
1 TITLE An updated phylogeny of the Alphaproteobacteria reveals ...
67
1 TITLE 1 An updated phylogeny of the Alphaproteobacteria reveals that the parasitic Rickettsiales 2 and Holosporales have independent origins 3 AUTHORS 4 Sergio A. Muñoz-Gómez 1,2 , Sebastian Hess 1,2 , Gertraud Burger 3 , B. Franz Lang 3 , 5 Edward Susko 2,4 , Claudio H. Slamovits 1,2* , and Andrew J. Roger 1,2* 6 AUTHOR AFFILIATIONS 7 1 Department of Biochemistry and Molecular Biology; Dalhousie University; Halifax, 8 Nova Scotia, B3H 4R2; Canada. 9 2 Centre for Comparative Genomics and Evolutionary Bioinformatics; Dalhousie 10 University; Halifax, Nova Scotia, B3H 4R2; Canada. 11 3 Department of Biochemistry, Robert-Cedergren Center in Bioinformatics and 12 Genomics, Université de Montréal, Montreal, Quebec, Canada. 13 4 Department of Mathematics and Statistics; Dalhousie University; Halifax, Nova Scotia, 14 B3H 4R2; Canada. 15 16 *Correspondence to: Department of Biochemistry and Molecular Biology; Dalhousie 17 University; Halifax, Nova Scotia, B3H 4R2; Canada; 1 902 494 2881, [email protected] 18
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of 1 TITLE An updated phylogeny of the Alphaproteobacteria reveals ...

1
TITLE 1
An updated phylogeny of the Alphaproteobacteria reveals that the parasitic Rickettsiales 2
and Holosporales have independent origins 3
AUTHORS 4
Sergio A. Muñoz-Gómez1,2, Sebastian Hess1,2, Gertraud Burger3, B. Franz Lang3, 5
Edward Susko2,4, Claudio H. Slamovits1,2*, and Andrew J. Roger1,2* 6
AUTHOR AFFILIATIONS 7
1 Department of Biochemistry and Molecular Biology; Dalhousie University; Halifax, 8
Nova Scotia, B3H 4R2; Canada. 9
2 Centre for Comparative Genomics and Evolutionary Bioinformatics; Dalhousie 10
University; Halifax, Nova Scotia, B3H 4R2; Canada. 11
3 Department of Biochemistry, Robert-Cedergren Center in Bioinformatics and 12
Genomics, Université de Montréal, Montreal, Quebec, Canada. 13
4 Department of Mathematics and Statistics; Dalhousie University; Halifax, Nova Scotia, 14
B3H 4R2; Canada. 15
16
*Correspondence to: Department of Biochemistry and Molecular Biology; Dalhousie 17
University; Halifax, Nova Scotia, B3H 4R2; Canada; 1 902 494 2881, [email protected] 18

2
ABSTRACT 19
The Alphaproteobacteria is an extraordinarily diverse and ancient group of bacteria. 20
Previous attempts to infer its deep phylogeny have been plagued with methodological 21
artefacts. To overcome this, we analyzed a dataset of 200 single-copy and conserved 22
genes and employed diverse strategies to reduce compositional artefacts. Such 23
strategies include using novel dataset-specific profile mixture models and recoding 24
schemes, and removing sites, genes and taxa that are compositionally biased. We 25
show that the Rickettsiales and Holosporales (both groups of intracellular parasites of 26
eukaryotes) are not sisters to each other, but instead, the Holosporales has a derived 27
position within the Rhodospirillales. A synthesis of our results also leads to an updated 28
proposal for the higher-level taxonomy of the Alphaproteobacteria. Our robust 29
consensus phylogeny will serve as a framework for future studies that aim to place 30
mitochondria, and novel environmental diversity, within the Alphaproteobacteria. 31
KEYWORDS: Holosporaceae, Holosporales, mitochondria, origin, Finniella inopinata, 32
Stachyamoba, Peranema, photosynthesis, Rhodospirillales, Azospirillaceae, 33
Rhodovibriaceae. 34

3
INTRODUCTION 35
The Alphaproteobacteria is an extraordinarily diverse and disparate group of bacteria 36
and well-known to most biologists for also encompassing the mitochondrial lineage 37
(Williams, Sobral, and Dickerman 2007; Roger, Muñoz-Gómez, and Kamikawa 2017). 38
The Alphaproteobacteria has massively diversified since its origin, giving rise to, for 39
example, some of the most abundant (e.g., Pelagibacter ubique) and metabolically 40
versatile (e.g., Rhodobacter sphaeroides) cells on Earth (Giovannoni 2017; Madigan, 41
Jung, and Madigan 2009). The basic structure of the tree of the Alphaproteobacteria 42
has largely been inferred through the analyses of 16S rRNA genes and several 43
conserved proteins (Garrity 2005; Lee et al. 2005; Rosenberg et al. 2014; Fitzpatrick, 44
Creevey, and McInerney 2006; Williams, Sobral, and Dickerman 2007; Brindefalk et al. 45
2011; Georgiades et al. 2011; Thrash et al. 2011; Luo 2015). Today, eight major orders 46
are well recognized, namely the Caulobacterales, Rhizobiales, Rhodobacterales, 47
Pelagibacterales, Sphingomonadales, Rhodospirillales, Holosporales and Rickettsiales 48
(the latter two formerly grouped into the Rickettsiales sensu lato), and their 49
interrelationships have also recently become better understood (Viklund, Ettema, and 50
Andersson 2012; Viklund et al. 2013; Rodríguez-Ezpeleta and Embley 2012; Wang and 51
Wu 2014, 2015). These eight orders were grouped into two subclasses by Ferla et al. 52
(2013): the subclass Rickettsiidae comprising the order Rickettsiales and 53
Pelagibacterales, and the subclass Caulobacteridae comprising all other orders. 54
The great diversity of the Alphaproteobacteria itself presents a challenge to deciphering 55
the deepest divergences within the group. Such diversity encompasses a broad 56
spectrum of genome (nucleotide) and proteome (amino acid) compositions (e.g., the 57
A+T%-rich Pelagibacterales versus the G+C%-rich Acetobacteraceae) and molecular 58
evolutionary rates (e.g., the fast-evolving Pelagibacteriales, Rickettsiales or 59
Holosporales versus many slow-evolving species in the Rhodospirillales) (Ettema and 60
Andersson 2009). This diversity may lead to pervasive artefacts when inferring the 61
phylogeny of the Alphaproteobacteria, e.g., long-branch attraction (LBA) between the 62
Rickettsiales and Pelagibacterales, especially when including mitochondria (Rodríguez-63
Ezpeleta and Embley 2012; Viklund, Ettema, and Andersson 2012; Viklund et al. 2013; 64

4
Luo 2015). Moreover, there are still important unknowns about the deep phylogeny of 65
the Alphaproteobacteria (Williams, Sobral, and Dickerman 2007; Ferla et al. 2013), for 66
example, the divergence order among the Rhizobiales, Rhodobacterales and 67
Caulobacterales (Williams, Sobral, and Dickerman 2007), the monophyly of the 68
Pelagibacterales (Viklund et al. 2013) and the Rhodospirillales (Ferla et al. 2013), and 69
the precise placement of the Rickettsiales and its relationship to the Holosporales 70
(Wang and Wu 2015; Martijn et al. 2018). 71
Systematic errors stemming from using over-simplified evolutionary models (which often 72
do not fit complex data as well by, for example, not accounting for compositional 73
heterogeneity across sites or branches) are perhaps the major confounding and limiting 74
factor to inferring deep evolutionary relationships; the number of taxa and genes (or 75
sites) can also be important factors. Previous multi-gene tree studies of the 76
Alphaproteobacteria were compromised by at least one of these problems, namely, 77
simpler or less realistic evolutionary models (because they were not available at the 78
time; e.g., Williams, Sobral, and Dickerman 2007 used the simple WAG+Γ4 model that 79
cannot account for compositional heterogeneity across sites), poor or uneven taxon 80
sampling (because the focus was too narrow or few genomes were available; e.g., 81
Williams, Sobral, and Dickerman 2007 had very few rhodospirillaleans and no 82
holosporaleans; Georgiades et al. 2011 included only 42 alphaproteobacteria with only 83
one pelagibacteralean) or a small number of genes (because the focus was 84
mitochondria; e.g., Rodríguez-Ezpeleta and Embley 2012 used 24 genes; Wang and 85
Wu 2015 relied on 29 genes; Martijn et al. 2018 also used 24 genes; or because only a 86
small set of 28 compositionally homogeneous genes was used, e.g., Luo 2015). The 87
most recent study on the phylogeny of the Alphaproteobacteria, and mitochondria, 88
attempted to counter systematic errors (or phylogenetic artefacts) by reducing amino 89
acid compositional heterogeneity (Martijn et al. 2018). Even though some deep 90
relationships were not robustly resolved, these analyses suggested that the 91
Pelagibacterales, Rickettsiales and Holosporales, which have compositionally-biased 92
genomes, are not each other’s closest relatives (Martijn et al. 2018). A resolved and 93
robust phylogeny of the Alphaproteobacteria is fundamental to addressing questions 94
such as how streamlined bacteria, intracellular parasitic bacteria, or mitochondria 95

5
evolved from their alphaproteobacterial ancestors. Therefore, a systematic study of the 96
different biases affecting the phylogeny of the Alphaproteobacteria, and its underlying 97
data, is much needed. 98
Here, we revised the phylogeny of the Alphaproteobacteria by using a large dataset of 99
200 conserved single-copy genes and employing carefully designed strategies aimed at 100
alleviating phylogenetic artefacts. We found that amino acid compositional 101
heterogeneity, and more generally long-branch attraction, were major confounding 102
factors in estimating phylogenies of the Alphaproteobacteria. In order to counter these 103
biases, we used novel dataset-specific profile mixture models and recoding schemes 104
(both specifically designed to ameliorate compositional heterogeneity), and removed 105
sites, genes and taxa that were compositionally biased. We also present three draft 106
genomes for endosymbiotic alphaproteobacteria belonging to the Rickettsiales and 107
Holosporales: (1) an undescribed midichloriacean endosymbiont of Peranema 108
trichophorum, (2) an undescribed rickettsiacean endosymbiont of Stachyamoeba 109
lipophora, and (3) the holosporalean ‘Candidatus Finniella inopinata’, an endosymbiont 110
of the rhizarian amoeboflagellate Viridiraptor invadens (Hess, Suthaus, and Melkonian 111
2015). Our results provide the first strong evidence that the Holosporales is unrelated to 112
the Rickettsiales and originated instead from within the Rhodospirillales. We incorporate 113
these and other insights regarding the deep phylogeny of the Alphaproteobacteria into 114
an updated taxonomy. 115
RESULTS 116
The genomes and phylogenetic positions of three novel endosymbiotic 117
alphaproteobacteria (Rickettsiales and Holosporales) 118
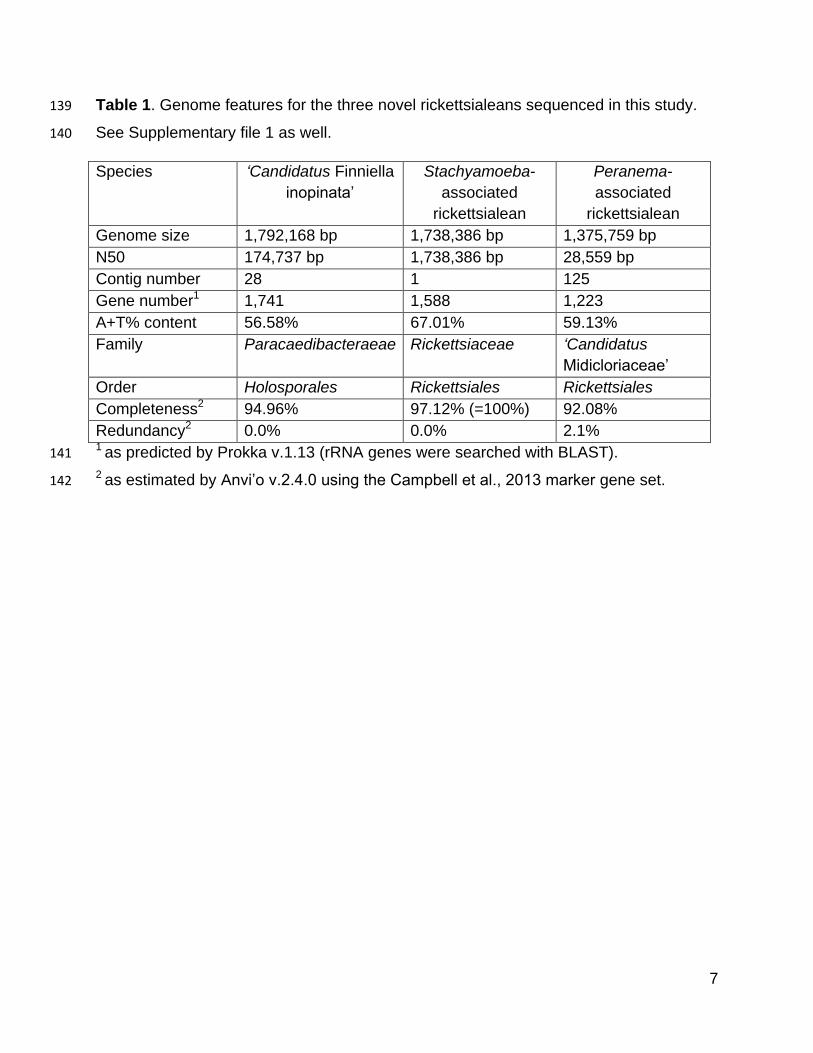
We sequenced the genomes of the novel holosporalean ‘Candidatus Finniella 119
inopinata’, an endosymbiont of the rhizarian amoeboflagellate Viridiraptor invadens 120
(Hess, Suthaus, and Melkonian 2015), and two undescribed rickettsialeans, one 121
associated with the heterolobosean amoeba Stachyamoeba lipophora and the other 122
with the euglenoid flagellate Peranema trichophorum. The three genomes are small with 123
a reduced gene number and high A+T% content, strongly suggesting an endosymbiotic 124
lifestyle (Table 1). Comparisons of their rRNA genes show that these genomes are truly 125

6
novel, being considerably divergent from other described alphaproteobacteria. As of 126
February 2018, the closest 16S rRNA gene to that of the Stachyamoeba-associated 127
rickettsialean belongs to Rickettsia massiliae str. AZT80, with only 88% identity. On the 128
other hand, the closest 16S rRNA gene to that of the Peranema-associated 129
rickettsialean belongs to an endosymbiont of Acanthamoeba sp. UWC8, which is only 130
92% identical. Phylogenetic analysis of both the 16S rRNA gene and a dataset that 131
comprises 200 single-copy conserved marker genes (see below) confirm that each 132
species belongs to different families and orders within the Alphaproteobacteria 133
(Supplementary file 1 and Figure 2-figure supplement 1). ‘Candidatus Finniella 134
inopinata’ belongs to the recently described ‘Candidatus Paracaedibacteraceae’ in the 135
Holosporales (Hess, Suthaus, and Melkonian 2015), whereas the Stachyamoeba-136
associated rickettsialean belongs to the Rickettsiaceae, and the Peranema-associated 137
rickettsialean belongs to the ‘Candidatus Midichloriaceae’, in the Rickettsiales.138
7
Table 1. Genome features for the three novel rickettsialeans sequenced in this study. 139
See Supplementary file 1 as well. 140
Species ‘Candidatus Finniella
inopinata’
Stachyamoeba-
associated
rickettsialean
Peranema-
associated
rickettsialean
Genome size 1,792,168 bp 1,738,386 bp 1,375,759 bp
N50 174,737 bp 1,738,386 bp 28,559 bp
Contig number 28 1 125
Gene number1 1,741 1,588 1,223
A+T% content 56.58% 67.01% 59.13%
Family Paracaedibacteraeae Rickettsiaceae ‘Candidatus
Midicloriaceae’
Order Holosporales Rickettsiales Rickettsiales
Completeness2 94.96% 97.12% (=100%) 92.08%
Redundancy2 0.0% 0.0% 2.1% 1 as predicted by Prokka v.1.13 (rRNA genes were searched with BLAST). 141
2 as estimated by Anvi’o v.2.4.0 using the Campbell et al., 2013 marker gene set.142

8
Compositional heterogeneity appears to be a major confounding factor affecting 143
phylogenetic inference of the Alphaproteobacteria 144
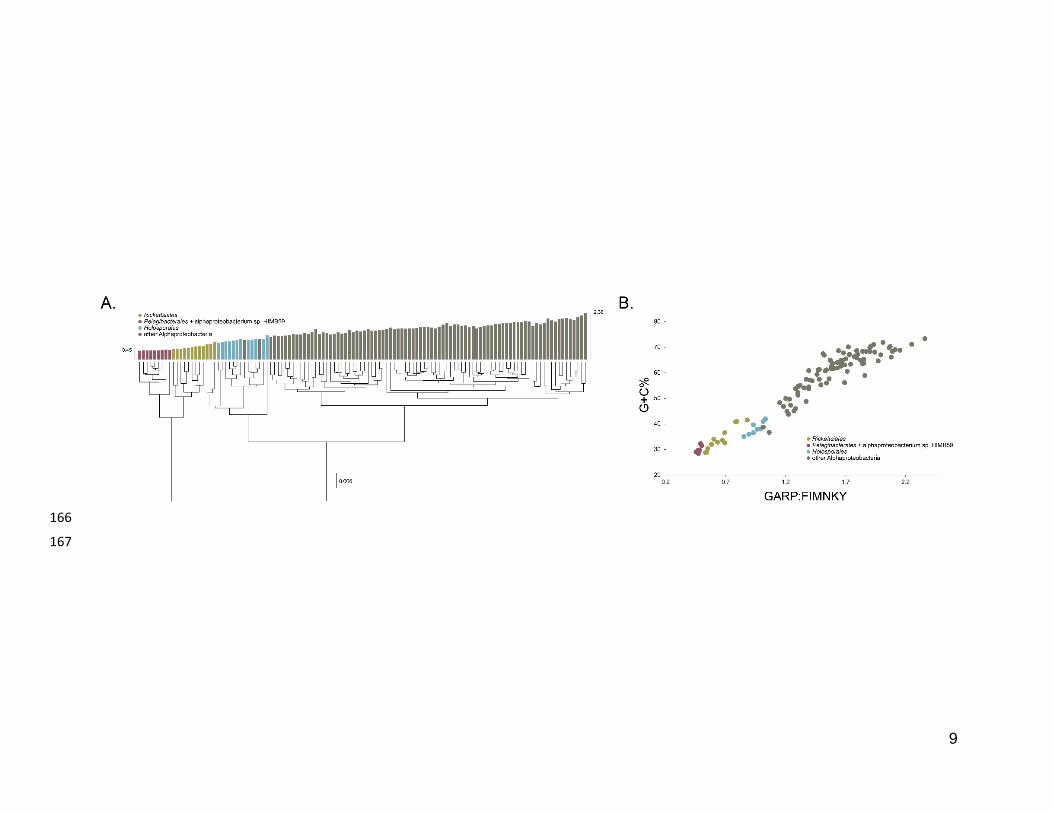
The average-linkage clustering of amino acid compositions shows that the Rickettsiales, 145
Pelagibacterales (together with alphaproteobacterium sp. HIMB59) and Holosporales 146
are clearly distinct from other alphaproteobacteria. This indicates that these three taxa 147
have divergent proteome amino acid compositions (Figure 1A). These taxa also have 148
the lowest GARP:FIMNKY amino acid ratios in all the Alphaproteobacteria (Figure 1A; 149
GARP amino acids are encoded by G+C%-rich codons, whereas FIMNKY amino acids 150
are encoded by A+T%-rich codons. Proteomes that have low GARP:FIMNKY ratios are 151
compositionally biased and therefore come from A+T%-rich genomes); the 152
Pelagibacterales (including alphaproteobacterium sp. HIMB59) being the most 153
divergent, followed by the Rickettsiales and then the Holosporales. Such biased amino 154
acid compositions appear to be the consequence of genome nucleotide compositions 155
that are strongly biased towards high A+T%—a scatter plot of genome G+C% and 156
proteome GARP:FIMNKY ratios shows a similar clustering of the Rickettsiales, 157
Pelagibacterales (including alphaproteobacterium sp. HIMB59) and Holosporales 158
(Figure 1B). This compositional similarity in the proteomes of the Rickettsiales, 159
Pelagibacterales (plus alphaproteobacterium sp. HIMB59) and Holosporales, which also 160
turn out to be the longest-branching alphaproteobacterial groups in previously published 161
phylogenies (e.g., Wang and Wu 2015), could be the outcome of either a shared 162
evolutionary history (i.e., the groups are most closely related to one another), or 163
alternatively, evolutionary convergence (e.g., because of similar lifestyles or 164
evolutionary trends toward small cell and genome sizes).165
9
166
167

10
Figure 1. Compositional heterogeneity in the Alphaproteobacteria is a major factor that 168
confounds phylogenetic inference. There are great disparities in the genome G+C% 169
content and amino acid compositions of the Rickettsiales, Pelagibacterales (including 170
alphaproteobacterium sp. HIMB59) and Holosporales with all other alphaproteobacteria. 171
A. A UPGMA (average-linkage) clustering of amino acid compositions (based on the 172
200 gene set for the Alphaproteobacteria) shows that the Rickettsiales (brown), 173
Pelagibacterales (maroon), and Holosporales (light blue) all have very similar proteome 174
amino acid compositions. At the tips of the tree, GARP:FIMNKY amino acid ratio values 175
are shown as bars. B. A scatterplot depicting the strong correlation between G+C% 176
(nucleotide compositions) and GARP:FIMNKY ratios (amino acid composition) for the 177
120 taxa in the Alphaproteobacteria (and outgroup) shows a similar clustering of the 178
Rickettsiales, Pelagibacterales (including alphaproteobacterium sp. HIMB59) and 179
Holosporales.180

11
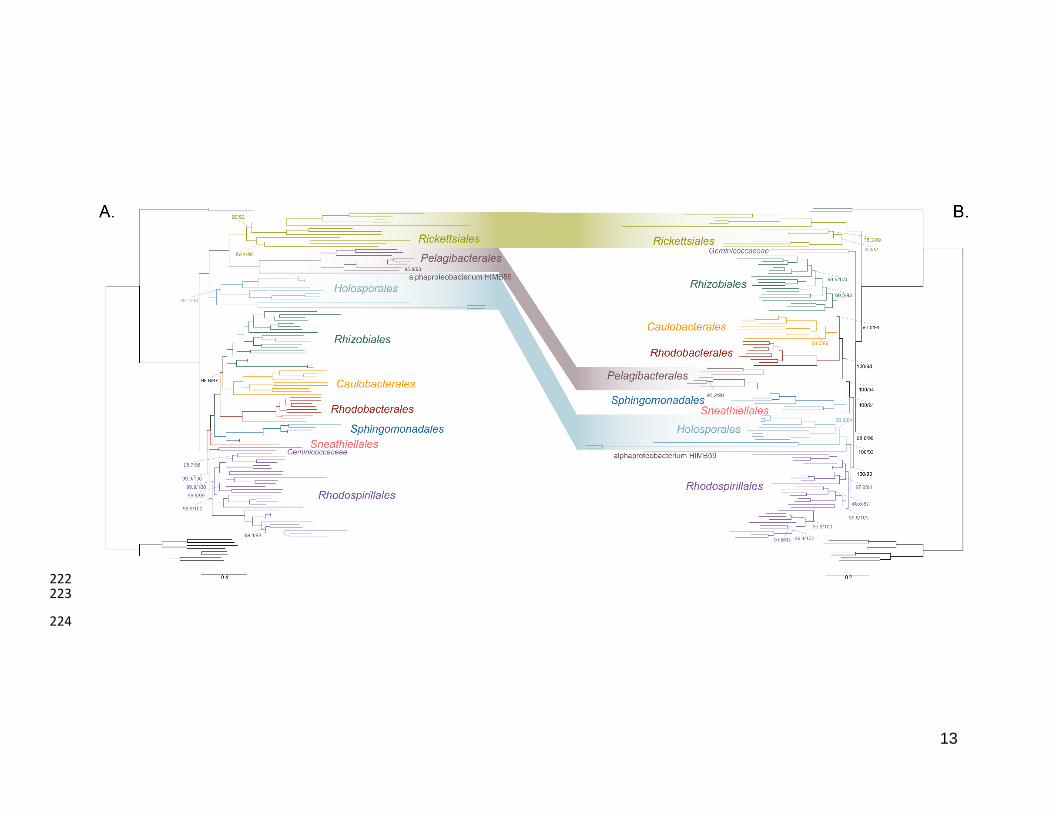
As a first step to discriminate between these two alternatives, we used maximum 181
likelihood to estimate a tree on a dataset that comprised 200 single-copy and rarely 182
laterally-transferred marker genes for the Alphaproteobacteria (as determined by Phyla-183
AMPHORA; see Methods for more details; Wang and Wu 2013) under the site-184
heterogenous model LG+PMSF(ES60)+F+R6. The resulting tree united the 185
Rickettsiales, Pelagibacterales (with alphaproteobacterium sp. HIMB59 at its base) and 186
Holosporales in a fully supported clade (Figure 2A; see Figure 2-figure supplement 1 for 187
labeled trees). The clustering of these three groups is suggestive of a phylogenetic 188
artefact (e.g., long-branch attraction or LBA); indeed, such a pattern resembles the one 189
seen in the tree of proteome amino acid compositions (see Figure 1A). This is because 190
the three groups have the longest branches in the Alphaproteobacteria tree and have 191
compositionally-biased and fast-evolving genomes (see Figure 2). If evolutionary 192
convergence in amino acid compositions is confounding phylogenetic inference for the 193
Alphaproteobacteria, methods aimed at reducing compositional heterogeneity might 194
disrupt the clustering of the Rickettsiales, Pelagibacterales and Holosporales. 195
To further test whether the clustering of the Rickettsiales, Pelagibacterales and 196
Holosporales is real or artefactual we used several different strategies to reduce the 197
compositional heterogeneity of our dataset (see Figure 2-figure supplement 2 for the 198
diverse strategies employed). When removing the 50% most compositionally-biased 199
(heterogeneous) sites according to ɀ (a novel metric that measures amino acid 200
compositional disparity at a site; see Methods), the clustering between the Rickettsiales, 201
Pelagibacterales, alphaproteobacterium sp. HIMB59 and Holosporales is disrupted 202
(Figure 2B; see also Figure 2-figure supplement 3). The new more derived placements 203
for the Pelagibacterales, alphaproteobacterium sp. HIMB59 and Holosporales are well 204
supported (further described below), and support tends to increase as compositionally-205
biased sites are removed (Supplementary file 2-table S1). Furthermore, when each of 206
these long-branching and compositionally-biased taxa is analyzed in isolation (i.e., in 207
the absence of the other), and compositionally heterogeneity is further decreased, new 208
phylogenetic patterns emerge that are incompatible, or in conflict, with their clustering 209
(Figure 2-figure supplement 4 and Figure 3-figure supplement 1-5). Various strategies 210
to reduce compositional heterogeneity, such as removing the most compositionally-211

12
biased sites, recoding the data into reduced character-state alphabets, or using only the 212
most compositionally homogeneous genes, converge to very similar phylogenetic 213
patterns for the Alphaproteobacteria in which the clustering of the Rickettsiales, 214
Pelagibacterales, alphaprotobacterium sp. HIMB59 and Holosporales is disrupted; the 215
Pelagibacterales, alphaproteobacterium sp. HIMB59 and Holosporales have much more 216
derived phylogenetic placements (e.g., Figure 3, Figure 2-figure supplement 4 and 217
Figure 3-figure supplement 1-5). On the other hand, removing fast-evolving sites does 218
not disrupt the clustering of these three long-branching groups (Supplementary file 2-219
table S2), suggesting that high evolutionary rates per site are not a major confounding 220
factor when inferring the phylogeny of the Alphaproteobacteria.221
13
222 223
224

14
Figure 2. Decreasing compositional heterogeneity by removing compositionally-biased 225
sites disrupts the clustering of the Rickettsiales, Pelagibacterales (including 226
alphaprotobacterium sp. HIMB59) and Holosporales. All branch support values are 227
100% SH-aLRT and 100% UFBoot unless annotated. A. A maximum-likelihood tree 228
inferred under the LG+PMSF(ES60)+F+R6 model and from the untreated dataset which 229
is highly compositionally heterogeneous. The three long-branching orders, the 230
Rickettsiales, Pelagibacterales (including alphaprotobacterium sp. HIMB59) and 231
Holosporales, that have similar amino acid compositions form a clade. B. A maximum-232
likelihood tree inferred under the LG+PMSF(ES60)+F+R6 model and from a dataset 233
whose compositional heterogeneity has been decreased by removing 50% of the most 234
biased sites according to ɀ. In this phylogeny the clustering of the Rickettsiales, 235
Pelagibacterales and Holosporales is disrupted. The Pelagibacterales is sister to the 236
Rhodobacterales, Caulobacterales and Rhizobiales. The Holosporales, and 237
alphaproteobacterium sp. HIMB59, become sister to the Rhodospirillales. The 238
Rickettsiales remains as the sister to the Caulobacteridae. See Figure 2-supplement 239
figure 1 for taxon names. See Figure 2-figure supplement 3 for the Bayesian consensus 240
trees inferred in PhyloBayes MPI v1.7 under the CAT-Poisson+Γ4 model. See also 241
Figure 2-figure supplement 2 and 4-7. 242
243

15
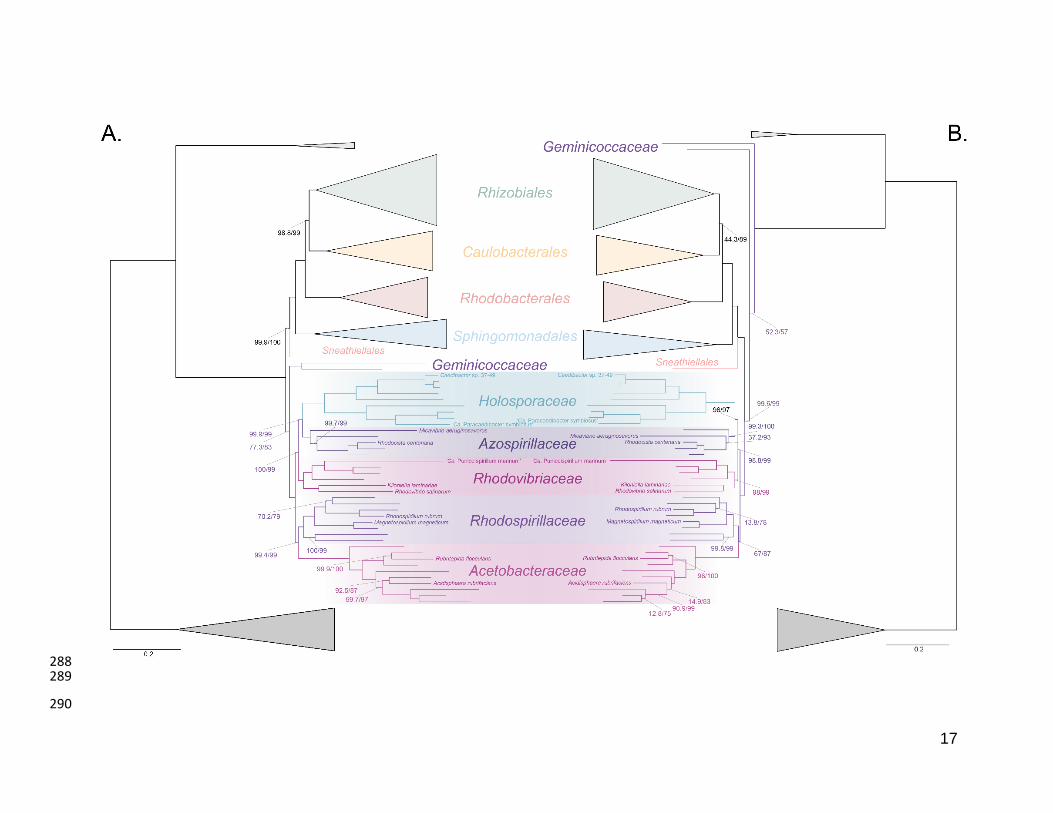
The Holosporales is unrelated to the Rickettsiales and is instead most likely derived 244
within the Rhodospirillales 245
The Holosporales has traditionally been considered part of the Rickettsiales sensu lato 246
because it appears as sister to the Rickettsiales in many trees (e.g., Hess, Suthaus, and 247
Melkonian 2015; Montagna et al. 2013; Santos and Massard 2014). It is exclusively 248
composed of endosymbiotic bacteria living within diverse eukaryotes, and such a 249
lifestyle is shared with all other members of the Rickettsiales (with the possible 250
exception of a recently reported ectosymbiotic rickettialean; see Castelli et al. 2018). 251
When we decrease, and then account for, compositional heterogeneity, we recover tree 252
topologies in which the Holosporales moves away from the Rickettsiales (e.g., Figure 253
2B, Figure 2-figure supplement 4B and 4D). For example, the Holosporales becomes 254
sister to all free-living alphaproteobacteria (the Caulobacteridae) when only the 40 most 255
homogeneous genes are used (Figure 2-figure supplement 4D) or when 10% of the 256
most compositionally-biased sites are removed (Supplementary file 2-table S1). When 257
compositional heterogeneity is further decreased by removing 50% of the most 258
compositionally-biased sites, the Holosporales becomes sister to the Rhodospirillales 259
(Figure 2B and Supplementary file 2-table S1; and see also Figure 2-figure supplement 260
4B). 261
Similarly, when the long-branching and compositionally-biased Rickettsiales, 262
Pelagibacterales, and alphaproteobacterium sp. HIMB59 (plus the extremely long-263
branching genera Holospora and ‘Candidatus Hepatobacter’) are removed, after 264
compositional heterogeneity had been decreased through site removal, the 265
Holosporales move to a much more derived position well within the Rhodospirillales 266
(Figure 3A, Figure 3-figure supplement 1B, 1C and Figure 3-figure supplement 6). If the 267
very compositionally-biased and fast-evolving Holospora and ‘Candidatus Hepatobacter’ 268
are left in, the Holosporales are pulled away from its derived position and the whole 269
clade moves closer to the base of the tree (Figure 3-figure supplement 7). The same 270
pattern in which the Holosporales is derived within the Rhodospirillales is seen when 271
these same taxa are removed, and the data are then recoded into four- or six-character 272
states (Figure 3B, Figure 3-figure supplement 6 and Figure 3-figure supplement 8). 273

16
Specifically, the Holosporales now consistently branches as sister to a subgroup of 274
rhodospirillaleans that includes, among others, the epibiotic predator Micavibrio 275
aeruginosavorus and the purple nonsulfur bacterium Rhodocista centenaria (the 276
Azospirillaceae, see below) (Figure 3). This new placement of the Holosporales has 277
nearly full support under both maximum likelihood (>95% UFBoot; see Figure 3) and 278
Bayesian inference (>0.95 posterior probability; see Figure 3-figure supplement 6).Thus, 279
three different analyses independently converge to the same pattern and support a 280
derived origin of the Holosporales within the Rhodospirillales: (1) removal of 281
compositionally-biased sites (Figure 3A), (2) data recoding into four-character states 282
using the dataset-specific scheme S4 (Figure 3B and Figure 3-figure supplement 7), 283
and (3) data recoding into six-character states using the dataset-specific scheme S6 284
(Figure 3-figure supplement 8); each of these strategies had to be combined with the 285
removal of the Pelagibacterales, alphaproteobacterium sp. HIMB59, and Rickettsiales to 286
recover this phylogenetic position for the Holosporales.287
17
288 289
290

18
Figure 3. The Holosporales (renamed and lowered in rank to the Holosporaceae family 291
here) branches in a derived position within the Rhodospirillales when compositional 292
heterogeneity is reduced and the long-branching and compositionally-biased 293
Rickettsiales, Pelagibacterales, and alphaproteobacterium sp. HIMB59 are removed. 294
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. A. A 295
maximum-likelihood tree, inferred under the LG+PMSF(ES60)+F+R6 model, to place 296
the Holosporaceae in the absence of the Rickettsiales, Pelagibacterales, and 297
alphaproteobacterium sp. HIMB59 and when compositional heterogeneity has been 298
decreased by removing 50% of the most biased sites. The Holosporaceae is sister to 299
the Azospirillaceae fam. nov. within the Rhodospirillales. B. A maximum-likelihood tree, 300
inferred under the GTR+ES60S4+F+R6 model, to place the Holosporaceae in the 301
absence of the Rickettsiales, Pelagibacterales, and alphaproteobacterium sp. HIMB59, 302
and when the data have been recoded into a four-character state alphabet (the dataset-303
specific recoding scheme S4: ARNDQEILKSTV GHY CMFP W) to reduce compositional 304
heterogeneity. This phylogeny shows a pattern that matches that inferred when 305
compositional heterogeneity has been alleviated through site removal. See Figure 3-306
figure supplement 6 for the Bayesian consensus trees inferred in PhyloBayes MPI v1.7 307
and under the and the CAT-Poisson+Γ4 model. See also Figure 3-figure supplement 1-308
5 and 7-8.309

19
A fourth independent analysis further supports a derived placement of the Holosporales 310
nested within the Rhodospirillales. Bayesian inference using the CAT-Poisson+Γ4 311
model, on a dataset whose compositional heterogeneity had been decreased by 312
removing 50% of the most compositionally-biased sites but for which no taxon had been 313
removed, also recovered the Holosporales as sister to the Azospirillaceae (see Figure 314
2-figure supplement 3). 315
The Rhodospirillales is a diverse order and comprises five well-supported families 316
The Rhodospirillales is an ancient and highly diversified group, but unfortunately this is 317
rarely obvious from published phylogenies because most studies only include a few 318
species for this order (Williams, Sobral, and Dickerman 2007; Georgiades et al. 2011; 319
Ferla et al. 2013). We have included a total of 31 Rhodospirillales taxa to better cover 320
its diversity. Such broad sampling reveals trees with five clear subgroups within the 321
Rhodospirillales that are well-supported in most of our analyses (e.g., Figure 2B and 3). 322
First is the Acetobacteraceae which comprises acetic acid (e.g., Acetobacter 323
oboediens), acidophilic (e.g., Acidisphaera rubrifaciens), and photosynthesizing 324
(bacteriochlorophyll-containing; e.g., Rubritepida flocculans) bacteria. The 325
Acetobacteraceae is strongly supported and relatively divergent from all other families 326
within the Rhodospirillales. Sister to the Acetobacteraceae is another subgroup that 327
comprises many photosynthesizing bacteria, including the type species for the 328
Rhodospirillales, Rhodospirillum rubrum, as well as the magnetotactic bacterial genera 329
Magnetospirillum, Magnetovibrio and Magnetospira (Figure 3). This subgroup best 330
corresponds to the poorly defined and paraphyletic Rhodospirillaceae family. We amend 331
the Rhodospirillaceae taxon and restrict it to the clade most closely related to the 332
Acetobacteraceae. As described above, when artefacts are accounted for, the 333
Holosporales most likely branches within the Rhodospirillales and therefore we suggest 334
the Holosporales sensu Szokoli et al. (2016) be lowered in rank to the family 335
Holosporaceae (containing e.g., Caedibacter sp. 37-49 and ‘Candidatus 336
Paracaedibacter symbiosus’), which is sister to the Azospirillaceae (Figure 3). The 337
Azospirillaceae fam. nov. contains the purple bacterium Rhodocista centenaria and the 338
epibiotic (neither periplasmic nor intracellular) predator Micavibrio aeruginosavorus, 339

20
among others. The Holosporaceae and the Azospirillaceae clades appear to be sister to 340
the Rhodovibriaceae fam. nov. (Figure 3), a well-supported group that comprises the 341
purple nonsulfur bacterium Rhodovibrio salinarum, the aerobic heterotroph Kiloniella 342
laminariae, and the marine bacterioplankter ‘Candidatus Puniceispirillum marinum’ (or 343
the SAR116 clade). Each of these subgroups and their interrelationships—with the 344
exception of the Holosporaceae that branches within the Rhodospirillales only after 345
compositional heterogeneity is countered—are strongly supported in nearly all of our 346
analyses (e.g., see Figure 2B and 3). 347
The Geminicoccaceae might be sister to all other free-living alphaproteobacteria (the 348
Caulobacteridae) 349
The Geminicocacceae is a recently proposed family within the Rhodospirillales 350
(Proença et al. 2017). It is currently represented by only two genera, Geminicoccus and 351
Arboriscoccus (Foesel et al. 2007; Proença et al. 2017). In most of our trees, however, 352
Tistrella mobilis is often sister to Geminicoccus roseus with full statistical support (e.g., 353
Figure 2B and 3A, but see Figure 3-figure supplement 6 for an exception) and we 354
therefore consider it to be part of the Geminococcaceae. Interestingly, the 355
Geminicoccaceae tends to have two alternative stable positions in our analyses, either 356
as sister to all other families of the Rhodospirillales (e.g., Figure 2A and 3A), or as sister 357
to all other orders of the Caulobacteridae (i.e., representing the most basal lineage of 358
free-living alphaproteobacteria; Figure 2B and Figure 2-figure supplement 3B, Figure 3B 359
and Figure 3-figure supplement 6, or Figure 2-figure supplement 4B, Figure 3-figure 360
supplement 1C, Figure 3-figure supplement 2B-D, Figure 3-figure supplement 3B, 3D, 361
and Figure 3-figure supplement 5C). Our analyses designed to alleviate compositional 362
heterogeneity, specifically site removal and recoding (without taxon removal), favor the 363
latter position for the Geminicoccaceae (Figure 2B and 3B). Moreover, as 364
compositionally-biased sites are progressively removed, support for the affiliation of the 365
Geminicoccaceae with the Rhodospirillales decreases, and after 50% of the sites have 366
been removed, the Geminicoccaceae emerges as sister to all other free-living 367
alphaproteobacteria with strong support (>95% UFBoot; Supplementary file 2-table S1). 368
In further agreement with this trend, the much simpler model LG4X places the 369

21
Geminicocacceae in a derived position as sister to the Acetobacteraceae (Figure 2-370
figure supplement 5), but as model complexity increases, and compositional 371
heterogeneity is reduced, the Geminicoccaceae moves closer to the base of the 372
Alphaproteobacteria (Figure 2A and 3A). Such a placement suggests that the 373
Geminicoccaceae may be a novel and independent order-level lineage in the 374
Alphaproteobacteria. However, because of the uncertainty in our results we opt here for 375
conservatively keeping the Geminicoccaceae as the sixth family of the Rhodospirillales 376
(Figure 3A). 377
Other deep relationships in the Alphaproteobacteria (Pelagibacterales, Rickettsiales, 378
alphaproteobacterium sp. HIMB59) 379
The clustering of the Pelagibacterales (formerly the SAR11 clade) with the Rickettsiales 380
and Holosporales is more easily disrupted than that of the Holosporales, either when 381
long-branching (or compositionally-biased) taxon removal is performed to control for 382
compositional attractions or not. The removal of compositionally-biased sites (from 30% 383
on; 16,320 out of 54,400 sites; see Supplementary file 2-table S1, Figure 2B, Figure 2-384
figure supplement 3B and Figure 3-figure supplement 4B), data recoding into four-385
character states (Figure 3-figure supplement 4C), and a set of the most compositionally 386
homogeneous genes (Figure 3-figure supplement 4D), all support a derived placement 387
of the Pelagibacterales as sister to the Rhodobacterales, Caulobacterales and 388
Rhizobiales. Attempts to account for compositional heterogeneity both across sites 389
(e.g., Rodríguez-Ezpeleta and Embley 2012; Viklund, Ettema, and Andersson 2012; 390
Viklund et al. 2013, and Martijn et al. 2018) and taxa (e.g., Luo et al. 2013; Luo 2015) 391
tend to disrupt the potentially artefactual clustering of the Pelagibacterales and the 392
Rickettsiales (in contrast to the studies of e.g., Williams, Sobral, and Dickerman 2007; 393
Thrash et al. 2011; and Georgiades et al. 2011 that did not account for compositional 394
heterogeneity). The Caulobacterales is sister to the Rhizobiales, and the 395
Rhodobacterales sister to both (e.g., Figure 2B and 3). This is consistent throughout 396
most of our results and such interrelationships become very robustly supported as 397
compositional heterogeneity is increasingly alleviated (Supplementary file 2-table S1). 398
The placement of the Rickettsiales as sister to the Caulobacteridae (i.e., all other 399

22
alphaproteobacteria) remains stable across different analyses (see Supplementary file 400
2-table S1, and also Figure 2B and Figure 3-figure supplement 2); this is also true when 401
the other long-branching taxa, the Pelagibacterales, alphaproteobacterium sp. HIMB59 402
and Holosporales, and even the Beta- Gammaproteobacteria outgroup, are removed 403
(see Figure 3-figure supplement 2 and Figure 3-figure supplement 3). Yet, the 404
interrelationships inside the Rickettsiales order remain uncertain; the ‘Candidatus 405
Midichloriaceae’ becomes sister to the Anaplasmataceae when fast-evolving sites are 406
removed (Supplementary file 2-table S2), but to the Rickettsiaceae when 407
compositionally-biased sites are removed (Supplementary file 2-table S1). The 408
placement of alphaproteobacterium sp. HIMB59 is uncertain (e.g., see Figure 2 and 409
Figure 2-figure supplement 3, and Figure 2-figure supplement 4 and Figure 3-figure 410
supplement 5; in contrast to Grote et al. 2012); taxon-removal analyses suggest that 411
alphaproteobacterium sp. HIMB59 is sister to the Caulobacteridae (Figure 3-figure 412
supplement 5), but the inclusion of any other long-branching group immediately 413
destabilizes this position (e.g., see Figure 2 and Figure 2-figure supplement 2, and 414
Figure 2-figure supplement 4). This is consistent with previous reports that suggest that 415
alphaproteobacterium sp. HIMB59 is not closely related to the Pelagibacterales (Viklund 416
et al. 2013; Martijn et al. 2018). 417
DISCUSSION 418
We have employed a diverse set of strategies to investigate the phylogenetic signal 419
contained within 200 genes for the Alphaproteobacteria. Specifically, such strategies 420
were primarily aimed at reducing amino acid compositional heterogeneity among taxa—421
a phenomenon that permeates our dataset (Figure 1). Compositional heterogeneity is a 422
clear violation of the phylogenetic models used in our, and previous, analyses, and 423
known to cause phylogenetic artefacts (Foster 2004). In the absence of more 424
sophisticated models for inferring deep phylogeny (i.e., those that best fit complex data), 425
the only way to counter artefacts caused by compositional heterogeneity is by removing 426
compositionally-biased sites or taxa, or recoding amino acids into reduced alphabets 427
(e.g., see Susko and Roger 2007; Heiss et al. 2018; Viklund, Ettema, and Andersson 428
2012). A combination of these strategies reveals that the Rickettsiales sensu lato (i.e., 429

23
the Rickettsiales and Holosporales) is polyphyletic. Our analyses suggest that the 430
Holosporales is derived within the Rhodospirillales, and that therefore this taxon should 431
be lowered in rank and renamed the Holosporaceae family (see Figure 2B and 3). The 432
same methods suggest that the Rhodospirillales might indeed be a paraphyletic order 433
and that the Geminicoccaceae could be a separate lineage that is sister to the 434
Caulobacteridae (e.g., Figure 2B). These two results, combined with our broader 435
sampling, reorganize the internal phylogenetic structure of the Rhodospirillales and 436
show that its diversity can be grouped into at least five well-supported major families 437
(Figure 3). 438
In 16S rRNA gene trees, the Holosporales has most often been allied to the 439
Rickettsiales (Montagna et al. 2013; Hess, Suthaus, and Melkonian 2015). The 440
apparent diversity of this group has quickly increased in recent years as more and more 441
intracellular bacteria living within protists have been described (e.g., Hess, Suthaus, and 442
Melkonian 2015; Szokoli et al. 2016; Eschbach et al. 2009; Boscaro et al. 2013). An 443
endosymbiotic lifestyle is shared by all members of the Holosporales and is also shared 444
with all those that belong to the Rickettsiales. Thus, it had been reasonable to accept 445
their shared ancestry as suggested by some 16S rRNA gene trees (e.g., Montagna et 446
al. 2013; Santos and Massard 2014; Hess, Suthaus, and Melkonian 2015). Apparent 447
strong support for the monophyly of the Rickettsiales and the Holosporales recently 448
came from some multi-gene trees by Wang and Wu (2014, 2015) who expanded 449
sampling for the Holosporales. However, an alternative placement for the Holosporales 450
as sister to the Caulobacteridae has been reported by Ferla et al. (2013) based on 451
rRNA genes, by Georgiades et al., (2011) based on 65 genes, by Schulz et al., (2015) 452
based on 139 genes, as well as by Wang and Wu (2015) based on 26, 29, or 200 genes 453
(see the supplementary information in Wang and Wu, 2015). This placement was 454
acknowledged by Szokoli et al. (2015), who formally established the order 455
Holosporales. Most recently, Martijn et al. 2018, who used strategies to reduce 456
compositional heterogeneity, and similarly to Wang and Wu (2015), recovered a number 457
of placements for the Holosporales within the Alphaproteobacteria; however these 458
different placements for the Holosporales were poorly supported. Here we provide 459
strong evidence for the hypothesis that the Holosporales is not related to the 460

24
Rickettsiales, as suggested earlier (Georgiades et al. 2011; Ferla et al. 2013; Szokoli et 461
al. 2016). The Rickettsiales sensu lato is polyphyletic. We show that the Holosporales is 462
artefactually attracted to the Rickettsiales (e.g., Figure 2A), but as compositional bias is 463
increasingly alleviated (through site removal and recoding), they move further away 464
from them (Figure 2B). The Holosporales is placed within the Rhodopirillales as sister to 465
the family Azospirillaceae (Figure 3). The similar lifestyles of the Holosporales and 466
Rickettsiales, as well as other features like the presence of an ATP/ADP translocase 467
(Wang and Wu 2014), are therefore likely the outcome of convergent evolution. 468
A derived origin of the Holosporales has important implications for understanding the 469
origin of mitochondria and the nature of their ancestor. Wang and Wu (2014, 2015) 470
proposed that mitochondria are phylogenetically embedded within the Rickettsiales 471
sensu lato. In their trees, mitochondria were sister to a clade formed by the 472
Rickettsiaceae, Anaplasmataceae and ‘Candidatus Midichloriaceae’, and the 473
Holosporales was itself sister to all of them. This phylogenetic placement for 474
mitochondria suggested that the ancestor of mitochondria was an intracellular parasite 475
(Wang and Wu, 2014). But if the Holosporales is a derived group of rhodospirillaleans 476
as we show here (see Figure 3), then the argument that mitochondria necessarily 477
evolved from parasitic alphaproteobacteria no longer holds. While the sisterhood of 478
mitochondria and the Rickettsiales sensu stricto is still a possibility, such a relationship 479
does not imply that the two groups shared a parasitic common ancestor (i.e., a parasitic 480
ancestry for mitochondria). The most recent analyses done by Martijn et al. (2018) 481
suggest that mitochondria are sister to all known alphaproteobacteria, also suggesting 482
their non-parasitic ancestry. Our study, and that of Martijn et al., thus complement each 483
other and support the view that mitochondria most likely evolved from ancestral free-484
living alphaproteobacteria (contra Sassera et al. 2011; Wang and Wu 2014, 2015). 485
The order Rhodospirillales is quite diverse and includes many purple nonsulfur bacteria 486
as well as all magnetotactic bacteria within the Alphaproteobacteria. The 487
Rhodospirillales is sister to all other orders in the Caulobacteridae, and has historically 488
been subdivided into two families: the Rhodospirillaceae and the Acetobacteraceae. 489
Recently, a new family, the Geminicoccaceae, was established for the Rhodospirillales 490

25
(Proença et al. 2017). However, some of our analyses suggest that the 491
Geminicoccaceae might be sister to all other Caulobacteridae (e.g., Figure 2B and 3B). 492
This phylogenetic pattern, therefore, suggests that the Rhodospirillales may be a 493
paraphyletic order. The placement of the Geminicoccaceae as sister to the 494
Caulobacteridae needs to be further tested once more sequenced diversity for this 495
group becomes available; if it were to be confirmed, the Geminicoccaceae should be 496
elevated to the order level. Whereas the Acetobacteraceae is phylogenetically well-497
defined, there has been considerable uncertainty about the Rhodospirillaceae (e.g., 498
Ferla et al., 2013), primarily because of poor sampling and a lack of resolution provided 499
by the 16S rRNA gene. We subdivide the Rhodospirillaceae sensu lato into three 500
subgroups (Figure 3). We restrict the Rhodospirillaceae sensu stricto to the subgroup 501
that is sister to the Acetobacteracae (Figure 3). The other two subgroups are the 502
Rhodovibriaceae and the Azospirillaceae; the latter is sister to the Holosporaceae 503
(Figure 3). 504
Based on our fairly robust phylogenetic patterns, we have updated the higher-level 505
taxonomy of the Alphaproteobacteria (Figure 4). We exclude the Magnetococcales from 506
the Alphaproteobacteria class because of its divergent nature (e.g., see Figure 1 in 507
Esser, Martin, and Dagan 2007 which shows that many of Magnetococcus’ genes are 508
more similar to those of beta-, and gammaproteobacteria). In agreement with its 509
intermediate phylogenetic placement, we endorse the Magnetococcia class as 510
proposed by Parks et al. (2018). At the highest level we define the Alphaproteobacteria 511
class as comprising two subclasses sensu Ferla et al. (2013), the Rickettsidae and the 512
Caulobacteridae. The former contains the Rickettsiales, and the latter contains all other 513
orders, which are primarily and ancestrally free-living alphaproteobacteria. The order 514
Rickettsiales comprises three families as previously defined, the Rickettsiaceae, the 515
Anaplasmataceae, and the ‘Candidatus Midichloriaceae’. On the other hand, the 516
Caulobacteridae is composed of seven phylogenetically well-supported orders: the 517
Rhodospirillales, Sneathiellales, Sphingomonadales, Pelagibacterales, 518
Rhodobacterales, Caulobacterales and Rhizobiales. Among the many species claimed 519
to represent new order-level lineages on the basis of 16S rRNA gene trees (Cho and 520
Giovannoni 2003; Kwon et al. 2005; Kurahashi et al. 2008; Wiese et al. 2009; Harbison 521

26
et al. 2017), only Sneathiella deserves order-level status (Kurahashi et al. 2008), since 522
all others have derived placements in our trees and those published by others (Williams 523
et al. 2012; Bazylinski et al. 2013; Venkata Ramana et al. 2013; Harbison et al. 2017). 524
The Rhodospirillales order comprises six families, three of which are new, namely the 525
Holosporaceae, Azospirillaceae and Rhodovibriaceae (Figure 4). This new higher-level 526
classification of the Alphaproteobacteria updates and expands those presented by Ferla 527
et al. (2013), the ‘Bergey’s Manual of Systematics of Archaea and Bacteria’ (Garrity 528
2005; Whitman 2015), and ‘The Prokaryotes’ (Rosenberg et al. 2014). The classification 529
scheme proposed here could be partly harmonized with that recently proposed by Parks 530
et al., (2018) by elevating the six families within the Rhodospirllales to the order level; 531
the phylograms by Parks et al., (2018), however, are in conflict with those shown here 532
and many of their proposed taxa are as well. 533

27
534
Figure 4. A higher-level classification scheme for the Alphaproteobacteria and the 535
Magnetococcia classes within the Proteobacteria, and the Rickettsiales and 536
Rhodospirillales orders within the Alphaproteobacteria. 537
538

28
CONCLUSIONS 539
We employed a combination of methods to decrease compositional heterogeneity in 540
order to disrupt artefacts that arise when inferring the phylogeny of the 541
Alphaproteobacteria. This is an example of the complex nature of the historical signal 542
contained in modern genomes and the limitations of our current evolutionary models to 543
capture these signals. A robust phylogeny of the Alphaproteobacteria is a precondition 544
for placing the mitochondrial lineage. This is because including mitochondria certainly 545
exacerbates the already strong biases in the data, and therefore represents additional 546
sources of artefacts in phylogenetic inference (as seen in Wang and Wu, 2015 where 547
the Holosporales is attracted by both mitochondria and the Rickettsiales). The robust 548
phylogenetic framework developed here will serve as a reference for future studies that 549
aim to place mitochondria and novel not-yet-cultured environmental diversity within the 550
Alphaproteobacteria. 551
TAXON DESCRIPTIONS 552
Rickettsidae emend. (Alphaproteobacteria) Rickettsia is the type genus of the subclass. 553
The Rickettsidae subclass is here amended by redefining its circumscription so it 554
remains monophyletic by excluding the Pelagibacterales order. The emended 555
Rickettsidae subclass within the Alphaproteobacteria class is defined based on 556
phylogenetic analyses of 200 genes which are predominantly single-copy and vertically-557
inherited (unlikely laterally transferred) when compositionally heterogeneity was 558
decreased by site removal or recoding. Phylogenetic (node-based) definition: the least 559
inclusive clade containing Anaplasma phagocytophilum HZ, Rickettsia typhi Wilmington, 560
and ‘Candidatus Midichloria mitochondrii’ IricVA. The Rickettsidae does not include: 561
Pelagibacter sp. HIMB058, ‘Candidatus Pelagibacter sp.’ IMCC9063, 562
alphaproteobacterium sp. HIMB59, Caedibacter sp. 37-49, ‘Candidatus Nucleicultrix 563
amoebiphila’ FS5, ‘Candidatus Finniella lucida’, Holospora obtusa F1, Sneathiella 564
glossodoripedis JCM 23214, Sphingomonas wittichii, and Brevundimonas subvibrioides 565
ATCC 15264. 566
Caulobacteridae emend. (Alphaproteobacteria) Caulobacter is the type genus of the 567
subclass. The Caulobacteridae subclass is here amended by redefining its 568

29
circumscription so it remains monophyletic by including the Pelagibacterales order. The 569
emended Caulobacteridae subclass within the Alphaproteobacteria class is defined 570
based on phylogenetic analyses of 200 genes which are predominantly single-copy and 571
vertically-inherited (unlikely laterally transferred) when compositionally heterogeneity 572
was decreased by site removal or recoding. Phylogenetic (node-based) definition: the 573
least inclusive clade containing Pelagibacter sp. HIMB058, ‘Candidatus Pelagibacter 574
sp.’ IMCC9063, alphaproteobacterium sp. HIMB59, Caedibacter sp. 37-49, ‘Candidatus 575
Nucleicultrix amoebiphila’ FS5, ‘Candidatus Finniella lucida’, Holospora obtusa F1, 576
Sneathiella glossodoripedis JCM 23214, Sphingomonas wittichii, and Brevundimonas 577
subvibrioides ATCC 15264. The Caulobacteridae does not include: Anaplasma 578
phagocytophilum HZ, Rickettsia typhi Wilmington, and ‘Candidatus Midichloria 579
mitochondrii’ IricVA 580
Azospirillaceae fam. nov. (Rhodospirillales, Alphaproteobacteria) Azospirillum is the 581
type genus of the family. This new family within the Rhodospirillales order is defined 582
based on phylogenetic analyses of 200 genes which are predominantly single-copy and 583
vertically-inherited (unlikely laterally transferred). Phylogenetic (node-based) definition: 584
the least inclusive clade containing Micavibrio aeruginoavorus ARL-13, Rhodocista 585
centenaria SW, and Inquilinus limosus DSM 16000. The Azospirillaceae does not 586
include: Rhodovibrio salinarum DSM 9154, ‘Candidatus Puniceispirillum marinum’ IMCC 587
1322, Rhodospirillum rubrum ATCC 11170, Terasakiella pusilla DSM 6293, Acidiphilium 588
angustum ATCC 49957, and Elioraea tepidiphila DSM 17972. 589
Rhodovibriaceae fam. nov. (Rhodospirillales, Alphaproteobacteria) Rhodovibrio is the 590
type genus of the family. This new family within the Rhodospirillales order is defined 591
based on phylogenetic analyses of 200 genes which are predominantly single-copy and 592
vertically-inherited (unlikely laterally transferred). Phylogenetic (node-based) definition: 593
the least inclusive clade containing Rhodovibrio salinarum DSM 9154, Kiloniella 594
laminariae DSM 19542, Oceanibaculum indicum P24, Thalassobaculum salexigens 595
DSM 19539 and ‘Candidatus Puniceispirillum marinum’ IMCC 1322. The 596
Rhodovobriaceae does not include: Rhodospirillum rubrum ATCC 11170, Terasakiella 597

30
pusilla DSM 6293, Rhodocista centenaria SW, Micavibrio aeruginoavorus ARL-13, 598
Acidiphilium angustum ATCC 49957, and Elioraea tepidiphila DSM 17972. 599
Rhodospirillaceae emend. (Rhodospirillales, Alphaproteobacteria) Rhodospirillum is the 600
type genus of the family. The Rhodospirillaceae family is here amended by redefining its 601
circumscription so it remains monophyletic. The emended Rhodospirillaceae family 602
within the Rhodospirillales order is defined based on phylogenetic analyses of 200 603
genes which are predominantly single-copy and vertically-inherited (unlikely laterally 604
transferred). Phylogenetic (node-based) definition: the least inclusive clade containing 605
Rhodospirillum rubrum ATCC 11170, Roseospirillum parvum 930l, Magnetospirillum 606
magneticum AMB-1 and Terasakiella pusilla DSM 6293. The Rhodospirillaceae does 607
not include: Rhodocista centenaria SW, Micavibrio aeruginoavorus ARL-13, ‘Candidatus 608
Puniceispirillum marinum’ IMCC 1322, Rhodovibrio salinarum DSM 9154, Elioraea 609
tepidiphila DSM 17972, and Acidiphilium angustum ATCC 49957. 610
Holosporaceae (Rhodospirillales, Alphaproteobacteria) Holospora is the type genus of 611
the family. The Holosporaceae family as defined here has the same taxon 612
circumscription as the Holosporales order sensu Szokoli et al., (2016), but it is here 613
lowered to the family level and placed within the Rhodospirillales order. The new family 614
rank-level for this group is based on the phylogenetic analysis of 200 genes, which are 615
predominantly single-copy and vertically-inherited (unlikely laterally transferred), when 616
compositionally heterogeneity was decreased by site removal or recoding (and coupled 617
to the removal of the long-branching taxa Pelagibacterales and Rickettsiales). The 618
family contains three subfamilies (lowered in rank from a former family level) and one 619
formally-undescribed clade, namely, the Holosporodeae, and ‘Candidatus 620
Paracaedibacteriodeae’, ‘Candidatus Hepatincolodeae’, and the Caedibacter-621
Nucleicultrix clade. 622
METHODS 623
Genome sequencing 624
Cultures of Viridiraptor invadens strain Virl02, the host of ‘Candidatus Finniella 625
inopinata’, were grown on the filamentous green alga Zygnema pseudogedeanum strain 626

31
CCAC 0199 as described in (Hess and Melkonian 2013). Once the algal food was 627
depleted, Viridiraptor cells were harvested by filtration through a cell strainer (mesh size 628
40 µm to remove algal cell walls) and centrifugation (~1,000 g for 15 min). For short-629
read sequencing, DNA extraction of total gDNA was carried out with the ZR 630
Fungal/Bacterial DNA MicroPrep Kit (Zymo Research) using a BIO101/Savant FastPrep 631
FP120 high-speed bead beater and 20 uL of proteinase K (20 mg/mL). A sequencing 632
library was made using the NEBNext Ultra II DNA Library Prep Kit (New England 633
Biolabs). Paired-end DNA sequencing libraries were sequenced with an Illumina MiSeq 634
instrument (Dalhousie University; Canada). (number of reads=3,006,282, read 635
length=150 bp). For long-read sequencing, DNA extraction was performed using a 636
CTAB and phenol-chloroform method. Total gDNA was further cleaned through a 637
QIAGEN Genomic-Tip 20/G. A sequencing library was made using the Nanopore 638
Ligation Sequencing Kit 1D (SQK-LSK108). Sequencing was done on a portable 639
MinION instrument (Oxford Nanopore Technologies). (total bases=191,942,801 bp, 640
number of reads=73,926, longest read=32,236 bp, mean read length=2,596 bp, mean 641
read quality=9.4). 642
Peranema trichophorum strain CCAP 1260/1B was obtained from the Culture Collection 643
of Algae and Protozoa (CCAP, Oban, Scotland) and grown in liquid Knop media plus 644
egg yolk crystals. Total gDNA was extracted following Lang and Burger (2007). A 645
paired-end sequencing library was made using a TruSeq DNA Library Prep Kit 646
(Illumina). DNA sequencing libraries were sequenced with an Illumina MiSeq instrument 647
(Genome Quebec Innovation Centre; Canada). (number of reads=4,157,475, read 648
length=300 bp). 649
Stachyamoeba lipophora strain ATCC 50324 cells feeding on Escherichia coli were 650
harvested and then broken up with pestle and mortar in the presence of glass beads (< 651
450 µm diameter). Total gDNA was extracted using the QIAGEN Genomic G20 Kit. A 652
paired-end sequencing library was made using a TruSeq DNA Library Prep Kit 653
(Illumina). DNA sequencing libraries were sequenced with an Illumina MiSeq instrument 654
(Genome Quebec Innovation Centre; Canada). (number of reads=35,605,415, read 655
length=100 bp). 656

32
Genome assembly and annotation 657
Short sequencing reads produced in an Illumina MiSeq from Viridiraptor invadens, 658
Peranema trichophorum, and Stachyamoeba lipophora were first assessed with 659
FASTQC v0.11.6 and then, based on its reports, trimmed with Trimmomatic v0.32 660
(Bolger, Lohse, and Usadel 2014) using the options: HEADCROP:16 LEADING:30 661
TRAILING:30 MINLEN:36. Illumina adapters were similarly removed with Trimmomatic 662
v0.32 using the option ILLUMINACLIP. Long sequencing reads produced in a Nanopore 663
MinION instrument from Viridiraptor invadens were basecalled with Albacore v2.1.7, 664
adapters were removed with Porechop v0.2.3, lambda phage reads were removed with 665
NanoLyse v0.5.1, quality filtering was done with NanoFilt v2.0.0 (with the options ‘--666
headcrop 50 -q 8 -l 1000’), and identity filtering against the high-quality short Illumina 667
reads was done with Filtlong v0.2.0 (and the options ‘--keep_percent 90 --trim --split 500 668
--length_weight 10 --min_length 1000’). Statistics were calculated throughout the read 669
processing workflow with NanoStat v0.8.1 and NanoPlot v1.9.1. A hybrid co-assembly 670
of both processed Illumina short reads and Nanopore long reads from Viridiraptor 671
invadens was done with SPAdes v3.6.2 (Bankevich et al. 2012). Assemblies of the 672
Illumina short reads from Peranema trichophorum and Stachyamobea lipophora were 673
separately done with SPAdes v3.6.2 (Bankevich et al. 2012). The resulting assemblies 674
for both Viridiraptor invadens and Peranema trichophorum were later separately 675
processed with the Anvi’o v2.4.0 pipeline (Eren et al. 2015) and refined genome bins 676
corresponding to ‘Candidatus Finniella inopinata’ and the Peranema-associated 677
rickettsialean were isolated primarily based on tetranucleotide sequence composition 678
and taxonomic affiliation of its contigs. A single contig corresponding to the genome of 679
the Stachyamoeba-associated rickettsialean was obtained from its assembly and this 680
was circularized by collapsing the overlapping ends of the contig. Gene prediction and 681
genome annotation was carried out with Prokka v.1.13 (see Table 1). 682
Dataset assembly (taxon and gene selection) 683
The selection of 120 taxa was largely based on the phylogenetically diverse set of 684
alphaproteobacteria determined by Wang and Wu (2015). To this set of taxa, recently 685
sequenced and divergent unaffiliated alphaproteobacteria were added, as well as those 686

33
claimed to constitute novel order-level taxa. Some other groups, like the 687
Pelagibacterales, Rhodospirillales and the Holosporales, were expanded to better 688
represent their diversity. A set of four betaproteobacteria and four 689
gammaproteobacterial were used as outgroup (see Figure 2-figure supplement 6 for 690
taxon names; see Supplementary file 2-table S3 for accession numbers). 691
A set of 200 gene markers (54,400 sites; 9.03% missing data, see Figure 2-figure 692
supplement 6) defined by Phyla-AMPHORA was used (Wang and Wu 2013). The genes 693
are single-copy and predominantly vertically-inherited as assessed by congruence 694
among them (Wang and Wu 2013). In brief, Phyla-AMPHORA searches for each 695
marker gene using a profile Hidden Markov Model (HMM), then aligns the best hits to 696
the profile HMM using hmmalign of the HMMER suite, and then trims the alignments 697
using pre-computed quality scores (the mask) previously generated using the 698
probabilistic masking program ZORRO (Wu, Chatterji, and Eisen 2012; Wang and Wu 699
2013). Phylogenetic trees for each marker gene were inferred from the trimmed multiple 700
alignments in IQ-TREE v1.5.5 (Minh, Nguyen, and von Haeseler 2013; Nguyen et al. 701
2015) and under the model LG4X+F model. Single-gene trees were examined 702
individually to remove distant paralogues, contaminants or laterally transferred genes. 703
All this was done before concatenating the single-gene alignments into a supermatrix 704
with SequenceMatrix v 1.8 (Vaidya, Lohman, and Meier 2011). Another smaller dataset 705
of 40 compositionally-homogenous genes (5,570 sites; 5.98% missing data) was built 706
by selecting the least compositionally heterogeneous genes from the larger 200 gene 707
set according compositional homogeneity tests performed in P4 (Foster 2004; see 708
Supplementary file 2-table S4 for a list of the 40 most compositionally homogenous 709
genes). This was done as an alternative way to overcome the strong compositional 710
heterogeneity observed in datasets for the Alphaproteobacteria with a broad selection of 711
taxa. In brief, the P4 tests rely on simulations based on a provided tree (here inferred for 712
each gene under the model LG4X+F in IQ-TREE) and a model (LG+F+G4 available in 713
P4) to obtain proper null distributions to which to compare the X2 statistic. Most 714
standard tests for compositional homogeneity (those that do not rely on simulate the 715
data on a given tree) ignore correlation due to phylogenetic relatedness, and can suffer 716
from a high probability of false negatives (Foster 2004). 717

34
Variations of our full set were made to specifically assess the placement of each long-718
branching and compositionally-biased group individually. In other words, each group 719
with comparatively long branches (the Rickettsiales, Pelagibacterales, Holosporales, 720
and alphaproteobacterium sp. HIMB59) was analyzed in isolation, i.e., in the absence of 721
other long-branching and compositionally-biased taxa. This was done with the purpose 722
of reducing the potential artefactual attraction among these groups. Taxon removal was 723
done in addition to compositionally-biased site removal and data recoding into reduced 724
character-state alphabets (for a summary of the different methodological strategies 725
employed see Figure 2-figure supplement 2). 726
Removal of compositionally-biased and fast-evolving sites 727
As an effort to reduce artefacts in phylogenetic inference from our dataset (which might 728
stem from extreme divergence in the evolution of the Alphaproteobacteria), we removed 729
sites estimated to be highly compositionally heterogeneous or fast evolving. The 730
compositional heterogeneity of a site was estimated by using a metric intended to 731
measure the degree of disparity between the most %AT-rich taxa and all others. Taxa 732
were ordered from lowest to highest proteome GARP:FIMNKY ratios; ‘GARP’ amino 733
acids are encoded by %GC-rich codons, whereas ‘FIMNKY’ amino acids are encoded 734
by %AT-rich codons. The resulting plot was visually inspected and a GARP:FIMNKY 735
ratio cutoff of 1.06 (which represented a discontinuity or gap in the distribution which 736
separated the long-branching and compositionally-biased taxa Pelagibacterales, 737
Holosporales and Rickettsiales from all others) was chosen to divide the dataset into 738
low GARP:FMINKY (or %AT-rich) and higher GARP:FIMNKY (or ‘GC-rich’) taxa (Figure 739
2-figure supplement 7). Next, we determined the degree of compositional bias per site 740
(ɀ) for the frequencies of both FIMNKY and GARP amino acids between the %AT-rich 741
and all other (‘GC-rich’) alphaproteobacteria. To calculate this metric for each site the 742
following formula was used: 743
ɀ = (𝜋𝐹𝐼𝑀𝑁𝐾𝑌%AT-rich − 𝜋𝐹𝐼𝑀𝑁𝐾𝑌%GC-rich) + (𝜋𝐺𝐴𝑅𝑃%GC-rich − 𝜋𝐺𝐴𝑅𝑃%AT-rich)
where 𝜋𝐹𝐼𝑀𝑁𝐾𝑌 and 𝜋𝐺𝐴𝑅𝑃 are the sum of the frequencies for FIMNKY and GARP 744
amino acids at a site, respectively, for either ‘%AT-rich’ or ‘%GC-rich’ taxa. According to 745
this metric, higher values measure a greater disparity between %AT-rich 746

35
alphaproteobacteria and all others; a measure of compositional heterogeneity or bias 747
per site. The most compositionally heterogeneous sites according to ɀ were 748
progressively removed using the software SiteStripper (Verbruggen 2018) in increments 749
of 10%. We also progressively removed the fastest evolving sites in increments of 10%. 750
Conditional mean site rates were estimated under the LG+C60+F+R6 model in IQ-751
TREE v1.5.5 using the ‘-wsr’ flag (Nguyen et al. 2015). 752
Data recoding 753
Our datasets were recoded into four- and six-character state amino acid alphabets 754
using dataset-specific recoding schemes aimed at minimizing compositional 755
heterogeneity in the data (Susko and Roger 2007). The program minmax-chisq, 756
which implements the methods of Susko and Roger (2007), was used to find the best 757
recoding schemes—please see Figure 3, Figure 2-figure supplement 4 and Figure 3-758
figure supplement 1-6, and Figure 3-figure supplement 8 legends for the specific 759
recoding schemes used for each dataset. The approach uses the chi-squared (X2) 760
statistic for a test of homogeneity of frequencies as a criterion function for determining 761
the best recoding schemes. Let 𝜋𝑖 denote the frequency of bin 𝑖 for the recoding 762
scheme currently under consideration. For instance, suppose the amino acids were 763
recoded into four bins, 764
RNCM EHIPTWV ADQLKS GFY. 765
Then 𝜋4would be the frequency with which the amino acids G, F or Y were observed. 766
Let 𝜋𝑖𝑠 be the frequency of bin 𝑖 for the 𝑠th taxa. Then the X2 statistic for the null 767
hypothesis that the frequencies are constant, over taxa, against the unrestricted 768
hypothesis is 769
𝑡𝑠 = ∑ (𝜋𝑖𝑠 − 𝜋𝑖)2
𝑖𝑠/𝜋𝑖
The X2 statistic provides a measure of how different the frequencies for the 𝑠th taxa are 770
from the average frequencies. The maximum 𝑡𝑠 over 𝑠 is taken as an overall measure of 771
how heterogeneous the frequencies are for a given recoding scheme. The minmax-772

36
chisq program searches through recoding schemes, moving amino acids from one bin 773
to another, to try to minimize the 𝑚𝑎𝑥 𝑡𝑠 (Susko and Roger 2007). 774
Phylogenetic inference 775
The inference of phylogenies was primarily done under the maximum likelihood 776
framework and using IQ-TREE v1.5.5 (Minh, Nguyen, and von Haeseler 2013; Nguyen 777
et al. 2015). ModelFinder in IQ-TREE v1.5.5 (Kalyaanamoorthy et al. 2017) was used to 778
assess the best-fitting amino acid empirical matrix (e.g., JTT, WAG, and LG), on a 779
maximum-likelihood tree, to our full dataset of 120 taxa and 200 conserved single-copy 780
marker genes (see Supplementary file 2-table S5 and Supplementary file 2-table S6). 781
We first inferred guide trees (for a PMSF analysis) with a model that comprises the LG 782
empirical matrix, with empirical frequencies estimated from the data (F), six rates for the 783
FreeRate model to account for rate heterogeneity across sites (R6), and a mixture 784
model with 60 amino acid profiles (C60) to account for compositional heterogeneity 785
across sites—LG+C60+F+R6. Because the computational power and time required to 786
properly explore the whole tree space (given such a big dataset and complex model) 787
was too high, constrained tree searches were employed to obtain these initial guide 788
trees (see Figure 2-figure supplement 6 for the constraint tree). Many shallow nodes 789
were constrained if they received maximum UFBoot and SH-aLRT support in a 790
LG+PMSF(C60)+F+R6 analysis. All deep nodes, those relevant to the questions 791
addressed here, were left unconstrained (Figure 2-figure supplement 6). The guide 792
trees were then used together with a dataset-specific mixture model ES60 to estimate 793
site-specific amino acid profiles, or a PMSF (Posterior Mean Site Frequency Profiles) 794
model, that best account for compositional heterogeneity across sites (Wang et al. 795
2018). The dataset-specific empirical mixture model ES60 also has 60 categories but, 796
unlike the general C60, was directly estimated from our large dataset of 200 genes and 797
120 alphaproteobacteria (and outgroup) using the methods described in (Susko, 798
Lincker, and Roger 2018; ModelFinder (Kalyaanamoorthy et al. 2017) suggests that the 799
LG+ES60+F+R6 model is the best-fitting model; the R6 model component, however, 800
considerably increases computational burden; see Supplementary file 2-table S6 and 801
Supplementary file 2-table S7). Final trees were inferred using the 802

37
LG+PMSF(ES60)+F+R6 model and a fully unconstrained tree search. Those datasets 803
that produced the most novel topologies under maximum likelihood were further 804
analyzed under a Bayesian framework using PhyloBayes MPI v1.7 and the CAT-805
Poisson+Γ4 model (Lartillot and Philippe 2004; Lartillot, Lepage, and Blanquart 2009). 806
This model allows for a very large number of classes to account for compositional 807
heterogeneity across sites and, unlike in the more complex CAT-GTR+Γ4 model, also 808
allows for convergence to be more easily achieved between MCMC chains. PhyloBayes 809
MCMC chains were run for at least 10,000 cycles until convergence between the chains 810
was achieved and the largest discrepancy (i.e., maxdiff parameter) was ≤ 0.4 (except 811
for the untreated dataset analyzed in Figure 2-figure supplement 3A; see 812
Supplementary file 2-table S8 for several summary statistics for each PhyloBayes 813
MCMC chain, including discrepancy and effective sample size values). A consensus 814
tree was generated from two PhyloBayes MCMC chains using a burn-in of 500 trees 815
and sub-sampling every 10 trees. 816
Phylogenetic analyses of recoded datasets into four-character state alphabets were 817
analyzed using IQ-TREE v1.5.5 and the model GTR+ES60S4+F+R6. ES60S4 is an 818
adaptation of the dataset-specific empirical mixture model ES60 to four-character 819
states. It is obtained by adding the frequencies of the amino acids that belong to each 820
bin in the dataset-specific four-character state scheme S4 (see Data Recoding for 821
details). Phylogenetics analyses of recoded datasets into six-character state alphabets 822
were analyzed using PhyloBayes MPI v1.7 and the CAT-Poisson+Γ4 model. Maximum-823
likelihood analyses with a six-state recoding scheme could not be performed because 824
IQ-TREE currently only supports amino acid datasets recoded into four-character 825
states. 826
Other analyses 827
The 16S rRNA genes of ‘Candidatus Finniella inopinata’, and the presumed 828
endosymbionts of Peranema trichophorum and Stachyamoeba lipophora were identified 829
with RNAmmer 1.2 server and BLAST searches. A set of 16S rRNA genes for diverse 830
rickettsialeans and holosporaleans, and other alphaproteobacteria as outgroup, were 831
retrieved from NCBI GenBank. The selection was based on Hess et al., (2016), Szokoli 832

38
et al., (2016) and Wang and Wu (2015). Environmental sequences for uncultured and 833
undescribed rickettsialeans were retrieved by keeping the 50 best hits resulting from a 834
BLAST search of our three novel 16S rRNA genes against the NCBI GenBank non-835
redundant (nr) database. The sequences were aligned with the SILVA aligner SINA 836
v1.2.11 and all-gap sites were later removed. Phylogenetic analyses on this alignment 837
were performed on IQ-TREE v1.5.5 using the GTR+F+R8 model. 838
A UPGMA (average-linkage) clustering of amino acid compositions based on the 200 839
gene set for the Alphaproteobacteria was built in MEGA 7 (Kumar, Stecher, and Tamura 840
2016) from a matrix of Euclidean distances between amino acid compositions of 841
sequences exported from the phylogenetic software P4 (Foster 2004; 842
http://p4.nhm.ac.uk/index.html). 843
Data availability 844
The genome of ‘Candidatus Finniella inopinata’, endosymbiont of Peranema 845
trichophorum strain CCAP 1260/1B and endosymbiont of Stachyamoeba lipophora 846
strain ATCC 50324 were deposited in NCBI GenBank under the BioProject 847
PRJNA501864. Raw sequencing reads were deposited on the NCBI SRA archive under 848
the BioProject PRJNA501864. Multi-gene datasets as well as phylogenetic trees 849
inferred in this study were deposited at Mendeley Data under the DOI: 850
http://dx.doi.org/10.17632/75m68dxd83.1. 851
ACKNOWLEDGEMENTS 852
Sergio A. Muñoz-Gómez is supported by a Killam Predoctoral Scholarship and a Nova 853
Scotia Graduate Scholarship. This work was supported by Natural Sciences and 854
Engineering Research (NSERC) Discovery Grants 2016-06792 to A.J.R, RGPIN/05754-855
2015 to C.H.S, RGPIN/05286-2014 to G.B., and RGPIN-2017-05411 to B.F.L. We thank 856
Jon Jerlström Hultqvist and Gina Filloramo (both at Dalhousie University) for advice on 857
long-read sequencing with the Nanopore MinION. We also thank Camilo A. Calderón-858
Acevedo for advice about taxonomic issues, and Franziska Szokoli for reading and 859
commenting on a late version of this manuscript. Bruce Curtis (Dalhousie University) 860
and Peter G. Foster (Natural History Museum of London) kindly provided technical help 861

39
with bioinformatics and with the software P4, respectively. We thank Joanny Roy, 862
Georgette Kiethega, Matus Valach, and Shona Teijeiro (all at the Université de 863
Montréal), and Drahomira Faktora (University of South Bohemia), for help with the 864
culturing, DNA preparation and sequencing of the endosymbiont of Peranema 865
trichophorum. Some of the genome data used in this study were produced by the US 866
Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration 867
with the user community. 868
COMPETING INTERESTS 869
No competing interests declared by the authors. 870
REFERENCES 871
Bankevich, Anton, Sergey Nurk, Dmitry Antipov, Alexey A. Gurevich, Mikhail Dvorkin, Alexander 872 S. Kulikov, Valery M. Lesin, et al. 2012. “SPAdes: A New Genome Assembly Algorithm 873 and Its Applications to Single-Cell Sequencing.” Journal of Computational Biology 19 (5): 874 455–77. https://doi.org/10.1089/cmb.2012.0021. 875
Bazylinski, Dennis A., Timothy J. Williams, Christopher T. Lefèvre, Denis Trubitsyn, Jiasong 876 Fang, Terrence J. Beveridge, Bruce M. Moskowitz, et al. 2013. “Magnetovibrio 877 Blakemorei Gen. Nov., Sp. Nov., a Magnetotactic Bacterium (Alphaproteobacteria: 878 Rhodospirillaceae) Isolated from a Salt Marsh.” International Journal of Systematic and 879 Evolutionary Microbiology 63 (5): 1824–33. https://doi.org/10.1099/ijs.0.044453-0. 880
Bolger, Anthony M., Marc Lohse, and Bjoern Usadel. 2014. “Trimmomatic: A Flexible Trimmer 881 for Illumina Sequence Data.” Bioinformatics 30 (15): 2114–20. 882 https://doi.org/10.1093/bioinformatics/btu170. 883
Boscaro, Vittorio, Sergei I. Fokin, Martina Schrallhammer, Michael Schweikert, and Giulio 884 Petroni. 2013. “Revised Systematics of Holospora-like Bacteria and Characterization of 885 ‘Candidatus Gortzia Infectiva’, a Novel Macronuclear Symbiont of Paramecium 886 Jenningsi.” Microbial Ecology 65 (1): 255–67. https://doi.org/10.1007/s00248-012-0110-887 2. 888
Brindefalk, Björn, Thijs J. G. Ettema, Johan Viklund, Mikael Thollesson, and Siv G. E. 889 Andersson. 2011. “A Phylometagenomic Exploration of Oceanic Alphaproteobacteria 890 Reveals Mitochondrial Relatives Unrelated to the SAR11 Clade.” PLoS ONE 6 (9): 891 e24457. https://doi.org/10.1371/journal.pone.0024457. 892
Castelli, Michele, Elena Sabaneyeva, Olivia Lanzoni, Natalia Lebedeva, Anna Maria Floriano, 893 Stefano Gaiarsa, Konstantin Benken, et al. 2018. “The Extracellular Association of the 894 Bacterium ‘Candidatus Deianiraea Vastatrix’ with the Ciliate Paramecium Suggests an 895 Alternative Scenario for the Evolution of Rickettsiales.” BioRxiv, November, 479196. 896 https://doi.org/10.1101/479196. 897
Cho, Jang-Cheon, and Stephen J. Giovannoni. 2003. “Parvularcula Bermudensis Gen. Nov., 898 Sp. Nov., a Marine Bacterium That Forms a Deep Branch in the Alpha-Proteobacteria.” 899 International Journal of Systematic and Evolutionary Microbiology 53 (Pt 4): 1031–36. 900 https://doi.org/10.1099/ijs.0.02566-0. 901
Eren, A. Murat, Özcan C. Esen, Christopher Quince, Joseph H. Vineis, Hilary G. Morrison, 902 Mitchell L. Sogin, and Tom O. Delmont. 2015. “Anvi’o: An Advanced Analysis and 903

40
Visualization Platform for ‘omics Data.” PeerJ 3 (October): e1319. 904 https://doi.org/10.7717/peerj.1319. 905
Eschbach, Erik, Martin Pfannkuchen, Michael Schweikert, Denja Drutschmann, Franz Brümmer, 906 Sergei Fokin, Wolfgang Ludwig, and Hans-Dieter Görtz. 2009. “‘Candidatus 907 Paraholospora Nucleivisitans’, an Intracellular Bacterium in Paramecium Sexaurelia 908 Shuttles between the Cytoplasm and the Nucleus of Its Host.” Systematic and Applied 909 Microbiology 32 (7): 490–500. https://doi.org/10.1016/j.syapm.2009.07.004. 910
Esser, Christian, William Martin, and Tal Dagan. 2007. “The Origin of Mitochondria in Light of a 911 Fluid Prokaryotic Chromosome Model.” Biology Letters 3 (2): 180–84. 912 https://doi.org/10.1098/rsbl.2006.0582. 913
Ettema, Thijs J. G., and Siv G. E. Andersson. 2009. “The Alpha-Proteobacteria: The Darwin 914 Finches of the Bacterial World.” Biology Letters 5 (3): 429–32. 915 https://doi.org/10.1098/rsbl.2008.0793. 916
Ferla, Matteo P., J. Cameron Thrash, Stephen J. Giovannoni, and Wayne M. Patrick. 2013. 917 “New RRNA Gene-Based Phylogenies of the Alphaproteobacteria Provide Perspective 918 on Major Groups, Mitochondrial Ancestry and Phylogenetic Instability.” PLoS ONE 8 919 (12): e83383. https://doi.org/10.1371/journal.pone.0083383. 920
Fitzpatrick, David A., Christopher J. Creevey, and James O. McInerney. 2006. “Genome 921 Phylogenies Indicate a Meaningful Alpha-Proteobacterial Phylogeny and Support a 922 Grouping of the Mitochondria with the Rickettsiales.” Molecular Biology and Evolution 23 923 (1): 74–85. https://doi.org/10.1093/molbev/msj009. 924
Foesel, Bärbel U., Anita S. Gössner, Harold L. Drake, and Andreas Schramm. 2007. 925 “Geminicoccus Roseus Gen. Nov., Sp. Nov., an Aerobic Phototrophic 926 Alphaproteobacterium Isolated from a Marine Aquaculture Biofilter.” Systematic and 927 Applied Microbiology 30 (8): 581–86. https://doi.org/10.1016/j.syapm.2007.05.005. 928
Foster, Peter G. 2004. “Modeling Compositional Heterogeneity.” Systematic Biology 53 (3): 929 485–95. 930
Garrity, George, ed. 2005. Bergey’s Manual of Systematic Bacteriology: Volume 2 : The 931 Proteobacteria. 2nd ed. Springer US. //www.springer.com/gp/book/9780387950402. 932
Georgiades, Kalliopi, Mohammed-Amine Madoui, Phuong Le, Catherine Robert, and Didier 933 Raoult. 2011. “Phylogenomic Analysis of Odyssella Thessalonicensis Fortifies the 934 Common Origin of Rickettsiales, Pelagibacter Ubique and Reclimonas Americana 935 Mitochondrion.” PLoS ONE 6 (9): e24857. https://doi.org/10.1371/journal.pone.0024857. 936
Giovannoni, Stephen J. 2017. “SAR11 Bacteria: The Most Abundant Plankton in the Oceans.” 937 Annual Review of Marine Science 9 (January): 231–55. https://doi.org/10.1146/annurev-938 marine-010814-015934. 939
Grote, Jana, J. Cameron Thrash, Megan J. Huggett, Zachary C. Landry, Paul Carini, Stephen J. 940 Giovannoni, and Michael S. Rappé. 2012. “Streamlining and Core Genome 941 Conservation among Highly Divergent Members of the SAR11 Clade.” MBio 3 (5): 942 e00252-12. https://doi.org/10.1128/mBio.00252-12. 943
Harbison, Austin B., Laiken E. Price, Michael D. Flythe, and Suzanna L. Bräuer. 2017. 944 “Micropepsis Pineolensis Gen. Nov., Sp. Nov., a Mildly Acidophilic 945 Alphaproteobacterium Isolated from a Poor Fen, and Proposal of Micropepsaceae Fam. 946 Nov. within Micropepsales Ord. Nov.” International Journal of Systematic and 947 Evolutionary Microbiology 67 (4): 839–44. https://doi.org/10.1099/ijsem.0.001681. 948
Heiss, Aaron A., Martin Kolisko, Fleming Ekelund, Matthew W. Brown, Andrew J. Roger, and 949 Alastair G. B. Simpson. 2018. “Combined Morphological and Phylogenomic Re-950 Examination of Malawimonads, a Critical Taxon for Inferring the Evolutionary History of 951 Eukaryotes.” Royal Society Open Science 5 (4): 171707. 952 https://doi.org/10.1098/rsos.171707. 953

41
Hess, Sebastian, and Michael Melkonian. 2013. “The Mystery of Clade X: Orciraptor Gen. Nov. 954 and Viridiraptor Gen. Nov. Are Highly Specialised, Algivorous Amoeboflagellates 955 (Glissomonadida, Cercozoa).” Protist 164 (5): 706–47. 956 https://doi.org/10.1016/j.protis.2013.07.003. 957
Hess, Sebastian, Andreas Suthaus, and Michael Melkonian. 2015. “‘Candidatus Finniella’ 958 (Rickettsiales, Alphaproteobacteria), Novel Endosymbionts of Viridiraptorid 959 Amoeboflagellates (Cercozoa, Rhizaria).” Applied and Environmental Microbiology 82 960 (2): 659–70. https://doi.org/10.1128/AEM.02680-15. 961
Kalyaanamoorthy, Subha, Bui Quang Minh, Thomas K. F. Wong, Arndt von Haeseler, and Lars 962 S. Jermiin. 2017. “ModelFinder: Fast Model Selection for Accurate Phylogenetic 963 Estimates.” Nature Methods 14 (6): 587–89. https://doi.org/10.1038/nmeth.4285. 964
Kumar, Sudhir, Glen Stecher, and Koichiro Tamura. 2016. “MEGA7: Molecular Evolutionary 965 Genetics Analysis Version 7.0 for Bigger Datasets.” Molecular Biology and Evolution 33 966 (7): 1870–74. https://doi.org/10.1093/molbev/msw054. 967
Kurahashi, Midori, Yukiyo Fukunaga, Shigeaki Harayama, and Akira Yokota. 2008. “Sneathiella 968 Glossodoripedis Sp. Nov., a Marine Alphaproteobacterium Isolated from the Nudibranch 969 Glossodoris Cincta, and Proposal of Sneathiellales Ord. Nov. and Sneathiellaceae Fam. 970 Nov.” International Journal of Systematic and Evolutionary Microbiology 58 (Pt 3): 548–971 52. https://doi.org/10.1099/ijs.0.65328-0. 972
Kwon, Kae Kyoung, Hee-Soon Lee, Sung Hyun Yang, and Sang-Jin Kim. 2005. “Kordiimonas 973 Gwangyangensis Gen. Nov., Sp. Nov., a Marine Bacterium Isolated from Marine 974 Sediments That Forms a Distinct Phyletic Lineage (Kordiimonadales Ord. Nov.) in the 975 ‘Alphaproteobacteria.’” International Journal of Systematic and Evolutionary Microbiology 976 55 (5): 2033–37. https://doi.org/10.1099/ijs.0.63684-0. 977
Lang, B. Franz, and Gertraud Burger. 2007. “Purification of Mitochondrial and Plastid DNA.” 978 Nature Protocols 2 (3): 652–60. https://doi.org/10.1038/nprot.2007.58. 979
Lartillot, Nicolas, Thomas Lepage, and Samuel Blanquart. 2009. “PhyloBayes 3: A Bayesian 980 Software Package for Phylogenetic Reconstruction and Molecular Dating.” 981 Bioinformatics 25 (17): 2286–88. https://doi.org/10.1093/bioinformatics/btp368. 982
Lartillot, Nicolas, and Hervé Philippe. 2004. “A Bayesian Mixture Model for Across-Site 983 Heterogeneities in the Amino-Acid Replacement Process.” Molecular Biology and 984 Evolution 21 (6): 1095–1109. https://doi.org/10.1093/molbev/msh112. 985
Lee, Kyung-Bum, Chi-Te Liu, Yojiro Anzai, Hongik Kim, Toshihiro Aono, and Hiroshi Oyaizu. 986 2005. “The Hierarchical System of the ‘Alphaproteobacteria’: Description of 987 Hyphomonadaceae Fam. Nov., Xanthobacteraceae Fam. Nov. and Erythrobacteraceae 988 Fam. Nov.” International Journal of Systematic and Evolutionary Microbiology 55 (Pt 5): 989 1907–19. https://doi.org/10.1099/ijs.0.63663-0. 990
Luo, Haiwei. 2015. “Evolutionary Origin of a Streamlined Marine Bacterioplankton Lineage.” The 991 ISME Journal 9 (6): 1423–33. https://doi.org/10.1038/ismej.2014.227. 992
Luo, Haiwei, Miklós Csűros, Austin L. Hughes, and Mary Ann Moran. 2013. “Evolution of 993 Divergent Life History Strategies in Marine Alphaproteobacteria.” MBio 4 (4): e00373-13. 994 https://doi.org/10.1128/mBio.00373-13. 995
Madigan, Michael T., Deborah O. Jung, and Michael T. Madigan. 2009. “An Overview of Purple 996 Bacteria: Systematics, Physiology, and Habitats.” In The Purple Phototrophic Bacteria, 997 1–15. Advances in Photosynthesis and Respiration. Springer, Dordrecht. 998 https://doi.org/10.1007/978-1-4020-8815-5_1. 999
Martijn, Joran, Julian Vosseberg, Lionel Guy, Pierre Offre, and Thijs J. G. Ettema. 2018. “Deep 1000 Mitochondrial Origin Outside the Sampled Alphaproteobacteria.” Nature 557 (7703): 1001 101–5. https://doi.org/10.1038/s41586-018-0059-5. 1002

42
Minh, Bui Quang, Minh Anh Thi Nguyen, and Arndt von Haeseler. 2013. “Ultrafast 1003 Approximation for Phylogenetic Bootstrap.” Molecular Biology and Evolution 30 (5): 1004 1188–95. https://doi.org/10.1093/molbev/mst024. 1005
Montagna, Matteo, Davide Sassera, Sara Epis, Chiara Bazzocchi, Claudia Vannini, Nathan Lo, 1006 Luciano Sacchi, Takema Fukatsu, Giulio Petroni, and Claudio Bandi. 2013. “‘Candidatus 1007 Midichloriaceae’ Fam. Nov. (Rickettsiales), an Ecologically Widespread Clade of 1008 Intracellular Alphaproteobacteria.” Applied and Environmental Microbiology 79 (10): 1009 3241–48. https://doi.org/10.1128/AEM.03971-12. 1010
Nguyen, Lam-Tung, Heiko A. Schmidt, Arndt von Haeseler, and Bui Quang Minh. 2015. “IQ-1011 TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood 1012 Phylogenies.” Molecular Biology and Evolution 32 (1): 268–74. 1013 https://doi.org/10.1093/molbev/msu300. 1014
Parks, Donovan H., Maria Chuvochina, David W. Waite, Christian Rinke, Adam Skarshewski, 1015 Pierre-Alain Chaumeil, and Philip Hugenholtz. 2018. “A Standardized Bacterial 1016 Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life.” Nature 1017 Biotechnology 36 (10): 996–1004. https://doi.org/10.1038/nbt.4229. 1018
Proença, Diogo N., William B. Whitman, Neha Varghese, Nicole Shapiro, Tanja Woyke, Nikos 1019 C. Kyrpides, and Paula V. Morais. 2017. “Arboriscoccus Pini Gen. Nov., Sp. Nov., an 1020 Endophyte from a Pine Tree of the Class Alphaproteobacteria, Emended Description of 1021 Geminicoccus Roseus, and Proposal of Geminicoccaceae Fam. Nov.” Systematic and 1022 Applied Microbiology, December. https://doi.org/10.1016/j.syapm.2017.11.006. 1023
Rodríguez-Ezpeleta, Naiara, and T. Martin Embley. 2012. “The SAR11 Group of Alpha-1024 Proteobacteria Is Not Related to the Origin of Mitochondria.” PLoS ONE 7 (1): e30520. 1025 https://doi.org/10.1371/journal.pone.0030520. 1026
Roger, Andrew J., Sergio A. Muñoz-Gómez, and Ryoma Kamikawa. 2017. “The Origin and 1027 Diversification of Mitochondria.” Current Biology 27 (21): R1177–92. 1028 https://doi.org/10.1016/j.cub.2017.09.015. 1029
Rosenberg, Eugene, Edward F. DeLong, Stephen Lory, Erko Stackebrandt, and Fabiano 1030 Thompson, eds. 2014. The Prokaryotes: Alphaproteobacteria and Betaproteobacteria. 1031 4th ed. Berlin Heidelberg: Springer-Verlag. 1032 //www.springer.com/gp/book/9783642301964. 1033
Santos, Huarrisson Azevedo, and Carlos Luiz Massard. 2014. “The Family Holosporaceae.” In 1034 The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, edited by Eugene 1035 Rosenberg, Edward F. DeLong, Stephen Lory, Erko Stackebrandt, and Fabiano 1036 Thompson, 237–46. Berlin, Heidelberg: Springer Berlin Heidelberg. 1037 https://doi.org/10.1007/978-3-642-30197-1_264. 1038
Sassera, Davide, Nathan Lo, Sara Epis, Giuseppe D’Auria, Matteo Montagna, Francesco 1039 Comandatore, David Horner, et al. 2011. “Phylogenomic Evidence for the Presence of a 1040 Flagellum and Cbb3 Oxidase in the Free-Living Mitochondrial Ancestor.” Molecular 1041 Biology and Evolution 28 (12): 3285–96. https://doi.org/10.1093/molbev/msr159. 1042
Susko, Edward, Léa Lincker, and Andrew J. Roger. 2018. “Accelerated Estimation of Frequency 1043 Classes in Site-Heterogeneous Profile Mixture Models.” Molecular Biology and Evolution 1044 may026. https://doi.org/10.1093/molbev/msy026. 1045
Susko, Edward, and Andrew J. Roger. 2007. “On Reduced Amino Acid Alphabets for 1046 Phylogenetic Inference.” Molecular Biology and Evolution 24 (9): 2139–50. 1047 https://doi.org/10.1093/molbev/msm144. 1048
Szokoli, Franziska, Michele Castelli, Elena Sabaneyeva, Martina Schrallhammer, Sascha 1049 Krenek, Thomas G. Doak, Thomas U. Berendonk, and Giulio Petroni. 2016. 1050 “Disentangling the Taxonomy of Rickettsiales and Description of Two Novel Symbionts 1051 (‘Candidatus Bealeia Paramacronuclearis’ and ‘Candidatus Fokinia Cryptica’) Sharing 1052

43
the Cytoplasm of the Ciliate Protist Paramecium Biaurelia.” Applied and Environmental 1053 Microbiology 82 (24): 7236–47. https://doi.org/10.1128/AEM.02284-16. 1054
Thrash, J. Cameron, Alex Boyd, Megan J. Huggett, Jana Grote, Paul Carini, Ryan J. Yoder, 1055 Barbara Robbertse, Joseph W. Spatafora, Michael S. Rappé, and Stephen J. 1056 Giovannoni. 2011. “Phylogenomic Evidence for a Common Ancestor of Mitochondria 1057 and the SAR11 Clade.” Scientific Reports 1 (June): 13. 1058 https://doi.org/10.1038/srep00013. 1059
Vaidya, Gaurav, David J Lohman, and Rudolf Meier. 2011. “SequenceMatrix: Concatenation 1060 Software for the Fast Assembly of Multi-gene Datasets with Character Set and Codon 1061 Information.” Cladistics 27 (2): 171–80. https://doi.org/10.1111/j.1096-1062 0031.2010.00329.x. 1063
Venkata Ramana, V., S. Kalyana Chakravarthy, E. V. V. Ramaprasad, V. Thiel, J. F. Imhoff, Ch 1064 Sasikala, and Ch V. Ramana. 2013. “Emended Description of the Genus 1065 Rhodothalassium Imhoff et Al., 1998 and Proposal of Rhodothalassiaceae Fam. Nov. 1066 and Rhodothalassiales Ord. Nov.” Systematic and Applied Microbiology 36 (1): 28–32. 1067 https://doi.org/10.1016/j.syapm.2012.09.003. 1068
Verbruggen, Heroen. 2018. “SiteStripper Version 1.03.” http://www.phycoweb.net/software. 1069 Viklund, Johan, Thijs J. G Ettema, and Siv G. E Andersson. 2012. “Independent Genome 1070
Reduction and Phylogenetic Reclassification of the Oceanic SAR11 Clade.” Molecular 1071 Biology and Evolution 29 (2): 599–615. https://doi.org/10.1093/molbev/msr203. 1072
Viklund, Johan, Joran Martijn, Thijs J. G. Ettema, and Siv G. E. Andersson. 2013. “Comparative 1073 and Phylogenomic Evidence That the Alphaproteobacterium HIMB59 Is Not a Member 1074 of the Oceanic SAR11 Clade.” PLoS ONE 8 (11): e78858. 1075 https://doi.org/10.1371/journal.pone.0078858. 1076
Wang, Huai-Chun, Bui Quang Minh, Edward Susko, and Andrew J. Roger. 2018. “Modeling Site 1077 Heterogeneity with Posterior Mean Site Frequency Profiles Accelerates Accurate 1078 Phylogenomic Estimation.” Systematic Biology 67 (2): 216–35. 1079 https://doi.org/10.1093/sysbio/syx068. 1080
Wang, Zhang, and Martin Wu. 2013. “A Phylum-Level Bacterial Phylogenetic Marker Database.” 1081 Molecular Biology and Evolution 30 (6): 1258–62. 1082 https://doi.org/10.1093/molbev/mst059. 1083
———. 2014. “Phylogenomic Reconstruction Indicates Mitochondrial Ancestor Was an Energy 1084 Parasite.” PLoS ONE 9 (10): e110685. https://doi.org/10.1371/journal.pone.0110685. 1085
———. 2015. “An Integrated Phylogenomic Approach toward Pinpointing the Origin of 1086 Mitochondria.” Scientific Reports 5 (January): 7949. https://doi.org/10.1038/srep07949. 1087
Whitman, William B., ed. 2015. Bergey’s Manual of Systematics of Archaea and Bacteria. Wiley. 1088 https://onlinelibrary.wiley.com/doi/book/10.1002/9781118960608. 1089
Wiese, Jutta, Vera Thiel, Andrea Gärtner, Rolf Schmaljohann, and Johannes F. Imhoff. 2009. 1090 “Kiloniella Laminariae Gen. Nov., Sp. Nov., an Alphaproteobacterium from the Marine 1091 Macroalga Laminaria Saccharina.” International Journal of Systematic and Evolutionary 1092 Microbiology 59 (2): 350–56. https://doi.org/10.1099/ijs.0.001651-0. 1093
Williams, Kelly P., Bruno W. Sobral, and Allan W. Dickerman. 2007. “A Robust Species Tree for 1094 the Alphaproteobacteria.” Journal of Bacteriology 189 (13): 4578–86. 1095 https://doi.org/10.1128/JB.00269-07. 1096
Williams, Timothy J., Christopher T. Lefèvre, Weidong Zhao, Terry J. Beveridge, and Dennis A. 1097 Bazylinski. 2012. “Magnetospira Thiophila Gen. Nov., Sp. Nov., a Marine Magnetotactic 1098 Bacterium That Represents a Novel Lineage within the Rhodospirillaceae 1099 (Alphaproteobacteria).” International Journal of Systematic and Evolutionary 1100 Microbiology 62 (10): 2443–50. https://doi.org/10.1099/ijs.0.037697-0. 1101

44
Wu, Martin, Sourav Chatterji, and Jonathan A. Eisen. 2012. “Accounting for Alignment 1102 Uncertainty in Phylogenomics.” PloS One 7 (1): e30288. 1103 https://doi.org/10.1371/journal.pone.0030288. 1104
1105

45
SUPPLEMENTARY FIGURE LEGENDS 1106
Figure 2-figure supplement 1. A labeled version showing taxon names for Figure 2. 1107
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. 1108
Figure 2-figure supplement 2. A diagram of the strategies and phylogenetic analyses 1109
employed in this study. 1110
Figure 2-figure supplement 3. Bayesian consensus trees inferred with PhyloBayes 1111
MPI v1.7 and the CAT-Poisson+Γ4 model. Branch support values are 1.0 posterior 1112
probabilities unless annotated. A. Bayesian consensus tree inferred from the full dataset 1113
which is highly compositionally heterogeneous. B. Bayesian consensus tree inferred 1114
from a dataset whose compositional heterogeneity has been decreased by removing 1115
50% of the most biased sites according to ɀ. See Figs. 2A and 2B for the most likely 1116
trees inferred in IQ-TREE v1.5.5 and the LG+PMSF(C60)+F+R6 model. 1117
Figure 2-figure supplement 4. Maximum-likelihood trees to assess the placements of 1118
the Holosporales, Rickettsiales, Pelagibacterales and alphaproteobacterium sp. 1119
HIMB59 when all four groups are included. Branch support values are 100% SH-aLRT 1120
and 100% UFBoot unless annotated. A. A tree that results from the analysis of the 1121
untreated dataset. B. A tree that results from the analysis of a dataset from which the 1122
50% most compositionally-biased sites have been removed. C. A tree that results from 1123
the analysis of a dataset that has been recoded into the four-character state recoding 1124
scheme S4 (recoding scheme: RNCM EHIPTWV ADQLKS GFY). D. A tree that results 1125
from the analysis of a dataset that only comprises the 40 most compositionally 1126
homogeneous genes. 1127
Figure 3-figure supplement 1. Maximum-likelihood trees to assess the placement of 1128
the Holosporales in the absence of the Rickettsiales, Pelagibacterales and 1129
alphaproteobacterium sp. HIMB59. Branch support values are 100% SH-aLRT and 1130
100% UFBoot unless annotated. A. A tree that results from the analysis of the untreated 1131
dataset. B. A tree that results from the analysis of a dataset from which the 50% most 1132
compositionally-biased sites have been removed. C. A tree that results from the 1133
analysis of a dataset that has been recoded into the four-character state recoding 1134
scheme S4 (recoding scheme: ARNDQEILKSTV GHY CMFP W). D. A tree that results 1135
from the analysis of a dataset that only comprises the 40 most compositionally 1136
homogeneous genes. 1137
Figure 3-figure supplement 2. Maximum-likelihood trees to assess the placement of 1138
the Rickettsiales in the absence of the Holosporales, Pelagibacterales, and 1139
alphaproteobacterium sp. HIMB59. Branch support values are 100% SH-aLRT and 1140
100% UFBoot unless annotated. A. A tree that results from the analysis of the untreated 1141
dataset. B. A tree that results from the analysis of a dataset from which the 50% most 1142
compositionally-biased sites have been removed. C. A tree that results from the 1143
analysis of a dataset that has been recoded into the four-character state recoding 1144
scheme S4 (recoding scheme: PY RNMF GHLKTW ADCQEISV). D. A tree that results 1145

46
from the analysis of a dataset that only comprises the 40 most compositionally 1146
homogeneous genes. 1147
Figure 3-figure supplement 3. Maximum-likelihood trees to assess the placement of 1148
the Rickettsiales in the absence of the Holosporales, Pelagibacterales, 1149
alphaproteobacterium sp. HIMB59 and the Beta-, and Gammaproteobacteria outgroup. 1150
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. A. A 1151
tree that results from the analysis of the untreated dataset. B. A tree that results from 1152
the analysis of a dataset from which the 50% most compositionally-biased sites have 1153
been removed. C. A tree that results from the analysis of a dataset that has been 1154
recoded into the four-character state recoding scheme S4 (recoding scheme: RNMF 1155
GHLKTW ADCQEISV PY). D. A tree that results from the analysis of a dataset that only 1156
comprises the 40 most compositionally homogeneous genes. Branch support values 1157
are 100% SH-aLRT and 100% UFBoot unless annotated. 1158
Figure 3-figure supplement 4. Maximum-likelihood trees to assess the placement of 1159
the Pelagibacterales in the absence of the Holosporales, Rickettsiales and 1160
alphaproteobacterium sp. HIMB59. Branch support values are 100% SH-aLRT and 1161
100% UFBoot unless annotated. A. A tree that results from the analysis of the untreated 1162
dataset. B. A tree that results from the analysis of a dataset from which the 50% most 1163
compositionally-biased sites have been removed. C. A tree that results from the 1164
analysis of a dataset that has been recoded into the four-character state recoding 1165
scheme S4 (recoding scheme: EGIV ARNDQHKMPSY LFT CW). D. A tree that results 1166
from the analysis of a dataset that only comprises the 40 most compositionally 1167
homogeneous genes. 1168
Figure 3-figure supplement 5. Maximum-likelihood trees to assess the placement of 1169
alphaproteobacterium sp. HIMB59 in the absence of the Holosporales, Rickettsiales and 1170
Pelagibacterales. Branch support values are 100% SH-aLRT and 100% UFBoot unless 1171
annotated. A. A tree that results from the analysis of the untreated dataset. B. A tree 1172
that results from the analysis of a dataset from which the 50% most compositionally-1173
biased sites have been removed. C. A tree that results from the analysis of a dataset 1174
that has been recoded into the four-character state recoding scheme S4 (recoding 1175
scheme: RLKMT ANDQEIPSV CW GHFY). D. A tree that results from the analysis of a 1176
dataset that only comprises the 40 most compositionally homogeneous genes. 1177
Figure 3-figure supplement 6. Bayesian consensus trees inferred with PhyloBayes 1178
MPI v1.7 and the CAT-Poisson+Γ4 model. Branch support values are 1.0 posterior 1179
probabilities unless annotated. A. Bayesian consensus tree inferred to place the 1180
Holosporales in the absence of the Rickettsiales and the Pelagibacterales and when 1181
compositional heterogeneity has been decreased by removing 50% of the most biased 1182
sites according to ɀ. B. Bayesian consensus tree inferred to place the Holosporales in 1183
the absence of the Rickettsiales and the Pelagibacterales and when the data have been 1184
recoded into a four-character state alphabet (the dataset-specific recoding scheme S4: 1185
ARNDQEILKSTV GHY CMFP W) to reduce compositional heterogeneity. See Figs. 2A 1186

47
and 2B for the most likely trees inferred in IQ-TREE v1.5.5 and the 1187
LG+PMSF(C60)+F+R6 and GTR+ES60S4+F+R6 models, respectively. 1188
Figure 3-figure supplement 7. Maximum-likelihood trees to assess the placement of 1189
the Holosporales when the fast-evolving Holospora and ‘Candidatus Hepatobacter’ are 1190
also included in the absence of the Rickettsiales, Pelagibacterales and 1191
alphaproteobacterium sp. HIMB59. Branch support values are 100% SH-aLRT and 1192
100% UFBoot unless annotated. 1193
Figure 3-figure supplement 8. Bayesian consensus tree inferred to place the 1194
Holosporales in the absence of the Pelagibacterales, alphaproteobacterium sp. 1195
HIMB59, and Rickettsiales, and when the data have been recoded into a six-character 1196
state alphabet (the dataset-specific recoding scheme S6: AQEHISV RKMT PY DCLF 1197
NG W) to reduce compositional heterogeneity. Branch support values are 1.0 posterior 1198
probabilities unless annotated. 1199
Figure 2-figure supplement 5. Maximum-likelihood tree from the untreated dataset 1200
from which no taxon has been removed and analyzed under simpler LG4X model. In 1201
this tree, derived from an analysis using a model that does not account for 1202
compositional heterogeneity across sites, the Geminicoccaceae has a more derived 1203
placements within the Rhodospirillales as sister to the Acetobacteraceae. 1204
Figure 2-figure supplement 6. Constraint tree, used for IQ-TREE analyses, labeled 1205
with taxon names and also degree of missing data per taxon. Magnetococcales in gray; 1206
Rickettsiales in brown; Pelagibacterales in maroon; Holosporales in light blue; 1207
Rhizobiales in green; Caulobacterales in orange; Rhodobacterales in red; Sneathiellales 1208
in pink; Rhodospirillales in purple; Beta- and Gammaproteobacteria in black. 1209
Figure 2-figure supplement 7. GARP:FIMNKY ratios across the proteomes of the 120 1210
alphaproteobacteria and outgroup used in this study. 1211
1212
1213
1214
SUPPLEMENTARY FILES 1215
Supplementary file 1. A 16S rRNA gene maximum-likelihood tree of the Rickettsiales 1216
and Holosporales that phylogenetically places the three endosymbionts whose 1217
genomes were sequenced in this study: (1) ‘Candidatus Finniella inopinata’ 1218
endosymbiont of Viridiraptor invadens strain Virl02, (2) an alphaproteobacterium 1219
associated with Peranema trichophorum strain CCAP 1260/1B, and (3) an 1220
alphaproteobacterium associated with Stachyamoeba lipophora strain ATCC 50324. 1221
Branch support values are SH-aLRT and UFBoot. 1222
1223

48
1224
Supplementary file 2-table S1. Ultrafast bootstrap (UFBoot) variation for several 1225
clades discussed in this study as compositionally-biased sites, according to ɀ, are 1226
progressively removed in steps of 10%. 1227
Supplementary file 2-table S2. Ultrafast bootstrap (UFBoot) variation for several 1228
clades discussed in this study as the fastest sites are progressively removed in steps of 1229
10%. 1230
Supplementary file 2-table S3. GenBank assembly accession numbers for the 120 1231
alphaproteobacterial and outgroup genomes used in this study. 1232
Supplementary file 2-table S4. A list of the least compositionally heterogeneous genes 1233
out of the 200 single-copy and vertically-inherited genes used in this study. 1234
Supplementary file 2-table S5. Model fit of amino acid replacement matrices as 1235
components of simple models that do not account for compositional heterogeneity 1236
across sites. Models are ordered from lowest to highest BIC. -LnL: log-likelihood; df: 1237
degrees of freedom or number of free parameters; AIC: Akaike information criterion; 1238
AICc: corrected Akaike information criterion; BIC: Bayesian information criterion. 1239
Supplementary file 2-table S6. Model fit of amino acid replacement matrices as 1240
components of complex models that account for compositional heterogeneity across 1241
sites. Models are ordered from lowest to highest BIC. -LnL: log-likelihood; df: degrees of 1242
freedom or number of free parameters; AIC: Akaike information criterion; AICc: 1243
corrected Akaike information criterion; BIC: Bayesian information criterion. 1244
Supplementary file 2-table S7. Model fit of LG+ES60+F for which the model 1245
component that accounts for rate heterogeneity across sites varies. Models are ordered 1246
from lowest to highest BIC. -LnL: log-likelihood; df: degrees of freedom or number of 1247
free parameters; AIC: Akaike information criterion; AICc: corrected Akaike information 1248
criterion; BIC: Bayesian information criterion. 1249
Supplementary file 2-table S8. Several summary statistics for the PhyloBayes MCMC 1250
chains run for each analysis under the CAT-Poisson+Γ4. 1251
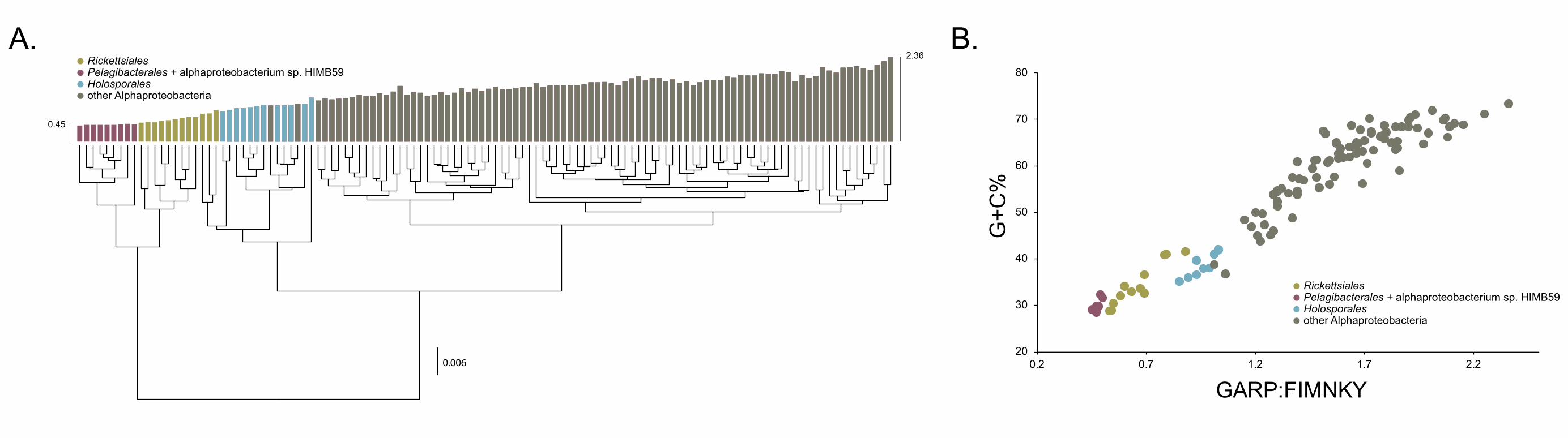
B.
20
30
40
50
60
70
80
0.2 0.7 1.2 1.7 2.2
G+C
%
GARP:FIMNKY
A.
0.45
2.36
0.006
RickettsialesPelagibacterales + alphaproteobacterium sp. HIMB59Holosporalesother Alphaproteobacteria
RickettsialesPelagibacterales + alphaproteobacterium sp. HIMB59Holosporalesother Alphaproteobacteria
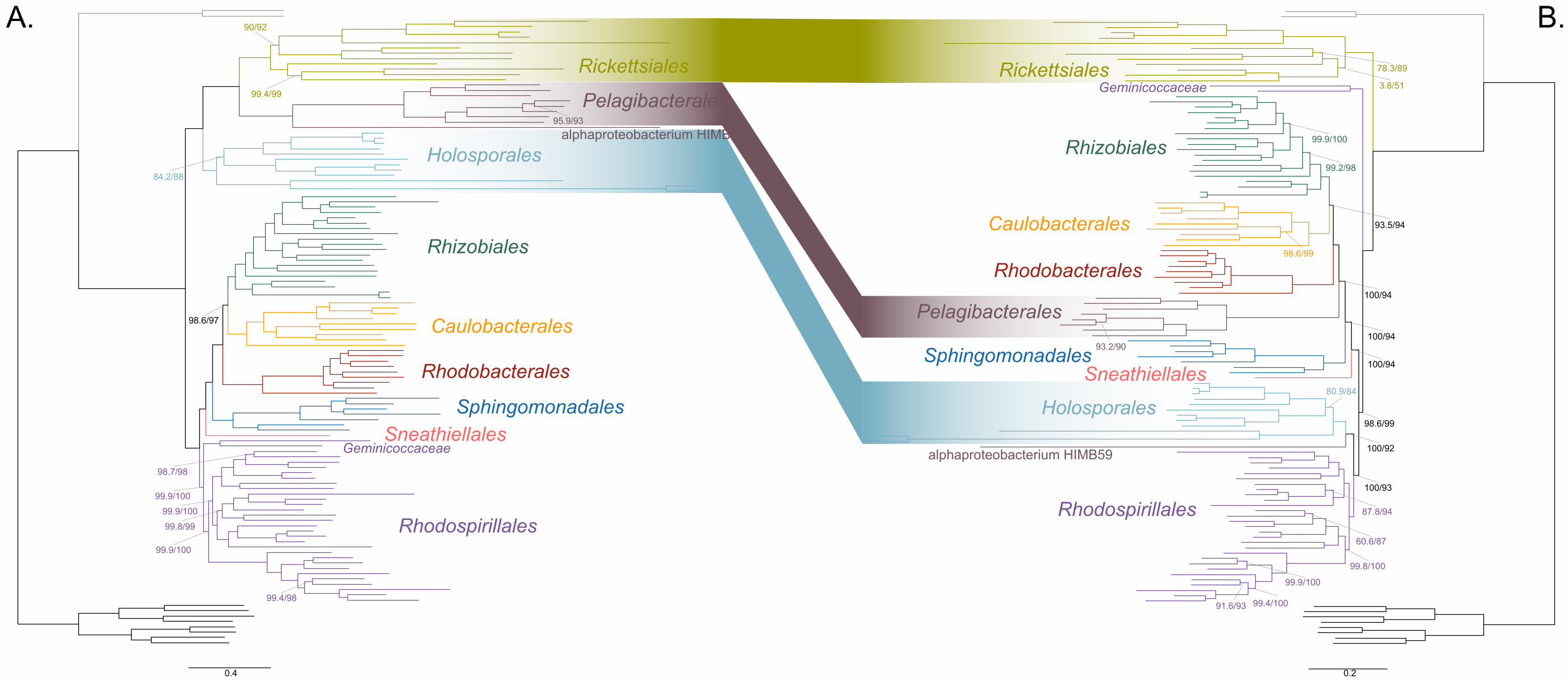
99.4/99
99.9/100
90/92
84.2/88
99.9/100
99.8/99
98.7/98
95.9/93
99.4/98
98.6/97
99.9/100
99.4/100
80.9/84
100/94
3.8/51
99.8/100
100/94
99.9/100
100/92
78.3/89
98.6/99
60.6/87
93.5/94
100/94
98.6/99
87.8/94
93.2/90
100/93
99.2/98
91.6/93
99.9/100
0.4 0.2
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
RhodospirillalesRhodospirillales
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Holosporales
Holosporales
Pelagibacterales
Pelagibacterales
Rickettsiales Rickettsiales
A. B.
Geminicoccaceae
Geminicoccaceae
alphaproteobacterium HIMB59
alphaproteobacterium HIMB59
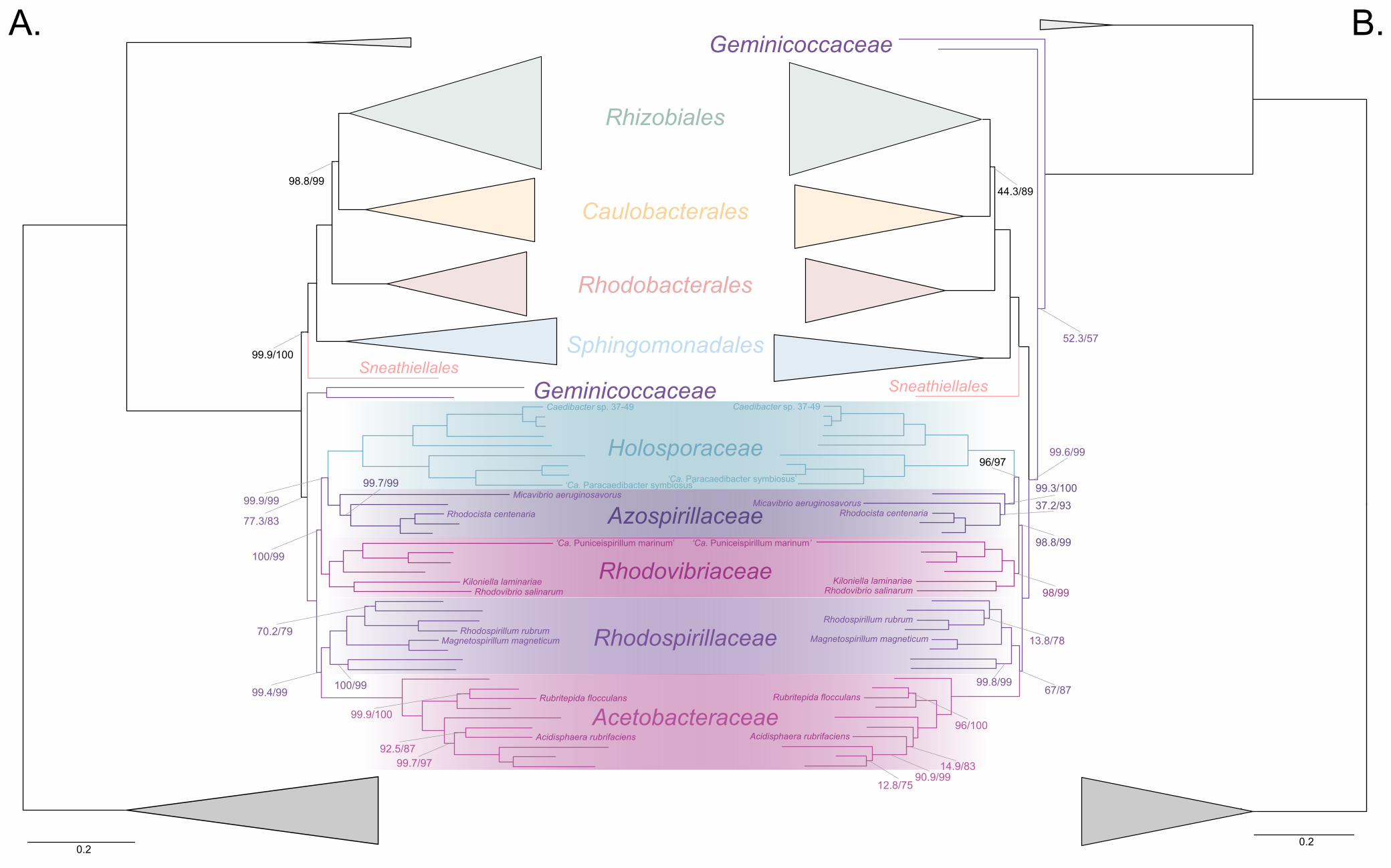
44.3/89
52.3/57
100/100
99.3/100
12.8/75
98.8/99
96/97
67/87
14.9/83
99.6/99
99.8/99
96/100
37.2/93
13.8/78
98/99
90.9/99
100/99
77.3/83
99.9/100
99.9/99
100/99
92.5/87
100/100
98.8/99
99.7/97
70.2/79
99.7/99
99.9/100
99.4/99
Holosporaceae
Azospirillaceae
Rhodovibriaceae
Rhodospirillaceae
Acetobacteraceae
0.20.2
A. B.
Geminicoccaceae
Geminicoccaceae
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Sneathiellales
Rhodospirillum rubrumRhodospirillum rubrum
Rhodocista centenaria Rhodocista centenaria
Micavibrio aeruginosavorusMicavibrio aeruginosavorus
Rhodovibrio salinarum Rhodovibrio salinarumKiloniella laminariaeKiloniella laminariae
‘Ca. Puniceispirillum marinum’ ‘Ca. Puniceispirillum marinum’
Magnetospirillum magneticum Magnetospirillum magneticum
Acidisphaera rubrifaciens Acidisphaera rubrifaciens
Rubritepida flocculans Rubritepida flocculans
Caedibacter sp. 37-49 Caedibacter sp. 37-49
‘Ca. Paracaedibacter symbiosus’‘Ca. Paracaedibacter symbiosus’
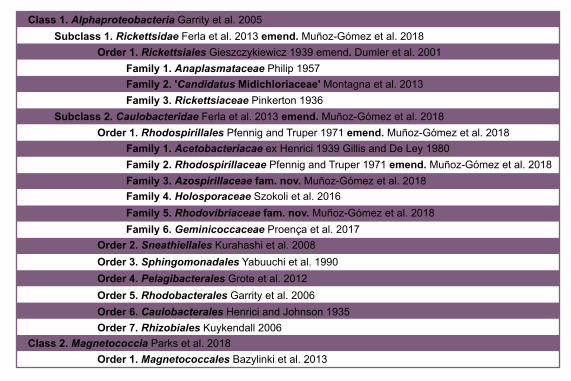
Class 1. Alphaproteobacteria Garrity et al. 2005Subclass 1. Rickettsidae Ferla et al. 2013 emend. Muñoz-Gómez et al. 2018
Order 1. Rickettsiales Gieszczykiewicz 1939 emend. Dumler et al. 2001Family 1. Anaplasmataceae Philip 1957
Family 3. Rickettsiaceae Pinkerton 1936Family 2. 'Candidatus Midichloriaceae' Montagna et al. 2013
Subclass 2. Caulobacteridae Ferla et al. 2013 emend. Muñoz-Gómez et al. 2018Order 1. Rhodospirillales Pfennig and Truper 1971 emend. Muñoz-Gómez et al. 2018
Family 1. Acetobacteriacae ex Henrici 1939 Gillis and De Ley 1980Family 2. Rhodospirillaceae Pfennig and Truper 1971 emend. Muñoz-Gómez et al. 2018Family 3. Azospirillaceae fam. nov. Muñoz-Gómez et al. 2018Family 4. Holosporaceae Szokoli et al. 2016Family 5. Rhodovibriaceae fam. nov. Muñoz-Gómez et al. 2018Family 6. Geminicoccaceae Proença et al. 2017
Order 2. Sneathiellales Kurahashi et al. 2008Order 3. Sphingomonadales Yabuuchi et al. 1990Order 4. Pelagibacterales Grote et al. 2012Order 5. Rhodobacterales Garrity et al. 2006Order 6. Caulobacterales Henrici and Johnson 1935Order 7. Rhizobiales Kuykendall 2006
Class 2. Magnetococcia Parks et al. 2018Order 1. Magnetococcales Bazylinki et al. 2013

Rhodovibrio salinarum DSM 9154
Methylovorus glucosetrophus SIP3-4
Bartonella quintana RM-11
alphaproteobacterium AAP81b
Candidatus Arcanobacter lacustris
Methylocystis sp. SC2
Octadecabacter antarcticus 307
Caenispirillum salinarum AK4
Belnapia moabensis DSM 16746
Candidatus Midichloria mitochondrii IricVA
Candidatus Odyssella thessalonicensis L13
Chelativorans sp. J32
Hirschia baltica ATCC 49814
Thermopetrobacter sp. TC1
endosymbiont of Peranema
Caedibacter sp. 38-128
Magnetofaba australis IT-1
Sphingomonas wittichii
alphaproteobacterium L41A
Geminicoccus roseus DSM 18922
Nitrosospira multiformis ATCC 25196
Oceanicaulis sp. HTCC2633
Candidatus Hepatobacter penaei
Xanthobacter autotrophicus Py2
Magnetococcus marinus MC-1
Acidiphilium angustum ATCC 35903
Polymorphum gilvum SL003B-26A1
Candidatus Finniella lucida
Candidatus Paracaedibacter symbiosus
Caedibacter varicaedens
Spongiibacter tropicus DSM 19543
Rhodocista sp. MIMtkB3
Anaplasma phagocytophilum HZ
Rhodomicrobium vannielii ATCC 17100
Rhodobacter sphaeroides 2.4.1
Rhodospirillum rubrum ATCC 11170
Candidatus Pelagibacter ubique HTCC8051
Pelagibacterium halotolerans B2Pseudovibrio sp. FO-BEG1
endosymbiont of Stachyamoeba
Hyphomonas neptunium ATCC 15444
Thalassobaculum salexigens DSM 19539
Jannaschia sp. EhC01
Thalassospira profundimaris WP0211
Elioraea tepidiphila DSM 17972
Ralstonia solanacearum GMI1000
Methylocella silvestris BL2
Candidatus Pelagibacter ubique HTCC1002
Commensalibacter intestini A911
Maricaulis maris MCS10
Kordiimonas gwangyangensis DSM 19435 - JCM 12864
Gluconacetobacter diazotrophicus PA1 5
Enterobacter soli ATCC BAA-2102
alphaproteobacterium HIMB59
Ketogulonicigenium vulgare WSH-001
Oceanibaculum indicum P24
Terasakiella pusilla DSM 6293
Sneathiella glossodoripedis JCM 23214
Roseomonas cervicalis ATCC 49957
Phaeospirillum fulvum MGU-K5
Rhodopseudomonas palustris TIE-1
Burkholderia thailandensis E264
Candidatus Puniceispirillum marinum IMCC1322
Tistrella mobilis KA081020-065
Brevundimonas subvibrioides ATCC 15264
Wolbachia endosymbiont of Culex quinquefasciatus Pel
Parvularcula bermudensis HTCC2503
Congregibacter litoralis KT71
Phenylobacterium zucineum HLK1
alphaproteobacterium AAP38
Orientia tsutsugamushi Boryong
Roseibium sp. TrichSKD4
Paracoccus denitrificans PD1222
alphaproteobacterium HIMB114
Asticcacaulis excentricus CB 48
Granulibacter bethesdensis CGDNIH1
Rubellimicrobium thermophilum DSM 16684
alphaproteobacterium Q-1
Wolbachia endosymbiont of Onchocerca ochengiEhrlichia canis Jake
Acidisphaera rubrifaciens HS-AP3
Kiloniella laminariae DSM 19542
Caedibacter sp. 37-49
Alteromonas lipolytica
alphaproteobacterium SCGC AAA288-N07
Roseospirillum parvum strain 930IPararhodospirillum photometricum DSM 122
alphaproteobacterium BAL199
Hyphomicrobium denitrificans ATCC 51888
Meganema perideroedes DSM 15528
Candidatus Paracaedibacter acanthamoebae isolate PRA3
Magnetospirillum magneticum AMB-1
Sagittula stellata E-37
Rhodospirillum centenum SW
Gluconobacter oxydans H24
Candidatus Jidaibacter acanthamoeba
Candidatus Caedibacter acanthamoebae
Parvibaculum lavamentivorans DS-1
Neorickettsia sennetsu Miyayama
Candidatus Nucleicultrix amoebiphila FS5
Rickettsia typhi Wilmington
Holospora undulata HU1
Rhizobium leguminosarum bv. trifolii WSM1689
Holospora obtusa F1
Methylobacterium extorquens AM1
alphaproteobacterium IMCC14465
Micavibrio aeruginosavorus ARL-13
Rubritepida flocculans DSM 14296
alphaproteobacterium Mf105b01
Ahrensia sp. R2A130
Candidatus Pelagibacter sp. IMCC9063
Pelagibacter sp. HIMB058
alphaproteobacterium RS24
alphaproteobacterium HIMB5
Ruegeria sp. ANG-R
Zymomonas mobilis sub mobilis ATCC 10988
Inquilinus limosus DSM 16000
alphaproteobacterium SCGC AAA280-P20
alphaproteobacterium LLX12ACitromicrobium sp. JLT1363
90/92
84.2/88
99.9/100
99.9/100
95.9/93
98.7/98
99.9/100
99.4/99
99.4/98
98.6/97
99.8/99
alphaproteobacterium BAL199
Methylobacterium extorquens AM1
Rhodobacter sphaeroides 2.4.1Paracoccus denitrificans PD1222
Zymomonas mobilis sub mobilis ATCC 10988
Inquilinus limosus DSM 16000
Gluconobacter oxydans H24
Hirschia baltica ATCC 49814
Candidatus Odyssella thessalonicensis L13
alphaproteobacterium HIMB59
Oceanibaculum indicum P24
alphaproteobacterium HIMB5
Hyphomicrobium denitrificans ATCC 51888
Methylocella silvestris BL2
Candidatus Caedibacter acanthamoebae
Octadecabacter antarcticus 307
alphaproteobacterium Mf105b01alphaproteobacterium RS24
Oceanicaulis sp. HTCC2633
Acidisphaera rubrifaciens HS-AP3
alphaproteobacterium Q-1
Maricaulis maris MCS10
Acidiphilium angustum ATCC 35903
endosymbiont of Peranema
Magnetospirillum magneticum AMB-1Terasakiella pusilla DSM 6293
Parvibaculum lavamentivorans DS-1
alphaproteobacterium L41A
Orientia tsutsugamushi Boryong
Holospora undulata HU1
Wolbachia endosymbiont of Culex quinquefasciatus Pel
Phenylobacterium zucineum HLK1
Tistrella mobilis KA081020-065
Rhizobium leguminosarum bv. trifolii WSM1689
Rhodocista sp. MIMtkB3
Methylocystis sp. SC2
alphaproteobacterium SCGC AAA288-N07
Rickettsia typhi Wilmington
alphaproteobacterium HIMB114
Geminicoccus roseus DSM 18922
alphaproteobacterium SCGC AAA280-P20
alphaproteobacterium LLX12A
Methylovorus glucosetrophus SIP3-4Burkholderia thailandensis E264
Candidatus Nucleicultrix amoebiphila FS5
Pelagibacter sp. HIMB058
Belnapia moabensis DSM 16746
Sagittula stellata E-37
Micavibrio aeruginosavorus ARL-13
Roseibium sp. TrichSKD4
alphaproteobacterium AAP38
Bartonella quintana RM-11
Polymorphum gilvum SL003B-26A1
Ketogulonicigenium vulgare WSH-001
Roseomonas cervicalis ATCC 49957
Rhodomicrobium vannielii ATCC 17100
Rhodovibrio salinarum DSM 9154
Ehrlichia canis Jake
Phaeospirillum fulvum MGU-K5
alphaproteobacterium IMCC14465
Citromicrobium sp. JLT1363
Hyphomonas neptunium ATCC 15444
Rhodospirillum centenum SW
Gluconacetobacter diazotrophicus PA1 5
Spongiibacter tropicus DSM 19543
Jannaschia sp. EhC01
Kiloniella laminariae DSM 19542
Ahrensia sp. R2A130
Candidatus Puniceispirillum marinum IMCC1322
Candidatus Jidaibacter acanthamoeba
Roseospirillum parvum strain 930I
Rubritepida flocculans DSM 14296
Commensalibacter intestini A911
Candidatus Midichloria mitochondrii IricVA
Thermopetrobacter sp. TC1
Neorickettsia sennetsu Miyayama
Candidatus Paracaedibacter symbiosus
Parvularcula bermudensis HTCC2503
Candidatus Pelagibacter sp. IMCC9063
Thalassobaculum salexigens DSM 19539
Rubellimicrobium thermophilum DSM 16684
Anaplasma phagocytophilum HZ
Chelativorans sp. J32
Ruegeria sp. ANG-R
Elioraea tepidiphila DSM 17972
Alteromonas lipolytica
Pseudovibrio sp. FO-BEG1
Sneathiella glossodoripedis JCM 23214
alphaproteobacterium AAP81b
Candidatus Pelagibacter ubique HTCC8051
Kordiimonas gwangyangensis DSM 19435 - JCM 12864
Pelagibacterium halotolerans B2
Candidatus Finniella lucida
Asticcacaulis excentricus CB 48
Meganema perideroedes DSM 15528
Candidatus Arcanobacter lacustris
Rhodopseudomonas palustris TIE-1
Congregibacter litoralis KT71
Nitrosospira multiformis ATCC 25196
Thalassospira profundimaris WP0211
Candidatus Paracaedibacter acanthamoebae isolate PRA3
endosymbiont of Stachyamoeba
Brevundimonas subvibrioides ATCC 15264
Caenispirillum salinarum AK4
Ralstonia solanacearum GMI1000
Magnetococcus marinus MC-1
Xanthobacter autotrophicus Py2
Caedibacter sp. 38-128
Holospora obtusa F1
Pararhodospirillum photometricum DSM 122
Candidatus Hepatobacter penaei
Candidatus Pelagibacter ubique HTCC1002
Magnetofaba australis IT-1
Rhodospirillum rubrum ATCC 11170
Caedibacter varicaedens
Caedibacter sp. 37-49
Granulibacter bethesdensis CGDNIH1
Sphingomonas wittichii
Wolbachia endosymbiont of Onchocerca ochengi
Enterobacter soli ATCC BAA-2102
100/92
99.9/100
100/94
99.8/100
78.3/89
99.2/98
100/93
93.5/94
100/94
87.8/94
91.6/9399.4/100
93.2/90
80.9/84
99.9/100
3.8/51
100/94
60.6/87
98.6/99
98.6/99
0.4 0.2
120 taxa&
200 genesdataset
Site removalin steps of 10%
by compositional
biasby rate
Recodinginto S4
Set of 40 mostcompositionally
homogeneous genes
Taxonremoval
Phylogeneticanalyses
Phylogeneticanalyses
Phylogeneticanalyses
Phylogeneticanalyses
Taxonremoval
Taxonremoval
Phylogeneticanalyses
Phylogeneticanalyses
Phylogeneticanalyses
Taxonremoval
Phylogeneticanalyses
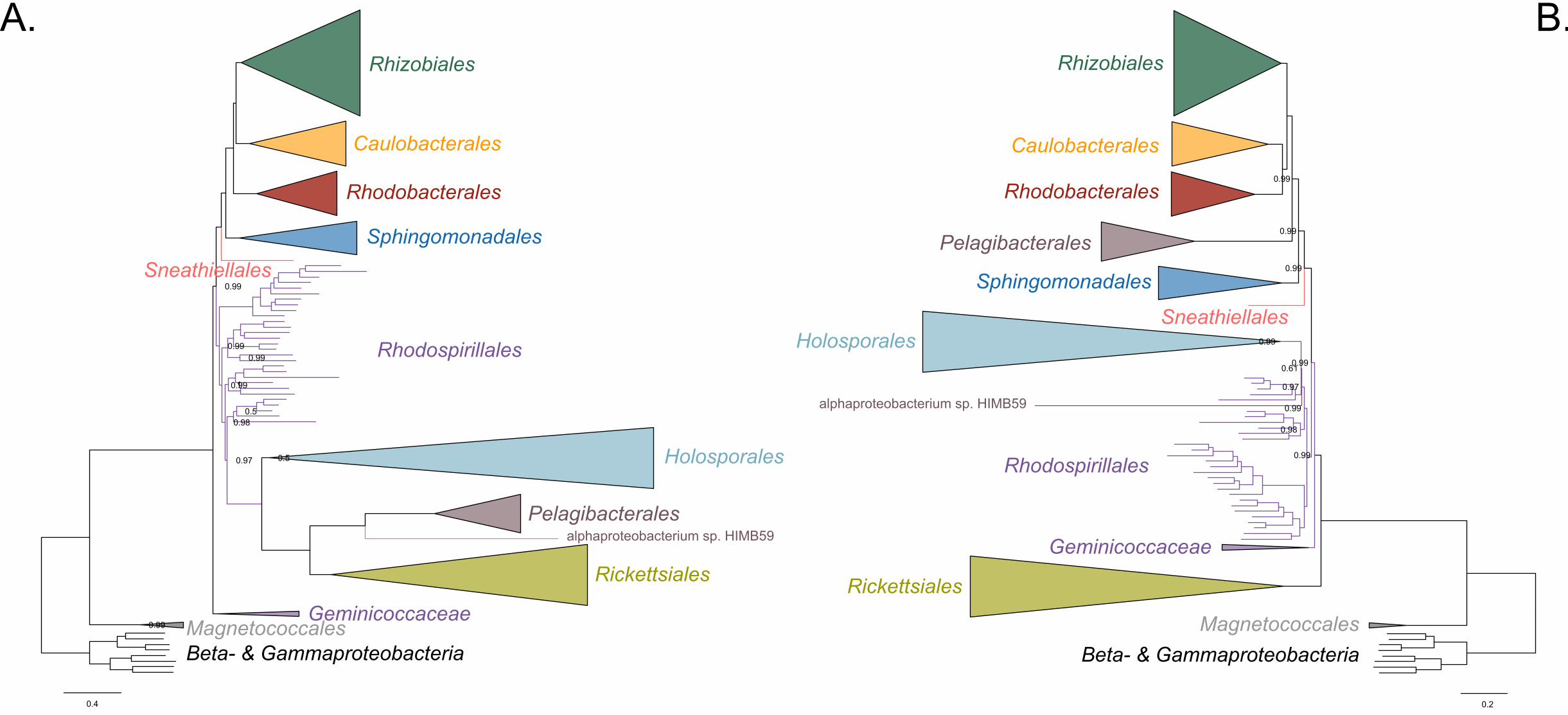
0.2
alphaproteobacterium sp. HIMB59
0.99
0.97
0.99
0.99
0.98
0.99
0.61
0.99
0.99
0.99
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Magnetococcales
Holosporales
Beta- & Gammaproteobacteria
Rickettsiales
Pelagibacterales
A. B.
0.4
0.99
0.99
0.99
0.50.98
0.99
0.99
0.97 0.5
1
alphaproteobacterium sp. HIMB59
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Holosporales
Beta- & Gammaproteobacteria
Rickettsiales
Pelagibacterales
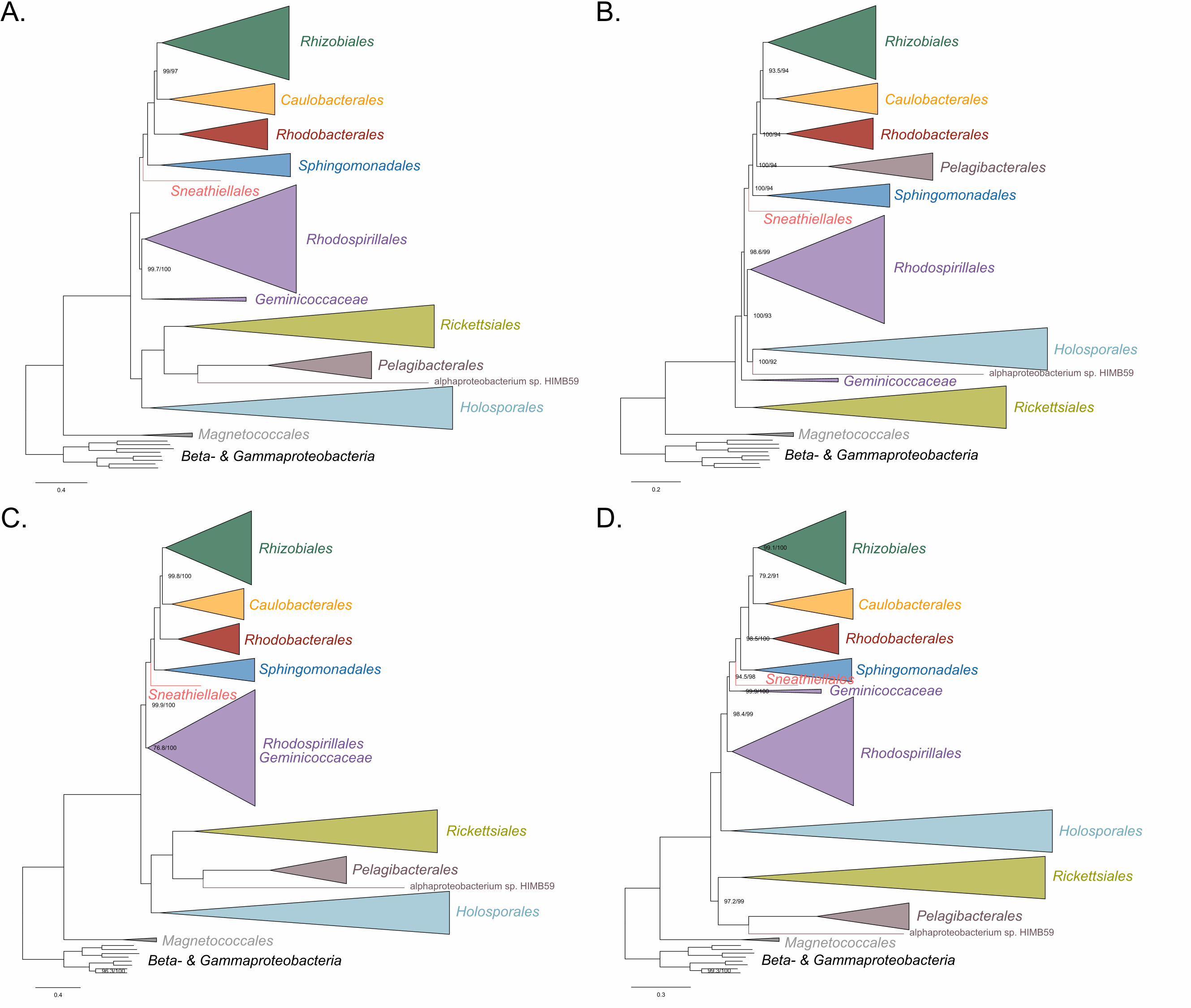
0.4
alphaproteobacterium sp. HIMB59
99.7/100
99/97
0.2
alphaproteobacterium sp. HIMB59
93.5/94
100/94
100/94
100/93
100/94
100/92
98.6/99
0.4
alphaproteobacterium sp. HIMB59
99.9/100
96.3/100
99.8/100
76.8/100
0.3
alphaproteobacterium sp. HIMB59
99.3/100
94.5/98
97.2/99
99.9/100
99.1/100
98.5/100
98.4/99
79.2/91
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Pelagibacterales
Holosporales
MagnetococcalesBeta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Holosporales
Rickettsiales
MagnetococcalesBeta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
RhodospirillalesGeminicoccaceae
Rickettsiales
Pelagibacterales
Holosporales
MagnetococcalesBeta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Pelagibacterales
Holosporales
MagnetococcalesBeta- & Gammaproteobacteria
Pelagibacterales
A. B.
C. D.
0.3
Holospora obtusa F1alphaproteobacterium sp. HIMB59
Holospora undulata HU1Candidatus Hepatobacter penaei
98.5/97
100/97
100/96
100/97
95.1/82
100/46
99.4/99
100/5299.2/100
36.9/88
64.7/50
100/97
97.7/46
99.2/98
85.3/82
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Rhodospirillales
Sneathiellales
Geminicoccaceae
Rickettsiales
Magnetococcales
Pelagibacterales
Beta- & Gammaproteobacteria
Holosporales
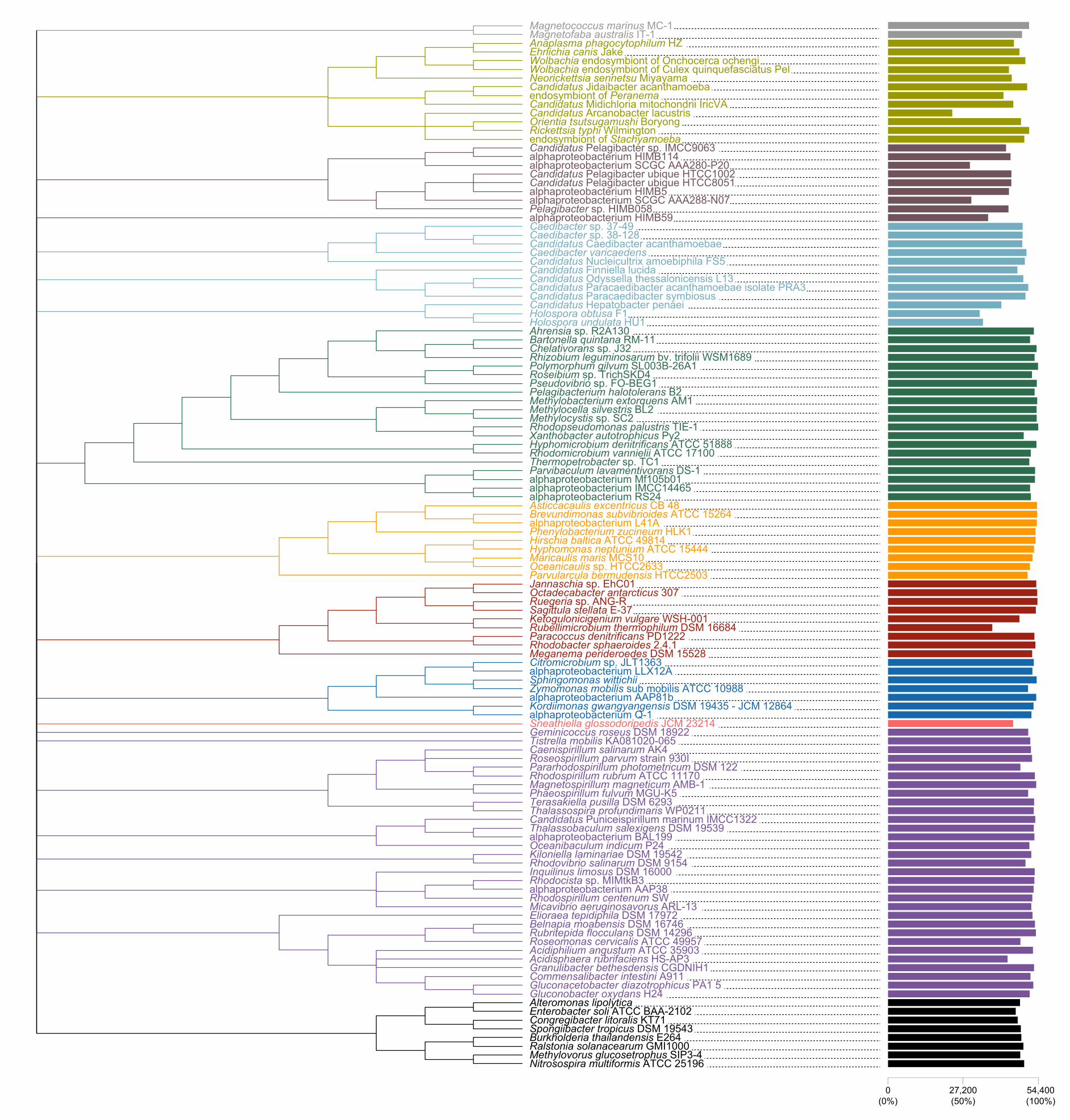
Rhodobacter sphaeroides 2.4.1
alphaproteobacterium HIMB59
Commensalibacter intestini A911
alphaproteobacterium SCGC AAA280-P20
Parvularcula bermudensis HTCC2503
alphaproteobacterium AAP38
Caedibacter sp. 37-49
Hirschia baltica ATCC 49814
Roseospirillum parvum strain 930I
Thermopetrobacter sp. TC1
Zymomonas mobilis sub mobilis ATCC 10988
Oceanibaculum indicum P24
Pseudovibrio sp. FO-BEG1
Nitrosospira multiformis ATCC 25196
Rhizobium leguminosarum bv. trifolii WSM1689
Octadecabacter antarcticus 307
Rubellimicrobium thermophilum DSM 16684
Rubritepida flocculans DSM 14296
alphaproteobacterium SCGC AAA288-N07
Candidatus Odyssella thessalonicensis L13
Candidatus Pelagibacter sp. IMCC9063
Pelagibacterium halotolerans B2
alphaproteobacterium Q-1
Candidatus Puniceispirillum marinum IMCC1322
Pararhodospirillum photometricum DSM 122
Ehrlichia canis Jake
Rhodocista sp. MIMtkB3
Burkholderia thailandensis E264
Rhodomicrobium vannielii ATCC 17100
Thalassospira profundimaris WP0211
alphaproteobacterium HIMB114
Candidatus Pelagibacter ubique HTCC8051
Candidatus Hepatobacter penaei
Magnetospirillum magneticum AMB-1
Ralstonia solanacearum GMI1000
Caedibacter varicaedens
alphaproteobacterium BAL199
Gluconobacter oxydans H24
Candidatus Paracaedibacter acanthamoebae isolate PRA3
Candidatus Pelagibacter ubique HTCC1002
Magnetofaba australis IT-1
Caedibacter sp. 38-128
Ruegeria sp. ANG-R
Sneathiella glossodoripedis JCM 23214
Methylobacterium extorquens AM1
Tistrella mobilis KA081020-065
Candidatus Jidaibacter acanthamoeba
Citromicrobium sp. JLT1363
Methylovorus glucosetrophus SIP3-4
Holospora obtusa F1
Kordiimonas gwangyangensis DSM 19435 - JCM 12864
Methylocystis sp. SC2
Holospora undulata HU1
Jannaschia sp. EhC01
alphaproteobacterium AAP81b
Oceanicaulis sp. HTCC2633
alphaproteobacterium Mf105b01
Spongiibacter tropicus DSM 19543
Phenylobacterium zucineum HLK1
Caenispirillum salinarum AK4
Rhodovibrio salinarum DSM 9154
Rhodopseudomonas palustris TIE-1
Magnetococcus marinus MC-1
alphaproteobacterium LLX12A
Maricaulis maris MCS10
Candidatus Finniella lucida
Chelativorans sp. J32
Meganema perideroedes DSM 15528
Ketogulonicigenium vulgare WSH-001
Bartonella quintana RM-11
Roseomonas cervicalis ATCC 49957Acidiphilium angustum ATCC 35903
Kiloniella laminariae DSM 19542
Methylocella silvestris BL2
Anaplasma phagocytophilum HZ
Candidatus Nucleicultrix amoebiphila FS5
Gluconacetobacter diazotrophicus PA1 5
Hyphomonas neptunium ATCC 15444
Paracoccus denitrificans PD1222
Elioraea tepidiphila DSM 17972
Rickettsia typhi Wilmington
Wolbachia endosymbiont of Culex quinquefasciatus Pel
Ahrensia sp. R2A130
Xanthobacter autotrophicus Py2
Sagittula stellata E-37
Terasakiella pusilla DSM 6293
Candidatus Caedibacter acanthamoebae
Enterobacter soli ATCC BAA-2102
Roseibium sp. TrichSKD4
Acidisphaera rubrifaciens HS-AP3
Inquilinus limosus DSM 16000
alphaproteobacterium RS24
endosymbiont of Peranema
endosymbiont of Stachyamoeba
Alteromonas lipolytica
Candidatus Paracaedibacter symbiosus
Granulibacter bethesdensis CGDNIH1
Phaeospirillum fulvum MGU-K5
Parvibaculum lavamentivorans DS-1
Asticcacaulis excentricus CB 48
Wolbachia endosymbiont of Onchocerca ochengiNeorickettsia sennetsu Miyayama
alphaproteobacterium L41A
alphaproteobacterium IMCC14465
Thalassobaculum salexigens DSM 19539
Candidatus Midichloria mitochondrii IricVA
Brevundimonas subvibrioides ATCC 15264
Orientia tsutsugamushi Boryong
Belnapia moabensis DSM 16746
Congregibacter litoralis KT71
Geminicoccus roseus DSM 18922
Rhodospirillum rubrum ATCC 11170
Micavibrio aeruginosavorus ARL-13Rhodospirillum centenum SW
Hyphomicrobium denitrificans ATCC 51888
Sphingomonas wittichii
Candidatus Arcanobacter lacustris
Polymorphum gilvum SL003B-26A1
Pelagibacter sp. HIMB058alphaproteobacterium HIMB5
54,400(100%)
27,200(50%)
0(0%)
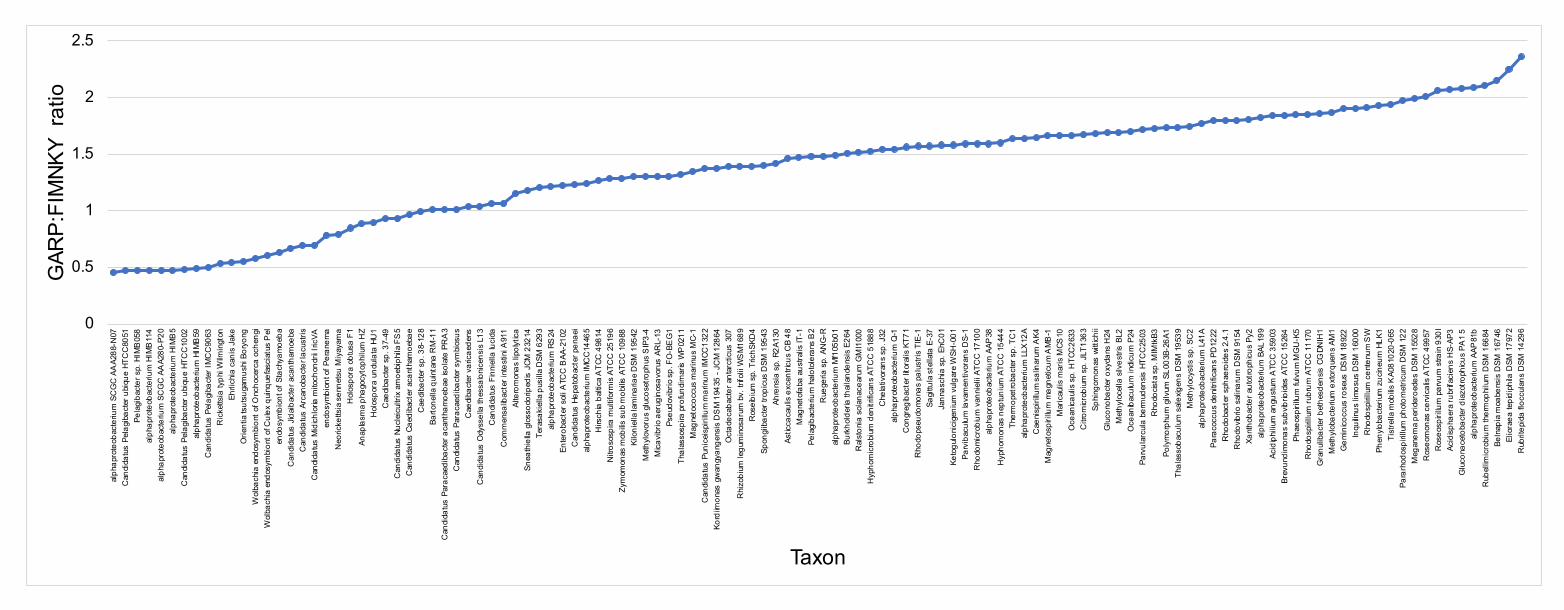
0
0.5
1
1.5
2
2.5
alph
apro
teob
acte
rium
SC
GC
AA
A288
-N07
Can
dida
tus
Pela
giba
cter
ubi
que
HTC
C80
51Pe
lagi
bact
er s
p. H
IMB
058
alph
apro
teob
acte
rium
HIM
B11
4al
phap
rote
obac
teriu
m S
CG
C A
AA2
80-P
20al
phap
rote
obac
teriu
m H
IMB
5C
andi
datu
s Pe
lagi
bact
er u
biqu
e HT
CC
1002
alph
apro
teob
acte
rium
HIM
B59
Can
dida
tus
Pela
giba
cter
IMC
C90
63R
icke
ttsia
typh
i Wilm
ingt
onEh
rlich
ia c
anis
Jak
eO
rient
ia ts
utsu
gam
ushi
Bor
yong
Wol
bach
ia e
ndos
ymbi
ont o
f Onc
hoce
rca
oche
ngi
Wol
bach
ia e
ndos
ymbi
ont o
f Cul
ex q
uinq
uefa
scia
tus
Pel
endo
sym
bion
t of S
tach
yam
oeba
Can
dida
tus
Jidai
bact
er a
cant
ham
oeba
Can
dida
tus
Arca
noba
cter
lacu
stris
Can
dida
tus
Mid
ichlo
ria m
itoch
ondr
ii Iric
VAen
dosy
mbi
ont o
f Per
anem
aN
eoric
ketts
ia s
enne
tsu
Miy
ayam
aH
olos
pora
obt
usa
F1An
apla
sma
phag
ocyt
ophi
lum
HZ
Hol
ospo
ra u
ndul
ata
HU1
Cae
diba
cter
sp.
37-
49C
andi
datu
s Nu
clei
cultr
ix a
moe
biph
ila F
S5
Can
dida
tus
Caed
ibac
ter a
cant
ham
oeba
eC
aedi
bact
er s
p. 3
8-12
8Ba
rtone
lla q
uint
ana
RM
-11
Can
dida
tus
Para
caed
ibac
ter a
cant
ham
oeba
e iso
late
PR
A3
Can
dida
tus
Para
caed
ibac
ter s
ymbi
osus
Cae
diba
cter
var
icae
dens
Can
dida
tus
Ody
ssel
la th
essa
loni
cens
is L
13C
andi
datu
s Fi
nnie
lla lu
cida
Com
men
salib
acte
r int
estin
i A91
1Al
tero
mon
as li
polyt
icaSn
eath
iella
glo
ssod
orip
edis
JCM
232
14Te
rasa
kiel
la p
usill
a D
SM 6
293
alph
apro
teob
acte
rium
RS
24En
tero
bact
er s
oli A
TCC
BAA
-210
2C
andi
datu
s He
pato
bact
er p
enae
ial
phap
rote
obac
teriu
m IM
CC
1446
5H
irsch
ia b
altic
a A
TCC
498
14N
itros
ospi
ra m
ultif
orm
is A
TCC
251
96Zy
mom
onas
mob
ilis s
ub m
obilis
ATC
C 10
988
Kilo
niel
la la
min
aria
e D
SM 1
9542
Met
hylo
voru
s gl
ucos
etro
phus
SIP
3-4
Mica
vibr
io a
erug
inos
avor
us A
RL-
13Ps
eudo
vibr
io s
p. F
O-B
EG
1Th
alas
sosp
ira p
rofu
ndim
aris
WP
0211
Mag
neto
cocc
us m
arin
us M
C-1
Can
dida
tus
Puni
ceis
piri l
lum
mar
inum
IMCC
1322
Kord
iimon
as g
wan
gyan
gens
is D
SM
194
35 -
JCM
128
64O
ctad
ecab
acte
r ant
arct
icus
307
Rhi
zobi
um le
gum
inos
arum
bv.
trifo
lii W
SM
1689
Ros
eibi
um s
p. T
richS
KD4
Spon
giib
acte
r tro
picu
s D
SM 1
9543
Ahre
nsia
sp.
R2A
130
Astic
caca
ulis
exc
entri
cus
CB
48M
agne
tofa
ba a
ustra
lis IT
-1Pe
lagi
bact
eriu
m h
alot
oler
ans
B2
Rue
geria
sp.
AN
G-R
alph
apro
teob
acte
rium
Mf1
05b0
1Bu
rkho
lder
ia th
aila
nden
sis
E264
Ral
ston
ia s
olan
acea
rum
GM
I100
0H
ypho
mic
robi
um d
enitr
ifica
ns A
TCC
518
88C
hela
tivor
ans
sp. J
32al
phap
rote
obac
teriu
m Q
-1C
ongr
egib
acte
r lito
ralis
KT7
1R
hodo
pseu
dom
onas
pal
ustri
s TI
E-1
Sagi
ttula
ste
llata
E-3
7Ja
nnas
chia
sp.
EhC
01Ke
togu
loni
cige
nium
vul
gare
WSH
-001
Parv
ibac
ulum
lava
men
tivor
ans
DS-
1R
hodo
mic
robi
um v
anni
elii
ATC
C 1
7100
alph
apro
teob
acte
rium
AAP
38H
ypho
mon
as n
eptu
nium
ATC
C 1
5444
Ther
mop
etro
bact
er s
p. T
C1
alph
apro
teob
act e
rium
LLX
12A
Cae
nisp
irillu
m s
alin
arum
AK4
Mag
neto
spiri
llum
mag
netic
um A
MB
-1M
aric
aulis
mar
is M
CS10
Oce
anica
ulis
sp.
HTC
C26
33C
itrom
icrob
ium
sp.
JLT
1363
Sphi
ngom
onas
witt
ichii
Glu
cono
bact
er o
xyda
ns H
24M
ethy
loce
lla s
ilves
tris
BL2
Oce
anib
acul
um in
dicu
m P
24Pa
rvul
arcu
la b
erm
uden
sis H
TCC
2503
Rho
docis
ta s
p. M
IMtk
B3Po
lym
orph
um g
ilvum
SL0
03B-
26A1
Thal
asso
bacu
lum
sal
exig
ens
DSM
195
39M
ethy
locy
stis
sp.
SC
2al
phap
rote
oba c
teriu
m L
41A
Para
cocc
us d
enitr
ifica
ns P
D12
22R
hodo
bact
er s
phae
roid
es 2
.4.1
Rho
dovib
rio s
alin
arum
DSM
915
4Xa
ntho
bact
er a
utot
roph
icus
Py2
alph
apro
teob
acte
rium
BAL
199
Acid
iphi
lium
ang
ustu
m A
TCC
359
03Br
evun
dim
onas
sub
vibrio
ides
ATC
C 15
264
Phae
ospi
rillu
m fu
lvum
MG
U-K
5R
hodo
spiri
llum
rubr
um A
TCC
1117
0G
ranu
libac
ter b
ethe
sden
sis C
GDN
IH1
Met
hylo
bact
eriu
m e
xtor
quen
s AM
1G
emin
icocc
us ro
seus
DS
M 1
8922
Inqu
ilinus
lim
osu s
DSM
160
00R
hodo
spiri
llum
cen
tenu
m S
WPh
enylo
bact
eriu
m z
ucin
eum
HLK
1Ti
stre
lla m
obilis
KA0
8102
0-06
5Pa
rarh
odos
piril
lum
pho
tom
etric
um D
SM 1
22M
egan
ema
perid
eroe
des
DSM
155
28R
oseo
mon
as c
ervi
calis
ATC
C 4
9957
Ros
eosp
irillu
m p
arvu
m s
train
930
IAc
idisp
haer
a ru
brifa
cien
s H
S-AP
3G
luco
nace
toba
cter
dia
zotro
phicu
s PA
1 5
alph
apro
teob
acte
rium
AAP
81b
Rub
ellim
icro
bium
ther
mop
hilu
m D
SM 1
6684
Beln
apia
moa
bens
is D
SM 1
6746
Eli o
raea
tepi
diph
ila D
SM 1
7972
Rub
ritep
ida
flocc
ulan
s D
SM 1
4296
GAR
P:FI
MNK
Y ra
tio
Taxon
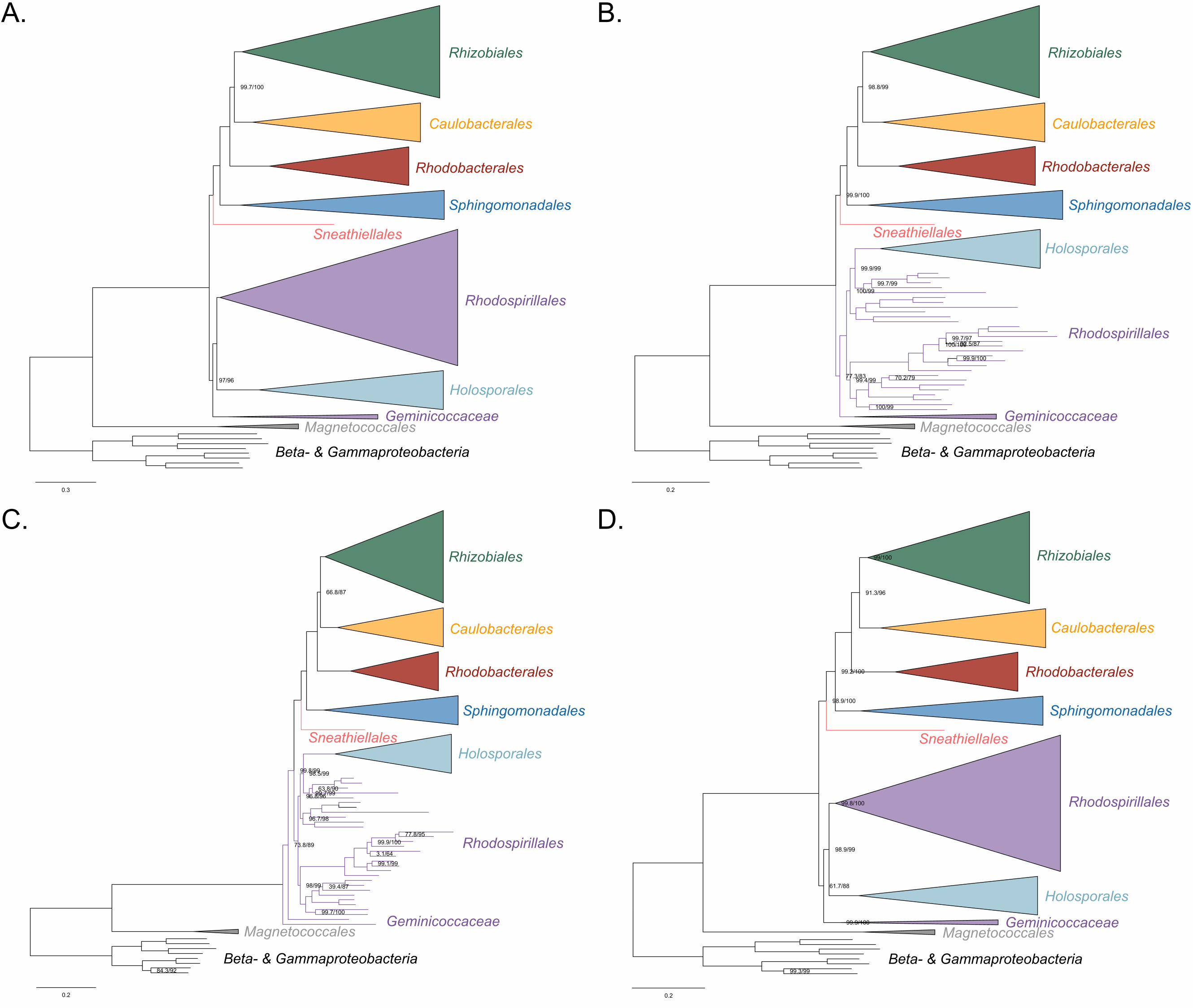
0.3
99.7/100
97/96
0.2
77.3/83
99.9/99
98.8/99
100/100
99.4/99 70.2/79
99.9/100
99.9/100
99.7/97
100/99
92.5/87
99.7/99
100/99
0.2
3.1/64
39.4/87
66.8/87
99.9/10077.8/95
84.3/92
99.1/99
98.5/99
63.8/90
98/99
99.7/99
73.8/89
99.7/100
99.8/99
96.8/96
96.7/98
0.2
99.2/100
61.7/88
91.3/96
99.9/100
98.9/100
98.9/99
99/100
99.3/99
99.8/100
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Holosporales
Magnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Holosporales
Magnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Holosporales
Magnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Holosporales
Magnetococcales
Beta- & Gammaproteobacteria
A. B.
C. D.
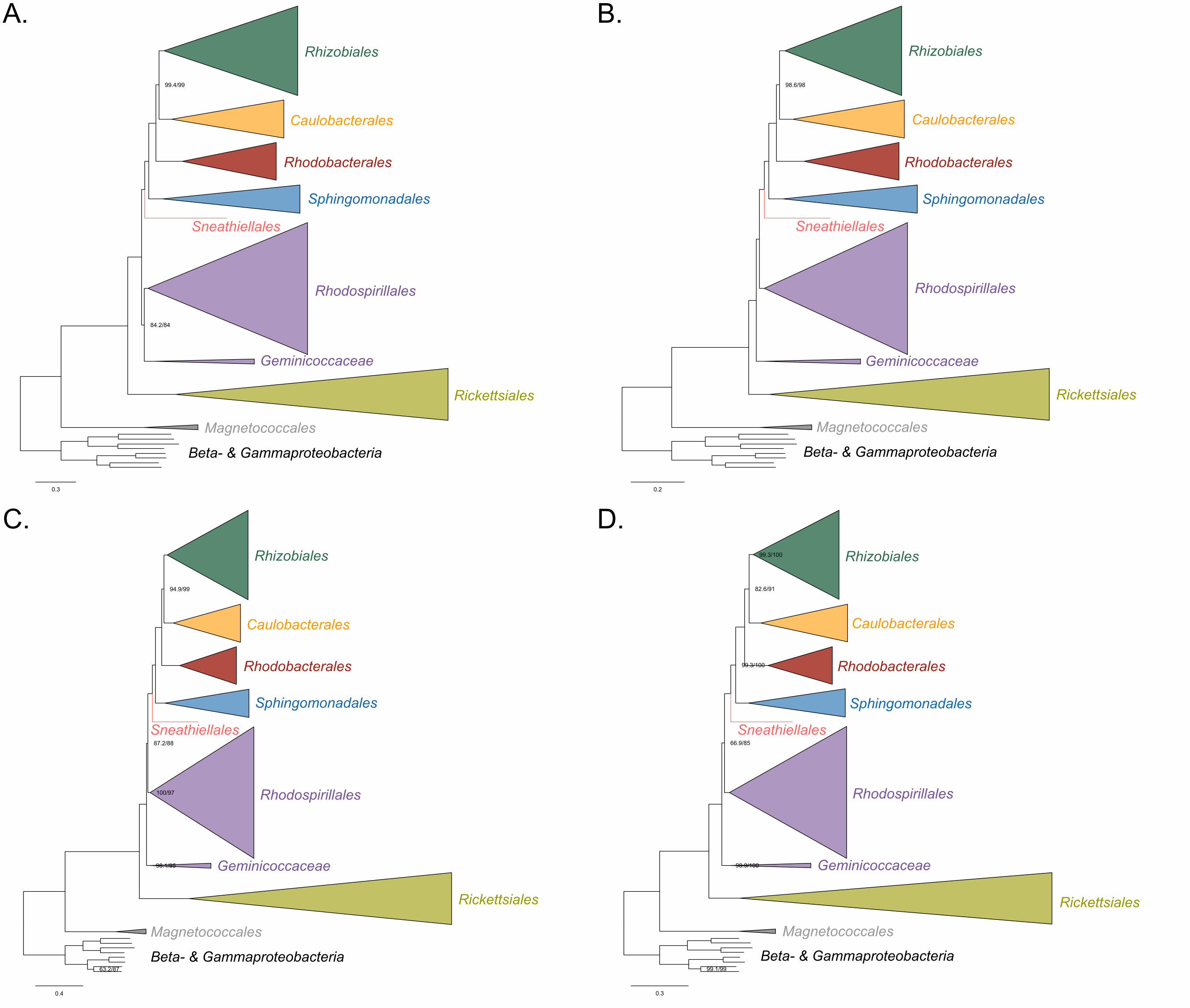
0.3
99.4/99
84.2/84
0.2
98.6/98
0.4
98.1/95
100/97
94.9/99
87.2/88
63.2/87
0.3
99.3/100
82.6/91
99.3/100
98.9/100
66.9/85
99.1/99
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Magnetococcales
Beta- & Gammaproteobacteria
Sneathiellales
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Magnetococcales
Beta- & Gammaproteobacteria
Sneathiellales
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Magnetococcales
Beta- & Gammaproteobacteria
Sneathiellales
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Rhodospirillales
Geminicoccaceae
Rickettsiales
Magnetococcales
Beta- & Gammaproteobacteria
Sneathiellales
A. B.
C. D.
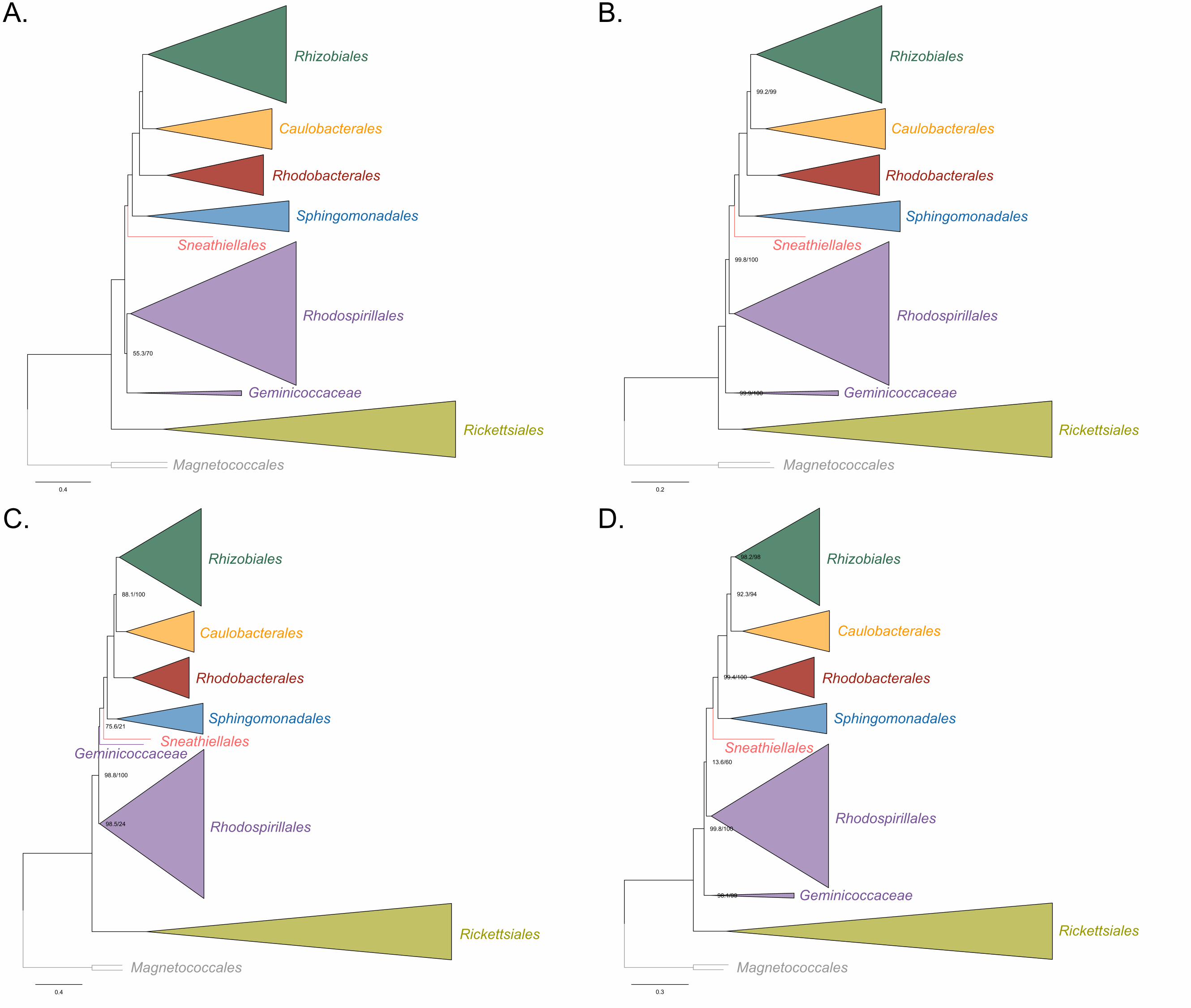
0.4
55.3/70
0.2
99.2/99
99.8/100
99.9/100
0.4
98.5/24
75.6/21
98.8/100
88.1/100
0.3
13.6/60
99.8/100
98.1/99
92.3/94
98.2/98
99.4/100
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Magnetococcales
Rickettsiales
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Magnetococcales
Rickettsiales
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Rhodospirillales
Geminicoccaceae
Magnetococcales
Rickettsiales
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Magnetococcales
Rickettsiales
A. B.
C. D.
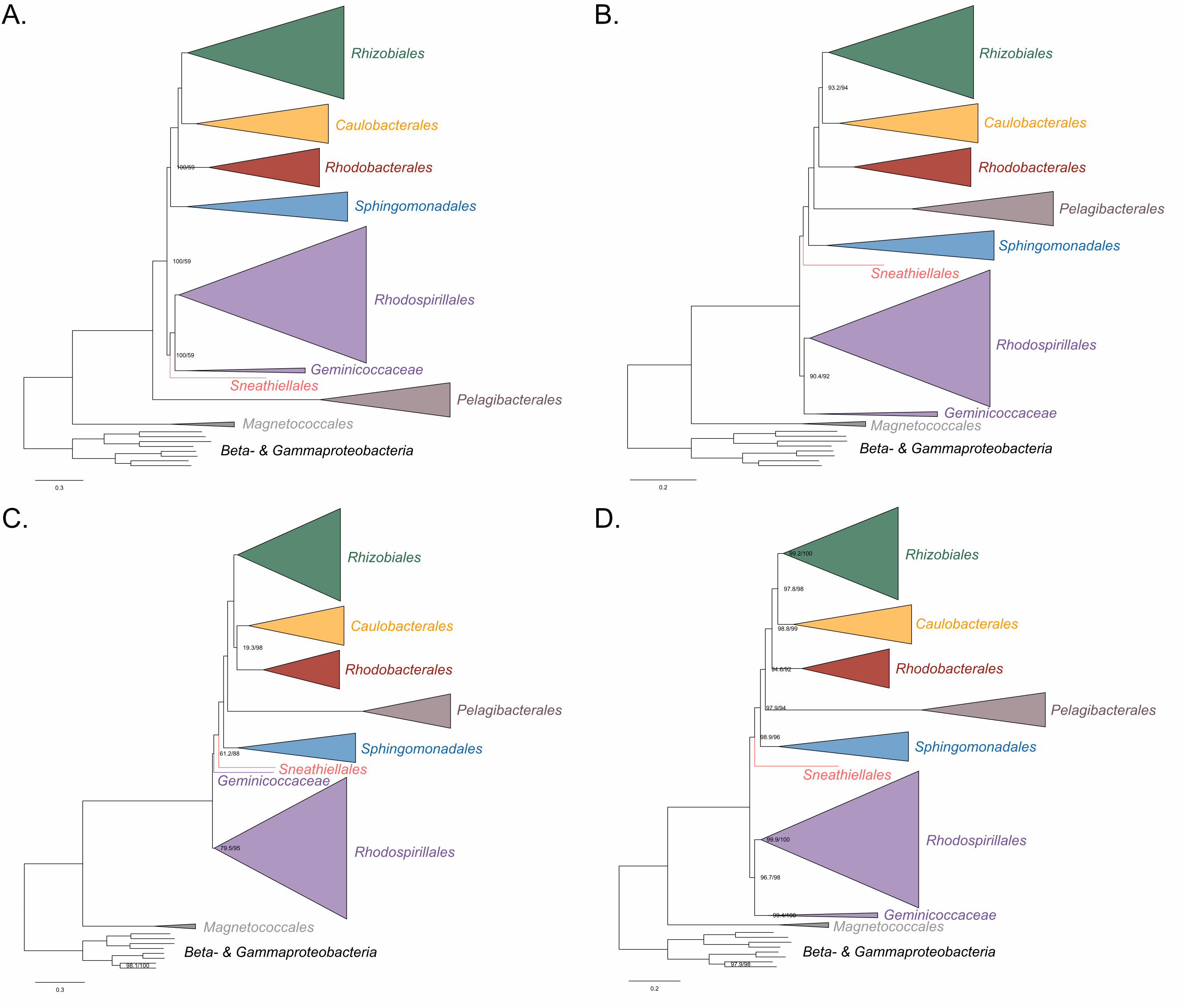
0.3
100/59
100/59
100/59
0.2
93.2/94
90.4/92
0.3
61.2/88
19.3/98
98.1/100
79.5/95
0.2
97.8/98
99.9/100
94.6/92
98.9/96
96.7/98
99.2/100
99.4/100
97.9/94
97.9/98
98.8/99
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Pelagibacterales
Magnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Pelagibacterales
MagnetococcalesBeta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae
Pelagibacterales
Magnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
SphingomonadalesSneathiellales
Rhodospirillales
Geminicoccaceae
Pelagibacterales
Magnetococcales
Beta- & Gammaproteobacteria
A. B.
C. D.
0.3
alphaproteobacterium sp. HIMB59
99.8/100
0.2
alphaproteobacterium sp. HIMB59
99.2/99
99.8/97
99.5/97
0.3
alphaproteobacterium sp. HIMB59
97.4/91
99.8/100
89.1/100
86.4/90
95.6/99
0.2
alphaproteobacterium sp. HIMB59
99.1/91
87.8/86
99.1/100
95.2/96
99.5/99
99.9/100
98.4/100
98.4/99
99.9/96
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Beta- & Gammaproteobacteria
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Beta- & Gammaproteobacteria
A. B.
C. D.
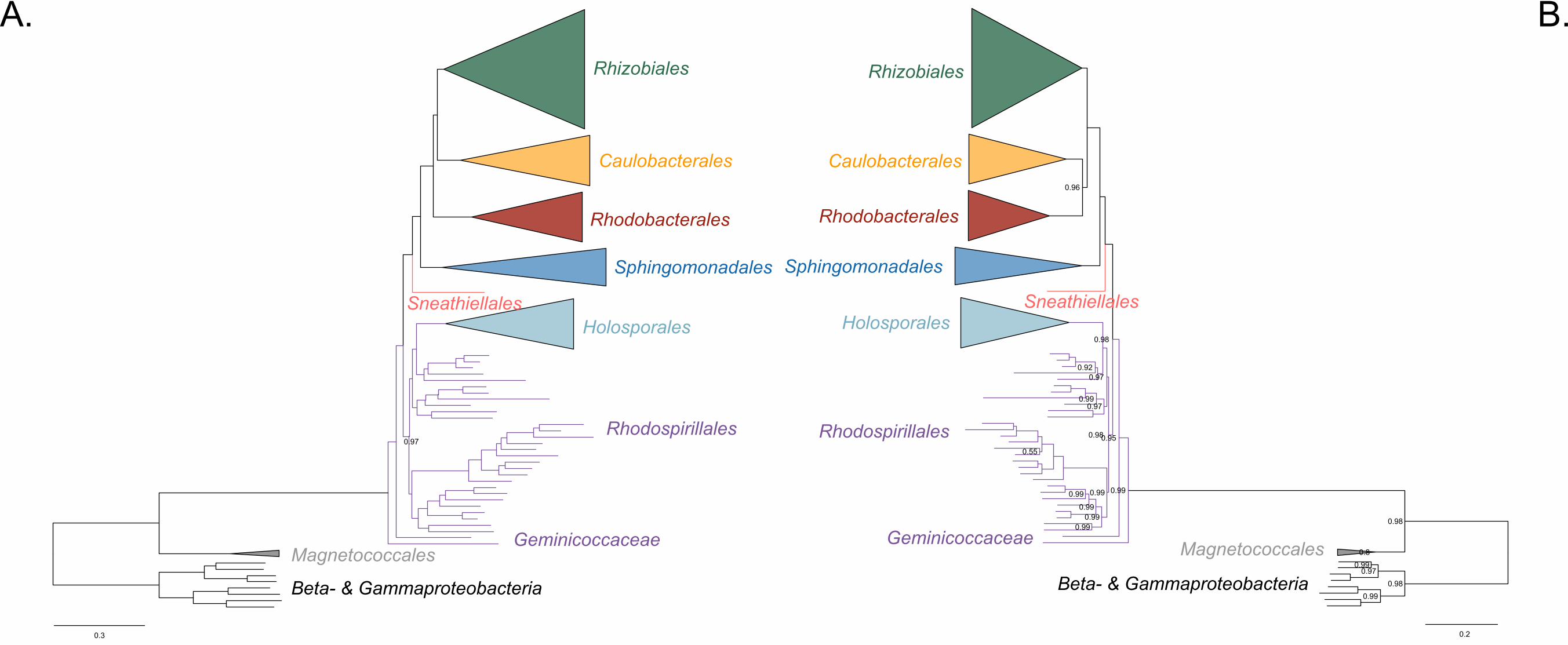
0.3
0.97
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Holosporales
Beta- & Gammaproteobacteria
0.2
0.99
0.99
0.98
0.99
0.97
0.99
0.97
0.99
0.99
0.99
0.98
0.92
0.55
0.99
0.99
0.98
0.95
0.8
0.96
0.98
0.97
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
Geminicoccaceae Magnetococcales
Holosporales
Beta- & Gammaproteobacteria
A. B.
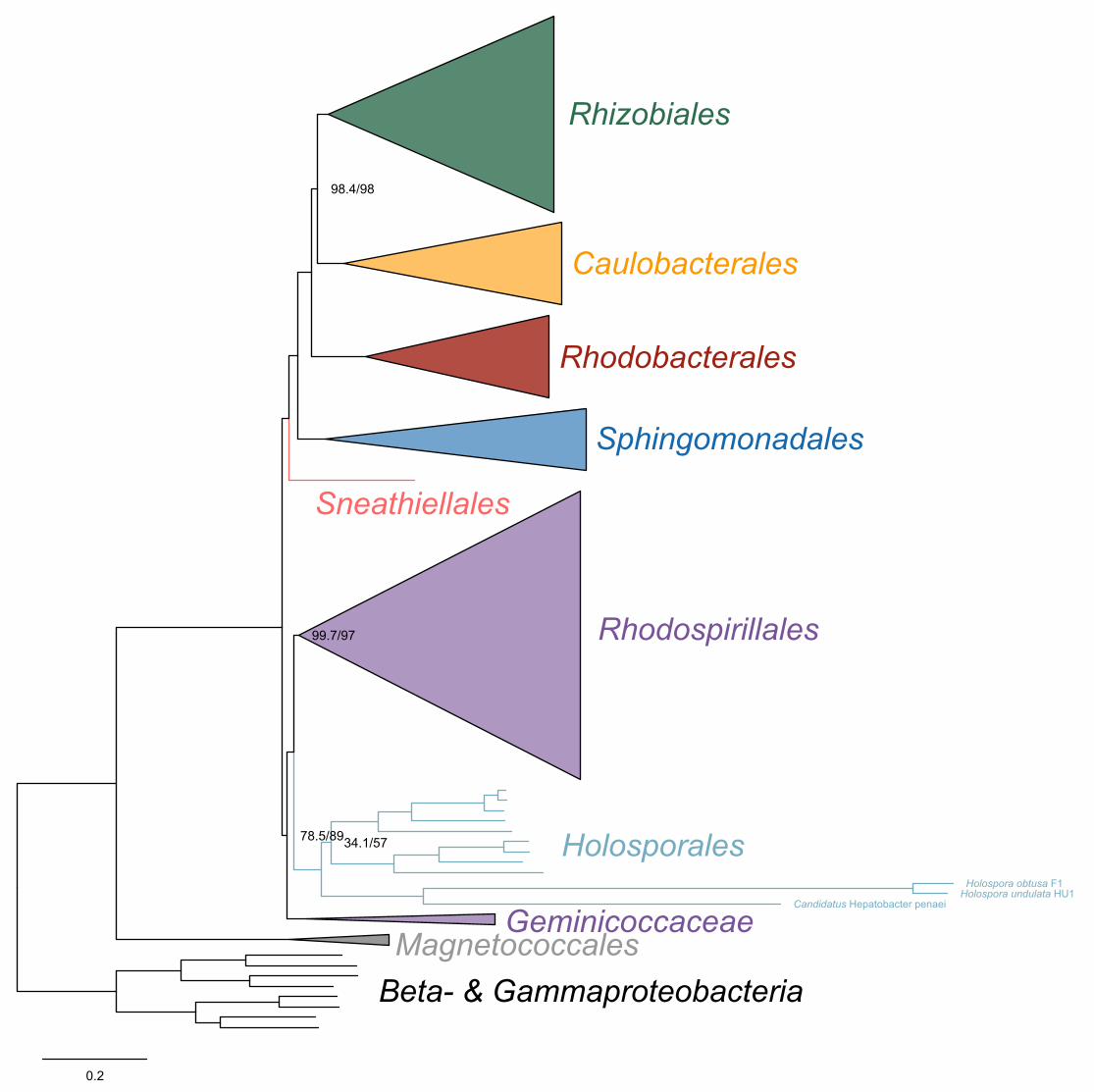
0.2
Holospora undulata HU1Candidatus Hepatobacter penaei
Holospora obtusa F1
98.4/98
34.1/57
99.7/97
78.5/89
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Beta- & Gammaproteobacteria
Holosporales
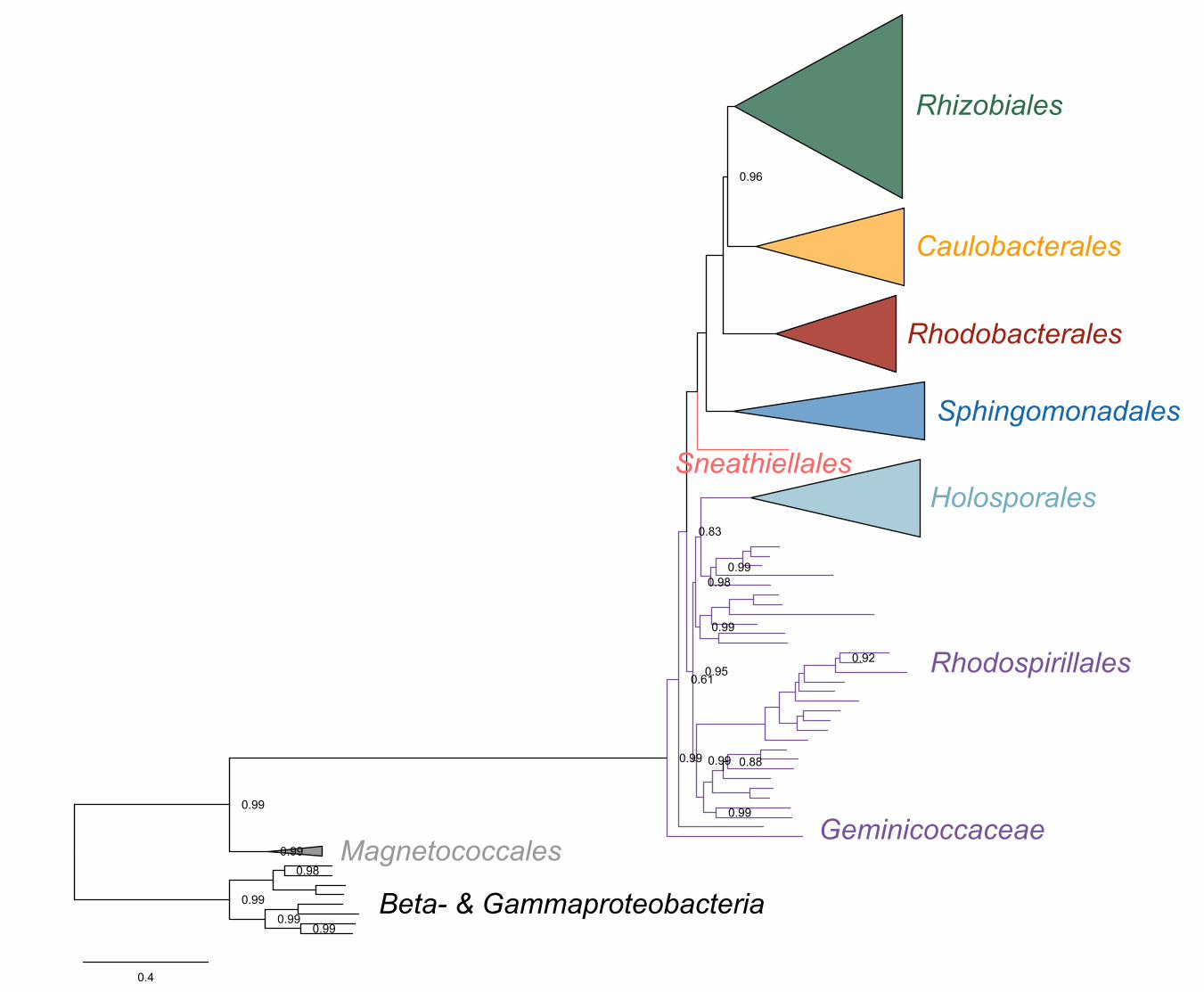
0.4
0.99
0.99
0.96
0.98
0.98
0.99
0.61
0.99
0.95
0.99
0.99
0.99
0.99
0.88
0.99
0.83
0.92
0.99
Rhizobiales
Caulobacterales
Rhodobacterales
Sphingomonadales
Sneathiellales
Rhodospirillales
GeminicoccaceaeMagnetococcales
Holosporales
Beta- & Gammaproteobacteria


![Turning Back [updated 6.5.2015]](https://static.fdokumen.com/doc/165x107/6335f35102a8c1a4ec01fd86/turning-back-updated-652015.jpg)

















