







Bahasa
Halaman
Hukum


LOW LYING STATES OF LITHIUM HYDRIDE
By
ROBERT MICHAEL PREDNY
A DISSERTATION PRESENTED TO THE GRADUATE COUNOL OF
THE UNIVERSITY OF FLORIDA
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE
DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
1969

Dedicated to
rrry Parents
wife and family

ACKNOWLEDJMENT S
I would like to express my appreciation to Dr. Yngve
Ohm and Dr. Darwin W. Smith for their encouragement and inter
est in directing this research.
I am also indebted to Dr. Harvey Michels for the sugges
tion or this problem and also the computer program to carry
it out. His helpful discussions were indispensable.
The assistance of the members of the Quantum Theory
Project was greatly appreciated.
I am particularly grateful to the Computing Center.
Without its generous support this study could not have been
completed.
I especially thank my wife, Faye, for her interest and
the long hours she spent in typing this manuscript.
iii

TABLE OF CONTENTS
Page
ACKNOWLEIXiME'NTS • • • • • • • • • • • • • • • • • iii
LIST OF TABLES • • • • • • • • • • • • • • • • • vi
LIST OF FIGURES
Chapter
• • • • • • • • • • • • • • • • • viii
I. INTRODUCTION. • • • • • • • • • • • • • • 1
II.
III.
1.1. Review of the Experimental Properties of Lithium Hydride • • • •
1.2. Theoretical Calculations • • • • • •
MEl'HOD OF CALCULATION • • • • • • • • • •
COMPUTATIONAL PROCEDURES ••••••• • •
3.1. Basis Orbitals. • • • • • • • • • • •
3.2. Configurations •• • • • • • • • • • •
1
10
17
25
25
27
3.3. Spin Functions • • • • • • • • • • • • • 31
'IV. LOWEST STATES OF 3~+, 371 AND 1 71' SD1lfflRY • • • • • • • • • • • • • • • • • • 32
4.1. The Lowest Lithium Hydride 3~+ State •• 32
4. 2. The Lowest Lithium Hydride 3rr state • • 36
4.3. The Lowest Lithium Hydride 1 1T state. • 39
V. POTENTIAL CURVES FOR LOW LYDm LITHIUM HYDRIDE STATES • • • • • • • • • • • • • • • • • • • 43
5.1. Basis Orbitals. • • • • • • • • • • • 43
5. 2. The Lithium Hydride l r + States • • • • 46
iv

5.3. The Lithium Hydride 3 L+ States ••••• • 59
5.4. The Lithium Hydride 3TT States •••••• o9 l 5.5. The Lithium Hydride TT States •••••• 75
5.o. The Lithium Hydride x2 f + State •••••• 81
VI. DISCUSSION AND SUMMARY
APPENDIX I • • • • • • • • • • • • • • • • • • • • • • • 88
APPENDIX II • • • • • • • • • • • • • • • • • • • •
BIBLIOGRAPHY - • • • • • • • • • • • • • • • • • • •
BIOGRAPHICAL SKETCH. • • • • • • • • • • • • • • •
V
• 93
109

LIST OF TABLES
TABLE
1. SPECTROSCOPIC CONSTANTS. • • • • • • • • • •
2. BASIS SLATER TYPE ORBITALS FOR THE LOWEST 3 ~ + SI'ATE OF LITHTC!M HYDRIDE •
3. ENERGY FOR THE LOWEST 3 2.. + SI'ATE OF LITHIUM HIDRIDE • • • • • • • • • • •
4. BASIS SLATER TYPE ORBITALS FOR THE LOWEST 3 rr srATE OF LITHIUM HYDRIDE •
5. ENERGIES FDR THE LOWEST 3rr srATE OF LITHitn.lf HYDRIDE • • • • • • • • • • •
6. BASIS SLATER TYPE ORBITALS FDR THE LOWEST 1 TI' 6'TATE OF LITHIUM HIDRIDE •
7. ENmGY FOR THE LOWEST l 7T srATE OF LITHIUM HYDRIDE • • • • • • • • • • •
8. BASIS SLATER TYPE ORBITALS FDR THE CALCULATIONS ON THE LOW LYJNG STATES
• • •
• • • •
• • • •
• • • •
• • • •
• • • •
Page
2
33
34
37
38
40
OF LITHIU11 HD)RJDE • • • • • • • • • • • • • • 44
9. POTENI'IAL ENERGIES FOR THE l i + SfATES OF LITHIUM HYDRIDE ••••••••••• • • • • 48
10. 125 CONFIGURATIONAL WAVEFUNCTION FOR THE 1~ + S'fATES OF LITHIUM HYDRIDE • • • • • • 50
ll. POTENTIAL ENERGIES FOR THE l~ + srATFS OF LITHml HYDRilJE USING AN EXTENDED BASIS SEI'. • 55
12. POTENTIAL EN:rnGIF.S FDR THE 3~ + srATES OF LITHIUM HYDR1DE •••••••••••••••• 60
13. 125 CONFIGURATIONAL WAVEI<"'UNcrimr FOR THE 3 L + STATES OF LITHIUM HYDRIDE • • • • • • • • 62
l4. COEFFICIENTS OF THE ~IN+OONFIGURATICNS OF THE SECOND AND THIRD ~ SI'ATES • • • • • • • 66
vi

1.,. POTENI'IAL ENERGIES FOR THE J 2.. + STATES OF LITHIUM HYDRIDE USING AN EXTENDED BASIS SET. • • • •
lo. POTENI'IAL ENEIDIES ltUR THE 3TT STATES OF
17.
18.
LITHIUM HYDRIDE • • • • • • • • • • • • •
125 CONFIGURATIONAL WAVEFtJNm'IDN FOR THE JTT STATES OF LITHIUM HYDRIDE ••••••
POTEm IAL ENE.FOIES FOR THE l 7T STATF..S OF LITHIUM HYDRIDE • • • • • .. • • • • • • •
19. f5 CONFIGURATIONAL WAVEFUNCTION FOR THE TI STATES OF LITHIUI1 HYDRIDE ••••••
20. POTENTIAL ENEIDY OF THE 2 r + STATE OF
• • • • • •
• • • • • •
• • • • • •
• • • • • •
• b1
• 70
• 72
• 1b
• 78
LITHIUM HYDRIDE PLUS • • • • • • • • • • • • • • • • • 82
21. ~5 CONFIGURATIDNAL WAVEF01lCTION FOR THE r + STATE OF LITHIUM HYDRIDE PLUS • • • • • • • • • • 84
22. CONFIGURATIONS AND OOEFFICIENTS OF THE WAVEFUNCTION USED FOR THE LOWF.ST 3~ + STATE OF LITHIUM HYDRIDE • • • • • • • • • • • • • • • • • • • 94
2J. CONFIGURATIONS AND COEFFICIENTS 0) THE WAVEFUNC!'ION USED FOR THE l.OWEST TT STATE OF LITHIUM HYDRIDE • • • • • • • • • • • • • • • • • • 99
24. CONFIGURATIONS AND COEFFICIENrS OF THE WAVEFUNCI'ION USED FOR THE LOWEST l7t STATE OF LITHIUM HYDRIDE ••••• •• •••••• • • • • •• 104
25. COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVE.FUNCTION FOR THE LITHIU1 HYDRIDE lL+ STATES ••• 110
2o. COEFFICIENTS OF THE J2.5 CONFIGURATIONAL WAVE.FUNCTION FOR THE LI'llIIUM HYDRIDE .3 J:+ STATES ••• llo
27. COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVEFUNCTION FOR THE LITHIUM HYDRIDE 371 STATES ••• 122
28. OOEFFICIENTS OF THE 125 OONFIGURATIONAL WAVEFUNCTION :FOR THE LITHIUM HYDRIDE 1 TT STATES ••• 128
29. COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVEFUNCTION FOR THE LITHitM HYDRIDE PLUS 2 ~ + STATE • • • • • • • • • • • • • • • • • • • .134
vii

LIST OF FIGURES
l. POTENTIAL ENERGY CURVF.S FOR THE l ~ + srAT:ES OF LITHIUI1 HYDRIDE • • • • • • • • • • • • • • • • 49
2. POTENTIAL ENERGY CURVES FOR THE 3 ~ + srATES OF LITHIUM HYDRIDE • • • • • • • • • • • • • • • • 61
.3. POTENTIAL ENERGY CURVES FOR THE 37f STATES OF LITHIUM HYDRIDE • • • • • • • • • • • • • ••• 71
4. POTENTIAL ENERGY CURVES FOR THE l 7r srATE.S OF LITHIUM HYDRIDE ••••••••• • • • • ••• 77
,. POTENTIAL ENERGY CURVE. FOR THE 2 ~ + STATE OF LITHIUM HYDRIDE PLUS • • • • • • • • • • • • • 83 .
viii

CHAPTER I
INTRODUm'ION
l.l. Review of the Experimental Properties
of Lithium Hydride
The experimental potential energy curves for diatomio
molecules are obtained from their absorption spectra . Diatomic
lithium hydride or deuteride gas is obtained by combining Li
metal and excess H2 or n2 gas at temperatures of 1000° C.
When the reaction has stopped, the absorption spectrum is
obtained by irradiating the gas in the UV region. Exposure
times of 2 hours or more are used. Crawford and Jorgensen(!)
investigated the absorption spectra of LiH and LiD between
3200 and 4300 i. Thirty-five bands of the 1 ~ -1 Z:: spectrum
were measured for LiD and 28 bands for LiH. Two emission
bands were also obtained for Lillo Vibrational and rotational
constants we"'."8 d~tennined for the xl~+ and A1 l+ states.
Their potential energy curves were obtained for low lying vibra
tional. levels by the use of Dunham's molecular potential energy
function. Crawford and Jorgensen's values for the spectroscopic
constants of these states are listed by Herzberg.( 2) The values
are given in Table 1. The values !'or the energy of the separated
1

2
TABLE 1
SPEm'ROSOOPIC OONSTANTS
MOLECULAR STATE (LiH)
x1r+ Alr_+ Bl7T
De(eV)4 2.,154 1.0765 0.035
D0 (ev)• 2.4288 1.o602 0.0227
Energy-( a. u.) -8.0704 -7.9496 -7.9113
Re(a.u.) 3.015b 4.906b 4.494b
:Energy-( a.u.) Sep. Atomsc -7-97865 -7.91074 -7-91074
Te (cm-1) 0 26,516.2b 34912a
Vibrational Constants
we (cm-1) 1405.65b 234.41b 215.5a
WeXe (cm-1) 23.20b -28.95b 42.4a
Rotational Constants
Be (cm-1) 7.5131b 2.8186b 3.383&
D<e (cm-1) 0.2132b -0.078.31b 0.9868
Dipole Moment (D) -5.882d
a. see ref. (7) b. see ret. (1) and (2) · (4) c. calculated from Moore's tables (3) and Pekeris d. see ref. (14)

atoms were calculs.t.(lld from Moore's tables<3) and the results
of Pekeris. (4)
ihe first excited state of LiH was found to exhibit
anomalous behavior as seen by its abnomal. band spectrum con
stants. This A1Z:. + state's unusual shape was explained by
Mulliken<5) as being due to its Li+u"- character at large
internuclear distances. In this analysis the ground state,
i.e. x1r+ state, has mainly ionic Li+ii- character around its
equilibrium. internuclear separation,while the All,+ state is
predo~in.antly Li+u"- at large internuclear separations. A plot(5)
of these two states and a curve of the Li+Ir interaction showed
that the ionic Li +ir curve crosses the A1 E + curve at large
uiternuclear distances. The x1r + state dissociates into the
112s(1s22s) plus H2s(ls) states. The A1 ~ + state goes into the
ti2P(ls22p) plus H2s(ls) atomic states. Rosenbaum< 6) used Klein's
method to obtain the experimente.l potential energy curve for the
A1!_ + state. He found that this curve crosses the coulombic
curve of Li+Ii.
Velasco(7) analyzed the UV absorption spectrum of LiH
between 2000 and 3200 X. He found a new band system between the
ground state and an excited 11T state. From the breaking off of
the rotational structure of the Bl TT -x1 2.. + system bands he deter
mined vecy accurate dissociation limits for the B1 7T state. There
fore he has bean able to obtain accurate dissociation energies of
the three experimentally observed states of lithium hydride. The
dissociation energy, D8 , for the x1 2_ + state is 2.5154:o.0002ev.
3

For the A1 2+ state D8 is 1.0765!0.0002ev and 0.035!0.00leV for
the B1 TT state. The spectroscopic constants for the B1 TT state
are given in Table 1.
Potential energy curves for the x1 r +, A1 l. + and B1 TT states
of L1H haTe been calculated by Fallon, Vanderslice and Mason< 8)
using the lcy'dberg-Klein-Rees (RKR) method. The curve for the
A1 I, + istate using this method also .has been obtained by
Singh and Jain. ( 9) Reduced potential curves tor the states of
LiH have been obtained by Jenc(lO) using the rum method and
compared to theoretical calculations. He concluded that the
anomalous behavior of the A1 ~ + state of LiH was due to effects
which wruld not be accOllllted for in the usual adiabatic approx
imation. Krupenie, et al.(ll) estimated long range attractive
potentials for the x12. + state of Lill by extrapolation of .func
tions fitted to the mm potential curves. They also estimated
curves for the lowest 3t+ state. Using the collision integrals
computed from the potentials, they calculated values for the
diffusion coefficient and viscosity of gaseous LiH systems. Halmann
and Laulicht(l2) have computed Frank-Condon factors based on rum
potential functions. These factors control the distribution of
intensity between vibrational bands.
KlempererC13) and his coworkers studied the inf'rared
absorption spectrum of gaseous LiH. In order to obtain an
appreciable absorption it is necessary to work at temperatures
greater than looOO c. They analyzed the P and R branches of the
0-1 and 1-2 ba.'lds. From the line intensities a dipole derivative
4

ot l.8!0.3 was calculated. Wharton, Gold and Klemperer(l.4)
used molecular beam resonance methods to obtain values f'or the
dipole manent by analyzing the J•l, Mj•O, to J•l, IMjl •l
transition. They obtained values for the dipole moment, quad
ru.pole coupling constant;, spin rotation constant arxl. rotational.
magnetic moment by adjustment of constants of the effective
Hamiltonian to tit the spectrum. The lowest vibrational. level
has a dipole moment of -5.882:!:o.003 debyes for LiH and -5.868:!: + 0.003 for LiD. The rotational magnetic moment is -0.654-
+ (15) 0.007nm. for LiH and -0.272-0.005 for LiD. Lawrence, et al.
observed resonances of the rotational magnetic moments in molecular
beams ot LiH and LiD. Their rotational. magnetic moments agreed
+ with those above. They obtained a dipole moment of -5.9-0.5
debyes with the polarity of Li +H- e Rothstein ( 16) aJ..so used
molecular beam techniques to obtain dipole moments and nuclear
hyperfine interaction constants.
Under standard con:litions, lithium hydride is a white
crystalline solid. Recent reviews of its properties are given
in references (17) to (20). It is generally prepared(l9, 20)
b;y the reaction between liquid lithium and excess hydrogen gas
at high temperatures. A typical reaction temperature is 750°
with a run time around 48 hoursf 20) . The crystal has the NaCl
type structure. It consists ot two ions and four electrons in
the primitive unit cell. The space group is ~5 or Fm3m. The
most recent crystallographic data on lithium hydride :are given
b:, Staritzky and Walker( 2l) who reported that the lattice constant
5

•
of LiH is 4.0834!0.000:5 i and that of LiD is 4.o684!0.000:5 i. They also investigated the refractive index.
flle solid has a melting point of 961° Kand a latent heat
or fusion of 4,900:!:700 cal/mole. This value was obtained by
Messer<22 > from freezing point lowerings. The standard heat of
formation of LiH is -21.666!0.026 kcal/mole and that of LiD
is -21.7a4:o.021 kcal/mole. These results were obtained by
Gunn and Green <23> from the heat of hydrolysis of Li and Lill
at 2.5.00!0.04° C,using a calorimetric bomb. They also calculated
the crystal energy- of Lili to be 217.76 kcal/mole and that of LiD
to be 218.76 kcal/mole.
The ionic character of lithium hydride was demonstrated
by Moers< 24) in a study of the electrical coniuctivity of molten
and solid LiII. The H2/LilI electrode was of interest recent1y( 25)
1n regard to fuel cells. Johnson, et a1.< 26) determined thenno
d;ynarnic properties from emf measurements on solid LiH between
675 and 885° K. Using the heat capacity data of Vogt~ 27 ) they
obtained the following results for a temperature of 298° K;
AHr0 a-21.79!0.29 kcal/mole,~Gf0 ~16.16!0.o, kcal/mole and
A O 8 + 4 _, / . (28) uSr ""-1 .9-0. Cd..1/deg.mole. Kelley and Kl.Ilg obtained
the value -16.3 cal/deg.mole for 6Sr O •
Numerous other thermal properties of LiH,_ for example,
dissociation pressure of H2, heat capacity of solid and liquid
LiH, coefficient of thermal expansion, etc., are presented in
references (19) and (29). Compressibility studies(JO) of LiH
did not show any evidence of a phase change up to a pressure
of 240 kilo bars.
6

Lithium hydride reacts readily with water. Machin and
Tompkins(3l) studied the kinetics or the reaction between J¼O
vapor and crystalline LiH in the temperature range 0-121° C.
The chemical nature of the product is determined by the amount
or water vapor. The product is predominantly 1120 when only
enough H20 is present to react with a single surface layer or
LiH. With more H20 the product is almost exclusively LiOH. The
rate controlling step in the production of 112 from the reaction
is the formation or the oxide.
LiH will also react readily with tm atomspheric gases
02 and n2 at elevated temperatures. With N2 the products,
~, Li2NH or Li3N may be formed. At high temperature LiH
will react violently with the halogens. With ammonia, lithium
amids ·will ba fo:rmad. And depending on the tempe~atu.,"""e the reac
tion with 002 will form either the thiosulfate or the sulfide.
Lithium hydride reacts with alcohols with the formation of
lithium alcoholates. It is also slightly soluble in ether and
other polar organic solvents. Due to its solubility and the
fact that it is a strong reducing agent it can be used for
organic reduction. Recently it has been used in polymerization
reactions.
Schlesinger and otheraC32) investigated the use or Lili
in producing other hydrides by hydrogen exchange reactions,
i.e. LiH + MX~MH + LiX. Lill has been used to produce SiJ\ and
B2H6 from the respective halides. L1A1H4 is a more useful com
pound for hydride synthesis and it is also quite soluble in ether.
7

8
It is formed by the reaction between lithiwn hydride and
aluminum chloride. The majority of the applications of LiH to
chemical synthesis are through LiA1H4 since this product is more
reactive and soluble than LiH i tsel:f.
Alkali hydride crystals are considered to be ionic in
character. The amount of ionicity in solid lithium hydride has
been investigated and found to be around 80%. Both Lwidquist(33)
and Hurst(34) calculated the cohesive energy an:i structure
factors for crystall.ine lithium hydride,assuming complete ioniza
tion. Morita and Takahash1(3.5) in a calculation of the cohesive
energy took into account the covalent character of LiH, using the
method of semi-localized crystal orbitals. All of them calculated
values within ±25 kcal/mole of the experimental value. Phillips
and Harris(36) obtained values of the crystal structure factors
which they used to determine electron density distributions.
They found that an ionic model with overlaps gave the best
agreement with tba experimental data. Calder, et al. 0 7) per
formed X-ray and neutron diffraction analysis of LiH in order to
obtain structure factors am electron distributions. The theo
retical. results of Waller and Lundquist03) and Hurst<34) colli)ared
well with their results. They indicated that the Li+ ion was
largely unaffected by the crystal field wh:i.le the r ion had a
pronoun.cad contraction as suggested by Lundqul.st. <33) They also
predicted that bonding in LiH is between 80 and 100,% ionic from
the ·x-ray data.
Brodsky and Burstein08) analyzed the infrared (IR) renec
tion spectra of single crystal LiH and LiD. Using a shell model

tor vibrating ions they calculated the static dielectric constant
and Szigeti effective charge. They obtained a value of (0.53!
0.02)e for the Szigeti effective charge which compares well
with the experimental value of 0.52e by Filler and Bu.rste1rJ39) .
Verble, Warren and Yarnell (40) studied the lattice dynamics of
LiH and LiD. They measured phonon dispersion curves using the
techniques of coherent inelastic scattering of themal neutrons.
These curves were then used to fit several rigid ion and shell
models. The best fit was obtained with a seven parameter shell
model. The results they· obtained indicated that the bonding
is 88% ionic. A Szigeti effective charge of 0.516e was calcula
ted. .Also, a small forbidden band exists between the acoustic
and optic branches in LiH but not in LiD. Jaswal and Harczy-(41)
used a def'omation dipole model to calcu.lsta the vibrational
spectra of LiH and LiD. They obtained a gap in the frequency
spectrum of Lili bounded by 9.83 X 1ol3 and 11.2 X 1ol3 rad/sec.
while the frequency spectrum for LiD showed no gap.
The optical absorption spectrum of thin films of solid
Lill in the ultraviolet (UV) (42) shows a sharp band at 2517 j
while for LiD the bcmd lies at 2482 X. The infrared absorption
spectrum(20,43) of these films has two bands at 11.0 and 17.2JA,
tor LiH and at 17.0 and 22.4~for Li.D. The fundamental absorp
tion is the 17 .2 p. band for Lili and 22.!i JA- band for LiD.
Color centers in lithium hydride crystals have been studied
by Pretzel and his coworker/44) and also by Dvinyaninov and
Gavrilov$45) Pretzel, et al. have folllld an F band at 2.4eV and
9

a V1 bam at 3.5eV and discussed their formation in detail. The
F band is produced by the formation of electrons while the Vi
band most probably results trom a H2
molecule trapped in an
anion site. They have also studied Li colloid bands and impur
i ty absorption bands.
1.2. Theoretical Calculations
Lithium hydride has been the subject of many theoretical
calculations since the first treatment by Knipp. (h6) Dile to
its simplicity with only four electrons, two of which are
bonding and two are in a closed inner shell, Lili has been used
as a test case for many theoretical procedures. Karo and_
01Bon<47) carried out the first extensive calculation of the
pvtantial energy curves of the x1 ! + and 1..1 I_+ states. Thay
used configuration interaction ( CI) in the framework of both the
valence bond (VB) and salt-consistent field molecular orbital
(SCF-MO) methods. They considered internuclear separations from
2.0 to 8.0 a.u. For the ground state they obtained a D8 •1.669eV
with Re•3.245 a.u. and a dipole moment,µ., of -6.0SD. For the
first excited state they calculated that De~0.8487eV at Raa4.90 a.u.
and µ.•+3.44D. Karo(4B) also performed electron population anal
ysis of his wavefunctions. His results disagreed with Mulliken1s(5)
prediction that the A 1 ~ + state 1s predominantly Li +H- at large
internuclear separations. Karo found that the strongest inter
action 1s between nonpolar configurations.
Ehbing<49) used a set of 10 orbitals in elliptical
coordinates ani 53 configurations in a SCF-CI study of the rr +
10

state at 2.99 a.u. His value for the energy was -8.04).28 a.u.
an:l -5.96D for the dipole moment;. In a calculation on the
ground state of Lili Matsen and Browne ( 50) obtained an energy ot
-8.04379 a.u. at 3.075 a.u. arxl a dipole moment of -5.51D using
21 basis Slater type orbitals (ST0's) and 20 configurations.
Harris am TaylorC5l) studied the potential curve for the x1 r +
state using open shell techniques with CI and an elliptic orbital
basis.
Kahalas and Nesbet<52) carried out a calculation on LiH
using the HF-Roothaan method. They also considered configuration
interaction. Besides the energy and dipole mo~nt th97 reported
a value of 35.3 kc/sec for the quadrupole coupling constant of
D in LiD. Using a HF-Roothaan wave.function, Cade and nu/53)
calculated e value of -6.002D for the dipole moment of Li.H.
They (54) also obtained the best Hartree-Fock energy for the
LiH x1 1 + state. They used 16 basis SI'O' s with optimized orbital
exponents and obtained analytic SCF wavefunctions from a solution
11
of the HF-Roothaan equations. Their calculated energy was -7.98731 a.u.
at an internuclear separation of J.015 a.u. The spectroscopic
constants obtained from their potential curve agreed well with
the experimental values. An ionization potential of 7.02ev
was calculated for Lill. They also presented a very extensive
review of the calculations on lithium hydride.
The best ene.rgies of Lill were obtained by Browne and
Mats~n(55) and Bender and Davidson.(56) Browne and MatsenC55)
used a mixed Slater and elliptic type orbital basis. The

Slater type orbitals were used to represent the lithium inner
core orbitals, and the elliptic orbitals, the boming orbitals.
Their basis consisted of 11 sro• s and 10 elliptic orbitals and
their wavefunction had up to 28 terms. Using a VB-CI method
they calculated an energy or -8.0:561 a.u. at R•J.046 a.u., a
dissociation energy of 2.J4eV, and a dipole moment of 5.9,3D.
They also evaluated spectroscopic constants and obtained a
quadrupole coupling constant of .34.2 kc/sec for Din LiD.
Bender and Davidson<56) used an iterative natural orbital
procedure to obtain an energy of -8.0606 a.u. at an intermiclear
distance or 3.0147 a.u. They calculated a dipole moment of
5.96500. They used .32 elliptic orbital basis functions and 45
configurations consisting of 12 c:r, 6 n and 5 o molecular orbitals.
Brown and Shull(57) obtained accurate potential energy
and dipole moment curves for low lying 1f + states of LiH
between internuclear separations of 1.0 and 10.0 a.u. Their
wavef'u.nction consisted of 69 configurations and 25 elliptic basis
orbitals which were taken fran two optimized sets, one for the
xl~+ state and the other for the Al~+ state. They calculated
34 points on the potential curves and obtained an energy of
-B.0556 a.u. at 3.060 a.u. with a dipole moment of ..;5.B9D for
the ground state and -7.9372 a.u. at 4.928 a.u. and +3.96n
respectively for the first excited state. A munerical vibra
tional and rotational analysis was performed for both states
and the spectroscopic parameters obtained agreed well with
experiment. They computed natural spin orbitals for these states
12

13
and analyzed the results in tenns of three zero order config
urations; Li(2s)H(2s), Li(2P)H(2S) and Li+(ls)H(ls). Their
results indicated that the x1 Z:. + state has predominantly Li +ucharacter but the H- ion is strongly polarized. They found th.at
a large unbalance of charge at small intermclear distances, R,
was responsible for the large equlibriwn internuclear separation
for the AlL+ state. At R less than 5.5 a.u. the covalent
Li(2s)H(2s) and Li(2P)H( 2s) are predominant. They also pre
sented results for the potenti. al curves of the second aDi third
excited 12, + states, but there was no Li Js or 3p character in
their basis set, so their results are only qualitative. The
second excited state has a metastable equilibrium at 3.70 a.u.
and another minimum around 10.0 a.u. which is due to the stabil-
1 ty of the Li +n- configurations. The third l r + excited state
has a minimum about 7 .50 a.u.
TaylorC5B) performed a calculation on the lowest 3 L + state
of LiH. He found this state to be repulsive in nature. Csizmadia,
et al.(59) using a group orbital methcxi carried out calculations
on the first three 1l+ states am. the first two 3.z:.+ states.
They did not fim any minimum in the third 1 L + and second 3 ~ +
states. However, they did find a hump in the lowest 3 L. + state
at 7 .o a. u.
Recently, Bender and Davidson( 60) obtained results on many
of. the low lying states of lithium hydride. They reported cal
culations on tre first six l~+ states, the first five JL+
states, the first three 37r and 1 TI states am the lowest 3 ~

and l ~ states. They used a basis of 23 STO I a and a SCF-CI
procedure to obtain the potential energy and dipole moment
curves between internuclear separations of 1.5 and 6.o a.u.
The energy of the x1~ + state was -8.0036 a.u. at 3.00 a.u.
They found that the third 1 ~ + state had a metastable equilib
rium around 4.0 a.u. while the fourth l~+ state was repulsive 1
in energy in this range. The fifth z. + state was bound at
4.0 a.u. The only bolll'ld 3~+ states were the third and fourth
states which had minimums between 4.0 and 4.5 a.u. Their cal
culations on the lowest 1 TT state, which was found to be bound
experimentally, ( 7) predicted it to be repulsive. The other
1 TT , 3n , 1,6 and 3 .D. states which they reported had minimums
around 4. 0 a. u. They also tabulated oscillator strengths
r or the various transitions. With an extended basis set of 52
ST01s and 939 configurations they used the SCF wavefunction in
a natural orbital analysis. An energy of -8.05998 a.u. and a
dipole moment of 5.B529D were obtained as compared to the exper
imental values of -8.0705 a.u. and 5.8JD.
Browne( 6l) obtained a binding energy of o.104!0.016ev for
LiH+ molecule ion. He used a mixed STO am elliptic orbital
basis and configuration interaction to calculate the potential
energy curve. The minimum was found at 4.25 a.u. and an energy
of -7.780848 a.u. was calculated with a 20 term wavefu.nction.
The ionization potential of LiH was estimated to be between
7.81 and 7.91ev.
The purpose of this paper iB the investigation of low

' ,
15
lying states of diatomic lithium hydride. In particular low
lying singlet an::l triplet sigma and pi states are considered.
These states' potential energy curves are accurately con:puted in
the range of internuclear separations between 1.0 and 10.0 a.u. 1 + l +
Except for the r 2,__ and A L states, this is the most accurate
calculation on these Lili states. Also, the molecular ioniza
tion limit, i.e. the lowest Lili+ 2 z_+ state, is studied in
order to determine Rydberg states<2) of LiH. Rydberg states of
Lili have not been studied previously. A Rydberg series of molec
ular states has electronic transitions analogous to those fer
atoms. These states have similar potential curves ani equi
librium internuclear separations.
A valence bond or atomic orbital (AO) configuration inter
action procedure is used for the calculations. Matsen an:l
Browne<50) presented the advantages of the AO-CI procedure over
the self-consistent field molecular orbital method. For a given
basis set and a full CI, both the AO-CI and SCF-MJ-CI methods
give the same results. However, with tha large basis sets needed
for an accurate calculation, only a limited CI is possible.
Within the restriction of a limited CI the atomic orbital
approach has certain advantages. The initial self-consistent
step is not required, which leads to more versatility in choice
of configurations since the l-1) 1s obtained from this step may not
be optimum for a limited CI. The atomic orbital approach goes
smoothly into the proper united atom (R=-0) and separated atans
(R"' 00) atanic states with only a few configurations. This

16
makes the choices of optimum orbital exponents for the basis .
orbitals easier since the calculations can be carried in f'rom
these eDi points. Also, open shell configurations and states
which do not have closed shell singlet symmetry are less diffi
cult to treat by the atomic orbital approach.
The basis orbitals used are Slater type orbitals. This
basis set is obtained .from the separated atom states and united
atom states. The optimized STO I s f'rom these atomic states are
combined to give the molecular basis. Slater type orbitals are
not as good as elliptic orbitals in representing bonding orbitals
in the molecule. However, this disadvantage is overcome with a
large con.figuration interaction treatment. Also, STO's provide
a better description of atomic-like orbitals and therefore give
better results at large internuclear separations. For the pro
cedure used here, they only need to be optimized for the limit
mg atomic states rather than at a number of internuclear
separations for each molecular state which would be desirable
for elliptic orbitals.
These calculations were carried out on an IBM 360/.50
computer using a diatomic molecule program of H.H. Michels.
This program was developed by Michels and F. Harris and mod
ified for use on the IBM 360/.50 conputer by the author.

CHAPTER II
METIDD OF CALCULATIDN
The methods used in this calculation have been previoua
ly reported by Harris, Taylor, Michels, etc.< 62-66) In
this procedure, the Born-Oppenheimer approxirnation( 67, 68)
is used to separate the electronic am nuclear motion. The
Schrodinger _equation for the electrcnic motion is
L-.C['f+ vcR,,~jfc~v,)~[cib<fcf.,SJ (2.1) . }
Both the potential energy, E(~), and the wavefunction,
Cf(rj,s), depend on the nuclear separations, Ri. The po
tential, V(i\_,i\), also includes the nuclear repulsion,
~ ZaZbe2/ .... . a<o Rab• The energy, E(R.), is used as the potential l.
energy f'uncticn for the nuclear motion. The wavefuncticn,
<f(rj,sj), is antisymmetric with respect to the exchange of
the coordinates of any two electrons. The j th electrcn I s
space and spin coordinates are given by rj .and sj respective
ly.
In the configuration interaction method, the energy is
optimized with respect to a wavefunction which consists of a
linear combination of configuration functions. This wavefunc-
17

tion is of the form
CtJ ~x lu T(~s) =: L ck T k (.f, s).
~.::.)
(2.2)
The wave.function, lrk(r,s), is the kth configuration am. the
cics fom the set of expansion coefficients. The k sum is
over all the configurations to be included. The coefficients
listed in the various tables are the "normalized" expansion
coefficients, ( 66) 8k, which are defined by ck~ where Sick
is the normalization integral S ~k~ ~ J t . In tems of
the 8i(, the wavefunction, lf (r,s), is given by an expansion
over nomalized con!'igurations, i.e.
'fc~, s') = L ck ~kc~, s) ~ \ (2.J)
= 2. ak [ 'f~ c~, s)/s:k ]~ k:
For an expansion using a finite nwnber of configuration
tenns this approximate wavefunction is not an exact solution
to Schrodinger1 s equation, H't' =E 'f. For a diatomic molecule,
the H!uniltonian H, is given by tJ "1
H = f. .h, + l21 ½, t + ZA ½ (2,4)
where N is the number of electrons in the system, R is the
internuclear separation and
18

19
where A and B refer to the two nuclei. In order to obtain the
best wavefunction, tbs coefficients, <;c, are detennined by
applying the variation principle to the expectation value of
the Hamiltonian,
S4'.y-(~\s) Hlfc;,s)J.~~s =Stpc'f,s)EPn\s)J~J~
=-Lc~tck H.,e~-= 2..cf ck E 511e ~k ,Q,k
(2.6)
where
S LP/en~ c-r )Af and
(2.7)
This procedure leads to the following set 0£ secular equations
(2 •. 8)
Non-trival solutions 0£ this system of equations exist only
when their determinant van~hes
det / H 9-k - E S ~~ I ::: 0. (2.9)
The roots, Ei' obtained are upper bounds ( 69) to the true energies
of the i states considered. The lowest root is more accurate
than the other roots.
In the valence bond or atomic orbital approach, each
configuration function,~k(r,s), is given by an antisynmie
trized product of space and spin orbitals( 65, 7o, 7l)

(2.10)
where * is the antisymmetrizer and c}k(r) is a spatial
f'imction coITesponding to the kth configuration. The spin
f'imction, ®m( S), is the mth spin eigenfunction of s2 and
Sz• The spatial function, Pie(~), is a product of om electron
functions N
P~ c~J == 7f J,.w. ct; J ;.:1 (2.11)
where N is the number of electrons of the system.
The antisymmetrizer 54- is a sum of the permutations over
the N electrons
J :=. {N n-Y"-L c-1)'f' P. p (2.12)
Using this in equation (2.10) results in the following
expansion for o/k(r,s)
<+>ie(f $) = ( N ~)¼-L C-l)f p C pf) P @'m (5) ) p ~
-l/4 J. = (N n 21- 2. u~W-(P)p~C?~) ej. (SJ.
}-l p (2.13)
Ujm(P) is the spin representation matrix:( 7l) of the per
mutation P defined by( 7l) ,l
p @'Mcs')=J U~'MCP') ®·(SJ ~~ 1 J- a- '
20

where the j sum is over the set of spin eigenfunctions ot
s2 and Sz•
In order to obtain the secular determinant the matrix
elements or an operator, fl, are needed. The matrix elements
are given by( 62 )
½.i = S <f/ (f,S) e-i Ci\ S) Jf ,H
=cN1r12, L u . cv1)u (R) .,..,~ P,R "tr..1- } 'Y\
X s~rcs)0~(s)c\s SP,JPi)&pp(R~)~~-~ ~ ,. (2.15)
'l'he sum over all the permutations, P, is a Hennitian operator
and since the operators of interest are totally symmetric
with respect to the electrons, one has P et"" t}'p. The spin
f'unctions, ®1 , a..-c com~truct-ed to be orthogonal. Using these
tacts and the following property of the unitary matrices,
Uici(P), t1mzi(PR)a t' Um1(P) U1n(R), equation (2.15) reduces toC 62)
Lu%~ CQ)~co) Q .
(2.16)
where
Q==P1
R a~ &~(Q') =ST:C?)&p(qZ)jt_ For the spatial integrals of the overlap operators,
the following resultsC 62 ) are obtained
S<o ') == S cf*c~> cp cot)~~ N :::: JI Ci- I j,.;) c2.11i
21

where qi is the ind.ex of the electron which the permutation.,
Q., puts in the place of the 1th electron. The one electron
overlap integral is defined by
(2.18)
For the Hamiltonian operator H., the following equationC62)
is obtained
where
and
(,;_ti 1/" l k ~) = S ,¢/ c iD 1ztHil ~;,_ ~ c fi) ~c-f,. J~ :f; ~ f "-. · (2.21)
Defining ( ~ I ~ ) t ')' - (; f ;h_ l 1-') / ( t I 1-> and ( A } / Y{' / ~ ~ / :_ ( A ~ l K) ~ J J /{;. / k) ( j_ } J} equation (2.19) becomes< 62 )
N N
H(QJ=S(Q'>F~ -1-Lc;.1_i__11_.J'+L <•~l¼--/i; is]. 4;;1 i.;,i-21 (2.22)
The basic integrals (i/j)., (i/h/j)., and (ij/ ~) can be
evaluated( 62 , 71) with the existing integral programs.
22

The bases used for the one electron spatial .f'tmction,
~(:j), are Slater type orbitals (ST0 1a)
rl ( t) ::: p (-N <'~ . ,()'\ v' Y\~ --1 0 l'M.~ I( I'"\ ) y.;.,. 1- ex \A.;.,}+-" ?Yl." r~ J, i- 1,9_. cos tt~
} A (2.23)
in spherical coordinates, or
¢/ f 1-l= ex p(-S, ~i-). '1/-' m, ~) r,-i ~~"'}~os t1i! (2.24)
with mixed spherical and elliptical coordinates< 65) where
r j Di -1 P11 lmi. I ( cos 8 j) can be expressed in terms of ~ j and 'lj• The orbital exponent b1=! )i,.,O(iR/2, where R is the
interzmclear separation, and the llj_, 11 and !llj_ refer to the
appropriate quantum numbers.
The important spin representation matrices, Ujm(P), for
N electrons can be constructed from those for N-1 electrons(7o, 7i)
or from the coefficients< 62 ) of the terms containing the electron
spins, CX and f->, when the spin basis .functions are constructed
using the genealogical. method.(70,7l) This procedure automat
ically gives orthogonal linearlJ" independent spin basis functions.
For a three electron doublet there are two linearly independent
The four electron singlet state also has two basis functions
which are
23

The three spin bases for the four electron triplet are
-1~
8>1 Cs" s .:i, s 3 ,S '+ > = ( 2) ::i. l d (J ex. ex. - f cl ol.o() , \¼ ~ ~ Cs 1, s~,s,,s~') -==- ( b ) ~c r:;J..t.2.rx._r;;J._ ta r:J....DZcJ..- :2d--ctf,. ol )
1- r c2.21) -V.:2...
and @1[sl,s.,,i,1s'tJ := ( 12) ( ~f ex.ct tfcx..,_,1.cx -;2_ e>Zcx.ot-.r5 ).
24

CHAPTER III
OOMPUTATIONAL PROCEDURES
3.1. Basis Orbitals
Diatanic molecules are considered to have tvo limiting
points: the separated ato~ with infinite internuclear separa
tion, i.e. R=tDO, and the united atom with zero separation,
i.e. RaO. Each molecular state therefore, in the context of
these limiting separations, connects to particu1ar atomic states.
In the case of lithium hydride the united atom is beryllium
am the separated atoms are lithium and ~rogen. As an
example, the ground x1 ~ + state of LiH necessarily has an
united atom limit of the lowest Be2S(ls22s2) state and a
separated atoms limit of the lowest Li2S(l.s22s) state and the
lowest H2s(ls) state.
IdeallJr, the parameters of the basis orbitals for the
t"l+ state of LiH would vary with internuclear separation, R,
from those appropriate tor Be2s(1s22s2) at R-0 and those ror
Li2S(ls22s) and II2s(la) at R11 00. Computati<nally, it would be
too expensive to optimize these parameters at each internuclear
separation; then-efore, it would be convenient to use a fixed
basis set. For the purposes of . this calculat.ion the basis set
used consists of Slater type orbitals (ST0 1s) optimized for each
of the limiting atomic states and combined to give a basis set
25

26
for the Lill molecular state. These STO's are optimized in the
sense that the energy is minimized with respect to variation of
their orbital exponents, 5 . In the range from R=l.O a.u. to 10.0 a.u. the LiH SID basis
set would consist of the corresponding Li an:l H STO's plus the
outer shell orbitals for the correct Be states. The inner core
Be orbitals would not be needed, since within this range the
internuclear separation is large enough so that the molecular
inner core orbitals are mainly Li in character. This is equiv
alent to assuming that the lithium-hydrogen separation is mt so
close that the inner core electrons on Li are strongly perturbed
by the hydrogen nucleus.
In this study, the above basis consists of the following
Slater type orbitals obtained from the appropriate atomic states.
The inner core orbitals are the ls, ls', 2p0 , 2p+ and 2p_ STO's
obtained from the appropriate lithium separated atom state.
The Li ls' STO is used to accrunt for radial correlation in the
inner core and the Li 2p STO' s for angular correlation. The outer
shell orbitals consist of a 2s and/or 2p outer orbital, etc.,
from the Li atanic state and 2s, ls and 2p outer orbitals from
the Be atomic state. The orbitals on hydrogen are a ls and a
2p. This H 2p STO is used to account for polarization of the hy
drogen atom.
In the above notation the bar over an orbital indicates
that the orbital is used in a different m~mer than suggested
by its orbital designation. For example, in a simple orbital

description of lithium, i.e. Li2S(ls22s) or Li2P(ls22p), the
ls orbital is used for the two inner coro electrons while the
2s or 2p orbital is used for the outer electron. In other
words, its quantum munber n indicates that the orbital is used to
describe the nth shell. In the standard notation an orbital
is designated by ns, np, etc. The bar over an orbital indicates
that this orbital is not used in the nth electron shell. The
2p STO's are used to obtain angular correlation in the inner
core of the appropriate atom. This is indicated by their rel
atively large orbital exponents. The beryllium ls sro has an
orbital exponent which is appropriate for an outer shell orbital.
In the beryllium atondc state calculation it is used to give a
node to the 2s STO and therefore has the same orbital exponent.
27
The hydrogen 2p orbital can be considered to form a limar
combination with the hydrogen ls orbital. The resulting hybrid
ized orbital is used to represent the polarizability of the hydrogen
electron. The orbital exponent of this 2p orbital is set equal to
that of the ls orbital.
The procedures for obtaining the appropriate sro bases
for the beryllium and 11 thium atontlc states are listed and
discussed in detail by the author in reference (72). The atomic
energies obtai..~ed were 99.6% of the experimental value or better
with the configuration interaction wave.functions presented.
3.2. Configurations
For the purposes of constructing the configurations for
the c. I. wave.function, the following grouping of orbitals was

28
used. The molecular inner core orbitals grouped togethEr were
- - -the lithium atomic orbitals ls, ls', 2p0 , 2p+' and 2p_ am the
· beryllium orbital. ls. The Be ls orbi ta1 was arbitrarily includ~d
in this group. The outer shell orbitals used to represent lith
ium in Li.H consisted ot the lithium outer shell atondc orbitals,
2s, 2p, etc., and the beryllium orbitals 2s an1. 2p. The last
groq> consisted of the hydrogen ls, 2p0 , 2p +, and 2p_ STO' s.
In describing the configurations, the inner core orbitals
used to represent the two inne:nnost electrons on lithium in LiH
will be denoted by i, 11 , etc., the outer orbitals on Li by o, o',
etc., and the hydrogen orbitals by h, h', etc. The primary covalent
type configuration derived from physical intuition wruld coo.sist of
two electrons in the Li inner core, one in a Li outer orbital and
one in a hydrogen orbital, i.e. ii' oh. Similarily, primary
+ -Li H type configurations will be represented by i 11 h h' and
Li-H+ type by i i I o o 1 • These starting configurations can be
seen from physical ir,tuition to represent the grCWld state. The
trutt gi•otuld state can be thought of as a mixture of three ideal-
+n- • i- + ized descriptions, i.e • . covalent LiH, ionic Lin ani ionic L H.
The state will be represented as a superposition of these classes
of configurations. From each of these initial configurations
three other types can be formed by substitution. One type will be
formed by substituting an Li imler core orbital for an outer
shell o orbital. The second will have one of the i inner core orbit
als in a configuration replaced by an o outer shell orbital in order
to represent a single excitation. The third will have both i inner

core orbitals replaced by an o outer shell orbital to give a
dou.ble excitation.
29
The computer program has a limit of 125 configurations. Due
to the large basis sets used in the calculations, many more than
125 configurations are possible. Therefore, some must be elimi
nated to give a limited CI wavefunction. Considering that from its
properties lithium hydride is somewhere between being covalent LiH
az¥i. ionic Li+H-, the important initial configurations should be of
the LiH aJXl Li+Ir primary types. From physical intuition the most
important covalent LiH ccnfiguration would have two electrons in
lithium la-type orbitals for the inner core, and one electron in
a lithium 2s~type orbital, and one in a hydrogen ls-type orbital
for the outer shell. The most important ionic Li +H- configurations
would have a similar inner care, but would have two electrons in
~drogen ls-type orbitals for the outer shell.
With these configurations as a starting point, a limited
search through the configuration list was performed in order to
obtain the best 125 configurations. In general grrups of config
urations, for example, thcs e of the (LilaBe2s )be type where b runs
over the other outer shell orbital.a and cover the hydrogen basis
orbitals, were added to the starting configurations and tmir
effect on the energy observed. Unless trey lowered the energy
those configurations with the smallest coefficients in the wave
function were eliminated. About 20 to 30 configurati ans were
eliminated and other different groups of configurations tried.
This procedure was repeated until a comprehensive sampling

30
of configurations was covered. A set of configurations was chosen
from those tried in the described procedure. This set was varied
slightly among these chosen configurations. The final set used
was the one which gave the best energy. This procedure was used
at an internuclear separation of either 2.0 a.u. or 4.0 a.u.
since with the basis orbitals used the wavefunction is less
accurate at small internuclear separations. The final set of
configurations obtained was then used for the energy calculations
at the other internuclear separations.
It must be noted that this procedure does not guarantee
the best 125 configurations. The contribution of any config
uration to the energy depends on the other configurations in the
wave.tunction. Therefore, to conduct a conplete search would be
prohibitively time consuming. The magnitude of a configuration's
coefficient in the wavefunction does not directly give the
configuration's contribution to the energy. Two approximately
linearly dependent configurations would have nearly equal and per
haps large coefficients but together would not contribute more to
the lowering of the energy than one of them alone. For the case
of non-orthogonal basis orbitals, approximate linear dependencies
are always present to some degree and must be checked.
Partial energies analogous to those for an orthogonal
basis were tried but were not fom1d to be useful since they
varied too much with different configurational wavetunctions.
This expression was given by 1 _
"'~ . r<..-.x
E,;. == L C~C;: Hk;/sk .-f Ciecp_Hie.Q/s 0 k=t >- k,.t::1 /. k;,.,.

Jl
where the i..T1dex k runs over a truncated part of the CI expan
sion of about 40 configurations.
Although the procedure described for choosing configurations
has many inadequacies in obtaining the best possible 125 configur
ations, it does lead to a qualitative measure of the importance of
various types of groups of configurations without very great expense.
The maximum energy difference between the worst and best set of
configurations used during the search was about 0.006 a.u.
3. J. Spin Functions
The spin states considered in this calculation are:
the three electron doublet, the four electron singlet and the
four electron triplet. The three electron doublet and four
electron singlet have two linearly independent spin functions.
The four electron triplet has three linearly independent spin
functions. In the computer program only the G\ spin function
is used. For example, in the case of four electrc:1 singlet the
LilsLils'Li2sHls configuration with the spin function would
have singlet coupling; while the LilsLi2sLils 1llls configuration
with the @1 spin function would have triplet coupling. This
would be equivalent to the½( B 1-J.f@2) spin function. The
functions E)i, and ½( G>1 -./J G2) are linearly independent and
therefore can be used as a basis for this spin state. The results
for the three electron doublet and the four electron triplet are
similar.< 72 ) The spin representation matrices are constructed
from the ®1 spin functions.

CHAPTER IV
LOWEST STATES OF J 2. +, 3]1 and 1 Jr SD1METRY
4.1. The Lowest Lithium Hydride 3 E+ State
The lowest LiH 3~+ state connects with the lithium
2s(ls22s) plus eydrogen 2s(ls) atomic states in the separated
atom limit and the beryllium .3p(ls22s2p) atcmic state in the
united atom limit. Therefore, the basis orbitals used to des
cribe the lithi1.m1 atom in LiH 3~ + consist of those far tm
Li2S(ls22s) ~tate plus the 2s, ls and 2p ST0 1 s for the BeJP
(ls22s2p) state. These basis orbitals used for the LiH Ji+
state are listed in Table 2. Their parameters are given at an
internuclear separation of 2.0 a. u. At this internuclear separa
tion the basis STO I s for LiH have the same scaling as for the
atomic states.
This state was found to be repulsive in energy. The
energies calculated and the various intemuclear separations
used are given in Table J. The configurations and their coef
ficients in the wavefunction are arbitrarily listed at an inter
nuclear separation of 4.0 a.u. in Table 22 of .Appendb: I. The
majority of low lying states of LiH have minimwnB at"ound 4.0 a.u.
The energy at R= 00 is obtained from a calculation of the
Li2S(ls22s) state using the basis STO' s in Table 2 excluding those
for hydrogen.
.32
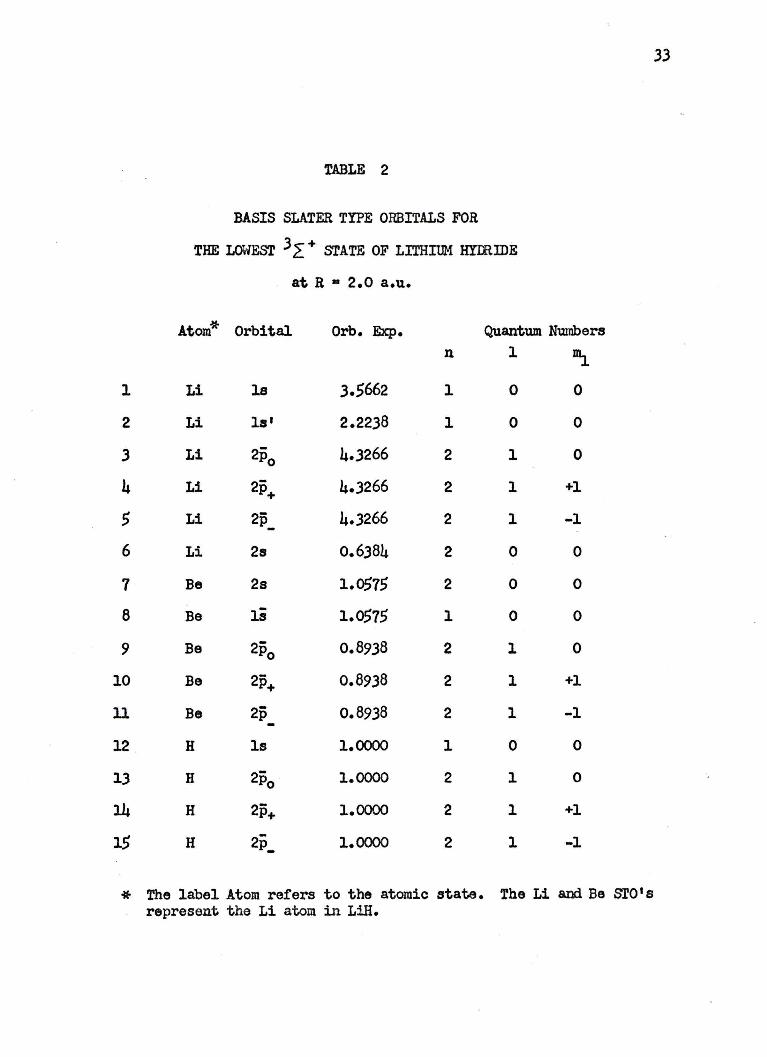
33
TABLE 2
BASIS SLATER TYPE ORBITALS FOR
THE LOWEST 3 L + srATE OF LITHIUM HYIRIDE
at R • 2.0 a.u.
Atom* Orbital Orb. Exp. Quantum Numbers n 1 ~
1 Li ls 3.5662 1 0 0
2 Li ls' 2.2238 1 0 0
3 Li 2po 4.3266 2 1 0
4 Li 2p+ 4.3266 2 1 +l
5 Li 2p_ 4.3266 2 1 -1
6 Li 2s o.6J84 2 0 0
7 Be 2s 1.0575 2 0 0
8 Be ls 1.0575 1 0 0
9 Be 2po 0.8938 2 l 0
10 Be 2p+ 0.8938 2 l +l
11 Be 2p 0.8938 2 l -1
12 H ls 1.0000 1 0 0
13 H 2po 1.0000 2 l 0
14 H 2i5+ 1.0000 2 1 +l
15 H 2p_ 1.0000 2 l -1
* The label Atom refers to the atomic state. The Li and Be ST0 I s represent the Li atom in LiH.

R(a.u.)
1.0
2.0
3.0
4.0
5.0
6.o
7.0
a.o 00
TABLE .3
ENERGY FOR THE LOWEST 3E° STATE
OF LrI'HIUM HYDRIDE
Electronic Energy (a.u.)
-10.289798
-9.346228
--8.931927
-8.699008
-8.557275
-8.462710
-8.394165
-8.)41880
-1.967903
.34
Potential Energy (a.u.)
-1.2897979
-7.8462279
-7.9319268
-7.9490082
-7.9572745
-7.9627102
-1.9655932
-7.9668797
-1.9679025

From the magnitude of the coefficients of the configur
ations in Table 22, one finds that those with the largest values,
i.e. greater than 0,10, are all covalent type configurations.
These important configurations suggest that the inner core
electrons are represented by the linear combination c111 ls+
~Li la'+ c3Be ls. The outer core lithium electrons are
described by the following orbitals: Li 2s (which is most
important), Be 2s and Be 2p0 • over the range of internuclear
separations considered the order of importance of inner core
combinations is 1 2, 2 8, 2 2, l 8, l 1, 8 8, 4 5, and 3 3.
These numbers designate orbitals in Table 2. At large inter
nu.clear separations covalent configurations involving the Li 2s
orbital. are the most important in 8JJY' group. For example, the
coefficient of the l 2 6 12 configuration has a magnitude of 1.3
from R .. 2.0 to 8.0 a.u. Configurations involving the Be 2s
orbital also have large magnitudes. Configurations involving
the Ee 2p increased in importance with decreasing internuclear
separation. The coefficient of the 12912 configuration varies
from 0.008 at 8.0 a.u. to 0.44 at 2.0 a.u. Of course the hydrogen
H ls orbital is the most important. The H 2p orbitals are less
important but allow for polarization of b;ydrogen. The magnitude
of the coefficient of the configurations involving H 2p generally
increases with decreasing R until about 2.0 a.u. At small inter
nuclear separation, i.e. at 2.0 and 1.0 a.u.,the ionic type con
figurations also have large magnitudes.
A second calculation was perfomed by adding to the basis
Li 2p0 , Li 2p+, and Li 2p_ sro•s with orbital exponents of
35

0.5237 at 2.0 a.u. The wavefunctian consisted of 125 configur
ations and a limited search through configurations was performed.
The: energy or the lowest root at an internuclear separation ot
4.0, 6.o and 8.o a.u. is -7.9528961, -7.9633712, and -7.8244385
a.u. respectively. The energy of the next root is -7.8244385,
-7.8795953, and -7.8946733 a.u. respectively.
4.2. The Lowest Lithium Hydride 3rr State
The lowest 3 TT state of lithium hydride cormects to the
lowest Li2P(ls22p) state plus H2S(ls) state in the separated
atom limit and the Ba3P(ls22s2p) state in the united ~tan limit.
Its ST0 basis therefore is corrposed of those functions obtained
from a calculation of the Li2P(ls22p) state, the 2s, ls and 2p
ST0's fran the Be3P(ls22s2p) state and ls plus 2p functions for
hydrogen. The ST0 basis is given in Table 4 tor an internuclear
separation of 2.0 a.u.
This 3 lf state is bound with its minimum lying around
4.0 a.u. The potential energy calculated. at the various
intermiclear separations is given in Table 5. For Ra4.0 a.u.,
E(OO)-E(R) equals 0.0064044 a.u. or 0.1743 e.v. The energy at
R• 00 is calculated using the basis of Table 4 minus the hydrogen
sro•s. The configurations and their coefficients for this wave
function at the minimum of R•4.0 a.u. are given in Table 23 of
Appendix I.
As seen f'rom the most important configurations in the
wavefunction of Tabla 23, i.e. those with coefficients greater
than 0.10, the most significant inner core canbinations are
36
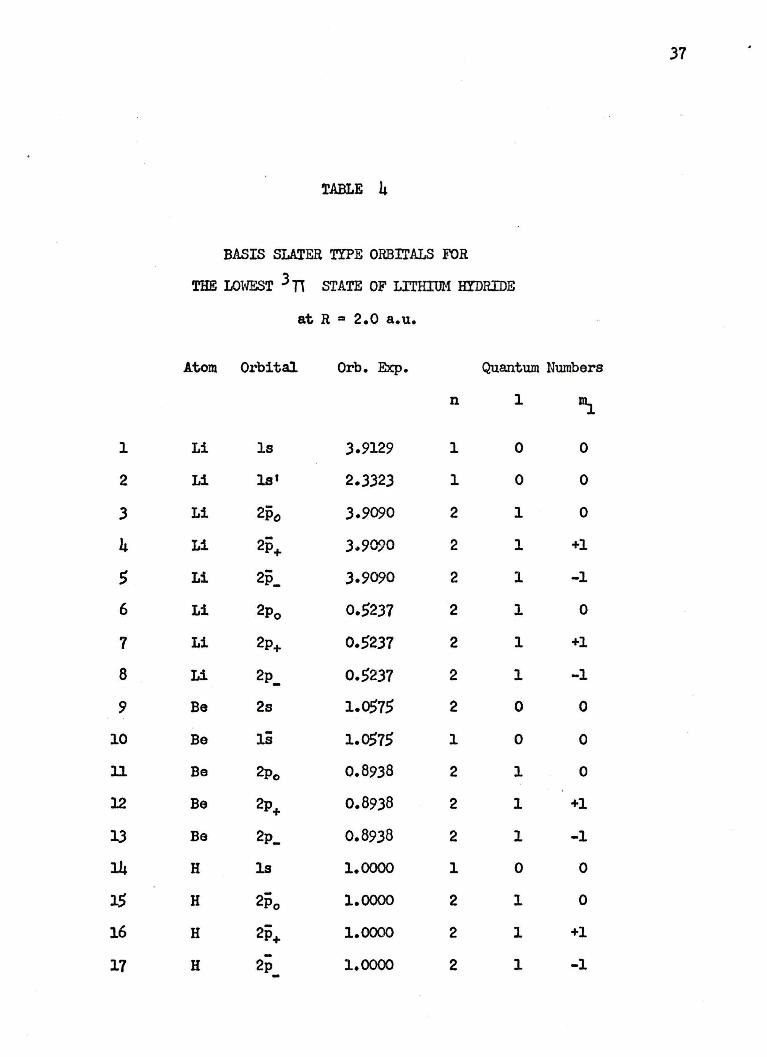
37
TABLE 4
BASIS SLATER TYPE ORBrrALS FOR
THE LOWli'~T J TI STATE OF LITHIUM HYDRIDE
at R .. 2.0 a.u.
Atom Orbital Orb. Eicp. Quantum Numbers
n 1 ~
1 Li ls J.9129 1 0 0
2 Li ls' 2.3323 1 0 0
3 Li 2pc 3.9090 2 1 0
4 Li 2p+ 3.9090 2 l +l
s Li 2-p_ 3.9090 2 1 -1
6 Li 2po 0.5237 2 l 0
7 Li 2p+ 0.,237 2 1 +l
8 Li 2p_ 0.,237 2 1 -1
9 Be 2s 1.057, 2 0 0
10 Be ls 1.057, l 0 0
11 Be 2po 0.8938 2 l 0
12 Be 2p+ 0.8938 2 1 +l
lJ Be 2p_ 0.8938 2 1 -1
14 H ls 1.0000 1 0 0
l5 H 2po 1.0000 2 l 0
16 H 2p+ 1.0000 2 1 +l
-17 H 2p 1.0000 2 1 -1 -

R(a.u.)
1.0
2.0
2.~
3.0
3.5
4.0
4.S
,.o
5.5
6.o 7.0
a.o 00
TABLE 5
ENERGIES FOR THE LOWEST 3 lT STATE
OF LITHIUM HYDRIDE
Electronic Potential
-10.254330 -7.2.543299
-9.)18081 -1.8180807
-9.077003 -7.8779934
-8.900073 -7.9000729
-8.763800 -1.9066515
-8.657260 -7.9072597
-8.572645 -7.9059785
-8.504451 -7.9044510
-8.448664 -7.9032091
-8.402311 -7.9023110
-8.329924 -7.9013525
-8.275974 -7.9009743
-1.900855 -1.9008553
38
Eoo - Ea
-o.64652.54
-0.0827746
-0.0228619
-0.0007824
+0.0058022
+0.0064044
+0.0051232
+0.0035957
+0.0023538
+0.0014557 . +0.0004972
+0.0001190

39
11., 12.,110., 22,210, and 10 10. The numbers designate the
orbitals in Tabla 4. Orbital 7, the Li 2p+ STO, ccmbined with
the orbital 14, the H ls STO, is the daninant outer smll com
binaticn throughout most of the range of R considered especial.ly
at large R. As the internuclear separation decreases, orbital
12, the Be 2p+ STO, also becomes significant. This also occurs
for a lesser extent for covalent configurations containing the
-H 2p ST0 1s orbitals 1.5, 16, and 17. At very small R, i.e.
1.0 or 2.0 a.u., ionic type ccnfigurations become significant as
judged from the magnitude of their coefficients, especially those
with an outer shell of the type 9 12 or 10 12, which correspond
to the mai.Ii configurations for the Be3P(ls22s2p) state.
4· • .3. The Lowest LithiUlll Hydride 1 TT State
The lowest 1 TI state of lithium hydride connects with the
united atom Be1P(ls22s2p) state and the separated atom Li2P
(1s22p) plus n2s(ls) states which are singlet coupled. Therefore
the basis ST0 1s for this Lili 1TI state consist of the basis
orbitals used for the Li2P(ls22p) state, the outer shell 2s,
ls and 2p orbitals for the Be1P(1s22s2p) state plus ls and
2p ST0 1s for n2s(ls). The basis set is given in Table 6.
As seen by the energies in Table 7, the LiH 1 71 sta"te
was calculated repulsive in energy although vecy flat at inter
nuclear separations greater than 4. 0 a. u. The em rgy for R=OO is
obtainad from the calculated energy of the Li2P(ls22p) state using
the basis STO's in Table 8, omitting the H orbitals, plus 0.50 a.u.
which is tm energy of the H2S(ls) state. The configurations
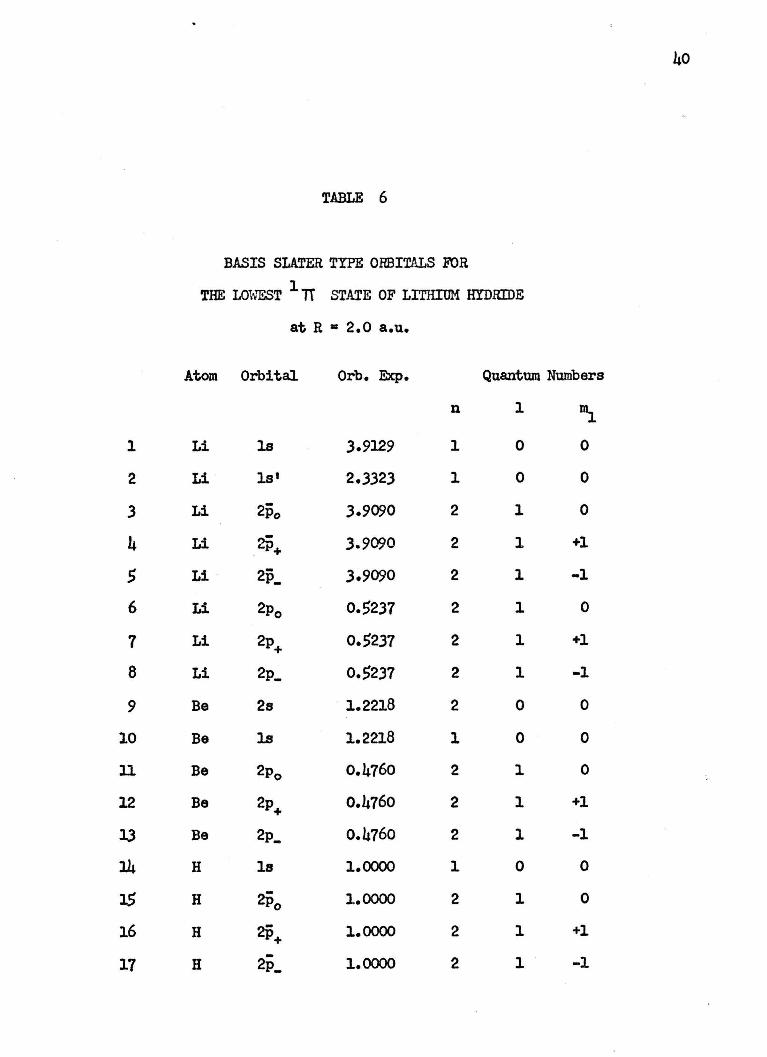
40
TABLE 6
BASIS SLATER TYPE ORBITALS FOR
THE LOWEST 1 1T STATE OF LITHIUM HYDRIDE
at R • 2.0 a.u.
Atom Orbital Orb. Exp. Quantum Numbers
n 1 1\ l Li ls 3.9129 1 0 0
2 Li ls' 2.3323 1 0 0
3 Li 2i5o 3.9090 2 l 0
4 Li 2p+ 3.9oc;o 2 1 +l
5 Li · 2-p_ 3.9090 2 1 -1
6 Li 2po 0.5237 2 l 0
7 Li 2p+ 0.5237 2 1 +l
8 Li 2p_ 0.5237 2 1 -1
9 Be 2s · 1.2218 2 0 0
10 Be ls 1.2218 1 0 0
11 Be 2po 0.4760 2 l 0
12 Be 2p+ 0.4760 2 1 +l
lJ Be 2p_ 0.4760 2 1 -1
14 H ls 1.0000 1 0 0
15 H 2po 1.0000 2 1 0
16 H 2p 1.0000 2 l +l +
17 H 2p_ 1.0000 2 1 -1

R(a.u.)
1.0
2.0
3.0
4.0
s.o 6.o 7.0
a.o 00
TABLE 7
ENERGY FOR THE LOWEST 1 TT STATE
OF LITHIUM HYDRIDE
Electronic Energy (a.u.)
-10.212022
-9.296431
-8.884393
-8.648280
-8.S00286
-8.400598
-8.329226
-8.275685
-7.900779
Potential Energy (a.u.)
-1.2120221
-7.7964313
-7.8843933
-7.8982803
-1.9002859
-7.9005977
-7.9006549
-7-9006845
-7.9007787

composing the wavefunction for the 1 TT state and their coef
ficients are listed in Table 24 of Appendix I.
If one compares the orbital exponents of the Li 2p orbitals,
numbers 6, 7, and 8, with those of the Be 2p orbitals, numbers ll,
12, am 13, it will be noticed that they are close in magnitude.
A slight improvement would have been made in this calculation by
using orbital exponents for the Be 2p sro' s which differ more
. lTT from those for the Li 2p ST0 1s. A calculation for this LIB
state at R•4.o a.u. with the basis for the LIB 3TT state given
in Table 4 gave an energy :llllprovement of 0.0004412 a.u.
The important configurations of the wavefunction will be
considered to be those with coefficients greater than 0.10.
AB seen from Table 24, the main configurations are of the covalent
type with inner core ST0 1 s Li ls, Li ls', Be ls and outer shell
STO' s Li 2p+J Be 2p+ and H ls. Moat of these are of the primary
type with two electrons in izmer core orbitals. Also of signif
icance are the singly excited configurations where an inner core
orbital is replaced by a Be 2s sro. At large internuclear sep
aration only the configurations with the Li 2p+ ST0 have large coef
ficients. However, as R decreases, configurations with the Be 2P+
STO also become important. At internuclear separation less than
4.0 a.u. the coefficients of ionic configurations become large as
- - + well as some configurations with H 2p ST0 1s. At 1.0 a.u. the Li H
configurations with large coefficients are those which correspond
to the important configurations in the Be¾>(1s22s2p) atanic state
calculation.

CHAPTER V
POTENI'IAL CURVES FOR LOW LYING LITHIUM HYilUDE STATF.s
$.1. Basis Orbitals
For the purposes of econonzy- in calculating the integrals,
the same basis set was used in the calculations for all the
states considered in this chapter. The basis STO' s are listed
in Table 8 with parameters for an internuclear separation of
2.0 a.u. The inner core lithium orbitals, ls, ls', 2p0
, 2p+
and 2p_, were obtained from a calculation on the Li2S(ls22s) state
where an optimization of the orbital exponents was performed.
The Li 2s STO was also obtained from this calculation. The
ensrg;y of this calculation was -7.46b.517 a.u. The Li 3a STO
was obtained by optimizing the orbital exponents of the 6 pre
viously mentioned orbitals and the Ja sro in a calculation of
the Li2S(ls2Js) state. The energy of the Li2S(ls23s) state from
this calculation was -7.342461 a.u. and the energy of the
Li2S(ls22s) state was -7.4ob733 a.u. The Li 2p0 STO was obtained
by optimizing the orbital exponents of the basis set in a
calculation of the Li2P(ls22p) state. The energy obtained
for the Li2P(ls22p) state was -7.399894 a.u. The Li 3p0
STO's
orbital exponent was obtained in a calculation of the Li2P(ls23p)
43
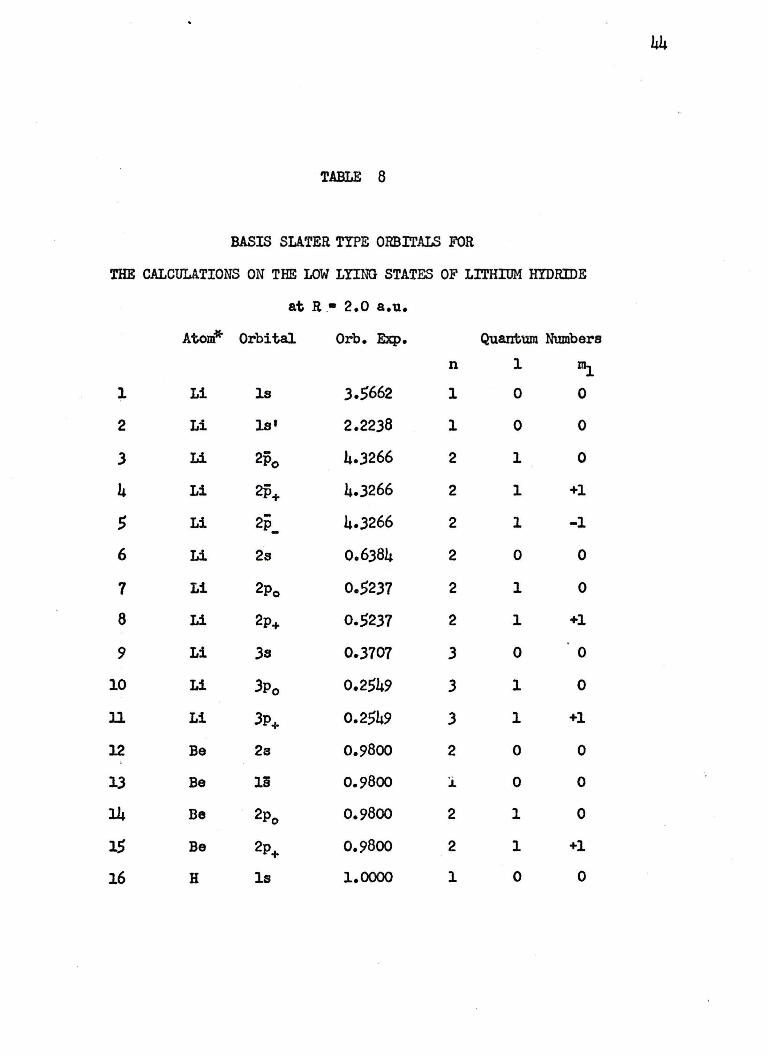
44
TABLE 8
BASIS SLATER TYPE ORBITALS FOR
THE CALCULATIONS ON THE LOW LYING STATES OF LITHIUM HYDRIDE
at R .• 2.0 a.u.
Atom* Orbital Orb. Exp. Quantum Numbers
n l Illi l Li ls 3.5662 l 0 0
2 Li ls' 2.2238 l 0 0
3 Li 2i5o 4.3266 2 l 0
4 Li 2i5+ 4.3266 2 l +l
5 Li 2p_ 4 • .3266 2 l -1
6 Li 2s 0.6384 2 0 0
7 Li 2Po 0.52.37 2 l 0
8 Li 2P+ 0.5237 2 l +l
9 Li Js 0.3707 3 0 0
10 Li 3p0 0.2549 3 l 0
ll Li .3P+ 0.2549 3 l +l
12 Be 2s 0.9800 2 0 0
13 Be ls 0.9800 :.i. 0 0
14 Be 2po 0.9800 2 l 0
15 Be 2p+ 0.9800 2 l +l
16 H ls 1.0000 l 0 0

TABLE 8 Continued
Atom* Orbital Orb. Exp. Quantum Numbers
n 1 ~
17 H 2po 1.0000 2 1 0
18 H 2-p+ 1.0000 2 1 +l
19 H 2p_ 1.0000 2 1 -1
* The label Atom refers to the atomic state. The Li am Be STO's are used to represent Li in Lill.

state. This calculation was similar to that for the Li2S(ls23s)
state. The energy calculated tor the Li2P(ls2Jp) state was
-1.328178 a.u.
The orbital exponents of the Be 2s and Be ls 5'T0 1s were
restricted to be the same since the ls SI'O was used to represent
a node for the 2s STO. The Be 2s, ls and 2p0
sro•s were obtained
from a ca1culation of the Be1S(ls22s2) state in which the orbital
exponents of these outer shell orbitals were optimized while hold
ing the parameters of the inner core oribtala fixed. These inner
core orbitals were similar to those used for the lithiwn calcula
tions and obtained by optimization in a calculation of the Be+2
1s(ls2) state.
46
The orbital exponents of the Li 2P+, Li 3P+ and Be 2p+ sro•s
are the same as those of the Li 2p0 , Li Jp0 , and Be 2p0
STO's. The
orbital exponent of the H ls, 2p0 , 2p and 2p STO' s were set + -
equal to 1.0000. Due to computer limitations it was not possible
to include the Li 2p , Li Jp and Be 2p STO's in the basis set. - - -Therefore covalent configurations with n coupled bonding, i.e.
(Lils)2Li2p+H2p_ and (Lils)2Li2p_H2P+,could not be included in the
t state calculations. It was tound that inclusion of this type
of configuration lowered the energy less than 0.0001 a.u. There
fore neg1ecting l'\Coupled configurations was not a poor approxima
tion.
5.2. The Lithiwn Hydride 1~ + states
The four lowest LiH 1L. + states were considered in this
calculation. In the separated atom limit these states connect
with the ti2s(ls22s), Li2P(ls22p), Li2s(ls2Js) and Li2P(ls2Jp)
states plus the H2s(ls) state. Experimentally, the separated

atom limits are -7.97865, -7.91074, -7.85469 and -7.83775 a.u.
respectively. These values are obtained from Moore's tables(3)
aid the results of Pekeris(4).
The potential energies obtained are listed 1n Table 9, and
the potential curves are presented in Figure 1. The energies at
R• 00 are obtained from a calculation of the Li atomic states
using the basis of Table 8 without the H sro• s. The H2S(ls)
47
energy ot 0.50 a.u. is added to the Li energies to obtain the values
tor R• 00. The binding energies of the Ill+ and the A1 2,+ states
are estimated to be 2.223.SeV and 0.9453eV. The farm of the
coulombic potential curve for Li +il- was obtained f'ran Mulliken~ 5 )
This curve is only qualitatively accurate and is included for
illustrative purposes. The equation is
The configuration list for these wavefunctions is given in
Table 10. The coefficients obtained for the various states
are arbitrarily given at 4.0 a.u. in Table 25 of Appendix II.
The wavefunctions used are complicated and hard to analyze
especially at small internuclear separations where a great many
configurations have large coefficients. In general, at large R
the most important conf'igurat ion in any group is the covalent
configuration w1 th the outer shell Li orbital. corresponding to the
molecular state's correct separated atom limit.
,As discussed by Mulliken(5) and seen in Figure 1, the shape
of the potential curve of the ground state of Lill resembles quite
strongly the Li+If" curve. Therefore, it is likely that the X1 .L +

R(a.u.)
1.0
2.0
3.0
4.0
5.0
6.o
7.0
a.o 9.0
10.0
00
TABLE 9
POTENTIAL ENERGIES FOR THE l t, + srATES
OF LITHIUM HYDRIDE
Potential Energy- (a.u.)
Roots
l 2 3
-1.325096 -7.232410 -7.087392
-7.973947 -1.829859 -7-729413
-8.049230 -7-920797 -7.834002
-8.033040 -7.934385 -7.846038
-8.006858 -7.934916 -7.846061
-7-985806 -7.93lll7 -7.846955
-7.974577 -7.923322 -7.849919
-7.970032 -7.914498 -1.853511
-7.968415 -1.907738 -7.855652
-7.967852 -1.903676 -1.855696
-7.967513 -1.900176 -7.845041
48
4
-1.063031
-7.709346
-7.815646
-1.830755
-7.828962
-7.827148
-7.828379
-7.830698
-7.832463
-1.833616
-7.826779

-1.70
-7.80
-. ::s . C'IS ........
~ -1.90 H Q)
~
-8.oo
-8.10 o.o 1.0 2.0
../
/
' .,.,/ , ____ .,
3.0 4.0
.,,,,,.
5.0
-__ __. . ..-- --
q
2. --- -- - -L._-;11-- - >,. "
,- 1 ,,,,,,., ,,.,---- ----....-----
6.0 7.0 B.o 9 •. 0 10.0 Internuclear Separation (a.u.)
FIGURE 1. POTENTIAL ENERGY CURVES FOR THE lL + STATES
OF LITHIUM HYDRIDE
00
No.
1
2
.3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
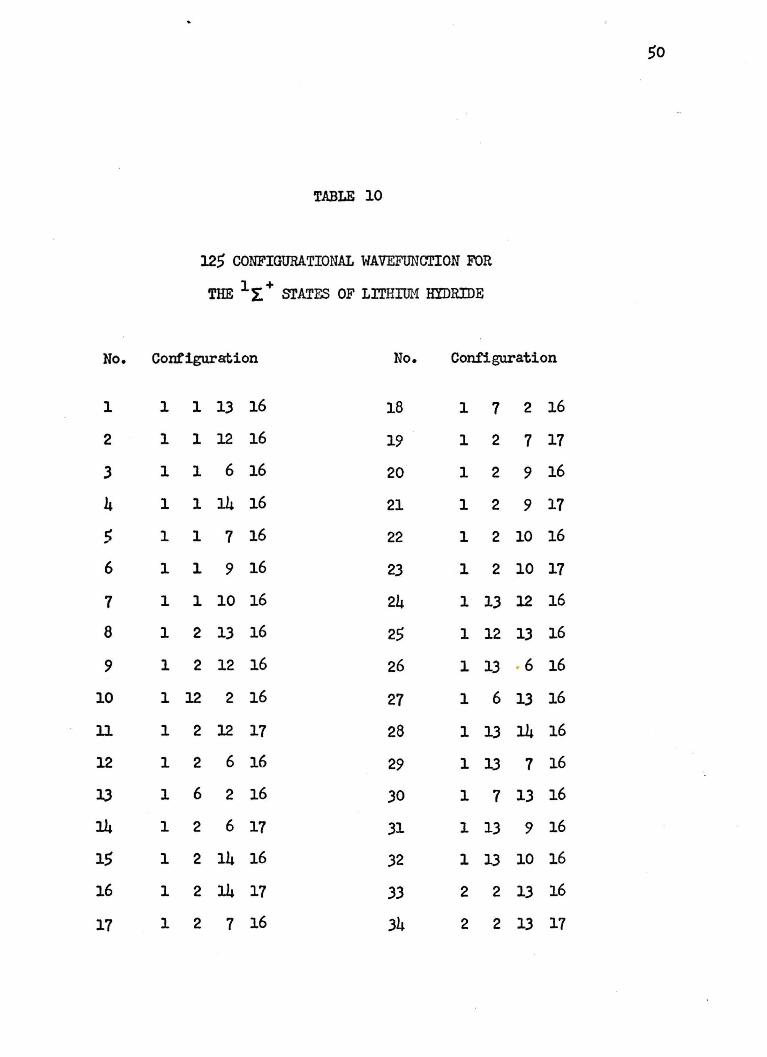
TABLE 10
125 CONFIGURATION.AL WAVEFUNCTION FOR
THE l~+ STATES OF LITHIUM HYDRIDE
Conf"igura:tion No. Configuration
1 l 13 16 18 l 7 2 16
1 l 12 16 19 l 2 7 17
l 1 6 16 20 1 2 9 16
l 1 14 16 21 l 2 9 17
1 1 7 16 22 l 2 10 16
1 l 9 16 23 1 2 10 17
1 1 10 16 24 1 13 12 16
1 2 13 16 25 1 12 13 16
1 2 12 16 26 1 13 • 6 16
l 12 2 16 27 1 6 1.3 16
1 2 12 17 28 1 13 14 16
1 2 6 16 29 1 13 7 16
1 6 2 16 30 1 7 13 16
1 2 6 17 31 1 13 9 16
1 2 14 16 32 l 13 10 16
l 2 14 17 33 2 2 13 16
1 2 7 16 34 2 2 13 17
50

51
TABLE 10 Continued
No. Configuration No. Configuration
35 2 2 12 16 56 13 13 14 16
36 2 2 12 17 59 13 13 7 16
37 2 2 6 16 60 13 13 9 16
38 2 2 6 17 61 13 13 10 16
39 2 2 14 16 62 3 3 12 16
40 2 2 7 16 63 3 3 6 16
1.il 2 2 7 17 64 3 3 7 16
42 2 2 9 16 65 3 3 9 16
43 2 2 10 16 66 3 3 10 16
44 2 13 12 16 67 4 5 13 16
45 2 12 13 16 68 4 5 12 16
46 2 13 6 16 69 4 5 6 16
47 2 6 13 16 70 4 5 7 16
48 2 13 14 16 71 4 5 9 16
49 2 14 13 16 72 4 5 10 16
50 2 13 7 16 73 12 12 1 16
51 2 7 13 16 74 12 12 2 16
52 2 13 9 16 75 12 12 13 16
53 2 13 10 16 76 l 12 6 16
54 13 13 1 16 77 1 6 12 16
55 13 13 2 16 78 l 12 14 16
56 13 13 12 16 79 1 12 7 16
57 13 13 6 16 80 2 12 6 16
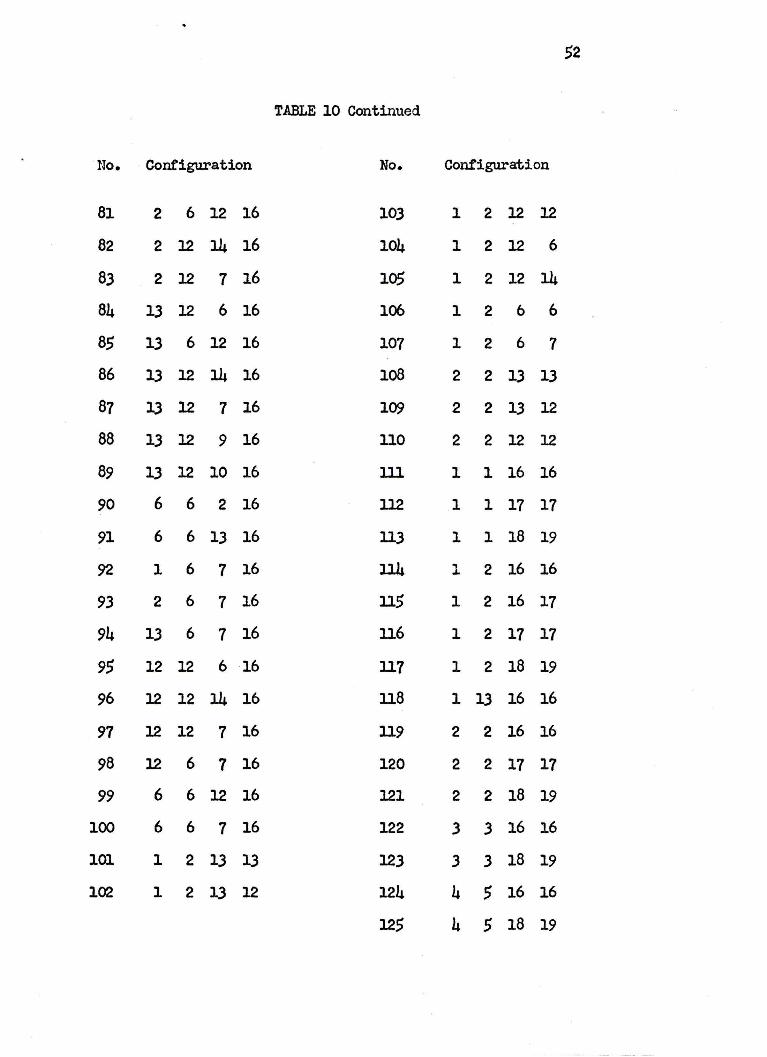
52
TABLE 10 Continued
No. Configuration No. Configuration
81 2 6 12 16 103 1 2 12 12
82 2 12 14 16 104 1 2 12 6
83 2 12 7 16 105 1 2 12 14
84 13 12 6 16 106 1 2 b 6
85 13 6 12 16 107 1 2 6 1
86 13 12 14 16 108 2 2 13 13
87 lJ 12 7 16 109 2 2 13 12
88 13 12 9 16 ll0 2 2 12 12
89 13 12 10 16 lll 1 1 16 16
90 6 6 2 16 112 .1 1 17 17
91 6 6 13 16 ll3 1 1 18 19
92 1 6 7 16 ll4 ., 2 16 16 .L
93 2 6 1 16 ll5 1 2 16 17
94 13 6 1 16 U6 1 2 17 17
95 12 12 6 16 ll7 l 2 18 19
96 12 12 14 16 ll8 1 lJ 16 16
97 12 12 7 16 ll9 2 2 16 16
98 12 6 7 16 120 2 2 17 17
99 6 6 12 16 121 2 2 18 19
100 6 6 7 16 122 3 3 16 16
101 1 2 lJ lJ 123 3 3 18 19
102 1 2 lJ 12 124 4 5 16 16
125 4 5 18 19

state has a large amount of Li+H- character in this region
where the two curves resemble each other so closezy-. This can
also be seen by looking at the ionic Li~- type configurations in
the wavefunction. For the x1 r+ state, they have a maximum
importance at 3.0 a.u. and then gradually trail off. For the
A1~ + state their importance gradually increases to a maximum
around 6. 0 a. u. For the next two 1 ~ + states tmse Li~- config
urati ans become important at large interzmclear separations.
The potential energies of Table 9 for the third 1 L + and
l~+ fourth £- states indicate that these states are bound at R•lO. O
a.u. since the energies at 10.0 a.u. lie below the calculated
separated atom limits. In fact, at 10.0 a.u. the third l~+
state lies below the experimental separated atom limit of -7.85469
a.u. A metastable equilibrium is also observed in the fourth lL+
state at 4.0 a.u. However, it is felt that this is due to inter
action between third and fourth wave.functions in this range of R
and iua;r not be the case pb;ysically. Brown and Shull(57) observed
a metastable equilibrium in the third 1 L + state at 3. 70 a.u. and
conjectured that there is another equilibrium around 10.0 a.u.
They- found a minimum in the fourth 1~+ state at 7.50 a.u.
However, their basis set did not include an:, lithium Js and 3p
character and therefore is only roughly qualitative. Bemer and
Davidson< 60) calculated the potential curves of these states
between 1.5 and 6.0 a.u. They found that the third lL.+ state
has a metastabi.e equilibrium at 4. 0 a. u. and that the fourth 1 L. +
state is repulsive in this range. They also fo,md that the
S3
fourth excited state, i.e. the 1 .Z:+ state, has a minimum around 4.0
a.u.

In order to study the third and fourth 1 r_ + states in
more detail at large R, a double precision integral program
was used. This program gives more accurate results at these
large interzmclear separations. Also, a Li 3<1<, and a Li 4s
sro were added to the basis set with orbital exponents 0.3333
and 0.2500 respectively which were detemined using Slater's
rules. Twenty configurations which seemed to have the smallest
effect on the energy were replaced by appropriate covalent con
figurations containing the 3do and 4s STO' s. Also, when Brown
and Shull 1 s potential curves for the first two l.L + states
were compared with the curves obtained from this calculation,
it was found that this calculation is much poorer than theirs
in two regions. For the xl~ + state this region was from 1.0
to 6.0 a.u. and for the A1 2_ + state from 5.0 to 9.0 a.u. These
are the regions in which the LiT character of these states has
its greatest importance. Therefore, the wavefunction used in
this calculation does not have enough Li +u- chara.:.;ter. In order
to test this conjecture a hydrogen ls' sro with orbital exponent
0.60oo was added to the basis set of Table 8 along with the Li
3do and 4s sro 1 s. Eight of the configurations involving 3d0
54
and 4s were replaced by ionic LiY type configurations involv
ing the H ls I sro. Another set of calculations was perfomed
using this H ls I orbital. The potential energy results for these
two states are given in Table 11. Tha energies between 8.0 and
12.0 a.u. and for R=oo were obtamed using the double precision
integral program, while those between 3.0 and 6.o a.u. were
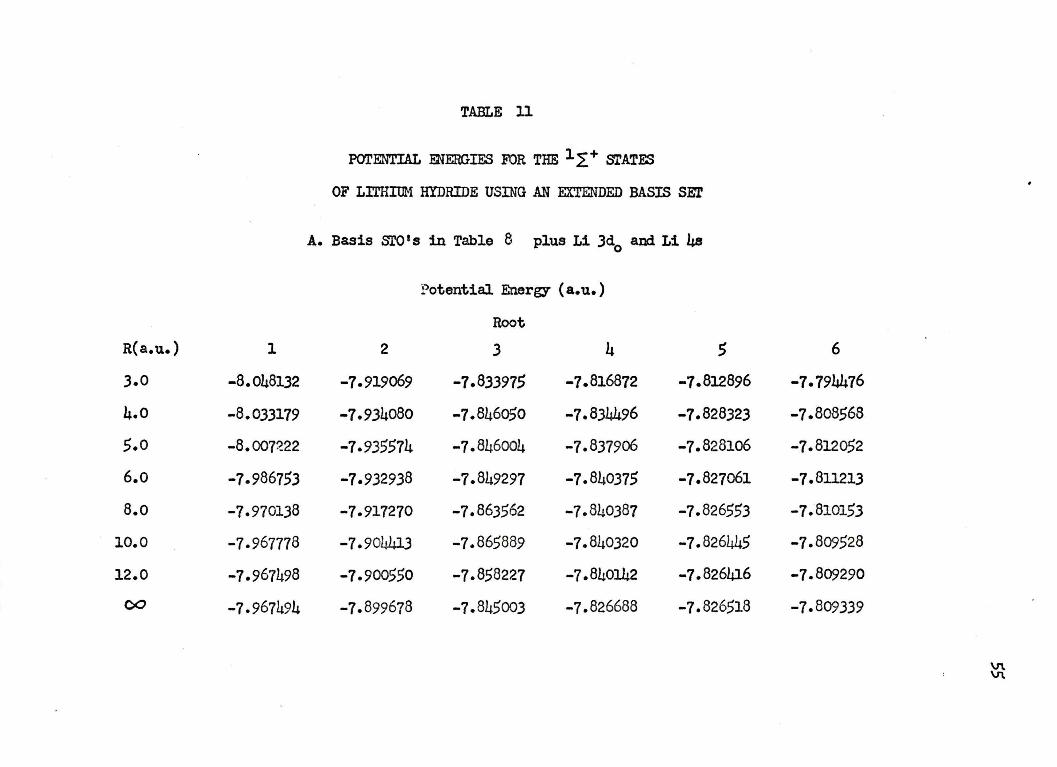
TABLE 11
POTENTIAL :ENERGIES FOR THE lL+ srATES
OF LITHIUM HYDRIDE USING AN EXTENDED BASIS SE?
A. Basis sro I s in Table 8 plus Li Jd0
and Li 4s
Potential Energy ( a. u.)
Root
R(a.u.) l 2 3 4 5 6
3.0 -8.048132 -7-919069 -7.833915 -7.816872 -1.812896 -7-794476
4.0 -8.033179 -7.934080 -7.846050 -7.834496 -7.828323 -7.808568
5.0 -8.007~22 -7.935574 -7.846004 -1.837906 -7.828106 -7.812052
6.o -7.986753 -7.932938 -7.849297 -7.840375 -1.827061 -7.8ll213
8.o -7.970138 -7-917270 -7.863562 -7.840387 -7.826553 -7.810153
10.0 -1.967778 -7.904413 -7.865889 -7.840320 -7.826445 -7.809528
12.0 -7-967498 -7.900550 -7.858227 -7.840142 -7.826416 -7.809290
00 -7.967494 -1.899678 -7.845003 -1.826688 -7.826518 -1.809339
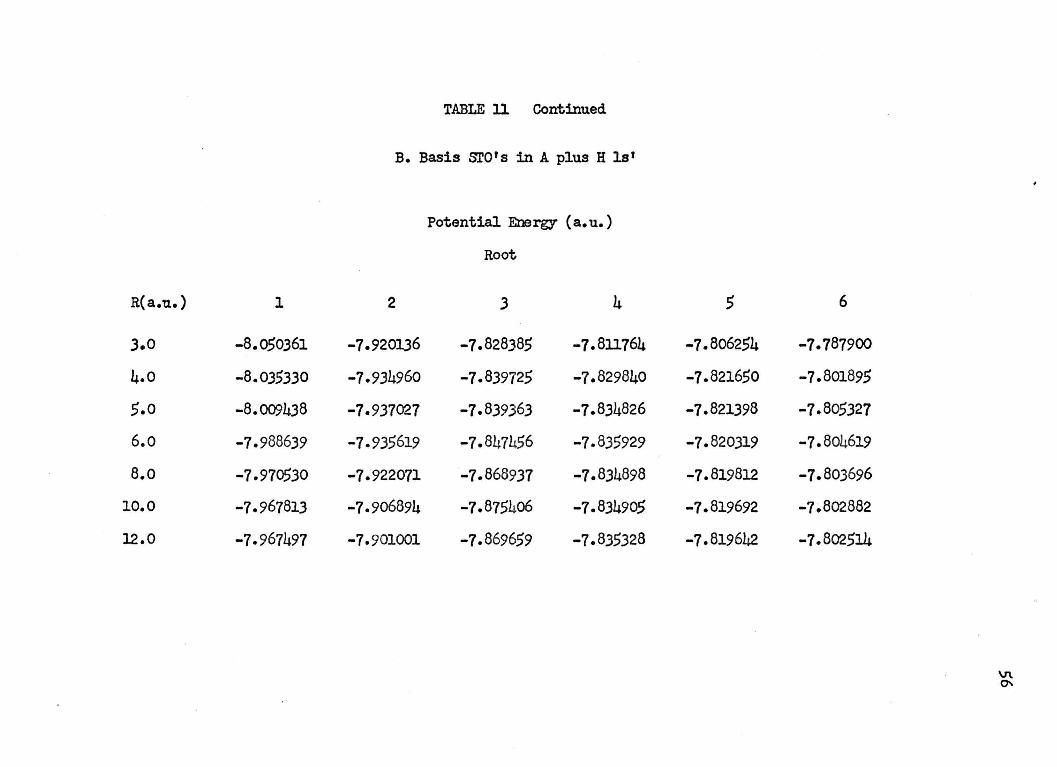
TABLE 11 Continued
B. Basis sro•s in A plus H ls'
Potential Energy ( a. u.)
Root
R(a.u.) 1 2 3 4 5 6
3.0 -8.050361 -1.920136 -7.828385 -7.811764 -7.806254 -7.787900
4.0 -8.035330 -7.934960 -7.839725 -7.829840 -7.821650 -7.801895
5.0 -8.009438 -1.931021 -1.839363 -7.834826 -1.821398 -7.805327
6.o -1.988639 -7-935619 -7.847456 -7.835929 -7.820319 -7.804619
8.o -1.910530 -7.922071 -1.868937 -7.834898 -7.819812 -1.803696
10.0 -1.967813 -7-906894 -7.875406 -7.834905 -7.819692 -7.802882
12.0 -7.967497 -7-901001 -7.869659 -7.835328 -7.819642 -7.802514

51
obtained using the single precision version. With the druble
precision integral program at 4.0 a.u., and the wavef'unctiion
. of Table 11, part A, the energies obtained were -8.033175,
-7.934073, -7.846154, -7.834288, -7.828192 and -7.810720 a.u.
respectively. Except for the sixth root, the results obtained
with the two integral programs agree vecy well.
In comparing these two sets of calculations for the x1 !. +
state, it is evident that while their energies are almost identical
for ~8.0 a.u., the energy obtained using the H ls' orbital is
better between 3.0 airl 6.0 a.u. For the A1f+ state the energies
of the two calculations are almost the same at 4. 0 a. u. and
12.0 a.u. but the calculation using the H 1s 1 STO is much better
between 5.0 and 10.0 a.u. The enorgy for the third 1 L.+ state
is also better with the H ls I orbital at R 2: 8. 0 a. u. Both
calculations indicate that the third 1 L + state is bound at
10.0 a.u. Also, a vecy slight metastable equilibrium is now
observed in the third 1 ~ + state at 4.0 a.u. instead of the
fourth as in Table 9. For the higher l~+ states the calcula
tions without the H ls' orbital give lower energies since these
states do not have strong Li~ character. The results indi-
1-c+ cate that the fourth , state is bound between 6.0 and 8.0
a.u. The fif'th and sixth states seem to have minimums between
4.0 and 5.0 a.u •
. These results suggest that for low lying Lili l l + states
the bonding and shape of their potential curves are determined to
some extent by the Li +n- character in these states. This char-

acter accounts for the equilibrium internuclear separation and
1 + depth of the potential curve of the x-_t state. The second,
third and fourth 1 L + states all have min:iJnums at large inter
nuclear separations and the importance of the Li+Ir configura
tions of these states increases at these large R. In view of
this fact, it saems that generally the Li+H- character is
important where the Li+H- curve is near the potential curve of
these states and this character accounts for the above behavior.
Mulliken(,) first used this analysis to accrunt for the shape
of these potential curves. With higher excited states this
character would occur at extremely large internuclear separ
ations. Therefore, these states should be Rydberg states with
minimums around 4.0 a.u. The Rydberg limit would be the LiH+
x2 2_ + state which has a equilibrium internuclear separation at
4.25 a.u. according to Browne.< 61) The separated atan limit
of the Li+Ir state is estimated to be -7.80802 a.u. from the
electron affinity of n< 2) and the ionization potential of 11.(3)
This lies between the separated atom limits of Li2F(ls24f') plus
H2S(ls) and 112s(1s25s) plus H2s(ls) which have energies of
-7.81175 and -7.80415 a.u. respectively. Any 11+ state with
separated t1to."ll limits above that of Li +H- would not be influ
enced by this ionic state.
The binding energies obtained for the x1 L+ state and
Alz:+ are estimated to be 2.2248eV and l.Ol63eV from the best
calculations of Table 11 as canpared to the experimental values
of 2.5154eV and l.0765eV respectively. The binding energies for
58

59
the third l r+ and fourth 1 z:+ states are 0.827JeV and 0.J728eV
respectively as obtained from their lowest, calculated energies.
The binding energies of the fifth and sixth 1 2.. + states whi. ch
seem to be Rydberg states are estimated to be 0.049leV and
0.0738eV respectively. For the transition between the first
two l~+ states, Te equals 25,089.0 cm-1 compared to the exper
imental value of 26,516.2 cm-1•
5.J. The Lithiwn Hydride 3 r+ States
The first four li thiwn hydride 3 2.. + states are considered
in this calculation. These states connect with the Li2S(ls22s),
Li2P(ls22p), -Li2S(ls2Js) or Li2P(ls2Jp) plus H2S(ls) separated
atom states. The calculated potential energies are given in
Table 12 and the potential curves are presented in Figure 2.
The configuration list for the wavefunctions is given in Table
13. The coefficients of the configurations in these wavefunc
tions are given at 4.0 a.u. in Table 26 o~ Appendix II.
The lowest 3 ~ + state arises from the Li 2 S( ls22s) atomic
state and is repulsive in energy. The second JL.+ state is also
repulsive. It arises from the Li2P(1s22p) plus H2s(ls) separated
atom states and becomes strongly repulsive in energy between 4.0
and 6.0 a.u. At 4.0 a.u. it levels off before rising sharply.
The third 3 ~ + state has a shallow minimum at 5. 0 a. u. Fran
Figure 2 there see~s to be an avoided curve crossing between the
second and third 3 r_+ states between 4.0 and 5.0 a.u. An
avoided crossing would be indicated by an analysis of the wave
function. Although potential energy curves of states of the same

R(a.u.)
1.0
2.0
3.0
4.0
5.0
6.o
7.0
a.o 9.0
10.0
(X)
TABLE 12
POTENTIAL ENERGIES FOR THE 3 I:+ STATES
OF LITHIUM HYDRIDE
Potential Energy (a.u.)
Roots
1 2 3
-7.257414 -7.122746 -7.098189
-7.845943 -1.150506 -1.718697
-1-935399 -7.842231 -7.822275
-7-952601 -7.851053 -7.839600
-7.959172 -7.86ll84 -7.847245
-1.963361 -7.880403 -7.844216
-7.965724 -7.890ll6 -7.842676
-1.966859 -7.894990 -7.842244
-7.967332 -7.897424 -7.842387
-7.967510 -7.898615 -7.842773
-7.967513 -7.900176 -7.845041
60
4
-1.037354
-7.680528
-1.110582
-1.808986
-7.829753
-1.828983
-7.826754
-7.824595
-7.823024
-7.822235
-1.828362
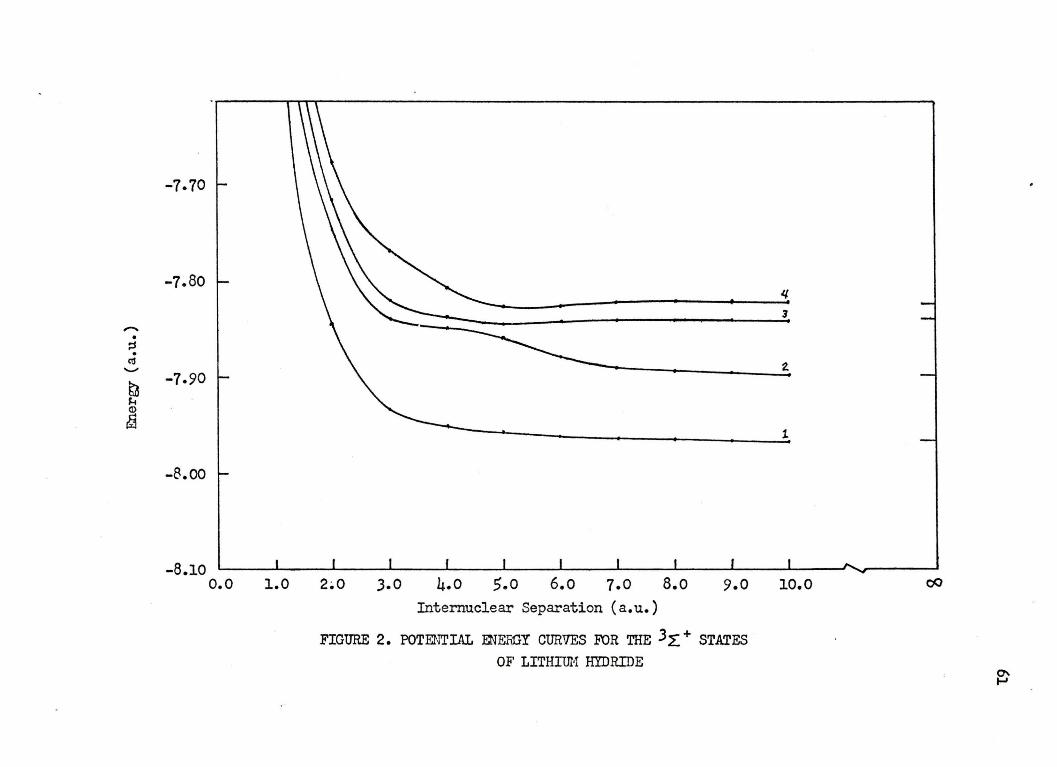
-• ::s •
<IS ........
6'.3 S.. (I)
~
-1.10
-7.80
-1.90
-8.00
-8.10 o.o 1.0 3.0 4.0 5.o 6.o 8.0 9.0
Internuclear Separation (a.u.)
FIGURE 2. POTENTIAL ENEiiGY CURVES FOR THE 3 L + STATES
OF LITHIUM HYDRIDE
" 3
2.
1
10.0

No.
l
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
TABLE 13
125 CONFIGURATIONAL WAVEFUNCTION FOR
THE 3L + STATES OF LITHIUM HYDRIDE
Configuration No. Configuration
l 1 2 16 18 1 2 6 17
l 1 13 16 19 1 2 14 16
l 1 12 16 20 l 2 14 17
1 1 6 16 21 l 2 7 16
1 1 14 16 22 1 2 7 17
1 1 7 16 23 1 2 9 16
l 1 9 16 24 1 2 9 17
1 l 10 16 25 l 2 10 16
l 2 13 16 26 l 2 10 17
1 13 2 16 27 l 13 12 16
1 2 13 17 28 l 12 13 16
1 2 12 16 29 1 1.3 6 16
l 12 2 16 .30 l 6 13 16
l 2 12 17 31 1 13 7 16
· 1 2 6 16 32 1 13 9 16
l 6 2 16 .33 1 13 10 16
l 16 6 2 34 2 2 1 16
62

63
TABLE 13 ContiDUed
No. Contigurat:1.on No. Conf'igu.ration
35 2 2 13 16 58 lJ 13 1 16
36 2 2 13 17 59 13 13 2 16
37 2 2 12 16 60 lJ 13 12 16
38 2 2 12 17 61 13 13 6 16
39 2 2 6 16 62 13 lJ 14 16
40 2 2 6 17 63 13 13 7 16
41 2 2 14 16 64 13 13 9 16
42 2 2 7 16 6.5 13 13 10 16
43 2 2 7 17 66 3 3 13 16
44 2 2 9 16 67 ··3 3 12 16
4.5 2 2 9 17 68 3 3 6 16
46 2 2 10 16 69 3 3 7 16
47 2 2 10 17 70 3 3 9 16
48 2 l.3 12 16 7l 3 3 10 16
49 2 12 13 16 72 4 5 13 16
50 2 13 6 16 73 4 5 12 16
51 2 6 13 16 74 4 5 6 16
.52 2 16 6 13 15 4 5 7 16
53 2 13 6 17 76 4 5 9 16
54 2 13 14 16 77 4 5 10 16
55 2 13 7 16 78 12 12 1 16
56 2 13 9 16 79 12 12 2 16
51 2 13 10 16 80 12 12 13 16
64
TABLE 13 Continued
No. Configuration No. Con!igaration
81 1 12 6 16 105 6 6 12 16
82 1 6 12 16 106 1 2 13 12
83 1 ]2 7 16 107 1 2 13 6
84 2 12 6 16 108 1 2 1.3 14
85 2 6 12 16 l()C) 1 2 13 7
86 2 12 7 16 no 1 2 12 6
87 2 12 9 16 lll 1 2 12 14
88 2 12 10 16 112 1 2 12 7
89 13 12 6 16 ll3 1 2 6 14
90 13 6 12 16 ll4 1 2 6 7
91 13 12 14 16 n, 2 2 1.3 12
92 13 12 7 16 ll6 2 2 13 6
93 13 12 9 16 ll7 2 2 13 14
94 13 12 10 16 ll8 2 2 12 6
95 6 6 2 16 ll9 2 2 12 14
96 6 6 13 16 120 2 2 12 7
97 1 6 7 16 121 2 2 6 14
98 2 6 7 16 122 2 2 6 7
99 13 6 7 16 123 1 l 16 17
100 13 6 9 16 124 l 2 16 17
101 12 12 6 16 125 2 2 16 17
102 12 12 7 16
103 12 12 9 16
104 12 6 7 16

65
synnnetry do oot cross, their wavefunctions may. In the region of
the avoided crossing, the wavefunction of the lower state becomes
that of the upper state and vice versa. For example, some of the
main configurations in the second and third J Z + states are given
in Table 14 along with their coefficients at 4.0, 5.0 am 6.0 a.u.
The switching of magnitude of these coefficients at 4.0 am 5.0 a.u.
indicates an avoided curve crossing. About 102 out of 12.5 ccni'ig
urations switch the magnitude of their coefficients between the
second and the third 3~+ states from 4.0 to 5.0 a.u. The major
ity of c<Xl.figurations which do not change involve the lithium
3p0
orbital. One can state,from observing the coeffic:l.ents
of the configurations for the 3~ + state, that the wavefunction
of the secon:l 32, + state at ,5.o a.u. strongly resembles the wave
fu.L'1.ction or the third 3z. + state at 4.0 a.u. and vice versa. This
would indicate that a curve crossing has taken place.
The fourth root obtained from this calculation rises rapidly
for R less than 5.0 a.u. This disagrees with the results obtained
by Bender and Davidson. (60) Therefore, this calculation was re
peated with Li 3d0 and Li 4a STO's with orbital exponents of
0.333.3 and 0.2500 respectively added to the basis set. The
energies obtained are given in Table 15. At 5.0 am 6.0 a.u.
the character of the outer lithium orbital of the fourth root
is Jp. At smaller internuclear separations of 3.0 and 4.0 a.u.
the configurations in the fourth root containing the Jp STO
decrease in magnitude while those in the sixth root increase.
At ,3.0 a.u. the fourth 32.+ state contains strong 3d character.
This state has a minimum araind 4.0 a.u. The fifth 3~+ state
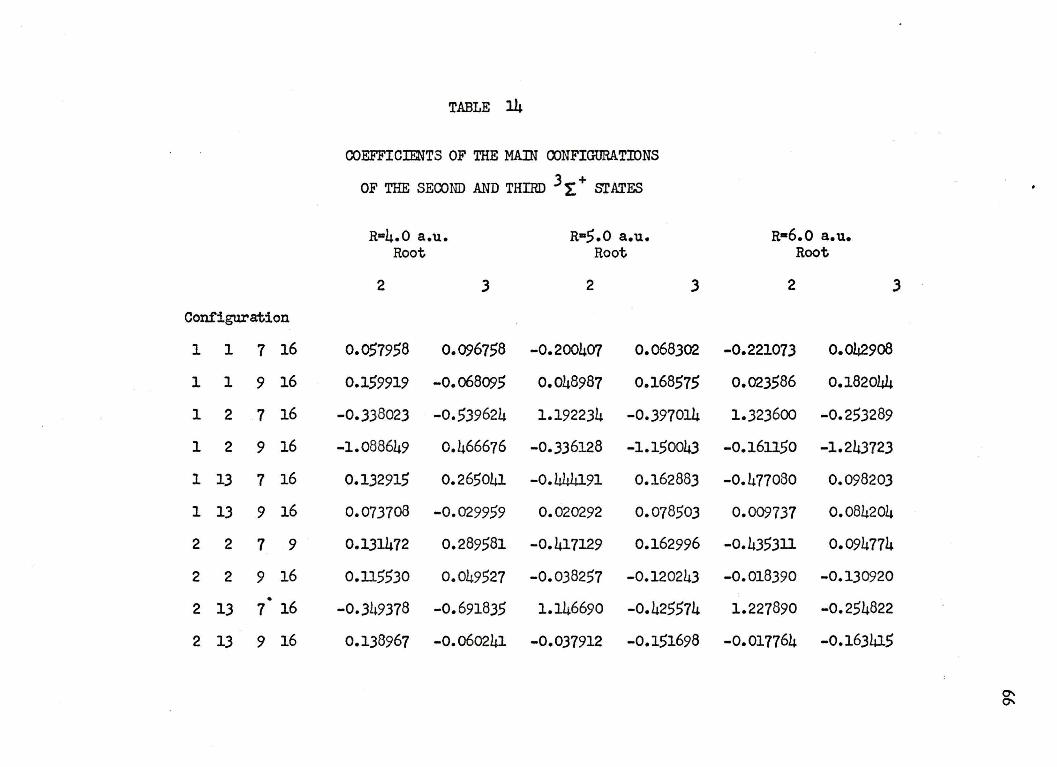
TABLE 14
COEFFICIFNTS OF THE MAIN OONFIGURATIONS
OF THE SEOOND AND THIRD 3 l: + srATES
R•4.0 a.u. R•5.0 a.u. R•6.0 a.u. Root Root Root
2 3 2 3 2 3
Configuration
1 1 7 16 0.057958 0.096758 -0.200407 0.068302 -0.221073 0.0429o8
1 l 9 16 0.159919 -0.068095 0.048987 0.168575 0.023586 0.182044
1 2 7 16 -0.338023 -0.539624 1.192234 -0.397014 1.323600 -0.253289
1 2 9 16 -1.088649 0.466676 -0.336128 -1.150043 -0.161150 -1.243723
l 13 7 16 0.132915 0.265041 -0.444191 0.162883 -0.477080 0.098203
1 13 9 16 0.073708 -0.029959 0.020292 0.078503 0.009737 0.084204
2 2 7 9 0.131472 0.289581 -0.417129 0.162996 -0.435311 0.094774
2 2 9 16 0.115530 0.049527 -0.038257 -0.120243 -0.018390 -0.130920
2 13 7 • 16 -0.3h9378 -0.691835 1.146690 -0.425574 1.227890 -0.254822
2 13 9 16 0.138967 -0.060241 -0.037912 -0.151698 -0.017764 -0.163415

TABLE 15
POTENTIAL ENERGIES FOR THE 3 L+ STATES
OF LITHIUM HYDRIDE USING AN EXTENDED BASIS SEr
Potential Energy ( a. u.) Root
R(a.u.) 1 2 3 4 5 6
3.0 -7.935270 -7.842564 -7.824803 -7.816025 -7.796752 -7.756968
4.0 -7.952681 -7.851989 -7.849158 -7.831905 -7.810431 -7.780805
5.0 -7.959302 -7.869541 -7.848113 -7.83llll -7.813350 -7.801399
6.o -7-963416 -7.883772 -7.845357 -7.829005 -1.813080 -7.804662
10.0 -1.967385 -7.898732 -7.843317 -1.826603 -7.8ll914 -7.808145
00 -7.967494 -1.899678 -7.845003 -1.826688 -7.826518 -1.809339

has a minimum around ,.o a.u. The main configurations for this
state are those containing the Li 4s sro. Those containing the
Li 3<lo increase at large R. At 10.0 a.u. this state lies higher
than its separated atan limit which is the Li 2n(ls23d) state.
Evidently, there are not enough configurations containing the
68
Li 3d0
sro in this wavefunction. The sixth 3 ~ + state is repul
sive and rises sharply around 5.0 a.u. The wavefunctions for these
3L+ states are complicated; however, from the results obtained
above, there would seem to be curve crossings between the fourth,
fifth and sixth 3 ~+ states.
Considering that the Lili 3 t+ states arising from the
Li2P(ls22p) and Li3P(ls2Jp) states are similar it may be plausible
to generalize such behavior to all 3 L + states arising from sim-
ilar atomic lithium states. These states have an outer orbital
whose value for 1 quantum number is odd. Therefore, the potential
energy curves for the 3 2. + states may be explained using the above
facts. The 3L+ states arising fran lithium atomic states that
have outer orbitals with even 1 tend to be bound and perhaps Ryd
berg states of LiH+. However, 3 ~ + states arising from lithium
atomic states that have outer orbitals with odd 1 are repulsive
and rise sharply around 6.o a.u. Therefore at small R, i.e.
between 2.0 and 6.0 a.u.,the shape of the various potential energy
curves are determined by interaction between these two types of
states. The shapes would therefore be accounted for by avoided curve
crossings.
Three bound 3 L+ states were observed. These are the third,
fourth and perhaps the fifth state. Their binding energies are

69
estimated to be 0.1131, 0.1420 and O.OJ9loV respectively from
the best results obtained.
5.4. The Lithium Hydride 37i States
The f'irst two lithium hydride 37t states are considered.
These states connect to the Li2P(ls22p) or Li2P(ls23p) plus
H2s(ls) atomic states in the separated atom limit. The inter
nuclear separations considered and the potential energies ob
tained are presented in Table 16. The potential energy curves
are given in Figure 3. The configuration list for this calcula
tion is given in Table 17 and the coefficients for the wavefunc
tion at 4.0 a.u. are listed in Table 27 of Appendix II.
The most important configurations in the first 3TT state are
the covalent configurations involving the Li 2P+ sro. For the
second 31f state the configurations involving the Li 3P+ and H
ls sro 1s have the largest magnitude although those involving Li 2p+
are also large at all the internuclear separations considered.
The first 37f state is the lowest bound triplet state of
lithium hydride. It has a minimum around 4.0 a.u. with an
estimated binding energy of O.lBOOeV. The second 31T state is bound
at 4.0 a.u. with a calculated binding energy of 0.0792eV. Bender
and Davidson( 60) also observed that these two states were bound, as
well as the third 37T state, with equilibrium internuclear separa
tions near 4.0 a.u. The fact that the Lili+ 2Z:. + state is
bound at 4.25 a.u. would indicate that the LIB 31f states were
Rydberg states with the dissociation limit Lili+. The binding
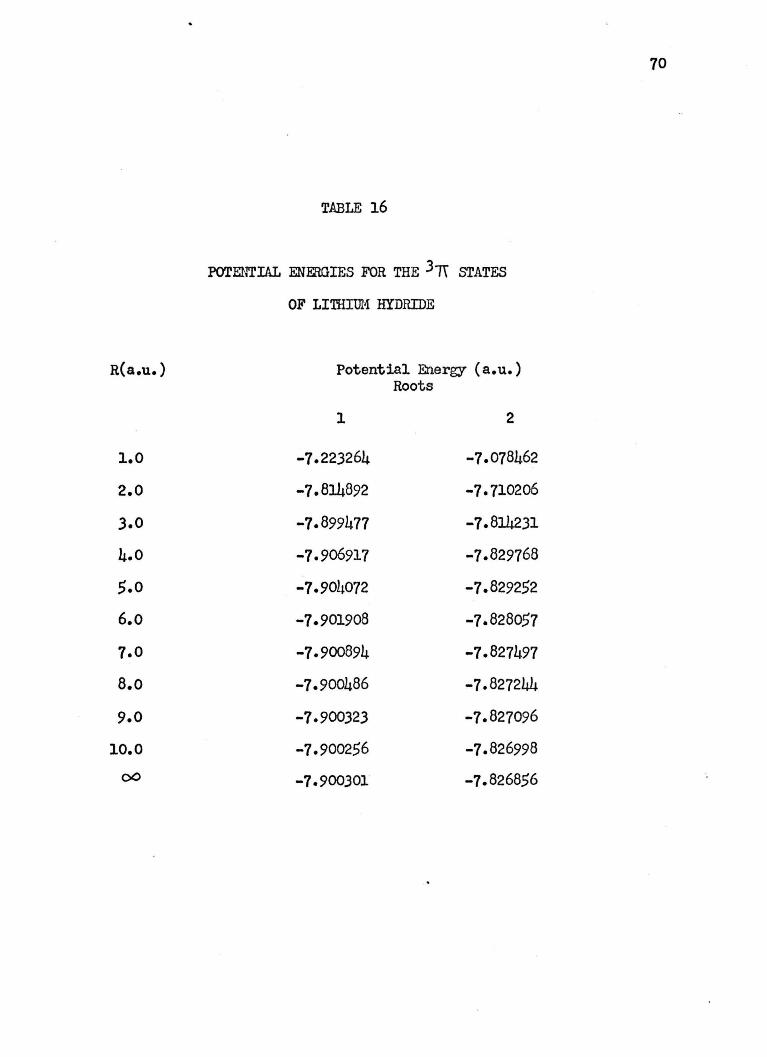
R(a.u.)
1.0
2.0
3.0
4.0
,.o
6.o
7.0
a.o 9.0
10.0
00
TABLE 16
POTENTIAL ENERGIES FOR THE 3n srATES
OF LITHill1 HYDRIDE
Potential Energy (a.u.) Roots
1 2
-7.223264 -7.078462
-7.814892 -1.710206
-7.899477 -7.814231
-7-906917 -7.829768
-7.904072 -7.829252
-1.901908 -7.828057
-7-900894 -7.827497
-7-900486 -7.827244
-7-900323 -1.827096
-7.900256 -1.826998
-7.900301 -7.826856
70

-• :::1 • cu .........
~ f-t Q)
~
-7.60
-1.10
-7.80
-7-90
-8.oo o.o 1.0
z.
1
2.0 3.0 4.0 5.0 6.0 1.0 a.o 9.0 10.0 Internuclear Separation (a.u.)
FIGURE 3. POTENTIAL ENERGY CURVES FUR THE 3,r STATES
OF LITHIUM HYDRIDE
00

No.
l
2
3
4
5
6
7
8
9
10
ll
12
lJ
14
15
16
17
TABLE 17
125 CONFIGURATION.AL WAVEFUNC'l'ION FOR
THE 31T STATES OF LITHIUM HYDRIDE
Configuration No. Configuration
l l 13 18 18 1 15 2 16
l l 12 18 19 l 2 15 17
l 1 - 6 18 20 1 2 7 18
1 l 14 18 21 1 2 8 16
1 1 15 16 22 1 8 2 16
1 l 15 17 23 1 16 8 2
1 l 7 18 24 l 2 8 17
l l 8 16 25 l 2 9 18
1 l 8 17 26 l 2 10 18
l l ll 16 27 l 2 ll 16
1 l ll 17 28 l 2 11 17
1 2 4 16 29 1 13 15 16
l 2 13 18 .30 1 13 8 16
1 2 12 18 31 l 8 ]J 16
1 2 6 18 32 1 ]J 8 17
1 2 14 18 33 1 ]J 11 16
l 2 15 16 34 2 2 l 18
72
73
TABLE 17 Continued
No. Configuration No. Configar ation
35 2 2 4 16 58 2 13 11 17
36 2 2 13 18 59 13 l3 15 16
37 2 2 12 18 6o l3 l3 8 16
38 2 2 6 18 61 13 13 8 17
39 2 2 14 18 62 13 13 11 16
40 2 2 15 16 63 3 3 15 16
41 2 2 15 7 64 3 3 8 16
42 2 2 7 18 65 3 3 8 17
43 2 2 8 16 66 3 3 11 16
44 2 2 8 17 67 4 5 15 16
45 2 2 11 16 68 4 5 8 16
46 2 2 11 17 69 4 5 8 17
47 2 13 4 16 70 4 5 ll 16
48 2 4 13 16 71 4 5 11 17
49 2 13 15 16 72 1 12 15 16
50 2 15 13 16 73 1 12 8 16
51 2 13 15 17 74 1 l2 11 16
52 2 13 8 16 15 2 12 l5 16
53 2 8 13 16 76 2 15 12 16
54 2 16 8 13 77 2 12 8 16
55 2 JJ 8 17 78 2 8 12 16
56 2 13 11 16 79 2 12 8 17
51 2 ll 13 16 80 2 12 ll 16
74
TABLE 17 Continued
No. Configuration No. Configuration
81 13 12 15 16 104 1 2 13 15
82 13 12 8 16 105 1 2 13 8
83 13 12 8 17 106 1 2 12 15
84 13 12 11 16 107 1 2 12 8
85 1 6 15 16 108 1 2 6 15
86 1 6 8 16 109 1 2 6 8
87 1 6 11 16 no 1 2 14 15
88 2 6 15 16 lll 1 2 14 8
89 2 6 8 16 112 1 2 7 15
90 2 6 8 17 113 1 2 7 8
91 2 6 11 16 114 2 2 1 8
92 13 6 15 16 115 2 2 13 15
93 13 6 8 16 116 2 2 13 8
94 13 6 8 l'j 117 2 2 12 15
95 13 6 11 16 - ll8 2 2 12 8
96 12 12 15 16 119 2 2 6 8
91 12 12 8 16 120 2 2 14 8
98 12 12 11 16 121 2 2 1 8
99 12 6 15 16 122 1 1 16 18
100 12 6 8 16 123 1 2 16 18
101 12 6 11 16 124 1 2 17 18
102 6 6 8 16 125 2 2 16 18
103 6 6 11 16

energies of these states ~re less than 0.2sV and the potential
curves are very similar in appearance to that of Ll.Ir".
5.5. The Lithium Hydride l7r states
Calculations were performed on the first two LiH 1 TT
states. These states connect to the separated atom limits of
Li2P(ls22p) plus u2s(ls) and Li2P(ls23p) plus H2s(la) respec
tively. The internuclear separations considered and the pot
ential energies obtained are listed in Table 18. The potential
curves are presented in Figure 4. The configurations used for
these wavefunctions are listed in Table 19. Their coefficients
are given in Table 28 of Appendix II.
The most important configurations in the first 1TI state
are those involving the Li 2p+ and H ls ST0 1s. For the second
1 7r the configurations with the largest magnitude are the covalent
configurations containing the Li 3P+ STO. Those involving the
Li 2p+ orbital are also large for this state.
The calculated potential curve for the first 17f state
is repulsive in energy but extremely flat. Experimentally, this
state has been studied by VelascoC7) who found it to be bound with
a binding energy of O.OJ5eV and an equilibrium internuclear separ
ation of 4.49 a.u. Bender and Davidson1s(60) calculation which
included a Li 3d STO, also predicted this state to be repulsive.
Therefore, probably more inner core correlation is needed in order
to obtain a brund state. The second 1 lT state is found to be bound
with a minimum between 4.0 and ,.o a.u. and an approximate binding
15
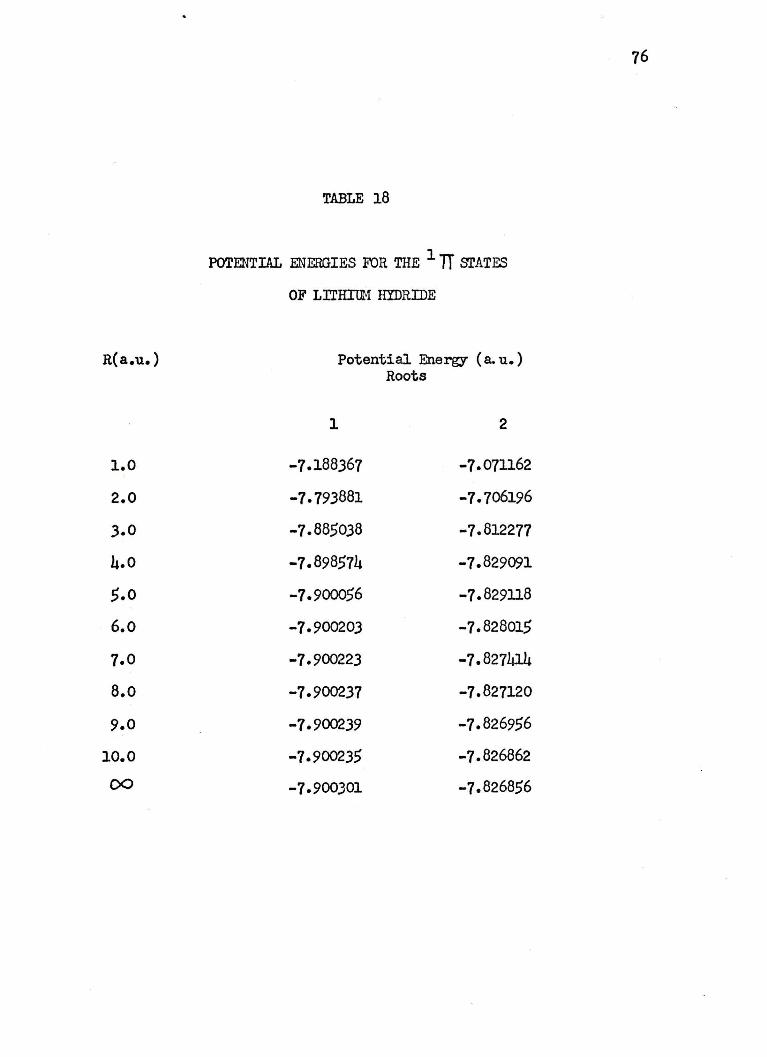
R(a.u.)
1.0
2.0
3.0
4.0
5.0
6.o
1.0
8.o
9.0
10.0
00
TABLE 18
POTENTIAL ENERGIES FOR THE l 1T STATE:S
OF LTIHit11 HYDRIDE
Potential Energy ( a. u. ) Roots
l 2
-1.188367 -7.07ll62
-1.793881 -1.106196
-7.885038 -7.812277
-7.898574 -7.829091
-1-900056 -7.829ll8
-1.900203 -1.828015
-7.900223 -7.827414
-7-900237 -7.827120
-1.900239 -7.826956
-7-900235 -1.826862
-7-900301 -7.826856
76
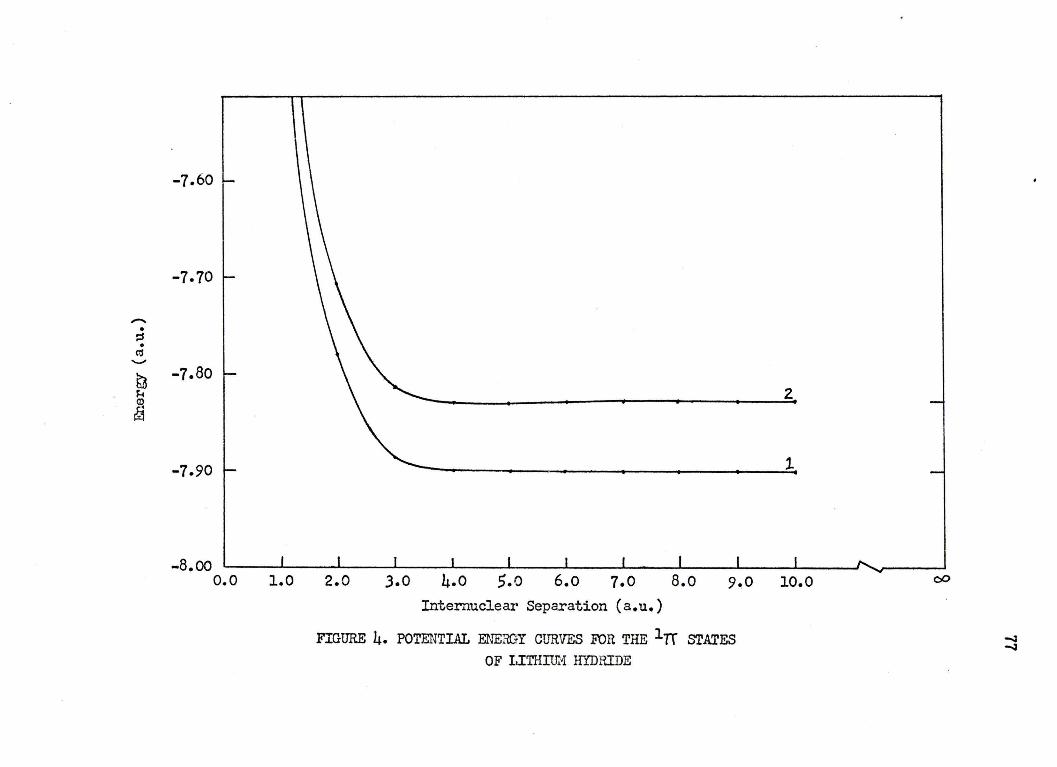
-• ::s . <ii -k3 H 0)
~
-7.60
-1.10
-7.80
-7.90
-8.oo o.o 1.0 2.0 3.0 4.0 5.o 6.o 7.0 8.0
Internuclear Separation (a.u.)
FIGURE 4. POTENTIAL ENERGY CURVES FOR THE 1TT STATES
OF LITHIUM HYDIUDE
2
1
c,O

No.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
TABLE 19
125 CONFIGURATIONAL WAVEFUNCTION FOR
THE 1 Ti STATES OF LITHIUM HYDRIDE
Configuration No. con:tigurat.ion
1 1 13 18 18 l 2 15 16
1 1 12 18 19 1 15 2 16
1 1 6 18 20 1 2 1.5 17
1 1 14 18 21 1 2 7 18
1 1 15 16 22 1 2 8 16
1 1 1.5 17 23 1 8 2 16
l 1 7 18 24 1 2 8 17
1 1 8 16 25 1 2 9 18
1 l 8 17 26 l 2 10 18
1 l 11 16 27 1 2 11 16
1 l 11 17 28 l 2 11 17
l 2 4 16 29 l 13 15 16
1 4 2 16 30 1 13 8 16
1 2 13 18 31 1 8 13 16
1 2 12 18 32 1 13 8 17
1 2 6 18 33 1 13 11 16
1 2 14 18 34 2 2 1 18
78
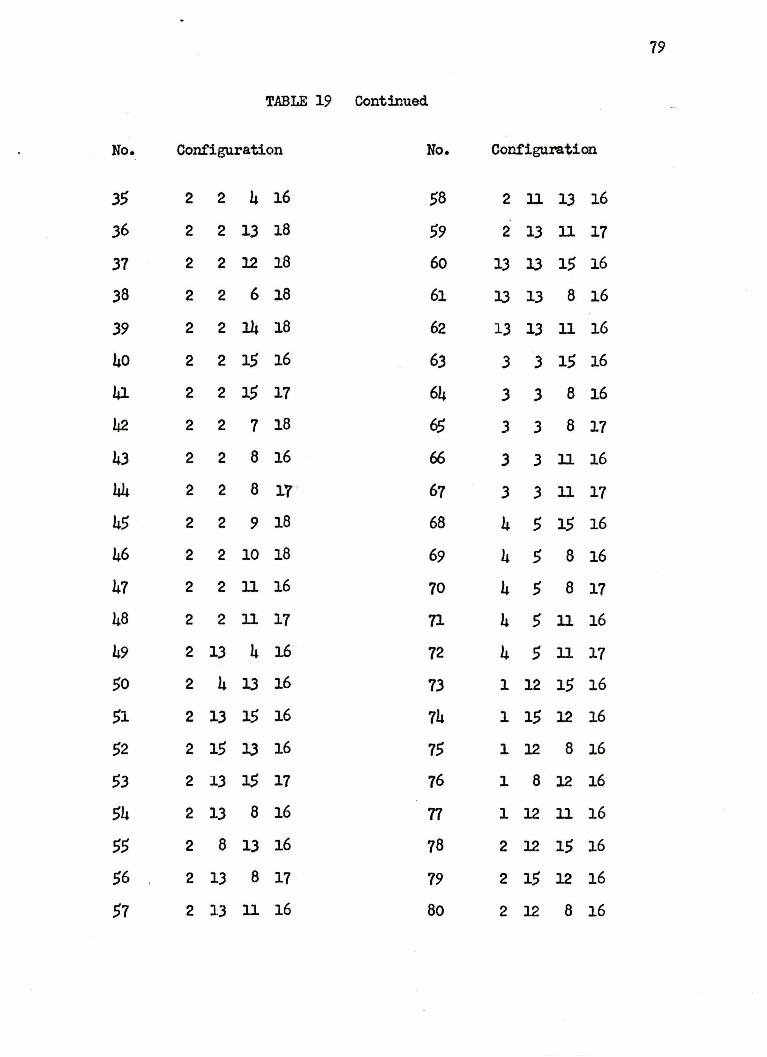
19
TABLE 19 Continued
No. Configuration No. Configuration
35 2 2 4 16 58 2 11 13 16
36 2 2 13 18 59 2 13 11 17
37 2 2 12 18 60 13 l3 15 16
38 2 2 6 18 61 l3 13 8 16
39 2 2 14 18 62 13 13 11 16
40 2 2 15 16 63 3 3 15 16
41 2 2 15 17 64 3 3 8 16
42 2 2 7 18 65 3 3 8 17
43 2 2 8 16 66 3 3 11 16
44 2 2 8 l,f' 67 3 3 11 17
45 2 2 9 18 68 4 5 15 16
46 2 2 10 18 69 4 5 8 16
47 2 2 11 16 70 4 5 8 17
48 2 2 11 17 71 4 5 11 16
49 2 13 4 16 72 4 5 11 17 50 2 4 l3 16 73 1 12 15 16
51 2 13 15 16 74 l 15 12 16
52 2 15 l.3 16 15 1 12 8 16
53 2 l.3 15 17 76 l 8 12 16
54 2 13 8 16 77 l 12 11 16
55 2 8 13 16 78 2 12 15 16
56 2 13 8 17 79 2 15 12 16
57 2 13 ll 16 80 2 J2 8 16
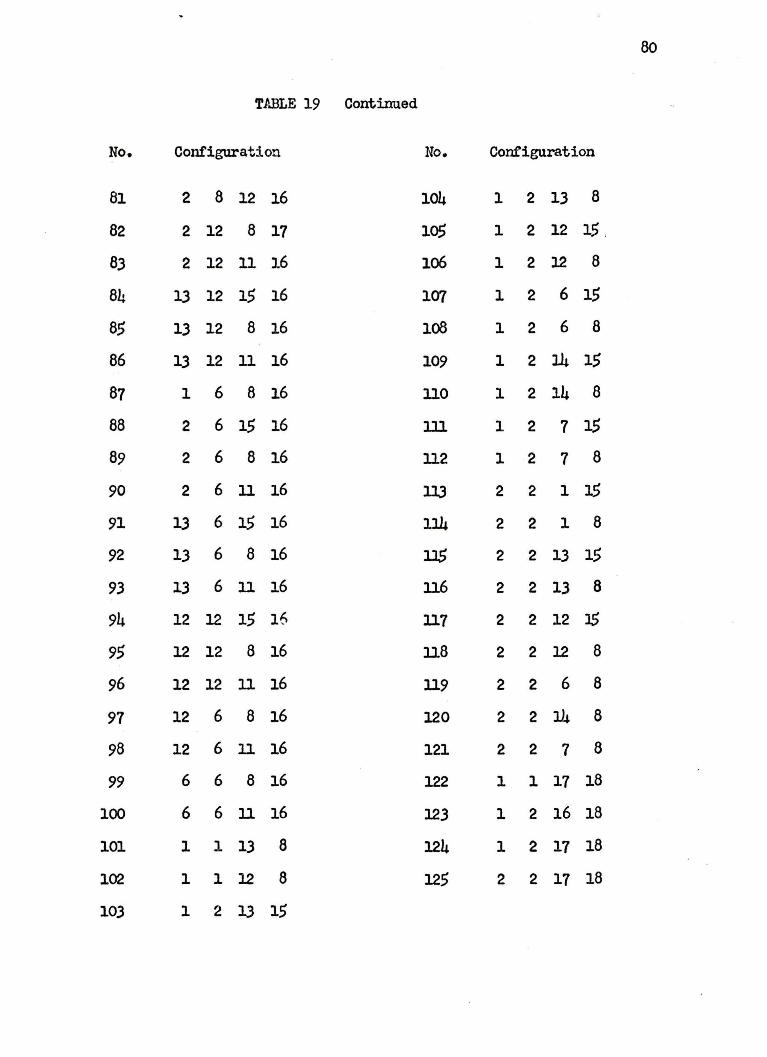
80
TABLE 19 Continued
No. Configuration No. Configuration
81 2 8 12 16 104 1 2 13 8
82 2 12 8 17 105 l 2 12 15 .
83 2 12 11 16 lo6 1 2 12 8
84 13 12 15 16 107 1 2 6 15
85 13 12 8 16 lo8 1 2 6 8
86 13 12 11 16 109 1 2 14 15
87 1 6 8 16 no l 2 14 8
88 2 6 15 16 lll 1 2 7 15
89 2 6 8 16 112 1 2 7 8
90 2 6 ll 16 113 2 2 1 15
91 13 6 15 16 114 2 2 1 8
92 13 6 8 16 J.J5 2 2 13 15
93 13 6 11 16 ll6 2 2 13 8
94 12 12 15 16 ll7 2 2 12 l5
95 12 12 8 16 118 2 2 12 8
96 12 12 11 16 ll9 2 2 6 8
91 12 6 8 16 120 2 2 14 8
98 12 6 11 16 121 2 2 1 8
99 6 6 8 16 122 1 1 17 18
100 6 6 ll 16 123 1 2 16 18
101 l l 13 8 124 1 2 17 18
102 1 1 12 8 125 2 2 17 18
103 l 2 13 15

energy of O. 0615eV. Bender and Davidson also fotmd the third
17T state to be bound at 4.0 a.u. The equilibrium interm.iclear
separations of these states are aromid 4.0 a..u. and the potential
energy curves are all shallow. This wouJ.d indicate that the
LiH 1 TI states are also Rydberg states with the Lili+ x2~ + state
as the limit.
5.6. The Lithium Hydride Plus x2L+ State
The lowest lithium hydride plus ground 2 r + state was
considered in this calculation. The potential energies calcu
lated are presented in Table 20 and the potential curve is
given in Figure 5. The configuration list used is given in Table
21 and their coefficients are listed in Table 29 of Appendix II.
This state connects to the Li+ 1s(ls2) plus II2s(ls) sep
arated atom states. The energy of the Li+ 1s(ls2) atomic state
is calculated using the basis in Table 8. and 32 configurations.
An energy of -7.27133 a.u. as compared to an experimental value
of -7 .28049 a.u. is obtained. The Lili+ x2 z:_+ state is calcula
ted to have an estimated binding energy of 0.1098eV and an
equilibrium internuclear separation aromid 4.0 a.u. This result
agrees with the values obtained by Browne( 6l) who calculated
dissociation energies of o.104!0.016eV and an equilibrium inter
m.iclear separation of 4.25 a.u. BrO'Wlle 1s energy, -7.780848 a.u.
at Re,is better than the result obtained here. However, the
energies of the potential curve are more accurate in this calcula
tion.
81

R(a.u.)
1.0
2.0
3.0
4.0
,.o
6.o
1.0
B.o
9.0
10.0
00
TABLE 20
POTENTIAL ENERGY OF THE 2 I:+ STATE
OF LITHIUM HYDRIDE PLUS
Potential Energy (a.u.)
-7.074723
-7.670684
-7-762942
-7-775364
-7. 774647
-7-773139
-7-772273
-7.771859
-7.771650
-7-771535
. -7. 771328
82
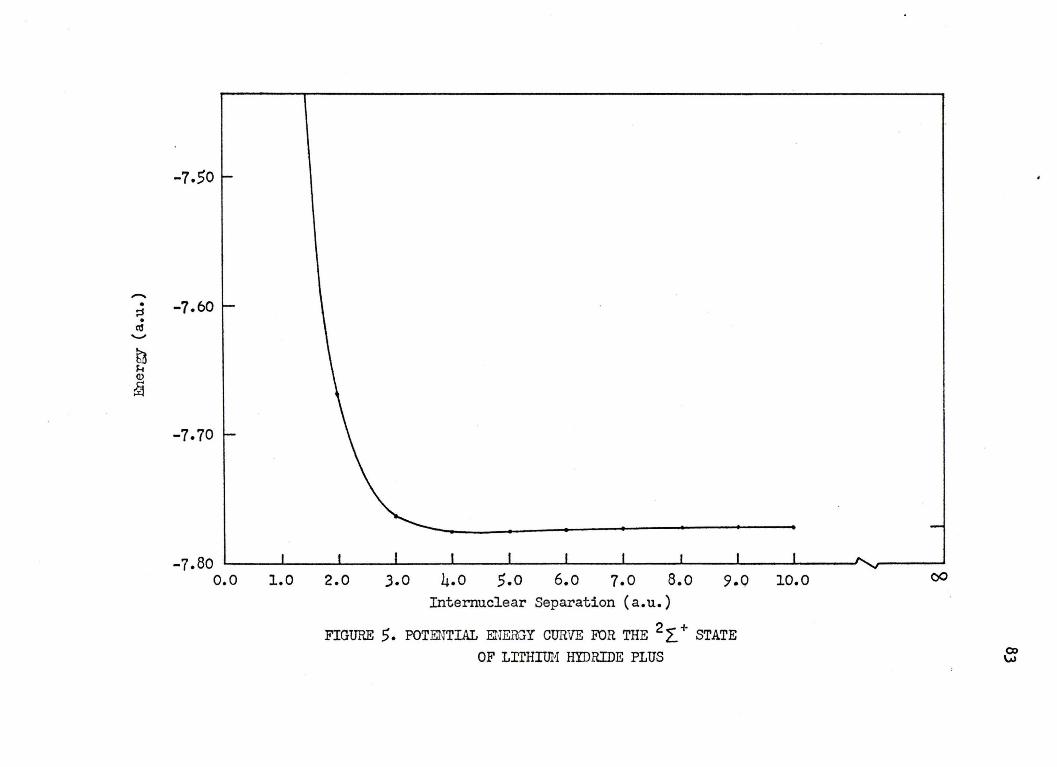
-• ~ • (1S
'-"
k3 r-. Q)
~
-7.50
-7.60
-1.10
-7.80 o.o 1.0 2.0 .3.0 4.0 5.0 6.o 8.0 9.0 10.0
Internuclear Separation (a.u.)
FIGURE 5. POTENTIAL EHEffiY CURVE FOR THE 2 L + STATE
OF 1rrHnn1 HYDRIDE PLUS

No.
1
2
3
4
5
6
7
8
9
10
11
12
lJ
14
15
16
17
TABLE 21
125 CONFIGURATIONAL WAVEFUNCTION FOR
THE 2t.+ STATE OF LITHIUM HYDRIDE PLUS
Configuration No. Configuration
1 1 16 18 3 3 17
1 1 17 19 4 , 16
1 2 16 20 4 , 17
l 2 17 21 l 12 16
1 13 16 22 1 16 12
l 16 13 23 l 12 17
l 13 17 24 l 6 16
2 2 16 25 1 16 6
2 2 17 26 l 6 17
2 4 19 27 l 14 16
2 5 18 28 l 16 14
2 13 16 29 l 14 17
2 16 13 30 l 7 16
2 13 17 31 1 16 7
13 13 16 32 l 7 17
13 13 17 33 1 9 16
3 3 16 34 2 12 16
84
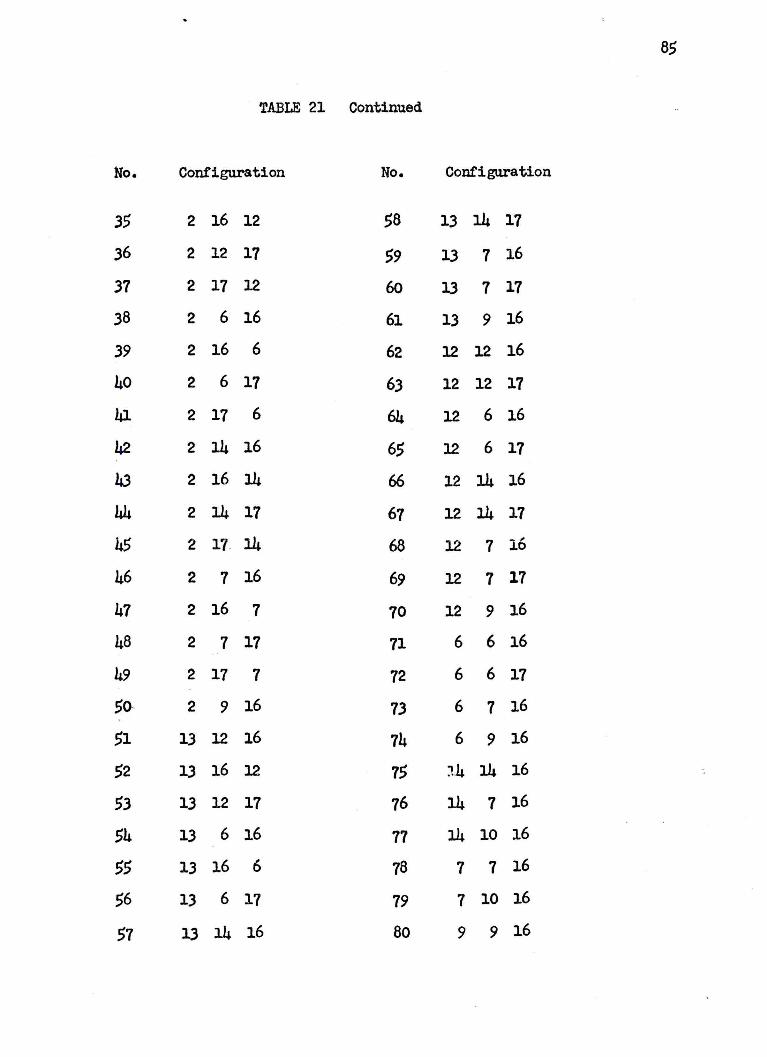
85
TABLE 21 Continued
No. Configuration No. Configuration
35 2 16 12 58 13 14 17
36 2 12 17 59 13 7 16
31 2 17 12 60 l3 7 17
38 2 6 16 61 13 9 16
39 2 16 6 62 12 12 16
40 2 6 17 63 12 12 17
la. 2 17 6 64 12 6 16
42 2 14 16 65 12 6 17
43 2 16 14 66 12 14 16
44 2 14 17 67 12 14 17
45 2 17. 14 68 12 7 - , J.O
46 2 7 16 69 12 7 17
47 2 16 7 70 12 9 16
48 2 7 17 71 6 6 16
49 2 17 7 72 6 6 17
50- 2 9 16 73 6 7 16
51 13 12 16 74 6 9 16
52 13 16 12 75 14 14 16
53 13 12 17 76 14 7 16
54 13 6 16 77 l4 10 16
55 13 16 6 78 7 7 16
56 13 6 17 79 7 10 16
57 13 14 16 80 9 9 16
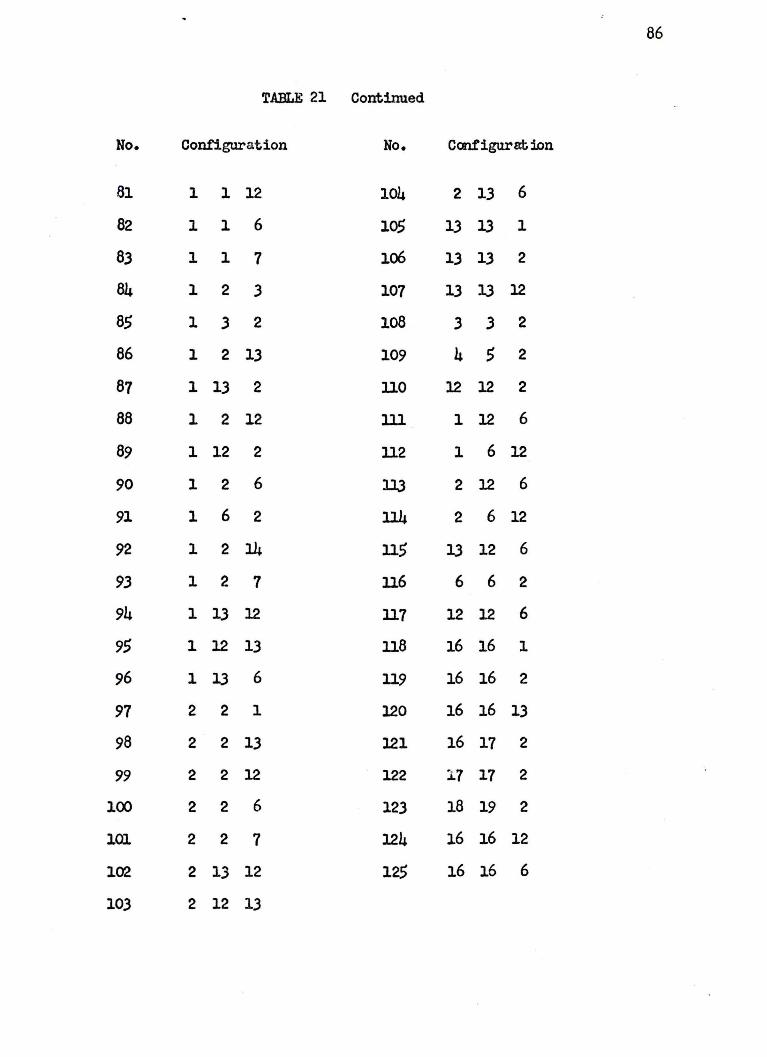
86
TABLE 21 Continued
No. Configuration No. Ccnf'igurat ion
81 1 1 12 104 2 13 6
82 l l 6 105 13 13 l
83 1 1 7 lo6 13 13 2
84 1 2 3 107 13 13 l2
85 1 3 2 108 3 3 2
86 l 2 13 109 4 5 2
87 l 13 2 ll0 12 12 2
88 l 2 12 lll 1 12 6
89 l 12 2 ll2 l 6 12
90 l 2 6 lJJ 2 12 6
91 1 6 2 ll4 2 6 12
92 l 2 14 n, 13 12 6
93 1 2 7 ll6 6 6 2
94 1 13 12 ll7 12 12 6
95 l 12 13 ll8 16 16 1
96 l 13 6 ll9 16 16 2
91 2 2 1 120 16 16 13
98 2 2 13 121 16 17 2
99 2 2 12 122 17 17 2
100 2 2 6 123 18 19 2
101 2 2 7 124 16 16 12
lO'l 2 13 12 125 16 16 6
103 2 12 13

. This state has not been observed experimentally. The lithium
hydride ground state would ionize to give this molecular ion state.
From the results obtained the ionization potential would be approx
imately 7.45l8eV. This state 'WOuld be the ionization limit of
the Icy-dberg states. The equilibrium internuclear separations of
these LiH Rydberg states would lie around 4.0 a.u. and their
potential energy curves would be similar to that of LiH+.
87

CHAPTER VI
DISCUSSION AND SUMMA.RY
Potential energy curves have been obtained for low
lying states of diatomic lithium hydride and the ground
x2 ~ + state of LiH+. Large STO bases and 125 configurational
interaction wavefunctions were used in this study. The sro
bases were obtained from optimized basis sets for the appro
priate united and separated atom states. Except for the LiH
ground x1~ + state and the first excited Alf+ state this
investigation is the most accurate study of the potential
energy curves of the low lying lithium hydride states con
aio.ered. Only three LiH states, i.e. the x1 L +, A1 2+, and
131-TI, have been observed experimentally.
All of the 1 ~ + states studied are bound. The bin.ding
energies of the calculated x1z:_ +, A1 Z:., + states are 89 and 88%
of the experimental values respectively. The next two 11 +
states are bound at large internuclear separations around
1O.O and 8.o a.u. respectively, due perhaps to their Li+H
character at large R. The fifth and sh-th 1 ~ + states are
bound between 4.0 and 5.0 a.u. These and higher l~+ states
are probably Rydberg states with an equilibrium internuclear
separation, Req, around 4.0 a.u.
88

69
Brown and Shull(57) have obtained more extensive curves
for the x1 ~+ and A1 ~+ states. Also, their results for the
x1 L. + state were better around the equilibrium internuclear
separation. The best energy obtained by the author was -8.054571
a.u. at 3.0 a.u. using 132 configurations and a 20 sro basis as
compared to -8.05549 a.u. by Brown and Shull. The sro basis
was similar to that of Table 8 except that the outer Li 3s, Jp,
etc. sro•s wero not present. Also, inner core Li Jp sro 1s with
the same orbital exponents as the Li 2p STO I s and a H ls I and
2p' sro with orbital exponents of o.6000 were added to the
basis.
An estimate of the accuracy of 1 2. + curves can be made
by comparing the energies of Table 9 with the experimental
results at Req and R= oo. At R= 00 the x1 L+ and A1L+ states
are approximately 0.J0eV higher than the experimental vaJ.ues.
At their Req values, the x1 L.. + and A 1 ~+ states lie 0. 58 am
0.40eV, respective~r, above the experimental energies. Since
these states have different character at different internuclear
separations their spectroscopic constants are needed for a
more accurate analysis. A comparison with the experimental
energies also shows that the higher 1 Z: + states are also in error
by 0.JOeV at R .. co. However, due to the fact that the basis
does not take into acccunt correlation in their outer shells
these higher 1 z.+ states are probably less accurate at smaller
R than the xl~+ and A1 ~+ states. It can al.so be stated with
certainty that the third and fourth 1 L + states are bound around

8.0 to 10.0 a.u. since the lowest energies of these states
both lie below the experimental energies of their respective
separated atom limits and the energies obtained nru.st be upper
bound to the true values. Also, the integrals calculated
are accurate at the large R considered and other LiH states
go correctly to their separated atom limits.
The lowest Jr+ state is repulsive in energy. The second
J L+ state is also repulsive but shows a sharp rise in energy
between 4.0 and 6.0 a.u. The third, fourth and fifth JL.+
states are bound between 4.0 and 5.0 a.u. The sixth JL+
state also rises sharply between 4.0 and 6.0 a.u. Evidence of
an avoided curve crossing is found between the second and third
3L+ states around 4.0 a.u. and perhaps between the fourth,
fifth and sixth 3~+ states at larger R. Therefore, the pot
ential energy curves for the 3 L + states are complicated by
interactions between them at R 6.0 a.u. In cases where there
is strong interacti~n between states of the same symmetry, the
Born-Oppenheimer approximation and the concept of a potential
energy curve is no longer accurate.< 2)
The lowest bound triplet state is the lowest 37t state
which is bound at 4.0 a.u. The second 31f state is also
observed to be bcund at 4.0 a.u. This agrees with the results
of Bender and Davidson< 60) who also found that the third JTT
state has a minimum around 4.0 a.u.
The B1 1T state is found to be extremely flat but repul
sive. This state is found experimentally( 7) to have binding
90

energy of 0.0J5eV. Evidently, more correlation is needed in
the wavefunction. The nex:t 1 11 state is bound between 4.0
am. 5.0 a.u. Bender and Davidson found the third 1 Tf state
to be bound around 4.5 a.u. and also strong evidence of :inter
action between the second and third 1 TT states.
A comparison or the energy of the B1 7T state with the
experimental value at Req and Ra 00 shows that they differ
by 0.JOeV This value can be taken as a rough estimate of the
error in the other Rydberg states and the repulsive curves.
It must be remembered, however, with the bases used, the results
are less accurate at small internuclear separations. Also,
1£ the wavefunction for a particular state does not have the
correct character in a certain region of internuclear separ
ations, such as the Li+iI- character needed for the first four
lL.+ states, the results will be less accurate in this region.
The x2 ~ + state of LiH+ is calculated to have a binding
energy of 0.llOeV with a minimum near 4.0 a.u. The ionization
potential of LiH is estimated to be 7.45eV fran the difference
in energy between the Lili x1 2._ + state and the Lili+ x2 2_ + state.
Bender and Davidson have found that the lowest 1 [),, and 3 b..
states are botmd with minimums between 4.0 and 4.5 a.u. The
shapes and minimums of the potential cuwes of the 1 TT , 1 b. ,
3 7f and 3 ~ states and the higher 1 ~ + states are very sim-
Uar to that of the LiH+ x2 ~ + state. This indicates that
these states are .lcy'dberg states of lithiwn hydride.
Not many transitions are likely to be observed in the
diatontf..c Lili system. Transitions between the ground x1 2+
91

92
state and third or fourth 12+ state are not very probable
since their equilibrium internuclear separations differ greatly
from that of the ground state. Transitions to higher lz+
states are possible if these states are bound between 4.0 and
5.0 a.u. Transitions between the ground state and the l TT
states are also likely to occur. Icy-dberg series of transitions
occur between the ground state and 11 + or 1 TT states which
arise from Li2P(ls2np) atomic states. other Rydberg series
occur from the Al~+ or E1TI states. Since singlet-triplet
transitions are forbidden and the lowest triplet state is re
pulsive, not many transitions are likely to be observed between
triplet states. Bands involving triplet states are most likely
to originate from the lowest 37T state.

APPENDIX I
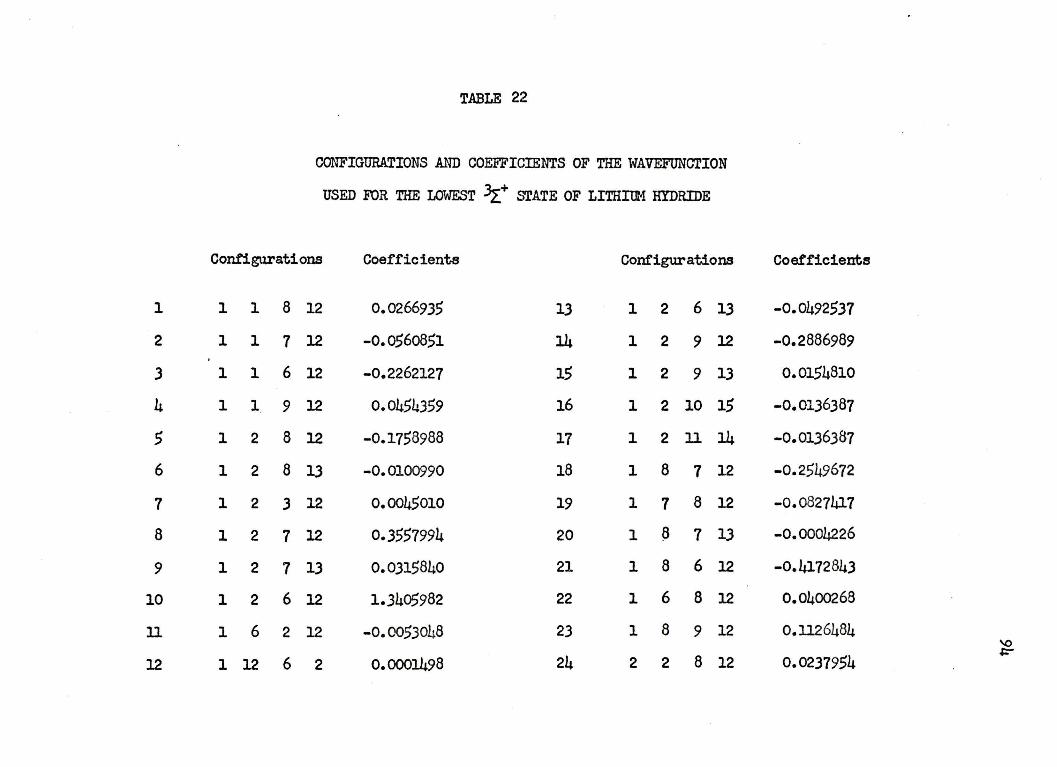
1
2
3
4
5
6
7
8
9
10
11
12
TABLE 22
CONFIGURATIONS AND COEFFICIENTS OF THE WAVEFUNCTION
USED FOR THE LOWEST Jr+ STATE OF LITHitM HYDRIDE
Con:f'igurati ons Coefficients Configurations
1 1 8 12 0.0266935 13 1 2 6 13
1 1 1 12 -0.0560851 14 1 2 9 12
1 1 6 12 -0.2262127 15 1 2 9 13
1 1 9 12 0.0454359 16 1 2 10 15
1 2 8 12 -0.1758988 17 1 2 11 14
1 2 8 13 -0.0100990 18 1 8 1 12
1 2 3 12 0.0045010 19 1 1 8 12
1 2 7 12 0.3557994 20 1 ~ 7 13
1 2 1 13 0.0315840 21 1 8 6 12
1 2 6 12 1.3405982 22 1 6 8 12
1 6 2 12 -0.0053048 23 1 8 9 12
1 12 6 2 0.0001498 24 2 2 8 12
Coefficients
-0.0492537
-0.2886989
0.0154810
-0.0136387
-0.0136387
-0.2549672
-0.0827417
-0.0004226
-0.4172843
0.0400268
0.1126484 'O
0.0237954 .i:-
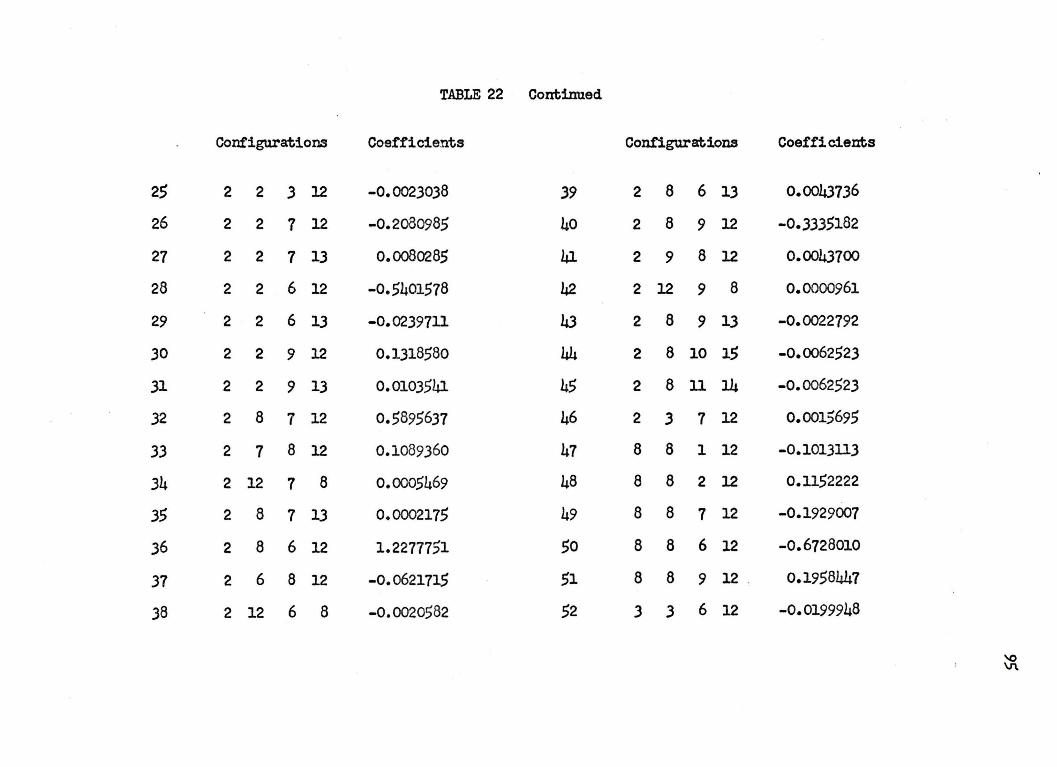
TABLE 22 Continued
Configurations Coefficients Configurations Coeffi cient;s
25 2 2 3 12 -0.0023038 39 2 8 6 13 0.0043736
26 2 2 7 12 -0.2080985 40 2 8 9 12 -0.3335182
27 2 2 7 13 0.0080285 41 2 9 8 12 0.0043700
28 2 2 6 12 -0.5401578 42 2 12 9 8 0.0000961
29 2 2 6 13 -0.0239711 43 2 8 9 13 -0.0022792
30 2 2 9 12 0.1318580 44 2 8 10 15 -0.0062523
31 2 2 9 13 0.0103541 45 2 8 11 14 -0.0062523
32 2 8 7 12 o.5895637 46 2 3 7 12 0.0015695
33 2 7 8 12 0.1089360 47 8 8 1 12 -0.1013113
34 2 12 7 8 0.0005469 48 8 8 2 12 0.1152222
35 2 8 7 13 0.0002175 49 8 8 7 12 -0.1929007
36 2 8 6 12 1.2277751 50 8 8 6 12 -0.6728010
31 2 6 8 12 -0.0621715 51 8 8 9 12 . 0.1958447
38 2 12 6 8 -0.0020582 52 3 3 6 12 -0.0199948
'O V\
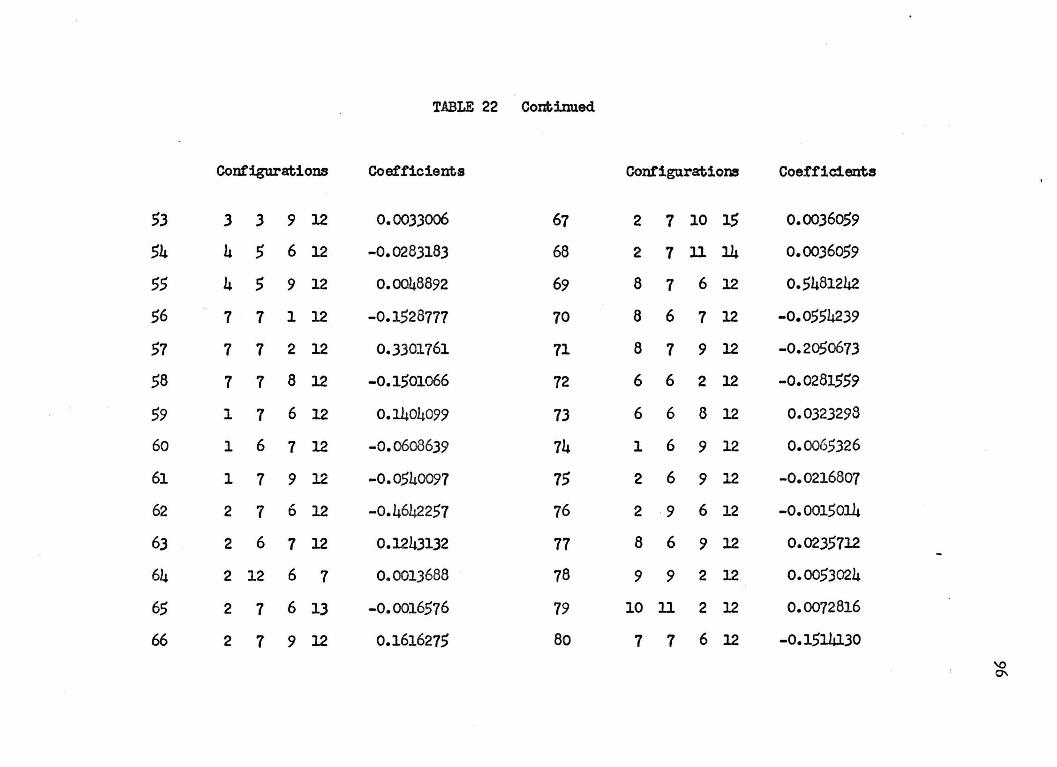
TABLE 22 Continued
Configurations Coefficients Configurations Coefficients
53 3 3 9 12 0.0033006 67 2 7 10 1.$ 0.00360,$9
.54 4 .5 6 12 -0.0283183 68 2 7 11 14 0.0036059
55 4 .5 9 12 0.0048892 69 8 7 6 12 o.5481242
56 7 7 1 12 -0.1528777 10 8 6 1 12 -0.0554239
51 1 7 2 12 0.3301761 71 8 7 9 12 -0.2050673
58 7 7 8 12 -0.1501066 72 6 6 2 12 -0.0281559
59 l 7 6 12 0.1404099 73 6 6 8 12 0.0323298
60 1 6 1 12 -0.0608639 74 1 6 9 12 o.0065326
61 l 1 9 12 -0.0540097 75 2 6 9 12 -0.0216807
62 2 7 6 12 -0.4642257 76 2 9 6 12 -0.0015014
63 2 6 7 12 0.1243132 11 8 6 9 12 0.0235712
64 2 12 6 1 0.0013688 78 9 9 2 12 0.0053024
65 2 7 6 13 -0.0016576 19 10 11 2 12 0.0072816
66 2 7 9 12 0.1616275 80 7 1 6 12 -0.1514130 '-0
°'
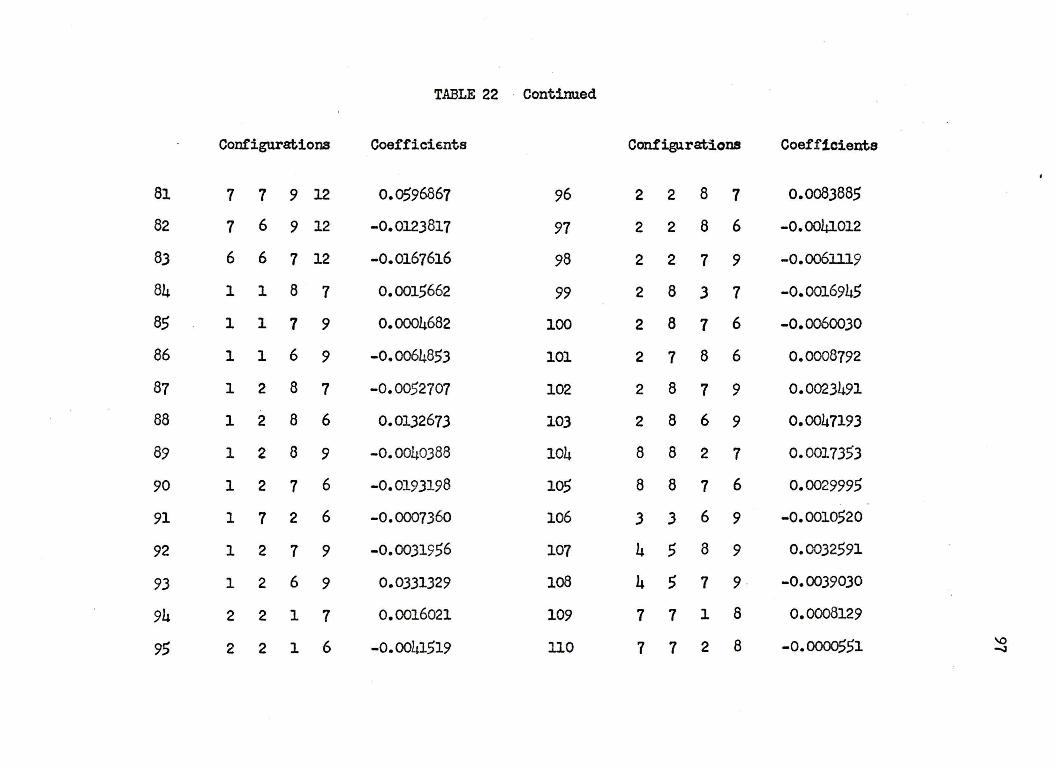
TABLE 22 · Continued
Configurations CoefficiEnts Configurations Coefficients
81 7 7 9 12 0.0596867 96 2 2 8 7 o.oo83885
82 7 6 9 12 -0.0123817 97 2 2 8 6 -0.0041012
83 6 6 7 12 -0.0167616 98 2 2 7 9 -0.006lll9
84 1 1 8 1 0.0015662 99 2 8 3 1 -0.0016945
85 1 1 7 9 0.0004682 100 2 8 1 6 -0.0060030
86 1 1 6 9 -0.0064853 101 2 1 8 6 0.0008792
87 1 2 8 7 -0.0052707 102 2 8 7 9 0.0023491
88 1 2 8 6 0.0132673 103 2 8 6 9 0.0047193
89 1 2 8 9 -0.0040388 104 8 8 2 1 0.0017353
90 1 2 1 6 -0.0193198 105 8 8 1 6 0.0029995
91 1 1 2 6 -0.0007360 106 3 3 6 9 -0.0010520
92 1 2 1 9 -0.0031956 107 4 5 8 9 0.0032591
93 1 2 6 9 0.0331329 108 4 5 1 9 -0.0039030
94 2 2 1 7 0.0016021 109 1 1 1 8 0.0008129
95 2 2 1 6 -0.0041519 llO 1 7 2 8 -0.0000551 \0 ~

TABLE 22 Continued
Configurations Coefficients
-lll 10 11 2 8 -0.0006460
112 7 7 2 9 0.0011563
113 2 7 6 9 -0.0006758
114 8 7 6 9 -0.0003369
115 12 12 2 8 -0.0012231
116 1 2 12 13 0.0282231
117 2 2 12 13 0.0103671
118 2 8 12 13 0.00lll39
119 14 15 2 8 -0.0002199
120 12 12 1 9 0.0010385
121 12 12 2 7 0.0010888
122 12 13 2 7 0.0002033
123 12 2 13 7 0.0004135
124 12 13 2 6 -0.0009174
12.5 12 2 13 6 -0.0016738 "° 0)

Configurations
1 l l 10 16
2 l l 9 16
3 l 1 ll 16
4 1 l 12 14
5 1 l 7 14
6 1 l 7 15
7 1 2 10 16
8 1 2 9 16
9 1 2 ll 16
10 1 2 12 14
ll 1 12 2 14
12 1 2 12 15
TABLE 23
CONFIGURATIONS AND COEFFICIENTS OF THE WAVEFUNCTION
USED FOR THE LOWEST JTT STATE OF LITHIUM HYDRIDE
Coefficients Configurations
0.0061788 13 l 2 6 16
-O.Oll8624 14 1 2 7 14
-0.0054767 15 1 2 7 15
0.0059459 16 1 10 12 14
0.1595967 17 l 10 7 14
-0.0065657 18 1 7 10 14
-0.0369713 19 2 2 1 16
o.o605217 20 2 2 10 16
0.0142526 21 2 2 9 16
-0.0346271 22 2 2 4 14
-0.0008593 23 2 2 11 16
-0.0155563 24 2 2 12 14
Coefficients
0.0063277
-0.8323309
0.0481456
0.0029845
0.1895050
0.0019983
-0.0089587
-0.0439125
0.0613864
0.0009688
0.0027119
0.0103988
TABLE 23 Continued
Configurations Coefficients Configurations Coefficients
25 2 2 12 15 -0.0163285 39 2 15 7 10 -0.0012665
26 2 2 6 16 0.0171882 40 2 3 12 14 0.0015893
27 2 2 7 14 -0.1464073 41 2 3 7 15 -0.0016861
28 2 2 7 15 0.0328826 42 2 4 7 17 0.0010527
29 2 10 4 14 -0.0076909 43 10 10 12 14 0.0279999
30 2 . 4 10 14 0.0048835 44 10 10 7 14 0.3746655
31 2 10 12 14 -0.0423977 45 10 10 7 15 -0.01J6ll9
32 2 12 10 14 0.0112690 46 3 3 12 14 0.0011326
33 2 14 12 10 0.000.5468 47 3 3 7 14 0.0218374
34 2 10 12 15 0.0016757 48 4 5 12 14 0.0014508
35 2 10 7 14 -0.5795247 49 4 5 7 14 0.0307450
36 2 7 10 14 -0.0117930 50 1 9 12 14 -0.0042930
37 2 14 7 10 -0.0001208 51 1 9 7 14 -0.0710728
38 2 10 7 15 0.0107678 52 1 14 7 9 -0.0001122 I-'
8

TABLE 23 Contirmed
Configurations Coefficients Configurations Coefficients
53 2 9 4 14 0.0025038 67 2 15 7 ll 0.0003880
54 2 9 12 14 0.0146437 68 2 6 7 14 0.0000763
55 2 12 9 14 o.oo458h3 69 9 9 12 14 0.0046537
56 2 14 12 9 -0.0003719 70 9 9 7 14 0.0851890
51 2 9 7 14 0.2340378 71 9 9 7 15 -0.0048898
58 2 9 7 15 -0.0064742 72 1 1 10 7 -0.0026846
59 2 15 7 9 0.0010986 73 1 1 10 12 0.0028626
60 10 9 12 14 -0.0213323 74 1 1 9 7 -0.0010018
61 10 9 7 14 -0.3333986 15 1 1 j 7 -0.0013238
62 10 9 7 15 0.0149061 76 l 1 6 7 0.0006009
63 1 11 7 15 -0.0020402 77 1 2 10 7 0.0242567
64 2 ll 12 14 -0.0004055 78 1 10 2 7 0.0014727
65 2 ll 7 14 O.Ooo6153 79 1 2 10 12 -0.0241227
66 2 14 7 ll -0.0000393 80 1 2 9 7 -0.0080595 b .....

TABLE 23 Continued
Configuration Coefficients Configurations Coefficients
81 1 9 2 7 -0.0012002 96 2 2 ll 12 -0.0072878
82 1 2 9 12 0.0195243 97 2 2 6 7 -0.0232298
83 1 2 3 7 0.0009144 98 2 10 9 7 0.003JJ.hO
84 1 2 11 7 0.0286169 99 2 9 10 7 -0.0001625
85 1 2 12 6 -0.0047011 100 2 10 9 12 -0.0015013
86 1 2 6 7 -0.0212063 101 2 9 10 12 -0.0002022
87 1 10 9 7 -0.0022711 102 2 10 11 7 0.0165882
88 2 2 1 7 0.0126810 103 10 10 1 7 -0.0006116
89 2 2 1 12 -0.0062956 104 10 10 2 7 -0 .. 0022434
90 2 2 10 7 O.Ol.49644 105 10 10 ll 7 -0.0073127
91 2 2 10 12 -0.0126451 106 10 3 9 7 0.00010n
92 2 2 9 7 -0.0164796 107 3 3 9 7 -0.0010065
93 2 2 9 12 0.0154993 108 3 3 ll 7 -0.0023520
94 2 2 3 7 0.0018880 109 3 3 6 7 0.0013327 t-'
95 2 2 11 7 0.0279527 110 4 5 2 7 0.0011556 2

TABLE 23 Continued
Configurations Coefficients
lll 4 5 9 7 -0.0019220
112 4 5 11 7 -0.0034282
113 4 5 6 ·7 -0.0018139
114 9 9 2 12 -0.0004988
115 9 9 10 7 0.0013125
116 2 9 ll 7 -0.0060879
117 10 9 11 7 0.0039263
118 1 2 14 16 0.0205018
119 1 2 15 16 0.0012693
120 14 14 1 7 -0.0006583
121 14 14 2 7 -0.0000050
122 14 1 15 7 -0.0005948
123 14 15 2 7 -0.0000157
124 14 15 -0.0000869 ...,
2 12 a 125 16 17 2 7 0.0002066
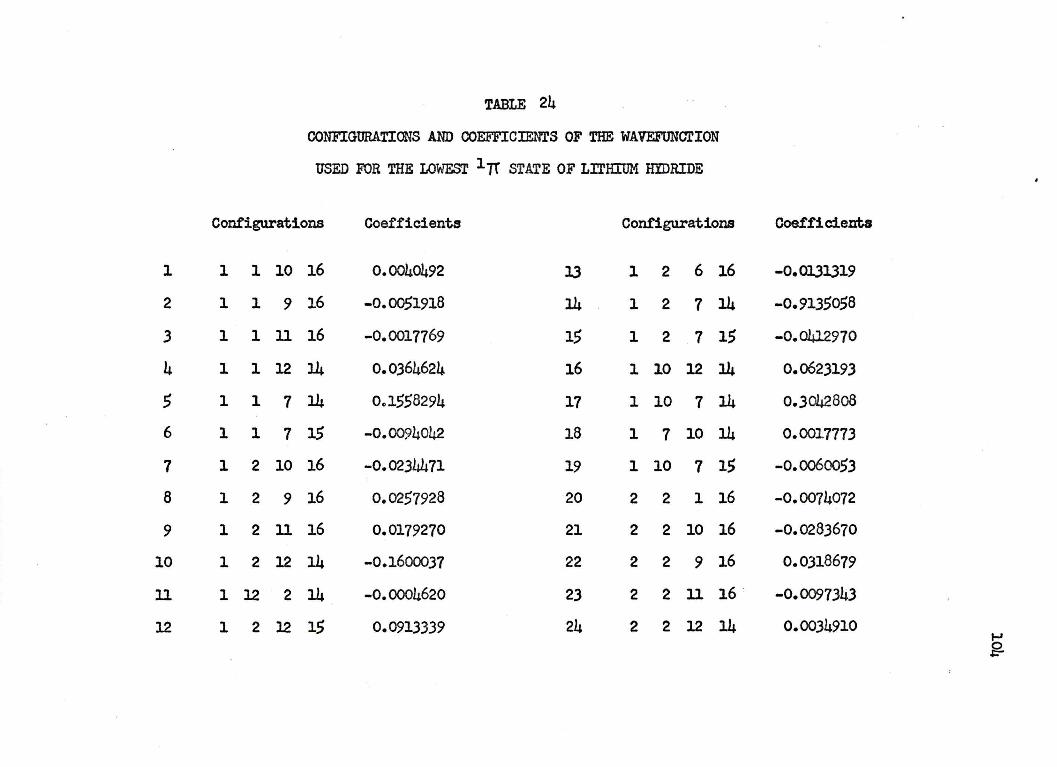
1
2
3
4
5
6
7
8
9
10
11
12
TABLE 24
CONFIGURATIONS AND COEFFICIENTS OF THE WAVEFUNGI'ION
USED FOR THE LOWEST l7t STATE OF LrrHIUM HIDRIDE
Configurations Coefficients Configurations
l 1 10 16 0.0040492 l3 1 2 6 16
l 1 9 16 -0.0051918 14 1 2 7 14
1 l 11 16 -0.0017769 15 1 2 7 15
1 1 12 14 0.0364624 16 1 10 12 14
1 1 7 14 0~1558294 17 1 10 7 14
1 1 7 15 -0.0094042 18 1 7 10 14
l 2 10 16 -0.0234471 19 1 10 7 15
1 2 9 16 0.0257928 20 2 2 1 16
1 2 11 16 0.0179270 21 2 2 10 16
1 2 12 14 -0.1600037 22 2 2 9 16
1 12 2 14 -0.0004620 23 2 2 11 16
1 2 12 15 0.0913339 24 2 2 12 14
Coef'ficie.ata
-0.0131319
-0.9135058
-0.0412970
o.o623193
0.3042808
0.0017773
-0.0060053
-0.0074072
-0.0283670
0.0318679
-O.CXYJ7343
0.0034910 ..., g.
TABLE 24 Contirmed
Configurations Coefficients Contiguratioll!I Coefficients
25 2 2 l?. 15 0.1244982 39 2 3 12 l4 0.0000136
26 2 2 6 16 0.0151948 40 2 3 7 15 -0.0017421
27 2 2 7 14 0.2236998 41 2 4 7 17 0.0009942
28 2 2 7 15 -0.1200208 42 10 10 9 16 -o.oono65
29 2 10 9 16 0.0016453 43 10 10 12 14 0.1507784
30 2 10 4 14 -0.0043773 44 10 10 12 15 -0.0184809
31 2 4 10 14 0.0062023 45 10 10 7 14 0.7069831
32 2 10 12 14 -0.2199755 46 3 3 12 14 0.0044805
33 2 12 10 14 0.0029467 47 3 3 1 14 0.0187947
34 2 10 12 15 0.0101693 48 4 5 12 14 0.0067267
35 2 10 7 14 -1.1953680 49 4 5 7 14 0.0259702
36 2 7 10 14 -0.0114888 50 9 9 2 16 -0.0002597
37 2 10 7 15 0.0256709 51 1 9 12 14 . -0.0082)80
38 2 7 10 15 0.0009817 52 1 9 7 14 -0.1181352 ~
5l
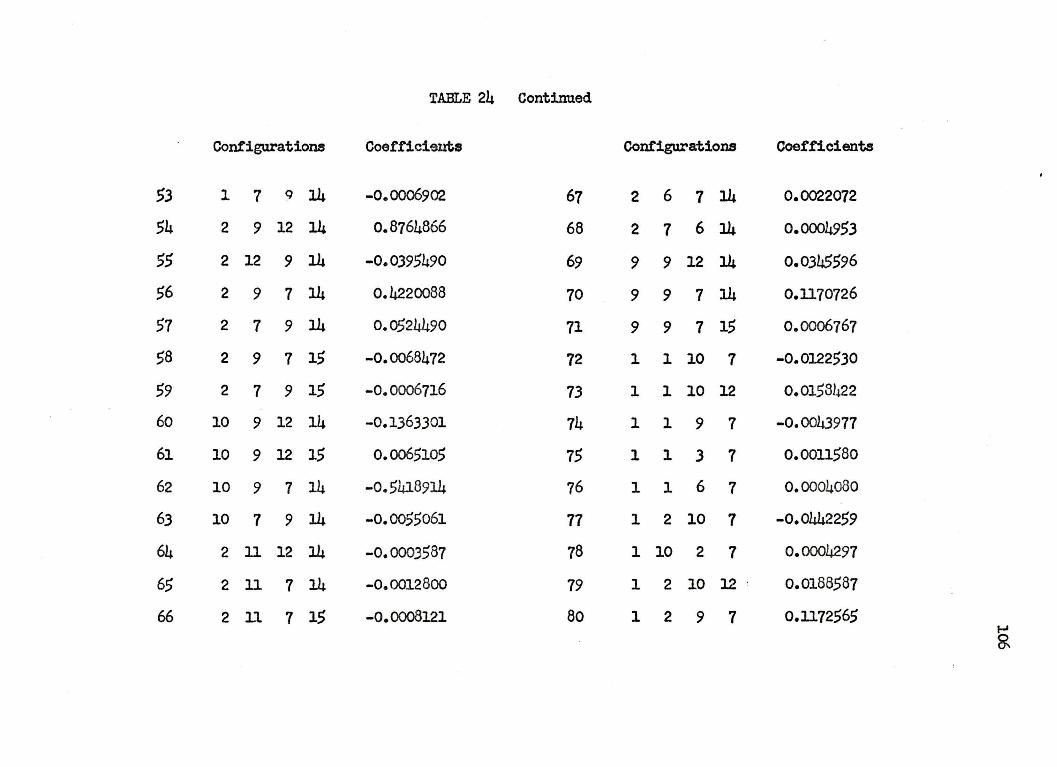
TABLE 24 Continued
Configurations Coefficients Conf'igurations Coefficients
53 1 7 Q 14 -0.0006902 67 2 6 7 14 0.0022072
54 2 9 12 14 o.8764866 68 2 7 6 14 0.0004953
55 2 12 9 14 -0.0395490 69 9 9 12 14 0.0345596
56 2 9 7 14 0.4220088 70 9 9 7 14 O.ll70726
51 2 7 9 14 0.0524490 71 9 9 7 15 o.ooo6767
58 2 9 7 15 -0.0068472 72 1 1 10 7 -0.0122530
59 2 7 9 15 -0.0006716 73 1 1 10 12 O.Ol58li22
60 10 9 12 14 -0.1363301 74 1 1 9 7 -0.0043977
61 10 9 12 15 o.oo65105 75 1 1 3 7 0.0011580
62 10 9 7 14 -0.5418914 76 1 1 6 7 0.0004080
63 10 7 9 14 -0.0055061 77 1 2 10 7 -0.0442259
64 2 11 12 14 -0.0003587 78 1 10 2 1 0.0004297
65 2 11 7 14 -0.0012800 79 1 2 10 12 . 0.0188587
66 2 ll 7 15 -0.0008121 80 1 2 9 7 0.1112565 t-' ~
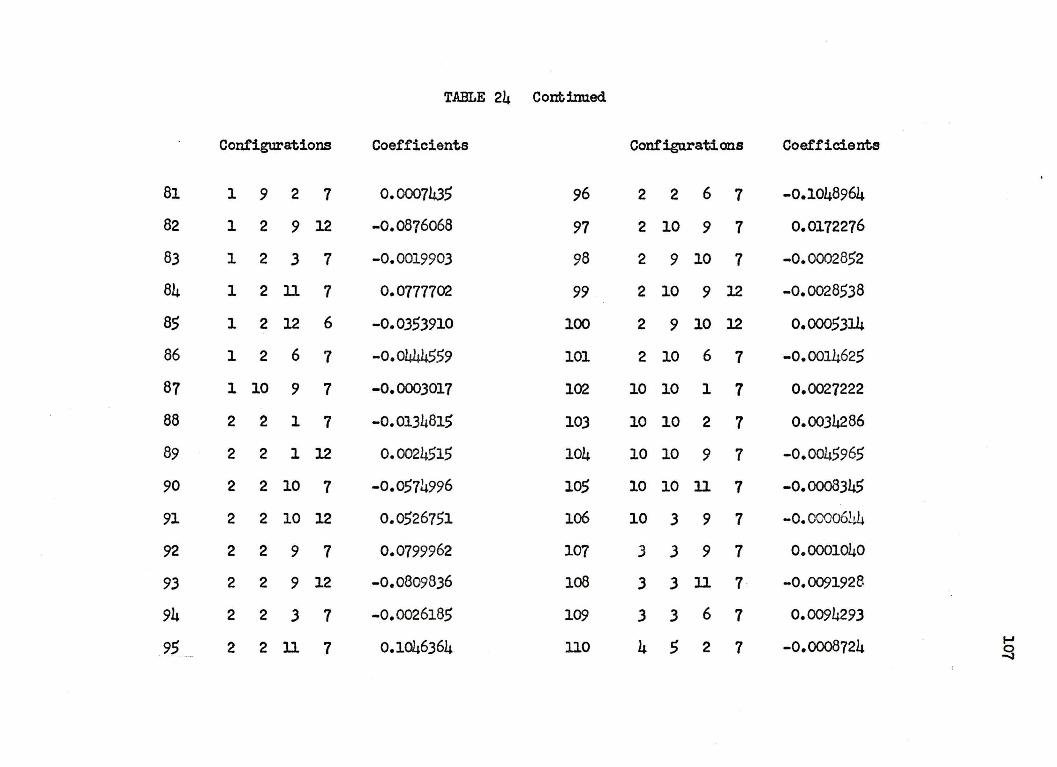
TABLE 24 Continued
Configurations Coefficients Configurations Coefficients
81 1 9 2 7 0.0007435 96 2 2 6 7 -0.1048964
82 1 2 9 12 -0.0876068 91 2 10 9 7 0.0172276
83 1 2 3 7 -0.0019903 98 2 9 10 7 -0.0002852
84 1 2 11 7 0.07777(12. 99 2 10 9 12 -0.0028538
85 1 2 12 6 -0.03.53910 100 2 9 10 12 0.0005314
86 1 2 6 7 -0.0444559 101 2 10 6 7 -0. OOJ.4625
87 1 10 9 7 -0.0003017 1(12. 10 10 1 7 0.0027222
88 2 2 1 7 -0.0134815 103 10 10 2 7 0.0034286
89 2 2 1 12 0.00245'15 104 10 10 9 7 -0.0045965
90 2 2 10 7 -0.0574996 10, 10 10 11 7 -0.0008345
91 2 2 10 12 0.0526751 106 10 3 9 7 -o.oooo6Li.4
92 2 2 9 7 0.0799962 107 3 3 9 7 0.0001040
93 2 2 9 12 -0.0809836 lo8 3 3 11 7 -0.0091928
94 2 2 3 7 -0.0026185 109 3 3 6 7 0.0094293
95 2 2 11 7 0.1046364 110 4 5 2 7 -0.0008724 I-' ~
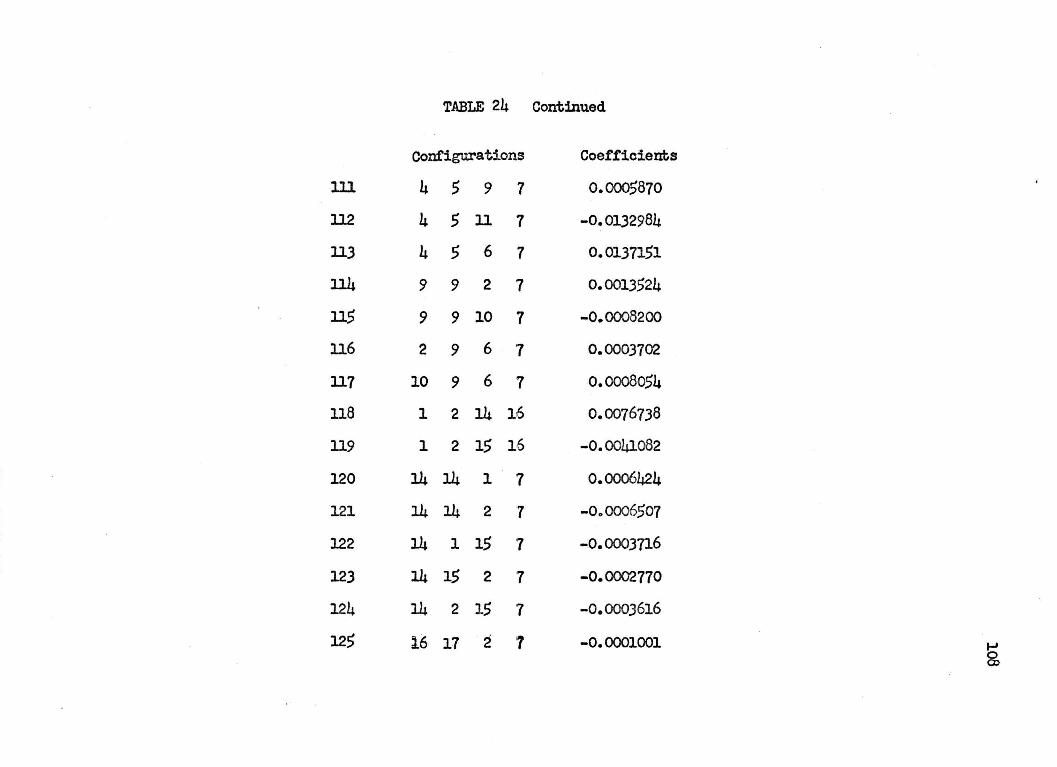
TABLE 24 Continued
Configurations Coefficients
111 4 5 9 7 0.000,870
l12 4 5 11 7 -0.0132984
ll3 4 5 6 7 0.0137151
114 9 9 2 1 0.0013524
115 9 9 10 1 -0.0008200
116 2 9 6 1 0.0003702
117 10 9 6 7 0.0008054
118 1 2 14 16 0.0076738
119 1 2 15 16 -0.004lo82
120 14 14 1 7 0.0006424
121 14 14 2 ·7 -000006507
122 14 1 15 7 -0.0003716
123 14 15 2 7 -0.0002770
124 14 2 15 1 -0.0003616
125 16 17 2 1 -0.0001001 .... 8

APPENDIX II
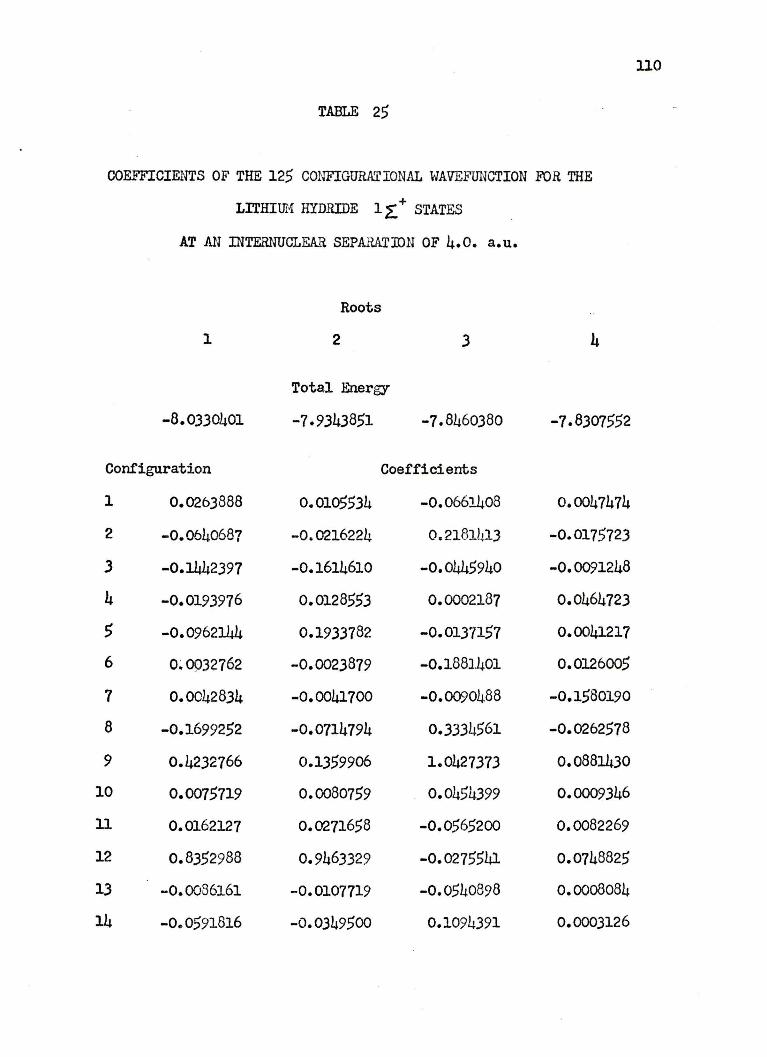
TABLE 2.5
COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVEFUNCTION FOR THE
LrrHIU1'1 H'IDRIDE 1 ~ + STATES
AT AN INTERNUCLEAR SEPAHATJDN OF 4.0. a.u.
Roots
1 2 3 4
Total Energy
-8.0330401 -7-9343851 -7.8460380 -7.8307552
Configuration Coefficients
1 0.0263888 0.010.5534 -0.0661408 0.0047474
2 -0.0640687 -0.0216224 0.218lhl3 -0.0175723
3 -0.1442397 -0.1614610 -0.0445940 -0.0091248
4 -0.0193976 0.0128553 0.0002187 0.0464723
5 -0.0962144 0.1933782 -0.0137157 0.0041217
6 0~0032762 -0.0023879 -0.1881.401 0.0126005
7 0.0042834 -0.0041700 -0.0090488 -0.1580190
8 -0.1699252 -0.0714794 0.3334561 -0.0262578
9 0.4232766 0.1359906 1.0427373 0.0881430
10 0.0075719 0.0080759 0.0454399 0.0009346
ll 0.0162127 0.0271658 -0.0565200 0.0082269
12 0.8352988 0.9463329 -0.0275541 0.0748825
13 -0.0036161 -0.0107719 -0.0.540898 0.0008084
14 -0.0591816 -0.0349500 0.1094391 0.0003126
llO
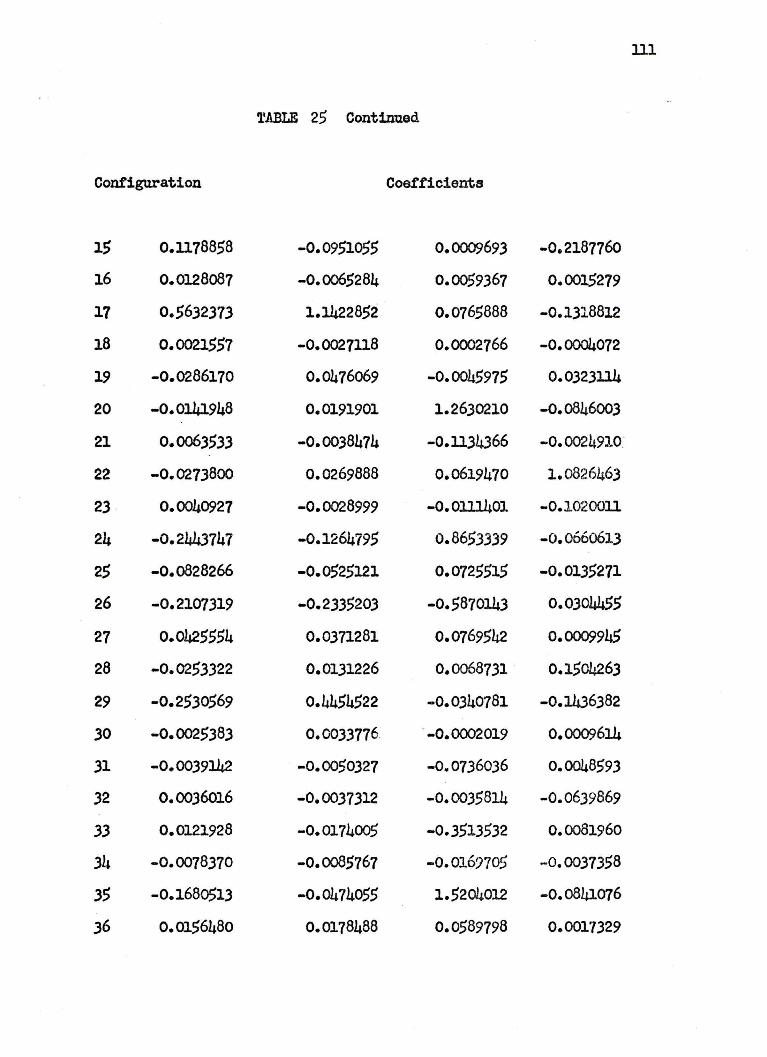
lll
1'AJ3LE 25 Continued
Configuration Coefficients
15 0.1178858 -0.0951055 0.0009693 -0.2187760
16 0.0128087 -0.0065284 0.0059367 0.0015279
17 0.$632373 1.1422852 0.0165888 -0.1318812
18 0.0021557 -0.0027ll8 0.0(X)2.766 -0.0004072
19 -0.0286170 0.0476069 -0.0045915 0.0323114
20 -0.0141948 0.0191901 1.2630210 -o.0846003
21 o.0063533 -0.0038474 -0.1134366 -0. 0024910.
22 -0.0273800 0.0269888 0.0619470 1.0826463
23 o.0040927 -0.0028999 -O.Ol.11401 -0.10-ioon
24 -0.2443747 -0.1264795 o.8653339 -0.0660613
25 -0.0828266 -0.0525121 0.0125515 -0.013,271
26 -0.2107319 -0.2335203 -0.5870143 0.030445,
27 0.0425.554 0.0371281 0.0769542 0.0009945
28 -0.0253322 0.0131226 0.0068731 · 0.1$'04263
29 -0.2530569 0.4454522 -0.0340781 -0.1436382
30 -0.0025383 0.0033776. --0.0(X)2.019 0.0009614
31 -0.0039142 · -o. 00.50327 -0.0736036 o.0048593
32 0.0036016 -0.0037312 -0.0035814 -0.0639869
33 0.0121928 -0.0174005 -0.3513532 0.0081960
34 -0.0078370 -0.0085767 -0.0169705 -0.0037358
35 -0.1680513 -0.0474055 1.5204012 -0.0841076
36 0.0156480 0.0178488 0.0589798 0.0017329
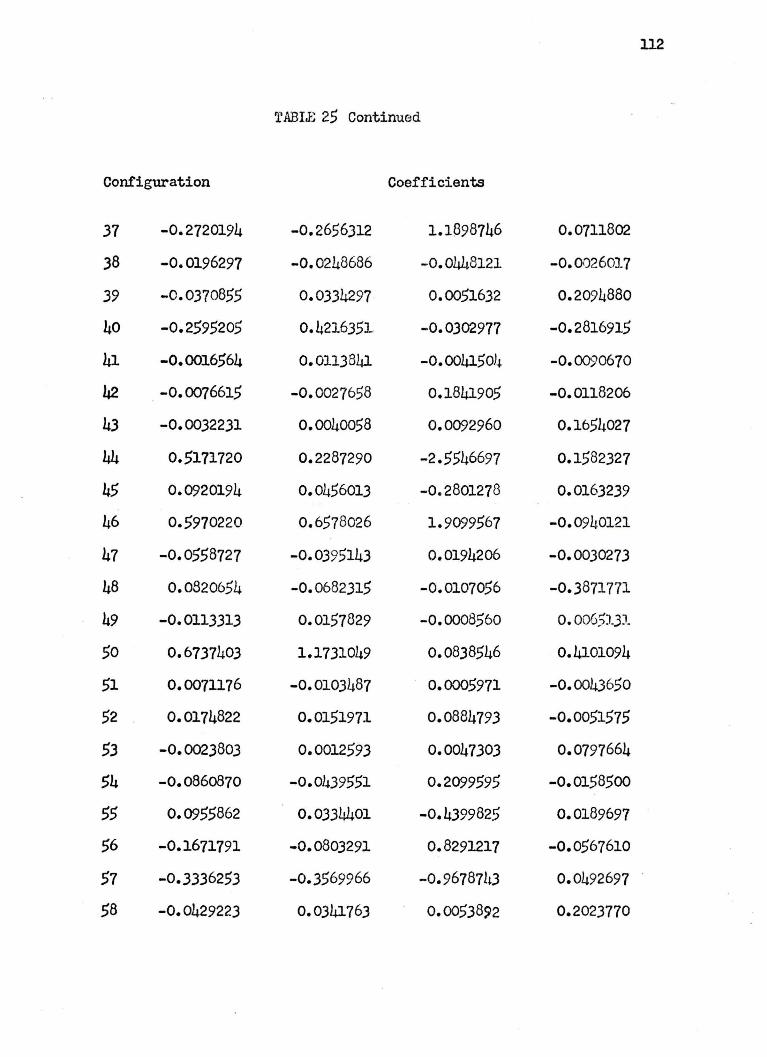
ll2
TABLE 25 Continued
Configuration Coefficients
37 -0.2720194 -0.2656312 1.1898746 0.0711802
38 -0.0196297 -0.0248686 -0.0448121 -0.0026017
39 -0.0370855 0.0334297 0.0051632 0.2094880
40 -0.2595205 0.421635:L -0.0302977 -0.2816915
41 -0.0016564 0.0113841 -0.0041504 -0.0090670
42 -0.0076615 -0.0027658 0.1841905 -0.0118206
43 -0.0032231 0.0040058 0.0092960 0.1654027
44 0.5171720 0.2287290 -2.5546697 0.1582327
45 0.0920194 0.0456013 -0.2801278 0.0163239
46 0.5970220 0.6578026 1.9099567 -0.0940121
47 -0.0558727 -0.0395143 0.0194206 -0.0030273
48 0.0820654 -0.0682315 -0.0107056 -0.3871771
49 -0.0113313 0.0157829 -0.0008560 0.0065131
50 0.6737403 1.1731049 0.0838546 0.4101094
51 0.0071176 -0.0103487 0.0005971 -0.0043650
52 0.0174822 0.0151971 0.0884793 -0.0051575
53 -0.0023803 0.0012593 0.0047303 0.0797664
54 -0.086o870 -0.0439551 0.2099595 -0.0158500
55 0.0955862 0.0334401 -0.4399825 0.0189697
56 -0.1671791 -0.0803291 0.8291217 -0.0567610
57 -0.3336253 -0.3569966 -0.96787h3 0.0492697
58 -0.0429223 0.0341763 0.005)892 0.2023770
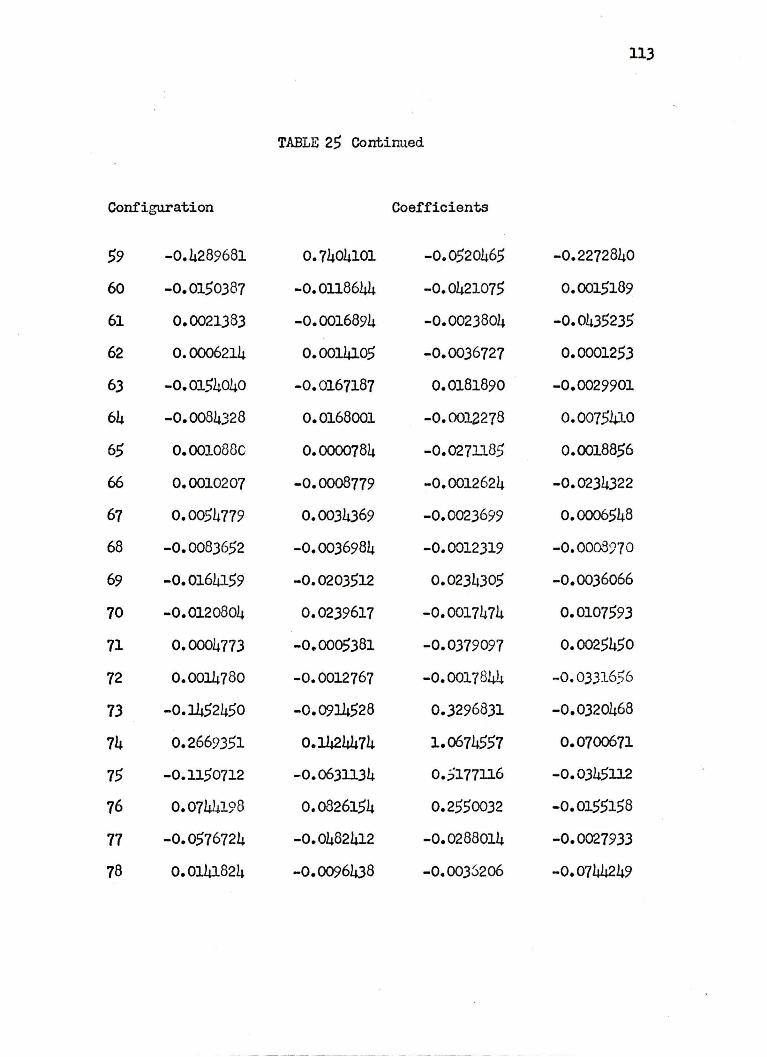
113
TABLE 25 Contirmed
Configuration Coefficients
59 -0.4289681 0.7404101 -0.0520465 -0.2272840
60 -0.0150387 -O.Oll8644 -0.0421075 0.0015189
61 0.0021383 -0.0016894 -0.0023804 -0.0435235
62 0.0006214 0.0014105 -0.0036727 0.0001253
63 -0.0154040 -0.0167187 0.0181890 -0.0029901
64 -0.0084328 0.0168001 -0.0012278 0.0075410
65 0.0010880 0.0000784 -0.0271185 0.0018856
66 0.0010207 -0.0008779 -0.0012624 -0.0234322
67 0.0054779 0.0034369 -0.0023699 0.0006548
68 -0.0083652 -0.0036984 -0.0012319 -o.ooos:no
69 -0.0164159 -0.0203512 0.0234305 -0.0036066
70 -0.0120804 0.0239617 -0.0017474 0.0107593
71 0.0004773 -0.0005381 -0.0379097 0.0025450
72 0.0014780 -0.0012767 -0.0017844 -0.03.31656
73 -0.1452450 -0.0914528 0.3296831 -0.0320468
74 0.2669351 0.1.h24474 1.0674557 0.0700671
75 -O.ll,50712 -0.0631134 0.5177116 -0.034.5112
76 0.0744198 o.o826154 0.2550032 -0.0155158
77 -0.0576724 -0.0482412 -0.0288014 -0.0027933
78 O.OJ.41824 -0.0096438 -0.0035206 -o.07w_a49

114
'!'ABLE 25 Continued
79 0.1402528 -0.2426648 0.0190643 o.0869297
80 -0.2531644 -0.2761684 -0.8604312 0.0422363
81 0.1000523 0.0720312 -0.1413969 0.0102632
82 -0.0341895 0.0281542 0.0050032 0.1768521
83 -0.3944873 0.6774129 -0.0476278 -0.2199378
84 0.3104043 0.3243362 o.8940498 -0.0041948
85 -0.0378873 -0.02682o6 0.0916156 -0.0054065
86 0.0380790 -0.0279030 -0.0052609 -0.1943496
87 0.5538764 -0.9446554 o.0652214 0.2563430
88 0.0058514 0.0045586 0.0123723 -0.0001995
89 -0.0008554 0.0006491 0.0007363 o.ol.44068
90 -0.0233746 -0.0252594 -0.0873642 0.0027054
91 0.01308n 0.0305485 0.0844360 -0.0021480
92 -0.0294641 0.0485515 -0.0032622 -0.0048623
93 0.0879222 -0.1458636 0.0089379 0.0161453
94 -0.1162156 0.1924423 -0.0123160 -0.0208725
95 -0.0993821 -0.0950310 -0.1639708 0.0059739
96 -0.0098808 0.0061391 0.001.3744 0.0516542
97 -0.2039225 0.3446249 -0.0235170 -0.0800938
98 o.0895933 -0.1476303 0.0098885 0.0158876
99 -0.0168966 -0.0152966 .:..0.0213074 0.0000512
100 -O.Ol0685Q 0.0174745 -0.0013420 -0.0018109
101 0.0319674 0.0369218 0.0079342 0.00ll966
102 -0.0669328 -0.1005279 -0.0325759 -0.0050891
103 0.0214392 0.0772908 0.0124342 0.0069015

ll5
TABLE 25 Continued
Configuration Coefficients
104 0.0251298 0.0170822 0.0498680 -0.0000798
105 0.0025056 0.0251599 -0.0023091 0.0106974
106 -0.0428306 -0.0273083 -0.0313395 0.0046043
107 -0.0259258 0.0073324 0.0090460 0.0013069
108 -0.0478330 -0.0389176 0.0220485 -0.0000720
109 0.1039194 o.0841579 -0.0338383 -0.0025580
no -0.0623721 -0.0501604 0.0165524 0.0039244
lll -0.0312008 0.0175334 0.0056904 0.0108303
ll2 0.0054775 -0.0042877 0.0014473 0.0008919
ll3 0.0094624 -0.0036871 0.0000669 -0.0009237
ll4 0.2173553 -0.1212672 -0.0340561 -0.0159013
ll5 0.0199116 -0.0159525 -0.0057344 -0.0059246
ll6 -0.0534346 0.0315159 0.0036232 0.0135260
ll7 -0.0801366 0.0306611 -0.0003185 0.0077100
ll8 -0.0068136 0.0038718 0.0011238 0.0023523
ll9 0.0503284 -0.0286807 -0.0062468 -0.0152!'14
120 -0.0125658 0.0058415 -0.0006565 -0.0044196
121 -0.0191333 0.0074094 -0. OOOJ.145 0.0018571
122 -0.0051167 0.0028848 0.0008747 0.0017882
123 0.0019283 -0.0007385 0.0000081 -0.0001856
124 -0.0071817 0.0040523 0.0012327 0.0025124
125 0.0027267 -0.001041~7 0.OOOOll5 -0.0002626

ll6
TABLE 26
COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVEFUNCTION FUR THE
LITHIUM HYDRIDE 3 i: + STATES
AT AN INTERNUCLEAR SEPARATION OF 4.0. a.u.
Roots
1 2 3 4
Total Energy
7.952601 7.851C>532 7.8395999 7.7328257
Configuration Coefficients
1 -0.0188883 -0.0238635 0.0156086 -0.0118563
2 0.1014832 0.1639664 -0.0936894 0.0751097
3 -0.1386833 -0.3142631 0.1592624 -0.1369021
4 -0.1658108 0.1220262 0.0180989 -0.0198293
5 0.0047766 -0.0059366 -0.0042980 -0.0079303
6 0.0654314 0.0579581 0.0967580 -0.2148646
7 -0.0052760 0.1599187 -0.0680951 -0.0024609
8 -0.0019201 0.0380614 O.ll48580 0.1018341
9 -0.4461087 -0.5994639 0.36298~,1 -0.3227677
10 -0.0653997 -0.0407020 0,0382542 -0.0221.7j;3
11 -0.0022650 -0.0057617 0.0006208 -0.013lll3
12 0.6913762 1.2952432 -0.6875926 0,6790544
13 0.0789606 -0.0103452 -0.0219551 0.0167984
14 0.0235055 0.0459394 -0.0232090 0.0467197

117
TABLE 26 Continued
Configuration Coefficients
15 1.0297965 -0.3525982 -0.2570159 0.1586859
16 -0.0268730 0.0460238 -0.0108595 -0.0028957
17 0.0001396 0.0000)31 -0.0001047 o.0000615
18 -0.0434202 -0.0586078 0.0357129 -0.0242401
19 -0.0167169 0.0410974 0.0385103 0.0167473
20 0.0070280 0.0037545 -0.0130089 0.0607009
21 -0.4041060 -0.3380233 -0.5396237 1.3460951
22 0.0194818 0.0197744 0.0152'225 -0.0824758
23 0.0184167 1.0886489 0.4666757 o.oll4031
24 -0.0004016 0.0818200 . -0.0269850 0.0037716
25 · 0.0157689 -0.2602573 -0.7919133 -0.?096932
26 -0.0004056 0.0227465 0.0554472 0.0501817
27 -0.4322800 -0.8291306 o.4748825 -0.2650415
28 -0.2023,01 -0.0960439 0.1072025 -0.0602598
29 -0.2361002 0.5742337 -O.l.419911 -0.0132487
30 0.0771219 -0.0759722 0.0072796 0.0133988
31 0.1362057 0.1329151 0.2605409 -0.3769584
32 -0.0092359 0.0737080 -0.0299585 -0.0043227
33 -0.0007510 0.0192300 0.0543519 0.0487986
34 -0. 0431.i759 -0.0505369 0.0301576 -0.0328217
35 0.0942709 0.3670794 -0.1917200 0.0537052
36 -0.0101882 -0.0268030 0.0188622 -0.0109235

118
TABLE 26 Continued
Configuration Coefficients
37 -0.2669473 1.3226911 0.6368131 · -0.2574004
38 0.0225475 0.0506198 -0.0352085 0.0292187
39 -0.3192135 1.0079879 -0.2978982 -0.0106865
40 -0.0245280 -0.0354731 0.0244531 -0.0115956
41 0.0106556 0.0125481 0.0128062 -0.0213964
42 0.1165173 0.1314716 0.2895814 -0.2907909
43 0.0105317 0.0078919 0.0020806 -0.0135754
44 0.0018813 -0.1155295 0.0495274 -0.0027123
45 0.0007123 0.0353580 -0.0128215 0.0022729
46 0.0039175 -0.0210006 -0.0738882 -0.0702184
47 -0.0005910 0.0098568 0.0250363 0.0174152
48 0.5858855 2.1235385 1.0697071 0.5324224
49 0.1359620 0.2418183 -0.1508315 0.0591415
50 o.8416597 -1.6928902 0.4017660 o.o680507
51 -0.0710207 -0.0124634 0.0293626 -0.0186667
52 -0.0003340 -0.0002294 0.0001513 0.0001031
53 0.0009050 -0.0005712 0.0004787 -0.0018339
54 -0. 013!~645 -0.0048595 -0.0067552 0.0298214
55 -0.3492834 -0.3493783 ·-0.6918346 0.9689382
- 56 -0.0069663 -0.1389673 0.0602410. 0.0087452
51 -o.00002L8 -0.0463147 -0.1301345 -0.111+2285
58 -0.2444397 -0.2693873 0.1903388 -0.1027564
59 0.1582,388 0.3966706 -0.2213104 0.0912642
60 -0.1530332 -0.6918924 0.3346289 -0.1782213

119
TABLE 26 Cont,inued
Configuration Coefficients
61 -0..5029422 0.8987696 -0.2030476 -0.0450674
62 0.0047435 0.0032897 0.0035214 -0.0053650
63 0.2096680 0.2105915 o.4155584 -0.5769063
64 0.0042283 0.0554785 -0.0234780 -0.0068544
65 -0.0003063 0.0230524 o.o6JJ54h 0.0572444
66 0.0040704 0.0024688 -0.0017096 0.0031563
67 -0.0053546 -0.0032962 0.0016760 -0.0080082
68 -0.0170318 -0.0080523 0$0099907 -0.0037178
69 0.0060492 0.0034021 0.0052780 -0.0222960
70 -Oc0001211 0.0229354 -Oe00983h9 0.0001.456
71 -0.0004043 0.0058854 0.0167478 0.0154588
72 0.0057940 0.0034549 -0.0024892 0.0045759
73 -0.0076164 -0.0046232 0.0024487 -0.0114406
74 -0.0240701 -0.0ll4ll0 0.0140966 -0.0052234
75 0.0086311 0.0048131 0.0071.918 -0.0317544
76 -0.0001762 000324508 -0.0139061 0.0001985
77 -o.ooor,853 0.0033362 0.0236970 0.0218958
78 -0.2548742 -0.3216469 . 0.21.59264 -0.ll79183
19 0.3209737 0.9431477 -0.5040255 0.2375647
80 -0.1174168 -0.4587775 0.2315261 -o.n78998
81 0.0893538 -0.2586416 0.0729176 0.0080ll3
82 -0.0905024 0.0371163 0.0132402 -0.0254814

120
TABLE 26 Continued
Configuration Coefficients
83 -0.0707844 -0.0729532 -0.l.486555 0.1842327
84 -0.3384973 0.8155384 -0.2107166 -0.0356566
65 0.1294057 0.1198048 -0.0932942 0.0587421
86 0.1919564 0.1969113 0.3905110 -0.5100838
87 0.0184992 0.0l.41629 -0.0091726 0.0014779
88 -0.0005763 0.0089690 0.0252314 0.0240547
89 0.488ll52 -0.8878131 0.2185625 0.0428684
90 -0.0475055 -0.0741615 0.0471670 -0.0297181
91 -0.0016260 -0.0017lll -0.0016045 -0.0000845
92 -0.2447831 -0.2524331 -0.4974478 o.6488119
93 -0.0202534 -0.0043040 0.0044533 0.0010542
94 0.0003141 -0.0105556 -0.0288159 -0.0267387
95 -0.0420157 0.0899421 -0.0256793 -0.0017617
96 0.,0489689 -0.0829952 0.0228636 0.0004288
91 o.ol142o8 0.0l.15997 0.0239820 -0.0300282
98 -0.0328028 -0.0357330 -0.0700373 o.0869514
99 0.0370189 0.0403762 0.0781753 -0.0975609
100 -0.0027505 0.0025.318 -0.0005101 -0.0010022
101 -0.1537779 0.1865492 -0.0338830 -0.0262342
102 0.0776372 0.0812263 0.1597197 -0.1990745
103 0.0113541 -0.0134886 o.003s,65B 0.0018216
104 -0.0204079 -0.0218773 -0.0423461 o.0528495

121
TABLE 26 Continued
Configuration Cosf'!ic:ta11ts
105 -0.0268257 0.0225828 -0.0038689 -0.0030218
106 -0.0032190 0.0209311 -0.0070321 -0.0014058
107 0.0273925 -0.0427780 0.0058105 0.0070917
lo8 -0.0023442 -0.0035371 o. 0156.3.39 0.0139271,
109 -0. 0063363 0.0079596 -0.0412400 0.0116071
llO -0. 0286809 0.0296342 Oe0027750 -0.0071483
lll -0.0135063 0.0523717 -0.0445o87 -0.0369210
ll.2 0.0191916 -0.0291707 0.0832890 -0.0115361
113 o.o4ho463 -0.0574882 0.0303572 0.0169069
114 -0.0407853 0.0268170 -0.0484383 o.0085483
us 0. 0033674 -0.0218952 0.0074708 0 .. 0009116
ll.6 -0.0106151 0.0378395 -0.0140400 -0.0041947
ll.7 0.0018097 -0.05896ol 0.0254788 -0.0090470
ll.8 0. 0025514 -0.0154420 0.0091145 0.0020617
119 -0.0086301 0.09878.5'7 -0.0501154 0.0265002
120 0.0048813 -o.0066457 0.0190721 -0.0158965
121 0.0244203 -0.0521809 0.0243036 -000095366
122 -0.0191543 0.0100054 -0.0164916 0.0096650
123 -0.0033213 -0.0048134 0.0027629 -O.OJ.17418
124 0.0202976 0.0178858 -0.0348615 0.1146504
125 0.0051790 0.0011304 -0.0097162 0.02$4963

122
TABLE 27
COEFFICIENTS OF THE 125 CONFIGURATION.AL WAVEFUNCTION FOR THE
Ll'THIOM HYDRIDE 3 TT STATES .
AT AN INTEIDTaCLEAR SEPARt.TION OF 4.0 a.u.
Roots
1 2
Total Energy
-7.9069171 -7.8297676
Configuration Coef.f'icients
1 0. 0010978 -0. 00020.59
2 0. 0006931 0.0024772
3 0.0042.542 -0.0005693
4 0.0034468 0.0007850
5 -0.005.5032 -0.0072491
6 -Oe 0022785 -0.0012145
7 0. 0011400 0.000Cf967
8 -0.2414072 -O. lll9169
9 0.0120634 0. 0041324
10 -0.0013024 0.2759344
11· 0.0005786 -0.0123570
12 -0.0016672 -0.0015985
13 0. 0056980 0.0040J.u2
14 0.0110891 -O.Oll6712
15 -0.0382156 -0.0089243

123
TABLE 27 Continued
Contiguration Coefficients
16 -0.0089841 -0.0069345
17 0.0467373 0.0489718
18 -0.0013825 0.0001332
19 0.0162788 0.0195249
20 -0.0225342 -0.0023090
21 1.4238663 0. 6628978
22 0~0030551 0.0008921
23 0. 0000570 -0.0000166
24 -0.0726042 -0.0452362
25 0.0009940 0.0089503
26 0.0023160 0.0026671
27 O.Oll5418 1.6431907
28 -0.0043030 0.0978935
29 -0.0030551 -O.OD.6226
JO -0.5334562 -0.2468175
31 -0.0031479 -0.0013465
32 0.0057240 -0.0009941
33 0.0002013 0.5934870
34 O. OOJ.4053 0.0005589
35 0.0043619 0.0022922
36 0.0107419 0. 0091400
37 -0.0131879 -0.0150671

124
TABLE 27 Continued
Configuration Coef'ficients
38 0.0062193 0.0046719
39 -0.0007094 -0.0022725
40 -0.0427973 -0.0291748
41 0.0077826 0.0038297
42 -0.0060838 0.0002544
43 -0~4855324 -0.2222624
44 0.0007899 -0.0148274
45 -0.0054254 o.5594263
46 -0.0008711 0.5594263
47 0.001348.3 0.0006291
48 -0.0034919 -0.0014828
49 0.078!'495 0.0593421
50 -0.0125614 -o.0112c62
51 -0.0013367 -0 .. 0009346
52 1.3812234 0.6399731
53 0.0106897 0.0079563
54 0.0002524 -0.0000022
55 -0.0225680 0.00,2080
56 0.0110525 l.58603.52
57 -0.0025637 -0.0012403
58 -0.0000944 -0.0016490
59 -O.Oh5ll83 -0.0367.517
60 -0.0451183 -0.4000985

125
TABLE 27 Continued
Configuration Coe£ r i cierrt s
61 0.0115243 -0.0016486
62 -0.0060140 0.9903097
63 -0.0008685 -0.00082,36
64 -000205333 -0.0094620
65 0.0016371 -0.0002531
66 -0.0001419 0.0234624
67 -0.0010383 -0.0010672
68 -o. 0290950 -o.0134lo6
69 0.0022211 0.0010612
70 -0.0001755 0.0332170
71 0.0002899 -0.0032615
72 0.0060170 0.0091330
73 0.288fi850 0.1333896
74 0.0003760 -0.3248344
15 -0.0427492 -0.0308346
76 -0.0035701 0. 0028378
77 -0.7870071 -0.3665858
78 0.0014254 -0.0018704
19 0.0083046 -0.0014467
80 -0.0058083 0.9041769
81 0. 0487410 0.0407477
82 1.0922392 0.5078604

126
TABLE 27 Continued
Configuration Coefficients
8.3 -0.007672.3 0. 001094.3
84 o. 0058020 1.2471032
85 -0.000600.3 -0.0022781
86 -0.0560998 -0. 0255159
87 0.0000513 0.0626708
88 0.00793.39 0.00,0295
89 0.1637212 0.077051.3
90 -0.0019826 0.0003942
91 0. 0009635 -0.1876196
92 -0.0079144 -o.oo62189
93 -0. 2177282 -0.1020878
94 0.0017140 -0. 0002613
95 -0.0006081 0.2470734
96 -O.Ol.42043 -0.012.3070
91 -0.3968174 -0.185.399.3
98 -0.0013120 0. 4508328
99 0. 0039067 0.0032.39.3
100 0.1685354 0.0795715
101 -0.0000454 · -0.1897496
102 -0. 020.3231 -0.0097407
103 0.0001301 0. 0225071
104 -0.0096376 0.0253684
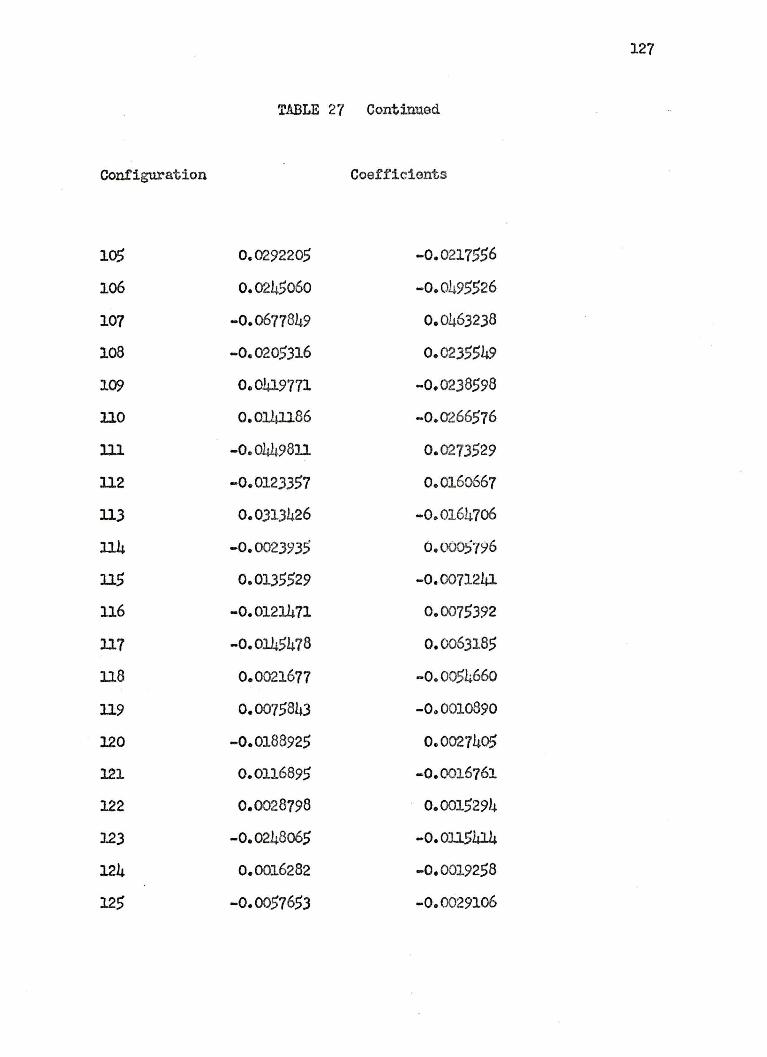
127
TABLE 27 Continued
Configuration Coefficients
105 0.0292205 -0.0217556
106 0. 0245060 -0.0495526
107 -0.0677849 0. 0463238
108 -0.0205316 0.0235549
109 0. 0419771 -0.0238598
110 0.0141186 -o.~66576
111 -O.Olw.9811 0.0273529
112 -0. 0123357 0.0160667
113 0. 0313426 -0.0164706
114 -0. 0023935 0. 000!?/96
115 0.0135529 -0. 0071241
116 -0. 0121471 0.0075392
117 -O.Ol.45478 0. 0063185
118 0.0021677 -0.0054660
119 0. 0075843 -0.0010890
120 -0.0188925 0.0027405
121 0.0116895 - 0. 0016761
122 0.0028798 0.001529h
123 -0.0248o65 -0. 0115414
124 0.0016282 -0. 0019258
125 -0. 0057653 -0.0029106

TABLE 28
COEFFICIENTS OF THE 125 CONFIGURATIONAL WAVEFUNCTION FOR THE
LITIIImi HYDRIDE l 7T STATES
AT AN INTERNUCLEAR SEPARATION OF 4. 0 a. u.
Roots
l 2
Total Energy
-7.8985744 -7.8290914
Configuration Coefficients
1 0.0003561 0.0022806
2 -0.0030564 -0.00~2553
3 0.0048011 0. 0015619
4 -0.0034500 -0.0002969
5 -0.0017996 0.0042715
6 -0.0039859 0.0018960
7 0.0038018 -0.0000164
8 -0.2462767 0.1061540
9 0.0120944 -0.0047746
10 -0.0076734 -0.2348258
11 0. 0009182 0.0122439
12 -0.0024864 0.0023799
13 -0.0012830 0.0009483
14 -0.0020385 -0.0091855
128

129
TABLE 28 Continued
Configuration Coeff'icienta
16 -0. 0355481 0. 0037679
17 0.0120217 -0.0010556
18 0.0352355 -0.0374471
19 0.0023lll -0.0016476
20 0. 0198814 -0.0211507
21 -0. 0179257 0.0022072
22 1.4568477 -o.6465U8
23 -0.00ll336 0.0000515
24 - 0.0678790 0.0483374
25 0.0031839 -0.0068254
26 0. 0031112 -0.0020307
27 0. 0486181 1.J.i..398704
28 -0.00770!µ. -0. 0982802
29 0.0053606 0.0002091
30 - 0.5350676 0.1722202
31 -0.0003669 0.0016825
32 0.00154o6 0. 0005519
33 -0. 0105330 . -0.3607801
34 0.0001354 -0.0017218
35 0.0046123 -0.0025749
36 0.0016487 -O.OOll.478
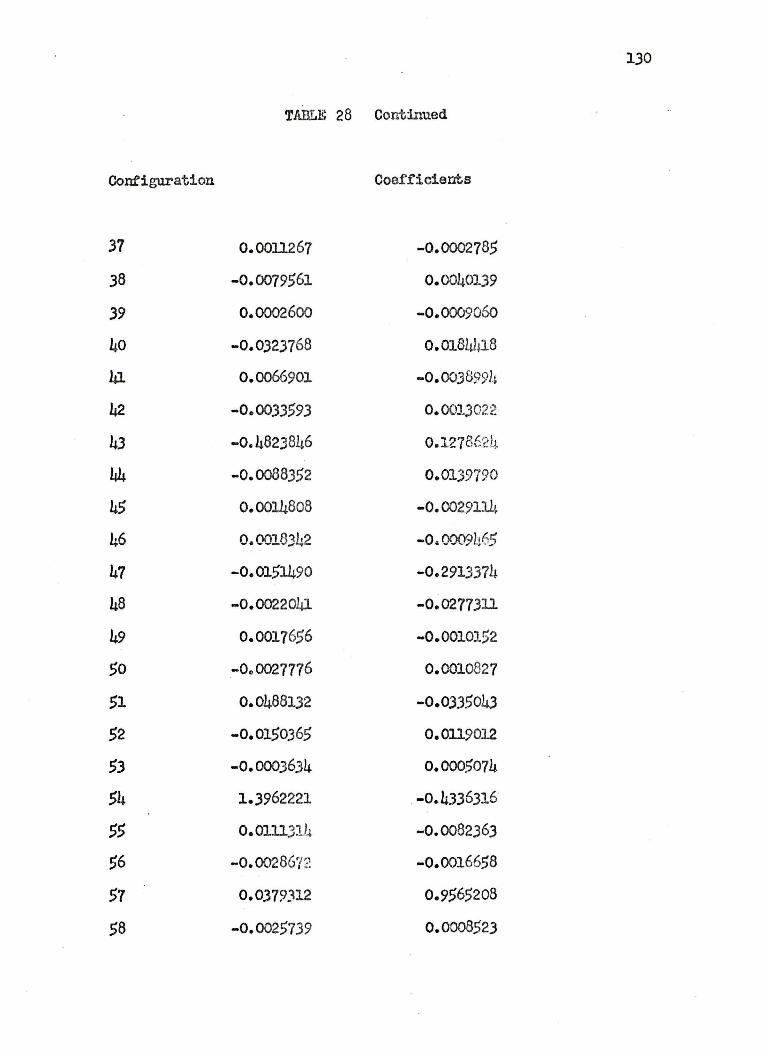
130
TABLE 28 Continued
Confi guration Coefficients
37 o.oon267 -0.0002785
38 -0.0079561 0.0040139
39 0.0002600 -0.0009060
40 -0.0323768 0.0184!µ.8
41 0.0066901 -o.003899h
42 -0.0033593 0. 0013022.
43 -0.4823846 0.12786211.
44 -0. 0088352 0.0139790
45 0.0014808 -0.0029114
46 0. 0018.342 - 0~0009L~65
47 -0. 0151490 -0. 291.337li
48 -0.0022041 -0. 0277.311
49 0.0017656 -0.0010152
50 -Oe 0027776 0.0010827
51 0.0488132 -0.033.5043
52 -o. 0150365 0.0119012
53 -0.0003634 o. 000.r,074
54 1.3962221 .-0. 4336316
55 0. 011131).. -0.0082363
56 -0. 00286'( :.:: -0.0016658
51 0.0.379312 0.9565208
58 -0. 0025739 0.0008523

131
TABLE 28 Continued
Configuration Coeffic.i.ents
59 0.0002225 0.0019391
60 -0.0235070 0.0193478
61 -0.8778998 0.2468797
62 -0.0200802 -0.5405228
63 -0.0006100 0.0007353
64 -0. 0201.,944 0.0102757
65 000010110 -0.0005244
66 -0.0008347 -0.0234892
67 0.0004030 0.0022347
68 -0.0006703 0.0009365
69 -0.0290463 0.0145589
70 0.0014.596 -0.0007535
71 -0.0011530 -0.0332425
72 Og0005698 0.0031901
73 -0.002~451 0.0004698
74 -0.0081967 0.0034840
75 0.2936804 -0.0703479
76 0.0042ll8 -0.0028793
77 0.0046723 0.0141001
78 -0.0212858 0.0123338
79 0.0040185 -0.0053831
Bo -0.801.4160 0.2051143
81 -0.0035468 0.0038558
82 O.OOll537 0.0001015

132
TABLE 28 Continued
Configuration Coefficients
83 -0.0178181 -0.4393820
84 0. 0188079 -0.0164961
85 1.1153902 -0.2496899
86 0.0185870 0.5291036
87 -0.0570366 0.0017409
88 0.0043014 -0.0004659
89 0.1671636 -0.0215743
90 0.0019221 0.0400142
91 -0.0024101 0.0010174
92 -0.2232593 0.0211223
93 -0.0012784 -0.0365303
94 -0.0025225 OaOOJ2820
95 -0~ 4071389 o.o6414o5
96 -0.0039372 -0.1277358
91 0.1731089 -0.0017076
98 - 0.0003982 -0.0059860
99 -0.0206123 - 0.0036528
100 0.0003056 0.0094232
101 -0.0076874 0.0001057
102 0.0082466 -0.0008564
103 0. 0149232 0.0130191
104 -0.0019923 -0.0199331
105 -0.0382853 -0.0268018
106 o.04n936 0.0445940
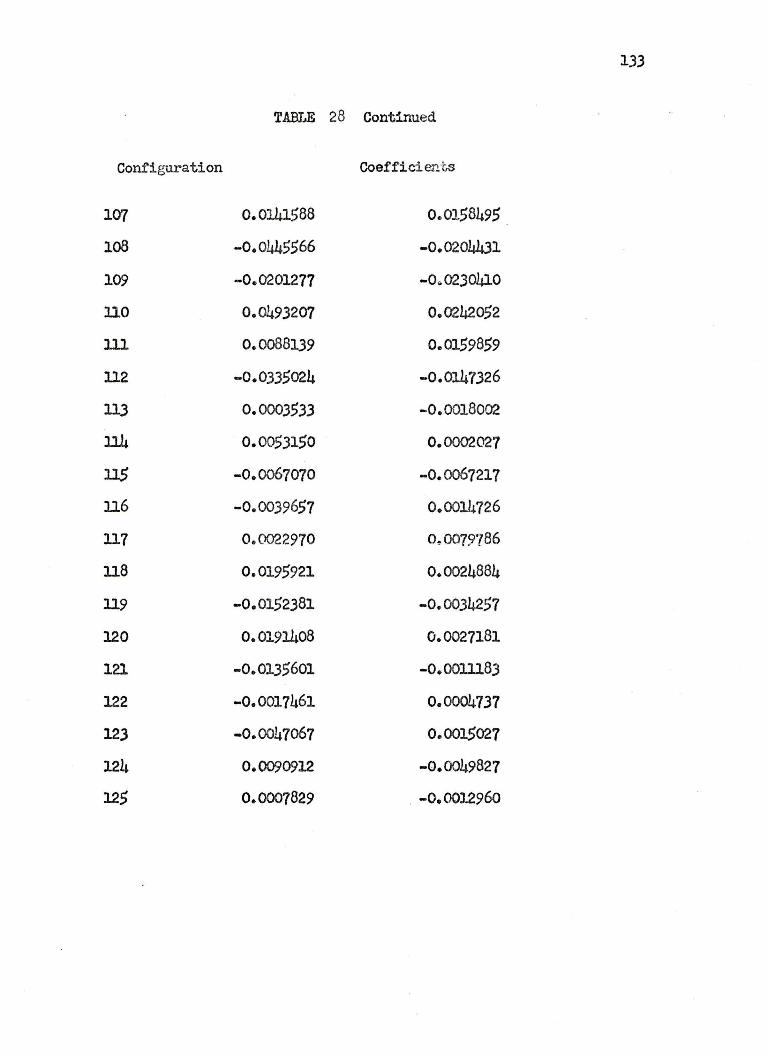
133
TABLE 28 Continued
Configuration Coeffidents
l(Jl 0.0141.588 0.,0158495
108 -0.0445566 -0.0204431
109 -0.0201277 -0~0230410
llO 0.0493207 0.0242052
lll 0.0088139 0.0159859
ll2 -0.0335024 -0.0147326
ll3 0.0003533 -0.0018002
114 0.0053150 0.0002027
11.5 -0.0067070 -0.0067217
ll6 -0.0039657 0.0014726
ll7 0.0022970 0~0079786
ll8 0.0195921 0.0024884
ll9 -0.01.52381 -0.0034257
120 0.0191408 0.0027181
121 -0.013.5601 -O.OOlll83
122 -0.0017461 0.0004737
123 -0.0047067 0.0015027
124 0.0090912 -0.0049827
125 0.0007829 . -0. 00)296o

TABLE 29
COEFFICIENTS OF THE 12.5 CONFIGURATIONAL WAVEFUNCTION FOR THE
LrrHIUM HYDRIDE PLUS 2 ~ + STATE
AT AN INTERNUCLEAR SEPARATION OF 4.0 a.u.
Total Energy -7.777.5364
Config. Coefficient Config. Coefficient
1 0.2663924 15 1.2112970
2 -0.0283708 16 -0.1019457
3 1.5831586 17 0.0211898
4 0.14.58196 18 -0.0024629
5 0.6519017 19 0.0301473
6 -0.0003638 20 -0.0035055
7 -O.o604220 21 -0.3911798
8 0.67,58196 22 0.0004215
9 -0.0637132 2.3 0.0.331609
10 0.0008752 24 0.0991201
ll 0. 0008752 25 -0.0001495
12 -0.018.5.573 26 -0.0066927
13 0.0014960 27 -0.0001409
14 0.1700576 28 0. 0000478
134

135
TABLE 29 Continued
Config. Coefficient. Conf'ig. Coefficient
29 -0.0013250 51 1.67CY2.766
30 -0.0004914 52 0.001370}
31 -0.0001920 53 0.1290326
32 0.0005207 54 o.4288826
33 -0.0127870 55 -0.0003451
34 1.1573330 56 -O.CY2.61173
35 -0.0025185 51 -0.0011322
36 -0.0986109 58 0.0030867
37 0.0004073 59 -0.0018912
38 -O.Jll.3464 60 -0.0012018
39 0.0004282 61 -0.0510053
40 0.0212907 62 o.6611432
hJ. 0.0002851 63 -0.0474384
h2 0.0017068 64 -o • .:;6047o6
43 -0.000:5431 65 O.CY2.04602
h4 -0.0058104 66 0.0007274
45 0.0015882 67 -0.0018307
46 0.0012794 68 0.0013399
47 0.0003857 69 0.0007539
48 0.0027228 70 0.0390429
49 -0.0009377 71 0.0553998
50 0.0420400 72 -0.0025125
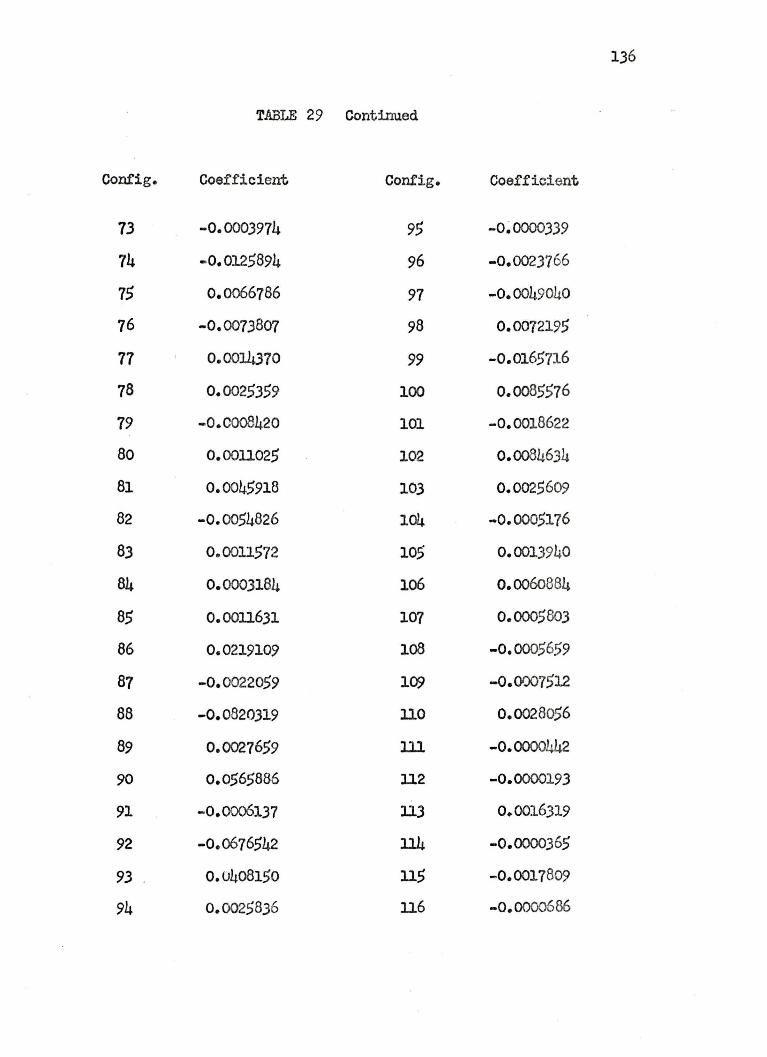
136
TABLE 29 Continued
Config. Coefficient Config. Coefficient
13 -0.0003974 95 -0~0000339
74 -0.0125894 96 -0.0023766
15 0.0066786 97 -0.0049040
76 -0.0073807 98 0.0072195
77 0.0014370 99 -0.0165716
78 0.0025359 100 o.0085576
19 -0.0008420 101 -0.0018622
80 0.0011025 102 o.0084634
81 0.0045918 103 0.0025609
82 -0.0054826 104 -0.0005176
83 O.OOll572 105 0.0013940
84 0.0003184 106 o.oo6o884
85 0.0011631 107 0.0005803
86 0.0219109 108 -0.0005659
87 -0.0022059 1C9 -0.0007512
88 -0.0820319 110 0.0028056
89 0.0027659 lll -0.0000442
90 0.0565886 112 -0.0000193
91 -0.0006137 113 0.0016319
92 -0.0676542 114 -0.0000365
93 0.0408150 115 -0.0017809
94 0.0025836 116 -0.0000686

137
TABLE 29 Continued
Con.fig. Coefficient
117 0.0008425
118 -0.0008887
119 0.0032856
120 -0.0035440
121 -0.0005315
122 0.0002810
123 -0.0002211+
124 o.00238o8
125 -0.0005343

BIBLIOGRAPHY
1. Crawford, F.H., and Jorgensen, T., Jr., Phys. Rev. l!I, 358, 932 (1935); !±2,, 745 (1936).
2. Herzberg, G., Spect.ra of Diatom1.c Molecules (D. Van Nostrand Co., Inc., Princeton, iJ .'J., 195'"0), 2nd Edition.
3. Moore, c.E., u.s. National Bureau of Standards, Gire. No. 467 (1958).
4. Pekeris, C.L., Phys. Rev. ll2, 1649 (1958); ll5, 1216 (1959); ~6, 143, 1470 (1962). -
s. Mulliken, R.s., Phys. Rev. 2,Q, 1028 (1936).
6. Rosenbaum, E.J., J. Cham. Phys.~' 16 (1938).
7. Velasco, R., Can. J. Phys.~, 1204 (1957).
8~ Fallonj R,J.; Va.~derslice, J.T., and Mason; E.A.; J. Chema Phys. ;g_, J.453 (1960); Jl, 944 (1960).
9. Singh, N.L., and Jain, D.C., Proc. Phys. Soc . 79, 274, 753 (1962); Can. J. Phys. 40, 520 (1962); Jain, D.C:, and Sah, ?., J. Chem. Phys . ~,-i553 (1963 ).
10. Jene, F. , Collectio11 Czech. Chem. Commun. 28, 2052 (1963); _gz, 2579 (1964); J. Nol. Spectry. ~, 63 (1966).
ll. Krupenio, P.H., Mason, E.A., and Vanderslice, J.T., J. Chem. Phys. 1,2, 2399 (1963).
12. Halmar..n, M.,and Laulicht, I., J. Chem. Phys. 46, 2684 (1967).
13. K1emperer, W., J. Chems Phys. 23, 2li52 (1955); Norris, W.G., and Klemperer, W., J . Chem. Phys. 28, 749 (1958); James , T.C., Norris, W.G., and Klemperer, w., J7 .Chem. Phys . 32, 728 (1960). -
l4. Wharton, L •. , Gold, L.P., and Klemperer, W., J. Chem. Phys. _ll, 1255 (1960); E_, 2149 (1962).
].5. Lawrence, T.R., Anderson, C.H., and Ramsey, N.F., Phys. Rev. !_30, 1865 (1963).
138

139
16. Roth.stein, E., U.S. Atomic Ener-gy Comm .. UCRL-17948 (1968).
17. Hurd, D.T., ChEw::t~try of the Hydrl.des (John Wiley and Sons Inc., New York, N.Y., 1952), p.27.
18. Messer, C.E., U.S. Atomic Energy Comm. NY0-9470 (1960); Davis, T.F., u.s. Atomic Energy COTJU. TID-3558 (1960); Roos, A., Chim. and L~d. (Paris)~' 86 (1961).
19. Gibb, T.R.P., Jr., and Messer, C.E., U.S. Atomic Energy Comm. Ni""()-3957 (1954).
20. Pretzel, F.E., Rupert, G.N., Mader, C.L., storms, E.K., Gritton, G.v., and Rushing, c.c., Phys. and Chem. Solids ~, 10 (1960).
21. staritzky, E., and Wa:Iker, D.I., Anal. Chem.~, 1055 (1956).
22. Messer, C.E., U.S. Atomic Energy Comm. NY0-8027 (1960).
23. Gunn, S.R.,and Green, L.G., J. Am. Chem. Soc. §.Q, 4782 (1958).
24. Moers, K., A. Anorg. Allgem. Chem.±!}, 179 (1920).
25. Plal'!lbeckj J.AQj Elder,. J.P., and. Laj_t.:,_nen, H.A,, J. ElectroChem. Soc. 113,931 (1966).
26. Johnson, C.E., Heinrich, R.R. , and Crouthamel, C.E~, J. Ph:.,s. Chem. 12,, 242 (1966).
27. Vogt, J.W., Tapco Report, NP-11888, Oct., 1961.
28. Kelley, K.K., and King, E.G., U.S. Bureau of Mines Bulletin 592 - (U.S. Government Printing Office, Washington, D.c., 1961).
29. Welch, F.H., U.S. Atomic Energy Comm. DC-61-3-73 (1961); U.S. Atomic Energy Comm. XDC-61-5-67 (1961); Field House, I.B., Hedge, J.C., end .i..ang, J.L., PB Rept. 151583, U.S. Govt. Research Repts. 31, 367 (1959); Messer, C.E., Damon, E.B., and Haybury, P.C., u.s. Atomic Energy C-omm. lfi0-3958 (1955); Heumann, F.K., and Salmon, O.N., U.S. Atoud.c Energy Comm. KAPL-1667 (1956).
30. Gross, D.T., McMillan.,. W.G., Michael, E.D., am Mas, C.P., Phys. Rev. 109, 1858 (1958); Weil, R.,and Lawson, A.W., J. Chem. Phys: 37, 2730 (1962); Stephens, D.R., and Lilley, E.M., J. Appl. Phys. l,2, 177 (1968). .
31. Machin, W.D., and Tompkins, F.c., Trans. Faraday Soc.~, 2205 (1966).

140
32. Schlesinger, H.I., Brown, H.C., Gilbreath, J.R., and Katz, J.J., J. Am. Chem. Soc. 75, 195 (1953); Finholt, A.c., Bond, A. c., Jr. 1 .;nd t1oblasinge:r.-_, H~ Lr J. Am. Chem. Soc • .22, 1199 (1947) •
.3.3. Waller, I., and Lundquiet, s.o., Arkiv fysik 7, 121 (195.3); Lundquist , S.O., Arkiv F'ysik 8, 177 (1954); woatin, A., Waller, I., and Lundquist, s.IT., Arkiv Fysik g, 371 (1962).
,34. Hurst, R.P., Phys. Rev. 114, 746 (1959). -.35. Morita., A .. , and Takahashi, K., Progr. Theoret. Phys. 19,
257 (1958). -
36. Phillips, T.J., and Harris, P.M., U.S. Dept. Com., Office Tech. Sen-. PB Rept. 156107 (1959).
37. Calder, R.S., Cochran, W., Griffiths, D., and Lowd.a, R.D., Phys. Chem. Solids Q, 621 (1962).
,38. Brodsky, M.H., and Burstein, E., J. Phys. Chem. Solids ~, 1655 (l.967) •
.39. Filler, A.s., and Burstein, E., Bull. Am. Phys. Soc. 2, 198 (1960).
40. Verble, JeL., Warren, tTeL~, and Yarnell, J.L~, Phys. Rev. 168, 980 (1968). -
41: Jaswal, s.s., and Harey, J.R., Phys. Rev. ,!I!, 1090 (1968).
42. Bach, F., and Bonhoetfer, K.F., z. Physik. Chem. B2.3, 256 (193.3); Kapuatinskii, A.F., Shsmovskii, L.M., andBayushkina, K.s., J. Phys. Chem. (u.s.s.R.) !Q, 620 (1937).
4.). Z:iJnmarman, W.B., and Montgomery, D.J., Phys. Rev. 120, 405 (1960). -
44.
45.
Pretzel, F.E., and Rushing, C.C., Phys . Chem. Solids 17, 232 ( 1961); Leuis, W .B. , and Pretzel, F. E. , Phys. Chem." Solids 19, 1.39 (1961); Pretzel, F.E., Gritton, G.V., Rushing-;-c.c., Friauf, RoJ., Lewis, W.B., and Wald.stein, P., Phys • Chem. Solids 2 3, 325 ( 19 62) ; Pretzel, F. E. , and Petty, R.L., Phys Hev. gz_, 777 (1962).
Dvinyaninov, B.L., and Gavrilov, F.F., Opt. Spiktrosk. 20, 74 (1966); Dvi.n,yaninov, B.L., Gavrilcv, F.Fo, ar:rl Smetanin, G.I., Opt. Spektrosk. 2.3, 247 (1967); Gavrilov, F.F., Dvinyaninov, B.L., stadukhlri, V.M., and Shulgin, B.V., Izv. Akad. Nauk SSSR, Ser. Fiz . J!, 2040 (1967) .

46. Knipp, J.K., J. Cham. Phys.~, 300 (1936).
47. Karo , A.M.? arrl Olson, A.R., J. Chem. Phys. 30, 1232, 1241 (1959). -
48. Karo, A.M., J. Chem. Phys. ;g, 182 (1959); E , 907 (1960).
49. Ebbing, D.D., J. Chem. Phys.~' 1361 (1962).
50. Matsen, F .A., and Browne, J.C., J. Phys. Chem. ~, 2332 (1962).
51. Harris, F.E., and Taylor, H.S., Physica J.Q, 105 (1964).
52. Kahalas, S.L. , and Nesbet, R.K., J. Chem Phys. 12, 529 (1963).
53. Cade, P.E., and Huo, W.M., J. Chem. Phys. ~' 1063 (1966).
54. Cade, P.E., and Huo, W.M., J. Chem. Phys. hl,, 614 (1967).
55. Browne, J.C., and Matsen, F.A., Phys. Rev. 135, A.1227 (1964).
56. Bender, C. F., and Davidson, E.R., J. Physe Chem. ]!2, 2675 (1966).
57. Brown, R.E., and Shull, H., Int. J. Quantum Chem.~, 663 (1968 ).
58. Taylor, H.S., J. Chem. Phys • .22, 3382 (1963).
59. Csizmadia, I.G., Sutcliffe, B.T. , and Barnett, M.P., Can. J. Chem. 42 , 1645 (1964).
60. Bender, C.F., and Davidson, E.R., J. Chem. Phys. !!2, 4222 (1968 ).
61. Browne, J.C., J. Chem Phys. g, 3495 (1964).
62. Harris, F.E., J. Chem. Phys. J_g, 3 (1960).
63. Taylor, H.S., and Harris, F.E., Mol. Ph.fa. 6, 186 (1963); J. Chem. Phys.~' 2591 (1963). -
64. Michels, H.H., and Harris, F.E., J. Chem. Phys. 39, 1465 (1963). - ,
65. Schneiderman, S.B., and Michels, H.H., J. Ghem. Phys.~, 3706 (1965).
66. Michels, H.H., J. Chem. Phys.!±!!, 3834 (1966).

67. Slater, J.C., ~~tum Theor,;r of Holecules and Solids, Vol. 1 (McGraw-H:iirBook Co., Inc., New York, N.Y., 1963).
68. Kotani, M., Ohno, K., and Kaya.ma, K., Handbuch Der Physik, Vol. XXXVTI/2, Molecules II (1961), p.39-J.42.
69. Hylleraas, E.A., and Undheim, B., z. Physik 65, 159 (1930); Mac.Donald, J.K.L., Phys . Rev. 43, 830 (1933); Shull, H., and Lowd.in, P.O., Phys . Rev. l.Io, 1466 (1958).
70. Paunz, R., Alternant Molecular Orbital Method (W.B. Saunders Co., Philadelphia, Pa., 1967) .
71. Kotani, M., Amemiya, A., Ishiguro, E., and Kimura, T., Tables of Molecular Integrals . (Haruzen Co., Tokoyo, 1955).
72. Predny, R.M., Master's Thesis, University of Florida . (1968).

BIOGRAPHICAL SKETCH
Robert Michael Predny was born November 2, 1941, at
Chicago, Illinois. He graduated from Lind.bloom High School in
Jm1e, 1959. He attended the Illinois Institute of Technology
and received the degree of Bachelor of Science in June, 1963.
He was awarded a National Defense Act Fellowship from September,
J.963 through August, 1966 to continue st,udies <lt the University
of Florida. He received the degree Master of Science in March,
1968. For the school term September, 1966 through June, 1969,
he has been a teaching and a graduate research assistant in the
ryepartment of Chemist~-.
aobert Michael is married to the former Faye Marie Krause
and they have a dauehter, Robin Michelle. He is a member of
Phi Lamba Upsilon Chemical Society.
143

This dissertation was prepared under the direction
of t he chairman of the candidate's supervisory committee
and has been approved by all members o~ t.hat committee.
It was submitted to the Dean of the College of Arts and
Sciences and to the Graduate Council, and was approved
as partial f'u.l:fillment of the requirements for the degree
of Doctor of Philosophy.
August, 1969
Dean,
Dean, Graduate School
;{ . .L; I ---r .. .1 .. , .z (! '-.f_. l c, L/ - c L-
• ) , J
Top Related
Copyright © 2022 FDOKUMEN

