






Bahasa
Halaman
Hukum


American Journal of Physiology H00986-2002R2 Final Accepted Version
Delayed Arteriolar Relaxation After Prolonged Agonist Exposure: Functional
Remodeling Involving Tyrosine Phosphorylation.
Michael A. Hill1, Simon J. Potocnik1, Luis A. Martinez-Lemus2,
and Gerald A. Meininger2
1Microvascular Biology Group, School of Medical Sciences, RMIT University, Bundoora,
Victoria 3083, Australia and 2Department of Medical Physiology, College of Medicine,
Texas A&M University, College Station, Texas 77843, USA.
Running Title: Functional Remodeling in Arterioles
Address for Correspondence:
Michael A. Hill, PhD.
Microvascular Biology Group
School of Medical Science
RMIT University
Bundoora, Victoria 3083
AUSTRALIA
Ph: 61-3-9925-7754
FAX: 61-3-9925-7628
e-mail: [email protected]
Copyright (c) 2003 by the American Physiological Society.
Articles in PresS. Am J Physiol Heart Circ Physiol (April 24, 2003). 10.1152/ajpheart.00986.2002

American Journal of Physiology H00986-2002R1 24/04/2003
2
Summary:
While arteriolar contraction is dependent on Ca2+- induced myosin phosphorylation other
mechanisms including Ca2+ sensitization and time-dependent phenomena such as cytoskeletal
and cellular reorganization may contribute to contractile events. We hypothesized that if
arteriolar smooth muscle exhibits time-dependent behavior that this may be manifested in
differences in relaxation following short and long-term exposure to contractile agonists.
Studies were conducted in isolated arterioles, pressurized to 70 mmHg. In initial experiments
(n = 10), rate of relaxation was measured following acute (5 min) or prolonged (4 hr)
exposure to 5µM norepinephrine (NE). Prolonged exposure to NE resulted in a significantly
(P < 0.05) increased time for relaxation in physiological salt solution. Rapid relaxation of
vessels exposed to NE for 4 hrs was observed following superfusion with 0 mM Ca2+ buffer
indicating that the alteration in relaxation was reversible and Ca2+ dependent. A similarly
impaired dilation was not observed with 4 hr exposure to KCl (75 mM). To determine
mechanisms contributing to the effects of prolonged NE exposure studies were performed in
the presence of the microtubule depolymerizing agent demecolcine (10µM) or a series of
tyrosine phosphorylation inhibitors. While demecolcine caused significant vasoconstriction
(P < 0.05) and potentiated NE vasoconstriction it did not prevent the effect of long-term NE
exposure on relaxation. Genistein, while having no effect on acute NE-induced contraction,
concentration-dependently inhibited prolonged NE constriction. Similarly, the Src (PP1) and
p42/44 MAP kinase (PD98059) inhibitors prevented maintenance of long-term NE
contraction. The data indicate that prolonged exposure to NE induces biochemical alterations
that impair relaxation following removal of the agonist. The contractile effects are Ca2+
dependent, involve tyrosine phosphorylation, but do not appear to involve the polymerization
state of the microtubule network.

American Journal of Physiology H00986-2002R1 24/04/2003
3
Key Words: arterioles; remodeling; myogenic; agonist; Ca2+; cytoskeleton, tyrosine
phosphorylation.

American Journal of Physiology H00986-2002R1 24/04/2003
4
Introduction
Contraction of arterioles in response to both agonists and increased intravascular
pressure has been shown to be largely dependent on the classical smooth muscle biochemical
pathway involving Ca2+-calmodulin induced myosin regulatory light chain phosphorylation
(for reviews see references 10, 16, 18 and 36). The requirement for these pathways has been
directly confirmed in studies of isolated arterioles in combination with fluorescence
measurements of intracellular Ca2+ and electrophoretic estimations of protein phosphorylation
(26, 33, 48, 49). However, it is now appreciated that other mechanisms including Ca2+
sensitization and time-dependent phenomena such as cytoskeletal and cellular reorganization
may contribute to contractile responsiveness (7, 15, 30, 38, 40, 41). These mechanisms may
support the Ca2+-myosin light chain phosphorylation pathway and, further, provide a
mechanism for maintained tension development at comparatively low energy cost. The latter
suggestion appears intuitively relevant to arterioles given that these vessels typically
demonstrate a maintained state of partial contraction or tone – a phenomenon important for
the establishment of vascular resistance and necessary for the function of vasodilators.
Historically, many studies examining pathways activated by contractile agonists have
been limited to relatively acute exposures to these agents. Whereas, studies aimed at
delineating events related to compensatory growth responses, such as remodeling in
hypertension, have often considered long-term responses to trophic stimuli including
catecholamines and angiotensin II (4, 5). Further, growth-related studies have been often
performed on cultured cells which typically exhibit phenotypic changes compared to cells in
the in vivo state. It is likely, however, that rather than distinct mechanisms there exists a
continuum of responses influenced by both the duration of exposure to the contractile
stimulus and possibly the contractile response itself. It has, therefore, been suggested that the
functional properties of smooth muscle are themselves altered by events such as

American Journal of Physiology H00986-2002R1 24/04/2003
5
reorganization of the cytoskeleton as the tissue undergoes adaptation to environmental factors
(15). As an extension of this we hypothesized that vascular remodeling may begin at a much
earlier time frame and be initially characterized by alterations in cytoskeletal properties
and/or the physical relationship between adjacent smooth muscle cells. This latter alteration
being considered a property of cell-cell and cell-matrix interactions. Support for the
occurrence of such rearrangements is provided by studies of cultured vascular cells exposed
to agonist and mechanical stimuli (8, 34).
With respect to the above general hypothesis it would be expected that if arteriolar
smooth muscle exhibits time frame-dependent changes in cytoskeletal or cellular arrangement
that this may be manifested in differences in the rate and extent of relaxation when
comparing vascular responses to short and long term exposure to a contractile agonist.
Therefore the aim of the present studies was to compare acute (5 min) and prolonged (4 hr)
exposures to contractile stimuli and to examine the involvement of candidate signaling
pathways.

American Journal of Physiology H00986-2002R1 24/04/2003
6
Materials and Methods:
Animal:
The studies used 59 male Sprague-Dawley rats weighing between 200 and 350g.
Before experiments, rats were housed in pairs in a dedicated animal facility with a 12:12 hr
light-dark cycle. During this period rats were allowed free access to a standard rat chow and
drinking water. All procedures were approved by the Animal Ethics and Experimentation
Committee at RMIT University.
Isolated Arteriole Preparation :
Rats were anesthetized with sodium pentothal (100mg/kg intraperitoneal) after which
the right and/or left cremaster muscle was exteriorized, excised from the animal and placed in
a cooled (4oC) chamber containing dissection buffer (3mM 3-N-morpholino propanesulfonic
acid (MOPS); 145mM NaCl; 5mM KCl; 2.5 mM CaCl2; 1mM MgSO4; 1mM NaH2PO4;
0.02 mM EDTA; 2mM pyruvate; 5mM glucose and 1% albumin) (12). Segments (1.2 – 2.2
mm in length) of the main intramuscular arteriole (1A) were dissected from the muscle as
previously described (26, 48). Vessel segments were then cannulated with glass
micropipettes, secured using 10-0 monofilament suture and mounted in a custom-designed
tissue chamber (volume 5 ml). The vessel preparations were then positioned on the stage of
an inverted microscope. The cannulated arterioles were continually superfused (0.5 - 4
ml/min.) with a physiologic buffer solution, containing, 111mM NaCl, 25.7mM NaHCO3,
4.9mM KCl, 2.5mM CaCl2, 1.2mM MgSO4, 1.2mM KH2PO4, 11.5mM glucose and 10mM 2-
N-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES). Vessel segments were
gradually pressurized to 70 mmHg and warmed to 34oC during a 60 minute equilibration
period. To be suitable for use in studies vessels had to be free of pressure leaks and develop

American Journal of Physiology H00986-2002R1 24/04/2003
7
spontaneous basal tone as evidenced by an active diameter between 40 and 65% of passive
diameter at 70 mmHg.
Measurement of vessel diameter (in µm) was performed on-line using an electronic
video caliper. Changes in vessel diameter following exposure to a given treatment were
typically normalized to passive (maximally dilated with 0 mM Ca2+ buffer containing 2 mM
EGTA) diameter at 70 mmHg to account for slight variation in absolute vessel diameters
observed between preparations.
In experiments requiring measurement of changes in intracellular Ca2+, vessels were
incubated (60 min, room temperature) with 2µM fura 2-acetoxy methylester (Molecular
Probes, Eugene, OR) in buffer containing 0.5% DMSO and 0.01% pluronic. The abluminal
surface of the vessel segment was exposed to the fura 2-AM solution to restrict dye loading
to the vascular smooth muscle layer (Meininger et al., 1991). The dye loading procedure was
followed by a 30 min washout period with the physiologic salt solution. Fura 2 loaded
vessels were exposed to epi-illumination (75W Xenon source) with light of alternating
excitation wavelengths (340 and 380 nm) using a motor-controlled filter wheel (20 Hz).
Fluorescence emission was transferred from the microscope to a photomultiplier tube
(Hamematsu, Bridgewater, NJ) coupled to a signal processor. (Texas A&M University,
College Station, TX). Simultaneous trans-illumination with wavelengths greater than 610 nm
provided a non-fluorescent image that enabled measurement of internal arteriolar diameter
while fluorescence data were collected. This procedure did not interfere with measurements
of Ca2+-related fluorescence. Fluorescence emission intensities were expressed as the
340/380 nm ratio to allow quantitative estimates of changes in arteriolar wall intracellular
Ca2+. Details of theses procedures have been presented in previous publications (17, 26, 48).

American Journal of Physiology H00986-2002R1 24/04/2003
8
Experimental Protocols:
Acute and Prolonged Exposure to Norepinephrine
After establishing baseline diameter, arterioles were exposed to norepinephrine (NE,
5µM) until a maximal contraction was achieved (approximately 5 min). The superfusate was
then rapidly changed and vessel diameter and time recorded as the NE was washed out.
Following return to basal diameter, or after 15 minutes superfusion with fresh physiological
salt solution (chosen as a predetermined surrogate endpoint), vessels were subsequently
exposed to adenosine (3 x 10-4M) and then 0 mM Ca2+ buffer containing 2 mM EGTA to
obtain a measurement of maximal passive diameter. Vessels were then superfused with Ca2+
containing buffer for 20 min to re-establish basal conditions. The exposure to NE was then
repeated with the exception that the contraction was maintained for 4 hr before washout as
described above. Dilator responses to adenosine and 0 mM Ca2+/2mM EGTA buffer were
similarly repeated.
Time control studies were conducted to determine whether vessels responded
similarly to acute NE exposures performed 4 hrs apart. A further set of control experiments
were conducted to determine whether the initial acute exposure to NE exerted a
preconditioning effect. In these experiments, relaxation responses to adenosine (10µM) were
compared prior to exposure to NE (that is in the presence of spontaneous myogenic tone
alone at 70 mmHg intraluminal pressure) and to a similar adenosine exposure following 4hrs
of NE (5µM) exposure. NE was washed out, as for the above experiments, prior to addition
of adenosine.
In a separate set of vessels (n = 7) loaded with Fura 2 (2 µM) the above protocol was
repeated to examine Ca2+ responses during the acute and prolonged NE exposures.
Experiments were performed in both the presence and absence of Fura 2 loading to determine
any possible buffering effects due to dye loading.

American Journal of Physiology H00986-2002R1 24/04/2003
9
Short and Long-term Exposure to KCl
To examine the effect of contraction per se the above studies were repeated in a
separate set of arterioles (n = 5) substituting KCl (75 mM) as the contractile agent. Short
and prolonged exposures to KCl were performed as described above.
Effect of Microtubule Depolymerization on Arteriolar Responses to Acute and Prolonged
Exposure to Norepinephrine
Previous studies in our laboratories had shown modulation of arteriolar
contractile responses to NE by the polymerization state of the microtubular network (37).
Therefore to examine the possible involvement of the microtubular network in the differences
in contractile responses to acute and prolonged NE exposure, experiments were performed in
the absence and presence of the depolymerizing agent demecolcine (10 µM). The response
of arterioles to acute NE (5 µM) exposure was studied in the absence of the agent after which
vessels were exposed to demecolcine for 60 min and the acute NE response repeated. The
prolonged response (4 hr) to NE was then determined in the continued presence of
demecolcine. Conditions for these experiments were established in previous studies (37, 38).
Effect of Tyrosine Phosphorylation Inhibition on Arteriolar Responses to Acute and
Prolonged Exposure to Norepinephrine
To examine whether tyrosine phosphorylation-dependent events contribute to the
differences in contractile responses to acute and prolonged NE exposure, experiments were
performed in the absence and presence of genistein (3, 10 and 30 µM). The response of
arterioles to acute NE (5 µM) exposure was studied in the absence of the inhibitor after which
vessels were exposed to genistein for 20 min and the acute NE response repeated. The

American Journal of Physiology H00986-2002R1 24/04/2003
10
prolonged response (4 hr) to NE was then determined in the continued presence of the
tyrosine kinase inhibitor. Only one concentration of genistein was examined in any given
arteriolar segment.
In addition to the studies of the non-specific tyrosine kinase inhibitor (genistein)
studies were conducted in the presence of the Src inhibitor PP1 (10 µM) and the MEK
inhibitor PD98059 (50 µM) to prevent activation of P42/44 MAP kinase. Studies were
performed according to the protocol used for genistein. The rationale for examining the
effects of these inhibitors was based on studies implicating the involvement of Src and MAP
kinase activation in both agonist and pressure-induced smooth muscle responses (24, 25, 46).
Drugs and Chemicals
Buffer salts and chemicals were obtained from Sigma unless otherwise stated. NE
(arterenol) was dissolved in 1mM ascorbic acid as a stock solution at 10-2M and diluted in
krebs solution to 5 µM in 100 ml quantities as required through out the protocol. Working
solutions of NE were protected from light and all solutions were kept below room
temperature (either on ice or on a ‘cold’ block). A stock solution of adenosine was prepared
in Krebs at a concentration of 10-2 M and added directly to the arteriole bath (5 ml) to achieve
a concentration of 3 x 10-4M. PD98059 (Biomol Research Laboratories, Plymouth Meeting,
PA, USA) dissolved in DMSO at a concentration of 10-1 M was stored at –20oC in 50 µl
aliquots and diluted to 50 µM in krebs containing DMSO (600µl/100ml) for each experiment.
Frozen 100 µl aliquots of PP1 (Biomol Research Laboratories, Plymouth Meeting, PA, USA)
in DMSO at a concentration of 10-2M were diluted in krebs to 10 µM for each experiment.
Demecolcine dissolved in DMSO (1 mM) was further diluted in Krebs to 10 µM.
Concentrations of norepinephrine, adenosine, genistein, PD98059, PP1 and
demecolcine used were based on previous studies (26,30, 32). In addition, PD98059 was

American Journal of Physiology H00986-2002R1 24/04/2003
11
shown to inhibit phosphorylation of p42/44 MAP kinase in response to epidermal growth
factor or increases in intraluminal pressure as determined by gel electrophoresis/western
transfer (data not shown).
Statistics and Data Handling
To account for differences in starting vessel diameters data has been normalized
according to the following equation: Diameter (% max) = (diameter under a given
experimental condition/diameter in 0mM Ca2+ buffer containing 2mM EGTA) X 100. All
measurements were taken at an intraluminal pressure of 70 mmHg and maximal diameters
were measured prior to prolonged NE exposure.
Differences in group data, for the effect of a given treatment, were determined using
analysis of variance (ANOVA). When the ANOVA indicated an overall significant change,
differences between individual groups were determined using the t-test. Results are
presented as the mean ± standard error of the mean (SEM) and differences were considered
significant when P < 0.05, n represents both the number of arteriole segments and rats
studied.

American Journal of Physiology H00986-2002R1 24/04/2003
12
Results:
Arterioles used in these studies had active diameters of 80.8 ± 2.4 µm at 70 mmHg in
the absence of agonists and/or inhibitors. Passive diameters, obtained in 0mM Ca2+/2mM
EGTA prior to agonist/inhibitor protocols, were 153.8 ± 1.8 µm. Diameter measurements are
subsequently presented as a percentage of this passive diameter and are termed % maximal
diameter.
Acute and Prolonged Exposure to Norepinephrine
Acute exposure to NE (5 µM) resulted in a rapid contraction from 46.9 ± 3.9% to 23.9
± 1.7% of maximal diameter which was subsequently maintained during the prolonged
exposure (26.9 ± 1.7 % of maximal diameter at 4 hrs; n.s. compared to acute exposure)
(Figures 1 and 2). Despite reaching a similar level of vasoconstriction, subsequent washout
of the mechanical response with fresh buffer was significantly (P < 0.05) delayed in arterioles
subjected to the prolonged NE exposure. In 10 arterioles acutely exposed to NE, baseline
diameter was reached in 2:17 ± 0:45 minutes after initiating washout with fresh buffer. In
contrast, 7 of the 10 vessels remained constricted for periods of greater than 15 minutes when
washout followed the prolonged (4 hr) NE exposure. After 4hr exposure to NE, arterioles
also exhibited an impaired relaxation response to topically applied adenosine (3 x 10-4M)
although full relaxation was attained on removal of extracellular Ca2+ (Figures 1 and 4).
Control experiments comparing acute responses to NE at time 0 and 4 hrs later
indicated that the delayed relaxation following the prolonged NE exposure was not due to
time-dependent changes in the preparation independent of the action of the α-agonist.
Similarly, an additional series of vessels (n = 6) were studied to examine the effect of 4 hr NE
exposure in the absence of an acute pre-exposure to the agonist to exclude any effect
resulting from pre-conditioning. Consistent with impaired relaxation following prolonged
exposure to NE, after 4 hrs superfusion with 5 µM NE vessels dilated to 78.3 ± 3.7 % of

American Journal of Physiology H00986-2002R1 24/04/2003
13
maximal diameter in response to adenosine (10-4M) compared to 92.2 ± 0.9 % prior to NE
exposure (p < 0.05). As above, arterioles exposed to NE for 4 hrs dilated to maximum when
superfused with 0 mM Ca2+/2mM EGTA buffer.
In contrast to NE, exposure to KCl (75 mM) did not impair subsequent relaxation
responses (Figure 2). Further, unlike the NE-induced responses, KCl contractions tended to
wane over the 4hr time period (Figure 2).
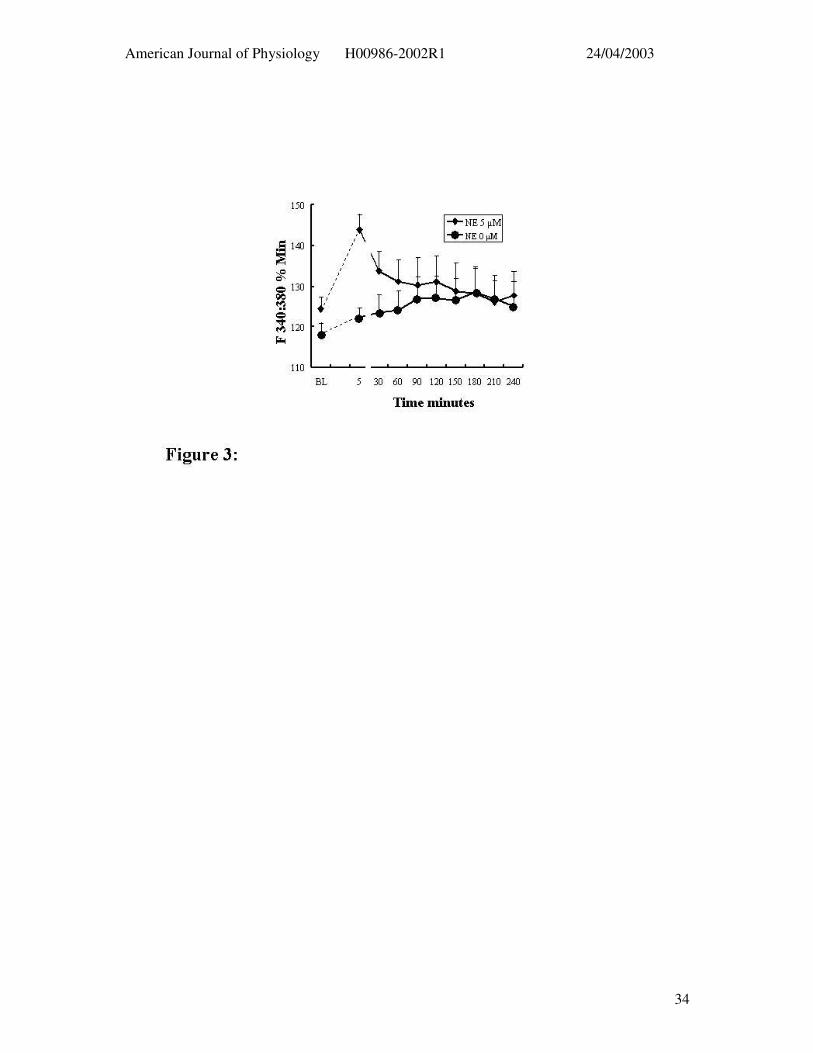
Vessels loaded with the Ca2+-sensitive dye Fura 2 showed a similar impairment in
relaxation following prolonged NE exposure. NE (5µM) caused a transient increase in Ca2+
that declined towards baseline over the period of prolonged agonist exposure despite the
maintained vasoconstriction. Comparison with time controls, not exposed to NE,
demonstrated that after 30 – 60 min exposure to the α-agonist that Ca2+i levels were not
statistically different from baseline (Figure 3).
The Ca2+ response did not appear to be a function of pre-exposure to NE or time as
peak changes were 178.9 ± 7.3 % (relative to values in 0 Ca2+ buffer) during the first NE
treatment compared to 183.0 ± 10.9 % in response to the second NE exposure (not
significantly different). Similarly, after 5 mins exposure to NE Ca2+ had plateaued to 139.4 ±
4.2 % and 134.2 ± 6.3 % (not significantly different) respectively.
Effect of Microtubular Depolymerization
One hour demecolcine (10 µM) treatment caused a decrease in baseline diameter and
potentiated NE-induced constriction as seen in our previous studies (36, 37) (see Table 1).
Despite the presence of the microtubule depolymerizing agent, arterioles maintained the
prolonged NE constrictor response and continued to show the impaired relaxation on wash
out of the agonist (Figure 5; Table 1).

American Journal of Physiology H00986-2002R1 24/04/2003
14
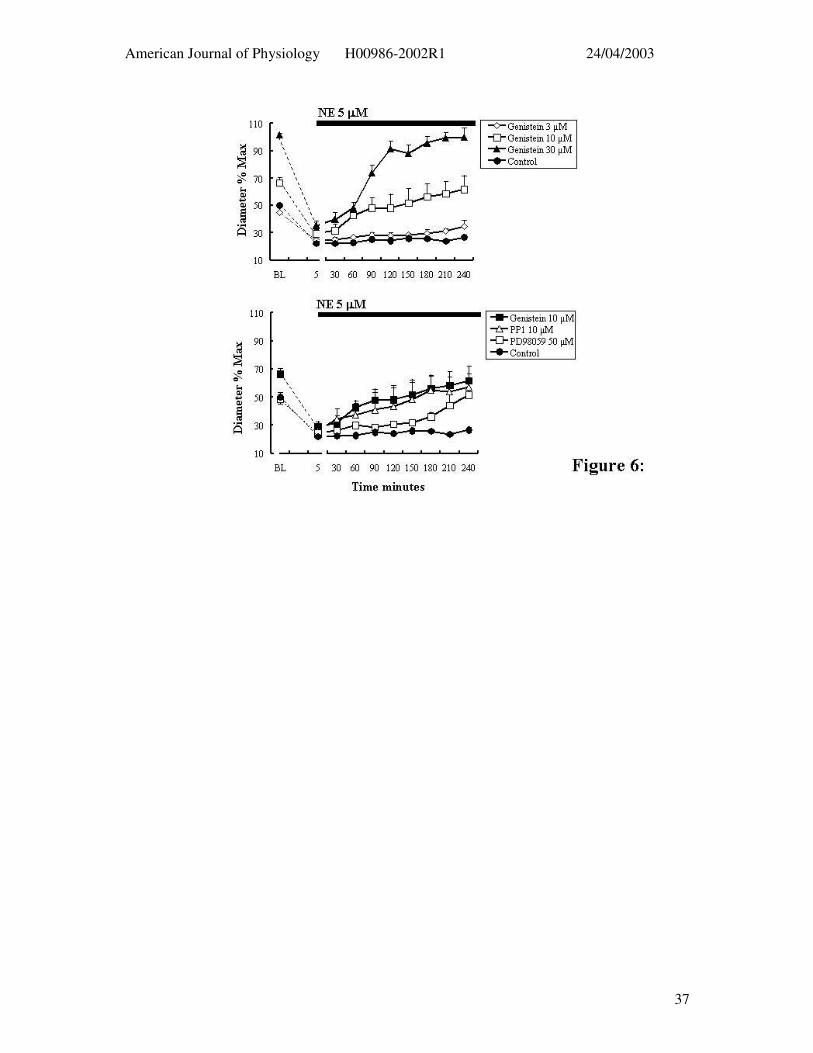
Effect of Tyrosine Kinase Inhibitors
The initial series of studies examined the effect of the general tyrosine kinase
inhibitor, genistein, at 3, 10 and 30 µM. Genistein caused a concentration-dependent loss of
basal tone but did not significantly inhibit the acute contractile response to NE (5µM) (Table
1). In contrast, genistein treatment resulted in a concentration-dependent loss of the NE-
induced constriction over time despite the continued presence of the agonist (Figure 6; Table
1).
The Src inhibitor, PP1 (10 µM, n = 4), did not affect either the basal level of
myogenic tone nor the acute contraction to NE (Figure 6b; Table 1). Similarly to genistein,
however, vessels treated with PP1 could not maintain the constrictor response to NE over the
4 hr time period (Figure 6b; Table 1). At approximately 1 hr into the 4 hr time course
arterioles had returned to control diameters. The MEK inhibitor, PD 98059 (50 µM, n = 6)
similarly showed little effect on the spontaneous level of myogenic tone or the acute
contractile response to NE (Figure 6b; Table 1). However, in the presence of PD98059 the
contraction to NE waned over the 4 hr exposure period (Figure 6b; Table 1).
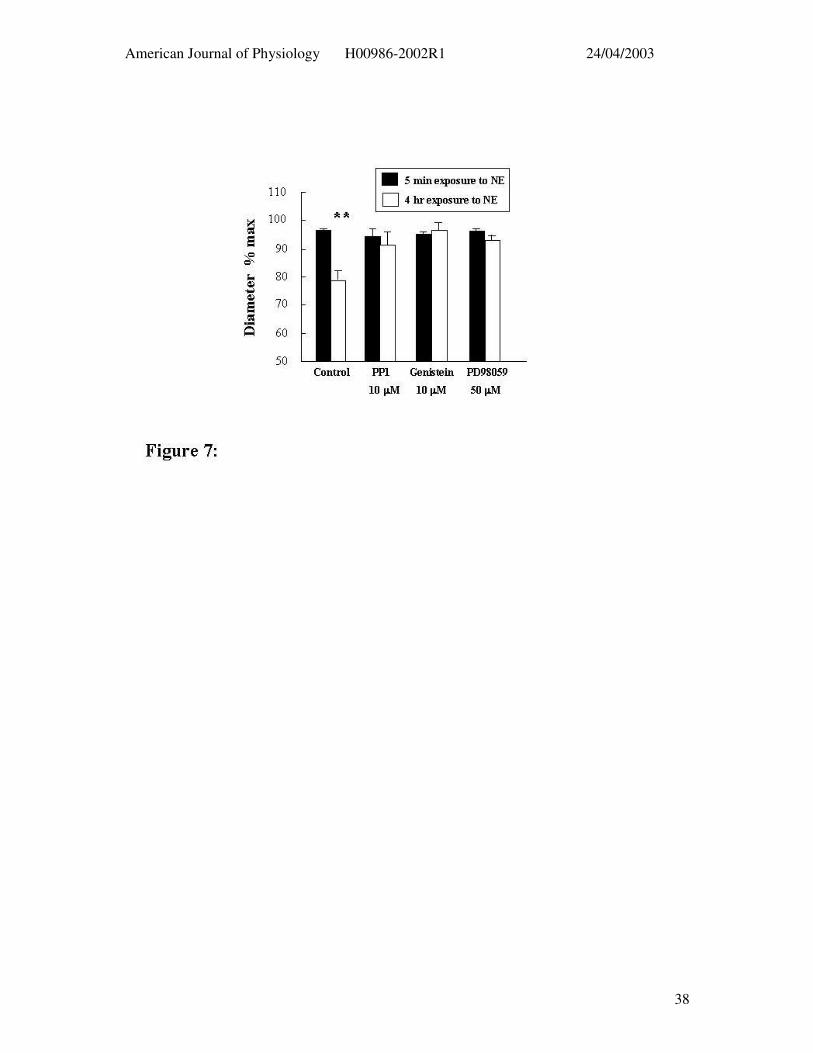
Time-dependent inhibition of the 4 hr NE constriction by the tyrosine phosphorylation
inhibitors prevented the possibility of determining the time to washout. Despite this
sufficient tone remained to assess adenosine (3 x 10-4 M) – induced dilation (Figure 7).
Control vessels exposed to NE for 4 hrs showed an impaired dilator response to adenosine
compared to the acute NE exposure while vessels treated with the inhibitors did not show an
attenuated adenosine response (Figure 7).

American Journal of Physiology H00986-2002R1 24/04/2003
15
Discussion:
The results of the present studies demonstrate that a 4 hr period of norepinephrine-
induced arteriolar constriction is sufficient to initiate phenomena that prolong the constrictor
response despite washout of the agonist. Such an effect was in contrast to that seen following
washout of an acute (5 min) NE -induced contraction. Further, the effect could not be
reproduced by a 4 hr exposure to KCl (75 mM) suggesting a possible requirement for
receptor binding and not contraction per se. Involvement of tyrosine phosphorylation is
suggested by inhibition of the potentiated adrenergic response by the broad spectrum tyrosine
kinase inhibitor, genistein, and the selective inhibitors PD98059 and PP1. As the impaired
relaxation response, following prolonged agonist exposure, was reversed by removal of
extracellular Ca2+ we have referred to the phenomenon as “functional remodeling”.
Control experiments were performed to demonstrate that changes in the ability to
relax following 4 hr NE treatment were not a function of either time or a pre-conditioning
phenomenon resulting from acute (5 min) exposure to the agonist. Thus, vessels showed
reproducible contractile responses to acute NE exposure applied at 4hr intervals and, further,
washout/relaxation was unaffected by this time period. A lack of a pre-conditioning effect
was shown by impaired relaxation to adenosine after 4hr continuous exposure to NE in a
group of vessels not subjected to any pre-exposure to the agonist. It was therefore concluded
that the impaired relaxation response was indeed a function of the prolonged agonist
exposure.
Remodeling has been used to describe, in particular, structural vascular adaptations
whereby alterations in the wall:lumen ratio are evident. Such remodeling can be hypotrophic,
eutrophic or hypertrophic depending on the growth response of the medial layer (27, 28, 29).
These classifications have resulted from the results of many earlier studies, especially those
related to changes in resistance vessel structure occurring in hypertension (1, 3, 14) and states

American Journal of Physiology H00986-2002R1 24/04/2003
16
of altered blood flow (6, 23). In the case of eutrophic remodeling it is believed that an initial
period of active vasoconstriction is followed by rearrangement of normal smooth muscle cells
to maintain a reduced diameter. Although not directly shown it is conceivable that the results
of the present study are consistent with an early stage in such a process. Interestingly, similar
signaling pathways (i.e. tyrosine phosphorylation events involving cSrc, and MAP) have been
implicated in both structural remodeling and the functional remodeling demonstrated in the
present studies (for example see references 20, 24, 25, 45). Similarly, support for early
agonist-induced remodeling events in tracheal smooth muscle involving tyrosine
phosphorylation has been provided by Gunst and colleagues (15, 42).
A possible explanation for the delayed relaxation response to the longer term
exposure to NE is that agonist binding initiates a pathway(s), in addition to Ca2+-calmodulin-
myosin light chain kinase mediated activation of myosin, that operate in a time-dependent
manner to influence the contraction/relaxation process. Such pathways may require either a
longer onset period, or time for washout, resulting in an altered time course in comparison to
those simply stimulated by a rise in Ca2+i (as might be expected for KCl-induced activation).
An obvious class of candidate mechanisms involves processes that alter the standard relation
between Ca2+ and contraction such as Ca2+ sensitization (40, 41). Ca2+ sensitization has been
demonstrated to occur in arteriolar smooth muscle and may involve tyrosine phosphorylation,
as tyrosine phosphatase inhibition, by pervanadate, causes constriction without a measurable
change in intracellular Ca2+ (31).
In addition to a Ca2+ sensitization process, a number of vasoactive agonists are known
to induce cytoskeletal rearrangement in various smooth muscle preparations (19, 20, 44, 45)
and modulate cell migration and attachment (20). Of possible relevance to our study, many
of these events have been shown to be dependent on tyrosine phosphorylation dependent
pathways. Of specific relevance to the present studies α1-adrenoceptor agonists, NE and

American Journal of Physiology H00986-2002R1 24/04/2003
17
phenylephrine, have been shown to activate tyrosine phosphorylation events including those
involving FAK, cSrc and p42 MAP kinase (2, 21, 47). Such studies have implicated these
signaling events in the adrenergic stimulation of non-selective cation currents, contraction
and mitogenic responses.
An interesting observation in the present study was that arteriolar constriction to KCl
(75 mM) was not maintained throughout the 4 hr observation period. Despite the continued
presence of KCl, arterioles had returned to control diameters by approximately 120 min. This
may indicate that maintained long-term constriction requires the involvement of signaling
mechanisms not activated by simple membrane depolarization. Similarly in the presence of
the non-selective tyrosine kinase inhibitor, genistein (10 and 30 µM), the NE constriction
tended to wane after approximately 60 - 90 minutes. This occurred despite genistein not
significantly affecting the acute contraction to NE (see Table 1). Thus it is tempting to
speculate that tyrosine phosphorylation – mediated events are required for maintenance of
prolonged arteriolar constriction. Further studies are required to determine whether these
events reflect the action of additional parallel contractile pathways (for example through thin
filament regulation, Ca2+ sensitization, cytoskeletal rearrangement etc) or form a support role
in maintaining the contractile pathways.
An obvious question relates to the physiological significance, or advantage, of an
apparent time-dependent switch in the mechanisms underlying a maintained arteriolar
contraction. It is conceivable that these additional pathways provide a mechanism where
there is maintained tension development at comparatively low energy cost. Such an
argument has previously been forwarded for the relevance of Ca2+ sensitization (40).
Regardless of the underlying mechanism this may confer an energetic advantage to smooth
muscles, such as are evident in the arteriolar wall, which exhibit a high level of maintained
tone.

American Journal of Physiology H00986-2002R1 24/04/2003
18
An additional consideration is that the functional and growth responses to
vasoconstrictors are often modulated by the generation of paracrine factors that exert either a
positive or negative effect on the vasomotor event. Of relevance to the results of the present
studies, DeFily et al. (11) have suggested that a component of the prolonged in vivo α1-
adrenergic-mediated constriction of canine coronary arterioles results from the release of
endothelin. Endothelin has been reported to modulate Ca2+ sensitivity via tyrosine
phosphorylation-dependent (35, 39) and independent (13) mechanisms in arterial smooth
muscle. Furthermore, prolonged in vivo administration of NE induces remodeling of small
arteries via a process dependent on endothelin (9). Consistent with these observations,
involvement of endothelin in the studies of DeFily was confirmed by experiments
demonstrating that either an ETA-receptor antagonist or an inhibitor of conversion of
prohormone to active endothelin prevented the sustained constriction to phenylephrine.
Interestingly, these investigators had previously shown that under in vitro conditions canine
coronary arterioles did not respond to α1-agonists suggesting that in vivo the source of
required endothelin was not restricted to the wall of the arteriole under study (22). It appears
likely that in the in vivo situation α1-agonist stimulation of cardiac myocyte endothelin
production contributed (42). With respect to the present data it was felt unlikely that in the
absence of significant parenchymal tissue, and in the presence of a superfusion system, that
sufficient quantities of a vasoconstrictor would accumulate to explain the prolonged
vasoconstrictor response.
In summary the results of the present study demonstrate that prolonged exposure of
arterioles to the contractile agonist NE leads to the stimulation of tyrosine phosphorylation-
dependent mechanisms which impair relaxation on washout of the agonist. Further, the
prolonged (over a 4 hr period) NE-induced contraction was totally prevented by inhibitors of
cSrc, MEK and general tyrosine phosphorylation suggesting that such events are critical to

American Journal of Physiology H00986-2002R1 24/04/2003
19
force maintenance in arterioles. It is suggested that these observations are consistent with an
early phase of the remodeling process and, in particular, represent a functional adaptation
whereby force is maintained by mechanisms supporting, or acting in parallel to, Ca2+-
mediated contraction. Future experiments are required to determine whether these results are
applicable to prolonged myogenic constriction resulting from a sustained increase in
intraluminal pressure. In addition, to make these observations relevant to common clinical
situations, such as in hypertension, interactions that occur between mechanical and
neurohumoral stimuli should be considered.

American Journal of Physiology H00986-2002R1 24/04/2003
20
Acknowledgements:
Studies described in this manuscript were supported by grants from the National
Health and Medical Research Council of Australia, the National Heart Foundation the Sir
Edward Dunlop Foundation.

American Journal of Physiology H00986-2002R1 24/04/2003
21
References:
1. Aalkjaer C, Heagerty AM, Petersen KK, Swales JD, and Mulvany MJ. Evidence for
increased media thickness, increased neuronal amine uptake, and depressed
excitation-contraction coupling in isolated resistance vessels from essential
hypertensives. Circ. Res. 61:181-186, 1987.
2. Albert AP, Aromolaran AS, and Large WA. Agents that increase tyrosine
phosphorylation activate a non-selective cation current in single rabbit portal vein
smooth muscle cells. J. Physiol. 530:207-217, 2001.
3. Baumbach GL and Heistad DD. Remodeling of cerebral arterioles in chronic
hypertension. Hypertension 13:968-972, 1989.
4. Boonen, HC, Daemen, MJ, Eerdmans, PH, Fazzi GE, van Kleef EM, Schiffers PM,
and De Mey JG. Mesenteric small artery changes after vasoconstrictor infusion in
young rats. J. Cardiovasc. Pharmacol. 22:388-395, 1993.
5. Brouwers-Ceiler DL, Nelissen-Vrancken HJ, Smits JF, and De Mey, JG. The
influence of angiotensin II-induced increase in aortic wall mass on compliance in rats
in vivo. Cardiovasc. Res. 33:478-484, 1997.
6. Buus CL, Pourageaud F, Fazzi GE, Janssen FG, Mulvany MJ, and De Mey JGR.
Smooth muscle cell changes during flow-related remodeling of rat mesenteric
resistance arteries. Circ. Res. 89:180-186, 2001.

American Journal of Physiology H00986-2002R1 24/04/2003
22
7. Cipolla MJ, Gokina NI, and Osol G. Pressure-induced actin polymerization in
vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J.
16:72-76, 2002.
8. Cowan DB, Lye SJ, and Langille BL. Regulation of vascular connexin43 gene
expression by mechanical loads. Circ. Res. 82:786-793, 1998.
9. Dao HH, Martens FM, Lariviere R, Yamaguchi N, Cernacek P, de Champlain J, and
Moreau P. Transient involvement of endothelin in hypertrophic remodeling of small
arteries. J. Hypertension 19:1801-1812, 2001.
10. Davis MJ, and Hill MA. Signaling mechanisms underlying the vascular myogenic
response. Physiol. Rev. 79:387-423, 1999.
11. DeFily DV, Nishikawa Y, and Chilian WM. Endothelin antagonists block a1-
adrenergic constriction of coronary arterioles. Am. J. Physiol. 276:H1028-H1034,
1999.
12. Duling BR, Gore RW, Dacey RG, Jr, and Damon DN. Methods for isolation,
cannulation, and in vitro study of single microvessels. Am. J. Physiol. 241:H108-
H116, 1981.
13. Evans AM, Cobban HJ, and Nixon GF. ETA receptors are primary mediators of
myofilament calcium sensitization induced by ET-1 in rat pulmonary artery smooth

American Journal of Physiology H00986-2002R1 24/04/2003
23
muscle: a tyrosine kinase independent pathway. Br. J. Pharmacol. 127:153-160,
1999.
14. Folkow B, Grimby G, and Thulesius O. Adaptive structural changes of the vascular
walls in hypertension and their relation to the control of the peripheral resistance.
Acta Physiol. Scand. 44:255-272, 1958.
15. Gerthoffer WT and Gunst S. Focal adhesion and small heat shock proteins in the
regulation of actin remodeling and contractility in smooth muscle. J.Appl. Physiol.
91:963-972, 2001.
16. Harder DR, Narayanan J, Gebremedhin D, and Roman RJ. Transduction of physical
force by the vascular wall: role of phospholipase C and cytochrome P-450 metabolites
of arachidonic acid. Trends Cardiovasc. Med. 5:7-14, 1995.
17. Hill MA, Zou H, Davis MJ, Potocnik SJ, and Price S. Transient increases in diameter
and [Ca2+]i following acute pressure increases are not obligatory for myogenic
constriction. Am. J. Physiol. 278:H345-H352, 2000.
18. Hill MA, Zou H, Potocnik SJ, Meininger GA, and Davis MJ. Arteriolar smooth
muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic
reactivity. J. Appl. Physiol. 91:973-983, 2001.
19. Hirshman CA and Emala CW. Actin reorganization in smooth muscle cells involves
Gq and Gi-2 activation of Rho. Am. J. Physiol. 277:L653-L661, 1999.

American Journal of Physiology H00986-2002R1 24/04/2003
24
20. Ishida T, Ishida M, Suero J, Takahashi M, and Berk BC. Agonist-stimulated
cytoskeletal reorganization and signal transduction at focal adhesions in vascular
smooth muscle cells require c-Src. J. Clin. Invest. 103:789-797, 1999.
21. Jin N, Siddiqui RA, English D, and Rhoades RA. Communication between tyrosine
kinase pathway and myosin light chain kinase pathway in smooth muscle. Am. J.
Physiol. 271:H1348-H1355, 1996.
22. Jones CJH, Kuo L, Davis MJ, and Chilian WM. α-Adrenergic responses of isolated
coronary microvessels. Basic Res. Cardiol. 90:61-69, 1995.
23. Langille BE, Bendeck MP, and Keely FW. Adaptations of carotid arteries of young
and mature rabbits to reduced carotid blood flow. Am. J. Physiol. 256:H931-H939,
1989.
24. Matrougui K, Eskildsen-Helmond YEG, Fiebeler A, Henrion D, Ley BI, Tegui A. and
Mulvany MJ. Angiotensin II stimulates extracellular signal-regulated kinase activity
in intact pressurized rat mesenteric resistance arteries. Hypertension 36:617-621,
2000.
25. Matrougui K, Tanko LB, Loufrani L, Gorny D, Levy BI, Tedgui A, and Henrion D.
Involvement of Rho-kinase and the actin filament network in angiotensin II-induced
contraction and extracellular signal-regulated kinase activity in intact rat mesenteric
resistance arteries. Arterioscler. Thromb. Vasc. Biol. 8:1288-1293, 2001.

American Journal of Physiology H00986-2002R1 24/04/2003
25
26. Meininger GA, Zawieja DC, Falcone JC, Hill MA, and Davey J. Calcium
measurement in isolated arterioles during myogenic and agonist stimulation. Am. J.
Physiol.261:H950-H959, 1991.
27. Mulvany MJ. Vascular remodeling of resistance vessels: can we define this?
Cardiovasc. Res. 41:9-13, 1999.
28. Mulvany MJ. Small artery remodeling and significance in the development of
hypertension. News Physiol. Sci. 17:105-109, 2002.
29. Mulvany MJ, Baumbach GL, Aalkjær C, Heagerty AM, Korsgaard N, Schiffrin EL,
and Heistad DD. Vascular remodeling. Hypertension 28:505-506, 1996.
30. Murphy TV, Spurrell BE, and Hill MA. Tyrosine phosphorylation following
alterations in arteriolar intraluminal pressure and wall tension. Am. J. Physiol.
281:H1047-H1056, 2001
31. Murphy TV, Spurrell BE, and Hill MA. Mechanisms underlying pervanadate-
induced contraction of rat cremaster muscle arterioles. Eur. J. Pharmacol. 442:107-
114, 2002.
32. Murphy, T.V., Spurrell, B.E. and Hill, M.A. Cellular signalling in arteriolar
myogenic constriction: involvement of tyrosine phosphorylation pathways. Clin. Exp.
Pharmacol. Physiol. 29:612-619, 2002.

American Journal of Physiology H00986-2002R1 24/04/2003
26
33. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, and Lederer WJ.
Relaxation of arterial smooth muscle by calcium sparks. Science 270:633-637, 1995.
34. Noria S, Cowan DB, Gotlieb AI, and Langille BL. Transient and steady-state effects
of shear stress on endothelial cell adherens junctions. Circ. Res. 85:504-514, 1999.
35. Ohanian J, Ohanian V., Shaw L, Bruce C, and Heagerty AM. Involvement of tyrosine
phosphorylation in endotheli-1-induced calcium-sensitization in rat small mesenteric
arteries. Br. J. Pharmacol. 120:653-661, 1997.
36. Osol G. Mechanotransduction by vascular smooth muscle. J. Vasc. Res. 32:275-292,
1995.
37. Platts SH, Falcone JC, Holton WT, Hill MA, and Meininger GA. Alteration of
microtubular polymerization modulates arteriolar vasomotor tone. Am. J. Physiol.
277:H100-H106, 1999.
38. Platts SH, Martinez-Lemus LA, and Meininger GA. Microtubule-dependent
regulation of vasomotor tone requires rho kinase. J. Vasc. Res., 39:173-182, 2002.
39. Sato A, Hattori Y, Sasaki M., Tomita F, Kohya T, Kitabatake A, and Kanno M.
Agonist-dependent difference in the mechanisms involved in Ca2+-sensitization of
smooth muscle of porcine coronary artery. J. Cardiovasc. Pharmacol. 35:814-821,
2000.

American Journal of Physiology H00986-2002R1 24/04/2003
27
40. Somlyo AP and Somlyo AV. Signal transduction and regulation in smooth muscle.
Nature 372:231-236, 1994.
41. Somlyo AP and Somlyo AV. Signal transduction by G-proteins, rho-kinase and
protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol.
522:177-185, 2000.
42. Tang DD and Gunst SJ. Roles of focal adhesion kinase and paxillin in the
mechanosensitive regulation of myosin phosphorylation in smooth muscle. J.Appl.
Physiol. 91:1452-1459, 2001.
43. Tiefenbacher CP, DeFily DV, and Chilian WM. Requisite role of cardiac myocyte in
coronary α1-adrenergic constriction. Circulation 98:9-12, 1998.
44. Togashi H, Emala CW, Hall IP, and Hirshman CA. Carbachol-induced actin
reorganization involves Gi activation of Rho in human airway smooth muscle cells.
Am. J. Physiol. 274:L803-L809, 1998.
45. Wang P and Bitar KN. Rho A regulates sustained smooth muscle contraction through
cytoskeletal reorganization of HSP27. Am. J. Physiol. 275:G1454-G1462, 1998.
46. Wesselman JP, Dobrian AD, Schriver SD, and Prewitt, R.L. Src tyrosine kinases and
extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases mediate

American Journal of Physiology H00986-2002R1 24/04/2003
28
pressure-induced c-fos expression in cannulated rat mesenteric small arteries.
Hypertension 37:955-960, 2001.
47. Zhong H and Minneman KP. Activation of tyrosine kinases by α1A-adrenergic and
growth factor receptors in transfected PC12 cells. Biochem. J. 344:889-894, 1999.
48. Zou H, Ratz PH, and Hill MA. Role of myosin phosphorylation and [Ca2+]i in
myogenic reactivity and arteriolar tone. Am. J. Physiol.269:H1590-H1596, 1995
49. Zou H, Ratz PH, and Hill MA. Temporal aspects of Ca2+ and myosin phosphorylation
during myogenic and norepinephrine-induced arteriolar constriction. J. Vasc. Res.
37:556-567, 2000.
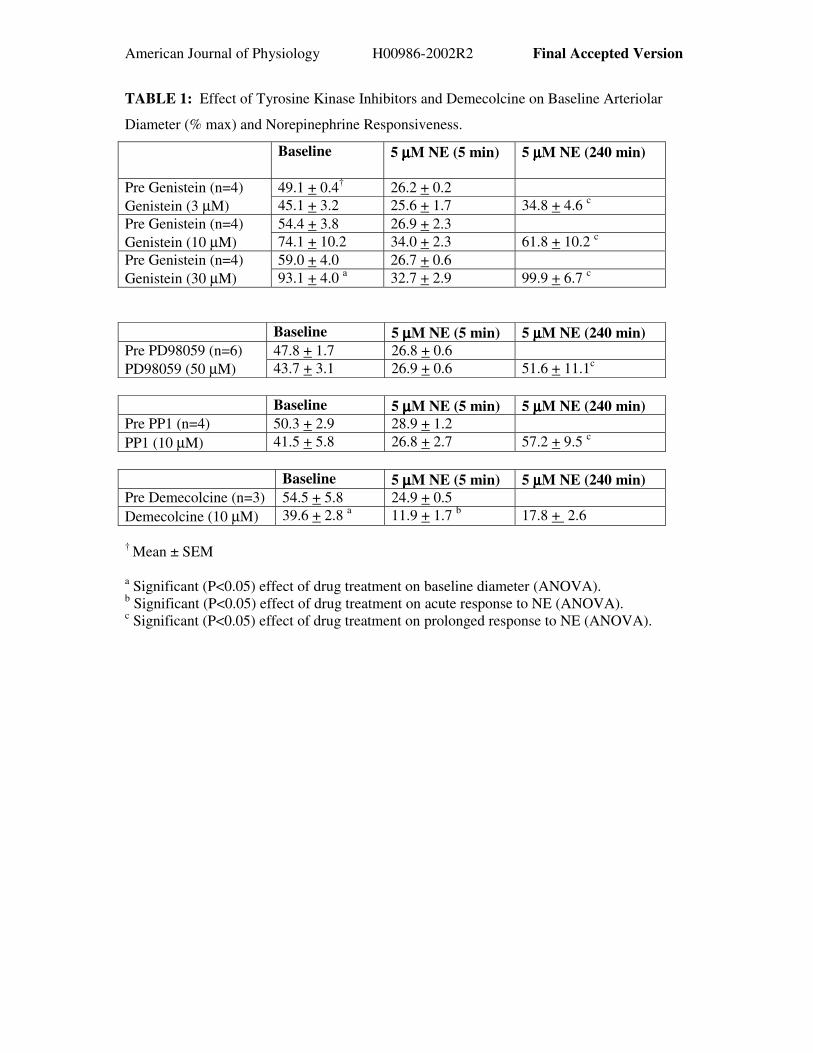
American Journal of Physiology H00986-2002R2 Final Accepted Version
TABLE 1: Effect of Tyrosine Kinase Inhibitors and Demecolcine on Baseline Arteriolar
Diameter (% max) and Norepinephrine Responsiveness.
Baseline 5 µµµµM NE (5 min) 5 µµµµM NE (240 min)
49.1 + 0.4† 26.2 + 0.2Pre Genistein (n=4)Genistein (3 µM) 45.1 + 3.2 25.6 + 1.7 34.8 + 4.6 c
54.4 + 3.8 26.9 + 2.3Pre Genistein (n=4)Genistein (10 µM) 74.1 + 10.2 34.0 + 2.3 61.8 + 10.2 c
59.0 + 4.0 26.7 + 0.6Pre Genistein (n=4)Genistein (30 µM) 93.1 + 4.0 a 32.7 + 2.9 99.9 + 6.7 c
Baseline 5 µµµµM NE (5 min) 5 µµµµM NE (240 min)47.8 + 1.7 26.8 + 0.6 Pre PD98059 (n=6)
PD98059 (50 µM) 43.7 + 3.1 26.9 + 0.6 51.6 + 11.1c
Baseline 5 µµµµM NE (5 min) 5 µµµµM NE (240 min)Pre PP1 (n=4) 50.3 + 2.9 28.9 + 1.2PP1 (10 µM) 41.5 + 5.8 26.8 + 2.7 57.2 + 9.5 c
Baseline 5 µµµµM NE (5 min) 5 µµµµM NE (240 min)Pre Demecolcine (n=3) 54.5 + 5.8 24.9 + 0.5Demecolcine (10 µM) 39.6 + 2.8 a 11.9 + 1.7 b 17.8 + 2.6
† Mean ± SEM
a Significant (P<0.05) effect of drug treatment on baseline diameter (ANOVA).b Significant (P<0.05) effect of drug treatment on acute response to NE (ANOVA).c Significant (P<0.05) effect of drug treatment on prolonged response to NE (ANOVA).

American Journal of Physiology H00986-2002R1 24/04/2003
30
Figure Legends:
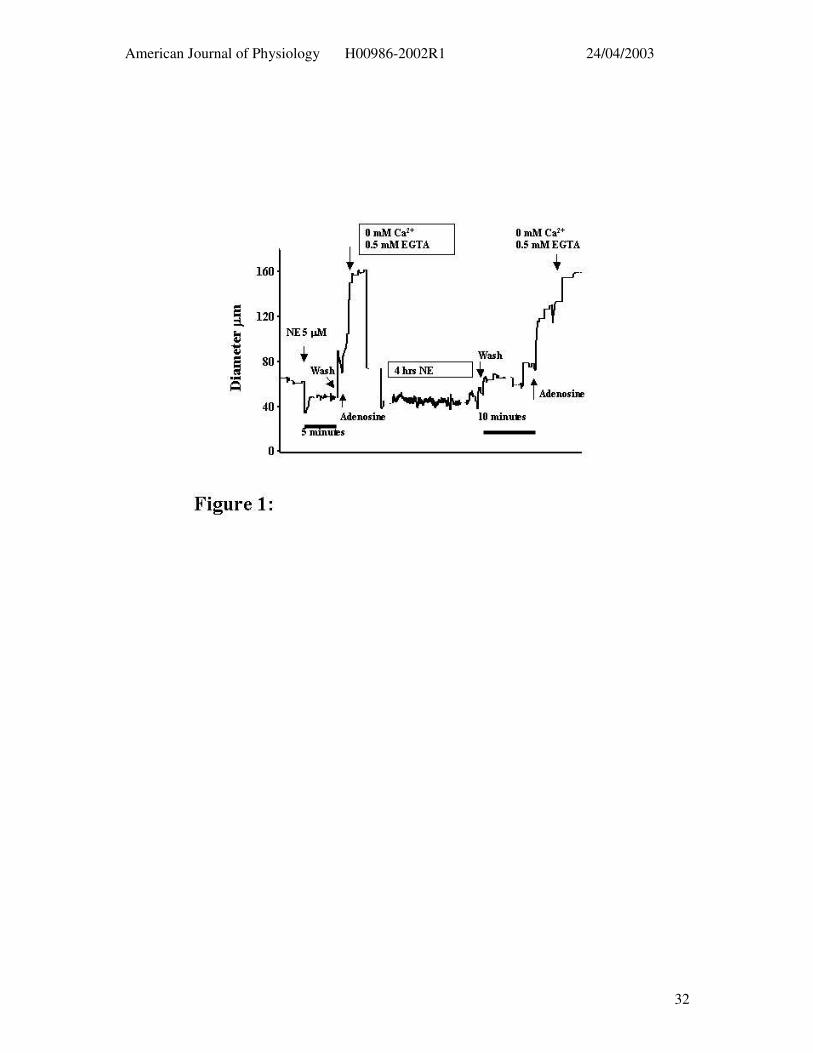
Figure 1: Example tracing illustrating the protocol used to contrast 5 min and 4 hr
exposures to contractile agents. The time scale has been omitted as a discontinuous scale has
been used in order to fit the entire protocol on one figure; 5 and 10 min time bars are shown
for the wash periods following acute and prolonged NE exposure, respectively.
Figure 2: The figure contrasts the 4hr constrictor responses to NE (5 µM, diamond
symbols, n = 10), KCl (75 mM, triangles, n=5) and control vessels exposed to buffer alone
(circles, n = 4). Results are presented as mean ± SEM; * P < 0.05 compared to baseline
(ANOVA).
Figure 3: Arteriolar smooth muscle Ca2+i changes during 4 hr exposure to NE (diamonds; n
= 7) as assessed using the Ca2+-sensitive dye fura 2 are shown in the lower panel. Time
controls (i.e. not exposed to NE) are shown by the circular symbols (n = 4). See Figure 2 for
diameter responses. Results are presented as mean ± SEM* P < 0.05 compared to baseline
(ANOVA).
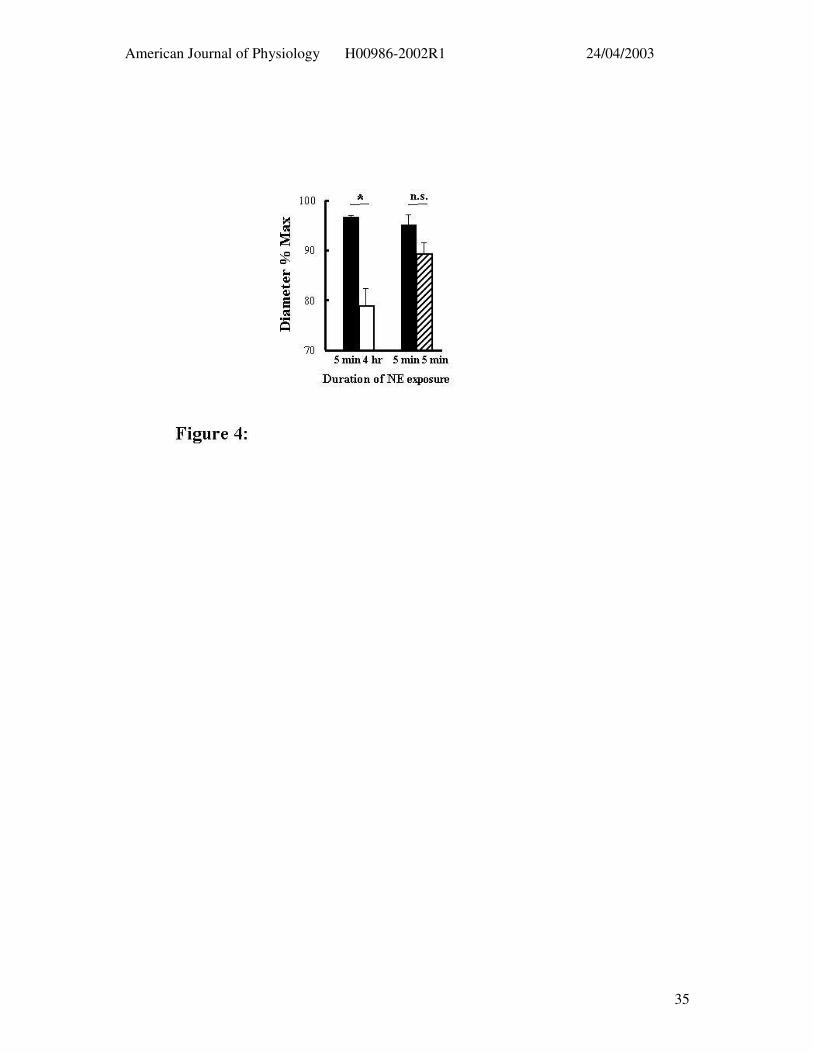
Figure 4: Effect of NE (5 µM) exposure on subsequent dilator responses to topical
adenosine (3 x 10-4 M). Greater relaxation responses were seen after 5 min exposure to NE
(solid bars, n = 5) compared to that following 4 hr NE superfusion (open bar). This was not
a function of time as a repeat acute exposure to NE at 4 hr (n = 4) was not associated with an
impaired response to adenosine (hatched bar). Results are presented as mean ± SEM; * P <
0.05; n.s. p = 0.2344.
.

American Journal of Physiology H00986-2002R1 24/04/2003
31
Figure 5: Effect of the microtubule depolymerizing agent, demecolcine (10 µM), on 5
min and 4 hr exposure to NE (5 µM). Demecolcine caused vasoconstriction and a potentiated
constrictor response to NE without preventing the delayed relaxation response (to both
washout and adenosine) observed following 4 hr NE exposure. BL = baseline; Aden =
adenosine (30µM); DC = demicholcine (10µM); and 0 Ca = 0mM Ca2+/2mM EGTA buffer.
Wash refers to the diameter obtained after extensive washout of NE with fresh buffer as
described in Materials and Methods; 30 – 240 refers to selected timepoints (in minutes)
during the prolonged exposure to NE. A discontinuous scale time scale was used in order to
fit the entire protocol on one figure. Results are presented as mean ± SEM; n = 3. See Table
1 for statistical comparisons relating to the effect of demecolcine on diameter and NE
responsiveness.
Figure 6: Effect of tyrosine phosphorylation inhibitors on 5 minute and 4 hour exposure
to NE (5 µM). The upper panel shows the effect of increasing concentrations of genistein (n
= 4) on 5 minute and 4 hour exposures to NE while the lower panel contrasts the effects of
PP1 (n = 4), PD98059 (n = 6) and genistein (n = 4). Results are presented as mean ± SEM.
Figure 7: Effect of tyrosine phosphorylation inhibitors on the ability of adenosine (3 x
10-4 M) to elicit dilation following 4 hrs exposure to NE (5 µM) (open bars). Control
responses (i.e. in the absence of inhibitors) after 5 min NE treatment for each group are
shown in the solid bars. Results are presented as mean ± SEM. ** P < 0.01 compared to
relaxation following 5 min exposure to NE.
American Journal of Physiology H00986-2002R1 24/04/2003
32

American Journal of Physiology H00986-2002R1 24/04/2003
33
American Journal of Physiology H00986-2002R1 24/04/2003
34
American Journal of Physiology H00986-2002R1 24/04/2003
35

American Journal of Physiology H00986-2002R1 24/04/2003
36
American Journal of Physiology H00986-2002R1 24/04/2003
37
American Journal of Physiology H00986-2002R1 24/04/2003
38

American Journal of Physiology H00986-2002R1 24/04/2003
39
Top Related
Copyright © 2022 FDOKUMEN

