
Thalassemia
23
FAKULTAS KEDOKTERAN UNIVERSITAS KRISTEN KRIDA WACANA Kampus II Ukrida Jl. Terusan Arjuna No.6 Jakarta 11510 Farah Farhanah binti Mansor NIM : 102009341 Email : [email protected] Thalassemia Beta Minor Bab I Pendahuluan Latar belakang Thalassemia merupakan sindrom kelainan yang diwariskan dan masuk ke dalam kelompok hemoglobinopati, yakni kelainan yang disebabkan oleh gangguan sintesis hemoglobin akibat mutasi di dalam atau dekat gen globin. Mutasi gen globin ini dapat menimbulkan dua perubahan rantai globin, yakni perubahan struktur rangkaian asam amino rantai globin tertentu(hemoglobinopati struktural), atau perubahan kecepatan sintesis atau kemampuan produksi rantai globin tertentu, disebut thalassemia. Hemoglobinopati yang ditemukan secara klinis, baik pada anak-anak atau orang dewasa, disebabkan oleh mutasi gen globin α atau β. Sedangkan, mutasi berat gen globin γ, ζ, dan ε dapat menyebabkan kematian pada awal gestasi. Thalassemia-β, terjadi akibat berkurangnya rantai globin-β (thalassemia-β + ) atau tidak diproduksi sama sekali rantai globin-β (thalassemia-β 0 ). Pada thalassemia-β minor, tampilan
description
belajar
Transcript of Thalassemia

FAKULTAS KEDOKTERAN UNIVERSITAS KRISTEN KRIDA WACANAKampus II Ukrida Jl. Terusan Arjuna No.6 Jakarta 11510
Farah Farhanah binti MansorNIM : 102009341
Email : [email protected]
Thalassemia Beta Minor
Bab I
Pendahuluan
Latar belakang
Thalassemia merupakan sindrom kelainan yang diwariskan dan masuk ke dalam kelompok
hemoglobinopati, yakni kelainan yang disebabkan oleh gangguan sintesis hemoglobin akibat
mutasi di dalam atau dekat gen globin. Mutasi gen globin ini dapat menimbulkan dua
perubahan rantai globin, yakni perubahan struktur rangkaian asam amino rantai globin
tertentu(hemoglobinopati struktural), atau perubahan kecepatan sintesis atau kemampuan
produksi rantai globin tertentu, disebut thalassemia. Hemoglobinopati yang ditemukan secara
klinis, baik pada anak-anak atau orang dewasa, disebabkan oleh mutasi gen globin α atau β.
Sedangkan, mutasi berat gen globin γ, ζ, dan ε dapat menyebabkan kematian pada awal
gestasi. Thalassemia-β, terjadi akibat berkurangnya rantai globin-β (thalassemia-β+) atau
tidak diproduksi sama sekali rantai globin-β (thalassemia-β0). Pada thalassemia-β minor,
tampilan klinis biasanya normal, hepatomegali dan splenomegali ditemukan pada sedikit
penderita.
Tujuan
Pembuatan makalah ini adalah bertujuan untuk mengetahui lebih lanjut mengenai penyakit
thalassemia-β minor. Ia juga bertujuan untuk mengetahui etiologi, patofisiologi dari penyakit
thalassemia-β minor. Disamping itu, makalah ini bertujuan untuk mengetahui pengobatan
yang terkini dan pencegahan yang dapat dilakukan pada pasien yang mengalami thalassemia-
β minor.

Bab II
Pembahasan Isi
2.1 Anamnesis
Untuk menegakkan diagnosis suatu penyakit atau kelainan pada sistem hemopoeisis
diperlukan kemampuan dan keterampilan melakukan anamnesis dan pemeriksaan organ-
organ yang terlibat dalam pembentukan sel darah. Anamnesis yang terarah diperlukan untuk
menggali lebih dalam dan luas keluhan utama pasien. Anamnesis merangkumi pertanyaan
seperti di bawah:
i.Keluhan Utama
- Apakah keluhan utama?
- Apakah ada keluhan pucat? Sejak kapan bermulanya keluhan?
- Gejala apa yang dirasakan pasien? Lelah, malaise, sesak napas, nyeri dada, atau tanpa
gejala?
- Apakah gejala tersebut muncul mendadak atau bertahap?
ii. Keluhan Penyerta
- Apakah ada keluhan lain?- Apakah ada keluhan perut kembung?- Tanyakan kecukupan makanan / ASI- Adakah tanda-tanda kehilangan darah dari saluran cerna (tinja gelap, darah per rektal,
muntah ‘butiran kopi’)?- Adakah sumber kehilangan darah lain?
iii. Riwayat Kelahiran
- Apakah ada sebarang komplikasi sebelum, saat dan setelah melahirkan?
iv. Riwayat Keluarga
- Adakah riwayat anemia dalam keluarga? Khususnya pertimbangkan penyakit sel sabit, thalassemia, dan anemia hemolitik yang diturunkan.

Gambar 1. Algoritma pendekatan diagnosis thalassemia1
2.2 Pemeriksaan Fisik
- Dilihat kesadaran pasien. Sakit ringan atau berat?
- Apakah kelihatan sesak napas atau syok akibat kehilangan darah akut?
- Tanda-tanda anemia (pucat, konjungtiva anemis, telapak tangan pucat)
- Adakah tanda-tanda ikterus?
- Diperiksa tanda-tanda vital
- Adakah tanda-tanda kerusakan trombosit (memar, petikie)?
- Adakah tanda-tanda leukosit abnormal atau tanda-tanda infeksi?
- Adakah hepatomegali, splenomegali atau massa abdomen?
2.3 Pemeriksaan Penunjang
i.Pemeriksaan darah tepi

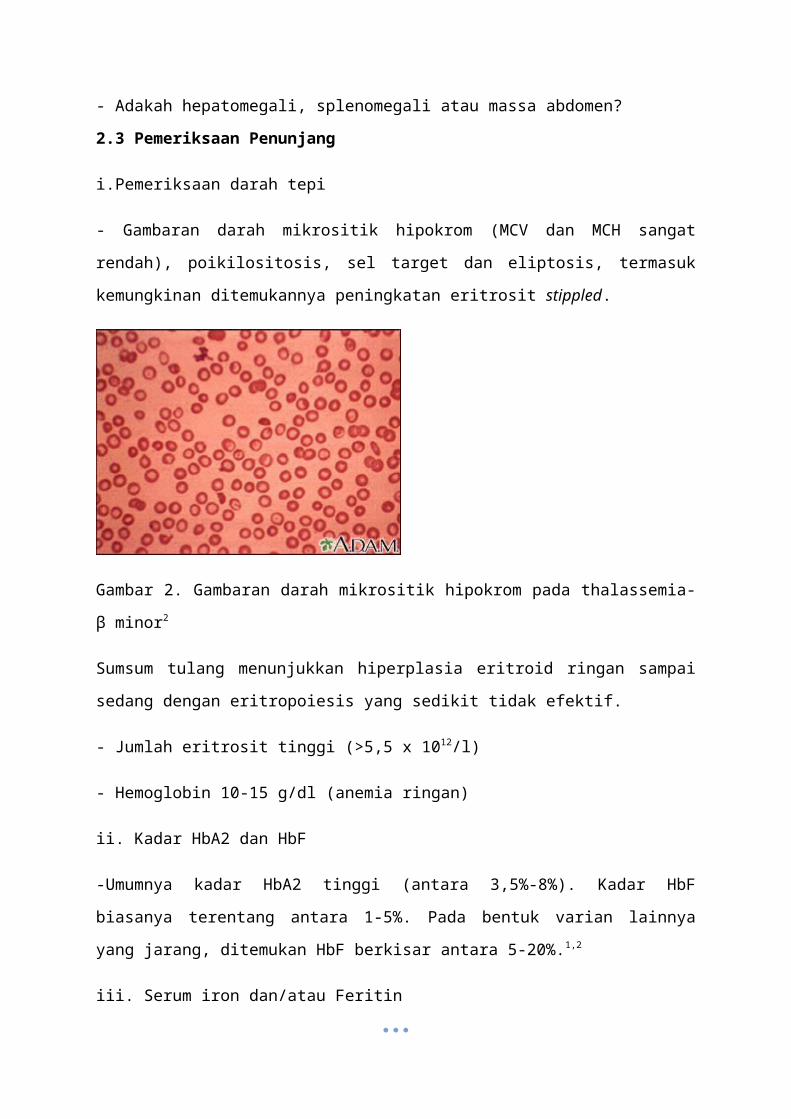
- Gambaran darah mikrositik hipokrom (MCV dan MCH sangat rendah), poikilositosis, sel
target dan eliptosis, termasuk kemungkinan ditemukannya peningkatan eritrosit stippled.
Gambar 2. Gambaran darah mikrositik hipokrom pada thalassemia-β minor2
Sumsum tulang menunjukkan hiperplasia eritroid ringan sampai sedang dengan eritropoiesis
yang sedikit tidak efektif.
- Jumlah eritrosit tinggi (>5,5 x 1012/l)
- Hemoglobin 10-15 g/dl (anemia ringan)
ii. Kadar HbA2 dan HbF
-Umumnya kadar HbA2 tinggi (antara 3,5%-8%). Kadar HbF biasanya terentang antara 1-
5%. Pada bentuk varian lainnya yang jarang, ditemukan HbF berkisar antara 5-20%.1,2
iii. Serum iron dan/atau Feritin
iv. Free Erythrocyte Protoporphyrin
v. Osmotic Fragility
Diagnosis prenatal untuk defek hemoglobin
Diagnosis prenatal tersedia dengan menggunakan DNA (vili korionik atau cairan amnion)
maupun darah janin. Karier harus diidentifikasi pertama kali (skrining dengan hitung darah
pada golongan etnik minoritas, pada konsultasi prakonsepsi, atau di klinik antenatal). Jika
seorang ibu merupakan karier, pasangannya harus diuji. Jika keduanya karier, terdapat satu
dalam empat kemungkinan bahwa janin homozigot, atau heterozigot ganda, dan satu dalam
dua kemungkinan bahwa janin adalah karier.
DNA fetal biasanya diamplifikasi dengan menggunakan reaksi rantai polimerase (polymerase
chain reaction, PCR) dan mutasi DNA dideteksi. Jika janin terkena dengan parah, pasangan
tersebut harus melakukan konsultasi, dan terminasi kehamilan, jika perlu, bisa ditawarkan.
2.4 Differential Diagnosis
1) Thalassemia α minor
Definisi
Sindrom thalassemia α biasanya disebabkan oleh delesi gen. Thalassemia-α minor/trait dapat
berupa bentuk homozigot-α+(-α/-α) atau heterozigot-α0(--/αα).3
Gambaran klinis
Sindrom ini menunjukkan tampilan klinis normal, anemia ringan dengan peningkatan eritrosit
yang mikrositik hipokrom. Pada saat dilahirkan, Hb Bart’s dalam rentangan 2-10%. Biasanya
pada penderita dewasa tidak ditemukan HbH(β4).3
2) Sickle Cell Anemia
Definisi
Penyakit sel sabit (anemia sel sabit homozigot) adalah anemia hemlitik kronik yang
disebabkan oleh mutasi titik pada gen globin-β yang menyebabkan substitusi valin terhadap
asam glutamat pada posisi keenam rantai globin-β. Hal ini menyebabkan HbS tidak larut pada
keadaan terdeoksigenasi. Rantai yang tidak larut mengkristal dalam sel darah merah yang
menyebabkan pembentukan sabit (sickling) dan oklusi vaskular.
Gambaran klinis
Gambaran klinis berupa anemia hemolitik berat yang diselingi oleh krisis. Gejala anemia
seringkali ringan jika dibandingkan dengan beratnya anemia karena Hb S relatif lebih mudah
melepaskan oksigen (O2) ke jaringan dibandingkan dengan Hb A, kurva disosiasi O2-nya
bergeser ke kanan.4

Ekspresi klinis Hb SS sangat bervariasi, beberapa pasien menjalani hidup yang hampir
normal, bebas dari krisis, tetapi pasien lain menderita krisis yang berat, bahkan pada masa
bayi dan dapat meninggal pada awal masa anak atau dewasa muda. Krisis dapat bersifat vaso-
oklusif, viseral, aplastik, atau hemolitik.4
Krisis vaso-oklusif dengan penyumbatan pembuluh kecil
-Disebabkan oleh peningkatan pembentukan sabit; presipitan yang sering adalah infeksi,
dehidrasi, asidosis, dan deoksigenasi. Nyeri abdomen disebabkan oleh infark yang mengenai
organ abdomen; nyeri tulang dapat terjadi pada punggung, pelvis, iga, dan tulang-tulang
panjang. Infark dapat mengenai sistem saraf pusat-menyebabkan stroke atau fits (serangan
tiba-tiba)-paru, limpa, atau ginjal. Pada anak-anak, ‘sindrom tangan-kaki’ disebabkan oelh
infark metafisis pada tulang kecil.
Krisis sekuestrasi viseral
Disebabkan oleh pembentukan sabit dengan kumpulan sel darah merah dalam hati, limpa,
atau paru. Sekuestrasi dalam paru, sebagian menyebabkan sindrom dada akut, mekipun infark
dan infeksi ikut berperan.
Krisis aplastik
-Krisis aplastik terjadi setelah infeksi oleh parvovirus B19. Hal ini menyebabkan penghentian
eritropoiesis sementara, yang pada orang sehat konsekuensinya tidak ada tetapi pada pasien
dengan harapan hidup sel darah merah yang berkurang, seperti Hb SS, dapat menyebabkan
anemia berat secara cepat yang memerlukan transfuse darah.
-Peningkatan kerentanan terhadap infeksi. fungsi splenik berkurang karena infark
menyebabkan autosplenektomi. Infeksi pneumokokal dapat menyebabkan pneumonia dan
meningitis. Infark mukosa usus merupakan predisposisi terjadinya infeksi Salmonella dan
osteomielitis dapat terjadi.
-Gambaran klinis lain meliputi batu empedu pigmen dengan kolesistitis, ulkus tungkai
kronik, nekrosis avaskular pada kaput femoris dan humeri atau tulang lainnya, kardiomiopati,
retinopati proliferative, dan nekrosis papiler ginjal (yang menyebabkan poliuria, kegagalan
untuk memekatkan urin, dan kecenderungan mengalami dehidrasi).4

Thalassemia-βminor
Thalassemia-α minor Anemia sel sabit
Etiologi Mutasi titik Delesi gen Mutasi titik pada gen globin-β (asam glutamat valin)
Manifestasi klinik Anemia ringan Anemia ringan/- Anemia hemolitik berat (gejala ringan jika dibandingkan dengan beratnya anemia), krisis
Hasil laboratorium -Hb 10-15g/dl-Eritrosit mikrositik hipokrom (MCV dan MCH sangat rendah)- Jumlah eritrosit tinggi (>5,5x1012/l)- kadar HbA2 tinggi (>3,5%) diagnosis pasti -rasio α/β meningkat
-Eritrosit mikrositik hipokrom (MCV dan MCH sangat rendah)- Jumlah eritrosit tinggi (>5,5x1012/l)- elektroforesis Hb normal- rasio α/β menurun
-Hb 6-9g/dl- sel sabit dan sel target dalam darah- elektroforesis Hb : Pada HbSS, HbA tidak terdeteksi, HbF(5-15%)
Tabel 1. Perbandingan antara thalassemia β minor, thalassemia α minor dan anemia sel sabit2-
4
2.5 Working Diagnosis
Thalassemia beta minor
Definisi : Thalassemia-β minor mempunyai genotip berupa heterozigot thalassemia-β,
seringkali disebut juga thalassemia-β trait. Fenotip kelainan ini secara klinis tidak
memberikan gejala(asimtomatik)
Manifestasi klinis
-Tampilan klinis normal. Dapat disertai dengan anemia ringan.
- Hepatomegali dan splenomegali ditemukan pada sedikit penderita.

Gambar 3. Manifestasi klinis pada thalasemia1
2.6 Etiologi
Mutasi gen, yakni bentuk delesi dan non delesi
1. Delesi gen globin-β
Paling sedikit 17 delesi yang berbeda yang hanya dijumpai pada thalassemia-β, namun
jarang dan tampaknya terisolasi, berupa kejadian tunggal (single event), kecuali delesi
619-bp pada ujung akhir 3’ gen-β lebih sering ditemukan, walaupun terbatas pada
populasi Sind dan Gujarat di Pakistan dan India. Delesi ini mencakup lebih kurang lebih
kurang 50% allel thalassemia-β. Bentuk homozigot delesi ini menghasilkan thalassemia-
β0. Heterozigot delesi ini menghasilkan peningkatan HbA2 dan HbF, sama yang dijumpai
pada bentuk mutasi lainnya thalassemia-β.1,2
2. Mutasi non delesi globin-β
Mutasi non delesi globin-β mencakup proses transkripsi, prosesing dan translasi, berupa
mutasi titik (point mutations):
- region promotor (promoter regions),
- mutasi transkrisional pada lokasi CAP (CAP sites, 5’-untranslated region)

- mutasi prosesing RNA: intron-exon boundaries, polyadenilation signal (Poly A signal),
splice site consensus sequences, cryptic sites in exons, cryptic sites in introns.
- mutasi yang menyebabkan translasi abnormal RNA messenger : inisiasi (initiation),
nonsense, dan mutasi frameshift.
3. Bentuk mutasi lainnya
- diwariskan secara dominan (dominantly inherited β thalassemias), varian globin tidak
stabil, thalassemia β tersembunyi, mutasi thalassemia β yang tidak terkait cluster gen
globin β, dan bentuk-bentuk bervariasi thalassemia β.
2.7 Epidemiologi
Thalassemia merupakan salah satu jenis anemia hemolitik dan merupakan penyakit keturunan
yang diturunkan secara autosomal yang paling banyak dijumpai di Indonesia dan Italia.
Thalassemia terdiri atas beberapa tipe. Di Indonesia lebih banyak ditemukan kasus
thalassemia beta. Insiden pembawa sifat Thalassemia di Indonesia berkisar antara 6-10%,
artinya dari setiap 100 orang 6-10 orang adalah pembawa sifat Thalassemia. Kalau sepasang
dari mereka menikah, kemungkinan untuk mempunyai anak penderita talasemia berat adalah
25%, 50% menjadi pembawa sifat (carrier ) talasemia, dan 25% kemungkinan bebas
thalassemia. Sebagian besar penderita thalassemia adalah anak-anak usia 0 hingga 18 tahun.
Faktor risiko
- Anak dengan orang tua yang memiliki gen thalassemia
- Anak dengan salah satu/kedua orang tua thalassemia minor
- Anak dengan salah satu orang tua thalassemia
- Resiko laki-laki atau perempuan untuk terkena sama
- Thalassemia Beta mengenai orang asli dari Mediterania atau ancestry (Yunani, Italia,
Ketimuran Pertengahan) dan orang dari Asia dan AfrikaPendaratan.
2.8 Patofisiologi
Pada thalassemia-β, di mana terdapat penurunan produksi rantai β, terjadi produksi
berlebihan rantai α. Produksi rantai globin γ, di mana pasca kelahiran masih tetap diproduksi
rantai globin α2γ2(HbF), tidak mencukupi untuk mengkompensasi defisiensi α2β2 (HbA). Hal
ini menunjukkan produksi rantai globin β dan rantai globin γ tidak pernah dapat mencukupi

untuk mengikat rantai α yang berlebihan. Rantai α yang berlebihan ini merupakan ciri khas
pada patogenesis thalassemia-β.
Rantai α yang berlebihan, yang tidak dapat berikatan dengan rantai globin lainnya, akan
berpresipitasi pada precursor sel darah merah dalam sumsum tulang dan dalam sel progenitor
dalam darah tepi. Presipitasi ini akan menimbulkan gangguan pematangan precursor eritroid
dan eitropoiesis yang tidak efektif, sehinga umur eritrosis menjadi pendek. Akibatnya, timbul
anemia.5
Anemia ini lebih lanjut lagi akan menjadi pendorong (drive) proliferasi eritroid yang terus
menerus (intense) dalam sumsum tulang yang inefektif, sehingga terjadi ekspansi sumsum
tulang. Hal ni kemudian akan menyebabkan deformitas skeletal dan berbagai gangguan
pertumbuhan dan metabolisme. Anemia kemudian akan ditimbulkan lagi (exacerbated)
dengan adanya hemodilusi akibat adanya hubungan langsung (shunting) darah akibat
sumssum tulang yang berekspansi dan juga oleh adanya splenomegali. Pada limpa yang
membesar makin banyak sel darah merah abnormal yang terjebak, untuk kemudian akan
dihancurkan oleh sistem fagosit.
Hiperplasia sumsum tulang kemudian akan meningkatkan absorpsi dan muatan besi.
Transfusi yang diberikan secara teratur juga menambah timbunan besi. Hal ini akan
menyebabkan penimbunan besi yang progresif di jaringan berbagai organ, yang akan diikuti
kerusakan organ dan diakhiri dengan kematian., bila besi ini tidak segera dikeluarkan.

Gambar 4. Patofisiologi thalasemia.5
2.9 Penatalaksanaan
Pasien dengan talasemia minor paling banyak mengalami anemia ringan (dengan sedikit
penurunan kadar hemoglobin dalam darah). Situasi ini bisa sangat erat menyerupai dengan
anemia ringan yang disebabkan oleh kekurangan zat besi. Namun, pasien dengan talasemia

minor memiliki kadar besi darah yang normal (kecuali mereka telah kekurangan zat besi oleh
sebab-sebab yang tertentu). Tidak ada pengobatan yang diperlukan untuk thalassemia minor.
Secara khusus, zat besi tidak perlu dan tidak disarankan.
Terapi preparat besi sebaiknya tidak diberikan kecuali memang dipastikan terdapat defisiensi
besi dan harus segera dihentikan apabila nilai Hb yang potensial pada penderita tersebut telah
tercapai. Diperlukan konseling pada semua penderita dengan kelainan genetik, khususnya
mereka yang memiliki anggota keluarga yang berisiko untuk terkena thalassemia berat.
2.10 Komplikasi
Gambar 5. Komplikasi thalasemia5
2.11 Pencegahan
Program pencegahan berdasarkan penapisan pembawa sifat thalassemia dan diagnosis
prenatal telah dapat menurunkan secara bermakna kejadian thalassemia mayor pada anak-
anak di Yunani, Siprus, Italia daratan dan Sardinia. Penapisan pembawa sifat thalassemia-β
lebih berdaya guna bila dikerjakan dengan penilaian indeks sel darah merah. Individu dengan

MCV dan MCH yang rendah dinilai konsentrasi HbA2-nya. Masalahnya timbul pada
penapisan individu dengan pembawa sifat thalassemia-α bersamaan (co-existent) dengan
thalassemia-α.
Prinsip Pedigree
Definisi Pedigree Analysis pemeriksaan yang teliti terhadap silsilah atau asal usul.
Beberapa Kegunaan Pedigree Analysis
1. Mengetahui bagaimana timbulnya suatu penyakit
- Setelah ditelaah lebih lanjut beberapa jenis penyakit atau kelainan akan menunjukkan adanya kejadian berulang yang dialami oleh lebih dari satu orang yang masih memiliki hubungan saudara satu sama lain. Berdasarkan pola yang ditunjukkan dari catatan silsilah keluarga (bagan riwayat keluarga/family tree), kita dapat memperkirakan sifat suatu penyakit. apakah penyakit tersebut bersifat diturunkan dari orang tua atau tidak diturunkan. 6
2. Mengetahui mekanisme atau pola penurunan penyakit
- Dari pola yang tampak dalam bagan riwayat keluarga dapat kita lihat pula mekanisme penurunan suatu penyakit.
3. Memperkirakan penetrance
- Penetrance adalah perkiraan berapa banyak penyakit tersebut akan timbul atau terjadi pada seseorang dengan kondisi gen tertentu.
4. Memperkirakan expressivity
- Expressivity adalah derajat beratnya manifestasi klinis suatu penyakit pada kondisi gen tertentu.
5. Sebagai Dasar Dari Konseling Genetik

Gambar 6. Simbol-simbol pada pedigree keluarga6
Dua salinan alel abnormal diperlukan untuk mengekspresikan trait autosomal resesif..
Secara umum, aturan berikut berlaku untuk pewarisan autosomal resesif:
Jika orang tua normal tetapi anak terkena, kedua orang tua adalah heterozigot. Rata-
rata, seperempat dari anak-anak mereka yang terkena, setengah darinya adalah
heterozigot, dan satu perempat adalah normal. Oleh karena itu, di kalangan anak-
anak, persentase untuk tidak mengembangkan kelainan (yaitu, menjadi normal atau
carrier) adalah tiga perempat, dan di antara anak-anak tidak terpengaruh, peluang
menjadi pembawa adalah dua pertiga.
Gambar 7. Pewarisan autosomal resesif5
Semua anak dengan orang tua yang terkena dan orang tua dengan genotip yang
normal mempunyai fenotip normal heterozigot.
Semua anak dengan ibubapa sebagai pengidap thalassemia turut mengalami hal yang
sama.

Pria dan perempuan mempunyai persentase yang sama untu terkena penyakit ini.
Heterozigot biasanya mempunyai fenotip normal tetapi membawa gen yang abnormal.
Kerabat lebih mungkin untuk membawa alel mutan yang sama, sehingga perkawinan
antar kerabat dekat (consanguinity) meningkatkan kemungkinan memiliki anak yang
terkena thalassemia. Dalam hubungan orangtua-anak atau adik-beradik, risiko
memiliki anak yang abnormal meningkat karena begitu banyak materi genetik mereka
adalah sama.7
Pada populasi tertentu, persentase heterozigot (pembawa) adalah tinggi karena efek
pendiri (yaitu, kelompok dimulai dengan beberapa anggota, salah satunya adalah
pembawa) atau karena pembawa memiliki keunggulan selektif (misalnya,
heterozigositas untuk anemia sel sabit sebagai pelindung terhadap malaria).7
2.12 Prognosis
Dubia. Prognosis tergantung pada tipe dan tingkat keparahan dari thalassemia. Seperti
dijelaskan sebelumnya, kondisi klinis penderita thalassemia sangat bervariasi dari ringan
bahkana simtomatik hingga berat dan mengancam jiwa, tergantung pula pada terapi dan
komplikasi yang terjadi.

Bab III
Penutup
Thalassemia-β minor adalah kelainan yang umum, biasanya tanpa gejala, seperti sifat
thalassemia-α ditandai oleh gambaran darah mikrositik hipokrom tetapi jumlah eritrosit tinggi
dan anemia ringan. Kelainan ini biasanya lebih berat dibandingkan sifat α; kadar Hb A2 yang
tinggi (>3,5%) memastikan diagnosis. Penyakit thalassemia-β minor harus dapat dideteksi
sejak awal lagi karena diagnosis memungkinkan dilakukannya konseling prenatal pada pasien
dengan seorang pasangan yang juga mempunyai kelainan haemoglobin yang nyata. Jika
keduanya membawa sifat thalassemia-β, sebanyak 25% anak berisiko untuk menderita
thalassemia mayor.
Kesimpulan
Hipotesis diterima. Pasien perempuan 16 tahun, sering lemah dan pingsan terutama saat haid,
mengkonsumsi rutin obat tambah darah namun keadaan tetap sama selain berpacaran dengan
sepupu yang sering mengeluh pusing dan pingsan mengidap thalassemia-β minor.

Daftar Pustaka
1. Atmakusuma D, Setyaningsih I. Dasar molekular thalassemia beta. Buku Ajar Ilmu
Penyakit Dalam 2009;5:1383-84
2. Medline Plus. Thalassemia minor. Edisi 5 Juli 2012. Diunduh dari
http://www.nlm.nih.gov/medlineplus/ency/imagepages/1499.htm, 16 September 2012
3. Mehta A, Hoffbrand V. Talasemia alfa. At A Glance Hematologi 2008;2:40-1
4. Hoffbrand AV, Pettit JE, Moss PAH. Anemia Sel Sabit. Kapita Selekta Hematologi
2005;4: 77
5. Rund D, Rachmilewitz E. Medical progress β-thalassemia. The New England Journal
of Medicine 2005;353:1135-46
6. The Merck Manual For Health Care Professionals. Overview of genetics ; pedigree
analysis. Edisi Mei 2007. Diunduh dari
http://www.merckmanuals.com/professional/special_subjects/general_principles_of_
medical_genetics/overview_of_genetics.html, 16 September 2012.
7. Campbell NA, Reece JB, Urry LA, Cain ML, Wasserman SA, Minorsky PV et al.
Biologi. Perilaku alel resesif 2010;8:298-9




















