






Bahasa
Halaman
Hukum


Condensed Matter Physics, ????, Vol. ?, No ?, ?????: 1–17
http://www.icmp.lviv.ua/journal
Regular article
Electronic properties of AlN crystal doped with Cr,Mn and Fe
S.V. Syrotyuk, V.M. ShvedLviv Polytechnic National University , 12 S. Bandera Str., 79013 Lviv, Ukraine
September 29, 2012
The spin-resolved electronic energy band spectra, as well as partial and total density of electronic statesof the crystal AlN, doped with Cr, Mn and Fe, have been evaluated within the projector augmented waves(PAW) approach by means of the ABINIT code. The Hartree-Fock exchange for correlated electrons is usedto describe the correlated orbitals in the PAW framework. The calculated one-electron energies for electronsof spin up and down are very different. We have found that all the considered crystals are ferromagnetic.
Key words: Electronic structure calculations; Strongly correlated electrons; Exact exchange for correlatedelectrons; Magnetic semiconductors; Projector augmented wave method
PACS: 71.55.Eq; 71.55.−i; 71.20.−b; 71.15.Mb; 71.27.+a; 75.30.±m
1. Introduction
Semiconductors doped with d-transition elements draw considerable attention of researchersdue to their possible applications in spintronics [1]. The discovery of ferromagnetism in them ledto an intensication of experimental and theoretical studies. The impurities of transition elementslead to a fundamental change in the electronic structure of semiconductors [2]. Crystal AlN with anadmixture of 3d-transition elements was investigated using the ultrasoft (USPP) pseudopotentialapproach within the LSDA theory [3] and in the LSDA + U approximation [4]. The partial andtotal densities of electronic states of the Mn-doped crystal AlN and co-doped with B, Si, and P,with spin up and down [5], have been evaluated recently by means of the LSDA KKR program [6].The parameter of the Coulomb energy U allows to take into account strong correlations betweenthe d-electrons of the transition element, and thus to improve the values of the electronic energyband spectrum of the crystal with impurity.
However, it is known that the Coulomb energy U depends on the system. This means that itis not transferable to the same crystal, but for example, with a vacancy [7]. In [7] also was foundinherent to LSDA + U approach the existence of several minima of the total energy functional,depending on the matrix of the initial occupation of the correlated orbitals.
The spin-resolved electronic energy band spectra, as well as partial and total density of elec-tronic states of the crystal AlN, doped with Cr, Mn and Fe, have been evaluated within theprojector augmented wave (PAW) approach [8, 9], implemented in the ABINIT [10] code. Theexact exchange, for 3d-electrons only, is used to improve the on-site correlations [11].
2. Calculation
The electronic structure calculations have been carried out using the PAW [9] method, in whichthe electron wave function with its full nodal structure |ψn(r)⟩ is represented in terms of the smooth
c⃝ S.V. Syrotyuk, V.M. Shved ?????-1

S.V. Syrotyuk, V.M. Shved
nodeless function |ψn(r)⟩ as|ψn(r)⟩ = τ |ψn(r)⟩, (2.1)
where τ is the transformation operator,
τ = 1 +∑a
∑α
(|ϕaα⟩ − |ϕaα⟩)⟨paα|, (2.2)
and |ϕaα(r)⟩ is the all-electron basis function, |ϕaα(r)⟩ is the pseudopotential basis function, and|paα(r)⟩ is projector function, which we calculated using the atompaw code [12]. The subscript αdenotes the quantum numbers, and the superscript a represents the atomic augmentation sphereindex.
We have generated the PAW functions for following valence basis states: 2s22p63s23p1 for Al,2s22p3 for N, 3s23p63d54s14p0 for Cr, 3s23p63d54s24p0 for Mn, and 3s23p63d64s24p0 for Fe. Theinclusion in the basis of 3s and 3p states of the Cr, Mn and Fe atoms slows down the calculation,but increases the precision and eliminates the problem of ghost states which can appear amongsolutions of the secular equation constructed on a minimal set of functions [13]. The radii of theaugmentation spheres are 1.6, 1.3, 1.9, 1.9, 1.9 a.u. for Al, N, Cr, Mn, Fe, respectively. Substituting(2.1), (2.2) to the Kohn-Sham equation
H|ψαk⟩ = |ψαk⟩εαk (2.3)
we obtain the system of linear equations
τ+Hτ |ψαk⟩ = τ+τ |ψαk⟩εαk, (2.4)
where α is a band number, k denotes the vector in rst Brillouin zone, and τ+ denes Hermitianconjugate of τ . The electron density in the PAW method is determined by the sum of three terms,
ρ(r) = ρ(r) +∑a
(ρa(r)− ρa(r)). (2.5)
The rst term is smooth pseudo-density, which is the Fourier series,
ρ(r) =∑nk
fnk|ψαk(r)|2 =1
Ω
∑G
ρ(G)eiGr (2.6)
where fnk is occupancy, weighted by the fractional Brillouin zone sampling volume. The terms ofthe density of electrons in the atomic spheres are determined from the coecients of the projectedpopulation of the states
W aαβ =
∑nk
fnk⟨ψαk|paα⟩⟨paβ |ψαk⟩ (2.7)
Hence, the contributions of the density in the atomic sphere can be written as
ρa(r) =∑αβ
W aαβφ
a∗
α (r)φaβ(r), ρ
a(r) =∑αβ
W aαβφ
a∗
α (r)φaβ(r). (2.8)
Exchange-correlation potential was calculated in the form of PBE0 [14] according to which theexchange-correlation energy
EPBE0xc [ρ] = EPBE
xc [ρ] +1
4(EHF
x [ψsel]− EPBEx [ρsel]), (2.9)
corresponds to the PBE exchange-correlation functional [15], and ψsel, ρsel represent the wavefunction and electron density of the selected electrons, respectively [16]. The last are the 3d-electrons of Cr, Mn and Fe.
The electronic energy bands and DOS have been evaluated by means of the ABINIT code [10].Integration over the Brillouin zone was performed on the Monkhorst-Pack [17] spatial grid of
?????-2

Electronic properties of AlN crystal doped with Cr, Mn and Fe
4 × 6 × 6. The lattice constant of the crystal AlN a = 4.36 Å, and the supercell parameterscontaining 16 atoms are 2 × 1 × 1. The iterations were performed to ensure calculating the totalenergy of the crystal with an accuracy of 10−8 Ha. Relativistic eects are taken into accountwithin the scalar relativistic approximation. The density of electronic states was evaluated fromthe equation
nβ(E) =∑αk
δ(E − Eαk)|P aαβ(k)|2, (2.10)
P aαβ(k) = ⟨paβ |ψαk⟩ =
∫drpa
∗
β (r−Ra)ψαk(r), (2.11)
where P aαβ(k) are the expansion coecients for both
|ψαk⟩ =∑β
P aαβ(k)|ϕaβ⟩, (2.12)
|ψαk⟩ =∑β
P aαβ(k)|ϕaβ⟩, (2.13)
smooth and true functions, respectively, within the augmentation sphere. As can be seen fromequation (2.9), the exchange-correlation energy functional depends on the density of electrons andon the selected wave function of strongly correlated electrons. First, the pseudo-wave function iscalculated from equations (2.4). Then from equation (2.1) is calculated the electron wave functionand obtained the density of electrons (2.5). Based on the last we nd the exchange-correlationenergy functional (2.9). The symmetry of the crystal is described by space group P-42m (number111) and Bravais lattice is tP (primitive tetragonal).
3. Electronic properties
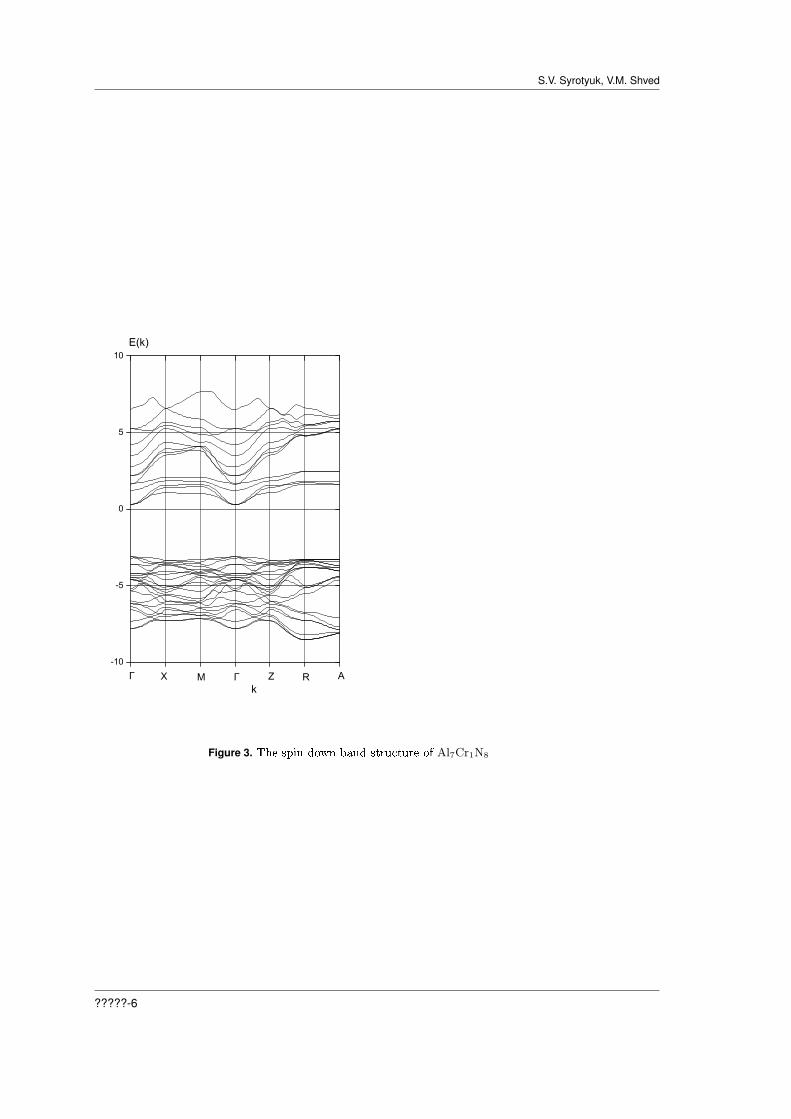
Figure 1 shows the partial and total density of electronic states of the crystal Al7Cr1N8 fora spin down. We see that the top of the valence band is formed predominantly with hybridizeds-states of Cr and p-states of N. The dispersion curves shown in Fig.3, indicate that for spindown our crystal is a semiconductor with a direct gap. It is substantially less than the gap Γ-X inthe usual crystal AlN. Aluminum nitride in the zinc-blende structure is an insulator with a wideindirect energy bandgap of 4.9 eV. Such a narrowing is explained by the fact that the crystal CrNis a Mott-Hubbard-type insulator with a small to negligible indirect band gap [18]. The bottom ofthe valence band is now at the point R, while in the the usual crystal AlN, it is at the point Γ.The Fermi level is located in the band gap, quite close to the bottom of the conduction band.
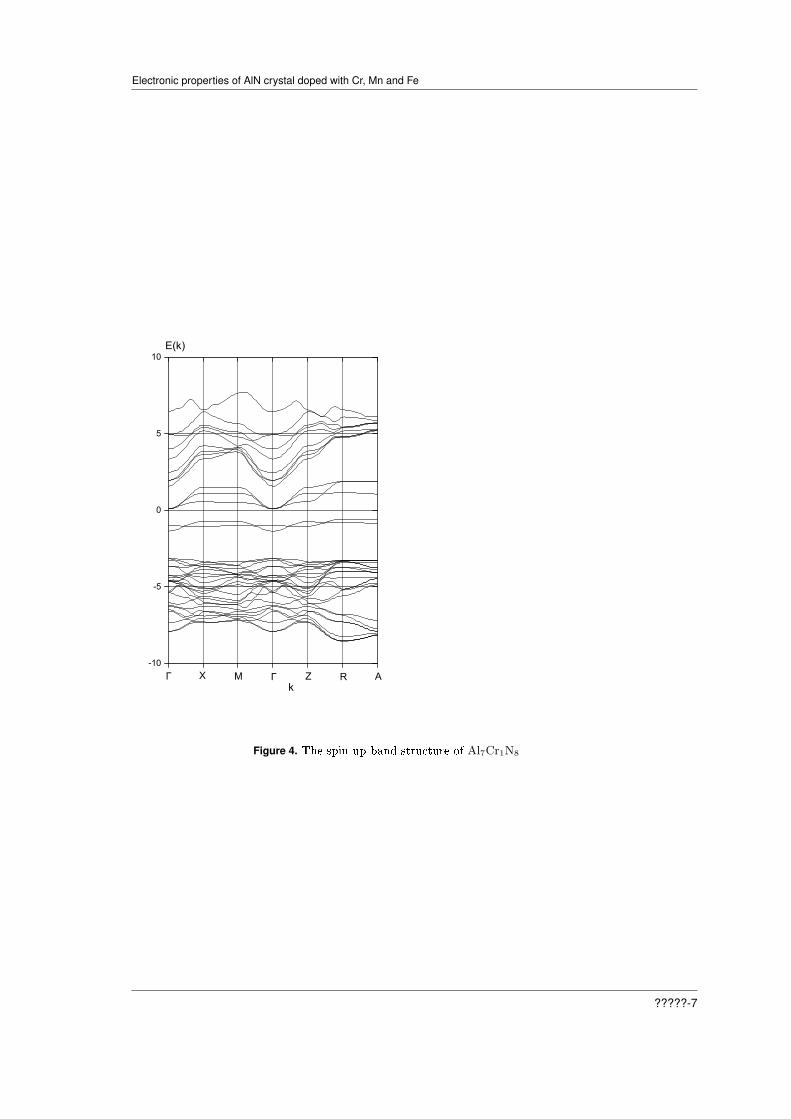
For spin up, we have a completely dierent picture, represented on Figures 2, and 4. The crystalAl7Cr1N8 is a semiconductor with indirect narrow gaps Γ-R and Γ-A. The Fermi level is now evencloser to the bottom of the conduction band. The top of the valence band is formed by hybridizedd-states of Cr, p-states of N and Al.
We turn to the analysis of the results of calculation for the crystal Al7Mn1N8, represented onFigures 5-8.
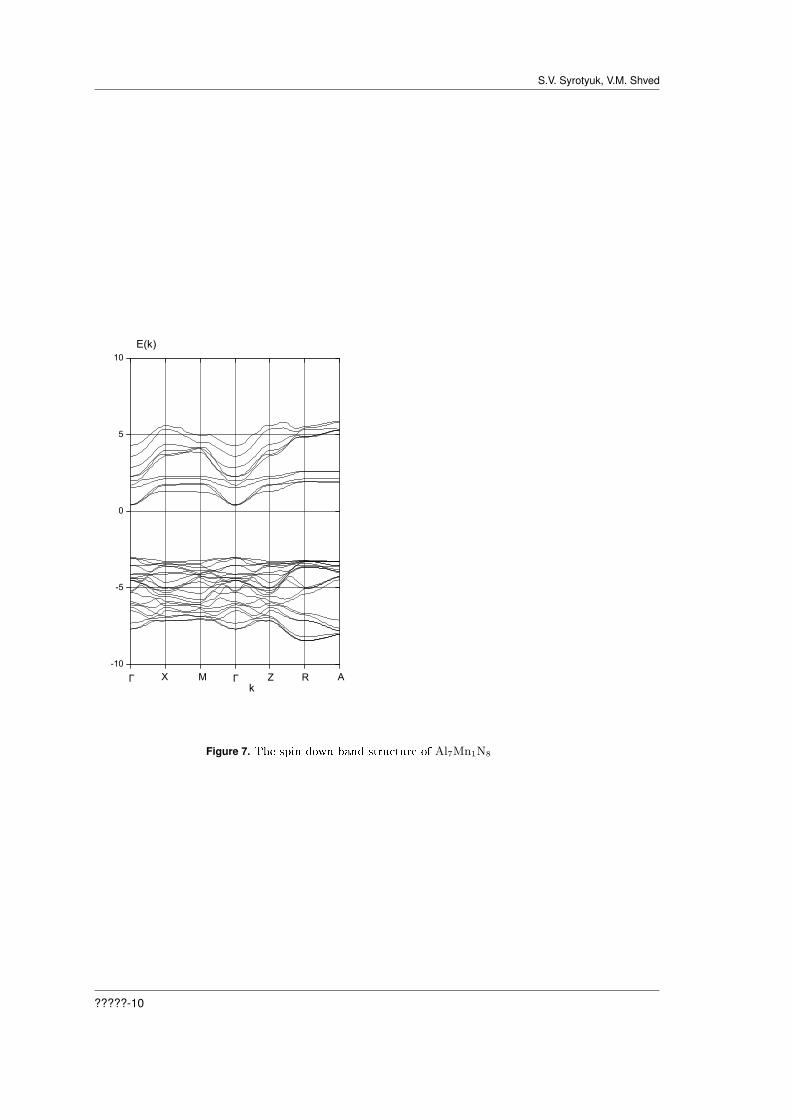
Figures 5 and 7 describe the electronic density of states and energy bands for spin down. FromFig.5 we see that the top of the valence band form the s-states of Mn and the p-states of N. Figure7 shows that the crystal is the direct band gap semiconductor, with bottom of the valence band atthe point R.
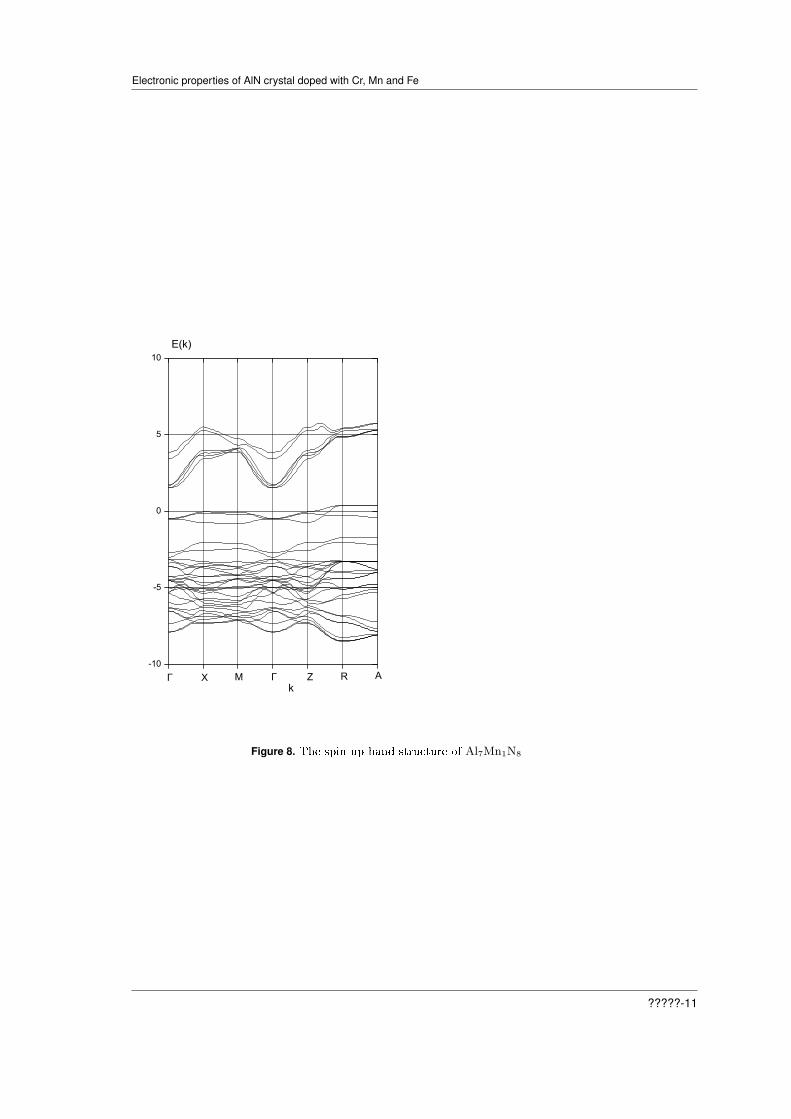
Figures 6 and 8 describe the electronic density of states and energy bands for spin up. Figure6 shows that the top of the valence band is formed by the p- and d-states of Mn and the p-statesof N, but the bottom of the conduction band form the s-states of N. And Figure 8 shows that thecrystal is the semiconductor with indirect gap Γ-ZR. The Fermi level is at the top of valence band.The presence of the spin up 3d states of Mn at Fermi level (Fig.8) and of the spin down ones inthe conduction band (Fig.6) qualitatively agrees with results, obtained within DFT+U approachfor Mn doped AlN [4].
?????-3

S.V. Syrotyuk, V.M. Shved
-10 -5 0 5
0,00,2 Al, s, dn
E-EF, eV
-10 -5 0 5
0,000,020,04 Al, p, dn
-10 -5 0 5
0,000,02 Al, d, dn
-10 -5 0 5
0,00,51,0
Cr, s, dn
-10 -5 0 5
0,00
0,05 Cr, p, dn
-10 -5 0 5
024 Cr, d, dn
-10 -5 0 5
0,00,51,0
N, s, dn
-10 -5 0 5
0,00,5 N, p, dn
-10 -5 0 5
0,0000,0050,010
N, d, dn
-10 -5 0 5
05
10n(E), 1/(eV·atom·spin)
tot, dn
Figure 1. The spin down partial, in 1/(eV·atom·spin), and total densities of states for Al7Cr1N8
?????-4

Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10 -5 0 5
0,00,20,4 Al, s, up
E-EF, eV
-10 -5 0 5
0,00,20,4 Al, p, up
-10 -5 0 50,00,2 Cr, s, up
-10 -5 0 5
0,00,20,4
Cr, p, up
-10 -5 0 5
020 Cr, d, up
-10 -5 0 5
0,00,2 N, s, up
-10 -5 0 5
0
5N, p, up
-10 -5 0 5
0,000,020,04 N, d, up
-10 -5 0 5
010
n(E), 1/(eV·atom·spin)
tot, up
Figure 2. The spin up partial, in 1/(eV·atom·spin), and total densities of states for Al7Cr1N8
?????-5
S.V. Syrotyuk, V.M. Shved
-10
-5
0
5
10
kARZMX
E(k)
Figure 3. The spin down band structure of Al7Cr1N8
?????-6
Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10
-5
0
5
10
ARZMX
k
E(k)
Figure 4. The spin up band structure of Al7Cr1N8
?????-7

S.V. Syrotyuk, V.M. Shved
-10 -5 0 5
0,00,10,2 Al, s, dn
-10 -5 0 5
0,0000,0050,010 Al, p, dn
-10 -5 0 5
0,0
0,5 Mn, s, dn
-10 -5 0 5
0,000,010,02
Mn, p, dn
-10 -5 0 5
012
n(E), 1/(eV·atom·spin)
E-EF, eV
Mn, d, dn
-10 -5 0 5
0,00,51,0 N, s, dn
-10 -5 0 5
0,0
0,1 N, p, dn
-10 -5 0 5
0
10 tot, dn
Figure 5. The spin down partial, in 1/(eV·atom·spin), and total densities of states for Al7Mn1N8
?????-8

Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10 -5 0 5
0,00,20,4 Al, s, up
n(E), 1/(eV·atom·spin)
E-EF, eV
-10 -5 0 5
0,0
0,2 Al, p, up
-10 -5 0 50,0
0,1 Al, d, up
-10 -5 0 5
0,0
0,2 Mn, s, up
-10 -5 0 5
0,00,51,0
Mn, p, up
-10 -5 0 5
01020
Mn, d, up
-10 -5 0 5
0,00,2 N, s, up
-10 -5 0 5
024
N, p, up
-10 -5 0 5
0,000,02 N, d, up
-10 -5 0 5
05
10 tot, up
Figure 6. The spin up partial, in 1/(eV·atom·spin), and total densities of states for Al7Mn1N8
?????-9
S.V. Syrotyuk, V.M. Shved
-10
-5
0
5
10E(k)
ARZMX
k
Figure 7. The spin down band structure of Al7Mn1N8
?????-10
Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10
-5
0
5
10E(k)
ARZMX
k
Figure 8. The spin up band structure of Al7Mn1N8
?????-11

S.V. Syrotyuk, V.M. Shved
-10 -5 0 5
0,00,10,2
Al, s, dn-10 -5 0 5
0,000,05 Al, p, dn
-10 -5 0 5
0,00
0,05 Al, d, dn
-10 -5 0 5
0,00,20,4 Fe, s, dn
-10 -5 0 5
0,000,050,10 Fe, p, dn
-10 -5 0 5
0
5Fe, d, dn
-10 -5 0 5
0,00,51,0
N, s, dn
-10 -5 0 5
012
N, p, dn
-10 -5 0 5
0,0000,0050,010
N, d, dn
-10 -5 0 5
010
n(E), 1/(eV·atom·spin)
E-EF, eV
tot, dn
Figure 9. The spin down partial, in 1/(eV·atom·spin), and total densities of states for Al7Fe1N8
?????-12

Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10 -5 0 5
0,0
0,5 Al, s, up
n(E), 1/(eV·atom·spin)
E-EF, eV
-10 -5 0 5
0,0
0,2 Al, p, up
-10 -5 0 5
0,00,10,2
Al, d, up
-10 -5 0 5
0,00,20,4
Fe, s, up
-10 -5 0 5
0,00,5 Fe, p, up
-10 -5 0 5
0
10 Fe, d, up
-10 -5 0 5
0,00,10,2 N, s, up
-10 -5 0 5
024
N, p, up
-10 -5 0 5
0,000,01 N, d, up
-10 -5 0 5
010 tot, up
Figure 10. The spin up partial, in 1/(eV·atom·spin), and total densities of states for Al7Fe1N8
?????-13
S.V. Syrotyuk, V.M. Shved
-10
-5
0
5
10E(k)
ARZMX
k
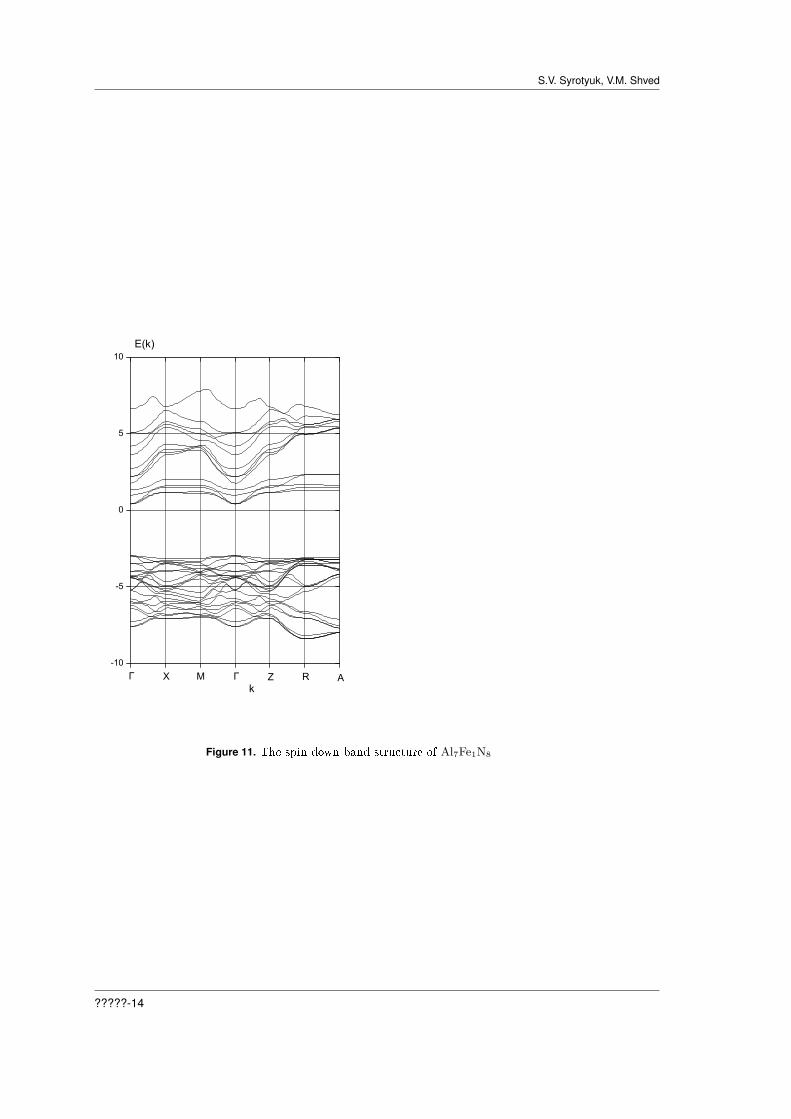
Figure 11. The spin down band structure of Al7Fe1N8
?????-14
Electronic properties of AlN crystal doped with Cr, Mn and Fe
-10
-5
0
5
10E(k)
ARZMX
k
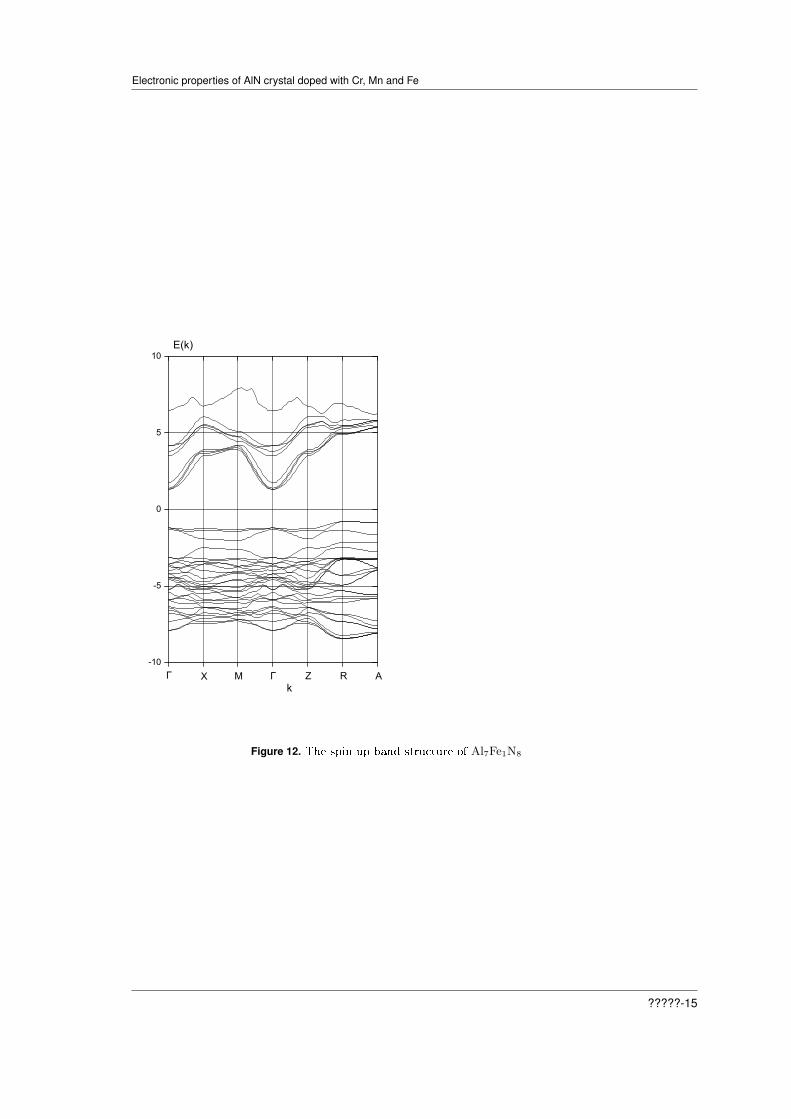
Figure 12. The spin up band structure of Al7Fe1N8
?????-15
S.V. Syrotyuk, V.M. Shved
Let us consider results of the calculation for the crystal Al7Fe1N8, represented on Figures 9-12.The calculation results for the spin down are presented in Figures 9 and 11. Figure 9 shows thatthe top of the valence band is formed by p-states of N, and the bottom of the conduction bandconsists predominantly of d-states of Fe. Figure 11 shows the direct band gap at the Γ point. TheFermi level is located near the bottom of the conduction band. The bottom of the valence band isat the point R.
The calculation results for the spin up are presented in Figures 10 and 12. From Fig.10 it isseen that the top of the valence band is formed by d-states of Fe and p-states of N. The bottomof the conduction band form the p-states of Al and s-states of N. From Fig.12 we see that thecrystal is semiconductor with indirect gaps Γ-R and Γ-A. The bottom of the valence band is atthe point R.
Table 1. The calculated electronic band gaps (for spin down, dn, for spin up, up, in eV) and spinmagnetic moments (m in µB) of crystals.
Al7Cr1N8 Al7Mn1N8 Al7Fe1N8
Ec − Ev,dn 3.36, direct 3.42, direct 3.35, directEc − Ev,up 0.66, indirect 1.51, indirect 2.13, indirectEc − EF,dn 0.27 0.38 0.39Ec − EF,up 0.09 1.51 1.30EF − Ev,dn 3.09 3.04 2.96EF − Ev,up 0.57 0.00 0.83
m, cell 1.3 2.7 3.3m, local 1.1 (Cr) 2.4 (Mn) 3.3 (Fe)
4. Conclusions
For the spin down all the crystals are the direct band gap semiconductors, with direct gap atthe point Γ. And for the spin up all the crystals are characterized by indirect band gap. All thecrystals show the ferromagnetic ordering. For AlCrN our result is conrmed by experimental dataon the revealed above room temperature ferromagnetism [19]. For AlMnN we observe a half-metallicbehavior in the sense that the Fermi level state density is nite for the majority spin and zero forthe minority spin, as it is seen from Figures 7 and 8. Fig.6 shows that at the Fermi level thereare the spin-polarized carriers. It is well matched with the results obtained in the work [20] withinthe LSDA approach. However, our calculation shows that the crystal AlCrN does not possessesthe half-metallicity predicted in the work [20]. We have calculated the electron energy spectrumand the density of electronic states in the formalism PAW GGA without accounting of the strongcorrelations of 3d-electrons. It was found that the state of half-metallicity is absent in the crystalAlCrN and existing in the crystals of AlMnN and AlFeN. Whereas for the latter crystal Figures 11and 12, obtained within the PAW PBE0 approach, show the absence of the half-metallic state. Aswe see from Table 1, values both direct and indirect gaps of crystals are very dierent. The largestcontributions to the density of states at the Fermi level provide the states of d- and p-symmetryof manganese and p-states of nitrogen. The dierence between the results obtained here with anaccount of the strong electron correlations, in the approach PBE0, and in theory LSDA, is due toinadequacy of the latter in the description of the wave functions and energy levels of 3d-electrons.
As a result, the calculation from rst principles of electronic and magnetic properties of theAlTN systems, where T is a transition element has been made taking into account the strongcorrelations 3d-electrons. The signicant narrowing of the interband gap was found for all systems.The calculated electron energy spectrum and the density of electronic states, as well as the valueof the Fermi energy are important parameters for the systems, which are considered as essentialcandidates for high-temperature electronics and optoelectronic applications [21].
?????-16

Electronic properties of AlN crystal doped with Cr, Mn and Fe
Another important factor is issue on maintenance of the ferromagnetic state at as much aspossible higher temperatures. However, it is already subject of a separate study.
References
1. Newman N., Wu S. Y., Liu H. X., Medvedeva J., Gu L., Singh R. K., Yu Z. G., Krainsky I. L.,Krishnamurthy S., Smith D. J., Freeman A. J., Schilfgaarde M. van, Phys. Stat. Sol. (a), 2006, 203,2729.
2. Tao Z.-K., Zhang R., Cui X.-G., Xiu X.-Q., Zhang G.-Y., Xie Z.-L., Gu S.-L., Shi Y., Zheng Y.-D.,Chin. Phys. Lett., 2008, 25, 1476.
3. Kaczkowski J., Jezierski A., Acta Physica Polonica A, 2009, 115, 275.4. Kaczkowski J., Jezierski A., Acta Physica Polonica A, 2009, 116, 924.5. Syrotyuk S.V., Shved V.M., Ukr. J. Phys., 2011, 56, 564.6. Akai H., Phys. Rev. Lett., 1998, 81, 3002.7. Jollet F., Jomard G., Amadon B., Phys. Rev. B, 2009, 80, 235109.8. Blochl P.E., Phys. Rev. B, 1994, 50, 17953.9. Tackett A.R., Holzwarth N.A.W., Matthews G.E., Computer Phys. Comm., 2001, 135, 348.10. Gonze X., Amadon B., Anglade P.-M., Beuken J.-M. et al., Computer Phys. Comm., 2009, 180, 258.11. Novak P., Kunes J., Chaput L., Pickett W.E., Phys. Stat. Sol. (b), 2006, 243, 563.12. Holzwarth N.A.W., Tackett A.R., Matthews G.E., Computer Phys. Comm., 2001, 135, 329.13. Holzwarth N.A.W., Matthews G.E., Dunning R. B., Tackett A.R., Zeng Y., Phys. Rev. B, 1997, 55,
2005.14. Ernzerhof M., Scuseria G.E., J. Chem. Phys., 1999, 110, 5029.15. Perdew J.P., Burke K., Ernzerhof M., Phys. Rev. Letters, 1996, 77, 3865.16. Tran E., Blaha P., Schwarz K., Novak P., Phys. Rev. B, 2006, 74, 155108.17. Monkhorst H.J., Pack J.D., Phys. Rev. B, 1976, 13, 5188.18. Zhang X.Y., Gall D., Phys. Rev. B 2010, 82, 045116.19. Song Y. Y., Quang P. H., Lim K. S., Yu S. C., J. Korean Phys. Soc., 2006, 48, 1449.20. Chen H., Zhang J.-F., Yuan H.-K., Commun. Theor. Phys., 2007, 48, 749.21. Zhao L., Lu P.-F., Yu Z.-Y., Guo X.-T., Ye H., Yuah G.-F., Shen Y., Liu Y.-M., Commun. Theor.
Phys., 2011, 55, 893.
Åëåêòðîííi âëàñòèâîñòi êðèñòàëà AlN, ëåãîâàíîãî
Cr, Mn òà Fe
Ñ.Â. Ñèðîòþê, Â.Ì. Øâåä
Íàöiîíàëüíèé óíiâåðñèòåò "Ëüâiâñüêà ïîëiòåõíiêà", âóë. Ñ. Áàíäåðè 12, 79013 ì. Ëüâiâ,Óêðà¨íà
Çàëåæíi âiä ñïiíà åëåêòðîííi åíåðãåòè÷íî¨ ñïåêòðè, à òàêîæ ïàðöiàëüíi é ïîâíi ùiëüíîñòi åëå-êòðîííèõ ñòàíiâ êðèñòàëà AlN, ëåãîâàíîãî Cr, Mn òà Fe, áóëè îòðèìàíi çà ìåòîäîì ïðîåêöiéíèõïàðöiàëüíèõ õâèëü (PAW) çà äîïîìîãîþ ïðîãðàìè ABINIT. Îáìiííèé ïîòåíöiàë Õàðòði-Ôîêàäëÿ ñêîðåëüîâàíèõ åëåêòðîíiâ âèêîðèñòîâó¹òüñÿ äëÿ îïèñó ñêîðåëüîâàíèõ îðáiòàëåé â ðàìêàõPAW. Ðîçðàõîâàíi îäíîåëåêòðîííi åíåðãi¨ äëÿ åëåêòðîíiâ çi ñïiíîì âãîðó i âíèç ¹ äóæå ðiçíi.Ìè âèÿâèëè, ùî âñi ðîçãëÿíóòi êðèñòàëè ¹ ôåðîìàãíåòèêàìè.
Êëþ÷îâi ñëîâà: ðîçðàõóíîê åëåêòðîííî¨ ñòðóêòóðè; ñèëüíî ñêîðåëüîâàíi åëåêòðîíè; òî÷íèé
îáìií äëÿ ñêîðåëüîâàíèõ åëåêòðîíiâ; ìàãíiòíi íàïiâïðîâiäíèêè; ìåòîä ïðîåêöiéíèõ
ïàðöiàëüíèõ õâèëü.
?????-17

Top Related
Copyright © 2022 FDOKUMEN

