







Bahasa
Halaman
Hukum


INFORM ATION TO USERS
This manuscript has been reproduced from the microfilm master. UMI films the text directfy from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be firom any type of conq)uter printer.
H ie quality of this reproduction is détendent upon the qnali^ of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins and inq)roper alignment can adversefy affect reproduction.
In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note wül indicate the deletion.
Oversize m aterials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each orig inal is also photographed in one exposure and is included in reduced form at the back of the book.
Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6” x 9" black and white photographic prints are available for any photogr^hs or illustrations appearing in this copy for an additional charge. Contact UMI direct^ to order.
UMIA Bell & Howell Information Company
300 North Zeeb Road. Ann Arbor. Ml 48106-1346 USA 313.'761-4700 800,'521-0600


LIQUID MOLDING OF TEXTILE REINFORCEMENTS: ANALYSIS OF FLOW INDUCED VOIDS AND EFFECT OF POWDER COATING ON
PREFORMING AND MOLDABÏLITY
DISSERTATION
Presented in Partial Fulfillment of the Requirements for the
Degree Doctor of Philosophy
in the Graduate School of The Ohio State University
By
Vivek Rohatgi
ooooooThe Ohio State Univesily
1995
Dissertation Committee:
Dr. L. James Lee
Dr. James F. Rathman
Dr. Kurt W. Koelling
Approved by
AdvisorDepartment of Chemical Engineering

OHI Number: 9612267
Copyright 1995 by Rohatgi, Vivek
All rights reserved.
DHI Microform 9612267 Copyright 1996, by DMI Company. All rights reserved.
This microform edition is protected against unauthorized copying under Title 17, United States Code.
UMI300 North Zeeb Road Ann Arbor, MI 48103

Copyright by
Vivek Rohatgi
1995

This dissertation is dedicated
to
My wife, Vidhi

ACKNOWLEDGMENTS
I would like to express my gratitude to my advisor. Dr. Ly James Lee for his advice
throughout this project. I would also like to acknowledge Drs. James Rathman and Kurt
Koelling as members of the dissertation committee for their valuable time, suggestions and
comments. I also wish to thank Mr. Jim Barron, Dr. Asjad Shafi and Dr. Dexter White of
Dow Chemical for the various useful discussions I had with them. My appreciation goes to
Mike Kukla and Shoujie Li for their technical assistance.
Finally, I wish to thank my parents, sister and my wife for their encouragement and
constant emotional support throughout this seemingly everlasting endeavor.
m

VTTA
Septem ber 26, 1966..........................................Bom, Patna, India
July 1985 - May 1989............................................ B. Tech, Chemical EngineeringInstitute of Technology, BHU Varanasi, India
September 1989 - September 1990.................. Polymer Engineering Research FellowDepartment of Chemical Engineering The Ohio State University Columbus, Ohio, USA
September 1990 - August 1991.......................Graduate Research AssociateDepartment of Chemical Engineering The Ohio State University
August 1991...........................................................M.S., Chemical EngineeringThe Ohio State University
September 1991 - present..................................... Graduate Research AssociateDepartment of Chemical Engineering The Ohio State University
Publications
"Influence of material and processing variables on resin-fiber interface in glass fiber reinforced polymeric composites", V. Rohatgi, M.S. Thesis, The Ohio State University (1991).
"Influence of processing and material variables on resin-fiber interface in liquid composite molding". Polymer Composites, 14 (2), April 1993, N. Patel, V. Rohatgi and L. J. Lee.
"Macro and microvoid formation in liquid composite molding", 9th ASM/ESD Conference, Dearborn, MI, November 1993, V. Rohatgi, N. Patel and L. J. Lee.
IV

"Permeability measurement of braided graphite fiber preforms and kinetic/rheological measurement and modeling of a BMI resin". Collaborative Core Research Program, Ohio Aerospace Institute, Cleveland, OH, January 1994, V. Rohatgi, M. Perry and L. J. Lee.
"Microflow analysis in resin transfer molding". Proceedings of the NSF Design and Manufacturing Grantees Conference, Jan. 1994, L. J. Lee, V. Rohatgi and N. Patel.
"Flow characterization and air entrapment and removal during impregnation of fiber reinforcements in liquid composite molding". Report No. ERC/NSM - P- 94-19, The Ohio State University, May 1994, V. Rohatgi, N. Patel and L. J. Lee.
"Microscale flow behavior and void formation mechanism during impregnation through a unidirectional stitched fiberglass mat". Polymer Engineering and Science, 35 (10), May 1995, N. Patel, V. Rohatgi and L. J. Lee.
"Effect of reactive tackifier on preforming and molding in RTM", AIChE Annual Meeting, November 1995, V. Rohatgi, S. Li and L. J. Lee.
"Experimental investigation of flow induced microvoids during impregnation of unidirectional stitched fiberglass mat". Polymer Composites, December 1995, V. Rohatgi, N. Patel and L. J. Lee.
Fields of Study
Major Field: Chemical Engineering
Minor Field: Polymers/Composites Science and Engineering1. Interfacial phenomena2. Two-phase flow in porous media3. Chemo-rheology of polymeric resins4. Preforming of powder coated textile reinforcements5. Material Characterization (Chemical, Physical, Thermal and
Mechanical) of polymeric resins & composites

TABLE OF CONTENTS
DEDICATION............................................................................................................................. ii
ACKNOWLEDGMENT............................................................................................................ iii
VITA .................................................................................................................................... iv
TABLE OF CONTENTS........................................................................................................... vi
LIST OF TABLES...................................................................................................................... xi
LIST OF FIGURES....................................................................................................................xii
CHAPTER PAGE
I. INTRODUCTION
1.1 Fiber Reinforced Polymer Composites...................................................................I
1.2 Polymer Composite Processes................................................................................ 5
1.2.1 Hand Lay - up and Spray - u p ...................................................................... 5
1.2.2 Prepreg Vacuum Bagging and Autoclaving............................................... 6
1.2.3 Filament Winding and Pultrusion................................................................6
1.2.4 Compression Molding of Sheet Molding Compound...............................7
1.2.5 Liquid Composite Molding (LCM)............................................................. 8
1.2.5.1 Resin Transfer molding (RTM )......................................................8
1.2.5.2 Structural Reaction Injection molding (SRIM)........................... 12
1.3 Resin Systems for LCM......................................................................................... 14
1.4 Reinforcements for LCM....................................................................................... 16
1.5 Tooling and Design Consideration for LCM ...................................................... 20
vi

1.6 Pumping/Dispensing Unit for L C M ....................................................................21
1.7 Scope of Study....................................................................................................... 22
II. LITERATURE REVIEW
2.1 Mold Filling and Fiber Wetting in LCM ............................................................ 29
2.1.1 Darcy's Law................................................................................................. 29
2.1.2 Equilibrium Contact Angle........................................................................31
2.1.3 Dynamic Contact Angle............................................................................. 32
2.1.4 Capillary Pressure.......................................................................................35
2.1.5 Wicking Phenomena.................................................................................. 38
2.2 Contact Angle and Surface Tension Measurements..........................................39
2.2.1 Direct Observation of Contact Angle....................................................... 39
2.2.2 Wetting Force Measurements................................................................... 43
2.2.3 Bundle Contact Angle................................................................................ 43
2.2.4 Surface Tension Measurements................................................................48
2.2.4.1 Capillary Rise M ethod...................................................................48
2.2.4.2 Ring M ethod...................................................................................49
2.2.4.3 Drop Volume and Drop Weight Methods................................... 49
2.2.4.4 Pendant Drop M ethod....................................................................50
2.2.4.5 Wilhelmy Technique...................................................................... 50
2.3 Void Formation Studies........................................................................................ 50
2.3.1 Experimental Studies on Void Formation................................................50
2.3.2 Modeling of Void Formation.....................................................................52
2.4 Measurement of Void Content..............................................................................58
2.4.1 Density Determination................................................................................58
2.4.2 Water Absorption........................................................................................ 59
vii

2.4.3 Micrography................................................................................................59
2.4.4 Confocal Scanning Optical Microscopy................................................. 61
2.4.5 Ultrasonic C - Scan.................................................................................... 61
2.4.6 Radiography................................................................................................ 62
2.5 Effect of Voids on Mechanical Properties............................................................62
2.6 Application of Polymer Powders in Composites................................................ 66
2.7 Tack and Drape Characteristics of Prepregs/Preforms...................................... 70
2.8 Modeling of Fiber Consolidation..........................................................................75
2.9 Rheo-kinetic Characterization of Bismaleimide Resins.................................... 77
in. ANALYSIS OF FLOW INDUCED VOIDS DURING FIBER IMPREGNATION
3.1 M aterials.................................................................................................................. 85
3.2 Instrumentation and Experimental Procedure.....................................................85
3.2.1 Liquid Properties and Contact Angle Measurements............................ 85
3.2.2 Flow Visualization of Macro and Micro V oids...................................... 89
3.3 Flow visualization of Macro voids Formation......................................................93
3.3.1 Axial Flow................................................................................................... 93
3.3.2 Transverse Flow........................................................................................ 101
3.4 Flow visualization of Micro voids Formation.................................................... 103
3.4.1 Axial Flow..................................................................................................103
3.4.2 Transverse Flow........................................................................................ 114
3.5 Mobilization of Macro and Microvoids..............................................................123
3.6 Vacuum Assisted Liquid Injection......................................................................124
V lll

IV. FIBER CONSOLIDATION AND SPRINGBACK IN POWDER (TACKIFIER) COATED PREFORMS
4.1 M aterials............................................................................................................... 125
4.2 Equipment and Experimental Procedure.......................................................... 125
4.2.1 Differential Scanning Calorimetry.........................................................125
4.2.2 Preforming Experiments......................................................................... 132
4.2.2.1 U-Shape Bending..........................................................................132
4.2.2.2 Vacuum Debulking...................................................................... 134
4.2.2.3 Lateral Compression.................................................................... 136
4.2.3 Scanning Electron Microscopy...............................................................139
4.2.4 Rheometrics Dynamic Analyzer.............................................................139
4.3 Results and Discussion....................................................................................... 142
4.3.1 Characterization of Reaction Kinetics...................................................142
4.3.2 Fiber Preforming ..................................................................................... 145
4.3.2.1 U-Shape Bending..........................................................................145
4.3.2.2 Vacuum Debulking.......................................................................148
4.3.2.3 Lateral Compression.................................................................... 154
4.3.3 Phenomenological Approach for Springback Control under Lateral Compression............................................................................................... 169
V. MECHANICAL PROPERTIES OF MOLDED COMPOSITES AND FLO\^^ CHARACTERISTICS OF TEXTILE REINFORCEMENTS
5.1 Effect of Voids on Fiberglass/UP Composites.................................................191
5.1.1 Dynamic Mechanical T est...................................................................... 192
5.1.2 Freeze-Thaw Cycling ..............................................................................197
5.1.3 Ultrasonic C-Scan ................................................................................... 199
IX

5.2 Mechanical Properties of Tackified Samples.................................................. 203
5.3 Flow Characteristics of Textile Reinforcements............................................. 212
5.3.1 Permeability of Fiber Preforms............................................................... 212
5.3.1.1 Braided Preforms......................................................................... 218
5.3.1.2 Tackified Woven Preforms.........................................................225
5.3.2 Effect of tackifier on fiber wetting in woven preform s....................... 229
5.3.2.1 Wicking Experiments...................................................................229
5.3.2.2 Measurement of capillary pressure vs. saturation....................231
VI. CONCLUSIONS AND RECOMMENDATIONS....................................................237
REFERENCES........................................................................................................................243
APPENDICES
A. Operational procedure for in-plane permeability measurements................... 250
B. Flexural properties of tackified (1 wt.%) BMI res in .......................................253

LIST OF TABLES
TABLE p a g e
1.1 Typical parts manufactured by RTM [Stark and Beitigam, 1987].............................13
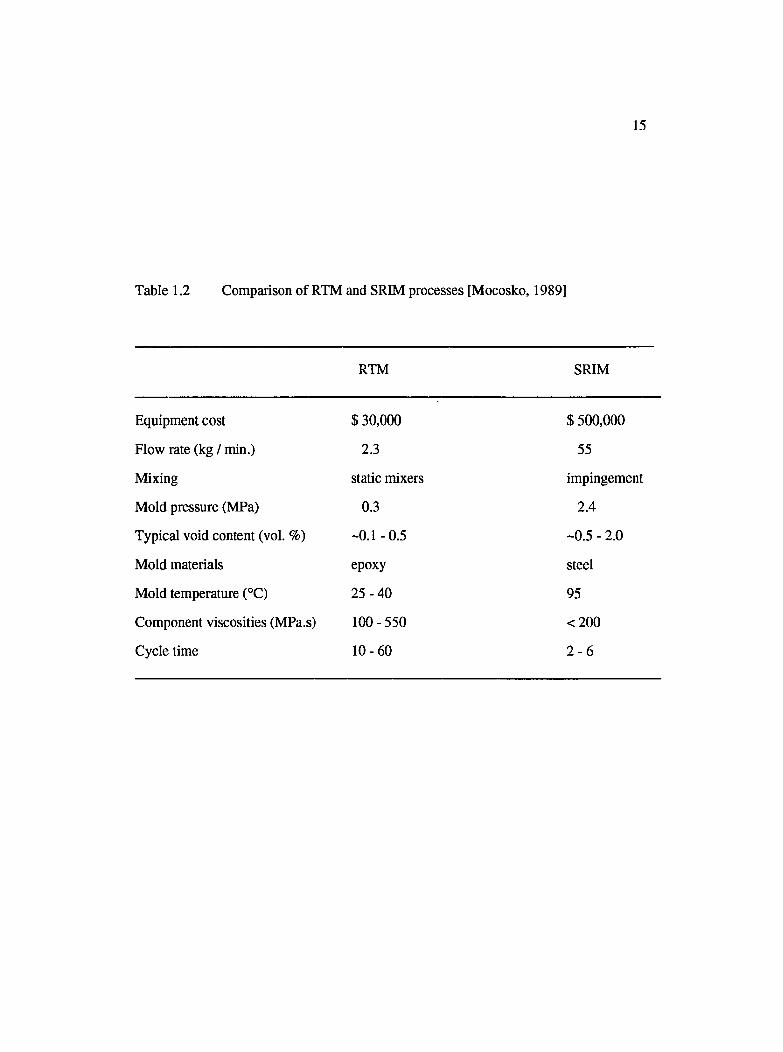
1.2 Comparison of RTM and SRIM processes [Mocosko, 1989].................................... 15
3.1 Room temperature properties of test liquids and equilibrium contact angles........ 90
4.1 Springback after vacuum debulking of BMI powder coated preforms..................153
4.2 Springback after lateral compression of BMI powder coated preforms............... 166
4.3 Springback after lateral compression of PMMA powder coated preform s.......... 168
5.1a Flexural properties of pure BMI resin.......................................................................205
5.1b Flexural properties of tackified BMI resin (a ~ 0 .5 ).............................................. 206
5.1c Flexural properties of tackified BMI resin (a ~ 0.6)............................................... 206
5.2a Flexural properties of BMI composite (higher tackifier cure)...............................211
5.2b Flexural properties of BMI composite (lower tackifier cure)................................211
5.3 Characteristics of braided preforms...........................................................................218
5.4 In-plane dry fiber permeabilities of 12k tow AS4 GP preforms............................ 224
5.5 Effect of tackifier concentration on in-plane permeability (BMI tackifier onsurface)........................................................................................................................... 228
B.l Flexural properties of tackified BMI resin (1 wt. % tackifier, a ~ 0.4)................254
B.2 Flexural properties of tackified BMI resin (1 wt. % tackifier, a ~ 0.5)................255
B.3 Flexural properties of tackified BMI resin (1 wt. % tackifier, a ~ 0.6)................256
XI

LIST OF FIGURES
FIGURE PAGE
1.1 Schematic of the resin transfer molding process.......................................................... 9
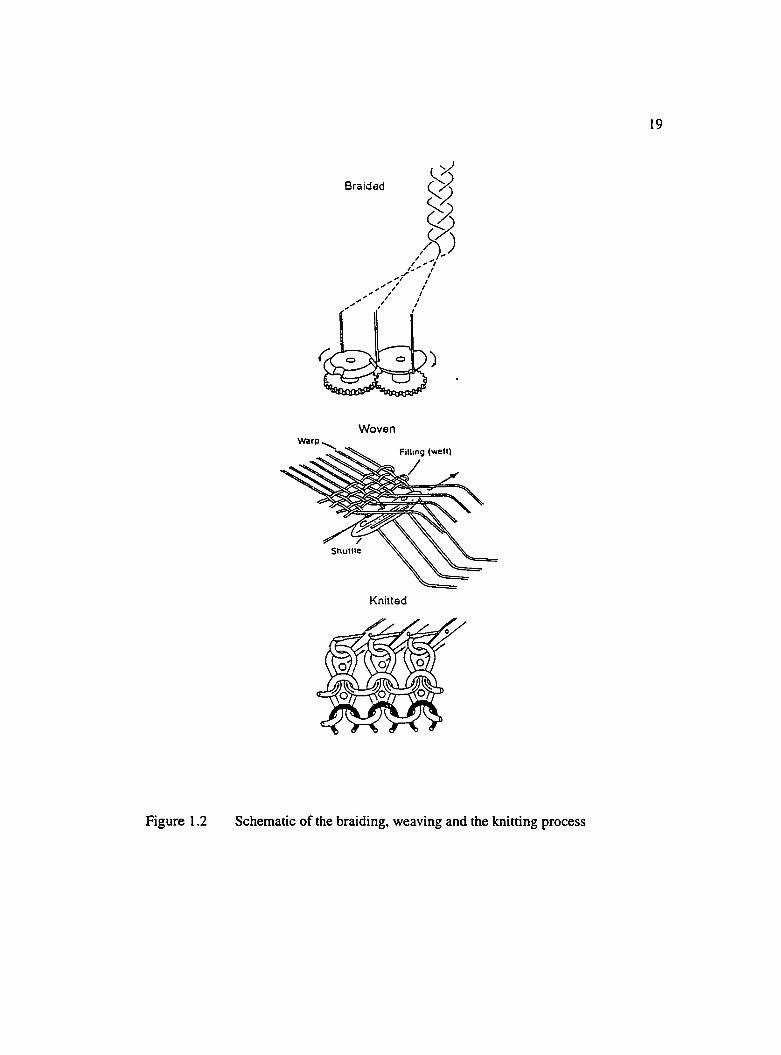
1.2 Schematic of the braiding, weaving and the knitting process....................................19
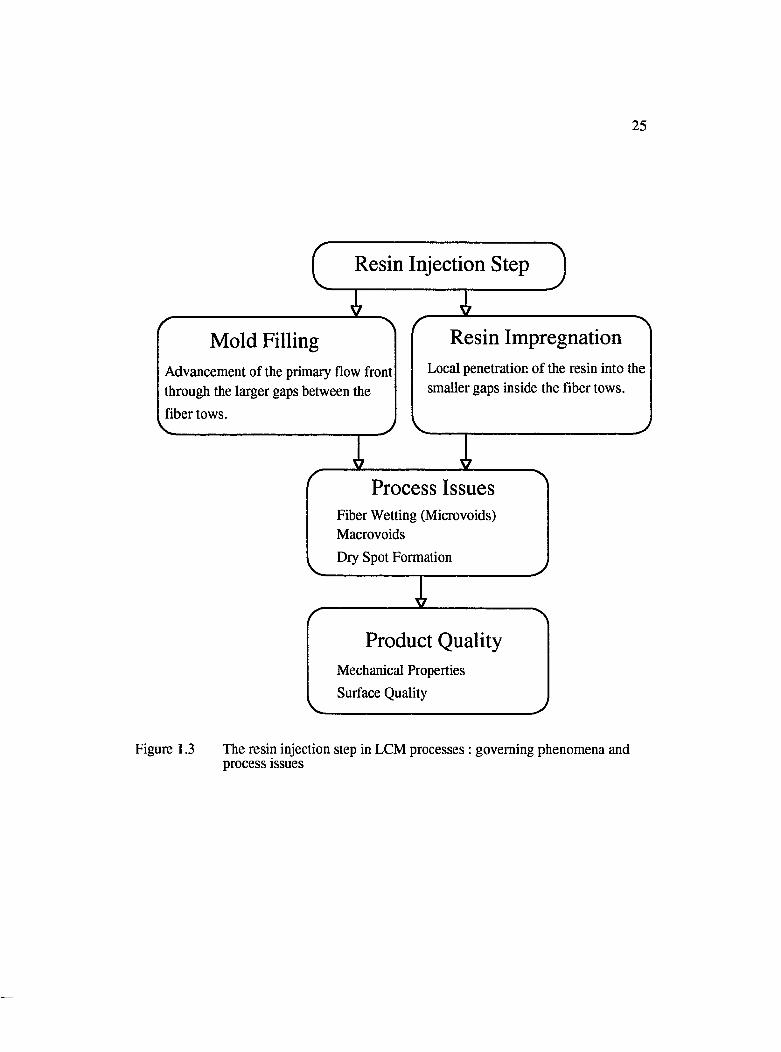
1.3 The resin injection step in LCM processes : governing phenomena and processissues............................................................................................................................... 25
1.4 Overview of resin transfer molding of polymer powder coated fiber preforms... 26
1.5 Fiber deformation modes : (a) U-shape bending and (b) lateral compression........ 28
2.1 Three phase equilibrium contact angle [Miller, 1977]............................................... 33
2.2 Schematic of the image analysis set - up for contact angle measurement of asessile drop [Neumann, 1992]...................................................................................... 40
2.3 Graph of Cosine 6 vs. 0 [Cahn's Manual DCA 322,1992].......................................42
2.4 Concept of the Wilhelmy technique [Miller, 1977]....................................................44
2.5 A typical curve illustrating the weight changes with time in a wickingexperiment [Hsieh and Yu, 1992]................................................................................47
2.6 Schematic of the flow front progression and the air entrapment processduring transverse flow [Pamas and Phelan, 1991].....................................................54
2.7 Schematic depiction of the channeling flow from larger to smallercapillary [Chan and Morgan, 1993 b ] ......................................................................... 56
2.8 Normalized shear strength as a function of void content[Feldgoise et al., 1991].................................................................................................. 65
2.9 Types of powder coating in composites [Cochran and Pipes, 1991]........................68
2.10 Schematic of the surface of a prepreg [Bonhomme, 1986]........................................ 73
2.11 Squeezing flow of a Newtonian liquid [Bonhomme, 1986] .....................................73
XU

2.12 Reaction mechanisms of a BMI resin : (a) Michael addition of diamine tobismaleimide and (b) bismaleimide homopolymerization....................................... 79
3.1 Schematic of the Dynamic Contact Angle Analyzer [DCA 322].............................87
3.2 Typical trace of force readings obtained in a surface tensionmeasurement experiment (e.g. UP resin)....................................................................88
3.3 Schematic of the flow visualization set-up; video assisted microscopy(VAM).............................................................................................................................94
3.4 Photograph showing the lead - lag at the flow front for axial flow:capillary number < 10"^............................................................................................... 95
3.5 Schematic of formation of macrovoids during axial flow .........................................98
3.6 Photograph of macrovoids trapped in the fiber m at....................................................99
3.7 Percent area macrovoids for flow along the fiber tows (axial flow).......................100
3.8 Percent area macrovoids for flow normal to the fiber tows (transverse flow) ... 102
3.9 Photographs showing the lead - lag at the flow front for axial flow and capillaiynumber > 10'^ : (a) overall view and (b) zoom v iew ............................................. 104
3.10 Schematic of formation of microvoids during axial flow ....................................... 105
3.11 Photograph of coagulated microvoids formed by joining of adjacent wickingstream s..........................................................................................................................107
3.12 Photographs of microvoids : (a) capillary number ~ 0.36 and(b) capillary number ~ 0.004....................................................................................108
3.13 Percent area microvoid content as a function of injection velocity for axialflow .............................................................................................................................. 109
3.14 Percent area macro and microvoids for axial flow : (a) silicone oil, 200 cs ,(b) DOP oil, (c) ethylene glycol and (d) master curve of all three liquids.........110
3.15 Photographs showing the optical transparency of composite samples :(a) good fiber wetting and (b) poor fiber wetting....................................................113
3.16 Percent area macro and micro voids during transverse flow of DOP oil.................115
3.17 Schematic of flow front progression and the formation of microvoids duringtransverse flow (mechanism I, capillary number < 10"2).......................................116
xin

3.18 Photograph illustrating lead-lag at the flow front during transverse flow(mechanism I, capillary number < 10'^)................................................................. 117
3.19 Photograph of microvoids formed during transverse flow of DOP oil :capillary number ~ 0.003 (2(X)X)...............................................................................118
3.20 Photographs showing the dynamics of microvoid formation and movement during transverse flow : (a) formation of microvoids, (b) and (c) movementwith change in shape and s iz e ................................................................................... 120
3.21 Schematic of the flow mechanism (II) for transverse flow : (a) an enlarged side view of flow in and around a fiber tow and (b) microvoid at the edgeof the fiber tow............................................................................................................. 122
4.1 Schematic of 6k, 4 HS woven fiber reinforcement................................................... 126
4.2 Structure of two components of Bismaleimide resin based tackifier...................... 127
4.3 Schematic of the Differential Scanning Calorimetry set-up.................................... 128
4.4 Scanning electron micrograph showing distribution of tackifier powder in"undebulked" fiber preform ........................................................................................133
4.5 Schematic of the U-shape bending device..................................................................133
4.6 Schematic of preform lay-up prior to debulking........................................................135
4.7 Schematic of the lateral compression device............................................................. 137
4.8 Calibration curve for the LVDT used in lateral compression experiments 138
4.9 Schematic of parallel plate set-up for theological measurements............................141
4.10 Scanning reaction rate profile for BMI tackifier........................................................143
4.11 Isothermal conversion profiles for BMI tackifier......................................................144
4.12 Glass transition temperatures of BMI tackifier under different cureconditions......................................................................................................................146
4.13 Springback in U-shape bending of fiber preforms as a function of tackifierconversion at different concentration levels.............................................................147
4.14 Photographs showing springback in U-shape bending of fiber preforms fordifferent debulking conditions................................................................................... 149
4.15 Photomicrograph showing powder coagulation and tackifier location forpreforms subjected to U-shape bending.................................................................... 150
XIV

4.16 Sintering of tackifier particles : (a) upon melting and (b) coagulation intodeformed droplets........................................................................................................ 151
4.17 Photomicrographs of the fiber preform with BMI tackifier applied usingthe solvent technique (a) low magnifications and (b) high magnification 152
4.18 Photomicrograph of surface of fiber preform with BMI tackifier and vacuumdebulked at94°C .......................................................................................................... 155
4.19 Consolidation behavior of BMI tackified preforms under lateral compression :(a) compacted thickness, (b) springback and (c) uncompacted thickness .......... 156
4.20 Viscoelastic behavior of tackified preforms : (a) change in strain as a function of time at constant pressure and (b) change in strain during springbackat zero stress..................................................................................................................158
4.21 Photomicrographs of surface of laterally compressed preforms with BMItackifier : (a), (b) showing interlayer coverage and (c) droplet possibly coexisting with a "manchon"......................................................................................160
4.22 Change in viscosity and viscoelastic properties of BMI tackifier as a function of temperature : (a) dynamic viscosity, (b) G* and (c) G' & G "................................ 163
4.23 Dynamic viscosity of PM M A...................................................................................... 167
4.24 Photomicrographs of surface of PMMA tackified preforms compressed at220°C : (a) low magnification and (b) high magnification.....................................170
4.25 Photomicrographs of surface of PMMA tackified preforms compressed at250°C : (a) low magnification and (b) high magnification.....................................171
4.26 Photomicrographs of surface of PMMA tackified preforms compressed at~287°C : (a) low magnification and (b) high magnification...................................172
4.27 Viscoelastic properties of PMMA : (a) G* and (b) G" and G "................................173
4.28 Phenomenological approach for springback control under lateralcompression..................................................................................................................174
4.29 Consolidation behavior of untackified fiber preform under lateral compression :(a) 8 layers and (b) 16 layers.......................................................................................176
4.30 Photomicrographs showing the consolidation of inter and intralayer gaps with increasing fiber volume fraction : (a) Vf = 0.32, (b) Vf = 0.45 and(c) Vf = 0.67............................................................................................................... 177
XV

4.31 Comparison of experimental vs. consolidation model for lateral compressionof 4HS preforms : (a) 8 layers and (b) 16 layers...................................................... 181
4.32 Viscoelastic properties of BMI tackifier under isothermal conditions :(a) 94°C, (b) 120°C and (c) 150°C............................................................................. 183
4.33 Overview of the phenomenological approach for springback control underlateral compression......................................................................................................186
4.34 Photomicrographs showing cross-section of laminates after springback :(a), (b) preforms with PMMA powder heated at ~287°C and (c) preformwith BMI tackifier vacuum debulked at 94°C ......................................................... 188
5.1 Torsion rectangular fixtures with the loaded sample [RDA InstructionManual, 1994]...............................................................................................................193
5.2 Drop in the dynamic stiffness of unidirectional stitched fiberglass mat reinforced UP composite samples with increased immersion times in hotwater : (a) no void, (b) ~ 7% macrovoid and (c) ~ 3% m icrovoid........................195
5.3 Formation of microcracks in unidirectional stitched fiberglass mat reinforced reinforced UP composite samples: (a) cracks on the surface, macrovoidsample and (b) cracks in the fiber tow, micro void sam ple.....................................198
5.4 Components of the SONOTEK Ultrasonic C - scan system................................... 199
5.5 Ultrasonic C - scan image of unidirectional stitched fiberglass mat reinforced UP composite samples : (a) Vg = 0.04 cm/sec., (b) Vg = 1.0 cm/sec. and(c) Vs = 3.9 cm/sec.................................................................................................... 201
5.6 Schematic of the 3-pt bending test............................................................................. 203
5.7 Configuration of the mold set-up for preparing clear castings...............................204
5.8 Comparison of mechanical properties of pure BMI resin and with 3 wt.%tackifier : (a) mean flexural strength and (b) mean % strain at break................... 208
5.9 Four clover leaf pattern indicative of residual microstresses at the tackifierparticle/resin matrix interface.................................................................................... 209
5.10 Scanning electron micrographs of the fracture surface : (a) pure BMI resinand (b) tackified BMI resin.........................................................................................210
5.11 Schematic of the in-house developed permeability set-up...................................... 213
XVI

5.12 Schematic of the Ashcroft Dead Weight Gauge Tester...........................................215
5.13 Calibration curves of the pressure transducers used for permeabilitymeasurements : (a) 100 psi range and (b) 500 psi range........................................ 216
5.14 Pressure rise vs. time curves for brmded preforms : (a) 3 layers [0/0/0](b) 2 layers [0/0] and (c) 2 layers [0/90] ...............................................................221
5.15 Pressure drop as a function of tackifier concentration and locationfor 4HS graphite fiber preforms................................................................................ 227
5.16 Permeability as a function of tackifier concentration for 4HS graphite fiberpreforms with BMI tackifier and vacuum debulked at 94°C.................................228
5.17 Comparison of wicking behavior of solvent and powder coated 4HS graphitefiber preforms.............................................................................................................. 230
5.18 Schematic of the centrifuge device.............................................................................232
5.19 Configuration of the fiber sample in the centrifuge device.................................... 235
5.20 Comparison of capillary pressure as a function of saturation for 4HSgraphite fiber samples with and without tackifier................................................... 236
xvii

CHAPTER I
INTRODUCTION
1.1 Fiber Reinforced Polymer Composites
The demand for light weight high strength materials in almost all walks of life has
brought about an increased usage of composite materials. Composites offer high strength
and stiffness, resistance to hostile environments as well as ability to be formed into
complex shapes.
Composite materials evolve from combining two or more physically distinct and
mechanically separable component materials. The key, however, lies in combining the
components in a synergistic way so as to enhance the properties of the final product.
Composite materials can include steel reinforced concrete, straw reinforced mud bricks,
linoleum, fiber reinforced ceramic / polymer or for that matter any other combination that
falls under the definition of a composite material. This text, however, will focus only on
methods, materials and applications of polymer composites, more specifically fiber
reinforced polymer composites.
Fiber reinforced polymer composites (FRP's) have been in use since World War II. In the
late 1940s and early 1950s, FRP's were used largely in the marine industry. However,
they have come a long way since then, and today, they are being used in a wide range of
applications. Polymer composites are used in the transportation industry (automotive,
tmck, ships and railway vehicles), in aerospace, defense and outer space applications, in
1

2
the recreational and sporting goods industries, in electronics, and in commercial
industries. As an example. Corvette, Fierro, and Avanti automobiles have had body
structures made of polymer composites for over 25 years. Another spectacular
application in the automotive industry is the sports car, the Dodge Viper that has all its
external panels with a total weight of about 77 kg made of polymer composite. In 1987,
Ford Motor Company completed a prototype replacing the 90 piece steel front stmcture
of an Escort automobile with a two piece composite structure [Johnson, 1987]. An all
composite chassis for Bugatti BE 110 automobile has been developed by Composites
Aquitaine, a French Company [High performance composites, 1993]. Another example
is the All Terrain Vehicle (Bandvagnen) designed and manufactured in Sweden.
Recently, a 175 ft tall polymer composite mast was fabricated for the Zeus luxury super
yacht, the largest boat of its type to be certified by the American Bureau of Shipping
[Stover, 1993]. Trains, such as the BART (the San Francisco Bay Area Transit System),
use composites for interior panels, and several trains have fully formed compartments
made of composites [Strong, 1989]. Rail vehicles in Europe are now also being made of
polymer composites. The Voyager, which circled the world without stopping, was an all
composite aircraft. Boeing's 767 commercial aircraft has over 30% of its structure, from
its nose landing gear doors to its vertical fin tip, based on polymer composite materials.
In October 1993, the first set of horizontal and vertical stabilizers were fabricated with
fiber reinforced plastics for Boeing's new 777 wide body passenger jet [Modern Plastics,
1994]. B - IB stealth bombers, DC - X missiles, and F -18 fighter jets make use of high
performance advanced polymer composites. The Solar and Heliospheric Observatory
(SOHO) spacecraft which was due to launch in July 1995, will have an all composite
Telescope Structure Assembly (TSA) mounted on it [McConell, 1993]. The sporting
goods and recreational industry uses composites for tennis racquets, golf clubs, baseball
bats, skis, snowmobiles etc. The most common uses for composites in electrical

3applications utilize the non conductive nature of composite materials. Typical examples
include printed circuit boards (PCBs), insulators, and radomes [Strong, 1989].
Commercial industries employ polymer composites for the manufacture of storage tanks,
bathtubs, kitchen sinks, compressed gas cylinders, medical equipment, building panels
etc. The success of polymer composites in today's globally competitive market is
primarily due to the large number of advantages they offer as compared to other
materials. The advantages are [Moritz, 1993]:
1. Higher strength / stiffness to weight ratio than most metals
2. Design flexibility
3. Thermal stability
4. Dimensional stability
5. Increased fatigue life
6. Improved corrosion and wear resistance
7. Finishing (Long lasting with minimum maintenance)
8. Parts consolidation &
9. Significant cost advantages (Low tooling costs)
As with all other materials, composites are not without their disadvantages. Perhaps the
greatest disadvantages are the lack of well - defined and easy to employ design rules and
lack of highly productive manufacturing methods. The resolution of these issues is the
overriding concept behind the extensive research efforts being directed towards
composite manufacturing processes, both in academia and in industry.
A fiber reinforced polymer composite consists of a fiber reinforcement, a matrix resin,
and an interface between the two. Fiber reinforcement provides strength and stiffness.
The matrix protects the reinforcement from adverse environmental effects and binds the
fibers, while the interface serves to transfer stress from the matrix to the fibers. Fiber

4reinforcement is usually glass, graphite, or kevlar fiber. In some cases, nylon and PET
fibers are also used. The matrix resin can be either a thermoplastic or a thermoset
polymer. These two polymer types differ in their respective intermolecular structures.
Thermoplastics are solids which can be softened and made to flow under the application
of heat and pressure. Upon cooling, the resin changes from a liquid back to a solid. The
process is thus reversible. Thermoplastics are widely gaining popularity because of their
ability to mold complex shapes, their ease of fabrication, and their cost effective
performance characteristics. The most common thermoplastic resins are polyethylene,
polypropylene, polystyrene, nylon, polycarbonate, thermoplastic polyester, etc.
Processing is usually accomplished by heating the material to soften it for molding. Once
the resin is molded to proper shape, it is cooled until hardened. In spite of the growing
popularity of thermoplastic resins, they are not so widely used in composite applications.
Some aerospace composites use a high temperature, semi - crystalline thermoplastic
called PEEK (polyether-ether-ketone), with graphite fiber reinforcements. Body panels
of the Saturn automobile, the newest venture of General Motors Corporation, also utilizes
some thermoplastic resins.
The majority of the composites manufactured today utilize reinforced thermosets. The
behavior of thermoset resins is very different from that of thermoplastic resins.
Thermosets are generally liquid resins which are heat activated (cured) resulting in an
irreversible cross - linking of the molecular structure. Once the resin is fully reacted and
solidified, it cannot be reformed to its original state. The most common thermosetting
polymers used are unsaturated polyesters, epoxies, vinyl esters, polyurethane's, phenolics,
and bismaleimides. Thermoset resins are more popular because they offer higher thermal
stability and improved heat resistance than thermoplastics.

1.2 Polymer Composite Processes
Fiber reinforced polymer composites are manufactured by a large number of processing
methods which include hand lay - up, spray - up, prepreg vacuum bagging and autoclave
curing, filament winding, pultrusion, compression molding of sheet molding compound
(SMC) and its derivatives (BMC, TMC etc.), and liquid composite molding (LCM),
which includes processes like resin transfer molding (RTM), structural reaction injection
molding (SRIM), and their variants. The following is a brief overview of the common
methods employed in the industry to manufacture fiber reinforced polymer composites.
However, since the objective of this study is the experimental investigation of the issues
related to the LCM process, it is discussed in much greater detail.
1.2.1 Hand Lay - up and Spray - up
Hand lay - up technique started in the forties and has been used since then to make
models, prototypes, and other parts that have low production volume. In this method,
layers of dry fiber mat are laid in the mold and the liquid resin is poured manually on
each layer. Entrapped air is removed by squeegees, rollers, and / or brush dabbing
[Schwartz, 1984]. The "lay - up" is made by building layer upon layer to obtain the
desired thickness. Curing is usually done at room temperature, and catalysts are often
added to speed up the reaction. This method uses mostly polyester resin and occasionally
epoxies. The reinforcement, however, is always fiberglass.
Hand lay - up technique is labor intensive and very time consuming. Thus, in an effort
to mechanize the hand lay - up process, spray - up technique was devised. In this
method, the fiber reinforcement (chopped rovings) and the catalyzed resin are
simultaneously deposited in the mold from a combination of a chopper and a spray gun.
Additional layers of the rovings and resin may be added to obtain the desired thickness.

6
Traditionally spray guns use pressurized air to spray the resin. More recently, "airless"
spray guns which dispense resin under hydraulic pressure through special nozzles have
been developed. They are preferred as they provide more controlled spray patterns and
reduce emission of volatiles [Moritz, 1993]. Typical applications of lay -u p /sp ray - up
processes include boat and boat hulls, truck roofs and housings, bathtubs, furniture etc.
1.2.2 Prepreg Vacuum Bagging and Autoclaving
In the prepreg method, wetting of the fibers occurs outside the mold. The fiber
reinforcement, usually arranged in a unidirectional tape or a woven fabric, is impregnated
with a partially cured resin. The resulting product is called a prepreg and is stored in a
freezer until molding when layers of prepregs are cut and laid into the mold. The prepreg
method allows a better control over the processing variables and thus, is a more precise
method than the hand lay - up method. However, the prepreg method usually involves
two additional steps, vacuum bagging and autoclaving. Vacuum bagging involves laying
pieces of prepreg and other materials onto a mold and enveloping the assembly with a
bag. Vacuum is pulled on the bag, which serves the dual purpose of compressing
(debulking) the prepreg plies and simultaneously withdrawing entrapped air. Further
debulking and curing is done in an autoclave pressure oven. Prepreg vacuum bagging /
autoclave curing is the traditional method for manufacturing high performance graphite /
epoxy composites for aerospace applications.
1.2.3 Filament Winding and Pultrusion
Filament winding draws fiber tows or bundles through a resin bath and wraps the
continuous tow onto a mandrel to form the part. Successive layers are added at the same
or different winding angles until the required thickness is reached. The mandrel is then
placed in an oven for curing. This process is called wet winding. Dry filament winding.

7which is less common, uses prepregs as the winding medium. Most standard composite
resins (polyesters, epoxy, phenolic etc.) can be used for filament winding. Continuous
reinforcements commonly used are glass (for price), carbon (for strength and modulus),
and aramid (for toughness and lightweight). Composite suspension leaf springs and
pressure vessels are usually filament wound [Strong, 1989].
Pultrusion is quite similar to filament winding in that it also involves passing a
continuous fiber reinforcement through a resin bath. However, instead of wrapping the
resin rich fibers onto a mandrel, the part is formed by passing the fibers through a heated
die. Resins and reinforcements used in filament winding are used in pultrusion too. The
largest market for pultruded parts is translucent building panels made from fiberglass and
polyester resin. Other applications include supports and panels for tmck trailers, door
supports for automobiles, ladder rails etc. [Strong, 1989].
1.2.4 Compression Molding o f Sheet Molding Compound
Sheet molding compound (SMC) is a complex composite of unsaturated polyester resin
to which thickeners, inorganic fillers, fiber reinforcements (usually chopped glass fibers),
catalyst, pigment, and other additives are added to form a paste like material. This paste
like material is stored for several days between layers of a carrier film, typically
polyethylene, until proper molding viscosity has been attained. Once ready, the carrier
film is removed from the charge, which is then molded in a heated matched metal mold
mounted in a hydraulic press [Moritz, 1993]. The SMC process is a reactive polymer
process in which part curing and shaping occur together. It is used extensively in the
automotive and truck industry. General Motors uses SMC for body panels of its AFV
minivans like the Pontiac Trans Sport, the Chevrolet Lumina, and the Oldsmobile
Silhouette. Each van uses approximately 320 lbs. of SMC [Wigotsky, 1989; Wood,

8
1988; Wood, 1990]. SMC is also used for roofs, rear decks, and outer door panels in
GM's redesigned F-body cars for 1993, the Chevrolet Camaro and Pontiac Firebird.
1.2.5 Liquid Composite Molding
The emergence of liquid composite molding processes (LCM) in recent years is an
excellent example of the proverbial saying, " Necessity is the mother of invention."
Liquid composite molding processes like RTM and SRIM are closed mold processes
which were developed due to the increasing desire to fabricate net or near - net shaped
disparate parts into a single unit at much lower costs, with much lower cycle times when
compared to other conventional composite molding processes. Lower costs are achieved
due to the lower energy requirements and the potential for high automation of the
process. Another major impetus for the development of LCM processes came from
restrictions imposed on chemical emissions from lay -up /sp ray - up techniques. Today,
more investment is being made in LCM than any other reinforced plastic process.
1.2.5.1 Resin Transfer Molding (RTM)
In recent years RTM has become a popular and effective fabrication technique for
producing a wide variety of composite parts. A schematic of the RTM process is shown
in Figure 1.1. The modus operandi consists of four basic steps, viz., loading the dry fiber
reinforcement into a preheated cavity followed by resin injection, resin curing, and
demolding. So, the process in itself is quite "easy" to understand. However, there are
certain "not so easy" issues involved in each of these steps that make the overall process
more complex in actual practice. The following is a brief description of the various steps
and the issues involved in RTM.

FIBERLOADING
RESININJECTION
CUREREACTION
COMPOSITEDEMOLDING
Figure 1.1 Schematic of the resin transfer molding process

10
Prior to loading the fiber reinforcement into the mold cavity, often some preparation is
needed. The entire tool surface (top and bottom halves of the mold) needs to be cleaned
and polished to give a smooth surface finish to the molded part. Several coatings of the
mold release agent are applied to the tool surface to prevent sticking and facilitate
demolding of the part. Gel coat is also often applied for the same reasons. Fiber
reinforcements also require some preparation; they are either stitched, woven, or braided
in different patterns. The prepared fiber reinforcement is called a preform. Other
methods of preforming rely on thermoforming/debulking fibers which contain
binder/tackifier. Conforming the fiber preform to snug fit the tool cavity is a more
difficult task than it might appear to be. To date, preforming is still done using an
empirical approach involving a lot of trial and error. Wrinkle formation, fiber buckling,
thickness reduction, and springback are some of the technical difficulties that arise when
the preform is made to conform to the tool cavity with a complicated geometry. While
wrinkle formation and fiber buckling are undesirable from the point of view of the
mechanical strength of the preform, thickness reduction and springback may lead to gaps
or spaces between the preform and the tool surface. This may cause channeling or race
tracking of the resin preventing complete wet-out of the fiber reinforcement. Incomplete
wetting of the fibers results in the formation of "dry spots" and "voids", which are
detrimental both to the surface quality and the mechanical strength of the molded part.
After loading the fiber preform into the mold, the mold halves are closed and clamped.
The mold containing the preform is reheated to a set temperature. Sometimes vacuum is
puUed on the mold at this stage to assist mold filling and purging of any entrapped air.
The next step in RTM is the injection of the liquid thermosetting resin into the preheated
tool cavity containing the preform. When required, packing and bleeding is done after
resin injection to ensure better impregnation of the fibers. Both one component and two

1 1
component resin systems are used. With two component systems, the components are
kept in separate tanks until injection [Chavka and Johnson, 1991; Johnson, 1990]. Just
prior to the resin injection, components are transferred to a static mixer where they are
thoroughly mixed. The mold and the fibers are usually kept at a higher temperature than
the incoming resin. Because of this temperature difference, there is an exchange of heat
from the fibers and tool surface to the incoming resin. The resin thus gets heated up
resulting in lowering of its viscosity. Low resin viscosity keeps the mold filling pressure
and mold clamping forces low and facilitates resin flow through the preform. In some
instances, the liquid resin is preheated before injection to further reduce its viscosity.
Resin is injected either at a constant flow rate or at a constant inlet pressure. Injection
pressures are usually low, so low cost tooling material like epoxy can be used. When the
resin flows out of the outlet vent, it marks the completion of the mold filling and resin
injection is stopped and the outlet vent is closed. Packing is accomplished by continuing
resin injection with the outlet valves closed. After an equilibrium pressure is attained in
the mold, the outlet vents are opened to allow some of the resin to bleed. This sequence
of packing and bleeding may be repeated several times. Although effective in getting rid
of the trapped volatiles, the drawbacks of packing and bleeding steps are that, first, the
cycle time is increased, and secondly, it results in unacceptable quantities of scrap resin.
Thus, packing and bleeding cannot be done when there is a constraint on the cycle time
and when the resin cost is prohibitive (~ $ 30 - $ 50 per pound). Incomplete displacement
of air, coupled with mechanical entrapment of voids during the resin injection step, is one
of the most serious and the least understood problems in RTM. Another reason for void
formation is the evaporation of volatile species in the resin dunng curing. Low molecular"
weight components of the resin itself may also be volatile at the curing temperature. In
addition, resins which cure by a condensation process, e.g., phenolics and some
polyimides, evolve volatiles by chemical reaction during cure [Judd and Wright, 1978].

12For most resin systems however, the main reason for void formation is mechanical
entrapment. The information available on the micro mechanics of this process is very
little compared to what is known about wetting and void formation in autoclave type
processes. It should be pointed out that in some applications it is desirable to have voids.
This is typically true when a premium is put on light weight and the composite is not
expected to perform structurally, as in foam core panels. In general however, voids are
an undesirable material defect which should be minimized or eliminated whenever
possible [Ghiorse, 1993].
After the resin injection step, the resin is cured during which the mold temperature is set
at a higher temperature in order to drive the reaction to completion. The length of the
cure cycle depends on several factors which include resin type, catalyst type and amount
used, part thickness, and curing temperature. Problems during the cure cycle include
incomplete and non - uniform cure in the part. These problems occur either due to low
cure temperature, localized variations in mold heating, localized heat generation due to
reaction exotherm, or a combination of all of these. When required, post curing is done at
a temperature higher than the cure temperature to achieve greater conversion. Post curing
may be done after the part has been demolded. The part however, must develop "green
strength" (strength a composite exhibits after resin gelation, but prior to complete cure)
before it can be demolded. Premature demolding results in inferior mechanical
properties. Typical parts manufactured using RTM are shown in Table 1.1
1.2.5.2 Stmctural Reaction Injection Molding (SRIM)
One of the variants of RTM is SRIM. The steps involved in the SRIM process are similai'
to RTM. However, there are some key differences in the resin injection and curing steps.
SRIM is a much faster process than RTM. Resin injection and curing steps get over in a

Table 1.1 Typical parts manufactured by RTM [Stark and Beitigam, 1987]
Use Part
Industrial........................Solar collectors Fan blades Water tanks
Recreational................... ................... Canoe paddlesTelevision antennae Snowmobiles
Construction................... ....................SeatingBathtubs Roof sections
Aerospace....................... ....................Airplane wing ribsCockpit hatch covers Airplane escape doors Fuselage
Automobile....................Leaf springs Side panels Bus shelters

14
matter of seconds as opposed to several minutes in the case of RTM. SRIM resins are
two component thermosetting liquid resins. They are highly reactive in comparison to
RTM resins and require very fast, high pressure impingement mixing to achieve thorough
mixing before injecting into the mold. Because of the high injection pressures used, steel
molds held together by a hydraulic press are used. The cure reaction is mixing activated
and is complete shortly after the resin reaches the outlet vent. Thus, no packing or
bleeding is possible, and if there is any air present, it remains trapped. After curing
reaction is complete, the part is removed from the mold and the process is completed.
Generally, no post cure is carried out. Typical applications of SRIM include electric
scooter frames [Ohmura et al., 1993], automotive bumper beams, instrument panels, load
floors and cross members [Babbington et al., 1990]. Table 1.2 compares the features of
the SRIM process with those of RTM [Mocosko, 1989].
1.3 Resin Systems for LCM
The selection of a resin system is primarily based on the cost and performance
requirements of the end - use application [Stark and Beitigam, 1987]. A resin system
includes, apart from the resin, other components like the curing agent, catalysts, fillers,
pigments, promoters, and inhibitors. An ideal resin is one which can stay at very low
viscosity for a long period and yet cure quickly. A long pot life allows resin injection at
lower pressures, improves fiber wet - out, and yields faster cycle times. Once cured, the
resin should have good structural and mechanical properties. Structural properties
include shrinkage characteristics, microcrack resistance, etc. Flexural modulus, tensile
strength, impact strength, and strength after impact, are some of the mechanical
properties that are taken into consideration while choosing a resin system. Other
considerations include chemical resistance, electrical properties, and fire characteristics.
15
Table 1.2 Comparison of RTM and SRIM processes [Mocosko, 1989]
RTM SRIM
Equipment cost $ 30,000 $ 500,000
Flow rate (kg / min.) 2.3 55
Mixing static mixers impingement
Mold pressure (MPa) 0.3 2.4
Typical void content (vol. %) -0.1 -0.5 -0.5 - 2.0
Mold materials epoxy steel
Mold temperature (°C) 25-40 95
Component viscosities (MPa.s) 100-550 <200
Cycle time 10-60 2 - 6

16
Thermoset RTM resins can be divided in general into two broad categories, viz.
aerospace and non - aerospace type resins. Aerospace resins include high performance,
high cost epoxies, bismaleimides, phenolics, and polyimides. Non - aerospace
applications use low cost epoxies, unsaturated polyester, vinyl ester, and hybrid resins
like blends of unsaturated polyesters and isocyanates. Typical SRIM resins are
polyurethanes, polyurethane/isocyanurates, polyurethane/polyester IPNs, and
polyurethane/urea hybrids [Lee, 1989; Mocosko, 1989].
1.4 Reinforcements for LCM
As with the resin system, the selection of the appropriate reinforcements is primarily
governed by the cost and the performance requirements of the end use application.
However, there are several other important mechanical, processing, and fiber
characteristics that also influence the choice of reinforcement. Apart from its mechanical
strength, a fiber reinforcement is characterized by four additional attributes, viz. (1) bulk
factor, which is the ratio of the volume of the given mass of "loose" reinforcement to the
volume of the same mass after forming; (2) drapeability or the ability of a fabric to
conform to the contours of the mold cavity; (3) wash resistance, or the ability to resist
movement during resin injection; and (4) wettability, or the ability to allow maximum
access to the resin to all the pores in the reinforcement.
Predominant fiber materials are glass (E and S types), graphite and kevlar. Glass fibers
are often used in parts with lower cost and performance requirements. This encompasses
most of the non - aerospace type applications. Graphite fibers provide the best property
performance with respect to their weight, and are mostly used in aerospace applications
where reduced weight and high performance characteristics are dominant factors. Kevlar
fibers are used in high temperature applications (upto 400 °C), and where high impact

17strength is required. Typical applications include aircraft and missile products, helmets,
bicycle frames, etc.
In order to enhance the physical and chemical interaction between the fibers and the
matrix resins, glass fibers are often treated with sizings. A typical commercial sizing may
include a film former, a silane coupling agent, lubricants, and additives such as anti -
statics, defoamers, and surface tension reducers [Plueddemann, 1974]. Surface
modification of graphite fibers is usually done by plasma treatment or chemical etching.
Kevlar fibers are used as is without any surface treatment.
Finally, the manner in which the fiber reinforcements are held together to form a fiber
mat or a preform is important in many ways. Many single fiber filaments are brought
together in the form of a bundle also called a roving or a tow. The most common method
for holding the fiber tows and maintaining the orientation is by using a continuous stitch.
Benefits of stitching include better interlaminar shear properties, damage tolerance, and
fiber alignment [Stark and Beitigam, 1987]. However, the use of stitches has one serious
disadvantage. Stitches interfere with the flow and result in the formation of voids. This
was found to be the case in the experiments that were conducted in this study. To avoid
this problem, other ways have been devised to hold the fiber tows. These include
weaving, knitting, and braiding the fiber tows in different patterns to obtain a self
supporting structure. Typical fiber reinforcements are formed as random chopped
strands, random continuous strands, unidirectional or bidirectional stitched mats,
unidirectional rovings, bidirectional wovens, and various other combinations using
stitching, weaving, knitting, braiding and filament winding. Weaving is done by
interlacing fiber tows of one set over and under the fiber tows of the other set. Depending
on the under over pattern, different harness types are obtained, e.g., 3 HS, 4 HS, 8 HS,
etc. The main characteristic of woven fibers is the fonnation of crimp caused by the

18under and over weave pattern at the intersection of weft and warp fibers. In knitting, the
interlacing is done by loops formed between neighboring tows in one set. Knitted fabrics
eliminate crimp and result in less bulky reinforcement. In braiding, fibers are interlaced
over a mandrel. Typical braid patterns are either two over and two under or one over and
one under. Triaxial braiding is one technique for introducing unidirectional fibers into a
braid. Woven materials have good drapeability. Non woven knitted reinforcements have
even better drapeabilty and wet-out and carry load more evenly [Margolis, 1988].
Braiding is specially suited for parts with complex shapes and is the most economical
preforming method [Becker, 1990]. Figure 1.2 shows a schematic of the braiding,
weaving and the knitting process.
Other methods employed in the industry to maintain the shape of the preforms involve
the use of binders or tackifiers. Binders are more common in automotive industry while
the use of tackifiers is specially common in the aerospace industry. These are normally
thermoplastic or thermoset resins that are solid at room temperature and are randomly
sprayed on the preform in the form of a fine powder. The preform is then heated either
by thermoforming or by debulking depending upon whether the preforms have binder or
tackifier on them. The binder / tackifier melts or reacts upon heating and solidifies either
by reacting in the case of thermosets or by cooling in the case of thermoplastics, thus
imparting rigidity to the preform. The choice of a binder / tackifier is governed by the
compatibility with the matrix resin. Usually, the tackifying material is an advanced form
of the matrix resin which can co-cure with the resin. Typical examples are epoxy,
polyester, phenolic, and bismaleimides. The amount sprayed is usually about 4 to 7
percent by weight of the preform [Hansen, 1990].
Random spraying of the binder/tackifier on the preform causes high concentration in
localized areas which results in poor wetting due to an increased resistance to resin flow.
1 9
Braided
WovenW a rp
F illin g (w e lt)
Shuttle
Knitted
Figure 1.2 Schematic of the braiding, weaving and the knitting process

20
Recently, Shields and Colton [1993] proposed a method for improving the wettability of
powder coated preforms. Instead of applying the powdered resin onto the preform itself,
they applied it to the fiber tows using an electrostatic powder fusion coating process.
They found that spreading the fiber tows during the coating process resulted in a much
improved fiber wet - out. Powdered fiber tows were then woven to obtain the preforms.
There are several different ways of applying the powder on the fiber reinforcements. One
approach, which is also investigated in this study, utilizes spraying individual layers with
a powdered tackifying material, and then stacking up the layers one on top of the other.
When heat and pressure are applied, the tackifier powder bonds the layers together into
shape. This technique is especially suited for large parts with complicated geometries. It
facilitates easier handling of the preform as a single unit, reduces bulk factor, and
improves drapeability. Moreover, it also helps in better control of the prefomi shape and
thickness. Net-shape preforms are critical to the fabrication of high performance RTM
parts. Wrinkles in the reinforcing fabric are often created when oversized preforms are
compressed into the molding tool, ultimately resulting in structural failure of the
composite [Barron, 1995].
The powder technique has historically been used to manufacture thermoplastic matrix
based prepregs. Powder coating techniques for making prepregs mainly fall into two
main categories; 1) wet powder coating that involves impregnation of fibers from a slurry
of polymer powder, and 2) a dry coating process, typically performed in a fluidized bed
with or without the aid of electrostatic deposition [Hirt et al., 1990].
1.5 Tooling and Design Considerations for LCM
The workman's adage, "if you want to do the job right, you need the right tool," was
never more apt as it is for the growing field of LCM [Monks, 1993]. A typical LCM tool

21can be broken down into five major areas, viz. the injection port(s), the air vent(s), the
guide pins, the mold cavity, and the gasket. The injection port(s) and air vent(s) provide
resin access to the mold and a means for removing volatiles and trapped air from the part.
The guide pins ensure proper alignment of the mold halves. The mold cavity imparts the
desired shape to the part, while the gasket seals the mold and prevents resin leakage
[Stark and Beitigam, 1987]. Other important considerations include a smooth surface to
minimize sticking, good temperature control, and ease of part removal.
A variety of tooling material can be used for RTM. Very low cost, unsophisticated
plastic tools can be used for extremely low volumes and prototype work [Butryn, 1991].
For low to medium volume production, epoxy, nickel, aluminum or even laminated
plastic tools with heating elements and thermocouples can be used at a relatively low
tooling cost. Steel molds with sophisticated heating arrangements are required for high
volume production.
When sizing the stiffness and thickness of a mold which is to be bolted together, internal
mold pressures must be carefully calculated. Also, the mold must be rigid enough to
compress the lofted preform without tool distortion. In the case of metal molds, hardened
shear edges to trim excess reinforcement from the preform in the pinch - off areas as the
mold is closed, reduces post molding finishing time and also provides a good seal
[Johnson, 1987].
1.6 Pumping / Dispensing Unit for LCM
Pressure pot and metering / mixing units are the two basic choices for resin injection.
Pressure pot equipment uses air pressure or a gear pump to transfer resin from the pot to
the mold. The drawback of using pressure pot equipment is that it is limited to one
component resin systems. The metering / mixing unit is more versatile and is capable of

22handling two or more component resin systems. Metering is done by a positive
displacement pump, usually a piston type, which maintains a constant volumetric ratio
between the components. Mixing is done either by static mixers in the case of RTM or
by impingement mixers in the case of SRIM. A flushing system is also used to prevent
resin gelation in the transfer system [Stark and Beitigam, 1987].
1.7 Scope of Study
Although liquid composite molding processes like RTM and SRIM have been in use in
the last several years, RTM was quoted as "the new kid on the block" [Stover, 1993]. So,
although LCM processes are increasingly being used to make composite parts, some
skepticism still exists in the minds of the people, which prevents a wider application of
these processes to make even more diverse structural components. In addition, the
numerous choices available for fiber reinforcement, resin, tooling material, and
processing conditions make it even more difficult to have a complete and an updated
database. This could explain why relatively very few people have a thorough
understanding of all the relevant issues or know how to make the most effective use of it.
As mentioned in an earlier section, one of the most serious problems is inadequate fiber
wetting and mechanical entrapment of voids during the resin injection step. Although the
problem of void formation is generic to all polymer composite manufacturing processes,
it is most serious and the least understood problem in LCM processes. The reason for
this is stringent requirements for low cycle times coupled with the complex nature of
resin flow through the fiber reinforcement. In most of the traditional processes like
prepreg vacuum bagging and autoclaving and compression molding of sheet molding
compound, etc., the fibers and the resin are in contact with each other for a prolonged
period of time. This provides intimate contact between the fiber and the resin leading to
good interface wetting and bonding. LCM processes are different in the sense that the

23fibers are initially in an unimpregnated form, and it is the complete impregnation of the
fibrous network in the shortest possible time, that is the ultimate goal. The resin injection
step in LCM processes involves two types of flow which occur simultaneously. One is
mold filling or the advancement of the bulk flow front through the larger gaps between
the fiber tows of the preform, and the other is impregnation, the local penetration of the
resin into the smaller gaps within the fiber tows. During the injection of resin into the
mold, the resin must quickly fill the mold and wet all the individual fibers before much
reaction occurs. However, this does not always happen as the resin injection step is
completed very fast (in an order of seconds for SRIM and minutes for RTM). This gives
very little time for the resin to displace all the air out of the preform. Also, depending on
the fiber architecture, injection flow rate, and resin properties, voids are trapped during
resin injection. Presence of voids result in poor wetting, and, consequently, poor bonding
yielding composite parts with non - uniform mechanical strength and / or inferior surface
quality. Excess void content (> 1%) decreases the composite's durability and fatigue
resistance and increases its susceptibility to weathering and moisture absorption.
Consistent production of high strength and good surface quality composites by LCM is
difficult and requires a much better understanding of the controlling material and
processing variables. In an earlier work [Rohatgi, 1991], influence of some of the
material and processing variables on resin - fiber bonding was studied using single
filament composites. In that study, it was assumed that the single filament was
completely wetted by the resin with no voids at the interface. Hence, the observed
differences in the behavior of composite samples was attributed to the difference in the
degree of interface bonding. Since in actual practice, a preform consisting of thousands
of single filaments is used, perfect wetting is never achieved. There are always some
amount of voids formed during resin injection. However, a better understanding of the
process and the issues involved can help minimize the problem of void formation.

24Figure 1.3 shows the governing phenomena and the process issues involved in the resin
injection step. Understanding the two type of flows during the resin injection step and
the formation of voids was the objective of the first part of this study. A new technique,
video assisted microscopy (VAM), was developed for this purpose, which is described in
Chapter ffl. The effects of flow rate, flow pattern, and liquid properties were correlated
to the microscale flow behavior and the formation of macro and micro voids in
unidirectional stitched fiberglass mats. In the past, there have been very few studies that
have systematically investigated the influence of all these factors on formation of voids.
In this text, air pockets trapped in the larger gaps between the fiber tows are referred to as
macro voids, while those trapped in the smaller gaps within the fiber tows are referred to
as micro voids. The knowledge gathered was used to construct processability diagrams.
Such diagrams can be used for selection of important material, and processing variables
for manufacturing nearly void free composites.
Another objective of this work as discussed in Chapter IV, was to study the effectiveness
of using a reactive tackifier powder to obtain "net-shape" preforms and investigate some
of the associated side effects of using the same during mold filling and curing stages.
The idea of using a reactive tackifier powder for making fiber preforms is relatively new.
The current practice in the composite industry using this technique is to mold parts by
trial and error, which is evidently very ineffective and costly. Since this is the first
scientific study that looks into all aspects of the process in detail, the results obtained
would be very useful in developing guidelines to optimize the process. The study would
be also helpful in identifying the relative importance of the relevant processing variables
that affect the different stages of molding tackified preforms.
Figure 1.4 summarizes the processing variables investigated and their role in affecting the
various processing issues. A commercial Bismaleimide (BMI) resin was used as the
25
^ R esin Injection Step ^
Process IssuesFiber Wetting (Microvoids)MacrovoidsDry Spot Formation
Product QualityMechanical Properties Surface Quality
M old Filling
Advancement of the primary flow front through the larger gaps between the fiber tows.
R esin Impregnation
Local penetration of the resin into the smaller gaps inside the fiber tows.
Figure 1.3 The resin injection step in LCM processes : governing phenomena and process issues

P rocess variab les
Concentration of Size of tackifier Application technique Debulking temperaturetackifier powder powder
(e.g. powder vs. solvent) Debulking time
Powder Coagulation
Net-shape compaction Interply adhesion & Springback
Viscoelasticproperty
Rheo - kinetics
Dissolution of tackifier in the resin
Permeability Fiber wetting
Mechanical properties of composite
Figure 1.4 Overview of resin transfer molding of polymer powder coated fiber preformsWON

27tackifier material. The effect of using this powder coating on consolidation behavior and
fiber springback in graphite preforms (6k, 4HS) were studied under two different modes
of fiber deformation (Figure 1.5).
Chapter V focuses on the effect of voids and the use of tackifier on the mechanical
properties of molded composites. Accelerated water boil test was used in conjunction
with a torsion type experiment to monitor the drop in dynamic properties of samples with
voids. Experiments were also conducted to observe the microcrack formation upon
freeze - thaw cycling of composite samples containing voids. Three point bending tests
were done according to ASTM standard D790-92 for determination of flexural properties
of both unreinforced and reinforced tackified samples. The effect of tackifier on fiber
wetting and mold filling were investigated in this chapter using a dynamic contact angle
analyzer and a centrifugal device (which measures the capillary pressure) in conjunction
with permeability experiments. The latter was also used to characterize the flow behavior
of braided reinforcements which have a different fiber architecture than stitched
unidirectional and woven type of fiber preforms.
Finally, conclusions and recommendations are summarized in Chapter VI. The overall
scope of this study is therefore, to develop a better understanding and scientific
guidelines for effective molding of textile type of reinforcements for high performance
composite applications. Moreover, even though the focus of this study was Liquid
Composite Molding, some of the findings and concepts that evolved from this work are
directly applicable to other manufacturing processes like vacuum assisted resin infusion
(SCRIMP), filament winding, pultrusion, injection-compression, hot melt prepregging,
and powder coating of SMC panels, to name a few.
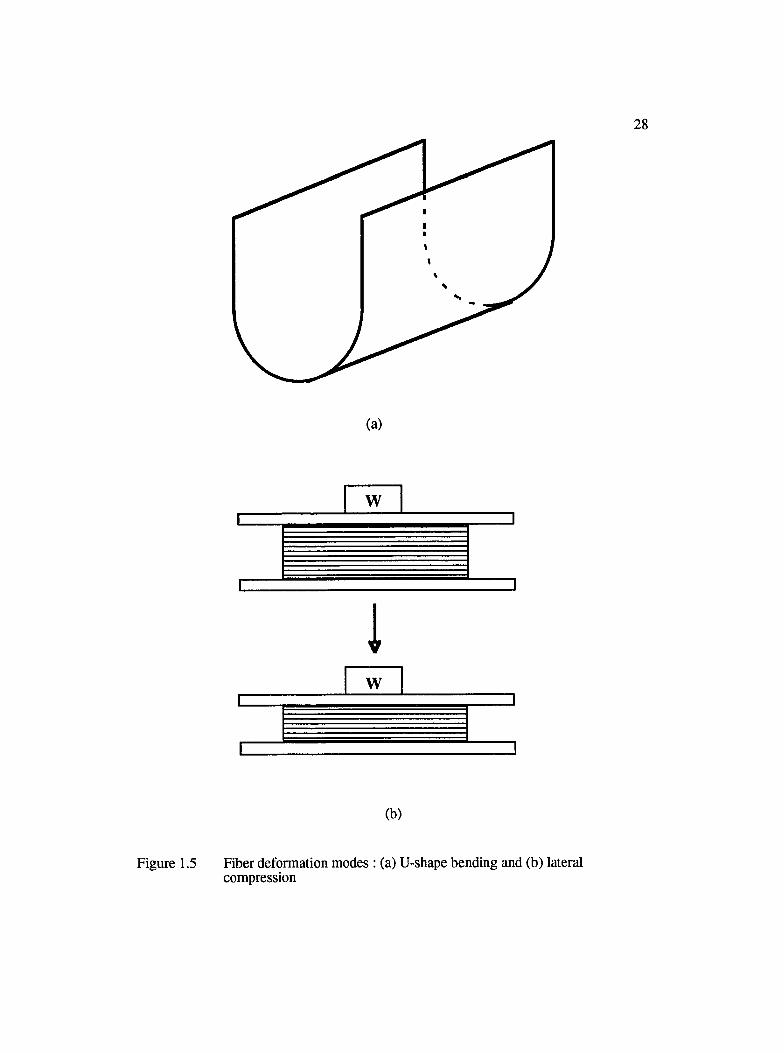
28
(a)
W
1
w
(b)
Figure 1.5 Fiber deformation modes : (a) U-shape bending and (b) lateral compression

CHAPTER H
LITERATURE REVIEW
2.1 Mold Filling and Fiber Wetting in LCM
The nature of flow of resin through the porous fiber preform in LCM is very much
similar to that of soil mechanics. In both processes, a wetting fluid flows through an
unsaturated, i.e., dry, porous medium, initially containing a non - wetting phase i.e. air.
However, one difference is that while soil mechanics is purely an infiltration type
process, LCM is not. The resin injection step in LCM processes consist of two types of
flow. One is macroflow or mold filling, which involves flow in the larger gaps between
the fiber tows. The other is microflow or resin impregnation, which involves infiltration
into the smaller gaps between the filaments of the fiber tows. Macro flow can be
considered to be an induced process in that it is initiated by an externally applied
mechanical pressure and is, thus, governed by viscous forces. Microflow, on the other
hand, can be considered to be a spontaneous process in which the driving force is the
cause of interaction between the liquid and solid phase. Microflow is mostly controlled
by the capillarity and the surface tension effects.
2.1.1 Darcy's Law
Darcy's law has often been used to model the steady state macroflow process. It states
that flow rate is directly proportional to the pressure gradient and is given by
29

30
where
Q is the flow rate
A is the cross - sectional area normal to the flow
K is the permeability of the preform (measure of the resistance to the flow)
|i is the liquid viscosity, and
1 is the pressure gradient
Based on Darcy’s law, the permeability, K, is a constant for a particular geometry of the
porous medium and is independent of flow rate and liquid properties. Certain
experiments, however, have shown otherwise, and K has been obsei’ved to vary with flow
rate and liquid properties [Dave and Houle, 1990; Foley and Gutowski, 1991]. This
discrepancy occurs because of the different rates of advancement of fluid between and
within the fiber tows. Darcy's law does not take into account any of the capillarity and
surface tension effects that govern the flow within the fiber tows. Thus, it becomes
evident from Equation 2.1 that Darcy's law alone cannot be used to predict the overall
flow process in LCM. To take into account the unsteady nature of the flow within the
fiber tows instead of taking permeability to be constant, it should be defined as:
K = Ki * Kr (2.2)
where
Ki is the intrinsic permeability
Kr is the relative permeability

31Ki is obtained from the Kozeny - Carman equation, and Kr varies from 0 to 1 depending
on the saturation of the porous medium [Dave and Houle, 1990]. In the following
sections, some of the theoretical and empirical models and correlations relevant to the
microflow process are discussed.
2.1.2 Equilibrium Contact Angle
When a liquid comes in contact with a solid surface, the equilibrium condition for wetting
is determined by the three phase equilibrium of solid, liquid, and vapor. As shown in
Figure 2.1, the equilibrium point of contact is described as the intersection of three
interfaces: solid - liquid, liquid - vapor and solid - vapor. The equilibrium condition is
given by the Young - Dupre' equation [Miller, 1977] as
Ysv ~ Y s l “ Y l v Cos0 (2.3)
where
Ysv is the solid surface energy
Ysl is the solid - liquid interface tension
Ylv is the liquid surface tension, and
Cos 9 is the cosine of the equilibrium contact angle 0
Contact angle is usually considered to be a measure of the wettability or degree of
wetting. Based on the magnitude of the contact angle, the liquid can be classified as
either one of the three, viz., spreading (6 = 0), wetting (O<0<9O°), or non - wettingO O
(90 <0<18O ). When a wetting liquid penetrates the empty capillaries, the solid - air
interface is replaced by the solid - liquid interface. The higher the value of the adhesion
tension ( Ylv Cos0), the more readily the wetting proceeds [Carino and Mollet, 1975]. In
general, in order to ensure wetting, the liquid phase should have a surface free energy or
surface tension lower than the solid surface free energy. Measuring the contact angles

32with liquids of known surface tension also provides a means of quantifying the
interaction between solids and liquids. The physical property that determines the extent
of this interaction is the work of adhesion between the solid and the liquid given by
[Kamath et a l, 1987]
W a = Y L v ( l + Cos0) (2.4)
2.1.3 Dynamic Contact Angle
When the liquid is in motion with respect to the solid or vice versa, the solid - liquid -
vapor interface is under transient conditions. The value of the contact angle then differs
from its equilibrium value and a dynamic contact angle develops. Even at very low
velocities, the dynamic contact angle is considerably larger than the equilibrium value.
Dynamic contact angle could be either advancing or receding depending on whether one
surface is advancing or receding over the other. Usually the values of the advancing and
receding contact angles are not the same except in cases of perfect wetting (Cos 0 = 1).
Advancing contact angles are greater than or equal to the receding contact angles. The
difference is termed as the contact angle hysteresis. Some of the sources of hysteresis
include surface roughness, surface heterogeneity, increased liquid penetration due to
diffusion, and surface deformation/relaxation effects [Domingue, 1992]. Since the nature
of LCM processes is to make the resin advance through the fiber preform advancing
contact angles are more relevant. Thus, any mention of the contact angle hereafter refers
to the advancing dynamic contact angle, unless stated otherwise.
The dynamic contact angle is influenced by the resin viscosity and surface tension.
Several studies have been undertaken to describe the effects of viscosity and surface
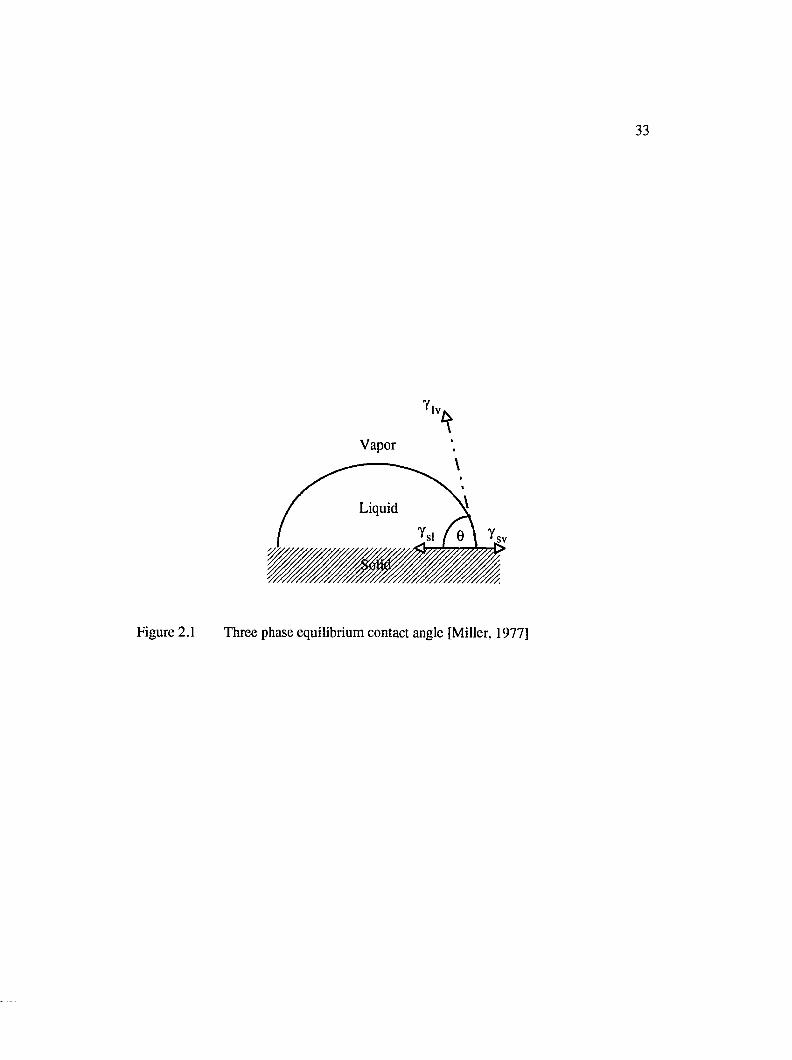
33
' I v ,
Vapor
Liquid
sv
Figure 2.1 Three phase equilibrium contact angle [Miller, 1977]

34
tension on the measured dynamic contact angle. This phenomenon is usually described
by the dimensionless capillary number (Ca#), defined as:
Ca# = ^ (2.5)Y l v
where
H is the viscosity of the impregnating resin, and
V is the relative velocity of the resin past the dry fibers
In general, the dynamic contact angle is reported to remain constant at sufficiently low
values of the capillary number, and then increase as Ca # increases. This was attributed
to the distortion of the meniscus shape due to viscous effects. The dependence of the
contact angle on the viscous drag was observed in the range of 10'^ < Ca # < 10*5 [Ahn
and Seferis, 1991].
Elmendorp and During [1991] obtained a relation between the dynamic contact angle and
the capillary number based on the precursor film model. This model explains the physics
behind the spreading of a liquid over a solid substrate. According to this model, the
liquid closest to the solid experiences the largest molecular attraction and so tends to
move faster than the bulk liquid. This causes a concavity in the liquid surface. Away
from the solid surface, the shape of the liquid surface, which determines the dynamic
contact angle, is governed by the balance of surface tension and viscous forces. The
correlation between the dynamic contact angle, equilibrium contact angle, and Ca # was
given as:
e / - e / = 53*C a# (2.6)

35Another correlation for dynamic contact angle may be described by the Friz equation for
resins with viscosities in the range of 10 -1000 mPas moving at velocities of the order of
0 .1 -1 0 cm/sec [Hayward and Harris, 1990]
tanOj = mV Y lv y
(2.7)
where
m and n are constants
Contact angle analysis is further complicated by the nature of the resin system and the
fiber surface. Resin systems usually consist of a mixture of compounds. Selective
adsorption of a certain species out of the resin or even a chemical reaction with the fiber
surface would create a composition gradient and may lead to changes in the contact
angle. In the same token, changes in fiber wettability would occur if the sizing or the
binder on the fiber interacts either physically or chemically with the resin. In addition to
these, chemical reaction and evaporation of volatile chemical species may also affect the
surface energies at the interface altering the dynamic contact angle. The effect of fiber
sizing on the contact angle was quantified in a recent study by Larson and Drzal [1992].
In the same study it was observed that evaporation of styrene from a vinyl ester resin
substantially increased the surface free energy of the resin and changed the fiber - liquid
interaction from favorable to unfavorable. Contamination of glass fibers with styrene
vapor and liquid resulted in an increase in contact angle, and zones of poor wetting with
increased porosity were observed in the molded composite [Hayward and Harris, 1990].
2.1.4 Capillary Pressure
In addition to the dynamic contact angle, another fundamental parameter to be considered
in flow through porous media is the capillary pressure (Pc). Capillary pressure is defined

36as the difference in the pressure between the wetting and the non - wetting phase at the
liquid - air interface.
Pc = Pnw - Pw = Ylv
r 1
1--------- (2 .8)
Equation 2.8 is the Laplace equation of capillarity. ri and r% are the principal radii of
curvature of the meniscus. The quantity is sometimes referred to as the mean curvature
of the interface, and depends on the shape and size of the capillary [Corey, 1986].
In soil mechanics, capillary pressure is termed as "suction". This is not surprising as it
provides the driving force for the liquid to impregnate the porous media. Capillary
pressure is a function of saturation. It is an increasing function of the non - wetting phase
saturation or, alternately, a decreasing function of the wetting phase saturation. The two
functions however, are not the same, and capillary pressure, like contact angle exhibits
hysteresis. Saturation is defined as the ratio of the volume of the fluid phase to the total
accessible pore volume. Thus, the capillary pressure is at the maximum when the fibers
are dry, and then decreases as the fibers get more and more wet or the wetting phase
saturation increases.
Assuming the filaments in the fiber tows to be cylindrical capillaries, a simple
relationship for evaluating the capillary pressure was obtained [Skartsis et al., 1992]:
(2.9)
where
d is a measure of the size of the capillary or the flow channel
The above equation is known as the Young - Laplace equation. The problem in using
Equation 2.9 is that it is very difficult to obtain realistic estimates for d. This is so

37because a typical fiber preform consists of a large number of capillaries of varying shapes
and sizes. One approach to estimate d utilizes the concept of hydraulic diameter, Dh.
This concept is identical to the one used to derive the Blake - Kozeny - Cannan equation
for flow through porous media [Skartsis et al., 1992]. In another approach, d was treated
as a function of porosity and the diameter of a single fiber (Df). In the same study,
another factor, the form factor (F), based on fiber alignment and flow direction, was
incorporated into the definition of the capillary pressure given by
P, = — Cos e (2.10)Df <})
where
<t> is the fiber porosity
For unidirectional fibrous preforms, it was found that F assumes a value of 4 for flow
along the fiber direction and a value of 2 for flow normal to the fiber direction. For
complex fiber alignment such as in woven fiber preforms, the study mentioned that F
could be determined from permeability measurements [Ahn and Seferis, 1991]. Typical
capillary pressures measured by Ahn et al. was in the order of 5 - 6 psi. Since in actual
production of composite parts by say an RTM process, injection pressures are usually of
the order 100 - 200 psi, one might argue the role played by the capillary pressure and the
research efforts being directed towards its determination. It should be noted that even
though the magnitude of the injection pressure or the applied mechanical pressure at the
inlet is many times higher than the capillary pressure, the situation is reversed at the flow
front. At the flow front, the mechanical pressure is zero, and it is the magnitude of the
capillary pressure that governs the resin impregnation. The importance of capillary
pressure is even more significant in low pressure processing, such as prepregging,
filament winding, and resin infusion processes [Ahn and Seferis, 1991].

38
2.1.5 Wicking Phenomena
Spontaneous liquid penetration of porous materials under the influence of capillary forces
is termed as wicking. The wicking phenomenon is commonly described by the theory of
Lucas and Washburn, which models the porous media as a bundle of cylindrical capillary
tubes and assumes a quasi steady creeping flow [Hodgson and Berg, 1988]. Substituting
the expression for Pc into the Hagen-Poiseuille equation, the following expression was
obtained to relate the rate of capillary penetration as a function of liquid properties and
effective capillary dimensions:
dh 1dt 8[ih
2yCos8- p g h *r‘ (2 .11)
Equation 2.11 was developed to model capillary flow in the vertical direction. Since
resin injection in LCM processes is done with the mold kept horizontally, the
gravitational forces can be neglected and Equation 2.11 reduces to
dh 1dt 8 |ih
2 y C os6 * r (2 .12)
Equation 2.12 can be readily integrated to yield the expression for distance traveled (h) as
a function of time (t).
h = ryCosG 2\i .
VF (2.13)
Although the above equation has been substantiated by wicking experiments with pure
liquids in several porous media, deviations have been obsei^ved for very short wicking
distances and in media which are swollen by the penetrating liquid. The theory also
shows some deviations for multi component penetrating liquids like suifactant solutions
[Hodgson and Berg, 1988]. The Lucas - Washburn theory would also not be applicable if

39the pore dimensions in the capillary are not uniform. An explanation for this is given by
Bayramli and Powell [1991]. In heterogeneous media, there is a fast axial motion of the
liquid in larger pores coupled with slower lateral motion of the liquid into the
neighboring smaller pores. The development of Equation 2.13 was obtained by
substituting the expression for capillary pressure in the Hagen - Poiseuille equation. The
presence of a meniscus at the liquid - air interface imposes a condition of plug flow at the
front of the liquid column, whereas use of Hagen - Poiseuille equation implies a parabolic
velocity distribution. This difference was compensated for by a fountain type motion of
fluids on both sides of the interface [Dullien, 1992].
2.2 Contact Angle and Surface Tension Measurements
Based on the discussion so far, it is evident that contact angle and surface tension play a
very important role in characterizing fiber wettability and penetration of resin into the
fibers. Nearly all the equations mentioned thus far incorporate these parameters. Thus,
their accurate determination becomes very important. The next section is therefore
devoted to describe some of the experimental techniques and the approaches that have
been used by previous researchers for contact angle and surface tension measurements.
2.2.1 Direct Observation Contact Angle
Most of the established experimental techniques for evaluating surface wetting properties
have been developed for use with flat surfaces. Under such conditions, the observation
and measurement of the contact angle is not difficult. Direct and photographic
measurements can be made using a microscope fitted with a camera. Nowadays,
sophisticated image analysis softwares are available for accurate measurement of contact
angles using the sessile drop technique. In this technique, a small hole is drilled in the
substrate using a sharp needle and a drop is made to grow from the bottom. Figure 2.2
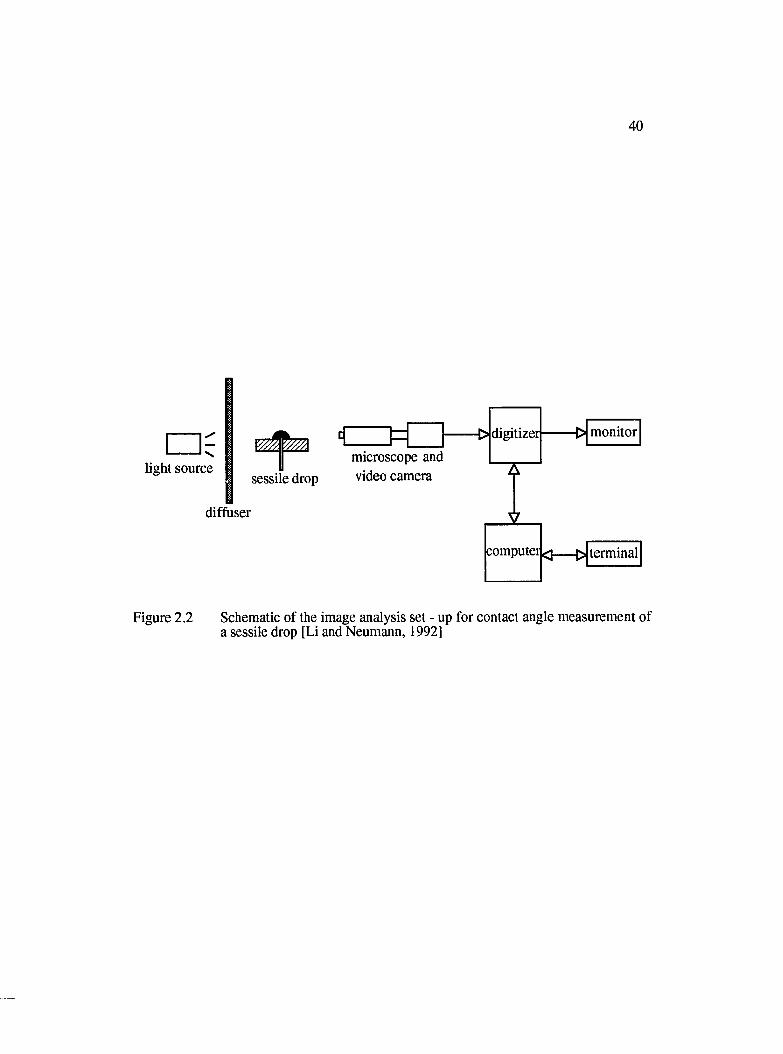
40
light sourcesessi e drop
microscope and video camera
diffuser
> monitordigitizer
computer < 3-----0 terminal
Figure 2.2 Schematic of the image analysis set - up for contact angle measurement of a sessile drop [Li and Neumann, 1992]

41
shows the block diagram of a typical digital image processing set - up for sessile drop
measurements. The video signal of the drop is transmitted to a digitizer which performs
the frame grabbing and digitization of the image to 512 by 512 pixels with 256 gray
levels each, where 0 represents black and 255 represents white. The digitized image is
transferred to the computer for image analysis and computation. Advancing contact
angles can also be obtained using this method by forming the drop using a motor driven
syringe.
Making contact angle measurements on single filaments is a much more difficult
experimental task. The direct approach requires that a drop of liquid be placed on a
horizontally mounted sample, and the contact angle be observed from a point in the same
horizontal plane and perpendicular to the long axis of the sample. However, this type of
experiment has several pitfalls. This is so because certain liquids have the tendency to
completely surround a single fiber and form a symmetrical unduloid shape, while others
can remain on one side of a fiber with a "clamshell" profile. In some instances, the same
liquid could be made to adopt either of these configurations depending upon how it was
deposited. Thus, this approach requires considerable precaution to make sure that the
true contact angle is observed and measured. The reason why such precision is required
is explained in the plot of Cos 0 as a function of 0 as shown in Figure 2.3. The plot
demonstrates the percentage uncertainty in the former due to a ± 1 ° error in measuring 0
between 0° and 90°.
Due to the inherent difficulties involved in accurate measurement of contact angles on
single fibers using direct methods, an indirect method was devised and successfully
adapted. This method is based on the Wilhelmy principle and is discussed next.
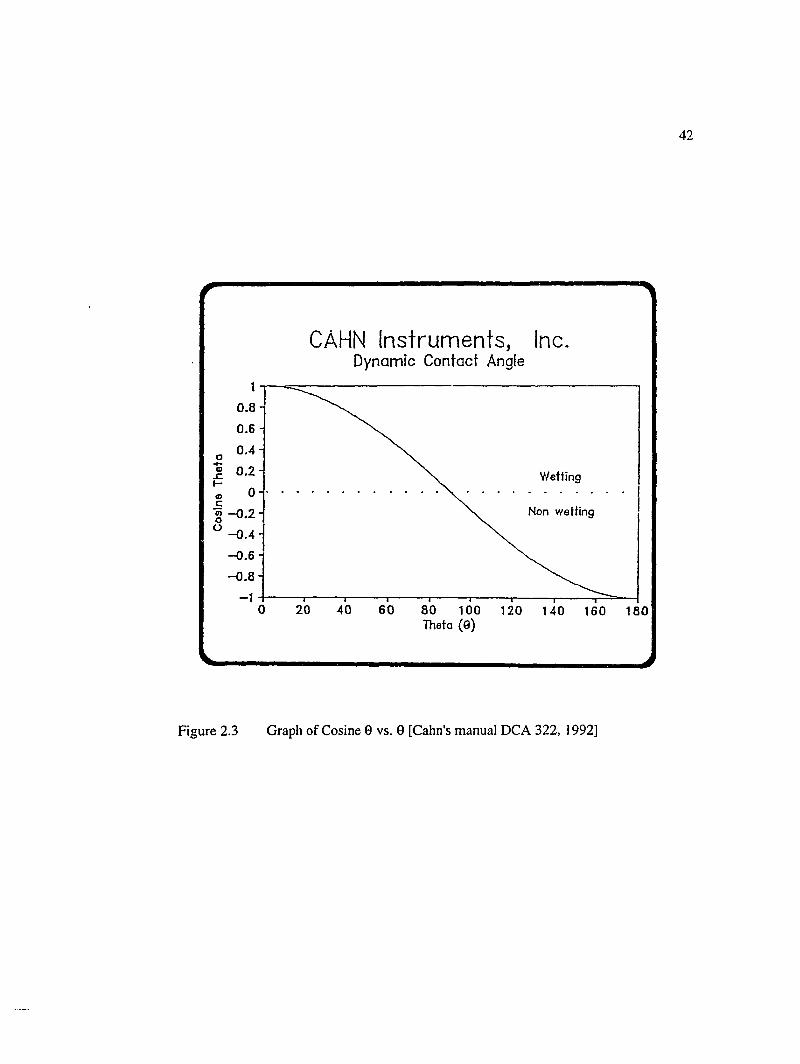
42
CÀHN Instruments, Inc.Dynamic Contact Angle
0 .8-1
0.6 -
o 0.4- 0 .2 -CJC
I -W ettin g
I -0.2-“ - 0 .4 -
— 0.6 -
- 0 . 8 -
Non w etting
0 20 40 60 80 100 120 140 160 180T h ê ta ( 0 )
Figure 2.3 Graph of Cosine 0 vs. 0 [Cahn's manual DCA 322, 1992]

43
2.2.2 Wetting Force Measurements
Since the development of the Wilhelmy technique, it has been applied extensively to
study the dynamic wetting behavior of fibers for various liquid / fiber systems. Lee and
Seferis [1988] used this technique to characterize the effect of surface treatment of carbon
fibers with a silicone oil and a low surface tension epoxy. This method is an indirect
method for determining contact angles as it does not involve direct observation of the
shape of the liquid surface on the solid. Rather, this technique involves measuring the
force that a liquid exerts on the fiber surface and calculating the contact angle based on
force values. According to the formula of Wilhelmy, the pull exerted on the fiber
inserted into a liquid is expressed by
Fw = P *Ylv *Cos6 (2.14)
P is the perimeter of the fiber along the three phase boundary line, and the other terms
have their usual meaning. If the perimeter and the surface tension are known, the contact
angle can be evaluated from Fw- The determination of the wetting force Fw can be
carried out by measuring the change in weight of a vertical fiber (with a microbalance)
that occurs when it is placed in contact with a liquid. Figure 2.4 illustrates this concept in
terms of the forces acting on the fiber before and after contact [Miller, 1977].
2.2.3 Bundle Contact Angle
For composite manufacturing processes, ideally the contact angle of fiber tows should be
measured instead of the contact angle with single fibers. Two different approaches have
been employed to do so. Chwastiak [1973] used wicking experiments to determine
bundle contact angles from changes in the surface free energy which occurs during the
wetting process. Another approach uses the same technique as that used for measuring
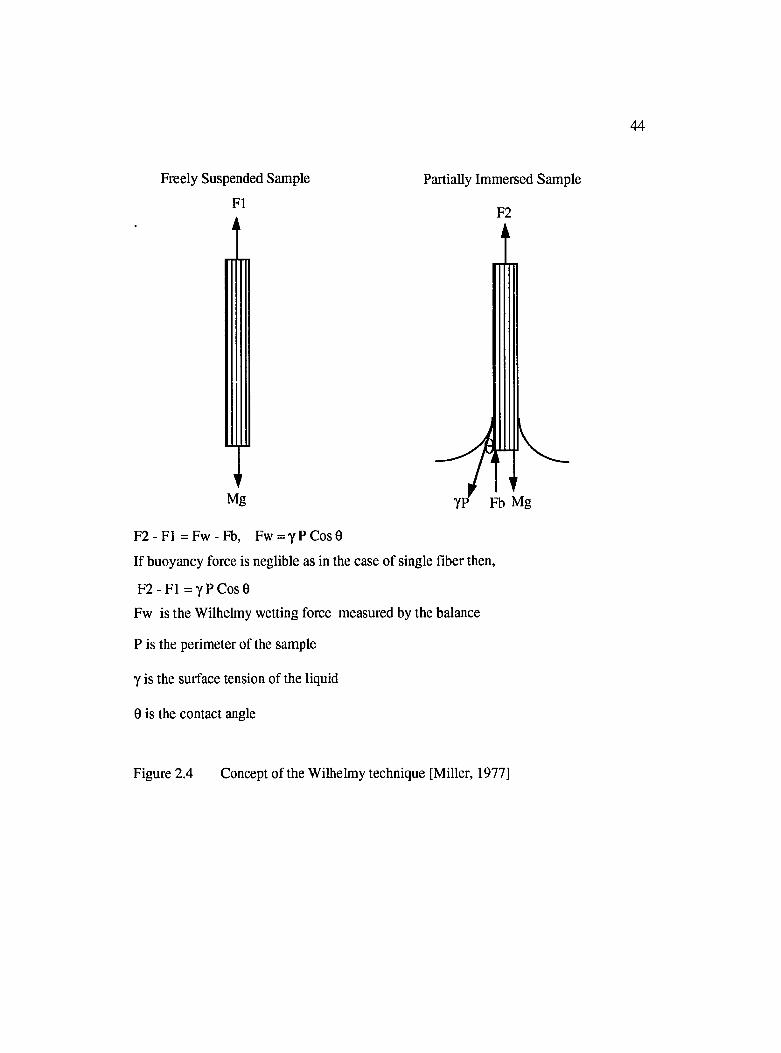
44
Freely Suspended Sample
FI
Partially Immersed Sample
F2
Mg
F 2 - F l = F w - F b , F w = y P C o s 9
If buoyancy force is neglible as in the case of single fiber then,
F2-F1 = y P C o s e
Fw is the Wilhelmy wetting force measured by the balance
P is the perimeter of the sample
Y is the surface tension of the liquid
0 is the contact angle
Fb Mg
Figure 2.4 Concept of tbe Wilhelmy technique [Miller, 1977]

45
contact angles with single fibers. The latter approach is much simpler. However, with
fiber tows, in addition to wetting, liquid uptake due to wicking also occurs. Thus, for
contact angle measurements, the two effects need to be decoupled.
Chwastiak obtained the following equation for change in the surface free energy by
neglecting the inertial term in comparison to the gravitational and viscous forces:
128(1Ay = ^ g P r A Ü ( m ) (2.15)
H is the total height of the fiber bundle, pf is the fiber density, df is the fiber diameter, p
is the liquid viscosity, (]) is the porosity, Kh is the hydraulic constant, V j is the total
volume of the bundle, Wf is the weight of the filaments in the fiber bundle, m is the
weight of the liquid wicked in time t, and X is the slope of the wicking data m vs. t on a
log - log plot.
All the terms on the right hand side of equation were readily obtained except for the
hydraulic constant. The porosity of the bundle was kept fixed by enclosing them tightly
in a tube. Thus, the accuracy of Equation 2.15 depends upon the accuracy with which the
hydraulic constant is determined. Once the free energy of wetting was obtained, the
contact angle was evaluated as follows
Cos0 = - ^ (2.16)Y lv
Hsieh and Yu [1992] used the Wilhelmy technique to determine the contact angle of
water on woven fabric strips. The experimental protocol used by them is explained as
follows. A strip of woven fabric was hung from a microbalance. The surface of the
liquid reservoir resting on a traveling stage was then brought close to the edge of the
fabric just enough to touch it. The force reading was recorded until a steady - state in the

46measurement was reached. The liquid surface was then pulled away from the strip and
the drop in the force readings was recorded until a steady state was achieved. Figure 2.5
is a schematic of a typical curve illustrating the weight changes during the experiment.
Point A to B is the zero baseline prior to the fabric - liquid contact. Point B to D shows
the force increase from the liquid contacting the fabric and the simultaneous wicking
action. The initial sharp force increase from point B to point C is mostly due to wetting
with some contribution from liquid uptake. The subsequent wicking is indicated by the
slower force change from point C to point D before reaching the steady state. Point D to
E records the separation process of the wetted fabric from the liquid. As the liquid level
moved away from the fabric edge, the liquid surface remained in contact with the lower
edge of the fabric. The slight increase in the force detection during this process was
attributed to the change in the fabric edge configuration and / or change in the contact
angle at the meniscus. At point F, the fabric is completely separated from the liquid. The
residual weight recorded indicates the total liquid retention (Wt) in the fabric.
The B - D section of the curve shown in Figure 2.5 results from simultaneous wetting and
wicking. The amount wicked into the fabric was obtained from Wt, The advance steady -
state force at point D was denoted as (A st) , and the wetting force was obtained as;
Fw = Ast -Wt (2.17)
The perimeter of the fabric was obtained from a similar experiment, but with a perfectly
wetting liquid with known surface tension, such as Hexadecane which made a zero
contact angle with the fabric. Once the perimeter of the fabric was known the contact
angle was calculated from the Wilhelmy equation after inputting the value of the surface
tension of water.

47
bû
-O
Time, sec.
Figure 2.5 A typical curve illustrating the weight changes with time in a wicking experiment [Hsieh and Yu, 1992]

48
2.2.4 Surface Tension Measurements
Surface tension of a liquid is a thermodynamic property, and for pure liquids depends
only on temperature, with respect to which it shows a monotonie decrease [Ahn and
Seferis, 1991]. Surface tension of polymer resins have been observed to change with
reaction. This was explained by Larson and Drzal [1992] as due to the evaporation of
volatile species. The following methods are used for its determination [Shaw, 1980].
2.2.4.1 Capillary Rise Method
This method is based on the measurement of the height of liquid column (h) in a capillary
of radius r immersed in a liquid. Since the measurements do not involve disturbance of
the liquid surface, slow time effects can be followed. Surface tension is given by the
expression
which for zero contact angle reduces to
(2 .1 9 )
where
Ap is the difference in the density of the liquid and vapor
For accuracy, corrections should be made for very narrow and wide capillaries. In
practice, the capillary rise method should only be used when the contact angle is zero,
owing to the uncertainty in measuring contact angles correctly. A variation of this
method is to measure the difference in capillary rise for capillaries of different sizes, thus
eliminating reference to the flat surface of the reservoir liquid. In this case, surface
tension is given by Equation 2.20:

49
A p £ iv ^ (2.20)2(ri-r,)
where
Ah is the difference in the height of the liquid meniscus in the two capillaries
2.2A.2 Ring Method
In this method the force required to detach a ring from a surface or interface is measured
either by suspending the ring from the arm of a balance or by using a torsion wire
arrangement (du Nouy tensiometer). The detachment force is related to the surface
tension by the expression
F is the puU on the ring, R is the mean radius of the ring, and p is a correction factor. To
ensure zero contact angle, platinum rings should be cleaned with a strong acid or by
flaming. The correction factor allows for the non vertical direction of the tension forces
and for the complex shape of the liquid supported by the ring at the point of detachment.
The value of P can be calculated from the equation of Zuidema and Waters [Shaw, 1980].
2.2.4.3 Drop Volume and Drop Weight Methods
Drops of liquid are allowed to detach themselves slowly from the tip of a vertically
mounted narrow tube, and either they are weighed or their volume is measured. At the
point of detachment
(2 .22)' 27ir 27ir

50
P is a correction factor which is required because on detachment, (a) the drop does not
completely leave the tip, and (b) the surface tension forces are seldom exactly vertical. P
is empirically shown to depend on the ratio
2.2.4A Pendant Drop Method
A pendant drop of the liquid is photographed, and its image is projected on to a graph
paper. From the dimensions of the drop, the surface tension can be computed using the
same image analysis software as that used in the sessile drop method for calculating
contact angles [Li and Neuman, 1992].
2.2.4.5 Wilhelmy Technique
In this method, a heat cleaned glass cover plate or a platinum plate is immersed vertically
in the liquid. The force exerted by the liquid on the plate is monitored using a
microbalance. If the perimeter of the plate is known, surface tension can be calculated
using the Wilhelmy Equation 2.14. The underlying assumption in this method is that the
plate makes a zero contact angle with the liquid whose surface tension is to be measured.
Wilhehny technique was used in this study, and details are given in the next chapter.
2.3 Void Formation Studies
2.3.J Experimental Studies on Void Formation
Bascom and Romans [1968] were first to report the formation of voids between the
filaments of fiber tows of glass fiber reinforced polyester resin composites produced by
filament winding. Microscopic observation of small composite samples showed 10^ to
10^ voids per cubic centimeter. They found that reducing the contact angle to zero, and

51causing the strands to oscillate as they passed through the resin markedly reduced the
number of voids.
Peterson and Robertson [1991 & 1992] studied the formation of voids in fiber rovings.
The primary factors in the generation of voids were attributed to the heterogeneity both
on bulk and local scales. They explained that heterogeneity on bulk scale results in
channeling causing void formation. Heterogeneity on the local scale results in trapping
of voids by coalescence of wicking streams. They also explained why changes in the
void size and distribution occurs during the mold filling process in terms of viscous
forces. The viscous action of the resin can free formerly trapped voids allowing them to
flow out with the resin. Viscous forces may also move a void lodged in a constriction by
reducing its size. Under static conditions, changes in void size occurs by coalescence due
to surface tension.
Mahale et al. [1992] used the refractive index matching technique and image analysis to
quantify void formation during radial impregnation of random continuous glass fiber
mats. They reported a critical value of capillary number (Ca # = 2.5 x 10"3), below
which void contents increased exponentially with decreasing capillary numbers. Above
this critical value negligible entrapment of voids was observed. Chen et al. [1993] used a
similar approach to visualize void formation using an oil with the same refractive index
as the fibers. Based on the sizes and location of the voids, they classified them as (1)
small cylindrical micro voids inside the fiber bundles, mesovoids encompassing several
filaments inside the fiber bundles, and spherical macro voids outside the fiber bundles.
They found that low viscosity, zero contact angle, high mold temperature, and high
pressure resulted in minimal void formation. Also, increasing the fiber volume fraction
reduced the amount of voids trapped. They reported that if capillarity is dominant,
surfactants can help in reducing voids by reducing the wetting contact angle.

52Hayward and Harris [1990] studied the effect of vacuum on void formation in glass fiber
reinforced polyester resin system. They found that resin injection with the assistance of
vacuum resulted in substantial improvements in composite quality (i.e., reduced void
formation). Their experiments showed that the effects of vacuum assistance occurred
only at the point of initial contact of the resin front with the glass preform. Subsequent
resin injection using vacuum did not lead to improvement in previously poorly wet - out
portions of the preform. A similar study on the effect of vacuum has also been perfonned
by Lundstrom et al. [1992]. They reported that most of the voids were concentrated to a
small area close to the flow front. With increasing vacuum, the maximum void content
and the size of the region of detectable voids decreased. The void volume fraction was
detemiined with optical microscopy and image analysis. Stabler et al. [1992] studied
drag induced void formation in braided graphite prefonn and epoxy system. They
observed that most of the voids were located in the indention regions where the fiber tows
crossed each other. They explained that voids were trapped because indention regions
were low pressure regions. Once the air bubble was lodged at the notch, it was very
difficult to move. They reported that for conditions of light mold surface waxing with
buffing, low initial bubble content, and mold vibration frequency of 10 Hz, void
formation was minimized.
2.3.2 Modeling o f Void Formation
Several theoretical models have been proposed for void formation. Although these
models are formulated using several simplifying assumptions and for simplified
geometry, they indicate some of the physical phenomena that takes place on the micro
scale.
Elmendorp and During [1990] developed a model for void formation during transverse
flow in aligned hexagonal array of fibers. The shape of the liquid surface advancing

53through the fiber bundles was determined based on the precursor film model. They
assumed that the viscous forces do no not influence the shape of the liquid surface other
than altering the value of the static contact angle. Their model predicted that if upon
coalescence of two flow fronts the dynamic contact angle was greater than tc/2, a void
would be formed downstream of the fiber. They reported that for capillary number
greater than 0.05, voids can be trapped due to dynamic effects. This implied a critical
impregnation velocity of 1.5 micron/sec. Also, at a particular volume fraction, void
content increased with increasing capillary number. The capillary number at which void
formation started (critical capillary number) decreased with increase in fiber volume
fraction.
Void formation during flow perpendicular to unidirectional fiber tows has been modeled
by Parnas and Phelan [1991]. Darcy's law was used to describe both macro and micro
flow. Consideration of capillary forces was neglected. The concept of this model is
based on the premise that as the advancing flow front encounters a fiber bundle, it fiows
around it, entrapping a pocket of air as it does so. After the front surrounds and bypasses
a fiber bundle, the fiber bundle is slowly impregnated with the fluid. The basis for the
assumed entrapment mechanism is that the interstitial space within the fiber bundles is
much smaller than the spaces between the fiber bundles that make up the preform. Thus,
the permeability of a fiber bundle, k%, is much less than the permeability of the space
between the bundles, ki. Considering the flow geometry depicted in Figure 2.6, when
ki/k2 equals 1.0, the flow front penetrates the fiber bundles at the same rate as it advances
through the space between the bundles and no air is trapped. However, as the fiber
bundles become successively less permeable and the ratio ki/k] tends to infinity, the rate
of fiber impregnation relative to the motion of the advancing front becomes negligible.
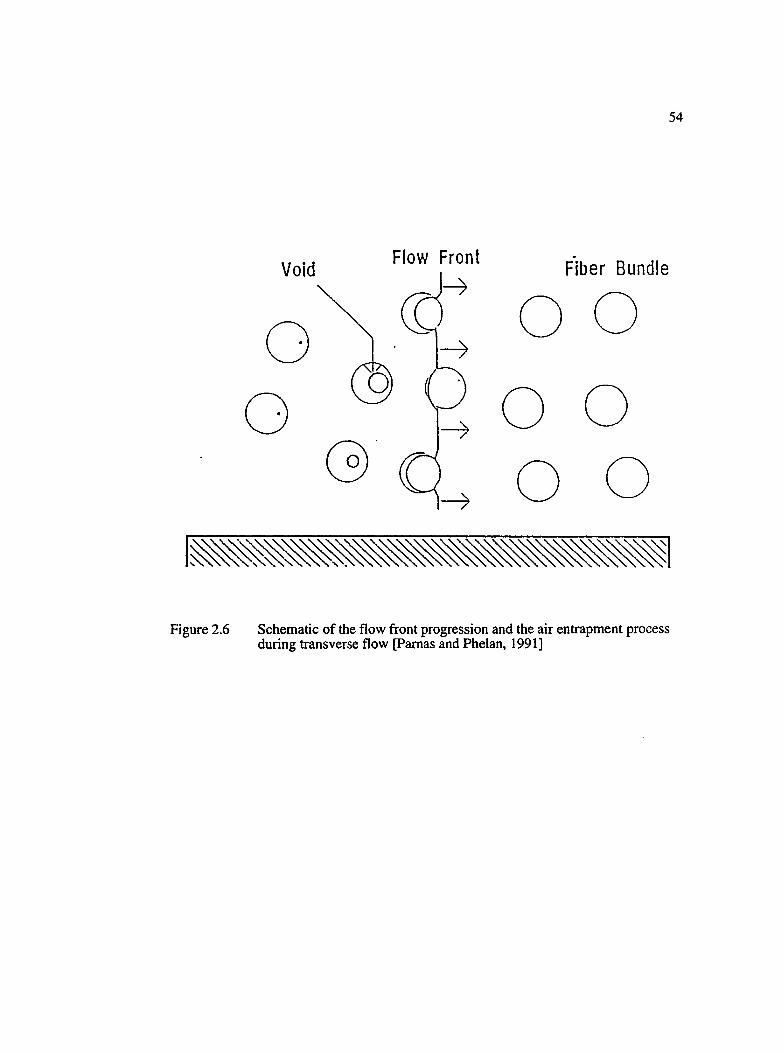
54
\/nîH F l o w Front F i b e r B u n d l e
Figure 2.6 Schematic of the flow front progression and the air entrapment process during transverse flow [Pamas and Phelan, 1991]

55
When a point is reached where the flow front splits into two upon reaching a fiber bundle
and then recoalesces downstream of the bundle before all the air in the bundles can be
forced out, air is trapped within the bundles and remains trapped.
Chan and Morgan [1992] have developed some simple models for void formation under
different scenarios. One of their models predicted void formation for axial flow in the
unidirectional fiber preform. Again Darcy's law was used to model resin flow. The flow
was characterized by a global flow front advancing through the larger pores between the
fiber bundles with subsequent radial penetration into the smaller pores within the bundles.
Because of the differences in time scales associated with the two types of filling, voids
were trapped at the flow front. Chan and Morgan [1993a] used a similar approach to
model void formation for circular, elliptical and rectangular cross - sectioned tows
oriented either along or normal to the global resin flow direction. The model was used to
estimate the void size and distribution in fabric preforms. Another work by Chan and
Morgan [1993b] focuses on the formation of microvoids within the fiber tows. Figure 2.7
depicts a schematic of two capillaries lying adjacent to each other, but of unequal sizes.
The model assumes that at time equals zero (t = 0), the resin front in the two capillaries
coincide in the axial direction. A net capillary pressure, which is the difference in
capillary pressures between the two fronts, exists. The net capillary pressure causes the
resin to flow forward in the smaller capillary. This forward flow leads to a transverse
flow of resin from the larger to the smaller capillary. The forward flow in the smaller
capillary is accompanied by a reverse flow in the larger capillary. They suggested that
this forward - reverse capillary flow leads to the possibility for void formation in two
ways. The forward moving resin front in the smaller capillary can merge with another
such forward moving front to enclose a void resulting in a void length of approximately L
(Figure 2.7). Voids can also form when the surrounding resin flows transversely across

56
II
U l
time = 0
time > 0
Figure 2.7 Schematic depiction of the channeling flow from larger to smaller capillary [Chan and Morgan, 1993b]

57
to enclose the void region left by the receding front in the larger capillary. This leads to a
void of length L].
Chen et al. [1993] also proposed a model based on the concept of two levels of porosity
of fiber mats. One outside the fiber tows, and the other within the fiber tows. Their
model includes liquid bypassing with initial air trapping, subsequent capillary invasion of
fiber bundle with air compression, and mobilization to explain air entrapment
phenomena. The concept of liquid bypassing and initial air trapping is similar to that of
Pamas and Phelan and Chan and Morgan. During flow, most of the liquid will pass
through the space between the bundles bypassing the space within the bundles. However,
some of the liquid will penetrate into the fiber bundles owing to capillary suction and the
external pressure. The amount of air entrapped within the fiber bundles will depend upon
the speed of the advancing flow front and the magnitude of the capillary forces. A
modified Lucas - Washburn type equation was suggested for liquid penetration given by
U =
where
Kb is the permeability of the fiber bundles
<t>b is the bundle porosity &
AP is the external pressure drop from the bulk flow
After initial air trapping, the model takes into account the effect of pressure difference
inside and outside the fiber bundle on the size of the trapped voids. Finally, the
mobilization step was modeled based on the relative magnitudes of the viscous and
capillary forces. The criteria for mobilization was that the viscous forces should be
greater than the capillary forces. An engineering approach was used to estimate the
overall void content given by Equation 2.24:

58
Void Fraction = (initial void size) x (fiber volume fraction) x P (2.24)
where
P = (compression factor) x (mobilization factor)
The compression factor was estimated by the air compression model. Mobilization
efficiency was determined empirically by fitting experimentally obtained void content vs.
capillary number.
2.4 Measurement of Void Content
Several methods have been used for the estimation of void content in composites. All of
them have limitations. A brief description of the salient features of each technique is
summarized below.
2.4.1 Density Determination
This method is used for quick estimates of void content as it is relatively simple to carry
out and does not require any sophisticated equipment. The void content is related to the
densities of the fiber, resin, and composite and to the volume fractions of the fiber and the
resin by the following expression:
Vv = V , - ( V f + V j (2.25a)
(2.25b)Pc P f Pr
V, W and p are volume, weight and density respectively. Subscripts c, f, r, and v denote
composite, fiber, resin and voids respectively. Precise knowledge of the void content,
therefore, requires accurate determination of the various densities and the resin and fiber
weight fractions. The densities are obtained from either the water buoyancy technique

59ASTM D792 or the density gradient technique ASTM D1505. The fiber / resin content is
usually obtained from chemical or thermal methods. The former involves acid digestion
of the resin, whilst in the latter, the resin is removed by thermal degradation and weight
changes are monitored by gravimetry. A variation of 0.1% in pc, Pf, pr, Vf or Vr results
in a variation of 2.5% in the estimated void content [Judd and Wright, 1978]. Also, this
technique only estimates the overall void content based on small samples, and provides
no information on the size, shape, location, or distribution of voids.
2.4.2 Water Absorption
This method requires the determination of the equilibrium water uptake of pure resin and
composite. Void volume is given as
Vv = Wc - Wr (2.26)
Wei s the water absorbed by the composite, and Wr is the water absorbed by the pure
resin. Its validity depends upon complete saturation of the voids. Analysis is also
complicated from the fact that resin swelling, hydrolysis, or leaching may occur. Thus,
this method is limited to cases where such reactions do not occur. The accuracy of this
method is estimated to be no greater than the density determination method.
2.4.3 Micrography
This technique is adapted from metallography in which a reflecting type microscope is
used for analyzing metallurgical specimens. A small section of the composite is cut and
mounted on a block, e.g., bakelite. It is then polished sequentially on silicon carbide
abrasive disks with increasing grit size. This is followed by polishing using a diamond or
alumina paste [Olsen et al., 1992]. In some cases the surface of the specimen may be
coated with a substance such as isopropylbiphenyl to reduce scattering of light from

6 0
surface imperfections. The prepared specimen is viewed under a microscope fitted with a
disc engraved with a fine grid. A void count is made by counting only those voids which
fall under grid intersections being counted. The use of void count method has been
reported at least in two instances by Kohn et al. [1968] and Feldgoise et al. [1991]. Void
content is given as
inn pVoid content (%) = (2.27)
?v is the number of grid points covered by the voids, and ?t is the total number of grid
points. The precision of this method has been studied before [Kohn et al., 1968]. The
number of measurements necessary to achieve a 95% confidence level so that the enor of
the mean will not exceed ±1% may be calculated from the equation:
== 4 == 4 s= (2.28)
Vx is the variance and s is the standard deviation. A statistical study of this technique
indicates that if uniform randomness of void distribution in the specimen is assumed.
Equation 2.28 can be applied to the number of grid points measured rather than the
number of specimens. This implies that a void count of only one specimen be made
provided that the void distribution and specimen selection are truly random. Practical
experience using this method has verified this argument. Micrography is more accurate
than the other two methods and also gives information on the shape and location of voids.
However, the specimen used for observing voids cannot be used for mechanical testing.
If the mechanical test is carried out first, then it makes it difficult to distinguish voids
from microcracks produced in the testing procedure.
Using optical microscopy to observe voids may require some preparation to improve the
contrast between the fibers and the resin. Etching and staining are common techniques

61
used successfully by metallurgists to enhance the microstructure [Guild and
Summerscales, 1993]. For glass fiber composites, dyeing the liquid can increase the
contrast. These methods, however, do not work well for carbon fibers. It has been shown
that depositing a uniform film of optically transparent material on the surface can
increase the contrast in carbon fiber composites. Once the film is deposited the sample is
illuminated under incident light. As the light passes through the films and is reflected
back from the composite, a phase change, which is dependent on the structure, takes
place. Thus interference occurs which results in a variation of color [Metcalfe and
Wilson, 1984].
2.4.4 Confocal Scanning Optical Microscopy {CSOM)
This is a relatively new technique that has demonstrated some promise in chaiacterizing
the resin fiber interface [Thomason and Knoester, 1990]. Unlike normal optical
microscopy, it is possible to focus on just one plane of the specimen. The information
from above and below the focus is excluded from the detector. Thus, by scanning the
sample under the laser beam and digitally storing the data obtained, it is possible to
reconstruct an optical section of the sample. Consequently, bulk specimens may be
imaged at different depths without any time consuming sample preparation. In principle,
CSOM can operate in transmission mode, reflection mode, or in fluorescence mode.
Thomason and Knoester used CSOM to observe the resin - fiber interface in
unidirectional glass fiber reinforced epoxy composites.
2.4.5 Ultrasonic C - Scan
This is the most widely used method in the industry for qualitative and quantitative non
destructive evaluation of flaws in composites. It can be applied in a number of ways.
The simplest is to transmit a short pulse of ultrasonic energy through the specimen and

6 2
measure the attenuation or "dB drop" caused by the passage of the ultrasonic pulse. This
technique has the advantage that it can assess the whole test piece rather than only a small
portion. Because voids are strong scatterers of ultrasonically generated elastic waves,
they cause a dramatic decrease in the amplitudes of the transmitted signal (dB drop) and
an increase in the ultrasonic attenuation. Hsu [1988] showed that void content (volume
%) in unidirectional and woven carbon fiber reinforced epoxy laminates was directly
proportional to the slope of the attenuation with respect to frequency. They found that
void contents determined from attenuation slope compared well with void contents
determined by acid digestion.
2.4.6 Radiography
Radiographic techniques have also been used in some instances. The samples are first
impregnated with molten sulfur and then radiographed using a tungsten target. A stereo
pair of radiographs is taken to facilitate examination of the void distribution throughout
the sample. It has been demonstrated that voids of micron size can be detected. The
drawback of this technique is the same as that for water absorption, and accuracy of the
results depends upon complete filling of voids with molten sulfur.
2.5 Effect of Voids on Mechanical Properties
Numerous researchers in the past have developed both empirical and theoretical
correlations between void content and various mechanical properties. Properties that
decrease the most due to voids are matrix dominated properties like the interlaminar shear
strength, transverse flexural strength, and transverse flexural modulus. Voids affect shear
properties of composites by creating localized areas of stress concentration. These areas
become nucléation sites for shear failure. Impact strength may, however, increase with
increasing void content [Judd and Wright, 1978]. This is attributed to the weakened

63bonding contribution to the formation of a more extensive yielding zone at the
propagating crack tip. Greater toughness then arises because of debonding and pulling
out of fibers and to a limited extent by delamination.
For a void free composite, the three factors which determine shear strength are the
ultimate shear strength of the reinforcement, the matrix material, and the bond at the resin
- fiber interface. In an ideal case, the lower bound on composite shear strength Xc is the
shear strength of the matrix, Xm. Chamis [1969] developed the following semi -
empirical relationship based on the assumption that shear strength of the composite was
related to the maximum shear strain in the matrix;
(2.29)Pv
where
Pf is the experimental - theoretical correction factor
(pm is the limiting matrix shear strain
Gc, Gm and Gf are the shear moduli of the composite, matrix, and fiber
respectively
Vv is the void volume
Pv is the void correction factor = --------------^ —
■ ^ k{\ - V J
Noyes and Jones [1968] developed Equation 2.30 for cases where voids have a
significant effect on failure characteristics
KV„11 + V,(K - 1)
(2.30)
K is an empirical constant, and Vf is the fiber volume fraction.

64
Greszczuk [1967] has developed equations to correlate the shear strength of composites
with spherical and cylindrical voids to the shear strength with no voids [Bowles and
Frimpong, 1992].
for spherical voids6V„
7C(1 - V,)(2.31a)
for cylindrical voids — = 1 -X
4V..7C(1 - V,)
(2.31b)
From Equations 2.31a and 2.31b, it can be readily seen that for the same void content,
cylindrical voids causes a greater drop in the shear strength than spherical voids. Figure
2.8 shows the plot of Equations 2.29, 2.30, and 2.31 (a) normalized against the shear
strength values at zero void content. Ghiorse [1992] measured the interlaminar shear
strength, flexural strength and flexural modulus as a function of void content in graphite
fiber/epoxy systems. Interlaminar shear strength was measured using a three point
loading fixture in accordance with ASTM D - 2344. Flexural strength and modulus were
measured using three point flexural tests in accordance with ASTM D - 790. He reported
that for every 1% increase in void content upto 5 %, there was a 9.7% drop in the
interlaminar shear, 10.3% drop in the flexural strength and a 5.3% drop in the flexural
modulus. Bowles and Frimpong [1992] obtained similar results for the interlaminar shear
and flexure strength as a function of void content in a graphite fiber/polyimide matrix
system.
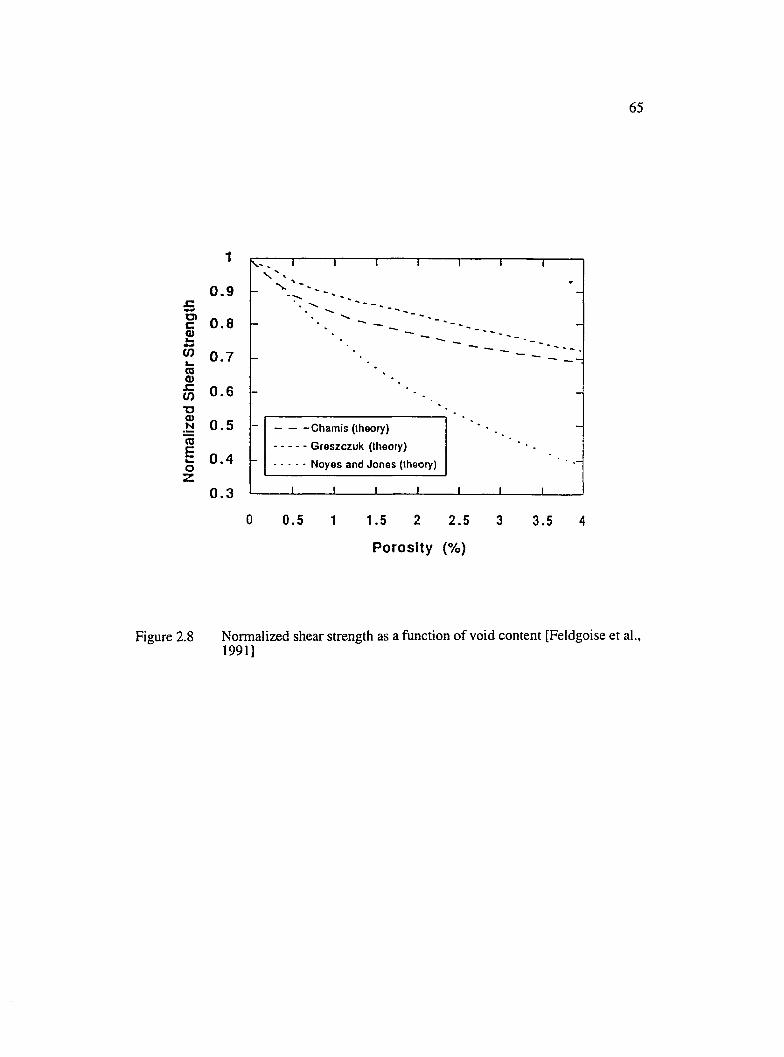
65
o>c2toswTJ<uNmEoz
1
0.90.8
0.70.6
0.50.40.3
■v
C ham is (Iheory)
G reszczu k (Iheory)
■ ■ • N oyés and Jo n e s (theory)
0.5 1 1.5 2 2.5
Porosity (%)
3.5 4
Figure 2.8 Normalized shear strength as a function of void content [Feldgoise et al., 1991]

6 6
Fan [1993] developed Equation 2.32 to predict the flexural strength as a function of void
content.
F = F„(Fv+V,)
1 • MIp.JI" (1 - F. - V.) J
(2.32)
where
F is the flexural strength of the composite
Fo is the flexural strength of the composite with no voids
Fv is the fiber volume fraction
P r and pm are the densities of the fiber reinforcement and the resin matrix
respectively
Vv is the void content (vol. %)
This theoretical correlation was found to agree quite well with the experimental results
obtained by Ghiorse [1992]. The suggested formula, Equation 2.32, has also been found
to be quite accurate for the prediction of reduction in the modulus of elasticity.
2.6 Application of Polymer Powders in Composites
One of the earliest references to the incorporation of powdered resin in fiber tows is the
patent by Price [1973]. Many other workers have subsequently discussed or patented
different versions of the technique [Gibson and Manson, 1992]. The idea of using
powders was developed to provide an alternative means to produce composites from
thermoplastic resins that are difficult to prepreg due to their high melt viscosity or due to
the need to use high boiling point precursor solvents which are difficult to remove.

67Several different forms of powder coated composites are available. Figure 2.9 shows
three of them. These include sheathed materials, melt fused materials and binder
materials. This nomenclature is based on the method used to hold the powder on the
fiber. In a sheathed system, the polymer powder is impregnated into the fiber tow, and
then the tow is coated with molten polymer to hold the powder on the fibers.
The hot melt system utilizes a fluidized bed to impregnate the tow and then a heat source
to melt the particles to fuse them onto the fiber. Successful impregnation of powders
relies on pins or rollers to open up and spread the fiber tow in order to present as large an
area as possible for the incorporation of powder into the strand. However, in alternative
versions of the process, an air knife [Muzzy et al., 1990] and a venturi [de, Jager G, 1987]
have been proposed to open up the yarn. Some studies describe the incorporation of the
powder into the tow solely in terms of mechanical effects while others have emphasized
the importance of electrostatic attraction [Throne and Soh, 1990]. Once the tows have
been filled with the resin, the next step is to pass them through an oven to melt the
powder and complete the impregnation process. Infra-red heating has been found to be
useful, especially in the early stages of heating in order to achieve a high rate of
temperature increase. Typical oven residence times are in the range of 10 -100 seconds.
The binder system uses a polymer to bind the powder particles on the fibers. This
method produces the most flexible and drapeable prefrom. However, complete removal
of the binder can become difficult. In this process, the polymer powder (having a
particle size < 20 pm) is mixed with a binder in a solution. The fibers are then passed
through a bath containing this solution, where they are coated with the polymer powder.
The amount of polymer powder deposited on the fibers depend upon the powder
concentration in the solution and the rate at which the fibers are pulled through the bath.
Cochran and Pipes [1991] studied the feasibility of this approach for making Poly-ether-

6 8
Sheathed Melt Fused Binder
Figure 2.9 Types of powder coating in composites [Cochran and Pipes, 1991]

69
ether-ketone (PEEK) powder coated preforms. In another study by Vodermayer et al.
[1993], impregnation of carbon fiber rovings was investigated using a similar approach,
but with an aqueous polymer powder dispersion process. The advantage of using an
aqueous dispersion being lesser health hazards. However, there is still the need to
remove the water after impregnation of the fiber rovings. Thus, dry powder coating of
fiber tows is often the preferred approach as it circumvents a lot of inherent difficulties
associated with removal of liquids, resulting in faster line speeds.
There are several types of thermoplastic powder suitable of use in powder impregnation
technique. Candidate resin include the high performance materials, PEEK,
polyethersulfone (PES), polyphenylene sulfide (PPS) and polyetherimide (PEI), but there
is also considerable potential for using the commodity "engineering thermoplastics", such
as nylon 12 and polypropylene.
Some composite applications require the use of textile type (stitched, knitted, woven,
etc.) preforms for improved mechanical properties. However, to obtain good mechanical
properties, it becomes necessary to obtain full consolidation of bulky textile type
preforms. Consolidation is an important issue as bulk factors (the ratio of preform
thickness to final part thickness) are on the order of 5:1 for 2D textiles and 3:1 for 3D
textiles [Hugh et al., 1993]. To facilitate consolidation, polymer powders based preforms
were developed. Full consolidation of powder coated textile preforms entails a two-step
process. It involves an initial debulking step to obtain the wetting needed for intimate
contact of resin and fiber, followed by final "net-shape" consolidation. Initial debulking
is done by any of the common forming methods such as, rubber molding, hydroforming,
diaphragm forming, and matched die molding. Final consolidation is usually
accomplished by standard autoclave or heat-press procedures. Several studies focusing
on the flexural rigidity and consolidation behavior of thennoplastic powder coated

70preforms have been undertaken in recent years at the NASA Langley Research Center
[Hirt et al., 1990 & Hugh et al., 1993]. In aerospace industry, usually a reactive material
(tackifier) is used for consolidation of fiber preforms. The reactive material can either be
uncatalyzed or catalyzed thermoset resin with generally the same cure chemistry as the
matrix resin. Some of the common methods of tackifier application utilize veils, solvent
spray and powder [Kittelson and Hackett, 1994]. Veils can be placed between adjacent
plies of broad goods followed by fusing the ply stacks with heat and pressure to form a
preform. Tackifiers can also be applied from solvents by spraying onto each broad good.
However, for the same reasons as in the case of thermoplastic resins, the preferred
approach is to use powders which can be applied onto the broad goods by a sifter type
apparatus. Application of heat and pressure causes the powder to melt and flow between
the layers of the fiber preform. Since the powder concentration typically ranges from 4 to
7% by weight of the fibers, tackified preforms can be thought of as very low content
prepregs. Again, one of the important issues in using thermoset powder coated preforms
is the need for significant debulking during the consolidation process. It is important to
note that since the density of the fibers is nearly twice that of the resin, 4 to 7 weight
percent of tackifier equates to about 8 to 14 percent of the total resin in the finished part.
Thus, tackifier can become a major component in the matrix resin and can significantly
affect the mechanical properties of the molded part. It is because of this reason, that the
chemistry of the tackifiers is chosen so as to be compatible with the matrix resin.
2.7 Tack and Drape Characteristics of Prepregs /Preforms
One of the major concerns in using prepregs and preforms is insufficient tack and poor
drape characteristics. Tack is the property that allows the layers of the fiber mat to stick
to one another so that the whole fiber stack can be handled as a single unit with reduced
slippage between the layers. Drape refers to the ability to conform to the tool surface.

71Often prepregs and preforms need to be deformed in order to fit the shape and curvature
of the tool surfaces. If the elastic energy stored during this deformation is high, the fibers
will overcome the adhesion forces due to tack during relaxation, and the prepreg/preform
will "springback" to its undeformed state. The springback phenomenon is also referred to
as non-conformance to the tool surface [Gutowski and Bonhomme, 1988], and is a
manifestation of the elastic stresses stored in the fibers.
Several attempts have been made to measure and model the phenomenon of tack
adhesion. Although there is no universally accepted technique for measuring tack,
various methods exist ranging from the primitive to the more sophisticated. One of the
older methods of measuring tack involves sticking a piece of prepreg on a vertical steel
surface, and stating that the tack is high enough if the prepreg does not fall in thirty
seconds [Meissonnier, 1988]. The newer methods derived from two main ASTM
standards [D 2979-71 & D 3121-73] are used mostly by the pressure sensitive adhesives
industry [Bonhomme, 1986].
Meissonnier [1988] developed a simple test to study tack and drape characteristics of
bismaleimide resin based graphite fiber prepregs. The experimental technique used was
similar to the flatwise compressive/tensile test. Prepreg samples were glued to the
crossheads of an Instron machine using special tabs, and subjected to a compression-
tension cycle. First, the samples were brought in contact with a constant cross-head
speed, held in contact under a constant load for a given "hold-time", and then pulled apart
in the opposite direction with the same speed till the samples came apart. The significant
parameters of the test were : rate of loading and unloading, the maximum pressure, hold
time and temperature. The measured quantities were : tension modulus, energy absorbed
during tension loading, strain and creep compliance under the maximum pressure during

72hold time. Tack was measured by calculating the work of detachment per unit volume
(Ev) by integrating the area under the stress-strain curve (Equation 2.33):
Ev = Jq"“ ct. de (2.33)
where
a is the stress e is the strain
Creep compliance, C(t) was used to describe the drape behavior of the prepregs, and was
obtained by plotting strain versus time during hold time at the maximum pressure
(Equation 2.34). The faster a material creeps, the lesser is the elastic energy stored during
deformation, and less likely it will overcome the tack adhesion to springback [Seferis and
Meissonnier, 1989].
C(t) = ^ (2.34)a
Bonhomme [1986] observed that the surface of the prepreg tape is textured with many
small asperities (Figure 2.10). Good tack adhesion then depends upon how effectively
these asperities grow under combined effects of applied pressure and surface tension
forces to establish sufficient contact area with the tool surface or to a previously applied
layer of prepreg. Bonhomme assumed each asperity peak as a small circular resin patch,
and modeled its growth using squeezing flow approach. However, decrease in surface
area due to coagulation of resin patches was not considered. Furthermore, assuming that
the resin behaves as a Newtonian fluid. Equation 2.35 was derived for the geometry
shown in Figure 2.11.
P - Pb - ^ (2 3 5 )

73
actual surface texture
idealizedsurfacetexture
resin patch model
Figure 2.10 Schematic of the surface of the prepreg [Bonhomme, 1986]
H(t)777777777
R(t)
Figure 2.11 Squeezing flow of a newtonian liquid [Bonhomme, 1986]

74
Equation 2.35 was then rewritten in terms of the applied force with pressure at the
boundary pb = 0 (i. e. neglecting the surface tension effects), the volume of the patch, V =
HA, and A = 7i r 2-
t2 . H,f p d t = - — iiV^ f H ' ^ d H (2.36)J 0 -TT Jt, H
For F = Fa = constant, ti = 0, t% = t. Hi = Hq, and H% = H, the following result was
obtained for the wetted area of the patch as a function of time
A(t) 1 + —
1
(2.37)
where
N is = 4 for a Newtonian fluid and
To is the time constant given by Equation 2.38
3 IX8 n F,
(2J8)
where
Ao and Hq are the starting area and height of the resin patch
For surface tension driven squeezing flow, the expression for area growth was derived
similar in form to Equation 2.37 by substituting patm - Pc for the boundary pressure py.
where• ,u -11 / 2 Yr Cos 0
P c is the capillary pressure ( = —*—^ ------)
However, for this case, N = 2 and time constant x is given by Equation 2.39:

X =4% yCosG y
75
(139)
2.8 Modeling of Fiber Consolidation
In order to better understand the "deconsolidation" or the springback phenomena in
composites, a conceptual mathematical model for transverse stiffness of a bundle of
confined fibers was developed by Gutowski [1985]. The model assumes that the
deflection behavior of a fiber is given by the strength of material solutions for a beam of
span length li and diameter d (Equation 2.40). Also, that the fiber is arched with a height Cl, but ^ is large and so the arch length and span length are very close.Cl
pAf
n Eld j c i ;
(2.40)
where
P is the applied pressure
Af is area of the fibers exposed to pressure p
E is the modulus of elasticity
I is the moment of inertia
C is a constant and depends on the assumed end conditions
5 is the beam deflection
The final equation arrived at for transverse stiffness was obtained as given by Equation
2.41 :
P = ^
J _
vVo Vf64 a p
I Vo
(2.41)
a y

76where
Vo is the initial fiber volume fraction
Va is the available fiber volume fraction
Vf is the fiber volume fraction for some deflection, 8
Equation 2.41, thus implies that at P = 0 at Vf = Vq, and
p = oo as Vf Va
Gauvin and Chibani [1988] proposed an empirical model given by Equation 2.42 to
correlate the compaction pressure (P) and the compressed thickness of the reinforcement
(h):
- ^ = AqP + Ai l n P + ^ + A3 (2.42)
where
h is the original reinforcement thickness
Ao, A i, A2 and A3 are the curve fitting parameters
Batch and Cuminskey [1990] developed a semi-empirical model based on the
compressibility behavior of one layer of reinforcement. A Hookean and non-Hookean
regime were suggested. Model equations arrived at by Batch and Cuminskey are given in
Equations 2.43 and 2.44:
P = k ( V f - V o ) (2.43)
k = ko
if Vf < Vf* ^cont.

77
and
J _ J _
k = ko Y ---- T " (2.44)
■ vT
if Vf > Vf* ^cont.
where
k is the fiber compaction spring constant
ko is the fiber compaction spring constant for the Hookean regime
T| is the fiber packing efficiency
V is the ultimate fiber fraction at maximum pressure
V f is the volume fraction at the transition between Hookean and non-Hookean cont.
Claus and Loos [1989] expressed the compaction behavior of textile prefoiTns in terms of
a logarithmic series. Their model is given by Equation 2.45:
d = ai + a2 In P + a3 (In P)2 + 3 4 (In P)3 + ........ (2.45)
where
d is the deflection of the fiber bed, and P the compaction pressure
The number of terms in the series were chosen to give the desired level of correlation
with experimental results.
2.9 Rheo-kinetic Characterization of Bismaleimide Resins
Bismaleimide resins are used mostly as matrix resins for high performance composite
applications. They exhibit low moisture absorption, excellent chemical stability, and
thermal and mechanical properties superior to most epoxy resins. Low moisture

78absorption comes from the fact that the imide functionality has lower capacity for
hydrogen bonding than either -OH or -NH2 containing polymers.
Due to the aromatic nature and high crosslink density of the cured network, the fully
cured resins are brittle. Thus, bismaleimide resins are often reacted with aromatic
diamines, which reduce the crosslink density by increasing the distance between the
crosslinks [Tungare and Martin, 1993]. However, the addition of diamines results in a
slight decrease in the thermal stability of bismaleimide resins.
Another approach to reduce brittleness is to create an interpenetrating network by
modifying the resin with a silicone monomer such as diphenylsilanediol. Properties of
silicone modified bismaleimide resins have been studied by Voit and Seferis [1987].
They observed that the silicone additive provided important morphological modification
to the resin matrix by improving impact toughness while, at the same time, retaining the
high temperature properties of the resin.
The curing behavior of bismaleimide resins using formulations containing different
stoichiometric ratios of 1,1' - (methylene - 4,1 - phenylene) bismaleimide and 4,4' -
methylenedianiline (MDA) was studied by Tungare and Martin [1992]. Their study
revealed that curing can take place via two reaction pathways (Figures 2.12 a and b).
They are amine addition to the maleimide double bond of the bismaleimide, which occurs
readily at low temperatures (> 75 °C), and the homopolymerization of the bismaleimide
double bonds, which occurs at high temperatures (> 155 °C). At lower temperatures, the
two reactions are in a sequential order, but at high temperatures, both of them occur
simultaneously.
The maleimide double bonds of the bismaleimide resin are weakened by the electron
withdrawing nature of the adjacent carbonyl groups, and hence, nucleophilic addition of

79amines to the maleimide double bonds occurs readily at low temperatures. The secondary
amine hydrogens resulting from the reaction of the primary amines are much less reactive
with maleimide double bonds. The melting point of 1,1'- (methylene - 4,1 - phenylene)
bismaleimide is 155 °C. In the molten state, the double bonds of the bismaleimide resin
(weakened by the electron withdrawing nature of the adjacent carbonyl groups) readily
react by free radical polymerization [Tungare and Martin, 1992]. Thus, at higher
temperatures ~ > 155° C, homopolymerization occurs by a thermally initiated, multistep,
radical mechanism which leads to crosslinking in the network.
As illustrated in Figure 2.12, the amine addition reaction causes extension of the network
chains, whereas the homopolymerization leads to chain extension and crosslinking in the
network. As a consequence of two reaction mechanisms, different curing conditions lead
to different types of network with different thermal, mechanical and rheological
properties.
N
OBismaleimide
-i- nh2— ■NH,
oDiamine
N-
O
.NH NH—
(a)
Figure 2.12 Reaction mechanisms of a BMl resin : (a) Michael addition of diamine to bismaleimide and (b) bismaleimide homopolymerization
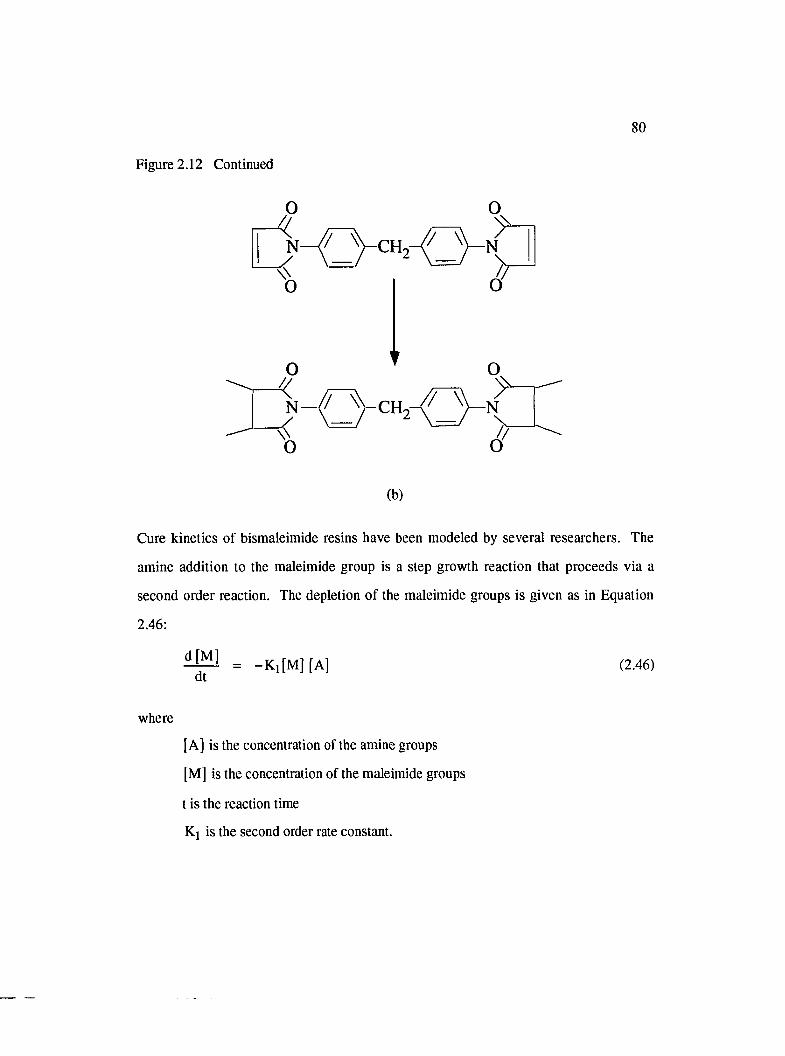
Figure 2.12 Continued
80
o
N
o
N- y \ '
OX
o> -
OX
I— ] S ^
(b)
Cure kinetics of bismaleimide resins have been modeled by several researchers. The
amine addition to the maleimide group is a step growth reaction that proceeds via a
second order reaction. The depletion of the maleimide groups is given as in Equation
2.46:
^ = - K , t M l [ A ]dt
(2.46)
where
[A] is the concentration of the amine groups
[M] is the concentration of the maleimide groups
t is the reaction time
Kj is the second order rate constant.

81
The maleimide and amine conversions, and a/^ are given by Equations 2.47 and
2.48:
_ K 1 - [ m ]
^ ■ [Mol
_ [Aq] - [A ]
^
(2.47)
(2.48)
where
[Mq] is the initial concentration of maleimide groups
[Aq]. is initial concentration of the amine groups
Equation 2.46 can be written in terms of the maleimide conversion, using Arrhenius
temperature dependence for Kj , as in Equation 2.49:
[M o f= Ai exp d t ‘
-E lRT
X [l -ajvi] 1 -
[Ao][Ao] (2.49)
The kinetic model developed by Tungare and Martin [1992] for BMI
homopolymerization has the following form:
d[M]dt
= k2t Ml -k2i[M]-k2p[M] Ml - k2p[M] M (2.50)
Ml
dtMl k2p[M] Ml
- k 2p
2Ml - k ] p Ml M (2.51)

82
M
dt= k2p[M] Ml ^2p M, M (2.52)
where
Ml
M
is the concentration of the maleimide radicals
is the concentration of the polymer formed
k 2 i is the reaction rate constant for the initiation step
k 2 p is the reaction rate constant of the propagation step
k 2 t is the reaction rate constant of the termination step
Tungare and Martin [1992] found that in the multistep homopolymerization, the chain
propagation step is an order of magnitude slower than the initiation and the termination
reactions and is the rate controlling step. The single step amine addition reaction occurs
much more rapidly than the chain propagation reaction during the homopolymerization of
BMI. This model, however, does not consider the interactions between the two reactions
and the diffusion effect on reaction rate.
The change in viscosity as a function of conversion for BMI resins has not been modeled
as extensively as the reaction kinetics. Meissonnier [1988] proposed an empirical model
for two different formulations of BMI (Kerimid 70003 and 70015) based on the WLF
(William-Landel-Ferry) equation given as:
A ( T - T g )L ogn(T) = Log^Tgo ■
B + T - T„(Z53)
where
|i is the viscosity
T is the temperature
Tg is the glass transition temperature of the resin at any conversion

83is the viscosity of the uncured resin at its Tg
Based on the relation derived by Bueche [1962], the Tg of the resin can be expressed as a
function of the advancement of the reaction, a , by Equation 2.54;
whereTgQ and Tg” are the glass transition temperatures of the uncured and fully cured
resin
Evolution of viscosity for n^h order reaction kinetics was then computed given any time
and temperature history by combining Equations 2.53, 2.54, 2.55 and experimentally determining Tg^ and Tg”
a ( t n ) = a ( t n . i ) + ( 1 - a ( t n - i ) ) " B n - t n - i ^ ^ ( 2 5 5 )
where
A is the pre-exponential factor
Ek is the activation energy
R is the gas constant
T is the temperature in (K)
Since this viscosity model is based on the overall curing kinetics, the model paiameters
are dependent on the resin composition. If the resin composition is changed, the
parameters have to be reevaluated.
For the BMI formulation used in this study. Equations 2.56 and 2.57 were found adequate
to describe the changes in viscosity and glass transition temperatures as a function of
overall conversion [Shafi, 1994].

84
[ 1 = Ho exp. (-----^ (2. 56)Otgel " GO
where
ttgel = 0.5 and7161
Ho = HooCxp.T(°K)
~ ^gp ^ Cl « T„^ 1 - C2 a
(2.57)Bp
whereTgo= 281.7 K
Cl and 0 2 are 0.218 and 0.793 respectively
In a companion study, the effect of cure conditions on reaction kinetics and viscosity
build up of different formulations of BMI resin has been studied in greater detail
[Srinivasan et al., 1995].

CHAPTER III
ANALYSIS OF FLOW INDUCED VOIDS DURING FIBERIMPREGNATION
3 .1 Materials
For flow visualization experiments, several non-reactive liquids having different surface
tensions and viscosities were used. These include water, DOP oil (diphenyl-octyl-
phthalate), silicone oils (dimethylpolysiloxane, Dow Coming), glycerin, and ethylene
glycol.
There are numerous choices of fiber reinforcements for LCM processes. However, most
of them can be characterized by two types of pores; one between the fiber bundles and the
other within the bundles. In this work, a unidirectional stitched fiberglass mat (CoFab
AO 108) was used as it was easier to analyze the observed flow behavior because of the
simple fiber architecture. It is a non-woven type of reinforcement with fiber bundles
oriented in one direction only, and held together by continuous stitches.
3 .2 Instrumentation and Experimental Procedure
3.2.1 Liquid Properties and Contact Angle Measurements
The viscosities and surface tensions of the experimental liquids were either measured or
obtained from handbooks. Viscosity measurements were made using the Brookfield
viscometer, while for surface tension and contact angle measurements, a Dynamic Contact
85

8 6
Angle Analyzer (DCA 322) was used. DCA 322 (Figure 3.1) is based on the Wilhelmy
principle. In this technique, a solid sample suspended from a balance is partially immersed
in a liquid and the force exerted by the liquid on the solid is monitored. To recall, the
Wilhelmy wetting force is given by
Fw = P ylCOs(0) (3.1)
where
P is the perimeter of the sample
is the surface tension of the liquid, and
0 is the contact angle made by the liquid on the sample
The value of Fw is obtained from the balance. Thus, it follows that if two of the three
quantities on the r.h.s. of Equation 3.1 are known, it can be solved for the third quantity.
For contact angle measurements, it was necessary to know the surface tension of test
liquids. For surface tension measurements, a heat cleaned glass cover slip of known
perimeter was used as the sample. It was suspended from the arm of the microbalance via
a hangdown wire. The test liquid was contained in a beaker resting on a motor driven
traveling stage. The liquid was slowly raised by moving the stage upwards. At the first
contact of the liquid with the sample, there was an increase in the force. The force then
decreased due to buoyancy as more and more of the sample was immersed in the liquid.
After a sufficient length of the sample was traversed by the liquid, the direction of the stage
was reversed to obtain the data for the receding cycle. The wetting force was obtained by
extrapolating the force reading to zero depth of immersion to account for the buoyancy
factor. This is a point when the sample first comes in contact with the liquid. The value of
the contact angle is assumed to be close to zero. This assumption is usually valid when
there is no hysteresis between the advancing and the receding cycles. Figure 3.2 shows a
typical trace of the curves obtained from a surface tension experiment. In this case, the
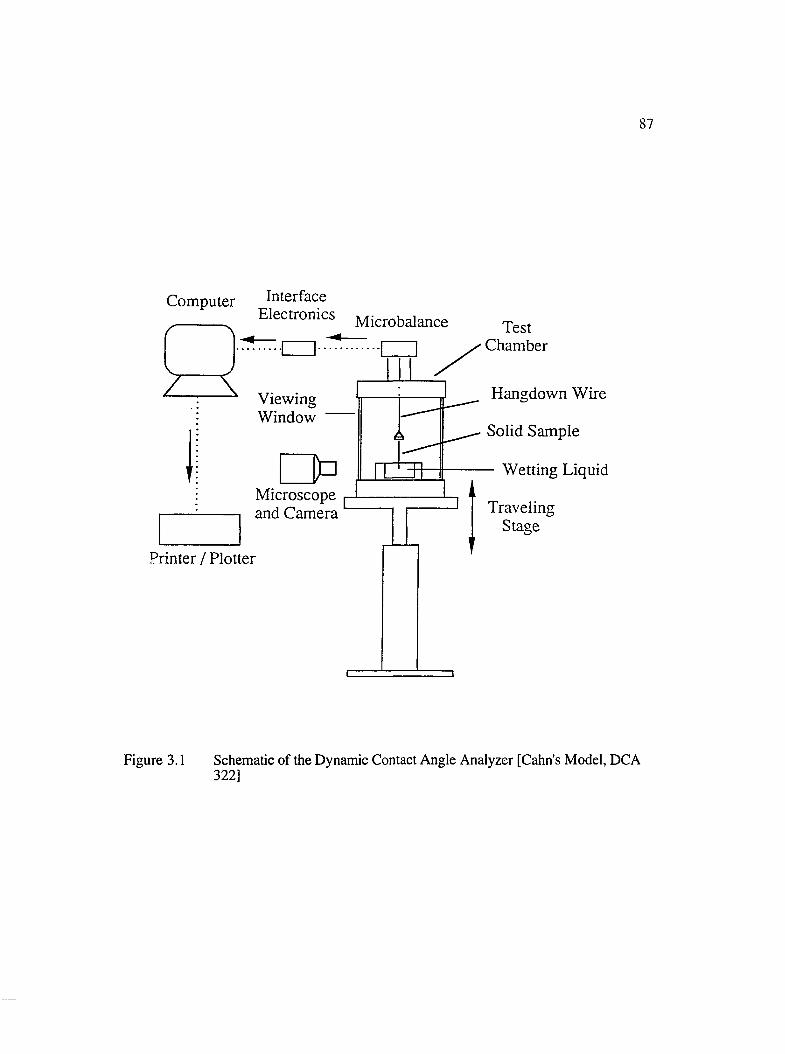
87
Computer Interface Electronics
/ \
V , J/ \
Microbalance
Printer / Plotter
ViewingWindow
Microscope and Camera
TestChamber
Hangdown Wire
Solid Sample
Wetting Liquid
TravelingStage
Figure 3.1 Schematic of the Dynamic Contact Angle Analyzer [Cahn’s Model, DCA 322]
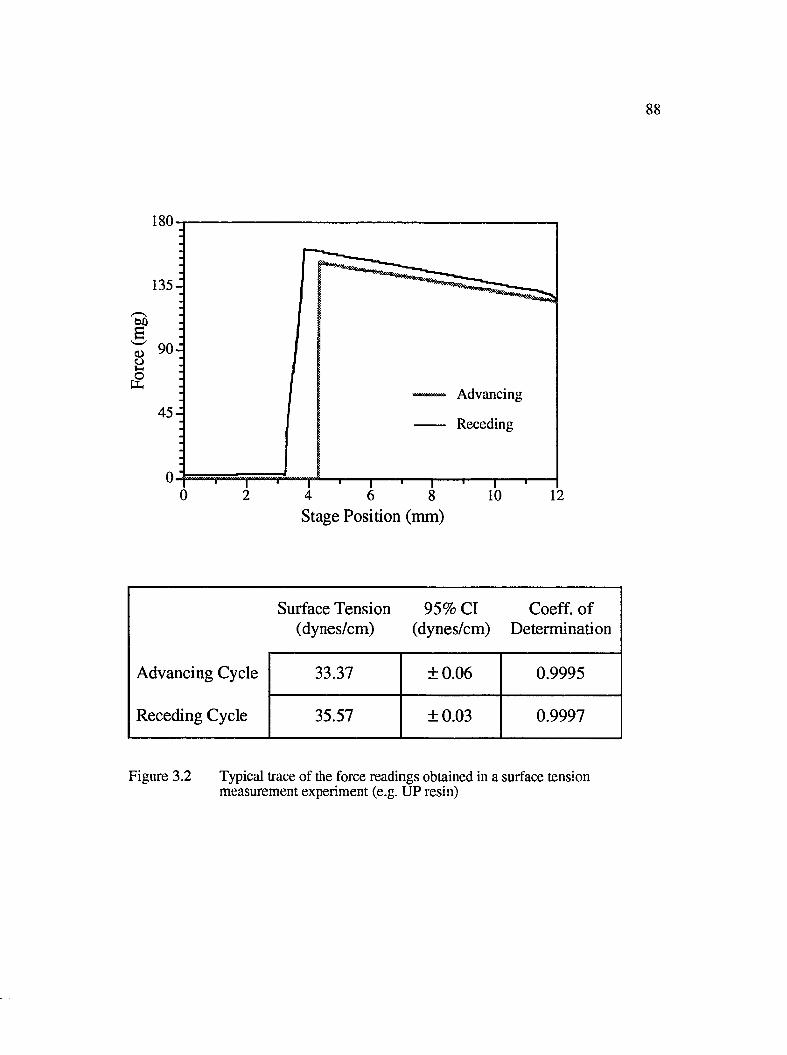
88
180
135-
B : ^ 9 0 -
I ;Advancing
Receding4 5 -
40 2 6 8 10 12Stage Position (mm)
Surface Tension 95% Cl Coeff. o f(dynes/cm) (dynes/cm) Determination
Advancing Cycle 33.37 ± 0 .0 6 0.9995
Receding Cycle 35.57 ±0 .03 0.9997
Figure 3.2 Typical trace of the force readings obtained in a surface tension measurement experiment (e.g. UP resin)

89
curve represents the force values from which the surface tension of unsaturated polyester
resin was calculated.
After obtaining the surface tension of test liquids, contact angles were measured in a similar
fashion. However, a single filament was used as the sample instead of the cover slip. The
perimeter of the filaments was measured using wetting liquids like Hexane and Hexadecane
which gave nearly zero contact angles. In a parallel study, contact angles between fiber
bundles and sample liquids were also measured [Patel et al., 1994]. The physical
properties of the liquids and the corresponding contact angles with the fibers are given in
Table 3.1.
3.2.2 Flow Visualization o f Macro and Micro Voids
A transparent acrylic mold with a light source underneath was used as a flow cell for
visualization experiments. The mold dimensions were 5.7" x 3.54" (1 x w). A rectangular
cavity having dimensions 3.35" x 1.97" (1 x w) was created using a lexan gasket. Strips of
fiber mat were cut so that its edges were flush with the side« of the rectangular cavity. This
was done to ensure that no race - tracking or channeling occurred from the sides. Also, a
gap of about 0.4" was kept between the fiber mat and the inlet so that the liquid reached the
fiber mat as a plug flow. Liquid was injected at a constant flow rate using a Harvard
apparatus infusion / withdrawal pump (Model 919). It is a positive displacement pump
with a piston cylinder type mechanism for injection. The highest superficial velocity
achieved in the mold was 3.9 cm/sec. Care was taken to remove all the air bubbles from
the source liquid and the transparent tube connecting the pump to the mold. Flow behavior
was observed for both flow along and normal to the fibers for different injection flow rates.
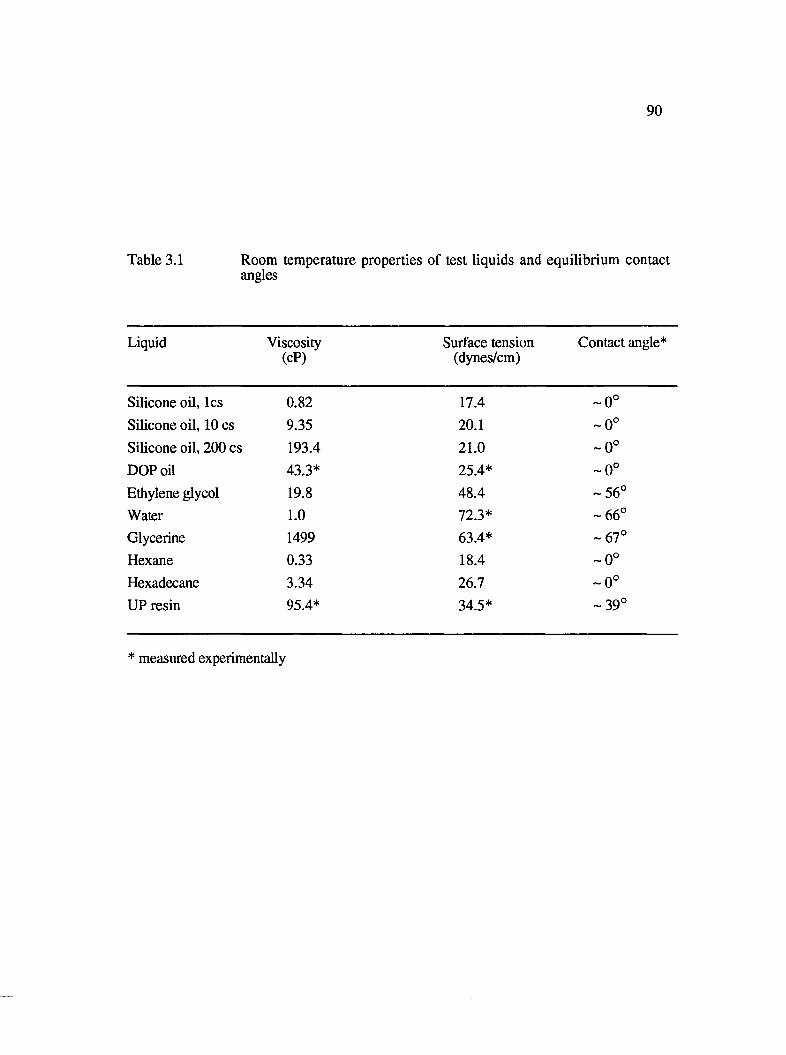
90
Table 3.1 Room temperature properties of test liquids and equilibrium contactangles
Liquid Viscosity(cP)
Surface tension (dynes/cm)
Contact angle*
Silicone oil, les 0.82 17.4 - 0 °Silicone oil, 10 cs 9.35 20.1 - 0 °Silicone oil, 200 cs 193.4 21.0 - 0 °
DOP oil 43.3* 25.4* - 0 °Ethylene glycol 19.8 48.4 -5 6 °Water 1.0 72.3* - 6 6 °Glycerine 1499 63.4* - 6 7 °Hexane 0.33 18.4 - 0 °Hexadecane 3.34 26.7 - 0 °UP resin 95.4* 34.5* -3 9 °
* measured experimentally

91
The fiber volume fraction for these experiments, as calculated from Equation 3.2, was
43%.
(3.2)1- f Vf ] = 1 -n
I V cJ t
where
(|) is the porosity
Vf is the volume of the fibers
Vc is the volume of the cavity
% is the surface density of the fiber mat (ML'2)
p is the density of the glass fiber (ML"3)
n is the number of layers of the fiber mat
t is the final gap or the cavity thickness
The flow pattern of the liquid through the fiber mat and the formation of macrovoids during
liquid injection were viewed on a 13" T.V. monitor using a CCD video camera (Cohu
Model 4915-2001) and recorded on an S-YHS recorder (Panasonic Model AG 1960).
Macrovoids were viewed at a magnification of 16 times. After liquid injection was
completed, the mold was scanned for voids. A representative number of photographs were
taken for each run and the area fraction of macrovoids was computed.
Observation of formation of microvoids in the smaller gaps (~ 1pm) between the 13pm
diameter filaments in the fiber tows posed quite a challenge. This is so because it required
a magnification of greater than 200 times to see the filaments and the trapped microvoids.
Achieving a high magnification was not a problem as most commercial microscopes offer
magnifications greater than or up to 1000 times. However, at such high magnifications,
the working distance between the sample and the objective lens is in the order of a few
millimeters. Thus, for proper focusing of the sample at high magnifications (> lOOX), the

92sample must be only a few millimeters from the lens. This is where the problem arises.
Such a requirement on the proximity of the sample to the lens makes it impossible with
most microscopes to put a flow cell under the lens and focus on the filaments of the fiber
mat. Thus, in - situ observation of formation and movement of microvoids during liquid
injection becomes impossible with conventional microscopes.
Several different approaches were tried to tackle the problem before finally meeting with
success. Some of the approaches tried included using an Infinity lens, a reflecting
microscope (used in metallurgical microscopy), scanning electron microscopy (SEM), and
confocal laser microscopy. Both metallurgical microscopy and confocal laser microscopy
have been used in the past to observe the microstructure of fiber reinforced polymer
composites. The major limitation of these techniques is that they are off - line techniques in
the sense that they cannot be used for in - situ observations.
After a lot of experimentation and unsuccessful attempts at solving the problem, a
combination of the Cohu video camera (having a 1/2" view detector area), the T.V.
monitor, and more importantly, a 6.5 X Zoom lens (D. O. Industries) with a 5X adapter
and a 2X eyepiece finally did the trick. With this set - up, high magnifications of greater
than 400 X were achieved at a working distance of more than an inch, hence the name,
video assisted microscopy (YAM). This allowed the transparent acrylic mold to be placed
between the Zoom lens and the brightfield/darkfield stage for on-line viewing of the
formation of microvoids during liquid injection.
One last problem that needed to be solved was to improve the contrast between the
transparent injection liquids and the translucent white glass fibers. Some techniques like
etching and staining have been tried by previous researchers to improve the contrast. In
this work, a tint of red dye was added to the liquids which helped in defining the edges of

93the voids more clearly. The amount added was well below the critical concentration of
about 1% and was just enough to improve the contrast without altering the physical
properties of the liquids. The brightfield/darkfield stage was used for the same purpose.
In the darkfield mode, the reflections at the glass-air interfaces disappear resulting in a
better contrast
As in the case of macrovoids, after liquid injection, a representative number of photographs
were taken for each run and the area fraction of microvoids was computed using the Point
count technique [Kohn et al., 1968; Feldgoise et al., 1991]. Thus, void (both macro and
micro) fraction is reported as percentage detected area voidage. The schematic of the flow
visualization set - up developed in this study is shown in Figure 3.3.
3.3 Flow visualization of Macrovoids Formation
3 .7 ./ Axial Flow
A t low flow rates (v ~ < 0.1 cm / sec.), it was observed that the liquid penetrated through
the fiber mat at unequal rates, as shown in Figure 3.4. The same behavior was observed at
even higher flow rates (v > 0.1 cm / sec.) for some liquids like water, which have very
high surface tension and vei"y low viscosity. Thus, in essence, it is the capillary number
that governs how the liquid flows through the fiber mat.
The capillary number is usually defined as the ratio of viscous force (product of viscosity
and velocity) to capillary force:
C a # = ^ 1 0 - - (3.3)Ylv
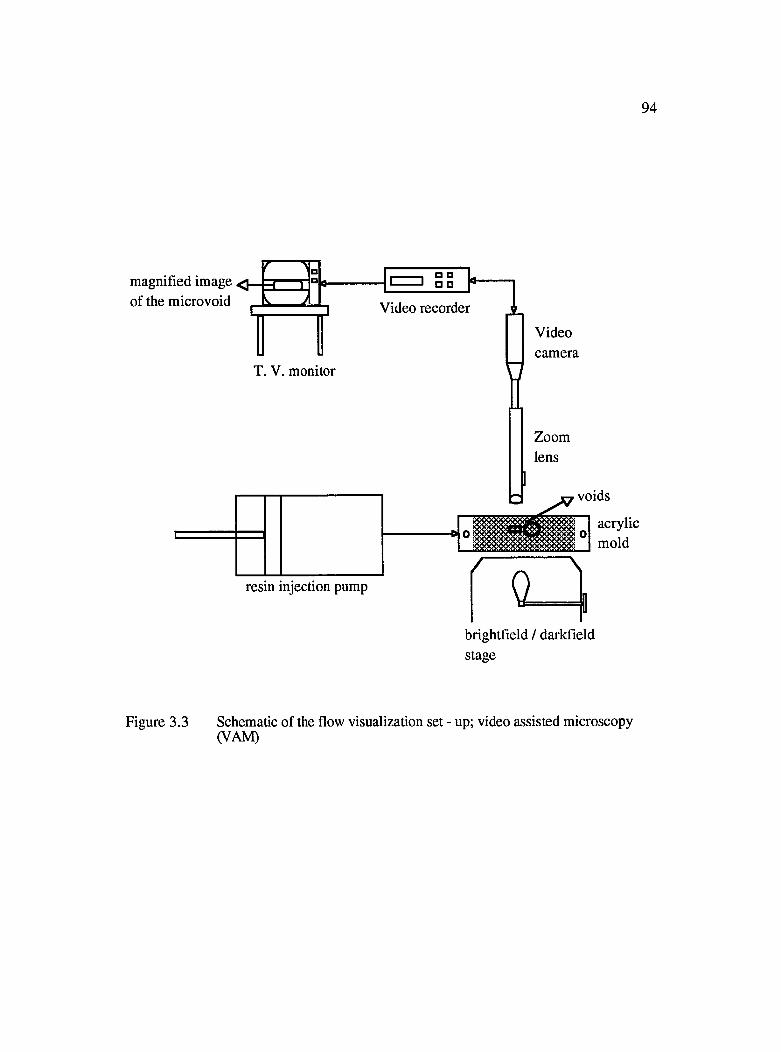
94
magnified image of the microvoid Video recorder
Videocamera
T. V. monitor
Zoomlens
voids
acrylicmold
resin injection pump
□ □
brightt'ield / darkfield stage
Figure 3.3 Schematic of the flow visualization set - up; video assisted microscopy (VAM)

95
Flow front leading in the tows
Flow direction
Figure 3.4 Photograph of the lead-lag at the flow front for axial flow ; capillary number < 10-^

96where
|i is the liquid viscosity in cP
V is the superficial velocity in cm/s
T l v is the surface tension of the liquid in dynes/cm
From Equation 3.3, it can be seen that capillary number is directly proportional to the
injection flow rate. Hence, for a given liquid, capillary number can be increased by
increasing the injection flow rate. The definition of capillary number as given by Equation
3.3, however, does not take into account an important material parameter, namely the
contact angle formed by the liquid meniscus with the walls of the capillary. Thus,
according to Equation 3.3, for different types of glass, graphite, and kevlar fibers, the
capillary number would be the same at a given flow rate for a particular liquid. This,
however, is not true. As the liquid penetrates an empty capillary, the solid - air interface is
replaced by the solid - liquid interface through the wetting process which depends on the
value of the adhesion tension. Adhesion tension (A), is a function of the surface tension of
the liquid and contact angle and is defined as [Carino and Mollet, 1975]:
A = Ys - Ysl = TLv * Cos (0) (3.4)
For wetting with a given liquid, the value of adhesion tension changes systematically with
changes in the solid surface characteristics due to changes in the contact angle [Miller,
1971]. Thus, a modified capillary number can be defined as:
C a#* = --------------^ 10~^ (3.5)Ylv Cos (0)
Incorporation of the contact angle generalizes the definition of the capillary number for any
type of liquid / fiber system. Thus, any mention of the capillary number hereafter refers to
the modified capillary number unless mentioned otherwise.

97At low capillary numbers, the capillary force dominates the viscous force and the liquid fills
the smaller pores first in preference to the larger pores. This results in a lead - lag type of
flow pattern, also known as fingering owing to its resemblance to the digits of the hand.
From flow visualization experiments it was observed that the lead - lag at the flow front
was responsible for the formation of surface macro voids at the stitches in the gaps between
the fiber tows.
The formation of macrovoids is explained by the schematic shown in Figure 3.5. When
the liquid first comes in contact with the fiber mat, it wicks in due to spontaneous
imbibition. At low capillary numbers, wicking always precedes the primary flow front
resulting in partial wetting of the fiber tows and the stitches. When the leading flow fronts
reach the double stitches, the liquid which is already present there due to wicking, exerts an
attractive force due to surface tension and transverse flow occurs. During transverse flow,
the adjacent leading flow fronts meet from opposite directions. If the lagging flow fronts
between the fiber tows do not pass the stitches before the transverse flow is completed,
macrovoids are trapped. The size and the number of macrovoids depend upon the extent of
lead - lag which, in turn, depends upon the ratio of viscous to capillary forces. Figure 3.6
is a photograph of surface macrovoids that remain trapped in the gaps between the fiber
tows after completion of the mold filling.
Figure 3.7 shows the percent area macrovoid content as a function of capillary number for
the different types of non - reactive liquids used in this study. Group 1 refers to the
various silicone oils and DOP oil. Group 2 refers to the data for ethylene glycol and Group
3 for water and glycerin. All the three groups of liquids show the same trend in the
macrovoid content as a function of capillary number. The macrovoid content decreases
exponentially with increasing capillary numbers with nearly zero void content beyond
capillary number ~ 10"3. This defines the critical capillary number.
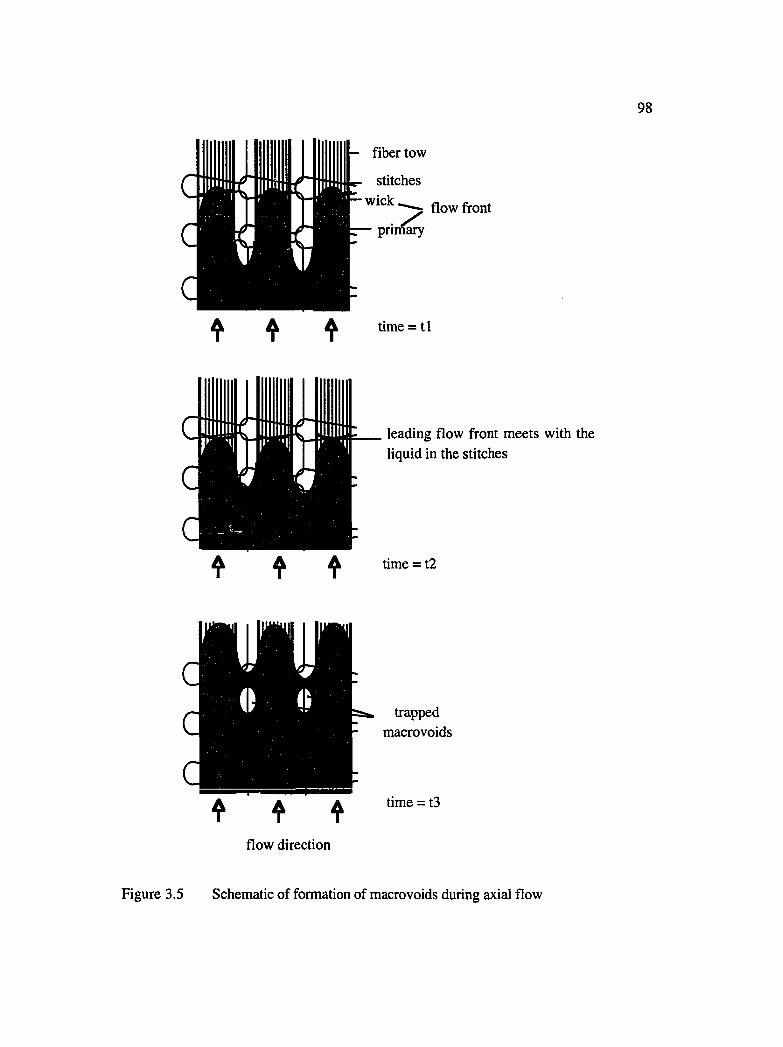
98
— fiber tow
stitches w ick^
p r ir i î^flow front
time = t l
leading flow front meets with the liquid in the stitches
time = t2
f f fflow direction
trappedmacrovoids
time = t3
Figure 3.5 Schematic of formation of macrovoids during axial flow

99
Macrovoids
Figure 3.6 Photograph of macrovoids trapped in the fiber mat
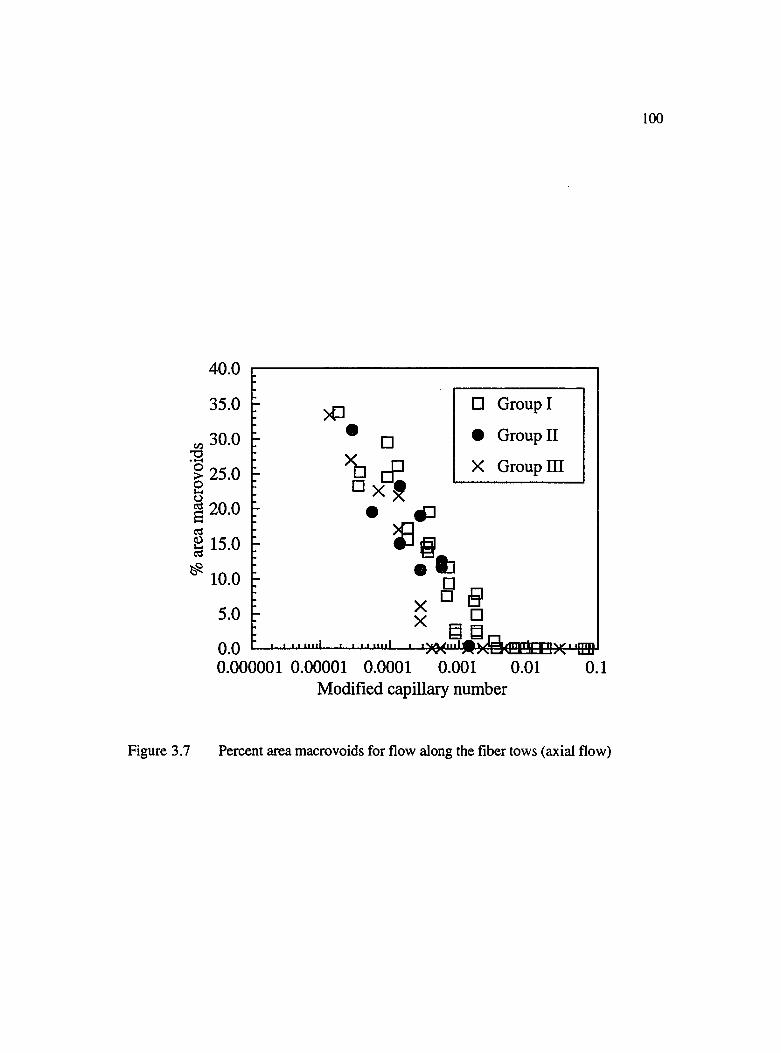
100
iII
> q□ Group I
• Group II
X Group III
40.0
35.0
30.0 P ^ o: X
25.0
20.0
15.0
10.0
5.0
0.0 0.000001 0.00001 0.0001 0.001 0.01 0.1
Modified capillary number
£
XX
I I m i l t t i - i IIuL
# ]
□ B □
Figure 3.7 Percent area macrovoids for flow along the fiber tows (axial flow)

101
The results shown in Figure 3.7 corroborate the reasoning given earlier for the formation of
macrovoids. At low capillary numbers, more macrovoids are formed because of the nature
of lead - lag at the flow front, wherein the liquid leads within the fiber tows and lags
between the tows. With increasing capillary numbers upto the critical capillary number, the
viscous forces become comparable to the capillary forces, reducing the extent of lead - lag,
and thereby minimizing the formation of macrovoids.
3.3.2 Transverse Flow
The nature and the mechanism of macrovoid formation during transverse flow in stitched
unidirectional fiber mat was found to be quite similar to that during axial flow. The liquid
led along the stitches and lagged in the tow between the stitches. The lead - lag however,
was not as strong as in the case of axial flow. Transverse flow occurred due to the flow of
liquid into the fiber tows and led to the formation of macrovoids. Figure 3.8 shows the
percent area macrovoid content during transverse flow. As in the case of axial flow, the
macrovoid content increases exponentially below a certain critical capillary number. For
transverse flow, this corresponds to the capillary number of ~ 3.0 x 10'3.
It was found that if the liquid was allowed to bleed at higher flow rates after the mold filling
was over, nearly all the macrovoids could be purged out from the fiber mat. However, this
does not seem to be a feasible solution for actual production of composite parts as it
involves wastage of expensive resin material. Also, since, both Figures 3.7 and 3.8 show
that no macrovoids are formed beyond the critical capillary number, it appears that the mold
filling should be carried out so that the capillary number is higher than the critical capillar}'
number. This indeed is true except for the fact that at high flow rates, another type of
voids, namely, the microvoids are formed within the fiber tows. These are not surface
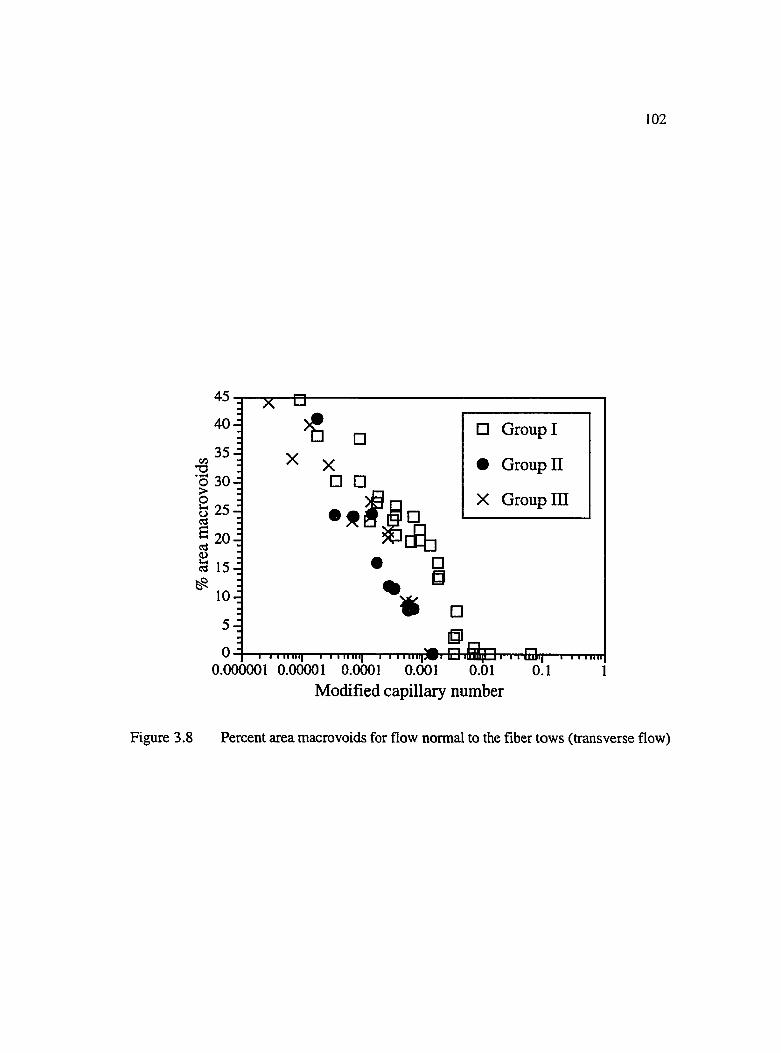
102
4 5
4 0 - j
3 5 - j
3 0 - :
2 5 J
2 0 - j
15-j
1 0 4
540
X X □ □• n i la
□ Group I
• Group II
X Group III
T I 11 iiii|0.001
□s
□0 ,
0.000001 0.00001 0.0001Modified capillary number
HBjl I I I I ITilfJ I III III!0.01 0.1
Figure 3.8 Percent area macrovoids for flow normal to the fiber tows (transverse flow)

103
voids and are more difficult to purge. The following sections describe formation of
microvoids at high flow rates for both axial and transverse flows.
3 .4 Flow Visualization of Microvoids Formation
3.4.1 Axial Flow
As in the case of macrovoids, the formation of microvoids during axial flow can be
correlated to the nature of lead - lag at the flow front. When the viscous force dominates
the capillary force, the nature of lead - lag at the flow front is reversed. Under such a
situation, the liquid leads in the gaps between the fiber tows and lags within the tows.
Figures 3.9 (a) and (b) depict the flow front wherein the silicone oil (200 cs.) is injected at
a superficial velocity of 3.9 cm / sec. No wicking was observed in this case. This is so
because at high flow rates, the primary flow front progresses at a rate equal to or faster than
the wicking rate. Also, the lead - lag is not very uniform. This is because of the non
uniformity in the gaps due to unequal spacing between the fiber tows.
A plausible mechanism to explain the formation of microvoids is shown by the schematic in
Figure 3.10. As can be seen in Figures 3.9 (a) and (b) and in the schematic in Figure 3.10,
the leading flow fronts diffuse inward from the larger gaps into the fiber tows. After
diffusing into the tows, the liquid meets with the lagging flow fronts moving in the flow
direction within the tows. When flow fronts meet from opposite directions, microvoids are
trapped. These microvoids may remain at the site of their formation or may move along
with the flow with gradual change in their shapes and sizes. The shape, size, and the
mobility of microvoids depend on the balance of the air pressure inside the microvoids,
surface tension forces, and the hydrodynamic pressure. Geometric factors like the non
uniformities within the fiber tows and stitches also play important roles in governing the
final shape and size of the microvoids.

104
Flow fron t leading between the tows
(a)
Leading flow fron t
Flow direction
Figure 3.9 Photographs showing the lead-lag at the flow front for axial flow and capillary number > 10"3 : (a) overall view and (b) zoom view

105
Q
d
fiber tow
fTi \stitches
— flow front
time = tl
trappedmicrovoids
time = t2
flow direction
Figure 3.10 Schematic of formation of microvoids during axial flow

106
It was observed that as the microvoids moved with the flow from a larger capillary to a
more constricted capillary, they were compressed in the lateral direction while their length
increased. In some instances due to surface tension effect, microvoids moving along
adjacent wicking streams merged together to form a larger void as shown in Figure 3.11.
Elongated, large cylindrical microvoids were formed at higher capillary numbers, while
much smaller microvoids were formed at comparatively lower capillary numbers. Also,
because of the 'round - up' type of mechanism for the formation of microvoids, most of
them were formed in the middle of the fiber tow. Figures 3.12 (a) and (b) show the
microvoids formed at Ca#* = 0.36 and ~ 0.004 respectively.
Figure 3.13 shows the percent area microvoid content as a function of velocity for silicone
oil (200 cs.), DOP oil, and ethylene glycol. All three liquids show an increase in the
microvoid content with an increase in injection velocities. The critical velocity (Vc), or the
velocity which marks the onset of formation of microvoids, was however, different for the
three liquids. The critical velocity was the least in the case of silicone oil and the highest
for ethylene glycol. These results show that for liquids with higher viscosities, microvoids
begin to form at much lower injection flow rates as compared to liquids with comparatively
lower viscosities. The percent area microvoid content was also plotted for the three liquids
as a function of capillary number. To get a better perspective of the range of capillary
numbers at which microvoids were formed, they are plotted along with the percent area
macrovoids as shown in Figures 3.14 (a), (b) and (c). All the three plots show that beyond
the critical capillary number (~ 2.0 x 10'3 for silicone oil and DOP oil and ~ 10'3 for
ethylene glycol), the microvoid content starts to increase and reaches nearly 5% at the
capillary number equal to 0.36. This was the highest capillary number that could be
achieved in the flow visualization experiments and corresponds to the injection velocity of
3.9 cm/scc. Figure 3.14 (d) shows that as in the case of macrovoids, microvoid content

107
I I
Microvoids I10 wm. s
Figure 3.11 Photograph of coagulated microvoids formed by joining of adjacent wicking streams
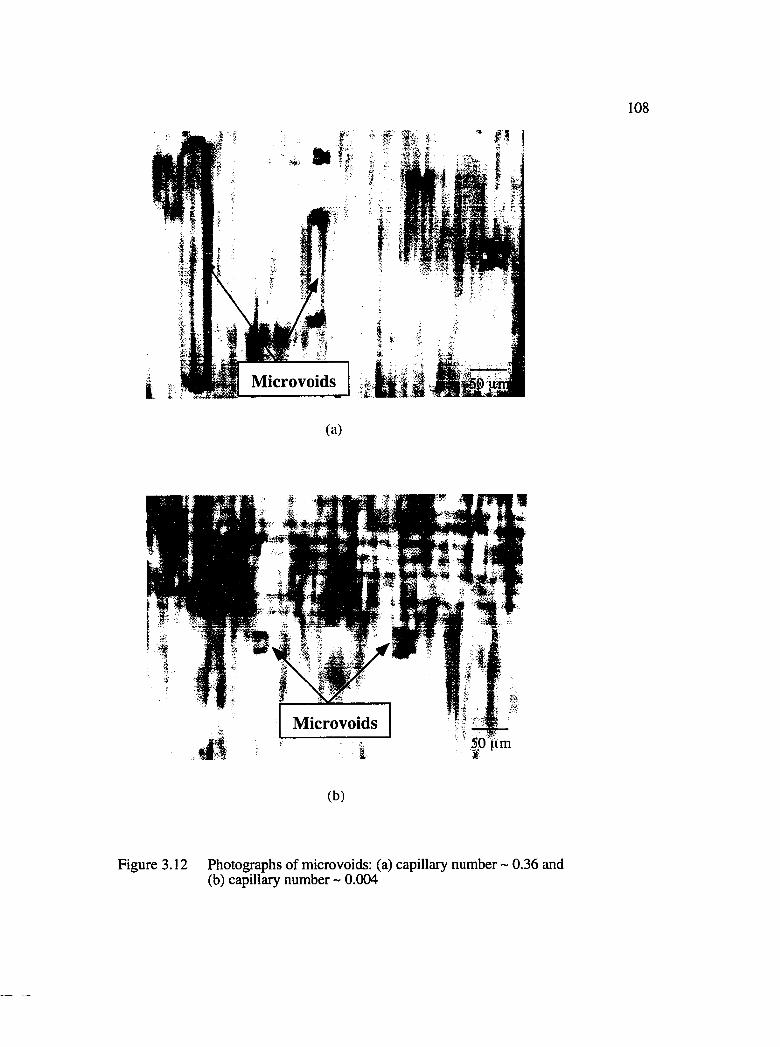
108
f
M icrovoids
(a)
M icrovoids
(b)
Figure 3.12 Photographs of microvoids: (a) capillary number ~ 0.36 and (b) capillary number ~ 0.004
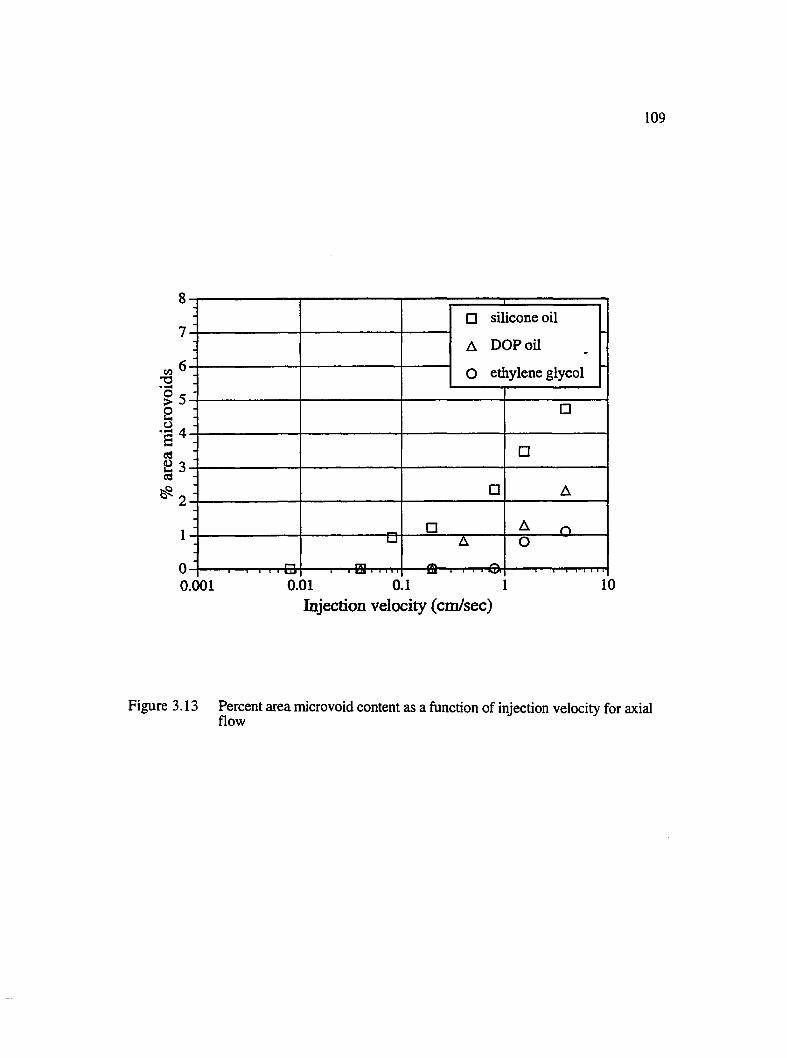
109
8q
7□ silicone oil
A DOP oil
O ethylene glycol
/
f i lCO ^ .
□
□as
^ o ■ □ A
1 □ ^ nX
A
----- Q A O
o.cK)1 0. 01 0 1 1Injection velocity (cm/sec)
Figure 3.13 Percent area microvoid content as a function of injection velocity for axial flow

110
18 .
16- □ □ macrovoids14- □12- □ ■ microvoids
c/3T3
OÜ 104e
I8-:6-i4-j
2-io J 0.0001
30.
12 204§ , | 1 5 ^
a 1 0 4cd
□
0.001-B il T-n n p
0.01- I — I I i l I I I
0.1
1816
• 1 4 ^■12 ‘I
§10864
42
0
Modified capillary number
(a)
A macrovoids
A microvoids
5 4
0 - j ------------1— I I I j h i i | r
0.0001 0.001
A A A ^ ....... .........1— I I ï n ii|0.01 0.1
4253
4 2 0 ^P
-15 •
410 §4 5
0
Modified capillary number
(b)
Figure 3.14 Percent area macro and microvoids for axial flow : (a) silicone oil, 200 cs and (b) DOP oil, (c) ethylene glycol and (d) master curves for all three liquids
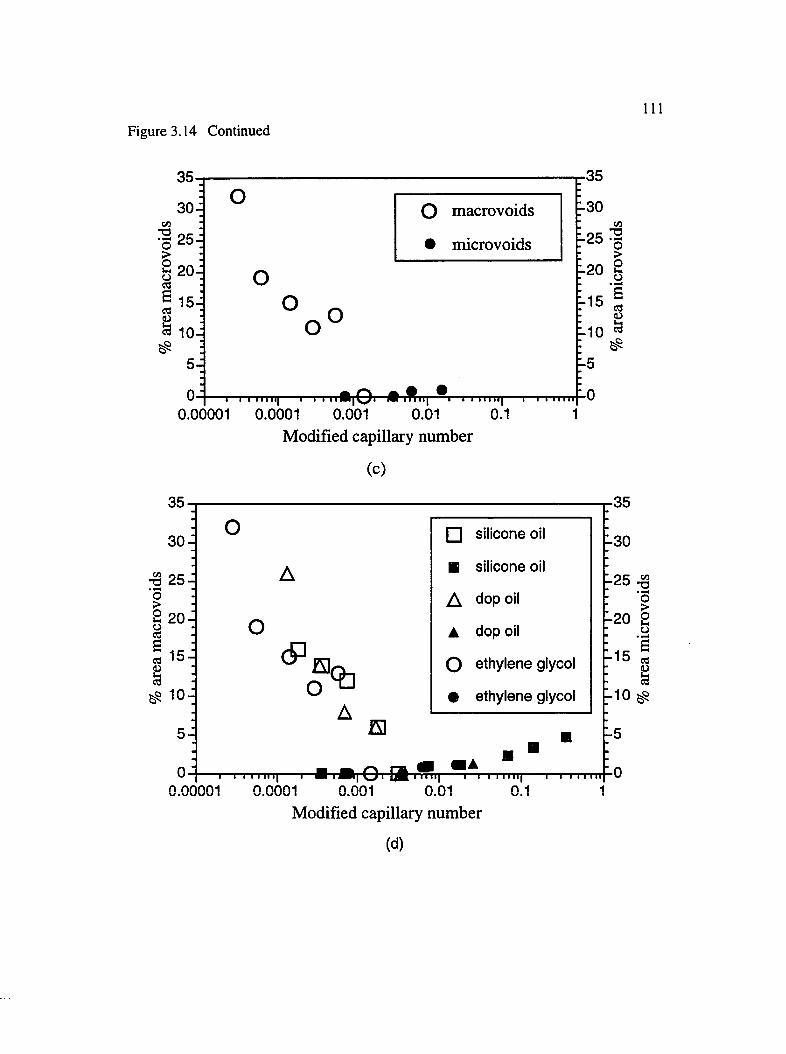
Figure 3.14 Continued111
CO1I§
353 0 :
2 5 -I
2 0 -i
1 5 :
1 0 :
5:0
0.00001
o o macrovoids
• microvoids
oo
o o
“I—I 1 111 lll0.0001 0.001 0.01
1— I I 111 l l l —
0.1T — I I M i l l
-20
35
30CA
25 1
I'i10 ^
50
Modified capillary number
(c)
35.
30 :
-Q 25: "o
§ 20-1
I i a . §
# 1 0 :
5:
0
O
d ?%
□ silicone oil
■ silicone oil
A dop oil
A dop oil
o e thy lene glycol
• e thy lene glycol
£3
I I I I M l i |
0.00001 0.00010 1 - *
0.001TTTTTf
0.011— I ' l 11 I I I ]
0.1
■35
-30
■25 ‘o
■20 2 s
I■10 ^
-5
■0
:15
Modified capillary number
(d)

1 1 2
for all the three liquids bunched together when plotted against the capillary number. The
plot also shows that both the macro and the microvoid content is nearly zero percent within
a certain range of the capillary numbers. Such plots can be used as effective tools for
designing optimal processing windows in actual production of composite parts to minimize
void content of either type.
It should be pointed out that the microvoid content reported here represents an average of
only those areas in the fiber tows with microvoids in them. Thus, the overall void content
would be lower. However, for structural parts, depending upon their end use, local void
content could be more significant than the overall void content because it is these localized
weak spots that become regions of stress concentration at the fiber / matrix interface and
provide opportunity for crack propagation under external loading. Apart from affecting the
structural integrity of the part, these localized voids can also cause optical inhomogeneity
due to the refractive index mismatch between the matrix and the reinforcing fibers. With
glass canopies of the fighter jets being replaced by fiber reinforced composite materials, a
great deal of effort is being directed towards making composite canopies as transparent as
the currently used polycarbonate sheets.
The effect of fiber wetting on optical transparency of composite samples can be seen from
Figures 3.15 (a) and (b). A grid was placed underneath the composite samples with good
and poor fiber wetting and photographs were taken under identical lighting conditions.
Although the sample with good fiber wetting (Figure 3.15 a) is not completely transparent,
the grid is more clearly visible when compared to the sample with poor wetting (Figure
3.15b).

113
(a)
(b)
Figure 3.15 Photographs showing the optical transparency of composite samples; (a) good fiber wetting and (b) poor fiber wetting

114
3.4.2 Transverse Flow
Formation of microvoids during transverse flow was also studied. It was observed that for
the same capillary number, the microvoid content was higher in the case of transverse flow
as compared to the axial flow. Also, unlike the case of axial flow, microvoids were formed
even at lower flow rates (capillary number -10"^). Figure 3.16 shows that for transverse
flow, there does not exist a range of capillary number within which the void content is
zero. Both macro and microvoids were formed across a broad range of capillary numbers.
The reason for this can be explained by two types of flow mechanisms, both of which can
lead to the formation of microvoids during transverse flow. However, depending on the
injection flow rate, more microvoids were formed by one mechanism than the other. At
lower flow rates, or for capillary number upto < ~10'2, Mechanism I shown by the
schematic in Figure 3.17 was more dominant. For the sake of clarity, the sequence of
events leading to microvoid formation is shown in three separate fiber tows. In reality, this
sequence occurs in each of the fiber tows as the flow progresses through the fiber mat.
When the main flow front or the primary flow front reaches the edge of the fiber tow, the
liquid first wicks into the stitches causing a lead-lag at the flow front (Figure 3.18).
Transverse flow or cross-flow then occurs wherein the liquid present in the stitches wicks
into the fiber tows by capillary action. When these wicking streams move towards each
other from opposite directions, they either collapse into one another or trap microvoids.
Microvoids are trapped if the surrounding hydrodynamic pressure gradient is smaller than
the sum of the capillary pressure due to surface tension forces and the air pressure inside
the voids. Figure 3.19 shows the photograph of microvoids trapped in the fiber tow
during transverse flow at a capillary number of 0.0033.
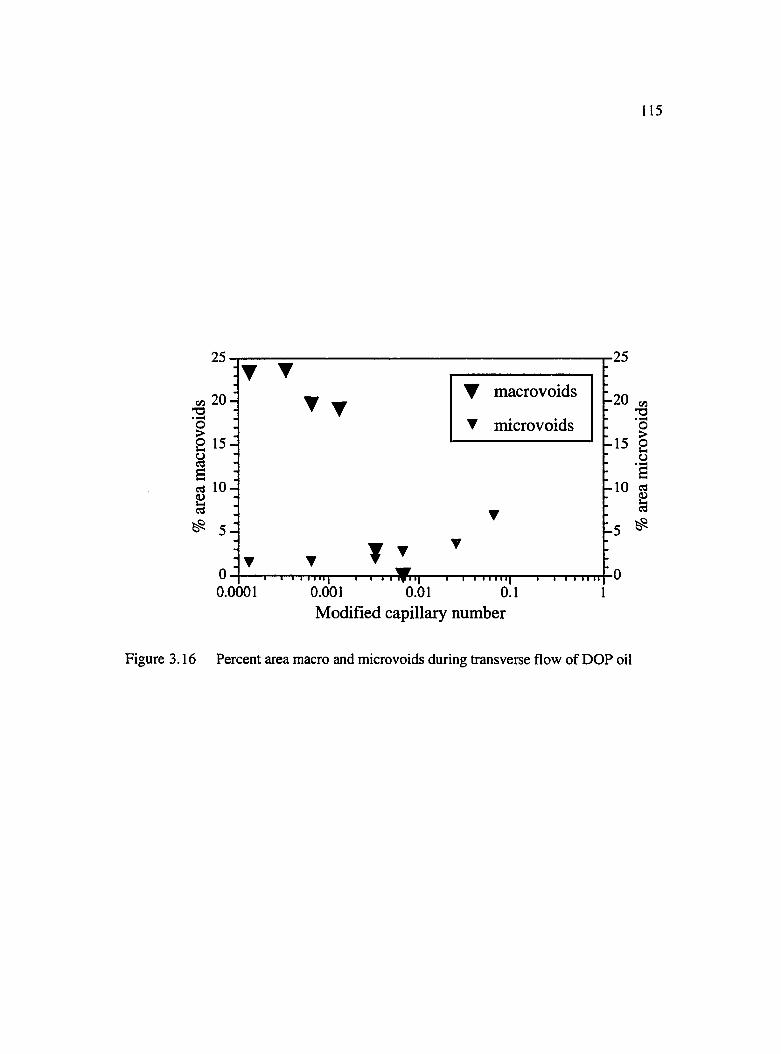
15
▼ macrovoids
▼ microvoids
10
0.0001Modified capillary number
Figure 3.16 Percent area macro and microvoids during transverse flow of DOP oil
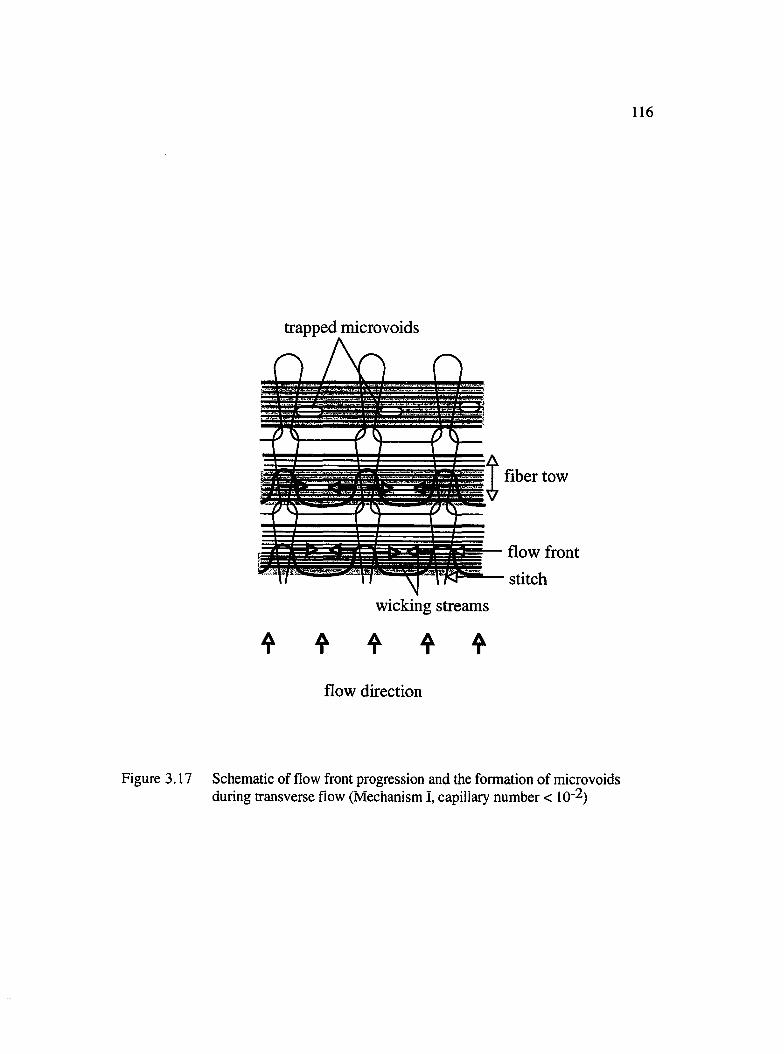
1 1 6
trapped microvoids
Îfiber tow
flow front
stitch
wicking streams
f f f f f
flow direction
Figure 3.17 Schematic of flow front progression and the formation of microvoids during transverse flow (Mechanism I, capillary number < 10'2)

117
flow front leading in the stitches
Flow direction
Figure 3.18 Photograph illustrating lead-lag at the flow front during transverse flow (Mechanism I, capillary number < 10‘2)

118
Microvoids
Figure 3.19 Photograph of microvoids formed during transverse flow of DOP oil capillary number ~ 0.003 (200X)

119
Figures 3.20 (a) through (c) show a sequence of photographs that illustrate the transverse
impregnation and the dynamics of microvoid formation and movement in the fiber tows.
Curved liquid menisci (Figure 3.20(a)) represent the wicking streams moving in opposite
directions within the capillaries. As the impregnation of the fiber tow continued, the liquid
menisci approached closer and microvoids were trapped (Figure 3.20 (b)). After being
formed, the microvoids (marked in figures by *) moved along with the flow with gradual
changes in their size and shape and stabilized once the surrounding forces reached an
equilibrium (Figure 3.20 (c)).
At higher flow rates or capillary numbers > ~10"2, the lead-lag decreases and the second
mechanism for microvoid formation becomes more dominant. Mechanism II is shown
schematically in Figure 3.21 (a). When the main flow front reaches the fiber tow, the
liquid flows around it, (macroflow) due to the higher permeability of the gaps between the
tows. The fiber tow is then subsequently impregnated by capillary action (microflow).
Thus, wetting in the fiber tow occurs behind the primary flow front. This means that the
fiber tow does not get completely saturated with the liquid as the primary flow front
progresses around the tow. If the microfiow does not displace all the air out of the fiber
tow before the primary flow front recoalesces downstream of the tow, air remains trapped
inside the tow. Similar explanation for this type of flow behavior has been suggested
earlier [Chen et al., 1993; Pamas and Phelan, 1991; Damani and Lee, 1990]. However the
actual formation of microvoids was not investigated. Most of the microvoids at higher
flow rates were formed near the edge of the tow as shown in Figure 3.21 (b).
More microvoids were formed by both mechanisms at higher flow rates as compared to
lower flow rates due to the difference in the time scales associated with the engulfment of
the fiber tow and the impregnation process. At higher flow rates, there is even lesser time
for the microflow to displace all the air out before recoalescence of the primary flow front

120
iSôl+im
(a)
ÈSoea*;
..
(b)
Figure 3.20 Photographs showing the dynamics of microvoid formation and movement during transverse flow : (a) formation of microvoids (b) and (c) movement with change in shape and size

Figure 3.20 Continued
121
(c)
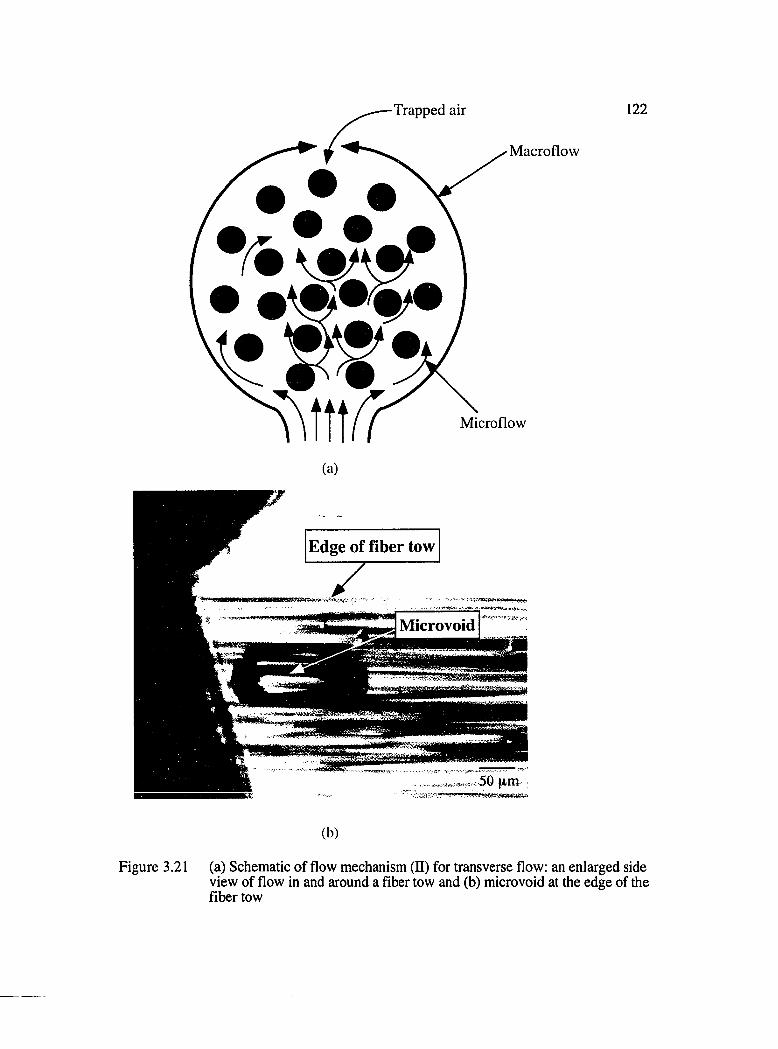
Trapped air 122
Macroflow
Microflow
(a)
Edge of fiber tow
7m m em T SIlM Microvoid
fSsStt
. 50 p,m
(b)
Figure 3.21 (a) Schematic of flow mechanism (II) for transverse flow: an enlarged sideview of flow in and around a fiber tow and (b) microvoid at the edge of the fiber tow

123
occurs. Also, at higher flow rates, the liquid progresses through the larger gaps at a much
faster rate, leaving the fiber tows even more poorly wetted.
3 .5 Mobilization of Macro and Micro voids
Flow visualization experiments also showed that for both axial and transverse flows,
microvoids formed during the mold filling stage were much more difficult to purge when
compared to the macrovoids. Microvoids remained trapped within the fiber tows even after
bleeding the liquid at much higher flow rates than at which they were formed. It is easier to
purge macrovoids as they lie in the path of much lesser flow resistance compared to the
microvoids. The ratio of the gap between the tows to that within the tows is in the order of
1000:1. So, when the liquid is pumped at higher flow rates during the bleeding stage,
most of the liquid flows through the path of least resistance, i.e. between the tows
bypassing the smaller gaps within the fiber tows. Thus, sufficient viscous drag due to the
hydrodynamic pressure is not generated to mobilize the microvoids.
The injection velocities needed to mobilize a void of length L, radius ry and lodged in a
capillary of radius rt can be estimated from the following correlation [Chen, 1993]:
(3.6)[tv _ r 2 * r, ^Ylv I n * r, ; l^LCos (0)^
where
P is the geometric factor (<1) and depends on the pore space configuration
Keff is the effective permeability
Based on Equation 3.6, the injection velocities required to mobilize stable macro and
microvoids would be in the order of 1 cm/sec and 100 cm/sec respectively for a fiber
volume fraction of about 50% and a liquid having a viscosity of 50 cP and a surface tension

124of 50 dynes/cm. Thus, much higher flow rates are required to completely remove the
microvoids as compared to macrovoids.
3 .6 Vacuum Assisted Liquid Injection
VALI (vacuum assisted liquid injection) was also tried in this study. It was found that both
the macro and the micro void contents were reduced considerably when the liquid was
injected into the fiber mat with the assistance of vacuum. Pulling vacuum during liquid
injection helped to increase the flow rate and so minimal macrovoids were formed.
Microvoid content was low because of reduction in pressure in the air pockets trapped
inside the fiber tows, resulting in improved tow impregnation. To obtain a part with 'zero'
voids however, requires a perfect vacuum applied uniformly in the mold cavity, and a very
good seal to prevent ;y leakage of vacuum. This may not be an easy task for a large mold
used in the actual production of composite parts. Moreover, the use of vacuum may not be
feasible when there are constraints on the cycle time (e.g. SRIM process), and in cases in
which the resin contains volatile species (e.g. UP and vinyl ester resins).

CHAPTER IV
FIBER CONSOLIDATION AND SPRINGBACK IN POWDER (TACKIFIER) COATED PREFORMS
4.1 Materials
A commercial bismaleimide (BMI) resin was used as the tackifier material, while graphite
preforms with satin weave (6k, 4HS) were used as the fiber reinforcement (Figure 4.1).
Although the exact formulation of the resin is proprietary, it is believed that the two major
components are 4, 4'-bismaleimido-diphenylmethane and 0, O' - diallyl bisphenol A
[Perry, 1993] whose structures are shown in Figure 4.2. A thermoplastic powder,
polymethyl-methacrylate (PMMA, Mw = 250, 000), was also used as a substitute for the
reactive BMI powder in some cases to decouple the effects of increase in modulus due to
reaction and tackifier location on springback of consolidated laminates.
4.2 Equipment and Experimental Procedure
4.2.1 Differential Scanning Calorimetry
TA Instruments' DSC 2910 was used to obtain the chemical reaction kinetics of BMI
tackifier. A DSC is designed on an enthalpy change method. It operates by recording the
difference in the energy required as a function of time to maintain the same programmed
temperature profile for the sample being studied and an empty reference container. Figure
4.3 shows the schematic of the set-up used. Test conditions and sample parameters are
125

Warp tows Fill tows
Gap between warp tows
Crossdirection
Machinedirection Crimps
Gap between fill tows
Figure 4.1 Schematic of 6k, 4HS woven fiber reinforcement

127
O OIl II
[ T ^ N —
a ü
4,4' - bismaleimido-diphenylmethane
CHz= CHCHj ^ CH3 / CHjCH = CH;
CH;
0 ,0 ' - diallyl Bisphenol A
Figure 4.2 Structure of two components of Bismaleimide resin based tackifier
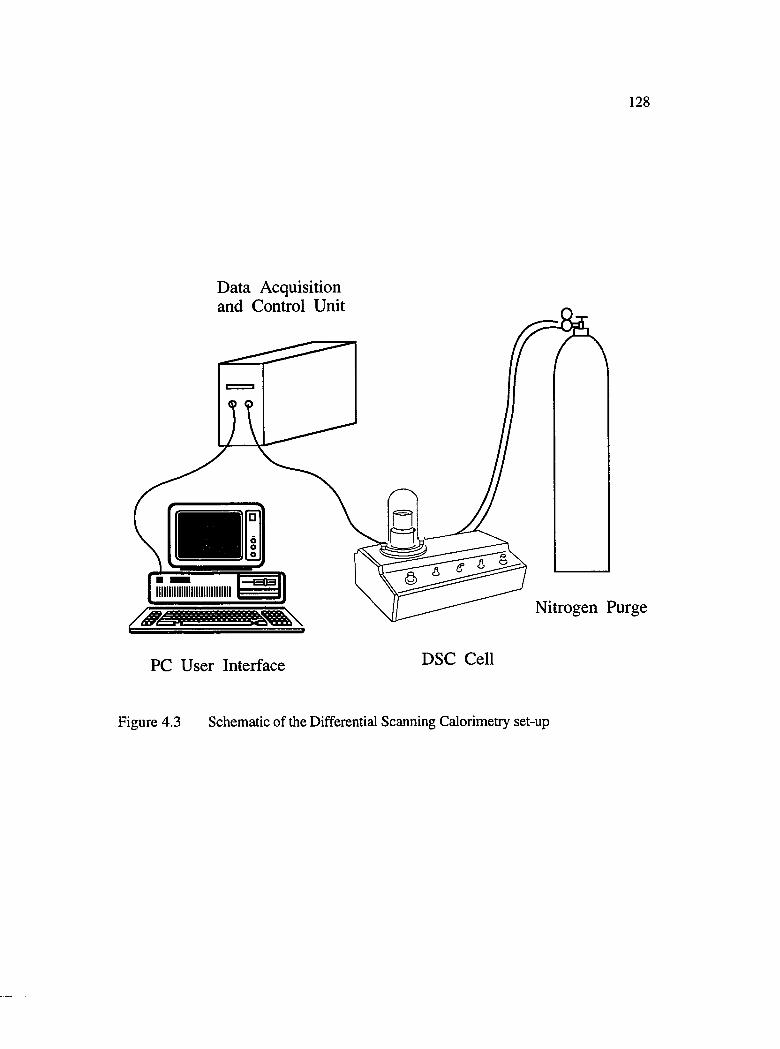
128
Data Acquisition and Control Unit
Nitrogen Purge
PC User Interface DSC Cell
Figure 4.3 Schematic of the Differential Scanning Calorimetry set-up

129
input into an interface computer before the start of each run. The computer is linked to the
data acquisition and control unit which is connected to the DSC cell. The DSC cell consists
of the sample and the reference pans placed on a cast constantan metal disc. Welded to the
base of the disc are a pair of chromel-alumel thermocouple wires. One set of wires
measures the temperature of the sample pan, while the other measures that of the reference
pan. An average temperature circuit measures and controls the temperature of the sample
and reference holders to conform to a predetermined time-temperature program. At the
same time, a temperature difference circuit compares the temperatures of the sample and
reference pans and proportions the power to the heaters under each pan so that the
temperature remains equal. When the sample undergoes a thermal transition, the power to
the heaters is adjusted to maintain thermal equilibrium between the pans, and a signal
proportional to the power difference appears as either an exothermic or an endothermie
peak [Billmeyer, 1984].
The operating temperature can be programmed either in the ramp mode (heat/cool at a fixed
rate), isothermal mode, step mode, or any combination of these. Hermetically sealed
aluminum pans capable of withstanding an internal pressure of 0.2 MPa (2 atm) were used.
These pans had an average volumetric capacity of about 10 mm^. Thus, depending on the
density of the material, the weight of the samples typically range from 5 to 20 mg.
To characterize the resin reaction kinetics, both scanning and isothermal experiments are
required. The scanning experiments are used to determine the total heat which can be
generated from a fresh sample. This is done by subjecting the sample under ramp
conditions twice. The second scan is done to get the baseline. A flat baseline indicates that
the material has reacted completely. By subtracting the baseline from the first curve, the
rate of reaction can be computed as a function of temperature. Integrating this curve over
the whole temperature range, the total heat of reaction can be obtained using Equation 4.1:

130
(4.1)
where, is the instantaneous heat generation rate and tf is the total time of reaction.
Next, using the total heat of reaction, AHtotal, the reaction rate and conversion (a) can
be computed using Equations 4.2 and 4.3:
1 r d H t ^dt AHtotai I dt J (4.2)
It should be noted here that dynamic scans only provide information about the ultimate heat
generated by a material, and do not give any insight into the reaction mechanism of
individual chemical species present in the material. Isothermal runs on the other hand, can
reveal the behavior of the reacting species and allow a better understanding of the kinetics
involved. The limitation however, is that isothermal tests are often halted if the reaction
becomes diffusion controlled (this occurs when the reaction temperature is below the glass
transition temperature of the resin), and thus, the reaction never goes to completion.
Typically, isotherm ally tested samples are further subjected to a series of dynamic scans in
order to determine the residual exotherm heat and the baseline. The final conversion at any
isothermal temperature is obtained by Equation 4.4:
“ rmal = (4.4)^ " to ta l
AHiso is the total area under the curve (computed from Equation 4.1) for the isothermal
reaction. The instantaneous reaction rate and conversion for isothermal tests can be obtained from Equations 4.2 and 4.3.

131MDSC (Modulated DSC) was used to measure the glass transition temperatures of the resin
cured under different conditions. This was done to see if the reaction pathway affected the
properties of the final network formed. MDSC offers several advantages over conventional
DSC. These include, increased sensitivity, improved resolution of closely occurring and
overlapping transitions and separation of reversing and non-reversing heat flow. In this
technique, a Liquid Nitrogen Cooling device (LNCA) is used to superimpose a sinusoidal
ripple (modulation) on a linear heating profile (underlying heating rate). The overall
heating profile thus depends on the underlying heating rate, the amplitude, and frequency
of modulation. The total heat flow comprises of two components as given by Equation 4.5
[MDSC Manual, 1993]. The reversing part being dependent on the heating rate and the
non-reversing dependent only on the absolute temperature.
^ [Cp + fR (t,T)] + fA (t,T) (4.5)
reversing non-reversingwhere
^ = heat flowf f . .= heating rate
Cp = sample heat capacity
t = time
fR (t, T) = kinetic response of any physical transition, and
fA (t, T) = kinetic response of any chemical transition
The MDSC software uses the Discrete Fourier Transformation (DPT) technique for signal
deconvolution to separate the non-reversing and reversing parts of the total heat flow. This
technique determines the measured amplitude of the sample temperature and heat flow
modulation by comparing the raw modulated data to a reference sine wave of the same
frequency. Any portion of the raw signal which follows the reference sine wave is the

132reversing part, and is "in phase" with the modulation. The remaining "out of phase"
portion constitutes the non-reversing part After determining the heat flow and temperature
amplitudes, the heat capacity is computed from Equation 4.6:
C p = -K ., *Qamp
\^am p j * ( ^ )
where
Cp = heat capacity Kcp = heat capacity constant Qamp = heat flow amplitude Tamp = temperature amplitude
Period = modulation period
Reversing heat flow is calculated by multiplying the heat capacity by the underlying heating
rate. This is then subtracted from the total heat flow to yield the non-reversing heat flow.
4.2.2 Preforming Experiments
4.2.2.1 U-Shape Bending
Figure 4.4 shows the initial distribution of the BMI powder prior to the preforming stage.
Eight layers of these tackified woven graphite fiber mats were placed between two plates
(Figure 4.5), which were then bent into a U shape and then held in that position with a
clamp. The gap between the two plates was controlled to keep the fiber volume fraction
equal to 55%. The whole bending device was then placed for different intervals of time in
a preheated oven set at a desired temperature. After a certain conversion of the tackifier
was achieved (obtained from DSC experiments), the oven was shut off and the samples
were allowed to cool down to room temperature. The preform was then taken out of the
bending device, and springback angles were measured.
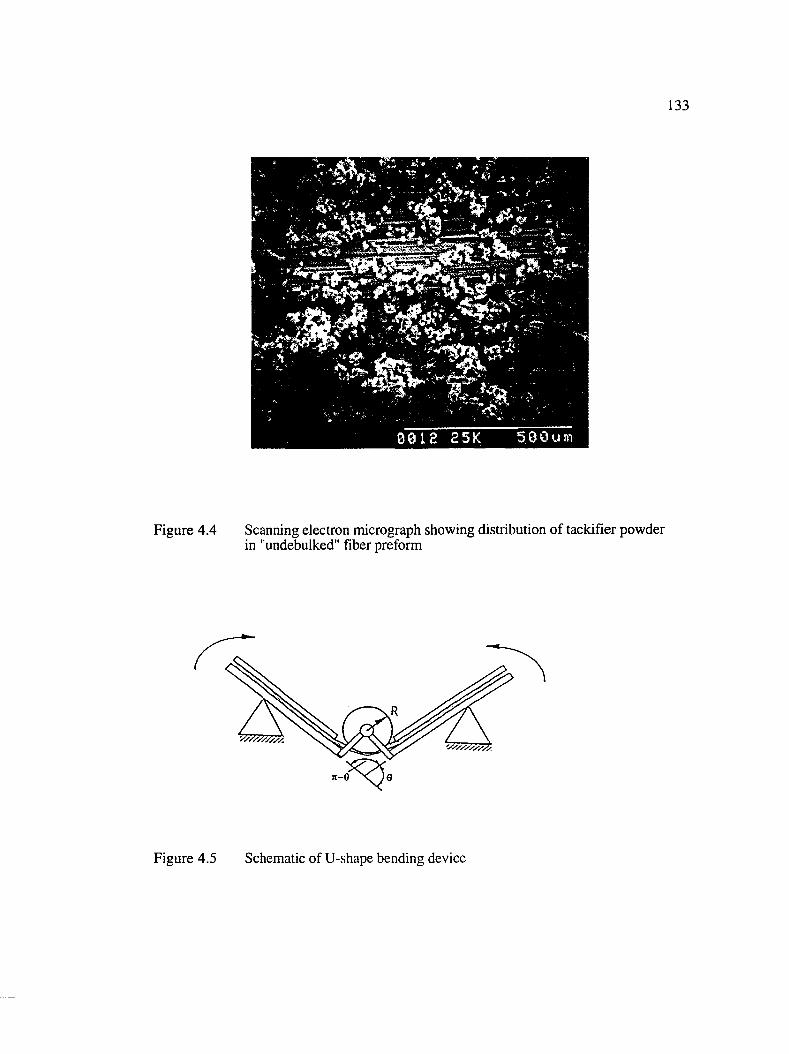
1 3 3
5 0 0 U m0 0 1 2
Figure 4.4 Scanning electron micrograph showing distribution of tackifier powder in "undebulked" fiber preform
Figure 4.5 Schematic of U-shape bending device

134
4.2.2.2 Vacuum Debulking
Vacuum debulking involves placing the fiber stack in a vacuum bag and pulling vacuum on
the sample. Prior to starting the debulking process however, certain preparatory steps are
needed. First, the desired amount of tackifier was applied to each layer. The layers were
stacked one on top of the other as illustrated schematically in Figure 4.6, and the whole
stack was then sandwiched between two Teflon release plies (Release Ease 234 TFP). The
"sandwich" was placed in the center of an aluminum plate. The temperature of the fiber
stack was controlled using a thermocouple connected to an Omega on-off controller. A
strip of jute fabric was placed as a runner from the vacuum tube to the ply stack. Tacky
tape was placed around the edges of the aluminum plate and around the vacuum tube and
thermocouple wire. The whole assembly was then sealed with an "envelope". The
envelope material was VAC-PAK HS -8171 co-extruded nylon 6 film with additives for
heat stabilization up to 205°C. To ensure a good vacuum seal, the envelope was pressed
against the Tacky Tape using a metal roller caster. The vacuum bag assembly was then
placed in a preheated oven and the vacuum hose connected to a vacuum pump. A vacuum
of 29 in Hg was pulled for different amounts of time at a constant temperature of 94°C.
The sample was then allowed to cool to room temperature, with the vacuum still being
pulled during the cooling period. After cool down, vacuum was stopped, the sample taken
out and the thickness of the fiber stack measured using vernier calipers. The samples were
left overnight and the thickness measured again. Springback was calculated by taking the
difference in sample thickness between the two readings. Thus, the values obtained and
reported here for vacuum debulking experiments reflect the long term relaxation behavior of
compressed fibers, rather than instantaneous springback.
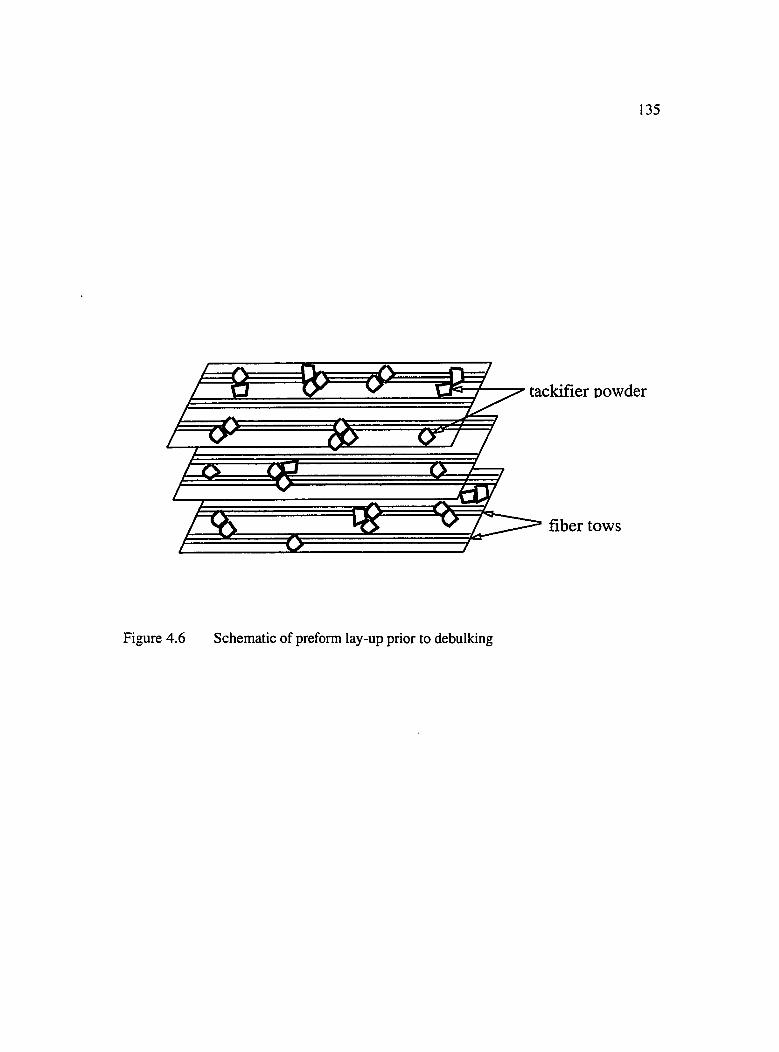
135
tackifier powder
fiber tows
Figure 4.6 Schematic of preform lay-up prior to debulking

136
4.2.2.B Lateral Compression
The experimental set-up that was devised to study the consolidation behavior and
springback of fiber preforms under lateral compression is shown in Figure 4.7. Mode of
fiber deformation in lateral compression is similar to vacuum debulking.
The only difference being that the fibers are consolidated by using a pneumatic force rather
than pulling vacuum. The press used is a one plunger dilatometer with a pneumatically
operated piston whose movement was controlled by toggling the lever on the three way
pneumatic valve. The position of the piston under different pressures was followed by an
LVDT (Trans-Tek, 0245-0000, 1-6). The data acquisition was an ADC-1 16 channel
system with a 40 gain amplifier on the first 8 channels (Remote Measuring Systems)
connected to a Macintosh computer SE/30. A nitrogen tank was used as the pressure
source. Calibration of the LVDT was performed using shims having different thicknesses.
Figure 4.8 shows the calibration curve for the LVDT used. The resolution of the LVDT
was 0.001 inch. Eight square pieces (5.08 cm x 5.08 cm) of the fiber preform were placed
in between two smooth metal plates with uniform thicknesses. Also, in order to minimize
heat loss through conduction, a wooden plate was placed between the lower metal plate and
the base of the press. The fiber preform along with the metal and wooden plates were then
covered by an aluminum box connected to a temperature controlled heat gun. This
assembly served as an oven. The temperature of the fiber mat was monitored using a
thermocouple attached to a digital temperature read-out device. Experiments were
conducted under different isothermal conditions. For experiments conducted at 94°C,
curing was done for 15 minutes. Consolidation was done under 30 psi gauge pressure (~
17 psi on the preform, as the c.s.a of the pneumatic cylinder was 14.52 cm2) with different
amounts of the tackifier, and as a function of pressure for untackified preforms. For high
temperature runs, after the curing cycle, the samples were cooled down to 38°C.
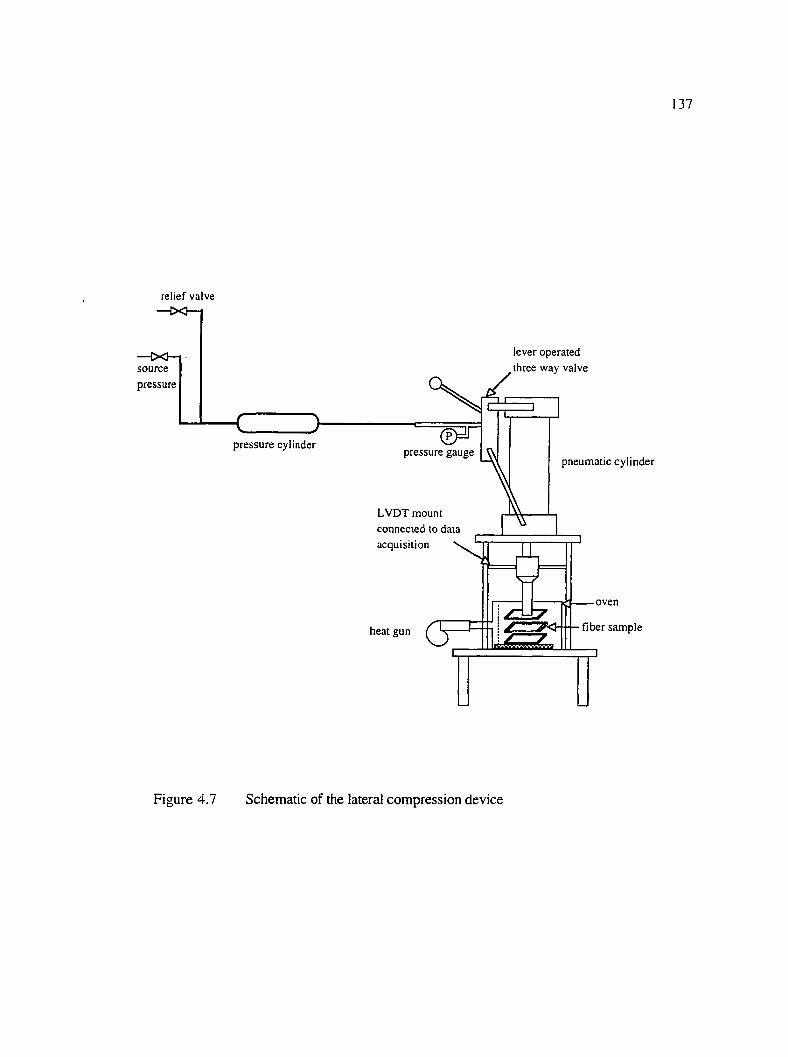
137
relief valve
lever operated three way valvesource
pressure
pressure gaugepressure cylinderpneumatic cylinder
LVDT mount connected to data acquisition N
:----- oven
fiber sampleheat gun
Figure 4.7 Schematic of the lateral compression device
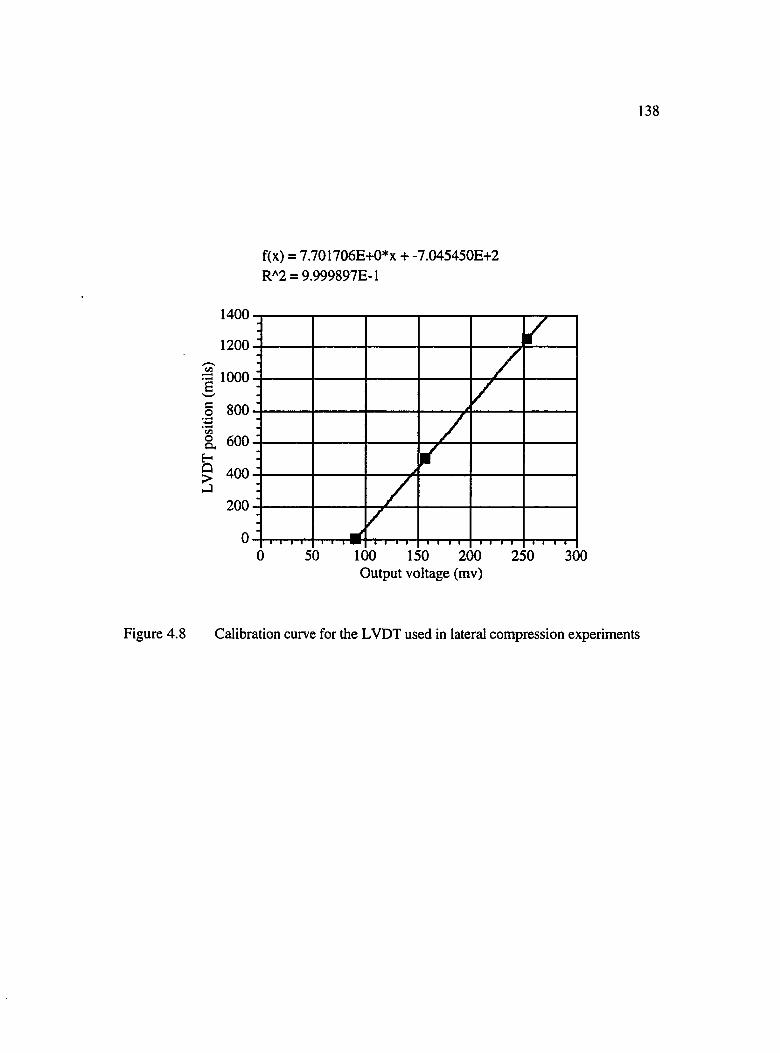
f(x) = 7.701706E+0*x + -7.045450E+2 RA2 = 9.999897E-1
1 3 8
1400
1200
= 1000
800
400
200
100 150 200 250Output voltage (mv)
300
Figure 4.8 Calibration curve for the LVDT used in lateral compression experiments

139
Springback was then obtained by taking the difference in the laminate thickness without
and with applied pressure.
4.2.3 Scanning Electron Microscopy
A Hitachi S-510 scanning electron microscope with a resolution of 70 Â , magnification
upto 150,000X, and an accelerating voltage of 25 KV was used to observe the distribution
of the tackifier powder in the fiber preform and to qualitatively describe the consolidation
and springback phenomenon. A carbon based ink was used to glue the sample onto the
holder. All samples were gold coated to provide a conductive layer. Photomicrographs
were taken using an installed Polaroid camera.
4.2.4 Rheometrics Dynamic Analyzer
A dynamic analyzer developed by Rheometrics, Inc. (a modified RDA II) was used to
monitor the changes in viscosity and viscoelastic properties G’ (elastic or storage modulus)
and G" (viscous or loss modulus) of BMI and PMMA powders as a function of
temperature and time.
The spectrometer part of the RDA consists of three subsystems: the actuator, transducer
and environmental control subsystems. The control computer synchronizes, generates, and
directs test instructions to, and processes raw data received from, all subsystems. The user
inputs commands such as type of mode, test sequence, strain rate, etc. in the test
control/analysis station (computer) which are transferred to the actuator controller through
the control computer. The actuator subsystem is comprised of electro-mechanical
components that apply a precise deformation to the test sample. The transformer "reads"
the response of the sample in mechanical energy form and converts it to electrical energy
before sending it to the control computer from where it is then transferred to the personal

140computer. The environmental control subsystem controls the temperature of the sample by
forced convection [RDA II Owner's Manual, 1990].
The RDA can operate both in the steady shear as weU as the dynamic mode. Steady shear
is employed to foUow the rheological changes of the polymer melt, while dynamic mode is
used to measure the viscoelastic properties. Also, there are four different types of fixture
geometries that can be used for testing the various different properties of the sample. These
include the parallel plate, cone and plate, concentric cylinder (couette flow), and rectangular
torsion. In this study, experiments were conducted under the dynamic mode using parallel
plate geometry (Figure 4.9). The actuator controls the angular displacement (6) or the
angular velocity (to) of the bottom plate according to the pre-specified conditions imposed
by the user. The relevant equations for testing under dynamic mode and for parallel plate
geometry are given by Equations 4.7 through 4.10:
K , = I (4.7)
Ky0 = —^ (4.8)
Y
where
Ky = strain constant
R = radius of the plate (mm)H = gap between the plates (mm)8 = actuator angular displacement in radians y = input strain
2* G
where
Kx = stress constant
Gc = 980.7 dynes/gm (98.07 Pa/gm)
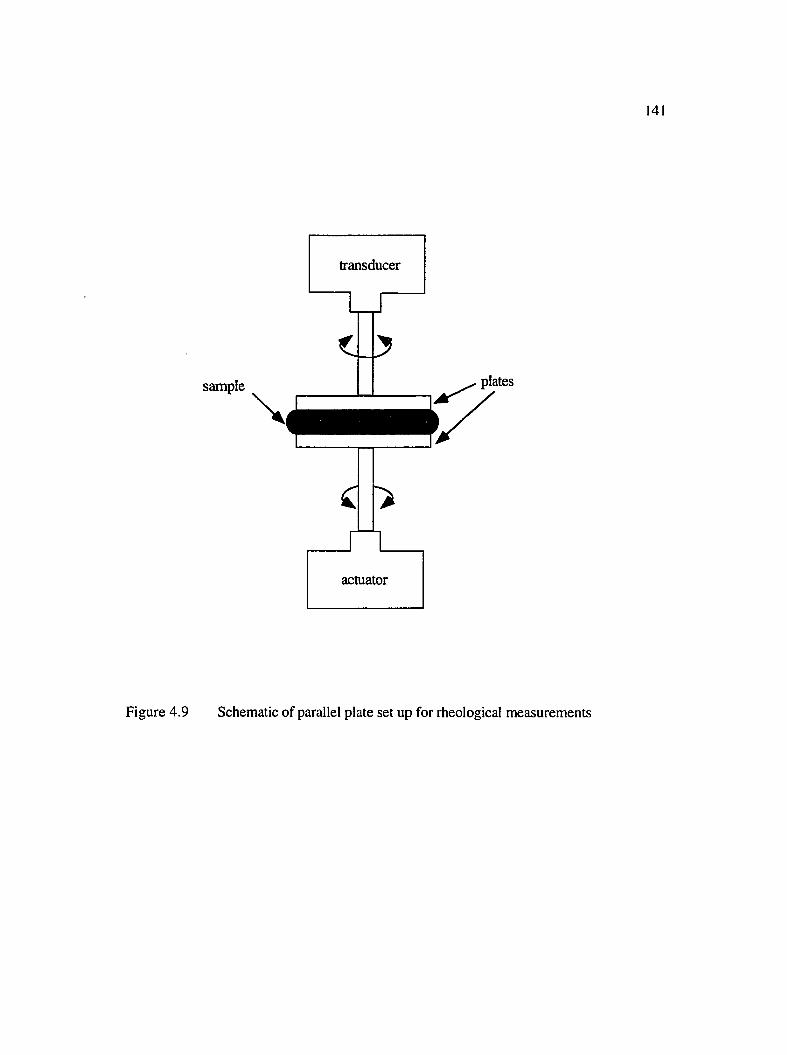
141
transducer
sample
\ iplates
actuator
Figure 4.9 Schematic of parallel plate set up for rheological measurements

142
The transducer measures the torque (M) on the upper plate which can be converted to stress
(t ), using the following equation:
X = M * Kx (4.10)
where
M = transducer torque in gm-cm
The viscoelastic properties (G' and G" ) are calculated from the values of shear stress and
strain from Equations 4.11 and 4.12 respectively.
G' = cos 5 ^ -1 (4.11)
G" = s i n ô f - l (4.12)y y )
where
S = phase angle between stress and strain
4.3 Results and Discussion
4.3.1 Characterization o f Reaction Kinetics
The scanning reaction rate profile and the isothermal conversion profiles for the BMI
tackifier used in this study are shown Figures 4.10 and 4.11 respectively. Scanning was
done from room temperature to 350°C at 5°C/min. The two reaction peaks in Figure 4.10 is
indicative of more than one reaction occurring. The first small peak at 125°C is probably
due to amine addition, while the second peak at 225°C due to BMI homopolymerization.
The isothermal conversion profiles shown in Figure 4.11 were obtained using Equation
4.4. With increasing temperatures, the reaction proceeds much faster and conversion
increases more rapidly with time. However, in all cases, the conversion profiles leveled
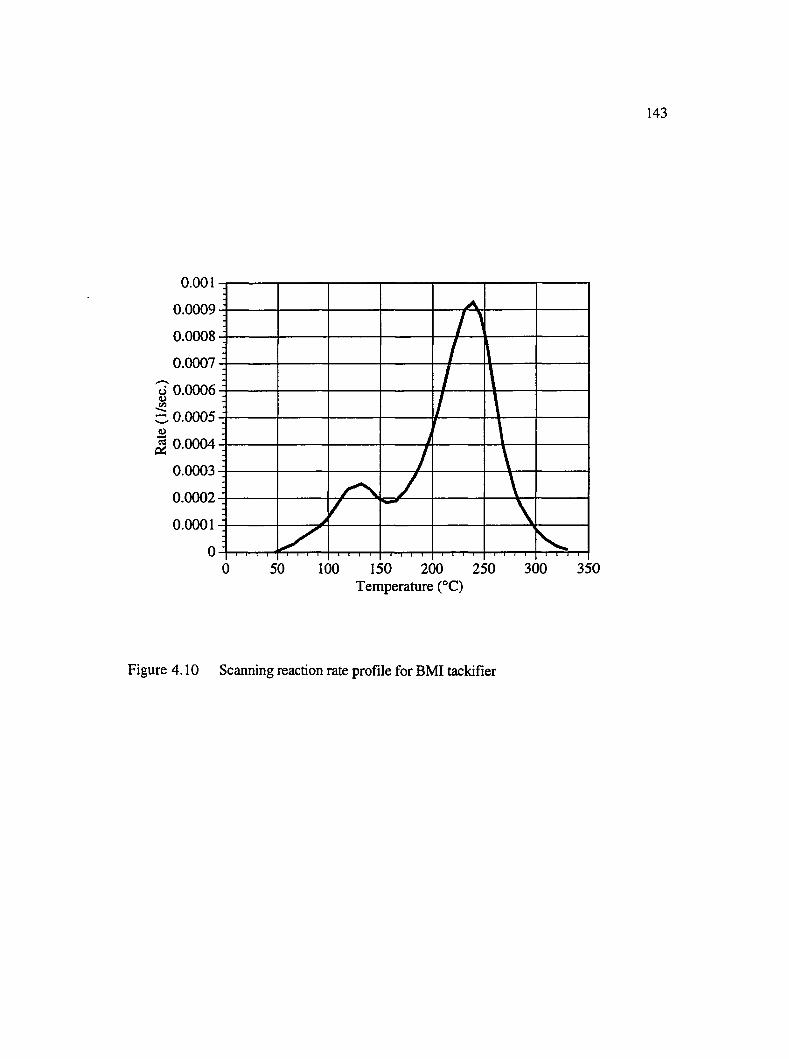
1 4 3
0.001
0.0009
0.0008
V
I 0.00044
0.00034
0.00024
0.00014
150 200 250 300Temperature (°C)
350100
Figure 4.10 Scanning reaction rate profile for BMI tackifier
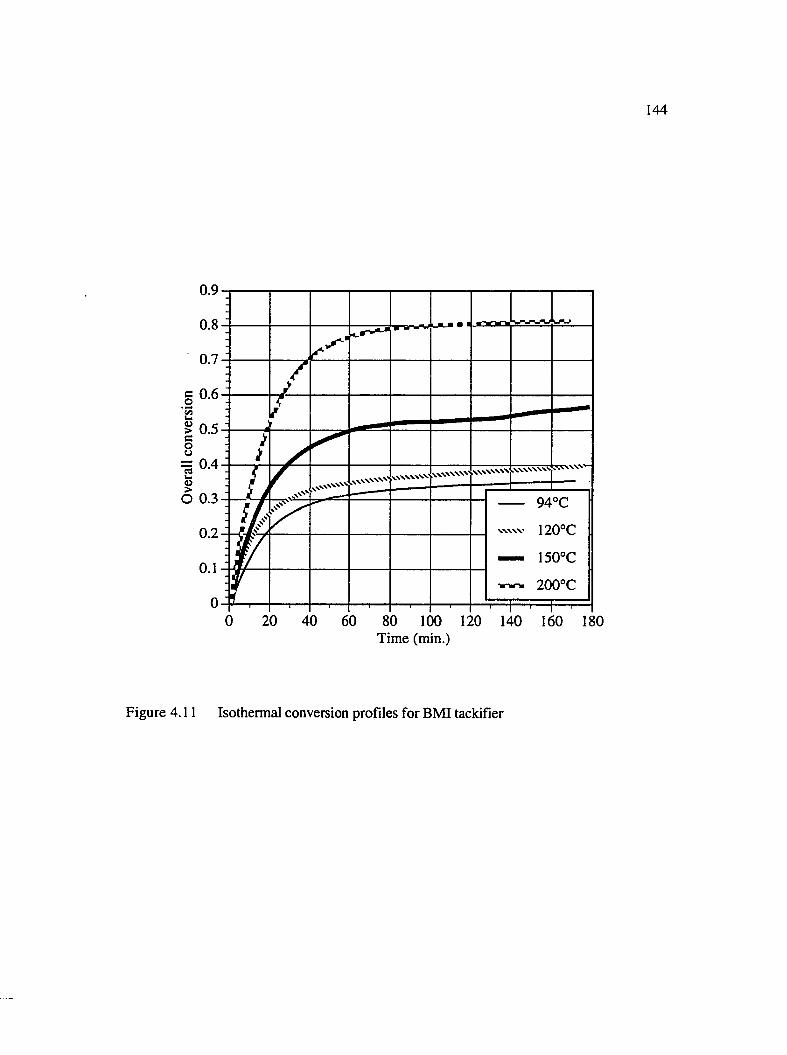
144
0.9
o2
§u& 4 : ,W N N V V V \< V .W W V '.U ^I
o 94°C
120°C
150°C
200°C
0 20 40 60 80 100 120 140 160 180Time (min.)
Figure 4.11 Isothermal conversion profiles for BMI tackifier

145
off indicating slowing down of the reaction due to diffusion controlled reaction kinetics. It
should be mentioned here that since the starting material is already cured to -35% , the
conversions reported here represent the amount reacted from the remaining unreacted 65%.
In cases where more than one reaction occur, different final products are formed depending
upon the cure history. This is also substantiated by Figure 4.12 which shows different
ultimate Tg (glass transition temperature) values for three different temperature profiles.
TPl represents scanning at 5°C/min. from room temperature to 350°C, TP2 is an isothermal
run at 130°C for 8 hours followed by an isothermal run at 220°C for 3 hours, and TP3
represents an isothermal run at 220°C for 3 hours and 20 minutes. The point of intersection
of the tangent to the lines on the heat capacity curves is usually taken as the glass transition
temperature, and is marked by an arrow in Figure 4.12.
4.3.2 Preforming Experiments
4.3.2.1 U-shape bending
Figure 4.13 shows the effect of tackifier concentration and degree of cure on the amount of
recovery or springback. As shown in the plot, at any particular concentration level,
increasing the degree of cure reduces the amount of springback. Also, for the same
conversion, increasing the tackifier concentration results in lower springback. Increasing
the concentration from 3 wt.% to 8 wt.% reduced the conversion required for no
springback, from .57 to .35. Ideally, for the tackifier to be soluble in the incoming matrix
resin during mold filling, the conversion should be less than .25 (of the tackifier) as the gel
conversion of the fresh BMI resin is around 0.5. Once the resin gels, the highly cross -
linked network formed does not permit the diffusion of significant amount of the solvent,
thereby drastically decreasing the solubility.
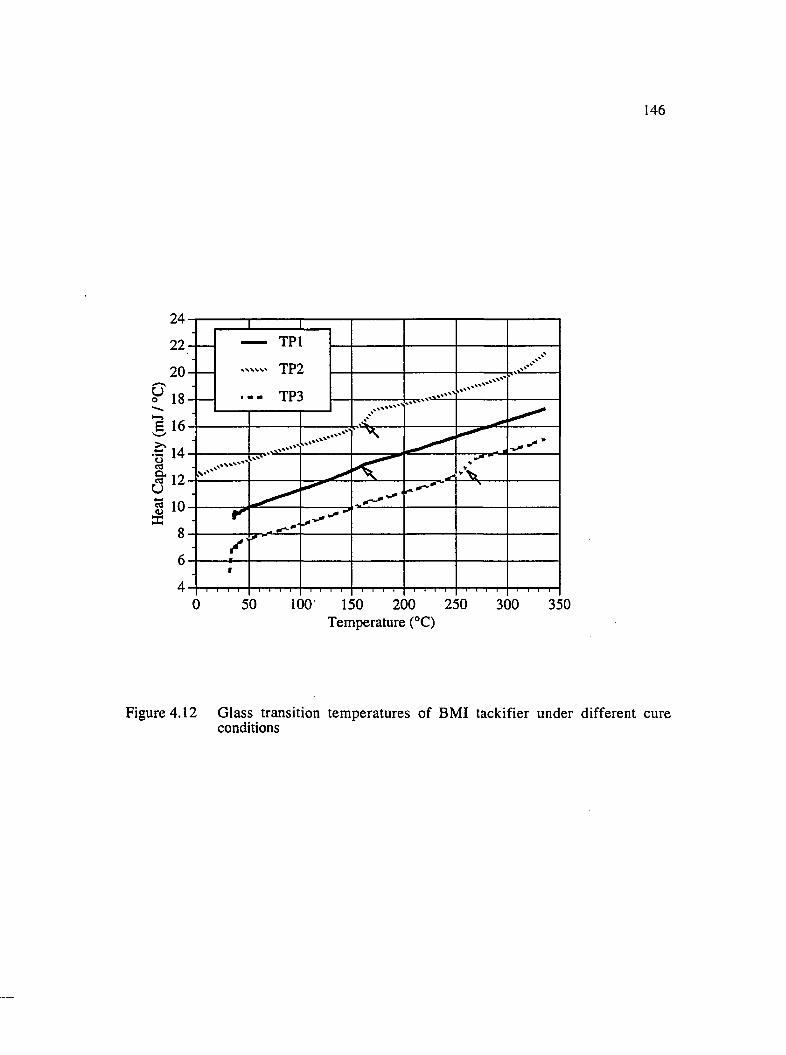
146
TPI
TP2
TP3
E 16
•n 14
12
S 10
0 50 100 150 200 250 350300Temperature (°C)
Figure 4.12 Glass transition temperatures of BMI tackifier under different cure conditions
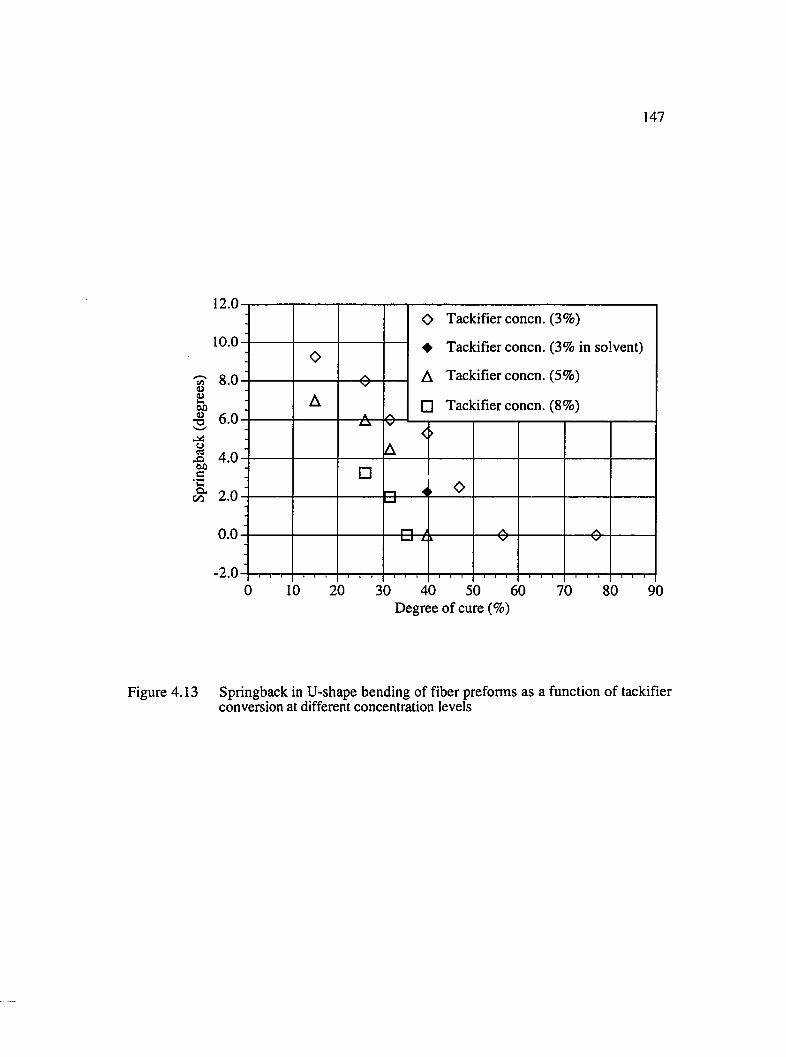
1 4 7
III
12. 0 -
10.0
8.0
6.0
4.0
2.0
0.0
- 2 . 0 -
- O Tackifier concn. (3%)
♦ Tackifier concn. (3% in solvent)
A Tackifier concn. (5%)
□ Tackifier concn. (8%)
o
A-A- A...
<A
>
- □n ^ . 0LI
n A
----1—1—1— 1 1 1 1 1 1
1—1 L k
" 1 "T" 1
V
1 1 1 1
----
1 1 10 10 20 30 40 50 60 70 80 90
Degree of cure (%)
Figure 4.13 Springback in U-shape bending of fiber preforms as a function of tackifier conversion at different concentration levels

148
This implies that an even higher tackifier concentration (> 8 wt. %) is required to achieve
zero springback at conversions less than .25. The trade-off is that at higher concentrations,
the preform becomes "boardy" and stiff, which is undesirable. Another side effect of
increasing tackifier concentration discussed in Chapter V, is reduced permeability due to
less free volume available for resin flow. Figures 4.14 (a) through (c) are photographs of
the front view of the preforms depicting springback under different cure conditions.
Figure 4.15 shows the photomicrograph of the surface of the U-shaped preform. When
compared with the initial distribution of the tackifier (Figure 4.4), this figure exhibits
substantial powder coagulation. The reason for this can be attributed to coalescence of
adjacent tackifier particles during melting and subsequent coagulation due to spreading of
liquid droplets. Coalescence of powder particles upon melting is known as sintering, and
eventually all contiguous particles flow into a deformed sphere representing a minimization
of surface and gravitational energy. The sintering phenomena and formation of deformed
droplets over a heated glass substrate are shown in Figures 4.16 (a) and (b).
An interesting result that was observed from these experiments was that when 3 wt. % of
the tackifier was applied dissolved in acetone, springback reduced from 5.3° to 2.2°. One
reason for this can be attributed to the increase in the elastic modulus of the fibers from a
more uniform coating of the tackifier on the filament surface (Figure 4.17). Springback
then decreases because the resistance to bending increases with increase in the tensile
modulus.
4.3.2.2 Vacuum Debulking
The results from vacuum debulking experiments are shown in Table 4.1. Both solvent and
powder methods were used to apply the tackifier powder on the fiber preforms.
Springback was observed to be the least when all the layers were soaked together in
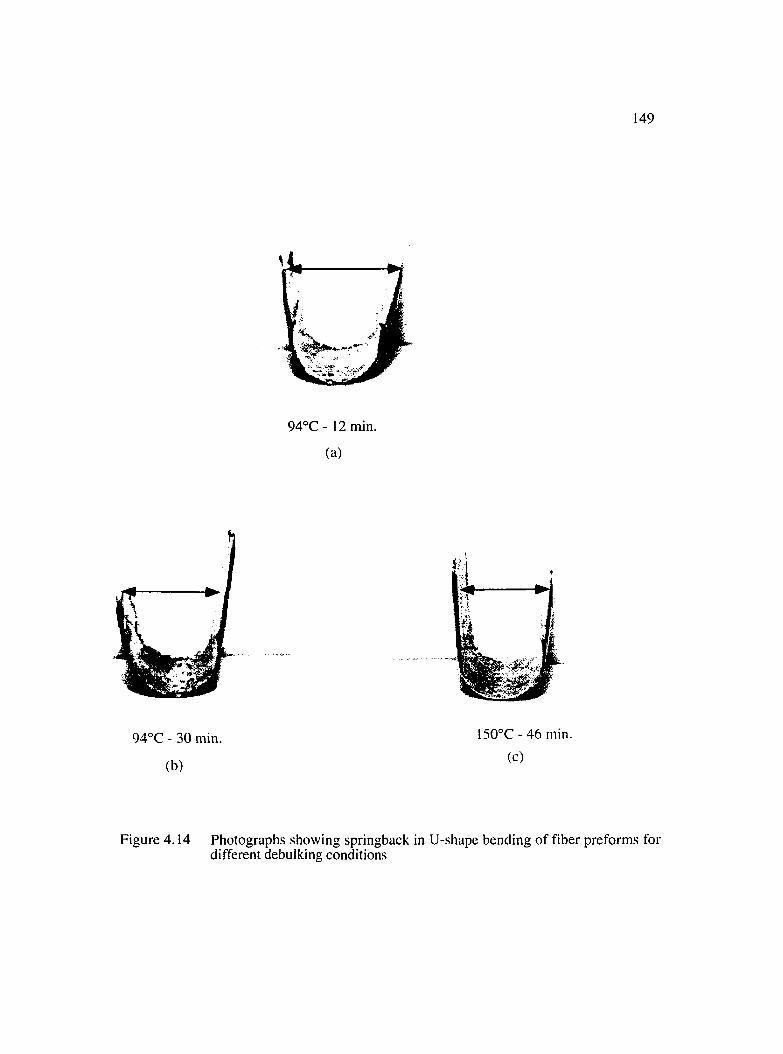
1 4 9
94°C- 12 min.
(a)
94°C - 30 min.
(b)
150°C-46m in.
(c)
Figure 4.14 Photographs showing springback in U-shape bending of fiber preforms for different debulking conditions

150
Figure 4.15 Photomicrograph showing powder coagulation and tackifier location for preforms subjected to U-shape bending

151
(a)
(b)
Figure 4.16 Sintering of tackifier particles : (a) upon melting and (b) coagulation into deformed droplets

152
(b)
FiEUi 4.17 Photomicmgraphs of the fiber preform with BMI tackifier applied using thesolvent technique (a) low magnification and (b) high magnification

Table 4.1 Springback after vacuum debulking of BMI powder coated preforms
Experimentalconditions
Tackifier application technique
Thickness before debulking
(mm)
Thickness after debulking
(mm)
Thickness, overnight (mm)
8 layers
Tackifier concn. 3 wt%
-30" Hg vacuum
&94 °C, 30 min.
Solvent
individuallayers
4.087 2.109 2.526
sb = 0.417
all layers together
3.119 2.158 2.197
sb = 0.039
Powder
Run 1 3.688 2.258 2.379
sb = 0.121
Run 2 3.785 2.158 2.224
sb = 0.066 KJ\W

154
solvent. For fiber layers soaked individually and stacked one on top of the other upon
drying, springback was found to be the maximum. Powder technique gave intermediate
results. However, it should be mentioned here that interply adhesion was the strongest for
the powder technique, whUe individual layers showed no bonding at all. Adhesion for all
the layers together, lay in between the two extremes. Photomicrograph (Figure 4.18) of
powder coated debulked sample shows that as in the case of U-shape bending, the tackifier
remains on the surface. This explains why good adhesion is achieved between the layers.
The solvent however dissolves the tackifier and therefore it does not remain on the surface,
but rather forms a coating on the filaments. So while interply adhesion is bad, springback
control is good. Thus, both "interlayer" and "intralayer" area coverage seem to be
important in order to have acceptable interply adhesion and to minimize springback.
4.S.2.3 Lateral Compression
The effect of tackifier concentration on preform consolidation and springback under lateral
compression is plotted in Figures 4.19 (a) through (c). The compacted thickness of the
preform increased with increasing tackifier concentration, but as in the case of bending
experiments, the amount of springback decreased. As a result, the final uncompacted
thickness (sum of compacted thickness + springback) leveled off in the concentration range
studied. The change in strain as a function of time under constant stress. Figure 4.20 (a)
and during springback. Figure 4.20 (b) shows the viscoelastic nature of the tackified fiber
preforms.
Lateral compression of powder coated laminates (3 wt. %) was also carried out by heating
the samples from room temperature to 120°C and 150°C. Since intralayer coverage in
vacuum debulked sample gave good springback control, and photomicrographs of 94°C
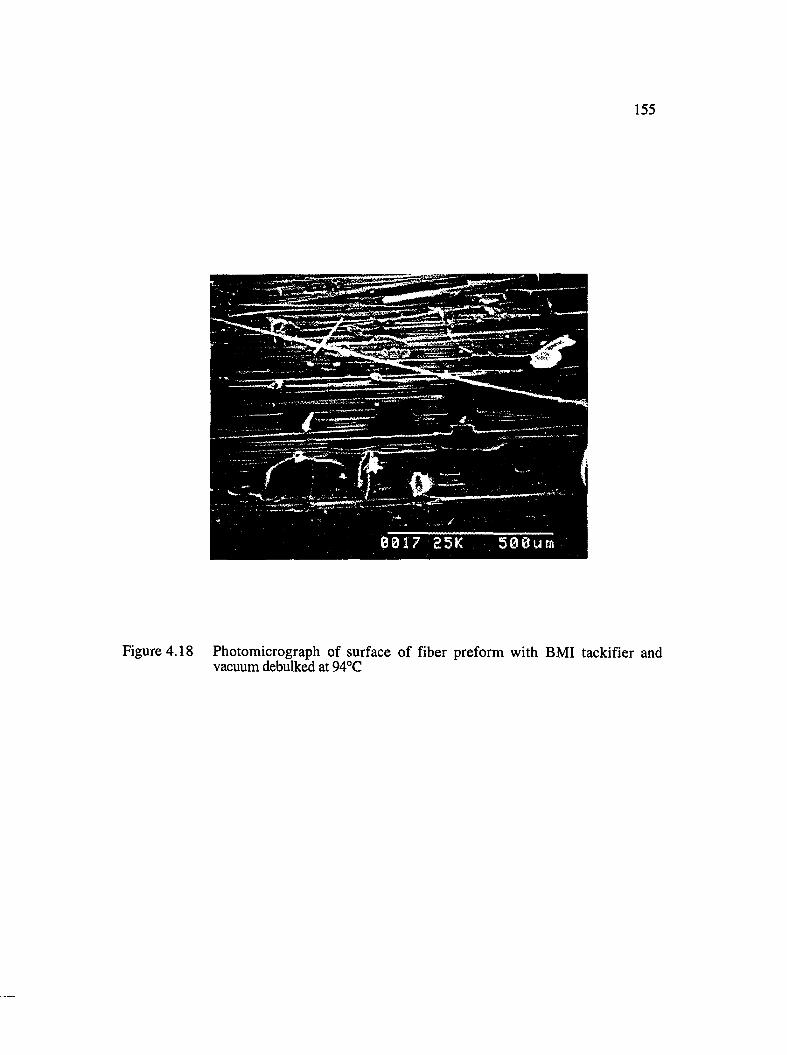
155
Figure 4.18 Photomicrograph of surface of fiber preform with BMI tackifier and vacuum debulked at 94°C
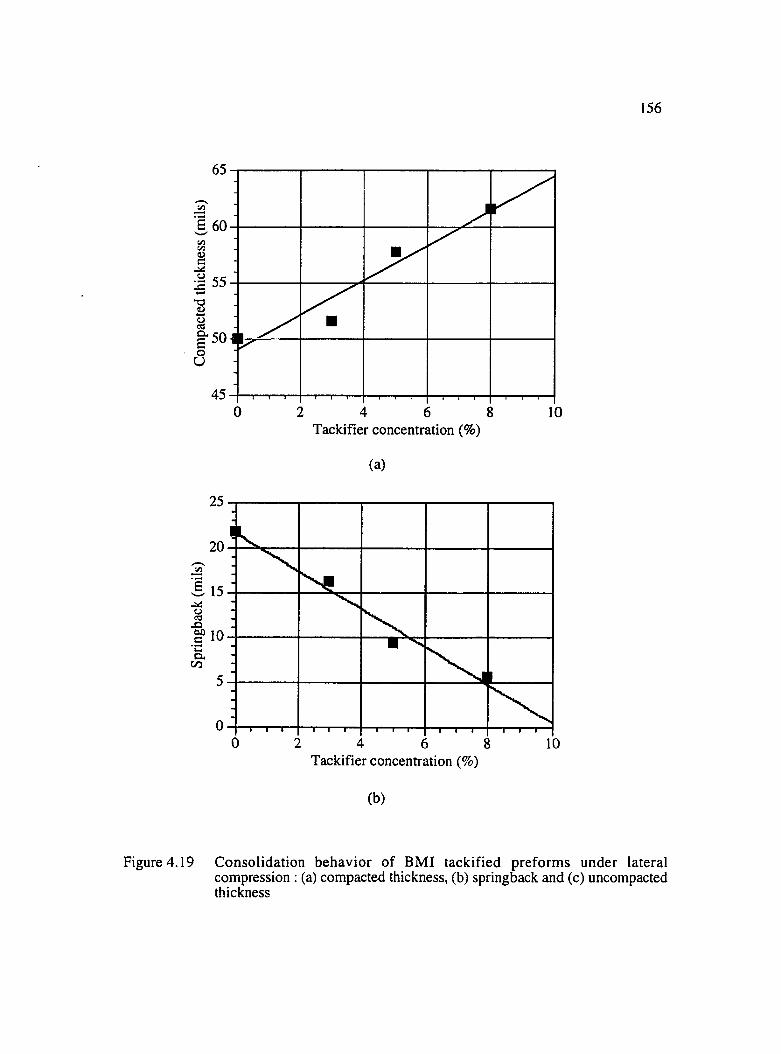
1 5 6
§ 6 0(/)
Tackifier concentration (%)
(a)
25
20
15
5
0
Tackifier concentration (%)
(b)
Figure 4.19 Consolidation behavior of BMI tackified preforms under lateral compression : (a) compacted thickness, (b) springback and (c) uncompacted thickness
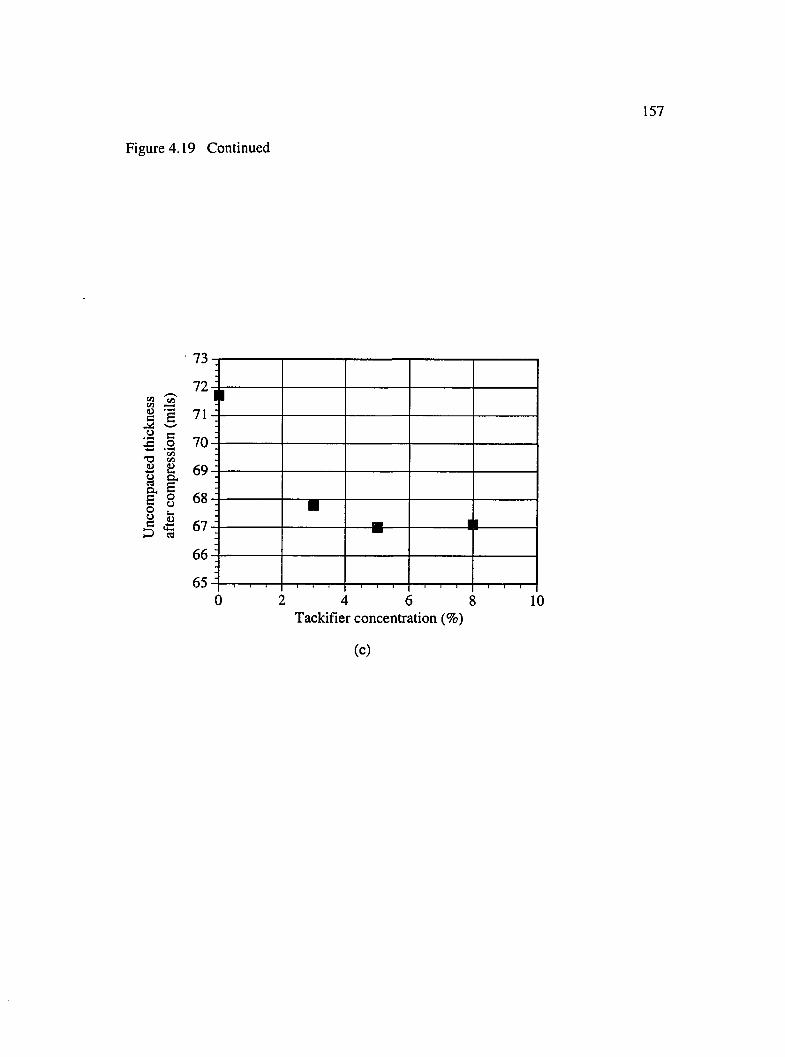
157
Figure 4.19 Continued
1I•5 I
II
73 n
79 -!
71 '1
70^
68^
67^H
1I■
65^c) : () 1(
Tackifier concentration (%)
(c)
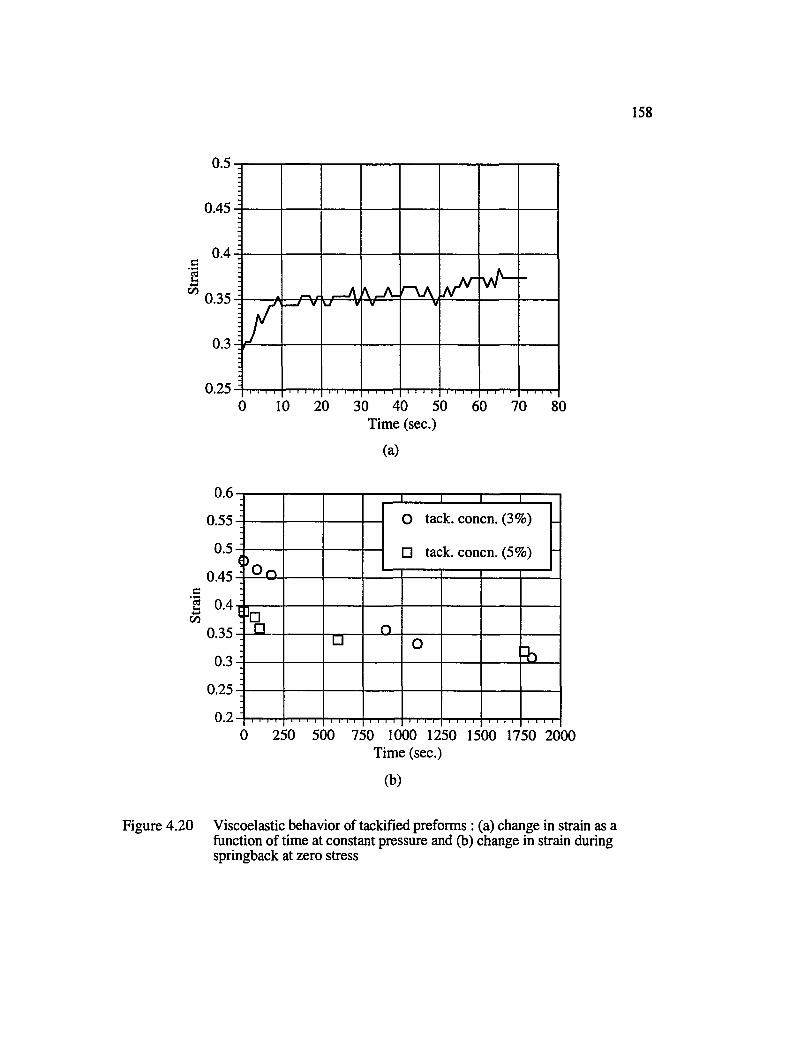
158
0.5
0.45 H
0.4-^IüCO
0.25-0 10 20 30 40 50 60 70 80
Time (sec.)
(a)
c
I00
0.6
0.55
0.5
0.45
0.4
0.35
0.3
0.25
0.2
1 1 1 O tack, concn. (3%)
:u lacK. con cn. '0)
------ W-
ou 0=b
:
0 250 500 750 1000 1250 1500 1750 2000Time (sec.)
(b)
Figure 4.20 Viscoelastic behavior of tackified preforms : (a) change in strain as a function of time at constant pressure and (b) change in strain during springback at zero stress

159
run showed the powder to be on the surface, the idea was to reduce the viscosity by rapid
heating, and thereby causing it to go inside the fiber tows. However, as shown in Figures
4.21 (a) and (b), the tackifier remains on the surface, and covers most of the interlayer
area. Droplets possibly coexisting with a "manchon" (sheath) covering a portion of the
filaments were also observed as shown in Figure 4.21 (c).
The phenomenon of spreading of liquid droplets on horizontal fibers (thin cylinders) was
investigated by Brochard [1986]. He found that droplets will spread out and wet the
filament surface, if the spreading coefficient, S, is larger than a threshold value. Sc given
by Equation 4.13:
S , = | y ( f ) ï (4.13)
For carbon fibers, - 10^ & — ~ 10‘2D y
where
Y is the surface tension of the liquid
b is the radius of the fiber
a is the molecular length of the liquid
Below Sc, the droplet does not spread, and the fiber remains dry. Above Sc, the droplet
completely spreads out into a manchon. At S = Sc, there exists an equilibrium between the
droplet and the manchon. Thus, a lower resin surface tension or a lower resin-substrate
interfacial tension would aid in increasing the wetted surface area coverage.
The spreading coefficient, S, depends upon the respective interfacial energies of the solid,
liquid and air, and is given by Equation 4.14.

160
(a)
(b)
Figure 4.21 Photomicrographs of surface of laterally compressed preforms with BMI tackifier : (a), (b) showing interlayer coverage and (c) droplet possibly coexisting with a "manchon"
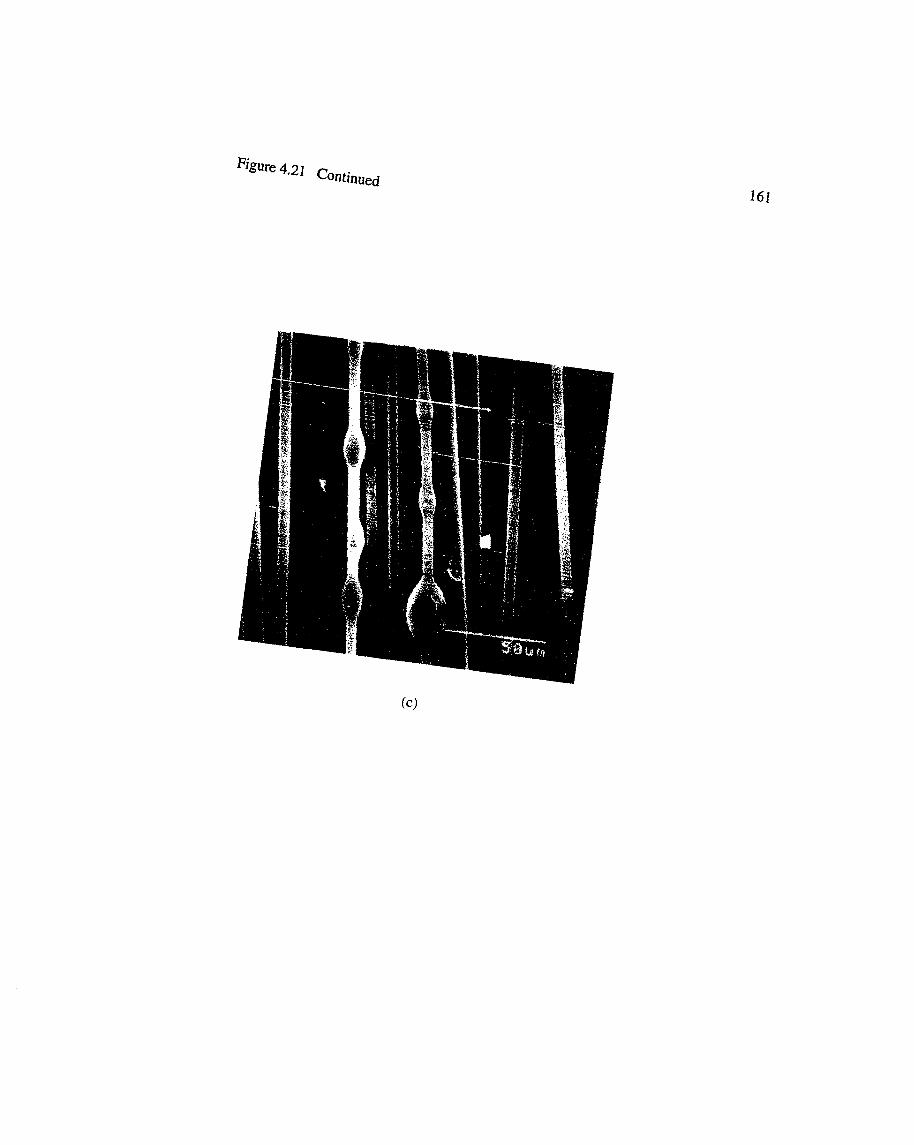
"'*“'* ‘'•21 C o n t i n u e
161
(c)

162
S = Ys - Ysl “ Ylv (4.14)
where
Ys is interfacial energy between solid/air
Ysl is interfacial energy between sobd/Uquid
Ylv is the bquid/air interfacial tension
Thus, if Ys, Ysl, and Ylv can be determined, the spreading criterion for a given fiber and
resin system can be obtained from Equations 4.13 and 4.14.
Moreover, the reason why the tackifier remains on the surface even on increasing the
temperature to 150 °C, can be explained from the plot of dynamic viscosity (|i*) shown in
Figure 4.22 (a). The dynamic viscosity was obtained at 5°C/minute since this
corresponded to the rate of increase in temperature in debulking experiments. As shown in
the plot, the dynamic viscosity first decreases with increase in temperature, and then there
is a rapid increase due to chemical reaction. Corresponding changes in the viscoelastic
modulus G*, O' (storage modulus) and G" (loss modulus) are shown in Figures 4.22 (b)
and (c). The cross-over point shown in Figure 4.22 (c) which typically marks the onset of
gelation occurs at 150°C.
At low frequencies and low shear rates, it has been established both theoretically and
experimentally [Middleman, 1968] that the relationship between dynamic viscosity |i* and
steady shear viscosity |l can be given by Equation 4.15:
lim |i* = limp. (4.15)
CO 0 ; y 0
Also, from continuum models of polymer melt rheology, this relationship originally
proposed by Cox and Merz [1958] holds true within experimental error even at non - zero
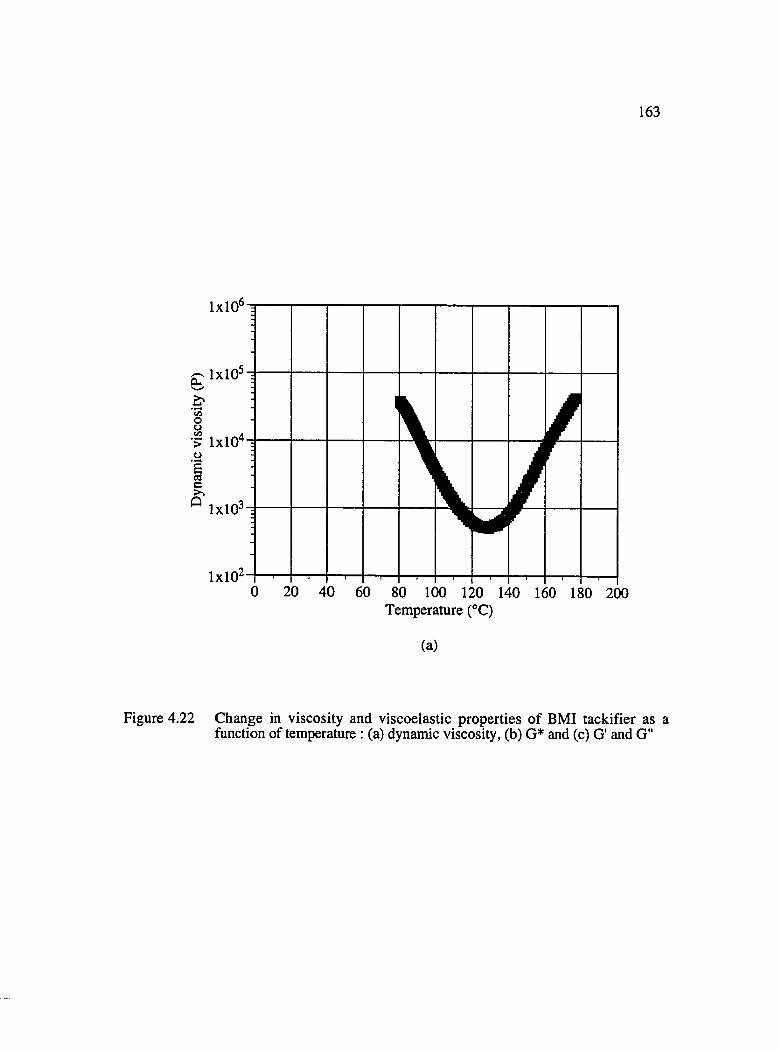
163
1x10
Temperature (°C)
(a)
Figure 4.22 Change in viscosity and viscoelastic properties of BMI tackifier as a function of temperature : (a) dynamic viscosity, (b) G* and (c) O' and G"
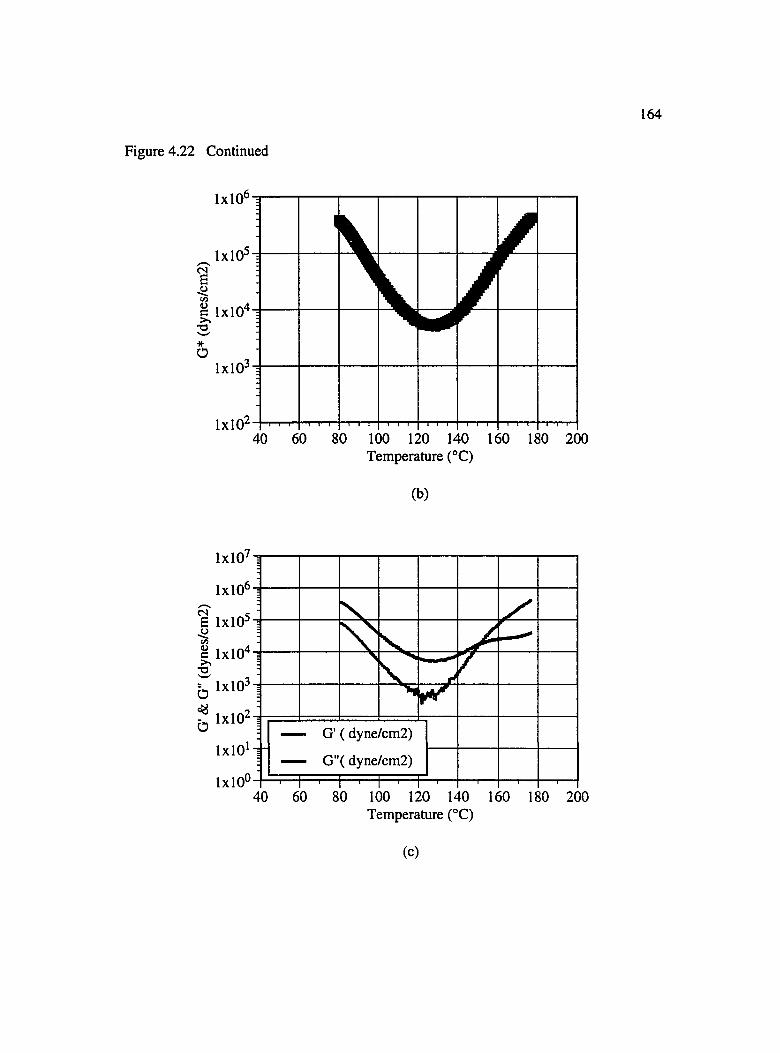
Figure 4.22 Continued
164
c IxlO'^
80 100 120 140 160 180 200Temperature (°C)
(b)
1x10'
c 1x10'
1x10G' ( dyne/cm2)
G"( dyne/cm2)1x10
1x10'
Temperature (°C)
(c)

165
frequencies and shear rates. Thus, before gel point, steady shear viscosity should follow
the same trend as the dynamic viscosity.
Springback results for 94°C, 120°C and 150°C runs are given in Table 4.2. Both 120°C
and 150°C ramp runs gave considerably lower springback than at 94°C. Although the
tackifier remains on the surface for all the three cases, the interlayer area coverage as shown
in Figures 4.18 and 4.21 (a), is higher at 120°C as compared to 94°C, due to increased
spreading rate owing to lower viscosity. The same is true for 150°C run since the resin
passes through its minimum viscosity before increasing again. Hahn and Jonach [Lange,
1984] also observed that the spreading rate of spherical epoxy drops, neglecting the effect
of gravitational forces (mass of drops < 1 0 0 mg) is inversely proportional to the melt
viscosity.
Springback control at higher temperatures therefore may come from combined effects of
increased modulus due to higher degree of cure and greater interlayer surface area coverage
due to lower viscosity. Both effects result in a greater "holding" force opposing the
relaxation of compressed elastic fibers.
In order to decouple the effects of increase in modulus due to chemical reaction, and
surface area coverage on springback control, PMMA powder was used at a concentration
of 3 wt.% at temperatures beyond its melting point. Lateral compression experiments were
conducted at 220°C, 250°C and 287°C to cover a broad range of melt viscosity (Figure
4.23). The results of lateral compression experiments are reported in Table 4.3. Lower
springback was achieved at 287°C as compared to the other two temperatures. Being a
thermoplastic powder, the modulus will be the same in all cases upon cooling to room
temperature, unlike the case of BMI powder. Thus, any observed differences in
springback values at different temperatures using PMMA powder should not come from
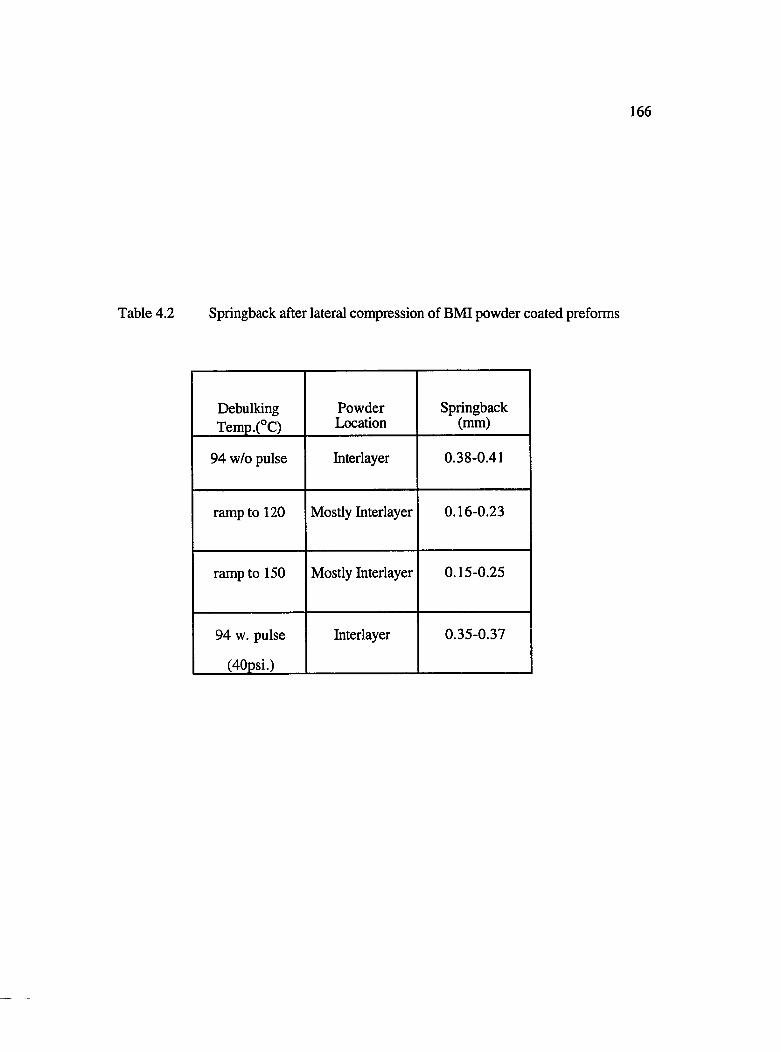
166
Table 4.2 Springback after lateral compression of BMI powder coated preforms
DebulkingTemp.(°C)
PowderLocation
Springback(mm)
94 w/o pulse Interlayer 0.38-0.41
ramp to 120 Mostly Interlayer 0.16-0.23
ramp to 150 Mostly Interlayer 0.15-0.25
94 w. pulse
(40psi.)
Interlayer 0.35-0.37
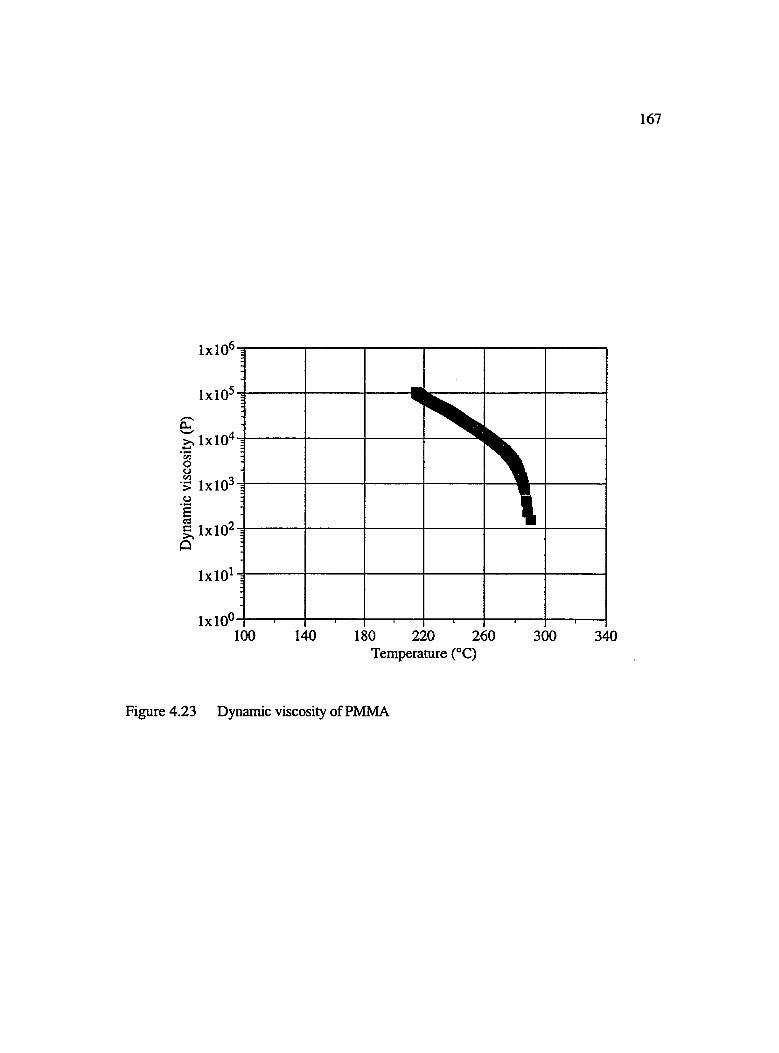
167
>. 1x10^
I 1x103
e ixiQZ
180 220 260 Temperature (°C)
340
Figure 4.23 Dynamic viscosity of PMMA
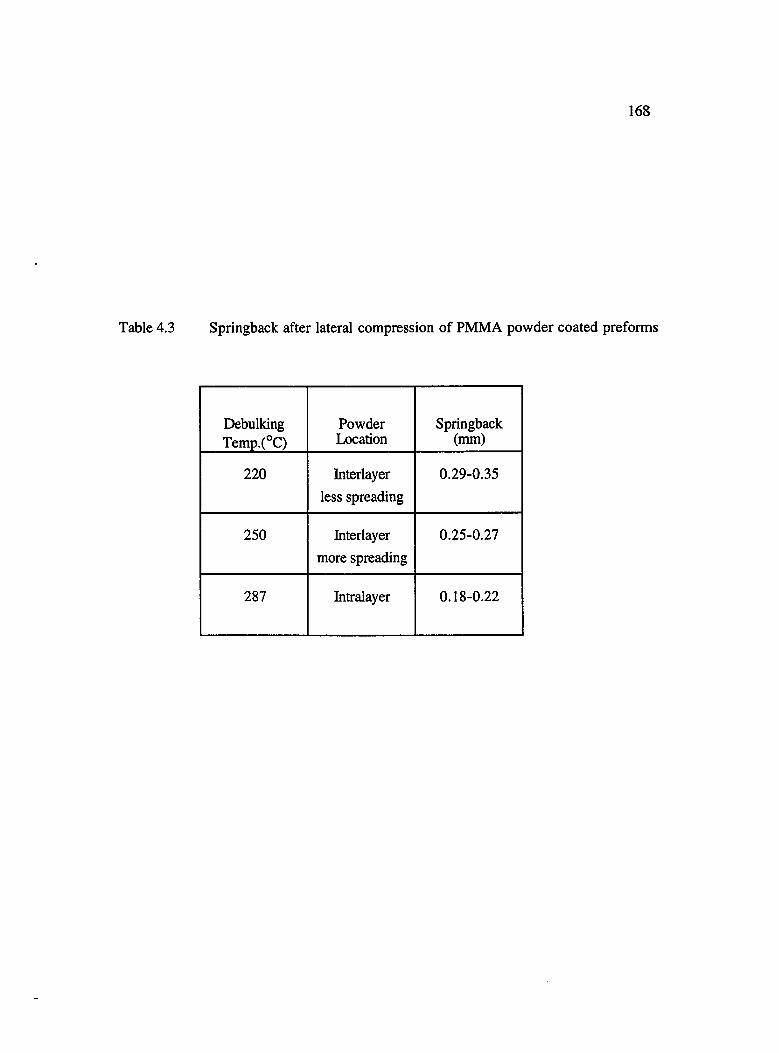
168
Table 4.3 Springback after lateral compression of PMMA powder coated preforms
DebulkingTemp.(°C)
PowderLocation
Springback(mm)
220 Interlayer less spreading
0.29-0.35
250 Interlayer more spreading
0.25-0.27
287 Intralayer 0.18-0.22

169differences in the modulus, but rather from differences in the wetted surface area coverage.
To see if this was true, photomicrographs for all the three cases were taken and are shown
in Figures 4.24 through 4.26. At 220°C, the powder remains on the surface as coagulated
"chunks" with no intralayer coverage as shown in Figures 4.24 (a) and (b). Figures 4.25
(a) and (b) for 250°C also show that the polymer melt remains on the surface. However,
the springback for 250°C run is lower because of increased spreading due to lower
viscosity. At 287°C however, the polymer melt migrates from the surface to within the
fiber tows (Figures 4.26 a & b) and forms a coating on the filaments. This is due to the
decrease in the dynamic viscosity and viscoelastic modulus at temperatures around 287°C
as shown in Figures 4.23 and 4.27 (a) and (b). Further decrease in springback at 287°C is
because of the intraiayer surface area coverage of the individual filaments.
4.3.3 Phenomenological Approach for Springback Control under Lateral Compression
Based on the results obtained from U-shape bending and lateral compression experiments,
it can be hypothesized that springback will occur when the force exerted by the elastic fiber
preform is greater than the "holding force" provided by the tackifier. As shown
schematically in Figure 4.28, the latter depends on the product of the modulus (which is a
function of degree of cure for a reactive powder) and wetted surface area coverage. Thus,
increase in either the area coverage or the modulus can increase the holding force to contr ol
the amount of springback. The ratio of the driving and opposing forces for springback can
therefore be expressed as:
driving force _ , force exerted by the fiber preform .. .holding force “ resin modulus x surface area coverage^ ^
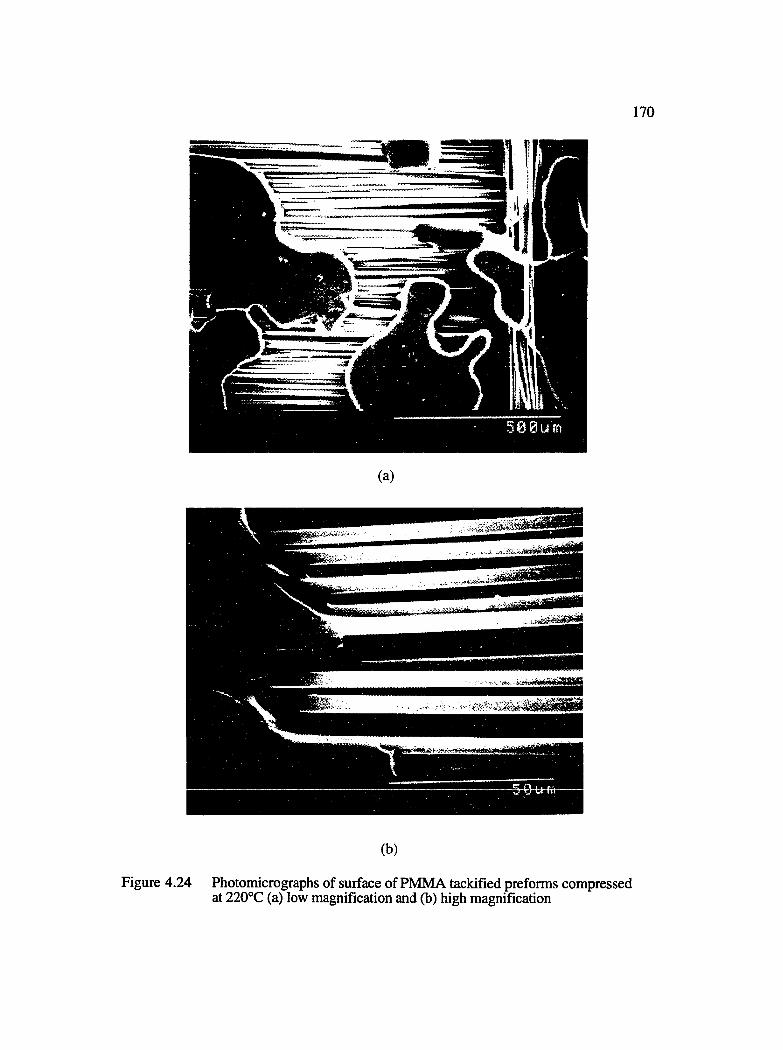
170
masm
(a)
5 0 0 urn
(b)
Figure 4.24 Photomicrographs of surface of PMMA tackified preforms compressed at 220°C (a) low magnification and (b) high magnification

171
(a)
1
(b)
Figure 4.25 Photomicrographs of surface of PMMA tackified preforms compressed at 250°C (a) low magnification and (b) high magnification

172
I «nwm
i
(a)
(b)
Figure 4.26 Photomicrographs of surface of PMMA tackified preforms compressed at ~287°C (a) low magnification and (b) high magnification
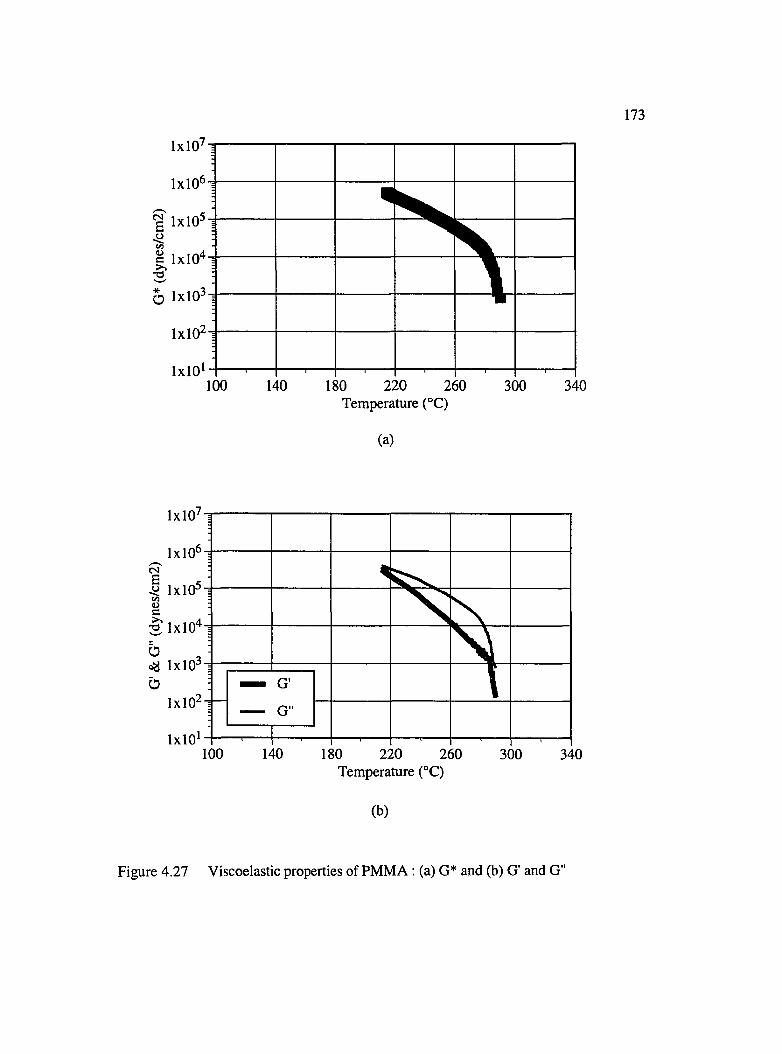
173
1x10
<N
C 1 x 1 0 ' 30 1 x 1 0
1 x 1 0220
Temperature (°C)100 180 260 340300
(a)
1 x 1 0 *100 140 220
Temperature (°C)180 260 300
(b)
Figure 4.27 Viscoelastic properties of PMMA : (a) G* and (b) O' and G"
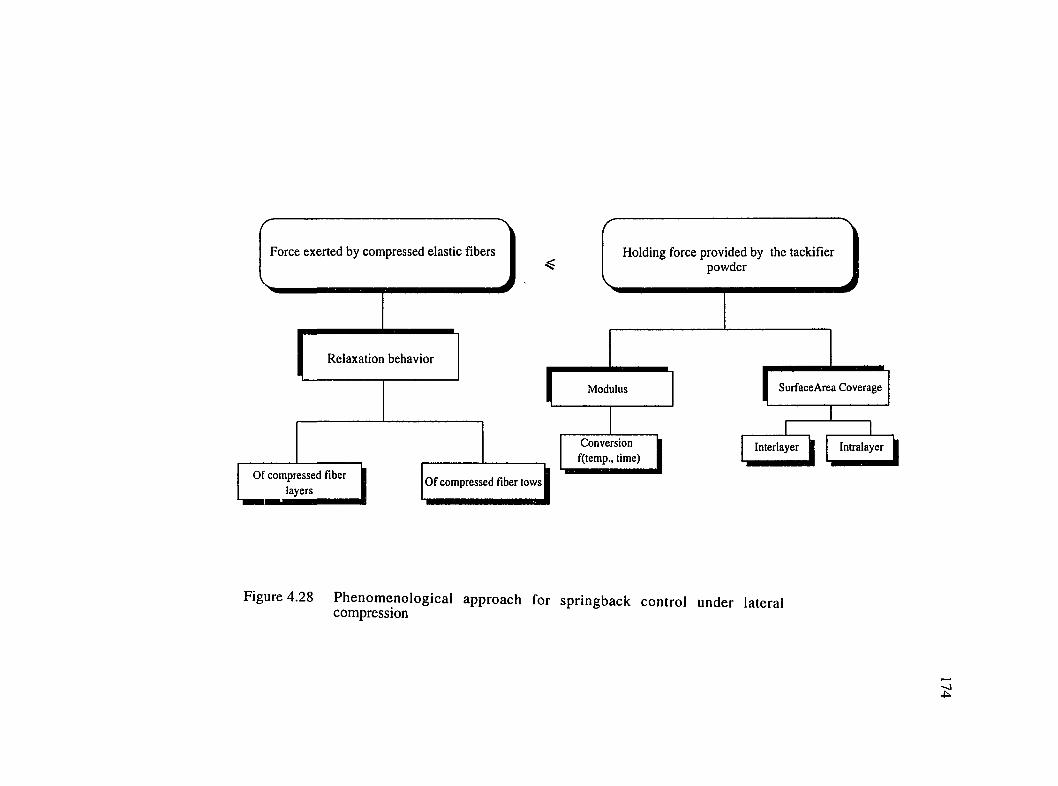
Force exerted by compressed elastic fibers Holding force provided by the tackifier powder
Relaxation behavior
SurfaceArea CoverageModulus
Conversion f(temp., lime)
Interlayer Intralayer
Of compressed fiber layers Of compressed fiber tows
Figure 4.28 Phenomenological approach for springback control under lateral compression
A

175
In order to estimate the magnitude of the force exerted by the compressed elastic fiber
preform, the compressibility behavior of untackified fibers was studied using the lateral
compression device shown in Figure 4.7. The underlying assumption in doing so was that
the force required to compress the fibers to a certain volume fraction should be similar to
that exerted by the fibers at the same volume fraction during the relaxation process. Of
course this assumption would not be valid for cases where there is significant hysteresis
between the compression and relaxation cycles.
Figures 4.29 (a) and (b) are the compressibility curves for 8 and 16 layers of fiber mats
respectively. Both plots show a similar trend of a non-linear increase in pressure with
increase in the fiber volume fraction beyond ~ 45%. Similar results have been obtained by
other researchers using both graphite fibers [Gutowski et al., 1987] and glass fibers [Kim
et al., 1990]. These plots also show that when the same sample was compressed the
second time, the curve shifted to the right. This indicates that a higher stress is required in
the first cycle, more so at the lower stress range, to obtain the same volume fraction as
compared to the second cycle. The reason for this is that after the first cycle, some of the
contact points do not revert back to their original state. This is also evident from the figures
which show a higher starting fiber volume fraction in the second cycle as compared to the
first. Thus, there is some hysteresis between the compression and relaxation cycles.
The non-linear increase in the pressure beyond Vf = 0.45 can be explained by the two level
consolidation theory proposed in this study, and is depicted in Figures 4.30 (a) through
(c). Figure 4.30 (a) is a micrograph of the cross-section of a composite sample (molded
using UP resin) with Vf = 0.32. Since a woven fiber reinforcement is used, each layer has
fiber bundles oriented both parallel and perpendicular to the cross-section. As can be seen
from Figure 4.30 (a), there is a large gap between the adjacent layers. With increasing
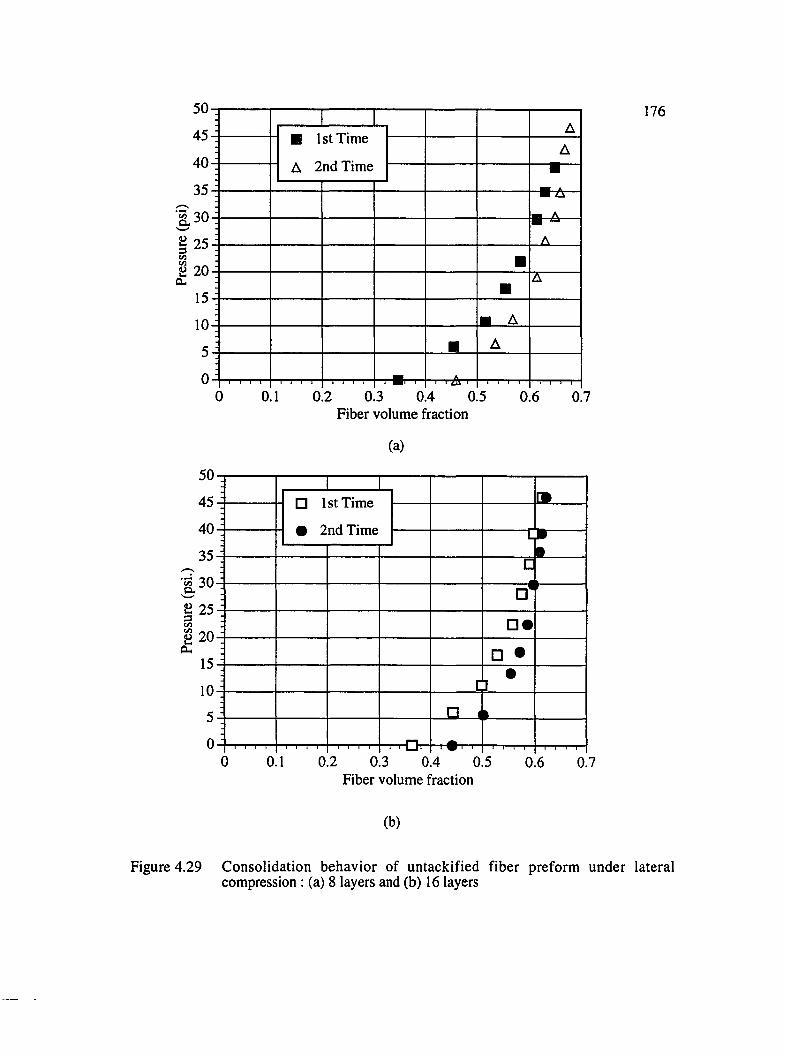
50
45-
40
35
U s o
il) 25-
20
15-
10 -
50
45-
40-
35-
•S30-
I 25-
S 20-
15-
10 -
5-
0 -
■ 1st Time
A 2nd Time
rA-0.1 0.2 0.3 0.4 0.5
Fiber volume fraction
(a)
0.6
I□ 1st Time
• 2nd Time
I I 1 I I I I I
[B
dr
□ •
0.1 0.2 0.3 0.4 0.5Fiber volume fraction
0.6
0.7
0.7
1 7 6
(b)
Figure 4.29 Consolidation behavior of untackified fiber preform under lateral compression : (a) 8 layers and (b) 16 layers
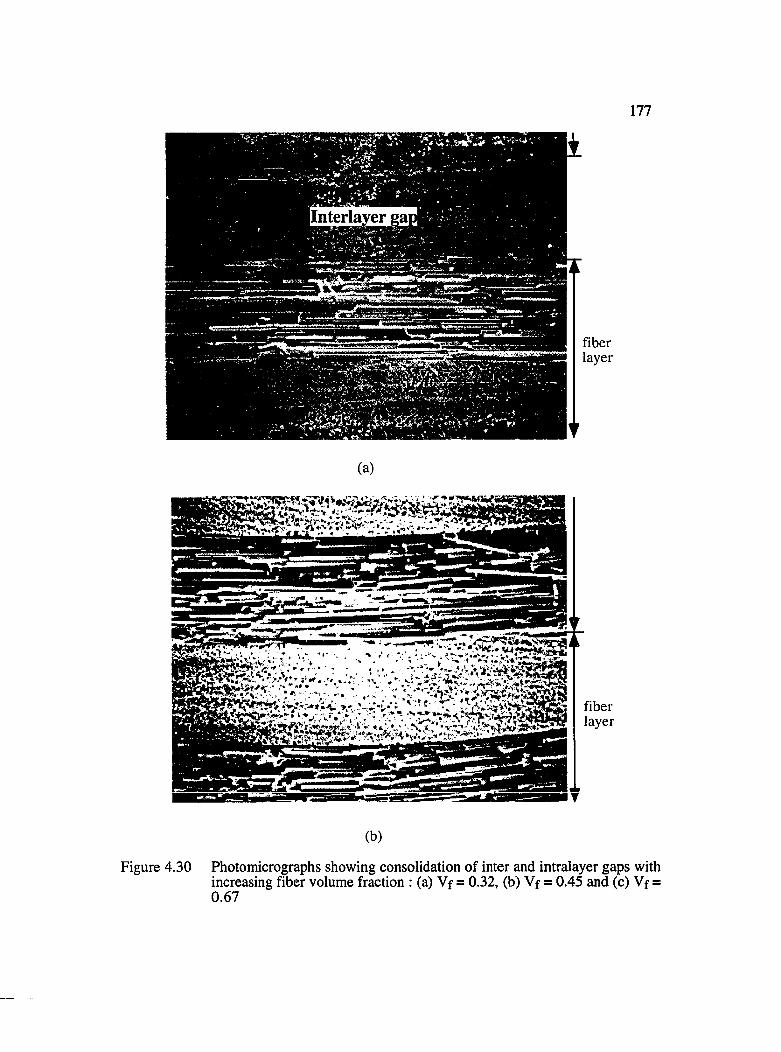
177
In terlaver ea
gijZ mZ wrr.
(a)
(b)
Figure 4.30 Photomicrographs showing consolidation of inter and intralayer gaps with increasing fiber volume fraction : (a) Vf = 0.32, (b) Vf = 0.45 and (c) Vf = 0.67
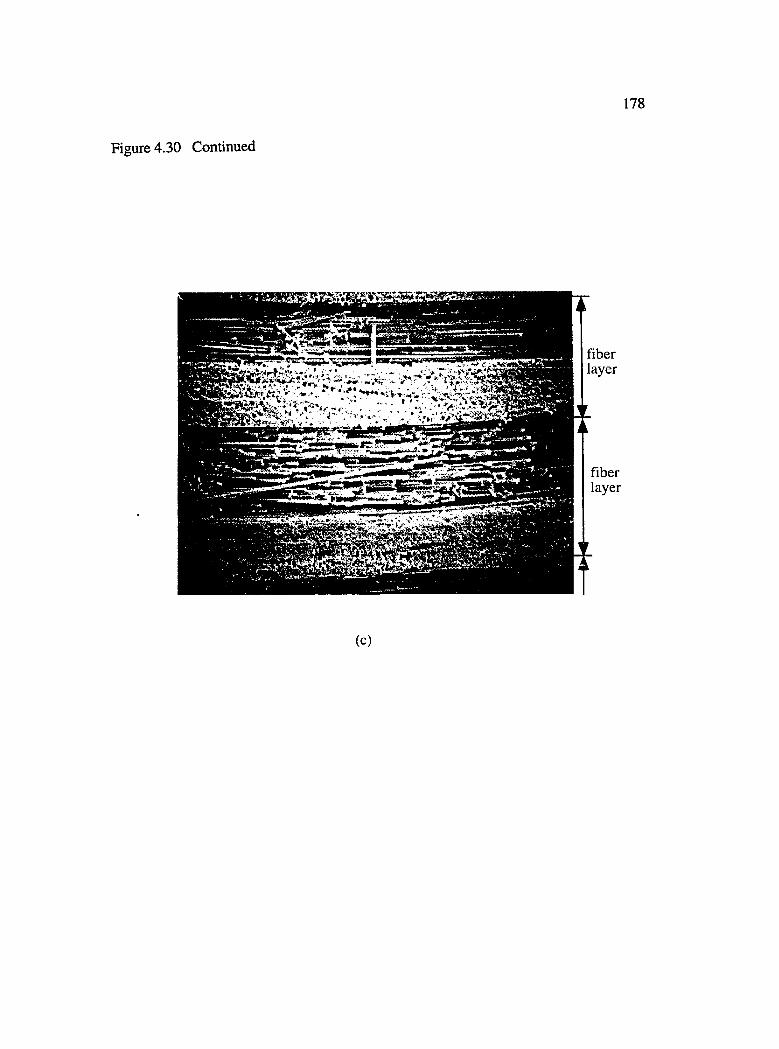
178
Figure 4.30 Continued
i f l B . H é
a
(c)

179
fiber volume fraction, which also represents the initial stage of fiber consolidation or inter
layer consolidation, these gaps disappear and fiber tows of adjacent layers start touching
each other (Figure 4.30 (b), Vf = 0.45). Since the resistance to compression of these inter
layer gaps is small, lower pressures are required. Compaction beyond V f= 0.45
represents the second stage of consolidation or intra-layer consolidation wherein the smaller
gaps between the filaments of the fiber bundles start to get compressed. Figure 4.30 (c)
shows considerable consolidation of the fiber tows at Vf = 0.67.
The consolidation behavior of various fiber preforms has been modeled by previous
researchers using different empirical correlations as mentioned in Chapter II. Gutowski et
al. [1987] developed a mechanistic model to describe the overall consolidation behavior as
a rapidly stiffening spring caused by an increase in the number of fiber-fiber contact points.
Another approach is to use the Finitely Extendable Non-Linear Elastic (FENE) spring
model which has a force law of the form expressed by Equation 4.18 [Hou, 1986]. Both
models however do not consider the two levels of consolidation and the hysteresis effect.
FENE model was chosen as the working model in this study because of its simplicity, and
also because it fitted the experimental data well.
Ff = Pf . A = k * (h (0) - h (i)) (4.18)'h (O ) - h ( i)
1 -
h (0) - m rf
where
Ff is the pressure on the preform
A is the nominal area of the preform
k is the spring constant
h (i) is the distance between the fiber layers at any time, t = ti
h (0) is the starting distance between the layers, t = to

m and rf are the number of layers and the radius of the tows in each layer
(h (0) - m rf) represents the maximum compressible distance achievable
n is an empirical constant
180
Also since
Vf (i) ^ MO)V f (0) h (i)
(4.19)
Substituting Equation 4.19 in Equation 4.18 yields the relationship between applied
pressure and fiber volume fraction as given by Equation 4.20:
P f = k f
1 -
Vf (i) - V f(0)Vf (i)
Vf (i) - Vf (0 )""Vf (i)
Vf": - V f (0)Y m
(4.20)
where
kf = kh(0)
Vf is the maximum fiber volume fraction achievable
The experimental data was fitted using Equation 4.20 for an assumed maximum fiber
volume fraction Vf” = 0.901, and are shown by solid lines in Figures 4.31 (a) and (b). k
and n are the model parameters and are obtained using non-linear regression. The values of
k and n for 8 layers and 16 layers are:
k = 12.7 & n = 1.35
k = 13.11 & n = 1.86
(for 8 layers)
(for 16 layers)
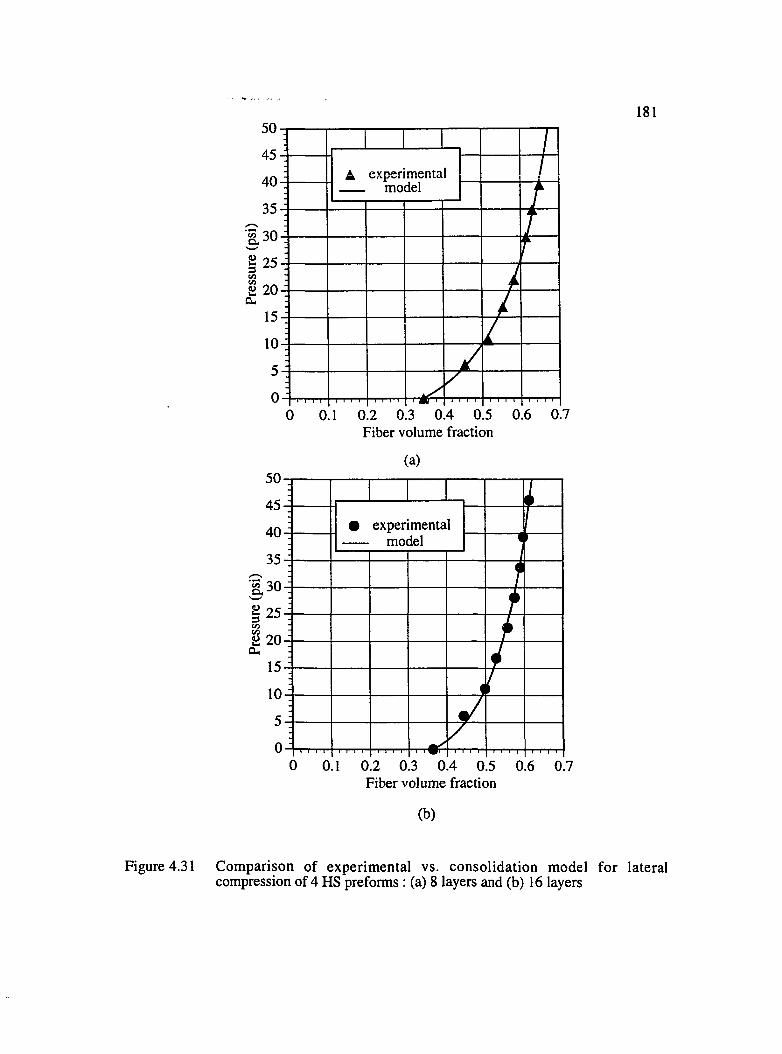
181
A experimental — model4 0 -
3
ICL
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7Fiber volume fraction
(a)
45-:# experimental — model40
8 304
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7Fiber volume fraction
(b)
Figure 4.31 Comparison of experimental vs. consolidation model for lateral compression of 4 HS preforms : (a) 8 layers and (b) 16 layers

182
Build up of viscoelastic modulus during preform consolidation can be obtained using RDA.
For EMI tackifier, changes in the viscoelastic properties with time at 94°C, 120°C and
150°C are shown in Figures 4.32 (a) through (c). Measurements were made using parallel
plates of 7.9 mm diameter at a frequency of 5 rad/sec. and a strain of 0.1. For both 94 °C
and 120°C, the values of G" are greater than O', which indicates that the resin did not reach
its gel point till 60 minutes for 94°C and 30 minutes for 120°C. However, Figure 4.32 (c)
shows a reversal in the trend for 150 °C, with G' being greater than G" from the beginning.
This is due to gelation which occurs within 20 seconds at 150°C. Before the gel point, the
material exhibits more liquid like characteristics, and hence G" is higher than G'. Beyond
gelation, G" levels off, and G' crosses over.
The wetted surface area coverage depends upon the tackifier location. Scanning electron
micrographs of the surface of tackified preforms showed that depending upon the
processing conditions, the area coverage could be both interlayer, i.e. between the layers,
or intralayer, i.e. within the fiber tows. The maximum interlayer coverage would be equal
to the nominal area of the preform, while the magnitude of the intralayer coverage would
depend upon the fraction of the total filament surface area covered. Since quantification of
the actual surface area coverage is difficult owing to the complex nature of spreading of a
viscoelastic melt over a porous substrate, the area coverage was studied qualitatively.
The criterion for intralayer coverage can be obtained by an order of magnitude analysis of
the forces favoring and resisting impregnation into the fiber tows. The main force favoring
impregnation into the tows would be the capillary pressure, Fc, while the resistance to
deformation would come from the force due to the viscoelastic modulus of the polymer
melt, Fg . Thus, the driving force, Fd for bundle impregnation (neglecting effects of
gravity and static friction) can be expressed by Equation 4.21.
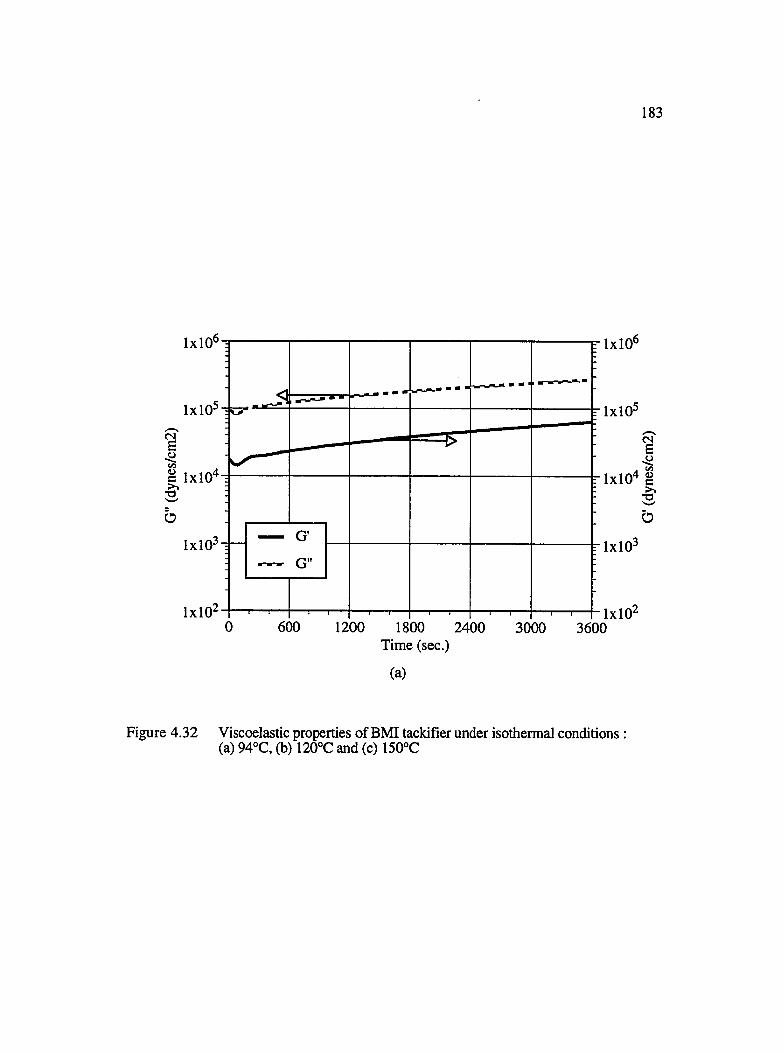
183
F 1x10'
TTTr 1x10
es
r lx lO '
r 1x10
1x10 1x100 600 1200 1800 2400 3000 3600
Time (sec.)
(a)
îêÜ
Figure 4.32 Viscoelastic properties of BMI tackifier under isothermal conditions :(a) 94°C, (b) 120°C and (c) 150°C

184
Figure 4.32 Continued
1x10' r i x i C
r 1x10(S
r lxlO'
1x10 r 1x10
1x10 1x100 200 400 600 800 1000 1200 1400 1600 1800
IÜ
Time (sec.)
(b)
1x10 F 1x10
1x10' rlx lO '
P 1x10
^ 1x10'
1x10 r 1x10
1x10 1x100 200 400 600 800 1000 1200 1400 1600 1800
Time (sec.)
(c)

185
Fd = 4 (Fc - Fg ) or Pn =% (Pc - G*) (4.21)
While values of the viscoelastic modulus can be obtained by using a RDA as shown earlier,
an estimate of the capillary pressure can be obtained by using Equation 2.10. Substituting
realistic values of the various parameters as given below, the capillary pressure can be
estimated to be ~ 10^ dynes/cm^.
F = 2.0
df = 10 |im
(|) = 0.45
Ylv = 40 dynes/cm
Cos 0 = 0.4
Based on this criterion, impregnation into the tows can be expected to be favored beyond
280°C for PMMA powder. This indeed is the case as observed from debulking
experiments. For BMI tackifier however, intralayer coverage was not observed. In case of
BMI, the viscoelastic modulus decreases then increases with increasing temperature
because of the chemical reaction. On the contrary, for PMMA, the viscoelastic modulus of
the melt continues to drop with increasing temperature. The driving force would therefore
continues to increase beyond 280°C, thereby favoring impregnation into the fiber tows.
Based on the experimental results, the overall phenomenological approach taken in this
study to successively control springback under lateral compression can be summarized in
Figure 4.33. For fiber preforms of a given architecture, the manipulated variables to
control springback are the tackifier modulus, and the surface area coverage. Tackifier
modulus can be increased by advancing the chemical reaction during the debulking stage.
However, due to limitations of the solubility of the tackifier in the matrix resin, this is not
desirable, and as discussed in the next Chapter, this could adversely affect the mechanical
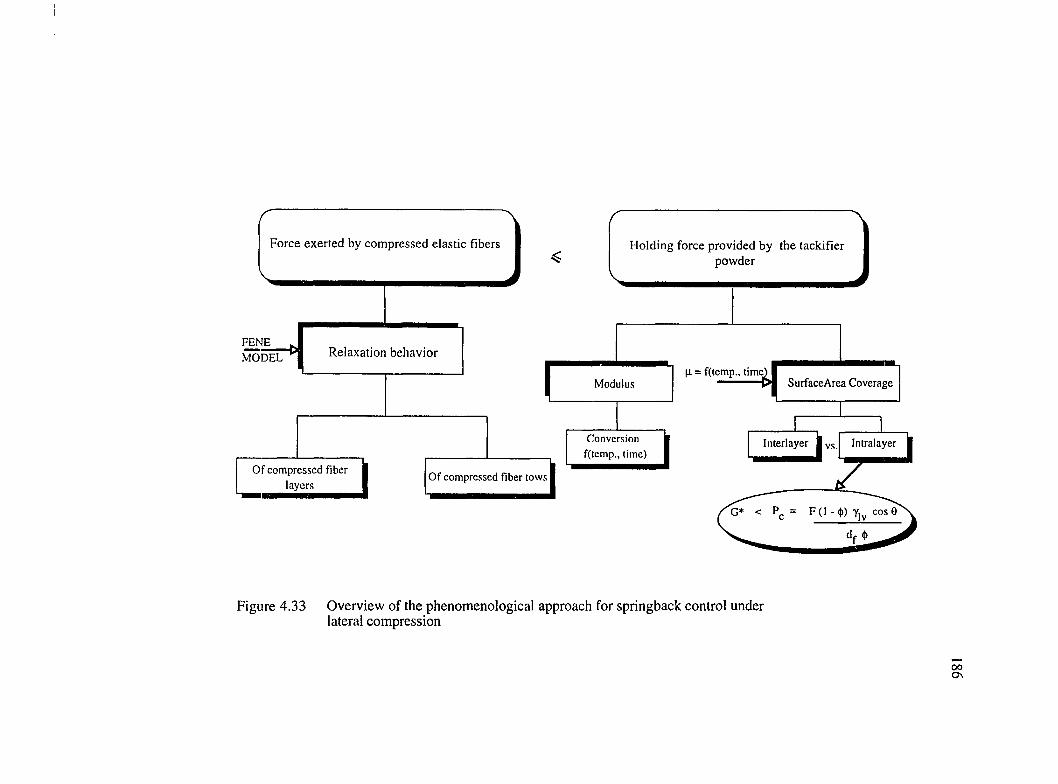
Force exerted by compressed elastic fibers Holding force provided by the tackifier powder
FEME
MODEL Relaxation behavior
SurfaceArea CoverageModulus
Conversion f(temp., time)
Interlayer Intralayervs.
Of compressed fiber layers Of compressed fiber tows
Figure 4.33 Overview of the phenomenological approach for springback control under lateral compression
00o\

187properties of the molded composite. Thus, a better alternative is to increase the surface area
coverage, and this can be achieved by increasing both the interlayer and intralayer
coverage, as there are two levels and consolidation, and consequently two levels of
springback.
Further proof o f this can be seen from Figure 4.34 (a), (b) and (c). These
photomicrographs represent the cross-section of the samples after springback, and figure
(a) and (b) corresponds to the case in which intralayer coverage was achieved using PMMA
powder at ~ 287°C. Figure (c) shows the cross-section of the sample with BMI tackifier at
94°C, and in which the tackifier remains on the surface. Figure (a) shows that the fiber
tows are compressed much more as compared to Figure (c). However in case of PMMA
heated at -287 °C, some gaps between the layers were observed (Figure b) showing
interlayer springback. These gaps are not present in the case of BMI, indicating that
springback results mostly from deconsolidation of fiber tows.
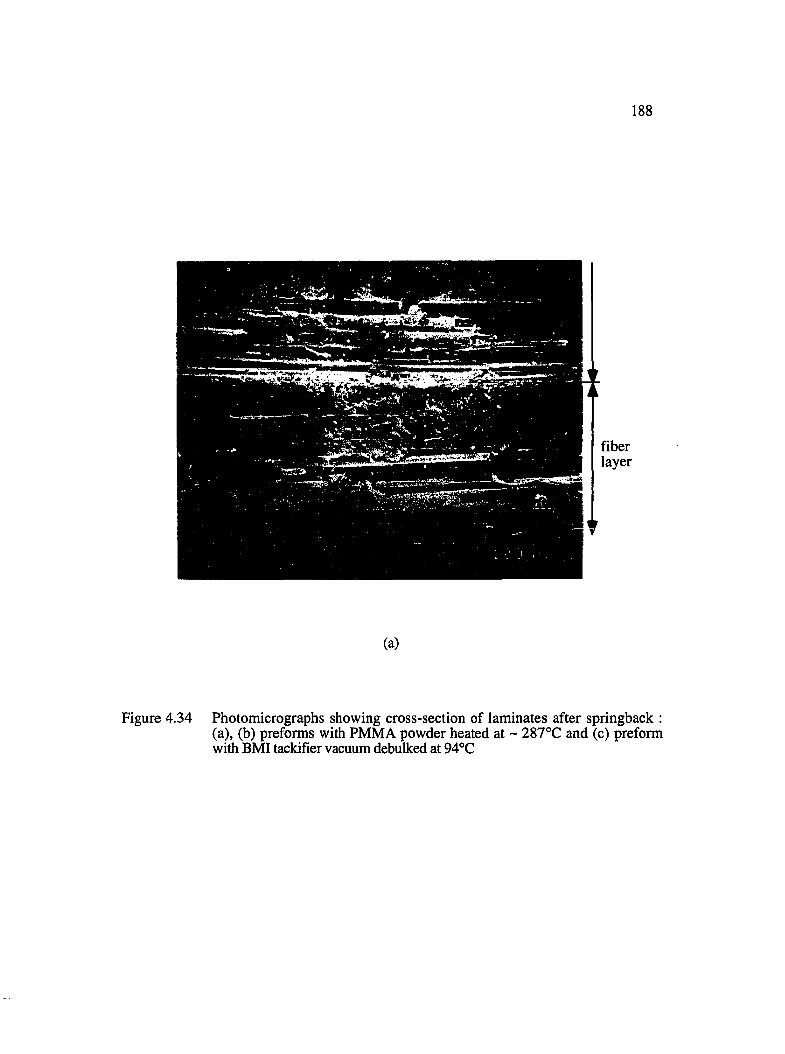
188
(a)
Figure 4.34 Photomicrographs showing cross-section of laminates after springback :(a), (b) preforms with PMMA powder heated at ~ 287°C and (c) preform widi BMI tackifier vacuum debulked at 94°C

189
Figure 4.34 Continued
gap between fiber tows
(b)

Figure 4.34 Continued
190
fiberlayer
(c)

CHAPTER V
MECHANICAL PROPERTIES OF MOLDED COMPOSITES AND PERMEABILITY CHARACTERISTICS OF TEXTILE REINFORCEMENTS
5 .1 Effect of Voids on Fiberglass/UP Composites
Stitched unidirectional fiberglass mat (CoFab AO 108) reinforced unsaturated polyester
(UP) resin composite samples were prepared using one layer of fiber reinforcement and
under similar conditions and same fiber volume fraction as mold filling experiments
described in Chapter III. The unsaturated polyester resin (Q 6585, Ashland Chemical)
used is a 1:1 mixture of propylene glycol and maleic anhydride containing 35% by weight
of styrene. The structure of the two constituents is shown below
O?
CH
~ C ” C H = CH“ C-------- O —Œ — CH 2 “O —
Maleic Anhydride Propylene Glycol
The unsaturated polyester resin has an average of 10.13 vinylene groups per polyester
molecule. The average molecular weight is 1580 gm/gm-mole, and the equivalent
molecular weight/(mole C=C) is 156 gm/gm mole. Additional styrene was added to make
the molar ratio of styrene to unsaturated polyester equal to 2.0.
191

192The initiator, USP 245 (2,5 dimethyl-2,5 bis (2-ethyl-hexanoyI-peroxy) hexane) was
added at 1% by weight of styrene and UP resin. Both unsaturated polyester resin and
styrene monomer were used as received without removing the inhibitor. In order to avoid
batch to batch variations, a large quantity of the resin (without the initiator) was prepared
and stored in the freezer. When required for molding experiments, a small amount was
taken out and the appropriate amount of initiator was added. Curing was done overnight at
130°C.
5.1.1 Dynamic Mechanical Test
A Rheometrics Dynamic Analyzer (RDA) was used in the torsion mode to monitor the drop
in the dynamic properties of composite samples immersed in hot water for up to 48 hours.
Consideration of dynamic properties become important when the structural configuration of
a part is dictated by flutter and vibration characteristics [Maymon et al., 1978].
Test samples used were rectangular strips having dimensions of 5.3 cm x 1.1 cm (1 x w).
Three strips were cut from composite samples with no voids, with ~ 3% microvoids and
with -7 % macrovoids. These strips were then immersed in hot water at 95°C successively
for 1 hr, 2 hrs, 4 hrs, 8 hrs and 48 hrs. After each immersion time, the sample was taken
out and placed between the top and bottom fixtures of the RDA as shown in Figure 5.1.
The top fixture was held stationary, while a cyclic twist was applied to the bottom fixture
during the experiment. All readings were taken at room temperature over a frequency
sweep from 0.1 rad/sec. to 25 rad/sec. at a maximum strain of 0.1%. The values of elastic
modulus G' were computed from Equations 4.10 and 4.11 along with 5.1:
Kx = 1000
t 3 + 1.8 ( - ) ________ w
wt-(5.1)
1 9 3
UPPERFIXTURE
RECTANGULARSAMPLE
ACTUALSAMPLELENGTH
COLLARDATAENTRY
SAMPLELENGTH
INSERT(SEE
NOTE).
LOWERFIXTURE
NOTE; S tandard Inserts are designed for the — . following sam ple th icknesses:j 0 .0 3 0 Inch (0 .7 6 2 m m |
0 .0 6 0 Inch (1 .5 2 m m l ) 0 .1 2 5 Inch (3 .17mm)
Figure 5.1 Torsion rectangular fixtures with the loaded sample [RDA Instruction Manual, 1994]

194where
Kx is the stress constant
t and w are the thickness and the width of the sample
The drop in the normalized elastic modulus or the dynamic stiffness of UP composite
samples with increased immersion times in hot water at 95°C are shown in Figures 5.2 (a),
(b) and (c). Figure 5.2 (a) shows the response of the sample with no voids, while Figures
5.2 (b) and (c) are for samples with ~ 7% macrovoids and ~ 3% microvoids, respectively.
Normalized values were obtained by taking the ratio of the elastic modulus of the fresh
sample to the elastic modulus of the same sample after specific intervals of immersion in
hot water. In torsion experiments, it is difficult to attribute the observed differences solely
to the different void content of the samples because artifacts may be induced due to slight
differences in the fiber orientation and fiber content. Thus, in order to avoid the influence
of any extraneous effects, only one sample was used and its drop in the stiffness values
was monitored with increased immersion times in hot water. Samples with different void
contents were compared by comparing the trend in the drop in the stiffness values. Based
on the results obtained for the three samples, all of them show a decrease in the percent
retention. This drop can be attributed to the diffusion of moisture into the samples. Under
ideal conditions, penetration of moisture in composites has been shown to follow the
classical Fickian process where diffusion is driven solely by the concentration gradient
[Wolff, 1993]. In actual practice, however, non-Fickian diffusion can occur in any one or
more o f the following three ways, viz., through the resin matrix, through the edges of the
sample, and via voids. Samples with voids show a greater drop in the retention values than
the sample with no voids. This is probably because of the greater amount of the moisture
absorbed by samples containing voids, leading to a more pronounced plasticization o f the
resin matrix and / or degradation of the resin - fiber interface.
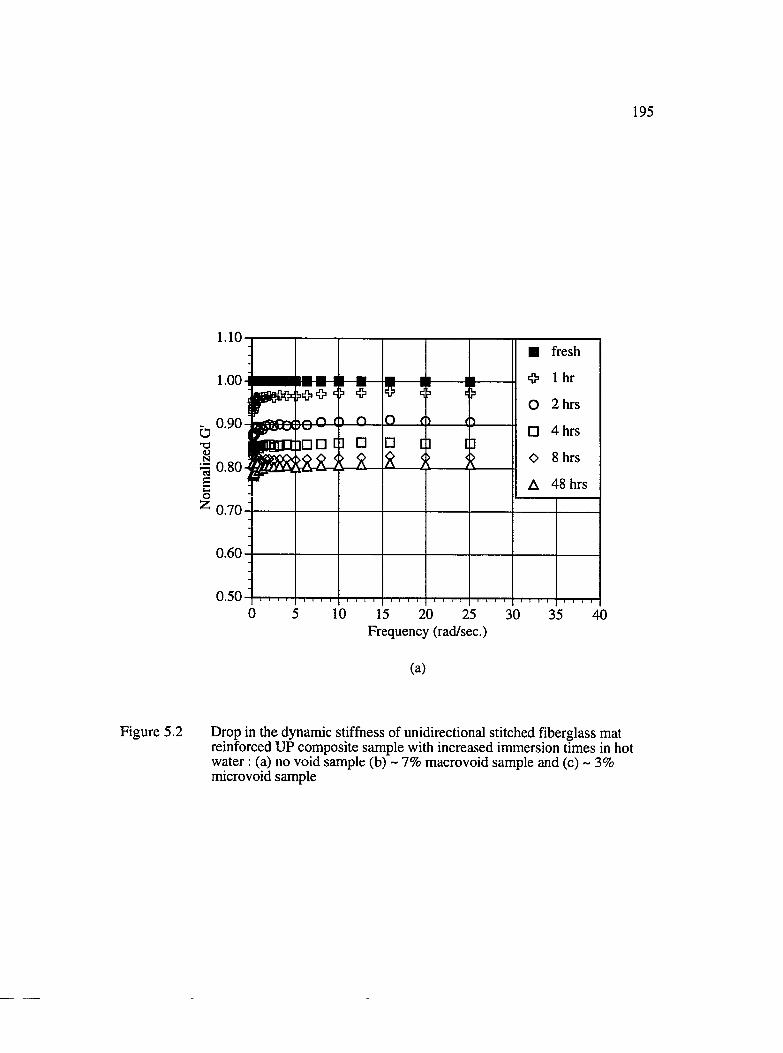
1 9 5
1. 10 -
1.00 -
^ 0 .9 0 -
1 ,
0.70
0.60
0.50-
-I H H 1 ■ ■ 1 1I
■ fresh
* 1 hr
O 2 hrs
□ 4 hrs
O 8 hrs
A 48 hrs
■> *
\ m O (
1 1=■
1 t
I>
]□ □ [ ] □ □ c] [> < \ !
]
:
-
---I'T 1 1 i l l 1 1' 1 1 1 -T-l 1"!0 5 10 15 20 25 30 35 40
Frequency (rad/sec.)
(a)
Figure 5.2 Drop in the dynamic stiffness of unidirectional stitched fiberglass mat reinforced UP composite sample with increased immersion times in hot water : (a) no void sample (b) ~ 7% macro void sample and (c) ~ 3% microvoid sample
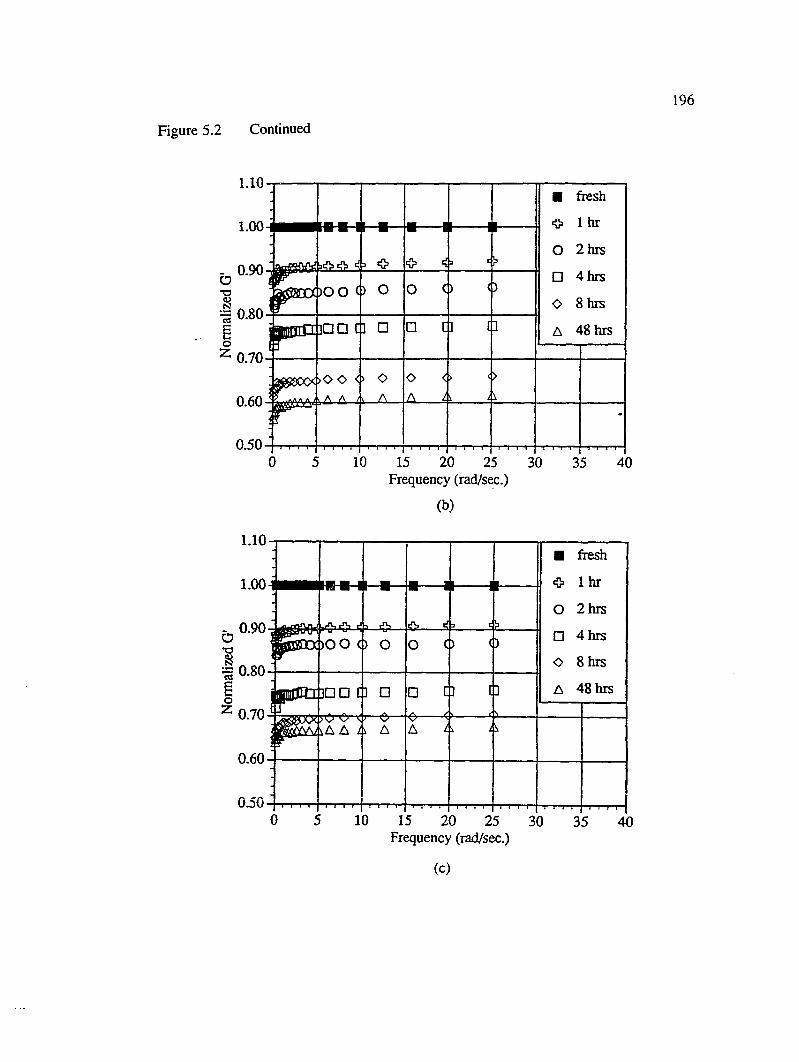
Figure 5.2 Continued
196
- 0.90
= 0.80
m-m
OO (p o
■4-
m □
o.Æ
*
□
< >
A
■ fresh
O 1 hr
O 2 hrs
□ 4 hrs
O 8 hrs
A 48 hrs
0.50 [ I I T I I I i I I )■! I - I I - j i r "i |"| I I I I j I - I I 1 I I - I I I I r - r I - I '
0 5 10 15 20 25 30 35 40Frequency (rad/sec.)
(b)
- 0.90
3 0.80
M - * - *
<gijSi A OO O (b O
□ □ CD □
A A A A
m m
■ fresh
O 1 hr
O 2 hrs
□ 4 hrs
O 8 hrs
A 48 hrs
10 15 20 25 30 35Frequency (rad/sec.)
40
(c)

197
5.1.2 Freeze-Thaw Cycling
Freeze - thaw cycling experiments were conducted to see the extent of degradation in
composite samples subjected to extreme temperature environments as in some applications.
Each freeze - thaw cycle consisted of immersing composite samples in hot water at 95°C for
12 hours followed by additional 12 hours in dry ice stored in a Dewar flask at about -40°C
cycling were observed under a transmission type optical microscope.
After about two weeks of freeze-thaw cycling of composite samples, microcracks or
fissures were observed in samples containing macro and microvoids. Figures 5.3 (a) and
(b) show these cracks on the surface of the macrovoid sample and within the fiber tow of
the microvoid sample. The formation of these microcracks can be explained as follows.
When composite samples are exposed to a moist environment, moisture diffuses in and if
voids are present, collects at the voids forming pockets of water. Thus, voids provide sites
for the accumulation of water inside the composite. At temperatures below freezing, like in
this experiment, the absorbed water expands. Depending upon the volume of water
absorbed at the voids, cracks of different sizes could be formed. Once the cracks are
formed, continued exposure to alternative hot and moist and cold environment can cause
more moisture to diffuse in and propagate the crack. Thus, for composites which are
fabricated to endure temperature extremes, it seems to be highly desirable to minimize the
void content.
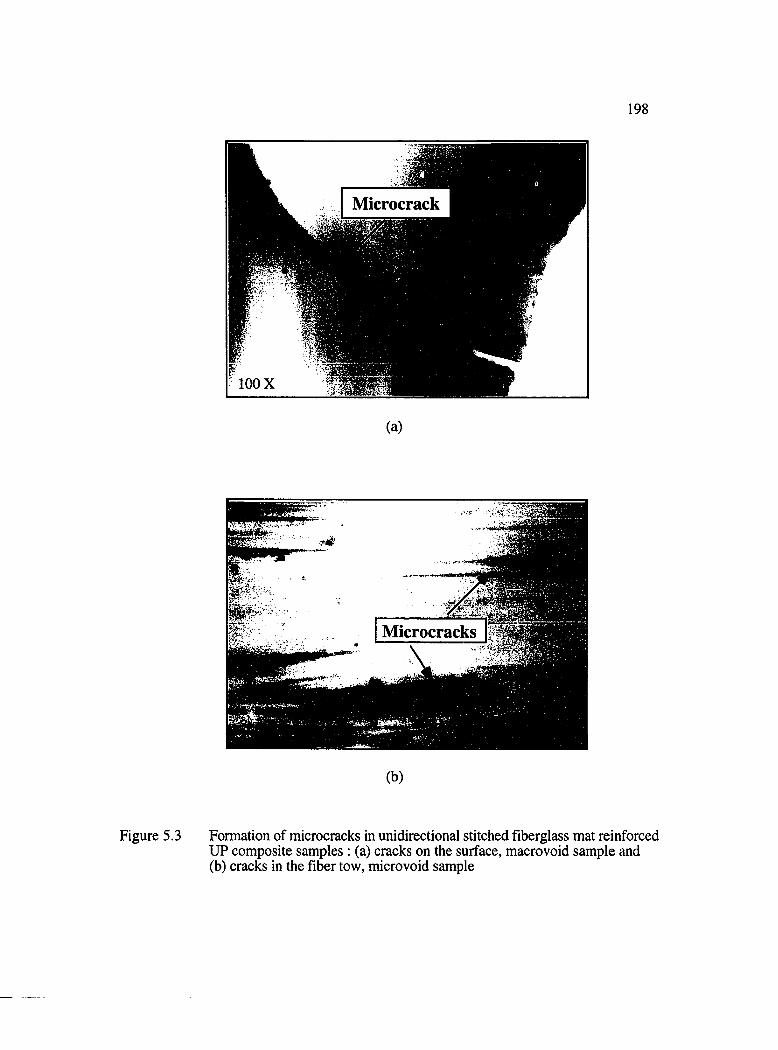
198
M icrocrack
100 X
(a)
M icrocracksm \
(b)
Figure 5.3 Formation of microcracks in unidirectional stitched fiberglass mat reinforced UP composite samples : (a) cracks on the surface, macrovoid sample and(b) cracks in the fiber tow, microvoid sample

1995.1.3 Ultrasonic C - Scan
In order to qualitatively characterize fiber wetting in cured composite samples prepared
under different processing conditions, ultrasonic C-scan imaging was employed. Figure
5.4 shows the components of the SONOTEK ultrasonic C - scan set - up that was used for
non-destructive detection of voids in composite samples. Ultrasonic C - scan facility was
made available by the cooperation of the Edison Welding Institute. This technique is based
on the transmission of sound waves through the composite sample placed in an immersion
tank filled with water. As shown in Figure 5.4, the set - up used consists of an
Z - axis manipulator
Computer Printer
\ \ HSE
Puiser / Receiver.
i
. ^ Motor
¥Immersion tank
Figure 5.4 Components of the SONOTEK Ultrasonic C - scan system
immersion tank resting on a support table. An aluminum frame lies on top of the tank to
support the mechanical system for scanning the specimen with a transducer. The ultrasonic
signals are generated and received by tbe puiser/receiver device which operates the
transducer mounted at the tip of the scanner. The operation is initialized by inputting the
desired scanning path for the transducer. Parameters such as scanning range, step and the

200threshold level above which the echo signals are to be sampled are all initialized. After the
required input information is entered, the scanner stepping motor control sequence is
activated. The motor controls the motion of the transducer in both X and Y directions. The
distance of the transducer from the composite specimen was adjusted by the Z axis
manipulator at the start of the experiment. All the electronics are interfaced with a
computer. The C - scan images were viewed on the computer screen, and hard copies were
obtained from the printer attached to the computer.
Ultrasonic C-scan images for unidirectional stitched fiberglass reinforced UP composites
molded using two layers of the reinforcement and using axial flow at different flow rates
are shown in Figures 5.5 (a), (b) and (c). Figure 5.5 (a) is the C-scan image of the
composite molded at a flow velocity of 0.04 cm/sec. For UP resin, this injection velocity
corresponds to a capillary number of 1.37 x 10'3. Based on the flow visualization
experiments using non - reactive liquids, this capillary number lies in the range where no
voids are formed within the tows. This seems to correlate nicely with the C-scan image in
which the tows appear as parallel dark bands. The uniform shading of the tows indicates
very little scattering of the transmitted signal characteristic of high level of impregnation of
fibers.
Figure 5.5 (b) is the C-scan image of the composite sample molded at a velocity of 1.0
cm/sec. This corresponds to a capillary number of 3.47 x 10'3 where some microvoids
start to form. These microvoids show up in the image as white spots within the fiber tows.
Figure 5.5 (c) shows a much different image than the other two. This is the scan of the
composite sample molded at a velocity of 3.9 cm/sec. The fiber tows are barely visible.
Also, the entire image has very light colors which show up as gray areas in this black and
white photograph.

201
iber tows
(a)
Figure 5.5 Ultrasonic C-scan image of unidirectional stitched fiberglass matreinforced UP composite samples ; (a) Vs = 0.04 cm/s, b) Vs = 1.0 cm/s and (c) Vs = 3.9 cnVs

Figure 5.5 Continued
202
Microvoids
(b)
(c)

203
Light colors represent lower amplitudes of the transmitted signal and a lot of scattering
which is characteristic of poor and variable degree of impregnation. Thus, Ultrasonic C-
scan imaging can be used as an effective qualitative tool for characterizing the degree of
fiber wetting in cured composite samples.
5 .2 Mechanical Properties of Tackified Samples
Flexural properties of neat resin plaques, resin co-cured with partially reacted tackifier
particles, and tackified preform reinforced BMI composites were measured according to the
procedure outlined in ASTM Standard D790-92 (Figure 5.6). Clear castings for the test
were prepared by pouring the degassed resin in a vertical glass mold, 30.48 cm x 30.48 cm
(Ixw). The thickness of the samples was controlled by using 0.125 cm thick aluminum
spacer. The mold configuration used is shown in Figure 5.7. Prior to pouring the resin, it
was degassed at 94°C - KX) °C for about an hour to ensure the removal of all the air bubbles
and volatiles. Cure cycle was 4 hours and 15 minutes at 180°C followed by 3 hours at
220°C. After cure, rectangular strips having dimensions of 6.35 cm x 1.27 cm x 0.125 cm
(1 X w X t) were cut using a diamond saw. The cross-head speed of the Instron machine
during the test was kept at 0.127 cm/min.
L/2 L/2X .
Support Span
Figure 5.6 Schematic of 3-point bending test

204
glass plate
(-1 Lspacer
gasket
clamps
Figure 5.7 Configuration of mold set-up for preparing clear castings
Two different methods were used to prepare samples of BMI resin with partially cured
tackifier particles. One of them involved mixing partially cured and ground tackifier
particles in the molten resin at a concentration of 3% by weight. In the other, about 1% of
the tackifier powder was sprinkled on the inside surfaces of the glass mold and then cured
to different extents before pouring in the resin. The results for the latter are reported in
Appendix B.
An Olympus polarizing microscope (Model BHSP 200) equipped with a sixth order Berek
compensator, and a magnification range from 40X to lOOOX was used to characterize the
residual microstresses around the tackifier particles embedded in the resin matrix. This was
done by observing the birefringence patterns under polarized light. Photographs were
taken using an installed Olympus PM-6 camera. Magnification was kept at lOOX.

205The results of the 3-point bending tests of unreinforced samples are shown in Tables 5.1
and 5.2. Tables 5.1 (a) is for pure BMI samples, while 5.1 (b) and (c) are for resin with 3
wt.% tackifier, and with tackifier conversions of 0.5 and 0.6 respectively. Flexural
strength values were evaluated using Equation 5.2:
3 P X LS =2 w X t
where
P is the load at break
L is the support span
w is the sample width
t is the sample thickness
Table 5.1(a) Flexural properties of pure BMI resin
(5.2)
Flexural Strength
(psi)
% Strain @ Break Modulus(psi)
26400.3 5.3 616150.62
28691.2 6.7 612504.97
24306.7 4.6 614750.07
28245.7 6.2 621653.09
25913.8 5.2 611901.89
Mean: 26711.5 5.6 615392.14
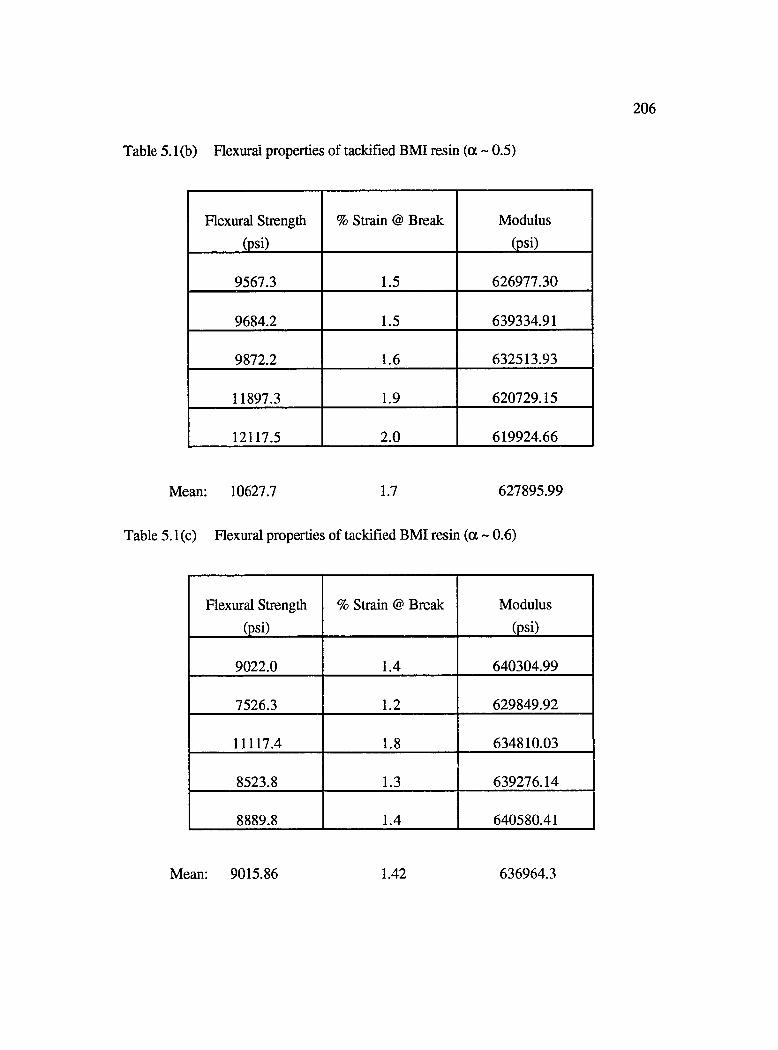
Table 5.1(b) Hexural properties of tackified BMI resin (a ~ 0.5)
206
Flexural Strength
(psi)
% Strain @ Break Modulus(psi)
9567.3 1.5 626977.30
9684.2 1.5 639334.91
9872.2 1.6 632513.93
11897.3 1.9 620729.15
12117.5 2.0 619924.66
;an: 10627.7 1.7 627895.99
c) Flexural properties of tackified BMI resin (a ~ 0.6)
Flexural Strength
(psi)
% Strain @ Break Modulus(psi)
9022.0 1.4 640304.99
7526.3 1.2 629849.92
11117.4 1.8 634810.03
8523.8 1.3 639276.14
8889.8 1.4 640580.41
Mean: 9015.86 1.42 636964.3

207As shown in Table 5.1 and in Figures 5.8 (a) and (b), both the mean flex strength and the
mean percent strain at break for pure resin samples were found to be higher than the
corresponding values of tackified resin samples. One reason for this could be due to the
insolubility of the tackifier particles in the matrix resin resulting in a two phase system.
Due to this heterogeneity, residual microstresses (Figure 5.9) develop probably due to the
mismatch in the thermal expansion and contraction of the two phases. Thus, when the
applied stress and the residual stress reach a certain critical level, premature fracture occurs.
The fracture surface of both pure resin (Figure 5.10 a) and tackified resin (Figure 5.10 b)
were also observed. The latter figure shows voids at the fracture surface which can also
explain the poor mechanical properties exhibited by the tackified resin samples. This
observed morphology is similar to the one observed in some rubber modified epoxy and
bismaleimide fracture surfaces [Voit and Seferis, 1987]. It was hypothesized that the
reason for this is due to cavitation, crazing, or debonding of the rubber particles that were
phase separated during the curing stage.
Tables 5.2 (a) and (b) compares the flexural strength, percent strain at break and modulus
of composite samples made from preforms coated with 3 wt.% tackifier powder debulked
at 145°C for 1 hour and 80°C for 20 minutes respectively to achieve two different cure
levels. The samples with lower degree of tackifier cure show slightly higher values for the
flexural strength, although the difference is not as drastic as in the case of unreinforced
sample. This is probably due to the masking effect of the fibers.
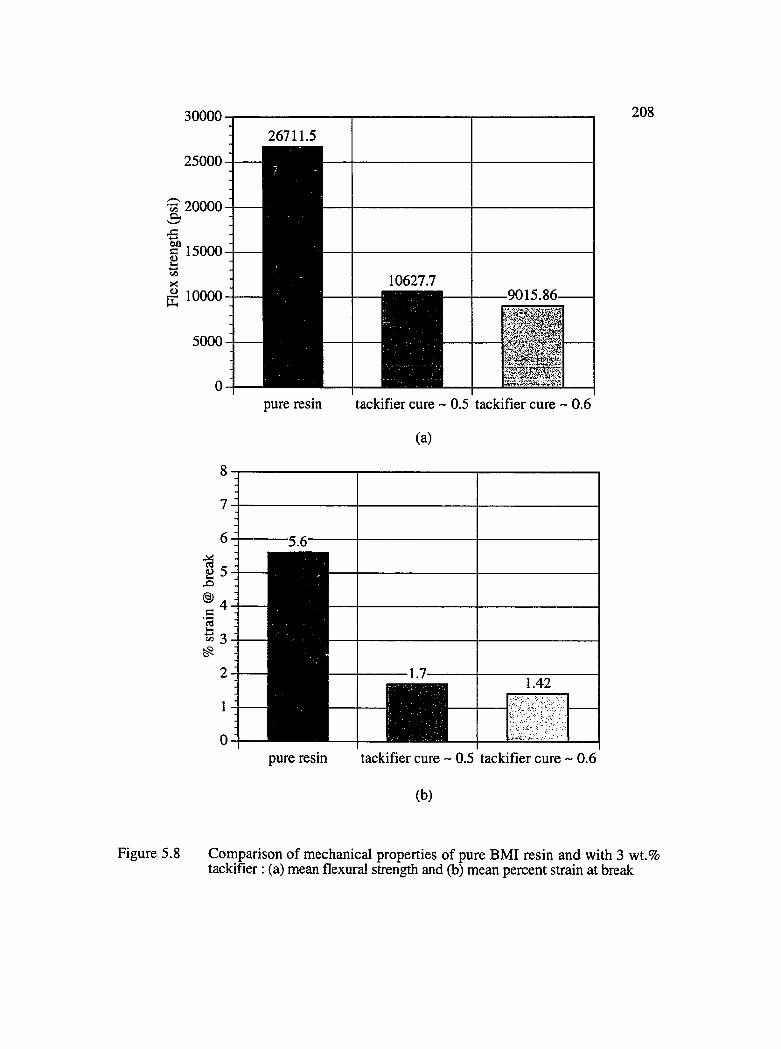
30000
25000
-a 20000 &.
c 15000II 10000
5000
26711.5
10627.79015.86-
pure resin
*
tackifier cure ~ 0.5 tackifier cure ~ 0.6
208
(a)
@1
pure resin tackifier cure ~ 0.5 tackifier cure ~ 0.6
(b)
Figure 5.8 Comparison of mechanical properties of pure BMI resin and with 3 wt.% tackifier ; (a) mean flexural strength and (b) mean percent strain at break

209
tackifie
matrix
Figure 5.9 Four clover leaf pattern indicative of residual microstresses at the tackifier particle/resin matrix interface

210
(a)
(b)
Figure 5.10 Scanning electron micrographs of the fracture surface : (a) pure BMI resin and (b) tackified BMI resin
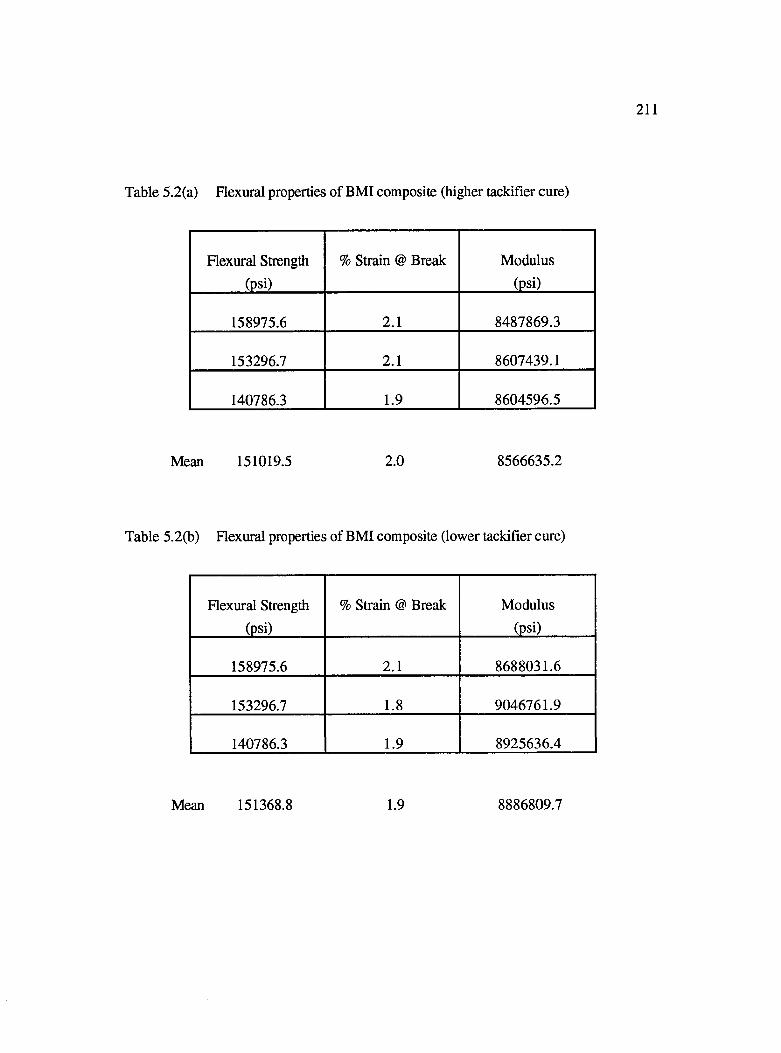
211
Table 5.2(a) Flexural properties of BMI composite (higher tackifier cure)
Flexural Strength % Strain @ Break Modulus
(psi) (psi)
158975.6 2.1 8487869.3
153296.7 2.1 8607439.1
140786.3 1.9 8604596.5
Mean 151019.5 2.0 8566635.2
Table 5.2(b) Flexural properties of BMI composite (lower tackifier cure)
Flexural Strength % Strain @ Break Modulus
(psi) (psi)
158975.6 2.1 8688031.6
153296.7 1.8 9046761.9
140786.3 1.9 8925636.4
Mean 151368.8 1.9 8886809.7

212
5 .3 Flow Characteristics of Textile Reinforcements
5.3.1 Permeability o f Fiber Preforms
Permeability measurement characterizes the flow resistance of a fiber reinforcement. Since
pressure drop and mold filling times during the resin injection stage are governed by
permeability values, their measurement becomes important from processing point of view.
Also, these values can vary over a wide range depending upon the fiber structure.
Moreover, fabrics with anisotropic structure would have varying permeability values in
different flow directions. Thus, permeability is a tensor quantity having both magnitude
and direction. The use of binder or tackifier material further complicates the issue, and
therefore warrants a systematic investigation.
Since permeability measurements of stitched unidirectional fiberglass mat were conducted
in a companion study [Wu, 1995], this work focuses on braided and 4HS tackified
reinforcements. The experimental set-up used for in-plane permeability measurements is
shown in Figure 5.11. Using a liquid of known viscosity and injecting it at a constant flow
rate, the pressure drop can be monitored as the liquid flows through the fabric.
Permeability values for an isotropic fiber mat can then be determined by Darcy's law given
by Equation 2.1. To recall, Darcy's law states that permeability is inversely proportional to
the pressure drop as given by Equation 5.3:
K = f ( ^ ) (5.3)AP~ T
where, all the terms have their usual meaning as defined earlier in this text.
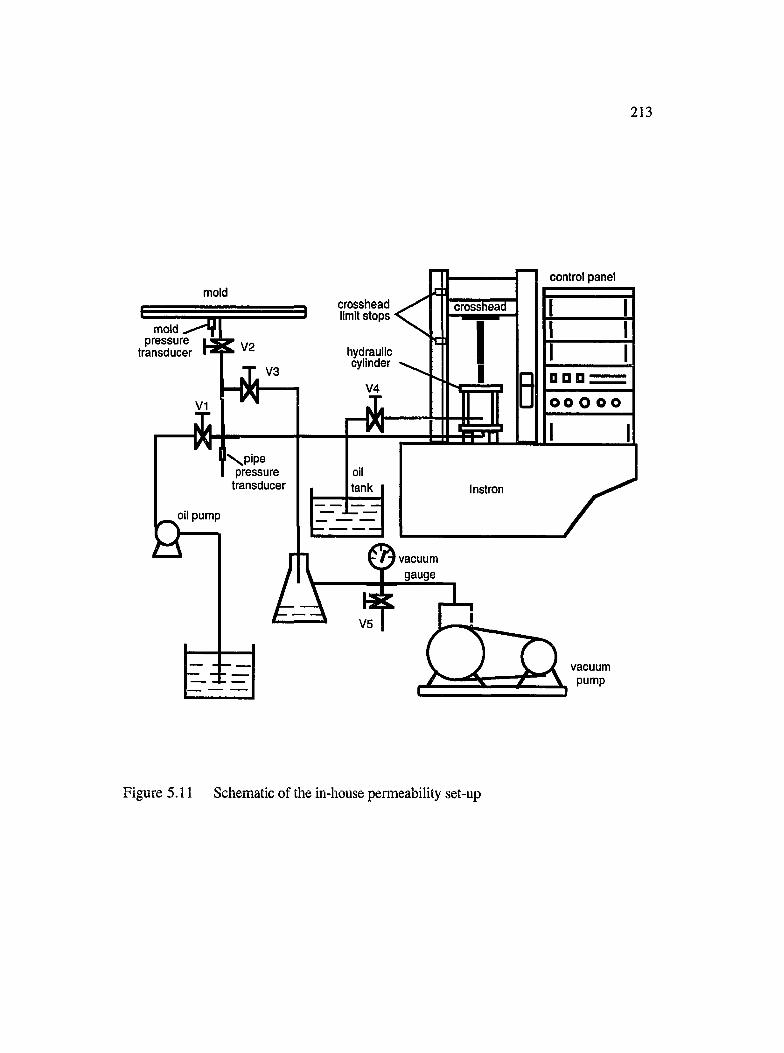
213
control panelmold
crosshead limit stops
crosshead
mold ^ pressure
transducer hydrauliccylinder
V4nonoo o o o
l \ p i p epressure
transducer tank Instron
oii pump
vacuumgauge
V5
vacuumpump
Figure 5.11 Schematic of the in-house permeability set-up

214
The pressure drop is from the inlet. Pin, to zero at the outlet which is open to the
atmosphere.
The test liquid used was Palatinol 7 IIP . This liquid is a mixture of isomers of 1 ,2 -
benzene dicarboxylic acid and is stable for applications below 250°C (BASF). Viscosity of
the oil was measured at several temperatures and curve fitted (Equation 5.4) using linear
regression [Perry, 1993].
|l = 107.6 - 0.835 T (°F) (5.4)
The thickness of the cavity required to achieve the specified fiber volume fraction was
calculated using Equation 3.2. Liquid was injected through the fiber samples at a constant
flow rate from a hydraulic cylinder using an Instron Universal Testing Machine (Model
1137). The nominal diameter, stroke and the volume of the cylinder were 8.26 cm, 7.62
cm and 408 cm3 respectively. The cylinder was filled with the test liquid using a gear
pump. Pressure was measured by two transducers, one at the inlet, and the other in the
mold an inch away from the inlet. Calibration of the pressure transducers was done using
an Ashcroft Dead Weight Gauge Tester shown in Figure 5.12. The pressure transducer
was mounted at the position E. Mineral oil was loaded in the reservoir A, and cylinder C
was filled by closing valve D, opening valve B, and slowly opening valve H. Next, valve
B to the reservoir was closed and valve D to the transducer at E was opened. Weights were
placed on the platform of piston F. Valve H was then closed to load the oil into cylinder G
and raise the weights about 5 cm above the platform. Readings were then taken using a
multimeter, and the sequence repeated for a range of weights. Calibration curves for two
of the pressure transducers are shown in Figures 5.13 (a) and (b). Details of the
operational procedures for permeability measurements are given in Appendix A.
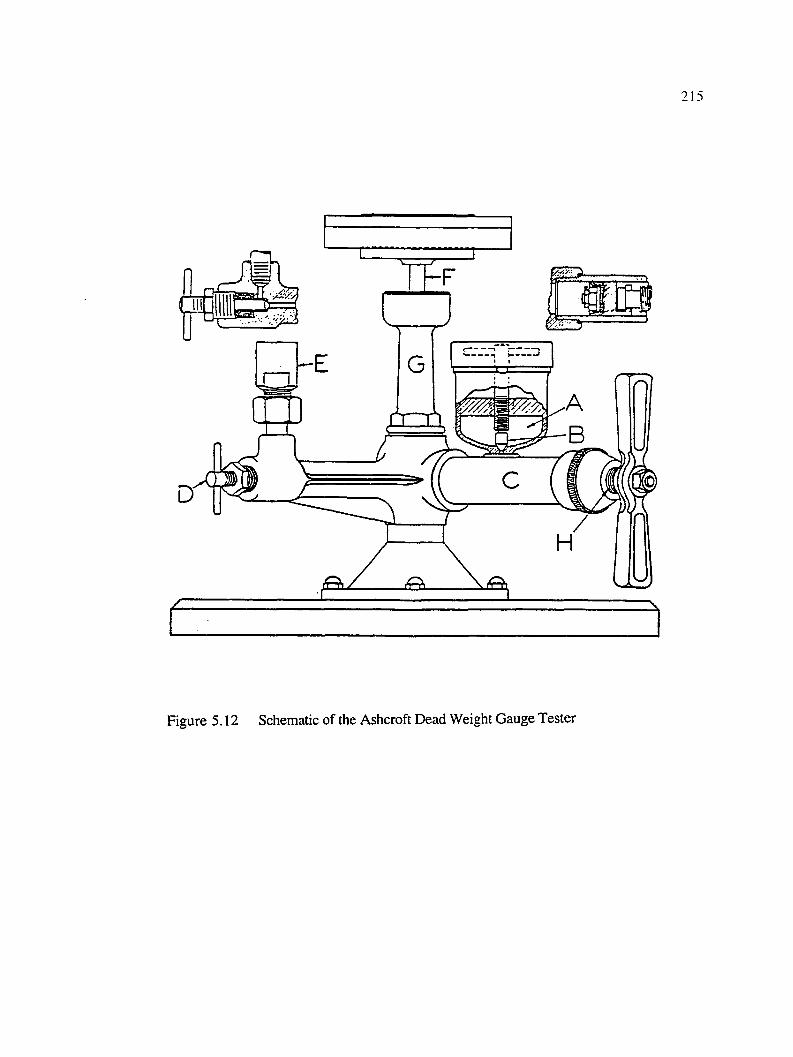
215
cm
Figure 5.12 Schematic of the Ashcroft Dead Weight Gauge Tester
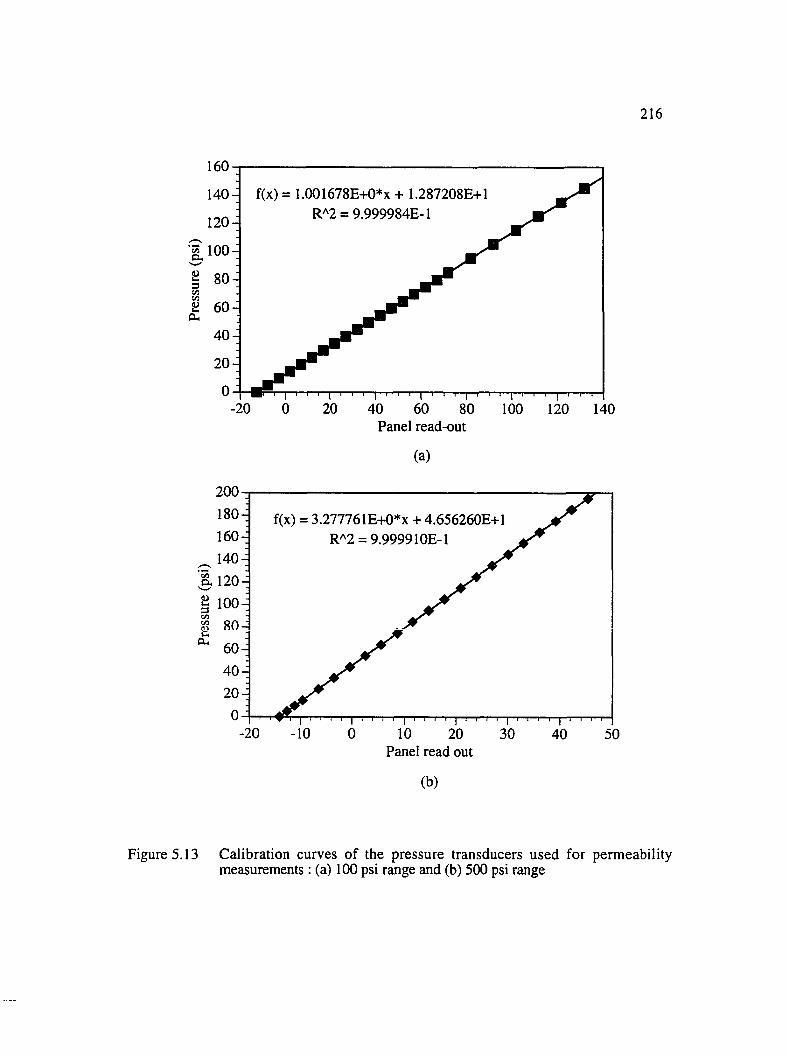
216
160
f(x)= 1.001678E+0*x+ 1.287208E+1 RA2 = 9.999984E-1
140-
120 -
•g 100-
8 0 -
6 0 -
4 0 -
20 -
60-20 0 20 40 80 100 120 140Panel read-out
(a)
7,00-180-^160
140 H
a 120^e3 100^
2 8 0 -O h 60^
40^20^
0-J
f(x) = 3.277761E+0*x + 4.656260E+1 RA2 = 9.999910E-1
I 1
10 20 Panel read out
(b)
Figure 5.13 Calibration curves of the pressure transducers used for permeability measurements : (a) 100 psi range and (b) 500 psi range

I l l
In cases, where fiber anisotropy exists, in-plane permeability values are obtained by a
radial flow visualization experiment and by using the set-up shown in Figure 5.11. For
example, injection of liquid in the center of an anisotropic preform results in an elliptical
flow front. The major axis of the ellipse defines the maximum principal direction
permeability, K%, while the minor axis defines the permeability in the y direction, namely
K y . The values of K x and K y then are determined from two relations. One is obtained
from the lengths of the major and minor axes of the fully developed ellipse as given by
Equation 5.5
length of minor elliptic axis K, ^length of major elliptic axis y
(5.5)
After the principal directions and the ratio of the permeabilities are known from flow
visualization, the second relation is obtained by measuring the pressure and relating it to the
flow rate, thickness of the cavity, geometry of the fiber mat and the viscosity of the liquid.
Equation 5.6 which is based on Darcy's law gives the relationship for calculating the
effective in - plane permeability from which the values of K x and K y are obtained [Wang
et al., 1992]:
Ke=(K^Ky)1 /2 Q pln(R/R;^) + lnû
P inlet
in"2 %
(5.6)
where
1( ^ ) i / : + lKy
' , R“„ xK1/2'
14- (5.7)
_0Ke is the effective permeability
R is the location of the flow front on the major axis (x direction)

218Rin is the radius of the inlet hole
h is the thickness of the cavity
5.3.1.1 Braided Preforms
The two types of braided graphite fiber reinforcements used were 6k tow, G30 - 500 and
12k tow, AS4 GP. 6k and 12k refer to 6000 and 12000 filaments per tow respectively.
Another difference between the two kinds of preforms was that the gaps between the fiber
tows for the 6k preform were much larger than for the 12k tow preform. Other
characteristics are shown in Table 5.3.
Table 5.3 Characteristics of braided preforms
6k G30-500 12k AS4 GP
No. of carriers 144 120
Ends/carrier 1 2
Preform diameter 3" 3.8"
Tow c.s.a 3.6 X 10‘4 in.2 7.54 X 10-4 in.2
Braid Angle ±45° ±45°
One method for calculating the fiber volume fraction for braided preforms is according to
Equation 3.2. The other method, which gives similar values is outlined in Equations 5.8
and 5.9 [Madge, 1993];

219
V f = - î ^ (5.8)
where
where
where
n = number of layers
Af = total fiber area in cross-section of braid
Ab = cross - sectional area of braided structure ( = t x 7 1 D)
t = ply thickness
D = preform diameter
^ ^ ^ M N ç A j , (5 ,9 )C O S 0
M = number of ends per carrier
Nc = number of carriers
Ay = tow cross - sectional area (c.s.a)
0 = tow braid angle
The results for pressure rise vs. time curves showed an S shape in the initial part for 6k
preforms, whereas Darcy's law predicts it to be linear. This discrepancy was due to
"system elasticity" which arose from the fact that the tygon tube used to convey the liquid
from the Instron machine to the mold was not totally incompressible. So, although the
liquid was pumped from the Instron at a constant flow rate, the flow rate might not have
been constant at the mold inlet thereby causing the artifact. The permeability equipment
was therefore modified by replacing the tygon tube by a metal tube. In addition, the
pipeline and the liquid were degassed prior to liquid injection to remove any trapped and
dissolved air. Permeability experiments for only 12k, AS4 GP preforms however were
conducted using the modified equipment, which are discussed next.

220Permeability measurements using unidirectional flow were tried, but were not very
successful. It is suspected that this was probably because the liquid flowed faster from the
sides of the cavity resulting in severe 'race -tracking'. This phenomenon seems to be more
pronounced when only a small number of fiber mats are used. Another reason could be the
"flexible" structure of the braided preforms. These preforms can be distorted more easily
as compared to other fiber types where the tows are held in place either by stitches or are
woven in such a way that their structural integrity is maintained. In order to circumvent the
problem of 'race - tracking', the radial flow method was used wherein the liquid was
injected through a small hole in the center of the circular fiber preforms. Fiber samples
with 10.16 cm diameter and 2.286 cm hole in the center were used for radial flow
measurements.
The pressure vs. time curves for three and two layers of fiber preforms are shown in
Figures 5.14 (a) through (c), and the permeability values are summarized in Table 5.4.
The orientation of the fiber layers are denoted in the parenthesis. As an example, [0/0/0]
denotes that each layer is oriented with its axis parallel to the other layer. In the case of
three layers, the porosity was 34%, while for two layers the porosity was 31%.
It is interesting to note that the injection pressure does not level off even after the liquid
reaches the edge of the fiber sample. One plausible explanation for this is that for braided
fiber preforms, the fiber structure is quite flexible and the preform is highly deformable.
Thus, during mold filling the orientation of the fiber tows may change, resulting in a
change of perm eability, and consequently, a change of the pressure drop.

221
120 -
100 -
80
a
ie
60
40
20
\
1
1
1
— Dry
— W etl
- - Wet2
1
111
-
11
1
— ■•■'■I , ■ ■ ■ ' V I 1 1 1 1 ■ ' 1 1 — 1 1 1- - - - - - - - - -
400 800 1200 Time (sec.)
1600 2000
(a)
Figure 5.14 Pressure rise vs. time curves for braided preforms : (a) 3 layers [0/0/0], (b) 2 layers [0/0] and (c) 2 layers [0/90]
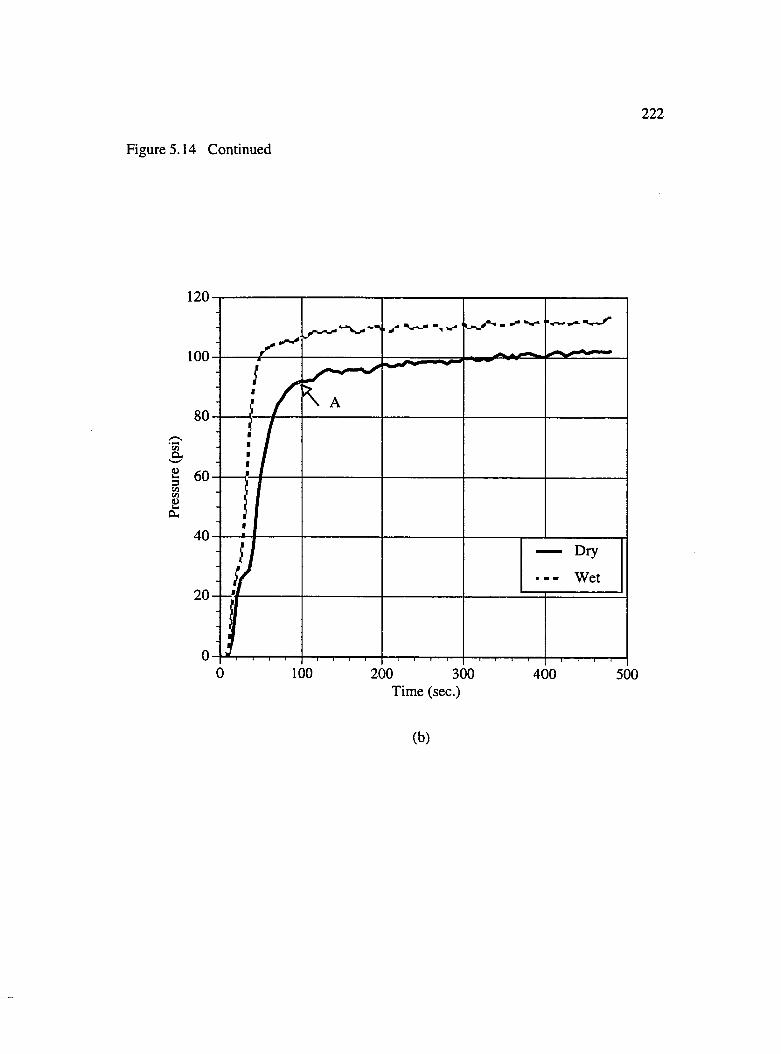
Figure 5.14 Continued
222
120
100
Cue
CL,
Dry
Wet
100 200 300 400 500Time (sec.)
(b)
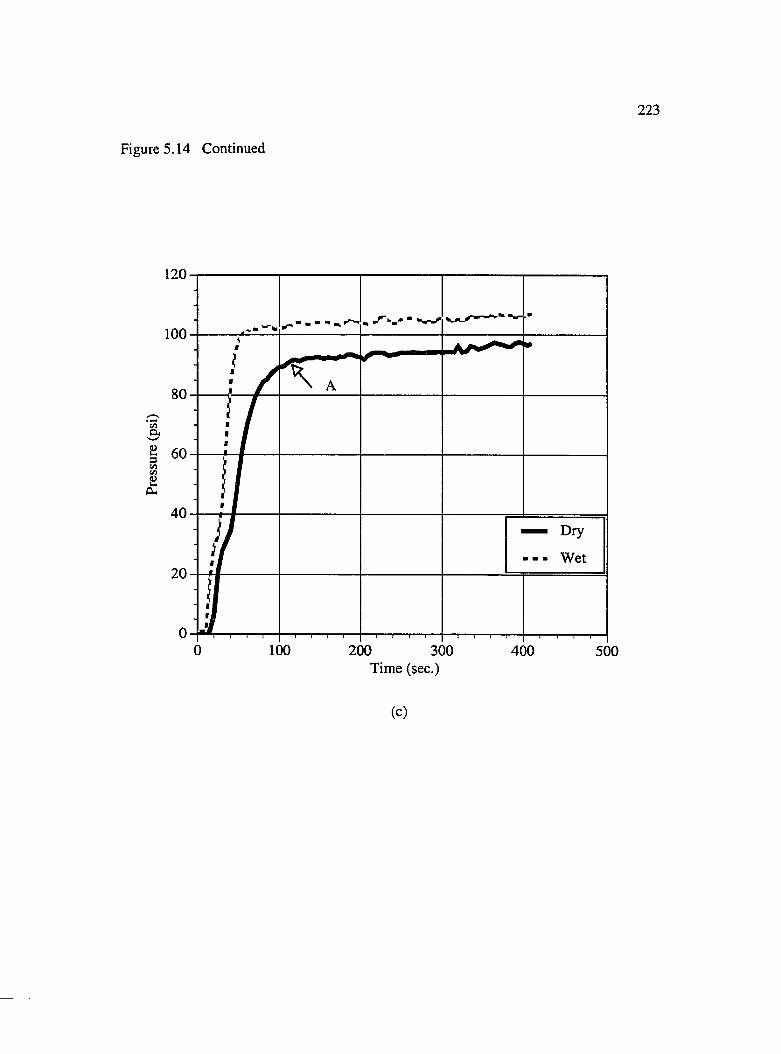
223
Figure 5.14 Continued
120
100
I40
Dry
Wet
100 200 300 400 500Time (sec.)
(c)
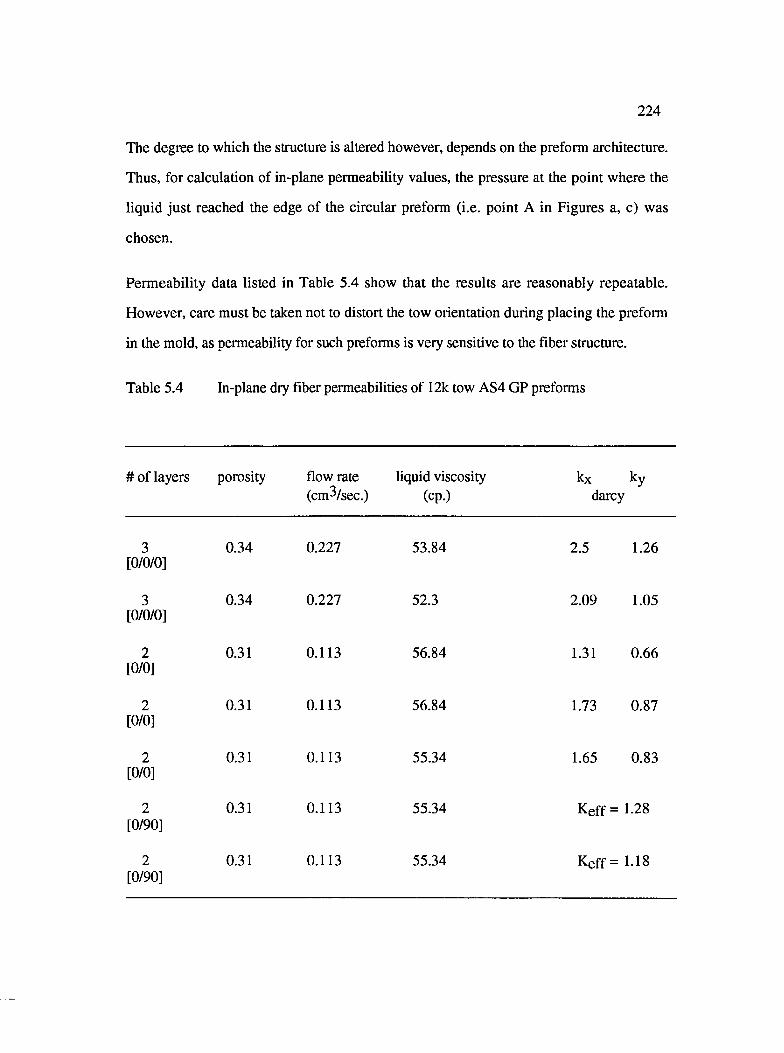
224
The degree to which tlie structure is altered however, depends on the preform architecture.
Thus, for calculation of in-plane permeability values, the pressure at the point where the
liquid just reached the edge of the circular preform (i.e. point A in Figures a, c) was
chosen.
Permeability data listed in Table 5.4 show that the results are reasonably repeatable.
However, care must be taken not to distort the tow orientation during placing the preform
in the mold, as permeability for such preforms is very sensitive to the fiber structure.
Table 5.4 In-plane dry fiber permeabilities of 12k tow AS4 GP preforms
# of layers porosity flow rate (cm^/sec.)
liquid viscosity (cp.)
k x k y
darcy
3[0/0/0]
0.34 0.227 53.84 2.5 1.26
3[0/0/0]
0.34 0.227 52.3 2.09 1.05
2[0/0]
0.31 0.113 56.84 1.31 0.66
2[0/0]
0.31 0.113 56.84 1.73 0.87
2[0/0]
0.31 0.113 55.34 1.65 0.83
2[0/90]
0.31 0.113 55.34 Keff = 1.28
2[0/90]
0.31 0.113 55.34 Keff = 1.18

225
5.3.1.2 Tackified Woven Preforms
As discussed in Chapter IV, preforming experiments showed that increasing the tackifier
concentration resulted in reduced springback. Also, better springback control was achieved
with intralayer surface area coverage or when the tackifier was inside the fiber tows. The
free volume available in the larger gaps between the fiber tows and in the smaller gaps
between the filaments of the fiber tows would decrease with increase in the tackifier
concentration. Thus, the actual free volume for resin flow in a tackified preform would be
lesser than the sample with no tackifier. This could result in a higher flow resistance or
decreased permeability for the tackified sample.
To see if this indeed was true, four different sets of samples were prepared to study the
effect of tackifier concentration and location on permeability of 4HS graphite fiber
preforms. One set comprised of preforms debulked at 94°C and having 3, 5 and 8 wt.% of
BMI tackifier (Set 1). The conditions of the second set were the same as the first set, but
were prepared using the solvent technique (Set 2). The third set of preforms were made
using the same concentrations as the other two, but with PMMA powder and at 287°C (Set
3). The control sample was the one with no tackifier (Set 4).
As in the case of braided preforms, radial flow was used instead of axial flow to measure
in-plane permeabilities of the tackified fiber preforms. To prepare the samples for the
experiment, 16 layers ((j) = .45) of the fiber were first cut into squares, 12.7 cm x 12.7 cm
(1 X w). The squares were then placed on a 10.16 cm diameter die cutter with a steel rule
knife edge [Grandon Manufacturing]. The die cutter and the stack of fibers were put
between two thick wooden plates and placed in a Carver Laboratory hydraulic hand press
to cut the circular fiber samples. A 1.905 cm steel punch was used to cut the center hole in

226the fiber stack. The fiber stack was then debulked using the vacuum debulking procedure
described in Chapter IV.
The pressure drop for the three sets and the control sample are shown in Figure 5.15. The
plot illustrates several interesting features. Firstly, for samples of Set 1, in which the
tackifier remains on the surface, the pressure drop increases with increasing tackifier
concentration. For samples of the other two sets in which the powder goes inside the fiber
tows, there is no significant change in the pressure drop with increasing tackifier
concentration. Secondly, the pressure drop for untackified sample is similar to samples of
Set 3. These results indicate that permeability is affected more by the blockage of the larger
gaps between the fiber tows as compared to the blockage of the smaller gaps within the
tows. Thirdly, the pressure drop values for Set 3 are much higher than that of Set 2, but
lower than samples of Set 1. The much lower pressure drop values obtained for samples
of Set 2 are due to the shrinking of fiber tows (due to the capillary effect of the solvent in
which the fibers were soaked in) causing an increase in the larger gaps between the tows,
and thereby decreasing the flow resistance.
Based on the increase in pressure drop for samples of Set 1, permeability values were
calculated using Darcy’s law, and are shown in Figure 5.16. Table 5.5 shows the percent
decrease in permeability with successive increase in tackifier concentration (or percent
decrease in porosity).
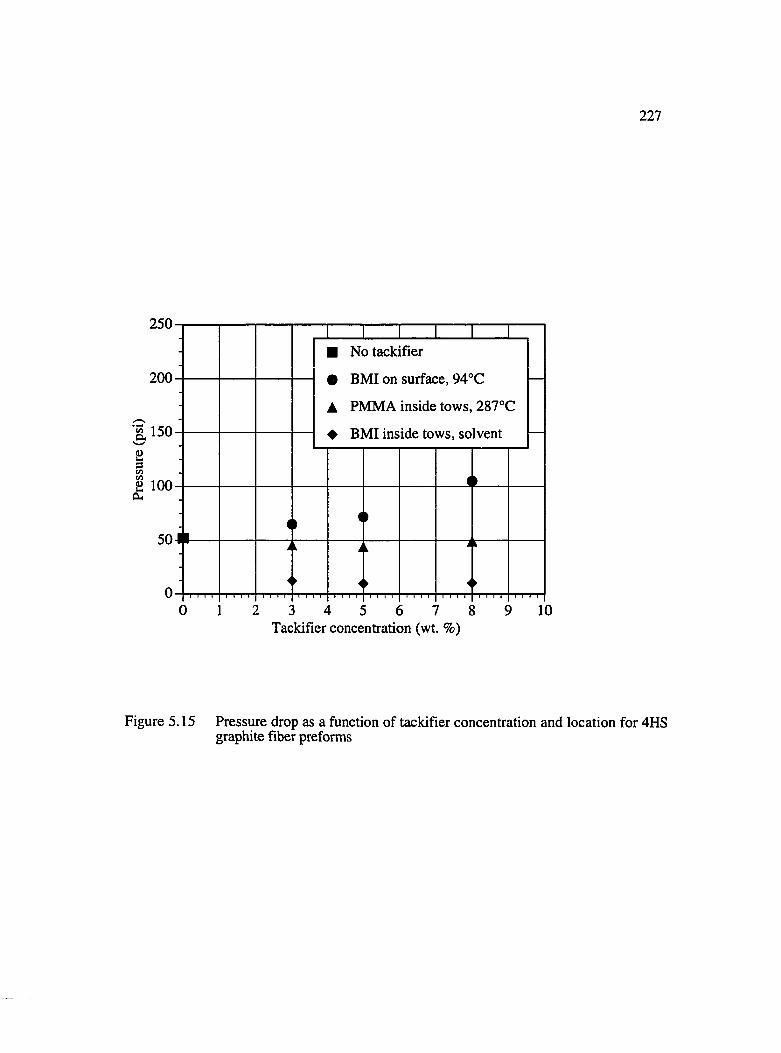
227
250
200
150
I 100.eu
50
0
I I I .. J. I ..I■ No tackifier
• BMI on surface, 94°C
▲ PMMA inside tows, 287°C
♦ BMI inside tows, solvent
5 6 7Tackifier concentration (wt. %)
10
Figure 5.15 Pressure drop as a function of tackifier concentration and location for 4HS graphite fiber preforms
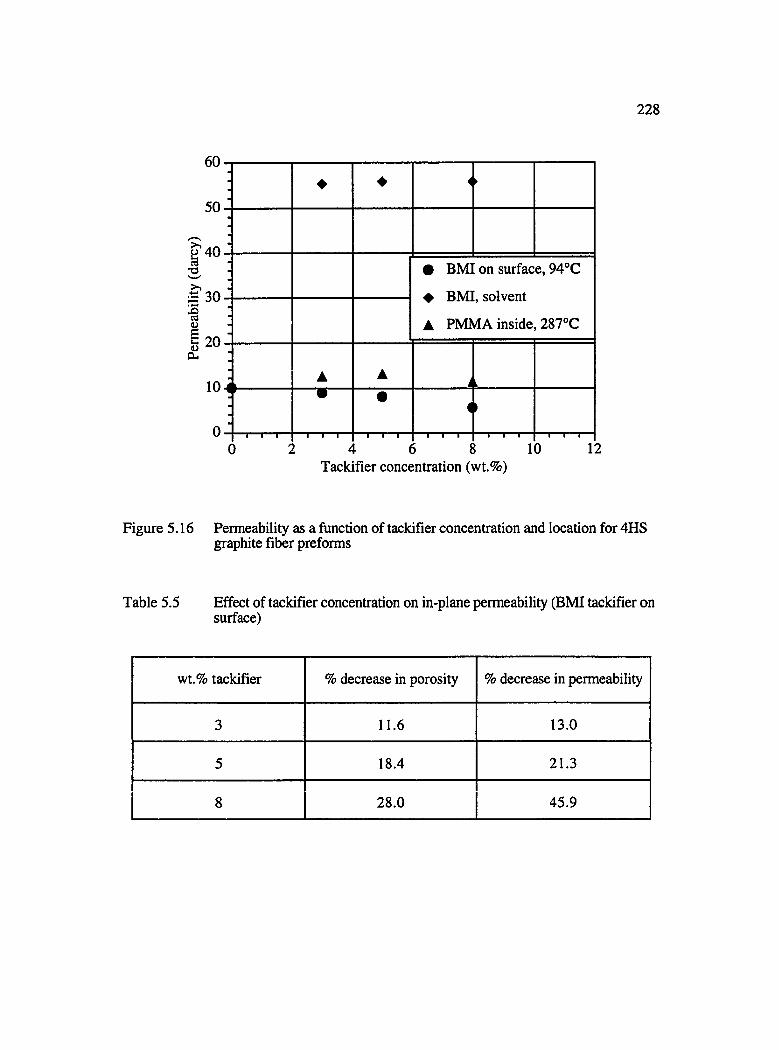
228
60
50
2 40IS 30
1eu
20
10
0
♦ <►
# BMI on surface, 94°C
A PMIklA inside 287°C
▲ A i 1'# •
1
0 :i (5 { 10 1Tackifier concentration (wt.%)
Figure 5.16 Permeability as a function of tackifier concentration and location for 4HS graphite fiber preforms
Table 5.5 Effect of tackifier concentration on in-plane permeability (BMI tackifier on surface)
wt.% tackifier % decrease in porosity % decrease in permeability
3 11.6 13.0
5 18.4 21.3
8 28.0 45.9

229
5 .3.2 Ejfect o f Tackifier on Fiber Wetting in Woven Preforms
5.3.2.1 Wicking Experiments
Dynamic Contact Angle Analyzer (DCA) described in Chapter III was used to study the
effect of tackifier application technique (powder vs. solvent) on wetting characteristics of
tackified preforms. This was done by comparing the amount of liquid retained by the fiber
preform treated by the two different techniques.
In these experiments, first a small piece of the fiber preform was placed on the balance loop
of the DCA. Next, the test liquid contained in a beaker resting on a traveling stage is
brought in contact with the edge of the preform. As the liquid starts to wick in, the force
changes are recorded on a computer. After the force reached the steady state, the fiber mat
was separated from the liquid. The force dropped until a steady state was achieved. Since
the initial weight of the preform was tared, the force readings recorded are the sum of the
Wilhelmy wetting force and the weight of the liquid wicked into the fiber tows. The final
steady state weight indicates the total liquid retention. Figure 5.17 shows the curves for
change in weight with time for the two samples. The segment AB is the baseline prior to
the contact between the fiber and the liquid. The segment BC shows an increase in force
readings. This is mostly due to wetting, with some contribution from liquid uptake. The
segment CD shows a slower change in force until it reaches a steady state. The segment
DE records the separation process of the wetted fabric from the liquid. The slight increase
in the force detection during this process was attributed to the change in the fabric edge
configuration and/or change in the contact angle at the meniscus. At point F, the fabric is
completely separated from the liquid. The residual weight recorded indicates the total liquid
retention (W,) in the fabric.

230
2003% in solvent180-3% powder
160-
140-
120 -
% 100- Ü :^ 8 0 -
6 0 -
4 0 -
20 -
0 500 1000 1500 2000 2500 3000 3500Time (sec.)
Figure 5.17 Comparison of wicking behavior of solvent and powder coated 4HS graphite fiber preforms

231
As shown in Figure 5.17 the liquid retention for the powder technique for 3 wt.% tackified
fiber preforms is greater than for the solvent technique. As liquid uptake due to wicking
occurs solely by capillary forces, the higher liquid retention in case of the powder sample
indicates more free volume in the capillaries for the liquid to wick in as compared to the
solvent sample. This is to be expected since the powder remains on the surface, while the
solvent causes the powder to go inside the fiber tows thereby blocking the interstitial
capillaries between the filaments of the fiber tows.
In order to study the effect of tackifier concentration, and the effect tackifier application
technique on wetting characteristics of fiber tows, a centrifugal device was used to
determine the relationship between capillary pressure, Pc and saturation, S. Since these
experiments involve starting with a completely wet sample, the ?c vs. S curves really
exhibit the drainage characteristics of the fiber samples rather than imbibition which occurs
during mold filling in Liquid Composite Molding. However, it has been observed by
several researchers for a wide variety of porous media, that although the magnitude of
capillary pressure at any saturation may be different for imbibition and drainage, the trend
remains the same. Thus, either curve can be used for comparative purposes. The
measurement technique and results obtained from centrifuge experiments are discussed
next.
5.3.2.2 Measurement o f Capillary pressure V5. saturation
The set up of the centrifuge device is shown in Figure 5.18 [Han, 1994]. It consists of a
metal container 44 cm in diameter and 30 cm in height mounted on a stand. The fiber
samples were placed on an aluminum beam connected to an axle which was rotated by a
multi speed motor. A wide range of angular velocities could be generated using this motor,
and the speed of rotation was measured by a Tachometer - Process Time Indicator. Fiber
232
fiber mat samples
multi-speed motor
gear box
Figure 5.18 Schematic of the centrifuge device

233
samples were kept in an acrylic mold 2.54 cm x 2.54 cm x 0.5 cm (1 x w x t) enclosed in a
plastic box with the front end open. The actual size of the cavity was varied using spacers
depending upon the number of fiber layers and porosity. A porous tissue was placed at the
front end to collect the liquid thrown out by the centrifugal forces during the experiment.
The experimental procedure for a typical run is as follows. The number of fiber layers
required to attain the desired porosity were cut, placed inside the mold and weighed. Test
liquid was then injected into the mold till the fibers were completely saturated. The mold
with the wet fibers was weighed again to determine the weight of the liquid at 100%
saturation. The mold was placed in the plastic box and then mounted on the aluminum
beam. During rotation, the centrifugal force tries to drive the liquid out of the pores, while
the capillary pressure tries to retain it. At equilibrium the two forces are balanced. Thus,
each rotation was carried out for several minutes till equilibrium was reached. After each
rotation at successively increasing speeds, the mold was taken out and weighed to
determine the amount of liquid remaining in the fiber mat. This process was repeated at
several speeds to obtain a relationship between saturation and capillary pressure which
were calculated according to Equations 5.10 and 5.11:
' =
where
m is the mass of the liquid retained at any speed of rotation
p is the density of the lest liquid
t is the cavity thickness
(|) is the porosity
Pc = (5.11)

234where
A p is the difference in the wetting and the non-wetting fluids
oi is the angular velocity
ri and X2 are the inner and outer radii of rotation (Figure 5.19)
The porosity (<])), for the different cases was calculated using Equation 5.12:
<t> = 1
/ ( \mf m,+I P f J I p. J (5.12)(2.54)- X t
where
mf is the mass of the fiber
Pf is the density of carbon fiber (~ 1.8 g/cm^)
mt is the mass of the tackifier
Pt is the density of the tackifier (~ 1.0 g/cm^)
Equation 5.11 is based on the assumption that at any cross-section of the sample normal to
the radius of rotation, the saturation is uniform [Collins, 1961]. Figure 5.20 shows the
capillary pressure vs. saturation curves for various samples. PMMA powder was used
because previous experiments described in Chapter IV showed that it was easier to control
the location of this powder as compared to BMI tackifier. All the curves show the same
trend of non linear increase in capillary pressure with decrease in saturation. At the start of
the experiment, capillary pressure is small because the saturation is high. With increasing
speeds of rotation, the liquid first starts to come out from the larger gaps and then from the
interstitial gaps between the filaments. This is why capillary pressure increases gradually
till a saturation of about 0.8 - 0.75, and then increases rapidly as the saturation decreases
further. So although the general trend is the same, the capillary pressure at any given
saturation is different for each condition under which the samples were made. Capillary
235
Fiber sample
Figure 5.19 Configuration of the fiber sample in the centrifuge device
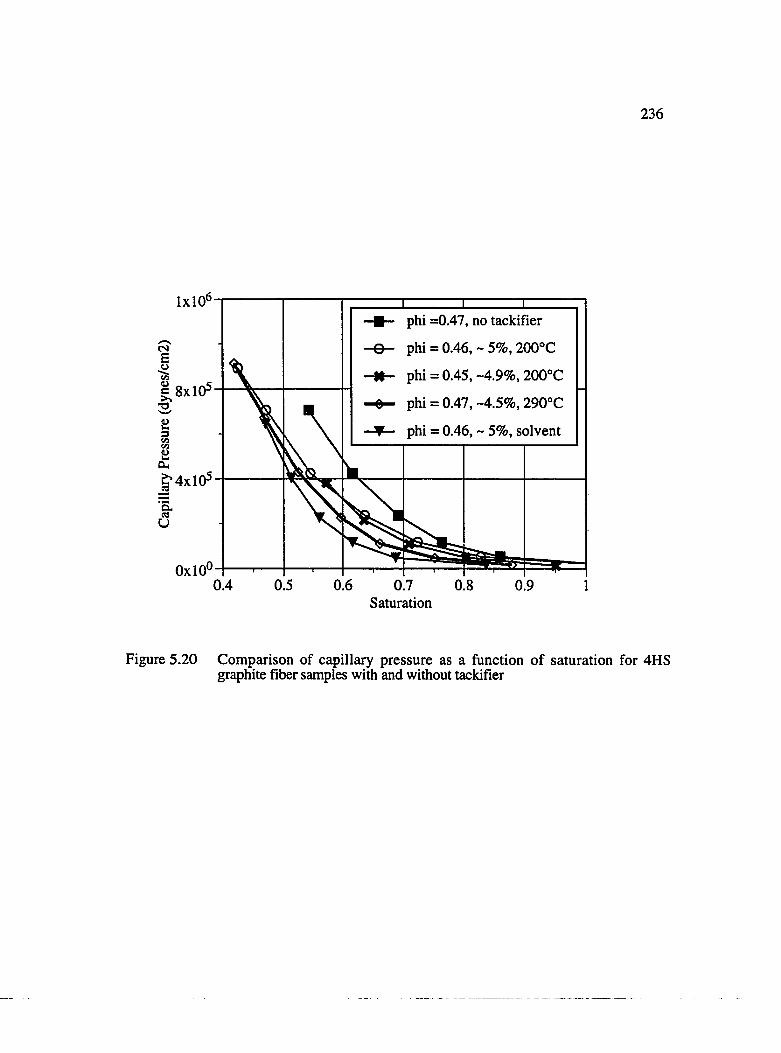
236
phi =0.47, no tackifier
phi = 0.46, - 5%, 200°C
phi = 0.45, -4.9% , 200“C
phi = 0.47, -4.5%, 290°C
phi = 0.46, - 5%, solvent
ë 8x105
\
0.6 0.7Saturation
Figure 5.20 Comparison of capillary pressure as a function o f saturation for 4HS graphite fiber samples with and without tackifier

237
pressure is the highest for the sample without any tackifier, followed by the ones in which
it remains on the surface (i.e. 200°C case), and then in which the tackifier goes inside the
tows (i.e. 290°C and solvent cases). These results indicate that since the capillary pressure
is lower, resin impregnation into fiber tows is hindered by the presence of tackifier. From
results obtained in Chapter III, it can be inferred that since the stability of a void depends
upon the relative magnitudes of hydrodynamic and capillary pressures, mobilization of
voids would be easier when the capillary pressure is lower. However, blockage of the
capillaries by the tackifier particles could provide hindrance to void movement. Thus, the
effect of both tackifier concentration and location on void formation and mobilization
should be further investigated.

CHAPTER VI
CONCLUSIONS AND RECOMMENDATIONS
6.1 Analysis of Flow Induced Voids
Air bubbles or voids are formed during the resin injection step in liquid composite molding
processes because of two simultaneous and competing flows. One is the flow through the
larger gaps between the fiber tows, and the other is the local penetration of the resin in the
fiber tows. A 2D flow visualization technique was therefore developed and successfully
implemented to observe the competing micro flows during liquid injection. From flow
visualization experiments, mechanisms of formation of macro voids and microvoids in
unidirectional stitched fiberglass preforms were proposed in this study. The results
obtained revealed that void formation during mold filling can be correlated to the relevant
processing variables by the dimensionless modified capillary number (Ca#*). Modified
capillary number depends on the liquid properties, injection flow rate, and liquid/fiber
contact angle. Large voids or macrovoids are formed between the fiber tows at low flow
rates corresponding to the capillary numbers < 10"3. Smaller voids or microvoids are
formed within the fiber tows at higher flow rates. Void formation for both axial and
transverse flows were studied. In both cases, two types of microflows are necessary for
macrovoid formation; one is fingering or the lead-lag at the flow front, and the other is the
transverse or cross flow. Mechanisms for the formation of microvoids is however
different for axial and transverse flow. During axial flow, microvoids are formed by a
'round - up' type of mechanism wherein the leading flow fronts loop back to meet the
238

239lagging flow fronts. In case of transverse flow, microvoids were formed by two types of
flow mechanisms, both of which resulted in microvoid formation across a wide range of
capillary numbers. For capillary number < ~10"2, microvoids were formed primarily due
to the lead-lag at the flow front, followed by cross-flow of the liquid leading in the stitches
into the fiber tows by capillary action. At higher capillary numbers (> ~10‘2), microvoids
were trapped mostly due to the macroflow around the fiber tow being completed before the
microflow could displace all the existing air witliin the tows.
For the same capillary number, void content was higher for transverse flow as compared to
the axial flow. Moreover, flow visualization experiments showed that for flow along the
fiber tows, minimal voids of either type were formed when the capillary number ranged
from 0.001 - ~ 0.005. However, no such process window was observed for transverse
flow. This indicates that in addition to the modified capillary number, fiber architecture and
flow pattern can also affect the selection of suitable molding conditions so as to minimize
the overall void content of a molded part. The size of trapped macro voids ranged from
IQ-l - 10 3 cm2, while that of microvoids ranged from 10"6 - lQ-4 cm2. Although,
vacuum assisted liquid injection helped to reduce the formation of both macro and
microvoids, the latter are more difficult to purge during regular mold filling.
Due to the complex nature of the microscale flow pattern, unidirectional stitched fiberglass
mat was chosen because of its simple architecture. However, as stated eai lier in Chapter 1,
fiber reinforcements with different structures and materials are being used in actual
production of composite parts, and is worth to carry out visualization experiments with
other fiber types in order to have a more complete database. Also, since the current
visualization set-up is limited to only one or two layers of the preform, a 3D visualization
and imaging technique should be developed to obtain information about formation of voids
in thicker fiber preforms. One approach for doing so would be to scan through the

240thickness of the preform using a laser source, much like the confocal scanning optical
microscopy.
Dynamic mechanical tests of aged composite samples showed that presence of macro and
micro voids result in reduced stiffness. Voids provide a path for diffusion of moisture
which causes plasticization of the resin matrix and degradation of the matrix/fiber interface.
Also, freeze-thaw cycling of voided composite samples showed significant microcrack
formation. Thus, it becomes imperative to minimize the formation of voids, and this
warrants a proper choice of the molding conditions. Although, the results obtained in this
study showed that voids indeed affect the properties o f the molded composite samples,
further investigation should be carried out to investigate which of the two kinds of voids
(macro or micro) have a more deteriorating effect on mechanical properties like flexural
strength and modulus, interlaminar shear and strength after impact.
6.2 Analysis of Powder Coating on Preforming and Moldability
A 4 HS type of textile reinforcement was also used in this study. The advantages of using
a woven preform includes better drapeability, and elimination of the use of stitches, a
structural inhomogeniety which results in formation of voids. The disadvantage however
is the need for considerable debulking or consolidation due to the high bulk factor. In
order to avoid high clamping pressures (one of the advantages of LCM over other
processes like SMC), a powder coating (tackifier) can be used to control deconsolidation or
springback of compressed fiber preforms. Since the current approach used in the
composite industry is to obtain "net-shaped" preforms by trial and error, which is evidently
very ineffective, the findings of this study will help in improving the understanding of the
issues involved in preforming and molding of tackified fabrics.

241The processing variables that were observed to affect dimensional changes in U-shape
bending and lateral compression of tackified preforms include powder concentration,
application technique (powder vs. solvent), location of application (between the layers vs.
within the fiber tows), and debulking temperature. A two level consolidation theory is
proposed to explain the mechanism of fiber springback, viz., interlayer consolidation or
compression of the gaps between the fiber tows, and intralayer consolidation or
compression of interstitial gaps within the fiber tows. Based on the experimental results, it
can also be hypothesized that fiber springback will occur when the force exerted by the
compressed elastic fibers upon release of an externally applied load, is greater than the
"holding" force provided by the tackifier. The latter depends upon the tackifier modulus
and the wetted surface area of the preform. Increasing the tackifier modulus and/or the area
coverage can therefore help in reducing the amount of springback. Experiments with a
reactive BMI powder and a non-reactive PMMA powder showed that area coverage can be
either interlayer or intralayer, depending upon the rheological properties of the viscoelastic
polymer melL A phenomenological approach was used in this study for springback control
under lateral compression. For a more quantitative analysis, models for increase in the area
coverage during the debulking stage and increase in the resin modulus as a function of
debulking temperature and time should be developed. For BMI resin, modeling of increase
in modulus as a function of conversion becomes quite complex due to two competing
reaction mechanisms, one leading to a linear polymer chain, and the other resulting in a
cross-linked network due to homopolymerization.
For the same tackifier concentration, better springback control is achieved from intralayer
coverage as compared to interlayer coverage. Increasing the degree of cure, and tackifier
concentration also resulted in lower springback. The drawback of advancing the chemical
reaction during the debulking stage comes from the limitations of tackifier solubility in the
incoming fresh resin during mold filling. Incomplete dissolution of the tackifier particles in

242the resin can adversely affect the mechanical properties due to residual microstresses at the
tackifier/resin interface.
Scanning electron micrographs (SEM) showed that during the debulking process, the
tackifier powder melts, coagulates and flows along the capillaries blocking the pore spaces
within the preform. Since mold filling is governed primarily by macro flow, blocking of
the larger gaps by the tackifier powder results in lower permeability or increased flow
resistance. Blocking of the interstitial pore spaces however affects fiber wettability by
lowering the capillary pressure which is a function of saturation. From results obtained in
Chapter III, it can be inferred that since the stability of a trapped void depends upon the
relative magnitudes of the hydrodynamic and capillary pressures, mobilization of
microvoids would be easier when the capillary pressure is lower. However, blockage of
the interstitial gaps could provide hindrance to void movement. Thus, it would be
interesting to further investigate how the tackifier concentration and location affects both the
formation and movement of macro and microvoids.

REFERENCES
K. J. Ahn and J. G. Seferis, Polymer Composites, 12 (3), 146, June (1991).
D. A. Babbington, J. K. Fielder, and D. P. Waszeciak, 33 ^ Annual PolyurethaneTechnical Marketing Conference, 620, Sept./Oct. (1990).
J. Barron, Dow Chemical Company, Freeport, Texas, Private Communication (1995).
G. L. Batch and S. Cuminskey, 4 5 * Annual Conference Composites Institute, The Society of Plastics Industry, Session 9-A (1990).
W. D. Bascom and J. B. Romans, Product and Development, 7 (3), 172 (1968).
E. Bayramli and R. L. Powell, Colloids and Surfaces, 56, 83 (1991).
W. Becker, SME Composites in Manufacturing, 9 (1990).
F. Brochard, Journal of Chem. Phys., 84 (8), 15 (1986)
F. W. Billmeyer, Textbook o f Polymer Science, 3rd ed., John Wiley and Sons, New York (1984).
L. R. L. Bonhomme, "Tack and Conformance o f Prepreg Tapes during Automatic Tape Layup ", M.S. Thesis, MIT, June (1986).
K. J. Bowles and S. Frimpong, Journal of Composite Materials, 26 (10), 1487 (1992).
F. Bueche, Physical properties o f polymers , Interscience publishers. New York (1962).
T. F. Butryn, 36th International SAMPE Symposium, 546 (1991).
L. Carino and H. Mollet, Powder Technology, 11, 189 (1975).
C. C. Chamis, NASA Technical Note D5367 (1969).
A. W. Chan and R. J. Morgan, SAMPE Quarterly, 23,48, April (1992).
A. W. Chan and R. J. Morgan, Proc. of 51®t SPE ANTEC, 844 (1993a).
243

244A. W. Chan and R. J. Morgan, SAMPE Quarterly, 24 (4), 49, July (1993b).
N. G. Chavka and C. F. Johnson, Proc. of a SM/ESD Adv. Composites Conference, 115 (1991).
Y. T. Chen, "Resin Transfer Molding o f Polycyanate: Chemorheology, Molding Experiment and Wetting Visualization", Ph. D., Dissertation, Department of Chemical Engineering, University of Minnesota (1993).
Y. T. Chen, H. T. Davis and C. W. Macosko, AIChE J., 41, 2261 (1995).
S. Chwastiak, Journal of Colloid and Interface Science, 42 (2), 298, February (1973).
S. J. Claus and A. C. Loos, Proc. of the American Society for Composites (1989).
R. Cochran and R. B. Pipes, 23rd International SAMPE Technical Conference, 1169(1991).
R. E. Collins, Flow o f Fluids through Porous Materials , Van Nostrand, Reinhold Publishing Corporation, New York (1961).
A. T. Corey, Mechanics o f Immiscible fluids in porous media. Water Resources Publishers (1986).
W. P. Cox and E. H. Merz Jr., Journal of Polymer Science, 28, 619 (1958).
S. G. Damani and L. J. Lee, Polymer Composites, 11 (3), 174 (1990).
R. Dave and S. Houle, Proc. of the American Society for Composites, 5 ^h Technical Conference, 539, June (1990).
J. Domingue, "The Phenomena o f Contact Angle Hysteresis", Cahn's Manual,(1992).
S. Dong and R. Gauvin, Polymer Composites, 14 (5), 414, October (1993).
F. A. L. Dullien, Porous media, flu id transport and pore structure, 2 ed.. Academic Press Inc., (1992).
J. J. Elmendorp and F. During, Proc. of 4 9 *h SPE ANTEC, 1361 (1991).
T. C. Fan, SAMPE Quarterly, 29 (4), 15, July/Aug. (1993).
S. M. Feldgoise, F. Foley, D. Martin and J. Bohan, 23^^ Intn'l SAMPE Technical Conference, 259, October (1991).
R. Gauvin and M. Chibani, 43^^ Annual Conference Composites Institute, The Society of Plastics Industry, Session 22-C (1988).

245
S. R. Ghiorse, SAMPE Quarterly, 24 (2), pp. 54, January (1993).
A. G. Gibson and J. A. Manson, Composites Manufacturing 3 (4), 223 (1992).
L. B. Greszczuk, Annual Conference Composites Institute, The Society of Plastics Industry, 22 (1967)
F. J. Guild and J. Summerscales, Composites, 24 (5), 383 (1993).
T. G. Gutowski, SAMPE Quarterly, 16 (4), 58 (1985).
T. G. Gutowski and L. Bonhomme, Journal of Composite Materials, 22, 204 (1988).
K. Han, Ph. D. Dissertation, The Ohio State University, Columbus, Ohio (1994).
M. J. Hannon, D. Rhum and K. F. Wissbrun, Journal of Coatings Technology, 48 (621), 42(1984).
R. S. Hansen, SME Technical Papers EM 90-214 (1990).
J. S. Hayward and B. Harris, Composites Manufacturing, 1 (3), 161, Sept. (1990).
High Performance Composites, 6, Nov ./Dec. (1993).
D. E. Hirt, J. M. Marchello and R. M. Baucom, 22nd International SAMPE Technical Conference, 360 (1990).
K. T. Hodgson and J. C. Berg, Journal of Colloid and Interface Science, 121 (1), 22 Jan. (1988).
Y. L. Hsieh and B. Yu, Textile Research Journal, 62 (11), 677 (1992).
D. Hsu, Proc. of the 46th sPE ANTEC, 1273 (1988).
M. K. Hugh, J. M. Marchello, R. M. Baucom and N. J. Johnston, 25th International SAPilPE Technical Conference, 812 (1993).
de. G. Jager, "Method and apparatus for applying powdered materials to filaments", WO 87/00563 (1987).
C. F. Johnson, Engineered Materials Handbook, 1, ASM, 564 (1987).
C. F. Johnson, Composite Materials Technology - Processes and properties , Ch 5, P. K. Mallick and S. Newman, Ed., Hanser Publishers, New York (1990).
N. C. W. Judd and W. W. Wright, SAMPE Journal, 14 (1), 10, Jan./Feb. (1978).
Y. K. Kamath, C. J. Dansizer, S. Hornsby and H. D. Weigmann, Textile Research Journal, 205, April (1987).

246J. L. Kittleson and S. C. Hackett, 39th International SAMPE Symposium, 83
(1994).
B. J. Kohn, A. G. Sands and R. C. Clark, Product and Development, 7 (3), 178 (1968).
Peter G. de Lange, Journal of Coatings Technology, 56 (717), 23 (1984).
B. K. Larson and L T. Drzal, Proc. of 50^^ SPE ANTEC, 752 (1992).
L. J. Lee, Fundamentals of reaction injection molding, Ch 4, 4 (2), Comprehensive Polymer Science, Pergamom Press (1989).
W. J. Lee and J. C. Seferis, Polymer Composites, 9 (1), 36 Feb. (1988).
D. Li, and A. W. Neumann, Journal of Colloid and Interface Science, 148 (1), 190, Jan. (1992).
T. S. Lundstrom, B. R. Gebart and C. Y. Lundemo, 4 7 ^ Annual Conference Composites Institute, The Society of Plastics Industry, Session 16-F, 1(1992).
J. Madge, General Electric Aircraft Engines Division, Cincinnati, OH, Private Communication (1994).
A. D. Mahale, R. K. Prud'homme and L. Rebenfeld, Polymer Engineering andScience, 32 (5), 319 (1992).
Cahn's Instruction Manual, Cahn Instruments, Inc., Cerritos, CA (1993).
J. M. Margolis, Proc. of 43*' Annual SPI Composites Institute Conf., Session 9-E (1988).
G. Maymon, R. P. Briley and L. W. Rehfield, Advanced Composite Materials - Environmental Effects , ASTM STP 658, 221 (1978).
V. P. McConnell, High Performance Composites, 22, Nov./Dec. (1993).
MDSC 2910, Modulated Differential Scanning Calorimeter Operator's Manual, TA Instruments, New Castle, Delaware (1993).
J. Meissonnier, "Processing Optimization o f Bismaleimide Matrix Composites", M.S. Thesis, University of Washington, January (1988).
B. L. Metcalfe and R. J. Wilson, Journal of Material Science Letters, 4, 235(1985).
S. Middleman, The Flow of High Polymers , Chapter 5, Interscience, N. Y. (1968)
B. Miller, Surface Characteristics o f Fibers and Textiles, Part II, Ch. 11,417, M. J. Schick, Ed., Marcel Dekker (1977).

247C. W. Mocosko, RIM : Fundamentals o f reaction injection molding, Hanser
Publisher (1989).
Modem Plastics, 24, January (1994).
R. Monks, Plastic Technology, 40, March (1993).
K. H. Moritz, The Composites Industry - An Overview (1993).
I. Muzzi, B. Varrughese and P. H. Yang, SPE ANTEC Proceedings, 48, (1990).
J. V. Noyés and B. H. Jones, AIAA, 68 (1968).
Y. Ohmura, Takao Uematsu, Tohru Imanara, Shoichi Sato, Takayoshi Imai, and Naomi Yamashita, Proc. of the 9^^ ASM/ESD Advanced Composite Conference, 69 (1993).
J. R. Olsen, D. E. Day and J. G. Stoffer, Journal of Composite Materials, 26 (8), 1181 (1992).
R. S. Pamas and F. J. Phelan Jr., SAMPE Quarterly, 53, Jan. (1991).
N. Patel, V. Rohatgi and L. J. Lee, Proc. of 49^^ Annual SPI Composites Institute Conf., Session 10-D (1994).
M. J. Perry, Ph. D. Dissertation, The Ohio State University, Columbus, Ohio(1993).
R. C. Peterson and R. E. Robertson, Proc. of the 7*^ ASM/ESD Advanced Composite Conference, 203 (1992).
R. C. Peterson and R. E. Robertson, Proc. of the 8^h ASM/ESD Advanced Composite Conference, 63 (1992).
E. P. Plueddemann, Ed., Composite materials: interfaces in Polymer Matrix Composites , vol. 6, Academic Press (1974).
R. V. Price, "Production of impregnated roving", US Patent 3742105 (1973).
Rheometrics Dynamic Analyzer, RDA II Owners Manual, Rheometrics Inc., Piscataway, NJ (1990).
V. Rohatgi, "Influence o f Material and Processing Variables on Resin - Fiber Interface in Glass Fiber Reinforced Polymeric Composites", M.S. Thesis, Department of Chemical Engineering, The Ohio State University, (1991).
V. Rohatgi, N. Patel and L. J. Lee, ERC Report ERC/NSM-P-94-19, May (1994).
A. J. Salem and R. S. Pamas, Proc. American Soc. Composites, 1012 (1991).

248M. M. Schwartz, Composite Materials Handbook, Ch 4, McGraw Hill Publication
(1984).
J. C. Seferis and J. Meissonnier, SAMPE Quarterly, 20 (3), 55 (1989).
A. Shafi, Dow Chemical Company, Freeport, Texas, Private Communication,(1994).
D. J. Shaw, Introduction to Colloid and Surface Chemistry, 3 ^ éd., Butterworths,(1980).
K. Shields and J. Colton, Polymer Composites, 14 (4), 341, Aug. (1993).
L. Skartsis, B. Khomami and J. L. Kardos, SAMPE Journal, 28 (5), 19, Sept./Oct. (1992).
R. Srinivasan, T. J. Wang and L. J. Lee, SPE ANTEC Proceedings, 53, (1995).
W. R. Stabler, G. B. Tatterson, R. L. Sadler and A. H. M. El-Sheikh, SAMPE Quarterly, 23 (2), 38 (1992).
E. B. Stark and W. V. Beitigam, Engineered Materials Handbook, 1, ASM, 168(1987).
D. Stover, High Performance Composites, 38, Nov. / Dec. (1993a).
D. Stover, High Performance Composites, 54, Sept./Oct. (1993b).
A. B. Strong, Fundamentals o f Composite Manufacturing: Materials, Methods and Applications, Society of Manufacturing Engineers, (1989).
J. L. Thomason and A. Knoester, Journal of Material Science Letters, 9, 258 (1990).
J. L. Throne and M.S. Soh, 35th International SAMPE Symposium, 2086 (1990).
A. V. Tungare and G. C. Martin, Journal of Applied Polymer Science, 46, 1125(1992).
A. V. Tungare and G. C. Martin, Polymer Engineering and Science, 33 (10), 614(1993).
A. M. Vodermayer, J. C. Kaerger and G. Hinrichsen, Composites Manufacturing, 4 (3), 123 (1993).
J. F. Voit and J. C. Seferis, Journal of Applied Polymer Science, 34, 1459 (1987).
T. J. Wang, C. H. Wu and L. J. Lee, Polymer Composites, 15 (4), 278 (1994).
V. Wigotsky, Plastics Engineering, 23, Sept. (1989).

249
E. G. Wolff, SAMPE Journal, 29 (3), 11, May/June (1993).
A.S. Wood, Modem Plastics, 50, October (1990).
A.S. Wood, Modem Plastics, 46, October (1988).
C. H. Wu, Ph. D. Dissertation, The Ohio State University, Columbus, Ohio (1995).

APPENDIX A
O PERATIO N A L PROCEDURE FOR IN-PLANE PERM EA BILITY
M EASUREM EN TS
250

251
A .l In-Plane Permeability Measurements
The following sequence of steps describe the procedure to be followed during a permeability experiment (see Figure B1 for details):
1) Degass completely the liquid to be used to remove any air bubbles or trapped volatiles. Turn on the INSTRON at least 30 minutes prior to starting the experiment. This is the recommended warm-up period.
2) Close valves V2 and V3, and open valves VI and V4.
3) Turn on the oil pump and let it circulate through the hydraulic system for 2 minutes.
4) After about two minutes of circulation, turn off the pump. Close valve VI and move the piston rod to the lowest position in order to drive out all the air bubbles in the hydraulic cylinder.
5) Repeat steps 2-4 several times until no air bubbles are observed.
6) With the piston in the lowest position, close valve V4.
7) Open valve V5 and turn on the vacuum pump. Then close valve V5 andopen valve V3 slowly to its maximum position. Hold the vacuum level at more than 28" for about 1 minute to purge all the air from the connecting tubings.
8) Close valve V3, open valve V5 and turn off the vacuum pump.
9) Set the upper and lower crosshead limit stops at appropriate positions.
10) Move the crosshead to the upper position.

252
11) Open valve VI and the oil pump. After the hydraulic cylinder is full of oil and the piston does not rise any further, turn off the oil pump. Close valveVI.
12) Move the crosshead down till it almost touches the piston rod.
13) Open valve V2 and let the oil flow through the inlet by moving the crosshead against the piston of the cylinder. This is done to purge any remaining air in the system. When no air bubbles are observed, stop the oil flow and wipe off the mold.
14) Set the desired velocity of the crosshead on the INS TRON panel.
15) Place the fiber sample into the mold. Close the mold and then tighten the bolts evenly so that the thickness of the mold is the same as that without the fiber sample. A vernier caliper can be used to check the thickness before and after loading the fiber sample. It is time now to start the experiment.
16) Watch closely the increase in pressure as the experiment progresses. If the pressure readings get close to the upper limit of pressure transducer range, change the velocity of the crosshead to a lower value to reduce the pressure drop, or stop the experiment by pressing the STOP button on the INSTRON panel.
17) Repeat liquid injections if "wet-fiber" permeability values are required.

APPENDIX B
FLEX URAL PRO PERTIES O F TA CK IFIED (1 wt. % ) BM I RESIN
253
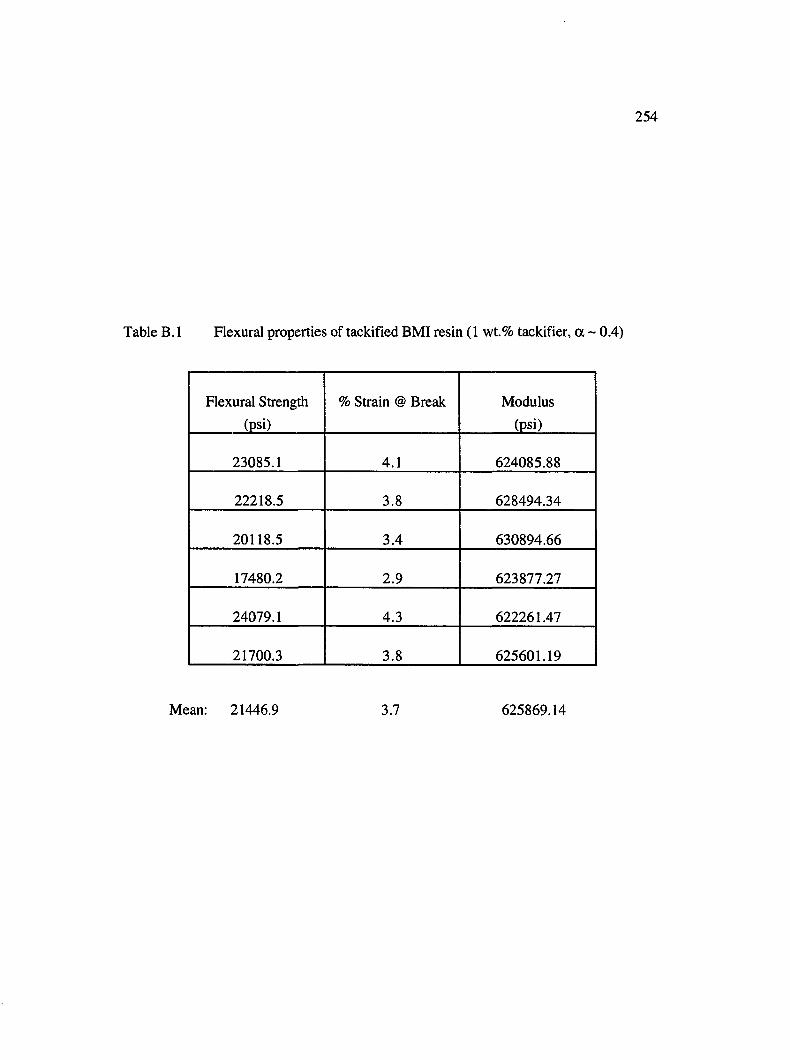
254
Table B. 1 Flexural properties of tackified BMI resin (1 wt.% tackifier, a ~ 0.4)
Flexural Strength (psi)
% Strain @ Break Modulus(psi)
23085.1 4.1 624085.88
22218.5 3.8 628494.34
20118.5 3.4 630894.66
17480.2 2.9 623877.27
24079.1 4.3 622261.47
21700.3 3.8 625601.19
Mean: 21446.9 3.7 625869.14
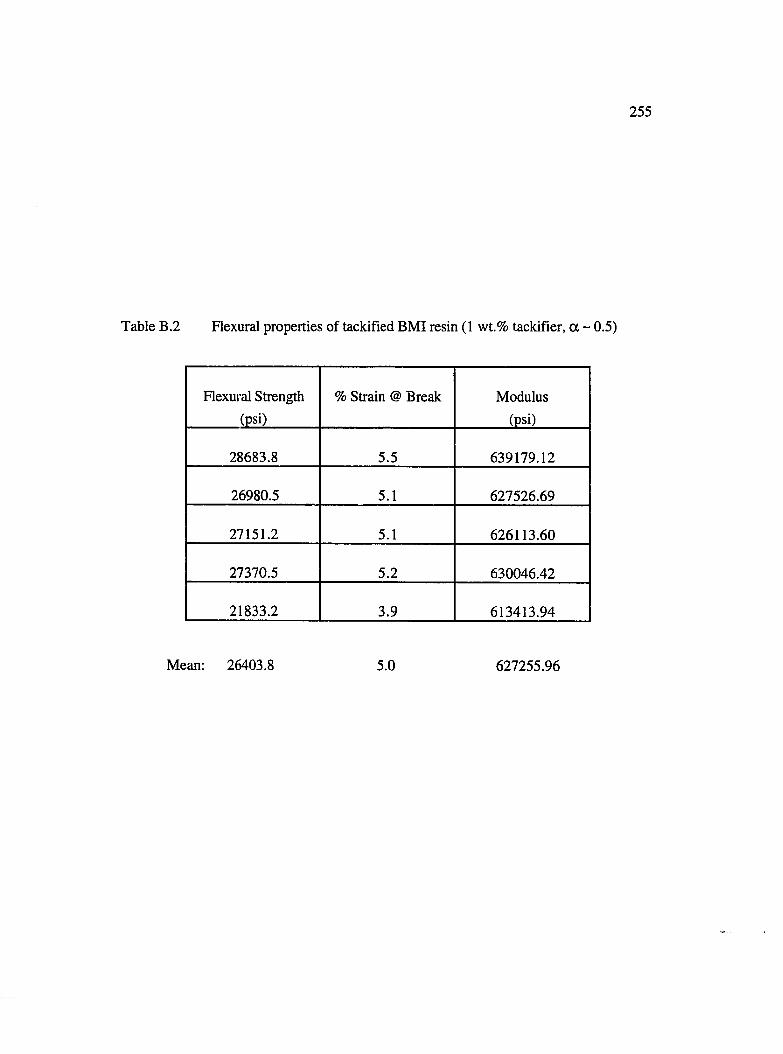
255
Table B.2 Flexural properties of tackified BMI resin (1 wt.% tackifier, a ~ 0.5)
Flexural Strength (psi)
% Strain @ Break Modulus(psi)
28683.8 5.5 639179.12
26980.5 5.1 627526.69
27151.2 5.1 626113.60
27370.5 5.2 630046.42
21833.2 3.9 613413.94
Mean: 26403.8 5.0 627255.96
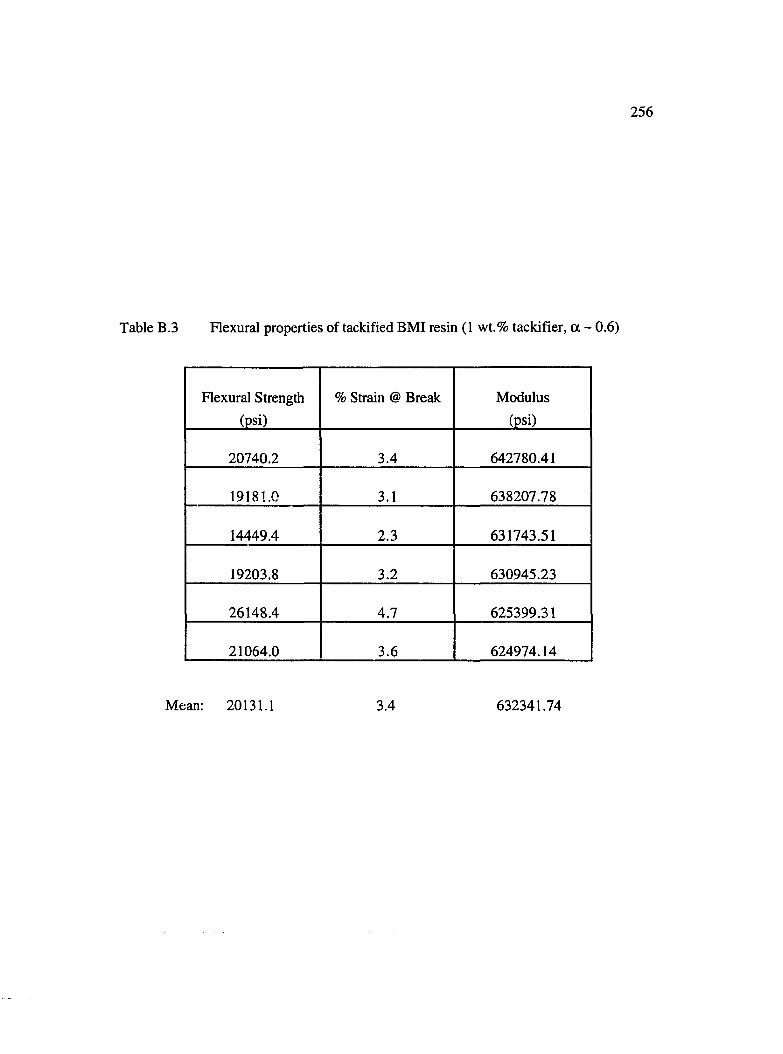
256
Table B.3 Flexural properties of tackified BMI resin (1 wt.% tackifier, a ~ 0.6)
Flexural Strength (psi)
% Strain @ Break Modulus
(psi)
20740.2 3.4 642780.41
19181.0 3.1 638207.78
14449.4 2.3 631743.51
19203.8 3.2 630945.23
26148.4 4.7 625399.31
21064.0 3.6 624974.14
Mean: 20131.1 3.4 632341.74
Top Related
Copyright © 2022 FDOKUMEN

