ltm 1
8
REGULASI PENDAFTARAN OBAT DI INDONESIA Sandra Monica (1206212432) Teknologi Bioproses Alur Registrasi Obat Tata laksana registrasi obat diatur oleh Badan POM dalam Keputusan Ka BPOM No. HK.00.05.3.1950 Tahun 2003 Tentang Kriteria dan Tata Laksana Registrasi Obat. Secara umum, registrasi obat dilakukan dalam dua tahapan, yaitu tahapan pra-registrasi yang bertujuan untuk menilai kelengkapan administrasi dari Industri Farmasi yang akan meregistrasi obat dan sekaligus menentukan kriteria registrasi dan jalur evaluasi, serta tahapan registrasi untuk menilai apakah obat tersebut layak mendapatkan ijin edar. Secara sistematis, dapat dilihat pada bagan berikut. Keterangan: 1. Pendaftaran oleh Industri Farmasi kepada kepala Badan POM, sekaligus tahapan pra-registrasi yaitu prosedur untuk menentukan jalur evaluasi dan kategori registrasi. Pada tahap pra-registrasi juga disertai dengan penyerahan dokumen pra-registrasi. 2. Pemberitahuan hasil pra-registrasi secara tertulis.
-
Upload
sandra-monica -
Category
Documents
-
view
230 -
download
3
description
ltm obat dan kosmetik
Transcript of ltm 1

REGULASI PENDAFTARAN OBAT DI INDONESIASandra Monica (1206212432)
Teknologi Bioproses
Alur Registrasi Obat
Tata laksana registrasi obat diatur oleh Badan POM dalam Keputusan Ka BPOM No. HK.00.05.3.1950 Tahun 2003 Tentang Kriteria dan Tata Laksana Registrasi Obat. Secara umum, registrasi obat dilakukan dalam dua tahapan, yaitu tahapan pra-registrasi yang bertujuan untuk menilai kelengkapan administrasi dari Industri Farmasi yang akan meregistrasi obat dan sekaligus menentukan kriteria registrasi dan jalur evaluasi, serta tahapan registrasi untuk menilai apakah obat tersebut layak mendapatkan ijin edar. Secara sistematis, dapat dilihat pada bagan berikut.
Keterangan:
1. Pendaftaran oleh Industri Farmasi kepada kepala Badan POM, sekaligus tahapan pra-registrasi yaitu prosedur untuk menentukan jalur evaluasi dan kategori registrasi. Pada tahap pra-registrasi juga disertai dengan penyerahan dokumen pra-registrasi.
2. Pemberitahuan hasil pra-registrasi secara tertulis.3. Pengajuan registrasi dengan menyerahkan berkas registrasi, mengisi formulir registrasi dan
disket, menyerahkan bukti pembayaran biaya evaluasi dan pendaftaran, serta hasil pra-registrasi.
4. Evaluasi berkas registrasi obat oleh Komisi Nasional Penilai Obat Jadi yang dibentuk oleh Badan POM.
5. Komisi Nasional Penilai Obat Jadi memberitahukan hasil evaluasi secara tertulis kepada Industri Farmasi pendaftar dan memberikan rekomendasi kepada kepala Badan POM.
6. Kepala Badan POM memberikan keputusan berupa pemberian ijin edar atau penolakan pemberian ijin edar. Keputusan ini disampaikan secara tertulis kepada Industri Farmasi yang

bersangkutan. Pemberian keputusan ini diberikan selambat-lambatnya berkisar antara 40-100 hari kerja (tergantung kategori dan jalur evaluasi) setelah menerima berkas registrasi yang lengkap.
7. Setelah mendapatkan ijin edar, Industri Farmasi yang bersangkutan boleh mulai memproduksi obat jadi tersebut untuk kemudian diedarkan.
8. Badan POM melaporkan pemberian ijin edar obat jadi kepada Menteri Kesehatan setiap satu tahun sekali.
Catatan
1. Jika pendaftar merasa keberatan terhadap hasil evaluasi KomNas Penilai Obat Jadi, dapat mengajukan permohonan dengar pendapat secara tertulis kepada Badan POM selambat-lambatnya 15 hari setelah pemberitahuan hasil evaluasi.
2. Jika pendaftar merasa keberatan terhadap penolakan pemberian ijin edar, maka boleh mengajukan permohonan peninjauan kembali kepada kepala Badan POM selambat-lambatnya 6 bulan setelah penolakan, dengan disertai data-data baru atau data yang pernah diajukan dilengkapi dengan justifikasi. Permohonan peninjauan kembali ini dapat dilakukan sampai 2 kali.
3. Kepala Badan POM dapat melakukan evaluasi kembali terhadap obat yang telah diberikan ijin edar untuk:
Obat dengan risiko efek samping lebih besar dibandingkan dengan efektifitasnya yang terungkap sesudah obat dipasarkan;
Obat dengan efektifitas tidak lebih baik dari plasebo; Obat yang tidak memenuhi persyaratan ketersediaan hayati/bioekivalensi
4. Kepala Badan POM dapat membatalkan ijin edar jika dikemudian hari terjadi salah satu dari hal-hal di bawah ini:
Berdasarkan penelitian atau pemantauan dalam penggunaannya setelah beredar tidak memenuhi kriteria.
Penandaan dan promosi menyimpang dari persetujuan izin edar. Selama 12 (dua belas) bulan berturut-turut obat yang bersangkutan tidak diproduksi,
diimpor atau diedarkan. Izin Industri Farmasi, Pedagang Besar Farmasi yang mendaftarkan, memproduksi atau
mengedarkan dicabut. Pemilik izin edar melakukan pelanggaran di bidang produksi dan/atau peredaran obat.
Berikut adalah alur registrasi obat online:

1. Membuat akun AeRo (Aplikasi e.Registrasi Obat)
(Sumber: http://aero.pom.go.id/)
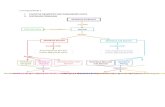
2. Melakukan Pra-Registrasi Obat Baru
(Sumber: http://aero.pom.go.id/)
3. Melakukan Registrasi Obat Baru
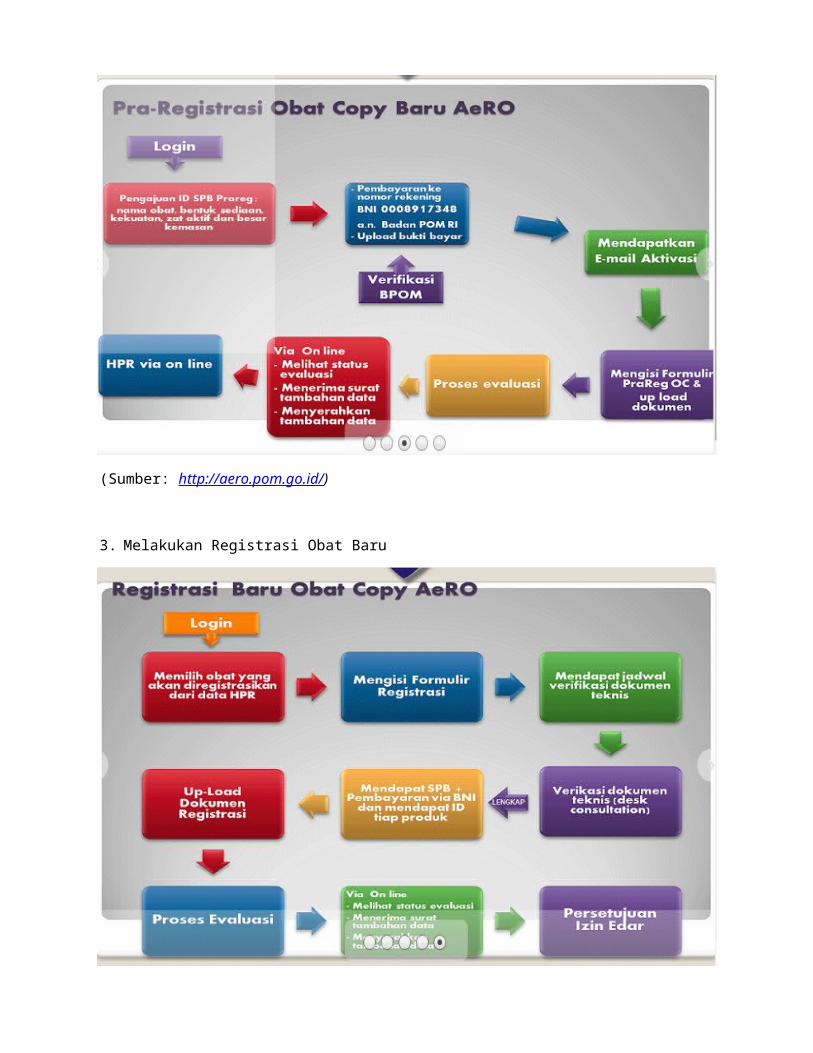
(Sumber: http://aero.pom.go.id/)
2.2. TAHAP PENGEMBANGAN DAN PENILAIAN OBAT
Tahap-Tahap Pengembangan dan Penilaian Obat
1. Meniliti dan skrining bahan obat.
2. Mensintesis dan meneliti zat/senyawa analog dari obat yang sudah ada dan diketahui efek farmakologinya
3. Meneliti dan mensintesis dan membuat variasi struktur
4. Dikembangkan obat alami dengan serangkaian pengujian yang dilaksanakan secara sistematik, terencana dan terarah untuk mendapatkan data farmakologik yang mempunyai nilai terapetik.
Tahap-Tahap Pengembangan dan Penilaian Obat dapat dilakukan dengan uji praklinik dan uji klinik.
Uji Praklinik: Suatu senyawa yang baru ditemukan (hasil isolasi maupun sintesis) terlebih dahulu diuji dengan serangkaian uji farmakologi pada hewan. Sebelum calon obat baru ini dapat dicobakan pada manusia, dibutuhkan waktu beberapa tahun untuk meneliti sifat farmakodinamik, farmakokinetik, farmasetika, dan efek toksiknya pada hewan uji.
Serangkaian uji praklinik yang dilakukan antara lain:

a. Uji farmakodinamik
Untuk mengetahui apakah bahan obat menimbulkan efek farmakologik seperti yang diharapkan atau tidak, titik tangkap, dan mekanisme kerjanya. Dapat dilakukan secara in vivo dan in vitro.
b. Uji Farmakokinetik
- Untuk mengetahui ADME (Absorpsi, Distribusi, Metabolisme dan Eliminasi
- Merancang dosis dan aturan pakai
c. Uji Toksikologi
- Mengetahui keamanannya
d. Uji Farmasetika
- Memperoleh data farmasetikanya, tentang formulasi, standarisasi, stabilitas, bentuk sediaan yang paling sesuai dan cara penggunaannya.
Uji Klinik Yaitu suatu pengujian khasiat obat baru pada manusia, dimana sebelumnya diawali oleh pengujian pada binatang atau pra klinik (Katzung, 1989).
Uji klinik Pada dasarnya uji klinik memastikan efektivitas, keamanan dan gambaran efek samping yang sering timbul pada manusia akibat pemberian suatu obat.
a) Uji Klinik Fase I
Fase ini merupakan pengujian suatu obat baru untuk pertama kalinya pada manusia. Yang diteliti disini ialah keamanan dan tolerabilitas obat, bukan efikasinya, maka dilakukan pada sukarelawan sehat, kecuali untuk obat yang toksik (misalnya sitostatik), dilakukan pada pasien karena alasan etik Tujuan fase ini adalah menentukan besarnya dosis maksimal yang dapat toleransi (maximally tolerated dose = MTD), yakni dosis sebelum timbul efek toksik yang tidak dapat diterima. Pada fase ini, diteliti juga sifat farmakodinamik dan farmakokinetiknya pada manusia. Hasil penelitian farmakokinetik ini digunakan untuk meningkatkan ketepatan pemilihan dosis pada penelitian selanjutnya. Uji klinik fase I dilaksanakan secara terbuka, artinya tanpa pembanding dan tidak tersamar, dengan jumlah subyek bervariasi antara 20-50 orang.
Pada fase ini obat dicobakan untuk pertama kalinya pada sekelompok kecil penderita yang kelak akan diobati dengan calon obat
Pada fase II awal, pengujian efek terapi obat dikerjakan secara terbuka karena masih merupakan penelitian eksploratif. Pada tahap biasanya belum dapat diambil kesimpulan yang mantap mengenai efek obat yang bersangkutan karena terdapat berbagai factor yang mempengaruhi hasil pengobatan, misalnya perjalanan klinik penyakit, keparahannya, efek placebo (Ganiswara, 1995).
Untuk membuktikan bahwa suatu obat berkhasiat, perlu dilakukan uji klinik komparatif yang membandingkannya dengan placebo; atau bila penggunaan placebo tidak memenuhi syarat etik, obat dibandingkan dengan obat standard yang telah dikenal. Ini dilakukan pada akhir fase II atau awal fase III, tergantung dari siapa yang melakukan, seleksi penderita, dan monitoring penderitanya. Untuk menjamin validitas uji klinik komparatif ini, alokasi penderita harus acak dan pemberian obat dilakukan secara tersamar ganda. Ini dsebut uji klinik acak tersamar ganda berpembanding.
Pada fase II ini tercakup juga penelitian dosis-efek untuk menentukan dosis optimal yang akan digunakan selanjutnya, serta penelitian lebih lanjut mengenai eliminasi obat, terutama

metabolismenya. Jumlah subjek yang mendapat obat baru pada fase ini antara 100-200 penderita (Ganiswara, 1995).
b ) Uji Klinik Fase II
Pada fase ini dicobakan pada pasien sakit. Tujuannya adalah melihat apakah obat ini memiliki efek terapi. Pada fase II awal, pengujian efek terapi obat dikerjakan secara terbuka karena masih merupakan penelitian eksploratif, karena itu belum dapat diambil kesimpulan yang mantap mengenai efikasi obat yang bersangkutan.
Untuk menunjukkan bahwa suatu obat memiliki efek terapi, perlu dilakukan uji klinik komparatif (dengan pembading) yang membandingkannya dengan plasebo; atau jika penggunaan plasebo tidak memenuhi persyaratan etik, obat dibandingkan dengan obat standar (pengobatan terbaik yang ada). Ini dilakukan pada fase II akhir atau awal, tergantung dari siapa yang melakukan, seleksi pasien, dan monitoring pasiennya. Untuk menjamin validasi uji klinik komparatif ini , alokasi pasien harus acak dan pemberian obat dilakukan secara tersamar ganda. Ini disebut uji klinik berpembanding, acak, tersamar ganda. Fase ini terjakup juga studi kisaran dosis untuk menetapkan dosis optimal yang akan digunakan selanjutnya(Ganiswara, 1995).
c ) Uji Klinik Fase III
-Pada manusia sakit, ada kelompok kontrol dan kelompok pembanding
- Cakupan lebih luas baik dari segi jumlah pasien maupun keragaman (misal : intra ras.
- Setelah terbukti efektif dan aman obat siap untuk dipasarkan.
Uji klinik fase III dilakukan untuk memastikan bahwa suatu obat-baru benar-benar berkhasiat (sama dengan penelitian pada akhit fase II) dan untuk mengetahui kedudukannya dibandingkan dengan obat standard. Penelitian ini sekaligus akan menjawab pertanyaan-pertanyaan tentang (1) efeknya bila digunakan secara luas dan diberikan oleh para dokter yang ‘kurang ahli’; (2) efek samping lain yang belum terlihat pada fase II; (3) dan dampak penggunaannya pada penderita yang tidak diseleksi secara ketat (Ganiswara, 1995).
Uji klinik fase III dilakukan pada sejumlah besar penderita yang tidak terseleksi ketat dan dikerjakan oleh orang-orang yang tidak terlalu ahli, sehingga menyerupai keadaan sebenarnya dalam penggunaan sehari-hari dimasyarakat. Pada uji klinik fase III ini biasanya pembandingan dilakukan dengan placebo, obat yang sama tapi dosis berbeda, obat standard dengan dosis ekuiefektif, atau obat lain yang indikasinya sama dengan dosis yang ekuiefektif. Pengujian dilakukan secara acak dan tersamar ganda.
Bila hasil uji klinik fase III menunjukan bahwa obat baru ini cukup aman dan efektif, maka obat dapat diizinkan untuk dipasarkan. Jumlah penderita yang diikut sertakan pada fase III ini paling sedikit 500 orang (Ganiswara, 1995).
d) Uji Klinik Fase IV
- Uji terhadap obat yang telah dipasarkan (post marketing surveilance)
- Memantau efek samping yang belum terlihat pada uji-uji sebelumnya
- Dug safety : drug mortality atau drug morbidity
- MESO : Monitoring Efek Samping Obat

Fase ini sering disebut post marketing drug surveillance karena merupakan pengamatan terhadap obat yang telah dipasarkan. Fase ini bertujuan menentukan pola penggunaan obat di masyarakat serta pola efektifitas dan keamanannya pada penggunaan yang sebenarnya. Survei ini tidak tidak terikat pada protocol penelitian; tidak ada ketentuan tentang pemilihan penderita, besarnya dosis, dan lamanya pemberian obat. Pada fase ini kepatuhan penderita makan obat merupakan masalah.Penelitian fase IV merupakan survey epidemiologic menyangkut efek samping maupun efektif obat. Pada fase IV ini dapat diamati (1) efek samping yang frekuensinya rendah atau yang timbul setelah pemakaian obat bertahun-tahun lamanya, (2) efektifitas obat pada penderita berpenyakit berat atau berpenyakit ganda, penderita anak atau usia lanjut, atau setelah penggunaan berulangkali dalam jangka panjang, dan (3) masalah penggunaan berlebihan, penyalah-gunaan, dan lain-lain. Studi fase IV dapat juga berupa uji klinik jangka panjang dalam skala besar untuk menentukan efek obat terhadap morbiditas dan mortalitas sehingga datanya menentukan status obat yang bersangkutan dalam terapi.
Referensi:
Registrasi Obat Online. 2015. Registrasi Obat Online. [ONLINE] Available at: http://aero.pom.go.id/. [Accessed 25 February 2015].