






Bahasa
Halaman
Hukum


Rapid upregulation of α4 integrin-mediated leukocyte adhesion by transforming growth factor-ß1
Rubén A. Bartolomé*, Francisco Sanz-Rodríguez*, Mar M. Robledo, Andrés Hidalgo and Joaquin Teixidó From the Centro de Investigaciones Biológicas, Department of Immunology, Madrid, Spain Running title: TGF-ß1 modulates α4 integrin adhesion Correspondence: Joaquin Teixidó. Centro de Investigaciones Biológicas, Department of Immunology, Velázquez 144, 28006 Madrid, Spain. Phone 34-91-5611800; Fax 34-91-5627518; e-mail [email protected] Keywords: α4 integrins, cell adhesion, TGF-ß1, SDF-1α *These authors equally contributed to this work. This work was supported by grant SAF99-0057 from Ministerio de Ciencia y Tecnología. Rubén A. Bartolomé is a recipient of a pre-doctoral fellowship from CSIC-Glaxo Wellcome. Francisco Sanz-Rodríguez and Andrés Hidalgo were recipients of pre-doctoral fellowships from Fundación Ramón Areces and the Comunidad de Madrid, respectively. Mar M. Robledo was a recipient of a post-doctoral fellowship from the Comunidad de Madrid. Andrés Hidalgo, present address: Department of Hematology, Mount Sinai School of Medicine, One Gustave L. Levy Place. New York, NY 10029.
MBC in Press, published on October 16, 2002 as 10.1091/mbc.E02-05-0275

The α4 integrins (α4ß1 and α4ß7) are cell surface heterodimers expressed mostly on
leukocytes that mediate cell-cell and cell-extracellular matrix adhesion. A characteristic feature
of α4 integrins is that their adhesive activity can be subjected to rapid modulation during the
process of cell migration. Here we show that transforming growth factor-ß1 (TGF-ß1) rapidly
(0.5-5 min) and transiently upregulated α4 integrin-dependent adhesion of different human
leukocyte cell lines and human peripheral blood lymphocytes (PBL) to their ligands VCAM-1
and CS-1/fibronectin. In addition, TGF-ß1 enhanced the α4 integrin-mediated adhesion of PBL to
TNF-α-treated human umbilical vein endothelial cells, indicating the stimulation of
α4ß1/VCAM-1 interaction. Although TGF-ß1 rapidly activated the small GTP-ase RhoA and the
p38 MAP kinase, enhanced adhesion did not require activation of both signalling molecules.
Instead, polymerization of actin cytoskeleton triggered by TGF-ß1 was necessary for α4 integrin-
dependent upregulated adhesion, and elevation of intracellular cAMP opposed this upregulation.
Moreover, TGF-ß1 further increased cell adhesion mediated by α4 integrins in response to the
chemokine SDF-1α. These data suggest that TGF-ß1 can potentially contribute to cell migration
by dinamically regulating cell adhesion mediated by α4 integrins.

3
INTRODUCTION
The α4 integrins (α4ß1 and α4ß7) are heterodimer cell adhesion receptors mainly
expressed on cells of hematopoietic origin that mediate cell-cell and cell-extracellular matrix
(ECM) interactions (reviewed in Lobb and Hemler, 1994; Hynes, 1992). VCAM-1 and the
alternatively-spliced CS-1 region of fibronectin constitute ligands for both integrins, whereas
α4ß7 can additionally interact with MAdCAM-1 (Lobb and Hemler, 1994). α4ß1 and α4ß7 play
key roles in leukocyte recruitment to inflammatory sites and in lymphocyte recirculation, and
α4ß1 function is required during hematopoiesis in the bone marrow (Springer, 1995; Butcher and
Picker, 1996; Arroyo et al., 1996; Butcher et al., 1999).
A characteristic feature of α4 integrins on most leukocytes is that its adhesive activity can
be upregulated by external stimuli leading to firm attachment. Several chemokines binding to
their G protein-coupled receptors, as well as cytokines whose receptors have tyrosine kinase
activity, have been previously demonstrated to rapidly and transiently increase α4 integrin-
dependent cell adhesion (Tanaka et al., 1993; Woldemar Carr et al., 1996; Grabovsky et al.,
2000; Lévesque et al., 1995). For instance, the chemokine SDF-1α upregulates α4 integrin-
mediated lymphocyte, hematopoietic progenitor and myeloma cell adhesion (Grabovsky et al.,
2000; Wright et al., 2002; Peled et al., 1999; Hidalgo et al., 2001; Sanz-Rodríguez et al., 2001).
The enhancement in adhesion was shown to be independent of changes in α4 surface expression,
and was suggested to be the result of variations in the avidity and/or affinity of these integrins for
their ligands.
Three mammalians isoforms of transforming growth factor-beta1 (TGF-ß1) have been
described, TGF-ß1, ß2 and ß3, encoded by different genes (Massagué, 1998). TGF-ß1 is a
multifunctional cytokine which regulates cell proliferation, differentiation and migration, and has

4
an important role in tissue recycling and repair (Massagué, 1998; Piek et al., 1999). In addition,
TGF-ß1 plays a pivotal role in tumor development, eliciting both positive and negative effects in
carcinogenesis (Massagué et al., 2000; Derynck et al., 2001; Wakefield and Roberts, 2002). TGF-
ß1 is a potent immunosuppresor (Letterio and Roberts, 1998), and its absence causes massive
leukocyte infiltration due to increased expression of inflammatory mediators and adhesion
molecules whose expression is negatively regulated by TGF-ß1 (Shull et al., 1992; Kulkarni et
al., 1993). TGF-ß1 exerts its biological functions through binding to a heteromeric cell surface
complex formed by types I and II TGF-ß receptors, which are transmembrane serine/threonine
kinases which are required for TGF-ß signalling (Derynck and Feng, 1997; Massagué, 1998).
Members of the Smad group of proteins mediate signalling from the receptors to the nuclei,
acting as intermediates for transcriptional regulation and cell cycle arrest (Derynck et al., 1998;
Piek et al., 1999; Massagué and Wotton, 2000; Miyazono et al., 2000).
TGF-ß1 controls the synthesis of several integrins (Ignotz and Massagué, 1988; Heino et
al., 1989; Parker et al., 1992; Wahl et al., 1993), which is detected after several hours of exposure
to the cytokine. On the other hand, TGF-ß1 can rapidly (minutes) and independently of Smads,
activate several signaling pathways including mitogen-activated protein (MAP) kinases
(Hartsough and Mulder, 1995; Hannigan et al., 1998; Sano et al., 1999; Hanafusa et al., 1999)
and small GTP-ases of the Rho subfamily (Mucsi et al., 1996; Bhowmick et al., 2001; Edlund et
al., 2002). The latter comprises a group of several proteins which are key regulators of the
organization of actin cytoskeleton and play important roles in cell migration (Hall, 1998).
In the present study we have investigated whether short exposure to TGF-ß1 could
influence α4-integrin dependent cell adhesion. A potential modulation of this adhesion by TGF-
ß1 could contribute to lymphocyte extravasation at sites of inflammation, as well as in the
trafficking of precursor cells in hematopoietic organs and in secondary lymphoid tissue.

5
MATERIALS AND METHODS
Cells and antibodies. Human peripheral blood lymphocytes (PBL) were isolated from buffy
coats by ficoll density gradient centrifugation (Biochrom KG, Berlin, Germany), followed by two
steps of cell adherence at 37ºC onto plastic flasks. Peripheral blood T lymphocytes (PBL-T) were
purified from PBL by immunomagnetic negative selection using a mixture of anti-CD19, anti-
CD16, and anti-CD14 mAb, following the method already described (Sancho et al., 1999). The
negatively-selected cell population was always more than 97% positive for CD3 expression, as
analyzed by flow cytometry. Isolated cells were washed and resuspended in adhesion medium
(RPMI/BSA 0.5%) for the assays. Human umbilical vein endothelial cells (HUVEC) were
isolated and cultured as previously described (Dejana et al., 1987). Cells were seeded on tissue
culture dishes coated with 0.5% gelatin and grown in Medium 199 (Biowhitaker, Verviers,
Belgium) supplemented with 20% fetal bovine serum (FBS) (Biowhittaker), 50 µg/ml endothelial
cell growth supplement (prepared from bovine brain), 100 µg/ml heparin (Sigma, St. Louis, MO)
and antibiotics, and used up to the second passage. The erythroleukemia K562, myeloid U937,
and lymphoid JY, RPMI 8866 and Jurkat human cell lines, were cultured in RPMI 1640 media
(Biowhitaker) supplemented with 10% FBS and antibiotics (complete medium). K562 α4
transfectants (Muñoz et al., 1996) were maintained in the same medium containing 1 mg/ml of
G418 (Calbiochem). The megakaryocytic leukemia-derived human cell line Mo7e was mantained
in complete medium and 5 ng/ml rhGM-CSF (R&D Systems, Abingdon, UK). The integrin anti-
α4 HP1/2, anti-CD19 (Bu12) and the control P3X63 mAb were gifts of Dr. Francisco Sánchez-
Madrid (Hospital de la Princesa, Madrid, Spain). Anti-CD16 and anti-CD14 were from
Pharmigen Transduction Laboratories (San Diego, CA).
α4 integrin ligands and cell adhesion assays. The recombinant FN-H89 fragment of fibronectin
which contains the CS-1 site and lacks the RGD central binding domain, was generated as

6
previously described (Mould et al., 1994). For adhesion to VCAM-1, we used the soluble 7-
extracellular domain recombinant human VCAM-1 (sVCAM-1) (R&D Systems). Before the
adhesion assays, cell lines were starved for 4 h by incubation in adhesion medium, without
detectable loss of viability as determined by trypan blue exclusion. PBL and PBL-T were used
directly after their isolation. Cells were labelled for 20 min at 37ºC with BCECF-AM (Molecular
Probes, Leiden, The Netherlands), washed and resuspended in adhesion medium, followed by
treatment with or without inhibitors or antibodies, and finally incubated with rhTGF-β1 (R&D
Systems). Cells (5-10x104 in 100 µl) were added in triplicates to 96-wells dishes (High-binding,
Costar, Cambridge, MA) coated with 2.5 µg/ml FN-H89 or 1-2 µg/ml of sVCAM-1. After
incubation for 10 min at 37ºC, or 2 min after a 15-sec centrifugation of plates, unbound cells
were removed by 3 washes with RPMI medium, and adhered cells quantified using a
fluorescence analyzer (POLARstar Galaxy, BMG Labtechnologies, Offenburg, Germany). For
cell adhesion to HUVEC, monolayers were treated for 10 h with or without 10 ng/ml of
recombinant human TNF-α (Peprotech, London, UK.). Cell adhesion to HUVEC was carried out
for 10 min at 37ºC, and unbound cells were processed and analyzed as indicated above. In
experiments using rhSDF-1α (R&D Systems), the chemokine was incubated with cells in
adhesion medium in the presence or in the absence of TGF-ß1. Inhibitors used in this study
included cytochalasin D, SB 203580, phenylarsine oxide, forskolin and 8-Br-cAMP (Sigma),
PD98059 (New England Biolabs, Beverly, MA), and okadaic acid (Calbiochem). Recombinant
C3 transferase was expressed and purified as previously described (Nobes and Hall, 1995). The
concentrations used for the different inhibitors were not cytotoxic, as measured in cell cycle
analysis by flow cytometry (not shown).
Actin polymerization assays. To determine the content of polymerized actin (F-actin) induced
by TGF-β1 treatment, 105 cells per condition were permeabilized, fixed and stained in a single

7
step by the addition of a 2x solution containing 0.5 mg/ml L-α-lysophosphatidylcholine (Sigma),
8% formaldehyde and 4 U/ml FITC-phalloidin (Molecular-Probes, Eugene, OR). Cells were
incubated at 22ºC for 10 min, washed twice with PBS and subjected to flow cytometry.
GTP-ase activity assays and MAP kinase activation. For GTP-ase assays we followed
essentially the method already reported (Robledo et al., 2001). The GST-C21 and GST-PAK-CD
fusion proteins were generated as described (Sander et al., 1998). To determine the effect of
TGF-ß1 on RhoA and Rac1 activation, cells (107 per condition) were first starved in adhesion
medium, followed by incubation at 37ºC in the same medium in the abscence or in the presence
of TGF-β1. Cells were washed in ice-cold PBS and incubated for 15 min at 4ºC in 200 µl of lysis
buffer (Robledo et al., 2001). Lysates were centrifuged, 15 µl of supernatant were kept for total
lysate samples, and the remaining 185 µl were mixed with fusion proteins precoupled to
glutathione-agarose beads. The beads and proteins bound to the fusion protein were washed in an
excess of lysis buffer, eluted in Laemmli sample buffer, and analyzed for bound Rac1 or RhoA
by Western blotting using monoclonal antibodies against human Rac1 (Pharmingen/Transduction
Laboratories, San Diego, CA) or RhoA (Santa Cruz Biotechnology, Santa Cruz, CA). For p38
MAP kinase activation, 107 cells were starved and subsequently incubated in the presence or in
the absence of TGF-ß1, followed by solubilization in 1 ml of RIPA lysis buffer (Sanz-Rodríguez
et al., 2001). After SDS-PAGE, immunoblots were blocked, incubated with anti-phospho-p38
antibodies (New England Biolabs), washed in T-PBS (0.05% Tween 20 in PBS), and further
incubated with HRP-conjugated secondary antibodies (DAKO A/S, Copenhagen, Denmark).
Blots were developped by a chemiluminiscent reaction and exposed to radiographic films. After
stripping and saturation of nonspecific protein binding sites, the same blots were reprobed with
anti-p38 antibodies (New England Biolabs) to test for total p38 protein content.
Statistical analysis. The results are expressed as the mean ±SD of data obtained from two or

8
more experiments each performed in triplicate. Statistical significance was determined using the
two-tailed Student's t-test.

9
RESULTS
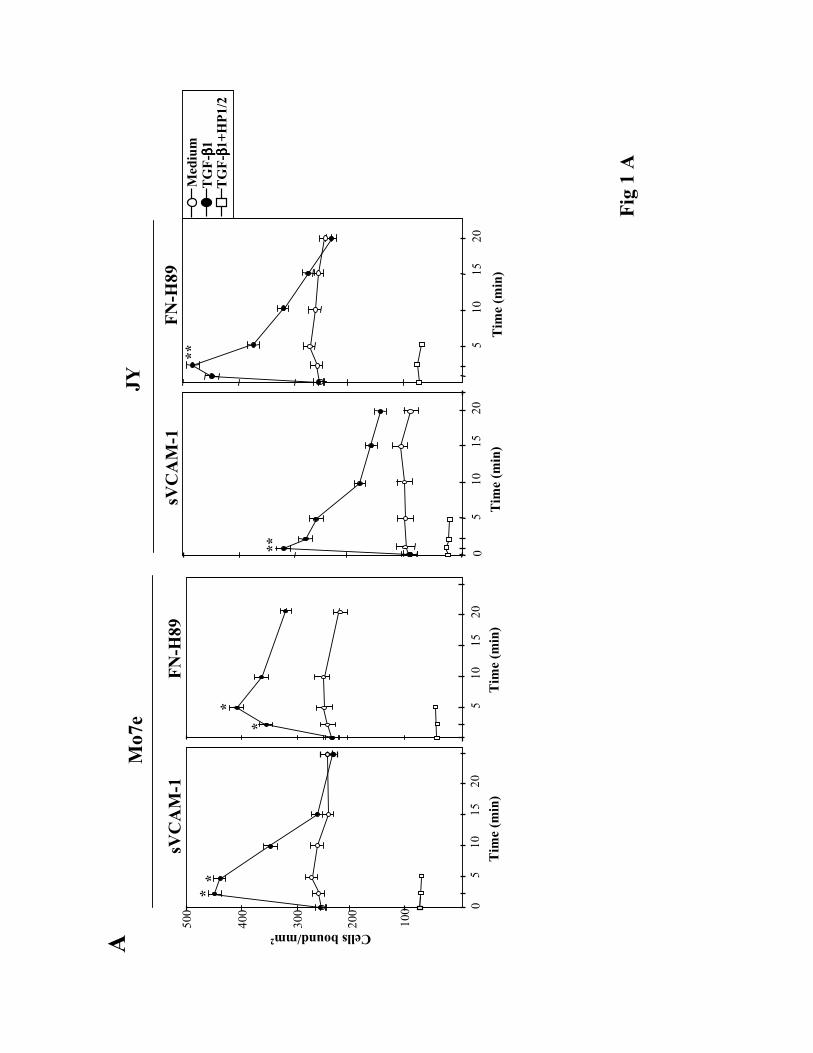
TGF-ß1 rapidly upregulates α4 integrin-mediated cell adhesion. To investigate whether
TGF-ß1 was capable of influencing α4 integrin-dependent cell adhesion we first used the Mo7e
and JY human cell lines as models for studies on α4ß1- and α4ß7-mediated adhesion,
respectively, as they exhibit low adhesive efficiencies to α4 integrin ligands (Lévesque et al.,
1995; Hidalgo et al., 2001; Chan et al., 1992). We pre-incubated these cells at 37ºC for short
periods of time with TGF-ß1, followed by their addition for 10 min to wells coated with α4
integrin ligands. TGF-ß1 rapidly (2.5-5 min) and transiently upregulated the attachment of Mo7e
cells to sVCAM-1, a soluble form of VCAM-1, and to FN-H89, a CS-1-containing fragment of
fibronectin (Fig. 1A, left). The anti-α4 HP1/2 mAb blocked the increased adhesion, indicating
that α4ß1 was mediating the enhancement in cell adhesion. Dose-response experiments showed
that concentrations of TGF-ß1 between 0.5-2 ng/ml were optimal for stimulation of adhesion,
whereas higher concentrations were without effect or slightly inhibitory (not shown). JY cells
displayed a low capability to attach to sVCAM-1, but TGF-ß1 rapidly (1 min) enhanced their
adhesion, as well as the attachment to FN-H89, whereas HP1/2 mAb blocked this adhesion (Fig.
1A, right) indicating the involvement of α4ß7. Additionally, TGF-ß1 upregulated the adhesion to
FN-H89 of the α4ß7+/α4ß1- RPMI 8866 cell line (not shown).
In cell lines which express α4ß1 and display high basal adhesion levels to α4 integrin
ligands (over 40% of the input), such as monocytic U937 cells, K562 cells expressing transfected
α4 (4M7 cells; Muñoz et al., 1996) (Fig. 1B) and Jurkat T cells (not shown), TGF-ß1 had a small
or no effect in their α4ß1-dependent adhesion. Parental K562 cells did not attach to FN-H89 or
sVCAM-1, and addition of TGF-ß1 did not induce their attachment to these ligands (not shown).
When we shortened the time of adhesion to 2 min after a short spin to place cells faster in
contact with α4 integrin ligands, we still detected a notable upregulation in Mo7e cell adhesion to

10
FN-H89 and sVCAM-1 after 2.5-5 min pre-incubation with TGF-ß1 (Fig. 2A), suggesting the
participation of early signalling events in the increased adhesion. To analyze if TGF-ß1 was
selectively targetting a cell population during its modulation of α4 integrin function, we pre-
incubated Mo7e cells with TGF-ß1 before attachment for 5 min to FN-H89, and subsequently,
non-bound cells were recovered and further pre-incubated with the cytokine before new adhesion
to FN-H89. Analysis of extent of cell adhesion in both cases revealed that TGF-ß1 upregulated
the attachment of both the initial and the non-bound cell population (Fig. 2B), suggesting that
TGF-ß1 did not exclusively targetted a determined cell population. When we examined whether
FN-H89-attached Mo7e cells which had not been previously in the presence of TGF-ß1, were still
capable of responding to TGF-ß1, we observed no significative changes in the levels of adhesion
compared with adhered cells incubated in medium alone (not shown).
Peripheral blood lymphocytes (PBL) express cell surface α4 integrins (α4ß1 and α4ß7),
whose adhesive activity has been shown to be the target of modulation by several chemokines
(Tanaka et al., 1993; Woldemar Carr et al. 1996; Pachynski et al., 1998; Grabovsky et al., 2000;
Wright et al., 2002). Therefore we used PBL, as well as T lymphocytes purified from PBL (PBL-
T), to study if TGF-ß1 could modulate their attachment to α4 integrin ligands. TGF-ß1 rapidly
and transiently augmented the attachment of PBL and PBL-T to sVCAM-1, which was blocked
by HP1/2 mAb, indicating the involvement of α4 integrins (Fig. 3, left). TGF-ß1 also increased
the adhesion of PBL and PBL-T to FN-H89 but to a lower extent compared to attachment to
sVCAM-1 (Fig. 3, right).
Control flow cytometry experiments addressed to study whether cell incubation with
TGF-ß1 altered α4 integrin surface expression showed that incubations up to 25 min with TGF-
ß1 did not influence the expression of α4 integrins on PBL and Mo7e cells (not shown).
As pro-inflammatory cytokines induce VCAM-1 expression in HUVEC (Osborn et al.,

11
1989), we incubated them with TNF-α and analyzed whether TGF-ß1 was capable of modulating
the subsequent adhesion of PBL and PBL-T. TGF-ß1 did not influence PBL (Fig. 4) or PBL-T
(not shown) adhesion to resting HUVEC. PBL and PBL-T adhered to higher levels upon TNF-α
treatment of HUVEC, and TGF-ß1 rapidly upregulated their α4ß1-dependent adhesion, as HP1/2
inhibited the increased adhesion. These results suggest that TGF-ß1-triggered upregulation of
PBL and PBL-T adhesion to HUVEC involves at least an increase in α4ß1/VCAM-1 interaction.
Activation of Rho GTP-ases and F-actin polymerization by TGF-ß1. Role in upregulation
of α4 integrin-dependent cell adhesion. To identify TGF-ß1-activated signalling routes which
could be involved in the increase in α4 integrin-mediated adhesion, we first tested on Mo7e cells
the effect of TGF-ß1 on the activation of the small GTPases RhoA and Rac1, key regulators of
the organization of actin cytoskeleton (Van Aelst and D'Souza-Schorey, 1997; Hall, 1998), as
they are activated by this cytokine in epithelial and fibroblastic cells (Mucsi et al., 1996; Atfi et
al., 1997; Bhowmick et al., 2001; Edlund et al., 2002 ). Mo7e cells were treated for different
times with TGF-ß1, following by cell solubilization and incubation of lysates with GST-fusion
proteins containing domains derived from Rho GTPase targets. For the detection of active RhoA
we used GST-C21, which contains the Rho binding domain of the Rho efector Rhotekin, and for
active Rac1 the CRIB (Cdc42/Rac interacting binding) domain of the Rac/Cdc42 effector
molecule PAK (GST-PAK-CD) was used. SDS-PAGE followed by Western blot using anti-
RhoA or anti-Rac1 antibodies showed that TGF-ß1 rapidly and transiently activated RhoA and
Rac1 (i.e.,GTP-loaded Rho A and Rac1) (Fig. 5A). The C3 transferase, an enzyme that
specifically ADP-ribosylates and blocks RhoA activation (Aktories et al., 1989), inhibited the
activation of Rho A triggered by TGF-ß1. Therefore, activation of RhoA and Rac1 by TGF-ß1 in
Mo7e cells takes place at times when cells become stimulated for increased α4 integrin-
dependent adhesion, raising the posibility that downstream signalling activated by these Rho

12
GTP-ases could participate in the upregulated adhesion.
We used C3 transferase to obtain some insight into a potential participation of RhoA
activation in TGF-ß1-triggered increase in Mo7e adhesion. C3 did not significantly alter the
enhancement in Mo7e adhesion to sVCAM-1 and FN-H89 in response to TGF-ß1 (Fig. 5B),
whereas it substantially inhibited the upregulation of α4 integrin-mediated cell adhesion in
response to the chemokine SDF-1α (Wright et al., 2002; Parmo-Cabañas et al. manuscript in
preparation). Moreover, Mo7e treatment with Y-27632, a specific inhibitor of the Rho
downstream effector ROCK, did not alter the increase in adhesion to FN-H89 in response to
TGF-ß1 (not shown). These results suggest that rapid activation of RhoA by TGF-ß1 in Mo7e
cells is not required for the increase in α4ß1-dependent adhesion.
In addition of activating RhoA and Rac1, TGF-ß1 rapidly (1-5 min) and transiently
triggered F-actin polymerization on Mo7e and JY cells (Fig. 6A, Left). Cell pre-incubation with
cytochalasin D, an agent that disrupts actin filaments, blocked the enhancement of F-actin
polymerization induced by TGF-ß1, whereas treatment with C3 resulted in partial inhibition (Fig.
6A, Right). Pre-incubation of PBL and Mo7e cells with cytochalasin D before adding TGF-ß1
substantially inhibited the subsequent TGF-ß1-stimulated upregulation of α4 integrin-dependent
cell adhesion (Fig. 6B), suggesting that a reorganization of the actin cytoskeleton was implicated
in the increased adhesion.
p38 MAP kinase activation by TGF-ß1 is not necessary for enhancement of α4 integrin-
dependent cell adhesion. TGF-ß1 triggered a rapid (1 min) phosphorylation of p38 MAP kinase
in Mo7e cells (Fig. 7A), which was sustained after 15 min incubation with the cytokine. Pre-
incubation of Mo7e cells with SB 203580, an inhibitor of p38 MAP kinase activation abrogated
the phosphorylation of this kinase induced by TGF-ß1, but it did not affect the increase in cell
adhesion to FN-H89 in response to TGF-ß1 was detected (Fig. 7B), indicating that activation of

13
p38 MAP kinases is not necessary for TGF-ß1-induced increase in adhesion. Moreover,
enhancement of α4ß1-dependent adhesion of Mo7e cells to FN-H89 and sVCAM-1 by TGF-ß1
was not influenced by pre-incubation with the MEK1 inhibitor PD98058, nor by the protein
phosphatase inhibitors okadaic acid and phenylarsine oxide (not shown).
Alterations in intracellular cAMP oppose the upregulation of α4 integrin-dependent cell
adhesion in response to TGF-ß1. We used forskolin, a direct activator of adenylate cyclase,
and 8-Br-cAMP, a cell-permeable cAMP derivative, to test whether alterations in the levels of
cAMP could affect the increase in TGF-ß1-triggered adhesion mediated by α4 integrins. Pre-
treatment of Mo7e cells with these two agents substantially inhibited the subsequent upregulation
of cell adhesion to sVCAM-1 and FN-H89 in response to TGF-ß1 (Fig. 8A), suggesting that
elevation of cAMP levels functionally opposed the signalling mechanisms involved in the α4
integrin-dependent enhanced adhesion.
As Cyt D inhibits both TGF-ß1-triggered F-actin polymerization and the increase in α4
integrin-dependent cell adhesion, together with the reported data showing that alterations in
cAMP levels affected the organization of actin cytoskeleton (Valitutti et al., 1993; Busca et al.,
1998; Dong et al., 1998), we tested the effect of 8-Br-cAMP in the induction of F-actin
polymerization by TGF-ß1. The results showed that 8-Br-cAMP substantially inhibited early (up
to 2.5 min) but not late (5 min or more) TGF-ß1-activated F-actin polymerization in Mo7e cells
(Fig. 8B). These data raise the possibility that interference with TGF-ß1-stimulated F-actin
polymerization by agents that affect intracellular cAMP levels could mediate the inhibitory
effects of these agents in upregulation of α4 integrin-dependent adhesion by TGF-ß1.
TGF-ß1 further increases SDF-1α-stimulated α4 integrin-dependent cell adhesion. We
previously reported that SDF-1α rapidly and transiently upregulated α4ß1-mediated adhesion of
Mo7e cells to sVCAM-1 and FN-H89 (Hidalgo et al., 2001). To study whether TGF-ß1 could

14
influence the increase in these adhesions, we incubated Mo7e cells with TGF-ß1 in the presence
or in the absence of SDF-1α, before subjecting them to adhesion to sVCAM-1 or FN-H89. As
expected, cell adhesion was notably upregulated by SDF-1α, and the presence of TGF-ß1 resulted
in a substantial further increase in the adhesion, that was totally blocked by HP1/2, indicating
involvement of α4ß1 in the stimulated adhesion (Fig. 9A). However, the additive effect of TGF-
ß1 in cell attachment was not observed when α4 integrin-mediated upregulation of adhesion by
SDF-1α reached 30% or more of the total cellular input in the assay, and was barely detected
after the effect of SDF-1α on stimulation of cell adhesion finished. It has also been demonstrated
that SDF-1α enhances the adhesion of T lymphocytes to VCAM-1 (Grabovsky et al., 2000).
Accordingly, SDF-1α triggered a large upregulation of T lymphocyte adhesion to sVCAM-1, and
TGF-ß1 induced a modest but statistically significant further increase in adhesion, whereas
HP1/2 blocked this adhesion (Fig. 9B). Furthermore, TGF-ß1 upregulated to a greater extent the
enhancement of T lymphocyte adhesion to FN-H89 in response to SDF-1α. Together these data
indicate that TGF-ß1 has an additive effect on the SDF-1α-induced increase in α4 integrin-
dependent cell adhesion.

15
DISCUSSION
A characteristic feature of α4 integrins is that their adhesive activity on leukocytes can be
rapidly modulated by external stimuli during the process of cell migration. Several chemokines
and cytokines binding to G protein-coupled receptors and tyrosine-kinase receptors, respectively,
have been previously demonstrated to trigger rapid and transient upregulation of α4 integrin-
dependent cell adhesion (Woldemar Carr et al., 1996; Grabovsky et al., 2000; Lévesque et al.,
1995; Peled et al., 1999; Hidalgo et al., 2001; Sanz-Rodríguez et al., 2001). In the present study
we show that TGF-ß1, a cytokine whose receptors are serine-threonine kinases, is also capable of
rapidly and transiently increase α4 integrin-mediated cell adhesion. Incubation with TGF-ß1 for
short periods of time (0.5-5 min) resulted in an enhancement in the subsequent adhesion of Mo7e
and JY cell lines, which express α4ß1 and α4ß7, respectively, to sVCAM-1 and the CS-1-
containing FN-H89 fragment of fibronectin. The upregulation of α4 integrin-mediated adhesion
was detected in adhesion assays as short as 2 min and was independent of changes in α4 cell
surface expression, suggesting that rapid TGF-ß1-triggered changes in α4 integrin avidity and/or
affinity for its ligands were responsible for the augmented adhesion. Cell exposure to TGF-ß1 for
longer than 30 min, or to concentrations of the cytokine of 4 ng/ml or higher, resulted in the
absence of adhesion-triggering effects or even produced a slight inhibition in cell attachment to
α4 integrin ligands.
Upregulation of cell adhesion by TGF-ß1 was detected in cell lines, such as the
mentioned Mo7e and JY, and cells displaying low levels of α4 integrin-dependent adhesion,
including whole PBL as well as PBL-T cells. Instead, cell lines showing high adhesion to α4
integrin ligands were refractory to TGF-ß1 adhesion modulatory effects. TGF-ß1 transiently
enhanced the adhesion of both PBL and PBL-T to sVCAM-1, and increased their attachment to
TNF-α-treated HUVEC, indicating the activation of the α4ß1/VCAM-1 adhesion pathway. TGF-

16
ß1 also augmented the adhesion of these cells to FN-H89, but to a lesser extent compared with
sVCAM-1.
Enhancement of adhesion by TGF-ß1 could either exclusively target a specific cell
population expressing α4 integrins activated enough to mediate a basal level of cell attachment
and TGF-ß1 producing a further activation, or it could target cells expressing non-active α4
integrins unable to mediate adhesion and TGF-ß1 activating them, or else a mixture of both
effects. The capability of unbound Mo7e cells recovered from TGF-ß1-stimulated adhesions, to
respond to this cytokine in a subsequent adhesion suggest that TGF-ß1 is not targetting a specific
cell population for its adhesion activating properties. However, whereas close to 30% of Mo7e
and JY cells in adhesion assays were cabable of enhancing their α4 integrin-dependent adhesion
in response to TGF-ß1, this percentage was reduced to less than 10% when using PBL or PBL-T.
At this point we do not know whether a particular lymphocyte subset or most lymphocytes are
potentially capable to respond to TGF-ß1 by stimulating their α4 integrin-mediated adhesion.
Several important signalling molecules are rapidly activated by TGF-ß1, including small
GTP-ases of the Rho family (Mucsi et al., 1996; Bhowmick et al., 2001; Edlund et al., 2002;
Hartsough and Mulder, 1995). We show here that TGF-ß1 rapidly and transiently activated
RhoA and Rac1 in Mo7e cells, coincident with the times of increased cell adhesion in response to
the cytokine. However, activation of RhoA by TGF-ß1 was found not to be required for α4
integrin-dependent upregulation of cell adhesion, as inhibition of RhoA activation by C3
transferase did not influence the enhanced adhesion. In addition to activate RhoA and Rac1, key
regulatory molecules of the actin cytoskeleton (Hall, 1998), TGF-ß1 triggered a transient increase
in cell F-actin polymerization, which was blocked by cytochalasin D and partially inhibited by
C3. Moreover, cytochalasin D substantially inhibited TGF-ß1-induced increase in α4 integrin-
mediated adhesion in PBL and Mo7e cells. These data suggest that a reorganization of actin

17
cytoskeleton in response to TGF-ß1, that is independent of RhoA activity, might result in an
enhancement in the avidity and/or affinity of α4 integrins for their ligands. As it has been
reported that Rac1 activation results in α4ß1 clustering in T lymphocytes, leading to increased
avidity and upregulation of T cell adhesion to fibronectin (D'Souza-Schorey et al., 1998), we can
not exclude that activation of Rac1 could represent a potential mediator response in the enhanced
α4 integrin-dependent adhesion by TGF-ß1.
The p38 MAP kinase represents an additional signalling molecule rapidly activated by
TGF-ß1 (Sano et al., 1999; Hanafusa et al., 1999). TGF-ß1 triggered a rapid phosphorylation of
p38 MAP-kinase in Mo7e cells, but inhibition of activation of this kinase, as well as that of ERK
MAP-kinase, did not influence the upregulation in α4 integrin-mediated adhesion, suggesting that
activation of MAP kinases was not necessary for the TGF-ß1-triggered increase in adhesion.
, , .. It has been earlier demonstrated that cell treatment with agents that increase
intracellular cAMP levels can inhibit TGF-ß1 actions (Daniel et al., 1987; Grainger et al., 1994;
Steiner et al., 1994). We show here that elevation of intracellular cAMP by incubating cells with
the cAMP analog 8-Br-cAMP, or with forskolin, notably reduced the upregulation of Mo7e cell
adhesion to sVCAM-1 and FN-H89 in response to TGF-ß1, and inhibited TGF-ß1-induced
increase in F-actin polymerization, indicating that these agents were altering at some point the
signalling leading to actin-regulated enhanced adhesion. Altogether, these results suggest that
TGF-ß1 might promote a rapid and actin-controlled spatial reorganization of α4 integrins in the
plasma membrane, potentially leading to the clustering of these integrins, and increased cell
adhesion as a result of an enhancement in the avidity for their ligands. It is possible that in the
absence of stimulation, actin fibers might hold α4 integrins in an inactive form, and
reorganization of actin by TGF-ß1 could contribute to a transient release towards an active
integrin conformation, which could reflect the mentioned clustering, but also to a conformation

18
displaying higher affinity for their ligands.
Several chemokines are potent activators of lymphocyte adhesion involving α4 integrin
adhesive activity (Tanaka et al., 1993; Woldemar Carr et al., 1996; Pachynski et al., 1998;
Grabovsky et al., 2000; Wright et al., 2002). For instance, SDF-1α bound to the surface of
endothelial cells can interact with CXCR4-bearing lymphocytes in the blood stream and trigger
their α4ß1-dependent firm adhesion which enables them to resist blood shear flow, before
initiating transendothelial migration (Grabovsky et al., 2000). TGF-ß1 is expressed by endothelial
cells and, similarly to SDF-1α, it can bind to proteoglycans and ECM proteins (Fava and
McClure, 1987; Murphy-Ullrich et al., 1992; Butzow et al., 1993; Hannan et al., 1988; Antonelli-
Orlidge et al., 1989) and therefore could also be displayed by these molecules on endothelial cells
in the lumen of blood vessels, as well as in the underlying extracellular matrices. Consequently,
lymphocyte extravasation could represent a process potentially modulated by TGF-ß1 by its
transient upregulation of α4 integrin-dependent cell adhesion. Additionally, we show here that
TGF-ß1 modestly, but significantly further augmented the number of PBL-T cells adhered to α4
integrin ligands during rapid SDF-1α activation of this adhesion. The additional enhancement in
adhesion to both sVCAM-1 and FN-H89 was much larger when we used Mo7e cells, indicating
the likelihood of TGF-ß1 additive effects to SDF-1α in the modulation of α4 integrin-dependent
cell adhesion. It is clear from these results that, compared to SDF-1α and possibly to other
chemokines, TGF-ß1 contribution to increase in lymphocyte adhesion mediated by α4 integrins is
low. However, the results presented here on time kinetics of activation of this adhesion by SDF-
1α and TGF-ß1 raises the possibility that this cytokine might prolong SDF-1α-activated adhesion
and hence contribute to cell migration.
In addition to its potential contribution to lymphocyte extravasation at sites of
inflammation, rapid activation of α4ß1-dependent adhesion by TGF-ß1 might also have a role in

19
the trafficking of precursor cells in hematopoietic organs, where cell motility between different
niches are thought to occur, allowing controlled maturation responses. Moreover, modulation by
TGF-ß1 of lymphocyte α4ß7-dependent adhesion within secondary lymphoid organs could also
contribute to their motility during lymphocyte immune surveillance functions. All the above
processes require a rapid and dynamic modulation of α4 integrin-dependent adhesion, and hence,
TGF-ß1 together with chemokines could mediate controlled adhesion/detachment cycles
allowing cell migration.
A previous work reported that TGF-ß1 was capable of enhancing T lymphocyte
attachment to whole fibronectin and that TGF-ß1 downmodulated SDF-1α-triggered increase in
adhesion (Brill et al., 2001). A possible explanation for the discrepancy with our results is that we
used much shorter times (maximum 7 min) of cell exposure to SDF-1α and TGF-ß1, whereas in
the other case T lymphocyte adhesion was measured after 1 h of incubation with both stimuli. As
mentioned above, we have observed that long incubations with TGF-ß1 results in a subsequent
reduction in α4 integrin-mediated cell adhesion, that could also explain these different results.
The present data indicate that different extracellular stimuli acting through different types
of membrane receptors, such as TGF-ß1 receptors and CXCR4, are capable of triggering the
activity of the same adhesion molecules, although with different kinetics and potencies. In
addition, the signalling pathways activated by these different stimuli have common molecular
points, including Rho GTP-ases and MAP kinases, as well as common cell responses, such as
polymerization of the actin cytoskeleton. Interfering with the induction of actin polymerization
triggered by TGF-ß1 and SDF-1α results in both cases in the inhibition of upregulation of α4
integrin-mediated cell adhesion, indicating that the actin reorganization response might represent
a common mechanism for enhanced avidity/affinity of α4 integrins for their ligands. Collectively,
the results presented here suggest that rapid modulation of α4 integrin-dependent cell adhesion by

20
TGF-ß1 could represent an additional stimulus contributing to the process of cell migration.

21
ACKNOWLEDGMENTS
We thank Drs. Francisco Sánchez-Madrid, Santiago Lamas and Angel Corbí for
providing reagents. Natalia Wright is acknowledged for preparation of fusion proteins.
FOOTNOTES
Abreviations.
CS-1: Connecting segment-1 of fibronectin
VCAM-1: Vascular cell adhesion molecule-1
HUVEC: Human umbilical vein endothelial cells
SDF-1α: Stromal cell-derived factor-1α
ECM: Extracellular matrix

22
REFERENCES
Aktories, K., Braunn, U., Rosener, S., Just, I., and Hall, A. (1989). The rho gene product
expressed in E.coli is a substrate of botulinum ADP-ribosyltransferase C3. Biochem. Biophys.
Res. Commun. 158, 209-213.
Antonelli-Orlidge, A., Saunders, K.B., Smith, S.R., and D’Amore, P.A. (1989). An activated
form of transforming growth factor beta is produced by cocultures of endothelial cells and
pericytes. Proc. Natl. Acad. Sci. USA 86, 4544-4548.
Arroyo, A.G., Yang, J.T., Rayburn, H., and Hynes, R.O. (1996). Differential rrquirements for
α4 integrins during fetal and adult hematopoiesis. Cell 85, 997-1008.
Atfi, A., Djelloul, S., Chastre, E., Davis, R., and Gespach, C. (1997). Evidence for a tole of
Rho-like GTPases and stress-activated protein knase/c-Jun N-terminal kinase (SAPK/JNK) in
transforming growth factor β-mediated signaling. J. Biol. Chem. 272, 1429-1432.
Bhowmick, N.A., Ghiassi, M., Bakin, A., Aakre, M., Lundquist, C.A., Engel, M.E., Arteaga,
C.L., and Moses, H.L. (2001). Transforming growth factor-β1 mediates epithelial to
mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12,
27-36.
Brill, A., Franitza, S., Lider, O., and Hershkoviz, R. (2001). Regulation of T-cell interaction
with fibronectin by transforming growth factor-β is associated with altered Pyk2
phosphorylation. Immunology 104, 149-156.
Busca, R., Bertolotto, C., Abbe, P., Englaro, W., Ishizaki, T., Narumiya, S., Boquet, P.,
Ortonne, J.P., and Ballotti, R. (1998). Inhibition of Rho is required for cAMP-induced
melanoma cell differentiation. Mol. Biol. Cell 9, 1367-1378.
Butcher, E.C., and Picker, L.J. (1996). Lymphocyte homing and homeostasis. Science 272,

23
60-66.
Butcher, E.C., Williams, M., Youngman, K., Rott, L., and Briskin, M. (1999). Lymphocyte
trafficking and regional immunity. Adv. Immunol. 72, 209-253
Butzow, R., Fukushima, D., Twardzik, D.R., and Ruoslahti, E. (1993). A 60-kD protein
mediates the binding of transforming growth factor-beta to cell surface and extracellular
matrix proteoglycans. J. Cell Biol. 122, 721-727.
Chan, B.M.C., Elices, M.J., Murphy, E., and Hemler, M.E. (1992). Adhesion to vascular cell
adhesion molecule-1 and fibronectin. Comparison of α4ß1 (VLA-4) and α4ß7 on the human
B cell line JY. J. Biol. Chem. 267, 8366-8370
Daniel, T.O., Gibbs, V.C., Milfay, D.F., and Williams, L.T. (1987). Agents that increase
cAMP accumulation block endothelial c-sis induction by thrombin and transforming growth
factor-β. J. Biol. Chem. 262, 11893-11896
Dejana, E., Colella, S., Languino, L.R., Balconi, G., Corbascio, G.C., and Marchisio, P.C.
(1987). Fibrinogen induces adhesion, spreading and microfilament organization of human
endothelial cells in vitro. J. Cell Biol. 104, 1403-1411.
Derynck, R., , and Feng, X.H. (1997). TGF-β receptor signaling. Biochim. Biophys. Acta. 1333,
F105-F150.
Derynck, R., Shang, Y., and Feng, X.H. (1998). Smads transcriptional activators of TGF-beta
responses. Cell 11, 737-740.
Derynck, R., Akhurst, R.J., and Balmain, A. (2001). TGF-β signaling in tumor suppression and
cancer progression. Nature Genetics 29, 117-129.
Dong, J.M., Leung, T., Manser, E., and Lim, L. (1998). cAMP-induced morphological changes
are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J. Biol.
Chem. 28, 22554-22562.

24
D'Souza-Schorey, C., Boettner, B., and Van Aelst, L. (1998). Rac regulates integrin-mediated
spreading and increased adhesion of T lymphocytes. Mol. Cell Biol. 18, 3936-3946
Edlund, S., Landstrom, M., Heildin, C.H., and Aspenstrom, P. (2002). Transforming growth
factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPasses
Cdc42 and RhoA. Mol. Biol. Cell 13, 902-914.
Fava, R.A., and McClure, D.B. (1987). Fibronectin-associated transforming growth factor. J.
Cell Physiol. 131, 184-189.
Grabovsky, V., S. et al. (2000). Subsecond induction of α4 clustering by immobilized
chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion
molecule 1 under flow conditions. J. Exp. Med. 192, 495-505.
Grainger, D.J., Kemp, R.P., Witchell, C.M., Weissberg, P.L., and Metcalfe, J.C. (1994).
Transforming growth factor β decreases the rate of proliferation of rat vascular smooth
muscle cells by extending the G2 phase of the cell cycle and delays the rise in cyclic AMP
before entry into M phase. Biochem. J. 299, 227-235.
Hall, A. (1998). Rho GTPases and the actin cytoskeleton. Science 279, 509-514.
Hanafusa, H., Ninomiya-Tsuji, M., Masuyama, N., Nishita, M., Fujisawa, J., Shibuya, H.,
Matsumoto, K., and Nishida, E. (1999). Involvement of the p38 mitogen-activated protein
kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem.
274, 27161-27167.
Hannan, R.L., Kourembanas, S., Flanders, K.C., Rogelj, S.J., Roberts, A.B., Faller, D.V., and
Klagsbrun, M. (1988). Endothelial cells synthesize basic fibroblast growth factor and
transforming growth factor beta. Growth Factors 1, 7-17.
Hannigan, M., Zhan, L., Ai, Y., and Huang, C-K. (1998). The role of p38 MAP kinase in TGF-
β1-induced signal transduction in human neutrophils. Biochem. Bioph. Res. Comm. 246, 55-58.

25
Hartsough, M.T., and Mulder, K.M. (1995). Transforming growth factor ß activation of
p44mapk in proliferating cultures of epithelial cells. J. Biol. Chem. 270, 7117-712416.
Heino, J., Ignotz, R.A., Hemler, M.E., Crouse, C., and Massagué, J. (1989). Regulation of
cell adhesion recpetors by transforming growth factor-beta. Concomitant regulation of
integrins that share a common beta 1 subunit. J. Biol. Chem. 264, 380-388.
Hidalgo, A., Sanz-Rodríguez, F., Rodríguez-Fernández, J-L-, Albella, R., Blaya, C., Wright, N.,
Prósper, F., Cabañas, C., Gutierrez-Ramos, J-C, and Teixidó, J.( 2001). The chemokine stromal
cell-derived factor-1α modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-
1 on bone marrow hematopoietic progenitor cells. Exp. Hematol.. 29:, 345-355.
Hynes. R.O. (1992). Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69, 11-
25.
Ignotz, R.A., and Massagué, J. (1987). Cell adhesion protein receptors as targets for
transforming growth factor-beta action. Cell 51, 189-197.
Kulkarni, A.B., Huh, C-G., Becker, D., Geiser, A., Lyght, M., Flanders, K.C., Roberts, A.B.,
Sporn, M.B., Ward, J.W., and Karlsson, S. (1993). Transforming growth factor β1 null mutation
in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 90,
770-774.
Letterio, J.J., and Roberts, A.B. (1998). Regulation of immune responses by TGF-beta. Annu.
Rev. Immunol. 16, 137-161.
Lévesque, J.-P., Leavesley, D.I., Niutta, S., Vadas, M., and Simmons, P.J. (1995). Cytokines
increase human hemopoietic cell adhesiveness by activation of very late antigen (VLA)-4 and
VLA-5 integrins. J. Exp. Med. 181, 1805-1815.
Lobb, R.R., and Hemler, M.E. (1994). The pathophysiologic role of α4 integrins In vivo. J. Clin.
Invest. 94, 1722-1728.

26
Massagué, J. (1998). TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753-791.
Massagué, J., Blain, S.W., and Lo, R.S. (2000). TGFbeta signaling in growth control, cancer,
and heritable disorders. Cell 103, 295-309.
Massagué, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling
system. EMBO J. 19, 1745-1754.
Miyazono, K., ten Dijke, P., and Heldin, CH. (2000). TGF-beta signaling by Smad proteins.
Adv. Immunol. 75, 115-157.
Mould, A.P., Askari, J.A., Craig, S.E., Garrat, A.N., Clements, J. and Humphries, M.J. (1994).
Integrin α4ß1-mediated melanoma cell adhesion and migration on vascular cell adhesion
molecule-1 (VCAM-1) and the alternatively spliced IIICS region of fibronectin. J. Biol. Chem.
269, 27224-27230.
Mucsi, I., Skorecki, K.L., and Goldberg, H.J. (1996). Extracellular signal-regulated kinase and
the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-betal
on gene expression. J. Biol. Chem. 271, 16567-16572.
Muñoz, M., Serrador, J., Sánchez-Madrid, F., and Teixidó, J. (1996). A region of the integrin
VLAα4 subunit involved in homotypic cell aggregation and in fibronectin but not VCAM-1
binding. J. Biol. Chem. 271, 2696-2702.
Murphy-Ullrich, J.E., Schultz-Cherry, S., and Hook, M. (1992). Transforming growth factor-
beta complexes with thrombospondin. Mol. Biol. Cell 3, 181-188.
Nobes, C.D., and A. Hall. (1995). Rho, rac and cdc42 regulate the assembly of multi-
molecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell
81, 53-62.
Osborn, L., Hession, C., Tizard, R., Vassallo, C., Luhowskyj, S., Chi-Rosso, G., and Lobb, R.
(1989). Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced

27
endothelial protein that binds to lymphocytes. Cell 59, 1203-1211.
Pachynski, R.K., Wu, S.W., Gunn, M.D., and Erle, D.J. (1998). Secondary lymphoid-tissue
chemokine (SLC) stimulates integrin α4β7-mediated adhesion of lymphocytes to mucosal
addressin cell adhesion molecule-1 (MAdCAM-1) under flow. J. Immunol. 161, 952-956.
Parker, C.M., Cepek, K.L., Russell, G.J., Shaw, S.K., Posnett, D.N., Schwarting, R., and
Brenner, M.B. (1992). A family of beta 7 integrins on human mucosal lymphocytes. Proc. Natl.
Acad. Sci. USA 89, 1924-1928.
Peled, A., Grabovsky, V., Habler, L., Sandbank, J., Arenzana-Seisdedos, F., Petit, I., Ben-
Hur, H., Lapidot, T., and Alon, R. (1999). The chemokine SDF-1 stimulates integrin-
mediated arrest of CD34+ cells on vascular endothelium under shear flow. J. Clin. Invest.
104, 1199-1211.
Piek, E., Heldin, CH., and Ten Dijke, P. (1999). Specificity, diversity, and regulation in TGF-
beta superfamily signaling. FASEB J. 12, 2105-2124.
Robledo, M.M., Bartolomé, R.A., Longo, N., Rodríguez-Frade, J.M., Mellado, M., Longo, I., van
Muijen, G.N.P., Sánchez-Mateos, P., and Teixidó, J. (2001). Expression of functional chemokine
receptors CXCR3 and CXCR4 on human melanoma cells. J. Biol. Chem. 276, 45098-45105.
Sancho, D., Yanez-Mo, M., Tejedor, R., and Sánchez-Madrid, F. (1999). Activation of
peripheral blood T cells by interaction and migration through endothelium: role of
lymphocyte function antigen-1/intercellular adhesion molecule-1 and interkeukin-15. Blood
93, 886-896.
Sander, E.E., van Delft, S., ten Klooster, J.P., Reid, T., van der Kamen R.A., Michiels, F.,
and Collard, J.G. (1998). Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes
either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J.
Cell Biol. 143, 1385-1398.

28
Sano, Y., Harada, J., Tashiro, S., Gotoh-Mandeville, R., Maekawa, T., and Ishii, S. (1999).
ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth
factor-beta signaling. J. Biol. Chem. 274, 8949-8957.
Sanz-Rodríguez, F., Hidalgo, A., and Teixidó, J. (2001). Chemokine stromal cell-derived factor-
1α modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and
VCAM-1. Blood 97, 346-351.
Shull, M.M, et al. (1992). Targeted disruption of the mouse transforming growth factor-β1
gene results in multifocal inflammatory disease. Nature 359, 693-699.
Springer, T.A. (1995). Traffic signals on endothelium for lymphocyte recirculation and leukocyte
emigration. Annu. Rev. Physiol. 57, 827-872.
Steiner, M.S., Wand, G.S., and Barrack, E.R. (1994). Effects of transforming growth factor
beta 1 on the adenylyl cyclase-cAMP pathway in prostate cancer. Growth Factors 11, 283-
290.
Tanaka, Y., Adams, D.H., Hubscher, S., Hirano, H., Siebenlist, U., and Shaw, S. (1993). T- cell
adhesion induced by proteoglycan-immobilized cytokine MIP-1. Nature 361, 78-82.
Valitutti, S., Dessing, M., and Lanzavecchia, A. (1993). Role of cAMP in regulating
cytotoxic T lymphocyte adhesion and motility. Eur. J. Immunol. 23, 790-795.
Van Aelst, L., and D’Souza-Schorey, C. (1997). Rho GTPases and signaling networks. Genes
Dev. 11, 2295-2322.
Wahl, S.M., Allen, J.B., Weeks, B.S., Wong, H.L., and Klotman, P.E. (1993) Transforming
growth factor beta enhances integrin expression and type IV collagenase secretion in human
monocytes. Proc. Natl. Acad. Sci. USA 90, 4577-4581.
Wakefield, L.M., and Roberts, A.B. (2002). TGF-beta signaling: positive and negative effects
on tumorigenesis. Curr. Opin. Genet. Dev. 12, 22-29.

29
Woldemar Carr, M.., Alon, R., and Springer, T.A. (1996). The C-C chemokine MCP-1
differentially modulates the avidity of β1 and β2 integrins on T lymphocytes. Immunity 4, 179-
187.
Wright, N., Hidalgo, A., Rodríguez-Frade, J-M., Soriano, S.F., Mellado, M., Parmo-Cabañas, M.,
Briskin, M.J., and Teixidó, J. (2002). The chemokine stromal cell-derived factor-1α modulates
α4ß7 integrin-mediated lymphocyte adhesion to MAdCAM-1 and fibronectin. J. Immunol. 168,
5268-5277.

30
Figure Legends
Figure 1. Modulation of α4 integrin-mediated adhesion by TGF-ß1 in human cell lines. BCECF-
AM-labelled cells were pre-incubated for the indicated times (A), or 2.5 min (B), in adhesion
medium with or without TGF-ß1 (1 ng/ml). Cells were then allowed to adhere for 10 min to
sVCAM-1- or FN-H89-coated wells. Some samples were pre-incubated with the anti-α4 HP1/2
mAb prior to exposure to TGF-ß1. Adhesions were quantified in a fluorescence analyzer and data
represent the mean ± SD of triplicate samples from representative results of at least three
independent experiments. **Adhesion was significantly stimulated, P<0.005, or * P<0.05,
according to the Student's two-tailed t test.
Figure 2. Characterization of α4ß1-mediated upregulation of cell adhesion by TGF-ß1. (A)
Mo7e cells were labelled with BCECF-AM, pre-incubated for the indicated times in adhesion
medium with or without TGF-ß1 (1 ng/ml), and allowed to adhere to FN-H89- or sVCAM-1-
coated wells for 2 min after a 15-sec plate centrifugation. (B) BCECF-labelled Mo7e cells were
pre-incubated for 5 min in adhesion medium in the presence or in the absence of TGF-ß1, and
allowed to adhere to FN-H89-coated wells for 5 min. Non-bound cells were recovered, again pre-
incubated for 5 min with or without TGF-ß1, and allowed to adhere to new FN-H89-coated wells
for 10 min. All adhesions were quantified in a fluorescence analyzer and data represent the mean
± SD of triplicate samples from representative results of at least two experiments. ***Adhesion
was significantly stimulated, P<0.001, or ** P<0.005, according to the Student's two-tailed t test.
Figure 3. Modulation of α4 integrin-mediated adhesion by TGF-ß1 in PBL and PBL-T.
BCECF-AM-labelled cells were pre-incubated for the indicated times in adhesion medium in the
presence or in the absence of TGF-ß1 (1 ng/ml). Cells were then allowed to adhere to sVCAM-1-
or FN-H89-coated wells for 10 min. Some samples were pre-incubated with the anti-α4 HP1/2
mAb prior to exposure to TGF-ß1. Adhesions were quantified in a fluorescence analyzer and data

31
represent the mean ± SD of triplicate samples from representative results of at least three
independent experiments. .
**Adhesion was significantly stimulated, P<0.005, or * P<0.05, according to the Student's two-
tailed t test.
Figure 4. Modulation by TGF-ß1 of α4 integrin-mediated PBL and PBL-T adhesion to HUVEC.
BCECF-AM-labelled cells were pre-incubated in adhesion medium for the indicated times with
or without TGF-ß1 (2 ng/ml), and allowed to adhere for 10 min to HUVEC monolayers, which
had been pre-treated for 10 h in the presence or in the absence of TNF-α. Some samples were
pre-incubated with the anti-α4 HP1/2 mAb prior to exposure to TGF-ß1. Adhesions were
quantified in a fluorescence analyzer and data represent the mean ± SD of triplicate samples from
representative results out of three independent experiments. **Adhesion was significantly
stimulated, P<0.005, or * P<0.05, according to the Student's two-tailed t test.

32
Figure 5. Activation of RhoA by TGF-ß1 is not required for upregulation of α4 integrin-
dependent cell adhesion. (A) TGF-ß1 activates RhoA and Rac1. Mo7e cells were pre-incubated
in adhesion medium in the presence or in the absence of C3 exozyme (50 µg/ml, 16 h) and
subsequently treated with TGF-ß1 (0.5 ng/ml) or with LPA (5 µM) for the stated times. Cells
were solubilized and lysates incubated with glutathione agarose-bound fusion proteins to detect
active RhoA and Rac1. Beads and proteins bound to fusion proteins were eluted in Laemli
sample buffer and analyzed for bound RhoA and Rac1 by Western Blot using anti-RhoA and
anti-Rac1 antibodies. Aliquots of the respective lysates served as controls for analyzing total
amounts of RhoA and Rac1. (B) Effect of C3 on the upregulation of α4 integrin-dependent cell
adhesion by TGF-ß1. Mo7e cells were exposed to C3 or adhesion medium alone (Control). After
labelling with BCECF-AM, cells were pre-incubated for 2.5 min in adhesion medium with or
without TGF-ß1 (1 ng/ml), and allowed to adhere to sVCAM-1- or FN-H89-coated wells for 10
min. Adhesions were quantified in a fluorescence analyzer and data represent the mean ± SD of
triplicate samples from representative results of at least two independent experiments.
Figure 6. Role of actin cytoskeleton in upregulation of α4 integrin-mediated cell adhesion by
TGF-ß1. (A) Left. Cells were incubated for the indicated times with TGF-ß1 (1 ng/ml) or medium
alone, stained with FITC-phalloidin and subjected to flow cytometry. Right. Cells were pre-
incubated with cytochalasin D (0.5 µg/ml, 30 min; Cyt D), with C3, or in adhesion medium alone
(Control), followed by exposure to TGF-ß1 for the stated times and processing for flow
cytometry as above. (B) BCECF-AM-labelled cells were treated for 30 min at 37ºC in adhesion
medium alone (Control) or with cytochalasin D (Cyt D; 0.5 µg/ml), pre-incubated for the stated
times in adhesion medium with or without TGF-ß1, and allowed to adhere to FN-H89-coated
wells for 10 min. Adhesions were quantified in a fluorescence analyzer and data represent the
mean ± SD of triplicate samples from representative results of three (PBL) or five (Mo7e)

33
independent experiments. **Adhesion was significantly inhibited, P<0.005, or * P<0.05 according
to the Student's two-tailed t test.
Figure 7. p38 MAP kinase activation by TGF-ß1 is not required for enhancement of α4 integrin-
dependent cell adhesion. (A) Mo7e cells were pre-incubated in adhesion medium for 45 min
with or without SB 203580 (13 µM), followed by treatment for the indicated times at 37ºC with
TGF-ß1 (1 ng/ml). Cells were solubilized and extracts subjected to immunoblotting using anti-
phospho-p38 MAP kinase antibodies. After stripping and saturating non-specific protein binding
sites, the same blots were reprobed with anti-p38 antibodies to check for total protein content.
Blots were developped by a chemiluminiscent reaction and exposed to a radiographic film. (B)
Mo7e cells were labelled with BCECF-AM, incubated for 45 min in adhesion medium with or
witout SB 203580 followed by incubation with TGF-ß1 (1 ng/ml). Cells were subjected to
adhesion to FN-H89 for 10 min at 37ºC. Adhesions were quantified in a fluorescence analyzer
and data represent the mean ± SD of triplicate samples from a representative result of three
experiments.
Figure 8. Alterations in intracellular cAMP oppose the upregulation of α4 integrin-dependent
cell adhesion in response to TGF-ß1. (A) Mo7e cells were labelled with BCECF-AM, incubated
for 1 h in adhesion medium without (Control) or with forskolin (35 µM) or 8-Br-cAMP (500
µM), following by incubation for 5 min with TGF-ß1 (1 ng/ml). Cells were added to wells coated
with sVCAM-1 or FN-H89 and subjected to adhesion for 2 min at 37ºC after a short spin.
Adhesions were quantified in a fluorescence analyzer and data represent the mean ± SD of
triplicate samples from a representative result of three independent experiments. **Adhesion was
significantly inhibited, P<0.005, or * P<0.05, according to the Student's two-tailed t test. (B)
Mo7e cells were treated with or without 8-Br-cAMP as above followed by incubation with TGF-
ß1 for the indicated times and staining with FITC-phalloidin and analysis by flow cytometry.

34
Figure 9. Role of TGF-ß1 in SDF-1α-stimulated α4 integrin-dependent cell adhesion. Mo7e (A)
or PBL-T (B) cells were labelled with BCECF-AM, pre-incubated for the indicated times with or
without TGF-ß1 (1 ng/ml), in the presence of SDF-1α (150 ng/ml; +SDF-1α) or in adhesion
medium alone (-SDF-1α) within the 2.5 or 5 min incubation with TGF-ß1. When SDF-1α
incubation was longer (5 min) compared to incubation with TGF-ß1 (2.5 min), then SDF-1α was
added before the cytokine. Cells were allowed to adhere for 2 min at 37ºC to sVCAM-1- or FN-
H89-coated wells after the plate was spun for 15 seconds. Some samples were pre-incubated with
the anti-α4 HP1/2 mAb before cytokine treatments. Non-bound cells were washed and extent of
adhesion measured in a fluorescence analyzer. Data represent the mean ± SD of triplicate samples
from a representative result of three experiments for each cell type. ***Adhesion was significantly
stimulated, P<0.001, or * P<0.05, according to the Student's two-tailed t test.
A
sVC
AM
-1
Tim
e (m
in)
Cells bound/mm2
100
200
300
400
500 0
510
1520
Tim
e (m
in)
510
1520
FN-H
89
TGF-
β βββ1M
ediu
m
TGF-
β βββ1+H
P1/2
Mo7
eJY
510
1520
FN-H
89
05
1015
20
Tim
e (m
in)
Tim
e (m
in)
sVC
AM
-1
**
**
***
*
Fig
1 A

U93
7B
Cells bound/mm2
250
500
750
1000 0
Medium TGF-ß1
TGF-ß1+HP1/2
FN-H
89
500
1000
1500
0
Medium TGF-ß1
TGF-ß1+HP1/2
sVC
AM
-120
00
K56
2-α ααα4 25
0
500
750
1000
0
FN-H
89
MediumTGF-ß1
TGF-ß1+HP1/2
500
1000
1500 0
sVC
AM
-1
MediumTGF-ß1
TGF-ß1+HP1/2
2000
Fig
1 B

Cells bound/mm2
AB
2 m
in a
dhes
ion
Medium
TGF-ββββ1-2.5 min
TGF-ββββ1-5 min
0
100
200
300
400
500
600
MediumTGF-ββββ1
MediumTGF-ββββ1
5 m
in
adhe
sion
Non
-bou
nd c
ells
,ne
w a
dhes
ion
***
**
0
100
200
300
400
500
600
***
***
0
100
200
300
***
FN-H
89sV
CA
M-1
TGF-ββββ1-5 min+HP1/2
TGF-ββββ1-5 min+HP1/2
TGF-ββββ1-5 min
Medium
FN-H
89
Fig
2 A
B

TGF-ββββ1-2.5 min+HP1/2
TGF-ββββ1-2.5 min
PBL-
T20
0 04080120
160 Medium TGF-ββββ1-5 min
sVC
AM
-1
0Ti
me
(min
)
Cells bound/mm2
50100
150
200
250
PBL
510
15
TGF-
β βββ1M
ediu
m
HP1
/2TG
F-β βββ1
+
20
PBL
0
100
200
300
400
500
515
20
Tim
e (m
in)
10
PBL-
T
050100
150
200
250
FN-H
89
TGF-
β βββ1M
ediu
m
HP1
/2TG
F-β βββ1
+
TGF-ββββ1-2.5 min
Medium TGF-ββββ1-5 min
TGF-ββββ1-2.5 min+HP1/2
***
**
**
Fig
3

0
50100
150
200
PBL
PBL-
T
0
100
200
300
400
500
Cells bound/mm2
Medium
TGF-ββββ1-2.5 min
TGF-ββββ1-5 min
TGF-ββββ1-5 min+HP1/2
Medium
Medium
-TN
F-α ααα
+TN
F-α ααα
TGF-ββββ1-2.5 min
TGF-ββββ1-5 min+HP1/2
TGF-ββββ1-5 min
TGF-ββββ1-2.5 min
TGF-ββββ1-5 min+HP1/2
TGF-ββββ1-5 min
*
**
**
**
Fig
4

0 1 2.5 5 15Time (min)
Active Rac1
Total Rac1
TGF-ββββ1
0 1 2.5 5 15
LPA
TGF-ββββ1
Active RhoA
Total RhoA
Time (min)
A
Cel
ls bo
und/
mm
²
Med
ium
Med
ium
TGF-ß1
TGF-ß1
0
50
100
150
200
250FN-H89
Control C3
0
150
300
450
600sVCAM-1
Med
ium
Med
ium
TGF-ß1
TGF-ß1
Control C3
B
Fig 5 A B
C3
1 2.5 5 15
Control

0
50
100
150
200
250
0
100
200
300
400
500
600
Med
iumBSA
Med
ium
TGF-ββββ1-2
.5 min
TGF-ββββ1-2
.5 min
TGF-β1β1β1β1-5
min
TGF-β1β1β1β1-5
min
Control Cyt D
Med
iumTGF-β
1β1β1β1-5
min
TGF-β1β1β1β1-5
min
Control Cyt D
BSA
Med
ium
Cel
ls bo
und/
mm
2
PBL Mo7e
B
AF-
actin
con
tent
(% o
f con
trol
)
Time (min) 50
80
100
120
140
160
10 15 20 25 5 10 15 20
Mo7e JY
* **
**
Fig 6 A B
TGF-ββββ1Medium
80
100
120
140
160 Mo7e
0 0.5 2.51 0 0.5 2.51 0 0.5 2.51 minControl Cyt D C3
TGF-ββββ1

BA
050100
150
200
250
Cells bound/mm²
TGF-β1β1β1β1-5 min
TGF-β1β1β1β1-5 min
BSA
Medium
Medium
Con
trol
SB20
3580
TG
F-ß1
(min
) 0
1
5
15
phos
pho-
p38
p38
0
1
5
15
SB20
3580
Med
ium
Fig
7

04080120
160
200
Cells bound/mm²
sVC
AM
-1
Med
ium
TG
F-ß1*
**
0
100
200
300
FN-H
89
Med
ium
TG
F-ß1**
**
Con
trol
8-B
r-cA
MP
Fors
kolin
75100
125
150
A
F-actin content (% of control)
Con
trol
8-B
r-cA
MP
B
0
1
2.
5
5
TG
F-ß1
(min
)
Fig
8 A
B

Mo7
eCells bound/mm²
-SD
F-1α ααα
+SD
F-1α ααα
-SD
F-1α ααα
+SD
F-1α ααα
sVC
AM
-1FN
-H89
Med
ium
TGF-ß1-5
min
TGF-ß1-5 m
in+HP1/2
TGF-ß1-2.5
min
TGF-ß1-5 m
in
TGF-ß1-2.5
min0
100
200
300
400
500
Med
ium0
100
200
300
400
500 Med
ium
TGF-ß1-5 m
in
Med
ium
TGF-ß1-5 m
in
TGF-ß1-5 m
in+HP1/2
***
Med
ium
TGF-ß1-5 m
in
Med
ium
TGF-ß1-5 m
in
***
***
1 m
in2.
5 m
in5
min
Med
ium TGF-ß1-5 m
in
TGF-ß1-2.5
min
Med
ium TGF-ß1-5 m
in
TGF-ß1-2.5
min
1 m
in2.
5 m
in5
min
A
***
Fig
9 A

0
200
400
600
800
0
100
200
300
400
500
sVC
AM
-1FN
-H89
PBL-
T
Med
ium
Med
ium
TGF-ß1 5 m
in
TGF-ß1-5 m
in+HP1/2
Med
ium
Med
ium
TGF-ß1-5 m
in+HP1/2
TGF-ß1 5 m
in
+SD
F-1α ααα
+SD
F-1α ααα
***
*
B
Fig
9 B
Top Related
Copyright © 2022 FDOKUMEN

