







Bahasa
Halaman
Hukum


DEVELOPMENT OF A MULTIFUNCTIONAL CATIONIC LIPID-
BASED SIRNA DELIVERY SYSTEM FOR THE TREATMENT OF
TRIPLE-NEGATIVE BREAST CANCER
Thesis by
MANEESH GUJRATI
In Partial Fulfillment of the Requirements
for the degree of Doctor of Philosophy
Dissertation Advisor: Zheng-Rong Lu
Department of Biomedical Engineering
Case Western Reserve University
August, 2015

CASE WESTERN RESERVE UNIVERSITY
SCHOOL OF GRADUATE STUDIES
We hereby approve the thesis/dissertation of
Maneesh Gujrati
candidate for the degree of Doctorate of Philosophy *.
(signed)
(date)
Zheng-Rong Lu
Efsthatios Karathanasis
Nicole Steinmetz
Julian Kim
May 6, 2015
* We also certify that written approval has been obtained for any proprietary
material contained therein.

© 2015
Maneesh Gujrati
All Rights Reserved

i
Table of Contents
Table of contents ................................................................................................. i
List of Tables ................................................................................................... viii
List of Figures .................................................................................................... ix
Acknowledgments .......................................................................................... xiv
List of Abbreviations ....................................................................................... xvi
Abstract ............................................................................................................ xix
Chapter 1: Introduction ...................................................................................... 1
1.1 The promise of gene therapy ...................................................................... 2
1.2 The RNA interference mechanism ............................................................. 3
1.3 The RNAi advantage .................................................................................. 5
1.3.1 A natural pathway ................................................................................ 5
1.3.2 A catalytic mechanism ......................................................................... 6
1.3.3 Target ‘undruggable’ targets ................................................................ 6
1.3.4 Upstream mechanism .......................................................................... 7
1.3.5 Enable drug discovery process ............................................................ 7
1.4 Opportunity for RNAi in cancer ................................................................... 8
1.4.1 Opportunity of RNAi-mediated gene therapy in triple-negative breast
cancer ........................................................................................................... 9
1.5 Considerations for systemic siRNA delivery ............................................. 11
1.6 Current approaches for siRNA delivery .................................................... 14
1.6.1 Targeted siRNA delivery .................................................................... 17
1.6.2 Chemically modified siRNA ................................................................ 20
1.6.3 Lipid-based siRNA delivery systems .................................................. 22

ii
1.6.4 Cationic liposomes ............................................................................. 23
1.6.5 Neutral liposomes .............................................................................. 24
1.6.6 Lipid-like siRNA nanoparticles……………………………….. ................ 26
1.67 Polymer-mediated siRNA delivery........................................................ 27
1.6.8 Theranostics ....................................................................................... 32
1.6.9 siRNA in clinical trials .......................................................................... 35
1.7 Objectives ................................................................................................. 39
Chapter 2: pH-Sensitive Cationic Lipid-Based siRNA Delivery Systems .... 42
2.1 Introduction ................................................................................................ 43
2.2 Endosomal escape: The rate-limiting step................................................. 45
2.3 pH-sensitive amphiphilicity ........................................................................ 46
2.4 Mechanism of endolysosomal escape ....................................................... 47
2.5 pH-sensitive lipids for siRNA delivery ........................................................ 49
2.6 Concluding remarks .................................................................................. 71
Chapter 3: Multifunctional Cationic Lipid-Based Nanoparticles Facilitate
Endosomal Escape and Reduction-Triggered Cytosolic siRNA Release ... 72
3.1 Introduction ................................................................................................ 73
3.2 Materials and Methods .............................................................................. 78
3.2.1 Synthesis of (1-aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-
ethyl)propionamide] ..................................................................................... 78
3.2.2. Preparation of ECO/siRNA Nanoparticles ......................................... 78
3.2.3 Nanoparticle Characterization ............................................................ 78
3.2.4. Entrapment Efficiency ....................................................................... 79
3.2.5 Heparin Displacement Assay ............................................................. 79
3.2.6 Gel Electrophoresis for siRNA Loading, Serum Protection, and
Glutathione-Mediated Nanoparticle Reduction ………………………………. 79

iii
3.2.7 Cell Culture ........................................................................................ 80
3.2.8 In Vitro Transfection Efficiency ........................................................... 81
3.2.9 Cytotoxicity ......................................................................................... 81
3.2.10 Flow Cytometry for Nanoparticle Cellular Uptake and Uptake Kinetics
Measurements ............................................................................................ 82
3.2.11 Protein Adsorption ............................................................................ 83
3.2.12 pH-Dependent Membrane Disruption Hemolysis Measurement ...... 84
3.2.13 Inhibition of Glutathione-Dependent Reduction with BSO ................ 84
3.2.14 Confocal Microscopy of Cellular Uptake of ECO/siRNA Nanoparticles
and Intracellular Release of siRNA ............................................................. 85
3.2.15 Immunofluorescence of Intracellular Trafficking of ECO/siRNA
Nanoparticles……………………………….. .................................................. 85
3.2.16 Statistical Analysis ........................................................................... 86
3.3 Results and Discussion ............................................................................. 86
3.3.1. Effect of N/P ratio on the physicochemical properties of ECO/siRNA
nanoparticles ............................................................................................... 86
3.3.2. Effect of N/P ratio on the biological properties of ECO/siRNA
nanoparticles ............................................................................................... 89
3.3.3. ECO/siRNA nanoparticles protect siRNA and promote cellular uptake
in the presence of serum proteins ............................................................... 93
3.3.4. ECO/siRNA nanoparticles are pH-sensitive and promote endosomal
escape ........................................................................................................ 96
3.3.5. Cytosolic reduction of ECO/siRNA nanoparticles is crucial for siRNA
release and RNAi activity ............................................................................ 99
3.4 Conclusions ............................................................................................. 102
3.3 Acknowledgments ................................................................................... 103

iv
Chapter 4: Silencing β3 Integrin by Targeted siRNA Nanoparticles Inhibits
EMT and Metastasis of Triple Negative Breast Cancer .............................. 104
4.1 Introduction .............................................................................................. 105
4.2 Materials and Methods ............................................................................ 107
4.2.1 Cell lines and reagents ..................................................................... 107
4.2.2 Preparation of ECO/siRNA nanoparticles ........................................ 107
4.2.3 Western blot analyses ...................................................................... 109
4.2.4 Flow cytometry for nanoparticle cellular uptake ............................... 109
4.2.5 Semi-quantitative real-time PCR analyses ....................................... 110
4.2.6 Invasion and proliferation assays ……………………………….. ........ 110
4.2.7 3-Dimensional (3D)-organotypic cultures ......................................... 111
4.2.8 Tumor growth and bioluminescent imaging (BLI) .............................. 111
4.2.9 In vivo therapeutic treatment ……………………………….. ............... 112
4.2.10 Immunofluorescence and immunohistochemical staining .............. 112
4.2.11 Statistical analyses ......................................................................... 114
4.3 Results .................................................................................................... 114
4.3.1 ECO/siβ3 nanoparticles induce sustained silencing of a3 integrin ... 114
4.3.2 ECO/siβ3 nanoparticles attenuate TGF-β-mediated EMT, invasion, and
proliferation ............................................................................................... 117
4.3.3 ECO/siβ3 nanoparticles attenuate outgrowth of murine and human
MECs in 3D-organotypic culture ............................................................... 119
4.3.4 Surface modification of ECO/siRNA nanoparticles with RGD peptide
promotes cellular uptake and sustains gene silencing .............................. 122
4.3.5 RGD-ECO/siβ3 nanoparticles inhibit pulmonary outgrowth of mouse
MECs in vivo ............................................................................................. 124

v
4.3.6 RGD-ECO/sia3 nanoparticles effectively inhibit primary tumor growth
and metastasis of malignant human MECs ……………………………….. .. 125
4.4 Discussion ............................................................................................... 132
4.5 Conclusions ............................................................................................. 135
4.6 Acknowledgments ................................................................................... 136
Chapter 5: Development of a pH-Cleavable PEG Surface Modification
Strategy to Overcome the PEG Dilemma in ECO/siRNA Nanoparticles .... 137
5.1 Introduction .............................................................................................. 138
5.2 Materials and Methods ............................................................................ 142
5.2.1 Cell Culture ...................................................................................... 142
5.2.2 Synthesis of mPEG(HZ)-mal and RGD-PEG(HZ)-mal ...................... 142
5.2.3 Preparation of PEG-modified ECO/siRNA nanoparticles ................. 144
5.2.4 Nanoparticle Characterization ........................................................... 145
5.2.5 pH-Dependent Membrane Disruption Hemolysis Measurement ....... 145
5.2.6 Flow Cytometry for Nanoparticle Cellular Uptake Measurements
……………………………….. ...................................................................... 146
5.2.7 Confocal Microscopy of Nanoparticle Uptake and Intracellular Release
of siRNA .................................................................................................... 147
5.2.8 In Vitro Luciferase Silencing Efficiency ............................................. 148
5.2.9 In Vivo Luciferase Silencing Efficiency ……………………………….. 149
5.2.10 Fluorescence Molecular Tomography ............................................. 149
5.2.11 Ex vivo flow cytometry and confocal microscopy ........................... 150
5.2.12 Statistical analyses .................................................................... 150
5.3 Results and Discussion ........................................................................... 150

vi
5.3.1 Surface modification of ECO/siRNA nanoparticles with pH-cleavable
PEG layer restores intrinsic pH-sensitive activity ...................................... 151
5.3.2 pH-cleavable RGD-PEG modification induces potent in vitro silencing
efficiency ................................................................................................... 155
5.3.3 Inclusion of hydrazone linkage enhances endosomal escape ......... 158
5.3.4 Targeted pH-sensitive nanoparticles exhibit potent and sustained in
vivo gene silencing .................................................................................... 162
5.3.5 Targeting ligand enhances tumor retention of nanoparticles by
promoting internalization within tumor cells ............................................... 163
5.4 Conclusions ............................................................................................. 168
5.5 Acknowledgments ................................................................................... 169
Chapter 6: Targeting eIF4E with a Dual pH-responsive siRNA Delivery
System to Overcome Drug Resistance in Triple-Negative
Breast Cancer ................................................................................................ 170
6.1 Introduction .............................................................................................. 171
6.2 Materials and Methods ............................................................................ 174
6.2.1 Cell Culture ....................................................................................... 174
6.2.2 Preparation of PEG-modified ECO/siRNA nanoparticles .................. 174
6.2.3 Semi-quantitative real-time PCR analyses ........................................ 175
6.2.4 Western blot analyses ...................................................................... 176
6.2.5 Cytotoxicity Assay ............................................................................ 177
6.2.6 In vivo tumor growth inhibition study ……………………………….. ... 177
6.2.7 Bioluminescent imaging ................................................................... 178
6.2.8 Toxicity, immune response, and pathology studies ........................... 178
6.3 Results and Discussion ........................................................................... 179

vii
6.3.1. Silencing eIF4E by RGD-targeted pH-cleavable PEG-modified
ECO/siRNA nanoparticles enhances sensitivity to paclitaxel in a drug-
resistance triple-negative breast cancer cell line ........................................ 179
6.3.2. Combination of siRNA targeting eIF4E and paclitaxel inhibits primary
tumor growth of drug-resistant MDA-MB-231 cells .................................... 182
6.3.3. Long-term systemic administration of RGD-PEG(HZ)-ECO/siRNA
nanoparticles elicits no chronic immune response or organ damage ......... 190
6.4 Conclusions ............................................................................................. 192
6.5 Acknowledgments ................................................................................... 192
Chapter 7: Future Work .................................................................................. 194
7.1 Summary of Work ................................................................................... 195
7.2 Long-term stability of ECO/siRNA nanoparticle formulations .................. 195
7.3 Treatment of late-stage, metastatic disease ............................................ 197
7.4 Alternative surface-modification strategies .............................................. 200
7.5 Synergy in co-delivery of siRNA and PTX ............................................... 202
Appendix ......................................................................................................... 205
A.1. Synthesis of ECO ................................................................................... 206
A.2. Synthesis of cRGD-PEG-maleimide ...................................................... 214
A.3. Synthesis of mPEG(HZ)-maleimide ....................................................... 216
A.4. Synthesis of cRGD-PEG(HZ)-maleimide ............................................... 217
References ...................................................................................................... 220

viii
List of Tables
Table 1.1. Examples of siRNA-based therapies against breast cancer .............. 11
Table 1.2. Examples of siRNA delivery systems for the treatment of cancers ... 15
Table 1.3. Select examples of siRNA therapies in clinical trials ......................... 38
Table 7.1. Considerations for nanoparticle delivery to specific sites ................ 199

ix
List of Figures
Figure 1.1. Synthetic siRNA-induced RNA interference ....................................... 5
Figure 1.2. Subtypes of breast cancer ............................................................... 10
Figure 1.3. Physiological barriers to the systemic delivery of siRNA
nanoparticles ...................................................................................................... 13
Figure 2.1. Structure and activity of DLinDMA-based cationic lipids .................. 51
Scheme 2.1. General structure of pH-sensitive cationic lipids for siRNA delivery
........................................................................................................................... 52
Figure 2.2. Structure and pH-sensitive activity of polydisulfide .......................... 54
Figure 2.3. Library of polymerizable surfactants exhibiting pH-sensitive
amphiphilicity for siRNA delivery ........................................................................ 58
Figure 2.4. Chemical structures and transfection efficiency of the spermine-
based library of nucleic acid carriers .................................................................. 61
Figure 2.5. Surface functionalization and self-assembly of EHCO to form
targeted siRNA nanoparticles ............................................................................. 63
Figure 2.6. In vivo efficacy of systemically administered RGD-targeted
EHCO/siRNA nanoparticles ................................................................................ 65
Figure 2.7. In vivo efficacy of intracranially-administered PEGylated
EHCO/siRNA nanoparticles ................................................................................ 66
Figure 2.8. Chemical structure and function of EKHCO and EHHKCO ............. 67
Figure 2.9. Chemical structures, hemolytic activity and silencing efficiency of
newly synthesized cationic lipid-based siRNA carriers ....................................... 70
Scheme 3.1. Formation of ECO/siRNA nanoparticles ........................................ 75

x
Scheme 3.2. Cellular trafficking of ECO/siRNA nanoparticles ........................... 76
Figure 3.1. Physicochemical evaluation of ECO/siRNA nanoparticles ............... 87
Figure 3.2. Biological activity of ECO/siRNA nanoparticles in U87 Glioblastoma
cells .................................................................................................................... 90
Figure 3.3. Activity of ECO/siRNA nanoparticles in serum-containing
conditions ........................................................................................................... 95
Figure 3.4. Evaluation of pH-sensitive hemolysis and endolysosomal escape of
ECO/siRNA nanoparticles .................................................................................. 98
Figure 3.5. Sensitivity of ECO/siRNA nanoparticles to reduction by endogenous
levels of glutathione .......................................................................................... 101
Figure 4.1. ECO/siβ3 nanoparticles induced sustained gene silencing of β3
integrin .............................................................................................................. 116
Figure 4.2. ECO/siβ3 nanoparticles attenuated TGF-β-mediated EMT, invasion
and proliferation ................................................................................................ 118
Figure 4.3. ECO/siβ3 nanoparticles attenuated 3D organoid outgrowth .......... 121
Figure 4.4. RGD modification of ECO/siRNA nanoparticles enhances uptake in
post-EMT breast cancer cells ........................................................................... 123
Figure 4.5. Pulmonary outgrowth of NME cells treated with the ECO/siRNA
treatment regimen ............................................................................................ 125
Figure 4.6. RGD-targeted ECO/siβ3 nanoparticles inhibited primary tumor
growth and EMT in mice after systemic administration ..................................... 128
Figure 4.7. RGD-ECO/siβ3 nanoparticles inhibited breast cancer metastasis and
primary tumor recurrence ................................................................................. 130
Schematic 5.1. pH-sensitive surface modification of ECO/siRNA nanoparticles
with RGD-PEG(HZ)-maleimide ......................................................................... 141

xi
Figure 5.1. pH-sensitivity of ECO/siRNA, PEG-ECO/siRNA, and PEG(HZ)-
ECO/siRNA nanoparticles ................................................................................ 153
Figure 5.2. Comparison of hemolytic activity of ECO, PEG-ECO, and PEG(HZ)-
ECO siRNA nanoparticles at pH levels corresponding to stages of intracellular
trafficking .......................................................................................................... 155
Figure 5.3. Cellular uptake and luciferase silencing efficiency of unmodified,
PEG-, PEG(HZ)-, RGD-PEG-, and RGD-PEG(HZ)-modified ECO/siRNA
nanoparticles .................................................................................................... 159
Figure 5.4. Confocal microscopy images of MDA-MB-231 cells incubated with
RGD-PEG-, and RGD-PEG(HZ)-modified ECO/siRNA nanoparticles .............. 160
Figure 5.5. Luciferase silencing efficiency after 48 hours in MDA-MB-231-luc
cells transfected with or without the endosomolytic agent chloroquine ............. 161
Figure 5.6. In vivo luciferase silencing efficiency following a single i.v. treatment
with various surface-modified ECO/siRNA nanoparticles ................................. 163
Figure 5.7. Tumor accumulation and retention of surface-modified ECO/siRNA
nanoparticles following i.v. administration ........................................................ 164
Figure 5.8. Flow cytometry and confocal microscopy analysis of single cell
suspensions obtained from primary MDA-MB-231 mammary fat pad tumors
following i.v. administration of various surface modified ECO/siRNA
nanoparticles .................................................................................................... 166
Figure 6.1. Evaluation of eIF4E mRNA and protein expression analysis in MDA-
MB-231 and MDA-MB-231.DR cells 5 days following treatment with RGD-
PEG(HZ)-ECO/siRNA nanoparticles ................................................................ 180
Figure 6.2. Dose-response curves as determined by MTT assay of MDA-MB-231
and MDA-MB-231.DR cells treated with varying concentrations of PTX following
prior treatment with RGD-PEG(HZ)-ECO/siRNA nanoparticles delivering sieIF4E
or siNS .............................................................................................................. 182

xii
Figure 6.3. In vivo efficacy of combination therapy involving PTX and RGD-
PEG(HZ)-ECO/sieIF4E nanoparticles quantified with bioluminescent
imaging ............................................................................................................. 185
Figure 6.4. Growth, size and final weight of tumors following combination therapy
involving PTX and RGD-PEG(HZ)-ECO/sieIF4E nanoparticles........................ 186
Figure 6.5. Quantification of mRNA expression of eIF4E, Survivin, Cyclin D1,
and VEGF following combination therapy involving PTX and RGD-PEG(HZ)-
ECO/sieIF4E nanoparticles .............................................................................. 189
Figure 6.6. Histological evaluation of liver, kidney, and primary tumor ............ 190
Figure 6.7. Immunogenicity of ECO and RGD-PEG(HZ)-ECO/siRNA
nanoparticles .................................................................................................... 191
Figure A1. Synthetic Procedure of ECO .......................................................... 206
Figure A2. Reaction scheme and 1H NMR of Intermediate (1)........................ 207
Figure A3. Reaction scheme and 1H NMR of Intermediate (2)........................ 208
Figure A4. Reaction scheme and 1H NMR of Intermediate (3)........................ 209
Figure A5. Reaction scheme and 1H NMR of Intermediate (4)........................ 210
Figure A6. Reaction scheme and 1H NMR of Intermediate (5)........................ 211
Figure A7. Reaction scheme and 1H NMR of Intermediate (6)........................ 212
Figure A8. Reaction scheme and 1H NMR of ECO ......................................... 213
Figure A9. Maldi-Tof Spectra of ECO .............................................................. 214
Figure A10. Synthetic procedure of cRGD-PEG3400-maleimide .................... 214
Figure A11. Maldi-tof and H1-NMR spectrum of cRGD-PEG3400-
maleimide .................................................................................................. 215/216

xiii
Figure A12. Synthetic procedure of mPEG5000(HZ)-Maleimide ..................... 216
Figure A13. H1 NMR spectrum of mPEG5000(HZ)-maleimide ....................... 217
Figure A14. Synthetic procedure of cRGD-PEG3400(HZ)-maleimide ............. 218
Figure A15. H1 NMR spectrum (Solvent: DMSO of cRGD-PEG3400(HZ)-
maleimide ......................................................................................................... 218
Figure A16. H1 NMR spectrum (Solvent: D2O) of cRGD-PEG3400(HZ)-
maleimide ......................................................................................................... 219

xiv
Acknowledgments
I would first like to acknowledge and thank my advisor, Dr. Zheng-Rong
Lu. Over the last four years, ZR consistently encouraged me to think both
critically and creatively about my research. I am grateful for the opportunity to
pursue my research interests with freedom and support. The guidance I received
from ZR during my Ph.D. was the main driver for my success and I could not
imagine having the same experience in another lab.
I would also like to thank each member of my Ph.D. committee for their
guidance and support: Efstathios Karathanasis, Nicole Steinmetz, and Julian
Kim. Each member played an integral role in shaping my research project, often
asking critical questions and encouraging me to approach my work from different
perspectives. I am grateful for the time and effort each put in to my personal and
professional development during my Ph.D.
I am extremely grateful for my fellow labmates, both past and present, for
their help along the way. I would especially like to thank Anthony Malamas who
took me under his wing when I first joined the lab. Additionally, I would like to
thank our collaborators Dr. Scott Welford and Dr. William P. Schiemann.
Lastly I would like to thank my funding support through the National
Science Foundation Graduate Research Fellowship (DGE-0951783).

xv
List of Abbreviations
ASO: antisense oligonucleotide
Bax: Bcl-2-associated X protein
BCA: bicinchorninic acid assay
Bcl-2: B-cell lymphoma 2
Bcl-xl: B-cell lymphoma-extra large
BLI: bioluminescent imaging
BN: bombesin
BSA: bovine serum albumin
BSO: buthionine-sulfoximine
ECO: (1-aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-ethyl)propionamide]
CA-IX: carbonic anhydrase-IX
CHO: Chinese hamster ovary cell line
CK-19: cytokeratin 19
CPP: cell-penetrating peptide
DLS: dynamic light scattering
DMEM: Dulbecco’s Modified Eagle Medium
DMSO: dimethyl sulfoxide
DNP: 2,4-dinitrophenol
DOGS: dioctadecylamidoglycylspermidine
DOPC: 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
DOPE: 1,2-Dioleoyl-sn-Glycero-3-Phosphoethanolamine
DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane

xvi
DOTMA: N-[1-(2,3 Dioleoyloxy)propyl]-N,N,N-trimethyl-ammonium methyl
sulphate
dsRNA: double-stranded RNA
E-cad: E-cadherin
eIF4E: eukaryotic translation initiation factor 4E
EMT: epithelial-mesenchymal transition
EpCAM: Epithelial cellular adhesion molecule
EPR: enhanced permeability and retention
ER: estrogen receptor
FACS: fluorescence-activated cell sorting
FBS: fetal bovine serum
FITC: Fluorescein isothiocyanate
FMT: fluorescence molecular tomography
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase
GFP: green fluorescent protein
GLUT-1: glucose transporter
GPCR: G protein-coupled receptor
GSH: glutathione
H&E: hematoxylin and eosin
HER2: human epidermal growth factor receptor 2
HIF-1α: Hypoxia-inducible factor 1α
HZ: hydrazone
IR: ionizing radiation
LAMP1: lysosomal-associated membrane protein 1
mRNA: messenger RNA

xvii
Maldi-TOF: matrix-assisted laser desorption/ionization time of flight
MMP: metalloproteinase
MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
MW: molecular weight
MWCO: molecular weight cutoff
N-cad: N-cadherin
NME: normal murine mammary gland cells
NMR: nuclear magnetic resonance
PAI-1: plasminogen activator inhibitor
PBS: phosphate buffered saline
pDNA: plasmid DNA
PEG: polyethylene glycol
PEI: polyethyleneimine
PET: positron emission tomography
pKa: acid dissociation constant
PLL: poly-L-lysine
PR: progesterone receptor
PS: phosphothionate
PTX: paclitaxel
RBC: red blood cells
RES: reticuloendothelial system
RGD: Arg-Gly-Asp peptide
RISC: RNA-induced silencing complex
RNAi: RNA Interference
RNase: ribonuclease

xviii
ROI: region of interest
siRNA: small interfering RNA
SFM: serum-free media
SLN: solid lipid nanoparticle
SNALP: stable nucleic acid-lipid particle
ssRNA: single-stranded RNA
TBE: Tris/Borate/EDTA
Tf: transferrin
TGF-β: transforming growth factor β
TNBC: triple-negative breast cancer
VEGF: vascular endothelial growth factor

xix
Development of a Multifunctional Cationic Lipid-Based siRNA Delivery System
for the Treatment of Triple-Negative Breast Cancer
Abstract
by
MANEESH GUJRATI
Triple-negative breast cancer (TNBC) is a subtype of breast cancer
associated with a poor prognosis and an aggressive clinical course. Due to the
lack of well-defined biomarkers, TNBCs are not amenable to currently available
targeted therapies, leaving systemic chemotherapy as the sole therapeutic
strategy. Unfortunately, disease relapse, metastasis, and drug resistance often
render standard small-molecule chemotherapy ineffective. Small interfering RNA
(siRNA) has garnered much attention as a promising avenue for cancer gene
therapy due to its ability to silence disease-related genes through the RNA
interference (RNAi) mechanism. Deeper insights into the molecular profile of
TNBCs have identified a number of novel gene targets well-suited for RNAi
therapy. We therefore aim to employ siRNA-mediated gene therapy to address
two critical risks associated with TNBCs: metastasis and drug resistance.
Effective gene silencing is contingent upon the delivery of siRNA
molecules into the cytosol of target cells and requires carefully designed
multifunctional delivery systems to overcome systemic delivery barriers. The
present work reports the development of a novel pH-sensitive cationic lipid
carrier (ECO) for targeted siRNA delivery. Optimization of the chemical structure
was performed to engender ECO/siRNA nanoparticles with two key features
necessary for efficient cytosolic siRNA delivery: i) pH-sensitive membrane
disruption and ii) glutathione-mediated siRNA release.

xx
As TNBCs are associated with an elevated risk of distant recurrence, our
first application was to exploit 3 integrin as a therapeutic target against
metastatic TNBC. Recently, 3 integrin has been coupled to epithelial-
mesenchymal transition (EMT) and metastasis. We therefore hypothesized that
functional disruption of 3 integrin may inhibit metastasis. Treatment of
metastatic TNBC cells with ECO/si3 nanoparticles in vitro effectively silenced 3
integrin expression, attenuated EMT and cell invasion, restored cytostasis, and
inhibited organoid outgrowth within a 3D cellular culture system. Systemic
administration of RGD-PEG-modified ECO/si3 nanoparticles in vivo alleviated
primary tumor burden, and more importantly, significantly inhibited metastasis
and disease relapse, thereby validating 3 integrin as a viable therapeutic target
and the in vivo delivery capacities of ECO/siRNA nanoparticles.
The major cause of metastatic treatment failure is multidrug resistance to
standard chemotherapies. Therefore, we next investigated siRNA targeting of the
eukaryotic translation initiation factor 4E (eiF4E) to improve the susceptibility of
drug-resistant TNBC cells to paclitaxel. In parallel, an RGD-targeted pH-
cleavable PEG surface modification strategy (RGD-PEG(HZ)) was designed.
Multimodal imaging was used to demonstrate the pH-cleavable strategy exhibited
superior in vivo gene silencing compared to the non-cleavable formulations.
Silencing of eiF4E in a drug-resistant TNBC cell line was sufficient to re-sensitize
cells to paclitaxel. Importantly, treatment with RGD-PEG(HZ)-ECO/sieIF4E
nanoparticles in combination with paclitaxel resulted in significant regression of
the primary tumor when compared to either therapy alone. These results
substantiate the advantage of combining nanoparticle-mediated eIF4E silencing
with small-molecule chemotherapy to provide a promising therapeutic solution to
drug-resistant TNBCs.

1
Chapter 1
Introduction
Adapted in part from Gene Therapy of Cancer 2013, 3rd Edition, p. 47-65
Maneesh Gujrati and Zheng-Rong Lu

2
1.1 The promise of gene therapy
Today, breast cancer remains a significant health risk worldwide. Within
the United States, breast cancer is the second leading cause of cancer-related
deaths amongst women, accounting for 14% of all cancer-related deaths (1). The
lifetime risk for developing breast cancer is 1 in 8 for women in the U.S.
Significant improvements have been achieved in reducing the morbidity
associated with certain classes of breast cancers, with the overall breast cancer
death rate falling steadily over the past several decades. According to National
Cancer Institute, death rates associated with breast cancer have been falling on
average 1.9% each year from 31.4 per 100,000 women in 1975 to 21.5 per
100,000 per women in 2011 (2). Furthermore, the overall 5-year relative survival
rate for breast cancers has improved from 75.2% in 1975 to 90.6% in 2011 (2).
While the overall trends are indeed promising, stark differences exist between
the various breast cancer subtypes and even more so between localized and
metastatic disease. In fact, the 5-year relative survival for women with localized
disease is 98.5% but falls dramatically to 25% for those suffering from
metastases due to a lack of therapeutics that can specifically address this
segment of the disease (3).
Breast cancer also imposes a significant socioeconomic burden within the
healthcare system where it accounts for 20% of all cancer-related expenditure
(4,5). In 2014, the United States spent over 17 billion dollars alone towards the
treatment of breast cancer (5). Importantly, the treatment of late-stage,
metastatic and terminal disease is significantly higher than the original treatment

3
phase. The reasons for this are twofold: i) costs for terminal disease are on
average eight-times most costly than the original treatment phase; and ii) many
of the costly targeted therapies are used with little to no therapeutic benefit.
Breast cancers are now understood to be segmented across a spectrum
of various subtypes, each with their own molecular profiles and susceptibilities to
therapy. Conventional small-molecule chemotherapies, such as anthracyclines
and taxanes, are commonly used as the first line of defense against breast
cancer. These drugs utilize various mechanisms to impede cell division, thereby
killing rapidly proliferating cells. However, these agents lack the ability promote
accumulation within the tumor to enhance bioavailability and target only
malignant cells. As a consequence, this class of therapy harbors an array of
severe side effects, including peripheral neuropathy, amongst others. A deeper
understanding of the molecular mechanisms and profiles of breast cancer has
allowed the development of a novel generation of gene therapies able to
delineate benign from malignant cells and exert selective targeting of cancer cells
to substantially reduce systemic toxicity by direct targeting of disease-related
genes. The advent of gene therapies against novel molecular targets has the
potential to substantially improve the overall disease outcome for highly
aggressive breast cancers.
1.2 The RNA interference mechanism
RNA interference (RNAi) is a relatively new and highly promising
technology for gene therapy that represents an entirely novel approach to the

4
treatment paradigms of many diseases. Widely considered one of the most
promising and rapidly advancing frontiers in medicine, the discovery of RNAi was
awarded the 2006 Nobel Prize for Physiology or Medicine, being heralded as “a
major scientific breakthrough that happens once every decade or so”. RNAi is a
gene silencing process in which double-stranded RNA (dsRNA) interferes with
the expression of a gene through a homologous sequence shared with the
dsRNA (6,7). RNA silencing occurs when the RNase III enzyme Dicer initiates
the cytoplasmic breakdown of dsRNA into small interfering RNA (siRNA) (8).
These siRNA fragments, generally 20-25 nucleotides in length, incorporate into
the RNA-induced silencing complex (RISC) (9,10). Once incorporated into the
RISC, the siRNA molecule is unwound into a single-stranded RNA (ssRNA)
where the sense strand ssRNA is then degraded (11). The RISC containing
sequence specific antisense strand of ssRNA is then able to seek out and target
mRNA that is complementary to the antisense strand (12). Cleavage of mRNA
occurs at nucleotide position 10 and 11 on the complementary antisense strand,
relative to the 5’-end (13). To further amplify gene silencing, the RISC complex
can continue on to destroy additional mRNA targets, allowing the therapeutic
effect to last for up to 7 days in rapidly proliferation cells and for several weeks in
non-diving cells (14).

5
Figure 1.1. Synthetic siRNA-induced RNA interference. RNAi can be induced by
introducing synthetic double-stranded siRNA. The siRNA associates with the
multiprotein complex RISC and the sense strand is degraded by the protein Argo-2 in
the RISC. The remaining antisense strand, serves as a guide to recognize the
corresponding mRNA, after which Argo-2 cuts and degrades the target mRNA
1.3 The RNAi Advantage
1.3.1 A natural pathway
The fact that RNAi is a highly conserved, natural biological pathway
involved in the regulation of gene expression in all mammalian cells has
propelled the notion of it being used as a safe and effective therapeutic platform.
While naturally occurring, a key advantage of RNAi-based therapy is that siRNA

6
molecules can be chemically synthesized and delivered into cells to elicit the
desired silencing effect. The direct introduction of synthetic siRNA into cells can
bypass the Dicer mechanisms to trigger gene silencing (14). Synthetic siRNA
molecules have already been shown to block specific expression of endogenous
and heterologous genes in several mammalian cell lines. Further, long-term gene
silencing can be achieved without interrupting endogenous microRNA pathways
through multiple administrations of synthetic siRNAs (15). With an mRNA
sequence in hand these synthetic siRNA molecules can be readily designed to
be both potent and highly selective.
1.3.2 A catalytic mechanism
A single molecule of siRNA is needed to active the RISC to initiate RNAi-
mediated silencing of the target mRNA. Specifically, once the siRNA molecule
degrades one mRNA, it can go on and continue to silencing a large number of
mRNA molecules, unlike other oligonucleotide-based technologies, such as
antisense therapy, which directly bind to their mRNA targets via Watson-Crick
base pairing at a stoichiometric one-to-one ratio of silencing molecule to target
mRNA (16). Therefore, siRNA-mediated RNAi requires a significantly lower
cytosolic dose than other antisense drugs.
1.3.3 Target ‘undruggable’ targets
Clinically available small-molecule medicines are limited in their
therapeutic scope to certain classes of drug targets: GPCRs, ion channels,
enzymes, nuclear hormone receptors (17,18). Similarly, protein-based drugs,

7
such as monoclonal antibodies, are currently restricted to cell surface receptors
and circulating proteins. In contrast, siRNA molecules can be synthetically
constructed to target any gene so long its mRNA transcript is known, thus
enabling the targeting of proteins that have until now been considered
“undruggable” by current therapies (19,20). The power of RNAi can also be
harnessed in diseases caused by mutations in a single allele. In such a case, the
siRNA molecule can be designed to target the specific disease-causing allele to
spare the normal allele from silencing. Accordingly, RNAi-based therapies can
regulate the expression of any disease-implicated gene.
1.3.4 Upstream mechanism
Currently available small molecules and antibody-based therapies
inactivate their protein targets via direct binding, but are not able to actively
eliminate the protein. The RNAi mechanism operates upstream of protein
synthesis to allow for the prevention of disease-causing proteins from being
translated in the first place. Unlike other antisense technologies, the RNAi
mechanism interferes only with translation and not with DNA transcription. As
siRNA does not interact with chromosomal DNA, the lack of DNA interaction
reduces the possibility of adverse gene alterations found in DNA-based gene
therapy (16).
1.3.5 Enable drug discovery process
As siRNA sequences can be synthetically designed based of the
knowledge of the mRNA target transcript, the identification of candidate siRNA

8
molecules can be more straightforward. The emergence of high-throughput gene
expression profiling of cancer cells has enabled the facile identification of genes
implicated with cancers. The application of RNAi towards these genes creates a
systematic approach to discover new drug targets by allowing loss of function
studies to elucidate their exact role. For drug discovery, RNAi has the greatest
impact on target validation and identification, a process by which the roles of
specific genes are functionally linked to a disease. Using RNAi to knockdown a
gene, it can be determined if the gene is involved in a certain pathway or
mechanism and whether it would serve as a viable target for small molecule
therapy.
1.4 Opportunity for RNAi in cancer
The discovery of RNAi and subsequent use of siRNA to exploit this
mechanism has revealed new opportunities for the development of novel
therapeutic systems to treat previously incurable diseases. It has been reported
that synthetic siRNAs are able to knock down targets in various diseases in vivo,
including hepatitis B virus, human papillomavirus, ovarian cancer, bone cancer,
hypercholesterolaemia, and liver cirrhosis, amongst others (21–25). The ability of
siRNA to target any gene with a complementary sequence, makes it a potentially
potent therapeutic, especially for cancer (20). Over the past several decades
many important genes associated with different cancers have been identified
along with their mutations and pathways through which they can be
characterized (26). As a genetic disease, cancer is a well suited candidate for
siRNA-mediated gene therapy. Already, a number of siRNAs have been

9
developed against dominant oncogenes, malfunctionally regulated oncogenes,
and viral oncogenes involved in carcinogenesis. In addition, siRNAs have been
studied as a therapeutic means of silencing target molecules crucial for tumor-
host interactions and tumor resistance to chemo- or radiotherapy. The resulting
silencing of these critical cancer-associated target proteins by siRNAs has led to
significant apoptotic and/or antiproliferative effects (27).
1.4.1 Opportunity for RNAi-mediate gene therapy in triple-negative breast
cancer
Breast cancer is a heterogeneous disease comprised of multiple subtypes
with different molecular profiles that govern cellular morphologies, clinical
aggressiveness, and response to therapy (Figure 1.2) (28). Triple-negative
breast cancer (TNBC), which contribute to 15% of diagnosed breast cancer
cases, is immunohistochemically negative for estrogen receptors (ER),
progesterone receptors (PR), and lacks the overexpression of the human
epidermal growth factor receptor 2 (HER2). Clinically, TNBCs exhibit a poor
prognosis with a devastatingly aggressive clinical course. For other breast cancer
subtypes, the positive expression of these biomarkers have allowed for the
development of molecularly targeted therapies: tamoxifen is effective against
ER+ breast cancers and Herceptin is targeted against HER2+ breast cancers. As
TNBCs lack such hormone makers and are not amenable to targeted therapies,
small-molecule chemotherapy, although limited in safety and efficacy, has
emerged as the standard of care.

10
Deeper insights into the molecular makeup of TNBCs have identified a
number of novel targets which, while not appropriate for targeting via a small-
molecular inhibitor or protein-based antibody, are well-suited for RNAi-based
therapy. A summary of genes being explored within breast cancers for siRNA-
based therapies and their current status is summarized in Table 1.1 (29).
Common approaches include targeting pathways involved in oncogenesis, tumor
cell survival and proliferation, induction of apoptosis, angiogenesis,
invasion/metastasis, and chemoresistance.
Figure 1.2. Subtypes of breast cancer

11
Table 1.1. Examples of siRNA-based therapies against breast cancer
1.5 Considerations for systemic siRNA delivery
While RNAi holds great potential for cancer therapy, many issues remain
to be resolved in order to develop this technology into a viable bedside treatment
option. The main challenge for clinical translation of RNAi is the efficient delivery
of siRNA therapeutics into target cells. Safe and efficient delivery systems are
needed to protect siRNA from enzymatic degradation during the process of in
vivo delivery and to effectively transfect target cells. Significant progress has
been made recently on the design and development of safe and effective siRNA
delivery systems. Recently reported siRNA delivery systems aimed at various
cancers are summarized in Table 1.2 (30).
Function Target gene BC subtype Cell system Tumor model Therapeutic effects
Cell cycle and proliferation Plk1 TNBC MDA-MB-435s s.c. Induction of apoptosis and inhibition of proliferation
TNBC MDA-MB-435s Orthotopic -
Erα Luminal A MCF-7 Orthotopic -
E2F3 TNBC BT20, LY-2 - -
Akt2 TNBC MDA-MB-231 - Increased mitochondrial volume
AAT TNBC MDA-MB-231 - -
DNMTs Luminal B BT474 Orthotopic Restored tumor suppressor gene expression
RhoA/RhoC TNBC MDA-MB-231 s.c. Reduction of cell metastasis
Orai3 Luminal A MCF-7 - Regulate cell survival and Ca2+ entry
ATM TNBC MDA-MB-231 Orthotopic Regulate DNA repair and cell cycle checkpoints
OPN TNBC MDA-MB-231 s.c. Inhibit angiogenesis
HER2/neu Luminal A and B SK-BR-3, BT-474, MCF-7, - -
TNBC MDA-MB-468
hPRLR Luminal A MCF-7 - Down regulation of cyclin D1
Cell death and survival Mcl-1 TNBC MDA-MB-435WT s.c. Reduced tumor volume when co-delivered with RPS6KA5
TNBC MDA-MB-435R s.c.
Bcl-2 NER2/neu+ N202.1A -
Survivin TNBC MDA-MB-231 - Block angiogenesis
BORIS TNBC MDA-MB-231 - -
Angiogenesis VEGF Luminal B SK-BR-3 - -
ARNT2 Luminal A MCF-7 - Affect HIF-1-regulated metabolism
53BP1 Luminal A, TNBC MCF-7, MDA-MB-231 - Inhibit invasion and metastasis
Chemosensitization P-gp Luminal A MCF-7/A s.c. Efflux transporter
Clusterin Luminal A MCF-7 -
RPN2 Luminal A MCF-7-ADR - Inhibit glycosylation of P-gp to decrease membrane localization
KIF14 TNBC MDA-MB-231, HCC1937 Orthotopic Enhance chemosensitivity to docetaxel
TLN1 TNBC MDA-MB-231, HCC38 Orthotopic Enhance chemosensitivity to docetaxel
EMT, tumor invasion and metastasis PAI-1 TNBC MDA-MB-231 - Inhibit ECM remodeling and angiogenesis
Stat3 TNBC (mouse) 4T1 - Inhibit cell proliferation, survival and tumor angiogenesis
AnxA1 Basal-like BLBC - Inhibit EMT
Smad2 Luminal A and B SK-BR-3 and MCF-7 - Inhibit EMT
COX-2 TNBC MDA-MB-231 - In vivo imaging
MLF2 TNBC Orthotopic Reduced primary tumor growth and lung metastasis
RPL39 TNBC Orthotopic Reduced primary tumor growth and lung metastasis

12
In order to harness the potent therapeutic effects of RNAi, siRNA must
first be efficiently delivered to target tissue and cells. Systemic siRNA delivery
from the bloodstream to the cytoplasm of the target cells has to overcome
numerous challenges in the delivery process (Figure 1.3). In the body, naked
siRNA is susceptible to degradation by endogenous enzymes. Its large size (~13
kDa) and negative charge prevent siRNA molecules from crossing cellular
membranes (31). To address these issues, siRNA is commonly complexed into
nanoparticles to protect against degradation and to facilitate cellular uptake.
Once administered into the bloodstream, the siRNA nanoparticles must traverse
the circulatory system while avoiding kidney filtration, phagocytosis, aggregation
with serum proteins, and enzymatic degradation (32). Phagocytic cells, such as
macrophages and monocytes, pose a significant immunological barrier in both
the bloodstream and the extracellular matrix of tissues. Responsible for
removing foreign materials as protection against viral infection, phagocytes are
also efficient at removing therapeutic agents from the body (31). Another barrier
to systemic siRNA delivery in vivo is the transport of the delivery vehicle from the
bloodstream across the vascular endothelial barrier. Generally, only molecules
less than 5 nm in diameter will cross the capillary endothelium, and those larger
ones will remain in circulation until they are taken up by tissues or organs and
cleared from the body. Some tissues, such as the liver, spleen, and in certain
cases, tumors, allow larger molecules up to 200 nm in diameter, the size of
typical drug delivery nanocarriers, to pass (33).

13
Figure 1.3. Physiological barriers to the systemic delivery of siRNA nanoparticles.
A systemically injected siRNA nanoparticle must avoid filtration, phagocytosis, and
degradation in the bloodstream, be transported across the vascular endothelial barrier,
diffuse through the extracellular matrix, be taken up by the cell, escape the endosome,
and unpackage and release the siRNA to the RNA interference machinery.
Once the complexed siRNA exits the bloodstream, is must diffuse through
the extracellular matrix creating further opportunity for uptake and clearance by
phagocytic cells. The extracellular matrix is a dense network of polysaccharides
and fibrous proteins that creates a resistance against the transport of delivery
vehicles and may interrupt the drug delivery process (34). Entry into the target
cells involves endocytosis wherein the siRNA will be trafficked through various

14
endocytic pathways. Once the siRNA delivery systems have been taken up into
the target cells, the next step in the delivery process is endosomal release,
where siRNA escapes from the endosomal compartments (35). If the siRNA
complexes do not escape from the endosome, they will be trafficked through an
endocytic pathway into lysosomes where the acidic environment will degrade the
siRNA cargo (36,37). To avoid this complication, delivery systems have been
designed to disrupt the endolysosomal membranes and release the siRNA
payload into the cytosol, a concept to be further explored in Chapter 2. Finally,
the siRNA must be released from the carrier so that it can be incorporated into
the RNAi silencing complex.
An ideal delivery system for siRNA should exhibit the following
characteristics: i) biocompatible (non-cytotoxic and non-immunogenic); ii)
biodegradable; iii) protect siRNA cargo from enzymatic degradation; iv) minimal
non-specific tissue uptake; v) tissue and target cell specificity; and vi) efficient
cytosolic siRNA release.
1.6 Current approaches for siRNA delivery
Various delivery vehicles have been developed providing the desired
properties listed above for the in vivo delivery of siRNA. Delivery vehicles
generally consist of cationic polymers or lipid-like materials and take advantage
of the anionic nature of siRNA to form complexes through ionic interactions.
These complexes provide protection against nuclease attack and facilitate
cellular uptake of siRNA via endocytosis. Although the overall positive charge of

15
the carrier-siRNA complexes improve cellular uptake due to favorable interaction
with the negatively charged cellular membrane, many of the cationic agents used
are cytotoxic, thus limiting their clinical potential (38–41). Many design strategies
of delivery vehicles have been developed to improve properties such as stability,
cellular delivery, and biocompatibility. Strategies ranging from the direct chemical
modification of siRNA to different non-viral vehicles have been designed and
evaluated. Numerous physicochemical and biological functions have been
introduced to the delivery systems to protect siRNA from degradation, enhance
cellular uptake, facilitate cytosolic siRNA release and allow site-specific delivery
(42). The development of biocompatible, efficient and specific siRNA delivery
systems to the target cancer cells is the key to translate siRNA into a bedside
therapeutics.
Table 1.2. Examples of siRNA delivery systems for the treatment of cancers

16
Systemic siRNA delivery is a convenient approach for cancer treatment.
However, systemic delivery of naked siRNA experiences rapid renal clearance
due to its small size (17,43). It has been shown that intravenous injection of
naked siRNA results in its accumulation in the kidney and urinary bladder within
the first 5 minutes of administration (44). Pharmacokinetics of siRNA can be
altered by complexing it with carriers to form nanoparticles to prevent rapid
clearance from circulation. Prolonged circulation of the siRNA delivery systems
would allow preferential accumulation of the delivery system in solid tumor due to
the enhanced permeability and retention (EPR) effect. The EPR effect is caused
by the improper formation of the rapidly growing tumor vasculature, which causes
it to be more permeable to large molecules and nanoparticles (45).
Systemic administration of nanoparticles often results in increased
accumulation in the liver, spleen, kidneys and lungs, the organs of the
reticuloendothelial system (RES) (46,47). The surface property of a delivery
system is a critical parameter for minimizing non-specific tissue uptake and
achieving target-specific siRNA delivery because it affects the interaction of the
delivery system with the surrounding environment. A positively charged surface
can readily associate with the negatively charged cellular membrane to facilitate
uptake, including non-specific uptake (48). However, negatively charged serum
proteins in the blood plasma will complex with the positively charged surfaces
rendering the delivery systems ineffective. Hydrophilic biocompatible
polyethylene glycol chains (PEG) are often used to modify the surface of the
delivery systems to mask their positive surface and, consequently, to avoid

17
aggregation in serum and to minimize non-specific tissue and cellular uptake
(49). PEGylation also plays an important role in protecting the delivery systems
against the immune system and accompanying phagocytes. PEG forms a barrier
around nanoparticles creating steric stabilization and protection from the
physiological surroundings (50). By altering the length and structure of the PEG
chain, the stabilization, protective properties and particle size can be optimized
for each delivery system (51). Targeting agents, mainly peptides and proteins,
can be readily conjugated to the modified surface with a PEG spacer to achieve
target-specific siRNA delivery.
Finally, the safety of siRNA delivery systems is the most important
parameter to consider as even the most efficient delivery systems will be deemed
useless if they elicit significant local or systemic toxicity. Early delivery vehicles
studied, namely viral vectors, induced an immune response and thus were found
to be toxic (52). To avoid stimulating an immune response, synthetic lipid and
polymer systems have been developed. Although non-viral delivery systems can
avoid induction of a significant immune reaction from the body, cationic carriers,
especially cationic polymers, can cause serious toxic side effects after systemic
administration. Biodegradable polycations containing environmentally sensitive
linkages have been prepared to reduce the toxicity of the carriers (53). Surface
modification of siRNA nanoparticles can also reduce the toxicity of the cationic
carriers.
1.6.1 Targeted siRNA delivery

18
Targeted siRNA delivery systems for cancer are a promising new class of
experimental therapeutics with the potential to revolutionize medicine with their
increase efficiency and relative low toxicity compared to conventional drugs. The
EPR effect allows siRNA nanoparticles to passively accumulate within tumors
following system administration. However, as siRNA needs to be internalized by
the cancer cells, the EPR effect alone may not provide the necessary means to
ensure a therapeutic effect. Therefore, a strategy of active targeting to cancer
cells is commonly used to enhance specific uptake in cancer cells. Active
targeting involves the direct interaction of a targeting agent with a specific
biomarker or receptor expressed on the surface of the target cells (54–56).
Various cell- and tissue-specific targeting agents, including various
antibodies, peptides or aptamers, have been identified and used for siRNA
delivery (57). When selecting a targeting agent, it is important that the cellular
receptor can be readily internalized after ligand binding and then re-expressed on
the cell surface to allow repeated targeting along with preventing prolonged
ligand-binding disruption. The selected targeting agent should also bind to its
respective receptor with high specificity and affinity. Delivery vehicles equipped
with targeting ligands are able to target and interact with specific cells to enhance
cellular uptake via receptor-mediated endocytosis. Further, since the majority of
the chemical conjugations of these ligands occur on the carrier and not the
nucleic acids of siRNA, the ability of siRNA for gene silencing will not be affected.
Many surface modification techniques allow multiple ligands to be attached to the
delivery vehicle. These multi-functional modification strategies combine ligands

19
as a means of potentially overcoming sequential delivery barriers. The
combination of targeting ligands and endosomal escape ligands can be used to
elicit cell surface binding and receptor mediated endocytosis and to promote
delivery to the cytosol and avoid endosomal-lysosomal degradation, respectively
(58,59). Cell-penetrating peptides (CPPs) are sometime employed to enhance
uptake and can increase cell membrane translocation of high molecular weight
cargo, including siRNA (60).
Recent studies have shown that some targeted siRNA delivery systems
may not produce more favorable biodistribution or enhanced tumor uptake that
non-targeted siRNA nanoparticles, but the former still result in more efficient
gene silencing efficiency in cancer cells (61). For example, luciferase-specific
DOTA-conjugated siRNA molecules were labeled for positron emission
tomography (PET) imaging with 64Cu. The labeled siRNA was then complexed
with cyclodextrin-containing polycation nanoparticles both with and without
transferrin (Tf) as a targeting ligand. The siRNA-nanoparticle complexes were
administered to mice bearing tumors expressing luciferase. PET imaging
revealed that the attachment of the Tf targeting ligand to the surface of the
nanoparticles had a negligible impact on the biodistribution compared to the non-
targeted nanoparticles. Both Tf-targeted and non-targeted nanoparticles were
found to have similar tumor localization kinetics and similar tumor accumulation 1
day after injection, thus accumulation was not correlated to the Tf receptor status
of the tumor cell. However, bioluminescent imaging (BLI) revealed that while
tissue distribution for the targeted and non-targeted nanoparticles was similar,

20
the targeted nanoparticles more effectively suppressed tumor luciferase
expression 1 day after injection. The results revealed that a higher portion of the
siRNA was able to localize in the target cells and thus actively function within the
tumor cells when delivered using the Tf-targeted nanoparticles. Thus, the
greatest advantage of using targeted delivery systems for siRNA comes from
targeted delivery systems being able to deliver more functional siRNA into tumor
cells than non-targeted systems due to receptor-mediated endocytosis (62).
1.6.2 Chemically modified siRNA
siRNA can be chemically modified to alter the physicochemical properties
and to increase the in vivo stability for improved circulation time. Chemical
modifications at various positions along the siRNA duplex have been made to
confer both nuclease resistance in addition to reducing nonspecific activation of
the immune system (63). A common modification is the replacement of the
phosphodiester group with a phosphothionate (PS) at the 3’-end (64). The
introduction of phosphorothioate backbone linkages at the 3’-end of the RNA
strands can inhibit enzymatic degradation by reducing siRNA’s susceptibility to
exonucleases (17). The conversion of 2’-OH into either an O-methyl group (2’-O-
Me), a fluoro (2’-F) group, or a 2-methoxyethyl (2’-O-MOE) group leads to the
decrease of hydrolysis associated with 2’-OH and an increase in half-life and
RNAi activity in cells cultured with plasma (65). The modification of siRNA with
2,4-dinitrophenol (DNP) lead to a heightened nuclease resistance along with an
increase in membrane permeability of the modified siRNA (66).

21
There may be concerns that direct siRNA modification will compromise
gene knockdown efficiency because RNAi evolved using native siRNA, and
altered siRNA may impair its recognition by the RNAi machinery. While it has
been shown that siRNA modified with boranophosphate improved resistance to
degradation, modification at the center position of the antisense strand reduced
RNAi activity (67). The degradation of modified siRNA into molecules not found
naturally in vivo may also raise concerns of introducing harmful byproducts
during this method of siRNA delivery. Furthermore, the requirement of siRNAs to
use cellular machinery for their mechanism of action limits the extent of chemical
modification. As a result, focus has been placed on creating delivery systems in
which unmodified siRNA will be loaded as a cargo for targeted systemic delivery.
1.6.3 Lipid-based siRNA delivery systems
Lipid-based transfection reagents are the most commonly used approach
for the in vitro delivery of nucleic acids to cells. Liposomes have been developed
for effective drug delivery for systemic siRNA delivery. Liposomes form
spontaneously in aqueous environments when a lipid bilayer forms a sphere with
an aqueous core. For example, a set of polar head-groups can form the outer
layer of the nanocomplex, while another set of polar head-groups faces the
interior hydrophilic core where the siRNA payload is held (68). Liposomes can
also form an amorphous structure in which the lipids and nucleic acids of the
siRNA are interspersed throughout. The liposome design can be easily tailored
as multiple lipid types can be incorporated creating flexibility in the physical and
chemical properties (69,70).

22
Several cationic lipid-based delivery systems have been developed for the
in vivo delivery of siRNA for cancer therapy. Lipid composition, siRNA-to-lipid
ratio, particle size and assembly process are the key parameters to optimize
when developing these delivery systems. Although cationic lipids are one of the
most popular and effective nucleic acid delivery vehicles, concerns regarding
their safety profile for therapeutic use exist for some (71). Both in vitro and in vivo
toxicity has been reported for certain cationic lipid siRNA nanoparticles, and
certain synthetic agents have been shown to increase off-target effects of siRNA
(71,72). Consequently, recent interest has focused on developing nontoxic lipid
delivery vehicles. Nevertheless, cationic lipids offer adequate protection against
siRNA degradation by nucleases, improve cell membrane penetration and
reduce siRNA renal clearance, making them a promising siRNA delivery vehicle.
1.6.4 Cationic liposomes
Liposomes have traditionally been popular and widely used delivery
systems because of their good safety profiles and a superior payload compared
to other delivery materials (73). Cationic liposomes have been developed for
delivering nucleic acids, including DNA and RNA. Cationic lipids, such as 1,2-
dioleoyl-3-trimethylammonium-propane (DOTAP) and N-[1-(2,3
Dioleoyloxy)propyl]-N,N,N-trimethyl-ammonium methyl sulphate (DOTMA), along
with helper lipids, such as 1,2-Dioleoyl-sn-Glycero-3-Phosphoethanolamine
(DOPE), are often used to form cationic liposomes and complex with negatively
charged DNA and siRNA, resulting in high in vitro transfection efficiency (74).
While efficient for in vitro nucleic acid delivery, cationic liposomes have had

23
limited success for in vivo gene downregulation due to non-specific tissue uptake
and siRNA release. Cationic liposomes may also interact with serum proteins,
lipoproteins and the extracellular matrix, which cause premature release of the
siRNA cargo. Further, cationic liposomes can activate the complement system of
the innate immune system, which causes the rapid clearance by activated
macrophages of the RES. The surface of cationic liposomes can be modified
with molecules such as PEG to minimize non-specific tissue uptake and to
enhance circulation half-life by eliminating aggregation and clearance by the
RES. An extended circulation half-life allows for sustained availability to take
advantage of the EPR effect, resulting in increased delivery to target sites. To
promote cellular uptake, targeting agents can be incorporated to enable targeted
delivery into a specific cell type. The combination of surface modification along
with active targeting has been demonstrated with a PEGylated liposome modified
with antibodies against receptors for transferrin or insulin for targeted siRNA
delivery to the brain in both mouse and monkey models (71,75).
Cationic immunoliposomal siRNA delivery nanoparticles modified with
anti-transferrin receptor single-chain antibody fragment have been developed to
silence human epidermal growth factor receptor-2 (HER-2) on human breast
carcinoma cells. In a mouse xenograft model, repeat intravenous administrations
of the immunoliposomes lead to inhibition of HER-2 expression. The results of
the in vivo delivery also demonstrated its ability to re-sensitize breast cancer cells
to chemotherapeutic drugs (76,77). Another cationic liposome composed of
dioctadecylamidoglycylspermidine (DOGS) has been used for gene silencing in

24
breast cancer cells (78,79). The lipoplexes composed of the cationic liposomes
complexed with the siRNA payload exhibited low cytotoxicity and mediated high
uptake of cyclin D1-specific siRNA by MCF-7 breast cancer cells in the presence
of serum proteins. They accumulated within specific cytoplasmic compartments
in the periphery of the nucleus. The cationic liposomes were also found to be
effective delivery vehicles of siRNA specific for plasminogen activator inhibitor
type 1 (PAI1) to triple-negative breast carcinoma MDA-MB-231 cells (80).
1.6.5 Neutral liposomes
Neutral liposomes have also been used for systemic siRNA delivery. The
neutral lipid 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) forms
liposomes with a size around 65 nm and a 65% siRNA encapsulation efficiency
in (DOPC)-based neutral liposomes (81). The neutral liposomes containing anti-
EphA2 siRNA have been tested for the treatment of ovarian cancer in animal
models. The liposomes were administered intravenously in mice bearing human
ovarian tumor xenografts. The liposomal EphA2-targeting siRNA inhibited
ovarian tumor growth following treatment. When delivered in concert with
paclitaxel, a significant reduction in the growth of ovarian cancers was observed
in the mouse tumor model (82). The intraperitoneal administration of the
liposomal EphA2-targeting siRNA was also evaluated and found to inhibit ovarian
cancer growth by a similar degree compared to intravenous administration in the
mouse tumor model. The DOPC-based neutral liposomes have additionally been
used for the delivery of other anti-cancer siRNAs, including those targeting focal-
adhesion-kinase, interleukin-8, and β-2 adrenergic receptor, in ovarian cancer

25
mouse models (83–85). The liposomes can deliver siRNA in vivo into tumors 10-
and 30-times more effectively than cationic liposomes (DOTAP) and naked
siRNA, respectively. Twice weekly intravenous injections of DOPC-liposomes at
a dose of 150 μg/kg/day resulted in substantial reduction in the expression of the
target genes (e.g., EphA2, FAK, neuropilin-2, IL-8 or Bcl-2) and tumor size in
mice with different human cancers, including subcutaneous xenografts and
orthotopic tumor models (82). A single injection of DOPC-liposomes (150 μg/kg,
iv. or i.p) leads to inhibition of target protein expression for over 4 days in tumors
in mice. DOPC-based liposomes did not cause any detectable distress or toxicity
and were found to be safe in mice and in nonhuman primates. These liposomes
do not exhibit any toxicity to normal cells including fibroblasts, bone marrow and
hematopoietic cells, making them highly attractive for further development.
1.6.6 Lipid-like siRNA nanoparticles
In addition to liposomal formulations, lipid siRNA nanoparticles have been
used in the systemic delivery of siRNA. Examples include stable nucleic acid-
lipid particles (SNALPs) and cationic solid lipid nanoparticles (SLN). SNAPLs are
constructed from a bilayer composed of a mixture of cationic and fusogenic lipids
to enable cellular uptake and endosomal release of the siRNA cargo. They have
been used to carry chemically modified siRNA targeting to HBV RNA as liver
cancer therapeutics in mouse tumor models as well as successfully deliver
intravenously administered siRNA (2.5 mg/kg) into monkeys with marked
inhibition of apolipoprotein B protein up to 11 days. siRNA delivered by SNALPs
exhibited extended half-life in the liver and resulted in a 95% reduction in HBV

26
serum titers. Delivery by SNALPs did not illicit an immune response and the
reduction in HBV DNA was sustained for up to 7 days following the last dose
administration (86). Solid lipid nanoparticles (SLNs) reconstituted from natural
components of protein-free low-density lipoproteins have also been used to
deliver siRNA. SLNs were prepared using a modified solvent-emulsification
method and composed of cholesteryl ester, triglyceride, cholesterol, dioleoyl
phosphatidylethanolamine (DOPE), and 3-beta-[N-(N’,N’-dimethylamino
ethane)carbamoyl]-cholesterol (DC–cholesterol). These carriers provide efficient
target gene silencing and serum stability, with a minimal level of cytotoxicity.
1.6.7 Polymer-mediated siRNA delivery
Polymer-based delivery systems have been investigated for delivering
plasmid DNA for some time, and more recently polymers have also been
developed for siRNA delivery. Similar to lipid-based systems, polymeric siRNA
delivery systems typically involves a cationic moiety as the core of the carrier.
Since 1990, a wide array of cationic polymers has been developed, initially for
the delivery of plasmid DNA, and some of them have been adapted for siRNA
delivery. While siRNA differs from plasmid DNA in terms of molecular weight,
charge ratio, stability and mechanism of action, they are both nucleic acids and
share similar characteristics applicable to their delivery in vivo. Therefore, much
of what has been learned while developing polymeric-based delivery systems for
DNA can be used in the development of these systems for siRNA (84).

27
Cationic polymers can be classified as synthetic or natural polymers.
Examples of synthetic cationic polymers include linear or branched
polyethyleneimine (PEI), poly-L-lysine (PLL), and cyclodextrin-based polycations.
Examples of natural cationic polymers include chitosan, atelocollagen, and
cationic polypeptides. The advantage of cationic polymers comes from the
simplicity and ease in which complexes can be formed between the polymer and
siRNA. Unlike cationic liposomes, which require a lengthy, multiple step process
for assembly, cationic polymers can complex with anionic siRNA by merely
mixing the two together.
Natural cationic polymers are often employed as they are considered both
biocompatible and biodegradable. Chitosan is the most commonly used natural
cationic polymer for nucleic acid delivery. Chitosan/siRNA nanoparticles have
been administered intranasally to endogenously enhanced green fluorescent
protein (GFP)-transgenic mice. The chitosan/(anti-GFP siRNA) nanoparticles
mediated knockdown of endogenously enhanced green fluorescent protein in
bronchiole epithelial cells (85). Chitosan-coated polyisohexylcyanoacrylate
(PIHCA) nanoparticles have also been used for the delivery of RhoA-specific
siRNA in a mouse model (87).
For siRNA, stable complexation with cationic polymers is an essential part
of successful delivery and transfection. Parameters such as
hydrophobicity/hydrophilicity, polymer molecular weight, and charge density can
be adjusted to achieve optimal complexation with siRNA. The N/P ratio, defined
as the ratio of protonatable polymer amine groups to nucleic acid phosphate

28
groups, is another important parameter that affects the electrostatic binding
between siRNA and cationic polymers (88). When designing nanoparticles as
delivery vehicles for siRNA delivery, an optimal stability condition should be met
such that the nanoparticles will remain chemically stable as to protect against
enzymatic degradation during the process of delivery yet vulnerable to polymer
coating rupture in the endosomal compartment. Minimizing the propensity for
nanoparticle aggregation through either electrostatic or steric stabilization can
achieve physical stability (88). Polymers hold major promise because their unit-
by-unit construct allows optimization and fine-tuning of the properties needed for
efficient transfection and release of siRNA (89). Although typically encapsulation
occurs through electrostatic interactions, some polymers can encapsulate siRNA
using hydrophobic interactions (90). Cationic nanoparticles are believed to
facilitate cellular uptake through associating with the negatively charged
membrane of the target cells. However, in vivo, negatively charged serum
proteins, such as albumin, lead to nanoparticle aggregation. Generally, 150 nm is
the considered size limit for cellular uptake by non-specific endocytosis (91).
Therefore, aggregates of nanoparticles exceeding this size will disrupt cellular
uptake and will not be internalized (92).
The most extensively used cationic polymers, PEI, come in a wide range
of molecular weights and have many protonable amino groups, giving a high
cationic charge density at physiological pH. siRNA forms complexes with PEI
through electrostatic interactions between the phosphate groups of siRNA and
the amino groups of PEI (88). PEI has been shown to have a superior

29
transfection efficiency compared to other polymers and has buffering capability
and proton sponge effect in the acidic endosome, thus releasing the siRNA load
into the cytoplasm (93). Despite these advantages, PEI had been found to be
cytotoxic and induce cell death through mechanisms such as necrosis and
apoptosis in a variety of cell lines (94,95). The cytotoxicity of PEI depends on
both molecular weight and degree of branching, higher molecular weight and
increased branching increase cytotoxicity (96,97). By removing uncomplexed PEI
remaining after the complexation reaction with siRNA, the toxicity can be
reduced. While these purified complexes improved the toxicity profile of the
delivery system, the complexes had to be administered at higher concentrations
to achieve the same level of transfection as their unpurified, cytotoxic counter
partners (94).
Introducing PEG chains to form block copolymers is one strategy used to
reduce the cytotoxicity of high molecular weight PEI. Biodegradable systems
synthesized from PEI and PEG increase the overall charge densities of the
system which in turn reduces the cytotoxicity (95). Further, PEG chains can form
a layer of protection against interactions with degradation enzymes and other
serum proteins and increase the circulation time of siRNA in vivo (96). A PEG
and PLL copolymer was developed and found that a higher molecular weight PLL
will produce prolonged siRNA circulation. Cyclodextrin-containing polycations
can self-assemble with siRNA to form nanoparticles approximately 50 nm in
diameter. These polymer-based nanoparticles form a delivery system where the
terminal imidazole groups of the cyclodextrin-containing polycation assist in the

30
intracellular trafficking and subsequent release of nucleic acids. This system can
be conjugated with PEG chains containing targeting ligands to interact
specifically with cell-surface receptors to create a targeted in vivo delivery system
(96).
Polymeric siRNA delivery systems have been tested as therapeutics for
treating many types of cancer. The complexes of a linear low-molecular weight
PEI with siRNA targeting the HER-2 receptor were administered in a mouse
model with SKOV-3 ovarian carcinoma xenograft. PEI/siRNA complexes
resulted in substantial downregulation of HER-2 and, consequently, significant
inhibition of tumor growth in the animal model (97). A nontoxic copolymer blend
of PEI and PEG, PEI-g-PEG, have been used to deliver siRNA specific for the
signaling peptide of the secretory clusterin, a prosurvival factor that protects cells
from ionizing radiation (IR) injury as well as chemotherapeutic agents for
sensitizing breast cancer cells (98). The siRNA nanoparticles have shown in vitro
suppression of both basal and IR-induced secretory clusterin expression, and
increased susceptibility to IR exposure in human MCF-7 breast cancer cells. This
study demonstrated the ability of the siRNA nanoparticles to mediate the
knockdown of specific cytoprotective factors for DNA repair, antiapoptotosis and
free radical scavenging and to enhance the sensitivity of cancer cells to radiation
therapy, and potentially to chemotherapy (51).
Low molecular weight PEI has been used to prepare biodegradable
cationic polymers for siRNA delivery for cancer treatment. PEI was reacted with
PEG diacrylate (PEI-alt-PEG) to synthesize a degradable poly(ester amine)

31
copolymer. An siRNA targeting against Akt1 complexed with the PEI-alt-PEG
copolymers was delivered through a nose-only inhalation system for the
treatment of lung cancer in a mouse model with urethane-induced lung cancer.
Akt plays a key role in cancer by stimulating cell proliferation, inhibiting
apoptosis, and modulating the protein translation (99,100). Amplification of genes
encoding Akt isoforms has been found in many tumors and dominant negative
alleles of Akt have been reported to block cell survival and to induce an apoptotic
response (101). After a delivery regimen for 4 weeks, the Akt1 siRNA-poly(ester
amine) complexes were successful in downregulating Akt-related signals.
Aerosol-delivered Akt1 siRNA nanoparticles decreased about 80% of the Akt1
protein expression specifically in the lungs and significantly inhibited the
progression of lung cancer in different mouse models. Recent findings also
suggest that aerosol delivery of Akt1 siRNA may be useful for the treatment of
chemotherapy- or radiotherapy-resistant lung cancer. Combination therapy with
other classical chemotherapeutic treatments may enhance the therapeutic
efficacy(102,103).
Polymeric siRNA delivery systems have been used in conjunction with
anticancer drugs for cancer treatment in animal models. For example, cationic
core-shell nanoparticles have been self-assembled from a biodegradable
amphiphilic copolymer poly{(N-methyldietheneamine sebacate)-co-
[(cholesteryloxocarbonylamidoethyl)methylbis(ethylene) ammonium bromide]
sebacate} (P(MDS-co-CES)). Enhanced siRNA transfection with the co-delivery
of paclitaxel was demonstrated in vivo in a 4T1 mouse breast cancer model. The

32
co-delivery of paclitaxel with Bcl-2-targeted siRNA increased cytotoxicity in MDA-
MB-231 human breast cancer cells (104).
1.6.8 Theranostics
Theranostics combines diagnostic and therapeutic strategies to diagnose,
treat and monitor the response to the therapy (105). Cancer is a heterogeneous
disease and current treatments are effective only at select stages and for only a
limited subpopulation of the patient pool (106). Traditionally, imaging and therapy
systems have been developed and investigated independently. Theranostics
allows the improved understanding of how each system works and provides a
comprehensive and coordinated platform for the diagnosis, treatment and
efficacy monitoring of cancer (107). In this way, imaging of the disease can be
done before, during and after treatment to track the delivery of therapeutics and
the response of the disease to the delivered therapeutics.
Recent advances in cancer biology have revealed the extent of the
molecular heterogeneity in cancers of the same type, among the cells of the
same tumor and between primary tumors and their metastatic foci. This
molecular diversity, coupled with the ability of cancer cells to develop multidrug
resistance, emphasizes the insufficiencies of monotherapy. The integration of
biomedical imaging with therapeutics, such as siRNA, has a potential to address
the challenges of cancer heterogeneity and adaption. Molecular imaging can be
used as a means of assessing the cellular phenotypes present in the tumor of
interest to guide the selection of an appropriate target-specific therapy. Repeated

33
molecular imaging can then be used to tailor targeting strategies and treatments
as the tumor evolves in response to the initial therapy (108).
In addition to providing information regarding the molecular makeup of
tumors, biomedical imaging can be used to study many facets of systemic siRNA
delivery to study and determine pharmacokinetics, delivery efficiency and
pharmacodynamics of siRNA delivery systems. siRNA delivery systems can be
labeled with imaging agents to provide real-time tracking of the delivery vehicles,
validating targeting efficacy, monitoring the expression pattern of molecular
targets and early feedback of the therapeutic efficacy before detection of any
anatomic changes (109).
siRNA has been incorporated into dextran coated superparamagnetic
nanoparticles for the simultaneous noninvasive imaging and delivery of siRNAs
to tumors. The coated nanoparticles were also modified with Cy5.5 near-infrared
fluorescence dye and MPAP membrane translocation peptides. A GFP targeting
siRNA was used to target a reporter gene GFP. Cellular uptake and silencing
efficiency of the complexes were investigated in vitro in 9L-GFP glioma tumor
cells. Fluorescence results indicated that these nanoparticle complexes were
able to localize within the cells, predominantly in perinuclear regions. Further, the
theranostic nanoparticles resulted in significant suppression of GFP fluorescence
in the 9L-GFP cells, while in control 9L-RFP cells, RFP fluorescence remained
unchanged. The feasibility of the superparamagnetic nanoparticle-complexes to
act as both imaging and delivery platform was then tested in mice implanted with
9L-GFP and 9L-RFP tumors with in vivo contrast enhanced MRI after

34
intravenous injection. The superparamagnetic nanoparticles were characterized
by their strong T2 magnetic susceptibility effects. The accumulation in tissue
was reflected by a decrease in T2 relaxation time, which results in a loss of
signal on MR images. It was found that the tumors appeared bright on T2 images
taken before injection of the complexes. After administration, images showed a
significant drop in signal decrease in the tumors as a result of MN-NIRF-siGFP
accumulation, indicating strong accumulation of the theranostic nanoparticles in
tumor (110). The therapeutic efficacy of theranostic nanoparticles was tested
with an anti-survivin siRNA in an animal tumor model. Survivin, a member of the
inhibitor of apoptosis protein (IAP) family, is expressed solely in most human
neoplasms making it an ideal therapeutic target (111). The theranostic
nanoparticles with siSurvivin were administered systemically to nude mice
bearing human colorectal carcinoma tumors. Silencing efficiency was confirmed
by RT-PCR. Transcript level of survivin in tumors treated with the nanoparticles
was 97% lower than control tumors. Additionally, levels of tumor-associated
apoptosis and necrosis were higher compared to controls (111).
The effectiveness of theranostic nanosized siRNA complexes can be
improved by conjugating tumor-specific ligands. Paramagnetic nanoparticles
were coupled with a fluorescent Cy5 dye component, siRNA and a PEGylated
cyclic Arg-Gly-Asp (RGD) peptide. RDG conjugated nanoparticles bound strongly
to αvβ3 integrin, which is overexpressed on endothelial cancer cells and were
readily internalized into cells by receptor-mediated endocytosis. MRI and
fluorescence imaging non-invasively revealed that the targeted nanoparticles

35
reached the cytoplasm of the target cells, possibly through the endolysosomal
pathway. Once in the cytosol, the disulfide bond was cleaved by a glutathione to
release the siRNA from the nanoparticle. The free siRNA then went on to
incorporate into the RISC to initiate posttranscriptional gene-silencing (112).
1.6.9 siRNA in clinical trials
Since 2004, a handful of siRNA therapies have undergone various
degrees of clinical evaluation (113–115). Table 1.3 summarizes select examples
of clinical trials involving siRNA. siRNA has been investigated for the treatment
of ocular, retinal, and respiratory diseases. Many of the trials employ local
administration of siRNA or delivery of naked siRNA, due to the target tissue
being easily accessible by localized delivery methods. For example, intravitreal
injection was used to delivery PF-655 (Quark Pharmaceuticals) as a means to
target RPT801 for the treatment of age-related macular degeneration and
diabetic macular edema. SYL040012 targeting β2 adrenergic receptor was
formulated into an eye drop for treatment against glaucoma.
Exploration into the application of siRNA towards cancer involving active
delivery strategies have begun to enter into the clinical phase of development.
Most notably, the siRNA therapeutic CALAA-01 (Calando Pharmaceuticals) was
the first targeted systemic siRNA delivery system in clinical trial for cancer
treatment to be terminated. The delivery system was comprised of four
components: (1) a cyclodextrin-based cationic linear polymer, (2) an siRNA that
silences the expression of the M2 subunit of ribonucleotide reductase (RRM2),

36
(3) PEG chains to increase the in vivo stability of the siRNA nanoparticles, (4) a
human transferrin protein (Tf) targeting ligand conjugated to the surface of the
nanoparticles to engage transferrin receptors overexpressed on the surface of
cancer cells. The four components formed a stable targeted siRNA delivery
system through complexation and self-assembly. The transferrin-tagged CALAA-
01 nanoparticles mediated efficient cancer cell targeting and intracellular uptake.
Initially, three patients with metastatic melanoma received doses of 18, 24, and
30 mg m-2 intravenously. One patient received an additional dose 1 month after
the final dose of the first cycle. Immunohistochemistry and staining was
conducted on biopsied tissue samples and compared with archived tissue
samples. The study showed that nanoparticles localized intracellularly in tumor
tissue and not in the surrounding epidermis. Tumor RRM2 mRNA levels were
measured by quantitative real time reverse-transcriptase polymerase chain
reaction (qRT–PCR) and a reduction in RRM2 mRNA was observed in post-
treatment samples (116). While not explicitly stated, the reasons for terminating
the clinical development may have stemmed from an observed rapid clearance
and activation of an acute inflammatory cytokine response (117).
ALN-VSP01 (Alnylam Pharmaceuticals’), a SNALP-based delivery system,
targets both VEGF and kinesin spindle proteins for the treatment of liver and
other solid tumor cancers (ALN-VSP02). Following delivery, 50% of patients with
hepatocellular carcinoma demonstrated an anti-VEGF effect. Another SNALP
based therapy, ALN-TTR01, was able to significantly knockdown levels of serum
transthyretin in patients with transthyretin-related amyloidosis. Many

37
pharmaceutical and biotechnology companies also have a number of siRNA
candidates in development, inevitably lending to an increase in the number of
siRNA-based therapies reaching clinical trials (118).

38
Table 1.3. Select examples of siRNA therapies in clinical trials
Clin
ical
ap
plic
atio
nN
ame
Ind
icat
ion
(s)
Gen
e ta
rget
(s)
Spo
nso
rSt
atu
s
Occ
ula
r an
d r
etin
alQ
PI-
10
07
Op
tic
atro
ph
yC
asp
ase
2Q
uar
k P
har
mac
euti
cals
Ph
ase
I
TD1
01
Pac
hyo
nyc
hia
co
nge
nit
a
Ke
rati
n K
6a,
K6
b, K
16
, K1
7
mu
tati
on
sP
ach
yon
ych
ia C
on
gen
ita
Pro
ject
Ph
ase
I co
mp
lete
d
AG
N2
11
74
5
Age
-rel
ated
mac
ula
r d
egen
erat
ion
ch
oro
idal
neo
vasc
ula
riza
tio
nV
EGFR
1A
llerg
an, S
irn
a Th
erap
euti
cs In
c.P
has
e I/
II co
mp
lete
d
SYL0
40
01
2G
lau
com
aβ
2 a
dre
ner
gic
rece
pto
rSy
len
tis
Ph
ase
I, II
Bev
asir
anib
Mac
ula
r d
egen
erat
ion
VEG
FO
pko
Hea
lth
, In
cP
has
e II,
co
mp
lete
d
Can
cer
NC
T00
67
25
42
Met
asta
tic
mel
ano
ma
LMP
2, L
MP
7, M
ECL1
, MA
RT-
1,
tyro
sin
ase,
gp
10
0, M
AG
E-3
Du
ke U
niv
ersi
ty M
edic
al C
ente
rP
has
e I
AP
N4
01
Solid
tu
mo
rs (
Mel
ano
ma,
Kid
ney
, Pan
crea
tic,
oth
er)
E3 u
biq
uit
in li
gase
Cb
l-b
Wak
e F
ore
st U
nvi
vers
ity,
Ap
erio
n
Bio
logi
csP
has
e I
ATU
02
7A
dva
nce
d s
olid
tu
mo
rsP
KN
3Si
len
ce T
her
apet
uic
s G
mb
HP
has
e I
CA
LAA
-01
Solid
tu
mo
rs
M2
su
bu
nit
of
R2
Cal
and
o P
har
mac
euti
cals
Ph
ase
I, t
erm
inat
ed
siG
12
D L
OD
ERP
ancr
eati
c C
ance
rM
uta
nt
KR
AS
Sile
nse
ed
Ltd
.P
has
e I
siR
NA
-Ep
hA
2-D
OP
CA
dva
nce
d, r
ecu
rren
t ca
nce
rEp
hA
2M
D A
nd
erso
n C
ance
r C
ente
rC
urr
entl
y e
nro
llin
g
TKM
-08
03
01
Pri
mar
y an
d s
eco
nd
ary
liver
can
cer
Po
lo-l
ike
kin
ase
1
NC
IP
has
e I
DC
R-M
YCH
epat
oce
llula
r C
arci
no
ma
MYC
Dic
ern
a P
har
mac
euti
cals
Inc
Ph
ase
I/II
Kid
ney
QP
I-1
00
2/I
5N
PA
cute
kid
ney
inju
ryp
53
Qu
ark
Ph
arm
., In
c.Te
rmin
ated
, Ph
ase
I
QP
I-1
00
2/I
5N
PD
elay
ed g
raft
fu
nct
ion
kid
ney
tra
nsp
lan
tp
53
Qu
ark
Ph
arm
., In
c.A
ctiv
e, P
has
e I,
II
QP
I-1
00
2/I
5N
PK
idn
ey in
jury
acu
te r
enal
fai
lure
p5
3Q
uar
k P
har
m.,
Inc.
Co
mp
lete
d, P
has
e I
SPC
36
49
Hep
atit
is C
vir
us
miR
-12
2Sa
nta
ris
Ph
arm
Act
ive
, Ph
ase
II
pH
IV7
-sh
I-TA
R-C
CR
5R
ZH
IV
HIV
Tat
pro
tein
, HIV
TA
R R
NA
,
hu
man
CC
R5
Cit
y o
f H
op
e M
edic
al
Cen
ter/
Ben
ite
cA
ctiv
e, P
has
e 0
ALN
-RSV
01
RSV
in v
olu
nte
ers
RSV
nu
cleo
cap
sid
Aln
ylam
Ph
arm
.C
om
ple
ted
, Ph
ase
II
ALN
-RSV
01
RSV
in lu
ng
tran
spla
nt
pat
ien
tsR
SV n
ucl
eoca
psi
dA
lnyl
am P
har
m.
Co
mp
lete
d, P
has
e I
ALN
-RSV
01
RSV
in lu
ng
tran
spla
nt
pat
ien
tsR
SV n
ucl
eoca
psi
dA
lnyl
am P
har
m.
Act
ive
, Ph
ase
IIA
nti
vira
l

39
1.7 Objectives
Objective 1: Evaluation of the effect of N/P ratio on physicochemical and
biological properties ECO/siRNA nanoparticle system
The physicochemical properties of a nanoparticle can directly influence the
biological efficacy of such systems both in vitro and in vivo. Chapter 3 focuses
on understanding the factors that govern how ECO and siRNA assemble into
nanoparticles with a specific interest in how the N/P ratio can directly influence a
wide array of parameters. Extensive physicochemical and biological
characterization of the ECO/siRNA nanoparticles reveals how the N/P ratio can
be easily altered to create a tunable system. Further, the intrinsic properties of
the ECO lipid structure are explored to fully characterize the multifunctional
properties exhibited by the ECO/siRNA nanoparticles. The results demonstrate at
the optimal N/P ratio, properties such as siRNA binding efficiency, nanoparticle
stability, silencing efficiency and cytotoxicity are balanced and the nanoparticles
exhibit pH-dependent membrane disruption and glutathione-mediated siRNA
decomplexation to promote endolysosomal escape and cytosolic siRNA release,
respectively.
Objective 2: Assess the therapeutic efficacy of peptide-targeted ECO/siRNA
nanoparticles delivering Integrin β3-specific siRNA on metastatic triple-
negative breast cancer
Functional disruption of integrin β3 has shown potential in vitro as a means to
inhibit TGF-β-mediated epithelial-mesenchymal transition, the seminal event in
the metastasis of breast cancer cells. In chapter 4, ECO/siRNA nanoparticles

40
delivering integrin β3-specific siRNA were used to evaluate the potential of RNAi-
based therapy against metastatic triple-negative breast cancer. The effect of
nanoparticle-mediated integrin β3 silencing was evaluated in vitro within the
context of the events that comprise the metastatic cascade. The ECO/siRNA
nanoparticles were modified with an RGD-targeting ligand anchored with a PEG
spacer to allow for the systemic administration of the nanoparticles in vivo. These
targeted nanoparticles were shown to enhance cellular uptake within post-EMT
breast cancer cells. Two animal models were used to determine the effect of
integrin β3 silencing on i) outgrowth within the pulmonary environment and ii)
primary tumor growth and development of metastases. RGD-targeted ECO/siβ3
nanoparticles were found to reduce primary tumor burden and pulmonary
outgrowth while also inhibiting metastasis.
Objective 3: Design a pH-cleavable PEG-moiety to overcome the PEG
dilemma
PEGylation of non-viral gene delivery systems often leads to an increase in
lysosomal degradation of the nucleic acid cargo and a decrease in functional
efficacy. To improve the efficacy of the targeted and PEGylated ECO/siRNA
nanoparticles, a pH-cleavable PEG system was created in Chapter 5 using the
acid-labile hydrazone linkage. In vitro characterization revealed the hydrazone
linkage restored the important pH-sensitive endolysosomal escape ability and
improved gene silencing. In vivo, noninvasive bioluminescent imaging (BLI) and
fluorescence molecular tomography (FMT) is used to monitor the in vivo tumor
targeting and functional activity of the siRNA nanoparticles and provide crucial

41
insights into the in vivo behavior of the targeted and pH-cleavable siRNA
nanoparticles.
Objective 4: Determine therapeutic efficacy of eIF4E silencing in
combination with paclitaxel using dual pH-responsive ECO/siRNA
nanoparticles in drug-resistant triple-negative breast cancer
Chapter 6 explores the combination of RNAi-based therapy against eIF4E with
the small-molecule chemotherapeutic paclitaxel in drug-resistance triple-negative
breast cancer. Using the RGD-targeted pH-cleavable PEGylated ECO/siRNA
nanoparticles, siRNA directed against eIF4E was used to sensitize drug-resistant
breast cancer cells to paclitaxel therapy in vitro. In vivo, a combination of
nanoparticle-delivered siRNA and paclitaxel was more effective in reducing tumor
growth than either therapy alone in a primary mammary fat pad tumor model.
The results highlight the potential of such combination RNAi- and small-
molecule-based therapeutic regimens to combat highly aggressive triple-negative
breast cancers.

42
Chapter 2
pH-Sensitive Cationic Lipid-Based siRNA Delivery Systems

43
2.1 Introduction
To date, a large number of nanoparticles systems have been explored to
overcome the obstacles and improve the silencing efficiency of siRNA delivery.
The early development of delivery vehicles for siRNA-based applications relied
heavily upon systems originally created for the intracellular delivery of DNA
(67,114,119–121). Initially, those materials developed for DNA delivery were
used for RNA delivery; however, siRNA delivery and DNA delivery differ in
regards to several important aspects (122). The molecular weight of siRNA is
approximately 13 kDa while the molecular weight of DNA molecules used in gene
therapy is typically several hundred times larger (123). As the size of a nucleic
acid/nanoparticle complex is in part dictated by the size of the nucleic acid cargo
in relation to the nanoparticle carrier, those materials optimized for DNA delivery
may not necessarily be ideal for siRNA delivery (124–126). Additional differences
include the site of action within the cell and the stability of the respective
molecules. To initiate RNAi-mediated gene silencing, siRNA molecules must be
delivered into the cytosol, unlike DNA which must localize within the nucleus.
Further, RNA molecules have an increased susceptibility to hydrolysis and
therefore, extra care must be taken to ensure sufficient packing and protection of
the RNA cargo from the harsh and dynamic physiological environments (40,127).
The field of lipid-based synthetic transfection began with the development
of N-[1-(2,3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride (DOTMA) as
the first cationic lipid-based gene delivery system (128–130). DOTMA contains a
trimethylammonium polar head (quaternary amino group), a glycerol-like

44
backbone with two linkage bonds, and two oleyl lipid hydrocarbon tails. An early
study based off the success of DOTMA aimed to identify the structural
characteristics of lipid-based systems critical for a high transfection efficiency
(131). The lipid-based analogues synthesized varied in backbone structure,
aliphatic chain composition, and linkage bonds. In vivo luciferase silencing data
following intravenous administration of the accompanying derivative structures
was used to propose a framework to classify the features common to those lipids
found most effective for in vivo DNA delivery (131). These features included: i) a
cationic head group and its neighboring aliphatic chain being in a 1,2-relationship
on the backbone; ii) an ether bond for bridging the aliphatic chains to the
backbone, and iii) paired oleyl chains as the hydrophobic anchor into the lipid
assembly.
Recent work has focused on optimizing materials to create delivery
systems specifically for the application of siRNA delivery (84,132–134). To do so,
an expanded library of 1200 lipid-based materials has been developed and
characterized specifically for siRNA delivery applications (135). Within the library
of structures, the following parameters were systematically adjusted to enable
the study of structure-function relations: i) hydrophobic tail chain length and
saturation; ii) linkage group between hydrophobic tail and amino group; iii)
primary R group on amine; and iv) post-synthetic quaternization of the amino
group. A similar framework was created and identified the following properties as
ideal: i) greater than two amines per head group; ii) amide bonds between the

45
amine ‘core’ and acyl tails; iii) greater than two acyl chains; iv) acyl chains
between 8 and 12 carbon atoms; and v) a least one secondary amine.
Regardless of DNA or siRNA as the cargo, nucleic acid delivery systems
must be designed to: i) protect the nucleic acid cargo from degradation during
systemic circulation; ii) ensure accumulation within the target site to avoid non-
specific uptake in healthy, non-target tissue; iii) promote cellular uptake and
endolysosomal escape; iv) release nucleic acids intracellularly. As such, cationic
lipid systems developed for siRNA delivery should invariably consist of three
main structural domains: 1) a cationic head group; 2) backbone and linker
groups; 3) hydrophobic tail groups. Careful design and characterization of
cationic lipid structures will allow for the fine-tuning of physicochemical properties
and biological activity to achieve maximum intracellular siRNA delivery and RNAi
activity.
2.2 Endosomal escape: The rate-limiting step
Following internalization, siRNA delivery vehicles encounter a unique set
of intracellular obstacles unlike those experienced during systemic circulation
and extracellular transport. SiRNA-based nanoparticles are typically internalized
via endocytosis, whereupon they will be trafficked through a series of maturing
acidic vesicles leading to the lysosomes (136,137). SiRNA systems must
therefore be designed to escape these compartments and avoid exposure to the
degradative enzymes contained within the harsh acidic environment of late-
endosomes and lysosomes. Non-targeted siRNA nanoparticles with a net
cationic surface charge can non-specifically interact with the negatively-charged

46
lipids of the cell membrane to promote internalization, hypothesized to be
through a combination of adsorptive pinocytosis and direct lipid membrane fusion
(138). Nanoparticles decorated with targeting ligands will mediate specific
interactions with receptors expressed on the cell-surface to facilitate receptor-
mediated endocytosis (139,140). While the exact mechanism of internalization
may vary depending on the physicochemical properties of the siRNA delivery
system, targeted or non-targeted nanoparticles will become localized within
endocytic vesicles (141).
The first vesicle in the trafficking pathway, the endosome, can fuse with
other sorting vesicles to transport the internalized content back to the cellular
membrane and induce exocytosis (142,143). In fact, recent work using a cationic
lipid-based siRNA nanoparticle system revealed that up to 70% of the
internalized siRNA underwent exocytosis (144). Endosomal vesicles not involved
in recycling will continue to be trafficked through the endocytic pathway to late-
endosomes (145). Late-endosomes engage ATPase proton-pump enzymes to
rapidly acidify the vesicle environment to a pH of 5-6 before maturing into
lysosomes with a pH of 4.5 and containing degradative enzymes (146). If trapped
inside these acidic vesicles, any siRNA or nucleic acid load will be degraded. As
such, an emphasis on designing siRNA delivery systems capable of escaping the
fate of exocytosis and/or lysosomal degradation is the key to achieving efficient
target gene knockdown.
2.3 pH-sensitive Amphiphilicity

47
A desirable feature of siRNA delivery systems is tunable pH-sensitive
amphiphilicity. Amphiphilicity is described as the characteristic of chemical
structures to contain both hydrophilic and lipophilic domains (147). Amphiphiles
can self-assemble in an aqueous environment, either alone or with the help of
co-lipid helpers, and can associate with nucleic acids (ie., siRNA, DNA,
microRNA) to form cationic-lipid/siRNA complexes (148). Such complexes
enhance the protection and the structural integrity of the siRNA cargo against
RNases.
As the siRNA carrier system progresses through the cellular trafficking
pathway, it will encounter an increasingly acidic environment as mentioned
above. Therefore, specific pH-sensitive amphiphilicity within an acidic
environment resulting in structural changes to enhance interactions with the lipid
walls of vesicles to initiate membrane disruption should be achieved to trigger
endolysosomal membrane destabilization and subsequent escape. Minimal
amphiphilicity should be observed at physiological pH to minimize non-specific
membrane disruption and cytotoxic effects as the siRNA carrier travels through
systemic circulation.
2.4 Mechanism of endolysosomal escape
While the precise mechanism by which cationic lipid-based siRNA delivery
systems mediate endolysosomal escape has yet to be entirely elucidated, two
main hypotheses exist to explain the observed event: i) the proton sponge effect
and ii) lamellar phase transition.

48
The proton sponge effect is a proposed mechanism for delivery materials
that contain amines with pKa values that range between 7 and 5, thus matching
the pH range encountered during endolysosomal trafficking (39,149). According
to the proton sponge effect, following internalization, the amine groups become
protonated as the endosome acidifies. These amines possess a buffering
capability that results in an influx of protons and chloride ions to create an
osmotic imbalance. As water enters the endosomes to counter this effect, the
endosomal vesicle inflates causing it to rupture and ultimately release its
contents into the cytosol. As such, primary, secondary and tertiary and imidazole
amines of various pKa’s are often incorporated into nucleic acid delivery systems
for their buffering capabilities (150).
Initially, it was argued that complexes arranged in the inverted hexagonal
phase had a greater transfection efficiency compared to those in the lamellar
phase (151,152). However, conflicting data has disputed that a direct correlation
exists between the complex structure and transfection efficiency (153,154). More
recently it has been suggested that the dynamic evolution of a complex’s
structure upon interactions with anionic cellular lipids is the critical factor that
determines the extent of transfection efficiency (151). The lamellar phase
transition occurs with a class of fusogenic cationic-based lipids that form into
non-bilayer formations under aqueous environments (155–157). These lipids can
adopt the inverted hexagonal phase characterized by a long, cylindrical, and
inverted micelle structure. Complexes comprised from such lipids that undergo a
transition from lamellar to an inverted hexagonal phase (lamellar-to-hexagonal

49
inverted phase transition) can fuse with the anionic lipid membranes of
intracellular vesicles to release their nucleic acid cargo into the cytosol. To
achieve the transition, the lipid must contain unsaturated hydrocarbon chains.
Studies have demonstrated that cationic-lipid based systems containing
unsaturated lipids have superior gene silencing efficiency compared to systems
containing saturated lipids. The unsaturation of the hydrophobic lipid tail directly
influences the fusogenic potential of the delivery system.
Dioleoylphosphatidylethanolamine (DOPE) is a lipid with fusogenic capabilities
and is commonly incorporated into delivery systems as a helper lipid to mediate
lamellar-to-hexagonal transition and improve the transfection efficiency
(158,159).
2.5 pH-Sensitive Lipids for siRNA Delivery
Lipids containing fusogenic capabilities and pH-sensitive, or ionizable,
domains are commonly used to improve the transfection efficiency of siRNA
delivery systems. The first developed systems for siRNA using pH-sensitive lipids
demonstrated that the pKa of the amino group governs the endosomal buffering
capability and facilitates endosomal escape. Ionizable lipids, capable of
protonation in an acidic environment, with a pKa of 6.7, were found to be highly
efficient in RNAi-mediate gene knockdown (156). A series of novel pH-sensitive
cationic lipids with varying degrees of saturation were developed: zero (1,2-
distearyloxy-N,N-dimethyl-3-aminopropane (DSDMA)), one 1,2-dioleyloxy-N,N-
dimethyl-3-aminopropane (DODMA)), two (1,2-dilinoleyloxy-N,N-dimethyl-3-
aminopropane (DLinDMA)), or three (1,2-dilinolenyloxy-N,Ndimethyl-3

50
aminopropane (DLenDMA)) double bonds per alkyl chain. Each lipid was then
incorporated into stable nucleic acid lipid particles to assess the impact of lipid
tail saturation on gene silencing efficiency, as more unsaturation within the acyl
chains may enhance the cone shape of the lipids to improve membrane
destabilization induced by the cationic lipids. It was reported that increasing the
saturation within the hydrocarbon chains from 0 to 2 resulted in greater
transfection efficiency, however, 3 double bonds led to no significant
improvement.
Accordingly, DLinDMA was used for further development as a pH-
sensitive cationic lipid siRNA delivery system by optimizing the linker region
connecting the hydrophobic and hydrophilic domains through the introduction of
groups exhibiting different rates of chemical or enzymatic stability. Introduction of
a ketal ring linker domain into DLinDMA (DLin-K-DMA) resulted in the best in vivo
silencing efficiency when compared to analogue lipids containing ester, alkoxy,
carbamate or thioether linkages. Next, the composition of the amine-based head
group was reevaluated using DLin-K-DMA by varying the number of ionizable
groups to study the downstream effect on overall pKa and functionality. Further
structural refinements in the head group design included addition of the following
groups: piperazino, morpholino, trimethylamine and bis-dimethylamino. The
dimethylamino group of the original DLin-K-DMA lipid was found to be superior to
any explored structural modification. As a final point of optimization, the distance
between the amino-based head group and the dioxolane linker domain was
varied through the introduction of methylene groups (Figure 2.1A). The distance
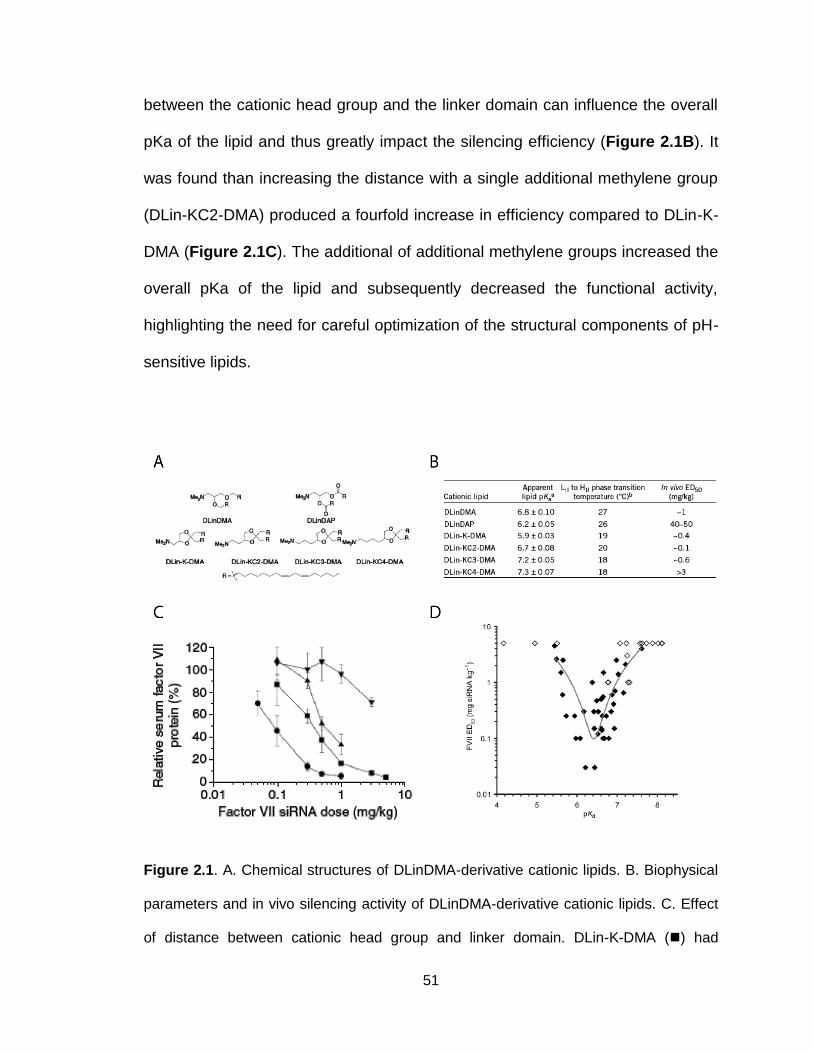
51
between the cationic head group and the linker domain can influence the overall
pKa of the lipid and thus greatly impact the silencing efficiency (Figure 2.1B). It
was found than increasing the distance with a single additional methylene group
(DLin-KC2-DMA) produced a fourfold increase in efficiency compared to DLin-K-
DMA (Figure 2.1C). The additional of additional methylene groups increased the
overall pKa of the lipid and subsequently decreased the functional activity,
highlighting the need for careful optimization of the structural components of pH-
sensitive lipids.
Figure 2.1. A. Chemical structures of DLinDMA-derivative cationic lipids. B. Biophysical
parameters and in vivo silencing activity of DLinDMA-derivative cationic lipids. C. Effect
of distance between cationic head group and linker domain. DLin-K-DMA () had

52
additional methylene groups added between the DMA headgroup and the ketal ring
linker to generate DLin-KC2-DMA (), DLin-KC3-DMA () and DLin-KC4-DMA (). The
activity of PFV formulations of each lipid was assessed in the mouse Factor VII model.
D. Plot of in vivo hepatic gene silencing activity vs. pKa in mice. The library of cationic
lipids were formulated in LNPs and subjected to an ED50 analysis and plotted against
their pKa.
A follow-up study conducted a comprehensive evaluation of 53 ionizable
lipid derivatives of DLin-KC2-DMA to correlate the pKa of the head group with the
intracellular siRNA release profile (160). Indeed, a compelling correlation
between pKa of the polar head group and the siRNA silencing efficiency was
reported. Within the library of 53 structures, the pKa’s ranged from 4.17 to 7.16.
Only those lipids with pKa’s ranging from 6.2-6.5 were able to elicit in vivo gene
silencing (Figure 2.1D). Such findings serve as a value design guide for the
future development of pH-sensitive cationic lipids for siRNA delivery.
Scheme 2.1. General structure of pH-sensitive cationic lipids for siRNA delivery

53
Over the years, our group has synthesized a number of rationally
designed polymerizable surfactants with pH-sensitive amphiphilicity for DNA and
siRNA delivery applications. All carriers are designed to contain the following
three domains with each playing an integral role in the formation of stable and
effective siRNA nanoparticles: i) a protonatable amino head group, ii) a cysteine-
based linker residue, and iii) geminal lipid tail groups (Scheme 2.1). The
protonable amine groups are comprised of various combinations of primary,
secondary, tertiary and aromatic amino groups which can electrostatically
complex with siRNA. The lipid tail groups introduce lipophilicity to further stabilize
the siRNA nanoparticles through hydrophobic interactions in an aqueous
solution. The pH-sensitive amphiphilicity will allow the carrier to alter its
amphiphilic structure when exposed to the acidic environment of the
endolysosomal pathway, resulting in membrane disruption and subsequent
endolysosomal escape. It is crucial that siRNA delivery carriers have selective
amphiphilic behavior such that high membrane disruption is achieved at the
endolysosomal pH and low disruption activity is exhibited at the physiological pH.
Alteration of the amine composition of the head group and the structure of the
lipophilic tails tunes the pH-sensitive amphiphilicity to improve the selectivity of
endolysosomal membrane disruption and enhance the gene silencing efficiency.
The inclusion of cysteine-based residues serves to stabilize the nanoparticles
through autoxidation-mediated polymerization of free thiols into disulfide bonds
and allow for surface modification with PEG and/or targeting moieties via facile
thiol-maleimide chemistry. Further, while the disulfide bonds remain stable during
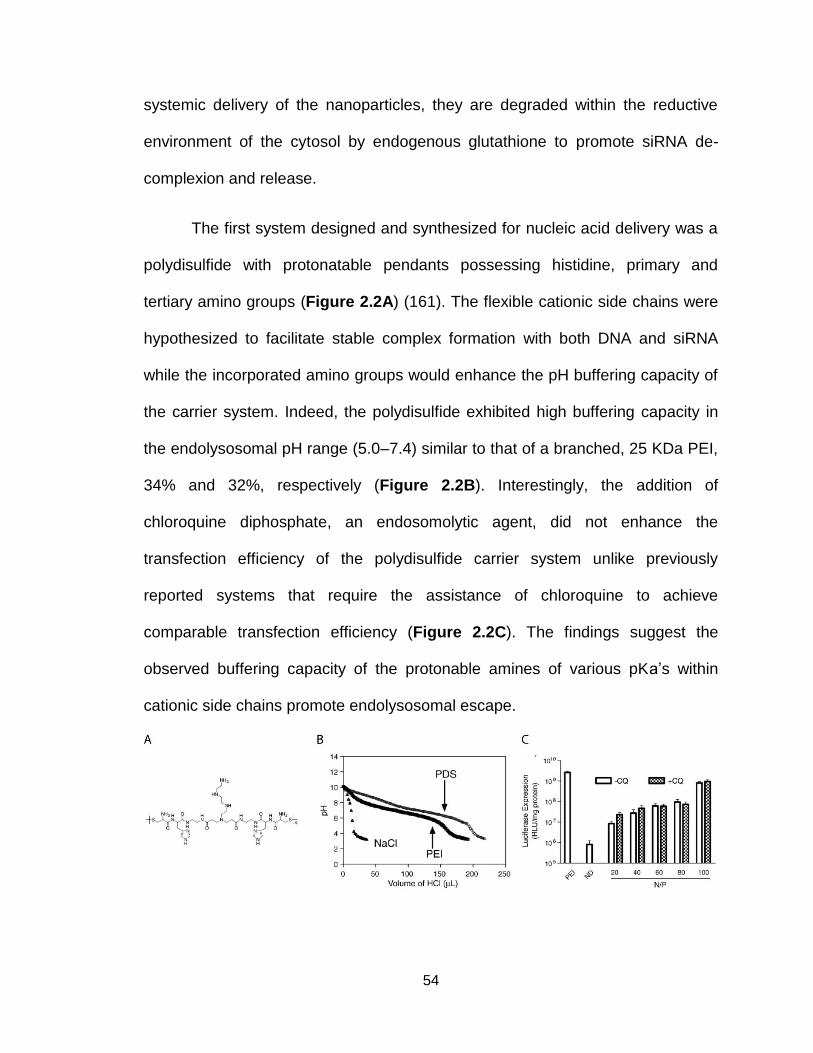
54
systemic delivery of the nanoparticles, they are degraded within the reductive
environment of the cytosol by endogenous glutathione to promote siRNA de-
complexion and release.
The first system designed and synthesized for nucleic acid delivery was a
polydisulfide with protonatable pendants possessing histidine, primary and
tertiary amino groups (Figure 2.2A) (161). The flexible cationic side chains were
hypothesized to facilitate stable complex formation with both DNA and siRNA
while the incorporated amino groups would enhance the pH buffering capacity of
the carrier system. Indeed, the polydisulfide exhibited high buffering capacity in
the endolysosomal pH range (5.0–7.4) similar to that of a branched, 25 KDa PEI,
34% and 32%, respectively (Figure 2.2B). Interestingly, the addition of
chloroquine diphosphate, an endosomolytic agent, did not enhance the
transfection efficiency of the polydisulfide carrier system unlike previously
reported systems that require the assistance of chloroquine to achieve
comparable transfection efficiency (Figure 2.2C). The findings suggest the
observed buffering capacity of the protonable amines of various pKa’s within
cationic side chains promote endolysosomal escape.

55
Figure 2.2. A) Structure of polydisulfide, B) Acid titration profiles of NaCl, PEI and
polydisulfide (PDS), C) transfection efficiency of PDS/DNA complexes in the absence or
presence of chloroquine (CQ) compared with PEI and naked DNA (161).
A small library of polymerizable surfactants exhibiting pH-sensitive
amphiphilicity for siRNA delivery was synthesized using combinatorial solid-
phase chemistry (162). The pH-sensitive amphiphilic cell membrane disruption
was evaluated in rat red blood cells at pH values corresponding to the
environment of the endolysosomal trafficking pathway. The siRNA delivery and
luciferase silencing efficiency was determined in a human U87-luc glioblastoma
cell line engineered to stably express firefly luciferase. Ten distinct structures
were designed and synthesized to create the library of polymerizable surfactants
each with a unique combination of a protonable amine-based head group, dual
peptide linker groups, and geminal, distal lipophilic tail groups to form
nanoparticles with siRNA through charge-charge complexation, lipophilic
condensation, and oxidative polymerization (Figure 2.3A). For the head group,
ethylenediamine (E), triethylenetetramine (T), pentataethylenehexamine (P), and
spermine (S) were selected. These polyamines differ in the number of amino
groups with various pKas to adjust the pH-sensitive amphiphilic behavior of the
carriers. Histidine (H), glycine (G) and cysteine (C) were selected for the peptide
linkage. Fatty acids of different chain lengths and degrees of saturation, including
lauric acid (L), oleic acid (O), and steric acid (St), were used as the hydrophobic
tail groups.

56
The pH-sensitive amphiphilic cell membrane disruption of the library was
evaluated using a hemolysis assay (Figure 2.3B). The results revealed varying
pH-dependent hemolytic activities governed by differences in their structural
composition. Generally, all surfactants exhibited lower hemolytic activity at pH
7.4 when compared with pHs 6.5 and 5.4. Histidine groups contain an imidazole
ring with a pKa of 6 and can become protonated when the environmental pH is
below 6. Additionally, when the imidazole ring is protonated, the histidine residue
can contribute towards the fusogenic properties of the lipid carrier (162). For
surfactants containing histidine residues with the same lipophilic tail groups, the
hemolytic activity increased with the number of protonable amino groups at pHs
7.4 and 6.5. This suggests that increasing the number of protonable amino
groups contributes to an increased head group charge and higher amphiphilicity
at neutral pH. The hemolytic activity was also dependent on the composition of
the lipophilic tail groups. Interestingly, carriers containing the same head group
but unsaturated oleoyl groups exhibited less hemolysis than those with saturated
lipophilic tails at pHs 7.4 and 6.5. By selecting a combination of head group and
lipophilic tail groups that exhibit low hemolysis at pH 7.4 and 6.5 and high
hemolytic activity at 5.4, the specific pH-sensitive amphiphilicity can be finely
tuned. Based on this criterion, EHCO, with its combination of primary, tertiary and
aromatic amine groups in tandem with the unsaturated oleic acid lipid tail groups,
demonstrated specific hemolytic activity only at pH 5.4 with negligible hemolysis
at other pH levels.

57
It has been well documented that the stoichiometric relationship between
cationic lipid charge and anionic RNA charge in the form of lipid-to-siRNA (N/P)
ratio can influence a number of parameters including carrier shape, size, cellular
trafficking, and silencing efficiency. One study using aminoglycoside-based lipids
found that when formulated with low N/P ratios the siRNA lipoplexes formed were
small, stable but anionic. Increasing the N/P ratio led to neutral lipoplexes that
were unstable and aggregated into large complexes [46]. By increasing the N/P
ratio even further, the lipoplexes formed were small, stable and carried a net
positive charge. However, such trends heavily depend on the chemical
composition of the lipid. Along these lines, EHCO was used to study the impact
of N/P ratio on the complexation and formation of siRNA/lipid nanoparticles
(Figure 2.3C). When mixed and incubated with siRNA for 30 minutes,
nanoparticle formation was observed with an N/P ratio as low as 0.5 with a
diameter of 200 nm. Increasing the N/P ratio to 4 increased the nanoparticle
diameter to 3 μm due to aggregation. When the N/P ratio was increased even
further to 10, the nanoparticle sized decreased to 151 nm. At an N/P of 10, the
other surfactants ranged in diameter between 160-210 nm.
The luciferase silencing efficiency and cytotoxicity was evaluated for all
surfactants at an N/P ratio of 10 in U87-luc cells (Figure 2.3D). EHCO resulted in
the highest luciferase silencing (88.4 ± 3.1%) of the surfactants (48% to 81%).
Importantly, all surfactants had high cell viability ranging from 78.6 ± 5.7% to 88.2
± 1.3%. Comparatively, the commercially available TransFast reagent induced
89.6 ± 5.6% luciferase silencing but had poor viability (57.6 ± 2.2%) and
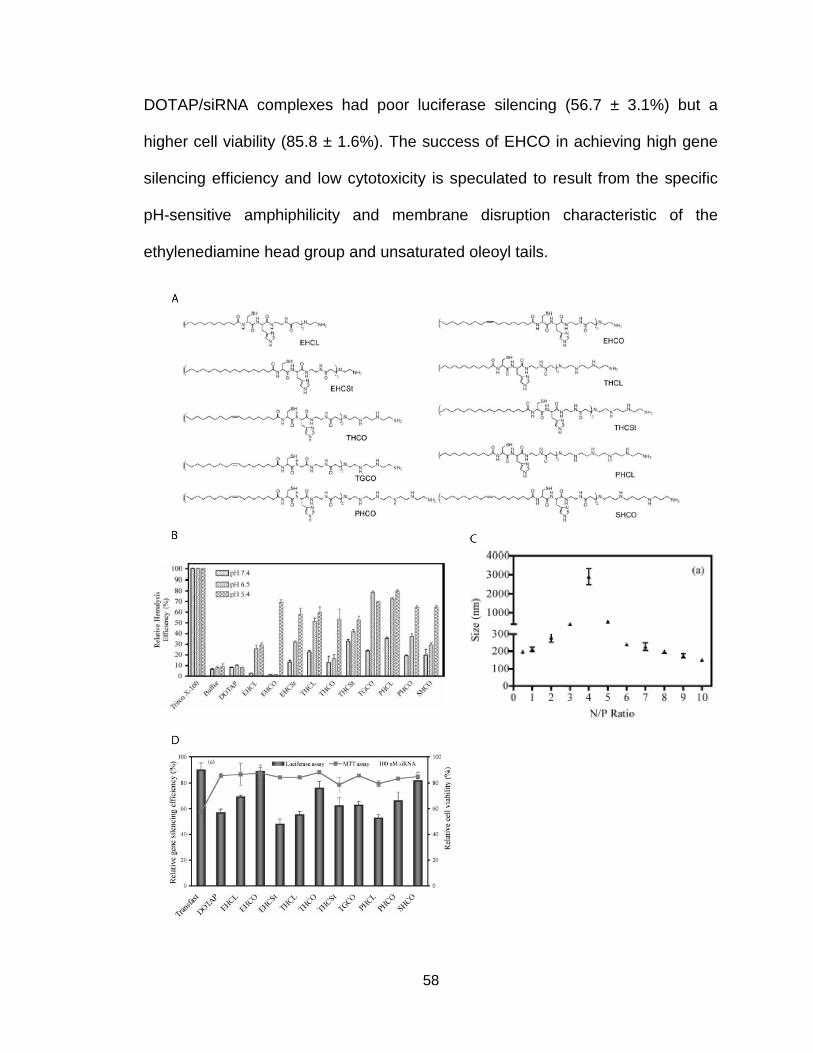
58
DOTAP/siRNA complexes had poor luciferase silencing (56.7 ± 3.1%) but a
higher cell viability (85.8 ± 1.6%). The success of EHCO in achieving high gene
silencing efficiency and low cytotoxicity is speculated to result from the specific
pH-sensitive amphiphilicity and membrane disruption characteristic of the
ethylenediamine head group and unsaturated oleoyl tails.

59
Figure 2.3. A) Chemical structures and abbreviated names of library of polymerizable
surfactants exhibiting pH-sensitive amphiphilicity for siRNA delivery, B) Hemolytic
activity of polymerizable surfactants and DOTAP at pHs 7.4, 6.5, and 5.4 with 1%Triton
X-100 and PBS as controls, C) Particle size of EHCO/siRNA complexes at different N/P
ratios, D) Luciferase silencing efficiency and cell viability of polymerizable surfactants
complexed with siRNA (100 nM). Transfact and DOTAP were used as controls (162).
Among the carriers developed in the library, SHCO also demonstrated
high transfection efficiency with minimal cytotoxicity and was chosen as the
parent compound for a new library of spermine-based protonable surfactants as
pH-sensitive multifunctional non-viral based delivery systems for both plasmid
DNA and siRNA (163,164). Eight new chemical structures were designed for
gene carriers (Figure 2.4A). Each carrier included spermine (S) as the
protonable head group to assist in packing the nucleic acid load. Spermine
naturally complexes with DNA through stabilization of their α-helix structure and
has commonly been incorporated into non-viral gene carrier systems. As before,
histidine (H) and cysteine (C) residues were included to allow fine-tuning of the
pH-sensitive behavior of the carriers and provide a means to stabilize and
functionalize the nanoparticles, respectively. Oleic acid (O) was used again for
the hydrophobic lipid tail groups to introduce flexibility and facilitate the phase
transition involved with endolysosomal escape. The inclusion L-lysine (K)
residues introduced a “Y” joint giving the lipid tails of the carriers an overall
wedge-shaped structure. β-alanine (A) was incorporated on the α-amino group of

60
the lysine and/or histidine residues to alter the distance between the two lipid
tails in an effort to understand the impact on transfection efficiency.
All the spermine-based carriers effectively complexed with pDNA via
charge-charge interactions at N/P ratios ≥ 2 with particle diameters ranging
between 90-130 nm for DNA complexes and 85-120 nm for siRNA complexes
(Figure 2.4B). The hemolytic activity of the carriers at two concentrations was
evaluated at pH 7.4 and 5.5. All carriers exhibited greater hemolytic activity at pH
5.5 compared with 7.4 and also as the concentration was increased from 16 to
40 μM (Figure 2.4C). SKACO demonstrated the most selective pH-sensitive
behavior with the largest increase of hemolytic activity as the pH decreased from
7.4 to 5.5. The incorporation of histidine within the structure again did not
improve the pH-sensitive hemolytic activity. It was observed that carriers with
histidine had lower pH-sensitive hemolytic activity and pDNA transfection
efficiency compared with those corresponding carriers that did not include
histidine (Figure 2.4D). In fact, carriers that included two histidine residues
(SHHKCO and SHHKACO) had even lower pDNA transfection efficiency than
those carriers with only one histidine (SKHCO and SKAHCO). For siRNA
delivery, SKAHCO (N/P 12, 50 nM siRNA) exhibited the highest silencing of
luciferase in U87 cells (84.6 ± 5.5%) (Figure 2.4E). Similarly as with pDNA
delivery, the histidine-containing carriers offered no distinct advantage over
carriers without histidine. The location of the lipid tails also had no clear impact
on the gene silencing efficiency of the spermine-based carriers. Based on these
data, the structural design of effective carriers for nucleic acids were found to be

61
different for pDNA and siRNA, as SKACO demonstrated the highest pDNA
transfection efficiency while SKAHCO had the greatest siRNA-mediated gene
silencing efficiency.

62
Figure 2.4. A) Chemical structures of the spermine-based library of nucleic acid carriers,
B) Particle size of pDNA (top) and siRNA (bottom) complexes at N/P ratio of 12, C)
Hemolytic activity of pDNA complexes formed with spermine-based carriers at N/P ratio
of 12 at pH 7.4 and 5.5. In vitro transfection efficiency of D) pDNA and E) siRNA
complexes at N/P ratio of 12 in U87 cells (163,164).
EHCO was chosen for further development for in vivo delivery due to the
observed high transfection efficiency and low cytotoxicity. Surface modification of
the siRNA nanoparticle surface is necessary for systemic in vivo delivery
applications to impart biocompatibility and avoid non-specific uptake by the
reticuloendothelial system (RES) of the liver and spleen (165,166). Polyethylene
glycol is a polymer commonly incorporated onto the surface, a process called
“PEGylation”, of nanoparticle systems to prolong blood clearance and reduce
non-specific tissue uptake. However, in doing so, PEGylation can also disrupt the
cellular uptake into the target cells (167). To overcome the inhibition of cellular
uptake, targeting ligands can be chemically modified to the distal end of PEG
moieties to restore cell-specific binding and internalization (139). The presence of
thiol groups in EHCO allows for facile and in situ conjugation of cell-specific
ligands to form a targeted siRNA delivery system using simple thiol-maleimide
chemistry (Figure 2.5A).
Bombesin (BN) receptors are found overexpressed on the surface of
various types of cancers, including ovarian and glioblastomas. A bombesin
peptide with high affinity towards BN receptors was used as a targeting ligand.

63
The peptide with a spacer of two 6-aminohexanoic acids was synthesized using
solid-phase chemistry and conjugated to a heterobifunctional NHS-PEG3400-
Maleimide to obtain BN-PEG-Mal. The peptide moiety was then conjugated to
EHCO through reaction of the maleimide group of BN-PEG-Mal with the thiol
groups of EHCO for 30 minutes at 2.5 mol% (168). An mPEG5000-Mal moiety was
used as a non-targeted control. Next, the BN-targeted EHCO/siRNA
nanoparticles were formed by self-assembly of EHCO with siRNA for an
additional 30 minutes through charge-charge complexation, hydrophobic
condensation and autoxidative polymerization.
Figure 2.5. A) Surface functionalization and self-assembly of EHCO to form targeted
siRNA nanoparticles, B) Confocal images of various EHCO/siRNA formulations with
rhodamine-red labelled anti-GFP siRNA to demonstrate (top) GFP silencing efficiency
and (bottom) cellular uptake (168).
Peptide modification of the EHCO/siRNA nanoparticles did not
significantly alter the nanoparticle size but promoted receptor mediated cellular
uptake and gene silencing. The cellular uptake and gene silencing was evaluated
in the Chinese Hamster Ovary (CHO)-d1EGFP cell line engineered to stably

64
express GFP and over-express BN receptors using a rhodamine-red labelled
anti-GFP siRNA (Figure 2.5B). Unmodified EHCO/siRNA nanoparticles were
readily internalized through non-specific interactions between the cationic
nanoparticle and the anionic cell membrane resulting in a strong intracellular red
fluorescent signal and reduced green fluorescence. Modification of EHCO/siRNA
nanoparticles with non-targeted mPEG (2.5 mol%) significantly reduced the
internalization of the nanoparticles and inhibited the gene silencing efficiency due
to a decrease in non-specific interactions with the cellular membrane. Peptide
modification with BN-PEG-Mal (2.5 mol%) to the surface of EHCO/siRNA
nanoparticles promoted robust receptor-mediated endocytosis, cellular
internalization, and gene silencing, as evident by the strong intracellular
rhodamine signal and a significant reduction of GFP signal.
The modification of EHCO/siRNA nanoparticles with targeting peptides
enables systemic in vivo delivery for therapeutic applications. Bombesin- and
RGD-targeted EHCO/siRNA nanoparticles, prepared in a similar manner as
described above, were used to deliver anti-HIF-1α siRNA (2.5 mg/kg) to regulate
tumor hypoxia in nude mice bearing human U87 glioblastoma xenografts (169).
RGD is commonly exploited for use as a targeting peptide due to its high affinity
and specificity towards αvβ3 integrin overexpressed in angiogenic blood vessels
and endothelial cancer cells. Intravenous injection of both BN- and RGD-targeted
EHCO/siRNA nanoparticles (RGD and BN) significantly suppressed tumor
growth in mice when compared to free siRNA (siRNA only) and non-targeted
PEGylated (mPEG) control groups (Figure 2.5A and B). PEI/siRNA complexes
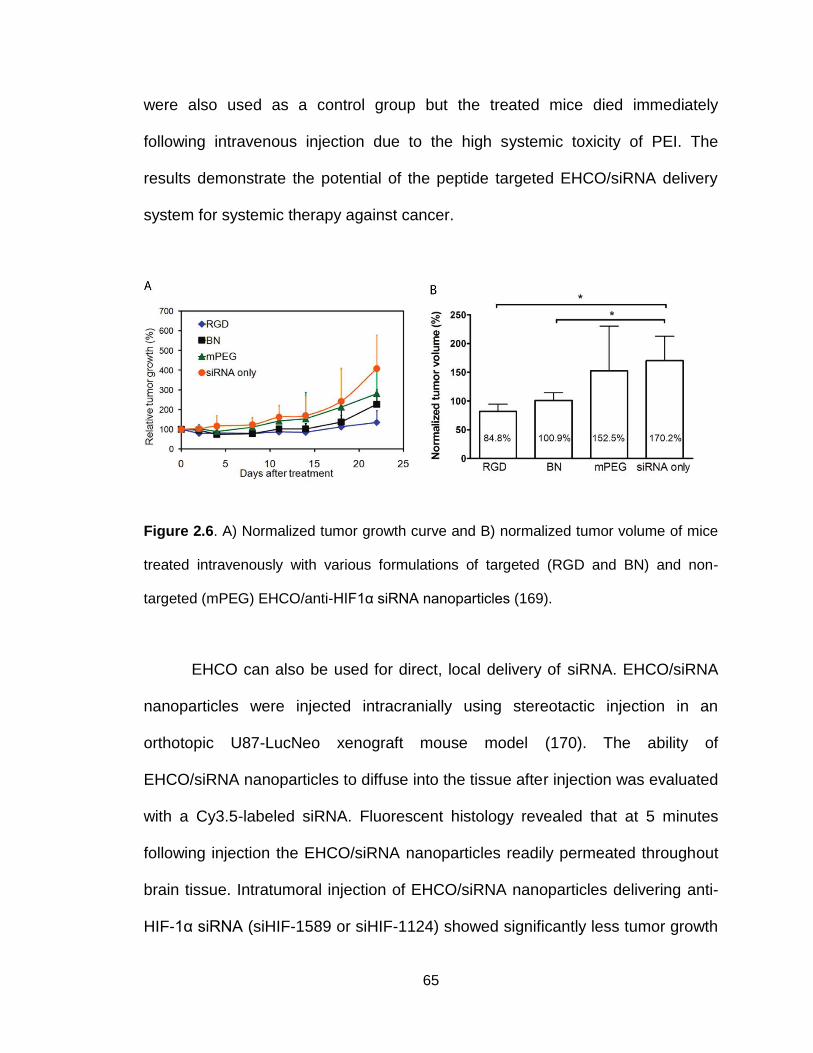
65
were also used as a control group but the treated mice died immediately
following intravenous injection due to the high systemic toxicity of PEI. The
results demonstrate the potential of the peptide targeted EHCO/siRNA delivery
system for systemic therapy against cancer.
Figure 2.6. A) Normalized tumor growth curve and B) normalized tumor volume of mice
treated intravenously with various formulations of targeted (RGD and BN) and non-
targeted (mPEG) EHCO/anti-HIF1α siRNA nanoparticles (169).
EHCO can also be used for direct, local delivery of siRNA. EHCO/siRNA
nanoparticles were injected intracranially using stereotactic injection in an
orthotopic U87-LucNeo xenograft mouse model (170). The ability of
EHCO/siRNA nanoparticles to diffuse into the tissue after injection was evaluated
with a Cy3.5-labeled siRNA. Fluorescent histology revealed that at 5 minutes
following injection the EHCO/siRNA nanoparticles readily permeated throughout
brain tissue. Intratumoral injection of EHCO/siRNA nanoparticles delivering anti-
HIF-1α siRNA (siHIF-1589 or siHIF-1124) showed significantly less tumor growth

66
than the non-specific control groups (siNeg or siGFP) (Figure 2.7A). PEGylated
EHCO/siRNA nanoparticles administered via an osmotic pump significantly
increased the survival of the HIF-1α knockdown group (PEG1589) compared to
the non-specific PEGylated EHCO control (PEGGFP) and non-PEGylated EHCO
(1589) (Figure 2.7B). Knockdown of HIF-1α resulted in robust downregulation of
HIF-1α transcriptional targets, including vascular endothelial growth factor
(VEGF), glucose transporter 1 (GLUT-1), c-MET, and carbonic anhydrase-IX
(CA-IX).
Figure 2.7. A) Normalized average radiance denoting tumor growth in mice injected
intracranially with EHCO formulated with various siRNAs, B) Survival curves of mice
treated with Alzet osmotic pumps delivering various formulations of EHCO/siRNA and
PEGylated EHCO/siRNA nanoparticles (170).
The structure of EHCO was modified further to create two new
multifunctional carriers for nucleic acid delivery with dual histidine residues:
EKHCO and EHHKCO (Figure 2.8A) (171). These two new carriers have the
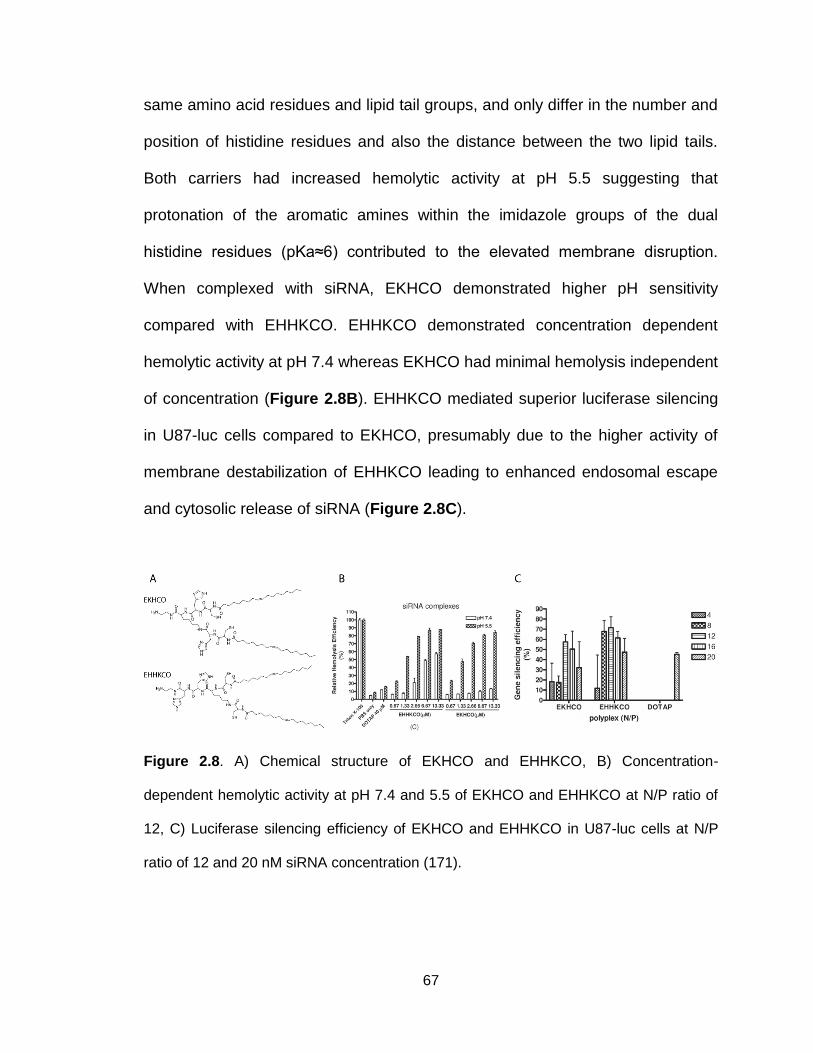
67
same amino acid residues and lipid tail groups, and only differ in the number and
position of histidine residues and also the distance between the two lipid tails.
Both carriers had increased hemolytic activity at pH 5.5 suggesting that
protonation of the aromatic amines within the imidazole groups of the dual
histidine residues (pKa≈6) contributed to the elevated membrane disruption.
When complexed with siRNA, EKHCO demonstrated higher pH sensitivity
compared with EHHKCO. EHHKCO demonstrated concentration dependent
hemolytic activity at pH 7.4 whereas EKHCO had minimal hemolysis independent
of concentration (Figure 2.8B). EHHKCO mediated superior luciferase silencing
in U87-luc cells compared to EKHCO, presumably due to the higher activity of
membrane destabilization of EHHKCO leading to enhanced endosomal escape
and cytosolic release of siRNA (Figure 2.8C).
Figure 2.8. A) Chemical structure of EKHCO and EHHKCO, B) Concentration-
dependent hemolytic activity at pH 7.4 and 5.5 of EKHCO and EHHKCO at N/P ratio of
12, C) Luciferase silencing efficiency of EKHCO and EHHKCO in U87-luc cells at N/P
ratio of 12 and 20 nM siRNA concentration (171).

68
To further explore how chemical modifications to the EHCO core structure
affect the physicochemical properties and performance of the carrier to deliver
siRNA, a new library of pH-sensitive amphiphilic cationic lipid carriers was
developed (Figure 2.9A) (172). Including EHCO, eight total carriers were
synthesized based on the three domain design framework: 1) a protonable
amino-based head group (E, ethylenediamine; S, spermine); 2) an amino acid-
based linker (C, cysteine; HC, histidine-cysteine); 3) two mono- or di-alkene
unsaturated fatty acid tails (O, oleic acid; Ln, linoleic acid). Two different
chemical scaffolds were incorporated into each of the three domains of the
delivery system to investigate: 1) the effect of the number of amines of the head
group; 2) the degree of unsaturation of the hydrophobic tail domain; and 3) the
role of the protonatable histidine residue of the linker group on RNAi-mediated
silencing efficacy.
Particle diameter decreased as the N/P ratio was increased for each of the
carriers although no trend was observed between carriers with differing head
and/or tail groups. The zeta potential increased as the N/P ratio was increased
for each of the carriers with spermine-based carriers exhibiting a significantly
quicker rise in surface charge at lower N/P ratios. For example, at an N/P ratio of
5, all ethylenediamine-based carriers carried a net anionic surface charge (-14.09
± 1.88 mV to -18.50 ± 2.43 mV) whereas spermine-based carriers were cationic
(24.02 ± 1.07 mV to 27.40 ± 1.25 mV). Spermine-based carriers were also more
efficient at condensing siRNA into nanoparticles compared to carriers containing
ethylenediamine. At an N/P ratio of 10, all carriers exhibited pH-dependent

69
membrane disruption (Figure 2.9B). However, increasing the number of amino
groups in the head domain improved hemolytic activity, even at pH 7.4. These
observations suggest that the two additional amino groups within spermine
enable stronger electrostatic interactions with siRNA and possess enhanced
protonation in acidic environments to facilitate greater membrane disruption.
However, the presence of additional protonable amines of histidine did not
translate to better hemolytic activity for both ethylenediamine and spermine-
based carriers, as those carriers possessing histidine domains exhibited weaker
membrane disruption. For this library of carriers, the incorporation of histidine is
hypothesized to decrease the overall pKa of the cationic carrier thereby reducing
the membrane disrupting ability. A similar observation was made for the
spermine-based library, for which the introduction of histidine groups offered no
distinct advantage (163,164). Accordingly, by altering the number and
composition of amino groups, and thus the overall pKa of the cationic carrier, the
degree of protonation and pH-sensitive behavior can be tuned.
Interestingly, hemolytic activity did not necessarily correlate to gene
silencing efficiency for the newly developed cationic lipid carriers (Figure 2.9C).
In fact, ECO and ECLn were found to have the greatest gene silencing efficiency
in HT29 and CHO cells despite not achieving the highest level of pH-sensitive
hemolysis at all 3 tested pHs. It may be that while spermine-based carriers
exhibited greater hemolysis, the superior ability to condense siRNA may inhibit
the cytosolic release of the siRNA cargo to decrease the silencing efficiency.
Previous work found that for a cationic lipid system the incorporation of two cis
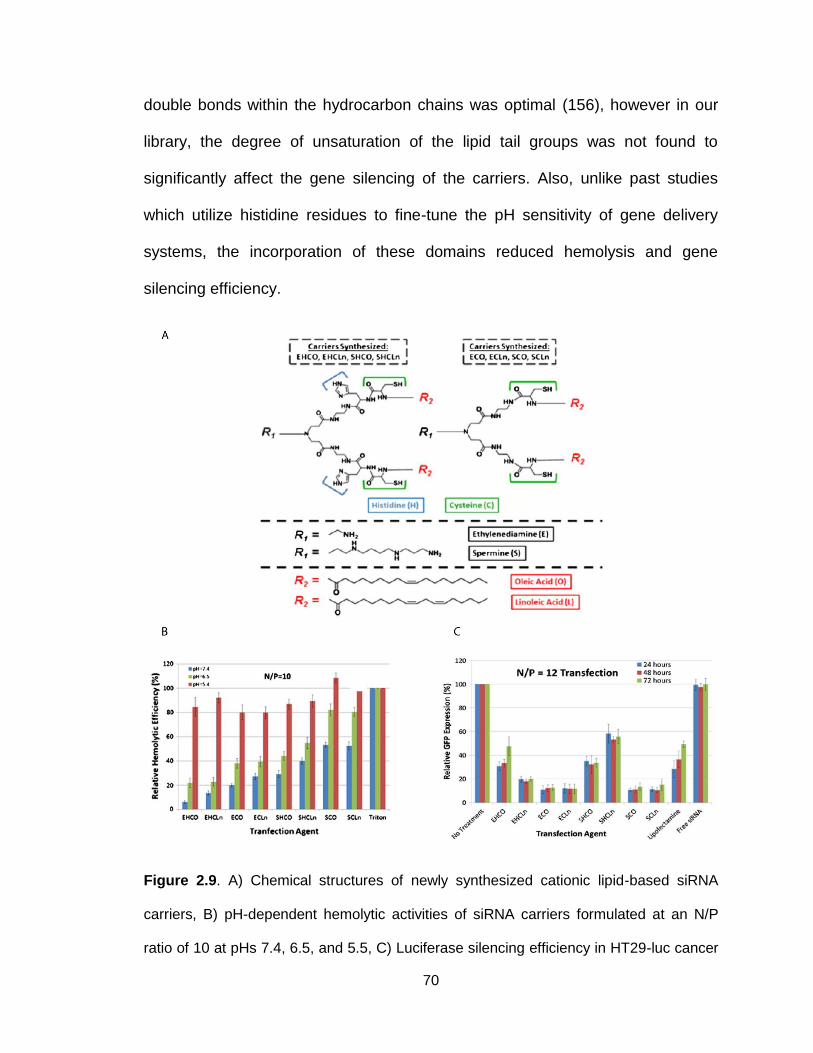
70
double bonds within the hydrocarbon chains was optimal (156), however in our
library, the degree of unsaturation of the lipid tail groups was not found to
significantly affect the gene silencing of the carriers. Also, unlike past studies
which utilize histidine residues to fine-tune the pH sensitivity of gene delivery
systems, the incorporation of these domains reduced hemolysis and gene
silencing efficiency.
Figure 2.9. A) Chemical structures of newly synthesized cationic lipid-based siRNA
carriers, B) pH-dependent hemolytic activities of siRNA carriers formulated at an N/P
ratio of 10 at pHs 7.4, 6.5, and 5.5, C) Luciferase silencing efficiency in HT29-luc cancer

71
cells at 100 nM siRNA concentration for all siRNA carriers formulated at an N/P ratio of
12 (172).
2.6 Concluding Remarks
From the newly designed library, ECO was chosen as the lead
multifunctional carrier for further development due to favorable luciferase
silencing in both cancerous and non-cancerous cell lines. A deeper
understanding and characterization of the relationship of how the
physicochemical properties of ECO/siRNA nanoparticles interplay with their
biological activity will be required. Additionally, exploration into surface
modification strategies to impart biocompatibility and tumor cell specificity will be
required before use in vivo. Reflecting upon the framework proposed by Akinc for
siRNA delivery systems (135), ECO met all the requirements except: i) greater
than two acyl chains and ii) acyl chains between 8 and 12 carbon atoms. Shorter
aliphatic chains ranging from 12 to 14 carbons in length have demonstrated
favorable transfection efficiencies compare to saturated chains greater than 14
carbons due to differences in fluidity. However, as discussed above, systems
with unsaturated chains exhibit superior silencing efficiency compared to
saturated analogues. Therefore, while the proposed framework may provide a
useful guide when developing new siRNA delivery, it should not be used
exclusively. From our experience, the most important parameters for
development of cationic lipid-based delivery systems are carefully tuning of the
pKa of the pH-sensitive, ionizable lipid structure and the ability of the lipid to
induce pH-sensitive membrane disruption.

72
Chapter 3
Multifunctional Cationic Lipid-Based Nanoparticles Facilitate Endosomal
Escape and Reduction-Triggered Cytosolic siRNA Release
Adapted from Molecular Pharmaceutics 2014, Vol. 11, p. 2734-2744
Maneesh Gujrati, Anthony Malamas, Tesia Shin, Erlei Jin, Yunlu Sun, Zheng-Rong Lu

73
3.1 Introduction
Over the past decade, small interfering RNA (siRNA) has been explored
intensely as a promising therapeutic candidate for gene therapy due to its ability
to regulate gene expression through RNA interference (RNAi). RNAi is an
endogenous regulatory mechanism reliant upon siRNA molecules to specifically
target and regulate the expression of a gene through the posttranscriptional
cleavage of the corresponding mRNA (173–175). As siRNA does not interact
with chromosomal DNA, the possibility of adverse gene alterations commonly
encountered with DNA-based gene therapies is greatly reduced. Further, through
the appropriate design of siRNA, it is possible to harness RNAi to silence nearly
any gene, offering a significantly broader therapeutic potential than conventional
small molecule-based therapies. The application of siRNA to exploit the RNAi
regulatory mechanism has revealed a host of new opportunities for the
development of novel therapeutics systems (20). Recently, numerous cancer-
associated genes have been identified, ranging from oncogenes to those genes
responsible for tumor-host interactions and tumor resistance against chemo- and
radiotherapy, positioning cancer as a well-suited candidate for siRNA-mediated
gene therapy (27).
While RNAi holds great potential as a promising therapeutic modality, a
number of extracellular and intracellular obstacles have restrained the
development and translation of RNAi-based technologies into the clinic
(176,177). These challenges include the degradation of naked siRNA within the
bloodstream by endogenous nucleases, low cellular uptake of siRNA due to its

74
anionic nature, immune response to naked siRNA, and rapid clearance by the
RES system, which occurs within minutes of intravenous administration (178).
Initial efforts to improve siRNA delivery relied upon viral delivery systems, as
viruses have been evolutionarily programmed to efficiently deliver their genetic
payload into host cells (179,180). Despite achieving high transfection efficiencies,
viral-based delivery systems often produce adverse immunogenic effects and are
associated with a high cost of production (181). To overcome such challenges,
various non-viral delivery vehicles, including liposomes, polycationic polymers,
conjugates, and cationic lipid-based nanoparticles have been developed (182–
185). These formulations, while achieving varying degrees of success in terms of
transfection efficiency, have encountered additional issues arising from
cytotoxicity, hemotoxicity, nanoparticle aggregation in serum, and poor
intracellular siRNA release from the delivery vehicles, thereby deeming them
unsatisfactory for clinical use (115,185).
As the barriers for siRNA delivery are many, it is important for a delivery
system to be multifunctional and address each of the major challenges. An ideal
siRNA delivery system will incorporate the siRNA payload and protect it from
degradation and clearance, allow for functionalization with targeting ligands to
improve delivery specificity, promote cellular uptake, provide a mechanism to
escape the fate of endosomal-lysosomal degradation and ultimately facilitate the
cytosolic release of the siRNA cargo into the target cells. To further improve the
siRNA delivery capability with these design requirements taken into
consideration, we recently developed a library of pH-sensitive amphiphilic lipid

75
carriers through solid-phase chemistry synthesis based off structural
modifications of a previously validated multifunctional carrier (168,169,172). Each
of the carrier designs was constructed to have three distinct regions of varying
composition: 1) a cationic head group; 2) cysteine-based functionalizable linkers;
and 3) a lipophilic region consisting of geminal lipid tails. We have shown that the
number of amino groups within the head group, the degree of unsaturation of the
lipid tail groups, and the structure and composition of the linker group have a
significant effect on various aspects of the delivery process, including cellular
uptake and gene silencing efficiency (162,172). Among these carriers, ECO ((1-
aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-ethyl)propionamide]) emerged
as a lead multifunctional carrier for further development because of its
effectiveness for mediating potent gene silencing in both cancerous and non-
cancerous cells (172).
Scheme 3.1. The formation of ECO/siRNA nanoparticles via electrostatic interactions
between the cationic head group and anionic siRNA, auto-oxidation of free thiol groups

76
within the cysteine residues to form disulfide crosslinks, and hydrophobic condensation
of lipid tail groups.
Scheme 3.2. ECO/siRNA nanoparticles facilitate cellular internalization resulting in
trafficking of the nanoparticles into the late endosomes. Within the late endosomes, the
pH-sensitive nature of ECO promotes endosomal escape. Once release into the cytosol,
endogenous glutathione (GSH) mediates reduction of disulfide bonds formed within
ECO/siRNA nanoparticles to release the siRNA cargo. Upon release, free siRNA is able
to initiate RNAi-induced gene silencing.
ECO is a cationic lipid containing three structural components
hypothesized to play a significant role and function: a protonable
ethylenediamine head group, two cysteine-based linker groups, and two oleic
acid lipid tails (Scheme 3.1). The ethylenediamine head group allows for the
electrostatic condensation of siRNA. The geminal oleic acid tails hydrophobically
aggregate in an aqueous environment to further condense the nanoparticles. The

77
free thiol groups of the cysteine residues can be autooxidized into reducible
disulfide linkages to further stabilize the nanoparticles. Additionally, the cysteine
residues can also provide a means to functionalize the carrier with targeting
moieties and/or biocompatible polymers, e.g. polyethylene glycol (PEG), to
improve biocompatibility and target–specific delivery (169). Finally, the structure
possesses pH-sensitive amphiphilicity, an essential ability for disrupting the
membrane of endosomal and lysosomal compartments to promote escape and
avoid degradation of the siRNA cargo within the acidic environment. Upon
successful escape, the disulfide bonds within the nanoparticle backbone are
designed to facilitate the release of siRNA in the reductive environment of the
cytosol (Scheme 3.2).
Herein, we report a comprehensive evaluation of the multifunctional
properties of ECO as a carrier for effective intracellular siRNA delivery.
ECO/siRNA nanoparticles were formed and characterized over a range of N/P
ratios. The physicochemical properties of the ECO/siRNA nanoparticles,
including serum stability, pH-sensitivity, and bio-reducibility, were determined in
correlation with intracellular siRNA delivery and gene silencing efficiency.
Further, the process of endosomal escape and mechanism of intracellular siRNA
release following cellular uptake of ECO/siRNA nanoparticles was investigated in
order to understand their gene silencing ability in U87 glioblastoma cancer cells.
The response to environmental stimulus, coupled with the superior gene
silencing and serum stability, is of particular interest and utility in overcoming the
delivery barriers against nanoparticle-mediated gene therapy.

78
3.2 Materials and Methods
3.2.1 Synthesis of (1-aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-
ethyl)propionamide]
The synthesis of (1-aminoethyl)imino-bis[N-(oleicyl-cysteinyl-1-
aminoethyl)propionamide] (ECO) was done by liquid-phase chemistry and is
described in full in the Appendix. The reaction scheme can be seen in Figure 1.
The ethylenediamine head group was synthesized first followed by the
cysteine/oleic acid tail groups. Once these two groups were synthesized they
were reacted together to arrive at the final ECO product. Each reaction
intermediate was confirmed through 1H NMR and Maldi-TOF (see Appendix).
3.2.2 Preparation of ECO/siRNA Nanoparticles
ECO/siRNA nanoparticles were prepared at N/P ratios between 6 and 20. ECO
and siRNA were diluted into equal volumes in nuclease-free water from stock
solutions of 2.5 mM in ethanol and 18.8 μM in nuclease-free water, respectively.
The equal volumes of ECO and siRNA were mixed followed by a 30-minute
incubation period at room temperature under gentle agitation.
3.2.3 Nanoparticle Characterization
The size and zeta potential of the ECO/siRNA nanoparticles at different N/P
ratios in PBS was determined by dynamic light scattering with a Brookhaven
ZetaPALS Particle Size and Zeta Potential Analyzer (Brookhaven Instruments).
Zeta potential measurements were repeated for nanoparticles incubated for 30
minutes in serum free, 10% and 50% serum media. To determine the pH-
sensitivity of ECO, ECO/siRNA nanoparticles were formulated and incubated in

79
PBS solutions at pH 7.4, 6.5, or 5.4 for 30 minutes prior to zeta potential
measurement.
3.2.4 Entrapment Efficiency
A Ribogreen assay (Molecular Probes) was used to quantify the entrapment
efficiency of siRNA within the ECO/siRNA nanoparticles (186). ECO/siRNA
nanoparticles were prepared at various N/P ratios at a final siRNA concentration
of 120 nM. Free siRNA following particle formation was detected using a
SpectraMax microplate reader (Molecular Devices) with an excitation of 500 nm
and emission of 525 nm. The entrapment efficiency of ECO/siRNA nanoparticles
was calculated in reference to a linear standard curve by dividing the complexed
siRNA concentration by the initial siRNA concentration and multiplying by 100%.
3.2.5 Heparin Displacement Assay
ECO/siRNA nanoparticles were prepared at an N/P ratio of 20 at a final siRNA
concentration of 120 nM and incubated for 30 minutes at 37⁰C with heparin
solutions of varying concentrations based on heparin/siRNA (w/w) ratio, ie. 0, 1,
2.5, 5. Following the incubation period, each sample, after the addition of loading
dye, was run on a 1% agarose gel containing ethidium bromide at 100 V for 25
minutes.
3.2.6 Gel Electrophoresis for siRNA Loading, Serum Protection, and
Glutathione-Mediated Nanoparticle Reduction
The ability of ECO to complex and condense siRNA was assessed by gel
electrophoresis. ECO/siRNA nanoparticles were prepared and 15 μL aliquots
mixed with 3 μL of loading dye (Promega) were loaded onto a 1% agarose gel

80
containing ethidium bromide. The gel was submerged in 0.5X Tris/Borate/EDTA
(TBE) buffer and run at 100 V for 25 minutes. Free siRNA was run as the control.
SiRNA bands were visualized using an AlphaImager ultraviolet imaging system
(Biosciences). For siRNA loading, ECO/siRNA complexes were prepared at N/P
ratios between 6 and 20 and run on the gel as described above. For the
assessment of glutathione-mediated nanoparticle reduction, ECO/siRNA
nanoparticles were incubated with 1 hour at 37°C in the presence of 5 mM
glutathione (GSH) (Sigma Aldrich). Following incubation, samples were loaded
onto a 1% agarose gel containing ethidium bromide and run in the same manner
as described. Serum protection of siRNA by the complexes was assessed by
incubation ECO/siRNA complexes in 50% serum at 37°C for 0.5, 1, 6, or 24
hours. At each intermittent time point, aliquots were taken and stored at -80°C.
After the final aliquot was taken at 24 hours, samples were incubated for 30
minutes with heparin at a heparin/siRNA (w/w) ratio of 5 to release the
complexed siRNA cargo and each sample was loaded on the 1% agarose gel
and run as described above. Free siRNA was also incubated in 50% serum for
0.5, 1, 6, or 24 hours and stored and run on the gel in a similar manner.
3.2.7 Cell Culture
Human glioblastoma U87 cells expressing a luciferase reporter enzyme (U87-
Luc) were obtained from ATCC (American Type Culture Collection) and cultured
in Dulbecco’s modified Eagle’s media (Invitrogen) and supplemented with 10%
fetal bovine serum (Invitrogen), 100 μg/mL streptomycin, and 100 unites/mL
penicillin (Invitrogen). The cells were maintained in a humidified incubator at

81
37⁰C and 5% CO2.
3.2.8 In Vitro Transfection Efficiency
U87-Luc cells were seeded in 24-well plates at a density of 2 x 104 cells and
allowed to grow for 24 hours. Transfections were carried out in serum-containing
(10% or 50% FBS) and serum free media with 40 nM anti-luciferase siRNA
concentration (Dharmacon: sense sequence: 5’-
CUUACGCUGAGUACUUCGAdTdT-3’, anti-sense sequence: 5’-
UCGAAGUACUCAGCGUAAGdTdT-3’). Following a 4 hour transfection period,
the media was replaced with fresh serum-containing media and the cells
continued to grow for an additional 72 hours. At 72 hours, the cells were rinsed
twice with PBS and lysed using the reporter lysis buffered provided in the
Promega Luciferase Assay kit. Following lysis, the cells were centrifuged at
10,000 g for 5 minutes and 20 μL cell lysate was transferred to a 96-well plate.
To quantify luciferase expression, 100 μL Luciferase Assay Reagent was added
to each well and the luminescence was read using a SpectraMax microplate
reader (Molecular Devices). Luciferase activity was normalized to the total
protein content measured from the cell lysate of each well using the BCA assay
(Thermo Scientific). Data was presented relative to the control, which received no
siRNA treatment. Lipofectamine RNAiMAX was used as a positive control and
was prepared per the manufacturer’s protocol (Life Technologies).
3.2.9 Cytotoxicity
U87 were transfected in 10% serum media with ECO/siRNA nanoparticles at an
siRNA concentration of 40 nM in a 96-well plate with a seeding density of 1 x 104

82
cells. After 48 hours, the MTT reagent (Invitrogen) was added to the cells for 4
hours followed by the addition of SDS-HCl and further incubation for 4 hours. The
absorbance of each well was measured at 570 nm using a SpectraMax
spectrophotometer (Molecular Devices). Cellular viability was calculated as the
average of the set of triplicates for each N/P ratio and was normalized relative to
the no treatment control.
3.2.10 Flow Cytometry for Nanoparticle Cellular Uptake and Uptake Kinetics
Measurements
Cellular uptake and intracellular delivery of ECO/siRNA nanoparticles was
evaluated quantitatively with flow cytometry. ECO/siRNA nanoparticles were
prepared with 40 nM AlexaFluor488-labelled siRNA (Qiagen). Approximately 2.5
x 104 U87 cells were seeded onto 12-well plates and grown for an additional 24
hours. The cells were transfected with ECO/siRNA nanoparticles in serum free,
10% or 50% serum media. After 4 hours, the transfection media was removed
and each well was washed twice with PBS. The cells were harvested by
treatment with 0.25% trypsin containing 0.26 mM EDTA, (Invitrogen) collected by
centrifugation at 1000 rpm for 5 min, resuspended in 500 μL of PBS containing
5% paraformaldehyde, and finally passed through a 35μm cell strainer (BD
Biosciences). Cellular internalization of ECO/siRNA nanoparticles was quantified
by the fluorescence intensity measurement of Alexa Fluor 488 fluorescence for a
total of 10,000 cells per each sample using a BD FACSCalibur flow cytometer.
Each N/P ratio was conducted in triplicate and the data presented represents the
mean fluorescence intensity and standard deviation.

83
Nanoparticle uptake kinetics was measured in a similar setup as
described above. ECO/siRNA nanoparticles were formulated with 40 nM Alexa
Fluor 488-labelled siRNA at an N/P ratio of 10. U87 cells were seeded in 24-well
plates at a density of 2 x 104 cells and allowed to grow for 24 hours.
Nanoparticles were administered in serum free, 10% or 50% serum media.
Nanoparticle uptake was measured at various time points up to 4 hours post-
transfection. At each time point, the cells were washed twice with PBS,
trypsinized, collected and fixed with 5% paraformaldehyde in PBS before
quantification of Alexa Fluor 488 fluorescence using a BD FACSCalibur flow
cytometer. The mean fluorescence of 10,000 cells was quantified for each
replica. Data presented represents the mean and standard deviation of three
replicas for each time point.
3.2.11 Protein Adsorption
ECO/siRNA nanoparticles were formulated at an N/P ratio of 10. To
quantify BSA protein adsorption, 500 μL of nanoparticle solution and 500 μL of
BSA solution at varying concentrations were added together, stirred and
incubated for 1 hour at 37⁰C. Nanoparticles were prepared such that the final
amine concentration for each condition was 150 μM. Serial dilutions of a stock
BSA solution (4 mg/mL) were carried out to achieve the various protein
concentrations: 2 mg/mL, 1 mg/mL, 0.5 mg/mL, 0.25 mg/mL, 0.125 mg/mL.
Following incubation, the ECO/siRNA nanoparticles were centrifuged at 10,000 g
for 20 minutes. The concentration of BSA was determined from the supernatant
using UV-vis spectroscopy on a SpectraMax spectrophotometer (Molecular

84
Devices) at 280 nm. A linear calibration curve from predetermined BSA
concentrations was used. Relative BSA adsorption was calculated by dividing the
amount of protein adsorbed for each BSA incubation concentration by the
amount of protein adsorbed for 0.125 mg/mL BSA.
3.2.12 pH-Dependent Membrane Disruption Hemolysis Measurement
The hemolytic activity was measured to determine the membrane-disruptive
ability of ECO/siRNA nanoparticles at pH levels corresponding to various stages
of intracellular trafficking. Red blood cells (RBCs) isolated from rats (Innovative
Research Inc.) were diluted 1:50 in PBS solutions at pH 7.4, 6.5, and 5.4.
ECO/siRNA nanoparticles were prepared at a volume of 100 μL and incubated
with an equal volume of the various RBC solutions in a 96-well plate at 37⁰C for 2
hours. Nanoparticles were prepared such that the final amine concentration for
each pH condition was 150 μM. Following incubation, samples were centrifuged
and the absorbance of the supernatants was determined at 540 nm. Hemolytic
activity was calculated relative to the hemolytic activity of 1% Triton X-100
(Sigma Aldrich), a non-ionic surfactant. Each pH was conducted in triplicate and
the data presented represents the mean and standard deviation.
3.2.13 Inhibition of Glutathione-Dependent Reduction with BSO
Intracellular glutathione (GSH) was depleted in order to establish the role of
cytosolic reduction of ECO/siRNA nanoparticles on gene silencing. U87 cells
were plated and prepared in the same manner as during transfection studies.
The cells were incubated overnight with 200 μM buthionine-sulfoximine (BSO)
obtained from Sigma Aldrich prior to transfection, which was carried out as

85
describe earlier with an N/P ratio of 10 and an anti-luciferase siRNA
concentration of 40 nM. Luciferase expression was quantified with a luciferase
assay and normalized with a BCA assay 48 hours post-transfection as described
above.
3.2.14 Confocal Microscopy of Cellular Uptake of ECO/siRNA Nanoparticles
and Intracellular Release of siRNA
Live cell confocal microscopy was used to assess the cellular uptake and
intracellular release of siRNA. Approximately 1 x 105 U87 cells were seeded onto
glass-bottom micro-well dishes. After 24 hours, the cells were stained with 5
µg/mL Hoechst 33342 (Invitrogen) and treated with ECO/siRNA nanoparticles in
10% serum media. Nanoparticles were formed at an N/P ratio of 10 and a 20 nM
siRNA concentration with an Alexa Fluor 488-labelled siRNA. Images were taken
using an Olympus FV1000 confocal microscope for up to 72 hours while the cells
were housed in a humidified weather station under 5% CO2.
3.2.15 Immunofluorescence of Intracellular Trafficking of ECO/siRNA
Nanoparticles
Following transfection with ECO/siRNA particles containing Alexa Fluor 647-
labelled siRNA (Qiagen), U87 cells were fixed at various time points with 4%
formaldehyde for 30 min at room temperature and permeabilized with 0.1%
Triton-X 100 (in PBS) for 5 minutes at room temperature. Cells were then
incubated in blocking buffer (2% BSA in PBS) for 1 hour. The primary antibody,
rabbit anti-Lysosomal-associated membrane protein 1 (LAMP1) (Abcam), was
added at 1μg/mL in blocking buffer and incubated at room temperature for 1

86
hour. The secondary antibody, Alexa Fluor 488 goat anti-rabbit IgG (Life
Technologies), was used at a 1:1000 dilution for 1 hour. Samples were
thoroughly washed with PBS and imaged using an Olympus FV1000 confocal
microscope.
3.2.16 Statistical Analysis
Experiments were performed in triplicate and presented as the mean and
standard deviation. Statistical analysis was conducted with ANOVA and two-
tailed Student’s t-tests using a 95% confidence interval. Statistical significance
was established only when p < 0.05.
3.3 Results and Discussion
3.3.1. Effect of N/P ratio on the physicochemical properties of ECO/siRNA
nanoparticles
The physicochemical properties of siRNA nanoparticles can have a direct
impact on the efficacy of intracellular siRNA delivery and gene silencing of the
delivery system (67,187,188). The understanding of these physicochemical
properties, including particle size, surface zeta potential, siRNA entrapment and
particle stability, in correlation with the intracellular siRNA delivery and gene
silencing efficiency is crucial for formulating a safe and effective siRNA delivery
system suitable for clinical development (189). The physicochemical properties
can be tailored based on the ratio of cationic and anionic charge (N/P ratio)
within the ECO/siRNA nanoparticles. The impact of N/P ratio on these
parameters was investigated for ECO/siRNA nanoparticles between an N/P ratio
of 6 and an N/P ratio of 20.

87
Figure 3.1. Physicochemical evaluation of ECO/siRNA nanoparticles. a) Effect of N/P
ratio on mean particle diameter and surface charge. b) siRNA entrapment within
nanoparticles determined by RiboGreen RNA quantitation assay over a range of N/P
ratios. c) Agarose gel retardation of ECO/siRNA nanoparticles compared to free siRNA
over a range of N/P ratios. d) Heparin displacement assay. ECO/siRNA nanoparticles
were prepared at N/P ratio of 20 and incubated for 30 minutes at 37⁰C with varying
amounts of heparin, based on heparin/siRNA (w/w) ratio.
The particle size of ECO/siRNA nanoparticles decreased while their zeta
potential increased as the N/P ratio increased (Figure 3.1a). The ability of ECO
to complex and entrap siRNA increased as a function of N/P ratio, from 82.1 ±
4.3 % at N/P=6 to 98.7 ± 5.0 % at N/P=20, as demonstrated by a RiboGreen
fluorescence-based assay (Figure 3.1b). The complexation of ECO with siRNA

88
was further validated through an agarose gel retardation assay (Figure 3.1c).
Compared to naked siRNA, a decrease in particle-bound siRNA migration as the
N/P ratio increased was observed. At an N/P ratio ≥ 14, the complexed siRNA
was completely prevented from migrating through the gel indicating that the
interactions between ECO and siRNA were strong enough to resist dissociation
during electrophoresis. Interestingly, at an N/P ratio ≥ 18, no siRNA signal was
observed in the loading well, suggesting that the negatively charged siRNA was
completely neutralized as ethidium bromide was not able to intercalate (190).
Some cationic polymers with high charge density, such as PEI, can form
inseparable complexes with siRNA such that the siRNA cargo cannot be
released once internalized into the cytosol (191). Therefore, it is important that
the interactions between the siRNA and carrier be stable during cellular uptake
but do not impede the cytosolic release of the siRNA. To study the electrostatic
interaction of the siRNA with ECO, ECO/siRNA nanoparticles were subject to
heparin displacement. Heparin is an anionic polysaccharide and a major
component of extracellular matrix that can compete with siRNA for binding to
disrupt ECO/siRNA complex stability (192). No decomplexation of siRNA from
the nanoparticles occurred at heparin/siRNA (w/w) ratio of 1. Partial
decomplexation of siRNA from the ECO/siRNA nanoparticles, as determined by
siRNA release on an agarose gel, occurred at heparin/siRNA (w/w) ratio of 2.5
while full decomplexation was observed at a ratio of 5 (Figure 3.1d).
These results suggest that the N/P ratio plays an essential role in
regulating size, charge, and ability of the ECO lipid carrier to complex siRNA into

89
stable nanoparticles. While particle size decreases, increasing the N/P ratio will
increase the zeta potential. Increased concentrations of amino groups enhance
the ability of ECO to condense the siRNA cargo by facilitating stronger ionic
interactions and compact particle formation.
3.3.2. Effect of N/P ratio on the biological properties of ECO/siRNA
nanoparticles
The N/P ratio significantly influences the physicochemical parameters of
ECO/siRNA nanoparticles, which can in turn influence the biological properties
and activity of the nanoparticles. The effect of the N/P ratio on the cellular
uptake, gene silencing and cytotoxicity of the siRNA nanoparticles was
investigated in vitro with U87 glioblastoma cells expressing a luciferase reporter
gene (U87-Luc). Cellular uptake of ECO/siRNA nanoparticles was determined
using an Alexa Fluor 488-labeled siRNA with flow cytometry in serum-free, 10%
serum, and 50% serum media (Figure 3.2a). Cellular uptake was found to
increase in an N/P ratio-dependent manner for all transfection conditions. Under
serum free conditions, Lipofectamine RNAiMAX (Lipofect.) mediated higher
cellular uptake than ECO for all N/P ratios. However, for 10% and 50% serum
conditions, ECO/siRNA nanoparticles at an N/P of 20 had enhanced cellular
uptake compared to Lipofectamine. A significant reduction in cellular uptake was
observed in 10% and 50% serum media for N/P ratios ≤ 12 when compared to
serum free media (p<0.05). At N/P ≥ 14, cellular uptake in all three transfection
conditions was not significantly different. As shown in the above study, high N/P
ratios resulted in an increase in both surface zeta potential and stability of the
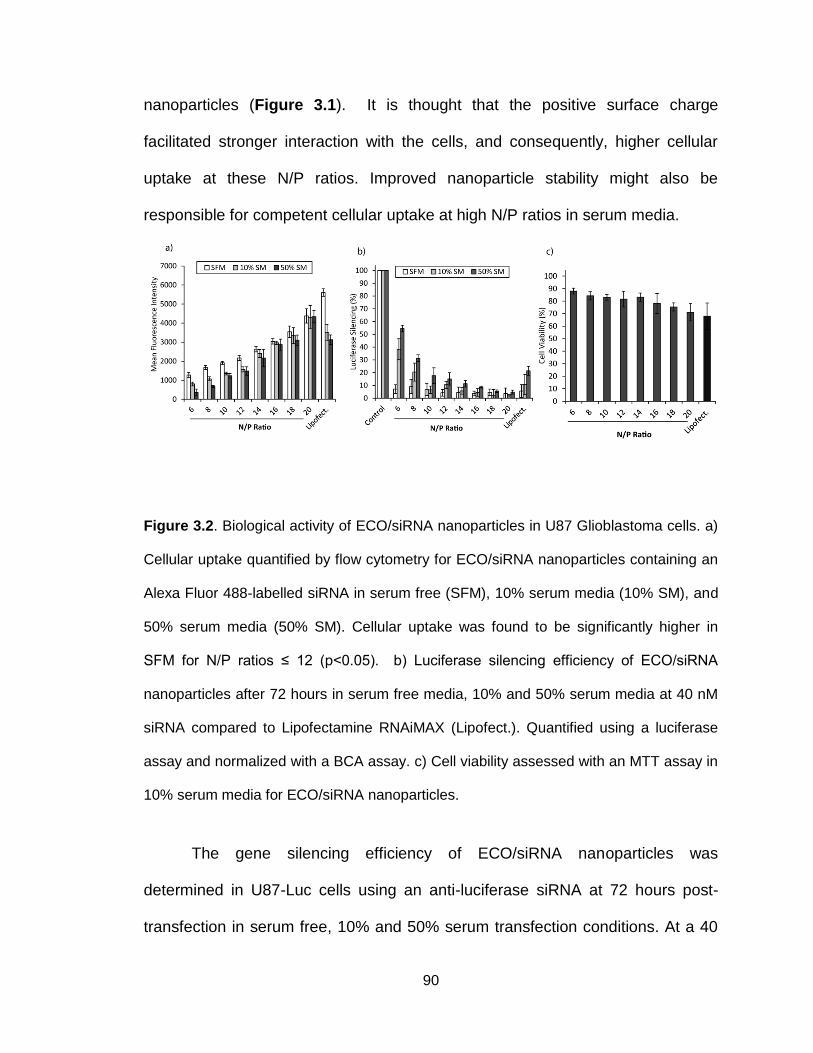
90
nanoparticles (Figure 3.1). It is thought that the positive surface charge
facilitated stronger interaction with the cells, and consequently, higher cellular
uptake at these N/P ratios. Improved nanoparticle stability might also be
responsible for competent cellular uptake at high N/P ratios in serum media.
Figure 3.2. Biological activity of ECO/siRNA nanoparticles in U87 Glioblastoma cells. a)
Cellular uptake quantified by flow cytometry for ECO/siRNA nanoparticles containing an
Alexa Fluor 488-labelled siRNA in serum free (SFM), 10% serum media (10% SM), and
50% serum media (50% SM). Cellular uptake was found to be significantly higher in
SFM for N/P ratios ≤ 12 (p<0.05). b) Luciferase silencing efficiency of ECO/siRNA
nanoparticles after 72 hours in serum free media, 10% and 50% serum media at 40 nM
siRNA compared to Lipofectamine RNAiMAX (Lipofect.). Quantified using a luciferase
assay and normalized with a BCA assay. c) Cell viability assessed with an MTT assay in
10% serum media for ECO/siRNA nanoparticles.
The gene silencing efficiency of ECO/siRNA nanoparticles was
determined in U87-Luc cells using an anti-luciferase siRNA at 72 hours post-
transfection in serum free, 10% and 50% serum transfection conditions. At a 40

91
nM siRNA concentration, gene silencing was dependent upon the N/P ratio,
although this trend was more evident in the presence of serum (Figure 3.2b).
High gene silencing efficiency was observed for the nanoparticles throughout the
N/P ratio range in serum free media: luciferase expression was inhibited to 7.2 ±
3.4 % for N/P=6 and 3.7 ± 3.3 % for N/P=20 at 72 hours post-transfection. In the
presence of 10% serum, luciferase silencing increased in an N/P dependent
manner from 38.41 ± 8.19 % luciferase expression for N/P=6 to 1.91 ± 0.97 %
luciferase expression for N/P=20 at 72 hours. Similarly for 50% serum, luciferase
silencing was less efficient for N/P ratios between 6 and 12 but was comparable
to serum free and 10% serum for N/P > 12. At N/P ≥ 10, ECO/siRNA
nanoparticles matched or exceeded the performance of Lipofectamine RNAiMAX
in their respective transfection conditions. It is interesting to note that in serum
free media, ECO/siRNA nanoparticles were equally as efficient at silencing
luciferase for N/P=6 as they were for N/P=20 despite a 4-fold difference in
cellular uptake. One possible explanation may be that the RNAi machinery
becomes saturated beyond a certain intracellular siRNA concentration (193,194).
Alternatively, it has been suggested that the efficiency of siRNA delivery via lipid
nanoparticles is limited by endocytic recycling, in which the siRNA nanoparticles
within the endocytic vesicles are expelled from the cytosol back into the
extracellular environment (144). For transfection conditions containing serum,
gene silencing efficiency correlated with cellular uptake due to significantly lower
cellular uptake at low N/P ratios compared to the serum free transfection
condition.

92
While a higher N/P ratio led to improved cellular uptake and gene
silencing, unwanted cytotoxic effects may arise as a result and should therefore
be monitored closely to ensure complete safety of a delivery system. The
cytotoxicity of the ECO carrier was evaluated using an MTT assay (Figure 3.2c).
Cell viability was evaluated 48 hours post-transfection and was found to
gradually decrease as the N/P ratio increased. Cell viability was especially
compromised at N/P > 14, which may be a result of increased positive charge
densities at high N/P ratios (195). Importantly, the overall viability of those cells
treated with ECO/siRNA nanoparticles at all N/P ratios remained higher than
those treated with Lipofectamine RNAiMAX.
Increased cellular uptake may be a direct consequence of the zeta
potential promoting interactions with the negatively charged cell membrane at
high N/P ratios. However, increased zeta potential will negatively influence the
biocompatibility of the delivery system. Additionally, low N/P ratios are not as
efficient at inducing gene silencing in the presence of serum which may be in part
contributed to reduced cellular uptake when compared to higher N/P ratios, but
also due to their lower siRNA entrapment efficiency and lower stabilities. In an
effort to optimize transfection conditions to maximize gene silencing while
minimizing cytotoxic effects in U87 glioblastoma cells, an N/P ratio of 10
appeared to be the best formulation of the ECO/siRNA nanoparticles and was
chosen for further studies. At an N/P of 10, ECO/siRNA nanoparticles averaged
112 nm in diameter, had a zeta potential of +18.2 mV, and silenced luciferase to
6.6% at 72 hours in 10% serum media while maintaining good cell viability.

93
3.3.3. ECO/siRNA nanoparticles protect siRNA and promote cellular uptake
in the presence of serum proteins
Serum proteins may lead to dissociation of the siRNA nanoparticles, and
premature release and degradation of siRNA (196). To address the question of
nanoparticle stability and siRNA protection from nuclease degradation, free
siRNA and ECO/siRNA nanoparticles (N/P=10) were incubated in PBS
containing 50% fetal bovine serum at 37 ⁰C for up to 24 hours. The agarose gel
chromatogram of the siRNA in both formulations at various time points of the
incubation revealed that free siRNA was prone to degradation within the first 30
minutes, and completely degraded by 6 hours, while siRNA within the
ECO/siRNA nanoparticles was preserved for at least 24 hours (Figure 3.3a). The
result suggests that ECO is able to sufficiently complex and pack siRNA into
stable nanoparticles such to protect the siRNA from enzymatic degradation in
serum.
Non-specific interaction of serum proteins with the ECO/siRNA
nanoparticles may also hinder membrane adsorption, block cellular entry, and
diminish the transfection efficiency, as has been demonstrated previously with
lipid-based nanoparticles (197,198). The kinetics of cellular uptake of
nanoparticles complexed with an Alexa Fluor 488-labelled siRNA was monitored
over the course of 4 hours in serum free, 10% and 50% serum media. While the
nanoparticle uptake is clearly higher in serum free media than in 10% and 50%
serum media (p<0.05), the cellular uptake under all transfection conditions
exhibits a similar biphasic trend (Figure 3.3b). This biphasic behavior has been

94
speculated to originate from an initial period where nanoparticles adhere to the
outer cell membrane before undergoing cellular entry and internalization (199).
Initial membrane adhesion is associated with slow nanoparticle internalization
until a steady state is achieved with a balanced rate of nanoparticle membrane
adhesion and internalization (200). The lower siRNA-associated fluorescence
signal observed in serum media was not due to siRNA degradation as the ECO
nanoparticles were effective in protecting the cargo siRNA (Figure 3.3a). The
difference in cellular uptake may then be in part due to the non-specific
interaction of serum proteins with nanoparticles and the consequent reduction of
zeta potential, diminishing the ability of the nanoparticles to adhere to the outer
cellular membrane and to undergo cellular internalization. This was confirmed
(Figure 3.3c) by the observation that ECO/siRNA nanoparticles had a reduced
zeta potential in 10% and 50% serum media compared to serum free media
(p<0.05). Cellular uptake and zeta potential was not found to be significantly
different between 10% and 50% serum transfection conditions suggesting that
serum protein binding to the ECO nanoparticles may reach a point of saturation.
To determine this, the binding of bovine serum albumin (BSA), the major protein
constituent of fetal bovine serum, to ECO/siRNA nanoparticles was quantified
following incubation over a range of BSA concentrations (Figure 3.3d). Indeed a
saturation point of BSA adsorption was observed for BSA concentrations ≥ 0.25
mg/ml.

95
Figure 3.3. a) Susceptibility to serum-degradation of free or complexed siRNA within
ECO nanoparticles. Samples were incubated in 50% serum for 0.5, 1, 6 and 24 hours.
Glutathione (5mM) was used to release complexed siRNA from ECO and the integrity of
siRNA cargo was evaluated with an agarose gel electrophoresis assay. b) Kinetics of
nanoparticle uptake with Alexa Fluor 488-labelled ECO/siRNA nanoparticles in U87 cells
in serum free (SFM), 10% and 50% serum media (10% SM and 50% SM). Levels of

96
cellular uptake of nanoparticles in SFM were found to be significantly higher than in 10%
and 50% SM for all time points (p<0.05). c) ECO/siRNA nanoparticles were formulated
at an N/P of 10 and the zeta potential was evaluated following incubation in either serum
free, 10%, or 50% serum media. Zeta potential of nanoparticles was found to be
significantly diminished by the presence of serum (p<0.05). d) Relative adsorption of
bovine serum albumin (BSA) to the ECO/siRNA nanoparticles after 1 hour incubation at
37⁰C as a function of BSA incubation concentration. e) Live-cell confocal imaging of
cellular uptake of ECO/siRNA nanoparticles in U87 cells and cytosolic distribution of
Alexa Fluor 488-labelled siRNA in 10% serum media. A dispersed siRNA-based
fluorescent signal is present 4 hours post-transfection and remains upwards of 72 hours.
The cellular uptake of ECO/siRNA nanoparticles in the presence of serum
was further investigated with confocal microscopy using an Alexa Fluor 488-
labeled siRNA (Figure 3.3e). Intracellular internalization and dispersed cytosolic
siRNA distribution was observed as early as 4 hours post-transfection. In
accordance with the observed sustained luciferase silencing (Figure 2b), the
dispersed signal intensity increased over time and persisted at least 72 hours
post-transfection. From these images, it is clear that even with a reduced zeta
potential, ECO/siRNA nanoparticles were effectively taken up by the cells in
serum and siRNA was released into the cytosol.
3.3.4. ECO/siRNA nanoparticles are pH-sensitive and promote endosomal
escape
Following successful internalization, one of the most crucial events for
effective intracellular siRNA delivery is the escape from the endosomal-

97
lysosomal pathway (201). It is imperative for the carrier to promote the escape
from such pathways for the purpose that siRNA must be available within the
cytosol to initiate RNAi. If the siRNA nanoparticles remain in these transport
vesicles, they will be at risk to lysosomal degradation (142). It has been proposed
that the multifunctional nanoparticles are able to escape endosomal-lysosomal
pathways with their ability to disrupt the membrane of the acidic endosomes and
lysosomes in a pH-sensitive manner (172,202). To validate this hypothesis, the
zeta potential and membrane disruption ability of ECO/siRNA nanoparticles at pH
levels of the extracellular (7.4) and endosomal and lysosomal environments (6.5
and 5.4) was studied. As the pH level decreased and became more acidic, amine
groups within the cationic head group of ECO become protonated and
consequently the zeta potential increased from 18.1 mV at pH=7.4, to 32.4 mV at
pH=6.5, to 49.5 mV at pH=5.4 (Figure 3.4a). The relative hemolytic activity of
ECO/siRNA nanoparticles in rat blood cells (RBCs), normalized to the hemolytic
activity of 1% Triton-X-100, was found to increase with acidity in a similar manner
through which maturing endocytic vesicles are acidified (Figure 3.4a). Hemolysis
of 48.5 ± 6.2 % at pH of 6.5 and 89.2 ± 5.4 % at pH of 5.4 demonstrated the
ability of these nanoparticles to interact with the membrane of late endosomes
and/or lysosomes in response to the pH changes. The low hemolytic activity of
ECO/siRNA nanoparticles at pH of 7.4 (12.5 ± 3.5 %) is consistent with the
observation that ECO/siRNA nanoparticles elicit a low cytotoxic effect on cells, as
minimal membrane disruption was observed.

98
Figure 3.4. a) Zeta potential measurements following incubation in PBS at various pH
levels demonstrate the pH-sensitivity of the ECO/siRNA nanoparticle. The zeta potential
was found to increase with increasing acidity. Hemolytic assay determined the pH-
dependent membrane-disruptive ability of ECO/siRNA nanoparticles increased
significantly (p < 0.05) with increasing acidity (pH= 7.4, 6.5, 5.4). Relative hemolytic
activity calculated with respect to the hemolytic activity of 1% Triton-X-100. b)
Immunofluorescence using an LAMP1-antibody (Alexa Fluor 488-labelled secondary
antibody) to stain for late endosomes reveals co-localization of ECO/siRNA (Alexa Fluor
647-labelled siRNA) nanoparticles occurs 2 hours post-transfection. At 4 hours, a
dispersed siRNA signal is present within the cytosol indicating that ECO/siRNA
nanoparticles are able to escape from late endosomes and release the siRNA cargo.
Intracellular trafficking of ECO/siRNA nanoparticles was further
determined using fluorescence confocal microscopy based on the localization of
an Alexa Fluor 647-labeled siRNA in respect to a specific marker for late

99
endosomes and lysosomes (anti-LAMP1). As shown, the ECO/siRNA
nanoparticles began interacting with the cell membrane with no visible co-
localization with LAMP1-stained vesicles within the first 5 minutes of transfection
(Figure 3.4b). At 30 minutes, areas of co-localization of nanoparticles and late
endosomes arose and co-localization increased at 2 hours, where the majority of
the siRNA-based fluorescent signal is coalescent with the vesicles that are
characteristic for late endosomes. By 4 hours, a dispersed siRNA signal
distribution appeared and the co-localization of the siRNA with LAMP1 was
diminished. This data suggests that the ECO/siRNA nanoparticles were trafficked
through the endosomal-lysosomal pathway to the late endosomes, whereupon
they were able to escape from the vesicles to release their cargo in the cytosol.
Although the exact pathways responsible for internalization and trafficking of the
nanoparticles have yet to be explored and defined, it has recently been
suggested that most nano-sized particles are trafficked to the lysosomes
regardless of their method of endocytosis (203). The result here suggests that,
irrespective of the endocytic pathway, the multifunctional carrier ECO can
promote effective early escape in the endosomal-lysosomal pathway, a key
feature responsible for its success in inducing gene silencing.
3.3.5. Cytosolic reduction of ECO/siRNA nanoparticles is crucial for siRNA
release and RNAi activity
Once escaped from the endosomal-lysosomal pathway, the final step of
the multi-stage process of intracellular siRNA delivery is to ensure the cytosolic
release of the siRNA cargo whereupon it will be available to bind to the RNA

100
induced silencing complex (RISC) and initiate RNAi. During nanoparticle
formation, the ECO/siRNA nanoparticles are stabilized by disulfide bonds. The
cleavage of these linkages within the reductive cytosolic environment, via
disulfide-thiol exchange initiated by endogenous glutathione, can facilitate the
release of the complexed siRNA (204,205). This bio-reducible functionality of
ECO was demonstrated by incubating nanoparticles at the physiological
concentration of glutathione (5 mM) for 1 hour at 37⁰C. Agarose gel
electrophoresis was used to evaluate whether the siRNA cargo could be
released in the presence of the reducing agent. In the absence of glutathione,
ECO successfully condensed siRNA into nanoparticles while in the presence of
glutathione, the siRNA cargo disassociated from the nanoparticles, indicating that
disulfide reduction by glutathione was sufficient to unpack the ECO/siRNA
nanoparticles (Figure 3.5a).
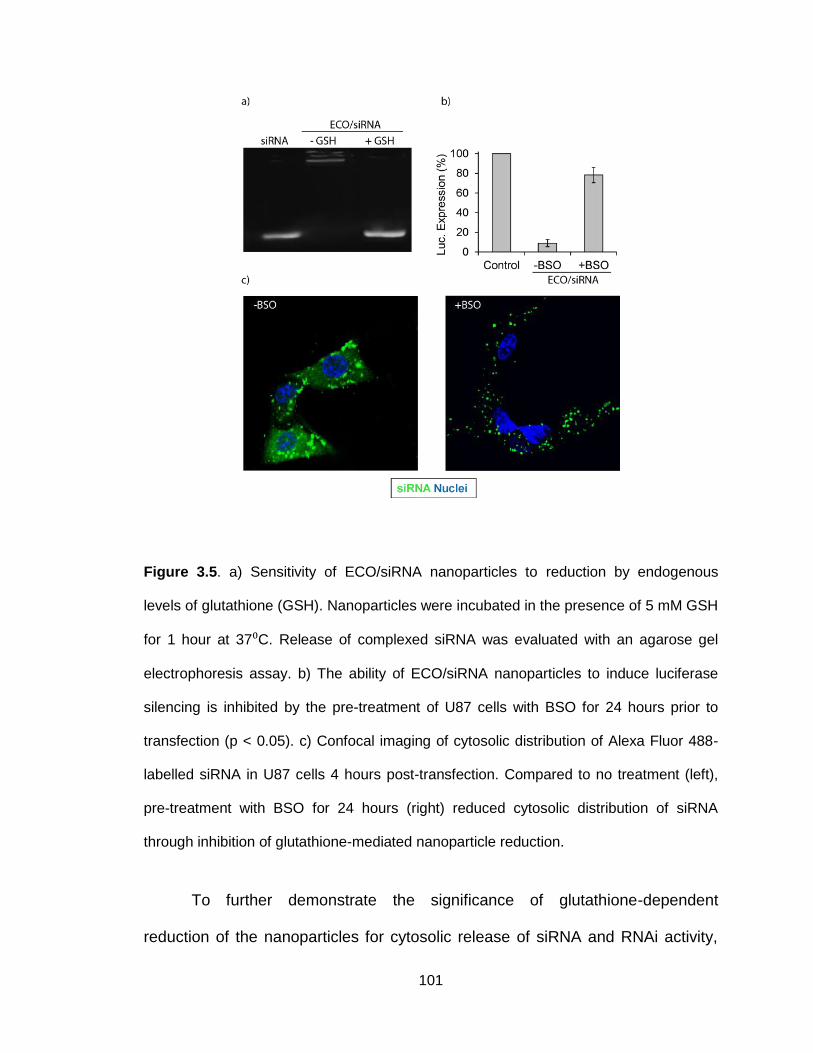
101
Figure 3.5. a) Sensitivity of ECO/siRNA nanoparticles to reduction by endogenous
levels of glutathione (GSH). Nanoparticles were incubated in the presence of 5 mM GSH
for 1 hour at 37⁰C. Release of complexed siRNA was evaluated with an agarose gel
electrophoresis assay. b) The ability of ECO/siRNA nanoparticles to induce luciferase
silencing is inhibited by the pre-treatment of U87 cells with BSO for 24 hours prior to
transfection (p < 0.05). c) Confocal imaging of cytosolic distribution of Alexa Fluor 488-
labelled siRNA in U87 cells 4 hours post-transfection. Compared to no treatment (left),
pre-treatment with BSO for 24 hours (right) reduced cytosolic distribution of siRNA
through inhibition of glutathione-mediated nanoparticle reduction.
To further demonstrate the significance of glutathione-dependent
reduction of the nanoparticles for cytosolic release of siRNA and RNAi activity,

102
U87-Luc cells were treated with buthionine sulfoximine (BSO) prior to
transfection with ECO/siRNA nanoparticles. BSO was implemented to deplete
intracellular glutathione by inhibiting γ-glutamylcystein synthetase, the enzyme
required to initiate glutathione synthesis (206,207). The ability of ECO/siRNA
nanoparticles to silence luciferase expression was significantly inhibited by the
BSO treatment (Figure 3.5b). Confocal microscopy further revealed that pre-
treatment of U87-Luc cells with BSO prevented the dispersed cytosolic
distribution of siRNA 4 hours post-transfection (Figure 3.5c). Unlike the
dispersed siRNA-associated fluorescence observed in the cytosol of untreated
cells (left), the fluorescence signal of the labeled siRNA in BSO-treated cells
remained punctate, indicative of intact nanoparticles (right). The result
demonstrate that the intracellular reduction of the nanoparticle, the final step in
the arduous intracellular delivery process of siRNA, plays a vital role and is a
requisite for achieving effective intracellular siRNA delivery and high gene
silencing efficiency of ECO/siRNA nanoparticles. The inclusion of the cysteine
residues within the structure of ECO is a key feature to stabilize the siRNA
nanoparticles and for cytosol-specific controlled siRNA release.
3.4 Conclusions
We have developed a cationic lipid-based ECO/siRNA nanoparticle delivery
system that demonstrates the multifunctionality critical for efficient intracellular
siRNA delivery and RNAi activity. The N/P ratio had a significant impact on the
size, zeta potential and siRNA complexation of the of ECO/siRNA nanoparticles.
Higher N/P ratio produced more compacted nanoparticles with improved siRNA

103
entrapment and increased zeta potential within the N/P ratio range of 6 – 20. In
serum free media, ECO/siRNA nanoparticles mediated high luciferase silencing
in U87-Luc while the silencing efficiency decreased at N/P < 10 under serum
media transfection conditions. For the U87 cell line, an N/P ratio of 10 was the
optimal formulation for balancing robust gene silencing with minimal adverse
cytotoxic effects. ECO/siRNA nanoparticles were effective to protect siRNA from
degradation in serum, able to escape from the late endosomes via their pH-
sensitive ability to induce membrane disruption at endosomal pH levels, and
released the siRNA payload in the reductive cytosolic environment through
cleavage of disulfide bonds within the nanoparticle. These functionalities of the
ECO/siRNA nanoparticles are critical for their capability to mediate efficient
intracellular siRNA delivery and effective gene silencing. The multifunctional
ECO/siRNA nanoparticles provide a promising platform delivery system for
efficient delivery of therapeutic siRNA in vitro and certainly warrant further
development and evaluation of their potential for in vivo delivery.
3.5 Acknowledgments
This material is based upon work partially supported by the National Science
Foundation Graduate Research Fellowship under Grant Number DGE-0951783
and the National Institutes of Health under Grant Number EB00489.

104
Chapter 4
Silencing β3 Integrin by Targeted siRNA Nanoparticles Inhibits EMT and
Metastasis of Triple Negative Breast Cancer
Adapted from Cancer Research 2015,
Maneesh Gujrati, Jenny Parvani, Margaret Mack, William Schiemann and Zheng-Rong
Lu

105
4.1 Introduction
Breast cancer is the second leading cause of cancer-related deaths
amongst women in the United States (208). The 5-year relative survival rate for
women with localized disease is 98.6%; however, for those diagnosed with
distant metastases, survival rates fall below 25%. Triple-negative breast cancer
(TNBC) is a highly aggressive subcategory of breast cancer that lack the
expression of estrogen receptor (ER) and progesterone receptor (PR), as well as
HER2 amplification. Currently, there is a lack of targeted therapies for TNBCs,
which renders chemotherapy as the standard of care (29,209,210). Although
TNBC patients are initially responsive to chemotherapy, most patients relapse,
and the recurrent tumor is usually highly metastatic and resistant to traditional
chemotherapy, which leads to a disproportionately large number of deaths
(29,209–211). Importantly, metastasis is associated with the aberrant activation
of epithelial-mesenchymal transition [EMT; (212–214)], which endows cancer
cells with elevated capabilities to invade and disseminate to distant sites (215).
Various molecular and microenvironmental factors can induce EMT, including
transforming growth factor- (TGF-). TGF- is a pleiotropic cytokine that
regulates virtually all aspects of mammary gland biology. TGF- induces the
dramatic upregulation of 3 integrin, which is essential for EMT and breast
cancer metastasis (216–218). Previous work has demonstrated that functional
disruption of 3 integrin inhibits TGF--mediated cytostasis, EMT, and invasion in
vitro (216)and reduces primary tumor burden in vivo (219). Given the essential
roles that 3 integrin plays in mediating breast cancer tumor progression, we

106
hypothesize that silencing 3 integrin expression has the potential to effectively
treat TNBC.
RNA interference (RNAi) is a natural biological mechanism for modulating
gene expression and can be exploited to effectively regulate the expression of 3
integrin in treating TNBC. Clinical application of RNAi requires efficient delivery of
therapeutic siRNA into the cytoplasm of target cells (172,220). We have recently
developed a multifunctional cationic lipid-based carrier, (1-aminoethyl)iminobis[N-
oleicylsteinyl-1-aminoethyl)propionamide] (ECO), which can effectively mediate
cytosolic siRNA delivery and facilitate efficient gene silencing in cancer cells
[Figure 4.1A; (172,221)]. ECO self-assembly with therapeutic siRNA forms
stable nanoparticles that can be readily functionalized with targeting moieties to
achieve targeted siRNA delivery to cancer cells. Considering the critical roles of
3 integrin in regulating EMT, proliferation and metastasis (222–224) and the
unmet need for targeted therapies tailored to TNBC (225), we sought to evaluate
silencing 3 integrin by targeted ECO/siRNA nanoparticles to treat metastatic
breast cancer.
The present study demonstrates the efficacy of ECO/si3 nanoparticles in
silencing 3 integrin expression and the consequent inhibition of TGF--mediated
EMT and invasion in breast cancer cells in vitro. The nanoparticles were modified
with a cyclic RGD peptide via a PEG spacer to improve biocompatibility and
systemic target-specific delivery of the therapeutic si3 in vivo (169). The efficacy
of the targeted ECO/si3 nanoparticles in alleviating primary tumor burden and

107
inhibiting TNBC metastasis was determined in tumor-bearing mice following
multiple intravenous injections.
4.2 Materials and Methods
4.2.1 Cell lines and reagents
MDA-MB-231 cells were obtained from ATCC (Manassas, VA) and
cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY)
supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY).
NME cells are Normal Murine Mammary Gland (NMuMG; obtained from ATCC)
cells that were engineered to overexpress EGFR by VSVG retroviral transduction
of particles encoded from a pBabe-EGFR construct as previously described
(226) and cultured in DMEM supplemented with 10% FBS and 10 μg/mL of
insulin. TGF-β stimulated NME cells are a model of TNBC, that display
aggressive, post-surgical primary tumor recurrence and metastatic phenotypes
(226,227). Both cell lines were engineered to stably express firefly luciferase by
transfection with pNifty-CMV-luciferase and selection with Zeocin (500 μg/ml;
Invitrogen, Carlsbad, CA). Cell lines were not independently authenticated. Early
passage cells were utilized for all cell and tumor work. The following siRNAs
were purchased from Integrated DNA Technologies (Coralville, IA): Mouse
integrin β3 sense: [GCUCAUCUGGAAGCUACUCAUCACT], Mouse integrin β3
antisense: [AGUGAUGAGUAGCUUCCAGAUGAGCUC], Human integrin β3
sense: [GCUCAUCUGGAAACUCCUCAUCACC], and Human integrin β3
antisense: [GGUGAUGAGGAGUUUCCAGAUGAGCUC].
4.2.2 Preparation of ECO/siRNA nanoparticles

108
The ECO lipid carrier was synthesized as described previously (172).
ECO (MW=1023) was dissolved in 100% ethanol at a stock concentration of 2.5
mM for in vitro experiments and 50 mM for in vivo experiments. The siRNA was
reconstituted in RNase-free water to a concentration of 18.8 μM for in vitro
experiments and 25 μM for in vivo experiments. For in vitro experiments, an
siRNA transfection concentration of 100 nM was used. ECO/siRNA nanoparticles
were prepared at an N/P ratio of 8 by mixing predetermined volumes of ECO and
siRNA for a period of 30 minutes in RNase-free water (pH 5.5) at room
temperature under gentle agitation to enable complexation between ECO and
siRNA. The total volume of water was determined such that the volume ratio of
ethanol:water remained fixed at 1:20. For RGD- and RAD-modified ECO/siRNA
nanoparticles, RGD-PEG3400-Mal or RAD-PEG3400-Mal (MW = 3,400 Da;
NANOCS, New York, NY) was first reacted with ECO in RNase-free water at 2.5
mol% for 30 minutes under gentle agitation and subsequently mixed with siRNA
in RNase-free water for an additional 30 min. RGD-PEG3400-Mal or RAD-
PEG3400-Mal were prepared at a stock solution concentration of 0.32 mM in
RNase-free water. Again, the total volume of water was determined such that the
volume ratio of ethanol:water remained fixed at 1:20. After the incubation, free
peptide derivative was removed from RGD- and RAD-modified ECO/siRNA
nanoparticles by ultrafiltration (Nanosep, MWCO = 100 K, 5000 × g, 5 min;
Sigma Aldrich, St. Louis, MO). Conjugation of RGD-PEG3400-Mal to ECO was
determined and confirmed through matrix assisted laser desorption ionization
time-of-flight (MALDI-TOF) mass spectrometry using a Bruker Autoflex III MALDI-

109
TOF instrument. The size of RGD-modified ECO/siRNA nanoparticles in PBS
was determined by dynamic light scattering with a Brookhaven ZetaPALS
Particle Size Analyzer.
4.2.3 Western blot analyses
Immunoblotting analyses were performed as previously described (228).
Briefly, NME and MDA-MB-231 cells were seeded into 6-well plates (1.5 × 105
cells/well) and allowed to adhere overnight. The cells were then incubated in the
absence or presence of TGF-β1 (5 ng/mL) for 3 d and then treated with
ECO/siRNA complexes for 4 h in complete growth medium. At each indicated
time point, detergent-solubilized whole cell extracts (WCE) were prepared by
lysing the cells in Buffer H (50 mM -glycerophosphate, 1.5 mM EGTA, 1 mM
DTT, 0.2 mM sodium orthovanadate, 1 mM benzamidine, 10 mg/mL leupeptin,
and 10 mg/mL aprotinin, pH 7.3). The clarified WCE (20 mg/lane) were
separated through 10% SDS-PAGE, transferred electrophoretically to
nitrocellulose membranes, and immunoblotted with the primary antibodies, anti-
3 integrin (1:1000; Cell Signaling) and anti--actin (1:1000; Santa Cruz
Biotechnology).
4.2.4 Flow cytometry for nanoparticle cellular uptake
Cellular uptake and intracellular delivery of ECO/siRNA and RGD-
ECO/siRNA nanoparticles was evaluated quantitatively using flow cytometry.
ECO/siRNA and RGD-ECO/siRNA nanoparticles were prepared with 40 nM
Alexa Fluor 488-labeled siRNA (Qiagen; Valencia, CA). Approximately 2.5 × 104
NME cells were seeded onto 12-well plates and grown for an additional 24 h. The

110
cells were transfected with ECO/siRNA nanoparticles in 10% serum media. After
4 h, the transfection media was removed and each well was washed twice with
PBS. The cells were harvested by treatment with 0.25% trypsin containing
0.26 mM EDTA (Invitrogen; Carlsbad, CA), collected by centrifugation at
1,000 rpm for 5 min, resuspended in 500 μL of PBS containing 5%
paraformaldehyde, and finally passed through a 35 μm cell strainer (BD
Biosciences; San Jose, CA). Cellular internalization of the nanoparticles was
quantified by the fluorescence intensity measurement of Alexa Fluor 488 for a
total of 1 × 104 cells per sample using a BD FACSCalibur flow cytometer. All the
experiments were performed in triplicate and the data represent mean
fluorescence intensity and standard deviation.
4.2.5 Semi-quantitative real-time PCR analyses
Real-time PCR studies were performed as described previously (228).
Briefly, NME or MDA-MB-231 cells (100,000 cells/well) were seeded overnight
onto 6-well plates and treated with TGF- (5 ng/mL) for 3 days upon delivery of
ECO nanoparticles with a non-specific siRNA or 3 integrin-specific siRNA. At
each indicated timepoint, total RNA was isolated using the RNeasy Plus Kit
(Qiagen, Valencia, CA) and reverse transcribed using the iScript cDNA Synthesis
System (Bio-Rad, Hercules, CA). Semi-quantitative real-time PCR was
conducted using iQ-SYBR Green (Bio-Rad) according to manufacturer’s
recommendations. In all cases, differences in RNA expression for each individual
gene were normalized to their corresponding GAPDH RNA signals.
4.2.6 Invasion and proliferation assays

111
Invasion assays were conducted as described previously (219). Briefly,
NME cells, unstimulated (Pre-) or stimulated with TGF- for 3 d (Post-EMT),
were treated with the ECO/siRNA complexes for an additional 2 d. The cells were
then trypsinized and their ability to invade reconstituted basement membranes (5
× 104 cells/well) was measured utilizing modified Boyden chambers, as
previously described (18). For the proliferation assay, the NME cells were
cultured (1 × 104 cells/well) in the presence (post-EMT) or absence (pre-EMT) of
TGF- (5 ng/mL) for 3 d and then treated with ECO/siRNA. Cell proliferation was
determined by 3H-thymidine incorporation as previously described (216).
4.2.7 3-Dimensional (3D)-organotypic cultures
3D-organotypic cultures utilizing the “on-top” method were performed as
described previously (228). NME or MDA-MB-231 cells, which were unstimulated
(Pre-EMT) or stimulated with TGF- (5 ng/mL) for 3 d (Post-EMT), were cultured
in 96-well, white-walled, clear bottom tissue culture plates (2,000 cells/well) with
50 L of Cultrex cushions (Trevigen, Gaithersburg, MD) in media supplemented
with 5% Cultrex. The cells were maintained in culture for 4 d with continuous
ECO/siRNA treatment every 2 d. Growth was monitored by bright-field
microscopy or bioluminescent growth assays (where indicated) using luciferin
substrate (229,230).
4.2.8 Tumor growth and bioluminescent imaging (BLI)
All the animal studies were performed in accordance with the Institutional
Animal Care and Use Committee for Case Western Reserve University. NME
cells (1 × 106 cells/mouse) were engineered to stably express firefly luciferase,

112
and were subsequently injected into the lateral tail vein of 4-6 week old, female
nude mice (nu/nu Balb/c background) after TGF-β stimulation (5 ng/mL) for 7 d.
Pulmonary outgrowth was monitored and determined as described previously
(28). MDA-MB-231 cells, also engineered to express firefly luciferase and
stimulated with TGF-β for 7 d, were engrafted into the mammary fat pad of
female nude mice. The primary tumor growth and metastatic burden were
monitored and determined as described above.
4.2.9 In vivo therapeutic treatment
The tumor bearing mice were intravenously injected with RGD or RAD modified
ECO/siRNA nanoparticles with a siRNA dose of 1.5 mg/kg every five days
starting at the day 17 after cancer cell inoculation. Surface modified ECO/siRNA
nanoparticles (N/P ratio of 8) were prepared directly before each treatment
according to the two-step formulation process described above based on the
siRNA dose (1.5 mg/kg siRNA, 18.6 mg/kg ECO, 2.5 mol% PEGylation of ECO
with either RGD- or RAD-functionalized PEG3400-maleimide). Each mouse (25 g
body weight) received on average the nanoparticles containing 37.5 μg siRNA,
464 μg ECO, and 47.7 μg of either RGD-PEG3400-mal or RAD-PEG3400-mal in
150 μL nuclease-free water at each injection.
4.2.10 Immunofluorescence and immunohistochemical staining
For visualization of the actin cytoskeleton, immunofluorescent analysis
was performed as previously described (14). NME cells (5 × 104 cells/well) were
plated onto glass-bottom confocal dishes and allowed to adhere overnight, after
which they were simultaneously stimulated with TGF-β (5 ng/mL) and treated

113
with ECO/siRNA nanoparticles, either siβ3 or siNS at 100 nM siRNA
concentration. After 48 and 72 h of simultaneous TGF-β stimulation and
nanoparticle treatment, the cells were washed with PBS, fixed with 4%
paraformaldehyde, permeabilized in 0.1% Triton-X 100, stained with Alexa Flour
488 phalloidin (25 μM; Invitrogen; Carlsbad, CA), and visualized under a
fluorescent confocal microscope.
For immunohistochemistry, primary tumor samples were embedded in
optimum cutting temperature (O.C.T.) compound (Tissue-TeK; Torrence, CA) in
preparation for cryostat sectioning and immediately frozen. The samples were
then sectioned, fixed in paraffin, and maintained at -80⁰C. The samples were
stained with H&E to evaluate the presence of tumor tissue. Briefly, the samples
were fixed in 10% formalin, rehydrated in 70% ethanol and rinsed in deionized
water prior to hematoxylin staining. Samples were then rinsed in tap water,
decolorized in acid alcohol, immersed in lithium carbonate and rinsed again in
tap water. Next, the eosin counterstain was applied and slides were dehydrated
in 100% ethanol, rinsed in Xylene and finally mounted on a coverslip with
Biomount. For immunofluorescence detection of fibronectin, the paraffin-
embedded slides were first deparaffinized using a series of washes in xylene and
decreasing concentrations of ethanol. Heat-induced antigen retrieval was
performed using a pressure cooker in sodium citrate buffer (10 mM Sodium
citrate, 0.05% Tween 20, pH 6.0) for 20 minutes. Following heat-induced antigen
retrieval, the samples were blocked in TBST solution containing donkey serum
and washed three times with TBST. The primary antibody (Abcam; Cambridge,

114
MA) was applied at dilution of 1:100 in blocking solution for 1 h followed by three
rinses with TBST. The Alexa Fluor 488 secondary antibody (Jackson; West
Grove, PA) was applied at a dilution of 1:5000 in blocking solution for 1 h
followed by three washes with TBST and counterstained with DAPI at a dilution
of 1:2500 in blocking solution. After washing with TBST and mounting in an anti-
fade mounting solution (Molecular Probes; Grand Island, NY), the samples were
imaged using a confocal microscope.
4.2.11 Statistical analyses
Statistical values were defined using unpaired Student’s t-test, with p < 0.05
considered to be statistically significant.
4.3 Results and Discussion
4.3.1 ECO/siβ3 nanoparticles induce sustained silencing of 3 integrin
We examined the ability of ECO/β3 integrin-specific siRNA nanoparticles
(ECO/siβ3) to silence β3 integrin expression in mouse NME breast cancer cells
(226,227) (see materials and methods), which are reminiscent of a basal-like
breast cancer cell line (227), and human MDA-MB-231 breast cancer cells, a
TNBC cell line (231). The expression of 3 integrin was elevated in both cell lines
after stimulation with TGF-β for 72 h (219). Subsequent treatment of the
stimulated cells with ECO/siβ3 nanoparticles resulted in the rapid loss of β3
integrin mRNA within the first 16 h following treatment (Figure 4.1B). 3 integrin
expression was reduced by ~75% and this downregulation was sustained for up
to 7 d in NME cells treated with TGF-β (Figure 4.1B and C). ECO/si3 treatment

115
of MDA-MB-231 cells reduced 3 integrin expression level to that of the
unstimulated cells (Figure 4.1B and D). Importantly, treatment with
ECO/nonspecific siRNA nanoparticles (ECO/siNS) failed to alter 3 integrin
expression in both cell lines (Figure 4.1B-D). Collectively, these results
demonstrate the ability of ECO/si3 nanoparticles to induce efficient and
prolonged silencing of 3 integrin expression in breast cancer cells.

116
Figure 4.1. ECO/siβ3 nanoparticles induced sustained gene silencing of β3 integrin. A,
ECO forms nanoparticles with siRNA through electrostatic interactions, disulfide cross-
linking and hydrophobic interactions. B, β3 integrin mRNA expression in quiescent or
TGF- stimulated (5 ng/mL, 72 hours) NME and MDA-MB-231 cells with the indicated
treatment groups at 100 nM siRNA by semi-quantitative real-time PCR (n=3, mean ± SE,
p0.01 for all time points beyond 8 hours). Western blot analysis of β3 integrin
expression in quiescent or TGF- stimulated (5 ng/mL, 72 hours) NME C, and MDA-MB-

117
231 d) cells at the indicated time points post-nanoparticle treatment with the indicated
treatment groups.
4.3.2 ECO/siβ3 nanoparticles attenuate TGF-β-mediated EMT, invasion, and
proliferation
Next, we investigated the effects of ECO/si3 nanoparticles on EMT, invasion,
and proliferation of breast cancer cells. Phalloidin staining of the actin
cytoskeletal architecture (222) revealed that quiescent NME cells displayed the
epithelial hallmark of densely packed and well-organized cortical actin network
(222), while those stimulated with TGF- exhibited dissolved junctional
complexes and acquired an elongated morphology consistent with stress fiber
formation that are characteristic of mesenchymal cells (Figure 4.2A). Treatment
of NME cells with ECO/siβ3 nanoparticles at the time of TGF-β stimulation
inhibited dissolution of the junctional complexes and stress fiber formation, while
treatment with ECO/siNS nanoparticles failed to impact TGF-β-induced
morphological changes (Figure 4.2A). Moreover, the phenotypic changes in
post-EMT cells were accompanied by alterations in the expression of EMT-
related genes (232). Silencing of β3 integrin with ECO/siβ3 nanoparticles
significantly reduced TGF--mediated upregulation of the mesenchymal markers,
N-cad and PAI-1, and inhibited TGF--mediated downregulation of the epithelial
markers, E-cad and CK-19 (Figure 4.2B and C). ECO/siNS nanoparticles did not
alter the effect of TGF- on the aforementioned EMT markers.
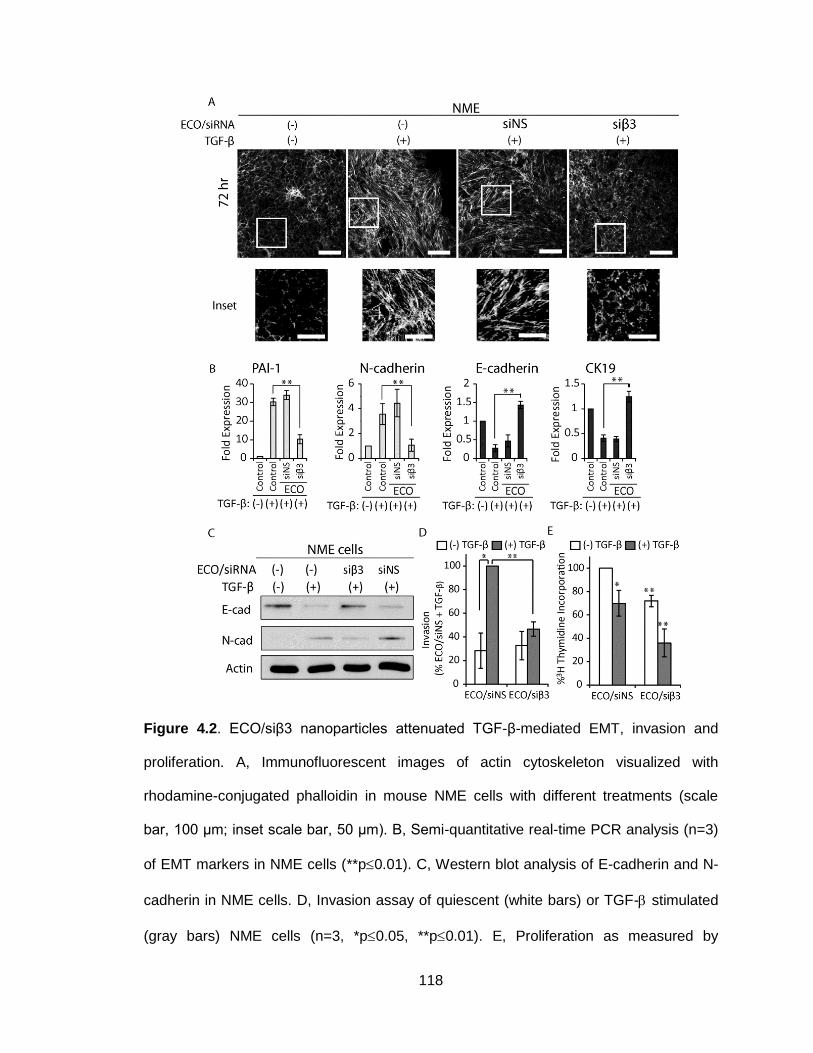
118
Figure 4.2. ECO/siβ3 nanoparticles attenuated TGF-β-mediated EMT, invasion and
proliferation. A, Immunofluorescent images of actin cytoskeleton visualized with
rhodamine-conjugated phalloidin in mouse NME cells with different treatments (scale
bar, 100 μm; inset scale bar, 50 μm). B, Semi-quantitative real-time PCR analysis (n=3)
of EMT markers in NME cells (**p0.01). C, Western blot analysis of E-cadherin and N-
cadherin in NME cells. D, Invasion assay of quiescent (white bars) or TGF- stimulated
(gray bars) NME cells (n=3, *p0.05, **p0.01). E, Proliferation as measured by

119
[3H]thymidine incorporation of either quiescent (white bars) or TGF- stimulated (gray
bars) NME cells (n=3, *p0.05, **p0.01). For all experimental groups, NME cells were
pre-treated with TGF- (5ng/mL; 72 hours) followed by ECO/siRNA nanoparticle
treatment using 100nM siRNA. For panels B, D-E, data represent mean SE. Results
for panels D-E are representative of three independent experiments.
TGF--mediated EMT is also associated with increased invasiveness
(232) and cell cycle arrest (233). TGF--stimulated NME cells treated with
ECO/siNS readily invaded reconstituted basement membrane, while ECO/siβ3
nanoparticles significantly inhibited invasion (Figure 4.2D). Conversely,
treatment of quiescent NME cells with ECO/siβ3 nanoparticles had no effect on
basal invasiveness, an event that is uncoupled from 3 integrin expression.
Previous studies demonstrate that parental NMuMG cells readily undergo
proliferative arrest when stimulated with TGF- (233). We found that the NME
cells override these cytostatic effects of TGF- (Figure 4.2E), while treatment
with ECO/si3 partially restores TGF--mediated cytostasis (Figure 4.2E).
Collectively, these findings indicate that ECO/si3 nanoparticle-mediated
silencing of β3 integrin attenuates TGF--induced EMT and invasion, and
partially restores TGF--mediated cytostasis.
4.3.3 ECO/siβ3 nanoparticles attenuate outgrowth of murine and human
MECs in 3D-organotypic culture
To study the effects of ECO/si3 nanoparticles in a physiologically relevant
system, we cultured NME and MDA-MB-231 cells in 3D-organotypic cultures to

120
recapitulate the elastic modulus of a distant metastatic site such as the
pulmonary microenvironment (234). This culture method presented additional
obstacles in the delivery and uptake of nanoparticles, since these organoids were
compact and surrounded by a dense matrix. Using confocal microscopy, we
confirmed that ECO/siRNA nanoparticles formulated with fluorescently-labeled
siRNA (AF-488) readily gained access to NME organoids by first penetrating into
the periphery within 30 min after treatment, and further dispersing throughout the
entirety of the organoid to reach a near-uniform distribution within 24 h (Figure
4.3A). The dispersion of ECO/siRNA nanoparticles into the inner cell layers of
the organoids suggests that ECO/siRNA uptake by these cells may result from
diffusion through intercellular spaces or through transcytosis (235). Fig. 3C and C
show that NME and MDA-MB-231 organoids stimulated with TGF-β exhibited
elevated growth as compared to their quiescent counterparts. Treatment with
ECO/si3 nanoparticles inhibited the growth of both quiescent and TGF--
stimulated NME and MDA-MB-231 organoids (Figure 4.3B and C) in comparison
to treatment with ECO/siNS nanoparticles. These results demonstrate the
effectiveness of ECO/si3 nanoparticles in attenuating the 3D outgrowth of post-
EMT breast cancer cells.
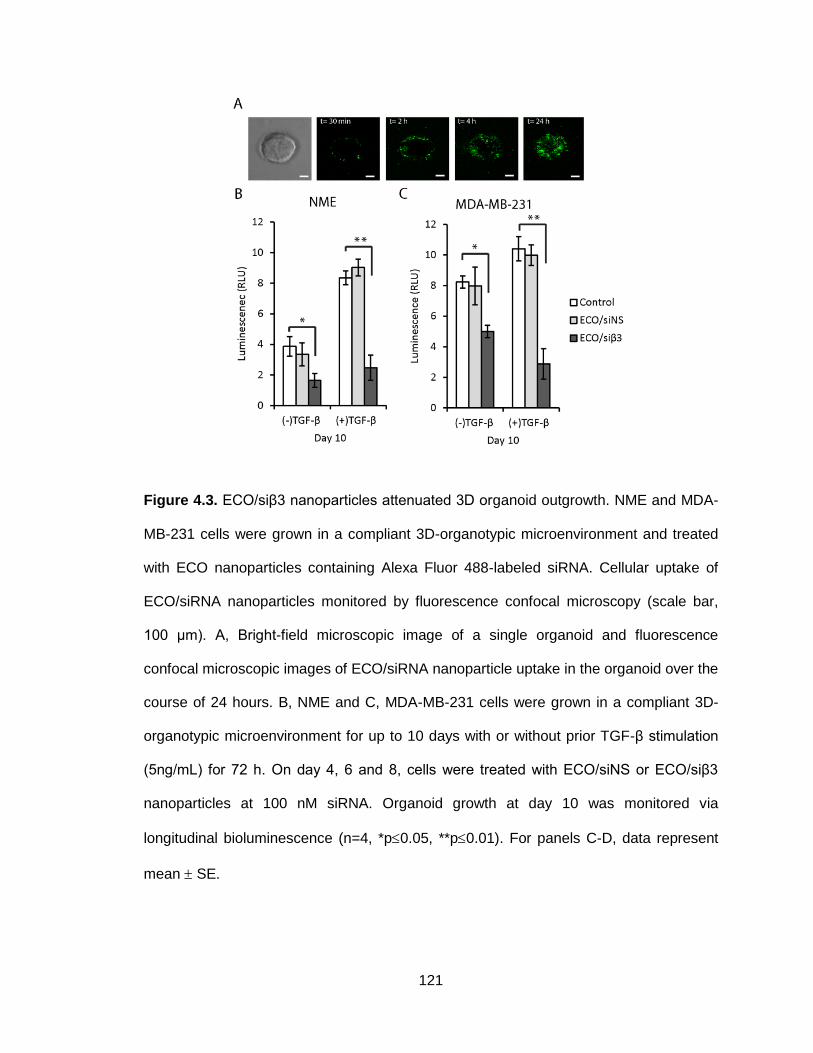
121
Figure 4.3. ECO/siβ3 nanoparticles attenuated 3D organoid outgrowth. NME and MDA-
MB-231 cells were grown in a compliant 3D-organotypic microenvironment and treated
with ECO nanoparticles containing Alexa Fluor 488-labeled siRNA. Cellular uptake of
ECO/siRNA nanoparticles monitored by fluorescence confocal microscopy (scale bar,
100 μm). A, Bright-field microscopic image of a single organoid and fluorescence
confocal microscopic images of ECO/siRNA nanoparticle uptake in the organoid over the
course of 24 hours. B, NME and C, MDA-MB-231 cells were grown in a compliant 3D-
organotypic microenvironment for up to 10 days with or without prior TGF-β stimulation
(5ng/mL) for 72 h. On day 4, 6 and 8, cells were treated with ECO/siNS or ECO/siβ3
nanoparticles at 100 nM siRNA. Organoid growth at day 10 was monitored via
longitudinal bioluminescence (n=4, *p0.05, **p0.01). For panels C-D, data represent
mean SE.

122
4.3.4 Surface modification of ECO/siRNA nanoparticles with RGD peptide
promotes cellular uptake and sustains gene silencing
An essential goal of in vivo siRNA delivery is to increase siRNA localization at the
disease site while minimizing its accumulation in non-target tissues (236). We
modified ECO/siRNA nanoparticles with a cyclic RGD peptide, which binds to
v3 integrin, or a non-targeting control cyclic RAD peptide via PEG spacers
(3,400 Da; (237) ) (Figure 4.4A and B). The size of RGD-modified ECO/siRNA
nanoparticles (RGD-ECO/siRNA) was 88.3 ± 5.2 nm, as determined by dynamic
light scattering (Figure 4.4C). Their cellular uptake was then examined in both
unstimulated and TGF-β-stimulated NME cells. TGF-β stimulation had no effect
on the cellular uptake of unmodified ECO/siRNA nanoparticles, while cellular
uptake of RGD-ECO/siRNA nanoparticles was robustly enhanced (Figure 4.4D
and E), leading to effective silencing of 3 integrin in TGF--treated cells (Figure
4.4F and G). Since v3 is a major receptor that recognizes the RGD targeting
peptide, we sought to determine whether β3 integrin silencing impacts cellular
uptake of RGD-ECO/siRNA nanoparticles. Although cellular uptake of RGD-
targeted nanoparticles was diminished upon β3 integrin silencing, uptake was
nonetheless elevated consistently, because of the presence of other receptors
for the peptide (Figure 4.4H;(238)). Taken together, these results show that
RGD-targeted ECO/siRNA nanoparticles efficiently promote cellular uptake and
robust gene silencing, particularly in post-EMT and metastatic breast cancer

123
cells.
Figure 4.4. RGD modification of ECO/siRNA nanoparticles enhances uptake in post-
EMT breast cancer cells. A, RGD-ECO/siRNA nanoparticles were prepared by modifying
the ECO/siRNA nanoparticles with RGD-targeted PEG ligand via thiol-maleimide
chemistry. B, Conjugation of RGD-PEG-maleimide to ECO was confirmed from MALDI-
TOF mass spectroscopy. The center of the bell was observed at m/z 5200 confirming
the conjugation of RGD-PEG-maleimide (m/z, 4100) to ECO (m/z, 1046). C, The size of
RGD-ECO/siRNA nanoparticles was determined by dynamic light scattering (DLS). NME

124
cells, with or without TGF-β stimulation (5ng/mL; 72 hours), were treated with ECO
nanoparticles with Alexa Fluor 488-labeled siRNA at 40 nM siRNA for 4 hours. NME
cells that were stimulated with TGF-β exhibited a higher cellular uptake of RGD-targeted
ECO/siRNA nanoparticles compared to the non-targeted ECO/siRNA nanoparticles as
confirmed by D, confocal microscopy (scale bar, 50 μm) and E, quantified by flow
cytometry (n=3, **p0.01). Quantitative analysis of β3 integrin mRNA levels following
treatment with siβ3 nanoparticles by real-time PCR (n=3, **p0.01) in F, NME and G,
MDA-MB-231 cells revealed RGD-targeted ECO/siRNA nanoparticles maintain gene
silencing. H, Cellular uptake in NME cells, both with and without TGF-β stimulation
(5ng/mL; 72 hours), was quantified by flow cytometry for RGD-ECO/siRNA nanoparticles
containing Alexa Fluor 488-labelled siRNA 4 hours after treatment. One group of TGF-β
stimulated NME cells (TGF-β + ECO/siβ3) was treated with ECO/siβ3 nanoparticles at
100 nM siRNA for 48 hours prior to cellular uptake with the RGD-targeted nanoparticles
to quantify the effect of β3 integrin silencing on targeted uptake (n=3, ±SE, *p0.05,
**p0.01). For panels C-F, data represent mean SE.
4.3.5 RGD-ECO/si3 nanoparticles inhibit pulmonary outgrowth of mouse
MECs in vivo
To evaluate the effect of 3 integrin silencing on pulmonary outgrowth, we
inoculated TGF--treated NME cells into the lateral tail vein of nude mice and
subsequently monitored pulmonary outgrowth. Systemic injections of RGD-
targeted ECO/si3 nanoparticles dramatically inhibited pulmonary outgrowth of
post-EMT NME cells (Figure 4.5), as compared to non-specific RAD-ECO/si3
and RGD-ECO/siNS treatment groups. These results demonstrate that RGD-
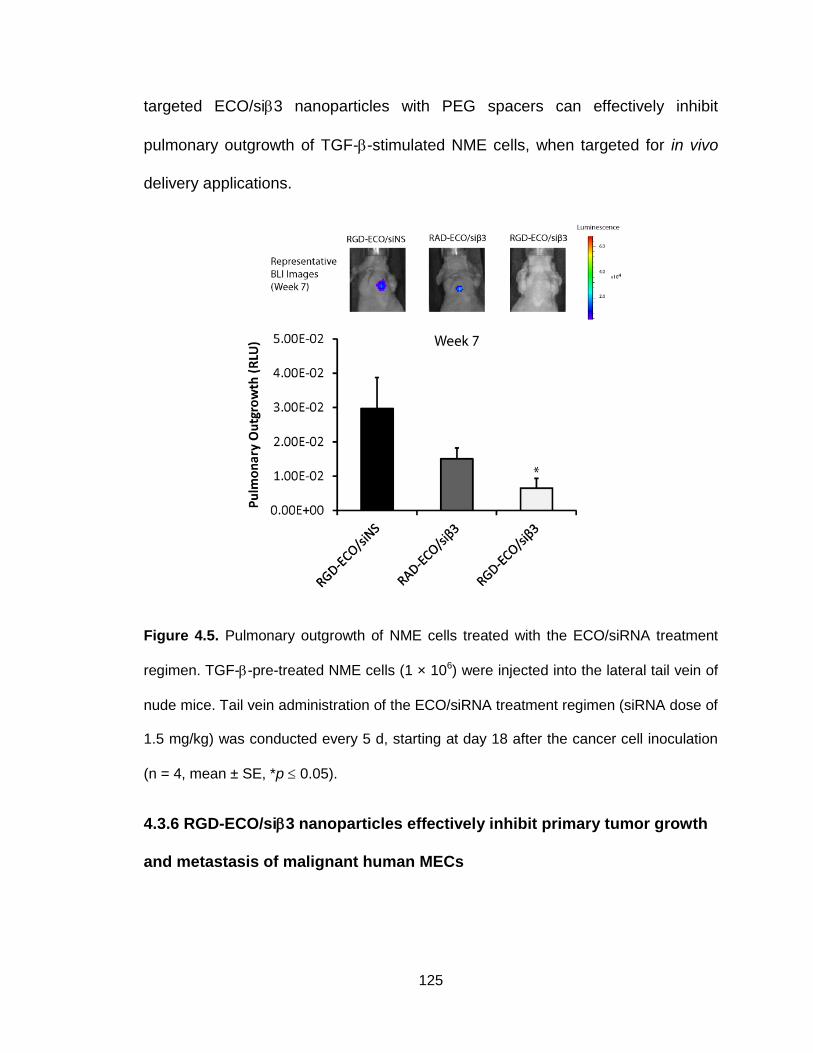
125
targeted ECO/si3 nanoparticles with PEG spacers can effectively inhibit
pulmonary outgrowth of TGF--stimulated NME cells, when targeted for in vivo
delivery applications.
Figure 4.5. Pulmonary outgrowth of NME cells treated with the ECO/siRNA treatment
regimen. TGF--pre-treated NME cells (1 × 106) were injected into the lateral tail vein of
nude mice. Tail vein administration of the ECO/siRNA treatment regimen (siRNA dose of
1.5 mg/kg) was conducted every 5 d, starting at day 18 after the cancer cell inoculation
(n = 4, mean ± SE, *p 0.05).
4.3.6 RGD-ECO/si3 nanoparticles effectively inhibit primary tumor growth
and metastasis of malignant human MECs

126
To further evaluate the in vivo effect of our targeted ECO/si3 nanoparticles,
MDA-MB-231 cells pretreated with TGF- were engrafted into the mammary fat
pad of nude mice. Mice were treated with RGD-ECO/si3 (1.5 mg/kg siRNA)
every 5 days, starting at day 17 (Figure 4.6A). Primary tumor burden was
monitored by bioluminescence imaging (BLI) and caliper measurements.
Compared to the untreated control, RGD-ECO/siNS or RAD-ECO/si3 treatment
groups, RGD-ECO/si3 treated mice exhibited significantly reduced primary
tumor burden (Figure 4.6B-D). The primary tumors were resected at week 9
(Figure 4.6A) and weighed. Figure 4.6E shows that RGD-ECO/si3 treatment
resulted in significantly reduced tumor weights as compared to the control
groups. Importantly, the therapeutic efficacy of RGD-ECO/si3 was reflected by
decreased mRNA expression of 3 integrin in the primary tumors, relative to that
in the control groups (Figure 4.6F). RAD-ECO/si3 treatment resulted in
marginally reduced 3 integrin expression (Figure 4.6F), which was consistent
with the marginally reduced primary tumor burden, which were not statistically
significant (Figure 4.6D and E). These data reflect partial uptake of the RAD-
ECO/si3 nanoparticles by primary tumors as a result of passive tumor
accumulation attributed to tumor vascular hyperpermeability. H&E staining of
tissue sections demonstrated similar histopathological patterns in RGD-
ECO/si3-treated and control groups, while untreated mice developed tumors
that were more vascularized than RGD-ECO/si3-treated tumors (Figure 4.6G).
Further immunostaining of tissue sections indicated that RGD-ECO/si3-treated

127
primary tumors exhibited decreased expression of a mesenchymal marker,
fibronectin (Figure 4.6H), which is associated with poor overall survival (239).

128

129
Figure 4.6. RGD-targeted ECO/si3 nanoparticles inhibited primary tumor growth and
EMT in mice after systemic administration. A, Schematic of targeted ECO/siRNA
nanoparticle treatment (1.5 mg siRNA/kg) schedule in vivo. Tumor growth was
monitored at the indicated time points B, and quantified by C, BLI (data represents mean
± SE, n=5, *p0.05, **p0.01), and D, caliper measurements (data represents mean ±
SE, n=5, *p0.05, **p0.01). E, Primary tumors were resected at week 9, and final tumor
weights of the indicated treatment groups were obtained (data represents mean ± SE,
n=5, **p0.01). F, Semi-quantitative real-time quantification of 3 integrin mRNA
expression from resected primary tumors of the indicated groups (data represents mean
± SE, n=5, **p0.01). G, H&E staining at 200X of primary tumors. H, H&E, DAPI and
fibronectin immunostaining of the indicated primary tumors (scale bar, 300 μm).

130

131
Figure 4.7. RGD-ECO/si3 nanoparticles inhibited breast cancer metastasis and
primary tumor recurrence. A, BLI images of mice at week 12 revealed differences in
metastasis and primary tumor recurrence for the different treatment groups after primary
tumor resection on week 9. B, Quantification of primary tumor recurrence (data
represents mean ± SE, n=5, *p0.05). C, Quantification of thoracic metastasis by BLI
(data represents mean ± SE, n=5, *p0.05, **p0.01). Mice were released from
ECO/siRNA therapeutic regimen at week 12. D, Representative BLI of mice on week 16.
E, Quantification of whole body tumors from D. (data represents mean ± SE, n=5,
*p0.05). F, Change in the body weight of mice bearing MDA-MB-231 primary tumors
across various treatment groups over the course of 16 weeks. The body weight was
measured weekly and reported as mean ± S.E. (n = 5) for each group. No significant
difference was observed between the various treatment groups at any time point.
RGD-ECO/si3 treatment resulted in robust inhibition of tumor metastases
(Figure 4.7A and C) and primary tumor recurrence (Figure 4.7B), as compared
to control groups at week 12 post-engraftment. Primary tumor recurrence was
evaluated by restricting the region of interest (ROI) of the bioluminescent image
to that of the area originally occupied by the resected primary tumor.
Interestingly, RAD-ECO/si3 treatment also mediated significant inhibition of
tumor metastases and primary tumor recurrence as compared to RGD-ECO/siNS
treatment, but to a lesser extent than RGD-ECO/si3. This decrease in the
efficacy of RAD-ECO/si3 could be attributed to the lack of specific targeting and
binding of the nanoparticles to the cancer cells. At 12 weeks post-engraftment,
the RGD-ECO/siβ3 group was released from nanoparticle treatment to evaluate

132
the lasting effects of therapeutic β3 integrin silencing on tumor recurrence and
metastases in comparison with the untreated control group. At 4 weeks post-
treatment release (16 weeks post-engraftment), the RGD-ECO/si3-treated mice
remained tumor-free, while the tumor burden of untreated mice continued to
increase (Figure 4.7D and E). Finally, throughout the entire course of treatment,
no significant difference was observed in the body weights across the different
treatment groups, demonstrating the low toxicity of the intravenously
administered, targeted and PEGylated ECO/siRNA nanoparticles (Figure 4.7F).
Collectively, these data highlight the effectiveness and safety of the systemic
administration of RGD-ECO/si3 nanoparticles for the inhibition of TNBC tumor
progression and metastases.
4.4 Discussion
Cancer metastasis involves a cascade of events, including EMT and local
invasion, intravasation, survival in circulation, extravasation, and outgrowth of
disseminated cells at the secondary site. Cancer cell EMT is considered to be a
critical step for the initiation of cancer metastasis. In order to alleviate metastasis,
it is essential to prevent EMT and to eliminate the dissemination and outgrowth of
cells that have already undergone EMT. Silencing EMT-related genes by RNAi
has the potential to revolutionize current treatment standards. β3 integrin has
been implicated as a powerful inducer of EMT (227,240), potentiating the
oncogenic effects of TGF-β by inducing invasion and metastases of MECs. Here,
we demonstrated that silencing the expression of 3 integrin with RGD-ECO/si3
nanoparticles prevented TGF--mediated EMT and inhibited TNBC metastasis.

133
Although functional disruption of β3 integrin has been shown to attenuate
TGF--mediated EMT and tumor progression, the utilization of β3 integrin siRNA
as a therapeutic regimen has been limited due to the lack of a clinically feasible
approach. In the present study, we demonstrated ECO to be a versatile, safe,
and effective siRNA delivery vehicle that forms stable RGD-targeted siRNA
nanoparticles for systemic siRNA delivery, which silenced 3 integrin expression,
and subsequently eliminated post-EMT cells responsible for metastases. The
inhibition of TGF--mediated EMT with ECO/si3 nanoparticles was evident by
the obstruction of TGF--mediated morphological changes, downregulation of
epithelial markers, and upregulation of mesenchymal markers in vitro. Moreover,
silencing 3 integrin also decreased invasiveness and reduced outgrowth of
breast cancer cells in 3D-culture and in vivo. The effectiveness of RGD-targeted
ECO/si3 nanoparticles for treating metastatic TNBC was evident by reduced
primary tumor burden in tumors with diminished expression of 3 integrin. More
importantly, ECO/si3 treatment abrogated metastases and primary tumor
recurrence after treatment release.
Several unique features of ECO and ECO/siRNA nanoparticles render the
delivery system effective for safe and systemic delivery of a therapeutic siRNA in
treating metastatic TNBC. ECO possesses pH-sensitive amphiphilicity, which is
essential to promote endo-lysosomal escape and avoid lysosomal siRNA
degradation. The amino groups within ECO become protonated when exposed to
the increasingly acidic environment of the endo-lysosomes, thereby increasing
the cationic charge of the nanoparticles to promote endo-lysosomal membrane

134
fusion for escape. Thiol groups of the cysteine residues are autoxidized into
disulfide bridges during nanoparticle formulation, which stabilize the
nanoparticles during circulation and become reduced by cytosolic glutathione,
resulting in siRNA release from ECO to initiate RNAi in cytoplasm (221). The thiol
groups also facilitate surface modification of ECO/siRNA nanoparticles with PEG
or targeting peptides. This modification enables targeted in vivo siRNA delivery
into tumor tissues and minimizes potential toxic side effects. This multi-
functionality uniquely resides within a simple small molecular lipid, making ECO a
versatile carrier for highly efficient cytosolic siRNA delivery. The formation of
targeted ECO/siRNA nanoparticles is straightforward and reproducible, and can
be readily scaled-up for clinical development.
We demonstrated that 3 integrin is a powerful therapeutic target for treating
metastatic TNBC, and that RGD-ECO/si3 nanoparticles are an effective vehicle
to systemically silence 3 integrin in post-EMT cancer cells. Specifically, RGD-
ECO/si3 may be beneficial for TNBC patients who currently lack targeted
treatments. Administering RGD-ECO/si3 nanoparticles in combination with
chemotherapy also has the potential to resensitize drug-resistant TNBC cells to
chemotherapy. Moreover, v3 integrin is highly expressed in the angiogenic
vasculature of many cancer types and the RGD peptide is a well-established
strategy for honing the delivery of therapeutics to the tumor (237,241). These
studies highlight the potential of our RGD-ECO/si3 regimen in targeting not only
the primary tumor, but also endothelial cells in angiogenic tumor vasculature for
treating breast cancer. However, β3 integrin silencing may cause potential side

135
effects in wound-healing, which remain to be addressed. Furthermore, ECO/siβ3
treatment of mice bearing established metastatic lesions yielded no therapeutic
effect (data not shown), therefore the proposed ECO/siβ3 regimen is likely most
effective at treating early stage breast cancer. Previous studies suggest that
established metastatic breast lesions utilize 1 integrin to sustain outgrowth
(242,243), which we currently hypothesize to limit the effectiveness of our RGD-
ECO/si3 regimen in the treatment of late stage metastatic breast cancer. Due to
the versatility of our multifunctional ECO/siRNA nanoparticles, targeted
nanoparticles with different targeting ligands and siRNAs, including 1 integrin
siRNA, can be readily formulated to address this issue. We are currently
exploring the effectiveness of dual 1 and 3 integrin targeting using our
ECO/siRNA nanoparticles (240).
4.5 Conclusion
These findings highlight the silencing of 3 integrin expression with our targeted
multifunctional ECO/siβ3 nanoparticles as a promising therapeutic strategy for
the effective treatment of metastatic TNBC associated with elevated β3 integrin.
Integrins have been commonly investigated as therapeutic targets in a multitude
of diseases. In cancers, integrins offer intriguing targets for drug therapy but
effective systemic methods to ablate their function or expression as a strategy to
block metastatic disease have been elusive. While antibodies and inhibitory
peptides have been used to inhibit integrins, RNAi-based technologies are more
attractive in terms of their therapeutic potential. SiRNA-mediated gene silencing

136
both eliminates the physical integrin protein to disrupt membrane and intracellular
interactions, disrupt integrin-related signaling pathways, and attenuate ligand-
dependent actions of integrin receptors. In summary, the peptide-targeted
ECO/siRNA nanoparticle delivery platform is a powerful tool to enable the safe
and efficient in vivo delivery of siRNAs.
4.6 Acknowledgments
Research support was provided, in part, by grants from the National Institutes of
Health to Z-R.L. (EB00489) and W.P.S. (CA129359 and CA177069), a National
Science Foundation Graduate Research Fellowship to M.D.G. (DGE-0951783)
and a Department of Defense Postdoctoral Fellowship to J.G.P (BC133808). We
thank Dr. Amita M. Vaidya for editing and proofreading the manuscript.

137
Chapter 5
Development of a pH-Cleavable PEG Surface Modification Strategy to
Overcome the PEG Dilemma in ECO/siRNA Nanoparticles

138
5.1 Introduction
Recently, we have developed (1-aminoethyl)iminobis[N-(oleicylcysteinyl-1-
amino-ethyl)propionamide] (ECO), a cationic lipid-based carrier that forms stable
nanoparticles with siRNA through ionic charge interactions (221,244). ECO
exhibits multifunctional properties necessary for the cytosolic delivery of siRNA
including pH-dependent membrane disruption and glutathione-mediated
decomplexation and siRNA release (221). While promising, cationic lipid-based
delivery systems exhibit poor pharmacokinetic profiles, short circulatory half-
lives, and suffer from significant RES uptake and clearance when delivered
systemically (73,245,246). Surface-modification with hydrophilic polyethylene
glycol (PEG) polymer chains can impart biocompatibility by masking the cationic
charge of the ECO/siRNA nanoparticles and prolong the pharmacokinetic
behavior of the nanoparticle complexes in vivo (247–249). The terminal groups of
PEG chains can be readily functionalized to reactive groups to allow for facile
covalent coupling to the surface of nanoparticles or targeting peptides. For
example, cysteine residues within the structure of ECO allow modification of the
nanoparticle surface with maleimide-functionalized PEG moieties via simple thiol-
maleimide chemistry. However, the steric hindrance provided to the nanoparticle
surface can negatively impact the gene silencing performance by inhibiting
cellular uptake within the target cells, leading to a significant loss of therapeutic
activity (250,251). It has also been observed that PEGylation can alter the
intracellular trafficking of non-viral gene delivery systems, inhibit endolysosomal

139
escape, and promote endosomal degradation, a generalized phenomenon
referred to as the ‘PEG dilemma’ (252,253).
To overcome the systemic barriers of PEGylated nanomedicines in vivo, a
wide array of stimuli-sensitive systems have been created to leverage various
physiological conditions specific to the disease or delivery site (254–257). Such
systems are engineered to undergo physicochemical changes in response to
endogenous cues, such as the pH gradients that exist at both the cellular and
tissue levels. Several strategies have been explored to improve the delivery
efficiency of PEGylated systems. Transient PEG-modifications have been widely
explored in previous work (258). The use of exchangeable PEG lipids, such as
PEG-ceramides, enables the time-dependent release of PEG from cationic lipid-
based systems to enhance intracellular delivery (259). Delivery systems
responsive towards a tumor-specific microenvironmental cue, such as proteases,
a reducing or acidic environment to affect drug delivery, are also in various
stages of development (260–262). PEG-modifications using disulfide or
orthoester linkages create a reducible system in which the PEG coat sheds in
response to the reductive cytosolic environment by endogenous glutathione
(263). Tumor-specific cleavable PEG systems have likewise been created for
enhanced delivery and efficiency into tumor cells. PEG-lipid systems composed
of the matrix metalloproteinase (MMP)-substrate peptide produce enzymatically
sensitive systems that are cleaved by MMPs that reside in the tumor extracellular
microenvironment (264,265).

140
Strategies dependent upon the exploitation of the acidic tumor
microenvironment have been explored (266–268). The pH of the extracellular
environment of tumor tissues (pH 6.5) is significantly lower than that of normal
tissue (pH 7.4) due to the elevated metabolic activity of tumor cells (269). As
such, cationic lipid-based delivery systems designed to shed their PEG coat in
response to the acidic pH can re-expose the positively charged surface to
improve tumor cell uptake (270). While this strategy improves cellular uptake,
such systems must still possess the ability to promote internalization and
facilitate endolysosomal escape to achieve effective cytosolic delivery of the
siRNA payload (271).
As PEGylation of cationic delivery systems is necessary for in vivo
delivery applications, a pH-cleavable surface modification strategy that can
attenuate cytotoxicity during systemic circulation and also promote internalization
and endolysosomal escape is desired. The present study focuses on the
development of a dual pH-sensitive and peptide-targeted siRNA delivery system.
The inclusion of the acid-labile hydrazone bond within a peptide-targeted PEG
moiety (RGD-PEG(HZ)-ECO/siRNA) created a pH-cleavable coating that sheds
from the core ECO/siRNA nanoparticle in response to the acidic endolysosomal
environment following uptake into tumor cells (Schematic 5.1). Once the core
ECO/siRNA nanoparticle has been re-exposed, the intrinsic pH-sensitive
amphiphilicity of ECO/siRNA nanoparticles enables endolysosomal membrane
disruption and escape to achieve potent gene silencing.
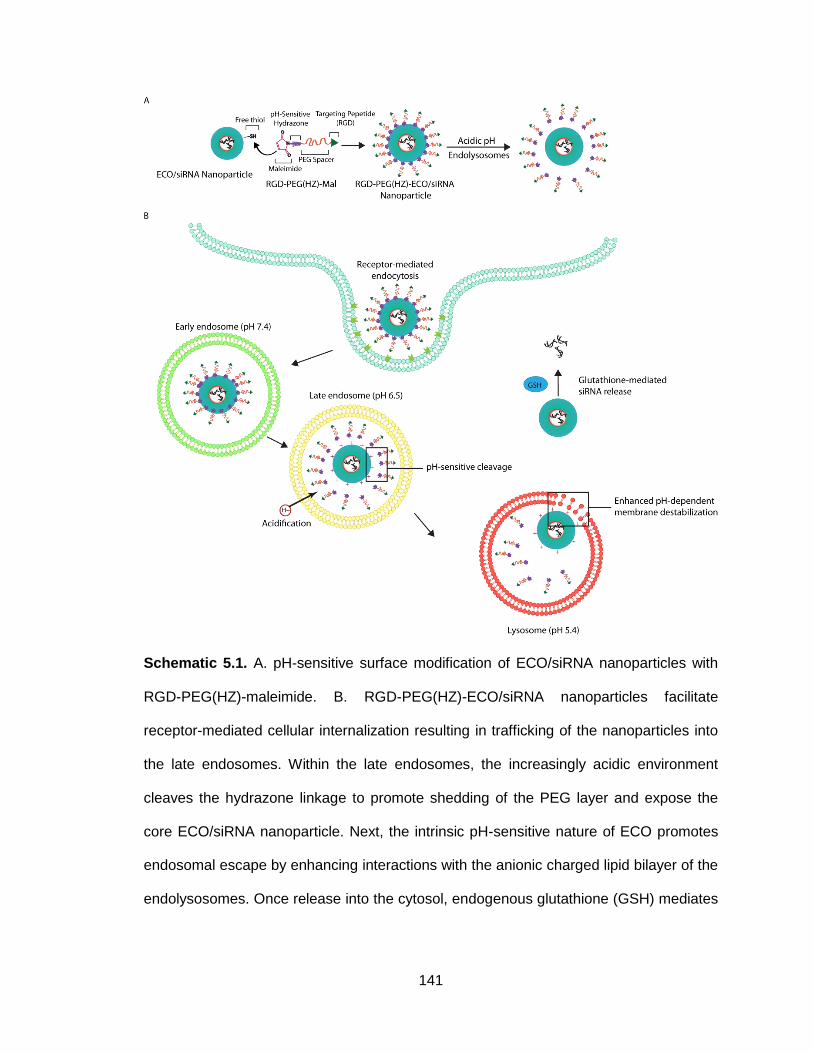
141
Schematic 5.1. A. pH-sensitive surface modification of ECO/siRNA nanoparticles with
RGD-PEG(HZ)-maleimide. B. RGD-PEG(HZ)-ECO/siRNA nanoparticles facilitate
receptor-mediated cellular internalization resulting in trafficking of the nanoparticles into
the late endosomes. Within the late endosomes, the increasingly acidic environment
cleaves the hydrazone linkage to promote shedding of the PEG layer and expose the
core ECO/siRNA nanoparticle. Next, the intrinsic pH-sensitive nature of ECO promotes
endosomal escape by enhancing interactions with the anionic charged lipid bilayer of the
endolysosomes. Once release into the cytosol, endogenous glutathione (GSH) mediates

142
reduction of disulfide bonds formed within ECO/siRNA nanoparticles to release the
siRNA cargo. Upon release, free siRNA is able to initiate RNAi-induced gene silencing.
5.2 Materials and Methods
5.2.1 Cell Culture: Human triple-negative breast cancer MDA-MB-231 cells
expressing a luciferase reporter enzyme (MDA-MB-231-Luc) were obtained from
ATCC (American Type Culture Collection) and cultured in Dulbecco’s modified
Eagle’s media (Invitrogen) and supplemented with 10% fetal bovine serum
(Invitrogen), 100 μg/mL streptomycin, and 100 unites/mL penicillin (Invitrogen).
The cells were maintained in a humidified incubator at 37⁰C and 5% CO2.
5.2.2 Synthesis of mPEG(HZ)-mal and RGD-PEG(HZ)-mal: NHS-PEG-SH
(MW 3400) and mPEG-HZ (MW 5000) were obtained from Nanocs, cRGDfk was
obtained from Peptides International. RGD-PEG-mal was prepared as previously
described (169). Full synthetic schemes and characterization are available within
the Appendix.
To synthesize the non-targeted, pH-cleavable PEG spacer, mPEG5000-
hydrazide (Laysan Bio) was reacted with N-4-acetylphenyl maleimide (APM).
First, 87.8 mg (MW=5000, 1 equivalent, 17.56 μmol) of the hydrazide-derivatized
mPEG5000 was dissolved in 10 mL DCM/MeOH (50/50) and 100 mg Na2SO4 was
added. Next, 11.7 mg (MW=215.2, 3.1 equivalent, 54.44 μmol) of APM was
dissolved in 1 mL DCM/MeOH (50/50) and added drop-wise into the mPEG-
hydrazide solution. After the addition of APM, acetic acid (1.77 μL of 34% v/v
solution in DCM, 0.6 equivalent, 10.54 μmol) was added. The reaction was
stirred for 24 hours at room temperature under nitrogen. After 24 hours, the

143
solution was precipitated into cold diethyl ether (3X) to obtain a purified product.
The H1 NMR spectrum (Solvent: CDCl3) of mPEG5000(HZ)-maleimide was used to
confirm the correct structure using the following characteristic peaks: 8.43 (s, 1H,
-NH-), 8.06 (d, 2H, in phenyl), 7.54 (d, 2H, in phenyl), 6.91 (2, 2H, two olefinic
protons of maleimide), 3.48-3.58 (m, 438H, PEG).
A three-step reaction was used to synthesize the cRGD-targeted pH-
cleavable PEG-hydrazone moiety (272–274). First, cRGDfk was first conjugated
to the heterobifunctional NHS-PEG3400-SH: 25 mg (MW=603.7, 2 equivalent, 41.4
μmol) of c(RGD)fk was dissolved in 5 mL DMF. Cyclic (RGD)fk was used at 2X
molar excess to the NHS-PEG3400-SH. Next, 70.4 mg (MW=3400, 1 equivalent,
20.7 μmol) of NHS-PEG3400-SH was dissolved in 1 mL of DMF and added drop-
wise into the c(RGD)fk/DMF solution. After addition, 100 μL of DIPEA was added
to the solution. The solution was stirred gently at room temperature for 4 hours.
The solution was precipitated into an excess of diethyl ether (3X) to remove
excess c(RGD)fk and obtain the purified cRGD-PEG3400-SH product. To ensure
free thiol availability, 100 mg of dithiothreitol (DTT) was added to the solution and
stirred overnight to reduce any disulfide bonds present in the synthesized cRGD-
PEG-SH. Free DTT was removed using a desalting spin column (1.8K MWCO).
The product was lyophilized, resuspended in chloroform and stored at -80⁰C.
Conjugation of cRGD to PEG was confirmed by an observed shift of ~600 in the
maldi-tof spectrum. To create a hydrazide-activated cRGD-PEG, 25.2 mg
(MW≈4000, 1 equivalent, 6.3 μmol) of cRGD-PEG-SH was dissolved 5 mL
chloroform and reacted with 4.25 mg (N-ε-maleimidocaproic acid) hydrazide

144
(EMCH, MW=225.24, 3 equivalents, 18.9 μmol), 4.39 μL of trimethylamine was
added to the reaction (TEA, 5 equivalents, 31.5 μmol). The reaction was carried
out for 4 hours at room temperature under stirring. After, the reaction solution
was purified using a spin column (1.8K MWCO). The product was lyophilized,
resuspended in chloroform and stored at -80⁰C. For the final step, 12 mg cRGD-
PEG(HZ) (1 equivalent, 2.84 μmol) was reacted with 1.2 mg N-4-acetylphenyl
maleimide (APM, 2 equivalent, 5.68 μmol) overnight at room temperature under
constant stirring. The reaction mixture was purified on a silica gel column using
chloroform:methanol mobile phase (9:1 v/v). The final product was concentrated
and lyophilized. The H1 NMR spectrum (Solvent: DMSO) of cRGD-PEG3400(HZ)-
maleimide was used to confirm the structure with the following characteristic
peaks: 8.48 (s, 1H, -NH-), 8.0 (d, 2H, in phenyl), 7.6-7.8 (m, cRGD), 7.45 (d, 2H,
in phenyl), 7.2 (2, 2H, two olefinic protons of maleimide), 3.1-3.5 (m, 304H,
PEG).
5.2.3 Preparation of PEG-modified ECO/siRNA nanoparticles: The ECO lipid
carrier was synthesized as previously reported (1, 2). ECO/siRNA nanoparticles
were prepared at an N/P ratio of 8, where N/P ratio denotes the ratio of cationic
charge from ECO to anionic charge of the siRNA. ECO (MW=1023) was
dissolved in 100% ethanol at a stock concentration of 2.5 mM for in vitro
experiments and 50 mM for in vivo experiments. The siRNA was reconstituted in
RNase-free water to a concentration of 18.8 μM for in vitro experiments and 50
μM for in vivo experiments. For in vitro experiments, an siRNA transfection
concentration of 100 nM was used. ECO/siRNA nanoparticles were prepared at

145
an N/P ratio of 8 by mixing predetermined volumes of ECO and siRNA for a
period of 30 minutes in RNase-free water (pH 5.5) at room temperature under
gentle agitation to enable complexation between ECO and siRNA. The total
volume of water was determined such that the volume ratio of ethanol:water
remained fixed at 1:20. For PEGylated nanoparticles, the PEG-derivative was
first reacted with ECO in RNase-free water at 2.5 mol% for 30 minutes under
gentle agitation and subsequently mixed with siRNA in RNase-free water for an
additional 30 min. Certain experiments called for conjugation at 1, 5, and 10
mol%. All PEG-derivatives were prepared at a stock solution concentration of
0.625 mM for in vitro experiments and 0.32 mM for in vivo experiments in RNase-
free water. Again, the total volume of water was determined such that the volume
ratio of ethanol:water remained fixed at 1:20. PEG-derivatives include: mPEG-
maleimide (Nanocs), mPEG(HZ)-maleimide, RGD-PEG-maleimide, RGD-
PEG(HZ)-maleimide.
5.2.4 Nanoparticle Characterization: The zeta potential of unmodified, mPEG-
and mPEG(HZ)-modified ECO/siRNA nanoparticle formulations at different pHs
in PBS was determined with a Brookhaven ZetaPALS Particle Size and Zeta
Potential Analyzer (Brookhaven Instruments). For each formulation, the
nanoparticles were diluted in PBS solutions at pH 7.4, 6.5, or 5.4. The zeta
potential measurement was taken at each indicated time point up to 4 hours.
Data represents the mean of three independently conducted experiments.
5.2.5 pH-Dependent Membrane Disruption Hemolysis Measurement: The
hemolytic activity was measured to determine the membrane-disruptive ability of

146
unmodified, mPEG- and mPEG(HZ)-modified ECO/siRNA nanoparticle
formulations at pH levels corresponding to various stages of intracellular
trafficking. Red blood cells (RBCs) isolated from rats (Innovative Research Inc.)
were diluted 1:50 in PBS solutions at pH 7.4, 6.5, and 5.4. ECO/siRNA
nanoparticles were prepared at a volume of 150 μL and incubated with an equal
volume of the various RBC solutions in a 96-well plate at 37⁰C for 2 hours.
Following incubation, samples were centrifuged and the absorbance of the
supernatants was determined at 540 nm. Hemolytic activity was calculated
relative to the hemolytic activity of 1% Triton X-100 (Sigma Aldrich), a non-ionic
surfactant. Each pH was conducted in triplicate and the data presented
represents the mean and standard deviation.
5.2.6 Flow Cytometry for Nanoparticle Cellular Uptake Measurements:
Cellular uptake and intracellular delivery of various ECO/siRNA nanoparticle
formulations include mPEG-maleimide, mPEG(HZ)-maleimide, RGD-PEG-
maleimide, RGD-PEG(HZ)-maleimide were evaluated quantitatively with flow
cytometry. The ECO/siRNA nanoparticle formulations were prepared with 25 nM
AlexaFluor647-labeled siRNA (Qiagen). Approximately 2.5 x 104 MDA-MB-231
cells were seeded onto 12-well plates and grown for an additional 24 hours. The
cells were transfected with each ECO/siRNA nanoparticle formulation in 10%
serum media. After 4 hours, the transfection media was removed and each well
was washed twice with PBS. The cells were harvested by treatment with 0.25%
trypsin containing 0.26 mM EDTA, (Invitrogen) collected by centrifugation at
1000 rpm for 5 min, resuspended in 500 μL of PBS containing 5%

147
paraformaldehyde, and finally passed through a 35μm cell strainer (BD
Biosciences). Cellular internalization of ECO/siRNA nanoparticles was quantified
by the fluorescence intensity measurement of AlexaFluor 647 fluorescence for a
total of 10,000 cells per each sample using a BD FACSCalibur flow cytometer.
Each formulation conducted in triplicate and the data presented represents the
mean fluorescence intensity and standard deviation.
For studying the endocytic trafficking pathway, the following inhibitors
were used 1 hour prior to transfection in MDA-MB-231 cells with the various
ECO/siRNA nanoparticles: 4⁰C, Cytochalasin D (5 μm/mL; Sigma Aldrich),
Genistein (200 μM; Sigma Aldrich), and Nocadozole (20 μM; Sigma Aldrich).
After 1 hour, the various ECO/siRNA nanoparticles formulated with the
fluorescent AF647-labeled siRNA, as descried above, were added to the cells.
After an additional 2 hours, the cells were harvested and processed as described
above. Similarly, cellular internalization of ECO/siRNA nanoparticles was
quantified by the fluorescence intensity measurement of AlexaFluor 647
fluorescence for a total of 10,000 cells per each sample using a BD FACSCalibur
flow cytometer. Each formulation conducted in triplicate and the data presented
represents the mean fluorescence intensity and standard deviation.
5.2.7 Confocal Microscopy of Nanoparticle Uptake and Intracellular Release
of siRNA: Live cell confocal microscopy was used to assess the cellular
uptake, endolysosomal escape, and intracellular release of siRNA.
Approximately 1 x 105 MDA-MB-231 cells were seeded onto glass-bottom micro-
well dishes. After 24 hours, the cells were stained for 30 minutes each with 5

148
µg/mL Hoechst 33342 (Invitrogen) and with 50 nM Lysotracker Green DND-26
(Molecular Probes). RGD-PEG- and RGD-PEG(HZ)- modified ECO/siRNA
nanoparticles were formed at an N/P ratio of 8 and a 25 nM siRNA concentration
with an AlexaFluor 647-labelled siRNA. Images were taken using an Olympus
FV1000 confocal microscope while the cells were housed in a humidified weather
station under 5% CO2.
5.2.8 In Vitro Luciferase Silencing Efficiency: MDA-MB-231-Luc cells were
seeded in 24-well plates at a density of 2 x 104 cells and allowed to grow for 24
hours. Transfections were carried out in 10% serum media with an N/P ratio of 8
and 100 nM anti-luciferase siRNA concentration (Dharmacon: sense sequence:
5’-CUUACGCUGAGUACUUCGAdTdT-3’, anti-sense sequence: 5’-
UCGAAGUACUCAGCGUAAGdTdT-3’). Following a 4 hour transfection period,
the media was replaced with fresh serum-containing media and the cells
continued to grow for up to 72 hours. For experiments using chloroquine (Sigma
Aldrich), transfections were conducted in a similar manner either with or without
100 μM chloroquine. As above, following a 4 hour transfection period, the media
was replaced with fresh serum-containing media and the cells continued to grow
for up to 48 hours. At each time point for luciferase silencing experiments, the
cells were rinsed twice with PBS and lysed using the reporter lysis buffered
provided in the Promega Luciferase Assay kit. Following lysis, the cells were
centrifuged at 10,000 g for 5 minutes and 20 μL cell lysate was transferred to a
96-well plate. To quantify luciferase expression, 100 μL Luciferase Assay
Reagent was added to each well and the luminescence was read using a

149
SpectraMax microplate reader (Molecular Devices). Luciferase activity was
normalized to the total protein content measured from the cell lysate of each well
using the BCA assay (Thermo Scientific). Data was presented relative to the
control, which received no siRNA treatment.
5.2.9 In Vivo Luciferase Silencing Efficiency: MDA-MB-231-Luc cells were
engrafted into the mammary fat pad of female nude mice (2 x 106 cells/mouse).
Once the tumors reached an average of 250 mm3, the mice were randomly
sorted into 5 groups (n=3): 1) PBS control, 2) PEG-ECO/siLuc, 3) PEG(HZ)-
ECO/siLuc, 4) RGD-PEG-ECO/siLuc, 5) RGD-PEG(HZ)-ECO/siLuc. All siRNA
nanoparticle variations were formulated at 1.0 mg/kg siRNA in a total injection
volume of 150 μL. All mice received a single intravenous tail vein injection of the
various nanoparticle formulations following bioluminescent imaging on day 0.
Expression of luciferase was quantified using bioluminescence imaging on day 0,
1, 3, 5, and 7. The bioluminescence signal intensity was quantified from a region
of interest (ROI) placed over the tumor area. Data was normalized to the average
signal intensity of day 0.
5.2.10 Fluorescence Molecular Tomography: Fluorescence imaging of siRNA
accumulation within primary MDA-MB-231 mammary fat pad tumors was
performed using the FMT 2500 quantitative fluorescence tomography system
(Perkin-Elmer). Mice were treated with an intravenous tail vein administration of
AlexaFluor 647-conjugated siRNA (1.0 mg/kg) with the various ECO nanoparticle
formulations in a total injection volume of 150 μL. The mice were imaged before

150
and after intravenous injection of the nanoparticles at 30 min, 1 h, 2 h, 4 h, 8 h,
and 24 h.
5.2.11 Ex vivo flow cytometry and confocal microscopy: Mice treated with AF
647-loaded ECO nanoparticles were sacrificed at 48 hours post-injection
whereupon the primary tumor was resected and disaggregated into single cell
suspensions using mechanical force and disaggregation solution as described
previously (61). The cell suspension was stained with FITC-conjugated mouse
MAb against human epithelial antigen (EpCAM) (HEA125; Miltenyi Biotec,
Auburn, CA) in the dark and on ice for 10 minutes. After staining, the cells were
washed and centrifuged, fixed with paraformaldehyde, and finally passed through
a 35μm cell strainer (BD Biosciences). Flow cytometry was conducted using the
fluorescein channel for HEA-FITC and Cy5 channel for AF647-conjugated siRNA
delivered by the ECO nanoparticles for a total of 10,000 cells per each sample
using a BD FACSCalibur flow cytometer. Gating within the fluorescein channel
was used to identify EpCAM (+) and EpCAM (-) populations. Each formulation
was conducted in triplicate and the data presented represents the mean
fluorescence intensity and standard deviation. Following flow cytometry, the cell
suspensions were examined under an Olympus FV100 confocal microscope.
5.2.12 Statistical analyses
Statistical values were defined using unpaired Student’s t-test, with p < 0.05
considered to be statistically significant.
5.3 Results and Discussion

151
5.3.1 Surface modification of ECO/siRNA nanoparticles with pH-cleavable
PEG layer restores intrinsic pH-sensitive activity
The chemical structure of ECO contains various amino groups such that it
carriers a net positive charge at neutral pH. These amine groups electrostatically
complex with the siRNA cargo and contribute towards the pH-sensitive
amphiphilic characteristic of the delivery system. The amino groups, across a
range of pKa’s, become protonated in an acidic environment to promote pH-
dependent membrane disruption to allow for endolysosomal escape. At an N/P
ratio of 8, ECO/siRNA nanoparticles exhibit a zeta potential of 22.3 ± 1.73 mV at
neutral pH. With increasing acidity, unmodified ECO/siRNA nanoparticles exhibit
a pH-sensitive and time-dependent increase in zeta potential correlating to the
protonation of the cationic ethylenediamine head group (Figure 5.1A). This
protonation is hypothesized to enhance the electrostatic interactions between the
ECO carrier and the anionic membrane lipids to promote bilayer destabilization to
enable escape into the cytosol. It was observed that modification of the siRNA
nanoparticle surface with mPEG3400 at a 2.5 mol% through thiol-maleimide
chemistry decreased the overall zeta potential to 12.3 ± 1.39 mV (Figure 5.1B).
While unmodified ECO/siRNA nanoparticles exhibited pH-sensitivity at pH 6.5
and 5.4, this behavior was attenuated in PEG-modified nanoparticles. After 4
hours of incubation in PBS solutions at pH 6.5 and 5.4, PEGylated ECO/siRNA
nanoparticles carried a zeta potential of 17.4 ± 1.1 mV and 18.6 ± 2.9 mV,
respectively, compared to 32.5 ± 2.7 mV and 39.8 ± 3.1 mV for unmodified
ECO/siRNA nanoparticles. This suggests that the surface aqueous phase formed

152
by PEGylation may impede the protonation of the cationic head group. This was
confirmed with the observation that the pH-sensitivity is further diminished
towards neutrality by increasing the PEG surface density to 10 mol% (Figure
5.1D).
The insertion of the acid-labile hydrazone linkage within the PEG moiety
created a pH-cleavable PEG layer to restore the pH-sensitivity of the core
ECO/siRNA nanoparticles (Figure 5.1C). Hydrazone is a well-characterized
linkage known to hydrolyze at pH levels corresponding to the environment of the
endolysosomal compartments (275). The hydrolysis of hydrazone occurs when
the –C=N nitrogen is protonated causing a nucleophilic attack of water and the
ultimate cleavage of the C-N bond. Previously, the hydrazone linkage has found
popularity as a means to conjugate chemotherapeutics, such as doxorubicin, to a
wide array of drug delivery systems and prodrugs to enhance intracellular
release (275,276). This particular hydrazone-modified PEG linkage has also
been used previously to create PEG(HZ)-phosphatidylethanolamine conjugates
capable of forming micelles (272). The hydrazone-based micelles were found to
be stable at physiological pH but highly sensitive to mildly acidic pHs, resulting in
robust degradation of micelles at pH 5.5. Similarly, the observed increase in zeta
potential at pH 6.5 and 5.4 presumably corresponds to the acid-catalyzed
hydrolysis of the hydrazone linkage and subsequent shedding of the PEG layer
whereupon the cationic ECO/siRNA core nanoparticle surface is exposed. Once
exposed, the ethylenediamine head group within the ECO/siRNA nanoparticles
becomes protonated. At pH 7.4, the zeta potential remains constant at ~12 mV

153
indicating that the PEG layer remains intact as a result of the hydrazone linkage
stability. Importantly, the stability of surface charge at pH 7.4 suggests the
ECO/siRNA nanoparticles will remain PEGylated within the bloodstream at the
normal physiological pH. Following exposure to pH 5.4, the hydrazone bond is
degraded to remove the PEG layer. In turn, the surface zeta potential of
PEG(HZ)-ECO/siRNA nanoparticles gradually increases and after 4 hours is
similar to that of the unmodified ECO/siRNA nanoparticles, 36.3 ± 1.9 mV and
39.8 ± 2.8 mV, respectively. However, the time to reach the maximum zeta
potential is prolonged, possibly due to the slower kinetics involved with the
hydrolysis of the hydrazone linkage.
Figure 5.1. Zeta potential of A) ECO/siRNA, B) PEG-ECO/siRNA, and C) PEG(HZ)-
ECO/siRNA nanoparticles incubated in PBS solutions at pH levels corresponding to
stages of intracellular trafficking (pHs 7.4, 6.5, 5.4). D) Increasing the PEGylation
surface density inhibits the pH-sensitive protonation of the ECO/siRNA nanoparticles.

154
We have previously shown that the protonation of the cationic head group
of ECO is directly coupled with the ability to induce pH-sensitive membrane
disruption, a key step for successful and efficient cytosolic delivery of siRNA
(221). To compare the effect of pH-cleavable to non-cleavable PEGylation, we
examined the pH-sensitive hemolytic activity of ECO/siRNA nanoparticles
modified with both PEGylation strategies (Figure 5.2A). Non-cleavable PEG-
ECO/siRNA nanoparticles had a significantly lowered ability to induce hemolysis
at pH 6.5 and 5.4 compared to unmodified ECO/siRNA nanoparticles. Again, this
is indicative of the PEG layer inhibiting the interactions with the lipid membrane
of the blood cells and also the protonation of the cationic head group of ECO. In
alignment, increasing the PEG surface density further inhibited the hemolytic
activity of the non-cleavable PEGylated nanoparticles (Figure 5.2B). Conversely,
the pH-cleavable PEG(HZ)-ECO/siRNA nanoparticles induced pH-sensitive
hemolysis on par with unmodified ECO/siRNA nanoparticles. As the hemolytic
activity was evaluated 2 hours following exposure of the red blood cells to the
nanoparticles, both formulations of nanoparticles would have reached similar
levels of protonation, as observed in Figure 1A and C, indicating the cleavage of
the hydrazone linkage and subsequent shedding of the PEG layer (Figure 5.1A
and C).

155
Figure 5.2. A) Comparison of hemolytic activity of ECO, PEG-ECO, and PEG(HZ)-ECO
siRNA nanoparticles at pH levels corresponding to stages of intracellular trafficking. B)
Increasing the PEGylation surface density inhibits the pH-sensitive hemolytic activity of
ECO/siRNA nanoparticles. Relative hemolytic activity was calculated with respect to the
hemolytic activity of 1% Triton-X-100.
5.3.2 pH-cleavable RGD-PEG modification induces potent in vitro silencing
efficiency
To facilitate ligand-specific uptake of the ECO/siRNA nanoparticles into
the target population of cells, a cyclic-RGD (RGD) peptide was conjugated to
both pH-cleavable and non-cleavable PEG moieties. Cellular uptake into MDA-
MB-231 human breast cancer cells was quantified using flow cytometry with
nanoparticles formulated with fluorescently labelled siRNA (Figure 5.3A). The
inclusion of the hydrazone linkage in both RGD-targeted and non-targeted
ECO/siRNA nanoparticles had no significant difference in the ability of the
nanoparticles to gain internalization into the cells when compared to the non-

156
cleavable counterparts. Unmodified ECO/siRNA nanoparticles exhibited robust
cellular uptake due to strong ionic interactions with the anionic cellular
membrane. PEGylation endows neutrality to the nanoparticle surface and
impedes such interactions with the cell surface. The addition of the RGD
targeting peptide enhanced the cellular uptake and internalization due to specific
binding of the RGD peptide to the αvβ3 integrin known to be present on the
cellular surface of the MDA-MB-231 cells (277).
Surface modification of nanoparticles has been shown to influence the
endocytic mechanism through which they are internalized by cells (142,278).
Further, the endocytic mechanism can dictate the fate of nanoparticles within the
intracellular trafficking pathways. By using a series of known pharmacological
inhibitors of various endocytic pathways (279), the primary mechanism of uptake
was elucidated for the various formulations of ECO/siRNA nanoparticles in MDA-
MB-231 cells: 1) incubation of cells at 4⁰C to inhibit energy-dependent endocytic
mechanisms, 2) cytochalasin D is generally classified as an inhibitor of
macropinicytosis/phagocytosis but recently has been implicated with inhibition of
clathrin- and caveolae-mediated pathways (280), 3) Genistein inhibits caveolae-
mediated endocytosis and 4) Nocodazole inhibits clathrin-mediated pathways.
PEGylated ECO/siRNA nanoparticles were found to be internalized primarily via
clathrin-mediate endocytosis while RGD-targeted nanoparticles entered primarily
via caveolae-mediated endocytosis (Figure 5.3B). Interestingly, unmodified
ECO/siRNA nanoparticles were found to rely on both energy-dependent and
independent mechanisms suggesting that the ECO lipid may be able to directly

157
fuse with the phospholipid membrane of cells. Clathrin- and caveolae-dependent
endocytosis of nanoparticles are often transferred to the lysosomes for
degradation, therefore, the ability of both RGD-targeted and non-targeted
nanoparticles to escape from the endolysosomal pathway is an important step for
achieving gene silencing. No significant difference was observed in endocytic
pathways between pH-cleavable and non-cleavable surface modifications (data
not shown).
The in vitro luciferase silencing efficacy of the surface modified
ECO/siRNA nanoparticles was evaluated in the MDA-MB-231-Luc cell line
(Figure 5.3C). We have shown that unmodified ECO/siRNA nanoparticles induce
potent and sustained gene silencing upwards of 95% due to their ability to readily
become internalized, escape from the endolysosomal pathway, and release the
cargo siRNA into the cytosol (172,221). Accordingly, ECO/siRNA nanoparticles
achieved 92.33 ± 2.08% luciferase silencing 72 hours post-treatment in MDA-
MB-231-Luc cells. The luciferase silencing efficiency was significantly inhibited
upon non-cleavable PEGylation of the nanoparticles (PEG-ECO/siRNA) to only
23.45 ± 1.52% after 72 hours. While decreased cellular uptake contributes to the
attenuated silencing efficiency, our hemolysis assay data suggests PEGylation
also interferes with the ability of the nanoparticles to escape from the
endolysosomal pathway (Figure 5.2). Indeed, the silencing efficiency significantly
increased to 47.86 ± 10.59% when the pH-cleavable PEG was used when
compared to PEG-ECO/siRNA. This observation was highlighted with the
addition of the RGD targeting peptide. Non-cleavable RGD-PEG-ECO/siRNA

158
nanoparticles only achieved 54.17 ± 10.01% luciferase silencing while the pH-
cleavable RGD-PEG(HZ)-ECO/siRNA formulation reached 83.64 ± 4.58%
luciferase silencing 72 hours post-treatment.
5.3.3 Inclusion of hydrazone linkage enhances endosomal escape
The ability of the hydrazone linkage to enable successful endolysosomal
escape was confirmed through live cell confocal imaging (Figure 5.4). MDA-MB-
231 cells were transfected with pH-cleavable and non-cleavable RGD-targeted
ECO nanoparticles formulated with a fluorescently labelled siRNA (red) while co-
stained with Lysotracker (green) to visualize the acidic compartments of the
endosomes and lysosome. Images taken after 10 minutes reveal that both RGD-
targeted nanoparticle formulations have similar interactions with the cellular
membranes, in accordance with similar levels of cellular uptake quantified in
Figure 5.3A. At 3 hours, both formulations exhibit strong co-localization (yellow)
with the late endosomes and lysosomes, consistent with the intracellular
trafficking of caveolae-mediated endocytosis. However, 6 hours post-treatment,
the non-cleavable RGD-targeted nanoparticles appear contained within the
endolysosomes as evident by the co-localization of the siRNA and Lysotracker
fluorescent signal. In contrast, the pH-cleavable RGD-targeted nanoparticles
appear to have successfully escaped from the endolysosomal pathway. Minimal
co-localization of the siRNA and Lysotracker signal is observed and the
dispersed siRNA signal within the cytosol indicates the siRNA has been released
from the nanoparticles, as seen with unmodified ECO/siRNA nanoparticles
(172,221). The inability of the non-cleavable RGD-targeted nanoparticles to

159
escape into the cytosol further validates the observed discrepancy in gene
silencing efficiency when compared with the pH-cleavable counterpart (Figure
5.3C).
Figure 5.3. A) Cellular uptake of unmodified, PEG-, PEG(HZ)-, RGD-PEG-, and RGD-
PEG(HZ)-modified ECO/siRNA nanoparticles quantified with flow cytometry using an
AF647-labeled siRNA. B) Cellular uptake of nanoparticle formulations using inhibitors for
the different mechanisms of endocytosis: 4⁰C: energy-dependent, Cytochalasin D:
macropinicytosis, phagocytosis, clathrin- and caveolae-dependent, Genistein: caveolae-
mediated, Nocodazole: clathrin-mediated. C) Luciferase silencing of unmodified, PEG-,
PEG(HZ)-, RGD-PEG-, and RGD-PEG(HZ)-modified ECO/siRNA nanoparticles in MDA-
MB-231-luc triple-negative breast cancer cells.

160
Figure 5.4. Confocal microscopy images of MDA-MB-231 cells incubated with RGD-
PEG-, and RGD-PEG(HZ)-modified ECO/siRNA nanoparticles at 10 min, 3 hr, and 6 hr.
DAPI, cell nucleus (blue); Lysotracker DND-26, lysosomes (green); siRNA, AF-647 (red).

161
The endosomolytic agent chloroquine that causes endosome rupture and
content release into the cytosol was used to further verify the role the hydrazone
linkage plays in promoting escape (281). For both targeted and non-targeted
nanoparticles, the silencing efficiency of pH-cleavable nanoparticles was not
affected by chloroquine whereas non-cleavable nanoparticles exhibited a
significantly enhanced silencing efficiency (Figure 5.5). The data suggests that
upon treatment with chloroquine, complexes modified with the non-cleavable
PEG were trapped within endolysosomes were then freed into the cytosol while
nanoparticles modified with the pH-cleavable PEG moiety had already escaped
from the endolysosomes.
Figure 5.5. Luciferase silencing efficiency after 48 hours in MDA-MB-231-luc cells
transfected with or without the endosomolytic agent chloroquine.

162
5.3.4 Targeted pH-sensitive nanoparticles exhibit potent and sustained in
vivo gene silencing
To study differences in the in vivo gene silencing efficiency of the
developed pH-cleavable surface modification strategy, the delivery of anti-
luciferase siRNA (1.0 mg/kg) following a single systemic administration of the
was investigated using bioluminescent imaging in a primary mammary fat pad
MDA-MB-231 breast cancer tumor model (Figure 5.6A and B). Unlike the in vitro
luciferase silencing findings, the non-targeted ECO/siRNA nanoparticles, both
pH-cleavable and non-cleavable formulations, had negligible silencing effect on
luciferase expression, suggesting minimal cellular uptake of the siRNA into the
tumor cells. In contrast, both RGD-targeted formulations induced luciferase
silencing to varying degrees for up to 7 days: the non-cleavable RGD-targeted
ECO achieved 52.04% luciferase silencing while the pH-cleavable RGD-targeted
formulation achieved 85.17% silencing compared to the no treatment control on
day 7. This trend is consistent with the enhancement of silencing efficiency
observed in vitro (Figure 5.3C).

163
Figure 5.6. In vivo luciferase silencing efficiency following a single i.v. treatment with
various surface-modified ECO/siRNA nanoparticles (1.0 mg/kg siRNA dose). A)
Quantification of bioluminescence signal from ROIs drawn over the tumor. B)
Representative BLI images of the different treatment groups.
5.3.5 Targeting ligand enhances tumor retention of nanoparticles by
promoting internalization within tumor cells
The tumor localization and subsequent delivery of functional siRNA
following systemic administration of the various siRNA nanoparticle formulations
was investigated. Fluorescence molecular tomography (FMT) enabled the
analysis of tumor accumulation and retention of the nanoparticle-delivered siRNA
over time. Mice received a single systemically administered dose of
fluorescently-labeled siRNA (AF 647) delivered by the various surface modified
ECO/siRNA nanoparticle formulations. Recent studies have demonstrated that
the presence of a targeting ligand does not impact the initial tumor accumulation
of the nanomedicine delivery system, but rather facilitates tumor cell targeting

164
resulting in longer tumor retention (34, 43). In alignment, longitudinal FMT
imaging studies revealed that the initial tumor localization of siRNA delivered by
the targeted and non-targeted nanoparticles was not significantly different for the
first 4 hours following systemic administration (Figure 5.7A and B). By 8 hours
however, RGD-targeted nanoparticles were observed to accumulate within the
tumor to a greater extent than the non-targeted formulations, regardless of
whether the PEG modification was pH-cleavable or not. At 24 hours, both RGD-
targeted nanoparticle formulations were retained within the tumor while the non-
targeted formulations appeared to be washed out, due in part to the inability of
the non-targeted nanoparticles to promote cellular internalization, as evident by
the minimal presence of siRNA signal from the tumor ROI.
Figure 5.7. Tumor accumulation and retention of surface-modified ECO/siRNA
nanoparticles following i.v. administration. A) Representative FMT images of a single
mouse from each treatment group over 24 hours post-treatment with nanoparticles
formulated with an AF647-tagged siRNA. An ROI was drawn over the area containing
the tumor. B) Quantification of fluorescence signal from each ROI.

165
Active targeting of the ECO/siRNA nanoparticles using the RGD peptide
may aid in the specific selection of cancer cells within the tumor site and promote
internalization of the nanoparticles to a greater extent inside the target cells
compared to non-target cells. To evaluate if selective uptake dependent upon cell
type occurred, ex vivo flow cytometry was used to quantify siRNA internalization
to study differences in cellular uptake of the targeted and non-targeted
nanoparticles between human tumor and murine stromal cells. At 48 hours,
tumors from the systemically treated mice were excised, disaggregated into
single cell suspensions, and stained for the epithelial cellular adhesion molecular
(EpCAM) using HEA-FITC to distinguish between the human MDA-MB-231
cancer cells and the murine stromal cells (61). FACS analysis of the cell
suspensions in the FITC channel revealed two distinct cellular populations:
EpCAM (+), human cancer cells and EpCAM (-), murine stromal cells (Figure
5.8A). Gating for EpCAM (-) cells in the AlexaFluor 647 channel revealed a
minimal shift in fluorescence between both targeted and non-targeted
nanoparticle formulations compared to the PBS negative control (Figure 5.8B). A
distinct shift was observed in EpCAM (+) cells for targeted nanoparticles
compared to both the non-targeted and PBS control groups, suggesting that the
RGD-targeted nanoparticles are internalized more efficiently and preferentially by
the human cancer cells (Figure 5.8C). Contour plots highlight the two distinct
EpCAM (+) and EpCAM (-) cellular populations (Figure 5.8D). While the siRNA
signal from non-targeted nanoparticle formulations was evenly distributed
throughout both populations, the siRNA signal in EpCAM (+) cells was markedly
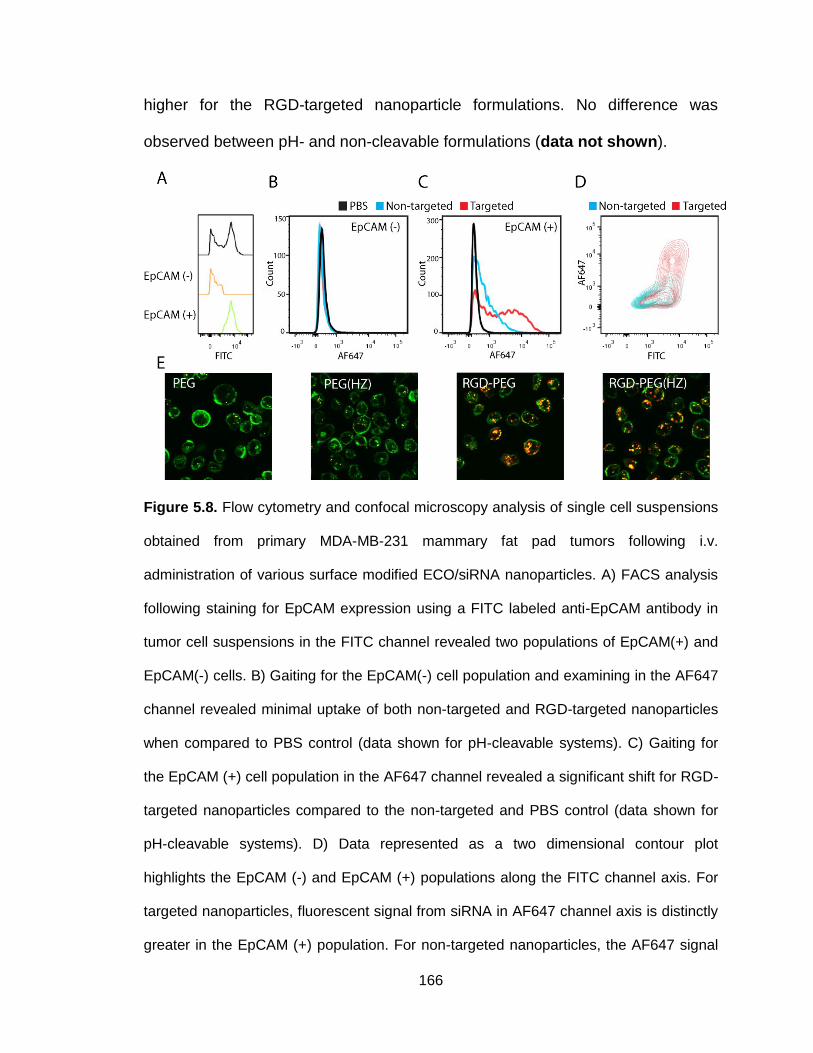
166
higher for the RGD-targeted nanoparticle formulations. No difference was
observed between pH- and non-cleavable formulations (data not shown).
Figure 5.8. Flow cytometry and confocal microscopy analysis of single cell suspensions
obtained from primary MDA-MB-231 mammary fat pad tumors following i.v.
administration of various surface modified ECO/siRNA nanoparticles. A) FACS analysis
following staining for EpCAM expression using a FITC labeled anti-EpCAM antibody in
tumor cell suspensions in the FITC channel revealed two populations of EpCAM(+) and
EpCAM(-) cells. B) Gaiting for the EpCAM(-) cell population and examining in the AF647
channel revealed minimal uptake of both non-targeted and RGD-targeted nanoparticles
when compared to PBS control (data shown for pH-cleavable systems). C) Gaiting for
the EpCAM (+) cell population in the AF647 channel revealed a significant shift for RGD-
targeted nanoparticles compared to the non-targeted and PBS control (data shown for
pH-cleavable systems). D) Data represented as a two dimensional contour plot
highlights the EpCAM (-) and EpCAM (+) populations along the FITC channel axis. For
targeted nanoparticles, fluorescent signal from siRNA in AF647 channel axis is distinctly
greater in the EpCAM (+) population. For non-targeted nanoparticles, the AF647 signal

167
is evenly distributed between EpCAM (-) and EpCAM (+) populations. E) Single cell
suspensions prepared from primary tumor disaggregates were examined under a
confocal microscope. Targeted nanoparticle formulations display a greater amount of
siRNA signal (red) in the EpCAM (+) (green) cells.
Following flow cytometry, the cell suspensions were observed under a
confocal microscope (Figure 5.8E). Microscopy analysis revealed both pH-
cleavable and non-cleavable RGD-targeted nanoparticle formulations were
readily internalized by EpCAM (+) cells whereas the non-targeted formulations
showed minimal uptake. While the targeted ECO/siRNA nanoparticles did not
initially transport the siRNA cargo to the tumor site more efficiently than non-
targeted nanoparticles according to the FMT data (Figure 5.7), it appears that
RGD peptide enabled cell-specific recognition and internalization of the siRNA.
The enhanced cellular uptake correlated to the prolonged retention of fluorescent
signal within the tumor ROI as determined by FMT (Figure 5.7) and also the
sustained luciferase silencing efficiency of the targeted nanoparticles (Figure
5.6). As internalization of siRNA from pH-cleavable and non-cleavable RGD-
targeted nanoparticle formulations appeared similar according to FMT and ex
vivo confocal microscopy, the improvement in silencing efficiency of the pH-
cleavable formulation may be a direct result of enhanced endolysosomal escape.
A surface modification strategy that combines PEGylation and active
targeting not only allows the ECO/siRNA nanoparticles to retain stealth
properties during circulation and accumulate at the tumor site by passive
targeting, but the PEG shielding moiety provides a tether for versatile

168
incorporation of various active targeting ligands. When further combined with the
pH-cleavable hydrazone linkage, the developed surface modification strategy
may significantly improve the therapeutic efficacy of the ECO/siRNA nanoparticle
delivery system compared to the non-targeted and non-cleavable systems.
Taken together with the in vivo luciferase silencing data, the differences in
silencing efficiency across all formulations suggest: i) targeted nanoparticles are
internalized to a greater extent by the tumor cells and ii) once internalized,
formulations with the pH-cleavable PEG modification are more effective in
delivering siRNA into the cytosol due to enhanced endolysosomal escape.
5.4 Conclusion
In summary, we have leveraged and incorporated the pH-cleavable
hydrazone linkage into a PEG spacer to enhance the transfection efficiency of
our PEGylated ECO/siRNA nanoparticle system both in vitro and in vivo. While
the non-cleavable PEG strategy displayed minimal gene knockdown, the pH-
cleavable PEG significantly enhanced RNAi activity, mediated by a restoration in
the ability to escape from the endolysosomes. When functionalized with an
RGD targeting peptide, the targeted and pH-cleavable PEG modification
achieved a silencing efficiency similar to that of unmodified ECO/siRNA
nanoparticles. The initial tumor accumulation of the non-targeted and targeted
nanoparticles was approximately equal within the first 4 hours of delivery;
however, RGD-targeted nanoparticles remained localized within the tumor area
to a greater extent over 24 hours. The greater uptake within tumor cells coupled

169
with the added pH-sensitivity allowed the pH-cleavable RGD-targeted
nanoparticles to achieve greater in vivo luciferase silencing activity compared to
the non-cleavable targeted formulation.
5.5 Acknowledgments
This material is based upon work partially supported by the National Science
Foundation Graduate Research Fellowship under Grant Number DGE-0951783
and the National Institutes of Health under Grant Number EB00489.

170
Chapter 6
Targeting eIF4E with a Dual pH-responsive siRNA Delivery System to
Overcome Drug Resistance in Triple-Negative Breast Cancer

171
6.1 Introduction
Breast cancer remains the leading diagnosis of cancer cases within
women in the United States (1, 2). Recently, the rate of mortality associated with
breast cancer has experienced a sharp decline, due in part to improved
diagnostic methods, the advent of targeted therapies, and refinements in the
dosing and formulation of systemic chemotherapies (284–286). As described in
Chapter 4, triple-negative breast cancer (TNBC) is a highly aggressive subtype
of breast cancer that accounts for approximately 10-15% of all diagnosed breast
cancer cases (287). TNBCs have limited treatment options due to their lack of
hormone targets, which typically include estrogen receptor (ER), progesterone
receptor (PR), and HER2-negative status (7, 8). Due to the lack of established
therapeutic targets, patients that present with TNBC are not candidates for
target-specific therapies directed at ER, PR, or HER2 receptors leaving systemic
chemotherapy as the sole therapeutic option. While initially responsive to
anthracycline- and taxane-based chemotherapy regimens, such as doxorubicin
and paclitaxel, TNBCs have a high incidence of drug resistance leading to
relapse and metastasis, leaving the 5 year survival rate at only 20% (29,208).
Therefore, there is an urgent need to uncover and validate therapeutic targets
that can circumvent drug resistance to improve the outcome of TNBC.
The eukaryotic translation initiation factor 4E (eIF4E) is the 5’ cap mRNA-
binding protein involved in the complex and multistep process of mRNA
translation, where it plays a rate limiting role for all cap-dependent translation
(288). It has been demonstrated that eIF4E exerts specific regulation over mRNA

172
targets with long 5’ untranslated regions rich in G/C content. Included within
these mRNAs are gene involved with cellular growth, proliferation, differentiation
and apoptosis (289). Within the context of cancers, eIF4E favors the selective
translation of a wide array of mRNAs implicated with tumorigenesis including: c-
Myc, cyclin D1, survivin, Bcl-2, Bcl-xL, VEGF, and MMP-9 (290,291). Such
control over the upregulation of downstream gene products suggests that eIF4E
overexpression can result in multiple modes of resistance to chemotherapeutic
agents.
Specifically within TNBCs, eIF4E expression correlates with overall and
disease-free survival; patients with elevated eIF4E expression exhibit a worse
prognosis (292–297). In fact, patients with stage I-III breast cancer and eIF4E
overexpression greater than 7-fold exhibit a greater rate of recurrence and
cancer-related death compared to patients with less than 7-fold overexpression
(298). Additionally, the expression of eIF4E may also play a predictive role in
surmising the response of TNBCs towards chemotherapy. A retrospective study
determined that patients whose tumors presented with low eIF4E expression
following neoadjuvant therapy had lower cancer recurrence compared with those
whose tumors exhibited high eIF4E overexpression (292). Therefore, knowledge
of eIF4E expression could help to guide future treatment regimens.
Previous studies have established that silencing of eIF4E in vitro induces
cell cycle arrest and enhances the chemosensitivity of MDA-MB-231 TNBC cells
to multiple chemotherapies, including paclitaxel, through an increase in the
Bax/Bcl-2 ratio involved in chemotherapeutic drug-induced apoptosis (299–301).

173
As such, the role of eIF4E as a potential cancer therapeutic has compelled the
exploration of therapies aimed to regulate eIF4E activity. Currently, there are a
handful of therapeutic strategies aimed to either directly or indirectly block eIF4E
expression or activity in various phases of pre-clinical and clinical development: i)
gene therapy using antisense oligonucleotides or siRNAs (302,303), ii) suicide
gene therapy (304), iii) hormone analog-4EBP fusion peptide (305), iv) inhibitory
small molecules (ie, ribavirin, 4EGI-1) (306,307).
Of particular interest, antisense or RNAi-based gene therapies have been
used to directly modulate the expression of eIF4E. The antisense oligonucleotide
LY2275796 has demonstrated that eIF4E silencing can reduce in vivo tumor
growth in PC-3 prostate and MDA-MB-231 breast cancer models (308). Despite
promising initial pre-clinical success, a Phase I clinical trial found that eIF4E
inhibition was not sufficient to achieve tumor inhibition (309). As human cancers
inevitably contain magnitudes more complexity compared to animal models, the
clinical data suggests that human cancers cells may bypass eIF4E-dependent
pathways due to the presence of redundant mechanisms, thus highlighting the
necessity for combination therapy.
While various attempts have been made to create eIF4E therapies,
siRNA-mediated RNAi strategies have yet to be employed in vivo. Further, to our
knowledge, the effect of eIF4E silencing in a drug-resistant TNBC cell line has
yet to be established or be translated into a viable systemically-administered
therapeutic regimen. As discussed in Chapter 5, a targeted, pH-cleavable
PEGylated siRNA delivery system may exhibit superior in vivo therapeutic

174
efficacy due to enhanced cytosolic delivery of siRNA into target cancer cells. The
present study focuses on leveraging the recently developed dual pH-sensitive
and peptide-targeted siRNA delivery system from Chapter 5 to silence eIF4E
and resensitize drug-resistant TNBC to paclitaxel therapy.
6.2 Materials and Methods
6.2.1 Cell Culture: Human triple-negative breast cancer MDA-MB-231 cells
expressing a luciferase reporter enzyme (MDA-MB-231-Luc) were obtained from
ATCC (American Type Culture Collection) and cultured in Dulbecco’s modified
Eagle’s media (Invitrogen) and supplemented with 10% fetal bovine serum
(Invitrogen), 100 μg/mL streptomycin, and 100 unites/mL penicillin (Invitrogen).
The cells were maintained in a humidified incubator at 37⁰C and 5% CO2. A
paclitaxel-resistant subline of the MDA-MB-231 cell line (MDA-MB-231.DR) was
induced by chronic exposure of MDA-MB-231 cells to 5 nM paclitaxel with
increasing concentration at each passage over 8 weeks to reach a final
concentration of 20 nM. Initially, cells were maintained at 5 nM paclitaxel (PTX)
with increasing concentration in increments of 5 nM after every other passage
over 8 weeks to reach a final concentration of 20 nM. Once resistance to
paclitaxel was confirmed, the MDA-MB-231.DR cells were maintained at 5 nM
PTX.
6.2.2 Preparation of PEG-modified ECO/siRNA nanoparticles: The ECO lipid
carrier was synthesized as described previously (221). ECO (MW=1023) was
dissolved in 100% ethanol at a stock concentration of 2.5 mM for in vitro
experiments and 50 mM for in vivo experiments. The siRNA was reconstituted in

175
RNase-free water to a concentration of 18.8 μM for in vitro experiments and 25
μM for in vivo experiments. For in vitro experiments, an siRNA transfection
concentration of 100 nM was used. ECO/siRNA nanoparticles were prepared at
an N/P ratio of 8 by mixing predetermined volumes of ECO and siRNA for a
period of 30 minutes in RNase-free water (pH 5.5) at room temperature under
gentle agitation to enable complexation between ECO and siRNA. The total
volume of water was determined such that the volume ratio of ethanol:water
remained fixed at 1:20. For RGD-PEG(HZ)-modified ECO/siRNA nanoparticles,
RGD-PEG(HZ)-Mal was first reacted with ECO in RNase-free water at 2.5 mol%
for 30 minutes under gentle agitation and subsequently mixed with siRNA in
RNase-free water for an additional 30 min. RGD-PEG(HZ)-mal was prepared at a
stock solution concentration of 0.32 mM in RNase-free water. Again, the total
volume of water was determined such that the volume ratio of ethanol:water
remained fixed at 1:20.
6.2.3 Semi-quantitative real-time PCR analyses: Real-time PCR studies were
performed as described previously. Briefly, MDA-MB-231 or MDA-MB-231.DR
cells (100,000 cells/well) were seeded overnight onto 6-well plates. The cells
were then treated with ECO nanoparticles with a non-specific siRNA or eiF4E-
specific siRNA (eIF4E: AAGCAAACCUGCGGCUGAUCU (GE Dharmacon) [33]).
At each indicated time point, total RNA was isolated using the RNeasy Plus Kit
(Qiagen, Valencia, CA) and reverse transcribed using the iScript cDNA Synthesis
System (Bio-Rad, Hercules, CA). Semi-quantitative real-time PCR was
conducted using iQ-SYBR Green (Bio-Rad) according to manufacturer’s

176
recommendations. In all cases, differences in RNA expression for each individual
gene were normalized to their corresponding GAPDH RNA signals.
Primer sequences:
eIF4E:
Sense 5’- CTACTAAGAGCGGCTCCACCAC-3’
Antisense 5’- TCGATTGCTTGACGCAGTCTCC-3’
VEGF:
Sense 5’-CCATGAACTTTCTGCTGTCTT-3’
Antisense 5’-ATCGCATCAGGGGCACACAG-3’
Survivin
Sense 5’-ATGGCCGAGGCTGGCTTCATC-3’;
Antisense 5’-ACGGCGCACTTTCTTCGCAGTT-3’
Cyclin-D
Sense, 5’-AGACCTGCGCGCCCTCGGTG-3’
Antisense, 5’-GTAGTAGGACAGGAAGTTGTTC-3’
GAPDH
Sense 5’-ACGGATTTGGTCGTATTGGGCG-3’;
Antisense 5’-CTCCTGGAAGATGGTGATGG-3’.
6.2.4 Western blot analyses: Immunoblotting analyses were performed as
previously described. Briefly, MDA-MB-231 and MDA-MB-231.DR cells were
seeded into 6-well plates (1.5 × 105 cells/well) and allowed to adhere overnight.
The cells were then treated with RGD-PEG(HZ)-ECO/siRNA complexes (N/P=8,
siRNA concentration of 100 nM) in complete growth medium. After 5 days,

177
detergent-solubilized whole cell extracts (WCE) were prepared by lysing the cells
in Buffer H (50 mM -glycerophosphate, 1.5 mM EGTA, 1 mM DTT, 0.2 mM
sodium orthovanadate, 1 mM benzamidine, 10 mg/mL leupeptin, and 10 mg/mL
aprotinin, pH 7.3). The clarified WCE (20 mg/lane) were separated through 10%
SDS-PAGE, transferred electrophoretically to nitrocellulose membranes, and
immunoblotted with the primary antibodies, anti-eIF4E (1:1000; Abcam) and anti-
-actin (1:1000; Santa Cruz Biotechnology).
6.2.5 Cytotoxicity Assay: Cytotoxicity assays were performed in a 96-well plate
as described previously, by seeding 2,000 MDA-MB-231 or MDA-MB-231.DR
cells/well. First, RGD-PEG(HZ)-modified ECO/siRNA nanoparticles were used to
transfect MDA-MB-231 or MDA-MB-231.DR with either siNS or sieIF4E for 48
hours. Next, the wells were washed twice with PBS and incubatiion with various
concentrations of PTX in fresh media. After 2 additional days, the MTT reagent
(Invitrogen) was added to the cells for 4 hours followed by the addition of SDS-
HCl and further incubation for 4 hours. The absorbance of each well was
measured at 570 nm using a SpectraMax spectrophotometer (Molecular
Devices). Cellular viability was calculated as the average of the set of triplicates
for each PTX concentration and was normalized relative to the no treatment
control. Drug resistance was confirmed with an MTT assay to determine the IC50
of paclitaxel. The IC50 was defined as the dose of drug required to inhibit cell
viability by 50%.
6.2.6 In vivo tumor growth inhibition study: For in vivo anti-tumor efficacy
studies, MDA-MB-231.DR cells (2 x 106 cells/mouse) were inoculated in the

178
mammary fat pads of female nu/nu mice. When the tumors reached and average
of 150 mm3, the mice were randomly sorted into 4 groups (n=5): 1) PBS control,
2) RGD-PEG(HZ)-ECO/siNS (1.5 mg/kg siRNA) + PTX (5 mg/kg), 3) RGD-
PEG(HZ)-ECO/sieIF4E (1.5 mg/kg siRNA) + PTX (5 mg/kg), 4) RGD-PEG(HZ)-
ECO/sieIF4E (1.5 mg/kg siRNA). RGD-PEG(HZ)-ECO/siRNA nanoparticles were
administered by intravenous injection into the lateral tail vein while PTX was
administered in 10% DMSO/PBS with an intraperitoneal injection. Tumor growth
was monitored by BLI and tumor size was monitored with caliper measurements.
Three days after the final treatment, the mice were sacrificed to harvest tumor
tissues.
6.2.7 Bioluminescent imaging: Longitudinal imaging of the mice was performed
using the Xenogen IVIS 100 imaging system. D-luciferin (Xenogen) was
dissolved in PBS (15 mg/mL), and 200 μL of the luciferin stock solution (15
mg/mL) was injected i.p. 5 minutes before measuring the light emission. Mice
were anesthetized and maintained under 2.5% isoflurane. Bioluminescent signals
were quantified using Living Image software (Xenogen) by drawing an ROI over
the tumor area.
.
6.2.8 Toxicity, immune response, and pathology studies: Female BALB/c
mice (Jackson Laboratories) were used to study the toxicity and immune
response of systemic treatment with ECO/siRNA and RGD-PEG(HZ)-
ECO/siRNA nanoparticles. Following 1, 3 and 5 injections (n=5 for each amount
of injections) spaced 5 days apart, blood was collected at 2 h and 24 h post-

179
injection. Plasma was isolated from blood samples using Microtainer tubes
(Becton Dickinson). To measure plasma cytokine levels, TNFα, IL-6, IL1-2, INFγ
were quantified by ELISA according to the manufacturer’s instructions
(Invitrogen).
6.3 Results and Discussion
6.3.1. Silencing eIF4E by RGD-targeted pH-cleavable PEG-modified
ECO/siRNA nanoparticles enhances sensitivity to paclitaxel in a drug-
resistance triple-negative breast cancer cell line
The ability of the pH-cleavable RGD-PEG(HZ)-ECO/siRNA nanoparticles
to mediate knockdown of eIF4E mRNA and protein was first evaluated in vitro.
To study the therapeutic consequence of eIF4E downregulation on drug-resistant
breast cancer cells, a paclitaxel-resistant subline of the MDA-MB-231 cell line
(MDA-MB-231.DR) was induced by chronic exposure of MDA-MB-231 cells to
paclitaxel. Initially, cells were maintained at 5 nM paclitaxel (PTX) with increasing
concentration in increments of 5 nM after every other passage over 8 weeks to
reach a final concentration of 20 nM. Once resistance to PTX was confirmed, the
MDA-MB-231.DR cells were maintained at 5 nM PTX. PTX-resistant cells were
found to upregulate eIF4E expression (Figure 6.1A and B). While a widely used
chemotherapeutic agent, PTX has been shown to activate signaling pathways
that both promote and inhibit cell death (310). The mechanism of action for PTX
involves the stabilization of microtubules to interfere with cell division and induce
a mitotic blockade. The inhibition of microtubule function upregulates the
proapoptotic family protein Bax and downregulates the antiapoptotic family

180
protein Bcl-2 (310). At the same time, PTX can also increase expression of
various survival factors and activate the phosphatidylinositol 3-kinase (PI3))/akt
pathway, an important regulation mechanism of cap-dependent translation.
Normally, binding of 4E-BP1 to eIF4E will prevent eIF4E from binding to eIF4G
and initiating cap-dependent translation. PTX has been found to diminish the
suppressive role of 4E-BP1 through hyperphosphorylation to reduce the
association affinity with eIF4E and promote its release (310). By doing so, eIF4E
activity is elevated through a regained association with eIF4G and initiation of
translation. Along these lines, treatment of MDA-MB-231 cells with PTX has been
demonstrated elsewhere to also increase eIF4E expression in a dose-dependent
manner (310). Importantly, treatment of both cell lines with sieIF4E delivered by
targeted and pH-cleavable nanoparticles was able to sustain potent eIF4E mRNA
and protein silencing for upwards of 5 days. Nanoparticles delivering a non-
specific siRNA induced no significant downregulation of eIF4E expression
suggesting that all silencing activity was due to the siRNA and not the ECO
carrier.

181
Figure 6.1. Evaluation of eIF4E mRNA and protein expression as determined by A)
qRT-PCR and B) western blot analysis in MDA-MB-231 and MDA-MB-231.DR cells 5
days following treatment with RGD-PEG(HZ)-ECO/siRNA nanoparticles (N/P=8)
delivering either sieIF4E or siNS (100 nM).
The therapeutic response of eIF4E silencing on enhancing the sensitivity
of breast cancer cells to paclitaxel was determined by quantifying the cell viability
after a combination of eIF4E silencing by the siRNA nanoparticles followed by
treatment with PTX (Figure 6.2). Cells were first treated with RGD-PEG(HZ)-
ECO/siRNA nanoparticles for 48 hours followed by treatment with varying
concentrations of PTX for an additional 48 hours. For all conditions, cell viability
decreased in a PTX concentration-dependent manner. The acquisition of
resistance to PTX in the MDA-MB-231.DR subline was evident by a shift in the
dose-response curve and the IC50. A clear re-sensitization of MDA-MB-231.DR
cells to PTX was observed when eIF4E was down-regulated using siRNA
delivered by RGD-PEG(HZ)-ECO/siRNA nanoparticles. For example, at 0.5
ng/mL PTX, treatment with PTX alone induced 44.6 ± 6.6 % viability in MDA-MB-
231 cells and 80.7 ± 2.9 % viability in the drug-resistant subline. When coupled
with silencing of eIF4E, viability dropped to 7.5 ± 5.4 % and 26.3 ± 3.6 % in MDA-
MB-231 and MDA-MB-231.DR, respectively, using the same concentration of
PTX.
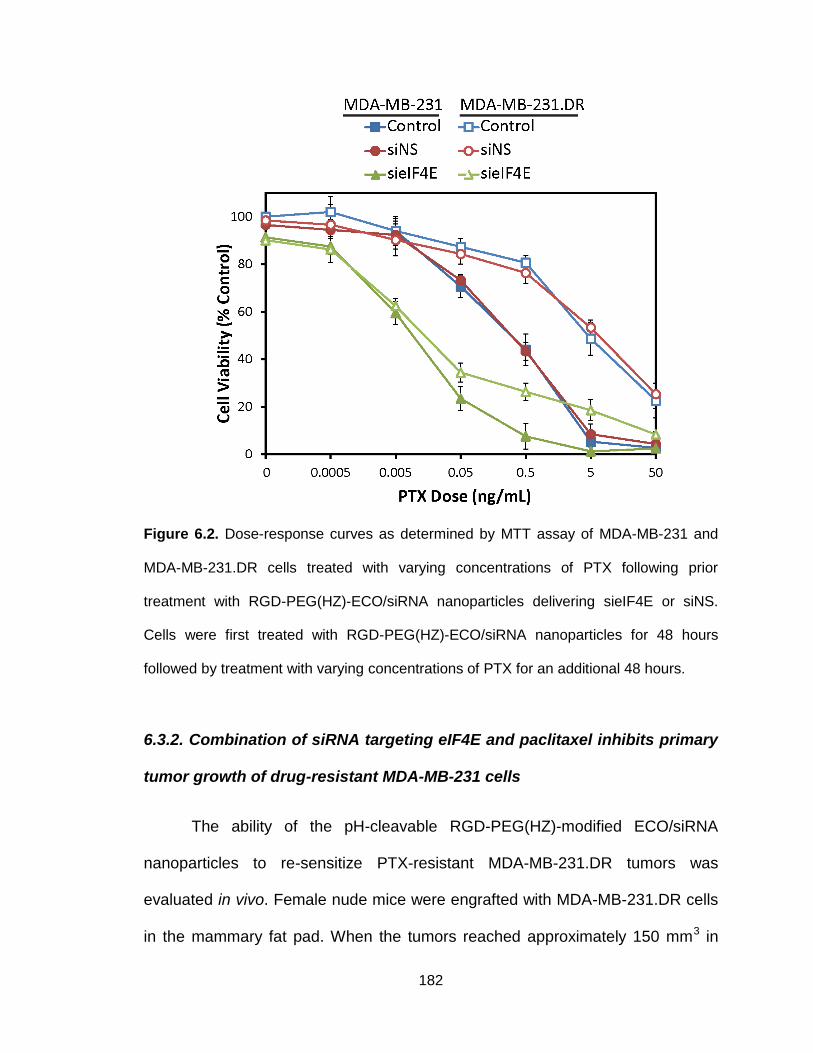
182
Figure 6.2. Dose-response curves as determined by MTT assay of MDA-MB-231 and
MDA-MB-231.DR cells treated with varying concentrations of PTX following prior
treatment with RGD-PEG(HZ)-ECO/siRNA nanoparticles delivering sieIF4E or siNS.
Cells were first treated with RGD-PEG(HZ)-ECO/siRNA nanoparticles for 48 hours
followed by treatment with varying concentrations of PTX for an additional 48 hours.
6.3.2. Combination of siRNA targeting eIF4E and paclitaxel inhibits primary
tumor growth of drug-resistant MDA-MB-231 cells
The ability of the pH-cleavable RGD-PEG(HZ)-modified ECO/siRNA
nanoparticles to re-sensitize PTX-resistant MDA-MB-231.DR tumors was
evaluated in vivo. Female nude mice were engrafted with MDA-MB-231.DR cells
in the mammary fat pad. When the tumors reached approximately 150 mm3 in

183
volume, the mice began receiving alternating treatments with siRNA
nanoparticles (1.5 mg/kg siRNA dose) and PTX (5 mg/kg) every 6 days.
Treatment with nanoparticles delivering non-specific siRNA (siNS) in combination
with PTX had no inhibitory effect on tumor growth. In fact, after several weeks of
treatment, the tumors of mice treated with siNS + PTX grew larger and more
rapidly compared to the no treatment control, as determined by bioluminescent
imaging and tumor volume measurements (Figure 6.3 and Figure 6.4). After 42
days of treatment, the no treatment control tumors were an average of 430.1 ±
37.8 mm3 in volume and weighed 404.9 ± 43.6 mg whereas siNS + PTX tumors
were 560.3 ± 46.2 mm3 in volume and weighed 715.3 ± 101.5 mg (Figure 6.4).
Interestingly, siNS + PTX-treated tumors exhibited elevated levels of eIF4E,
survivin, cyclin D1 and VEGF mRNA compared to the no treatment group,
suggesting that treatment of drug-resistance cancers with PTX can further drive
cancers to become more aggressive (Figure 6.5). This may occur through
activation of the (PI3K)/akt signaling pathway implicated with both cell survival
and drug resistance. As PTX relies upon apoptosis to induce tumor regression,
elevated expression of survivin, an antiapoptotic factor, may contribute to the
observed resistance to PTX therapy: experimental upregulation of survivin has
been shown to confer taxol resistance (311,312). Treatment of tumors with
nanoparticles delivering anti-eIF4E siRNA alone significantly attenuated tumor
growth to 240.4 ± 35.7 mm3 and 256.8 ± 33.5 mg at day 42. This finding is
supported by previously established data which demonstrates that silencing of
eIF4E induces cell growth arrest (308). Likewise, Phase I clinical evaluation of

184
the LY2275796 antisense oligonucleotide targeting eIF4E found only cytostasis,
and not anti-tumor activity, was achieved upon eIF4E downregulation (309). A
combination of sieIF4E and PTX significantly inhibited tumor growth and lead to
tumor regression, 50.2 ± 35.7 mm3 and 103.2 ± 67.4 mg. Significant knockdown
of eIF4E mRNA in addition to several downstream targets including the
antiapoptotic protein survivin, the oncogene cyclin D1, and the angiogenesis
factor VEGF was observed in both groups of mice treated with RGD-PEG(HZ)-
ECO/sieIF4E nanoparticles (Figure 6.5). By observing the downregulation of
these additional targets, the data suggests that targeting eIF4E may inhibit tumor
growth and resensitize cancer cells to PTX through multiple mechanism and
pathways. A downregulation of survivin may help suppress tumor growth by
increasing the susceptibility of cancer cells to apoptosis, particularly when
coupled with PTX therapy. Tumor growth may also be suppressed through a
reduction of angiogenesis, a consequence of VEGF downregulation. The mRNA
expression data in conjunction with the significant tumor regression observed for
the sieIF4E + PTX treatment group indicates that knockdown of eIF4E re-
sensitized the MDA-MB-231.DR cells to the cytotoxic effect of PTX.
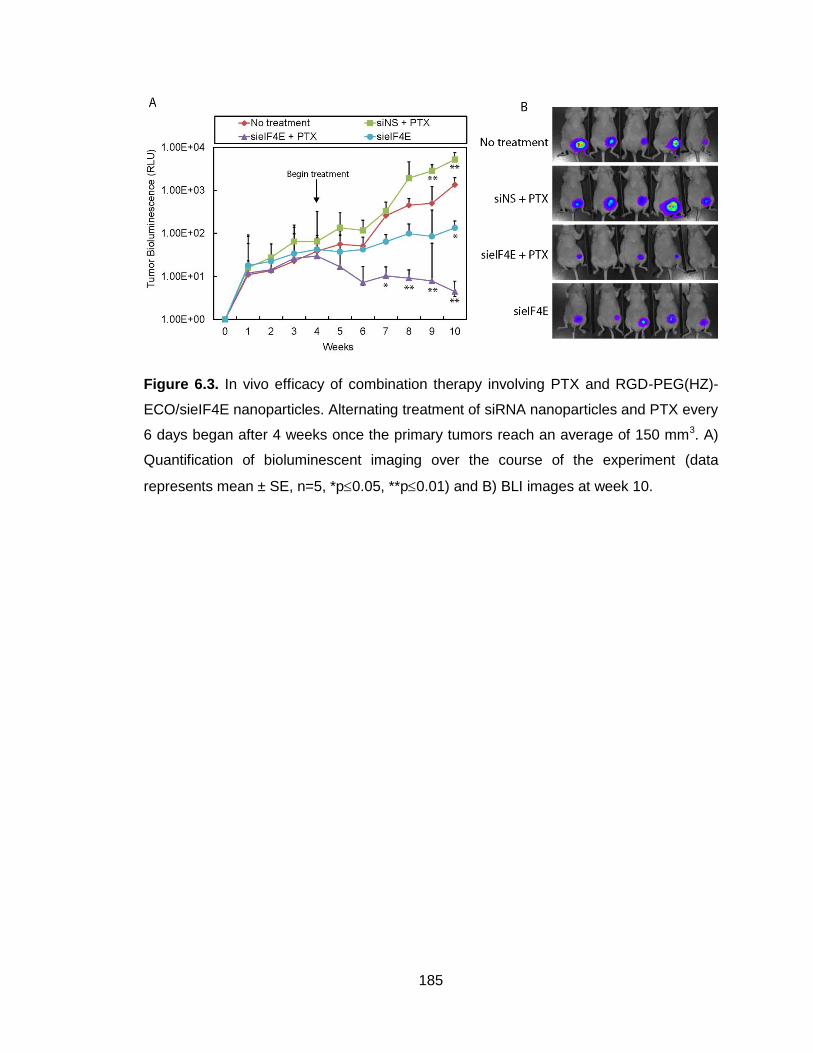
185
Figure 6.3. In vivo efficacy of combination therapy involving PTX and RGD-PEG(HZ)-
ECO/sieIF4E nanoparticles. Alternating treatment of siRNA nanoparticles and PTX every
6 days began after 4 weeks once the primary tumors reach an average of 150 mm3. A)
Quantification of bioluminescent imaging over the course of the experiment (data
represents mean ± SE, n=5, *p0.05, **p0.01) and B) BLI images at week 10.

186
Figure 6.4. In vivo efficacy of combination therapy involving PTX and RGD-PEG(HZ)-
ECO/sieIF4E nanoparticles. Alternating treatment of siRNA nanoparticles and PTX every
6 days began after 4 weeks once the primary tumors reach an average of 150 mm3. A)
Tumor growth was monitored using digital caliper measurements (data represents mean
± SE, n=5, *p0.05, **p0.01). B) Primary tumors were resected at week 10 and C) final
tumor weights were obtained (data represents mean ± SE, n=5, *p0.05, **p0.01).

187
A concern of an RNAi-based approach to eIF4E therapy is the implication
of non-specific silencing of eIF4E in healthy tissues. While the inclusion of the
RGD-targeting peptide can enhance selective uptake within tumor cells, as
demonstrated in Chapter 5, accumulation of the siRNA nanoparticles in non-
specific tissues may lead to eIF4E downregulation outside of the tumor. In the
current study, the expression of eIF4E following treatment with the siRNA
nanoparticles was not evaluated in other vital organs, however, it has been
reported elsewhere that no systemic toxicity was observed when 80%
knockdown of eIF4E was achieved in essential organs using antisense
technology (308). This may be explained by the phenomenon known as
oncogene addition whereby cancer cells are overly dependent on the expression
of a single gene for continued survival and proliferation. Under normal conditions,
eIF4E is likely to be inactive due to being bound to the inhibitory 4E-BPs (313). A
reduction in eIF4E levels would therefore have a minimal effect. Cancer cells
characterized with elevated eIF4E expression, however, are more dependent on
eIF4E expression than non-malignant cells or cancer cells with normal eIF4E
expression. The small-molecule inhibitor ribavirin, which inhibits eIF4E from
interacting with the 7-methylguanosine cap structure and 4EG1-1, disrupts the
assembly of eIF4E with eIF4F/eIF4G to inhibit translation, and can selectively
target the proliferation of cells overexpressing eIF4E (306). Additionally, studies
examining the global effect of eIF4E downregulation found that modulation of
eIF4E expression or function has negligible effects on global protein synthesis:
the translation of many growth factors is reliant upon eIF4E expression yet the

188
majority of cellular mRNAs can be translated with minimal eIF4E availability
(302).
Histopathological examination of liver and kidney tissues from mice
receiving the long-term PTX and nanoparticle treatments was performed by
hematoxylin and eosin (H&E) staining (Figure 6.6). No substantial toxicity or
tissue damage to the liver or kidney was observed across all treatment groups.
The absence of toxicity from treatment groups who received multiple
administrations of PTX may be attributed to the relatively low and infrequent
dosing: 5 mg/kg every 6 days compared to the conventional dose of 10 mg/kg.
Treatment groups who received sieIF4E treatment also revealed no structural
damage to the tissues, suggesting any systemic silencing of eIF4E harbors
minimal toxicity. Taken together, these data demonstrate the long-term safety of
a combinatory strategy of PTX and RGD-PEG(HZ)-ECO/sieIF4E nanoparticle
therapy.
Chemoresistance is a critical limitation in the treatment of patients with
TNBC, where the lack of effective targeted therapies has left small-molecule
based strategies as the sole option. Molecular targets exploited for targeted
therapy against drug-resistant TNBCs could contribute significantly to an
improved standard of care. While eIF4E has been explored as a therapeutic
target in TNBCs before, the presented study represents the first evaluation of
eIF4E in a drug-resistance TNBC cell line, therefore adding to the promising
body of work surrounding eIF4E. A distinct advantage of siRNA-based gene
therapy over ASO-mediated therapy is the ability to achieve therapeutic efficacy

189
with a much lower dose. ASOs require a stoichiometric one-to-one ratio of
silencing molecule to target mRNA, unlike siRNAs which can target and degrade
multiple mRNA targets with a single molecule. As such, pre-clinical studies using
ASOs have required a systemic dose of 25 mg/kg in order to suppress tumor
growth (302), whereas an siRNA dose of 1.5 mg/kg was able to achieve a similar
therapeutic effect. For clinical implementation of ASOs, although eIF4E mRNA
and protein expression was found to be down-regulated in tumor biopsies,
minimal tumor response was observed. Similarly, in our study sole targeting of
eIF4E with siRNA induced cytostasis of the tumors, resulting in stable disease.
Therefore, it may be that clinical translation of eIF4E therapy will require
complement treatment with other cytotoxic chemotherapeutics, similar to the
regimen proposed within this study, in order to fully exploit the effect of eIF4E is
silencing.
190
Figure 6.5. Semi-quantitative real-time quantification of eIF4E, Survivin, Cyclin D1, and
VEGF mRNA expression from the resected primary tumors (data represents mean ± SE,
n=5, *p0.05, **p0.0, #p>0.05).
Figure 6.6. Histological evaluation of liver, kidney, and primary tumor.
6.3.3. Long-term systemic administration of RGD-PEG(HZ)-ECO/siRNA
nanoparticles elicits no chronic immune response or organ damage
As nude mice bear an inhibited immune system, such animals are not a
suitable model to study potential immunogenic responses following systemic
administration of ECO/siRNA nanoparticles. Accordingly, immunocompetent
BALB/c mice were used to study the possible immune response following
repeated tail-vein injections for RGD-PEG(HZ)-modified ECO/siRNA
nanoparticles (Figure 6.7). Blood was collected at 2 h or 24 h following injections
following 1, 3 and 5 injections spaced 5 days apart. Systemic treatment with

191
unmodified ECO/siRNA nanoparticles elicited a robust activation of all cytokines
measured at both the 2 h and 24 h timepoints due to the high surface charge of
the nanoparticles. PEGylation in the form of RGD-PEG(HZ)-modification
significantly attenuated the immune response for all tested cytokines. While
cytokine levels increased at 2 h for IL-6, IL-12 and IFN-γ, the serum levels were
reduced to basal levels at 24 h, indicative of a transient response. Importantly,
serum levels were not compounded over the course of 5 repeated injections.
These data, in conjunction with pathological examination of the liver and kidneys
in Figure 6.6 are indicative of the long-term safety of PEGylated ECO/siRNA
nanoparticles.
Figure 6.7. Evaluation of immunogenicity of ECO and RGD-PEG(HZ)-modified
ECO/siRNA nanoparticles in mice following intravenous administration. At 2 h and 24 h
following each injection set, blood was collected and the plasma was isolated to be used
for cytokine ELISA measurements of TNF-α, IL-12, IFN-γ and IL-6 (data represents

192
mean ± SE, n=5, *p0.05, **p0.01). Solid and dotted lines indicate the mean ± SE
pertaining to baseline levels of each cytokine.
6.4 Conclusions
This study demonstrates that eIF4E is a promising therapeutic target to
address PTX-induced drug resistance in TNBCs. Furthermore, it confirms the
need to couple anti-eIF4E therapy with a secondary chemotherapeutic agent to
unlock the true potential of targeting eIF4E. Knockdown of eIF4E expression in
vitro re-sensitized PTX-resistant MDA-MB-231.DR cells to low doses of PTX.
When coupled with PTX therapy, delivery of an siRNA targeted against eIF4E
with the RGD-PEG(HZ)-ECO/siRNA nanoparticles resulted in robust silencing of
eIF4E and a significant regression of tumor growth. Efficient in vivo delivery of
siRNA was achieved through use of the targeted pH-cleavable PEG modification
on the ECO/siRNA nanoparticles, where a significant inhibition of tumor growth
was observed as early as after only 3 systemic treatments. We believe the
explored combinatory strategy may be useful for the treatment of drug-resistant
TNBCs. As eIF4E silencing has been demonstrated to sensitize cancers to many
types of chemotherapy drugs, the proposed delivery strategy may hold broad
applicability throughout cancer therapy. Future work should explore the ability of
eIF4E to re-sensitize cancers to additional chemotherapies, thus broadening the
therapeutic potential of this novel target.
6.5 Acknowledgments

193
This material is based upon work partially supported by the National Science
Foundation Graduate Research Fellowship under Grant Number DGE-0951783
and the National Institutes of Health under Grant Number EB00489.

194
Chapter 7
Future Work

195
7.1 Summary of Work
The aim of this thesis is to further the development of a platform cationic
lipid-based siRNA delivery system for cancer therapy, specifically for the
treatment of triple-negative breast cancers. While RNAi holds tremendous
therapeutic promise, insufficiencies in the delivery capabilities of siRNA-based
technologies have prevented clinical success. A recurrent theme throughout the
work is the focus on the multifunctionality required for cytosolic delivery of siRNA.
In Chapter 3, a rationally designed cationic lipid-based siRNA carrier, ECO, was
developed. Through a systematic exploration as to how the N/P ratio between
ECO and siRNA impacted physiochemical properties and biological activity, the
formulation of ECO/siRNA nanoparticles was optimized. Careful chemical design
of the lipid structure endowed ECO/siRNA nanoparticles with a host of
multifunctional properties. Specifically, ECO/siRNA nanoparticle exhibited pH-
sensitive amphiphilicity and glutathione-mediated reduction to allow for
endolysosomal escape and cytosolic release of the siRNA cargo, two rate-
limiting steps for siRNA delivery. ECO/siRNA nanoparticles were also designed
to enable facile surface modification with PEG and/or targeting ligands.
7.2 Long-term stability of ECO/siRNA nanoparticle formulations
With an eye towards commercialization, additional work should investigate
the ability to translate ECO/siRNA nanoparticle formulations into a clinical
product. In Chapter 2, the efficacy of ECO/siRNA nanoparticles in various
transfection conditions was described in detail. However, all data reported

196
throughout this thesis were from experiments using freshly prepared siRNA
nanoparticles. In the clinic, preparation of fresh formulations would require
extensive training to healthcare workers, and potentially introducing variability
between formulations. To prepare nanoparticles ready for clinical
implementation, the formulations should withstand storage for long periods of
time under controlled temperatures and conditions to maintain overall particle
size, shape and siRNA complexation (314).
Initial studies have begun to characterize the long-term stability of
unmodified and PEGylated ECO/siRNA nanoparticles. Both unmodified and
PEGylated ECO/siRNA nanoparticles have been demonstrated to maintain their
gene silencing efficiencies following short-term storage after lyophilization, further
evaluation of long-term storage of lyophilized nanoparticles is underway. When
stored in liquid solution at 4⁰C for 2 and 4 weeks, unmodified and RGD-
PEG(HZ)-modified nanoparticles were found to maintain silencing efficiencies of
59.17 ± 7.13% and 68.77 ± 8.67% at 2 weeks and 27.07 ± 9.22% and 21.43 ±
7.01% at 4 weeks. A thorough understanding of how the physicochemical
properties of both lyophilized and solution-stored ECO/siRNA nanoparticles are
maintained or altered following long-term storage would provide key insights into
possible formulation strategies used for the transportation and storage of
nanoparticles.

197
7.3 Treatment of late-stage, metastatic disease
In Chapter 4, RGD-targeted ECO/siRNA nanoparticles were employed to
deliver siRNA targeting integrin β3 to prevent metastasis of triple-negative breast
cancers through the inhibition of TGF-β-mediated EMT. Across two animal
models, RGD-targeted ECO/siβ3 nanoparticles demonstrated a significant ability
to inhibit the pulmonary outgrowth and metastasis of metastatic triple-negative
breast cancer cells. Importantly, over the course of 16 weeks, mice treated with
repeated systemic injections of PEGylated ECO/siRNA nanoparticles displayed
minimal toxicity, as determined by monitoring body weight. The RGD peptide is
known to primarily target αvβ3 integrins expressed on tumor-associated
vasculature. Targeting tumor vasculature and endothelial has become a popular
alternative to targeting tumor cells directly due to the potential to disrupt the
supply of blood, oxygen and essential nutrients to the tumor tissue. While robust
silencing of integrin β3 was observed in tumor tissue, it was not established
whether silencing of integrin β3 was observed in the tumor vasculature, and what
role, if any, that played in the inhibition of tumor growth and metastasis.
While moderate, but not statistically significant knockdown of integrin β3
was observed using the RAD-targeted ECO/siβ3 nanoparticles, mice treated with
the non-specific targeted nanoparticles exhibited attenuated primary tumor
growth and a significantly reduced primary tumor recurrence. Additional studies
establishing the in vivo target knockdown threshold and duration required to elicit
a desired phenotypic disease response will be critical for the design of dosing
regimens.

198
Although not shown, RGD-targeted ECO/siβ3 nanoparticles were
administered to mice bearing metastatic tumors in an attempt to treat late-stage
disease, although no efficacy was noted. Results from these experiments
suggest either i) RGD-targeted ECO/siRNA nanoparticles are unable to localize
to the metastatic sites or ii) integrin β3 may not be an appropriate target for late-
stage disease. Future work should assess the ability of RGD-targeted
ECO/siRNA nanoparticles to target and accumulate within metastatic lesions,
potentially through the use of whole mouse cryo-imaging techniques. While
nano-sized materials can readily gain vascular access to tumors through the
EPR effect, small metastases (<100 mm3 in size) typically do not exhibit the
extensive vascular networks displayed by larger, primary tumors (315). As such,
nanoparticles may require alternative targeting strategies to effectively and
selectively target metastatic sites. Depending on the metastatic site, the
physiochemical properties of ECO/siRNA nanoparticles may need to be altered
to achieve effective targeting to various organs as summarize in Table 7.1. By
optimizing the N/P ratio, as described in Chapter 3, the size and surface charge
of ECO/siRNA nanoparticles can be fine-tuned. Additional customization can
occur through use of different targeting strategies using the thiol-maleimide
chemistries.

199
Table 7.1. Considerations for nanoparticle delivery to specific sites. Adapted from (315).
If the siRNA nanoparticles are indeed able to localize to metastatic
lesions, alternate gene targets should be explored to be used alone or in concert
with integrin β3. Metastatic cells (post-EMT cells) are hypothesized to undergo a
reciprocal process, known as mesenchymal-epithelial transition (MET), at the
secondary site, in which the role of integrin β3 may become less important in cell
survival. Integrin β1 may serve as a promising starting point as expression of
integrin β1 has been established as necessary for metastatic outgrowth (219).
Further, dual inactivation of integrin β1 and β3 was more effective in reducing
breast cancer metastasis compared to sole targeting of integrin β1. Therefore,
co-delivery of siRNAs against integrin β1 and β3 may address late-stage disease
more effectively than targeting either integrin alone. Unlike other drug delivery
systems that are designed around the specific chemical properties of their drug
load (ie, hydrophobicity, molecular weight, release profile, etc), the platform
nature of ECO can be easily and quickly exploited to formulate nanoparticles with
Target site Nanoparticle size Surface characteristics Additional comments
Brain
5–100 nm: uptake efficiency
decreases exponentially with
size
Lipophilic moieties and neutral
charge enhance brain uptake
Leukocytes can take up nanoparticles in
circulation and then carry them to disease
sites in the brain
Lung>200 nm: particles are trapped
in lung capillariesPositive surface charge
Inhaled particles with low density (<0.4 g
per cm3) and of large size (>5 mm) are
also retained in the lung
Liver
<100 nm, to cross liver
fenestrae and target
hepatocytes. <100 nm
particles will be taken up by
Kupffer cells
No specificity neededLipid and lipid-like materials tend to
accumulate in the liver
Lymph nodes
6–34 nm: intra-tracheal
administration. 80 nm:
subcutaneous administration
Non-cationic, non-pegylated and
sugar-based particles
200 nm particles in circulation can be
taken up by leukocytes and trafficked to
lymph nodes
Bone Yet to be established
Compounds such as
alendronate and aspartic acid
adhere to bone and have been
used for bone targeting
Despite great importance, bone targeting
is under-researched

200
any siRNA and develop new targeting strategies using alternate targeting
peptides.
7.4 Alternative surface-modification strategies
In Chapter 5, a modified pH-cleavable PEGylation strategy was
developed to overcome the PEG dilemma, in which covalent PEGylation inhibits
cellular uptake, endolysosomal escape, and gene silencing efficiency.
Incorporation of a pH-cleavable hydrazone linkage into the RGD-targeted
PEG(HZ) moiety enhanced both endolysosomal escape and gene silencing
efficiency. While sustained gene silencing was observed in vivo using a
luciferase reporter gene, the FMT imaging study revealed only a fraction of the
injected nanoparticle dose accumulated within the tumor. A more robust
biodistribution and pharmacokinetic study could reveal the fate of the siRNA
nanoparticles following systemic administration.
A deeper understanding of the intratumoral distribution of siRNA delivered
by the ECO nanoparticles is also critical. Targeted nanoparticles may suffer from
a binding site barrier in which the targeted nanoparticles cannot penetrate deep
into the target tissue due to the high avidity of the targeting agent, a phenomenon
first observed in antibody-based therapeutics (316,317). While Chapter 5
showed specific internalization of siRNA into cancer cells, the question of how
deep into the tumor tissue the ECO/siRNA nanoparticles can penetrate has yet to
be answered. If the siRNA nanoparticles are found to be highly concentrated at
the periphery of the tumor tissue, optimization of the targeting ligand density

201
and/or the affinity of the ligand may enhance penetration. Again, re-optimization
of the N/P ratio may be necessary to alter the nanoparticle size and/or surface
charge, parameters that govern the tumor penetration capabilities.
Along these lines, it is not clear if the surface-modified ECO/siRNA
nanoparticles remain intact within the tumor tissue or if the siRNA cargo is first
release within the microenvironment before internalization. Comparison of gene
silencing efficiency using a nuclease-stabilized siRNA load with a non-stabilized
siRNA may allow investigation into this topic. Previous studies have
demonstrated that nuclease-stabilized siRNAs exert superior RNAi activity over
non-stabilized siRNA only in the presence of nucleases, similar to the nuclease-
rich extracellular environment (318). Similar levels and duration of gene silencing
for siRNA nanoparticles formulated with both types of siRNAs would be indicative
of siRNA release following cellular internalization. However, nanoparticles
formulated with nuclease-stabilized siRNA producing a more robust gene
silencing would suggest that the siRNA cargo is release extracellularly prior to
internalization.
An additional surface modification strategy could also be employed using
a pH-sensitive charge-conversion polymer to endow the siRNA nanoparticles
with extracellular pH-sensitivity while maintaining intracellular pH-sensitivity.
Such a modification would allow the siRNA nanoparticle to be delivered in a
“stealth” or “inactive” mode throughout circulation before becoming “active” within
the acidic tumor microenvironment (319,320). Extracellular pH-sensitivity is
important to address challenges with cellular uptake within the characteristically

202
acidic tumor microenvironment and to minimize non-specific uptake in non-target
tissues. While pH-sensitive delivery systems have been developed, these
systems are severely limited in their efficacy due to responding to only a single
pH condition, targeting either the tumor extracellular (pHe) or the intracellular pH
(pHi) condition: the delivery systems that respond to pHe often release the siRNA
payloads extracellularly, making them inefficient in silencing the target gene,
while those capable of responding to pHi cannot efficiently enhance the cellular
internalization of the delivery system.
At neutral pH, a charge-conversion polymer is anionic and can bind
electrostatically to the surface of cationic ECO/siRNA nanoparticles. Within the
tumor extracellular environment, the polymer layer would undergo charge-
conversion to become cationic and shed due to increased electrostatic repulsion
with the ECO/siRNA nanoparticles. Upon doing so, the cationic surface of the
nanoparticles would become re-exposed to facilitate tumor cell internalization. If
targeting ligands, such as the RGD-PEG(HZ) moiety developed in Chapter 5,
are functionalized to the nanoparticle surface through exploitation of free thiol
groups within the cysteine-based linker domain, they too will be re-exposed and
further enhance cellular targeting and uptake.
7.5 Synergy in co-delivery of siRNA and PTX
In Chapter 6, the benefits of combination therapy involving siRNA
targeted against eIF4E with the small-molecule paclitaxel against a drug-
resistance triple-negative breast cancer cell line were studied. Data gleaned from

203
an in vivo animal model demonstrated that the combination therapy was able to
re-sensitize drug-resistant cancer cells to PTX and significantly inhibit primary
tumor growth. While the siRNA was delivered using the surface-modification
strategy developed in Chapter 5, PTX was administered i.p. in a 10%
DMSO/PBS vehicle; the delivery of siRNA and PTX from the same delivery
vehicle has yet to be studied. The first step would be to determine if ECO can
effectively load PTX into a stable nanoparticle. With a highly hydrophobic
structure, it may be that PTX could be loaded into ECO/siRNA nanoparticles
through hydrophobic interactions with the oleic acid lipid tails. An alternate
strategy involving covalent attachment of PTX to ECO via a PEG linker could
also be explored. A thorough understand of how PTX incorporation into
ECO/siRNA nanoparticles would affect the physicochemical properties of the
siRNA nanoparticles, including siRNA binding efficiency, stability, pH-sensitive
hemolysis, and glutathione-mediated reduction would need to be evaluated.
While there are clear advantages to formulating PTX into a nanoparticle, such as
improved biodistribution, tumor targeting and a reduction of systemic toxicities, it
is unclear if formulation of siRNA and PTX into the same system would offer any
distinct advantage over their separate delivery.
The aforementioned studies will substantially enhance the understanding
of the ECO/siRNA platform delivery system and help to streamline the clinical
development of such a system. The key advantages of the ECO/siRNA delivery
system are provided by the chemical structure of the ECO lipid which generates
multiple levels of functionality and responsiveness to counter the complex

204
delivery barriers. The ability to easily customize the physicochemical properties
of the nanoparticles, alter the siRNA cargo towards targeting any gene, and
functionalize the surface with various modifications and targeting strategies will
allow the platform system to be configured to meet the needs of many diseases.
Through multidisciplinary collaborations, the ECO/siRNA delivery system has
already been used to sensitize human glioblastoma tumors to radiation therapy
and is currently under investigation targeting key genes against the Ebola virus.
Further development through continued collaboration between clinicians,
biologists and engineers will contribute to the already compelling body of
therapeutic data, expedite and streamline clinical translation, and ultimately
improve the disease outcomes of not only patients with triple-negative breast
cancers, but of all types.

205
Appendix

206
A.1. Synthesis of ECO
The synthesis of (1-aminoethyl)imino-bis[N-(oleicyl-cysteinyl-1-
aminoethyl)propionamide] (ECO) was done by liquid-phase chemistry and is
described below. The reaction scheme can be seen in Figure A1. The
ethylenediamine head group was synthesized first followed by the cysteine/oleic
acid tail groups. Once these two groups were synthesized they were reacted
together to arrive at the final ECO product. Each reaction intermediate was
confirmed through 1H NMR.
Figure A1. Synthetic Procedure of ECO
1. Synthesis of intermediate (1)
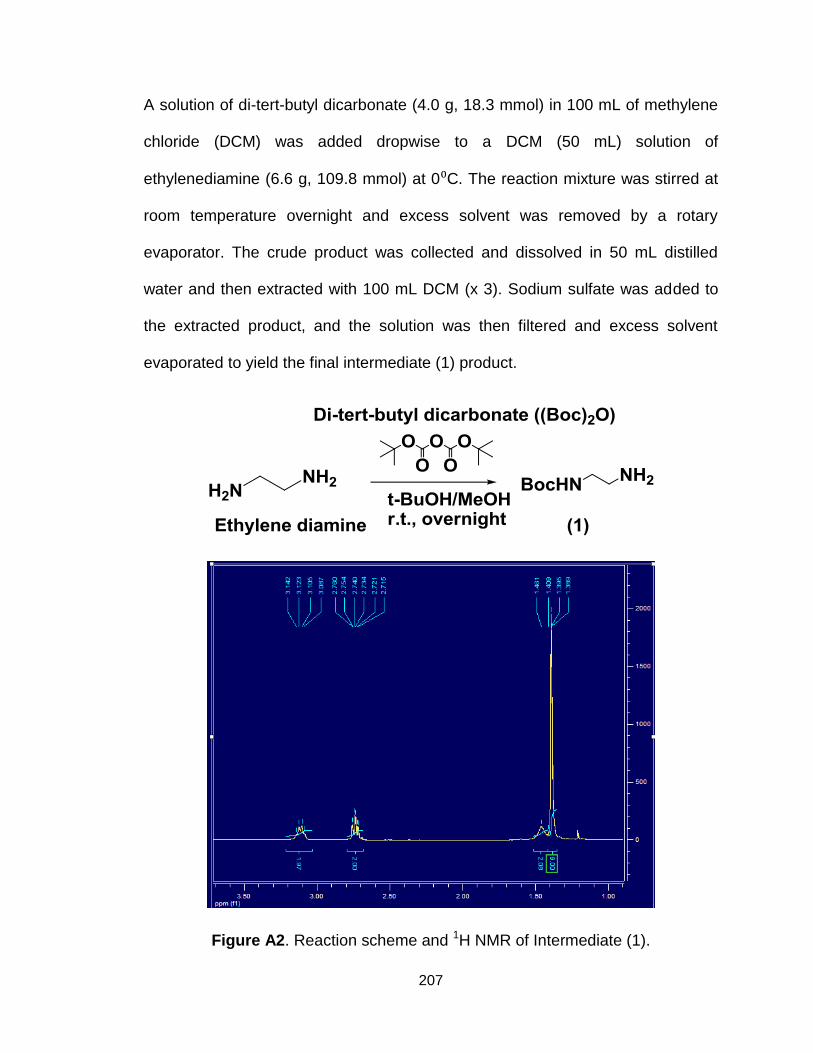
207
A solution of di-tert-butyl dicarbonate (4.0 g, 18.3 mmol) in 100 mL of methylene
chloride (DCM) was added dropwise to a DCM (50 mL) solution of
ethylenediamine (6.6 g, 109.8 mmol) at 0⁰C. The reaction mixture was stirred at
room temperature overnight and excess solvent was removed by a rotary
evaporator. The crude product was collected and dissolved in 50 mL distilled
water and then extracted with 100 mL DCM (x 3). Sodium sulfate was added to
the extracted product, and the solution was then filtered and excess solvent
evaporated to yield the final intermediate (1) product.
Figure A2. Reaction scheme and 1H NMR of Intermediate (1).

208
2. Synthesis of intermediate (2)
A solution of intermediate (1) (2.0 g, 12.5 mmol) in 10 mL of methanol was added
dropwise under nitrogen to a stirred solution of methyl acrylate (4.33 g, 50.9
mmol) in 20mL of methanol at 0⁰C. The resulting solution was allowed to warm to
room temperature and was stirred for 2 days. The solvent was removed under
reduced pressure using a rotary evaporator, dissolved in 30 mL of water and
extracted with 30 mL of DCM (x 3). The crude product was dried with sodium
sulfate followed by filtering of the solution. The filtered solution was placed in a
vacuum oven for 2 days to yield the final intermediate (2) product.

209
Figure A3. Reaction scheme and 1H NMR of Intermediate (2).
3. Synthesis of intermediate (3)
A solution of intermediate (2) (4.0 g, 12.05 mmol) in 20 mL of methanol was
added dropwise under nitrogen to a solution of ethylenediamine (45 g, 753 mmol)
in 30 mL of methanol over 1 hour at 0⁰C. The reaction mixture was allowed to
warm up to room temperature and stirred for 5 days. The solvent was removed
and the product was washed with 15 mL of diethyl ether (x 3).
Figure A4. Reaction scheme and 1H NMR of Intermediate (3).
4. Synthesis of intermediate (4)

210
Oleic acid (4.0 g, 14.2 mmol), N-hydroxysuccinimide (NHS) (1.85g, 15.75 mmol),
and N,N'-Dicyclohexylcarbodiimide (DCC) (3.71 g, 18.01 mmol) were added to
ethyl acetate (15 mL). The reaction was allowed to react overnight at room
temperature and the by-product, ducyclohexylurea (DCU), was removed by
centrifugation and filtering of the reaction solution. Excess solvent was
evaporated and product was dried over phosphorous pentoxide.
Figure A5. Reaction scheme and 1H NMR of Intermediate (4).
5. Synthesis of intermediate (5)
Intermediate (4) (4.87 g, 12.51 mmol) was then reacted with H-Cysteine(Trt)-OH
(5.08 g, 13.98 mmol) and N,N-diisopropylethylamine (DIPEA) (6 mL) in 60 mL of
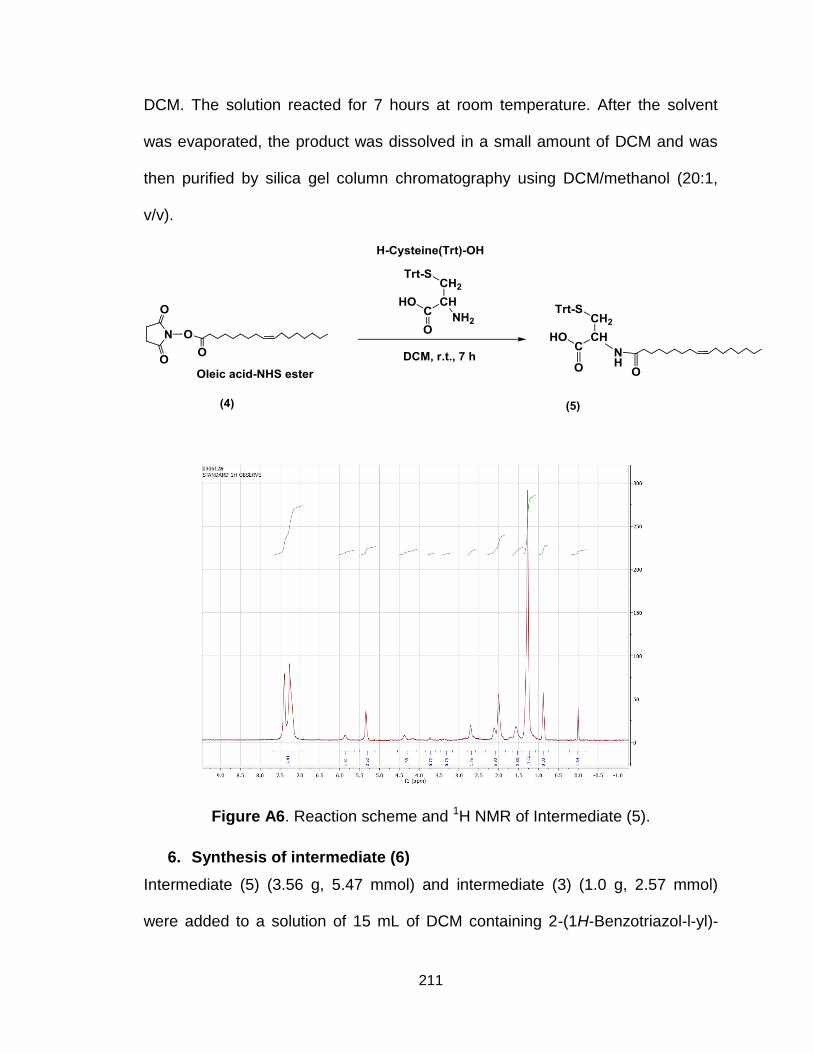
211
DCM. The solution reacted for 7 hours at room temperature. After the solvent
was evaporated, the product was dissolved in a small amount of DCM and was
then purified by silica gel column chromatography using DCM/methanol (20:1,
v/v).
Figure A6. Reaction scheme and 1H NMR of Intermediate (5).
6. Synthesis of intermediate (6)
Intermediate (5) (3.56 g, 5.47 mmol) and intermediate (3) (1.0 g, 2.57 mmol)
were added to a solution of 15 mL of DCM containing 2-(1H-Benzotriazol-l-yl)-

212
1,1,3,3-tetramethyluronium hyexafluorophosphate (HBTU, 2.15 g, 5.67 mmol)
and 1-hydroxybenzotriazole (HoBT, 0.76 g, 5.64 mmol). DIPEA (3 mL) was
added to the solution and stirred at room temperature overnight. The solvent was
removed and product was washed with ether (3 x 25 mL) to remove HBTU,
HoBT and DIPEA. The product was dissolved in water and extracted with DCM
to remove excess intermediate (5) and the organic phase was evaporated and
dried with sodium sulfate.
Figure A7. Reaction scheme and 1H NMR of Intermediate (6).
7. Final synthesis of ECO

213
Intermediate (6) was deprotected with DCM:TFA:triisobutylsilane:ethanedithiol at
a 7:2:1:1 ratio at room temperature for 30 minutes. The solvent was removed and
diethyl ether was added to the product and was precipitated and washed with
hexane to remove free protecting groups. The residue was dissolved again in
diethyl ether and re-dispersed in acetonitrile. The precipitate was collected,
dissolved in water and lyophilized to give the final product (1-
aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-ethyl)propionamide] (ECO).
Figure A8. Reaction scheme and 1H NMR of ECO.
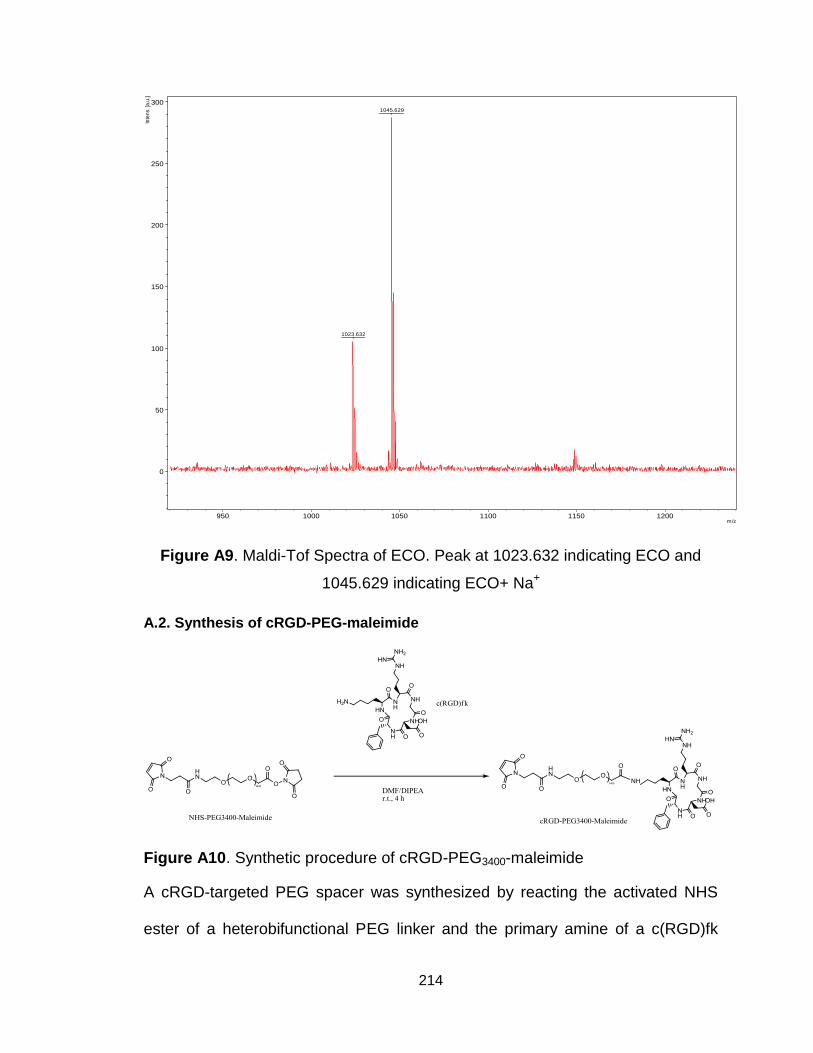
214
Figure A9. Maldi-Tof Spectra of ECO. Peak at 1023.632 indicating ECO and
1045.629 indicating ECO+ Na+
A.2. Synthesis of cRGD-PEG-maleimide
Figure A10. Synthetic procedure of cRGD-PEG3400-maleimide
A cRGD-targeted PEG spacer was synthesized by reacting the activated NHS
ester of a heterobifunctional PEG linker and the primary amine of a c(RGD)fk
1045.629
1023.632
0
50
100
150
200
250
300
Inte
ns.
[a.u
.]
950 1000 1050 1100 1150 1200m/z
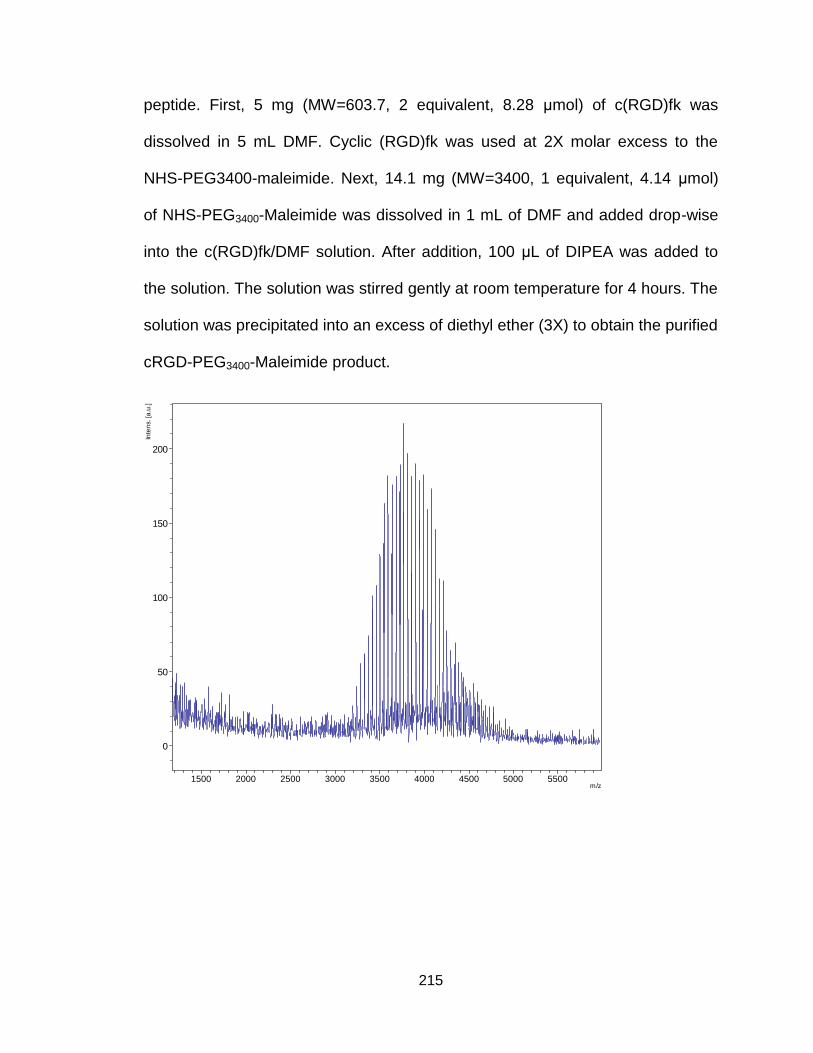
215
peptide. First, 5 mg (MW=603.7, 2 equivalent, 8.28 μmol) of c(RGD)fk was
dissolved in 5 mL DMF. Cyclic (RGD)fk was used at 2X molar excess to the
NHS-PEG3400-maleimide. Next, 14.1 mg (MW=3400, 1 equivalent, 4.14 μmol)
of NHS-PEG3400-Maleimide was dissolved in 1 mL of DMF and added drop-wise
into the c(RGD)fk/DMF solution. After addition, 100 μL of DIPEA was added to
the solution. The solution was stirred gently at room temperature for 4 hours. The
solution was precipitated into an excess of diethyl ether (3X) to obtain the purified
cRGD-PEG3400-Maleimide product.
0
50
100
150
200
Inte
ns. [a
.u.]
1500 2000 2500 3000 3500 4000 4500 5000 5500m/z

216
Figure A11. Maldi-tof and H1-NMR spectrum (Solvent: D2O) of cRGD-PEG3400-
maleimide.
A.3. Synthesis of mPEG(HZ)-maleimide
Figure A12. Synthetic procedure of mPEG5000(HZ)-Maleimide.

217
Figure A13. H1 NMR spectrum (Solvent: CDCl3) of mPEG5000(HZ)-maleimide.
Characteristic peaks: 8.43 (s, 1H, -NH-), 8.06 (d, 2H, in phenyl), 7.54 (d, 2H, in
phenyl), 6.91 (2, 2H, two olefinic protons of maleimide), 3.48-3.58 (m, 438H,
PEG).
A.4. Synthesis of cRGD-PEG(HZ)-maleimide

218
Figure A14. Synthetic procedure of cRGD-PEG3400(HZ)-maleimide.

219
Figure A15. H1 NMR spectrum (Solvent: DMSO of cRGD-PEG3400(HZ)-
maleimide. Characteristic peaks: 8.48 (s, 1H, -NH-), 8.0 (d, 2H, in phenyl), 7.6-
7.8 (m, cRGD), 7.45 (d, 2H, in phenyl), 7.2 (2, 2H, two olefinic protons of
maleimide), 3.1-3.5 (m, 304H, PEG).
Figure A16. H1 NMR spectrum (Solvent: D2O) of cRGD-PEG3400(HZ)-maleimide.

220
References
1. Howlander N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF. SEER cancer statistics review, 1975–2011. Bethesda, MD: National Cancer Institute; 2014.
2. Siegel, R., Ma, J., Zou, Z. and Jemal A. Cancer statistics, 2014. A Cancer Journal for Clinicians. 2014;64:9–29.
3. Dawood S, Hu R, Homes MD, Collins LC, Schnitt SJ, Connolly J, et al. Defining breast cancer prognosis based on molecular phenotypes: results from a large cohort study. Breast cancer research and treatment. Springer; 2011;126:185–92.
4. Murray CJL, Lopez AD. Measuring the global burden of disease. New England Journal of Medicine. Mass Medical Soc; 2013;369:448–57.
5. Mariotto AB, Yabroff KR, Shao Y, Feuer EJ, Brown ML. Projections of the cost of cancer care in the United States: 2010–2020. Journal of the National Cancer Institute. Oxford University Press; 2011;
6. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. nature. Nature Publishing Group; 2001;411:494–8.
7. Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Nature structural & molecular biology. Nature Publishing Group; 2004;11:214–8.
8. Sontheimer EJ. Assembly and function of RNA silencing complexes. Nature Reviews Molecular Cell Biology. Nature Publishing Group; 2005;6:127–38.
9. Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. nature. Nature Publishing Group; 2001;409:363–6.
10. Rand TA, Ginalski K, Grishin N V, Wang X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proceedings of the National Academy of Sciences of the United States of America. National Acad Sciences; 2004;101:14385–9.
11. Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. Elsevier; 2005;123:607–20.

221
12. Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. Elsevier; 2007;130:101–12.
13. Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. Elsevier; 2005;123:621–9.
14. Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. American Association for the Advancement of Science; 2002;297:2056–60.
15. John M, Constien R, Akinc A, Goldberg M, Moon Y-A, Spranger M, et al. Effective RNAi-mediated gene silencing without interruption of the endogenous microRNA pathway. Nature. Nature Publishing Group; 2007;449:745–7.
16. De Fougerolles A, Vornlocher H-P, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nature Reviews Drug Discovery. Nature Publishing Group; 2007;6:443–53.
17. Bumcrot D, Manoharan M, Koteliansky V, Sah DWY. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nature chemical biology. Nature Publishing Group; 2006;2:711–9.
18. Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nature reviews Genetics. 2007;8:173–84.
19. Kim S-S, Garg H, Joshi A, Manjunath N. Strategies for targeted nonviral delivery of siRNAs in vivo. Trends in molecular medicine. Elsevier; 2009;15:491–500.
20. Leung RKM, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacology & therapeutics. Elsevier; 2005;107:222–39.
21. Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proceedings of the National Academy of Sciences. National Acad Sciences; 2008;105:11915–20.
22. Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nature biotechnology. Nature Publishing Group; 2008;26:431–42.

222
23. Song E, Lee S-K, Wang J, Ince N, Ouyang N, Min J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature medicine. Nature Publishing Group; 2003;9:347–51.
24. NIU X, PENG Z, DUAN W, Wang H, Wang P. Inhibition of HPV 16 E6 oncogene expression by RNA interference in vitro and in vivo. International Journal of Gynecological Cancer. Wiley Online Library; 2006;16:743–51.
25. Halder J, Kamat AA, Landen CN, Han LY, Lutgendorf SK, Lin YG, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clinical Cancer Research. AACR; 2006;12:4916–24.
26. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. Nature Publishing Group; 2004;10:789–99.
27. Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Prospects of RNA interference therapy for cancer. Gene therapy. Nature Publishing Group; 2006;13:464–77.
28. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical Cancer Research. AACR; 2007;13:4429–34.
29. O’Toole S, Beith JM, Millar EK, West R, McLean A, Cazet A, et al. Therapeutic targets in triple negative breast cancer. Journal of clinical pathology. 2013;66:530–42.
30. Williford J-M, Wu J, Ren Y, Archang MM, Leong KW, Mao H-Q. Recent advances in nanoparticle-mediated siRNA delivery. Annual review of biomedical engineering. 2014;16:347–70.
31. De Fougerolles A, Novobrantseva T. siRNA and the lung: research tool or therapeutic drug? Current opinion in pharmacology. Elsevier; 2008;8:280–5.
32. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Molecular pharmaceutics. ACS Publications; 2008;5:505–15.
33. Scherphof GL. In vivo behavior of liposomes: Interactions with the mononuclear phagocyte system and implications for drug targeting. Targeted drug delivery. Springer; 1991. page 285–327.
34. Zamecnik J, Vargova L, Homola A, Kodet R, Sykova E. Extracellular matrix glycoproteins and diffusion barriers in human astrocytic tumours.

223
Neuropathology and applied neurobiology. Wiley Online Library; 2004;30:338–50.
35. Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. The Journal of clinical investigation. American Society for Clinical Investigation; 2007;117:3623.
36. Oliveira S, Van Rooy I, Kranenburg O, Storm G, Schiffelers RM. Fusogenic peptides enhance endosomal escape improving siRNA-induced silencing of oncogenes. International journal of pharmaceutics. Elsevier; 2007;331:211–4.
37. Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nature cell biology. Nature Publishing Group; 2003;5:410–21.
38. Boussif O, Lezoualc’h F, Zanta MAntoniet, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences. National Acad Sciences; 1995;92:7297–301.
39. Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. The journal of gene medicine. 2005;7:657–63.
40. Juliano R, Alam MR, Dixit V, Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic acids research. 2008;36:4158–71.
41. Aigner A. Nonviral in vivo delivery of therapeutic small interfering RNAs. Current opinion in molecular therapeutics. 2007;9:345–52.
42. Ozpolat B, Sood AK, Lopez‐Berestein G. Nanomedicine based approaches for the delivery of siRNA in cancer. Journal of internal medicine. Wiley Online Library; 2010;267:44–53.
43. Rappaport J, Hanss B, Kopp JB, Copeland TD, Bruggeman LA, Coffman TM, et al. Transport of phosphorothioate oligonucleotides in the kidney: Implications for molecular therapy. Kidney international. New York: Springer-Verlag; 1995;47:1462–9.
44. Santel A, Aleku M, Keil O, Endruschat J, Esche V, Fisch G, et al. A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene therapy. Nature Publishing Group; 2006;13:1222–34.

224
45. McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell type–specific delivery of siRNAs with aptamer-siRNA chimeras. Nature biotechnology. Nature Publishing Group; 2006;24:1005–15.
46. Braasch DA, Paroo Z, Constantinescu A, Ren G, Öz OK, Mason RP, et al. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorganic & medicinal chemistry letters. Elsevier; 2004;14:1139–43.
47. Bogdanov AA. Merging molecular imaging and RNA interference: early experience in live animals. Journal of cellular biochemistry. Wiley Online Library; 2008;104:1113–23.
48. Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GGM. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome–DNA complexes. Proceedings of the National Academy of Sciences. National Acad Sciences; 2003;100:1972–7.
49. Auguste DT, Furman K, Wong A, Fuller J, Armes SP, Deming TJ, et al. Triggered release of siRNA from poly (ethylene glycol)-protected, pH-dependent liposomes. Journal of Controlled Release. Elsevier; 2008;130:266–74.
50. Martina M-S, Nicolas V, Wilhelm C, Menager C, Barratt G, Lesieur S. The in vitro kinetics of the interactions between PEG-ylated magnetic-fluid-loaded liposomes and macrophages. Biomaterials. Elsevier; 2007;28:4143–53.
51. Mao S, Neu M, Germershaus O, Merkel O, Sitterberg J, Bakowsky U, et al. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly (ethylene imine)-graft-poly (ethylene glycol) block copolymer/SiRNA polyplexes. Bioconjugate chemistry. ACS Publications; 2006;17:1209–18.
52. Barquinero J, Eixarch H, Perez-Melgosa M. Retroviral vectors: new applications for an old tool. Gene therapy. Nature Publishing Group; 2004;11:S3–S9.
53. Vandenbroucke RE, De Geest BG, Bonné S, Vinken M, Van Haecke T, Heimberg H, et al. Prolonged gene silencing in hepatoma cells and primary hepatocytes after small interfering RNA delivery with biodegradable poly
(β‐amino esters). The journal of gene medicine. Wiley Online Library; 2008;10:783–94.
54. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nature Reviews Cancer. Nature Publishing Group; 2002;2:750–63.

225
55. Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, et al. Targeted nanoparticles for cancer therapy. Nano today. Elsevier; 2007;2:14–21.
56. Zureikat AH, McKee MD. Targeted therapy for solid tumors: current status. Surgical oncology clinics of North America. Elsevier; 2008;17:279–301.
57. Liu B. Exploring cell type-specific internalizing antibodies for targeted delivery of siRNA. Briefings in functional genomics & proteomics. Oxford University Press; 2007;6:112–9.
58. Sugita T, Yoshikawa T, Mukai Y, Yamanada N, Imai S, Nagano K, et al. Improved cytosolic translocation and tumor-killing activity of Tat-shepherdin conjugates mediated by co-treatment with Tat-fused endosome-disruptive HA2 peptide. Biochemical and biophysical research communications. Elsevier; 2007;363:1027–32.
59. Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, et al. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer research. AACR; 2007;67:2938–43.
60. Tseng Y-L, Liu J-J, Hong R-L. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Molecular pharmacology. ASPET; 2002;62:864–72.
61. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer research. 2006;66:6732–40.
62. Bartlett D, Su H. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proceedings of the National Academy of Science. 2007;104:15549–54.
63. Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nature medicine. Nature Publishing Group; 2005;11:263–70.
64. Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, et al. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense and Nucleic Acid Drug Development. Mary Ann Liebert, Inc.; 2003;13:83–105.

226
65. Czauderna F, Fechtner M, Dames S, AyguÈn H, Klippel A, Pronk GJ, et al. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic acids research. Oxford Univ Press; 2003;31:2705–16.
66. Hall AHS, Wan J, Shaughnessy EE, Shaw BR, Alexander KA. RNA interference using boranophosphate siRNAs: structure–activity relationships. Nucleic acids research. Oxford Univ Press; 2004;32:5991–6000.
67. Whitehead K a, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nature reviews Drug discovery. 2009;8:129–38.
68. Crooke ST. Antisense drug technology: principles, strategies, and applications. CRC Press; 2007.
69. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nature reviews Drug discovery. Nature Publishing Group; 2005;4:145–60.
70. Bedi D, Musacchio T, Fagbohun OA, Gillespie JW, Deinnocentes P, Bird RC, et al. Delivery of siRNA into breast cancer cells via phage fusion protein-targeted liposomes. Nanomedicine: Nanotechnology, Biology and Medicine. Elsevier; 2011;7:315–23.
71. Kim HS, Song IH, Kim JC, Kim EJ, Jang DO, Park YS. In vitro and in vivo gene-transferring characteristics of novel cationic lipids, DMKD (O, O’-dimyristyl-N-lysyl aspartate) and DMKE (O, O'-dimyristyl-N-lysyl glutamate). Journal of controlled release. Elsevier; 2006;115:234–41.
72. Rust DM, Jameson G. The novel lipid delivery system of amphotericin B: drug profile and relevance to clinical practice. Oncology nursing forum. 1997. page 35–48.
73. Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. Journal of Controlled Release. 2006;114:100–9.
74. Kim HS, Song IH, Kim JC, Kim EJ, Jang DO, Park YS. In vitro and in vivo gene-transferring characteristics of novel cationic lipids, DMKD (O, O’-dimyristyl-N-lysyl aspartate) and DMKE (O, O'-dimyristyl-N-lysyl glutamate). Journal of controlled release. Elsevier; 2006;115:234–41.
75. Pardridge WM. shRNA and siRNA delivery to the brain. Advanced drug delivery reviews. Elsevier; 2007;59:141–52.

227
76. Zhang Y, Zhang Y, Bryant J, Charles A, Boado RJ, Pardridge WM. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clinical Cancer Research. AACR; 2004;10:3667–77.
77. Pirollo KF, Zon G, Rait A, Zhou Q, Yu W, Hogrefe R, et al. Tumor-targeting nanoimmunoliposome complex for short interfering RNA delivery. Human gene therapy. Mary Ann Liebert, Inc. 2 Madison Avenue Larchmont, NY 10538 USA; 2006;17:117–24.
78. Lavigne C, Thierry AR. Specific subcellular localization of siRNAs delivered by lipoplex in MCF-7 breast cancer cells. Biochimie. Elsevier; 2007;89:1245–51.
79. Landen CN, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer research. AACR; 2005;65:6910–8.
80. Meryet-Figuières M, Resina S, Lavigne C, Barlovatz-Meimon G, Lebleu B, Thierry AR. Inhibition of PAI-1 expression in breast cancer carcinoma cells by siRNA at nanomolar range. Biochimie. Elsevier; 2007;89:1228–33.
81. Landen CN, Merritt WM, Mangala LS, Sanguino AM, Bucana C, Lu C, et al. Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer. Cancer biology & therapy. Taylor & Francis; 2006;5:1708–13.
82. Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nature medicine. Nature Publishing Group; 2006;12:939–44.
83. Merritt WM, Lin YG, Spannuth WA, Fletcher MS, Kamat AA, Han LY, et al. Effect of interleukin-8 gene silencing with liposome-encapsulated small interfering RNA on ovarian cancer cell growth. Journal of the National Cancer Institute. Oxford University Press; 2008;100:359–72.
84. Gary DJ, Puri N, Won Y-Y. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. Journal of controlled release : official journal of the Controlled Release Society. 2007;121:64–73.
85. Howard KA, Rahbek UL, Liu X, Damgaard CK, Glud SZ, Andersen MØ, et al. RNA interference in vitro and in vivo using a chitosan/siRNA nanoparticle system. Molecular Therapy. Nature Publishing Group; 2006;14:476–84.

228
86. De Martimprey H, Bertrand J-R, Fusco A, Santoro M, Couvreur P, Vauthier C, et al. siRNA nanoformulation against the ret/PTC1 junction oncogene is efficient in an in vivo model of papillary thyroid carcinoma. Nucleic acids research. Oxford Univ Press; 2008;36:e2–e2.
87. Pillé J-Y, Li H, Blot E, Bertrand J-R, Pritchard L-L, Opolon P, et al. Intravenous delivery of anti-RhoA small interfering RNA loaded in nanoparticles of chitosan in mice: safety and efficacy in xenografted aggressive breast cancer. Human gene therapy. Mary Ann Liebert, Inc. 2 Madison Avenue Larchmont, NY 10538 USA; 2006;17:1019–26.
88. Grayson ACR, Doody AM, Putnam D. Biophysical and structural characterization of polyethylenimine-mediated siRNA delivery in vitro. Pharmaceutical research. Springer; 2006;23:1868–76.
89. Khan A, Benboubetra M, Sayyed PZ, Wooi Ng K, Fox S, Beck G, et al. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: in vitro and in vivo studies. Journal of drug targeting. Informa UK Ltd UK; 2004;12:393–404.
90. Husmann M, Schenderlein S, Lück M, Lindner H, Kleinebudde P. Polymer erosion in PLGA microparticles produced by phase separation method. International journal of pharmaceutics. Elsevier; 2002;242:277–80.
91. Cherng J-Y, Talsma H, Verrijk R, Crommelin DJA, Hennink WE. The effect of formulation parameters on the size of poly ((2-dimethylamino) ethyl methacrylate)-plasmid complexes. European journal of pharmaceutics and biopharmaceutics. Elsevier; 1999;47:215–24.
92. Bishop NE. An Update on Non‐clathrin‐coated Endocytosis. Reviews in medical virology. Wiley Online Library; 1997;7:199–209.
93. Boussif O, Zanta MA, Behr J-P. Optimized galenics improve in vitro gene transfer with cationic molecules up to 1000-fold. Gene therapy. 1996;3:1074–80.
94. Boeckle S, Von Gersdorff K, Van der Piepen S, Culmsee C, Wagner E, Ogris M. Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. The journal of gene medicine. Wiley Online Library; 2004;6:1102–11.
95. Hunter AC. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Advanced drug delivery reviews. Elsevier; 2006;58:1523–31.

229
96. Thomas M, Klibanov AM. Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proceedings of the National Academy of Sciences. National Acad Sciences; 2002;99:14640–5.
97. Fischer D, Von Harpe A, Kunath K, Petersen H, Li Y, Kissel T. Copolymers of ethylene imine and N-(2-hydroxyethyl)-ethylene imine as tools to study effects of polymer structure on physicochemical and biological properties of DNA complexes. Bioconjugate chemistry. ACS Publications; 2002;13:1124–33.
98. Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene therapy. Nature Publishing Group; 2005;12:461–6.
99. Bettuzzi S. The new anti-oncogene clusterin and the molecular profiling of prostate cancer progression and prognosis. Acta Biomed Ateneo Parmense. 2003;74:101–4.
100. Sutton D, Kim S, Shuai X, Leskov K, Marques JT, Williams BRG, et al. Efficient suppression of secretory clusterin levels by polymer-siRNA nanocomplexes enhances ionizing radiation lethality in human MCF-7 breast cancer cells in vitro. International journal of nanomedicine. Dove Press; 2006;1:155.
101. Lawlor MA, Alessi DR. PKB/Akt a key mediator of cell proliferation, survival and insulin responses? Journal of cell science. The Company of Biologists Ltd; 2001;114:2903–10.
102. Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, et al. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer research. AACR; 1998;58:5667–72.
103. Xu C-X, Jere D, Jin H, Chang S-H, Chung Y-S, Shin J-Y, et al. Poly (ester amine)-mediated, aerosol-delivered Akt1 small interfering RNA suppresses lung tumorigenesis. American journal of respiratory and critical care medicine. Am Thoracic Soc; 2008;178:60–73.
104. Wang Y, Gao S, Ye W-H, Yoon HS, Yang Y-Y. Co-delivery of drugs and DNA from cationic core–shell nanoparticles self-assembled from a biodegradable copolymer. Nature materials. Nature Publishing Group; 2006;5:791–6.
105. Del Vecchio S, Zannetti A, Fonti R, Pace L, Salvatore M. Nuclear imaging in cancer theranostics. The quarterly journal of nuclear medicine and

230
molecular imaging: official publication of the Italian Association of Nuclear Medicine (AIMN)[and] the International Association of Radiopharmacology (IAR),[and] Section of the Society of. 2007;51:152–63.
106. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nature Reviews Cancer. Nature Publishing Group; 2005;5:161–71.
107. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature nanotechnology. Nature Publishing Group; 2007;2:751–60.
108. Wang Y, Shim MS, Levinson NS, Sung H-W, Xia Y. Stimuli-Responsive Materials for Controlled Release of Theranostic Agents. Advanced functional materials. 2014;24:4206–20.
109. Sumer B, Gao J. Theranostic nanomedicine for cancer. Nanomedicine. London: Future Medicine, c2006-; 2008;3:137–40.
110. Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In vivo imaging of siRNA delivery and silencing in tumors. Nature medicine. Nature Publishing Group; 2007;13:372–7.
111. Zaffaroni N, Pannati M, Diadone MG. Survivin as a target for new anticancer interventions. Journal of cellular and molecular medicine. Wiley Online Library; 2005;9:360–72.
112. Lee J, Lee K, Moon SH, Lee Y, Park TG, Cheon J. All‐in‐One Target‐Cell‐Specific Magnetic Nanoparticles for Simultaneous Molecular Imaging and siRNA Delivery. Angewandte Chemie. Wiley Online Library; 2009;121:4238–43.
113. Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer discovery. AACR; 2013;3:406–17.
114. Davidson BL, McCray PB. Current prospects for RNA interference-based therapies. Nature reviews Genetics. Nature Publishing Group; 2011;12:329–40.
115. Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnology journal. Wiley Online Library; 2011;6:1130–46.

231
116. Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Molecular pharmaceutics. ACS Publications; 2009;6:659–68.
117. Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R, et al. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proceedings of the National Academy of Sciences. National Acad Sciences; 2014;111:11449–54.
118. Wagle R, Ohtsuka M, Ling H, Pichler M. RNA interference in the clinics: where are we standing now? Future oncology (London, England). 2015;11:185–7.
119. Miele E, Spinelli G. Nanoparticle-based delivery of small interfering RNA: challenges for cancer therapy. International journal …. 2012;3637–57.
120. Burnett J, Rossi J. RNA-based therapeutics: current progress and future prospects. Chemistry & biology. 2012;19:60–71.
121. Zhu L, Mahato R. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert opinion on drug delivery. 2010;7:1209–26.
122. Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nature reviews Drug discovery. 2004;3:318–29.
123. Spagnou S, Miller A, Keller M. Lipidic carriers of siRNA: differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 2004;13348–56.
124. Mével M, Kamaly N, Carmona S, Oliver MH, Jorgensen MR, Crowther C, et al. DODAG; a versatile new cationic lipid that mediates efficient delivery of pDNA and siRNA. Journal of controlled release : official journal of the Controlled Release Society. Elsevier B.V.; 2010;143:222–32.
125. Ewert KK, Zidovska A, Ahmad A, Bouxsein NF, Evans HM, Mcallister CS, et al. Cationic Liposome – Nucleic Acid Complexes for Gene Delivery and Silencing : Pathways and Mechanisms for Plasmid DNA and siRNA. 2010;
126. Xu Y, Szoka F. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry. 1996;2960:5616–23.
127. Mintzer M, Simanek E. Nonviral vectors for gene delivery. Chemical reviews. 2008;259–302.

232
128. Zhang S, Zhao B, Jiang H, Wang B, Ma B. Cationic lipids and polymers mediated vectors for delivery of siRNA. Journal of controlled release : official journal of the Controlled Release Society. 2007;123:1–10.
129. Malone R. Cationic liposome-mediated RNA transfection. Proceedings of the …. 1989;86:6077–81.
130. Bouxsein N, McAllister C, Ewert K. Structure and gene silencing activities of monovalent and pentavalent cationic lipid vectors complexed with siRNA. Biochemistry. 2007;4785–92.
131. Ren T, Song Y, Zhang G, Liu D. Structural basis of DOTMA for its high intravenous transfection activity in mouse. Gene therapy. 2000;764–8.
132. Lu J, Langer R, Chen J. A novel mechanism is involved in cationic lipid-mediated functional siRNA delivery. Molecular pharmaceutics. 2009;6:763–71.
133. Larson SD, Jackson LN, Chen LA, Rychahou PG, Evers BM. Effectiveness of siRNA uptake in target tissues by various delivery methods. Surgery. 2007;142:262–9.
134. Garbuzenko OB, Saad M, Betigeri S, Zhang M, Vetcher A a, Soldatenkov V a, et al. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharmaceutical research. 2009;26:382–94.
135. Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nature biotechnology. 2008;26:561–9.
136. Parton RG, Simons K. The multiple faces of caveolae. Nature reviews Molecular cell biology. 2007;8:185–94.
137. Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nature reviews Molecular cell biology. 2005;6:112–26.
138. Hoekstra D, Rejman J. Gene delivery by cationic lipids: in and out of an endosome. Biochemical Society …. 2007;35:68–71.
139. Reddy J, Low P. Enhanced folate receptor mediated gene therapy using a novel pH-sensitive lipid formulation. Journal of Controlled Release. 2000;64:27–37.

233
140. Champion J, Mitragotri S. Role of target geometry in phagocytosis. … of the United States of America. 2006;2006.
141. Chithrani B, Chan W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano letters. 2007;
142. Iversen T-G, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today. Elsevier Ltd; 2011;6:176–85.
143. Jin H, Heller D, Strano M. Single-particle tracking of endocytosis and exocytosis of single-walled carbon nanotubes in NIH-3T3 cells. Nano letters. 2008;
144. Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nature biotechnology. Nature Publishing Group; 2013;31:653–8.
145. Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338:53–60.
146. Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochemical and biophysical research communications. 2007;353:26–32.
147. Davis J, Clare D, Hodges R, Bloom M. Interaction of a synthetic amphiphilic polypeptide and lipids in a bilayer structure. Biochemistry. 1983;5298–305.
148. Yang Y, Guo X, Lin W, Zhang L. Amphiphilic copolymer brush with random pH-sensitive/hydrophobic structure: synthesis and self-assembled micelles for sustained drug delivery. Soft Matter. 2012;454–64.
149. Sonawane ND, Szoka FC, Verkman a S. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. The Journal of biological chemistry. 2003;278:44826–31.
150. Creusat G, Rinaldi A. Proton sponge trick for pH-sensitive disassembly of polyethylenimine-based siRNA delivery systems. Bioconjugate …. 2010;994–1002.
151. Koynova R. An intracellular lamellar–nonlamellar phase transition rationalizes the superior performance of some cationic lipid transfection agents. Proceedings of the …. 2006;103:14373–8.

234
152. Koltover I, Salditt T, Rädler J, Safinya C. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science. 1998;281:78–81.
153. Wang L, Koynova R, Parikh H, MacDonald RC. Transfection activity of binary mixtures of cationic o-substituted phosphatidylcholine derivatives: the hydrophobic core strongly modulates physical properties and DNA delivery efficacy. Biophysical journal. 2006;91:3692–706.
154. Simberg D, Danino D, Talmon Y, Minsky a, Ferrari ME, Wheeler CJ, et al. Phase behavior, DNA ordering, and size instability of cationic lipoplexes. Relevance to optimal transfection activity. The Journal of biological chemistry. 2001;276:47453–9.
155. Tenchov BG, Wang L, Koynova R, MacDonald RC. Modulation of a membrane lipid lamellar-nonlamellar phase transition by cationic lipids: a measure for transfection efficiency. Biochimica et biophysica acta. 2008;1778:2405–12.
156. Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. Journal of controlled release : official journal of the Controlled Release Society. 2005;107:276–87.
157. Zuhorn IS, Bakowsky U, Polushkin E, Visser WH, Stuart MC a, Engberts JBFN, et al. Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Molecular therapy : the journal of the American Society of Gene Therapy. 2005;11:801–10.
158. Connor J, Huang L. pH-sensitive immunoliposomes as an efficient and target-specific carrier for antitumor drugs. Cancer research. 1986;3431–6.
159. Drummond D, Daleke D. Synthesis and characterization of N-acylated, pH-sensitive “caged”aminophospholipids. Chemistry and physics of lipids. 1995;75:27–41.
160. Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angewandte Chemie (International ed in English). 2012;51:8529–33.
161. Wang X-L, Jensen R, Lu Z-R. A novel environment-sensitive biodegradable polydisulfide with protonatable pendants for nucleic acid delivery. Journal of controlled release : official journal of the Controlled Release Society. 2007;120:250–8.

235
162. Wang X, Ramusovic S. Novel polymerizable surfactants with pH-sensitive amphiphilicity and cell membrane disruption for efficient siRNA delivery. Bioconjugate …. 2007;2169–77.
163. Xu R, Lu Z-R. Design, synthesis and evaluation of spermine-based pH-sensitive amphiphilic gene delivery systems: Multifunctional non-viral gene carriers. Science China Chemistry. 2011;54:359–68.
164. Xu R, Wang X, Lu Z-R. Intracellular siRNA delivery with novel spermine based surfactants. Chinese Science Bulletin. 2012;57:3979–84.
165. Nag OK, Awasthi V. Surface engineering of liposomes for stealth behavior. Pharmaceutics. 2013;5:542–69.
166. Schroeder A, Sigal A, Turjeman K, Barenholz Y. Using PEGylated nano-liposomes to target tissue invaded by a foreign body. Journal of drug targeting. 2008;16:591–5.
167. Kizelsztein P, Ovadia H, Garbuzenko O, Sigal A, Barenholz Y. Pegylated nanoliposomes remote-loaded with the antioxidant tempamine ameliorate experimental autoimmune encephalomyelitis. Journal of neuroimmunology. Elsevier B.V.; 2009;213:20–5.
168. Wang X-L, Xu R, Lu Z-R. A peptide-targeted delivery system with pH-sensitive amphiphilic cell membrane disruption for efficient receptor-mediated siRNA delivery. Journal of controlled release : official journal of the Controlled Release Society. Elsevier B.V.; 2009;134:207–13.
169. Wang X, Xu R, Wu X. Targeted systemic delivery of a therapeutic siRNA with a multifunctional carrier controls tumor proliferation in mice. Molecular Pharmaceutics. 2009;86:738–46.
170. Gillespie D, Aguirre M. RNA interference targeting hypoxia-inducible factor 1α via a novel multifunctional surfactant attenuates glioma growth in an intracranial mouse model. Journal of …. 2014;122:331–41.
171. Xu R, Wang X-L, Lu Z-R. New amphiphilic carriers forming pH-sensitive nanoparticles for nucleic acid delivery. Langmuir : the ACS journal of surfaces and colloids. 2010;26:13874–82.
172. Malamas AS, Gujrati M, Kummitha CM, Xu R, Lu Z-R. Design and evaluation of new pH-sensitive amphiphilic cationic lipids for siRNA delivery. Journal of Controlled Release. Elsevier B.V.; 2013;171:296–307.

236
173. Kurreck J. RNA interference: from basic research to therapeutic applications. Angewandte Chemie International Edition. Wiley Online Library; 2009;48:1378–98.
174. Hannon G. RNA interference. Nature. 2002;418:24–6.
175. Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double-stranded RNA in vitro. Genes & development. Cold Spring Harbor Lab; 1999;13:3191–7.
176. Sinn PL, Sauter SL, McCray PB. Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors–design, biosafety, and production. Gene therapy. Nature Publishing Group; 2005;12:1089–98.
177. Shegokar R, Al Shaal L, Mishra PR. SiRNA delivery: challenges and role of carrier systems. Die Pharmazie-An International Journal of Pharmaceutical Sciences. Govi-Verlag; 2011;66:313–8.
178. Shim MS, Kwon YJ. Efficient and targeted delivery of siRNA in vivo. FEBS journal. Wiley Online Library; 2010;277:4814–27.
179. Morris K V, Rossi JJ. Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene therapy. Nature Publishing Group; 2006;13:553–8.
180. Devroe E, Silver PA. Therapeutic potential of retroviral RNAi vectors. Expert opinion on biological therapy. Ashley Publications Ltd London, UK; 2004;4:319–27.
181. Somia N, Verma IM. Gene therapy: trials and tribulations. Nature Reviews Genetics. Nature Publishing Group; 2000;1:91–9.
182. Fenske DB, Chonn A, Cullis PR. Liposomal nanomedicines: an emerging field. Toxicologic pathology. Sage Publications; 2008;36:21–9.
183. Wang Y, Li Z, Han Y, Hwa Liang L, Ji A. Nanoparticle-based delivery system for application of siRNA in vivo. Current drug metabolism. Bentham Science Publishers; 2010;11:182–96.
184. Yuan X, Naguib S, Wu Z. Recent advances of siRNA delivery by nanoparticles. Expert opinion on drug delivery. Informa UK, Ltd. London; 2011;8:521–36.
185. Li S, Huang L. Nonviral gene therapy: promises and challenges. Gene therapy. Citeseer; 2000;7:31–4.

237
186. Jones LJ, Yue ST, Cheung C-Y, Singer VL. RNA quantitation by fluorescence-based solution assay: RiboGreen reagent characterization. Analytical biochemistry. Elsevier; 1998;265:368–74.
187. Sanvicens N, Marco MP. Multifunctional nanoparticles–properties and prospects for their use in human medicine. Trends in biotechnology. Elsevier; 2008;26:425–33.
188. Liu X, Howard KA, Dong M, Andersen MØ, Rahbek UL, Johnsen MG, et al. The influence of polymeric properties on chitosan/siRNA nanoparticle formulation and gene silencing. Biomaterials. Elsevier; 2007;28:1280–8.
189. Kong W-H, Sung D-K, Shim Y-H, Bae KH, Dubois P, Park TG, et al. Efficient intracellular siRNA delivery strategy through rapid and simple two steps mixing involving noncovalent post-PEGylation. Journal of Controlled Release. Elsevier; 2009;138:141–7.
190. Wu Y, Wang W, Chen Y, Huang K, Shuai X, Chen Q, et al. The investigation of polymer-siRNA nanoparticle for gene therapy of gastric cancer in vitro. International journal of nanomedicine. Dove Press; 2010;5:129.
191. Bettinger T, Carlisle RC, Read ML, Ogris M, Seymour LW. Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic acids research. Oxford Univ Press; 2001;29:3882–91.
192. Ruponen M, Rönkkö S, Honkakoski P, Pelkonen J, Tammi M, Urtti A. Extracellular glycosaminoglycans modify cellular trafficking of lipoplexes and polyplexes. Journal of Biological Chemistry. ASBMB; 2001;276:33875–80.
193. Wei J, Jones J, Kang J, Card A, Krimm M, Hancock P, et al. RNA-induced silencing complex-bound small interfering RNA is a determinant of RNA interference-mediated gene silencing in mice. Molecular pharmacology. ASPET; 2011;79:953–63.
194. Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. Nature Publishing Group; 2006;441:537–41.
195. Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. small. Wiley Online Library; 2008;4:26–49.

238
196. Katas H, Alpar HO. Development and characterisation of chitosan nanoparticles for siRNA delivery. Journal of controlled release. Elsevier; 2006;115:216–25.
197. Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Baldelli Bombelli F, et al. Physical− chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. Journal of the American Chemical Society. ACS Publications; 2011;133:2525–34.
198. Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proceedings of the National Academy of Sciences. National Acad Sciences; 2008;105:14265–70.
199. Lesniak A, Salvati A, Santos-Martinez MJ, Radomski MW, Dawson KA, Åberg C. Nanoparticle adhesion to the cell membrane and its effect on nanoparticle uptake efficiency. Journal of the American Chemical Society. ACS Publications; 2013;135:1438–44.
200. Wilhelm C, Gazeau F, Roger J, Pons JN, Bacri J-C. Interaction of anionic superparamagnetic nanoparticles with cells: kinetic analyses of membrane adsorption and subsequent internalization. Langmuir. ACS Publications; 2002;18:8148–55.
201. Kwon YJ. Before and after endosomal escape: roles of stimuli-converting siRNA/polymer interactions in determining gene silencing efficiency. Accounts of chemical research. ACS Publications; 2011;45:1077–88.
202. Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nature biotechnology. Nature Publishing Group; 2010;28:172–6.
203. Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. Elsevier Ltd; 2011;32:3435–46.
204. Won Y-W, Yoon S-M, Lee K-M, Kim Y-H. Poly (oligo-D-arginine) with internal disulfide linkages as a cytoplasm-sensitive carrier for siRNA delivery. Molecular Therapy. Nature Publishing Group; 2011;19:372–80.
205. Manickam DS, Li J, Putt DA, Zhou Q-H, Wu C, Lash LH, et al. Effect of innate glutathione levels on activity of redox-responsive gene delivery vectors. Journal of Controlled Release. Elsevier; 2010;141:77–84.

239
206. Drew R, Miners JO. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochemical pharmacology. Elsevier; 1984;33:2989–94.
207. Namgung R, Kim WJ. A Highly Entangled Polymeric Nanoconstruct
Assembled by siRNA and its Reduction‐Triggered siRNA Release for Gene Silencing. Small. Wiley Online Library; 2012;8:3209–19.
208. Siegel R, DeSantis C, Virgo K. Cancer treatment and survivorship statistics, 2012. A Cancer Journal for Clinicians. 2012;62:220–41.
209. Metzger-Filho O, Tutt A, De Azambuja E, Saini KS, Viale G, Loi S, et al. Dissecting the heterogeneity of triple-negative breast cancer. Journal of clinical oncology. American Society of Clinical Oncology; 2012;30:1879–87.
210. Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D’Incalci M, et al. Targeting triple negative breast cancer: is p53 the answer? Cancer treatment reviews. Elsevier; 2013;39:541–50.
211. Duffy MJ, McGowan PM, Crown J. Targeted therapy for triple‐negative breast cancer: Where are we? International Journal of Cancer. Wiley Online Library; 2012;131:2471–7.
212. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. Journal of mammary gland biology and neoplasia. Springer; 2010;15:117–34.
213. Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. The Journal of clinical investigation. American Society for Clinical Investigation; 2009;119:1417.
214. Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. Journal of mammary gland biology and neoplasia. Springer; 2010;15:253–60.
215. Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. Journal of mammary gland biology and neoplasia. Springer; 2010;15:169–90.
216. Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006;8:R42.

240
217. Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer research. AACR; 2007;67:3752–8.
218. Galliher-Beckley AJ, Schiemann WP. Grb2 binding to Tyr284 in TβR-II is essential for mammary tumor growth and metastasis stimulated by TGF-β. Carcinogenesis. Oxford Univ Press; 2008;29:244–51.
219. Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Molecular biology of the cell. 2013;24:3449–59.
220. Whitehead KA, Dahlman JE, Langer RS, Anderson DG. Silencing or stimulation? siRNA delivery and the immune system. Annual review of chemical and biomolecular engineering. Annual Reviews; 2011;2:77–96.
221. Gujrati M, Malamas A, Shin T, Jin E, Lu Z-R. Multifunctional cationic lipid-based nanoparticles facilitate endosomal escape and reduction-triggered cytosolic siRNA release. Molecular Pharmaceutics. 2014;11:2734–44.
222. Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11:R68.
223. Taylor MA, Davuluri G, Parvani JG, Schiemann BJ, Wendt MK, Plow EF, et al. Upregulated WAVE3 expression is essential for TGF-β-mediated EMT and metastasis of triple-negative breast cancer cells. Breast cancer research and treatment. Springer; 2013;142:341–53.
224. Balanis N, Yoshigi M, Wendt MK, Schiemann WP, Carlin CR. β3 Integrin–EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Molecular biology of the cell. Am Soc Cell Biol; 2011;22:4288–301.
225. Shastry M, Yardley DA. Updates in the treatment of basal/triple-negative breast cancer. Current Opinion in Obstetrics and Gynecology. LWW; 2013;25:40–8.
226. Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial–mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. Nature Publishing Group; 2010;29:6485–98.

241
227. Wendt MK, Taylor MA, Schiemann BJ, Sossey-Alaoui K, Schiemann WP. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor-β1 signaling in metastatic breast cancers. Breast Cancer Res. 2014;16:R24.
228. Taylor MA, Amin JD, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-β signaling in breast cancer cells. Neoplasia. Elsevier; 2011;13:406–IN2.
229. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer cell. Elsevier; 2005;8:241–54.
230. Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Molecular biology of the cell. Am Soc Cell Biol; 2011;22:2423–35.
231. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. American Society for Clinical Investigation; 2011;121:2750.
232. Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-β signaling during mammary tumorigenesis. Journal of mammary gland biology and neoplasia. Springer; 2011;16:127–46.
233. Wendt MK, Smith JA, Schiemann WP. p130Cas is required for mammary tumor growth and transforming growth factor-β-mediated metastasis through regulation of Smad2/3 activity. Journal of Biological Chemistry. ASBMB; 2009;284:34145–56.
234. Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nature Reviews Cancer. Nature Publishing Group; 2009;9:108–22.
235. Florence AT, Hussain N. Transcytosis of nanoparticle and dendrimer delivery systems: evolving vistas. Advanced drug delivery reviews. Elsevier; 2001;50:S69–S89.
236. Oh Y-K, Park TG. siRNA delivery systems for cancer treatment. Advanced drug delivery reviews. Elsevier; 2009;61:850–62.
237. Gasparini G, Brooks PC, Biganzoli E, Vermeulen PB, Bonoldi E, Dirix LY, et al. Vascular integrin alpha (v) beta3: a new prognostic indicator in breast cancer. Clinical Cancer Research. AACR; 1998;4:2625–34.

242
238. Polyak K. Heterogeneity in breast cancer. The Journal of clinical investigation. American Society for Clinical Investigation; 2011;121:3786.
239. Balanis N, Wendt MK, Schiemann BJ, Wang Z, Schiemann WP, Carlin CR. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. Journal of Biological Chemistry. ASBMB; 2013;288:17954–67.
240. Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Molecular biology of the cell. Am Soc Cell Biol; 2013;24:3449–59.
241. Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. British Journal of Cancer. Nature Publishing Group; 2004;90:561–5.
242. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic growth from dormant cells induced by a col-I–enriched fibrotic environment. Cancer research. AACR; 2010;70:5706–16.
243. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer research. AACR; 2008;68:6241–50.
244. Malamas AS, Gujrati M, Kummitha CM, Xu R, Lu Z-R. Design and evaluation of new pH-sensitive amphiphilic cationic lipids for siRNA delivery. Journal of controlled release : official journal of the Controlled Release Society. Elsevier B.V.; 2013;171:296–307.
245. Tseng Y-C, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Advanced Drug Delivery Reviews. Elsevier B.V.; 2009;61:721–31.
246. Li S-D, Huang L. Gene therapy progress and prospects: non-viral gene therapy by systemic delivery. Gene therapy. 2006;13:1313–9.
247. Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Advanced Drug Delivery Reviews. 2003;55:403–19.
248. Jokerst J, Lobovkina T, Zare R, Gambhir S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine. 2011;6:715–28.

243
249. Peracchia M, Fattal E, Desmaele D. Stealth® PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. Journal of Controlled Release. 1999;60:121–8.
250. Van Vlerken LE, Vyas TK, Amiji MM. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharmaceutical research. 2007;24:1405–14.
251. Li S, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. Journal of Controlled Release. 2010;145:178–81.
252. Hatakeyama H, Akita H, Harashima H. The Polyethyleneglycol Dilemma : Advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors. Biological and Pharmaceutical Bulletin. 2013;36:892–9.
253. Liu T, Thierry B. A solution to the PEG dilemma: efficient bioconjugation of large gold nanoparticles for biodiagnostic applications using mixed layers. Langmuir. 2012;28:15634–42.
254. Stuart M a C, Huck WTS, Genzer J, Müller M, Ober C, Stamm M, et al. Emerging applications of stimuli-responsive polymer materials. Nature materials. Nature Publishing Group; 2010;9:101–13.
255. Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. European journal of pharmaceutics and biopharmaceutics. Elsevier B.V.; 2009;71:431–44.
256. Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. Journal of controlled release. 2008;126:187–204.
257. Rapoport N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Progress in Polymer Science. 2007;32:962–90.
258. Petros R a, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nature reviews Drug discovery. Nature Publishing Group; 2010;9:615–27.
259. Rejman J, Wagenaar A, Engberts JBF., Hoekstra D. Characterization and transfection properties of lipoplexes stabilized with novel exchangeable polyethylene glycol–lipid conjugates. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2004;1660:41–52.

244
260. Choi JS, Mackay JA, Szoka FC. Low-pH-Sensitive PEG-Stabilized Plasmid-Lipid Nanoparticles : Preparation and Characterization. 2003;420–9.
261. Soppimath KS, Tan DC-W, Yang Y-Y. pH-Triggered Thermally Responsive Polymer Core-Shell Nanoparticles for Drug Delivery. Advanced Materials. 2005;17:318–23.
262. Zhang J. Pharmaco attributes of dioleoylphosphatidylethanolamine/cholesterylhemisuccinate liposomes containing different types of cleavable lipopolymers. Pharmacological Research. 2004;49:185–98.
263. Dong W, Kishimura A, Anraku Y, Chuanoi S, Kataoka K. Monodispersed Polymeric Nanocapsules : Spontaneous Evolution and Morphology Transition from Reducible Hetero-PEG PICmicelles by Controlled Degradation. 2009;33:3804–5.
264. Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Therapy. 2006;14:68–77.
265. Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y, et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials. Elsevier Ltd; 2011;32:4306–16.
266. Tomlinson R, Heller J, Brocchini S, Duncan R. Polyacetal-Doxorubicin Conjugates Designed for pH-Dependent Degradation. 2003;1096–106.
267. Shin J, Shum P, Thompson DH. A cid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG – lipids. 2003;91:187–200.
268. Kirpotin D, Hong K, Mullah N, Papahadjopoulos D, Zalipsky S. Liposomes with detachable polymer coating " destabilization and fusion of dioleoylphosphatidylethanolamine vesicles triggered by cleavage of surface-grafted poly ( ethylene glycol ). 1996;388:115–8.
269. Martin G, Jain R. Fluorescence ratio imaging measurement of pH gradients: calibration and application in normal and tumor tissues. Microvascular research. 1993;
270. Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. Journal of controlled

245
release : official journal of the Controlled Release Society. 2005;103:405–18.
271. Walker GF, Fella C, Pelisek J, Fahrmeir J, Boeckle S, Ogris M, et al. Toward synthetic viruses: endosomal pH-triggered deshielding of targeted polyplexes greatly enhances gene transfer in vitro and in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2005;11:418–25.
272. Kale A, Torchilin V. Design, synthesis, and characterization of pH-sensitive PEG-PE conjugates for stimuli-sensitive pharmaceutical nanocarriers: the effect of substitutes at the hydrazone linkage. Bioconjugate chemistry. 2007;18:363–70.
273. Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. Journal of controlled release. Elsevier B.V.; 2012;160:264–73.
274. Christie R, Anderson D, Grainger D. Comparison of hydrazone heterobifunctional cross-linking agents for reversible conjugation of thiol-containing chemistry. Bioconjugate chemistry. 2010;21:1779–87.
275. Prabaharan M, Grailer JJ, Pilla S, Steeber D a, Gong S. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials. Elsevier Ltd; 2009;30:5757–66.
276. Yoo H, Lee E, Park T. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. Journal of Controlled Release. 2002;82:17–27.
277. Haubner R, Wester H. Glycosylated RGD-containing peptides: tracer for tumor targeting and angiogenesis imaging with improved biokinetics. Journal of Nuclear Medicine. 2001;42:326–37.
278. Wilhelm C, Billotey C, Roger J, Pons J. Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials. 2003;24:1001–11.
279. Nam HY, Kwon SM, Chung H, Lee S-Y, Kwon S-H, Jeon H, et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. Journal of Controlled Release. Elsevier B.V.; 2009;135:259–67.

246
280. Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. 2008;105.
281. Olbrich C, Bakowsky U, Lehr C, Muller RH, Kneuer C. Cationic solid-lipid nanoparticles can efficiently bind and transfect plasmid DNA. Journal of Controlled Release. 2001;77:345–55.
282. Moulder, S, Hortobagyi G. Advances in the treatment of breast cancer. Clinical pharmacology and therapeutics. 2008;83:26–36.
283. Berry D, Cronin K, Plevritis S, Fryback D, Clarke L, Zelen M, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. The New England Journal of Medicine. 2005;353:1784–92.
284. Trends C, Report P. Image description. Cancer Trends Progress Report - 2007 Update End of image description. 2007;
285. Osbourne C. Steroid hormone receptors in breast cancer management. Breast Cancer Research Treatment. 1998;
286. Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26:3637–43.
287. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. A Cancer Journal for Clinicians. 2012;62:10–29.
288. Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clinical Cancer Research. 2010;16:4914–20.
289. Wendel H, Silva R, Malina A, Mills J, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes and Development. 2007;21:3232–7.
290. Konicek BW, Dumstorf CA, Graff JR. Targeting the eIF4F translation initiation complex for cancer therapy. Cell Cycle. 2014;7:2466–71.
291. Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nature Reviews. Nature Publishing Group; 2010;10:254–66.
292. Hiller DJ, Chu Q, Meschonat C, Panu L, Burton G, Li BDL. Predictive value of eIF4E reduction after neoadjuvant therapy in breast cancer. The Journal of surgical research. Elsevier Ltd; 2009;156:265–9.
293. McClusky DR, Chu Q, Yu H, DeBenedetti A, Johnson LW, Meschonat C, et al. A Prospective Trial on Initiation Factor 4E (eIF4E) Overexpression and

247
Cancer Recurrence in Node-Positive Breast Cancer. Annals of Surgery. 2005;242:584–92.
294. Byrnes KW, DeBenedetti A, Holm NT, Luke J, Nunez J, Chu QD, et al. Correlation of TLK1B in Elevation and Recurrence in Doxorubicin-Treated Breast Cancer Patients with High eIF4E Overexpression. Journal of the American College of Surgeons. 2007;204:925–33.
295. Holm N, Byrnes K, Johnson L, Abreo F, Sehon K, Alley J, et al. A Prospective Trial on Initiation Factor 4E (eIF4E) Overexpression and Cancer Recurrence in Node-Negative Breast Cancer. Annals of Surgical Oncology. 2008;15:3207–15.
296. Li B, Gruner J, Abreo F, Johnson L, Yu H, Nawas S, et al. Prospective Study of Eukaryotic Initiation Factor 4E Protein Elevation and Breast Cancer Outcome. Annals of Surgery. 2002;235:732–9.
297. Flowers A, Chu QD, Panu L, Meschonat C, Caldito G, Lowery-Nordberg M, et al. Eukaryotic initiation factor 4E overexpression in triple-negative breast cancer predicts a worse outcome. Surgery. Mosby, Inc.; 2009;146:220–6.
298. Li B, McDonald J, Nassar R, De Benedetti A. Clinical outcome in stage I to III breast carcinoma and eIF4E overexpression. Annals of surgery. 1998;227:756–61.
299. Zhou F, Yan M, Guo G, Wang F, Qiu H, Zheng F, et al. Knockdown of eIF4E suppresses cell growth and migration, enhances chemosensitivity and correlates with increase in Bax/Bcl-2 ratio in triple-negative breast cancer cells. Medical oncology (Northwood, London, England). 2011;28:1302–7.
300. Dong K, Wang R, Wang X, Lin F, Shen J-J, Gao P, et al. Tumor-specific RNAi targeting eIF4E suppresses tumor growth, induces apoptosis and enhances cisplatin cytotoxicity in human breast carcinoma cells. Breast Cancer Research and Treatment. 2008;113:443–56.
301. Oridate N, Kim H-J, Xu X, Lotan R. Growth inhibition of head and neck squamous carcinoma cells by small interfering RNAs targeting eIF4E or cyclin D1 alone or combined with cisplatin. Cancer Biology & Therapy. 2014;4:318–23.
302. Graff J, Konicek B. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. The Journal of …. 2007;117.

248
303. DeFatta R, Nathan C, De Benedetti A. Antisense RNA to eIF4E suppresses oncogenic properties of a head and neck squamous cell carcinoma cell line. The Laryngoscope. 2000;110:928–33.
304. Siegele B, Cefalu C, Holm N, Sun G, Tubbs J, Meschonat C, et al. eIF4E-targeted suicide gene therapy in a minimal residual mouse model for metastatic soft-tissue head and neck squamous cell carcinoma improves disease-free survival. The Journal of surgical research. 2008;148:83–9.
305. Culjkovic B, Borden KL. Understanding and Targeting the Eukaryotic Translation Initiation Factor eIF4E in Head and Neck Cancer. Journal of Oncology. 2009;2009:1–12.
306. Kentsis A, Topisirovic I. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proceedings of the National Academy of Science. 2004;101:18105–10.
307. Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67.
308. Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D, et al. eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling. Molecular cancer therapeutics. 2008;7:1782–8.
309. Hong DS, Kurzrock R, Oh Y, Wheler J, Naing A, Brail L, et al. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clinical Cancer Research. 2011;17:6582–91.
310. Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 through a Cdk1-dependent mechanism. Oncogene. 2005;24:4851–60.
311. Li F, Ambrosini G, Chu E, Plescia J. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:1–5.
312. Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. The Journal of biological chemistry. 2004;279:15196–203.
313. Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4914–20.

249
314. Kundu AK, Chandra PK, Hazari S, Ledet G, Pramar Y V, Dash S, et al. Stability of lyophilized siRNA nanosome formulations. International journal of pharmaceutics. Elsevier B.V.; 2012;423:525–34.
315. Schroeder A, Heller D a, Winslow MM, Dahlman JE, Pratt GW, Langer R, et al. Treating metastatic cancer with nanotechnology. Nature reviews Cancer. Nature Publishing Group; 2012;12:39–50.
316. Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nature materials. Nature Publishing Group; 2013;12:958–62.
317. Cheng Z, Zaki A Al, Hui J, Muzykantov V, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–10.
318. Hernandez FJ, Stockdale KR, Huang L, Horswill AR, Behlke M a, McNamara JO. Degradation of nuclease-stabilized RNA oligonucleotides in Mycoplasma-contaminated cell culture media. Nucleic acid therapeutics. 2012;22:58–68.
319. Yuan Y-Y, Mao C-Q, Du X-J, Du J-Z, Wang F, Wang J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Advanced materials (Deerfield Beach, Fla). 2012;24:5476–80.
320. Du J, Du X, Mao C, Wang J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. Journal of the American …. 2011;17560–3.
Top Related
Copyright © 2022 FDOKUMEN

