







Bahasa
Halaman
Hukum


Synergistic Activation of HIV-1 Expression byDeacetylase Inhibitors and Prostratin: Implications forTreatment of Latent InfectionSophie Reuse1., Miriam Calao1., Kabamba Kabeya2, Allan Guiguen1, Jean-Stephane Gatot1, Vincent
Quivy1, Caroline Vanhulle1, Aurelia Lamine3, Dolores Vaira4, Dominique Demonte1, Valerie Martinelli1,
Emmanuelle Veithen1, Thomas Cherrier5, Veronique Avettand6, Solene Poutrel3, Jacques Piette7, Yvan
de Launoit8, Michel Moutschen4, Arsene Burny1, Christine Rouzioux6, Stephane De Wit2, Georges
Herbein9, Olivier Rohr5, Yves Collette10, Olivier Lambotte3, Nathan Clumeck2, Carine Van Lint1*
1 Laboratory of Molecular Virology, Institut de Biologie et de Medecine Moleculaires (IBMM), Universite Libre de Bruxelles (ULB), Gosselies, Belgium, 2 Service des Maladies
Infectieuses, CHU St-Pierre, Universite Libre de Bruxelles (ULB), Bruxelles, Belgium, 3 Faculte de Medecine Paris-Sud, INSERM U802, Bicetre, France, 4 AIDS Reference
Center, University of Liege (ULg), Liege, Belgium, 5 Virology Institute, INSERM U575, Strasbourg, France, 6 Service de Virologie, EA3620, Universite Paris-Descartes, AP-HP,
Hopital Necker-Enfants-Malades, Paris, France, 7 Laboratory of Virology and Immunology, GIGA-R, University of Liege (ULg), Liege, Belgium, 8 Institut de Biologie de Lille,
Institut Pasteur de Lille, UMR 8117 CNRS, BP447, Universite de Lille 1, Lille, France, 9 Department of Virology, EA3186, IFR133, Franche-Comte University, Hopital Saint-
Jacques, Besancon, France, 10 Centre de Recherche en Cancerologie de Marseille, INSERM UMR 599, Marseille, France
Abstract
The persistence of transcriptionally silent but replication-competent HIV-1 reservoirs in Highly Active Anti-Retroviral Therapy(HAART)-treated infected individuals, represents a major hurdle to virus eradication. Activation of HIV-1 gene expression inthese cells together with an efficient HAART has been proposed as an adjuvant therapy aimed at decreasing the pool oflatent viral reservoirs. Using the latently-infected U1 monocytic cell line and latently-infected J-Lat T-cell clones, we heredemonstrated a strong synergistic activation of HIV-1 production by clinically used histone deacetylase inhibitors (HDACIs)combined with prostratin, a non-tumor-promoting nuclear factor (NF)- kB inducer. In J-Lat cells, we showed that thissynergism was due, at least partially, to the synergistic recruitment of unresponsive cells into the expressing cell population.A combination of prostratin+HDACI synergistically activated the 59 Long Terminal Repeat (5’LTR) from HIV-1 Major groupsubtypes representing the most prevalent viral genetic forms, as shown by transient transfection reporter assays.Mechanistically, HDACIs increased prostratin-induced DNA-binding activity of nuclear NF-kB and degradation ofcytoplasmic NF-kB inhibitor, IkBa . Moreover, the combined treatment prostratin+HDACI caused a more pronouncednucleosomal remodeling in the U1 viral promoter region than the treatments with the compounds alone. This morepronounced remodeling correlated with a synergistic reactivation of HIV-1 transcription following the combined treatmentprostratin+HDACI, as demonstrated by measuring recruitment of RNA polymerase II to the 5’LTR and both initiated andelongated transcripts. The physiological relevance of the prostratin+HDACI synergism was shown in CD8+-depletedperipheral blood mononuclear cells from HAART-treated patients with undetectable viral load. Moreover, this combinedtreatment reactivated viral replication in resting CD4+ T cells isolated from similar patients. Our results suggest thatcombinations of different kinds of proviral activators may have important implications for reducing the size of latent HIV-1reservoirs in HAART-treated patients.
Citation: Reuse S, Calao M, Kabeya K, Guiguen A, Gatot J-S, et al. (2009) Synergistic Activation of HIV-1 Expression by Deacetylase Inhibitors and Prostratin:Implications for Treatment of Latent Infection. PLoS ONE 4(6): e6093. doi:10.1371/journal.pone.0006093
Editor: Peter Sommer, Institut Pasteur Korea, Republic of Korea
Received October 15, 2008; Accepted May 7, 2009; Published June 30, 2009
Copyright: � 2009 Reuse et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: We acknowledge grant support from the Belgian Fund for Scientific Research (FRS-FNRS, Belgium), the Televie-Program of the FRS-FNRS, the Action deRecherche Concertee du Ministere de la Communaute Francaise (Universite Libre de Bruxelles, ARC program no. 04/09-309), the Programme d’Excellence ‘‘Cibles’’of the Region Wallonne, the Region Wallonne (Program WALEO 021/5110), the Internationale Brachet Stiftung, the Interreg III program (Intergenes project), theAgence Nationale de Recherches sur le SIDA (ANRS), the Sidaction and the Fondation pour la Recherche Medicale (FRM, France). S.R. is fellow of the Belgian Fondspour la Recherche dans l’Industrie et l’Agriculture (FRIA). M.C. is fellow of the Belgian Fonds pour la Recherche dans l’Industrie et l’Agriculture (FRIA) and of theTelevie-Program of the FRS-FNRS. A.G. is supported by a post-doctoral fellowship from the Region Wallonne (Program WALEO2616295). J.-S.G. is supported by apostdoctoral fellowship from the ULB (Universite Libre de Bruxelles, ARC program no. 04/09-309). C.V.L. is Directeur de Recherches of the FRS- FNRS. The fundershad no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected]
. These authors contributed equally to this work.
Introduction
HIV-1 infection can be treated effectively in many patients in
the developed world, using combinations of antiretroviral
therapeutics, called Highly Active Anti-Retroviral Therapy
(HAART). However, despite prolonged treatment with HAART,
the persistence of HIV-1 reservoirs harboring transcriptionally
silent but replication-competent proviruses represents the major
PLoS ONE | www.plosone.org 1 June 2009 | Volume 4 | Issue 6 | e6093

hurdle to virus eradication. These latently-infected cells are a
permanent source for virus reactivation and lead to a rebound of
the viral load after interruption of HAART. Therefore, current
anti-HIV-1 research efforts are increasingly focused on strategies
aimed at reducing the size of these persistent reservoirs of latent
HIV-1 by forcing viral gene expression. This kind of strategy
would allow latently-infected cells to die from viral cytopathic
effects or host cytolytic effector mechanisms following viral
reactivation, while the antiretroviral therapy would prevent
spreading of the infection by the neosynthetized virus [1,2].
Acetylation level of histone and non-histone proteins, controlled
by deacetylases (HDACs) and acetyltransferases (HATs), is a key
element regulating HIV-1 transcription. In agreement, we have
previously reported that treatment of latently HIV-1-infected cell
lines with HDAC inhibitors (HDACIs) induces viral transcription
and remodeling of the repressive nucleosome nuc-1, located
immediately after the HIV-1 transcription start site under latency
conditions [3,4]. Similar results were observed in transiently or
stably transfected HIV-1 Long Terminal Repeat (LTR) reporter
constructs [5,6,7], and on in vitro chromatin-reconstituted HIV-1
templates [8,9]. Based on these observations, administration of
HDACIs together with efficient HAART has been proposed as an
inductive adjuvant therapy for the decay of latent reservoirs
[10,11,12,13]. The Margolis group has reported that VPA
(valproic acid), in the presence of IL-2, induces rescue of
replication-competent HIV-1 from purified resting CD4+ T cells
obtained from HAART-treated patients with undetectable viral
load [14]. Later, in a clinical trial performed by same group, four
patients receiving HAART and the viral entry inhibitor enfuvirtide
were given VPA for three months, and a modest but significant
decrease in the frequency of latently-infected cells was noted in
three of the four patients [15]. However, given that at least two
studies have demonstrated that intensification of anti-HIV therapy
decreases the half-life of this population [16,17], it is unclear
whether VPA or intensification of HAART with enfuvirtide was
the critical factor for the decay of the latent reservoir. Recent
reports have failed to show a decay of resting CD4+ T cell
infection in patients who were prescribed VPA for clinical reasons
while receiving standard HAART [18,19,20]. These results led to
question the therapeutic potential of VPA, at least when used
alone, to reduce the size of the latent HIV-1 reservoirs.
We have previously demonstrated a strong synergistic activation
of HIV-1 promoter activity by the HDACI trichostatin A (TSA)
and the NF-kB inducer TNFa in the postintegration latency
model cell line U1 [3,21], suggesting that combinations of two
independent factors (NF-kB and chromatin) involved in HIV-1
reactivation from latency might be potent tools to decrease the
pool of latently-infected reservoirs. However, because of their
toxicity, the therapeutic use of TNFa and TSA is not possible.
Here, we investigated the HIV-1 reactivating potential of a
treatment combining HDACIs used in human clinical trials or
therapies (such as VPA and suberoylanilide hydroxamic acid
[SAHA]) and an NF-kB inducer with no tumor-promoting effects,
prostratin. This compound is an inducer of protein kinase C
activity which stimulates HIV-1 expression in latently-infected
lymphoid and myeloid cell lines and primary cells
[22,23,24,25,26,27,28,29] with minimal effects on the immune
system [26] and causing minimal cell cycle progression [26,29].
Moreover, prostratin also inhibits de novo HIV-1 infection via
posttranscriptional downregulation of the cellular HIV-1 receptors
CD4 and CXCR4 (CXC chemokine receptor 4) [24,27,30,31,32].
Therefore, the antimitogenic property of prostratin coupled with
its dual activity on HIV-1 infection (inhibition of viral infection
and upregulation of latent provirus expression), its relatively non-
toxic behavior and its potential widespread effect on different
HIV-1 reservoirs, make this compound a good candidate for viral
purging. Although the suitability of prostratin for use in humans is
unknown since preclinical testing in compliance with Food and
Drug Administration regulations is reportedly still underway
[12,33], preliminary pharmacokinetic studies are encouraging as
they show that 5 of 5 mice survived with no obvious effects at
100 mmol/Kg intragastric dose of prostratin with plasma concen-
trations reaching up to 1.42 mmol/L [24]. These results are not
surprising as plant extracts of Homalanthus nutans (a source of
prostratin) have already been used by the Samoan healers to treat
individuals with certain medical conditions such as hepatitis
[34,35].
Here, we demonstrated that a combination of prostratin+VPA
or prostratin+SAHA reactivated more efficiently than each
compound alone HIV-1 production both in several latently-
infected cell lines (U1 and J-Lat clones) and in CD8+-depleted
peripheral blood mononuclear cells (PBMCs) isolated from HIV-
1-infected patients receiving HAART and with undetectable viral
load. Mechanistically, HDACI increased prostratin-induced NF-
kB activation and potentiated nuc-1 remodeling. Moreover,
prostratin+HDACI combined treatment caused a synergistic
activation of HIV-1 transcriptional initiation and elongation.
Our results constitute a proof-of-concept for the coadministration
of two different types of therapeutically promising HIV-1 inducers
(one acting on the NF-kB pathway and the other acting on the
protein acetylation status) together with HAART as a therapeutic
perspective to decrease the pool of latent HIV-1 reservoirs.
Results
Synergistic activation of HIV-1 production by prostratinand clinically used HDACIs
HDACIs present several advantages for HIV-1 purging
strategies. They do not induce proliferation or activation of T
cells [36,37], act in a broad spectrum of cell lines and potently
repress CXCR4 chemokine receptor expression and function [38].
Moreover, whereas TSA has a high toxicity and a costly and
poorly efficient production [39], several HDACIs are safely
administered for other diseases [40,41,42,43,44,45] or tested in
clinical trials as anticancer drugs [46]. HDACIs are specific for
class I and II or class III HDACs and have been subdivided into
four distinct groups based on their structural characteristics:
hydroxamic acids; short-chain fatty acids; synthetic benzamide
derivatives; cyclic tetrapeptides/epoxides [39,46,47].
For various HDACIs, we determined an optimal concentration
in terms of both their cellular toxicity and their HIV-1 reactivation
potential by measuring cell viability (Figure S1 and Text S1) and
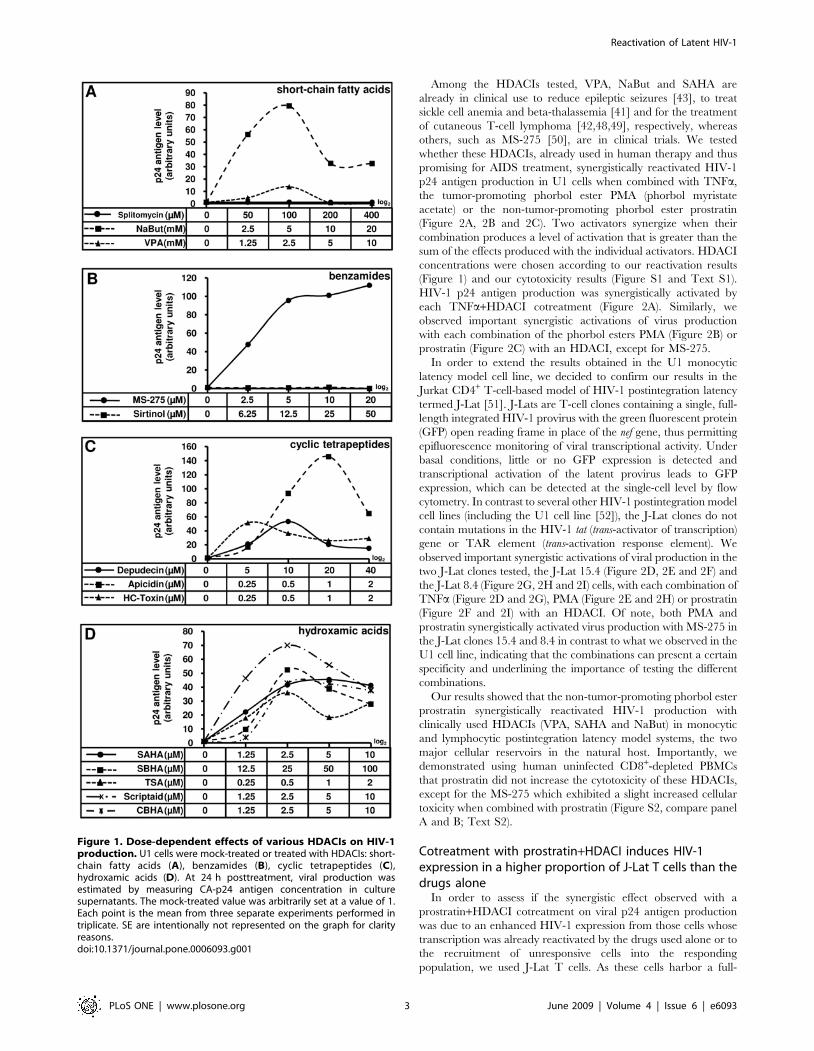
induction of p24 production in HDACI-treated U1 cells (Figure 1).
The latently HIV-1-infected monocytic cell line U1 is one of the
most studied cellular models of postintegration latency. All the
inhibitors of class I and II HDACs we tested increased core
antigen levels (p24) in the U1 cellular supernatants, independently
of the structural group the HDACIs belong to (Figure 1A, 1B, 1C
and 1D). In the short-chain fatty acid group, VPA was a weaker
inducer of HIV-1 p24 production than NaBut (sodium butyrate)
(Figure 1A). MS-275 induced viral p24 antigen production 112-
fold at a concentration of 20 mM (Figure 1B). Among the cyclic
tetrapeptide group, Apicidin at 1 mM exhibited the highest
reactivation of viral production (Figure 1C). All HDACIs from
the hydroxamic acid group were more efficient than TSA to
induce HIV-1 p24 antigen production (Figure 1D). Splitomycin
and Sirtinol, which are specific inhibitors of class III HDACs, had
no effect on p24 production (Figure 1A and 1B, respectively).
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 2 June 2009 | Volume 4 | Issue 6 | e6093
Among the HDACIs tested, VPA, NaBut and SAHA are
already in clinical use to reduce epileptic seizures [43], to treat
sickle cell anemia and beta-thalassemia [41] and for the treatment
of cutaneous T-cell lymphoma [42,48,49], respectively, whereas
others, such as MS-275 [50], are in clinical trials. We tested
whether these HDACIs, already used in human therapy and thus
promising for AIDS treatment, synergistically reactivated HIV-1
p24 antigen production in U1 cells when combined with TNFa,
the tumor-promoting phorbol ester PMA (phorbol myristate
acetate) or the non-tumor-promoting phorbol ester prostratin
(Figure 2A, 2B and 2C). Two activators synergize when their
combination produces a level of activation that is greater than the
sum of the effects produced with the individual activators. HDACI
concentrations were chosen according to our reactivation results
(Figure 1) and our cytotoxicity results (Figure S1 and Text S1).
HIV-1 p24 antigen production was synergistically activated by
each TNFa+HDACI cotreatment (Figure 2A). Similarly, we
observed important synergistic activations of virus production
with each combination of the phorbol esters PMA (Figure 2B) or
prostratin (Figure 2C) with an HDACI, except for MS-275.
In order to extend the results obtained in the U1 monocytic
latency model cell line, we decided to confirm our results in the
Jurkat CD4+ T-cell-based model of HIV-1 postintegration latency
termed J-Lat [51]. J-Lats are T-cell clones containing a single, full-
length integrated HIV-1 provirus with the green fluorescent protein
(GFP) open reading frame in place of the nef gene, thus permitting
epifluorescence monitoring of viral transcriptional activity. Under
basal conditions, little or no GFP expression is detected and
transcriptional activation of the latent provirus leads to GFP
expression, which can be detected at the single-cell level by flow
cytometry. In contrast to several other HIV-1 postintegration model
cell lines (including the U1 cell line [52]), the J-Lat clones do not
contain mutations in the HIV-1 tat (trans-activator of transcription)
gene or TAR element (trans-activation response element). We
observed important synergistic activations of viral production in the
two J-Lat clones tested, the J-Lat 15.4 (Figure 2D, 2E and 2F) and
the J-Lat 8.4 (Figure 2G, 2H and 2I) cells, with each combination of
TNFa (Figure 2D and 2G), PMA (Figure 2E and 2H) or prostratin
(Figure 2F and 2I) with an HDACI. Of note, both PMA and
prostratin synergistically activated virus production with MS-275 in
the J-Lat clones 15.4 and 8.4 in contrast to what we observed in the
U1 cell line, indicating that the combinations can present a certain
specificity and underlining the importance of testing the different
combinations.
Our results showed that the non-tumor-promoting phorbol ester
prostratin synergistically reactivated HIV-1 production with
clinically used HDACIs (VPA, SAHA and NaBut) in monocytic
and lymphocytic postintegration latency model systems, the two
major cellular reservoirs in the natural host. Importantly, we
demonstrated using human uninfected CD8+-depleted PBMCs
that prostratin did not increase the cytotoxicity of these HDACIs,
except for the MS-275 which exhibited a slight increased cellular
toxicity when combined with prostratin (Figure S2, compare panel
A and B; Text S2).
Cotreatment with prostratin+HDACI induces HIV-1expression in a higher proportion of J-Lat T cells than thedrugs alone
In order to assess if the synergistic effect observed with a
prostratin+HDACI cotreatment on viral p24 antigen production
was due to an enhanced HIV-1 expression from those cells whose
transcription was already reactivated by the drugs used alone or to
the recruitment of unresponsive cells into the responding
population, we used J-Lat T cells. As these cells harbor a full-
Figure 1. Dose-dependent effects of various HDACIs on HIV-1production. U1 cells were mock-treated or treated with HDACIs: short-chain fatty acids (A), benzamides (B), cyclic tetrapeptides (C),hydroxamic acids (D). At 24 h posttreatment, viral production wasestimated by measuring CA-p24 antigen concentration in culturesupernatants. The mock-treated value was arbitrarily set at a value of 1.Each point is the mean from three separate experiments performed intriplicate. SE are intentionally not represented on the graph for clarityreasons.doi:10.1371/journal.pone.0006093.g001
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 3 June 2009 | Volume 4 | Issue 6 | e6093
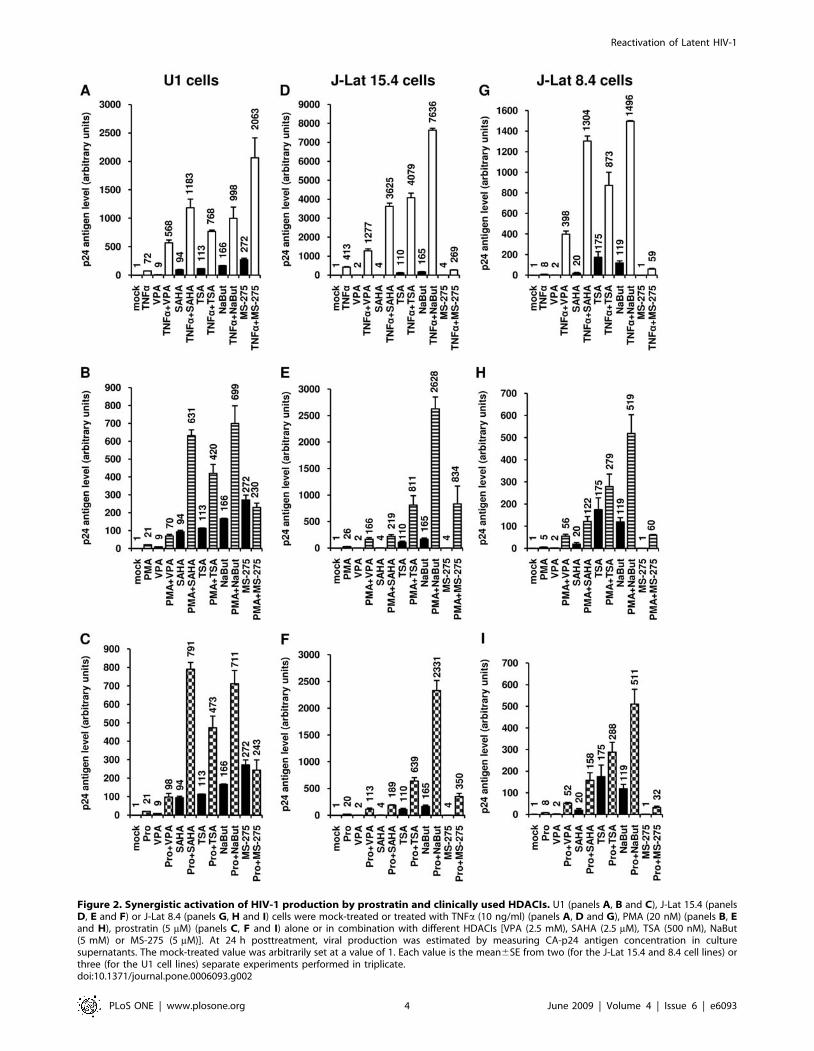
Figure 2. Synergistic activation of HIV-1 production by prostratin and clinically used HDACIs. U1 (panels A, B and C), J-Lat 15.4 (panelsD, E and F) or J-Lat 8.4 (panels G, H and I) cells were mock-treated or treated with TNFa (10 ng/ml) (panels A, D and G), PMA (20 nM) (panels B, Eand H), prostratin (5 mM) (panels C, F and I) alone or in combination with different HDACIs [VPA (2.5 mM), SAHA (2.5 mM), TSA (500 nM), NaBut(5 mM) or MS-275 (5 mM)]. At 24 h posttreatment, viral production was estimated by measuring CA-p24 antigen concentration in culturesupernatants. The mock-treated value was arbitrarily set at a value of 1. Each value is the mean6SE from two (for the J-Lat 15.4 and 8.4 cell lines) orthree (for the U1 cell lines) separate experiments performed in triplicate.doi:10.1371/journal.pone.0006093.g002
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 4 June 2009 | Volume 4 | Issue 6 | e6093
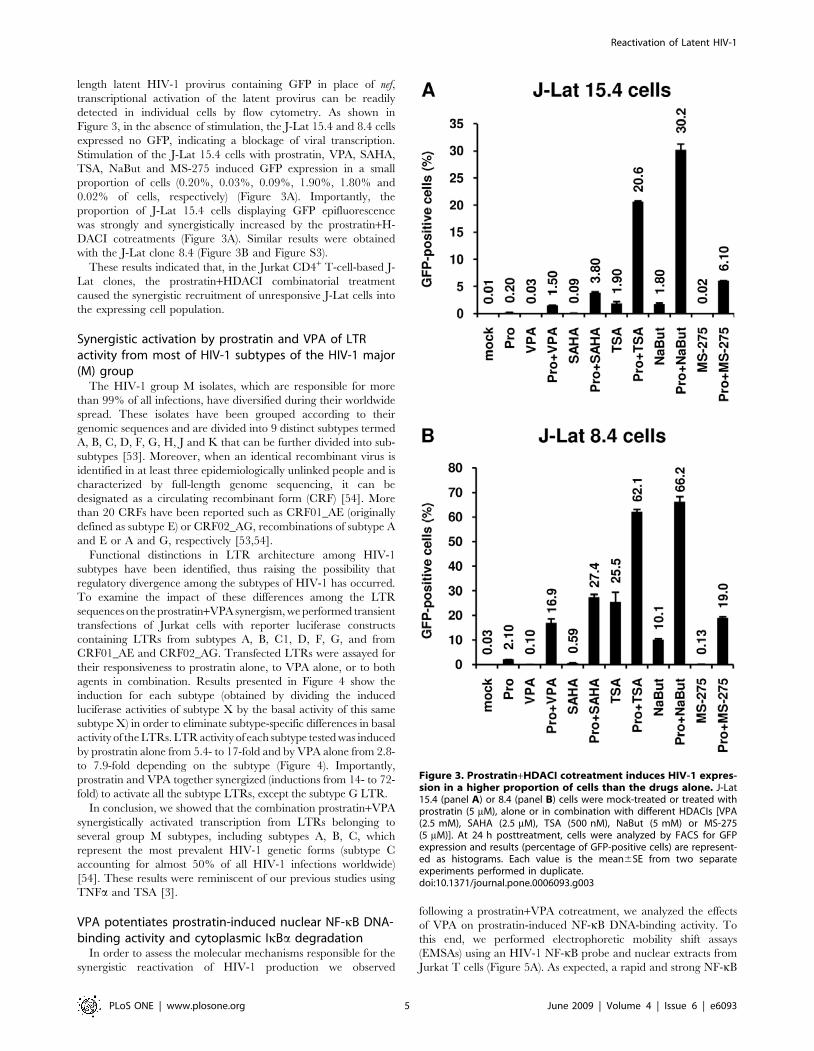
length latent HIV-1 provirus containing GFP in place of nef,
transcriptional activation of the latent provirus can be readily
detected in individual cells by flow cytometry. As shown in
Figure 3, in the absence of stimulation, the J-Lat 15.4 and 8.4 cells
expressed no GFP, indicating a blockage of viral transcription.
Stimulation of the J-Lat 15.4 cells with prostratin, VPA, SAHA,
TSA, NaBut and MS-275 induced GFP expression in a small
proportion of cells (0.20%, 0.03%, 0.09%, 1.90%, 1.80% and
0.02% of cells, respectively) (Figure 3A). Importantly, the
proportion of J-Lat 15.4 cells displaying GFP epifluorescence
was strongly and synergistically increased by the prostratin+H-
DACI cotreatments (Figure 3A). Similar results were obtained
with the J-Lat clone 8.4 (Figure 3B and Figure S3).
These results indicated that, in the Jurkat CD4+ T-cell-based J-
Lat clones, the prostratin+HDACI combinatorial treatment
caused the synergistic recruitment of unresponsive J-Lat cells into
the expressing cell population.
Synergistic activation by prostratin and VPA of LTRactivity from most of HIV-1 subtypes of the HIV-1 major(M) group
The HIV-1 group M isolates, which are responsible for more
than 99% of all infections, have diversified during their worldwide
spread. These isolates have been grouped according to their
genomic sequences and are divided into 9 distinct subtypes termed
A, B, C, D, F, G, H, J and K that can be further divided into sub-
subtypes [53]. Moreover, when an identical recombinant virus is
identified in at least three epidemiologically unlinked people and is
characterized by full-length genome sequencing, it can be
designated as a circulating recombinant form (CRF) [54]. More
than 20 CRFs have been reported such as CRF01_AE (originally
defined as subtype E) or CRF02_AG, recombinations of subtype A
and E or A and G, respectively [53,54].
Functional distinctions in LTR architecture among HIV-1
subtypes have been identified, thus raising the possibility that
regulatory divergence among the subtypes of HIV-1 has occurred.
To examine the impact of these differences among the LTR
sequences on the prostratin+VPA synergism, we performed transient
transfections of Jurkat cells with reporter luciferase constructs
containing LTRs from subtypes A, B, C1, D, F, G, and from
CRF01_AE and CRF02_AG. Transfected LTRs were assayed for
their responsiveness to prostratin alone, to VPA alone, or to both
agents in combination. Results presented in Figure 4 show the
induction for each subtype (obtained by dividing the induced
luciferase activities of subtype X by the basal activity of this same
subtype X) in order to eliminate subtype-specific differences in basal
activity of the LTRs. LTR activity of each subtype tested was induced
by prostratin alone from 5.4- to 17-fold and by VPA alone from 2.8-
to 7.9-fold depending on the subtype (Figure 4). Importantly,
prostratin and VPA together synergized (inductions from 14- to 72-
fold) to activate all the subtype LTRs, except the subtype G LTR.
In conclusion, we showed that the combination prostratin+VPA
synergistically activated transcription from LTRs belonging to
several group M subtypes, including subtypes A, B, C, which
represent the most prevalent HIV-1 genetic forms (subtype C
accounting for almost 50% of all HIV-1 infections worldwide)
[54]. These results were reminiscent of our previous studies using
TNFa and TSA [3].
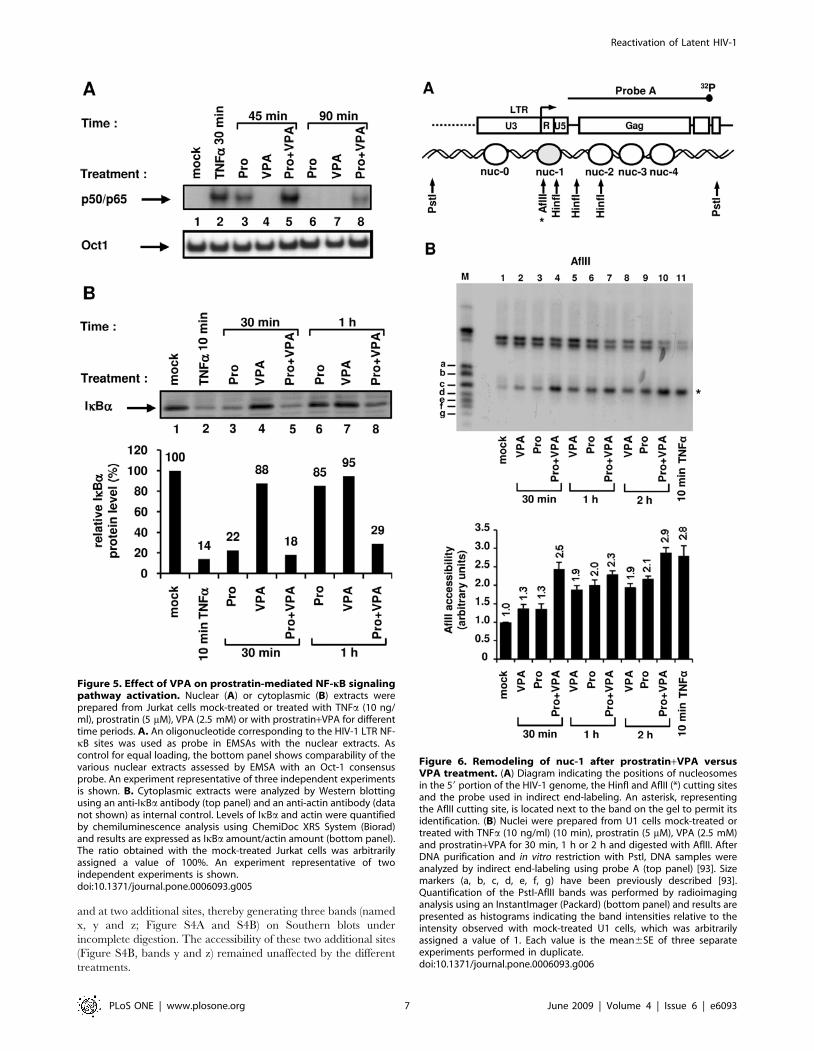
VPA potentiates prostratin-induced nuclear NF-kB DNA-binding activity and cytoplasmic IkBa degradation
In order to assess the molecular mechanisms responsible for the
synergistic reactivation of HIV-1 production we observed
following a prostratin+VPA cotreatment, we analyzed the effects
of VPA on prostratin-induced NF-kB DNA-binding activity. To
this end, we performed electrophoretic mobility shift assays
(EMSAs) using an HIV-1 NF-kB probe and nuclear extracts from
Jurkat T cells (Figure 5A). As expected, a rapid and strong NF-kB
Figure 3. Prostratin+HDACI cotreatment induces HIV-1 expres-sion in a higher proportion of cells than the drugs alone. J-Lat15.4 (panel A) or 8.4 (panel B) cells were mock-treated or treated withprostratin (5 mM), alone or in combination with different HDACIs [VPA(2.5 mM), SAHA (2.5 mM), TSA (500 nM), NaBut (5 mM) or MS-275(5 mM)]. At 24 h posttreatment, cells were analyzed by FACS for GFPexpression and results (percentage of GFP-positive cells) are represent-ed as histograms. Each value is the mean6SE from two separateexperiments performed in duplicate.doi:10.1371/journal.pone.0006093.g003
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 5 June 2009 | Volume 4 | Issue 6 | e6093
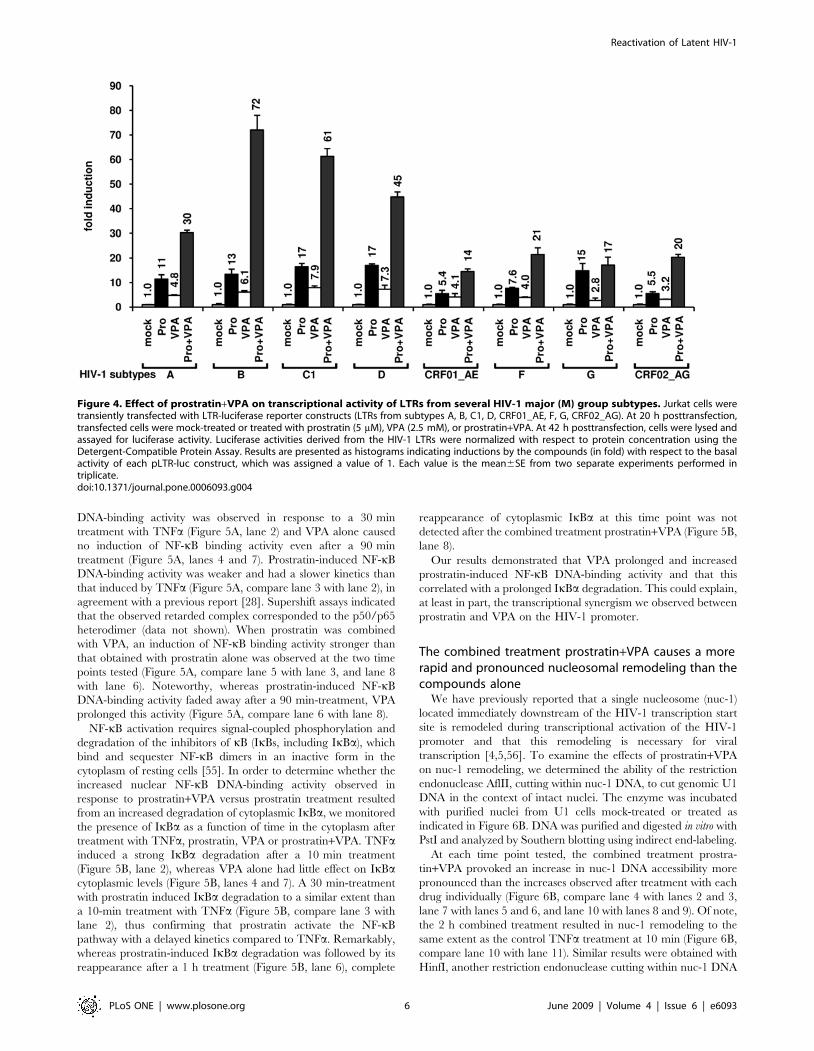
DNA-binding activity was observed in response to a 30 min
treatment with TNFa (Figure 5A, lane 2) and VPA alone caused
no induction of NF-kB binding activity even after a 90 min
treatment (Figure 5A, lanes 4 and 7). Prostratin-induced NF-kB
DNA-binding activity was weaker and had a slower kinetics than
that induced by TNFa (Figure 5A, compare lane 3 with lane 2), in
agreement with a previous report [28]. Supershift assays indicated
that the observed retarded complex corresponded to the p50/p65
heterodimer (data not shown). When prostratin was combined
with VPA, an induction of NF-kB binding activity stronger than
that obtained with prostratin alone was observed at the two time
points tested (Figure 5A, compare lane 5 with lane 3, and lane 8
with lane 6). Noteworthy, whereas prostratin-induced NF-kB
DNA-binding activity faded away after a 90 min-treatment, VPA
prolonged this activity (Figure 5A, compare lane 6 with lane 8).
NF-kB activation requires signal-coupled phosphorylation and
degradation of the inhibitors of kB (IkBs, including IkBa), which
bind and sequester NF-kB dimers in an inactive form in the
cytoplasm of resting cells [55]. In order to determine whether the
increased nuclear NF-kB DNA-binding activity observed in
response to prostratin+VPA versus prostratin treatment resulted
from an increased degradation of cytoplasmic IkBa, we monitored
the presence of IkBa as a function of time in the cytoplasm after
treatment with TNFa, prostratin, VPA or prostratin+VPA. TNFainduced a strong IkBa degradation after a 10 min treatment
(Figure 5B, lane 2), whereas VPA alone had little effect on IkBacytoplasmic levels (Figure 5B, lanes 4 and 7). A 30 min-treatment
with prostratin induced IkBa degradation to a similar extent than
a 10-min treatment with TNFa (Figure 5B, compare lane 3 with
lane 2), thus confirming that prostratin activate the NF-kB
pathway with a delayed kinetics compared to TNFa. Remarkably,
whereas prostratin-induced IkBa degradation was followed by its
reappearance after a 1 h treatment (Figure 5B, lane 6), complete
reappearance of cytoplasmic IkBa at this time point was not
detected after the combined treatment prostratin+VPA (Figure 5B,
lane 8).
Our results demonstrated that VPA prolonged and increased
prostratin-induced NF-kB DNA-binding activity and that this
correlated with a prolonged IkBa degradation. This could explain,
at least in part, the transcriptional synergism we observed between
prostratin and VPA on the HIV-1 promoter.
The combined treatment prostratin+VPA causes a morerapid and pronounced nucleosomal remodeling than thecompounds alone
We have previously reported that a single nucleosome (nuc-1)
located immediately downstream of the HIV-1 transcription start
site is remodeled during transcriptional activation of the HIV-1
promoter and that this remodeling is necessary for viral
transcription [4,5,56]. To examine the effects of prostratin+VPA
on nuc-1 remodeling, we determined the ability of the restriction
endonuclease AflII, cutting within nuc-1 DNA, to cut genomic U1
DNA in the context of intact nuclei. The enzyme was incubated
with purified nuclei from U1 cells mock-treated or treated as
indicated in Figure 6B. DNA was purified and digested in vitro with
PstI and analyzed by Southern blotting using indirect end-labeling.
At each time point tested, the combined treatment prostra-
tin+VPA provoked an increase in nuc-1 DNA accessibility more
pronounced than the increases observed after treatment with each
drug individually (Figure 6B, compare lane 4 with lanes 2 and 3,
lane 7 with lanes 5 and 6, and lane 10 with lanes 8 and 9). Of note,
the 2 h combined treatment resulted in nuc-1 remodeling to the
same extent as the control TNFa treatment at 10 min (Figure 6B,
compare lane 10 with lane 11). Similar results were obtained with
HinfI, another restriction endonuclease cutting within nuc-1 DNA
Figure 4. Effect of prostratin+VPA on transcriptional activity of LTRs from several HIV-1 major (M) group subtypes. Jurkat cells weretransiently transfected with LTR-luciferase reporter constructs (LTRs from subtypes A, B, C1, D, CRF01_AE, F, G, CRF02_AG). At 20 h posttransfection,transfected cells were mock-treated or treated with prostratin (5 mM), VPA (2.5 mM), or prostratin+VPA. At 42 h posttransfection, cells were lysed andassayed for luciferase activity. Luciferase activities derived from the HIV-1 LTRs were normalized with respect to protein concentration using theDetergent-Compatible Protein Assay. Results are presented as histograms indicating inductions by the compounds (in fold) with respect to the basalactivity of each pLTR-luc construct, which was assigned a value of 1. Each value is the mean6SE from two separate experiments performed intriplicate.doi:10.1371/journal.pone.0006093.g004
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 6 June 2009 | Volume 4 | Issue 6 | e6093
and at two additional sites, thereby generating three bands (named
x, y and z; Figure S4A and S4B) on Southern blots under
incomplete digestion. The accessibility of these two additional sites
(Figure S4B, bands y and z) remained unaffected by the different
treatments.
Figure 5. Effect of VPA on prostratin-mediated NF-kB signalingpathway activation. Nuclear (A) or cytoplasmic (B) extracts wereprepared from Jurkat cells mock-treated or treated with TNFa (10 ng/ml), prostratin (5 mM), VPA (2.5 mM) or with prostratin+VPA for differenttime periods. A. An oligonucleotide corresponding to the HIV-1 LTR NF-kB sites was used as probe in EMSAs with the nuclear extracts. Ascontrol for equal loading, the bottom panel shows comparability of thevarious nuclear extracts assessed by EMSA with an Oct-1 consensusprobe. An experiment representative of three independent experimentsis shown. B. Cytoplasmic extracts were analyzed by Western blottingusing an anti-IkBa antibody (top panel) and an anti-actin antibody (datanot shown) as internal control. Levels of IkBa and actin were quantifiedby chemiluminescence analysis using ChemiDoc XRS System (Biorad)and results are expressed as IkBa amount/actin amount (bottom panel).The ratio obtained with the mock-treated Jurkat cells was arbitrarilyassigned a value of 100%. An experiment representative of twoindependent experiments is shown.doi:10.1371/journal.pone.0006093.g005
Figure 6. Remodeling of nuc-1 after prostratin+VPA versusVPA treatment. (A) Diagram indicating the positions of nucleosomesin the 59 portion of the HIV-1 genome, the HinfI and AflII (*) cutting sitesand the probe used in indirect end-labeling. An asterisk, representingthe AflII cutting site, is located next to the band on the gel to permit itsidentification. (B) Nuclei were prepared from U1 cells mock-treated ortreated with TNFa (10 ng/ml) (10 min), prostratin (5 mM), VPA (2.5 mM)and prostratin+VPA for 30 min, 1 h or 2 h and digested with AflII. AfterDNA purification and in vitro restriction with PstI, DNA samples wereanalyzed by indirect end-labeling using probe A (top panel) [93]. Sizemarkers (a, b, c, d, e, f, g) have been previously described [93].Quantification of the PstI-AflII bands was performed by radioimaginganalysis using an InstantImager (Packard) (bottom panel) and results arepresented as histograms indicating the band intensities relative to theintensity observed with mock-treated U1 cells, which was arbitrarilyassigned a value of 1. Each value is the mean6SE of three separateexperiments performed in duplicate.doi:10.1371/journal.pone.0006093.g006
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 7 June 2009 | Volume 4 | Issue 6 | e6093

In order to further examine, at the chromatin level, the
molecular mechanisms involved in the prostratin+VPA synergism,
we analyzed levels of histone H4 acetylation (an activation mark)
in the nuc-1 region. To this end, we performed chromatin
immunoprecipitation (ChIP) assays using U1 cells mock-treated or
treated for different time periods with prostratin and VPA alone or
in combination (Figure S5 and Text S3). VPA induced nuc-1
histone H4 acetylation, a result in agreement with previous reports
[14,57]. Prostratin slightly and gradually induced histone H4
acetylation starting at the 20 min treatment up to the 2 h
treatment. This is in agreement with a previous study demon-
strating that during HIV-1 transcriptional activation by the
phorbol ester PMA, histone acetyltransferases are recruited to
the viral promoter via NF-kB [58]. Importantly, our results
indicated that, at each time point tested, the combined treatment
prostratin+VPA did not induce a level of acetylated histone H4
higher than the level observed after treatments with VPA alone
(Figure S5), suggesting that the more pronounced nuc-1
remodeling we observed by indirect end-labeling following
combined versus individual treatment was not due to a cooperative
induction of histone H4 acetylation.
These results demonstrated that the combined treatment
prostratin+VPA resulted in a more rapid and pronounced nuc-1
remodeling than the treatments with the compounds alone.
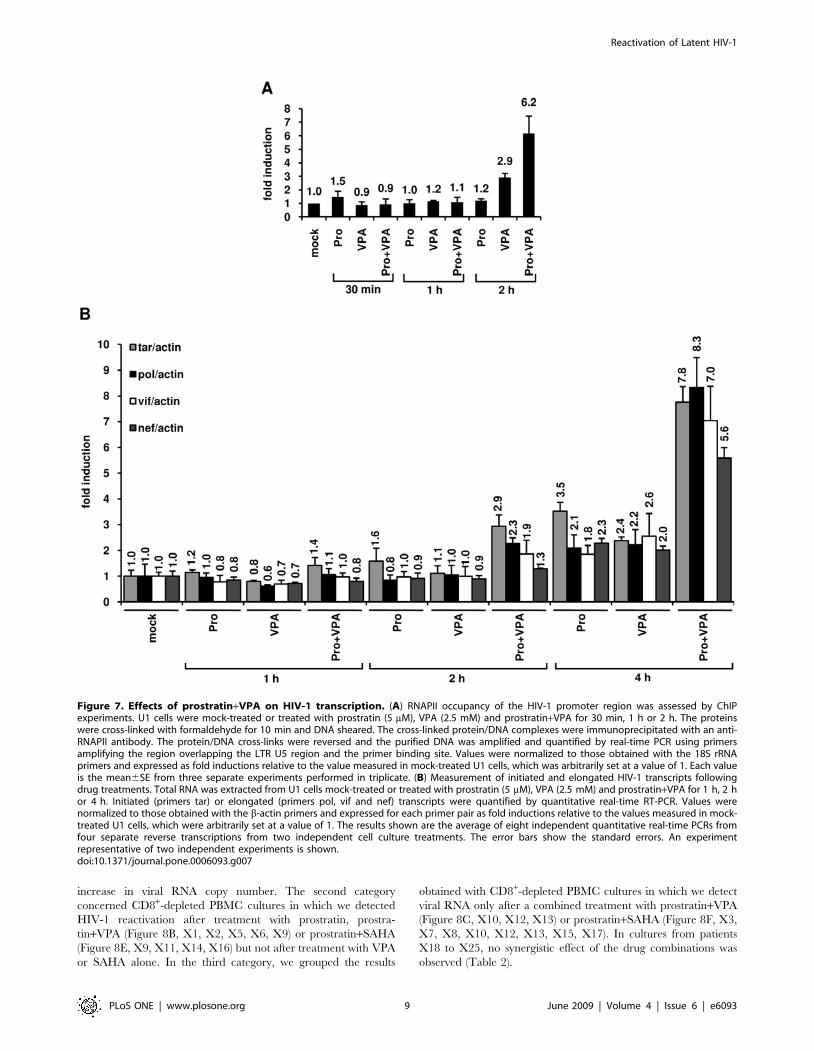
The combined treatment prostratin+VPA synergisticallyinduces transcription from the viral LTR
In order to test the effect of prostratin+VPA treatment on
transcriptional activation of the HIV-1 promoter, we first
monitored the RNA polymerase II (RNAPII) abundance in the
5’LTR region. To this end, we performed ChIP assays in U1 cells
after treatment with prostratin alone, VPA alone or prostra-
tin+VPA (Figure 7A). We used a primer pair overlapping the 39
extremity of the LTR U5 region and the 59 extremity of the leader
region allowing to specifically determine RNAPII occupancy at
the 5’LTR. After a 2 h combined treatment, we observed a
cooperative effect of prostratin and VPA on RNAPII recruitment
to the HIV-1 promoter and thus likely on transcription (Figure 7A).
To explore the functional relevance of this cooperative
recruitment, we quantified initiated and elongated HIV-1
transcripts in U1 cells mock-treated or treated with prostratin,
VPA or combination of both for different time periods (1 h, 2 h,
4 h). Initiated versus elongated HIV-1 transcripts were measured
by quantitative real-time RT-PCR with primer sets targeting four
different proviral regions: 1) TAR in the 59 60 bp; 2) pol gene
located roughly 2.9 kb downstream of the 5’LTR; 3) vif gene
located roughly 4.4 kb downstream of the 5’LTR; 4) nef gene,
located roughly 8.3 kb downstream of the HIV-1 LTR. Theses
primer sets allow analysis of transcriptional initiation and
elongation at progressive positions throughout the HIV-1 provirus.
As shown in Figure 7B, after a 4 h treatment with prostratin and
VPA alone, we observed increased relative amounts of both
initiated and elongated transcripts. Importantly, at the same time
point, the prostratin+VPA combined treatment caused a syner-
gistic accumulation of initiated and elongated transcripts
(Figure 7B). Indeed, the prostratin+VPA cotreatment induced a
7.8-fold increase in initiated transcripts and 8.3-fold (pol probe),
7.0-fold (vif probe) and 5.6-fold (nef probe) increases in elongated
transcripts compared to the mock-treated cells.
Altogether, our results measuring the RNAPII recruitment to
the HIV-1 promoter and the amount of transcripts demonstrated
that the prostratin+VPA synergism took place, at least in part, at
the transcriptional initiation and elongation levels and demon-
strated a temporal correlation between the degree of nuc-1
remodeling and the level of HIV-1 transcription.
Prostratin and HDACIs induce HIV-1 recovery in CD8+-depleted PBMCs and in HLA DR2 CD4+ T cells from HIV-1positive individuals with undetectable viral load
Our results suggested that a combination of prostratin+HDACI
(such as VPA, SAHA or NaBut) could have a higher potential than
the compounds alone in reactivating HIV-1 expression from cells
isolated from HIV-1-infected individuals with undetectable viral
load (,50 copies HIV-1 RNA/ml of plasma). To evaluate this
hypothesis, we purified PBMCs from blood of 52 selected patients
(Materials and Methods) and depleted them for CD8+ T cells,
CD8+ T cells having an antiviral activity [59]. Importantly, these
purified cells were cultured both in the absence of IL-2 and in the
absence of allogenic stimulation (i.e. coculture with PBMCs from
an uninfected individual) to avoid extensive nonspecific T-cell
activation and proliferation, and thus resulting in no amplification
of the genomic viral RNA level. After a one-day culture, cells were
mock-treated or treated with anti-CD2+anti-CD28 antibodies (as
a positive control), prostratin, HDACIs (VPA or SAHA) or with
combinations of prostratin+HDACIs. Six days after treatment, we
measured HIV-1 genomic RNA concentrations in culture
supernatants.
For 10 out of 52 CD8+-depleted PBMC cultures, we detected
viral RNA in the supernatant even in the absence of any
treatment. Therefore, these data were removed from our study.
Characteristics of the 42 remaining patients (named X1 to X42)
are presented in Table 1. This table also indicates, for each PBMC
culture, the combination of prostratin+HDACI tested (prostra-
tin+VPA and/or prostratin+SAHA) and the levels of rescue
obtained after anti-CD2+anti-CD28 treatment. These levels of
rescue showed that the 17 CD8+-depleted PBMC cultures,
presenting no viral outgrowth following treatment with prostratin
and HDACI(s) individually or in combination, exhibited no or
very weak levels of genomic viral RNA after anti-CD2+anti-CD28
costimulation compared to the levels observed with the PBMC
cultures presenting viral reactivation after drug treatments
(Table 1, compare patients X1 to X25 with X26 to X42). For
25 of the 42 PBMC cultures, we detected HIV-1 reactivation after
prostratin, VPA, SAHA and/or prostratin+HDACI exposure
(Tables 1 and 2, patients X1 to X25). We did not observe any
significant correlation between age, CD4 counts or length of
undetectable viremia, and the potency of our compounds to
reactivate HIV-1 from latency. In 17 of these 25 cultures,
prostratin alone reactivated viral expression, thereby confirming
its great potential as an HIV-1 replication inducer (Table 2, X1,
X2, X4 to X6, X9, X11, X14, X16, X18 to X25). VPA and
SAHA alone reactivated viral expression in only 3 cell cultures,
(Table 2, X4, X11, X14 and X4, X23, X24, respectively). In 8
cultures (Table 2, X3, X7, X8, X10, X12, X13, X15, X17), no
drug used alone could reactivate viral expression.
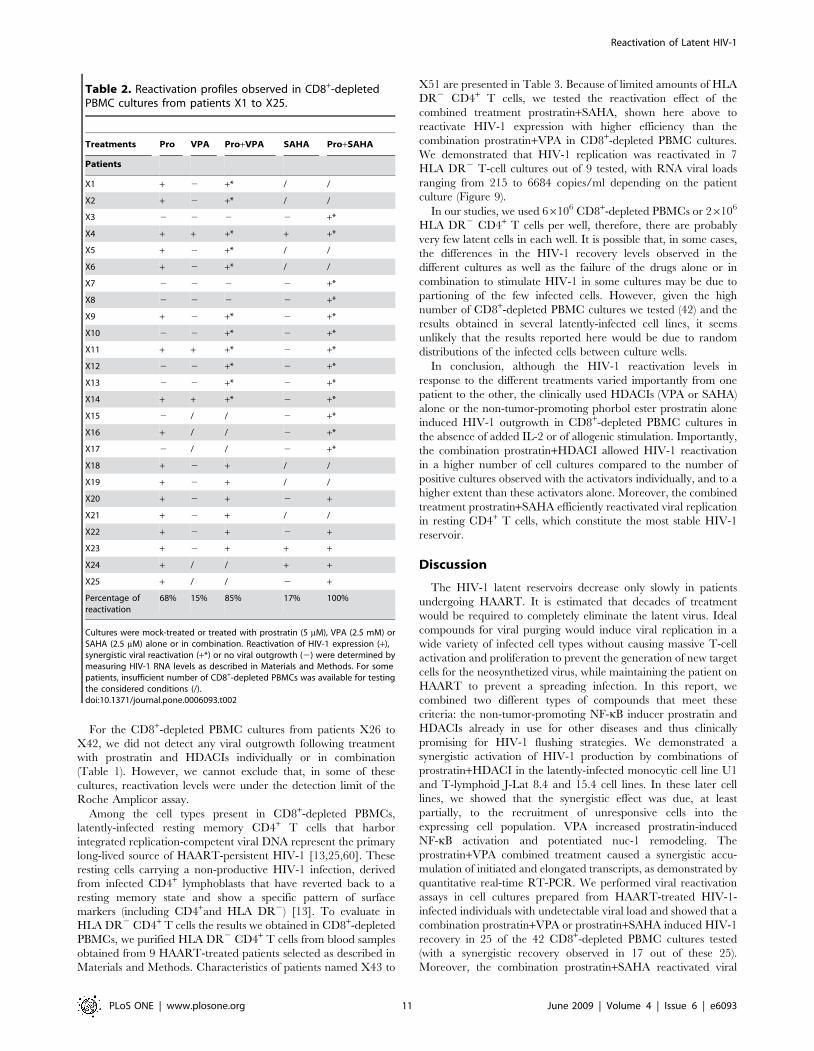
In 17 out of the 25 reactivated cultures, HIV-1 replication was
synergistically induced by a combination prostratin+HDACI
(Table 2, patients X1 to X17). These 17 CD8+-depleted PBMC
cultures can be subdivided in three categories for each
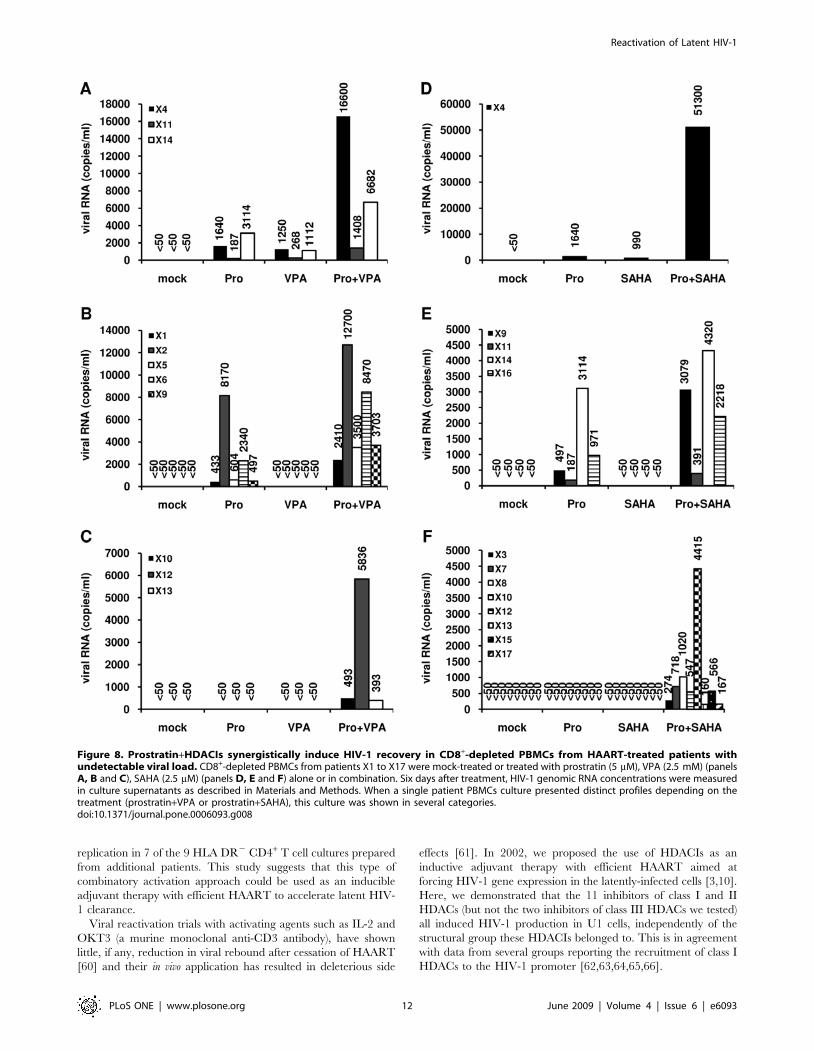
combination prostratin+HDACI (Figure 8A, 8B, 8C for prostra-
tin+VPA and Figure 8D, 8E, 8F for prostratin+SAHA). The first
category concerned 3 cultures for the combination prostra-
tin+VPA (Figure 8A, X4, X11, X14) and one culture for the
combination prostratin+SAHA (Figure 8D, X4). After treatment
with prostratin, VPA or SAHA alone, we detected HIV-1 genomic
RNA in the supernatants. After combined treatments with
prostratin+VPA or prostratin+SAHA, we measured a synergistic
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 8 June 2009 | Volume 4 | Issue 6 | e6093
increase in viral RNA copy number. The second category
concerned CD8+-depleted PBMC cultures in which we detected
HIV-1 reactivation after treatment with prostratin, prostra-
tin+VPA (Figure 8B, X1, X2, X5, X6, X9) or prostratin+SAHA
(Figure 8E, X9, X11, X14, X16) but not after treatment with VPA
or SAHA alone. In the third category, we grouped the results
obtained with CD8+-depleted PBMC cultures in which we detect
viral RNA only after a combined treatment with prostratin+VPA
(Figure 8C, X10, X12, X13) or prostratin+SAHA (Figure 8F, X3,
X7, X8, X10, X12, X13, X15, X17). In cultures from patients
X18 to X25, no synergistic effect of the drug combinations was
observed (Table 2).
Figure 7. Effects of prostratin+VPA on HIV-1 transcription. (A) RNAPII occupancy of the HIV-1 promoter region was assessed by ChIPexperiments. U1 cells were mock-treated or treated with prostratin (5 mM), VPA (2.5 mM) and prostratin+VPA for 30 min, 1 h or 2 h. The proteinswere cross-linked with formaldehyde for 10 min and DNA sheared. The cross-linked protein/DNA complexes were immunoprecipitated with an anti-RNAPII antibody. The protein/DNA cross-links were reversed and the purified DNA was amplified and quantified by real-time PCR using primersamplifying the region overlapping the LTR U5 region and the primer binding site. Values were normalized to those obtained with the 18S rRNAprimers and expressed as fold inductions relative to the value measured in mock-treated U1 cells, which was arbitrarily set at a value of 1. Each valueis the mean6SE from three separate experiments performed in triplicate. (B) Measurement of initiated and elongated HIV-1 transcripts followingdrug treatments. Total RNA was extracted from U1 cells mock-treated or treated with prostratin (5 mM), VPA (2.5 mM) and prostratin+VPA for 1 h, 2 hor 4 h. Initiated (primers tar) or elongated (primers pol, vif and nef) transcripts were quantified by quantitative real-time RT-PCR. Values werenormalized to those obtained with the b-actin primers and expressed for each primer pair as fold inductions relative to the values measured in mock-treated U1 cells, which were arbitrarily set at a value of 1. The results shown are the average of eight independent quantitative real-time PCRs fromfour separate reverse transcriptions from two independent cell culture treatments. The error bars show the standard errors. An experimentrepresentative of two independent experiments is shown.doi:10.1371/journal.pone.0006093.g007
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 9 June 2009 | Volume 4 | Issue 6 | e6093
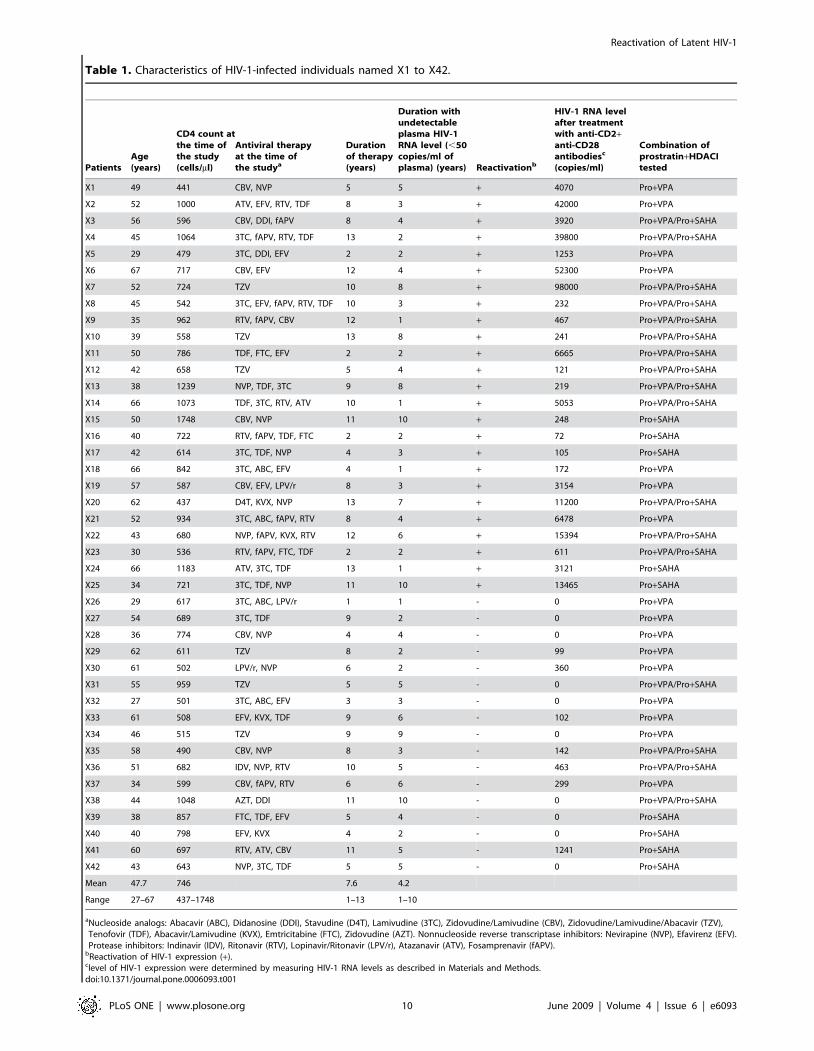
Table 1. Characteristics of HIV-1-infected individuals named X1 to X42.
PatientsAge(years)
CD4 count atthe time ofthe study(cells/ml)
Antiviral therapyat the time ofthe studya
Durationof therapy(years)
Duration withundetectableplasma HIV-1RNA level (,50copies/ml ofplasma) (years) Reactivationb
HIV-1 RNA levelafter treatmentwith anti-CD2+anti-CD28antibodiesc
(copies/ml)
Combination ofprostratin+HDACItested
X1 49 441 CBV, NVP 5 5 + 4070 Pro+VPA
X2 52 1000 ATV, EFV, RTV, TDF 8 3 + 42000 Pro+VPA
X3 56 596 CBV, DDI, fAPV 8 4 + 3920 Pro+VPA/Pro+SAHA
X4 45 1064 3TC, fAPV, RTV, TDF 13 2 + 39800 Pro+VPA/Pro+SAHA
X5 29 479 3TC, DDI, EFV 2 2 + 1253 Pro+VPA
X6 67 717 CBV, EFV 12 4 + 52300 Pro+VPA
X7 52 724 TZV 10 8 + 98000 Pro+VPA/Pro+SAHA
X8 45 542 3TC, EFV, fAPV, RTV, TDF 10 3 + 232 Pro+VPA/Pro+SAHA
X9 35 962 RTV, fAPV, CBV 12 1 + 467 Pro+VPA/Pro+SAHA
X10 39 558 TZV 13 8 + 241 Pro+VPA/Pro+SAHA
X11 50 786 TDF, FTC, EFV 2 2 + 6665 Pro+VPA/Pro+SAHA
X12 42 658 TZV 5 4 + 121 Pro+VPA/Pro+SAHA
X13 38 1239 NVP, TDF, 3TC 9 8 + 219 Pro+VPA/Pro+SAHA
X14 66 1073 TDF, 3TC, RTV, ATV 10 1 + 5053 Pro+VPA/Pro+SAHA
X15 50 1748 CBV, NVP 11 10 + 248 Pro+SAHA
X16 40 722 RTV, fAPV, TDF, FTC 2 2 + 72 Pro+SAHA
X17 42 614 3TC, TDF, NVP 4 3 + 105 Pro+SAHA
X18 66 842 3TC, ABC, EFV 4 1 + 172 Pro+VPA
X19 57 587 CBV, EFV, LPV/r 8 3 + 3154 Pro+VPA
X20 62 437 D4T, KVX, NVP 13 7 + 11200 Pro+VPA/Pro+SAHA
X21 52 934 3TC, ABC, fAPV, RTV 8 4 + 6478 Pro+VPA
X22 43 680 NVP, fAPV, KVX, RTV 12 6 + 15394 Pro+VPA/Pro+SAHA
X23 30 536 RTV, fAPV, FTC, TDF 2 2 + 611 Pro+VPA/Pro+SAHA
X24 66 1183 ATV, 3TC, TDF 13 1 + 3121 Pro+SAHA
X25 34 721 3TC, TDF, NVP 11 10 + 13465 Pro+SAHA
X26 29 617 3TC, ABC, LPV/r 1 1 - 0 Pro+VPA
X27 54 689 3TC, TDF 9 2 - 0 Pro+VPA
X28 36 774 CBV, NVP 4 4 - 0 Pro+VPA
X29 62 611 TZV 8 2 - 99 Pro+VPA
X30 61 502 LPV/r, NVP 6 2 - 360 Pro+VPA
X31 55 959 TZV 5 5 - 0 Pro+VPA/Pro+SAHA
X32 27 501 3TC, ABC, EFV 3 3 - 0 Pro+VPA
X33 61 508 EFV, KVX, TDF 9 6 - 102 Pro+VPA
X34 46 515 TZV 9 9 - 0 Pro+VPA
X35 58 490 CBV, NVP 8 3 - 142 Pro+VPA/Pro+SAHA
X36 51 682 IDV, NVP, RTV 10 5 - 463 Pro+VPA/Pro+SAHA
X37 34 599 CBV, fAPV, RTV 6 6 - 299 Pro+VPA
X38 44 1048 AZT, DDI 11 10 - 0 Pro+VPA/Pro+SAHA
X39 38 857 FTC, TDF, EFV 5 4 - 0 Pro+SAHA
X40 40 798 EFV, KVX 4 2 - 0 Pro+SAHA
X41 60 697 RTV, ATV, CBV 11 5 - 1241 Pro+SAHA
X42 43 643 NVP, 3TC, TDF 5 5 - 0 Pro+SAHA
Mean 47.7 746 7.6 4.2
Range 27–67 437–1748 1–13 1–10
aNucleoside analogs: Abacavir (ABC), Didanosine (DDI), Stavudine (D4T), Lamivudine (3TC), Zidovudine/Lamivudine (CBV), Zidovudine/Lamivudine/Abacavir (TZV),Tenofovir (TDF), Abacavir/Lamivudine (KVX), Emtricitabine (FTC), Zidovudine (AZT). Nonnucleoside reverse transcriptase inhibitors: Nevirapine (NVP), Efavirenz (EFV).Protease inhibitors: Indinavir (IDV), Ritonavir (RTV), Lopinavir/Ritonavir (LPV/r), Atazanavir (ATV), Fosamprenavir (fAPV).
bReactivation of HIV-1 expression (+).clevel of HIV-1 expression were determined by measuring HIV-1 RNA levels as described in Materials and Methods.doi:10.1371/journal.pone.0006093.t001
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 10 June 2009 | Volume 4 | Issue 6 | e6093
For the CD8+-depleted PBMC cultures from patients X26 to
X42, we did not detect any viral outgrowth following treatment
with prostratin and HDACIs individually or in combination
(Table 1). However, we cannot exclude that, in some of these
cultures, reactivation levels were under the detection limit of the
Roche Amplicor assay.
Among the cell types present in CD8+-depleted PBMCs,
latently-infected resting memory CD4+ T cells that harbor
integrated replication-competent viral DNA represent the primary
long-lived source of HAART-persistent HIV-1 [13,25,60]. These
resting cells carrying a non-productive HIV-1 infection, derived
from infected CD4+ lymphoblasts that have reverted back to a
resting memory state and show a specific pattern of surface
markers (including CD4+and HLA DR2) [13]. To evaluate in
HLA DR2 CD4+ T cells the results we obtained in CD8+-depleted
PBMCs, we purified HLA DR2 CD4+ T cells from blood samples
obtained from 9 HAART-treated patients selected as described in
Materials and Methods. Characteristics of patients named X43 to
X51 are presented in Table 3. Because of limited amounts of HLA
DR2 CD4+ T cells, we tested the reactivation effect of the
combined treatment prostratin+SAHA, shown here above to
reactivate HIV-1 expression with higher efficiency than the
combination prostratin+VPA in CD8+-depleted PBMC cultures.
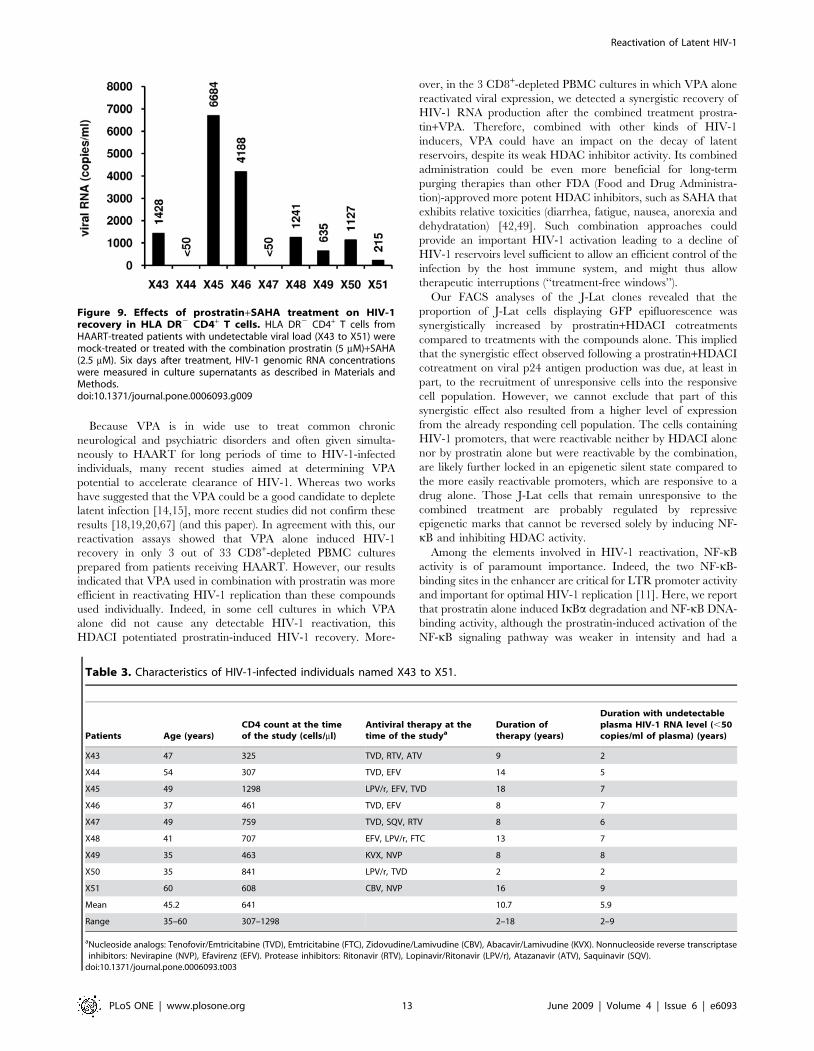
We demonstrated that HIV-1 replication was reactivated in 7
HLA DR2 T-cell cultures out of 9 tested, with RNA viral loads
ranging from 215 to 6684 copies/ml depending on the patient
culture (Figure 9).
In our studies, we used 66106 CD8+-depleted PBMCs or 26106
HLA DR2 CD4+ T cells per well, therefore, there are probably
very few latent cells in each well. It is possible that, in some cases,
the differences in the HIV-1 recovery levels observed in the
different cultures as well as the failure of the drugs alone or in
combination to stimulate HIV-1 in some cultures may be due to
partioning of the few infected cells. However, given the high
number of CD8+-depleted PBMC cultures we tested (42) and the
results obtained in several latently-infected cell lines, it seems
unlikely that the results reported here would be due to random
distributions of the infected cells between culture wells.
In conclusion, although the HIV-1 reactivation levels in
response to the different treatments varied importantly from one
patient to the other, the clinically used HDACIs (VPA or SAHA)
alone or the non-tumor-promoting phorbol ester prostratin alone
induced HIV-1 outgrowth in CD8+-depleted PBMC cultures in
the absence of added IL-2 or of allogenic stimulation. Importantly,
the combination prostratin+HDACI allowed HIV-1 reactivation
in a higher number of cell cultures compared to the number of
positive cultures observed with the activators individually, and to a
higher extent than these activators alone. Moreover, the combined
treatment prostratin+SAHA efficiently reactivated viral replication
in resting CD4+ T cells, which constitute the most stable HIV-1
reservoir.
Discussion
The HIV-1 latent reservoirs decrease only slowly in patients
undergoing HAART. It is estimated that decades of treatment
would be required to completely eliminate the latent virus. Ideal
compounds for viral purging would induce viral replication in a
wide variety of infected cell types without causing massive T-cell
activation and proliferation to prevent the generation of new target
cells for the neosynthetized virus, while maintaining the patient on
HAART to prevent a spreading infection. In this report, we
combined two different types of compounds that meet these
criteria: the non-tumor-promoting NF-kB inducer prostratin and
HDACIs already in use for other diseases and thus clinically
promising for HIV-1 flushing strategies. We demonstrated a
synergistic activation of HIV-1 production by combinations of
prostratin+HDACI in the latently-infected monocytic cell line U1
and T-lymphoid J-Lat 8.4 and 15.4 cell lines. In these later cell
lines, we showed that the synergistic effect was due, at least
partially, to the recruitment of unresponsive cells into the
expressing cell population. VPA increased prostratin-induced
NF-kB activation and potentiated nuc-1 remodeling. The
prostratin+VPA combined treatment caused a synergistic accu-
mulation of initiated and elongated transcripts, as demonstrated by
quantitative real-time RT-PCR. We performed viral reactivation
assays in cell cultures prepared from HAART-treated HIV-1-
infected individuals with undetectable viral load and showed that a
combination prostratin+VPA or prostratin+SAHA induced HIV-1
recovery in 25 of the 42 CD8+-depleted PBMC cultures tested
(with a synergistic recovery observed in 17 out of these 25).
Moreover, the combination prostratin+SAHA reactivated viral
Table 2. Reactivation profiles observed in CD8+-depletedPBMC cultures from patients X1 to X25.
Treatments Pro VPA Pro+VPA SAHA Pro+SAHA
Patients
X1 + 2 +* / /
X2 + 2 +* / /
X3 2 2 2 2 +*
X4 + + +* + +*
X5 + 2 +* / /
X6 + 2 +* / /
X7 2 2 2 2 +*
X8 2 2 2 2 +*
X9 + 2 +* 2 +*
X10 2 2 +* 2 +*
X11 + + +* 2 +*
X12 2 2 +* 2 +*
X13 2 2 +* 2 +*
X14 + + +* 2 +*
X15 2 / / 2 +*
X16 + / / 2 +*
X17 2 / / 2 +*
X18 + 2 + / /
X19 + 2 + / /
X20 + 2 + 2 +
X21 + 2 + / /
X22 + 2 + 2 +
X23 + 2 + + +
X24 + / / + +
X25 + / / 2 +
Percentage ofreactivation
68% 15% 85% 17% 100%
Cultures were mock-treated or treated with prostratin (5 mM), VPA (2.5 mM) orSAHA (2.5 mM) alone or in combination. Reactivation of HIV-1 expression (+),synergistic viral reactivation (+*) or no viral outgrowth (2) were determined bymeasuring HIV-1 RNA levels as described in Materials and Methods. For somepatients, insufficient number of CD8+-depleted PBMCs was available for testingthe considered conditions (/).doi:10.1371/journal.pone.0006093.t002
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 11 June 2009 | Volume 4 | Issue 6 | e6093
replication in 7 of the 9 HLA DR2 CD4+ T cell cultures prepared
from additional patients. This study suggests that this type of
combinatory activation approach could be used as an inducible
adjuvant therapy with efficient HAART to accelerate latent HIV-
1 clearance.
Viral reactivation trials with activating agents such as IL-2 and
OKT3 (a murine monoclonal anti-CD3 antibody), have shown
little, if any, reduction in viral rebound after cessation of HAART
[60] and their in vivo application has resulted in deleterious side
effects [61]. In 2002, we proposed the use of HDACIs as an
inductive adjuvant therapy with efficient HAART aimed at
forcing HIV-1 gene expression in the latently-infected cells [3,10].
Here, we demonstrated that the 11 inhibitors of class I and II
HDACs (but not the two inhibitors of class III HDACs we tested)
all induced HIV-1 production in U1 cells, independently of the
structural group these HDACIs belonged to. This is in agreement
with data from several groups reporting the recruitment of class I
HDACs to the HIV-1 promoter [62,63,64,65,66].
Figure 8. Prostratin+HDACIs synergistically induce HIV-1 recovery in CD8+-depleted PBMCs from HAART-treated patients withundetectable viral load. CD8+-depleted PBMCs from patients X1 to X17 were mock-treated or treated with prostratin (5 mM), VPA (2.5 mM) (panelsA, B and C), SAHA (2.5 mM) (panels D, E and F) alone or in combination. Six days after treatment, HIV-1 genomic RNA concentrations were measuredin culture supernatants as described in Materials and Methods. When a single patient PBMCs culture presented distinct profiles depending on thetreatment (prostratin+VPA or prostratin+SAHA), this culture was shown in several categories.doi:10.1371/journal.pone.0006093.g008
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 12 June 2009 | Volume 4 | Issue 6 | e6093
Because VPA is in wide use to treat common chronic
neurological and psychiatric disorders and often given simulta-
neously to HAART for long periods of time to HIV-1-infected
individuals, many recent studies aimed at determining VPA
potential to accelerate clearance of HIV-1. Whereas two works
have suggested that the VPA could be a good candidate to deplete
latent infection [14,15], more recent studies did not confirm these
results [18,19,20,67] (and this paper). In agreement with this, our
reactivation assays showed that VPA alone induced HIV-1
recovery in only 3 out of 33 CD8+-depleted PBMC cultures
prepared from patients receiving HAART. However, our results
indicated that VPA used in combination with prostratin was more
efficient in reactivating HIV-1 replication than these compounds
used individually. Indeed, in some cell cultures in which VPA
alone did not cause any detectable HIV-1 reactivation, this
HDACI potentiated prostratin-induced HIV-1 recovery. More-
over, in the 3 CD8+-depleted PBMC cultures in which VPA alone
reactivated viral expression, we detected a synergistic recovery of
HIV-1 RNA production after the combined treatment prostra-
tin+VPA. Therefore, combined with other kinds of HIV-1
inducers, VPA could have an impact on the decay of latent
reservoirs, despite its weak HDAC inhibitor activity. Its combined
administration could be even more beneficial for long-term
purging therapies than other FDA (Food and Drug Administra-
tion)-approved more potent HDAC inhibitors, such as SAHA that
exhibits relative toxicities (diarrhea, fatigue, nausea, anorexia and
dehydratation) [42,49]. Such combination approaches could
provide an important HIV-1 activation leading to a decline of
HIV-1 reservoirs level sufficient to allow an efficient control of the
infection by the host immune system, and might thus allow
therapeutic interruptions (‘‘treatment-free windows’’).
Our FACS analyses of the J-Lat clones revealed that the
proportion of J-Lat cells displaying GFP epifluorescence was
synergistically increased by prostratin+HDACI cotreatments
compared to treatments with the compounds alone. This implied
that the synergistic effect observed following a prostratin+HDACI
cotreatment on viral p24 antigen production was due, at least in
part, to the recruitment of unresponsive cells into the responsive
cell population. However, we cannot exclude that part of this
synergistic effect also resulted from a higher level of expression
from the already responding cell population. The cells containing
HIV-1 promoters, that were reactivable neither by HDACI alone
nor by prostratin alone but were reactivable by the combination,
are likely further locked in an epigenetic silent state compared to
the more easily reactivable promoters, which are responsive to a
drug alone. Those J-Lat cells that remain unresponsive to the
combined treatment are probably regulated by repressive
epigenetic marks that cannot be reversed solely by inducing NF-
kB and inhibiting HDAC activity.
Among the elements involved in HIV-1 reactivation, NF-kB
activity is of paramount importance. Indeed, the two NF-kB-
binding sites in the enhancer are critical for LTR promoter activity
and important for optimal HIV-1 replication [11]. Here, we report
that prostratin alone induced IkBa degradation and NF-kB DNA-
binding activity, although the prostratin-induced activation of the
NF-kB signaling pathway was weaker in intensity and had a
Figure 9. Effects of prostratin+SAHA treatment on HIV-1recovery in HLA DR2 CD4+ T cells. HLA DR2 CD4+ T cells fromHAART-treated patients with undetectable viral load (X43 to X51) weremock-treated or treated with the combination prostratin (5 mM)+SAHA(2.5 mM). Six days after treatment, HIV-1 genomic RNA concentrationswere measured in culture supernatants as described in Materials andMethods.doi:10.1371/journal.pone.0006093.g009
Table 3. Characteristics of HIV-1-infected individuals named X43 to X51.
Patients Age (years)CD4 count at the timeof the study (cells/ml)
Antiviral therapy at thetime of the studya
Duration oftherapy (years)
Duration with undetectableplasma HIV-1 RNA level (,50copies/ml of plasma) (years)
X43 47 325 TVD, RTV, ATV 9 2
X44 54 307 TVD, EFV 14 5
X45 49 1298 LPV/r, EFV, TVD 18 7
X46 37 461 TVD, EFV 8 7
X47 49 759 TVD, SQV, RTV 8 6
X48 41 707 EFV, LPV/r, FTC 13 7
X49 35 463 KVX, NVP 8 8
X50 35 841 LPV/r, TVD 2 2
X51 60 608 CBV, NVP 16 9
Mean 45.2 641 10.7 5.9
Range 35–60 307–1298 2–18 2–9
aNucleoside analogs: Tenofovir/Emtricitabine (TVD), Emtricitabine (FTC), Zidovudine/Lamivudine (CBV), Abacavir/Lamivudine (KVX). Nonnucleoside reverse transcriptaseinhibitors: Nevirapine (NVP), Efavirenz (EFV). Protease inhibitors: Ritonavir (RTV), Lopinavir/Ritonavir (LPV/r), Atazanavir (ATV), Saquinavir (SQV).
doi:10.1371/journal.pone.0006093.t003
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 13 June 2009 | Volume 4 | Issue 6 | e6093

delayed kinetics compared to that elicited by TNFa, in agreement
with a previous study [28]. Importantly, we showed that VPA
increased and prolonged prostratin-induced NF-kB DNA-binding
activity. This is consistent with the fact that acetylation/
deacetylation events regulate the NF-kB activating signaling
pathway at multiple levels, including the direct acetylation of
NF-kB [55]. Indeed, Lys221 acetylation enhances p65 DNA-
binding activity and, together with acetylation at Lys218, impairs
its assembly with IkBa, thereby impeding IkBa-dependent nuclear
export of the NF-kB complex and enabling prolongation of the
NF-kB response [55]. Moreover, the acetylated form of p50 binds
target sequences with higher affinity than the unacetylated form
does [55]. Noteworthy, the prolonged NF-kB DNA-binding
activity that we observed here was correlated with a prolonged
IkBa degradation. IkBa degradation is dependent upon its
phosphorylation by the IkB kinase (IKK) complex and its
subsequent ubiquitination. Thereby, VPA could prolong the
prostratin-induced IkBa degradation by prolonging the IKK
activity, as we have previously shown for the TNFa+TSA
combined treatment [21].
The nucleosome nuc-1 is positioned immediately downstream
of the HIV-1 transcription start and is remodeled upon
transcriptional activation by Tat and the HDACIs TSA or
Trapoxin in the absence of NF-kB stimulation [4,5]. These
observations suggest that nuc-1 plays a repressive role in HIV-1
transcription and that histone acetylation could be involved in
overriding this effect. Accordingly, we reported here that the
HDACI VPA induced nuc-1 histone H4 acetylation. Importantly,
we showed that the combination prostratin+VPA did not induce a
level of acetylated histone H4 higher than the level observed with
VPA alone. Therefore, the cooperative nuc-1 remodeling we
observed by indirect end-labeling following a prostratin+VPA
cotreatment was not due to a cooperative induction of histone H4
acetylation. Chromatin structure can be altered both by histone
posttranslational modifications (such as acetylation), which alter
histone/DNA or histone/histone interactions, and by ATP-
dependent chromatin remodeling complexes (such as SWI/SNF)
[68]. It has been previously reported that these SWI/SNF
complexes are recruited at gene promoters by transcription
factors, including NF-kB [69,70] and AP-1 [71,72], or by
acetylated histones [73]. Therefore, by inducing NF-kB and/or
AP-1 binding to the HIV-1 5’LTR [22,28,32], prostratin could be
involved in the remodeling of nuc-1 through the recruitment of
SWI/SNF by these transcription factors. This hypothesis could
explain the more pronounced nuc-1 remodeling we observed in
the postintegration latency model cell line U1 after the
prostratin+VPA cotreatment compared to the treatments by the
drugs alone.
We demonstrated a temporal correlation between the degree of
nuc-1 remodeling and the level of transcription in U1 cells by
showing that the prostratin+HDACI-induced increase in chroma-
tin remodeling at 2 h postinduction was correlated with an
increased RNAPII recruitment to the viral LTR at the same time
point and to an increased accumulation of both initiated and
elongated transcripts at 4 h postinduction. Expression of full-
length HIV-1 transcripts requires the action of the cellular kinase
P-TEFb, composed of a catalytic subunit, cyclin-dependent kinase
9 (Cdk9) and a regulatory subunit, cyclinT1. The absence or
inactivity of this protein, while generating short viral transcripts,
fails to support effective viral replication. P-TEFb mediates
transcriptional elongation of the bound polymerase complexes
by phosphorylating the C-terminal domain (CTD) of RNAPII.
Although P-TEFb is mainly recruited via the viral transactivator
Tat, other mechanisms that can ensure P-TEFb recruitment and
thereby transcriptional elongation have been described and can
account for signal-induced transcription elongation in Tat-
defective HIV-1 infected cells, such as the U1 cell line. Notably,
Sp1, p65 and histone acetylation can contribute to P-TEFb
recruitment to the HIV-1 promoter [74,75,76,77], via Brd4, a
positive regulatory component of P-TEFb, for p65 and histone
acetylation [75,76,77]. Brd4 is a bromodomain-containing protein
that recognizes acetylated histones [78] as well as acetylated p65
[75]. Altogether, these previous data could explain at the
molecular level our quantitative real-time RT-PCR results
showing that prostratin and VPA synergistically activated
transcriptional elongation in U1 cells. The prostratin+VPA
cotreatment could allow a more important recruitment of the
Brd4/P-TEFb complex to the HIV-1 promoter than the drugs
alone. In addition to activating the NF-kB signaling pathway,
prostratin up-regulates cyclinT1 expression, leading to an
increased association between cyclinT1 and cyclin-dependent
kinase 9 (Cdk9) which results in a more efficient P-TEFb kinase
activity [79].
Our reactivation assays suggested that when combined with
prostratin, SAHA could be a more potent HIV-1 reactivating
agent than VPA. Indeed, whereas a prostratin+VPA cotreatment
reactivated viral expression in 17 out of the 33 CD8+-depleted
PBMC cultures tested (11 presenting synergistic activation), a
combined treatment with prostratin+SAHA reactivated viral
expression in 18 out of 26 CD8+-depleted PBMC cultures (13
presenting synergistic activation) and in 7 out of the 9 HLA DR2
CD4+ T cell cultures we tested. Supporting our data, two very
recent studies, published during the revision of the present paper,
have reported that SAHA reactivates HIV-1 from latently-infected
cells [80,81]. SAHA is administered intravenously to patients with
refractory hematologic and solid tumors and has been approved
for use as a treatment of cutaneous T-cell lymphoma [42,48,49].
Because of its ability to promote HIV-1 expression, especially in
combination with prostratin, SAHA provides additional promise
for the decay of latent HIV-1 infection and deserves further
investigations.
Of note, in 17 out of 42 CD8+-depleted PBMC cultures, we did
not detect any viral outgrowth following treatment with prostratin
and HDACIs individually or in combination and no or very weak
viral outgrowth following an anti-CD2+anti-CD28 treatment
compared to the levels observed with the PBMC cultures
presenting viral reactivation after drug treatments. A possible
explanation to the variegated response to prostratin+HDACI
treatment observed in the CD8+-depleted PBMC cultures could
result from an ‘‘on-off switching’’ of transcriptional competence.
Local chromatin structure at the site of virus integration within the
host genome modulates the level of epigenetic HIV-1 transcrip-
tional repression as well as the ability of the virus to respond to
reactivating stimuli [82]. Indeed, some of the integrated HIV-1
DNA may be under strong epigenetic repression (through DNA
methylation and histone methylation and deacetylation) that
cannot be efficiently reversed by the drugs we used in this study
[7,83]. However, we cannot rule out the possibility that the viruses
present in the samples in which we could not detect any viral
reactivation were defective. Therefore, locus-specific transcrip-
tional repression might account, at least in part, for the
heterogeneous HIV-1 reactivation profiles we observed after
prostratin+HDACI cotreatment.
In conclusion, we report a proof-of-concept study for the
therapeutic potential coadministration of two different kinds of
HIV-1 activators (one acting on the NF-kB pathway and the other
acting on the protein acetylation status) aimed at efficient decay of
latent reservoirs in presence of HAART. However, we did not
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 14 June 2009 | Volume 4 | Issue 6 | e6093

observe reactivation of viral replication in all the patient cell
cultures tested, thereby emphasizing the importance to identify
other combinations involving SAHA, prostratin, prostratin-like
compounds with higher potency and/or other kinds HIV-1
inducers (such as inhibitors of histone- and DNA-methyltransfer-
ases). In this regard, a recent study has reported the practical
synthesis in gram quantities and at low cost of prostratin, 12-
deoxyphorbol-13-phenylacetate (DPP) and a variety of new 12-
deoxyphorbol analogs, which might be potentially superior clinical
candidates against latent HIV-1 [34].
Materials and Methods
Cell Lines and Cell CultureThe monocytic cell line U1 [84] and the T-lymphoid cell lines
Jurkat [85], J-Lat 8.4 and J-Lat 15.4 [51] were obtained from the
AIDS Research and Reference Reagent Program (National
Institute of Allergy and Infectious Diseases [NIAID], National
Institute of health [NIH]). All cell lines were grown in RPMI 1640
medium (Gibco-BRL) supplemented with 10% fetal bovine serum,
50 U/ml of penicillin, 50 mg/ml of streptomycin at 37uC in a
humidified 95% air/5% CO2 atmosphere.
ReagentsTNFa (catalog no. 300-01A) was purchased from Immuno-
source. SAHA (suberoylanilide hydroxamic acid) (catalog no. 270-
288-M001) was obtained from Alexis Biochemichals. Prostratin
(12-deoxyphorbol-13-acetate) (catalog no. PE 187-0001), HC-
Toxin (Cyclo(D-prolyl-L-alanyl-D-alanyl-L-2-amino-9,10-epoxy-
8-oxodecanoyl)) (catalog no. GR-320), SBHA (suberoyl bis-
hydroxamic acid) (catalog no. GR-323), Scriptaid (6-(1,3-dioxo-
1H,3H-benzo[de]isoquinolin-2-yl)-N-hydroxyhexanamide) (cata-
log no. GR-326) were provided by Tebu-Bio. NaBut (sodium
butyrate) (catalog no. B5887), VPA (2-propylpentanoic acid
sodium or valproic acid) (catalog no. P4543), Depudecin (D-
threo-D-ido-undeco-1,6-dienitol,4,5:8,9-dianhydro-1,2,6,7,11-
pentadeoxy) (catalog no. D5816), apicidin (cyclo[(2S)-2-amino-8-
oxodecanoyl-1-methoxy-L-tryptophyl-L-isoleucyl-(2R)-2-piperidi-
necarbonyl]) (catalog no. A8851), TSA (trichostatin A) (catalog no.
T8552) were purchased from Sigma-Aldrich. PMA (phorbol
myristate acetate) (catalog no. 524400), Splitomycin (1,2-dihy-
dro-3H-naphtho[2,1-b]pyran-3-one) (catalog no. 567750), MS-
275 (N-(2-Aminophenyl)-4-[N-(pyridine-3-ylmethoxycarbonyl)a-
minomethyl] benzamide) (catalog no. 382147), Sirtinol (2-[(2-
hydroxynaphthalen-1-ylmethylene)amino]-N-(1-phenethyl)benza-
mide) (catalog no. 566320), CBHA (m-carboxycinnamic acid bis-
hydroxamide) (catalog no. 382148) were obtained from Calbio-
chem. All compounds, resuspended and stored as recommended
by the manufacturer, were diluted immediately before use in cell
culture medium.
Virus production assays in U1 and J-Lat 8.4 or 15.4 celllines
HIV-1 production was measured in the U1 and J-Lat 8.4 or
15.4 cell line culture supernatants by determining CA-p24 antigen
concentration by an enzyme-linked immunosorbent assay (In-
nogenetics).
Transient transfection and luciferase reporter assaysJurkat cells were transfected with the pLTR (A, B, C1, D,
CRF01_AE, F, G, CRF02_AG) –luciferase reporter plasmids
(1000 ng) [86] using jetPEITM (POLYplus) according to the
manufacturer’s protocol. Cells were mock-treated or treated with
the different compounds as indicated, lysed and assayed for
luciferase activity as previously described [3].
EMSAsNuclear extracts were prepared and EMSAs with the HIV-1
NF-kB probe were performed as previously described [3]. As
loading controls, the same nuclear extracts were tested for binding
of Oct-1 (octamer-binding protein-1) to an Oct-1 consensus probe
(59-TGTCGAATGCAAATCACTAGAA-39).
Western blot analysesCytoplasmic extracts were prepared as previously described
[87]. Western blots were performed [87] with a rabbit antibody
against IkBa (1/1000 dilution; catalog no. 06-494; Upstate
Biotechnology) or against actin (1/1000 dilution; catalog
no.A2066; Sigma-Aldrich) and a peroxidase-conjugated goat
anti-rabbit IgG (1/3000 dilution; catalog no. 7074; Cell Signaling).
Indirect end-labeling techniqueThe indirect end-labeling experiments were performed as
previously described [4].
Study SubjectsWe selected 52 HIV-1-infected individuals at the St-Pierre
Hospital (Brussels, Belgium) and 9 additional patients at the
Bicetre Hospital (Bicetre, France) on the basis of the following
criteria: all volunteers were treated with HAART for at least 1
year, had an undetectable plasma HIV-1 RNA level (,50 copies/
ml) for at least 1 year and had a level of CD4+ T lymphocytes
higher than 300 cells/mm3 of blood. Characteristics (age, CD4+ T
cell count, antiviral regimens, duration of therapy, duration with
undetectable plasma HIV-1 RNA level) of patients from the St-
Pierre Hospital and from the Bicetre Hospital are presented in
Table 1 and 3, respectively.
Ethics StatementEthical approval was granted by the Human Subject Ethics
Committees of the Saint-Pierre Hospital and of the Bicetre
Hospital. All individuals enrolled in the study provided written
informed consent for donating blood.
Isolation of CD8+-depleted PBMCs and of HLA DR2 CD4+
T lymphocytesCD8+-depleted PBMCs were isolated from fresh whole blood
(50 ml) of the HIV-1-infected individuals described above by
adding RosetteSep human CD8 depletion mixture (StemCell
Technologies) to whole blood samples before density centrifuga-
tion on a Ficoll-Hypaque gradient (Pharmacia). HLA DR2 CD4+
T lymphocytes were isolated as previously described [88]. The
number of cells (CD8+-depleted PBMCs and HLA DR2 CD4+ T
lymphocytes) obtained after isolation varied depending on the
patients. In all cases, cells were washed with RPMI, resuspended at
26106 cells/ml of complete RPMI (RPMI, 10% fetal bovine
serum, supplemented with 50 U/ml of penicillin, and 50 mg/ml of
streptomycin). As a positive control of reactivation, CD8+-depleted
PBMCs were stimulated with anti-CD2+anti-CD8 antibodies used
at saturating concentrations [mAb 39C1.5 (rat IgG2a, anti-CD2)
in combination with mAb 6F10.3 (mouse IgG1, anti-CD2); anti-
CD28 mAbs (clone identification 28.2)] [89,90]. Activation by
anti-CD2+anti-CD28 monoclonal antibodies induces high-level,
long-lasting and monocyte-independent proliferation of resting T
cells, thymocytes and preactivated T cells [91,92].
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 15 June 2009 | Volume 4 | Issue 6 | e6093

Roche Amplicor quantitative assessments of HIV-1 RNAOne day after isolation, 66106 CD8+-depleted PBMCs or
26106 HLA DR2 CD4+ T cells were mock-treated or treated with
different compounds. Six days after treatment, culture superna-
tants were tested for quantitative HIV-1 RNA levels by using the
COBAS AmpliPrep/COBAS AMPLICOR HIV-1 MONITOR
Test version 1.5, according to the manufacturer’s instructions
(Roche Diagnostics) (lowest detection limit: 50 copies HIV-1
RNA/ml of plasma).
ChIP assaysChIP assays were performed using the ChIP assay kit (EZ ChIP
technology, Upstate). U1 cells were cross-linked after drug
treatments. To detect chromosomal flanking regions, pellets were
sonicated (Bioruptor sonicator) to obtain DNA fragments of 100–
400 nt. Chromatin immunoprecipitations were performed with an
antibody directed against RNAPII (catalog no. sc-899, Santa Cruz
Biotechnology). To test aspecific binding to the beads, a purified
IgG was used as a control for immunoprecipitation (catalog no. I-
1000, Vector Laboratories). Quantitative real-time PCR reactions
were performed using the MesaGreen qPCR mastermix (Euro-
gentec). Relative quantification using standard curve method was
performed for each primer pair and 96-well Optical Reaction
plates were read in an Applied Biosystems AbiPrism 7300 real-
time PCR instrument (Absolute Quantification Method). Fold
enrichments were calculated as percentages of input values
normalized to an unrelated genomic region (18S rRNA DNA)
and expressed as fold inductions relative to the values measured in
mock-treated U1 cells. Primer sequences used for quantification in
a region overlapping the LTR U5 region and the primer binding
site region (to avoid interference with the 3’LTR region) (FW, 59-
TGGAAAATCTCTAGCAGTGGC-39 and RV, 59-GAGTCC-
TGCGTCGAGAGATCT-39) and in the unrelated 18S rRNA
DNA region (FW, 59-TGGATACCGCAGCTAGGAATAA-39
and RV, 59-CCTCTTAATCATGGCCTCAGTTC-39) were
designed using the software Primer Express 2.0 (Applied
Biosystems).
RNA extractions and analyses of initiated and elongatedHIV-1 transcripts
Total RNA samples were isolated using the RNeasy Plus kit
(Qiagen) from 16106 U1 cells and digested with TURBO DNase
(TURBO DNA-freeTM kit, Ambion). First strand cDNA was
synthesized using SuperScript III Reverse Transcriptase (Invitro-
gen). Quantitative real-time PCR reactions were performed using
the MesaGreen qPCR mastermix (Eurogentec). Initiated tran-
scripts were detected with primers tar (FW, 59-GTTAGACCA-
GATCTGAGCCT-39 and RV, 59-GTGGGTTCCCTAGT-
TAGCCA-39). Elongated transcripts were detected with three
different sets of primers: pol (FW, 59-AATATGCAAGAAT-
GAAGGGTGC-39 and RV, 59-TGCTTTCTGTGGCTATTT-
TTTGTA-39), vif (FW, 59-ATGGCAGGTGATGATTGTGTG-
39 and RV, 59-CCATGTGTTAATCCTCATCCTGTC-39) and
nef (FW, 59-GCCTGGCTAGAAGCACAAGA-39 and RV, 59-
AGGTGTGACTGGAAAACCCAC-39). b-actin mRNA copies
were quantified with primers b-actin (FW, 59-GTCGA-
CAACGGCTCCGGC-39 and RV, 59-GGTGTGGTGCCA-
GATTTTCT-39) specific for a 239 bp region in the b-actin
mRNA. Quantitative real-time PCR reactions were performed for
each primer pair and 96-well Optical Reaction plates were read in
an Applied Biosystems StepOnePlus Real-Time PCR System
(Comparative Ct (DDCt) Quantification Method).
Flow cytometry analysesJ-Lat 8.4 or 15.4 cells were mock-treated or treated for 24 h
with the different compounds alone or in combination. Reco-
verded cells were washed twice in PBS, resuspended in PBS
containing 4% paraformaldehyde and fixed for 1 h. Cells were
next washed twice in PBS and resuspended in FACS buffer (PBS-
BSA 0.1%-NaN3 0.1%). The percentage of GFP-positive cells was
measured on a CXP cytometer (Cytomics FC 500, Beckman
Coulter) using CXP Software version 1.0 according to the
manufacturer’s instructions.
Supporting Information
Figure S1 Dose-response curves of U1 cellular viability after
HDACI treatment. U1 cells were mock-treated or treated with
increasing concentrations of an HDACI belonging to one of the
four structural families: short-chain fatty acids (A), benzamides (B),
cyclic tetrapeptides (C), hydroxamic acids (D). At 24 h posttreat-
ment, cellular viability was tested by measuring the mitochondrial
dehydrogenase activity with the WST-1 reduction assay. The
mock-treated value was arbitrarily set at a value of 100% of
cellular viability. Each point is the mean from three separate
experiments performed in triplicate. SE are intentionally not
represented on the graph for clarity reasons.
Found at: doi:10.1371/journal.pone.0006093.s001 (0.19 MB TIF)
Figure S2 Prostratin does not increase HDACI cytotoxicity in
CD8+-depleted PBMC cultures from uninfected individuals.
CD8+-depleted PBMCs were mock-treated or treated with VPA
(2.5 mM), SAHA (2.5 mM), TSA (500 nM), NaBut (5 mM), MS-
275 (5 mM), prostratin (5 mM) alone (A) or in combination (B). At
24 h posttreatment, cellular viability was tested by measuring the
mitochondrial dehydrogenase activity with the WST-1 reduction
assay. A value of 100% of cellular viability was arbitrarily assigned
to the mock-treated value (A) or to the prostratin-treated value (B).
Each value is the mean +/2 SE from three separate experiments
performed in triplicate.
Found at: doi:10.1371/journal.pone.0006093.s002 (0.12 MB TIF)
Figure S3 Prostratin+HDACI cotreatment induces HIV-1
expression in a higher proportion of cells than the drugs alone.
This figure shows as plots the same FACS results that are
presented as histograms in Figure 3B in the manuscript. J-Lat 8.4
cells were mock-treated or treated with prostratin (5 mM), alone or
in combination with different HDACIs [VPA (2.5 mM), SAHA
(2.5 mM), TSA (500 nM), NaBut (5 mM) or MS-275 (5 mM)]. At
24 h posttreatment, cells were analyzed by FACS for GFP
expression. The plots are representative of four independent
experiments obtained with J-Lat 8.4 cells. Similar results were
obtained with the J-Lat 15.4 T-cell clone (data not shown).
Found at: doi:10.1371/journal.pone.0006093.s003 (0.18 MB TIF)
Figure S4 The prostratin+VPA cotreatment causes a more rapid
and pronounced nucleosomal remodeling than the compounds
alone. (A) Diagram indicating the positions of nucleosomes in the
59 portion of the HIV-1 genome, the AflII and HinfI cutting sites
and the probe used in indirect end-labeling. Bold, lower case
letters are assigned to each HinfI cutting site (x, y and z) and are
located next to the bands on the gel to permit their identification.
(B) Nuclei were prepared from U1 cells mock-treated or treated
with TNFa (10 ng/ml) (30 min), prostratin (5 mM), VPA (2.5 mM)
and prostratin+VPA for 30 min, 1 h or 2 h and digested with
HinfI. After DNA purification and in vitro restriction with PstI,
DNA samples were analyzed by indirect end-labeling using probe
A (93). Size markers (a, b, c, d, e, f, g) have been previously
described (93).
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 16 June 2009 | Volume 4 | Issue 6 | e6093

Found at: doi:10.1371/journal.pone.0006093.s004 (0.69 MB TIF)
Figure S5 The prostratin+VPA cotreatment does not induce
levels of acetylated histone H4 higher than the levels observed
after the treatments with VPA alone. Acetylated H4 levels in the
nuc-1 region were assessed by ChIP experiments using U1 cells
mock-treated or treated with prostratin (5 mM), VPA (2.5 mM)
and prostratin+VPA for different periods of time. The proteins
were cross-linked with formaldehyde for 10 min and DNA
sheared. The cross-linked protein/DNA complexes were immu-
noprecipitated with an anti-Ac-H4 antibody. The protein/DNA
cross-links were reversed and the purified DNA was amplified and
quantified by real-time PCR using primers amplifying either the
nuc-1 region or the vif region. Fold enrichments in the nuc-1 and
vif regions were calculated as percentages of input values and
expressed as fold inductions relative to the value measured with
the nuc-1 primers in mock-treated U1 cells, which was arbitrarily
set at a value of 1. Each value is the mean +/2 SE from three
separate experiments performed in duplicate.
Found at: doi:10.1371/journal.pone.0006093.s005 (0.11 MB TIF)
Text S1 Supporting Information of Figure S1
Found at: doi:10.1371/journal.pone.0006093.s006 (0.08 MB
DOC)
Text S2 Supporting Information of Figure S2
Found at: doi:10.1371/journal.pone.0006093.s007 (0.07 MB
DOC)
Text S3 Supporting Information of Figure S5
Found at: doi:10.1371/journal.pone.0006093.s008 (0.08 MB
DOC)
Acknowledgments
We would like to thank the HIV-1-positive patients for their willingness to
participate in this study and Elodie Goudeseune who cares for the patients.
We thank Christelle Cardona, Noemie Stache and Adeline Melard for
excellent technical assistance. The following reagents were obtained
through the AIDS Research and Reference Reagent Program, Division
of AIDS, NIAID, NIH: the U1 cell line from Dr. T. Folks, the J-Lat Full
Length Clones (clones #8.4 and #15.4) from Dr. E. Verdin and the Jurkat
Clone E6-1 from Dr. A. Weiss.
Author Contributions
Conceived and designed the experiments: CVL. Performed the experi-
ments: SR MC AG JSG VQ CV AL DD VM EV TC VA SP JP YdL AB
CR OR YEC OL. Analyzed the data: SR MC OL CVL. Contributed
reagents/materials/analysis tools: YEC. Wrote the paper: SR MC CVL.
Performed measurements of HIV-1 genomic RNA concentrations: DV
GH. Performed patient selection: KK MM SDW OL NC. Performed the
isolation of HLA DR- CD4+ T lymphocytes: AL OL.
References
1. Bisgrove D, Lewinski M, Bushman F, Verdin E (2005) Molecular mechanisms of
HIV-1 proviral latency. Expert Review of Anti-infective Therapy 3: 805–814.
2. Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF (2007)
Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol5: 95–106.
3. Quivy V, Adam E, Collette Y, Demonte D, Chariot A, et al. (2002) Synergisticactivation of human immunodeficiency virus type 1 promoter activity by NF-
kappaB and inhibitors of deacetylases: potential perspectives for the develop-
ment of therapeutic strategies. J Virol 76: 11091–11103.
4. Van Lint C, Emiliani S, Ott M, Verdin E (1996) Transcriptional activation and
chromatin remodeling of the HIV-1 promoter in response to histone acetylation.Embo J 15: 1112–1120.
5. El Kharroubi A, Piras G, Zensen R, Martin MA (1998) Transcriptionalactivation of the integrated chromatin-associated human immunodeficiency
virus type 1 promoter. Mol Cell Biol 18: 2535–2544.
6. Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, et al. (1999) HIV-1 tat
transcriptional activity is regulated by acetylation. Embo J 18: 6106–6118.
7. Jordan A, Defechereux P, Verdin E (2001) The site of HIV-1 integration in thehuman genome determines basal transcriptional activity and response to Tat
transactivation. Embo J 20: 1726–1738.
8. Sheridan PL, Mayall TP, Verdin E, Jones KA (1997) Histone acetyltransferases
regulate HIV-1 enhancer activity in vitro. Genes Dev 11: 3327–3340.
9. Steger DJ, Eberharter A, John S, Grant PA, Workman JL (1998) Purified histone
acetyltransferase complexes stimulate HIV-1 transcription from preassemblednucleosomal arrays. Proc Natl Acad Sci U S A 95: 12924–12929.
10. Demonte D, Quivy V, Colette Y, Van Lint C (2004) Administration of HDACinhibitors to reactivate HIV-1 expression in latent cellular reservoirs:
implications for the development of therapeutic strategies. Biochem Pharmacol
68: 1231–1238.
11. Quivy V, De Walque S, Van Lint C (2007) Chromatin-associated regulation of
HIV-1 transcription: implications for the development of therapeutic strategies.Subcell Biochem 41: 371–396.
12. Margolis DM (2007) Confronting proviral HIV infection. Curr HIV/AIDS Rep4: 60–64.
13. Marcello A (2006) Latency: the hidden HIV-1 challenge. Retrovirology 3: 7.
14. Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM (2004) Coaxing
HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent
viral expression. Aids 18: 1101–1108.
15. Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, et al. (2005) Depletion
of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549–555.
16. Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, et al. (2007) Decayof the HIV reservoir in patients receiving antiretroviral therapy for extended
periods: implications for eradication of virus. J Infect Dis 195: 1762–1764.
17. Chun TW, Davey RT Jr, Engel D, Lane HC, Fauci AS (1999) Re-emergence of
HIV after stopping therapy. Nature 401: 874–875.
18. Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, et al. (2008)
Valproic acid without intensified antiviral therapy has limited impact on
persistent HIV infection of resting CD4+ T cells. Aids 22: 1131–1135.
19. Sagot-Lerolle N, Lamine A, Chaix ML, Boufassa F, Aboulker JP, et al. (2008)
Prolonged valproic acid treatment does not reduce the size of latent HIVreservoir. Aids 22: 1125–1129.
20. Siliciano JD, Lai J, Callender M, Pitt E, Zhang H, et al. (2007) Stability of the
latent reservoir for HIV-1 in patients receiving valproic acid. J Infect Dis 195:833–836.
21. Adam E, Quivy V, Bex F, Chariot A, Collette Y, et al. (2003) Potentiation of
tumor necrosis factor-induced NF-kappa B activation by deacetylase inhibitors isassociated with a delayed cytoplasmic reappearance of I kappa B alpha. Mol Cell
Biol 23: 6200–6209.
22. Bocklandt S, Blumberg PM, Hamer DH (2003) Activation of latent HIV-1
expression by the potent anti-tumor promoter 12-deoxyphorbol 13-phenylace-
tate. Antiviral Res 59: 89–98.
23. Biancotto A, Grivel JC, Gondois-Rey F, Bettendroffer L, Vigne R, et al. (2004)
Dual role of prostratin in inhibition of infection and reactivation of humanimmunodeficiency virus from latency in primary blood lymphocytes and
lymphoid tissue. J Virol 78: 10507–10515.
24. Kulkosky J, Culnan DM, Roman J, Dornadula G, Schnell M, et al. (2001)Prostratin: activation of latent HIV-1 expression suggests a potential inductive
adjuvant therapy for HAART. Blood 98: 3006–3015.
25. Kulkosky J, Sullivan J, Xu Y, Souder E, Hamer DH, et al. (2004) Expression of
latent HAART-persistent HIV type 1 induced by novel cellular activating
agents. AIDS Res Hum Retroviruses 20: 497–505.
26. Korin YD, Brooks DG, Brown S, Korotzer A, Zack JA (2002) Effects of
prostratin on T-cell activation and human immunodeficiency virus latency.J Virol 76: 8118–8123.
27. Gulakowski RJ, McMahon JB, Buckheit RW Jr, Gustafson KR, Boyd MR
(1997) Antireplicative and anticytopathic activities of prostratin, a non-tumor-promoting phorbol ester, against human immunodeficiency virus (HIV).
Antiviral Res 33: 87–97.
28. Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, et al. (2004) Prostratinantagonizes HIV latency by activating NF-kappaB. J Biol Chem 279:
42008–42017.
29. Brooks DG, Hamer DH, Arlen PA, Gao L, Bristol G, et al. (2003) Molecular
characterization, reactivation, and depletion of latent HIV. Immunity 19:
413–423.
30. Hezareh M, Moukil MA, Szanto I, Pondarzewski M, Mouche S, et al. (2004)
Mechanisms of HIV receptor and co-receptor down-regulation by prostratin:role of conventional and novel PKC isoforms. Antivir Chem Chemother 15:
207–222.
31. Witvrouw M, Pannecouque C, Fikkert V, Hantson A, Van Remoortel B, et al.(2003) Potent and selective inhibition of HIV and SIV by prostratin interacting
with viral entry. Antivir Chem Chemother 14: 321–328.
32. Rullas J, Bermejo M, Garcia-Perez J, Beltan M, Gonzalez N, et al. (2004)
Prostratin induces HIV activation and downregulates HIV receptors in
peripheral blood lymphocytes. Antivir Ther 9: 545–554.
33. Johnson HE, Banack SA, Cox PA (2008) Variability in content of the anti-AIDS
drug candidate prostratin in Samoan populations of Homalanthus nutans. J NatProd 71: 2041–2044.
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 17 June 2009 | Volume 4 | Issue 6 | e6093

34. Wender PA, Kee JM, Warrington JM (2008) Practical synthesis of prostratin,
DPP, and their analogs, adjuvant leads against latent HIV. Science 320:649–652.
35. Gustafson KR, Cardellina JH 2nd, McMahon JB, Gulakowski RJ, Ishitoya J, et
al. (1992) A nonpromoting phorbol from the samoan medicinal plant
Homalanthus nutans inhibits cell killing by HIV-1. J Med Chem 35: 1978–1986.
36. Marks PA, Richon VM, Breslow R, Rifkind RA (2001) Histone deacetylase
inhibitors as new cancer drugs. Curr Opin Oncol 13: 477–483.
37. Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, et al. (2001) Inhibitor of
histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral
and cutaneous T-cell lymphoma: a case report. Blood 98: 2865–2868.
38. Crazzolara R, Johrer K, Johnstone RW, Greil R, Kofler R, et al. (2002) Histone
deacetylase inhibitors potently repress CXCR4 chemokine receptor expression
and function in acute lymphoblastic leukaemia. Br J Haematol 119: 965–969.
39. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003)
Histone deacetylases (HDACs): characterization of the classical HDAC family.
Biochem J 370: 737–749.
40. Dover GJ, Brusilow S, Charache S (1994) Induction of fetal hemoglobin
production in subjects with sickle cell anemia by oral sodium phenylbutyrate.
Blood 84: 339–343.
41. Collins AF, Pearson HA, Giardina P, McDonagh KT, Brusilow SW, et al. (1995)
Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical
trial. Blood 85: 43–49.
42. Duvic M, Vu J (2007) Vorinostat: a new oral histone deacetylase inhibitor
approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs 16:
1111–1120.
43. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, et al. (2001) Histone
Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, MoodStabilizer, and Teratogen. J Biol Chem 276: 36734–36741.
44. Johannessen CU (2000) Mechanisms of action of valproate: a commentatory.
Neurochem Int 37: 103–110.
45. Tunnicliff G (1999) Actions of sodium valproate on the central nervous system.J Physiol Pharmacol 50: 347–365.
46. Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of
epigenetic (and more) treatments for cancer. Nat Rev Cancer 6: 38–51.
47. Yoo CB, Jones PA (2006) Epigenetic therapy of cancer: past, present and future.
Nat Rev Drug Discov 5: 37–50.
48. Marks PA (2007) Discovery and development of SAHA as an anticancer agent.
Oncogene 26: 1351–1356.
49. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval
summary: vorinostat for treatment of advanced primary cutaneous T-cell
lymphoma. Oncologist 12: 1247–1252.
50. Hess-Stumpp H, Bracker TU, Henderson D, Politz O (2007) MS-275, a potent
orally available inhibitor of histone deacetylases–the development of ananticancer agent. Int J Biochem Cell Biol 39: 1388–1405.
51. Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent
infection after acute infection of T cells in vitro. Embo J 22: 1868–1877.
52. Emiliani S, Fischle W, Ott M, Van Lint C, Amella CA, et al. (1998) Mutations in
the tat gene are responsible for human immunodeficiency virus type 1
postintegration latency in the U1 cell line. J Virol 72: 1666–1670.
53. Butler IF, Pandrea I, Marx PA, Apetrei C (2007) HIV genetic diversity:
biological and public health consequences. Curr HIV Res 5: 23–45.
54. Buonaguro L, Tornesello ML, Buonaguro FM (2007) Human immunodeficiency
virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and
therapeutic implications. J Virol 81: 10209–10219.
55. Calao M, Burny A, Quivy V, Dekoninck A, Van Lint C (2008) A pervasive roleof histone acetyltransferases and deacetylases in an NF-kappaB-signaling code.
Trends Biochem Sci 33: 339–349.
56. Verdin E, Paras P Jr, Van Lint C (1993) Chromatin disruption in the promoter
of human immunodeficiency virus type 1 during transcriptional activation.
Embo J 12: 3249–3259.
57. Jiang G, Espeseth A, Hazuda DJ, Margolis DM (2007) c-Myc and Sp1
contribute to proviral latency by recruiting histone deacetylase 1 to the human
immunodeficiency virus type 1 promoter. J Virol 81: 10914–10923.
58. Lusic M, Marcello A, Cereseto A, Giacca M (2003) Regulation of HIV-1 geneexpression by histone acetylation and factor recruitment at the LTR promoter.
Embo J 22: 6550–6561.
59. Chun TW, Justement JS, Moir S, Hallahan CW, Ehler LA, et al. (2001)
Suppression of HIV replication in the resting CD4+ T cell reservoir by
autologous CD8+ T cells: implications for the development of therapeuticstrategies. Proc Natl Acad Sci U S A 98: 253–258.
60. Blankson JN, Persaud D, Siliciano RF (2002) The challenge of viral reservoirs in
HIV-1 infection. Annu Rev Med 53: 557–593.
61. Hezareh M (2005) Prostratin as a new therapeutic agent targeting HIV viral
reservoirs. Drug News Perspect 18: 496–500.
62. Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, et al. (2007)
Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1
transcriptional silencing. Embo J 26: 412–423.
63. Imai K, Okamoto T (2006) Transcriptional repression of human immunode-
ficiency virus type 1 by AP-4. J Biol Chem 281: 12495–12505.
64. Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, et al. (2006) NF-
kappaB p50 promotes HIV latency through HDAC recruitment and repressionof transcriptional initiation. Embo J 25: 139–149.
65. He G, Margolis DM (2002) Counterregulation of chromatin deacetylation and
histone deacetylase occupancy at the integrated promoter of human immuno-
deficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1
activator Tat. Mol Cell Biol 22: 2965–2973.
66. Coull JJ, He G, Melander C, Rucker VC, Dervan PB, et al. (2002) Targeted
derepression of the human immunodeficiency virus type 1 long terminal repeat
by pyrrole-imidazole polyamides. J Virol 76: 12349–12354.
67. Steel A, Clark S, Teo I, Shaunak S, Nelson M, et al. (2006) No change to HIV-1
latency with valproate therapy. Aids 20: 1681–1682.
68. Hassan AH, Neely KE, Vignali M, Reese JC, Workman JL (2001) Promoter
targeting of chromatin-modifying complexes. Front Biosci 6: D1054–1064.
69. Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, et al. (2000) Ordered
recruitment of chromatin modifying and general transcription factors to the
IFN-beta promoter. Cell 103: 667–678.
70. Koutroubas G, Merika M, Thanos D (2008) Bypassing the requirements for
epigenetic modifications in gene transcription by increasing enhancer strength.
Mol Cell Biol 28: 926–938.
71. Henderson A, Holloway A, Reeves R, Tremethick DJ (2004) Recruitment of
SWI/SNF to the human immunodeficiency virus type 1 promoter. Mol Cell Biol
24: 389–397.
72. Watanabe H, Mizutani T, Haraguchi T, Yamamichi N, Minoguchi S, et al.
(2006) SWI/SNF complex is essential for NRSF-mediated suppression of
neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene 25:
470–479.
73. Singh M, Popowicz GM, Krajewski M, Holak TA (2007) Structural ramification
for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a
central ATPase of the SWI/SNF remodeling complex. Chembiochem 8:
1308–1316.
74. Choudhary SK, Archin NM, Margolis DM (2008) Hexamethylbisacetamide and
disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells.
J Infect Dis 197: 1162–1170.
75. Huang B, Yang XD, Zhou MM, Ozato K, Chen LF (2009) Brd4 coactivates
transcriptional activation of NF-kappaB via specific binding to acetylated RelA.
Mol Cell Biol 29: 1375–1387.
76. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, et al. (2005) The
bromodomain protein Brd4 is a positive regulatory component of P-TEFb and
stimulates RNA polymerase II-dependent transcription. Mol Cell 19: 523–534.
77. Yang Z, Yik JH, Chen R, He N, Jang MK, et al. (2005) Recruitment of P-TEFb
for stimulation of transcriptional elongation by the bromodomain protein Brd4.
Mol Cell 19: 535–545.
78. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K (2003) The double
bromodomain protein Brd4 binds to acetylated chromatin during interphase
and mitosis. Proc Natl Acad Sci U S A 100: 8758–8763.
79. Sung TL, Rice AP (2006) Effects of prostratin on Cyclin T1/P-TEFb function
and the gene expression profile in primary resting CD4+ T cells. Retrovirology
3: 66.
80. Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, et al. (2009)
Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide
hydroxamic acid. AIDS Res Hum Retroviruses 25: 207–212.
81. Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, et al. (2009)
Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells.
J Biol Chem 284: 6782–6789.
82. Katz RA, Jack-Scott E, Narezkina A, Palagin I, Boimel P, et al. (2007) High-
frequency epigenetic repression and silencing of retroviruses can be antagonized
by histone deacetylase inhibitors and transcriptional activators, but uniform
reactivation in cell clones is restricted by additional mechanisms. J Virol 81:
2592–2604.
83. Winslow BJ, Pomerantz RJ, Bagasra O, Trono D (1993) HIV-1 latency due to
the site of proviral integration. Virology 196: 849–854.
84. Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS (1987) Cytokine-
induced expression of HIV-1 in a chronically infected promonocyte cell line.
Science 238: 800–802.
85. Weiss A, Wiskocil RL, Stobo JD (1984) The role of T3 surface molecules in the
activation of human T cells: a two-stimulus requirement for IL 2 production
reflects events occurring at a pre-translational level. J Immunol 133: 123–128.
86. Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, et al.
(2000) Functional differences between the long terminal repeat transcriptional
promoters of human immunodeficiency virus type 1 subtypes A through G. J
Virol 74: 3740–3751.
87. Schoonbroodt S, Ferreira V, Best-Belpomme M, Boelaert JR, Legrand-Poels S,
et al. (2000) Crucial role of the amino-terminal tyrosine residue 42 and the
carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by
an oxidative stress. J Immunol 164: 4292–4300.
88. Lambotte O, Chaix ML, Gubler B, Nasreddine N, Wallon C, et al. (2004) The
lymphocyte HIV reservoir in patients on long-term HAART is a memory of
virus evolution. Aids 18: 1147–1158.
89. Ott M, Lovett JL, Mueller L, Verdin E (1998) Superinduction of IL-8 in T Cells
by HIV-1 Tat Protein Is Mediated Through NF-{kappa}B Factors. J Immunol
160: 2872–2880.
90. Costello R, Lipcey C, Algarte M, Cerdan C, Baeuerle PA, et al. (1993)
Activation of primary human T-lymphocytes through CD2 plus CD28 adhesion
molecules induces long-term nuclear expression of NF-kappa B. Cell Growth
Differ 4: 329–339.
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 18 June 2009 | Volume 4 | Issue 6 | e6093

91. Costello R, Cerdan C, Pavon C, Brailly H, Hurpin C, et al. (1993) The CD2 and
CD28 adhesion molecules induce long-term autocrine proliferation of CD4+ Tcells. Eur J Immunol 23: 608–613.
92. Pierres A, Lopez M, Cerdan C, Nunes J, Olive D, et al. (1988) Triggering CD 28
molecules synergize with CD 2 (T 11.1 and T 11.2)-mediated T cell activation.Eur J Immunol 18: 685–690.
93. Verdin E (1991) DNase I-hypersensitive sites are associated with both long
terminal repeats and with the intragenic enhancer of integrated human
immunodeficiency virus type 1. J Virol 65: 6790–6799.
Reactivation of Latent HIV-1
PLoS ONE | www.plosone.org 19 June 2009 | Volume 4 | Issue 6 | e6093
Top Related
Copyright © 2022 FDOKUMEN

