







Bahasa
Halaman
Hukum


EXPERIMENTAL CELL RESEARCH 238, 422–429 (1998)ARTICLE NO. EX973852
Serum Deprivation and Protein Synthesis Inhibition Induce TwoDifferent Apoptotic Processes in N18 Neuroblastoma Cells
J. Boix,1 J. Fibla,1 V.-J. Yuste, J. M. Piulats, N. Llecha, and J. X. Comella2
Molecular Neurobiology Group, Departament de Ciencies Mediques Basiques, Universitat de Lleida,Av. Rovira Roure, 44, 25198 Lleida, Catalonia, Spain
most neural populations, the extent of PCD by apoptosisN18 are murine neuroblastoma cells that underwent amounts around a 50% reduction in the initial number
cell death upon serum deprivation or inhibition of pro- of neurons. The basis of the phenomenon is a competi-tein synthesis by means of cycloheximide (CHX). In tion for gaining access to a limited supply of neuro-both cases, an ultrastructural morphology and an in- trophic factor provided by the target tissue of inner-ternucleosomal pattern of DNA fragmentation typical vation [2]. These trophic factor-dependencies and theof apoptosis were found. However, electron micros- apoptotic processes triggered upon trophic factor-depri-copy revealed abundant lipid vesicles in the cytoplasm vation have been described in cultures of neural [3–5],of CHX-treated cells that were not found in their se- as well as nonneural cells [6–8]. Alternatively, apoptosisrum-deprived counterparts. In addition, when both can be found in cells facing nonprogrammed stress stim-types of apoptotic cells were compared by means of uli such as g irradiation, genotoxic drugs, virus infec-flow cytometry and chromatin staining with propid- tion, or cytotoxic lymphocyte attacks. In these cases,ium iodide, the former showed consistently less fluo- macromolecular synthesis inhibition by means of CHXrescence than the latter. Therefore, in N18 cells, both
or actinomycin D does not prevent apoptosis and theirapoptotic processes seemed to differ at a structuraleffects range from neutral to detrimental. Moreover,level. At a functional level, we found that apoptosisCHX have proven to be able to trigger apoptosis in sev-was blocked by the protease inhibitor TLCK in CHX-eral cell lines [9]. It has been proposed that cells under-treated but not in serum-deprived cells. On the othergoing apoptosis when protein synthesis is inhibited,hand, we generated N18 clones that overexpressed Bcl-have already activated a program of apoptosis that is2 protein. After a period of 48 h we found that identicalheld in check by an inhibitory protein with an elevatedlevels of Bcl-2 protein were able to block apoptosis inturnover. Thus, the cell inability to synthesize this pro-serum-deprived but not in CHX-treated cells. In con-
clusion, two different biochemical pathways leading tein would release apoptosis and a paradigm of apop-to apoptosis seem to coexist in N18 neuroblastoma tosis release has been suggested [10, 11].cells. q 1998 Academic Press Apoptosis is a process of cell death essentially charac-
Key Words: apoptosis; neuroblastoma; cyclohexi- terized by a set of morphological events. Cytoplasmmide; protease inhibitors; Bcl-2. condensation and cell shrinkage with structural integ-
rity of mitochondria and lysosomes, chromatin compac-tion, membrane blebbing, and further cell fragmenta-tion into apoptotic bodies constitute the most acceptedINTRODUCTIONcell morphology of the process [12]. At a molecularlevel, a laddered pattern of DNA degradation causedApoptosis is a type of cell death involved in homeo-by the activation of endogenous endonucleases on astatic and pathogenic processes that are characteristicpreserved chromatin substructure seems to be the mostof multicellular organisms [1]. In many cases, these pro-unequivocal sign of apoptosis [13]. However, some genecesses seem to be developmentally regulated and requireproducts are becoming of major interest as regulatorscontinued RNA and protein synthesis in order to be ac-of apoptosis [14]. For instance a family of proteasescomplished. As a consequence, the concept of pro-related to interleukin-1b converting enzyme (ICE),grammed cell death (PCD) is applied. A remarkable ex-which activation seems to be required for apoptosis toample of PCD is found in vertebrates during the develop-
ment of the central and peripheral nervous system. In be executed [15, 16]. Another example is the family ofproteins with structural or functional homology to Bcl-2 [17]. Bcl-2 and some members of this family, when
1 Both researchers have contributed equally to this work and overexpressed, are able to block apoptosis [18–22]. Inshould be considered first coauthors.contrast, overexpression of the other members enhance2 To whom correspondence and reprint requests should be ad-
dressed. Fax: (34) 73702426. E-mail: [email protected]. apoptotic cell death [23–27]. It is demonstrated that
4220014-4827/98 $25.00Copyright q 1998 by Academic PressAll rights of reproduction in any form reserved.
AID ECR 3852 / 6i2d$$$261 01-09-98 08:25:15 ecal

423ANALYSIS OF TWO APOPTOTIC PROCESSES IN N18 CELLS
struct were transfected in N18TG2 cells by lipofection with the Lipo-these proteins interact and can form homo- or hetero-fectin reagent (GIBCO BRL/Izasa, Barcelona, Spain). Following G-dimers [28, 29]. It is known that Bcl-2 is an intracellu-418 selection, several cell clones were obtained and their Bcl-2 con-lar, membrane-bound protein, located in mitochondria, tent was determined by means of Western blotting. A highly ex-
endoplasmic reticulum, and nuclear envelope [30–32]. pressor clone was selected and named N18.JB15. The pcDNA3/bcl2-and the pcDNA3-lipofected pools were named N18.pcDNA3/bcl2 andResults have been reported involving Bcl-2 in cell pro-N18.pcDNA3, respectively. N18.pcDNA3 was used as the Bcl-2 non-tection from reactive oxygen species [33] and the con-expressing control in further experiments.trol of subcellular trafficking [34–36]. However, the
Quantification of cell death. In our experiments, two differentprecise mechanism, through which this family of pro- procedures were used to assess the proportion of cell death. Theteins exert their cellular functions is unknown. trypan blue staining procedure [44] and the flow cytometry method
reported by Darzynkiewicz and Li [45]. Values were obtained from,N18 is a neuronal cell line obtained from a mouseat least, three independent experiments and loaded into a Microsoftneuroblastoma [37] that has proved useful in the studyExcel data sheet. Means { SEM, diagrams, and other statisticalof ganglioside GM1 effects [38] and the characteriza-parameters were the result of processing all data through the afore-
tion of anandamide as an endogenous ligand for canna- mentioned software. The flow cytometer used in these experimentsbinoid receptors [39]. The N18TG2 subclone is defective was an EPICS XL from Coulter/Instrumentation Laboratories (Bar-
celona, Spain).in the enzyme HPRT (hypoxanthine phosphoribosyl-Electron microscopy. Healthy or apoptotic (1 1 106) cells from 60-transferase) and, as a consequence, allows the selection
mm culture plates were gently pelleted by centrifugation and washedof cell hybrids following its fusion to somatic cells. Hy-twice with PBS. Fixation was performed with 100 mM phosphatebrids of N18TG2 and embryonic neurons have been buffer (pH 7.4), containing 2.5% glutaraldehyde, for 30 min at 47C.
obtained in order to address experimental issues re- Then, pellets were rinsed twice with cold-PBS, postfixed in bufferedOsO4, dehydrated in graded acetone, and embedded in Durcupan ACMlated to developmentally regulated dependence on neu-resin (Fluka, Buchs, Switzerland). Ultrathin sections were obtained,rotrophic factors and apoptosis [40, 41]. Previous tomounted in copper grids, and counterstained with uranyl acetate andfurther studies with cell hybrids, we decided to analyzelead citrate. Specimens were studied with a Zeiss EM910 electron
the behavior of N18TG2 cells facing serum starvation, microscope. Control, serum-deprived, and CHX-treated cell prepara-macromolecular synthesis inhibition, or both simulta- tions were obtained from, at least, two independent experiments.neously. We found that cycloheximide was able to in- Analysis of DNA fragmentation. Cells from 60-mm culture dishes
(approximately 1 1 106) were collected in the culture medium, pel-duce apoptosis by a process dependent upon the activa-leted at 400g for 5 min, and rinsed twice with PBS. The pellet wastion of a putative TLCK-sensitive protease and notthen homogenized in 500 ml of a lysis buffer (100 mM CINa, 25 mMblocked by Bcl-2 overexpression. On the other hand, EDTA, 0.5% SDS, 10 mM Tris/CIH, pH 8.0) by means of an insulin
apoptosis by serum deprivation was prevented by Bcl- syringe. Proteinase K and DNase-free RNase were added to a finalconcentration of 100 and 10 mg/ml, respectively. Digestion was al-2 overexpression and not blocked by TLCK.lowed to proceed for 60 min at 507C. Then two consecutive phenol/chloroform/isoamyl alcohol (25:24:1) extractions were performed, fol-MATERIALS AND METHODS lowed by a precipitation with half a volume of 7.5 M ammoniumacetate and 2 vol of ethanol. The DNA was resuspended in TE (1
Cell culture conditions and reagents. N18TG2 cells were obtained mM EDTA, 10 mM Tris/CIH, pH 8.0) and its concentration measuredfrom Dr. Cashman (McGill University, Montreal, Quebec, Canada). by 260 nm absorptance. Equal DNA amounts were electrophoresedCells were routinely maintained at 37.27C in a saturating humidity in 1.5% agarose gels.atmosphere containing 95% air and 5% CO2. The growth medium
Protein extractions and Western blotting. Cells (in the range 0.5–was DMEM supplemented with 2 mM L-glutamine, antibiotics, and1 1 106) were detached from 60-mm culture dishes, pelleted at 400g10% heat-inactivated fetal calf serum (FCS), namely complete me-for 5 min, and washed twice with PBS at 47C. Then 250 ml of extrac-dium. Serum deprivation was performed by removing the completetion buffer (150 mM CINa, 5 mM EDTA, 1% Nonidet P-40, 1 mMmedium, rinsing twice with medium not containing FCS, and furtherPMSF, 20 mg/ml Leupeptin, 10 mg/ml Aprotinin in 50 mM Tris/CIH,culturing in this uncomplete medium for 24, 48, or 72 h. CHX waspH 7.2) was added. Pellets were gently disrupted by pipetting anddissolved in complete medium and was adjusted to the final concen-extraction let to proceed on ice for 5 min. Nuclear material wastrations referred to later in the text. Cells were placed in CHX-pelleted by centrifuging at 20,000g for 30 min and a clear superna-containing medium after being rinsed twice like their serum-de-tant, devoid of chromatin proteins, was obtained. These supernatantsprived counterparts. The extent of protein synthesis inhibition,were subjected to determination of total protein concentration bycaused by a 10 ng/ml–10 mg/ml range of CHX concentrations, wasmeans of the Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA).determined by culturing cells in the presence of trans-35S-label (aEqual amounts of protein were loaded into SDS–12% polyacrylamide35S-labeled Met and Cys mixture from ICN-Flow, Bucks, UK) andgel electrophoresis [46]. Proteins were electrotransferred from thefurther measurement of the label incorporated into protein by thegels to Immobilon-P filters (Millipore, Bedford, MA) and reacted withmethod of DiStefano et al. [42]. Benzamidine, Pepstatin, Leupeptin,the anti-human Bcl-2 monoclonal antibody, clone 124 (DAKO,PMSF (phenylmethylsulfonyl fluoride), and TLCK (N-tosyl-L-lysineGlostrup, Denmark). Immunoblots were further developed with anchloromethylketone) were dissolved first in DMSO and then addedanti-mouse IgG (Fab-specific) peroxidase conjugate from Sigma (St.to the culture medium. Final DMSO concentrations did not surpassLouis, MO) and ECL (Amersham, Buckinghamshire, UK).harmless 0.1% ratios. Culture dishes were purchased from Corning
(NY). Unless otherwise stated, all drugs and reagents were obtained RESULTSfrom Sigma (St. Louis, MO).
Cell transfections. MTbcl-2TKNeo plasmid was kindly supplied Serum Deprivation and CHX Treatment Induceby Dr. David L. Vaux [43] and its human bcl-2 cDNA insert was Apoptosis in N18 Cellsrecovered and placed under the control of a CMV constitutive pro-
N18 cells were deprived of serum for 24, 48, and 72 hmoter in the pcDNA3 expression vector (Invitrogen BV, Leek, TheNetherlands). Then the pcDNA3 vector and the pcDNA3/bcl-2 con- following the procedure reported under Materials and
AID ECR 3852 / 6i2d$$$262 01-09-98 08:25:15 ecal

424 BOIX ET AL.
cavity of the nuclear envelope was evidenced. The flat-tened profiles of the endoplasmic reticulum were ab-sent and seemed replaced by large, rounded, mem-brane-bounded vacuoles from which ribosomes weredetached. Some lipid droplets were also present. Mildchanges in mitochondrial structure indicated a certaindegree of damage that was not detected in control orserum-deprived cells. In a summary, the morphologicalpattern of apoptosis in CHX-treated cells could be dif-ferentiated from the serum-deprived one.
Characterization of Apoptosis in N18 Cells by MeansFIG. 1. Time course of N18 cell death as measured by trypanof Flow Cytometryblue-stained cells. Serum-deprived (,), 0.5 mg/ml CHX-treated (L),
and control-cultured N18 cells (h). Given values are the mean /In order to better quantify the apoptotic phenomenonSEM from three independent experiments.
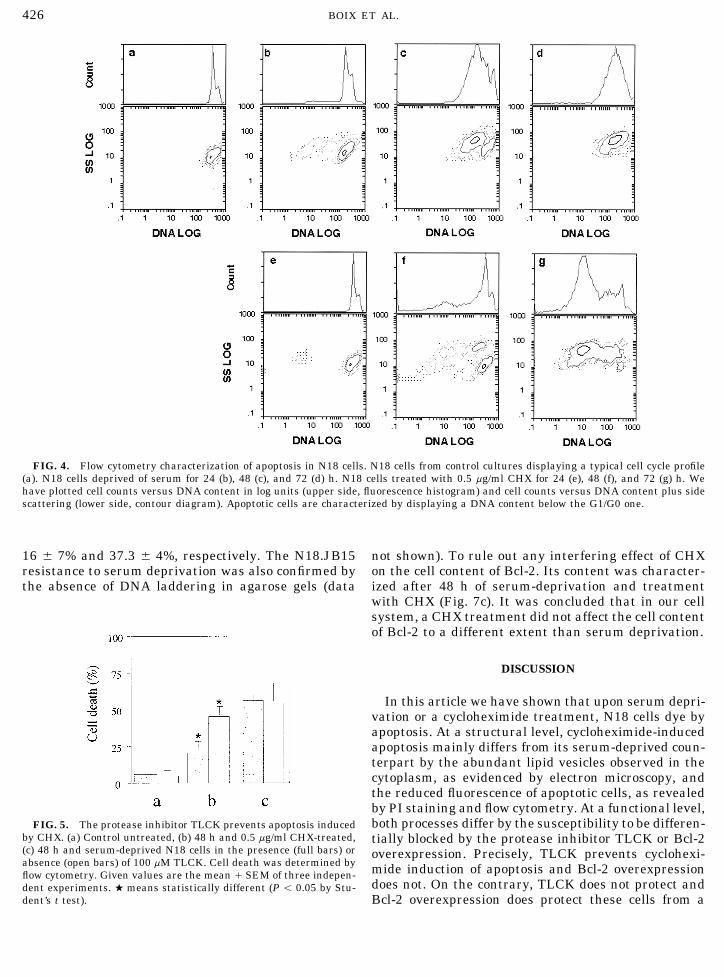
in cell populations, we decided to perform flow cytome-try studies in N18 cells. DNA was labeled with propid-ium iodide (PI) following the procedure referred to in
Methods section. Cell death was assessed by means of the procedures section. By distinguishing its DNA con-the trypan blue procedure. Dead cells rated 20.7 { 8.4, tent, this method evidenced the G1/G0, S, and G2/mito-67.1 { 13.5, and 93.7 { 4%, respectively. An analogous sis phases of the cell cycle in N18 cells (Fig. 4a). It istime course was determined by treating these cells with known that apoptotic cells and apoptotic bodies are0.5 mg/ml CHX and the ratios of cell death were 9.4 { 9, characterized by a DNA content below the G1/G0 one;45.7 { 11.6, and 59.9 { 14.3% (Fig. 1). At this CHX however, apoptotic bodies display a reduced size andconcentration, the incorporation of trans-35S-label into complexity that allows identification by forward versusprotein was determined after 24 h and accounted less side scatter measurements (FS/SS). In order to excludethan a 10% of the value displayed by untreated N18 cells. these and other undesirable particles, only those dis-
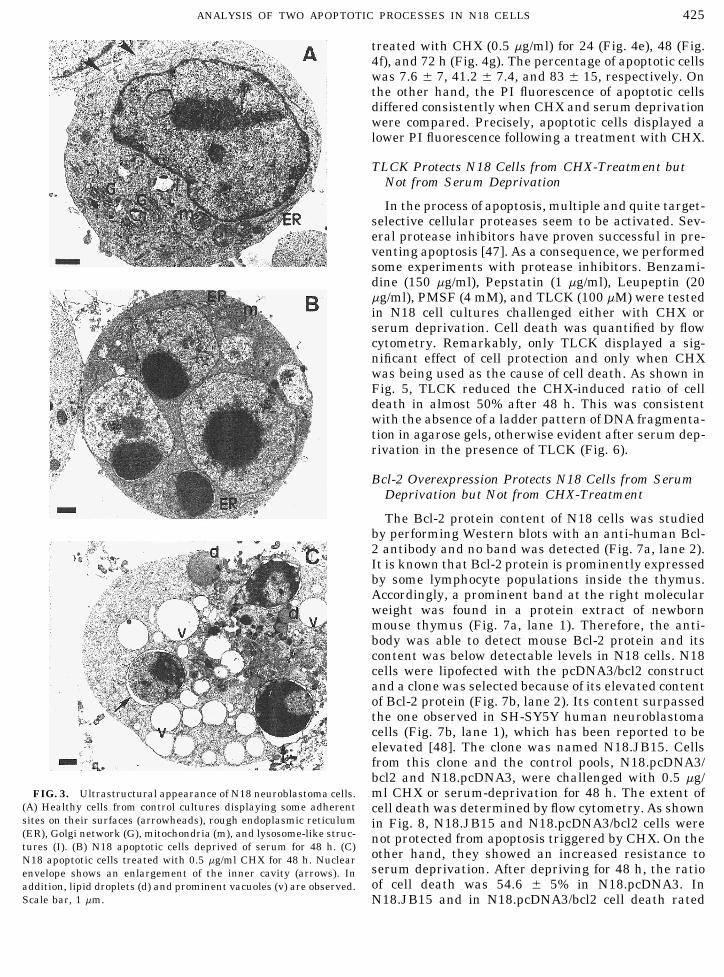
DNA from deprived, CHX-treated, and control, playing a FS/SS value typical of healthy N18 cells werehealthy cells was extracted and resolved in agarose analyzed. N18 cells were deprived of serum for periodsgels to better characterize the cell death process. The of 24 (Fig. 4b), 48 (Fig. 4c), and 72 h (Fig. 4d) and theobservation that deprived and CHX-treated cells exhib- percentage of apoptotic cells found to be 22.6 { 7, 53.2ited a ladder pattern of DNA fragmentation, which is { 15, and 81.6 { 15.2, successively. N18 cells were alsoconsidered a hallmark of apoptosis, suggested this typeof cell death (Fig. 2). At the beginnings, apoptosis wasand, to a great extent, continues to be a morphologicaldefinition. As a consequence, we decided to performelectron microscope studies (Fig. 3). We observed thatcontrol, healthy cells showed an spherical morphologywith the exception of some surface-flattened outlinesin which cells became adherent to other cells. The api-cal cell surface displayed also some poorly developedfinger-like processes. A large nucleus containing dis-crete amounts of peripherally located heterochromatinwith some internal aggregates was seen; however,loosely arranged euchormatin predominated. The cyto-plasm contained poorly developed rough endoplasmicreticulum, prominent Golgi profiles, some mitochon-dria, and lysosome-like structures. After serum depri-vation, N18 cells exhibited the characteristic ultra-structural pattern of apoptosis. Nucleus showed astrong structural transformation based on an extremechromatin compaction and segmentation. The densityof the cytoplasm matrix was increased but organellessuch as rough endoplasmic reticulum or mitochondriahad normal ultrastructural appearance. Following a
FIG. 2. DNA analysis in 1.5% agarose gels showing a laddertreatment with CHX, apoptotic changes in the nuclei pattern of DNA degradation characteristic of apoptosis. (a) N18 cellssimilar to those seen in serum-deprived cells were from control cultures. (b) N18 cells treated with 0.5 mg/ml CHX for
48 h. (c) N18 cells deprived of serum for 48 h.found but, in addition, an enlargement of the inner
AID ECR 3852 / 6i2d$$$262 01-09-98 08:25:15 ecal
425ANALYSIS OF TWO APOPTOTIC PROCESSES IN N18 CELLS
treated with CHX (0.5 mg/ml) for 24 (Fig. 4e), 48 (Fig.4f), and 72 h (Fig. 4g). The percentage of apoptotic cellswas 7.6 { 7, 41.2 { 7.4, and 83 { 15, respectively. Onthe other hand, the PI fluorescence of apoptotic cellsdiffered consistently when CHX and serum deprivationwere compared. Precisely, apoptotic cells displayed alower PI fluorescence following a treatment with CHX.
TLCK Protects N18 Cells from CHX-Treatment butNot from Serum Deprivation
In the process of apoptosis, multiple and quite target-selective cellular proteases seem to be activated. Sev-eral protease inhibitors have proven successful in pre-venting apoptosis [47]. As a consequence, we performedsome experiments with protease inhibitors. Benzami-dine (150 mg/ml), Pepstatin (1 mg/ml), Leupeptin (20mg/ml), PMSF (4 mM), and TLCK (100 mM) were testedin N18 cell cultures challenged either with CHX orserum deprivation. Cell death was quantified by flowcytometry. Remarkably, only TLCK displayed a sig-nificant effect of cell protection and only when CHXwas being used as the cause of cell death. As shown inFig. 5, TLCK reduced the CHX-induced ratio of celldeath in almost 50% after 48 h. This was consistentwith the absence of a ladder pattern of DNA fragmenta-tion in agarose gels, otherwise evident after serum dep-rivation in the presence of TLCK (Fig. 6).
Bcl-2 Overexpression Protects N18 Cells from SerumDeprivation but Not from CHX-Treatment
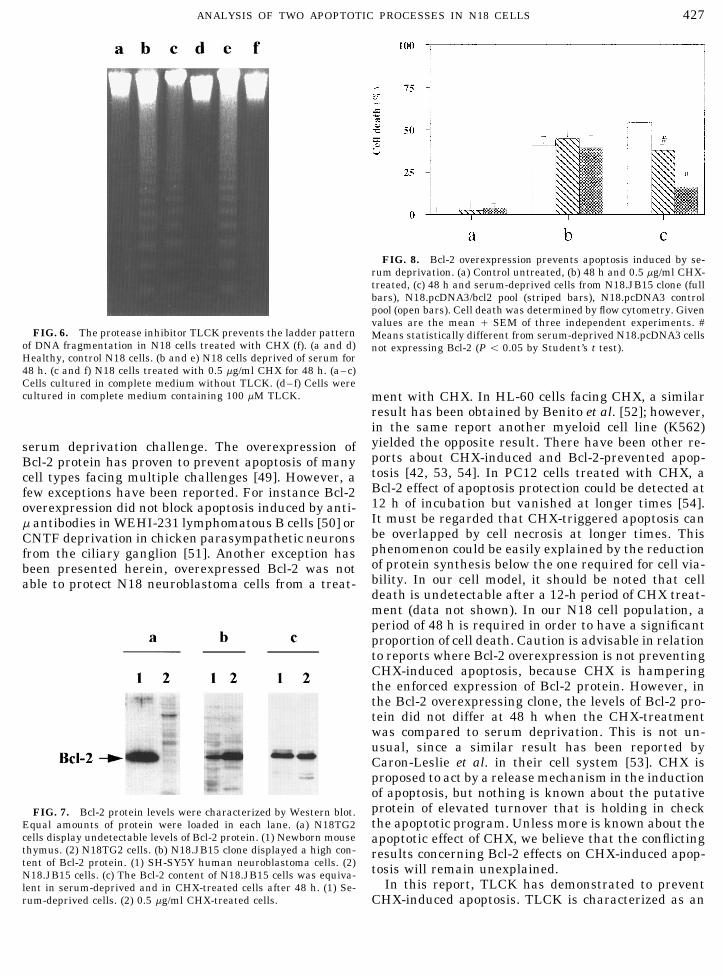
The Bcl-2 protein content of N18 cells was studiedby performing Western blots with an anti-human Bcl-2 antibody and no band was detected (Fig. 7a, lane 2).It is known that Bcl-2 protein is prominently expressedby some lymphocyte populations inside the thymus.Accordingly, a prominent band at the right molecularweight was found in a protein extract of newbornmouse thymus (Fig. 7a, lane 1). Therefore, the anti-body was able to detect mouse Bcl-2 protein and itscontent was below detectable levels in N18 cells. N18cells were lipofected with the pcDNA3/bcl2 constructand a clone was selected because of its elevated contentof Bcl-2 protein (Fig. 7b, lane 2). Its content surpassedthe one observed in SH-SY5Y human neuroblastomacells (Fig. 7b, lane 1), which has been reported to beelevated [48]. The clone was named N18.JB15. Cellsfrom this clone and the control pools, N18.pcDNA3/bcl2 and N18.pcDNA3, were challenged with 0.5 mg/ml CHX or serum-deprivation for 48 h. The extent ofFIG. 3. Ultrastructural appearance of N18 neuroblastoma cells.
(A) Healthy cells from control cultures displaying some adherent cell death was determined by flow cytometry. As shownsites on their surfaces (arrowheads), rough endoplasmic reticulum in Fig. 8, N18.JB15 and N18.pcDNA3/bcl2 cells were(ER), Golgi network (G), mitochondria (m), and lysosome-like struc- not protected from apoptosis triggered by CHX. On thetures (I). (B) N18 apoptotic cells deprived of serum for 48 h. (C) other hand, they showed an increased resistance toN18 apoptotic cells treated with 0.5 mg/ml CHX for 48 h. Nuclear
serum deprivation. After depriving for 48 h, the ratioenvelope shows an enlargement of the inner cavity (arrows). Inof cell death was 54.6 { 5% in N18.pcDNA3. Inaddition, lipid droplets (d) and prominent vacuoles (v) are observed.
Scale bar, 1 mm. N18.JB15 and in N18.pcDNA3/bcl2 cell death rated
AID ECR 3852 / 6i2d$$$263 01-09-98 08:25:15 ecal
426 BOIX ET AL.
FIG. 4. Flow cytometry characterization of apoptosis in N18 cells. N18 cells from control cultures displaying a typical cell cycle profile(a). N18 cells deprived of serum for 24 (b), 48 (c), and 72 (d) h. N18 cells treated with 0.5 mg/ml CHX for 24 (e), 48 (f), and 72 (g) h. Wehave plotted cell counts versus DNA content in log units (upper side, fluorescence histogram) and cell counts versus DNA content plus sidescattering (lower side, contour diagram). Apoptotic cells are characterized by displaying a DNA content below the G1/G0 one.
16 { 7% and 37.3 { 4%, respectively. The N18.JB15 not shown). To rule out any interfering effect of CHXon the cell content of Bcl-2. Its content was character-resistance to serum deprivation was also confirmed by
the absence of DNA laddering in agarose gels (data ized after 48 h of serum-deprivation and treatmentwith CHX (Fig. 7c). It was concluded that in our cellsystem, a CHX treatment did not affect the cell contentof Bcl-2 to a different extent than serum deprivation.
DISCUSSION
In this article we have shown that upon serum depri-vation or a cycloheximide treatment, N18 cells dye byapoptosis. At a structural level, cycloheximide-inducedapoptosis mainly differs from its serum-deprived coun-terpart by the abundant lipid vesicles observed in thecytoplasm, as evidenced by electron microscopy, andthe reduced fluorescence of apoptotic cells, as revealedby PI staining and flow cytometry. At a functional level,both processes differ by the susceptibility to be differen-FIG. 5. The protease inhibitor TLCK prevents apoptosis induced
by CHX. (a) Control untreated, (b) 48 h and 0.5 mg/ml CHX-treated, tially blocked by the protease inhibitor TLCK or Bcl-2(c) 48 h and serum-deprived N18 cells in the presence (full bars) or overexpression. Precisely, TLCK prevents cyclohexi-absence (open bars) of 100 mM TLCK. Cell death was determined by mide induction of apoptosis and Bcl-2 overexpressionflow cytometry. Given values are the mean / SEM of three indepen-
does not. On the contrary, TLCK does not protect anddent experiments. w means statistically different (P õ 0.05 by Stu-dent’s t test). Bcl-2 overexpression does protect these cells from a
AID ECR 3852 / 6i2d$$$263 01-09-98 08:25:15 ecal
427ANALYSIS OF TWO APOPTOTIC PROCESSES IN N18 CELLS
FIG. 8. Bcl-2 overexpression prevents apoptosis induced by se-rum deprivation. (a) Control untreated, (b) 48 h and 0.5 mg/ml CHX-treated, (c) 48 h and serum-deprived cells from N18.JB15 clone (fullbars), N18.pcDNA3/bcl2 pool (striped bars), N18.pcDNA3 controlpool (open bars). Cell death was determined by flow cytometry. Givenvalues are the mean / SEM of three independent experiments. #
FIG. 6. The protease inhibitor TLCK prevents the ladder pattern Means statistically different from serum-deprived N18.pcDNA3 cellsof DNA fragmentation in N18 cells treated with CHX (f). (a and d) not expressing Bcl-2 (P õ 0.05 by Student’s t test).Healthy, control N18 cells. (b and e) N18 cells deprived of serum for48 h. (c and f) N18 cells treated with 0.5 mg/ml CHX for 48 h. (a–c)Cells cultured in complete medium without TLCK. (d–f) Cells werecultured in complete medium containing 100 mM TLCK. ment with CHX. In HL-60 cells facing CHX, a similar
result has been obtained by Benito et al. [52]; however,in the same report another myeloid cell line (K562)yielded the opposite result. There have been other re-serum deprivation challenge. The overexpression ofports about CHX-induced and Bcl-2-prevented apop-Bcl-2 protein has proven to prevent apoptosis of manytosis [42, 53, 54]. In PC12 cells treated with CHX, acell types facing multiple challenges [49]. However, aBcl-2 effect of apoptosis protection could be detected atfew exceptions have been reported. For instance Bcl-212 h of incubation but vanished at longer times [54].overexpression did not block apoptosis induced by anti-It must be regarded that CHX-triggered apoptosis canm antibodies in WEHI-231 lymphomatous B cells [50] orbe overlapped by cell necrosis at longer times. ThisCNTF deprivation in chicken parasympathetic neuronsphenomenon could be easily explained by the reductionfrom the ciliary ganglion [51]. Another exception hasof protein synthesis below the one required for cell via-been presented herein, overexpressed Bcl-2 was notbility. In our cell model, it should be noted that cellable to protect N18 neuroblastoma cells from a treat-death is undetectable after a 12-h period of CHX treat-ment (data not shown). In our N18 cell population, aperiod of 48 h is required in order to have a significantproportion of cell death. Caution is advisable in relationto reports where Bcl-2 overexpression is not preventingCHX-induced apoptosis, because CHX is hamperingthe enforced expression of Bcl-2 protein. However, inthe Bcl-2 overexpressing clone, the levels of Bcl-2 pro-tein did not differ at 48 h when the CHX-treatmentwas compared to serum deprivation. This is not un-usual, since a similar result has been reported byCaron-Leslie et al. in their cell system [53]. CHX isproposed to act by a release mechanism in the inductionof apoptosis, but nothing is known about the putativeprotein of elevated turnover that is holding in checkFIG. 7. Bcl-2 protein levels were characterized by Western blot.the apoptotic program. Unless more is known about theEqual amounts of protein were loaded in each lane. (a) N18TG2
cells display undetectable levels of Bcl-2 protein. (1) Newborn mouse apoptotic effect of CHX, we believe that the conflictingthymus. (2) N18TG2 cells. (b) N18.JB15 clone displayed a high con- results concerning Bcl-2 effects on CHX-induced apop-tent of Bcl-2 protein. (1) SH-SY5Y human neuroblastoma cells. (2) tosis will remain unexplained.N18.JB15 cells. (c) The Bcl-2 content of N18.JB15 cells was equiva-
In this report, TLCK has demonstrated to preventlent in serum-deprived and in CHX-treated cells after 48 h. (1) Se-rum-deprived cells. (2) 0.5 mg/ml CHX-treated cells. CHX-induced apoptosis. TLCK is characterized as an
AID ECR 3852 / 6i2d$$$263 01-09-98 08:25:15 ecal

428 BOIX ET AL.
4. Comella, J. X., Sanz-RodrıB guez, C., Aldea, M., and Esquerda,irreversible inhibitor of trypsin-like serine proteases;J. E. (1994) J. Neurosci. 14, 2674–2686.however, it has also proven very effective at inhibiting
5. Greene, L. A. (1978) J. Cell Biol. 78, 747–755.the ICE-like protease activity that causes nuclear lam-6. Collins, M. K. L., Marvel, J., Malde, P., and Lopez-Rivas, A.ina degradation in cell-free systems of apoptosis [55].
(1992) J. Exp. Med. 176, 1043–1051.However, in the same report, the inhibition of the lam-7. Evan, G. I., Wyllie, A. H., Gilbert, C. S., Littlewood, T. D., Land,ina-protease did not prevent other nuclear apoptotic H., Brooks, M., Waters, C. M., Penn, L. Z., and Hancock, D. C.
phenomena like chromatin condensation and the (1992) Cell 69, 119–128.nucleosomal ladder formation. In our CHX-induced 8. Kelley, L. L., Koury, M. J., and Bondurant, M. C. (1992) J. Cell
Physiol. 151, 487–496.model of apoptosis, TLCK was not only able to prevent9. Colombel, M., Olsson, C. A., Ng, P.-Y., and Buttyan, R. (1992)cell death but the ladder pattern of DNA degradation
Cancer Res. 52, 4313–4319.as well. Therefore, another protease different from the10. Martin, S. J. (1993) Trends Cell Biol. 3, 141–144.lamina-one should be involved. In rat thymocytes, a11. Martin, S. J., Lennon, S. V., Bonham, A. M., and Cotter, T. G.TLCK-targeted protease has been reported to be acti-
(1990) J. Immunol. 145, 1859–1867.vated early in several apoptotic processes [56]. Our re-12. Kerr, J. F. R., Gobe, G. C., Winterford, C. M., and Harmon, B. V.sults favor the hypothetical scheme suggested by Ku-
(1995) in ‘‘Methods in Cell Biology’’ (L. M. Schwartz and B. A.mar and Harvey [47], which places a TLCK-sensitive Osborne, Eds.), Vol. 46, pp. 1–27, Academic Press, San Diego.protease early in the activation cascade of proteases 13. Bortner, C. D., Oldenburg, N. B. E., and Cidlowski, J. A. (1995)that characterizes apoptosis. Trends Cell Biol. 5, 21–26.
It is known that cells can undergo apoptosis facing 14. Steller, H. (1995) Science 267, 1445–1449.many different stimuli, but despite these diverse stim- 15. Martin, S. J., and Green, D. R. (1995) Cell 82, 349–352.uli a common apoptotic machinery seems to be trig- 16. Patel, T., Gores, G. J., and Kaufmann, S. H. (1996) FASEB J.
10, 587–597.gered to execute the process [57]. Genetic evidences17. Nunez, G., and Clarke, M. F. (1994) Trends Cell Biol. 4, 399–in Caenorhabditis elegans place Bcl-2 and an ICE-like
403.protease as central regulators of apoptosis [14]. In neu-18. Vaux, D. L., Cory, S., and Adams, J. M. (1988) Nature 335, 440–ral populations of vertebrates, the involvement of Bcl-
442.2 in the commitment of the apoptotic process has been19. Nunez, G., London, L., Hockenbery, D., Alexander, M., andproposed [58]. Similarly, the activation of a protease McKearn, J. P., and Korsmeyer, S. J. (1990) J. Immunol. 144,
cascade in the execution of apoptosis is, presently, an 3602–3610.accepted fact [58]. In N18 cells and depending on the 20. Hockenbery, D. M., Zutter, M., Hickey, W., Nahm, M., and Kors-procedure to induce apoptosis, we have identified two meyer, S. J. (1991) Proc. Natl. Acad. Sci. USA 88, 6961–6965.structurally different processes. This finding could be 21. Boise, L. H., Gonzalez-GarcıB a, M., Postema, C. E., Ding, L.,
Lindsten, T., Turka, L. A., Mao, X., Nunez, G., and Thompson,stimuli-related and quite irrelevant at a functionalC. B. (1993) Cell 74, 597–608.level. However, Bcl-2 and a TLCK-sensitive protease
22. Reynolds, J. E., Yong, T., Qian, L., Jenkinson, J. D., Zhou, P.,seem differentially involved in each process. In conclu-Eastman, A., and Craig, R. W. (1994) Cancer Res. 54, 6348–sion, we propose the existence of two distinct biochemi- 6352.
cal pathways involved in accomplishing apoptosis in 23. Oltvai, Z. N., Milliman, C. L., and Korsmeyer, S. J. (1993) CellN18 neuroblastoma cells. 74, 609–619.
24. Chittenden, T., Harrington, E. A., O’Connor, R., Flemington,C., Lutz, R. J., Evan, G. I., and Guild, B. C. (1995) Nature 374,The N18 neuroblastoma cell line was a generous gift from Dr. Neil733–736.R. Cashman (McGill University, Montreal, Quebec, Canada). MTbcl-
25. Farrow, S. N., White, J. H. M., Martinou, I., Raven, T., Pun,2TKNeo plasmid was kindly supplied by Dr. David L. Vaux (TheK.-T., Grinham, C. J., Martinou, J.-C., and Brown, R. (1995)Walter and Eliza Hall Institute of Medical Research, Parkville, Victo-Nature 374, 731–733.ria, Australia). We are very grateful to Dr. J. E. Esquerda for fruitful
26. Kiefer, M. C., Brauer, M. J., Powers, V. C., Wu, J. J., Umansky,discussions and technical assistance in electron microscopy. WeS. R., Tomei, L. D., and Barr, P. J. (1995) Nature 374, 736–739.thank also Mr. X. Calomarde for his image processing skills. Re-
search of the Molecular Neurobiology Group is being supported by 27. Yang, E., Zha, J., Jockel, J., Boise, L. H., Thompson, C. B., andSpanish FIS 94/1576 and CICYT PN.SAF97-0094 grants, European Korsmeyer, S. J. (1995) Cell 80, 285–291.Union BMH4-0010, Ajuntament de Lleida (Ajuts a la recerca) and 28. Yin, X.-M., Oltvai, Z. N., and Korsmeyer, S. J. (1994) Nature‘‘Generalitat de Catalunya’’ 1995SGR00266 grants. 369, 321–323.
29. Sato, T., Hanada, M., Bodrug, S., Irie, S., Iwama, N., Boise,L. H., Thompson, C. G., Golemis, E., Fong, L., Wang, H.-G., andREFERENCESReed, J. C. (1994) Proc. Natl. Acad. Sci. USA 91, 9238–9242.
30. Krajewski, S., Tanaka, S., Takayama, S., Schibler, M. J., Fen-1. Thompson, C. B. (1995) Science 267, 1456–1462. ton, W., and Reed, J. C. (1993) Cancer Res. 53, 4701–4714.2. Johnson, E. M., and Deckwerth, T. L. (1993) Annu. Rev. Neu- 31. Lithgow, T., van Driel, R., Bertram, J. F., and Strasser, A.
rosci. 16, 31–46. (1994) Cell Growth Differ. 5, 411–417.32. Gonzalez-GarcıB a, M., Perez-Ballestero, R., Ding, L., Duan, L.,3. Martin, D. P., Schmidt, R. E., DiStefano, P. S., Lowry, O. H.,
Carter, J. C., and Johnson, E. M. (1988) J. Cell Biol. 106, 829– Boise, L. H., Thompson, C. B., and Nunez, G. (1994) Develop-ment 120, 3033–3042.844.
AID ECR 3852 / 6i2d$$$264 01-09-98 08:25:15 ecal

429ANALYSIS OF TWO APOPTOTIC PROCESSES IN N18 CELLS
33. Hockenbery, D. M., Oltvai, Z. N., Yin, X.-M., Milliman, C. L., ter, T. G. (1996) in ‘‘Techniques in Apoptosis’’ (T. G. Cotter andS. J. Martin, Eds.), pp. 6–7, Portland Press, London, UK.and Korsmeyer, S. J. (1993) Cell 75, 241–251.
45. Darzynkiewicz, Z., and Li, X. (1996) in ‘‘Techniques in34. Meikrantz, W., Gisselbrecht, S., Tam, S. W., and Schlegel, R.Apoptosis’’ (T. G. Cotter and S. J. Martin, Eds.), pp. 89–94,(1994) Proc. Natl. Acad. Sci. USA 91, 3754–3758.Portland Press, London, UK.35. Ryan, J. J., Prochownik, E., Gottlieb, C. A., Apel, I. J., Merino,
46. Laemmli, U. K. (1970) Nature 227, 680–685.R., Nunez, G., and Clarke, M. F. (1994) Proc. Natl. Acad. Sci.47. Kumar, S., and Harvey, N. L. (1995) FEBS Lett. 375, 169–173.USA 91, 5878–5882.48. Reed, J. C., Meister, L., Tanaka, S., Cuddy, M., Yum, S., Geyer,36. Lam, M., Dubyak, G., Chen, L., Nunez, G., Miesfeld, R. L., and
C., and Pleasure, D. (1991) Cancer Res. 51, 6529–6538.Distelhorst, C. W. (1994) Proc. Natl. Acad. Sci. USA 91, 6569–49. Reed, J. C. (1994) J. Cell Biol. 124, 1–6.6573.50. Cuende, E., Ales-MartıB nez, J. E., Ding, L., Gonzalez-GarcıB a, M.,37. Augusti-Tocco, G., and Sato, G. (1969) Proc. Natl. Acad. Sci.
MartıB nez, A. C., and Nunez, G. (1993) EMBO J. 12, 1555–1560.USA 64, 311–315.51. Allsopp, T. E., Wyatt, S., Paterson, H. F., and Davies, A. M.38. Carlson, R. O., Masco, D., Brooker, G., and Spiegel, S. (1994)
(1993) Cell 73, 295–307.J. Neurosci. 14, 2272–2281.52. Benito, A., Grillot, D., Nunez, G., and Fernandez-Luna, J. L.39. Felder, C. C., Briley, E. M., Axelrod, J., Simpson, J. T., Mackie,
(1995) Am. J. Pathol. 146, 481–490.K., and Devane, W. A. (1993) Proc. Natl. Acad. Sci. USA 90,53. Caron-Leslie, L. M., Evans, R. B., and Cidlowski, J. A. (1994)7656–7660.
FASEB J. 8, 639–645.40. Linnik, M. D., Hatfield, M. D., Swope, M. D., and Ahmed, N. K.
54. Lindemboim, L., Haviv, R., and Stein, R. (1995) J. Neurochem.(1993) J. Neurobiol. 24, 443–446.64, 1054–1063.
41. Cashman, N. R., Durham, H. D., Blusztajn, J. K., Oda, K., Tab- 55. Lazebnik, Y. A., Takahashi, A., Moir, R. D., Goldman, R. D.,ira, T., Shaw, I. T., Dahrouge, S., and Antel, J. P. (1992) Dev. Poirier, G. G., Kaufmann, S. H., and Earnshaw, W. C. (1995)Dynam. 194, 209–221. Proc. Natl. Acad. Sci. USA 92, 9042–9046.
42. DiStefano, P. S., Schweitzer, J. B., Taniuchi, M., and Johnson, 56. Fearnhead, H. O., Rivett, A. J., Dinsdale, D., and Cohen, G. M.E. M., Jr. (1985) J. Cell Biol. 101, 1107–1114. (1995) FEBS Lett. 357, 242–246.
43. Vaux, D. L., and Weissman, I. L. (1993) Mol. Cell. Biol. 13, 57. Wertz, I. E., and Hanley, M. R. (1996) Trends Biochem. Sci. 21,7000–7005. 359–364.
58. Henderson, C. E. (1996) Neuron 17, 579–585.44. Gorman, A., McCarthy, J., Finucana, D., Reville, W., and Cot-
Received May 28, 1997Revised version received September 1, 1997
AID ECR 3852 / 6i2d$$$264 01-09-98 08:25:15 ecal
Top Related
Copyright © 2022 FDOKUMEN

