, et al. Cardiomyopathy as presenting sign of glycogenin-1
deficiency-report of three cases and review of the literature. Journal of inherited metabolic disease
2017;40:139-149.
28. Iijima H, Iwano R, Tanaka Y, et al. Analysis of GBE1 mutations via protein expression studies in
glycogen storage disease type IV: A report on a non-progressive form with a literature review.
Molecular genetics and metabolism reports 2018;17:31-37.
29. Paradas C, Akman HO, Ionete C, et al. Branching enzyme deficiency: expanding the clinical spectrum.
JAMA neurology 2014;71:41-47.
30. Mochel F, Schiffmann R, Steenweg ME, et al. Adult polyglucosan body disease: Natural History and
Key Magnetic Resonance Imaging Findings. Annals of neurology 2012;72:433-441.
31. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing
technologies. Nature reviews Genetics 2016;17:333-351.
32. Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for
translational research. Nature reviews Neurology 2016;12:294-309.
33. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a
large LGMD cohort of 4656 patients. Annals of clinical and translational neurology 2018;5:1574-1587.
34. Reddy HM, Cho KA, Lek M, et al. The sensitivity of exome sequencing in identifying pathogenic
mutations for LGMD in the United States. Journal of human genetics 2017;62:243-252.
35. Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome
sequencing for detecting exome variants. Proceedings of the National Academy of Sciences of the
United States of America 2015;112:5473-5478.
36. Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing
approaches cost-effective? A systematic review of the literature. Genetics in medicine : official journal
of the American College of Medical Genetics 2018;20:1122-1130.
37. Mantere T, Kersten S, Hoischen A. Long-Read Sequencing Emerging in Medical Genetics. Frontiers in
genetics 2019;10:426.
38. del Gaudio D, Yang Y, Boggs BA, et al. Molecular diagnosis of Duchenne/Becker muscular dystrophy:
enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative
genomic hybridization. Human mutation 2008;29:1100-1107.
39. Tattini L, D'Aurizio R, Magi A. Detection of Genomic Structural Variants from Next-Generation
Sequencing Data. Frontiers in bioengineering and biotechnology 2015;3:92.
40. Valipakka S, Savarese M, Johari M, et al. Copy number variation analysis increases the diagnostic yield
in muscle diseases. Neurology Genetics 2017;3:e204.
41. Johnson K, De Ridder W, Topf A, et al. Extending the clinical and mutational spectrum of TRIM32-
related myopathies in a non-Hutterite population. Journal of neurology, neurosurgery, and psychiatry
2019;90:490-493.

176
42. Zhou B, Ho SS, Zhang X, Pattni R, Haraksingh RR, Urban AE. Whole-genome sequencing analysis of
CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV
analysis. Journal of medical genetics 2018;55:735-743.
43. Merker JD, Wenger AM, Sneddon T, et al. Long-read genome sequencing identifies causal structural
variation in a Mendelian disease. Genetics in medicine : official journal of the American College of
Medical Genetics 2018;20:159-163.
44. La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis.
Nature reviews Genetics 2010;11:247-258.
45. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with
transcriptome sequencing. Science translational medicine 2017;9.
46. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a
joint consensus recommendation of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Genetics in medicine : official journal of the American College of
Medical Genetics 2015;17:405-424.
47. Brebner JA, Stockley RA. Polyclonal free light chains: a biomarker of inflammatory disease or
treatment target? F1000 medicine reports 2013;5:4.
48. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature
2016;537:347-355.
49. Dowling P, Murphy S, Ohlendieck K. Proteomic profiling of muscle fibre type shifting in neuromuscular
diseases. Expert review of proteomics 2016;13:783-799.
50. Gelfi C, Vasso M, Cerretelli P. Diversity of human skeletal muscle in health and disease: contribution of
proteomics. Journal of proteomics 2011;74:774-795.
51. Camargo A, Azuaje F, Wang H, Zheng H. Permutation - based statistical tests for multiple hypotheses.
Source code for biology and medicine 2008;3:15.
52. Deshmukh AS, Murgia M, Nagaraj N, Treebak JT, Cox J, Mann M. Deep proteomics of mouse skeletal
muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors.
Molecular & cellular proteomics : MCP 2015;14:841-853.
53. Beck M, Schmidt A, Malmstroem J, et al. The quantitative proteome of a human cell line. Mol Syst Biol
2011;7:549.
54. Kramer A, Green J, Pollard J, Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway
Analysis. Bioinformatics (Oxford, England) 2014;30:523-530.
55. Timmons JA, Szkop KJ, Gallagher IJ. Multiple sources of bias confound functional enrichment analysis
of global -omics data. Genome biology 2015;16:186.
56. Guttsches AK, Brady S, Krause K, et al. Proteomics of rimmed vacuoles define new risk allele in
inclusion body myositis. Annals of neurology 2017;81:227-239.
57. Li J, Yin C, Okamoto H, et al. Proteomic analysis of inclusion body myositis. Journal of neuropathology
and experimental neurology 2006;65:826-833.
58. Hutchinson DO, Jongbloed B. Two-dimensional gel electrophoresis in inclusion body myositis. Journal
of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2008;15:440-444.
59. Parker KC, Kong SW, Walsh RJ, et al. Fast-twitch sarcomeric and glycolytic enzyme protein loss in
inclusion body myositis. Muscle & nerve 2009;39:739-753.
60. Doppler K, Lindner A, Schutz W, Schutz M, Bornemann A. Gain and loss of extracellular molecules in
sporadic inclusion body myositis and polymyositis--a proteomics-based study. Brain pathology (Zurich,
Switzerland) 2012;22:32-40.
61. Roos A, Preusse C, Hathazi D, Goebel HH, Stenzel W. Proteomic Profiling Unravels a Key Role of
Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Frontiers in immunology
2019;10:1040.
62. Cardamone M, Darras BT, Ryan MM. Inherited myopathies and muscular dystrophies. Seminars in
neurology 2008;28:250-259.

177
63. Preisler N, Orngreen MC. Exercise in muscle disorders: what is our current state? Current opinion in
neurology 2018;31:610-617.
64. Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne Muscular Dystrophy:
retrospective analysis of 835 patients. Acta myologica : myopathies and cardiomyopathies : official
journal of the Mediterranean Society of Myology / edited by the Gaetano Conte Academy for the
study of striated muscle diseases 2012;31:121-125.
65. Kuperus E, Kruijshaar ME, Wens SCA, et al. Long-term benefit of enzyme replacement therapy in
Pompe disease: A 5-year prospective study. Neurology 2017;89:2365-2373.
66. High KA, Roncarolo MG. Gene Therapy. The New England journal of medicine 2019;381:455-464.
67. Roovers J, De Jonghe P, Weckhuysen S. The therapeutic potential of RNA regulation in neurological
disorders. Expert opinion on therapeutic targets 2018;22:1017-1028.
68. Nelson CE, Robinson-Hamm JN, Gersbach CA. Genome engineering: a new approach to gene therapy
for neuromuscular disorders. Nature reviews Neurology 2017;13:647-661.
69. Naddaf E, Barohn RJ, Dimachkie MM. Inclusion Body Myositis: Update on Pathogenesis and
Treatment. Neurotherapeutics : the journal of the American Society for Experimental
NeuroTherapeutics 2018;15:995-1005.
70. Mitsui E, Yoshida S, Shinoda Y, et al. Identification of ryuvidine as a KDM5A inhibitor. Scientific reports
2019;9:9952.
71. Vinogradova M, Gehling VS, Gustafson A, et al. An inhibitor of KDM5 demethylases reduces survival of
drug-tolerant cancer cells. Nature chemical biology 2016;12:531-538.

178

179
SUMMARY – SAMENVATTING

180
Primary muscle disorders comprise a large group of inherited and acquired diseases that
affect muscle structure, metabolism, or the function of muscle ion channels. Muscle
disorders presenting with slowly progressive muscle weakness constitute a group of
disorders eminently suited for a multi-level pattern recognition approach, clinically as well
as biologically. Given that only very few acquired muscle disorders present with slowly
progressive muscular weakness, an inherited muscle disorder (IMD) is typically suspected in
case of this clinical presentation. IMD constitute a clinically, genetically and
histopathologically very heterogeneous group of rare diseases with more than 160
genetically distinct entities identified, rendering the diagnostic process complex. Sporadic
inclusion body myositis (sIBM) constitutes a very relevant, clinically recognizable differential
diagnosis in patients over 50 years of age. A few other atypically presenting acquired muscle
disorders might also present slowly progressive muscle weakness, particularly idiopathic
inflammatory myopathies (IIM) with anti-HMGCR antibodies and the enigmatic, supposedly
very rare, putatively immune-mediated acquired myopathy, called sporadic late-onset
nemaline myopathy (SLONM).
The ultimate aim of this PhD thesis was to gain insights in pathomechanisms of both
inherited and acquired muscle disorders, clinically characterized by slowly progressive
muscle weakness and biologically ultimately by muscle degeneration. In this thesis I
particularly capitalized on ongoing revolutions in 1) genetics of IMD to identify patients with
very rare IMD and to study genotype-phenotype correlations and disease mechanisms of
these disorders; 2) proteomic studies of muscle, allowing an unbiased dissection of disease
signatures in both acquired and inherited muscle disorders.
This resulted in: 1) the identification of patients with rare IMD due to mutations in TRIM32,
BVES, VCP and other (novel) genes (still being studied), and the further unraveling of
pathomechanisms of these disorders; 2) unprecedented insights in the muscle proteome
and disease patterns of sIBM and VCP-related myopathy. Furthermore, we highlight the
unexpectedly high prevalence of patients with slowly progressing SLONM in whole exome
sequencing unsolved suspected IMD patient cohorts.
This PhD thesis nicely illustrates the relevance of studying both inherited and acquired
muscle disorders characterized by slowly progressive muscular weakness. We are on the
verge of fascinating and exciting times in myology research and clinical myology. Diagnosing
patients with rare IMD and dissecting disease mechanisms of muscle disorders will remain
highly relevant with regard to rapid translation of therapies to the clinics.

181
Spierziekten omvatten een grote groep van erfelijke en verworden aandoeningen die de
structuur, het metabolisme of de functie van ionenkanalen van de spier treffen. Spierziekten
die zich presenteren met traag progressieve spierziekte vormen een groep van
aandoeningen die uitermate geschikt zijn voor een benadering met patroonherkenning op
verscheidene niveaus, zowel klinisch als biologisch. Aangezien slechts weinig verworven
spierziekten zich presenteren met traag progressieve spierzwakte, wordt bij deze klinische
presentatie typisch de diagnose van een erfelijke spierziekte vermoed. Erfelijke spierziekten
vormen op zich ook een klinisch, genetisch en histopathologisch erg heterogene groep van
aandoeningen, met meer dan 160 verschillende genetische entiteiten, wat het diagnostisch
proces erg complex maakt. Sporadische ‘inclusion body’-myositis (sIBM) vormt een zeer
relevante en klinisch herkenbare differentiaaldiagnose in patiënten ouder dan 50 jaar.
Slechts enkele andere, zich atypisch presenterende, verworven spierziekten kunnen zich ook
met traag progressieve spierzwakte presenteren: inflammatoire myopathieën met anti-
HMGCR-antilichamen en de verondersteld uiterst zeldzame, enigmatische, waarschijnlijk
immuungemedieerde, verworven myopathie genaamd ‘sporadic late-onset nemaline
myopathy’ (SLONM).
Het ultieme doel van deze doctoraatsthesis was nieuwe inzichten verwerven in de
pathomechanismen van zowel erfelijke als verworven spierziekten, klinisch gekenmerkt
door traag progressieve spierzwakte en biologisch ultiem door spierdegeneratie. In deze
thesis maakte ik gebruik van aanhoudende revoluties in: 1) genetica van erfelijke
spierziekten met als doel het identificeren van patiënten met erg zeldzame spierziekten, het
bestuderen van genotype-fenotype correlaties en het ontrafelen van
ziektepathomechanismen; 2) proteomicsgebaseerde studies op spiermateriaal, met als doel
een onbevooroordeelde ontleding van ziektesignaturen van zowel erfelijke als verworven
spierziekten.
Dit resulteerde in: 1) de identificatie van patiënten met zeldzame erfelijke spierziekten t.g.v.
mutaties in TRIM32, BVES, VCP en andere (nieuwe) genen (die nog bestudeerd worden), en
een verdere ontrafeling van de ziektemechanismen van deze aandoeningen; 2) ongeziene
inzichten in het spierproteoom en ziektepatronen van sIBM en de VCP-gerelateerde
spierziekte. Hiernaast markeren we de onverwacht hoge prevalentie van patiënten met
traag progressieve SLONM in een cohorte van patiënten met het vermoeden van een WES-
onopgeloste erfelijke myopathie. Deze thesis illustreert de relevantie van het bestuderen
van zowel erfelijke als verworven spierziekten gekenmerkt door traag progressieve
spierzwakte. We staan aan de vooravond van erg fascinerende tijden in de
spierziektenresearch en –kliniek; het diagnosticeren en ontrafelen van deze ziekten zal
uiterst relevant blijven met het oog op snelle translatie van behandelingen naar de kliniek.

182

183
LIST OF COMMONLY USED
ABBREVIATIONS

184
ALS amyotrophic lateral sclerosis
ASO antisense oligonucleotide
bp base pairs
CADD Combined Annotation Dependent Depletion
CK creatine kinase
CNV copy number variant
DDR DNA damage response
DNA deoxyribonucleic acid
EM electron microscopy
EMG electromyography
ExAC Exome Aggregation Consortium
FDR false discovery rate
FLC free light chains
FSHD facioscapulohumeral dystrophy
FTD frontotemporal dementia
FVC forced vital capacity
GOF gain-of-function
GSD glycogen storage disorder
IBM inclusion body myopathy
IHC immunohistochemical
IIM idiopathic inflammatory myopathies
IMD inherited muscle disorder(s)
IPA Ingenuity Pathway Analysis
LGMD limb-girdle muscular dystrophy
LGMW limb-girdle muscular weakness
LOF loss-of-function
LRS long read sequencing
MGUS monoclonal gammopathy of unknown significance
MHC-I major histocompatibility complex I
MRI magnetic resonance imaging
MS mass spectrometry
MSP multisystem proteinopathy
MuSCs muscle stem cells
NCS nerve conduction studies
NGS next generation sequencing
NMD nonsense-mediated mRNA decay

185
OPMD oculopharyngeal muscular dystrophy
PCA principal component analysis
PDB Paget disease of bone
POPDC Popeye domain containing protein
sIBM sporadic inclusion body myositis
SLONM sporadic late-onset nemaline myopathy
SNP single nucleotide polymorphism
SNV single nucleotide variant
UZA Antwerp University Hospital
VCP valosin containing protein
VUS variant of unknown significance
WES whole exome sequencing
WGS whole genome sequencing

186

187
CURRICULUM VITAE

188
PERSONALIA
Name Willem De Ridder (MD)
Work address
Department of Neurology
Antwerp University Hospital (UZA)
Wilrijkstraat 10, B-2650 Edegem, BELGIUM
Phone number (+32) (0)3 265 17 81
Date of birth October 27th, 1989
Email [email protected]
Nationality Belgian
EDUCATION
Institute Period
Doctoral education program at the Antwerp Doctoral School University of Antwerp 2015-
Master after master in specialist medicine (neurology) University of Antwerp 2014-
Master in Medicine (magna cum laude (84%)) University of Antwerp 2010-2014
Clinical electrocardiography (post-academic education)
(summa cum laude)
University of Antwerp 2010-2011
Bachelor in Medicine (magna cum laude (84%)) University of Antwerp 2007-2010
Diploma of secondary school, majors in Latin-Mathematics Sint-Ursula Lyceum Lier 2001-2007
WORK EXPERIENCE
First year of neurology residency at the ZNA Middelheim hospital (Antwerp, Belgium): 01/08/2014 –
31/07/2015.
PhD student in the Neurogenetics Group, University of Antwerp (01/08/2015 – 31/08/2019), with
Prof. Dr. Peter De Jonghe and Prof. Dr. Jonathan Baets as doctoral supervisors.
Full time clinical residency in neurology at the University Hospital of Antwerp (Antwerp, Belgium):
01/09/2019 – present.
SKILLS
Specific experience in neuromuscular disorders: weekly consultations at the Neuromuscular
Reference Centre (NMRC) of the University Hospital of Antwerp since 01/08/2015.
Investigation of numerous patients with myopathies (solved and unsolved cases).
Nerve conduction studies and EMG: moderate experience
Single-fiber EMG (stimulated and voluntary): basic experience
Interpretation of Muscle MRI imaging:
Selected attendee of the 2nd MYO-MRI Training School in Paris (May 16-18).
Experience with regard to pattern recognition: retrospective and prospective interpretation
of MRI images of more than 400 patients up till now.

189
Myopathology:
Moderate general knowledge and experience.
Teaching by Prof. Dr. Jonathan Baets and Prof. Dr. Martin Lammens.
Specific experience with the evaluation of histological, histo-enzymological and
immunohistochemical stainings in inflammatory myopathies.
Molecular genetics:
Extensive experience with the interpretation of whole exome sequencing data and the
evaluation of the causality of variants in myopathies.
Proteomics and protein work:
Insights in the strengths, weaknesses and pitfalls of proteomic experiments.
Pathway level: experience with Ingenuity® Pathway Analysis (IPA®) for the study of pathway
enrichment.
Western blotting, interpretation, quantification and troubleshooting: moderate experience.
Metabolic myopathies:
Selected attendee of the Metabolic Myopathies focus course 2016 in Paris.
Basic experience with regard to the usefulness and interpretation of biochemical
investigations in suspected metabolic myopathies.
LANGUAGES
Dutch: native.
English: good.
French: good.
German: average.
PRESENTATIONS
2019 Poster presentation at the 2019 PNS Annual Meeting, June 22-25, 2019, Genua, Italy: ‘NARS
As A Candidate Gene In A Dominant CMT2 Family’
2019 Poster presentation at the MYOLOGY 2019 congress, March 25-28, Bordeaux, France:
‘Homozygosity of the autosomal dominant VCP p.Arg159His mutation’
2018 Poster presentation at the 15th international congress of neuromuscular diseases, July 6-
10, Vienna, Austria: ‘Homozygosity of the autosomal dominant VCP p.Arg159His mutation’
2018 Oral presentation at the 4th joint meeting Belgian-Dutch neuromuscular study club and
German reference center for neuromuscular diseases, Vaals, the Netherlands, May 25-26,
2018: ‘BVES loss-of-function mutations in LGMD2X with arrhythmia’
2016 Poster session at the EMBL–Wellcome Genome Campus Conference: Proteomics in Cell
Biology and Disease Mechanisms, September 14-17, Heidelberg, Germany: ‘Differential
proteomics to reveal protein signatures involved in the unexplained pathophysiology of
sporadic Inclusion Body Myositis’
2016 Active participation in the interactive session ‘Update on inflammatory myopathies’ at the

190
11th European Congress of Neuropathology in Bordeaux, France, July 7-9. (session together
with Prof. Dr. Martin Lammens, Prof. Dr. Werner Stenzel and Prof. Dr. Jan De Bleecker;
presentation of cases and images).
2016 Poster session at the 5th Internation Congress of Myology, Lyon, France, March 14-18,
2016: ‘LDB3 mutation in a distal weakness family’
2015 Selected speaker at the 97th Meeting of the Belgian-Dutch Neuromuscular Study Club,
UMC Utrecht, October 14, 2015: ‘LDB3 mutation in a distal weakness family’.
RELEVANT COURSES
2019 Boerhaave course: ‘'state of the art' behandeling van neuromusculaire aandoeningen’; 11th
of January, 2019, Amsterdam
2018 Boerhaave course: ‘Hoofd, schouders, knie en teen: diagnostiek van neuromusculaire
aandoeningen’; 12th of January, 2018, Amsterdam
2017 Selected attendee of the 20th Summer School of Myology of Paris, France, June 15-23,
2017
2016 Selected attendee of the Metabolic myopathies focus course in Paris, Frane, November 3-5,
2016. Active participation and presentation of a case concerning a patient with an unsolved
glycogenosis with polyglucosan bodies.
2016 Selected attendee of the 2nd MYO-MRI Training School in Paris (May 16-18). Plenary
presentation of multiple cases during the interactive sessions.
PARTICIPATION IN RELEVANT SCIENTIFIC CONFERENCES
2019 2019 PNS Annual Meeting, June 22-26, 2019, Genoa, Italy
2019 104th Meeting of the Belgian-Dutch Neuromuscular Study Club, April 3, 2019, Utrecht, the
Netherlands
2019 6th International Congress of Myology, March 25-28, 2019, Bordeaux, France
2018 103rd Meeting of the Belgian-Dutch Neuromuscular Study Club, October 10, 2019, Utrecht,
the Netherlands
2018 15th international congress of neuromuscular diseases, July 6-10, 2018, Vienna, Austria
2018 4th joint meeting Belgian-Dutch neuromuscular study club and German reference center
for neuromuscular diseases, Vaals, the Netherlands, May 25-26, 2018
2017 22nd International congress of the World Muscle Society, St. Malo, France, October 3-7,
2017
2017 100th Meeting of the Belgian-Dutch Neuromuscular Study Club, UMC Utrecht, April 5, 2017
2016 EMBL–Wellcome Genome Campus Conference: Proteomics in Cell Biology and Disease
Mechanisms, Heildelberg, Germany, September 14-17, 2016
2016 98th Meeting of the Belgian-Dutch Neuromuscular Study Club, UZA, Antwerpen, April 13,
2016
2016 11th European Congress of Neuropathology in Bordeaux, France, July 7-9, 2016

191
2016 5th Internation Congress of Myology, Lyon, France, March 14-18
2015 97th Meeting of the Belgian-Dutch Neuromuscular Study Club, UMC Utrecht, October 14,
2015
PUBLICATIONS
De Ridder W, Azmi A, Clemen CS, Eichinger L, Hofmann A, Schröder R, Johnson K, Töpf A, Straub V,
De Jonghe P, Maudsley S, De Bleecker JL, Baets J. A tale of the unexpected: multisystem
proteinopathy due to a homozygous p.Arg159His VCP mutation. Neurology. 2019
Hedberg-Oldfors C, De Ridder W, Kalev O, Böck K, Visuttijai K, Caravias G, Straub V, Töpf A, Baets J,
Oldfors A. Functional characterization of GYG1 variants in two patients with myopathy and
glycogenin-1 deficiency. Neuromuscular Disorders. 2019
De Ridder W*, Nelson I*, Asselbergh B, De Paepe B, Beuvin M, Ben Yaou R, Masson C, Boland A,
Deleuze JF, Maisonobe T, Eymard B, Symoens S, Schindler R, Brand T, Johnson K, Töpf A, Straub V, De
Jonghe P, De Bleecker JL, Bonne G†, Baets J†. Muscular dystrophy with arrhythmia caused by loss-of-
function mutations in BVES. Neurol Genet. 2019 (*These authors contributed equally to the
manuscript as first authors. †These authors contributed equally to the manuscript as last authors.)
Heytens K, De Ridder W, De Bleecker J, Heytens L, Baets J. Exertional rhabdomyolysis: Relevance of
clinical and laboratory findings, and clues for investigation. Anaesthesia and intensive care. 2019
Knuiman GJ, Kusters B, Eshuis L, Snoeck M, Lammens M, Heytens L, De Ridder W, Baets J, Scalco RS,
Quinlivan R, Holton J, Bodi I, Wraige E, Radunovic A, von Landenberg C, Reimann J, Kamsteeg EJ,
Sewry C, Jungbluth H, Voermans NC. The histopathological spectrum of malignant hyperthermia and
rhabdomyolysis due to RYR1 mutations. Journal of neurology. 2019
Johnson K*, De Ridder W*, Topf A, Bertoli M, Phillips L, De Jonghe P, Baets J, Deconinck T, Rakocevic
Stojanovic V, Perić S, Durmus H, Jamal-Omidi S, Nafissi S, Mongini T, Łusakowska A, Busby M, Miller
J, Norwood F, Hudson J, Barresi R, Lek M, MacArthur DG, Straub V. Extending the clinical and
mutational spectrum of TRIM32-related myopathies in a non-Hutterite population. J Neurol
Neurosurg Psychiatry. 2019 (*these authors contributed equally)
Johnson K, Bertoli M, Phillips L, Töpf A, Van den Bergh P, Vissing J, Witting N, Nafissi S, Jamal-Omidi
S, Łusakowska A, Kostera-Pruszczyk A, Potulska-Chromik A, Deconinck N, Wallgren-Pettersson C,
Strang-Karlsson S, Colomer J, Claeys KG, De Ridder W, Baets J, von der Hagen M, Fernández-Torrón
R, Zulaica Ijurco M, Espinal Valencia JB, Hahn A, Durmus H, Willis T, Xu L, Valkanas E, Mullen TE, Lek
M, MacArthur DG, Straub V. Detection of variants in dystroglycanopathy-associated genes through
the application of targeted whole-exome sequencing analysis to a large cohort of patients with
unexplained limb-girdle muscle weakness. Skeletal muscle. 2018

192
Østergaard ST, Johnson K, Stojkovic T, Krag T, De Ridder W, De Jonghe P, Baets J, Claeys KG,
Fernández-Torrón R, Phillips L, Topf A, Colomer J, Nafissi S, Jamal-Omidi S, Bouchet-Seraphin C,
Leturcq F, MacArthur DG, Lek M, Xu L, Nelson I, Straub V, Vissing J. Limb girdle muscular dystrophy
due to mutations in POMT2. J Neurol Neurosurg Psychiatry. 2017
PARTICIPATION IN CLINICAL STUDIES
Prospective Natural History Study of Patients With Myotubular Myopathy and Other CentroNuclear
Myopathies (NatHis-CNM)
ClinicalTrials.gov identifier: NCT03351270; participation as co-investigator
Early Phase Human Drug Trial to Investigate Dynamin 101 (DYN101) in Patients ≥ 16 Years With
Centronuclear Myopathies (Unite-CNM)
ClinicalTrials.gov identifier: NCT04033159; participation as co-investigator

193

194

195
ACKNOWLEDGEMENTS -
DANKWOORD

196
Vijf jaar geleden kreeg ik de kans om te starten aan de opleiding neurologie. Tijdens mijn
eerste, uiterst boeiende jaar als assistent neurologie in het AZ Middelheim, kreeg ik al zeer
snel voor mezelf de bevestiging dat neurologie inderdaad de juiste keuze was geweest.
Terwijl ik in dat jaar vooral de taak had om als clinicus te groeien, merkte ik al dat het niet
evident zou zijn om tijd te vinden voor wetenschap in een voltijds klinisch traject. Door mijn
ontluikende interesse in de wereld van de neuromusculaire aandoeningen, was ik reeds in
contact gekomen met Peter De Jonghe en Jonathan Baets. Ik besefte dat een PhD-traject
mijn beste kans was op verdere verdieping in de neuromusculaire aandoeningen, niet enkel
op wetenschappelijk vlak, doch ook klinisch. Ik ben hen dan ook enorm dankbaar om me
effectief de kans te geven om aan een PhD-traject te starten. Peter was tot dan toe op
wetenschappelijk gebied weinig actief op vlak van spierziekten, aangezien hij als
groepsleider van de Neurogenetics Group reeds researchprojecten rond een brede waaier
aan neurologische topics superviseerde. Hij had echter wel over een verloop van meer dan
15 jaar biosamples (DNA, spierbiopsiemateriaal) verzameld van een belangrijk aantal
patiënten met genetisch onopgeloste erfelijke spierziekten, alsook van patiënten met
zeldzame spierziekten zoals sporadic inclusion body myositis. Hij had het werk voortgezet
van Em. Professor J.J. Martin. Jonathan had zich verder ontwikkeld tot een expert op het
vlak van myopathologie, waartoe ik ook ingeleid werd door hem en Prof. Dr. M. Lammens.
Deze elementen maakten dat ik in de uitzonderlijke situatie terecht kwam om een
researchproject aan te vatten rond een unieke (quasi maagdelijke) patiëntencohorte en dat
toch alle nodige expertise aanwezig was. Ik ben hen vanzelfsprekend uiterst dankbaar voor
hun vooruitziendheid.
I would like to thank the members of the jury, Prof. Dr. Bart Loeys, Prof. Dr. Patrick Cras, Dr.
Umesh Badrising and Prof. Dr. Werner Stenzel, for their constructive comments and their
willingness to act as a jury member at my PhD defence.
Jonathan, ik ben je erg dankbaar voor de begeleiding die ik heb gekregen. Dit reeds van
meerdere maanden voor mijn afstuderen als master in de geneeskunde. Hoewel ik het
klinische veld van de neurologie nog verder moest verkennen, merkte ik al snel dat de
diagnostische puzzels binnen de neuromusculaire aandoeningen me ongelofelijk
aanspraken. Je hebt me begeleid bij het zoeken van mijn weg binnen de opleiding
neurologie. Het was een gouden raad om eerst een jaar brede klinische ervaring op te doen
alvorens in de diepte te duiken. Hierna kreeg mijn onderzoeksproject vorm onder de
raadgevingen van Peter en jou. Jullie hebben me de unieke kans gegeven om vier jaar full
time research te doen; dit natuurlijk met de nodige klinische blootstelling, waarbij ik de kans

197
kreeg om elk puzzelstukje zelf te leren leggen: van de kliniek, over de anatomopathologie
tot de genetica en de nodige functionele experimenten. Het is een unieke situatie om de
patiënt zelf te kunnen zien, elk mogelijk klinisch detail zelf te kunnen oppikken (‘deep
phenotyping’) en dan met deze finesses in de vingers ook wetenschappelijk in de diepte te
kunnen duiken. Ik heb me enorm geamuseerd tijdens de neuromusculaire raadplegingen,
niet alleen omdat deze materie zo boeiend is, maar ook dankzij je stijl als arts. Je leerde me
de semiologie van de neuromusculaire aandoeningen op een haast evocatieve manier te
vatten om zo Gordiaanse knopen te ontwarren. De raadplegingen werden doorspekt door
de nodige portie humor en zelfrelativering, wat toch helpt om zaken draaglijker te maken.
Het bestuderen van spierbiopten, waarbij kleuringen niet zelden iets weg hebben van
abstracte kunst, mondde niet zelden uit in haast poëtische, allegorische beschrijvingen, die
me hielpen om de werkelijke betekenis van afwijkingen écht in de vingers te krijgen. Ik besef
hiernaast heel goed dat het uniek is dat je – ondanks je drukke agenda – al mijn
hersenspinsels, schrijfsels en presentaties effectief op korte termijn nalas. Tot slot
apprecieer ik ook de oprechte bezorgdheid die je soms (tot vaak) uit wanneer ik broodnodig
een banaan moet eten. Ik had me geen betere mentor kunnen wensen.
Peter, zonder jou had ik nooit deze unieke kans kunnen krijgen. Hier zal ik je altijd dankbaar
voor blijven. Met al je ervaring had je een perfect plan uitgedacht voor een project met een
‘veilig’ luik rond de genetica van spierziekten en hiernaast een erg uitdagend (en meer
risicovol) luik rond sIBM. Voor mij persoonlijk is dit alleszins zeer goed uitgedraaid: ik heb op
heel veel vlakken erg veel ervaring kunnen opdoen – en het heeft ook de output opgeleverd
die nodig is voor een PhD-traject. Met je nuchtere en kritische blik heb je me vaak als
onervaren wetenschapper met de voeten op de grond gezet en heb me op de nodige
momenten ook geleerd geduldig te zijn. Je passie omtrent neuromusculaire aandoeningen
heeft helemaal op me afgestraald.
Tine, je hebt me als jonge, onvervaren wetenschapper al snel wat lessen geleerd en me
verder opgevoed. Ik denk dat je in het begin door mijn eerder introverte aard niet goed kon
inschatten wat je aan me had, maar ik denk dat ik je later toch heb kunnen duidelijk maken
hoeveel belang ik hechtte aan je raadgevingen en hoe dankbaar ik was voor het werk dat je
voor mij verzette. Ik ken weinig (geen?) mensen die zo gepassioneerd zijn door genetica als
jij, dat heb je me absoluut ook bijgebracht.
Iris en Elke, onze rotsen in de branding voor het NMRC, waar we altijd op kunnen rekenen.
Ik ben jullie enorm dankbaar voor jullie hulp bij het verzamelen van DNA-stalen van

198
patiënten en hun familieleden. Voor de (immer boeiende) huisbezoeken die we samen
gedaan hebben doorheen Vlaanderen. Ik kon steeds op jullie rekenen. Jullie lieve, zorgzame
aard apprecieer ik enorm, dit is een zegen voor onze patiënten – en ook ik kon ook steeds
op jullie rekenen, zeker ook bij nutritionele problemen.
Hannah en Danique, ik heb met jullie beiden een hele tijd het bureau gedeeld. Ik heb erg
veel gehad aan het uitwisselen van gedachten, soms ook aan debriefen over
struikelblokken. Danique, jou ben ik ook erg dankbaar voor de praktische hulp die je me
hebt geboden nadat we van Tine binnen de groep afscheid hadden moeten nemen. Ik zal
ook nooit vergeten hoe vaak je me gevoederd heb – en hier verdient Ron dus ook een
dankwoord!
Nina, jij bracht steeds een vrolijke, jeugdige noot (ik voelde me dan wel soms oud…). Verder
terugdenkend aan de tijd van de Neurogenetics Group: Sarah, Jolien, Inès, Tania, Katia,
Simona, bedankt voor deze tijd.
Jonathan DW, graag zou ik je hier ook vermelden. Het is erg leuk om de neuromusculaire
passie met jou te kunnen delen. Ik hoop oprecht dat je je weg in de neuromusculaire wereld
verder kan vinden. We hebben als ‘zaalbroeders’ reeds afgezien, maar ook heel wat plezier
gehad. Voor hulp bij mijn ontwikkeling als clinicus, helemaal in het begin (en natuurlijk ook
voor het plezier op de werkvloer), wil ik ook nog mijn andere (ex-)collega’s bedanken, in het
bijzonder Pegah, Massi, Lothar (die me mijn eerste EMG-stapjes leerde zetten), Géraldine.
Vincent, mijn eerste congres was samen met jou en ook voor jou was dit in zekere zin terug
een ‘eerste’ congres. Je hebt me hier – en ook op andere congressen – voorgesteld aan heel
wat prominente figuren uit het neuromusculaire veld. We hebben heel wat aangename
gesprekken gevoerd, niet enkel op congres, maar ook hier in Vlaanderen. Ik apprecieer je
erg als – integere -- wetenschapper en als mens.
Karim, I would like to thank you for all your help with the creation of the unique proteomic
dataset on sIBM. Thanks a lot for all the brainstorm sessions and teaching concerning mass
spectrometry and relevant lab-related matters. I really appreciate your kindness and your
willingness to help.
I would also like to thank Stuart Maudsley, who made it possible to establish this proteomic
dataset, as well as his group members that helped me in the lab.
Bob, bedankt om me bij te staan bij het luik microscopie en het afleveren van erg mooie
figuren. Het was en is erg aangenaam om met je samen te werken. Natacha en Safoura,
bedankt voor jullie hulp bij het vervaardigen van de talloze kleuringen die voor de
verscheidene projecten nodig waren.

199
Ik heb niet zelden raad en hulp gevraagd aan andere collega’s uit het V-gebouw, in het
bijzonder Elias en Sven. Bedankt dat ik steeds met vragen bij jullie terecht kon!
Last but not least… Hiernaast zou ik natuurlijk ook vooral Lise, mijn wederhelft, en mijn
familie willen bedanken voor alle steun die ik gekregen heb. Lise, tijdens deze periode van
vier jaar full time research, zette ik een geweldige stap in mijn leven door met jou te
trouwen. Dit was met voorsprong de mooiste dag van mijn leven. Mijn ouders, zus Anne en
schoonbroer Floris waren er steeds voor mij en hebben me niet alleen mentaal maar ook
praktisch heel erg gesteund. Zonder jullie steun zou ik het niet gekund hebben.
Ook de schoonfamilie zou ik willen bedanken voor de moral support en de interesse die ze
hadden in het werk dat ik leverde. Ten slotte zou ik ook nog mijn vrienden willen bedanken
voor hun steun, in het bijzonder Olivier, die steeds klaarstond om een luisterend oor te
bieden en om strategische plannen te bedenken.
Willem






















