
Ziegler-Natta catalysts for olefin polymerizations
68
Pergamon Prog. Polym. Sci., Vol. 20, 459-526, 1995 Copyright 0 1995 Elsevier Science Ltd Printed in Great Britain. All rights reserved 0079-6700/95 $29.00 0079-6700(94)00039-5 ZIEGLER-NATTA CATALYSTS FOR OLEFIN POLYMERIZATION: MECHANISTIC INSIGHTS FROM METALLOCENE SYSTEMS J. HUANG and G. L. REMPEL* Department of Chemical Engimeering, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada Abstract - The current development of the metallocene-based Ziegler-Natta catalysts has been reviewed. The discovery of these catalysts has offered the opportunity to obtain a deeper insight into the mechanism of Ziegler-Natta polymerizations. In this review, some mechanistic models for polymerization and stereoregulation, as well as the factors which affect the activity and stereo- specificity of the catalysts, have been discussed. The technology of olefin polymerization with the metallocene-based catalysts is in the early stage of commercialization. Using these catalysts, a large number of novel polymers with special properties have been obtained. Keywords - Metallocene, methylalumoxane, Ziegler-Natta, olefin polymerization, homogeneous catalysts, polyolefins, stereospecificity, mechanism, kinetics, zirconocenes. CONTENTS 1. Introduction 2. The Ziegler-Natta catalyst 2.1. The composition of Ziegler-Natta catalysts 2.2. Stereospecificity 2.2.1. Steric isomerism and tacticity 2.2.2. Stereochemical control by Ziegler-Natta catalysts 2.3. The mechanism of Ziegler-Natta polymerization 2.3.1. The Cossee mechanism 2.3.2. The trigger mechanism 2.3.3. Chain termination 2.4. The mechanism of stereoregulation in a-olefin polymerizations 2.4.1. Catalytic site control 2.4.2. Chain end control 2.5. Kinetics 2.6. The success of MgCl*-supported heterogeneous systems 2.7. Difficulties with theoretical studies 3. The history of the homogeneous Ziegler-Natta catalysts 4. The composition of homogeneous Ziegler-Natta catalysts 4.1. The metallocenes 4.2. The methylalumoxanes (MAO) 4.3. Cationic metallocene alkyl catalyst systems 5. Polymerization mechanism 5.1. The active center *Author to whom correspondence should be addressed. 461 461 461 462 462 462 463 463 464 466 466 467 467 467 469 469 469 471 471 472 474 475 475 459
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Ziegler-Natta catalysts for olefin polymerizations

Pergamon
Prog. Polym. Sci., Vol. 20, 459-526, 1995 Copyright 0 1995 Elsevier Science Ltd
Printed in Great Britain. All rights reserved 0079-6700/95 $29.00
0079-6700(94)00039-5
ZIEGLER-NATTA CATALYSTS FOR OLEFIN POLYMERIZATION: MECHANISTIC INSIGHTS
FROM METALLOCENE SYSTEMS
J. HUANG and G. L. REMPEL*
Department of Chemical Engimeering, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada
Abstract - The current development of the metallocene-based Ziegler-Natta catalysts has been reviewed. The discovery of these catalysts has offered the opportunity to obtain a deeper insight into the mechanism of Ziegler-Natta polymerizations. In this review, some mechanistic models for polymerization and stereoregulation, as well as the factors which affect the activity and stereo- specificity of the catalysts, have been discussed. The technology of olefin polymerization with the metallocene-based catalysts is in the early stage of commercialization. Using these catalysts, a large number of novel polymers with special properties have been obtained.
Keywords - Metallocene, methylalumoxane, Ziegler-Natta, olefin polymerization, homogeneous catalysts, polyolefins, stereospecificity, mechanism, kinetics, zirconocenes.
CONTENTS
1. Introduction 2. The Ziegler-Natta catalyst
2.1. The composition of Ziegler-Natta catalysts 2.2. Stereospecificity
2.2.1. Steric isomerism and tacticity 2.2.2. Stereochemical control by Ziegler-Natta catalysts
2.3. The mechanism of Ziegler-Natta polymerization 2.3.1. The Cossee mechanism 2.3.2. The trigger mechanism 2.3.3. Chain termination
2.4. The mechanism of stereoregulation in a-olefin polymerizations 2.4.1. Catalytic site control 2.4.2. Chain end control
2.5. Kinetics 2.6. The success of MgCl*-supported heterogeneous systems 2.7. Difficulties with theoretical studies
3. The history of the homogeneous Ziegler-Natta catalysts 4. The composition of homogeneous Ziegler-Natta catalysts
4.1. The metallocenes 4.2. The methylalumoxanes (MAO) 4.3. Cationic metallocene alkyl catalyst systems
5. Polymerization mechanism 5.1. The active center
*Author to whom correspondence should be addressed.
461 461 461 462 462 462 463 463 464 466 466 467 467 467 469 469 469 471 471 472 474 475 475
459

460 J. HUANG and G. L. REMPEL
5.2. The formation of the active center in metallocene/MAO systems 5.3. Polymerization mechanisms
53.1. Kaminsky’s model 5.3.2. Corradini’s model 5.3.3. Other considerations about the polymerization mechanism
5.4. The dependence of catalyst activity on the nature of the metallocene and metallocene-monomer interactions 5.4.1. Transition metal center 5.4.2. Cp ligands 5.4.3. Monomers
5.5. Functions of MAO 6. The mechanism of stereoregulation
6.1. Versatile stereoselectivity 6.1.1. Polymerization of ethylene 6.1.2. Polymerization of propylene and higher cr-olefins 6.1.3. Polymerization of cyclic olefins 6.1.4. Enantioselective cyclopolymerization of dienes
6.2. The mechanism of stereoregulation 6.2.1. Pino’s model 6.2.2. Corradini’s model 6.2.3. Brintzinger’s model
6.3. Other effects on the stereospecificity of the catalysts 6.3.1. Polymerization temperature 6.3.2. MAO concentration
7. Kinetics 7.1. The active species 7.2. Kinetic models
7.2.1. Ewen’s model 7.2.2. Chien’s model and parameter estimations 7.2.3. Hamielec’s model and computer modelling
7.3. The operational factors affecting catalyst activity 7 3.1. Polymerization temperature ( Tr) 7.3.2. The MAO concentration 7.3.3. Catalyst concentration 7.3.4. Solvents 7.3.5. Comonomer effect 7.3.6. Lewis acid and Lewis base catalyst modifiers
7.4. Catalyst deactivation 7.5. Molecular weight and molecular weight distribution
7.5.1. Chain transfer reactions 7.5.2. The molecular weight (MW) 7.5.3. Methods to increase the molecular weight 7.5.4. The molecular weight distribution (MWD)
8. Characterization of polymers 8.1. 13C-NMR studies
8.1.1. Steric defects in isotactic PP 8.1.2. Computer interpretation and simulation 8.1.3. Regioregularity and end-group analyses
8.2. The properties of the polymer 8.2.1. Anisotactic polypropylenes 8.2.2. Syndiotactic polypropylene (s-PP) 8.2.3. Other polymers
475 478 479 479 480
480 481 482 482 483 483 483 483 483 486 486 487 487 487 490 492 492 493 493 493 494 494 495 497 499 499 501 501 502 502 502 503 504 504 507 509 510 511 511 512 513 515 516 516 518 518

ZIEGLER-NATTA CATALYSTS 461
9. Theoretical significance and potential applications 519 9.1. Theoretical contributions - -
9.2. 9.3.
9.1.1. The origin of stereospecifrcity 9.1.2. The monometallic model 9.1.3. Migratory insertion 9.1.4. Cationic nature of the active center 9.1.5. Multiple active species in heterogeneous systems Benefit to kinetic studies Potential applications
9.4.
9.3.1. “Single-site” catalyst (SSC) technology 9.3.2. Difliculties hindering applications 9.3.3. The potential role in production of hybrid thermoplastic olefins Other homogeneous catalyst systems 9.4.1. CG catalyst 9.4.2. Other non-metallocene catalysts
10. Conclusions References
519 520 520 520 521 521 521 522 522 523 523 523 523 524 524 525
1. INTRODUCTION
In 1963 Karl Ziegler and Giulio Natta were awarded the Nobel Prize for Chemistry for their landmark discoveries of the polymerization catalysts named after them. The first Ziegler-Natta catalysts were discovered four decades ago. In 1953 Ziegler revealed that high density polyethylene was easily made at low pressure with binary mixture of metal alkyls and transition metal salts, and in the next year Natta demonstrated the ability of the same type of catalysts to form isotactic polymers from a-olefins. The discoveries changed polymer chemistry for ever, and provoked a worldwide research and develop- ment effort that culminated in many new commercial plastics and elastomers. The Ziegler-Natta catalyst has now joined the ranks of conventional cationic, anionic and radical initiators as one of the major methods available to initiate polymerizations, and it is doubtful that it can be challenged by any other catalyst for its versatility. Ziegler-Natta catalysts became prominent in a special period in the history of polymer science, a period that not only produced many new commercial polymers but also enhanced our basic knowledge of polymer properties and structure as well as polymerization processes.’
2. THE ZIEGLER-NATTA CATALYST
2.1. The Composition of Ziegler-Natta Catalysts
Generally speaking, the Ziegler-Natta catalyst is a complex formed by reaction of a transition metal compound (halide, or alkoxide, or alkyl or aryl derivative) of group IV- VIII transition metals with a metal alkyl or alkyl halide of Group I-III base metals.2 The former component is usually called the catalyst and the latter the cocatalyst. There are a very large number of patents involving every combination of pure or mixed metal alkyls with transition metal compounds, each claiming particular advantages. In practice, only a few group I-III metal alkyls are effective. Aluminum alkyls (such as AlEts, Al-i-Bq, AlEt2C1, AlEtCl, and AlEt20R) have been overwhelmingly preferred.3 Also, transition metal compounds containing titanium (Ti), vanadium (V), chromium (Cr) and, in special

462 J. HUANG and G. L. REMPEL
cases, molybdenum (MO), cobalt (Co), rhodium (Rh) and nickel (Ni) are primarily used.
Not long after Ziegler-Natta catalysts were discovered, it was found that electron donors could greatly affect the catalyst’s kinetic and stereochemical behavior. Electron donor compounds, such as amines, ethers and esters, have the potential of complexing and reacting with the components of the catalyst or the active centers. They have been used in controlled amounts in many Ziegler-Natta catalytic systems as a third component to increase catalyst activity and/or stereoselectivity.’
2.2. Stereospecifuity
Stereochemical control is one of the most important attributes of the Ziegler-Natta catalyst.
2.2.1. Steric Isomerism and Tacticity
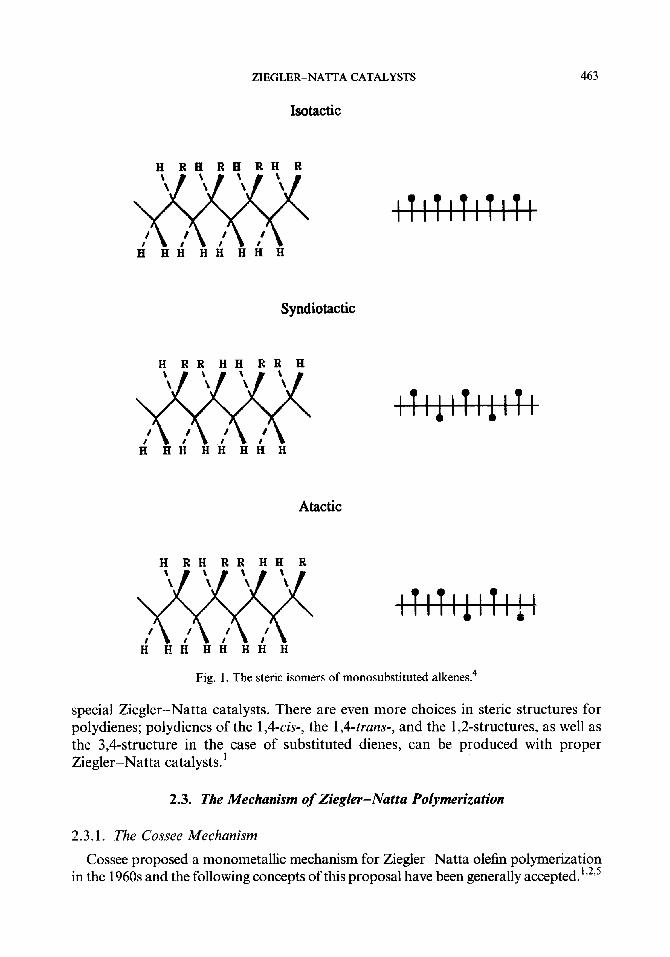
Steric isomerism is observed in the polymerization of alkenes whenever one of the carbon atoms of the double bond is at least monosubstituted. The polymeriza- tion of a monosubstituted ethylene, CH2=CHR (where R is any substituent group), leads to polymers in which every other carbon atom in the polymer chain is a pseudochiral center. Each pseudochiral center is a site of steric isomerism in the polymerization of CHz=CHR. Considering the main carbon-carbon chain of the polymer -(CH,-CHR ),- to be stretched out in its fully extended planar zigzag conformation, two different configurations are possible for each pseudochiral carbon since the R group may be situated on either side of the plane.4
The regularity in the configuration of successive pseudochiral centers determines the overall order of tacticity of the polymer chain. If the R groups on successive pseudo- chiral carbons are randomly distributed on the two sides of the planar zigzag polymer main chain, the polymer is termed atactic. An isotactic polymer structure occurs when the pseudochiral center in each repeating unit in the polymer chain has the same configuration, In this case, all the R groups will be located on one side of the plane of the carbon-carbon polymer chain. A syndiotactic polymer structure occurs when the configuration of the pseudochiral centers alternate from one repeating unit to the next with the R groups located alternately on the opposite sides of the polymer chain plane4 (see Fig. 1). For polymerizations of 1,2-disubstituted ethylenes and dienes, steric isomerism is quite complicated.
2.2.2. Stereochemical Control by Ziegler-Natta Catalysts
Ziegler-Natta catalysis provided for the first time stereochemical control of the polymerization process. By carefully selecting the combination of catalyst and cocatalyst, one is able to produce polymers with the desired steric structure.
The polyethylene produced in Ziegler-Natta polymerization is linear, which is characterized by the absence of long or short chain branching. For a-olefin polymer- ization, polyolefins of isotactic or syndiotactic structure can be obtained by using
ZIEGLER-NATTA CATALYSTS 463
ltltltltltl
Syndiotactic
H RR HH RR H
ITIIITIIITI
Atactic
ITITI I ITI I I “5’
Fig. 1. The steric isomers of monosubstituted alkenes.4
special Ziegler-Natta catalysts. There are even more choices in steric structures for polydienes; polydienes of the 1,4-c&, the 1,4-trans-, and the 1,2-structures, as well as the 3,4-structure in the case of substituted dienes, can be produced with proper Ziegler-Natta catalysts.’
2.3. The Mechanism of Ziegler-Natta Polymerization
2.3.1. The Cossee Mechanism
Cossee proposed a monometallic mechanism for Ziegler-Natta olefin polymerization in the 1960s and the following concepts of this proposal have been generally accepted.’ 1215

J. HUANG and G. L. REMPEL
Me
[7 = vacant coordination site @ = polymer chain
Fig. 2. Cossee mechanism for Ziegler-Natta olefin polymerization.6
2.3.1.1. The Active Center - The active center in Ziegler-Natta catalysts is the transition metal-carbon bond of the transition metal complex, which is formed by the interaction between two components of the catalytic system. The active complex has to contain at least one MT--C bond or MT--H bond (MT: transition metal). Furthermore, an open coordination place must be present or formed during the reaction.
2.3.1.2. Two-Step Mechanism - Polymerization takes place by two steps: (1) com- plexation of the monomer to the transition metal atom of the active center; (2) migratory insertion of the complexed monomer to the bond between the transition metal atom and first carbon atom of the polymer chain. Repetition of the processes is responsible for the chain growth.
In the Cossee mechanism (see Fig. 2) a vacant coordination site is generated initially, followed by olefin complexation. Formal migration of the polymer chain, P, and formation of the metal-carbon bond occur concertedly through a four-center transition state. This recreates a vacant coordination site at the site originally occupied by the polymer chain and the process continues; the growing polymer chain terminus flips from site to site.6
2.3.2. The Trigger Mechanism
Although the Cossee mechanism has been widely accepted, there are some problems which are very difficult to explain through this mechanism. For example, why the free, acidic coordination site of the active center is not attacked by Lewis bases such as esters present in the system; why the polymerization rate order relative to monomer concentration is higher than 1 .O; why isospecific polymerization seems to have a higher propagation rate than non-specific polymerization, and why the stereoregularity of the first inserted monomer seems to be lower than for the insertion of the rest.
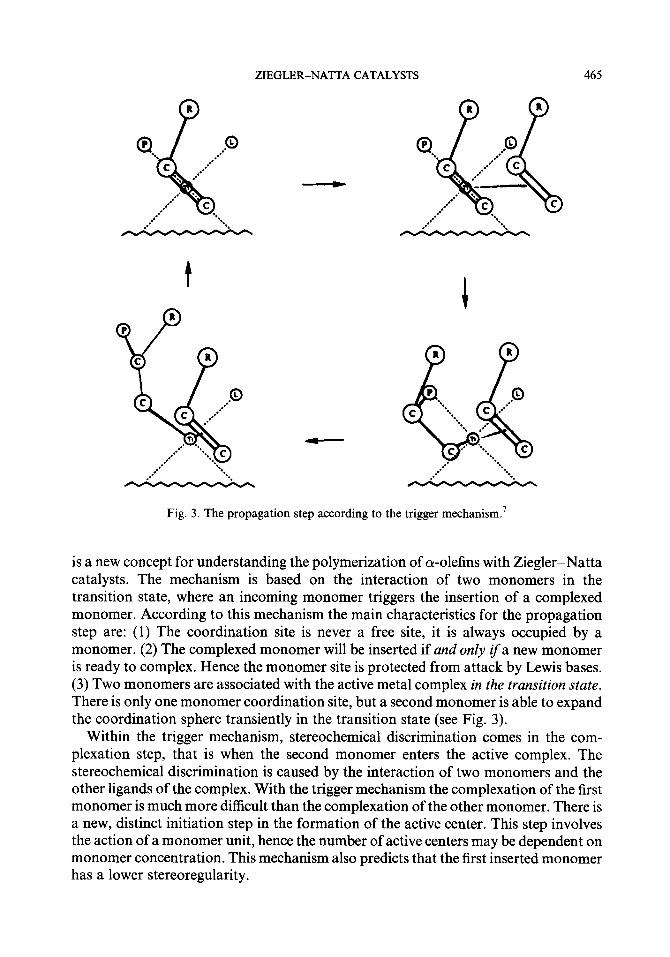
Recently the trigger mechanism was proposed by Ystenes.7 The trigger mechanism
ZIEGLER-NATTA CATALYSTS 465
Fig. 3. The propagation step according to the trigger mechanism.7
is a new concept for understanding the polymerization of a-olefins with Ziegler-Natta catalysts. The mechanism is based on the interaction of two monomers in the transition state, where an incoming monomer triggers the insertion of a complexed monomer. According to this mechanism the main characteristics for the propagation step are: (1) The coordination site is never a free site, it is always occupied by a monomer. (2) The complexed monomer will be inserted if and only if a new monomer is ready to complex. Hence the monomer site is protected from attack by Lewis bases. (3) Two monomers are associated with the active metal complex in the transition state. There is only one monomer coordination site, but a second monomer is able to expand the coordination sphere transiently in the transition state (see Fig. 3).
Within the trigger mechanism, stereochemical discrimination comes in the com- plexation step, that is when the second monomer enters the active complex. The stereochemical discrimination is caused by the interaction of two monomers and the other ligands of the complex. With the trigger mechanism the complexation of the first monomer is much more difficult than the complexation of the other monomer. There is a new, distinct initiation step in the formation of the active center. This step involves the action of a monomer unit, hence the number of active centers may be dependent on monomer concentration. This mechanism also predicts that the first inserted monomer has a lower stereoregularity.
466 J. HUANG and G. L. REMPEL
(a) by &elimination with H-transfer to monomer
7 A /
CHo-CH / I
c”,=ai
M j yt ----” M +
CHt \
CHt-cH~
@) by hydrogenation
R /
CHp-cilp U/l /,A
H
(c) by &elimination forming hydride
.‘c”; . . ..J” - R
A
CH*-cn/, +
M-H
R /
CHt=cH
+
M-H
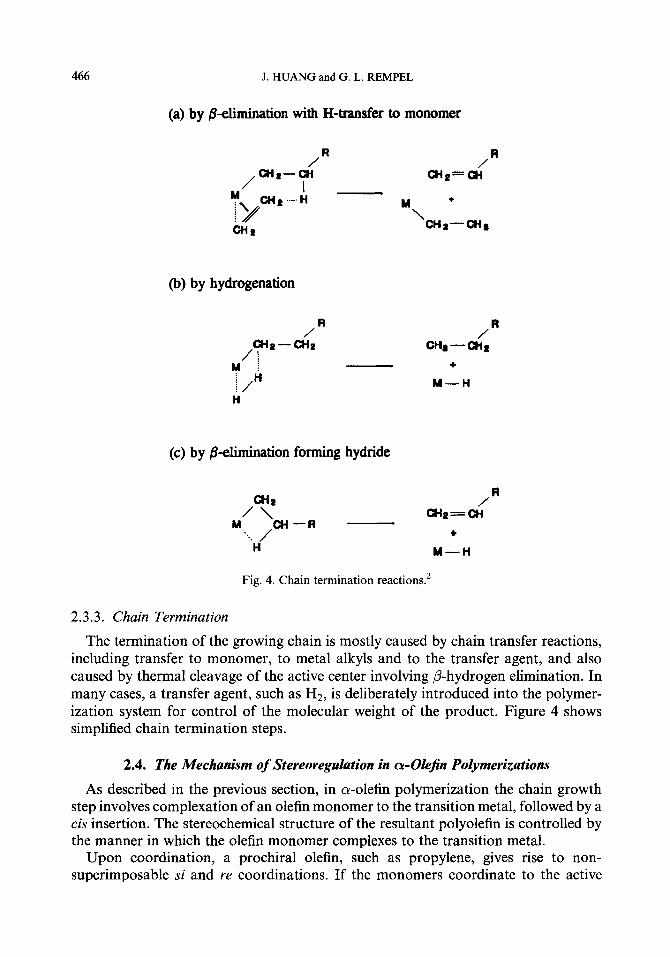
Fig. 4. Chain termination reactions2
2.3.3. Cha4n Termination
The termination of the growing chain is mostly caused by chain transfer reactions, including transfer to monomer, to metal alkyls and to the transfer agent, and also caused by thermal cleavage of the active center involving P-hydrogen elimination. In many cases, a transfer agent, such as HZ, is deliberately introduced into the polymer- ization system for control of the molecular weight of the product. Figure 4 shows simplified chain termination steps.
2.4. The Mechanism of Stereoregulation in cx-OleJin Polymerizations
As described in the previous section, in a-olefin polymerization the chain growth step involves complexation of an olefin monomer to the transition metal, followed by a cis insertion. The stereochemical structure of the resultant polyolefin is controlled by the manner in which the olefin monomer complexes to the transition metal.
Upon coordination, a prochiral olefin, such as propylene, gives rise to non- superimposable si and re coordinations. If the monomers coordinate to the active

ZIEGLER-NATTA CATALYSTS 461
center in si or ye position randomly, the resultant polymer will be atactic. The isotactic polymer is generated by a large series of insertions of all xi or all re coordinated monomers, while the syndiotactic polymer would be generated by alternate insertion of si and re coordinated monomers. The catalysts producing isotactic polymers are called isospecific, and those producing syndiotactic polymers syndiospecific.
The driving force for stereoregulation is steric in nature.lT8 That is, the stereo- specificity of a catalyst is determined by the difference in activation energy of the two coordination positions caused by steric interaction of the transition metal complex including the growing polymer chain with the incoming monomer. There exist two types of stereoregulation, detailed next.
2.4.1. Catalytic Site Control
Catalytic site control, which is also termed enantiomorphic site control, occurs mostly in heterogeneous catalyst systems. In these systems, the asymmetric nature of each active center forces the a-olefin to always add either in the si or in the re configuration, and thus isotactic chains are formed.
2.4.2. Chain End Control
Because the asymmetric feature of active centers is absent in most homogeneous systems, another driving force is permitted to occur, namely, steric interactions between the side groups of the last added and incoming monomers, for example, the methyl group of propylene. Chain end control can be either isotactic or syndiotactic depending on the exact steric interactions. If the interactions force the monomer to be inserted in opposite configurations after each growth step, syndiotactic polymer is formed. Chain end control usually occurs at very low reaction temperatures, when the polymer chain becomes rigid.
The Ziegler-Natta catalyst is also regioselective. In an isotactic a-olefin polymer- ization, 1,2-insertion, i.e. the unsubstituted carbon of the olefin attaches to transition metal, is favored.
Stereoregulation of Ziegler-Natta catalysts is not perfect. Stereoirregularity and regioirregularity normally exist in both isotactic and syndiotactic polymers to some extent. The former is caused by the insertion of monomers coordinated in opposite position, and the latter caused by 2,1- and 1,3_insertions. For isotactic polypropylene, there are two methods to describe the degree of isotacticity: (1) the weight percentage of the n-heptane insoluble fraction based on heptane extraction; (2) the content of mmmm pentad in the polymer based on NMR spectroscopic measurements. Studies on the distribution of steric defects in polymer chains provide important information about the mechanisms of stereochemical control.
2.5. Kinetics
The elucidation of the basic kinetic parameters that characterize the Ziegler-Natta polymerization is of prime importance. Only then can the origin of the obtained

468 J. HUANG and G. L. REMPEL
polymerization rate curves and the nature of the polymers that are formed in the presence of these catalysts be understood. A unified, universally accepted kinetic picture has not yet emerged, in spite of many excellent experimental and theoretical studies that have been made on this catalyst,
A famous kinetic model was proposed by Cossee in 1967.’ According to the two- step mechanism, the propagation reaction involves: (1) the complexing of a monomer (M) at the transition metal-carbon center (MT-P,),
MT-P, + Monomer & MT /pn
(1) k2 \ ;
Monomer (A) (M) (MA)
and (2) the insertion of the monomer into the transition metal-carbon bond,
ipn MT
\ ‘Monomer
(MA) (A’)
Under the steady-state condition, i.e. d[MA]/dt = 0,
wfl - kl k3 [Cl WI dt k#4] +kz +k3’
(2)
(3)
where [C] = [MA] + [A], and [A] = all vacant sites. Cossee suggested that step 2 is the rate-determining reaction, and k2 > kI [M] > k3.
Therefore, in terms of the Cossee’s mechanism, the polymerization rate R, is expressed as:
R, = WdkdClM~ (4) Since adsorption has been shown to be important to heterogeneous Ziegler-Natta
catalyst systems, many kinetic studies on heterogeneous systems have attempted to describe the polymerization by adsorption kinetics.’ The fraction of active centers with adsorbed monomer (0,) and the fraction with adsorbed Al cocatalyst (0,) are expressed in terms of Langmuir-Hinshelwood isotherms as:
where KA and KM are the equilibrium constants for adsorption of A and A4 with the surface of the transition metal salt crystals, respectively. The rate of polymerization is given by the relation:
R, = k&&. (7)

ZIEGLER-NATTA CATALYSTS 469
2.6. The Success of MgC&Supported Heterogeneous Systems
The Ziegler-Natta catalyst has been used in soluble, colloidal or heterogeneous form for both olefin and diene polymerizations. But before the 198Os, crystalline isotactic polyolefins could only be prepared with heterogeneous systems. Heterogeneous Ziegler-Natta catalyst systems have been a great success in commercial production of linear polyethylene and isotactic polypropylene and higher a-olefin polymers.
A representative of heterogeneous catalysts is the MgC12 supported titanium tetrachloride catalyst system, which was discovered in the mid-1970s and is termed “the second-generation” Ziegler-Natta catalyst. The system consists of a catalyst component, MgC12/Lewis Base/TiCb ternary mixture, and cocatalyst component, metal alkyl/Lewis base mixture. The chemical interaction between the catalyst and cocatalyst leads to initiation of polymerization. The catalyst is highly active and stereospecific. Catalyst efficiencies of 200,OOOg of polyethylene, and of over 40,OOOg of polypropylene per gram of titanium per hour have been reported, and isotacticities greater than 97% for propylene polymerization can be obtained.’ The MgC12 sup- ported catalysts brought about a remarkable simplification of polymerization and workup processes, and led to revolutionary developments for the commercial produc- tion of linear polyethylene and isotactic polypropylene.
2.7. Dificulties with Theoretical Studies
In contrast to the great successes in commercial production, reaching an in-depth knowledge in Ziegler-Natta catalysis has not been as rapid as we hoped for. Many important, fundamental questions about Ziegler-Natta catalysts remain unanswered despite decades of intensive research. A central difliculty is that these catalysts are heterogeneous and function in a ternary gas-polymer-catalyst or liquid-polymer- catalyst system. Few analytical probes are currently available that can provide a molecular-level view of the complex chemistry and physics occurring at each of the phase boundaries. Ziegler-Natta catalysts, in addition to being heterogeneous with respect to the number of phases present, are also heterogeneous with respect to the constitution of the active sites. Multiple sites, each having a different structure and reactivity, are often present and none may be considered to be completely characterized. This greatly compli- cates efforts to elucidate the details of Ziegler-Natta chemistry.‘o
More recently, however, homogeneous olefin polymerization catalysts have been developed. Although these catalysts are by no means simple, the fact that much of the chemistry of interest occurs in solution makes possible the application of powerful analytical methods such as nuclear magnetic resonance (NMR) spectroscopy. The catalysis can thus be related to the enormous database of organometallic model compounds and reactions.
3. THE HISTORY OF THE HOMOGENEOUS ZIEGLER-NATTA CATALYSTS
The first homogeneous Ziegler-Natta catalyst was discovered independently by Breslow and Natta in 1957.1’112 The catalyst, bis(cyclopentadienyl)titanium dichloride

470 J. HUANG and G. L. REMPEL
(CpzTiClz, Cp = n5-cyclopentadienyl) activated with alkylaluminum chloride (Al&Cl) exhibited a low polymerization activity (A) for ethylene, ca. A M lo4 g polyethylene (PE)/(mol Ti - h. atm), and none for propylene. It was found later that small amounts of impurities such as oxygen, ether and even moisture had a beneficial effect on the polymerization. Subsequently, small amounts of water were found to increase significantly the activity of the catalyst.13 The reaction between water and aluminum alkyls was shown to produce alkyl alumoxanes. In 1980 Kaminsky and coworkers2 used oligomeric methyl alumoxane (MAO) with group 4B metallocene compounds to obtain ethylene polymerization catalysts having extremely high activ- ities. For instance Cp2TiC12/MA0 has a polyethylene productivity of 9.3 x lo6 g PE/ (mol Ti - h - atm) at 20°C; the productivity is 9 x lo7 g PE/(mol Zr - h. atm) at 70°C with Cp2ZrC12/MA0. However, these catalysts are non-stereospecific produ- cing only atactic polypropylene, because of the symmetric feature of their active centers.
In the early 1980s Brintzinger and coworkers synthesized racemic ethylene-bridged bis(indenyl)zirconium dichloride, Et(Ind)2ZrC12, and racemic ethylene-bridged bis(4,5,6,7-tetrahydroindenyl)zirconium dichloride, Et(H41nd)2ZrC12,‘4 as well as their titanium analogues, Et(Ind)2TiC12 and Et(H41nd)2TiC12,‘5 which have both meSO and racemic configurations. The steric rigid chiral Et(Ind)2ZrC12 and Et(H41nd)2ZrC12 catalysts activated with MAO catalyzed the stereoselective polymer- ization of propylene with very high activities. It was the first time that the isotactic polyolefins were obtained from homogeneous Ziegler-Natta polymerizations. This finding demonstrated stereochemical control by the chiral ansa-indenyl ligands on the transition metal ion in the selection of one of the two enantiotopic faces (re or si) of a prochiral vinyl monomer in migratory insertion. As predicted, the meso- Et(Ind)2TiC12/MA0 system only produced atactic polypropylene. Since then a large group of ansa-metallocene compounds have been developed, each of them having unique catalytic activity and stereospecificity.
The discovery of the ansa-metallocene catalysts is believed to be of immense sig- nificance to the catalytic, organometallic and polymer sciences. The structure of a metallocene can be determined by X-ray diffraction. Modification of catalysts by variation of the ligands surrounding the active center permits a correlation of catalyst structure with catalytic activity and stereospecificity. Studies made on such metallo- cene compounds have increased our understanding of the molecular mechanism of stereochemical control in o-olefin polymerizations. It is believed that the true active species in the metallocene/MAO system are electrophilic 14-electron metallocene cations.” Most probably the cations do not exist freely, but rather are solvated in some way by MAO.”
More recently, cationic metallocene catalysts have received growing interest as model systems for homogeneous MAO activated metallocene catalysts. The research on this topic began with the work of Jordan et al., l7 who demonstrated in 1986 that the zirconium complex [Cp2ZrCH3(THF)]+[BPh4]- (Ph = phenyl) polymerizes ethylene in polar solvents. Subsequently, it has become possible to develop cationic metallocene catalyst systems which polymerize ethylene, propylene and higher a-olefins with

ZIEGLER-NATTA CATALYSTS 471
high activity even in non-polar solvents such as toluene. These systems are composed of Lewis base-free zirconocene cations with fluorinated or perfluorinated [BPhJ or suitably substituted carboranes as counterions.” The most promising system among them was developed by Chien et al19 The metallocene cation [Et(Ind)2Zr(CHs)]+B(C6F5)4, formed by the reaction of Et(Ind)2Zr(CH3)2 with tri- phenylcarbenium tetrakis(pentafluorophenyl)borate, PhsC+B(C,F&, showed a higher activity and similar stereospecificity when compared to the Et(Ind)2ZrC12/ MAO system.
Homogeneous metallocene based catalysts are of theoretical significance in studies of Ziegler-Natta polymerization. Compared to conventional heterogeneous systems in which a variety of active centers with different structures and activities usually coexist, homogeneous catalysts give very uniform catalytically active sites which possess controlled, well-defined ligand environments.3120 Therefore, the polymeriza- tion processes in homogeneous systems are often more simple, and kinetic and mechanistic analyses for these systems are greatly simplified. The recently developed base-free cationic metallocene is increasingly referred to as a “single-site” catalyst. The cationic metallocene catalyst affords a very simple and scientifically attractive system to investigate the details of stereoselective polymerization mechanisms.
4. THE COMPOSITION OF HOMOGENEOUS ZIEGLER-NATTA CATALYSTS
4.1. The Metallocenes
The main component of homogeneous Ziegler-Natta catalyst systems, the catalyst precursor, is the Group 4B transition metallocenes (titanocenes, zirconocenes and hafnocenes), which are characterized by two bulky cyclopentadienyl (Cp) or substi- tuted cyclopentadienyl ligands (CP’).~~ Two simple examples of these metallocenes are shown in Fig. 5.
These molecules have C2, symmetry. The two Cp rings in the molecules are not parallel. The Cp2M fragment is bent back with the centroid-metal-centroid angle 6J about 140” due to an interaction with the other two CI bonding ligands.22
GP Cl - ii - Cl
CpcTiC12 WWCH,),
Fig. 5. Structures of two metallocenes with Czu symmetry.

472 J. HUANG and G. L. REMPEL
Fig. 6. Structures of the Brintzinger catalysts.‘4
The chiral ansa-metallocenes, that is, metallocenes with two Cp’ ligands arranged in a chiral array and connected together with chemical bonds by a bridging group, were first synthesized by Brintzinger and coworkers. I4 The molecular structures of the two famous Brintzinger catalysts, Et(Ind)2ZrC12 and Et(H41nd)2ZrC12, are depicted in Fig. 6.
These two metallocenes have C, symmetry. An ansa-metallocene can have C,, C, or C1 symmetry depending upon the substituents on the two Cp’ rings and the structure of the bridging unit. A large number of ansa-metallocenes have since been synthesized by changing the transition metals (Ti, Zr or Hf) and substituents on the Cp rings, as well as the bridging groups. There are a wide variety of substituted Cp ligands. Among them methylcyclopentadienyl (MeCp), pentamethylcyclopentadienyl (Me&p), inde- nyl (Ind), tetrahydroindenyl (H41nd) and fluorenyl (Flu) ligands are most frequently used. The common bridging groups are ethylene (Et, -CH&H2-), dimethylsilene (Me$i, (CH&3=), isopropylidene (iPr, (CH,)&=), and ethylidene (CH,CH=). The bridging group not only provides a stereorigid conformation for the complex, but also dictates the distance between the transition metal atom and the Cp ligands and the bending angle 8, thus influencing catalyst activity and stereospecificity.
It is believed that a steric interaction of the Cp type ligands surrounding the active center with incoming monomer plays a key role in the stereoselectivity of the poly- merizations with these homogeneous catalysts. Changing the steric structure of the ligands in the metallocenes leads to the changes in steric structures of polyolefin products. Poly(a-olefins) of any steric structure (isotactic, syndiotactic and atactic) can be obtained simply by tailoring the stereorigid metallocene (catalyst precursor), basically according to the local symmetry.
4.2. The Methylalumoxanes (MAO)
MAO is the most important cocatalyst which activates the group 4B metallocenes in homogeneous Ziegler-Natta polymerization. Before the discovery of the MAO cocatalyst, the homogeneous Ziegler-Natta catalyst Cp2TiC12 was activated with alky- lahuninum chloride which led to poor catalyst activity. The use of MAO cocatalyst
ZIEGLER-NATTA CATALYSTS 473
Linear
cyclic
n=4-20
CH3 CH3
‘Al
I
/ t f 0-u 0-U / CH*
CHa n \ CHa
CHa
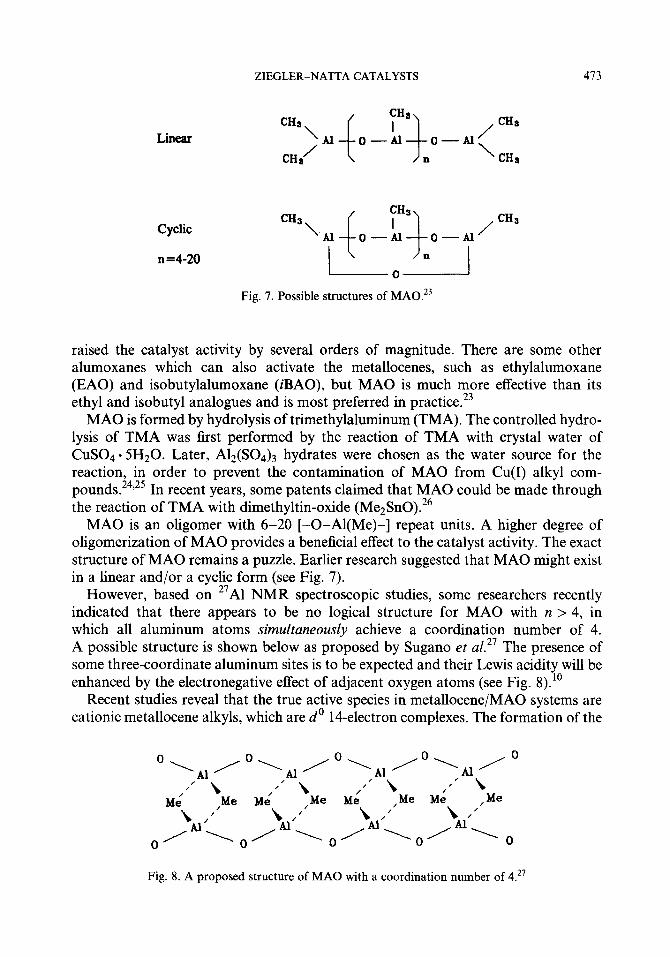
Fig. 7. Possible structures of MA0.23
raised the catalyst activity by several orders of magnitude. There are some other alumoxanes which can also activate the metallocenes, such as ethylalumoxane (EAO) and isobutylalumoxane (1’BAO), but MAO is much more effective than its ethyl and isobutyl analogues and is most preferred in practice.23
MAO is formed by hydrolysis of trimethylaluminum (TMA). The controlled hydro- lysis of TMA was first performed by the reaction of TMA with crystal water of CuSO4. 5H20. Later, Al2(SO4)3 hydrates were chosen as the water source for the reaction, in order to prevent the contamination of MAO from Cu(1) alkyl com- pounds.24125 In recent years, some patents claimed that MAO could be made through the reaction of TMA with dimethyltin-oxide (Me2Sn0).26
MAO is an oligomer with 6-20 [-0-Al(Me)-] repeat units. A higher degree of oligomerization of MAO provides a beneficial effect to the catalyst activity. The exact structure of MAO remains a puzzle. Earlier research suggested that MAO might exist in a linear and/or a cyclic form (see Fig. 7).
However, based on 27A1 NMR spectroscopic studies, some researchers recently indicated that there appears to be no logical structure for MAO with n > 4, in which all aluminum atoms simultaneously achieve a coordination number of 4. A possible structure is shown below as proposed by Sugano et aL2’ The presence of some three-coordinate aluminum sites is to be expected and their Lewis acidity will be enhanced by the electronegative effect of adjacent oxygen atoms (see Fig. 8)”
Recent studies reveal that the true active species in metallocene/MAO systems are cationic metallocene alkyls, which are do 14-electron complexes. The formation of the
Fig. 8. A proposed structure of MAO with a coordination number of 4.27

474 J. HUANG and G. L. REMPEL
catalytically active complex involves a series of reactions between metallocenes and MAO. For the halogen-containing metallocenes, a rapid alkylation of metallocene by MAO takes place first, and the active species arises from a methyl transfer reaction between the metallocene alkyls and MAO. The active species formed from these reactions is probably in combination with a poorly coordinating MAO anion. A detailed mechanism for the formation of the active center will be reviewed in Section 5.2.
4.3. Cationic Metallocene Alkyl Catalyst Systems
Active homogeneous catalysts can be obtained, in the absence of MAO, by allowing dialkyl metallocenes, such as Cp2Zr(CH&, to react with strongly Briinsted acidic salts in a 1 : 1 mole ratio. The true active species produced in situ is basically a 16electron cationic complex, such as [Cp2ZrCH$, which will be more or less affected by the counterion remaining in the system.
In attempts to synthesize more reactive base-free complexes, Lewis base adducts - [Cp#R(L)][BPhJ - have been extensively studied. Since the late 1980s many “cationic” metallocene alkyls, usually with BPh4 as the counterion, have been synthesized by the reactions of alkyl derivatives of the metallocene with BPh4 salts of RsNH, Ag, K and Cp2Fe.lg These reactive complexes were found to exhibit modest ethylene polymerization activity, and nil to moderate activity for propylene polymerization, because these preparations form undesirable side products which are Lewis bases or contain another transition metal. The activity of such systems is limited by the presence of a number of potential ligands, such as the solvent, the amine and, particularly, the [BPhJ anion, which compete effectively with the olefin monomer for a vacant coordination site on the metal center.28 Anion coordination can be virtually eliminated by using fluorinated anions, such as [B(C6F5)J, or carboranes. Incorporation of fluorine in [BPhJ was expected to suppress 7r-coordination of the benzene ring, therefore, the B(C6F& ion should be an inert non-complexing anion, and the reaction of its salt with metallocenes should result in reactive base-free cationic com- plexes.
Two procedures, which make use of this anion and avoid undesirable side products, have been reported. One method discovered by Marks et aZ.29 is:
Cp2Zr(CH3)2 + B(C6F5)3 -+ Cp2Zr+(CHs) + (C,F5)sB(CHs)-.
The other method was discovered by Chien et a1.19:
(8)
Et(Ind)2Zr(CH3)2+ Ph3C+B(C6F,); +
Et(Ind)2Zrf(CH3) + B(C6F5); + Ph,CCH3. (9)
In the latter system the Et(Ind)2Zrf(CH3) cation is extremely active and stereoselective in propylene polymerization and shows an unusual dependence of catalytic activity on the polymerization temperature (T,): its catalytic activity increases with a decrease in

ZIEGLER-NATTA CATALYSTS 475
Tp. The cation behaves like a “single-site” catalytic species at very low Tp, but probably exists in two or more conformationally isomeric states at high Tp. Experi- mental results are consistent with acceleration of the rate of conformational equilibria with increasing T,, to produce polypropylene of progressively lower stereoregularity and molecular weight (iwW).30
Chien also reported that a superior cationic catalyst was obtained from rac-
Me2Si(Ind)2ZrC12, Ph3C+B(C6F5)i, and triethylaluminum (TEA), which probably acts as an impurity scavenger. The catalyst polymerized propylene with an activity of 2 x lo9 g PP/(mol Zr - mol C3H6. h) at -20°C and 3 x 10’ g PP/(mol Zr - mol C3H6 -h) at -55°C. The isotacticity of the resulting polymers is 93.8 and 99.4% for Tp = -20 and -55°C respectively.30
5. POLYMERIZATION MECHANISM
5.1. The Active Center
With regard to the active center, there are two major aspects in which metallo- cene based homogeneous systems differ from conventional heterogeneous systems:
(1) All transition metal atoms form the active center in homogeneous systems. Direct measurement of the number of active centers has indicated that nearly 100% of the transition metal atoms are involved in the active centers, assuming that no more than one chain can grow at the same time on a given transition metal atom.3 (2) A homogeneous catalyst can be made as a “single-site”, that is, only a single type of active center is present in a homogeneous system under proper conditions. A “single-site” stereoselective catalyst can produce polymers with sharp melting transitions (T,), narrow distribution of molecular weights (MWD) with M,/M, 5 2, and uniform solubility characteristics.30
The active centers for homogeneous Ziegler-Natta catalyst systems are metallocene alkyl cations, i.e. cationic do 1Celectron complexes of the type [Cp,nii(R)]+ (M = Ti, Zr, Hf). Many researchers believe that the metallocene/MAO systems and the cationic metallocene systems probably share a common active species, although they are formed via different pathways.
5.2. The Formation of the Active Center in MetallocenelMAO Systems
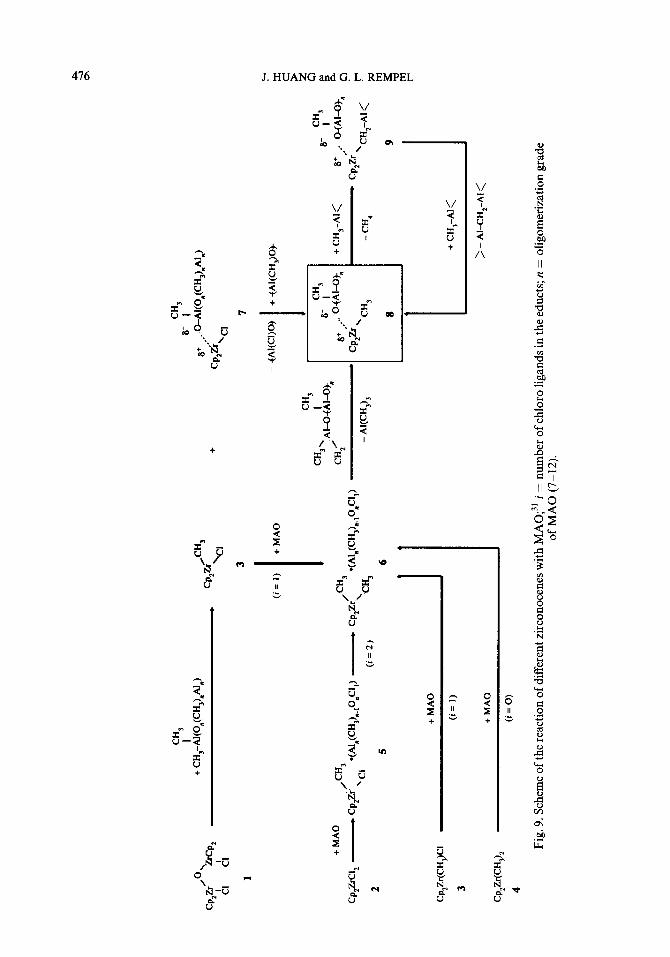
Kaminsky et al. presented a very detailed picture for the formation of active centers in metallocene/MAO systems (see Fig. 9).31 Four types of metallocenes with different a-bonding ligands, Cp2ZrC12, CpzZr(CHs)C1, Cp2Zr(CH3)2 and (Cp2ZrCQ20, are included.
Starting from Cp2ZrC12, the first step of the reactions after mixing the metallocene with MAO is the complexation and alkylation of Cp2ZrC12 with MAO:
476 J. HUANG and G. L. REMPEL
+

ZIEGLER-NA’ITA CATALYSTS 411
CpzZrClz + (Al(CHs)-0) n + Cp2ZrC12 - (Al(CH,)-0),
T1 Cp2Zr(CH3)C1 + Al,(CHs),_ tO,Cl + Cp2Zr(CH3)C1 - (Al,(CHs),_ iO,Cl).
(10)
In a subsequent alkylation Cp2Zr(CH3)2 is formed:
Cp,Zr(CHs)Cl + MAO - Cp2Zr(CH& + (Al,(CH&-iO,Cl). (11)
Cp2Zr(CH3)2 further reacts with MAO, forming compound (8) (see Fig. 9). Com- pound (8) contains the structural element Zr-O-Al.
-0 \
Al-CHs /
‘\ /Cm
Y + CHs
CP
\
Cp/zr\CH3 1 CHa
Al(CHs)s
+
CR3
I cp o--AI
\zr/ ‘o-
Cp/ ‘CH3
(12)
The Zr-0 bond has a polar character and could be of ionic nature. Compound 8 is believed to be the active species in the metallocene/MAO systems. Possibly, compound 8 exists in two different states that are in equilibrium:
C Ha
(j+ ..po&h =
i
CHa I
CP#w+
cp, Zr” Al(CH3)O’ -
,0 W-Oh
‘R Cp, Zr
‘R 1 (13)
The metallocene alkyl cation (at one side of the equilibrium) might be the true active center.
The reaction of the oxobridged zirconocene (compound 1 in Fig. 9) with MAO gives CpzZr(CHs)C1 as a first reaction product. The formation of this species must proceed by an alkylation step and cleavage of the Zr-0 bond.
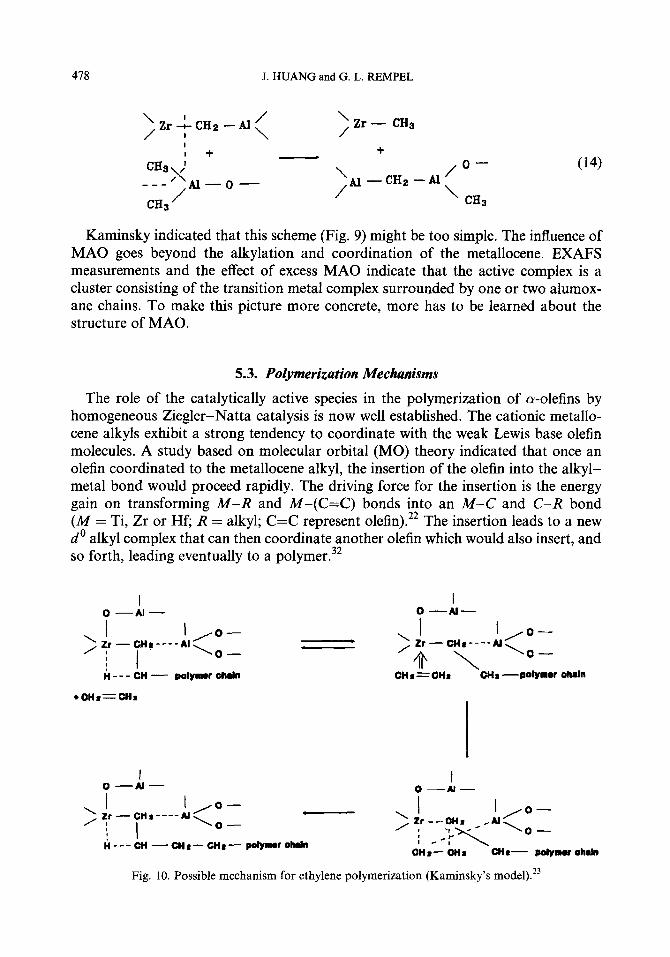
Evolution of methane was observed after metallocene was mixed with MAO. Methane is the product of a side reaction: condensation of the metallocene and MAO via a-hydrogen transfer. This side reaction is relatively fast under polymeriza- tion conditions. More than 50 moles of methane were eliminated per mole of zirconium in 2 h. The resulting Zr-CH2-Al structures seem to be inactive for poly- merization. The MAO present in the system could reactivate this structure to the active Zr-CH3 structure:
478 J. HUANG and G. L. REMPEL
\ Zr-kH2 --Al / I <
\ /
Zr - CHs
I +
/*- (14) -CH2 -Al
\ CBS
Kaminsky indicated that this scheme (Fig. 9) might be too simple. The influence of MAO goes beyond the alkylation and coordination of the metallocene. EXAFS measurements and the effect of excess MAO indicate that the active complex is a cluster consisting of the transition metal complex surrounded by one or two alumox- ane chains. To make this picture more concrete, more has to be learned about the structure of MAO.
5.3. Polymerization Mechanisms
The role of the catalytically active species in the polymerization of a-olefins by homogeneous Ziegler-Natta catalysis is now well established. The cationic metallo- cene alkyls exhibit a strong tendency to coordinate with the weak Lewis base olefin molecules. A study based on molecular orbital (MO) theory indicated that once an olefin coordinated to the metallocene alkyl, the insertion of the olefin into the alkyl- metal bond would proceed rapidly. The driving force for the insertion is the energy gain on transforming M-R and M-(C=C) bonds into an M-C and C-R bond (M = Ti, Zr or Hf; R = alkyl; C=C represent olefin).22 The insertion leads to a new do alkyl complex that can then coordinate another olefin which would also insert, and so forth, leading eventually to a polymer.32
I I O-AI- 0 --Al-
l > Lr
I -ala----AI
/O- ;I ‘o- i___CH- #MJlylnor chaill
l CH8=CHS
Ii---CH -WC-GHr- PdyfWOhJn
Fig. 10. Possible mechanism for ethylene polymerization (Kaminsky’s mode1).23
ZIEGLER-NATTA CATALYSTS 479
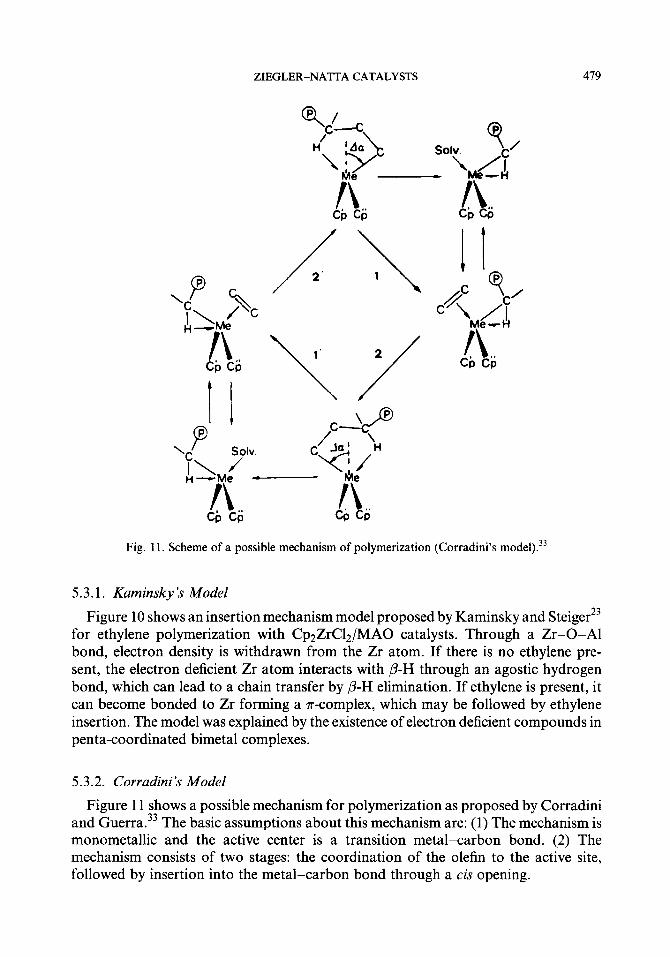
Fig. 11. Scheme of a possible mechanism of polymerization (Corradini’s model).33
5.3.1. Kaminsky ‘s Model
Figure 10 shows an insertion mechanism model proposed by Kaminsky and Steiger23 for ethylene polymerization with Cp2ZrC12/MA0 catalysts. Through a Zr-O-Al bond, electron density is withdrawn from the Zr atom. If there is no ethylene pre- sent, the electron deficient Zr atom interacts with P-H through an agostic hydrogen bond, which can lead to a chain transfer by ,0-H elimination. If ethylene is present, it can become bonded to Zr forming a 7r-complex, which may be followed by ethylene insertion. The model was explained by the existence of electron deficient compounds in penta-coordinated bimetal complexes.
5.3.2. Corradini’s Model
Figure 11 shows a possible mechanism for polymerization as proposed by Corradini and Guerra.33 The basic assumptions about this mechanism are: (1) The mechanism is monometallic and the active center is a transition metal-carbon bond. (2) The mechanism consists of two stages: the coordination of the olefin to the active site, followed by insertion into the metal-carbon bond through a cis opening.

480 J. HUANG and G. L. REMPEL
In Corradini’s model the active species is the Cp2A4R+ complex. The cationic character of the active species has been substantiated by the recent discovery of base-free cationic metallocene catalysts. According to a quantum mechanical ana- lyses for do complexes of the type Cp$VL (L = ligand),22 the best coordination of L may not be the one (most symmetrical and presumably sterically most favorable) along the symmetry axis which relates the two bent Cp rings. Energy minimum situations for a do complex and for L = H would correspond to angular deviations AWL x 65” from the symmetry axis. On the other hand, an additional stabilization of a geometry with C)I # 0” at the strongly unsaturated metal center could occur because of an agostic interaction of a hydrogen in the y-position. Corradini predicted that Q will differ from 0” whenever a monomer or solvent molecule becomes coordinated to the equatorial belt of the bent metallocene cation, together with the polymer chain. (Note that a geometry with Q # 0” makes the metal atom chiral in cation Cp’Cp”MR+ if the two cyclopentadienyl ligands Cp’ and Cp” are different.)
As seen in Fig. 11, at the end of each polymerization step the chain occupies the coordination site previously occupied by the alkene monomer. Hence, most consecu- tive polymerization steps correspond to a model obtained by exchanging the relative positions of the growing chain and of the monomer. This is extremely relevant for the stereospecific behavior of the catalysts.
5.3.3. Other Considerations About the Polymerization Mechanism
Some experimental results showed that the polymerization rate (RP) was between first and second order relative to monomer concentration.” They suggested that there is a process in monomer insertion which occurs from a his-olefin intermediate or transition state similar to the “trigger mechanism” proposed by Ystenes.7
5.4. The Dependence of Catalyst Activity on the Nature of the Metallocene and Metallocene-Monomer Znteractions
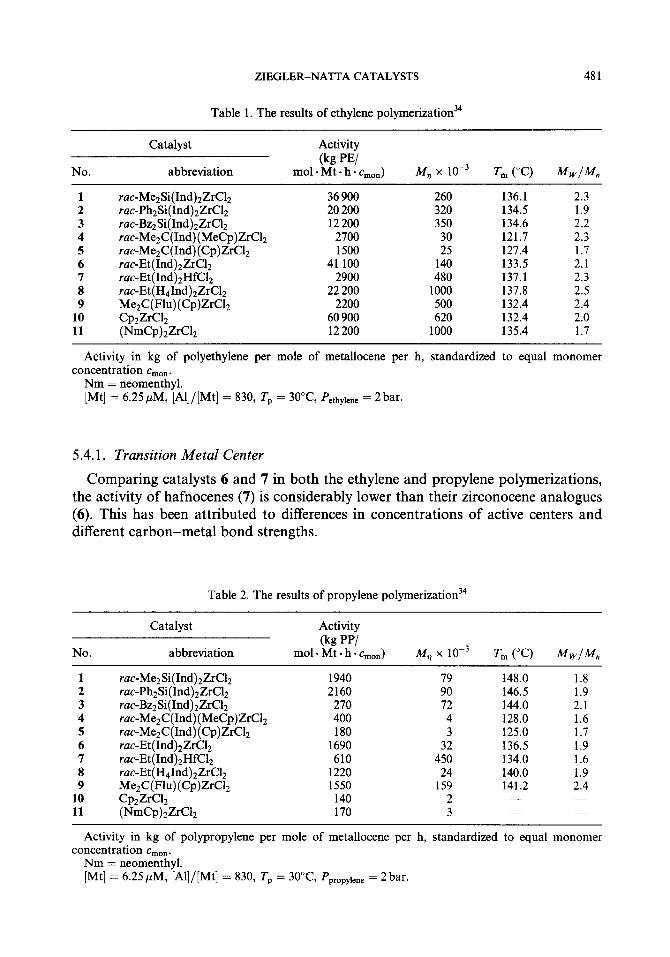
A comparative study of various catalysts was done by Kaminsky.34 In this study, ethylene and propylene were homopolymerized with 11 different metallocene catalysts activated with MAO under the same experimental conditions. The results, i.e. catalyst activity, viscosity-average molecular weight (M,), melting temperature (T,), and molecular weight distribution (M,/M,) are listed in Tables 1 and 2. This study combined with other reports has revealed the very complicated relationship of the catalyst activity with the nature of the metallocene, such as transition metal center and Cp ligand structures, and the interactions between metallocenes and monomers.
As seen in Tables 1 and 2, the complex trends in catalyst activity cannot be easily explained by simple arguments. Differences in the activities of two catalysts, all other circumstances being identical, can result from different concentrations of active species, from different propagation rates, or from a combination of both. And propagation rates are, in turn, affected by the rates of monomer coordination and monomer insertion which are influenced by steric or electronic factors. The following factors should be taken into consideration.
ZIEGLER-NATTA CATALYSTS 481
Table 1. The results of ethylene polymerization34
No.
Catalyst
abbreviation
Activity
(kg PE/ mol. Mt - h - cm,,) M,, x 1O-3 T, (“C) MwIM,
1 rue-MezSi(Ind)zZrC12 36 900 260 136.1 2.3 2 rac-PhzSi(Ind)2ZrC12 20 200 320 134.5 1.9 3 rat-BzzSi(Ind)zZrClz 12200 350 134.6 2.2 4 rat-MezC(Ind)(MeCp)Zr( 112 2700 30 121.7 2.3 5 rut-MezC(Ind)(Cp)ZrClz 1500 25 127.4 1.7 6 ruc-Et(Ind)zZrClz 41 100 140 133.5 2.1 7 ruc-Et(Ind)zHfClz 2900 480 137.1 2.3 8 ruc-Et(H41nd)2ZrC12 22 200 1000 137.8 2.5 9 MezC(Flu)(Cp)ZrClz 2200 500 132.4 2.4
10 CplZrC1z 60 900 620 132.4 2.0 11 ( NmCp)zZrClz 12 200 1000 135.4 1.7
Activity in kg of polyethylene per mole of metallocene per h, standardized to equal monomer concentration c,,,.
Nm = neomenthyl. [Mt] = 6.25,uM, [Al]/[Mt] = 830, Tp = 30°C Peth,,iene = 2bar.
5.4.1. Transition Metal Center
Comparing catalysts 6 and 7 in both the ethylene and propylene polymerizations, the activity of hafnocenes (7) is considerably lower than their zirconocene analogues (6). This has been attributed to differences in concentrations of active centers and different carbon-metal bond strengths.
Table 2. The results of propylene polymerization34
Catalyst Activity (kg PPI
No. abbreviation mol. Mt e h - cm,,) M,, x 1O-3 T, (“C) MwIW
1 ruc-MezSi(Ind)zZrC1, 1940 79 148.0 1.8 2 rue-Ph#i(Ind)zZrClz 2160 90 146.5 1.9 3 rue-BzzSi(Ind)zZrClz 270 72 144.0 2.1 4 rut-MezC(Ind)(MeCp)ZrClz 400 4 128.0 1.6 5 rut-MezC(Ind)(Cp)ZrClz 180 3 125.0 1.7 6 ruc-Et(Ind)2ZrC12 1690 32 136.5 1.9 7 rut-Et(Ind)zHfC12 610 450 134.0 1.6 8 ruc-Et(H41nd)zZrC12 1220 24 140.0 1.9 9 Me2C(Flu)(Cp)ZrClz 1550 159 141.2 2.4
10 Cp2ZrClz 140 2 11 (NmCp)zZrC& 170 3
Activity in kg of polypropylene per mole of metallocene per h, standardized to equal monomer concentration c,,,.
Nm = neomenthyl. [Mt] = 6.25 PM, [Al]/[Mt] = 830, Tp = 30°C Ppropyiene = 2 bar.

482 J. HUANG and G. L. REMPEL
5.4.2. Cp Ligands
The steric and electronic factors of the substituents on the Cp ligands affect the catalyst activities. Steric and electronic effects are always mixed and the overall effect on catalyst activity is dependent on the balance of the two. For the cases of electron donating substituents, Ewen*’ and Chien35 have reported the effects of alkyl substitution on the Cp ring for several zirconocenes, and the order of the activity was found to be (MeCp)2ZrC12 > (EtCp)2ZrC12 > (NmCp)2ZrC12 > Cp2ZrC12 > (Me5Cp)CpZrC12 > (Me5Cp)2ZrC12. These results were interpreted to mean that single alkyl substituents increase activity by electron donation enhancement of coordinated anionic propagation, whereas very bulky substituents decrease the activity due to steric hindrance. Similar results were shown by Kaminsky as noted in Tables 1 and 2; the effect of alkyl substitution in the Cp ring is activity enhancing when the electron releasing effect of alkyl substitution is decisive (comparing catalysts 4 and 5 in Tables 1 and 2), while the effect is activity decreasing when steric crowding is decisive (comparing systems 10 and 11 in Table 1). It was also reported by Pino that electron withdrawing substituents decrease polymerization activity and the molecular weight of polymer products.36 An increase in steric hindrance (comparing systems 1, 2 and 3 in Table 1) reduces the catalyst activities. The symmetry of the Cp ligands should also be taken into consideration. As shown in Table 1, the low activities of the three C-bridged species (4, 5 and 9) in ethylene polymerization are remarkable and cannot be explained with steric reasons, because the space volume between the Cp ligands available for catalysis is clearly larger than in bisindenyl systems like 1, the C-bridge being shorter than the Si-bridge, the Cp ligands thus forming a wider angle. In these cases the symmetric features of the ligands may be the cause.
5.4.3. Monomers
Comparing Tables 1 and 2, the order of succession of activity for these 11 catalysts in ethylene and propylene polymerization are totally different. (Ethylene: 10 > 6 > 1>8>2>3=11>7>4>9>5; Propylene: 2>1>6>9>8>7>4>3> 5 > 11 > 10.) Because these polymerizations were performed under identical condi- tions, the formation of active species should be the same in both series, and different propagation rates must be the reason for the different trends in activity in ethylene and propylene polymerization. In propylene polymerizations, systems lacking a high degree of stereospecificity (4, 5, 11 and 10) also show a poor activity. It seems that with the prochiral propylene monomer, a stereospecific mode of polymerization is strongly favored over the aspecific one. This factor is able to overcompensate electronic and steric influences coming from the metallocene structure. Moreover, the dramatic change for the highly syndiospecific system 9, which moves up in the activity scale from tenth for ethylene to the fourth for propylene among 11 systems, might indicate an energetic advantage of syndiospecific over isotactic polymerization. For the polymerization of higher a-olefins, the steric factor is expected to become more significant.

ZIEGLER-NATTA CATALYSTS 483
5.5. Functions of MAO
MAO undoubtedly acts as a methylating agent and an impurity scavenger, however, these are not its important roles. Other simple alkyl aluminum compounds can also alkylate metallocene chlorides and scavenge impurities, but they do not lead to high catalyst activities. MAO not only produces the cationic active species but also stabi- lizes the anion by formation of a “crown-alumoxane complex” analogous to crown- ether complexes of cations. If not stabilized the anion can attack the metallocene cation nucleophilically, which is the most common termination process in classical cationic polymerization.37 MAO can also take part in reactivation of the dormant species to active ones, as described in Section 5.2.
There are possible additional roles for MAO that reflect the large amount of it required for high activity and selectivity. The metallocene is probably surrounded by MAO even in the outer-sphere and thus prevents catalyst deactivation by bimole- cular processes between two metallocenes through either reductive elimination of PP chains or oxidative coupling forming hydrocarbon bridges between two transition metal atoms. Moreover, coordination of MAO should also lead to improved stereo- chemical control of catalysts.37
6. THE MECHANISM OF STEREOREGULATION
6.1. Versatile Stereoselectivity
The homogeneous metallocene based catalysts exhibit the ability to catalyze polymerizations with a variety of stereochemical control. Depending on the specific metallocene Cp ligands used, these systems present completely different stereospecific behaviors. It is feasible to produce polymers with desired stereostructures by changing the ligand structures of the metallocenes. According to the literature, these catalysts can catalyze the following polymerizations.
6.1.1. Polymerization of Ethylene
Homogeneous metallocene catalysts catalyze the homopolymerization of ethylene, producing linear polyethylene that is characterized by the absence of long- or short- branching.
The catalysts also catalyze the copolymerization of ethylene with higher a-olefins such as 1-butene, I-hexene and 1-octene, producing copolymers with evenly distrib- uted comonomer units along the polymer chain. The homogeneous metallocenes are the only systems capable of catalyzing the copolymerization of ethylene with cyclic olefins.
61.2. Polymerization of Propylene and Higher cr-OleJins
There are many types of polyolefins with different steric structures produced by using metallocene based catalysts.
484 J. HUANG and G. L. REMPEL
Fig. 12. Examples of non-stereospecific catalysts.
6.1.2.1. Atactic Polymers - The metallocenes, such as Cp2ZrC12 and meso- Et(Ind),TiCl,, have the same Cp (or Cp’) ligands and an achiral structure. These catalysts are non-stereospecific and furnish atactic polypropylene and other poly(o- olefins).
6.1.2.2. Isotactic Polypropylene - The stereorigid chiral ansa-metallocene catalysts, such as rac-Et(Ind)2ZrC12 (Fig. 6) and rac-MezSi(H41nd)2ZrC12, catalyze the isotactic polymerization of propylene. Before these catalysts were developed, isotactic polypropylene could only be made using heterogeneous Ziegler-Natta catalyst systems.
6.1.2.3. Syndiotactic Polypropylene - A syndiospecific catalyst, isopropylidene- (cyclopentadienylfluorenyl)zirconium dichloride, iPr(CpFlu)ZrClz, was designed by Ewen.38 The catalyst is not chiral, but has an internal plane of symmetry. The two Cp ligands are of very different sizes. The catalyst produces syndiotactic polypro- pylene.
w 0,o HSC
t CHs
Fig. 13. The iPr(CpFlu) ligand.38

ZIEGLER-NATTA CATALYSTS 485
H3C
t
CH3
CH3
Fig. 14. The iPr(MeCpFlu) ligand.3g
6.1.2.4. Hemiisotactic Polypropylene - Introduction of a methyl group into the Cp ring of iPr(CpFlu)ZrClz led to an amazing result: the new catalyst, iPr(3-MeCp- Flu)ZrClz (see Fig. 14) activated with MAO produces hemiisotactic polypropy- lene.39 (Hemi from the Greek, half.) It has become possible for the first time to generate hemiisotactic polypropylene through the direct polymerization of propy- lene. Before this catalyst was discovered, hemiisotactic polypropylene, the only polymer of hemitactic structure obtained at that time, was prepared by polymeriza- tion of 2-methylpentadiene.4 Since then, syntheses of hemiisotactic poly( 1 -butene) and poly(l-hexene) with iPr(3-MeCpFlu)ZrC12MA0 catalyst have also been reported.41
Hemitactic polymers represent a very interesting case in the field of macromolecular stereochemistry. Their typical feature is the coexistence of order and disorder: more precisely, order and disorder alternate along the chain in a well-defined way. In a hemitactic vinyl polymer, there are two alternating series of chemically equivalent stereogenic carbons: in the odd series the arrangement of the substituents follows a
Hemiisotactic:
Hemisyndiotactic:
Fig. 15. The structures of hemitactic polymers.40

486 J. HUANG and G. L. REMPEL
(1) (2)
Fig. 16. Cyclic olefins.
(3)
well-defined rule, while in the even series their arrangement is random. A polymer in which the positions of substituents at odd-numbered carbons are the same is defined as hemiisotactic, and a polymer in which substituents are related by a mirror glide plane is defined as hemisyndiotactk4’
6.1.2.5. Elastomeric Polypropylene - Using non-symmetric rat-[anti-ethylidene( l-q5- tetramethyl-cyclopentadienyl)( 1 -$-indenyl)titanium dichloride and its dimethyl derivative it is possible to produce elastomeric polypropylene.42 The polymer is considered to be a copolymer consisting of alternating blocks of stereoirregular/ amorphous and stereoregular/crystallizable PP segments. This structure is probably caused by the switching of the active site between two isomeric states. Monomer insertion in one site is less stereoselective than the other, and site switching occurs many times during the growth of polymer chains.
6.1.2.6. Optically Active Propylene Oligomer and Polymer - It is possible to separate the racemic mixture of Et(H41nd)2ZrC12 into pure enantiomers with the help of optically active binaphthol. The catalyst consisting of solely one enantiomer, such as S-Et(HJnd)zZrC12, is capable of producing optically active isotactic oligomers and polymers of propylene. So far, such a high degree of stereospecificity has only been achieved with this type of homogeneous catalyst.21
6.1.3. Polymerization of Cyclic Olejins
The homogeneous systems, such as Et(Ind)2ZrC12 and Et(H41nd)2ZrC12, allow the polymerization of cyclic olefins without promoting ring-opening reactions, which are typical for heterogeneous catalysts. The syntheses of isotactic homopolymers of cyclic olefins, such as cyclopentene (l), norbornene (2), and dimethanooctahydro- naphthalene (3) have been reported.31
6.1.4. Enantioselective Cyclopolymerization of Dienes
Cyclopolymerization of 1,5hexadiene using the optically active catalyst precursor, (RR)-Et(H41nd)zZr(BINOL) [BINOL = 1 ,I’-bi-2-naphtholate], yields optically active poly(methylene- 1,3cyclopentane) (PMCP) with a molar optical rotation of +5 1 .O”. Cyclopolymerization with (S,S)-Et(H41nd)2Zr(BINOL) affords the enantiomeric

ZIEGLER-NATTA CATALYSTS 487
PMCP with a molar optical rotation of -51.2”. Enantioselective cyclopolymerization represents a novel strategy for synthesis of optically active main-chain chiral polymers.43
6.2. The Mechanism of Stereoregulation
The stereoselectivity of these catalysts is believed to arise mainly from the steric interactions between the complexing monomer and Cp ligands of the active center and the last inserted monomer unit of the polymer chain. It may also be influenced by the electronic environment of the ligands.
Some mechanistic models have been proposed based on a correlation of the steric structures of metallocenes and the microstructures of the resulting polymer products.
6.2.1. Pino’s Model
An earlier model was proposed by Pino for polymerization of the propylene with the isospecific Et(H41nd)zZrC12/MA0 system. Although the exact structure of the active centers was unknown, Pino suggested that the essential features of the active center are, besides the two r-bonds between the Zr atom and the Cp ligands, a bond between the Zr atom and the MAO macromolecule, and a bond between the Zr atom and the last carbon atom of the growing polymer chain.
The cyclohexenyl group of one of the indenyl groups represents a ligand with a large steric hindrance to the incoming monomer, the two CH groups of the cyclopentadienyl ring of the other indenyl group represent the substituent with low steric hindrance to incoming monomer, and the alumoxane macromolecule represents a ligand with a large steric hindrance to the incoming monomer which renders the 1,24nsertion favored over the 2,1-insertion. The corresponding structure of the low energy transi- tion state leading to an isotactic dyad, and of the transition state with higher energy leading to a syndiotactic dyad are represented by r and s in Fig. 17, respectively. Since the transition state r is energetically favored, the catalyst is isospecific, and the stereoregulation is chiral site control. The meso Ti complex in the analogous Et(H41nd)2TiC12/MA0 system leads to atactic polymers. (See Fig. 17 t and u.)
6.2.2. Corradini’s Model
In the Corradini mode1,33 the active species is cationic. In addition to the aromatic Cp ligands of the precursor metallocenes, an incoming monomer molecule and a growing polymer chain are also coordinated to the metal in the models for the stage preceding monomer insertion. Calculations of the non-bonded interactions for the models suggested that the coordination of an additional ligand is unlikely. Thus MAO could not directly bond to the transition metal. The suggestion of cationic active species has been confirmed and accepted by other researchers.16’18,19
In a cationic active species there are two possible types of chirality, that is, chirality arising from coordinated ligands (type i) and the intrinsic chirality at the central metal atoms (type ii).
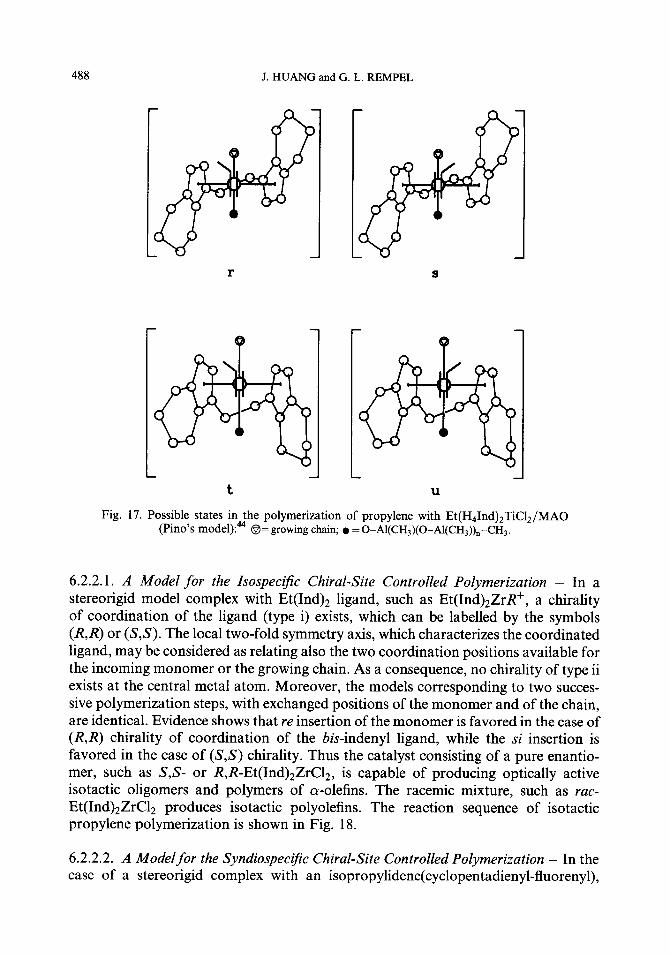
488 J. HUANG and G. L. REMPEL
Fig. 17. Possible states in. fhe polymeriz ,at ion 1 of propylene with Et(H41nd)2TiC12/MA0 (Pino’s model):* @= growing chain; l = 0-A1(CH,)(O_Al(CH,))._CH3.
U
6.2.2.1. A Model for the IsospeciJc Chiral-Site Controlled Polymerization - In a stereorigid model complex with Et(Ind), ligand, such as Et(Ind)2ZrR+, a chirality of coordination of the ligand (type i) exists, which can be labelled by the symbols (RJ?) or (S’S). The local two-fold symmetry axis, which characterizes the coordinated ligand, may be considered as relating also the two coordination positions available for the incoming monomer or the growing chain. As a consequence, no chirality of type ii exists at the central metal atom. Moreover, the models corresponding to two succes- sive polymerization steps, with exchanged positions of the monomer and of the chain, are identical. Evidence shows that re insertion of the monomer is favored in the case of (R,R) chirality of coordination of the his-indenyl ligand, while the si insertion is favored in the case of (S,S) chirality. Thus the catalyst consisting of a pure enantio- mer, such as S,S- or R,R-Et(Ind)2ZrC1,, is capable of producing optically active isotactic oligomers and polymers of a-olefins. The racemic mixture, such as rac- Et(Ind)2ZrClz produces isotactic polyolefins. The reaction sequence of isotactic propylene polymerization is shown in Fig. 18.
6.2.2.2. A Modelfor the SyndiospeciJic Chiral-Site Controlled Polymerization - In the case of a stereorigid complex with an isopropylidene(cyclopentadienyl-fluorenyl),

ZIEGLER-NA-ITA CATALYSTS 489
--I Fig. 18. The reaction sequence for isotactic propylene polymerization.6
iPr(CpFlu) ligand, there is no chirality of the coordination of the aromatic Flu and Cp ligands. A local symmetry plane characterizes the coordinated bridged ligand and relates also the two coordination positions available for the incoming monomer or the growing chain. However, the central metal atom is chiral (type ii), so that the two possible placements obtained by exchanging the coordination positions of the mono- mer and of the polymer chain are enantiomorphic. This kind of chirality can change during the polymerization reaction, and actually it will be inverted at most polymer- ization steps. If some enantioselectivity exists, the model complex has a tendency to be syndiospecific.
In the polymerization of propylene with iPr(CpFlu)ZrClJMAO catalyst, since the presence of the bulky fluorenyl group strongly reduces the room available for propy- lene complexation, the monomer can only approach the catalyst in two enantio- symmetric modes, with the re-face on one coordination site or with the si-face on the other. At each polymerization step, the coordination site moves from one side to the other of the catalyst, thus producing syndiotactic polypropylene.
The model can also explain why iPr(3-MeCpFlu)ZrClT produces hemiisotactic polymers. Introduction of a methyl group into the Cp ring of iPr(CpFlu)ZrC12 destroys the symmetry of the complex and the enantioselectivity of one side, at which the methyl group locates, of the catalyst. As a consequence, polymerization proceeds through two non-equivalent steps: on one side coordination occurs as in the syndiospecific catalyst iPr(CpFlu)ZrC12 while on the opposite side coordination is not controlled and addition of the monomer occurs in a random fashion. At the next step stereochemical control is again effective, being identical with that of the penultimate step; this fulfils the condition for production of a hemiisotactic polymer.

490
Fig. 19. An example of van
6.2.3. Brintzinger’s Model
der Waals’ outline of the ligand framework for the catalyst complex.45
Brintzinger and coworkers45’46 proposed a two-parameter (aperture/obliquity) model, for the correlation of chiral ansa-metallocene structures of C, symmetry with their properties as homogeneous Ziegler-Natta catalysts. Instead of analyzing individual conformers of the complete reaction complex, they considered dissected moieties separately and tried to define a rough measure for: (i) the cuneiform inner surface of the ansa-metallocene ligand framework, and (ii) the wedge-shaped outline of the “reaction complex” enveloping the coordinated reactants.
Brintzinger defined, as an inner cuneiform surface of the ligand framework, those two planes through the transition metal center which tangentially touch the ligand van der Waals’ surface at two ,8-substituents of each C5 ring. These two tangential planes intersect at an angle which is called the coordination gap aperture. The largest possible size of this angle in a given complex would provide a reasonable measure for the openness of its coordination site. The “edge” of the coordination gap, i.e. the intersection of the two tangential planes, is twisted relative to the plane bisecting the centroid-metal-centroid angle by an angle which is called the coordination gap obliquity. The obliquity angle for ansa-metallocene complexes can have either a posi- tive or negative sign, which is equivalent to S- or R-configuration, depending on the enantiomer considered. The coordination gap aperture is determined mainly by the ,& substituents at Cs rings and the structure of the bridging unit. The a-substituents at Cs rings have a minor effect. The obliquity sign descriptor depends only on the relative size of the two P-substituents at each C5 ring.
Based on a number of theoretical and experimental studies, Brintzinger assumed that the insertion transition state for a metallocene catalyst has a geometry close to that represented in Fig. 20. It is a four-membered metallocycle in which the migrating alkyl and olefin a-carbon atoms are attached to the metal center. The orientation of the substituents at this complex is controlled by the degree of partial sp3-hybridization

ZIEGLER-NATTA CATALYSTS 491
Fig. 20. Transition state geometry assumed for insertion of propylete into the Zr-Et bond of a zirconocene alkyl cation (bond distances in A).45
at the olefinic @terminus and by an agostic interaction of one of the H atoms at the migrating alkyl group. An important restriction of the transition state geometry results from the assumption that the olefin and polymer-chain substituents at the incipient C s . . C bond are oriented trans to each other.
The spatial requirements of any given transition state are determined by two planes through the metal center which tangentially touch the van der Waals’ spheres of the reaction complex at those two substituents which flank each side of the incipient C . .. C bond. The angle between these two planes is called the reaction complex aperture, which represents a reasonable measure for the spatial extension of the transition state under consideration. The two tangential planes define the reaction complex obliquity by the orientation of their intersection with respect to the ligand plane spanned by the metal center and two a-C atoms of the coordinated alkyl and olefin groups, respectively. The reaction complex obliquity is a measure for the chirality of the transition state considered. The aperture and obliquity angle of the reaction complex are determined by the transition metal and the monomer molecules.
The catalyst activity and stereoregularity are determined by the matching of the
--3A -
Fig. 21. Propylene-insertion state as in Fig. 20; projections parallel to both tangential planes (left) and parallel to the ZrC3 ligand plane (right).45
492 J. HUANG and G. L. REMPEL
parameters (aperture and obliquity) of the coordination gap and the reaction complex. A larger coordination gap, which accommodates the transition complex wedge with- out any particular steric strain and allows some mismatch of obliquity, will result in higher activity and lower stereoregularity. On the other hand, the very tight fit between reaction complex wedge and coordination gap is associated with a distinctly reduced activity and with a very high stereoregularity.
The analysis of the other metallocenes, e.g. of syndiospecific complexes with C, symmetry, revealed a different type of coordination gap asymmetry: while the obliquity would necessarily disappear in these cases, the coordination gap bisector plane is “vertically” kinked, relative to the centroid-metal-centroid bisector plane. Brintzinger suggested that the coordination gap kink angle should replace the obli- quity angle as the parameter for the model of syndiospecific metallocene catalysts.
6.3. Other E&cts on the Stereospecificity of the Catalysts
Besides the steric interaction of the active species and incoming monomer, there are many factors also affecting the stereospecificity of the catalysts.
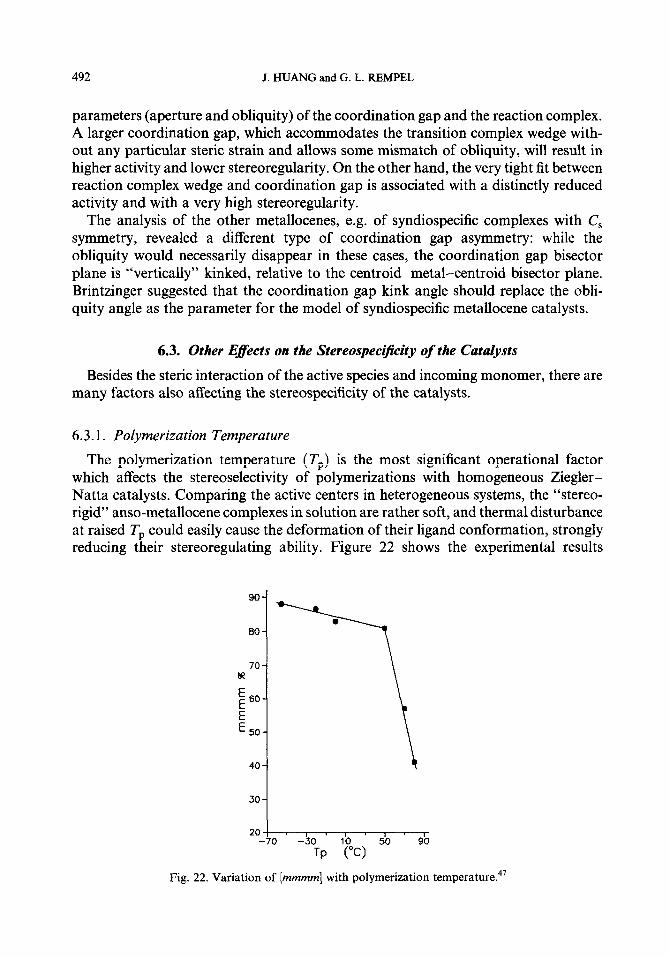
6.3.1. Polymerization Temperature
The polymerization temperature (Tp) is the most significant operational factor which affects the stereoselectivity of polymerizations with homogeneous Ziegler- Natta catalysts. Comparing the active centers in heterogeneous systems, the “stereo- rigid” anso-metallocene complexes in solution are rather soft, and thermal disturbance at raised Tp could easily cause the deformation of their ligand conformation, strongly reducing their stereoregulating ability. Figure 22 shows the experimental results
*O--1 TP (“C>
Fig. 22. Variation of [mmmm] with polymerization temperature.47

ZIEGLER-NA’ITA CATALYSTS 493
Table 3. The effect of [MAO] on mm and mmmm fractions of iso-PP produced at 30”C47
Catalyst system IWE-
ratio mm triad fraction
mmmm pentad fraction
Et(Ind)zZrClz/MAO 16400 0.885 0.835 Et(Ind)lZrClz/MAO 2500 0.872 0.818 Et(Ind)2ZrClz/MA0 1100 0.680 0.577 Et(H41nd)2ZrC12/MA0 75 000 0.949 0.921 Et(H41nd)2ZrClz/MA0 35 000 0.937 0.902 Et(H41nd)zZrC12/MA0 3500 0.932 0.899 Et(H41nd)2ZrC12/MA0 350 0.935 0.898
reported by Chien.47 Isotactic PP samples were made with Et(Ind)zZrCIJMAO catalyst, and the microstructures of the samples were determined by 13C-NMR and represented by the mmmm pentad content.
6.3.2. MAO Concentration
The MAO macromolecules surrounding the catalytic metallocene complexes can affect the stereoselectivity of the catalysts. Lowering of [MAO] results in iso-PP polymers containing lowered populations of homosteric sequences mm and mmmm as indicated by the data in Table 3.47
7. KINETICS
7.1. The Active Species
It should be noted that the activity values obtained directly from polymerization experiments are only apparent values which may be unsuitable for estimation of kinetic parameters, since more than one type of catalytically active species may exist in the same polymerization system, and not all active species take part in the poly- merization at the same time as many of them are temporarily dormant. In these homogeneous polymerization systems the active species could be in a series of dynamic equilibria, such as the equilibrium between the free active site and mono- mer-coordinated active site, between the active cationic site and dormant neutral site, and between one type of active species and the other, providing there is more than one type of active species present.
Based on the analyses of experimental results, some researchers have suggested that there are at least two types of active species, which have different activity and stereo- specificity, formed in the homogeneous catalyst systems. Chien37 indicated that in the case of the Et(H41nd)2ZrC12/MA0 system, two types of active species coexist in about equal amounts; one has more stereoselectivity, lo-20 times greater rate constant of propagation, and a factor of 5- 15 times faster chain transfer to MAO than the second type of active species. Metallocene complexes with different states of coordination with MAO are probably responsible for the various active species. Added evidence

494 J. HUANG and G. L. REMPEL
k c km kP
cp*Tii* + c ,d KPp$l [cl d . M CPP
\
CH=CH, _ CHFH-R + C
I
(3) (4)
C = [-AI(CHJ-O-1”
Fig. 23. Ewen’s kinetic model.*
for multiple active species involves the formation of elastic polypropylene, which is considered as a copolymer consisting of alternating blocks of stereoirregular and stereoregular PP segments. The reasonable explanation is that the catalytic species can exist in two isomeric states in equilibrium, one of which is stereospecific and the other which is not. The catalyst site can switch back and forth between the two states as propagation proceeds.42
Analyses of experimental results have indicated that in metallocene/MAO systems a large portion of the metal centers are dormant, and the catalytically active sites and the dormant sites are in equilibrium. The structure of the dormant site is unknown. It could be >M(R)-0-(Al(CHs,O), at the right side of the equilibrium shown in eq. (13) by Kaminsky,31 or as Mulhau dimerization. (See Section 7.4.) Chien R
t48 proposed the products of metallocene reported that for the Et(H41nd)*ZrC12/
MAO system, at [Al]/[Zr] > 3500 and Tp = 3O”C, two-thirds of the zirconocene became catalytically active. At low [Al]/[Zr] of 350, the total active species reduced to only 13%. Similar results were also reported by other researchers.48
Existence of multiple equilibria and multiple active species makes the kinetic study quite difficult. The polymerization rate (Rr) or the catalyst activity (A) obtained from experiments is only the apparent and average value under the assumption that all transition metal atoms become the active center. At present, the contribution of each type of active species to R, (or to A) cannot be distinguished.
7.2. Kinetic Models
7.2.1. Ewen’s Model
The first kinetic model for propagation in homogeneous systems was proposed by Ewen,* assuming that the propagation took place as shown in Fig. 23. This scheme, for the Cp2Ti(IV) polymerization of propylene, is representative of the kinetics for all of the polymerizations with Group 4B metallocenes.
Under pseudo-first-order conditions, the rate of polymerization can be expressed as:
R, = kobs&H&&][Cl, (15)
with
k ,,bsd = k,K$& + KM[C3H61 + &[‘I) (16)

ZIEGLER-NATTA CATALYSTS 495
and
KM[C3H6]+Kc[C]< 1. (17)
In the scheme, species 1 and 4 represent coordinatively unsaturated Ti(IV) complexes which are formally do 16-electron pseudotetrahedral species. Species 2 represents the catalyst in its resting state. Intermediate 3 is shown with the monomer coordinated at an al molecular orbital with the three non-Cp ligands and the transi- tion metal in a common plane.
This is a simplified model, which could explain the experimental results that showed polymerization rates vary linearly with the product of the monomer, metallocene and alumoxane concentrations at low monomer conversion with [Al] over a certain range.
7.2.2. Chien 3 Model and Parameter Estimations
Chien35’37 proposed a kinetic model for the systems in which more than one active species was present, and applied the model in the study of ethylene polymerization with Cp2ZrC12/MA0 catalyst and propylene polymerization with Et(Ind)2ZrC12/ MAO and Et(H.Jnd)2ZrC12/MA0 catalysts. The model assumes the presence of multiple active center types, chain transfer to MAO, chain transfer by P-H elimina- tion and first order deactivation reactions of active centers.
7.2.2.1. Polymerization Rate - For the system existing i types of active centers, the polymerization rate of the ith species is:
Rr,i = kp,i[Gl [Ml > (18) where kp,i and [CF] are the propagation rate constant and the concentration of the ith active species, respectively. The overall rate of polymerization is the sum of R,,is:
or, if [C;] cannot be determined,
R, = &&*I [Ml.
The total productivity P can be written as:
(20)
P = [M] C kp,i J [C,*]dt, i
Assuming that the catalytic species deactivate according to first order kinetics:
p(t) = [Ml ~kp,i[CFlo(l - eXP(-kc+>>. i
(21)
(22)
7.2.2.2. Estimation of Kinetic Parameters - [C*], is thought to be equal to the initial metal-polymer-bond concentration, [MPBlo, which can be calculated via the relation between the metal-polymer-bond concentration, [MPB], and polymerization

496 J. HUANG and G. L. REMPEL
Fig. 24. Variation of metal-polymer-bond concentration with yield of total polymer.35
yield, Y:
[MP~, = [MPB]~ + k$ Y&[M]. (23)
Since the [MPB] can be determined by the tritium radiolabelling method, a plot of [MPB], vs Y, extrapolated to Ys = 0 gives [MPB10 = [C*],. (See Fig. 24.) If the poly- mers produced with i th species can be separated from the others (Chien suggested that it can be done by solvent extraction in his study of propylene polymerization35) [C& can also be obtained from the [MPB]- Y plot for the i th fraction. The propagation rate constants kp,i and kp,avg can be calculated according to eqs. 18 and 19. In addition, the rate constants of chain transfer to MAO (/&‘) s can be obtained from the slope of the [MPB]-Y plots. The rate constant (kc) of chain transfer by ,0-H elimination, which is another main termination process, can be calculated from the MW data. The rates of chain transfer to MAO and chain transfer by P-H elimination are expressed as follows:
Rf = @[C*] [MAO], (24)
Rf = kt[C*]. (2%
The average degree of polymerization (DP) is:
thus:
DP = RP/ c & = ~~,~~*1~~1~1~~1~~*1~~~~1 + ktV*lL (26)
(DP)-’ = {(k;‘[MAO] + k$/k,}[M]-‘. (27)

ZIEGLER-NATTA CATALYSTS 497
Table 4. The rate constants for Et(H41nd)2ZrC12/MA0 in propylene polymerization37
[Al]/[Zr] ratio PP fraction kp,n (Me s1-l J&l (s-‘)
75 000 T 3500 T
c7
Gi
G
E 350 T
c7
c6
CS
E
400 0.085 970 0.015
1840 0.015 1370 0.026
80 0.003 130 0.0078
1480 0.047 2550 0.027 2590 0.041
97 0.0045 275 0.0027
[Zr] = l.OpM, Tp = 3O”C, [C3Hs] = 0.47 M (Pp = 1.7 atm). T: total, C,: C,-soluble fraction, E: ether-soluble fraction.
Since kp and @ can be determined through the [MPB]- Y plot, the value of kc can be obtained from the variation of number average degree of polymerization, DP, with
monomer concentration, i.e. from the slope of DF-[Ml-’ plot. Some kinetic parameters were reported by Chien for different polymerization
systems:
(1) Ethylene polymerization with Cp2ZrC12/MA0 system.35 Experimental conditions:
Tp = 70°C [Zr] = 48 PM, [MAO] = 51.4mM, P = 1.4atm.
Kinetic parameters:
k, = 168 (M s)-’ [C*] = 0.84 [Zr]
L$ = 0.012 s-l k[ = 0.28 s-‘.
MW and MWD at t = 1 h:
MW = 1100 MWD=2.7
kinetic chain lifetime - 4 s.
(2) Propylene polymerization with Et(H41nd)zZrC12/MA0 systems.37 The rate constants of polymerization for whole systems and for their fractions
were estimated. The results are listed in Table 4.
According to these data, Chien suggested that the system might have two types of active species (I and II), species I producing Cg- and C7-soluble fractions and species II producing C5- and ether-soluble fractions.
7.2.3. Hamielec’s Model and Computer Modelling
Hamielec et a1.4g recently proposed another kinetic model for ethylene polymeriza- tion with the Cp2ZrC12/MA0 system in a semi-batch reactor.

498 J. HUANG and G. L. REMPEL
7.2.3.1. Model Development - The assumptions made for the model are: (1) instanta- neous formation of sites of type I that produce low molecular weight polymer at 50°C; (2) irreversible first-order transition of these sites to sites of type II that produce high molecular weight polymer at 50°C; (3) first-order propagation with monomer and active sites; (4) chain transfer to monomer; (5) first-order deactivation of the sites of type II.
With these assumptions, the polymerization mechanism can be described as follows:
(1) Zr + MAO - CT Instantaneous formation of CT.
(2) G + 44 - Pl,l Instantaneous initiation of CT.
(3) C,l + M 2 &+1,1 Propagation of active species of type I.
(4) Pr,l ke - Pr,2 Transition of active species from type I to type II.
(5) 52 + A4 5 pr+1,2 Propagation of active species of type II.
(6) P,,, + M ktm’ - Qr,l + Pl,l Transfer to monomer-active species of type I.
(7) P&2 + M kmz - Qr,2 + P1,2 Transfer to monomer-active species of type II.
(8) 52 kd2 - Qr,2 Deactivation of active species of type II.
The ks, M, Q, and P, are, respectively, the rate constants, the monomer, dead polymer and living polymer.
The polymerization rate model was developed as follows:
RATE = 8, eekc’ + kc
kc82 [e-kdzl _ e-“‘] - kd2
(28)
where t is time, PE is ethylene pressure, and the lumped parameters 191 and 82 are defined as:
0, = k;lN;, 02 = k;2NTo, (29)
where k’s are the products of the ks and keq, and N;, is the initial number of active sites.
The number and weight average molecular weights of the whole polymer can be calculated by:
iv, = &I + Jl,l + h,2 + Jl,2
Lo,1 + Jo,1 + Lo,2 + 42 wn,
M
w
=L2,1 -tJ2,1 +L2,2+J2,2M
Ll,l + Jl,l + Ll,2 + 42 m’ (31)
where Mm is the molecular weight of the monomer. L,J and L,,2 are the moments of nth order for propagating polymer with site types I and II respectively, defined as:
L?I,l = c I-“& L,,2 = c %,2. (32)

ZIEGLER-NATTA CATALYSTS 499
Table 5. The experimental conditions and the estimated kinetic parameters4’
$1 k;z kc kd2 &ml k:m2
No. (2) (fd) (I!%!) (rnzr:06) (min - psi)-’ @in)-’ (min. psi)-’
1 71 13.08 21.08 0.035 22510 51 140 0.00591 0.0357 0.866 nla 2 50 13.08 21.08 0.650 835 1466 2.842 0.0366 0.8 0.011 3 71 6.04 21.08 0.231 3856 9649 0.00403 0.2569 0.120 nia 4 50 6.04 21.08 0.154 1961 5390 0.369 0.0218 0.160 0.0403 5 71 13.08 13.18 0.335 2582 2340 0.00666 0.0269 0.12 nla 6 50 13.08 13.18 0.160 3916 6618 0.0619 0.0530 0.095 0.0550 7 71 6.04 13.18 0.231 3536 2229 0.00474 0.0339 0.12 nla 8 50 6.04 13.18 0.142 2288 5650 0.250 0.0288 0.10 0.040
Jn,i and J,,* are the moments of nth order for dead polymer terminated at site types I and II, respectively, defined as:
Jn,l = c fQ,,l Jn,2 = ~rnQr,2, (33)
and r is the chain length.
7.2.3.2. Computer-Estimation of Kinetic Parameters - The estimation of kinetic para- meters employed computer-modelling technology. The approach is totally different from the conventional methods. The estimation procedures are: (1) Estimate para- meters 19,) 62, k, and kd2 by fitting the experimental data to the rate model (eq. 28). (2) Assign values for NT,, kiml and kirm2 until the best agreement between experimentally measured MW values and computer-calculated MW values is achieved. This step involves calculation of moments through 12 differential equations. A special program called LSODAR was employed to solve these equations. (3) Calculate the kkls and kL2s using the values of NT, obtained from step 2. Some of the results are shown in Table 5.
7.3. The Operational Factors Aficting Catalyst Activity
7.3.1. Polymerization Temperature ( Tp)
Tp is one of the most important operational factors for polymerizations. A detailed study of the Tp effect on catalyst activity was reported by Chien et a1.37 The propylene polymerizations initiated by rat-Et(H41nd)2ZrC12/MA0 and by rac-Et(Ind)2ZrC1z/ MAO proceeded at T,, of -55430°C. The polymerization activities increase mono- tonously with temperature. The activity data are shown in Table 6.
The Arrhenius plot of activity is linear over the entire range of Tp. The data points of the two catalyst systems fall on the same line. (See Fig. 25.) The overall activation energy for polymerization with both catalysts is 10.6 kcal mol-‘.
The strong dependence of the catalytic activity of zirconocene/MAO system on Tp was initially thought to be just the activation energy required for polymerization.
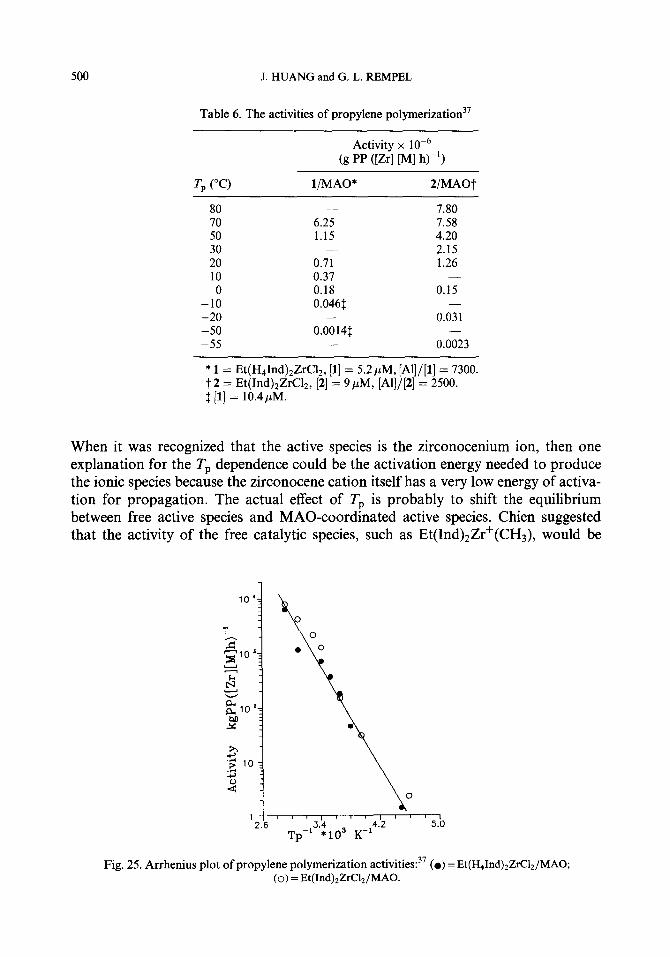
500 J. HUANG and G. L. REMPEL
Table 6. The activities of propylene polymerization37
Activity x 10m6 (g PP (lzrl WI W’)
Tp CC> l/MAO* Z/MAO?
80 70 50 30 20 10 0
-10 -20 -50 -55
6.25 1.15
- 0.71 0.37 0.18 0.046$
-
0.0014$ -
7.80 7.58 4.20 2.15 1.26
0.15
0.031
0.0023
* 1 = Et(H41nd)2ZrC12, [l] = 5.2pM, [Al]/[l] = 7300. t 2 = Et(Ind)2ZrC12, [2] = 9 PM, [All/[21 = 2500. $ [l] = 10.4pM.
When it was recognized that the active species is the zirconocenium ion, then one explanation for the Tp dependence could be the activation energy needed to produce the ionic species because the zirconocene cation itself has a very low energy of activa- tion for propagation. The actual effect of Tp is probably to shift the equilibrium between free active species and MAO-coordinated active species. Chien suggested that the activity of the free catalytic species, such as Et(Ind)2Zr+(CH3), would be
lfl~~,~~~I"'I 2.6 -13.4 4.2 5.0
TP *lo’ K-’
Fig. 25. Arrhenius plot of propylene polymerization activities:37 (0) = Et(H.+Ind),ZrCl,/MAO; (0) = Et(Ind)zZrC12/MA0.
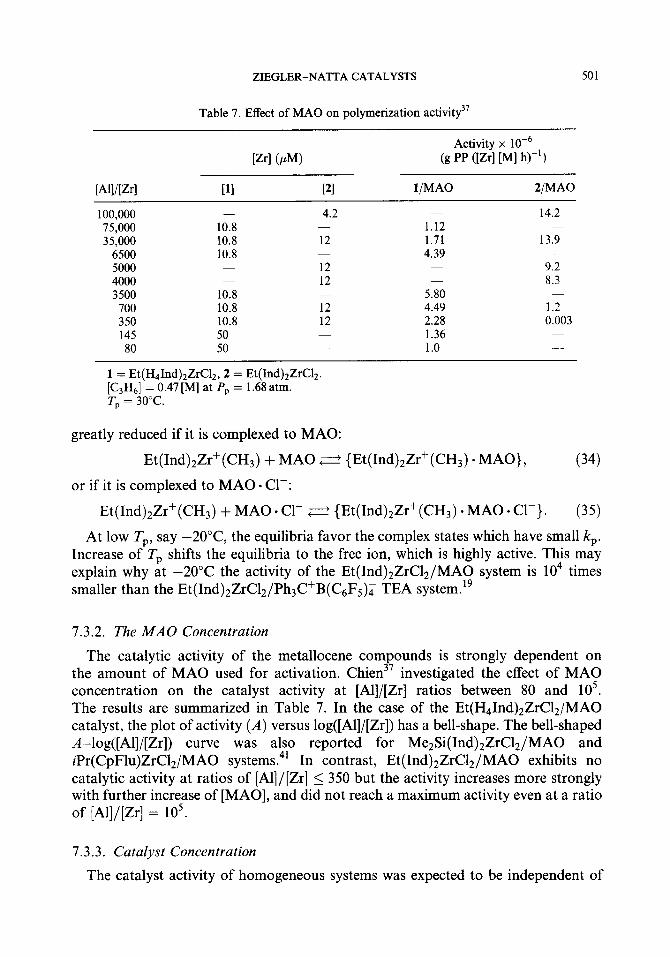
ZIEGLER-NATTA CATALYSTS 501
Table 7. Effect of MAO on polymerization activity3’
WI (CIW Activity x 1O-6
k PP (lzrl WI W’)
[AlI/tZrl 111 PI l/MAO 2/MAO
100,000 75,000 35,000
6500 5000 4000 3500 700 350 145 80
10.8 10.8 10.8
10.8 10.8 10.8 50 50
4.2 14.2 1.12
12 1.71 13.9 4.39
12 9.2 12 8.3
5.80 12 4.49 1.2 12 2.28 0.003
1.36 - 1.0
1 = Et(HJnd)2ZrC12, 2 = Et(Ind)zZrC1z. [C3H6] = 0.47[M] at Pp = 1.68atm. TP = 30°C.
greatly reduced if it is complexed to MAO:
Et(Ind)2Zr+(CH3) + MAO I {Et(Ind)2Zr+(CH3) *MAO},
or if it is complexed to MAO. Cl-:
(34)
Et(Ind)2Zr+(CHs) + MAO. Cl- I {Et(Ind)2Zr+(CH3) - MAO. Cl-}. (35)
At low Tp, say -20°C the equilibria favor the complex states which have small kp. Increase of Tp shifts the equilibria to the free ion, which is highly active. This may explain why at -20°C the activity of the Et(Ind)fZrClz/MAO system is lo4 times smaller than the Et(Ind)2ZrClz/Ph3C+B(C6F5>4 TEA system.”
1.3.2. The MAO Concentration
The catalytic activity of the metallocene compounds is strongly dependent on the amount of MAO used for activation. Chien37 investigated the effect of MAO concentration on the catalyst activity at [Al]/[Zr] ratios between 80 and 105. The results are summarized in Table 7. In the case of the Et(H41nd)zZrClz/MA0 catalyst, the plot of activity (A) versus log([Al]/[Zr]) has a bell-shape. The bell-shaped
A-WF4/[Zrl) curve was also reported for Me$i(Ind)2ZrC1:!/MA0 and iPr(CpFlu)ZrClJMAO systems.41 In contrast, Et(Ind)2ZrC12/MA0 exhibits no catalytic activity at ratios of [Al]/[Zr] 5 350 but the activity increases more strongly with further increase of [MAO], and did not reach a maximum activity even at a ratio of [Al]/[Zr] = 105.
7.3.3. Catalyst Concentration
The catalyst activity of homogeneous systems was expected to be independent of
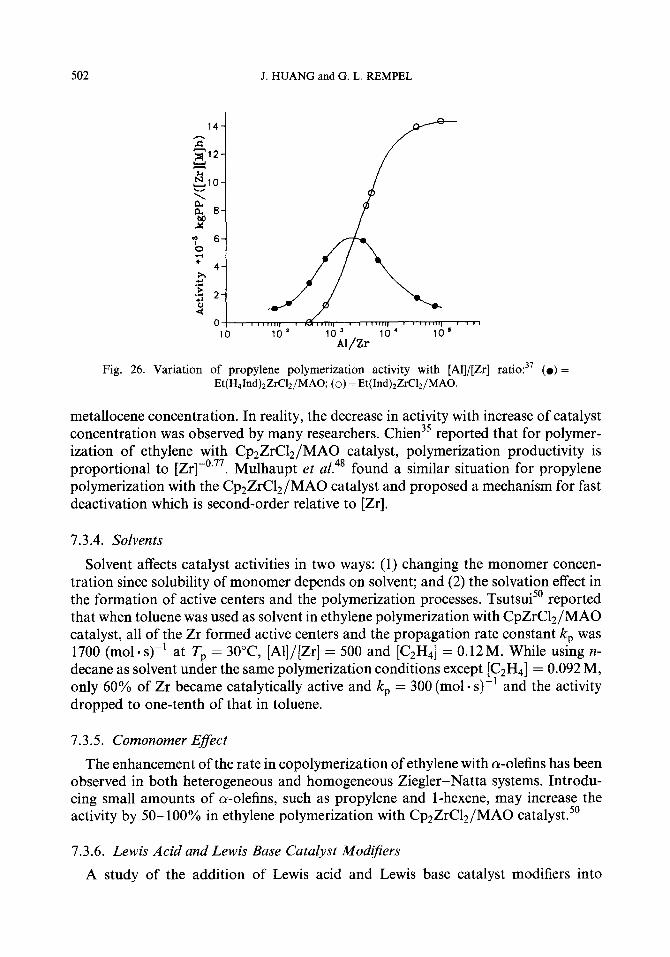
502 J. HLJANG and G. L. REMPEL
Fig. 26. Variation of propylene polymerization activity with [Al]/[Zr] ratio:37 (0) = Et(H41nd)2ZrCIz/MAO; (0) = Et(Ind)2ZrClz/MA0.
metallocene concentration. In reality, the decrease in activity with increase of catalyst concentration was observed by many researchers. Chien35 reported that for polymer- ization of ethylene with Cp2ZrC12/MA0 catalyst, polymerization productivity is proportional to [Zr]-0.77. Mulhaupt et aL4* found a similar situation for propylene polymerization with the Cp2ZrC12/MA0 catalyst and proposed a mechanism for fast deactivation which is second-order relative to [Zr].
7.3.4. Solvents
Solvent affects catalyst activities in two ways: (1) changing the monomer concen- tration since solubility of monomer depends on solvent; and (2) the solvation effect in the formation of active centers and the polymerization processes. Tsutsui” reported that when toluene was used as solvent in ethylene polymerization with CpZrC12/MA0 catalyst, all of the Zr formed active centers and the propagation rate constant !r, was 1700 (mol. s)-’ at Tp = 3O”C, [Al]/[Zr] = 500 and [CZH4] = 0.12 M. While using n- decane as solvent under the same polymerization conditions except [CZH4] = 0.092 M, only 60% of Zr became catalytically active and k, = 300 (mol. s)-’ and the activity dropped to one-tenth of that in toluene.
7.3.5. Comonomer Eflect
The enhancement of the rate in copolymerization of ethylene with a-olefins has been observed in both heterogeneous and homogeneous Ziegler-Natta systems. Introdu- cing small amounts of a-olefins, such as propylene and 1-hexene, may increase the activity by 50-100% in ethylene polymerization with Cp2ZrC12/MA0 catalyst.50
7.3.6. Lewis Acid and Lewis Base Catalyst Modljiers
A study of the addition of Lewis acid and Lewis base catalyst modifiers into

ZIEGLER-NATTA CATALYSTS 503
Cp2ZrC12/MA0, Et(H41nd)2ZrC12/MA0 and Et(Ind)zZrClz/MAO systems was reported by Mulhaupt et d4’ The addition of Lewis bases such as 2,2,6,6-tetra- methylpiperidine (TMP) reduces catalyst activities. This behavior is expected on the basis of complex formation of the Lewis bases with MAO and zirconocenes. Since MAO is present in large excess, MAO can scavenge a large portion of the Lewis base. When Lewis bases are present at higher concentration (Lewis base/Al molar ratio > 0.005) they compete successfully with propylene for the vacant coordination site at the zirconocene complexes, thus deactivating the catalysts.
Lewis acids such as borontrichloride only marginally affect propylene poly- merization, but it was unexpectedly found that trimethylboroxine (TMB), the cyclic anhydrides of boric acid, could substantially increase catalyst activity. Three- to five- fold catalyst productivities have been obtained when TMB is added at low TMB/Al molar ratios. At a higher TMB/Al ratio of approximately 0.1, the catalysts are severely poisoned.
“B-NMR spectroscopic data indicated that the oxygen atoms in TMB disappeared immediately after TMB was brought into contact with MAO. Some reactions may have occurred between TMB and MAO. It appears possible that boroxines eliminate a trace of trimethyl aluminum, a known catalyst poison, and form new Lewis acids which may promote the formation of cationic active zirconium sites. Also the in situ formation of heterogenized catalyst via MAO cross-linking may account for some increased activity.
7.4. Catalyst Deactivation
A slow catalyst deactivation was observed in almost all polymerizations with metallocene/MAO catalysts. The deactivation might be attributed to the formation of inactive species with a M-CH2-Al structure.31 Although these inactive species can be reactivated by reaction with MAO at high [MAO] - as MAO is consumed during the polymerization by side reactions, by impurities, by chain transfer and by recreating active sites - the regeneration of the active sites will decrease at lower [MAO], and it may not compensate for the loss of the active sites. Thus a polymerization rate decay occurs.
Mulhaupt et aL4’ found that there is another type of deactivation, which is fast and second-order relative to the active site concentration, in the Cp2ZrC12/MA0 system. The catalyst activation is completed within a few seconds after contacting zirconocene with MAO. After the maximum rate is reached, the second-order decay starts. It is fast as compared to the very slow subsequent decay. Mulhaupt proposed a kinetic scheme as shown below:
7 Q2Zr +MAO s Cp2ZrF + [MAO-X@] (36)
Dormant

504 J. HUANG and G. L. REMPEL
2c* 5 cz - c* dormant inactive
(37)
/ /*\ 2Cp2Zr + XCpzZr ZrCp26
‘P n ‘P/ ?I
X
Cp2Zr@ / 7\
‘P
+ Cp2Zr I Cp2Zr I ZrCp2P, Y@
n ‘Y ‘P/ ?I I
(38)
(39)
X, Y: Me, Cl, 0-AlMe-MAO; P: polymer.
The mechanism involves reversible second-order deactivation combined with a slower irreversible deactivation of the active and/or dormant zirconium sites. (See eq. 37.) The reversible conversion of active cationic zirconium sites into dormant neutral zirconium sites is shown in eq. 36. Most likely, the reversible second-order deactivation results from zirconocene dimerization as illustrated in eqs 38 and 39.
7.5. Molecular Weight and Molecular Weight Distribution
7.5 . 1. Chain Transfer Reactions
As in heterogeneous Ziegler-Natta catalyst systems, polymer chain growth is terminated by chain transfer reactions in homogeneous metallocene-based catalyst systems. Several types of transfer reactions which may occur in the homogeneous systems are listed as follows.
7.5.1.1. /?-Hydrogen Elimination - The metal center abstracts a H atom bonded to the P-C of the growing polymer chain, forming an M-H bond (M = transition metal) and leaving a polymer with an unsaturated end:
CH3 73
Zr-CH2-CH-P - CH2=C-P + Zr-H
P: polymer chain.
7.5.1.2. Chain Transfer by Monomer - P-Hydrogen elimination and olefin monomer insertion at the active center take place simultaneously without forming the M-H bond:

ZIEGLER-NA’M’A CATALYSTS 505
CH3
FH3 CH3 I
CH,-7-P /
Zr-CH,--&H-p 3 Zr / l!I li,C = kH-CH3
CH3
+ CHz=C-P + Zr-CH2-CH2-CH3.
7.5.1.3. Chain Transfer to MAO - The growing polymer chain attached to an active center exchanges with the methyl group of a MAO molecule, if MAO is used to activate catalyst, forming the M-CH3 bond in the active center and the Al-terminated polymer chain:
CH3 CH3
Zr-CH2-CH-P w Al-CH;!---CH-P + Zr-CH3
1 H+
CH3
\ CH-P
/ CH3
7.5.1.4. ,BCH3 Elimination - This is a newly discovered chain transfer mechanism in the polymerization of propylene. The metal center abstracts a CH3-group, instead of a H atom, at the ,0-C of the growing chain, thus forming a M-CH3 bond at the active center and leaving a polymer with an ally1 end?
7H3 7H3 7H3
Zr-CH2-CH-CH2-CH-P - Zr-CH3 + CH2=CH-CH2-CH-P.
Figure 27 was proposed by Rescon?’ to outline the catalytic cycles for propylene dimerization with (MeSCp)$%K12/MA0 (M = Zr, Hf). Here it is used to represent the propagation and termination reactions which could occur in the homogeneous Ziegler-Natta polymerization systems.
The chain transfer normally occurs via facile p-elimination (including transfer by monomer) in homogeneous Ziegler-Natta systems, which is the main reason for the much lower molecular weights of the produced polymers, compared with heterogeneous catalysts. According to analyses of the end-groups of copolymers obtained from ethylene-propylene copolymerization, Tsutsui52 concluded that for polymerization

506 J. HUANG and G. L. REMPEL
Fig. 27. Catalytic cycles for propylene polymerization.51
of propylene with Cp,ZrClJMAO catalysts, the chain transfer reaction would almost exclusively take place through a chain transfer by monomer mechanism.
Recently, Resconi et ~1.~~~~~ found that a different chain transfer mechanism, namely P-CH3 elimination, becomes viable and is actually the most important mechanism in propylene polymerization when (Me5Cp)#rV-type complexes (M = Zr, Hf) are used as catalyst precursors. A comparative study of propylene polymerizations with Cp2ZrClz, Cp,HfQ, (Me5Cp)2ZrC12, and (Me5Cp)2HfC12 catalysts was made based on 13C-NMR and GC-MS analyses of the dimer to pentamer fraction of polypropylene oligomers. The results are shown in Table 8. It seems that P-CH, elimination is a unique feature of the (Me,Cp),@-type catalysts. Chain transfer
Table 8. Percentage of different chain transfer mechanisms in propylene polymerization51
Metallocene TP P-H B-Me Al-tr
Cp,ZrClz 50 Cp,ZrC12 0 Cp,HfClz 50 (MeJpLZrCl2 50 We&pLZrC& 0 (MesCpLZrCl2 -40 (Me&pLHfClz 50 (MeKpLHfCl2 0
100
100 100
7.9 7.1
2 2.0
-
- 91.1 81.8 -
98 62.7
-
1 11.1
100 trace 35.3

ZIEGLER-NATTA CATALYSTS 507
to aluminum is only a minor termination mechanism at Tp = 50°C but it becomes the major one at low T,, (-40°C). In I-butene polymerization with (Me5Cp)2ZrC12 and (MeSCp)zHfClz catalysts, &Et transfer appeared to be inaccessible. A possible expla- nation is that an ethyl group is not easily accommodated in the reaction plane between the two Me$p ligands.
7.5.2. The Molecular Weight (MW)
The molecular weight of a polymer is a result of competition between chain growth and different chain termination reactions:
Mn = DP * Mm = (Rp/ C Rtr,i) Mm.
With conventional heterogeneous Ziegler-Natta catalysts molecular weights of polymers from ethylene and propylene polymerization are rather high, i.e. about 500,000, so that chain terminating agents like hydrogen have to be utilized. With homogeneous metallocene catalysts the situation is the opposite; many systems pro- duce only low molecular weight polymers, especially in stereospecific polymerizations.
7.5.2.1. The Dependence of MW on Metallocene Structure - Polymers produced with hafnocene catalysts have higher molecular weights than those with zirconocene and titanocene analogues, because the higher Hf-C bond energy suppresses the chain transfer reactions but also causes lower activity of the catalysts.
The effect of ligands on polymer molecular weight is very complicated. Usually, the MW of polymers can be increased by sterically and/or electronically suppressing chain transfer reactions.
(1) Steric effect: As mentioned previously, for most systems chain transfer occurs by P-H elimination. In order to reach the transition state for ,0-H elimination, it is necessary to rotate the polymer chain about the C(a)-C(p) bond so that the filled C(P)-H (T orbital overlaps with one of the vacant orbitals on the group 4B transi- tion metal atom. Steric effects can cause a significant increase in the energy of the transition state associated with P-H elimination, due to the non-bonded repulsion between the polymer chain and the periphery of the bulky ligand such as a fluorenyl ring. This effect can scale with the lateral dimensions of the carbocyclic ligand, increasing in the order Cp < Ind < Flu.” (2) Electronic effects: As the ligands around electron deficient Zr (or Hf, Ti) become more electron releasing, the thermodynamic driving force for ,0-H elimination diminishes. The electron releasing ability increases in the order Cp < Ind < Flu.” If the steric and electronic factors of ligands have opposite effects, the overall effect depends on which one is more pronounced.
The results obtained from Kaminsky’s comparative study (See Tables 1 and 2) show that for ethylene polymerization, there is no predominant structural pattern for a high MW system. The five systems with highest Mq include unbridged and bridged systems among the latter having C,- or C,-symmetry. For propylene polymerization, the
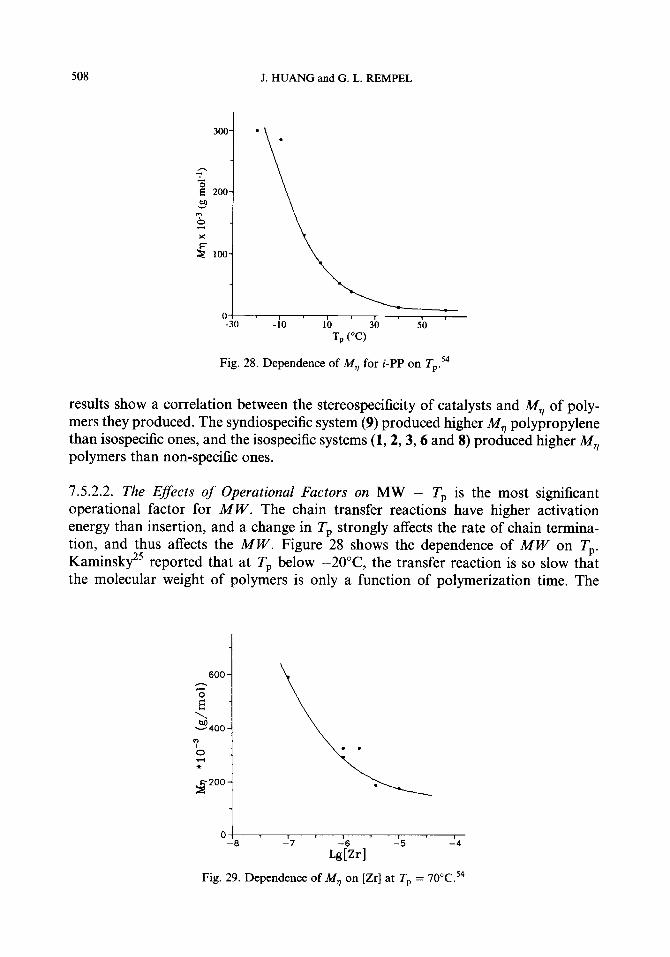
J. HUANG and G. L. REMPEL
300-
‘: s E ml-
s *? $: K
z loo-
01 -30 -10 10 30 50
T, (“‘2
.
Fig. 28. Dependence of M,, for i-PP on Tp.54
results show a correlation between the stereospecificity of catalysts and Mq of poly- mers they produced. The syndiospecific system (9) produced higher A&, polypropylene than isospecific ones, and the isospecific systems (1,2,3,6 and 8) produced higher A& polymers than non-specific ones.
7.5.2.2. The Eficts of Operational Factors on MW - TP is the most significant operational factor for MW. The chain transfer reactions have higher activation energy than insertion, and a change in TP strongly affects the rate of chain termina- tion, and thus affects the MW. Figure 28 shows the dependence of MW on TP. Kaminsk95 reported that at TP below -2O”C, the transfer reaction is so slow that the molecular weight of polymers is only a function of polymerization time. The
OC -0 -7
IA&X]
-5 -4
Fig. 29. Dependence of Mq on [Zr] at Tp = 70”C.54

ZIEGLER-NATTA CATALYSTS 509
\ e o FP
FP’ Si ~1
&
\ R’ zq
k2
211-e 3a.b
Fig. 30. Structures of specifically designed zirconocenes.56
concentration of metallocene catalyst is also significant to MW. As shown in Fig. 29, the average molecular weight decreases with the logarithm of increasing transition metal concentration.54 At low levels of [MAO], e.g. [Al]/[Zr] < 1000, an increase in [MAO] can significantly increase MW of polymer products. However, at high levels of [MAO], e.g. [Al]/ [Zr] > 10,000, further increases of [MAO] do not have much effect on MW.
7.5.3. Methods to Increase the Molecular Weight
Poor molecular weights for the polymer products is a major shortcoming with homogeneous metallocene catalyst systems. This limits these catalysts from being fully exploited for practical applications. There are some ways to improve MW of the polymer products:
(1) Using hafnocenes. As discussed above, replacing zirconocene catalysts with hafnocene analogues can increase the MW of polypropylene by one order of magnitude. (2) Reducing Tp. Since chain transfer reactions have higher activation energies than the propagation reaction, lowering Tp can efficiently suppress the transfer reactions, thus increasing MW. (3) Heterogenizing metallocene catalysts. Soga5’ reported that propylene poly- merization with the heterogeneous catalyst system Et(H41nd)2ZrC12/MAO/SiOz combined with Al(i-C4H9)s could produce polypropylene with a MW 4.5 times higher than that obtained with homogeneous Et(H41nd)2ZrC12/MA0 systems under the same conditions.
The drawback in using these methods for increasing MW is that they are always accompanied by decreases in catalyst activity.
7.5.3.1. High Molecular Weight Polypropylene Through Specifically Designed Zirco- nocene Catalysts - Recently, a technical breakthrough has been achieved in that specifically designed zirconocene catalysts have led to the production of polypropy- lenes with molecular weights far above Mw = 100,000 gmol-’ .56 The structures of the zirconocene catalysts of interest are shown in Fig. 30 and the results are listed in Table 9.
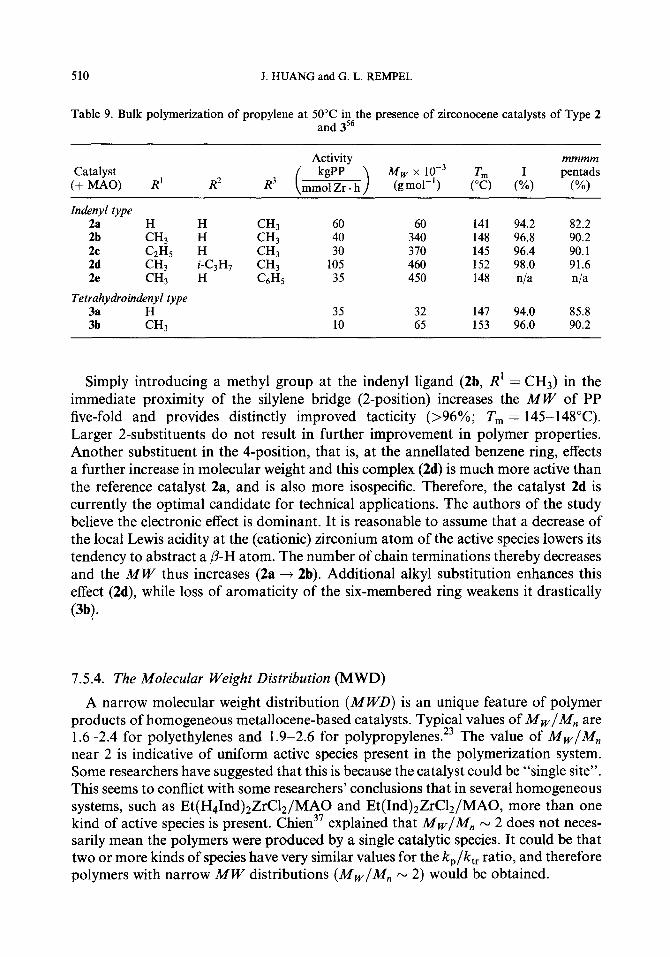
510 J. HUANG and G. L. REMPEL
Table 9. Bulk polymerization of propylene at 50°C in the presence of zirconocene catalysts of Type 2 and 356
Catalyst (+ MAO) R’ R2
Inaknyl type 2a :H, H CH3 60 60 141 94.2 82.2 2b H CH3 40 340 148 96.8 90.2 2c CzH5 H CH3 30 370 145 96.4 90.1 2d CH3 i-C3H7 CH3 105 460 152 98.0 91.6 2e CH3 H C6H5 35 450 148 nla nla
Tetrahydroindenyl type 3a H 35 32 147 94.0 85.8 3b CH3 10 65 153 96.0 90.2
Simply introducing a methyl group at the indenyl ligand (2b, R’ = CH3) in the immediate proximity of the silylene bridge (2-position) increases the MW of PP five-fold and provides distinctly improved tacticity (>96%; 7’, = 145-148°C). Larger 2-substituents do not result in further improvement in polymer properties. Another substituent in the 4-position, that is, at the annellated benzene ring, effects a further increase in molecular weight and this complex (2d) is much more active than the reference catalyst 2a, and is also more isospecific. Therefore, the catalyst 2d is currently the optimal candidate for technical applications. The authors of the study believe the electronic effect is dominant. It is reasonable to assume that a decrease of the local Lewis acidity at the (cationic) zirconium atom of the active species lowers its tendency to abstract a P-H atom. The number of chain terminations thereby decreases and the MFV thus increases (2a + 2b). Additional alkyl substitution enhances this effect (2d), while loss of aromaticity of the six-membered ring weakens it drastically
(3b).
7.5.4. The Molecular Weight Distribution (MWD)
A narrow molecular weight distribution (MWD) is an unique feature of polymer products of homogeneous metallocene-based catalysts. Typical values of Mw/M, are 1.6-2.4 for polyethylenes and 1.9-2.6 for polypropylenes.23 The value of M,/M,, near 2 is indicative of uniform active species present in the polymerization system. Some researchers have suggested that this is because the catalyst could be “single site”. This seems to conflict with some researchers’ conclusions that in several homogeneous systems, such as Et(H41nd)2ZrC12/MA0 and Et(Ind)2ZrC12/MA0, more than one kind of active species is present. Chien37 explained that Mw/M, - 2 does not neces- sarily mean the polymers were produced by a single catalytic species. It could be that two or more kinds of species have very similar values for the k,/k,, ratio, and therefore polymers with narrow MW distributions (M,/M, - 2) would be obtained.
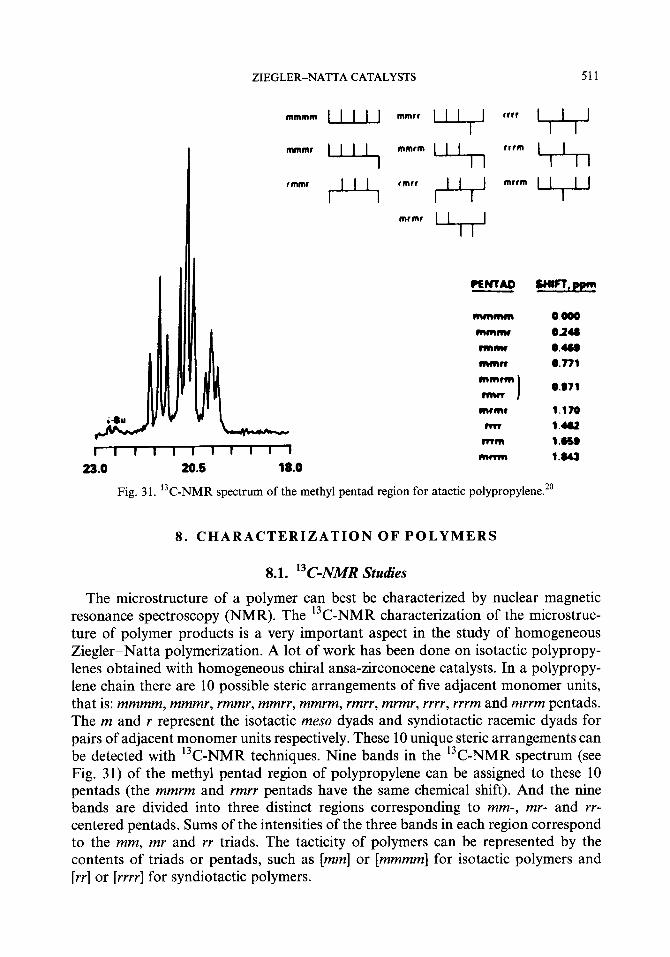
ZIEGLER-NATTA CATALYSTS 511
mmmm 1 1 1 1 ] mm” w “” w mmrm
rmrr
mrmr
t I I I II II II 1 28.0 20.5 18.0
9-l ‘Ifrn +-4 mrrm
w
mmmm mmmt rmml
mmn mmfm lJ mrmi
m?l
lllln
mwm
1 1
11 11
o.ooo o/u
0.0)
0.771
0971
1.17@ 142 1.65@ 1.843
Fig. 3 1. 13C-NMR spectrum of the methyl pentad region for atactic polypropylene.20
8. CHARACTERIZATION OF POLYMERS
8 1 13C-NA4R Studies . .
The microstructure of a polymer can best be characterized by nuclear magnetic resonance spectroscopy (NMR). The 13C-NMR characterization of the microstruc- ture of polymer products is a very important aspect in the study of homogeneous Ziegler-Natta polymerization. A lot of work has been done on isotactic polypropy- lenes obtained with homogeneous chiral ansa-zirconocene catalysts. In a polypropy- lene chain there are 10 possible steric arrangements of five adjacent monomer units, that is: mmmm, mmmr, rmmr, mmrr, mmrm, rmrr, mrmr, rrrr, rrrm and mrrm pentads. The m and r represent the isotactic meso dyads and syndiotactic racemic dyads for pairs of adjacent monomer units respectively. These 10 unique steric arrangements can be detected with 13C-NMR techniques. Nine bands in the 13C-NMR spectrum (see Fig. 31) of the methyl pentad region of polypropylene can be assigned to these 10 pentads (the mmrm and rmrr pentads have the same chemical shift). And the nine bands are divided into three distinct regions corresponding to mm-, mr- and rr- centered pentads. Sums of the intensities of the three bands in each region correspond to the mm, mr and rr triads. The tacticity of polymers can be represented by the contents of triads or pentads, such as [mm] or [mmmm] for isotactic polymers and [rr] or [rrrr] for syndiotactic polymers.
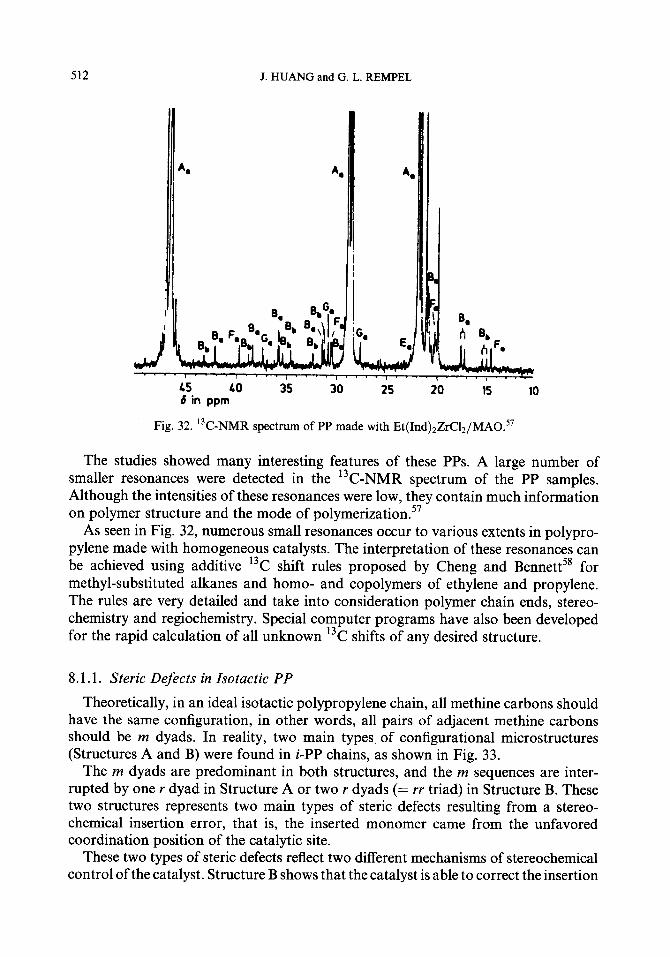
512 J. HUANG and G. L. REMPEL
4 E. u J
I....,.... . . . . . . . . . . . . . . . . . . .
45 40 is 30 25 20 6 in ppm
1’5 10
B.
Fig. 32. 13C-NMR spectrum of PP made with Et(Ind)2ZrC12/MA0.57
The studies showed many interesting features of these PPs. A large number of smaller resonances were detected in the 13C-NMR spectrum of the PP samples. Although the intensities of these resonances were low, they contain much information on polymer structure and the mode of polymerization.57
As seen in Fig. 32, numerous small resonances occur to various extents in polypro- pylene made with homogeneous catalysts. The interpretation of these resonances can be achieved using additive 13C shift rules proposed by Cheng and Bennett58 for methyl-substituted alkanes and homo- and copolymers of ethylene and propylene. The rules are very detailed and take into consideration polymer chain ends, stereo- chemistry and regiochemistry. Special computer programs have also been developed for the rapid calculation of all unknown i3C shifts of any desired structure.
8.1 .l. Steric Defects in Isotactic PP
Theoretically, in an ideal isotactic polypropylene chain, all methine carbons should have the same configuration, in other words, all pairs of adjacent methine carbons should be m dyads. In reality, two main types of configurational microstructures (Structures A and B) were found in i-PP chains, as shown in Fig. 33.
The m dyads are predominant in both structures, and the m sequences are inter- rupted by one r dyad in Structure A or two r dyads (= rr triad) in Structure B. These two structures represents two main types of steric defects resulting from a stereo- chemical insertion error, that is, the inserted monomer came from the unfavored coordination position of the catalytic site.
These two types of steric defects reflect two different mechanisms of stereochemical control of the catalyst. Structure B shows that the catalyst is able to correct the insertion

ZIEGLER-NATTA CATALYSTS 513
I 1 1 1 1 IIIIJ
Structure A Structure B
The equivalent dyad sequences:
mmmmrmmmm mmmmrrmmmm
Fig. 33. The main configurational microstructures in I’-PP chains.”
error by another Y placement immediately after the first Y dyad, so that the m sequences on both sides of the rr placement have the same relative configuration. This type of stereoregulation is termed enantiomorphic site control, because the chirality of the stereorigid catalytic site forces the incoming monomer back to its energetically favored coordination position. Enantiomorphic site control is most commonly seen in heterogeneous Ziegler-Natta polymerizations. Structure A is the result of another type of stereoregulation called chain-end control, which usually works in homogeneous systems where the chirality of the active site is absent and the monomer coordination is affected by the configuration of the last inserted monomer unit. If a stereochemical insertion error occurs, it is not corrected immediately, and the new configuration is maintained in subsequent propagation events until the next insertion error occurs.
The mechanism of stereochemical control of a catalyst can be distinguished by the study of 13C-NMR spectra of polymers. From Structure A, one can predict that the isotactic PP formed by the chain-end control will have three intense pentad bands in its 13C-NMR spectrum. Besides the most intense mmmm band, the other two bands should have similar intensity, i.e. [mmmr] = [mmrm]. According to Structure B, the isotactic PP formed by enantiomorphic site control should satisfy the following relations: [mr] = 2[ rr , and [mmmr] : [mmrr] : [mrrm] = 2: 2 : 1. i3C-NMR spectroscopy ]
provides a quantitative measurement of the fractions of the 10 pentads for a polymer sample, and thus gives accurate information about the mechanism of stereochemical control of the catalysts.
It was reported by many researcherss9 that the [mmmr] in many PP samples produced with Et(Ind)2ZrC1, and Et(H41nd)2ZrC12 catalysts is greater than [mmrr] and much larger than 2[mrrm]. This suggests that there are present many more single r insertion errors than would be expected for the enantiomorphic site control model. To explain this situation, the bi-catalytic site model and the dual control model have been pro- posed. The former assumes that part of the polymer is isotactic produced by isospecific species with the enantiomorphic site control and the rest is atactic formed by non- specific species. The latter assumes mixed stereoregulation in which the stereochemistry
is controlled by the enantiomorphic site and by the chain-end simultaneously.
8.1.2. Computer Interpretation and Simulation
Computer technology has been successfully applied in the interpretation and

514 J. HUANG and G. L. REMPEL
prediction of 13C-NMR spectra of the polymers. Different statistical models have been applied to a number of types of polypropylene.
Pioneering work in this area was started in the early 1960s when Bovey6’ and Fukurawa61 developed two simple models for chain-end control and enantiomorphic site control, respectively, based on the fractions of three triad placements, [mm], [mr], and [rr]. Later these statistical models were modified by Doi62l63 based on high resolu- tion 13C-NMR spectroscopy by which the contents of pentads could be detected.
8.1.2.1. The Chain-End Control Model - According to Bovey,60 in the case of chain- end control the fractions of the three triad placements are represented by the following relations:
[mm] = 0: (41)
[mr] = 2at(l -at) (42)
[rr] = (1 - a1)2,
where g1 is the probability that the adding propylene molecule takes the same configuration as the foregoing unit. If stereoregulation is chain-end controlled, the relation 4[mm][rr]/[mr]2 = 1 should hold. In a limiting case of g1 = 0.5, the catalytic site does not show any preference for m or r configuration, the absolute atactic PP with [mr] = 0.5 and [mm] = [rr] = 0.25 is obtained. The CJ~ can be calculated from experimentally measured triad fractions. On the other hand, if g1 is known, all the pentad intensities can be calculated using theoretical Bernoullian statistical equations for chain-end controlled stereospecific polymerizations.
8.1.2.2. The Enantiomorphic Site Control Model - The statistical model for the enantiomorphic site controlled propagation was proposed by Fukurawa.61 The triad fractions obey the following relations:
[mm] = 1 - 3a2( 1 - 02) (44)
[mr] = 2a2( 1 - q) (45)
[rrl = fl2(1 - a2>, (46)
where a2 is the probability of the monomer coordinating configuration preferred by the catalytic site. The relations 2[rr]/[mr] = 1 and 4[mm][rr]/[mr]2 = 1 are the test for the site control mechanism. The value of a2 can be calculated from [mm], [mr] and [rr] data if the enantiomorphic site control is proved.
Computer simulation (or so called statistical modelling) is a new technique dealing with complicated situations where the mechanism of stereochemical control cannot be clearly distinguished. Doing the modelling, one at first selects the statistical model and starting values of the parameters based on available knowledge, and uses a computer to carry out the computations and generate a simulated spectrum. Then according to the comparison of a simulated spectrum with a true one, the parameters will be adjusted and sent back to the computer, where the computations will be carried out over again. The process will be repeated several times until the best-fit of the simulated
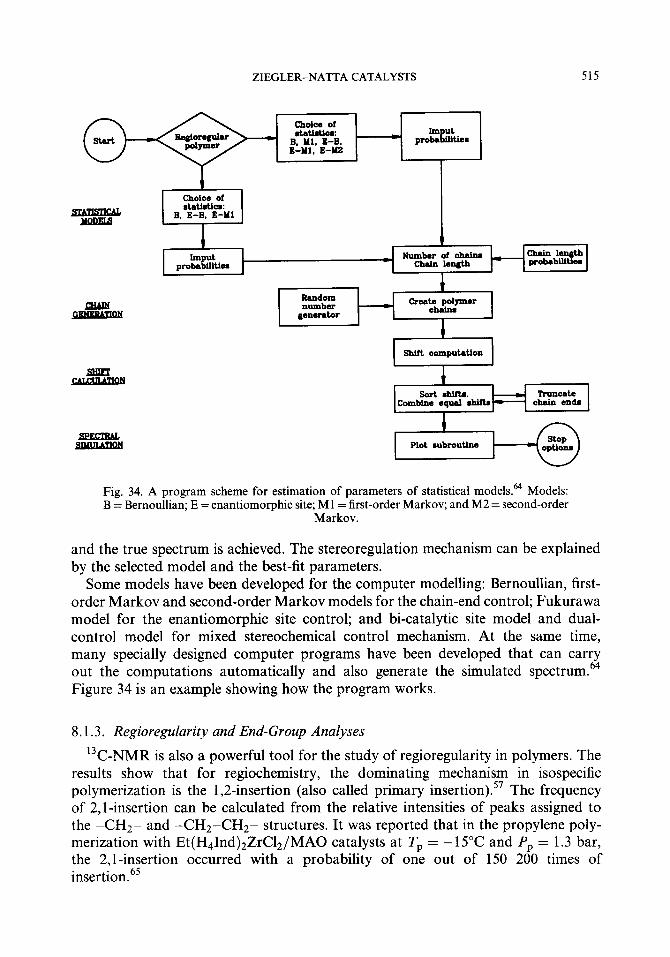
ZIEGLER-NA-ITA CATALYSTS 515
Yii%tc:f 8. Ml. E-k E-W. E-Y2
lmput * probabuxiel
Fig. 34. A program scheme for estimation of parameters of statistical models.64 Models: B = Bernoullian; E = enantiomorphic site; Ml = first-order Markov; and M2 = second-order
Markov.
and the true spectrum is achieved. The stereoregulation mechanism can be explained by the selected model and the best-fit parameters.
Some models have been developed for the computer modelling: Bernoullian, first- order Markov and second-order Markov models for the chain-end control; Fukurawa model for the enantiomorphic site control; and bi-catalytic site model and dual- control model for mixed stereochemical control mechanism. At the same time, many specially designed computer programs have been developed that can carry out the computations automatically and also generate the simulated spectrum.@ Figure 34 is an example showing how the program works.
8.1.3. Regioregularity and End-Group Analyses
13C-NMR is also a powerful tool for the study of regioregularity in polymers. The results show that for regiochemistry, the dominating mechanism in isospecific polymerization is the 1,2-insertion (also called primary insertion).57 The frequency of 2,1-insertion can be calculated from the relative intensities of peaks assigned to the -CH2- and -CH2-CH2- structures. It was reported that in the propylene poly- merization with Et(H41nd)2ZrC12/MA0 catalysts at Tp = -15°C and Pp = 1.3 bar, the 2,1-insertion occurred with a probability of one out of 150-200 times of insertion.65

516 J. HUANG and G. L. REMPEL
The 13C-NMR studies of polymer chain ends and oligomers provided information about the mechanism of chain transfer reactions. Chain transfers by P-hydrogen elimination, by monomer, and by P-methyl elimination in special cases have been reported.23)51s52
8.2. The Properties of the Polymer
8.2.1. Anisotactic Polypropylenes
8.2.1.1. Anisotacticity - No Ziegler-Natta catalyst is completely stereospecific. The best isospecific heterogeneous Ziegler-Natta catalyst produces polypropylene which contains configurational defects (2-5% racemic dyads). By common practice poly- propylene that is insoluble in refluxing n-heptane (T, 2 165”C, [mm] = 0.95 and Xc = 68%) is accepted as isotactic polypropylene (i-PP). (7’, = melting tem- perature; [mm] = mm triad content; and Xc = % crystallinity.) The properties of polypropylenes produced with soluble Et(Ind)2ZrC12/MA0 and Et(HJnd)2ZrC12/ MAO catalysts are significantly different in several respects from i-PP.
Chien35 reported that for PP produced with Et(Ind)2ZrC12/MA0 there is a gradual decrease of T, with Tp up to 2O”C, then T, drops rapidly with a further increase of Tp. The mmmm pentad content gradually changes from 0.86 to 0.81 between Tp of -55°C to +5O”C, then the mmmm content drops to 0.41 for Tp = 80°C. The polymers have excessively high solubility and are separable into fractions by solvent extraction (see Table 10). There are no n-heptane (C,) insoluble products at Tp > 70°C. The PPs are very heterogeneous with respect to microstructures according to solvent fractionation. The T, of the PP fraction decreases with decreasing rank of solvent (boiling point). The polymer fractions extracted with acetone or ether were brittle waxy substances characteristic of low T, and low X, polymers. The high solubility and low T, are due to the structural defects present along the main polymer chains.
The effect of Tp on polymer properties is much more severe in homogeneous catalyst systems than in heterogeneous systems, because the ansa-metallocene catalysts are not as stereorigid as the heterogeneous ones. There may be fluxional conformers for instance due to torsional twisting of the T5-ligands. There may also be dynamic
Table 10. Fractionation of anisotactic polypropylene by solvent extraction3’
Acetone
wt% of polymer soluble in
Ether Pentane Hexane Heptane
wt% of heptane
insoluble
80 22.5 34.8 17.5 25.2 0 0
70 16.4 32.0 15.3 36.3 0 0 50 6.5 13.6 2.4 46.4 13.6 17.5 20 2.4 4.7 2.8 2.6 12.9 74.6
-2: 2.1 4.3 2.4 2.9 13.9 74.4 1.8 4.2 2.2 3.0 13.5 75.3
-55 0.2 0.9 0.4 3.1 9.2 86.2
Catalyst: Et(Ind),ZrCl,/MAO; [Al]/[Zr] = 2500.

ZIEGLER-NATTA CATALYSTS 517
Fig. 35. Comparison between the models of packing of isotactic polypropylene in the o (on the left) and y (on the right) forms. Methyl groups which have identical joints in the two
modifications are indicated as black balls.@
dissociation/association of MAO and changes in the state of coordination. These fluctuations can alter stereochemical and/or regiochemical control during the propagation of a macromolecular chain.
The PP produced with homogeneous chiral ansa-metallocene catalysts is charac- terized by a low and broad T,, low homosteric sequence but appreciable Xc, which differs considerably from conventional i-PP and is referred to as anisotactic PP (uni-PP). The Greek prefix an has been deemed to be the most appropriate one to describe structures which deviate from the limiting structures.
8.2.1.2. Morphology of Anisotactic Propropylene - The crystallinity of ani-PP was measured by powder X-ray diffraction. The Xc of the ani-PP is around 60% compar- able to the Xc of 68% for i-PP obtained with MgClz-supported TiC13 catalysts. Unlike the highly isotactic PPs, which crystallize in the a-phase, the uni-PPs were found to crystallize either largely or predominantly in the y-phase.38Y59 Figure 35 shows the structures of PP crystals in Q- and y-phases.66
The features promoting y-crystallinity are comparatively short average dimensions of isotactic blocks, i.e. the presence of interruptions. As mentioned previously, there are two kinds of insertion errors, single r and double r, present in ani-PP chains, which could have important morphological consequences. In the second case, helical configurations are the same on each side of the rr placement, but in the first case, the two m sequences on each side of the single r placement have opposite helical configurations. That is, the single r error can invert the polymer chain from a clockwise helix to a counterclockwise helix (or vice versa). This may be the true reason why ani-PP favors the y-crystalline phase, as Natta and Corradini predicted that the y- modification becomes a preferred crystalline form if there is a discontinuity in helix configuration.67

518 J. HUANG and G. L. REMPEL
8.2.2. Syndiotactic Polypropylene (s-PP)
The synthesis of highly stereoregular and regioregular s-PP was first achieved by Ewen using iPr(CpFlu)ZrClz/MAO and iPr(CpFlu)HfClJMAO catalysts. Syndio- tactic polypropylene is a new plastic, it has a completely different property profile as compared to isotactic polypropylene. Recently Balbontin et aL6* studied the structure-property relations for this potentially interesting material. Polymer samples were made with iPr(CpFlu)ZrClz/MAO under six different sets of condi- tions. The weight average molecular weight (M,) of the s-PP samples ranged from 5.2 x lo4 to 26.6 x 104, higher than i-PP obtained with Et(Ind)2ZrC12/MA0 under similar conditions. And the molecular weight distribution is similar to isotactic poly- mers, M,/M,, = 2.2-2.9.
13C-NMR analysis showed that the polymers are highly syndiotactic. The fully syndiotactic pentads, [rrrr], are in the range of 81% (for the polymer obtained at Tp = 60°C in pentane) to 94% (for the polymer obtained at Tp = 0°C in toluene). No peaks due to head-to-head propylene sequences are detectable in any of the spectra for the s-PP samples, indicating that only defects due to stereoirregularity are present along the macromolecular chain. Furthermore, an absence of mmmm and mmmr pentads suggests a substantially random distribution of the isotactic units in an essentially syndiotactic chain. The polymers were found to be sterically homogeneous and the extraction fractionation was performed only by molecular weight.
According to Balbontin’s work, syndiotactic polypropylene has a higher melting point (T,): T, = 186°C for the sample made at Tp = O”C, Pp = 1.7 bar in toluene; T, = 166°C for the sample made at Tp = 80°C and Pp = 6 bar in toluene; and T, = 135°C for the sample made at Tp = 60°C and Pp = 4 bar in pentane. The stereo- regularity affects T, to a considerable extent, but the influence of the molecular weight on T, is negligible in the experimental range. Syndiotactic polypropylene is a highly crystalline polymer. It is less hard and rigid than isotactic propylene but more impact-resistant and transparent.69
8.2.3. Other Polymers
The homogeneous Group 4B metallocene-based Ziegler-Natta catalysts have shown great versatility in stereochemical control of the microstructure of poly- olefins. Many new types of polymers with special stereoregularities have been synthesized with metallocene catalysts and additional polymers are expected to appear in the near future. The polymers listed below are some examples, which possess unique properties and have a potential for practical applications.
(1) Atactic polypropylene: 100% atactic polypropylene is a viscous liquid or waxy and soft solid, and is reported to be appropriate for the blending of elastomers.23 (2) Elastomeric polypropylene: This thermoplastic elastomer is produced by the polymerization of propylene with non-symmetric ansa-titanocene/MAO catalysts. The polymer is considered as a stereoblock copolymer, comprised of multiple blocks of stereoregular, crystallizable PP and stereoirregular, amorphous PP segments, [(cry-PP),(am-PP),],. The polymers are uniform in MW. Below their T, the

ZIEGLER-NATTA CATALYSTS 519
materials exhibit characteristic properties of network elastomers, and above their Tm they become liquid. The physical cross-links are due to crystalline domains in these microphase-separated systems.” (3) Linear low density polyethylene [L-LDPE]: The titled polymer is produced by copolymerization of ethylene with higher o-olefins. Comonomers can be I-butene, or 1-hexene, or some long-chain a-olefins, such as 4-methyl-1-pentene and 1-octene. A notable feature of this copolymer made with homogeneous metallocene catalysts is the even distribution of the comonomer units over the whole polymer chain. It is much superior to those made with heterogeneous systems. By changing the types and/or the contents of comonomers, the density of the products can be well controlled.21171 (4) Ethylene-propylene copolymers: the copolymers are composed of evenly distributed ethylene and propylene units, whose ratio can be varied through the choice of polymerization parameters. The properties of the products can be mod- ified from plastic to elastic by changing the relative amounts of propylene units.72 (5) Polycycloalkenes: the homogeneous metallocene catalysts allow the polymeri- zation of cyclic olefins without ring-opening reactions. The isotactic homopolymers of cyclic olefins, such as cyclopentene, norbornene and dimethanooctahydro- naphthalene, show some interesting properties. The polymers are completely insoluble in common hydrocarbons, heat resistant up to temperatures above 4OO”C, and are highly crystalline. Copolymers of the cyclic olefin with ethylene are amorphous. These copolymers retain very high mechanical strengths up to their plasticization temperature and, through suitable adjustment to the composi- tion of the polymer, can be produced with a glass transition temperature of up to 250°C. The copolymers are highly transparent and their resistance to UV radiation and photochemical degradation is excellent. These materials may be used for optical discs and fiber optics.31,69 (6) Optically active polymers: there are two types of optically active polymers synthesized with homogeneous catalysts: (i) The optically active polyolefins - the catalyst (S,S)-Et(H41nd)2ZrC12 consisting of solely one enantiomer is capable of producing optically active isotactic oligomers and polymers of propylene.54 The polymers are formed predominantly in only one screw sense. (ii) The optically active poly(methylene-1,3-cyclopentane) - this polymer is synthesized via enantioselective cyclopolymerization of 1,Shexadiene catalyzed by (R,R)- or (S,S)-Et(H41nd)2ZrBINOL (BINOL = l,l’-bi-2-naphtholate). The enantioface selectivity reaches 91% at Tp = 23”C.43 The synthesis of optically active polymer materials has only been achieved with pure enantiomers of the soluble chiral stereo- rigid zirconocene catalysts.
9. THEORETICAL SIGNIFICANCE AND POTENTIAL APPLICATIONS
9.1. Theoretical Contributions
The discovery of homogeneous Group 4B metallocene/alumoxane catalysts has

520 J. HUANG and G. L. REMPEL
offered the opportunity to obtain a deeper insight into the mechanism of Ziegler- Natta polymerizations. Based on the results obtained from the study of homogeneous systems, the principles of the steric control mechanisms have been identified and metallocene structures have been correlated with polyolefln microstructures, molecular weight (MW) and molecular weight distributions (MWD). The clear correlation between catalyst structure and polymerization behavior promotes our understanding of the Ziegler-Natta catalysis at the molecular level. The practice of the synthesis of polymers with special stereotacticities by tailoring metallocene catalyst pecursors has greatly enriched our knowledge about the mechanisms of stereochemical control.
Some problems, which have puzzled researchers for decades, now seem to be solved from information obtained in the homogeneous system.
9.1.1. The Origin of StereospeciJicity
Natta suggested not long after the discovery of Ziegler-Natta catalysts that stereo- specificity is due to the chirality of the catalytically active center. The hypothesis was not experimentally proved until isotactic polypropylene was obtained with the soluble chiral stereorigid ansa-zirconocene/MAO catalyst.
9. I .2. The Monometallic Model
The question of whether the active site structure involves only the transition metal atom (the monometallic model) or both main-group metal and transition metal atoms (the bimetallic model) has long been a subject of debate. In the 1960s Cossee’ proposed that the active center is the transition metal-carbon bond, i.e. the monometallic model. In the 1970s Boor’ supported Cossee’s proposal according to the catalytic behavior of Phillips catalysts being somewhat similar to that of Ziegler-Natta catalysts. Although Cossee’s model has been generally accepted for years, the truly convincing evidence comes very recently from the discovery of cationic metallocene catalysts, these systems being free from main-group metals.
9.1.3. Migratory Insertion
Cossee’ suggested in his famous model that at the end of insertion step, the growing polymer chain migrates to the coordination site originally occupied by the monomer and recreates a vacant coordination site at the site originally occupied by the chain. As the polymerization process continues, the growing polymer chain terminus flips from side to side. This is also confirmed by the formation of hemitactic polymers. Due to the asymmetric structure of the catalyst, the two coordination sites are not equivalent, one being configuration-selecting and the other non-selecting. As the polymer chain shifts from site to site, the active center is alternatively specific or non-specific, thus forming hemitactic structures.
Cossee also suggested that the active center shows a tendency to rearrange the migrated polymer chain back to its original position. Rearrangement might occur,

ZIEGLER-NATTA CATALYSTS 521
but depends on its competition with the insertion reaction. If the rate of rearrangement is much lower than the rate of insertion of new monomer molecules, most consecutive polymerization steps correspond to polymerization models with the exchange of the relative positions of the growing chain and of the monomer. This is true for the case of hemispecific polymerization. On the other hand, if the rearrangement is comparable to monomer insertion, the catalyst with local symmetry similar to the hemispecific catalyst can now produce stereoregular-stereoirregular block copolymers. The formation of thermal elastic PP may be the case.
9.1.4. Cationic Nature of the Active Center
The concept that the active species of Ziegler-Natta catalysts are cationic complexes was first proposed by Shilov32 in the 1960s. For homogeneous systems, the hypothesis is definitely confirmed by the finding of Lewis base-free cationic metal- locene catalyst systems. The main question remaining is whether the active species of the classical heterogeneous Ziegler-Natta catalysts, too, are cationic. Zambelli73 indicated that at least four facts lead tentatively to suggest that this could be the case: (i) The same transition metals are involved in both homogeneous and hetero- geneous cases. (ii) The most efficient combinations of the classical Ziegler-Natta catalysts contain organometallic compounds of either Al or Be, which are more acidic than others and could abstract more efficiently an anion from the transition metal complex. (iii) Experimental results prove that the active species of the classical catalysts are electrophiles. (iv) Comparison indicates that propagation rate constants for heterogeneous catalysts are larger than those for homogeneous catalysts under similar conditions. It is unlikely that a neutral active species could be more active than a cationic one.
9.1.5. Multiple Active Species in Heterogeneous Systems
The narrow distribution of molecular weight obtained with homogeneous catalysts is in keeping with the assumption that the wide molecular weight distribution observed in polymers prepared with the heterogeneous catalysts is due to the existence of families of catalytic sites with different activity, stereospecificity and stability caused by tiny structural differences.
9.2. Benefit to Kinetic Studies
The polymerizations with homogeneous metallocene-based catalyst systems are no doubt the best systems for kinetic study of Ziegler-Natta polymerization. The kinetics are much simpler in homogeneous polymerization systems, especially in base-free cationic catalyzed polymerization systems, than those in heterogeneous systems. There is no practical difficulty in preparing the “single-site” catalyst (i.e. the catalyst has catalytic centers essentially of only one type).
The a-olefin polymerization with homogeneous metallocene catalysts offers a good opportunity to study the durability and deactivation of the catalysts, since the poly- merization systems remain homogeneous over a considerably long reaction period.72
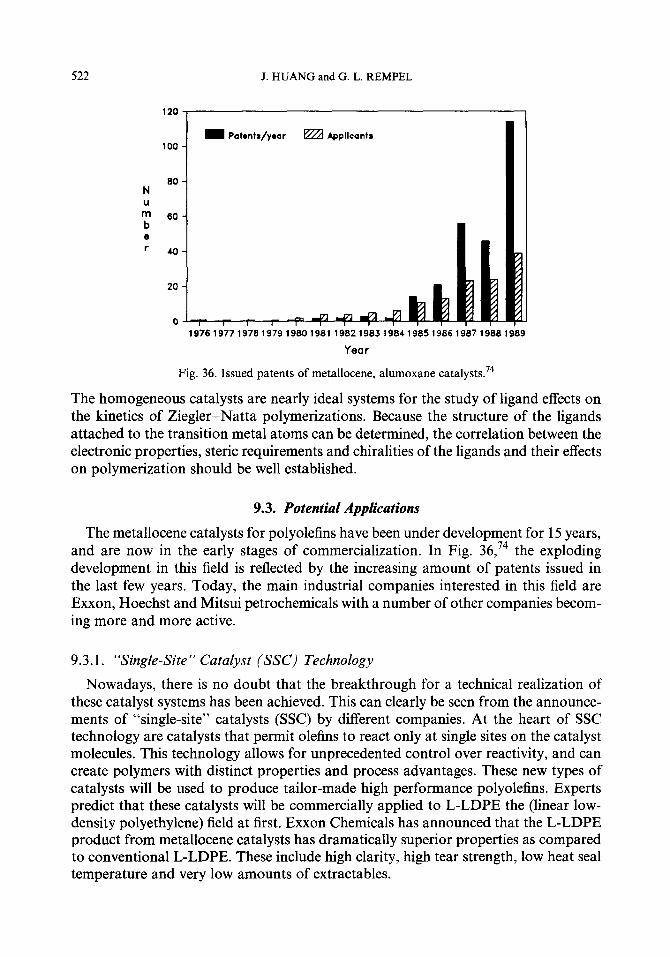
522 J. HUANG and G. L. REMPEL
120
i
m Pat*ntr/y*ar EQ Applicanta 100
80 N U m 60 1 __ b e r 40
20
197619771978197919801981 1982198319841985198619871988 5 198'
Year
Fig. 36. Issued patents of metallocene, alumoxane catalysts.74
The homogeneous catalysts are nearly ideal systems for the study of ligand effects on the kinetics of Ziegler-Natta polymerizations. Because the structure of the ligands attached to the transition metal atoms can be determined, the correlation between the electronic properties, steric requirements and chiralities of the ligands and their effects on polymerization should be well established.
9.3. Potential Applications
The metallocene catalysts for polyolefins have been under development for 15 years, and are now in the early stages of commercialization. In Fig. 36,74 the exploding development in this field is reflected by the increasing amount of patents issued in the last few years. Today, the main industrial companies interested in this field are Exxon, Hoechst and Mitsui petrochemicals with a number of other companies becom- ing more and more active.
9.3.1. “Single-Site” Catalyst (SSC) Technology
Nowadays, there is no doubt that the breakthrough for a technical realization of these catalyst systems has been achieved. This can clearly be seen from the announce- ments of “single-site” catalysts (SSC) by different companies. At the heart of SSC technology are catalysts that permit olefins to react only at single sites on the catalyst molecules. This technology allows for unprecedented control over reactivity, and can create polymers with distinct properties and process advantages. These new types of catalysts will be used to produce tailor-made high performance polyolefins. Experts predict that these catalysts will be commercially applied to L-LDPE the (linear low- density polyethylene) field at first. Exxon Chemicals has announced that the L-LDPE product from metallocene catalysts has dramatically superior properties as compared to conventional L-LDPE. These include high clarity, high tear strength, low heat seal temperature and very low amounts of extractables.

ZIEGLER-NATTA CATALYSTS 523
Metallocene catalysts offer many possibilities to synthesize novel polymers which have never before been produced by conventional Ziegler-Natta catalysts.
9.3.2. D@culties Hindering Applications
To make the commercial application of metallocene catalysts entirely successful, there are a number of serious problems which still need to be solved: (1) Structural defects and low molecular weights which cause poor thermal and mechanical proper- ties. Some researchers believe that to solve this problem, heterogenizing these catalysts (using gas phase or slurry processes instead of solution processes) is necessary for the control of the morphology of the polymer products. (2) The high cost of MAO. It is reported that there is a possibility to strongly reduce the amount of MAO needed to the same level as aluminum alkyls used in conventional processes. Developing MAO- free catalyst systems or partially replacing MAO with other cheaper chemicals could be another solution to this problem.
9.3.3. The Potential Role in Production of Hybrid Thermoplastic Olejins
The recently developed hybrid thermoplastic olefins cover a very broad range of products. They cannot be clearly identified as either polyethylene, polypropylene or elastomers, and their physical properties cover almost any combination of stiffness/ impact, ranging from a very flexible, high impact elastomeric grade to very high stiffness performance similar to engineering plastics.74 They provide one way to replace not only other commodity plastics, such as PVC and polystyrene, due to solution of some environmental problems, but also some engineering plastics, due to their better cost to performance relationship. These new polyolefins are strongly linked to the development of catalyst technology to derive specially formulated cata- lysts. The metallocene catalysts seem to meet many needs and may find applications in the production of hybrid thermoplastic olefins.
9.4. Other Homogeneous Catalyst Systems
It is worthwhile to mention that homogeneous Ziegler-Natta catalysts other than metallocene-based catalysts have also been developing rapidly in recent years. The catalyst precursors of these systems are non-metallocene organometallic compounds, such as monocyclopentadienyl derivatives.
9.4.1. CG Catalyst
A representative of monocyclopentadienyl catalysts is the CG catalyst (CG stands for constrained-geometry). This new type of homogeneous catalysts was developed by Dow Plastics.75 The catalyst system is based on group 4B transition metals, such as Ti, covalently bonded to a Cp group bridged with a heteroatom, such as nitrogen. The components are connected in such a way that a constrained cyclic structure is formed with the Ti center. The bond angle between the monocyclopentadienyl group, Ti center and heteroatom is less than 115”. Strong Lewis acid systems activate the catalyst to a highly efficient cationic form.

524 J. HUANG and G. L. REMPEL
The catalysts produce highly processable polyoiefins with a unique combination of narrow MWD and long-chain branches. Ethylene-octene copolymers produced with CG catalysts have useful properties across a range of densities and melting indexes. These novel copolymer families are called polyolefin plastomers (POP) and polyolefin elastomers (POE). POPS possess plastic and elastic properties. POEs have greater than 20 wt% octene comonomer units. Useful properties include high elasticity and cross- linkability.
The CG catalyst technology is not limited to the typical selection of C-Ca cu-olefins, but can include higher a-olefins. The open structure of the CG catalyst significantly increases the flexibility to insert higher a-olefin comonomers into the polymer structure. This technology also allows addition of vinyl-ended polymer chains to produce long-chain branching (LCB).
9.4.2. Other Non-Metallocene Catalysts
Recently, Giannetti et a1.76 reported that addition of MAO to Zr(CHfPh)d produces an ethylene polymerization catalyst having an activity comparable to that of metallocene-based systems. Subsequently, analogous catalytic systems were reported to promote partially isospecific polymerization of propylene, syndiospecific polymer- ization of styrene, and cis-1,Cpolymerization of butadiene. It is worth noting that metallocene-based catalysts are inactive for the polymerization of styrene and conjugated diolefins.
Zambelli et aZ.77 reported that the true active species of group 4B homoleptic hydrocarbyles, e.g. Zr(CHzPh)d, as well as monocyclopentadienyl compounds, e.g. CpZr(CH2Ph)3, are also cationic complexes analogous to those involved in metallocene-based catalytic systems. They successfully isolated the catalytic complex
[zr(CH,Ph>,l’[B(CH,Ph)(C6F5>31-, which is the first example of an isolated, catalytically active Cp-free group 4 cationic complex. The catalyst shows an activity of 2.5 x lo4 g PE/(mol Zr - ha atm) for ethylene polymerization at 50°C and 5 atm monomer pressure, and an activity of 1.5 x lo3 g PP/(mol Zr . h. atm) for propylene polymerization under similar conditions. The catalyst is partially isospecific (20% isotactic and 80% atactic).
Another type of non-metallocene homogeneous catalyst is transition metal alkoxides. Kakugo et a1.78 reported that a combination of metal alkoxide and MAO is a good catalyst for ethylene and propylene polymerization. The titanium complexes with a bidentate 2,2’-thiobis(6-tert-butyl-4-methylphenoxy) (TBP) ligand, such as (TBP)TiCl, or (TBP)Ti(OPr),, are extremely active toward olefins and styrene. The polymerization activities for ethylene and propylene are comparable to those of the Cp2ZrC12/MA0 catalyst. The catalysts produce stereoirregular polypropylene with very high molecular weight (>6 x 106) and highly syndiotactic polystyrene. Co- polymerization of ethylene and styrene with these catalysts gives highly alternating co- polymers. Thecatalystsare also active toward bothconjugated andnon-conjugateddienes.
10. CONCLUSIONS
The discovery of homogeneous metallocene-based Ziegler-Natta catalysts in the

ZIEGLER-NATTA CATALYSTS 525
1980s has opened up new possibilities to explore the mechanism of Ziegler-Natta catalysis and to fine-tune the stereochemical structure of polymer products. The particular significance of these discoveries is that the stereospecificity of the metallo- cene catalysts can be influenced by the substitution pattern on the Cp ligand. Using these metallocene catalysts, a large number of novel polymers with special properties have been obtained. Metallocene catalysts for polyolefins are now in the early stages of commercialization. The development of metallocene catalysts, or more widely homogeneous catalysts, may be a dominant force through the 1990s in the polyolefin industry.
1. 2. 3.
4. 5.
6. 7. 8. 9.
10.
11. 12. 13. 14.
15.
16.
17. 18. 19. 20.
21.
J. Boor, Ziegler-Natta Catalysts and Polymerizations, p. 1, Academic Press, New York (1979). H. Sinn and W. Kaminsky, Ad. Organomet. Chem. 18,99 (1980). P. J. T. Tait, Transition Metal and Organometallics as Catalysts for Olefin Polymerization (W. Kaminsky and H. Sinn Eds.), p. 315, Springer, Berlin (1988). G. Odian, Principles of Polymerization, p. 566, 2nd edn, John Wiley, New York (1981). K. S. Minsker, M. M. Karpasas and G. E. Zaikov, J. Macromol. Sci., Rev. Macromol. Chem. Phys. C27(1), 1 (1987). L. A. Castonguay and A. K. Rappe, J. Am. Chem. Sot. 114, 5832 (1992). M. Ystenes, Makromol. Chem., Macromol. Symp. 66, 71 (1993). J. A. Ewen, J. Am. Chem. Sot. 106,6355 (1984). P. Cossee, The Stereochemistry of Macromolecules (A. D. Ketley Ed.), p. 155, Dekker (1967). A. R. Siedle, W. M. Lamanna, R. A. Newmark, J. Stevens, D. E. Richardson and M. Ryan, Makromol. Chem., Macromol. Symp. 66,215 (1993). D. S. Breslow and N. R. Newburg, J. Am. Chem. Sot. 79,5072 (1957). G. Natta, P. Pino, G. Mazzanti and R. Lanzo, Chim. Znd. (Milan) 39, 1032 (1957). K. H. Reichert and K. R. Meyer, Makromol. Chem. 169, 163 (1973). F. R. W. P. Wild, M. Wasincionek, G. Huttner and H. H. Brintzinger, J. Organomet. Chem. 288,63 (1985). F. R. W. P. Wild, L. Zsolnai, G. Huttner and H. H. Brintzinger, J. Organomet. Chem. 232, 233 (1982). C. Pellecchia, A. Propo, P. Longo and A. Zambelli, Makromol. Chem., Rapid Commun. 12, 663 (1991). R. F. Jordan, C. S. Bajger, R. Willet and B. Scott, J. Am. Chem. Sot. 108,741O (1986). N. Herfert and G. Fink, Makromol. Chem., Rapid Commun. 14, 91 (1993). J. C. W. Chien, W.-M. Tsai and M. D. Rausch, J. Am. Chem. Sot. 113,857O (1991). J. A. Ewen, Catalytic Polymerization of Olefins (T. Keii and K. Soga Eds.), p. 271, Elsevier, Tokyo (1986). W. Kaminsky, A. Bark, R. Shiehl, N. Moller-Lindenhof and S. Niedona, Transition Metal and Organometallics as Catalysts for Olefn Polymerization (W. Kaminsky and H. Sinn Eds.), p. 291, Springer, Berlin (1988).
22. J. W. Lauher and R. Hoffmann, J. Am. Chem. Sot. 98, 1729 (1976). 23. W. Kaminsky and R. Steiger, Polyhedron 7(22/23), 2375 (1988). 24. E. Giannetti, G. M. Nicoletti and R. Mazzocchi, J. Polym. Sci., Polym. Chem. Ed. 23,2117 (1985). 25. W. Kaminsky, H. Sinn and R. Woldt, Makromol. Chem., Rapid Commun. 4,417 (1983). 26. K. Toshiro, M. Kitani, K. Sugimura and T. Ueda, Jpn Pat. JP 04,304,202 and JP 04,304,203. 27. T. Sugano, K. Matsubara, T. Fujita and T. Takahashi, J. Mol. Catal. 82, 93 (1993). 28. M. Bochmann and S. J. Lancaster, J. Organometal. Chem. 434, Cl (1992). 29. X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Sot. 113,3613 (1991). 30. J. C. W. Chien, M.-W. Tsai, Makromol. Chem., Macromol. Symp. 66, 141 (1993). 31. W. Kaminsky, A. Bark and R. Steiger, J. Mol. Catal. 74, 109 (1992). 32. F. S. Dyachkowskii, A. K. Shilova and A. E. Shilov, J. Polym. Sci. C16, 2336 (1967). 33. P. Corradini and G. Guerra, Prog. Polym. Sci 16, 239 (1991).
REFERENCES

526 J. HUANG and G. L. REMPEL
34.
35. 36. 37. 38.
39.
40. 41. 42.
43. 44.
45. 46.
47.
48. 49. 50. 51.
52. 53. 54. 55. 56.
57. 58. 59. 60. 61. 62. 63. 64.
65. 66. 67.
68. 69. 70.
W. Kaminsky, R. Engehausen, K. Zoumis, W. Spaleck and J. Rohrmann, Makromol. Chem. 193, 1643 (1992). J. C. W. Chien and B. P. Wang, J. Polym. Sci., Polym. Chem. Ed. 28, 15 (1990). N. Piccolrovazzi, P. Pino, G. Consiglio, A. Sironi and M. Moret, Organometl. 9, 3098 (1990). J. C. W. Chien and R. Sugimoto, J. Polym. Sci., Polym. Chem. Ed. 29,459 (1991). J. A. Ewen, M. J. Elder, R. I. Jones, S. Curtis and H. N. Cheng, Catalytic Olejn Polymerization (T. Keii and K. Soga Eds.), p. 439, Elsevier, Tokyo (1990). J. A. Ewen, M. J. Elder, R. I. Jones, L. Haspeslagh, J. L. Atwood, S. G. Bott and K. Robinson, Makromol. Chem., Macromol. Symp. 48149,253 (1991). M. Farina, G. D. Silvestro and P. Sozzani, Prog. Polym. Sci. 16,219 (1991). N. Herfert and G. Fink, Makromol. Chem., Macromol. Symp. 66, 157 (1993). G. N. Babu, R. A. Newmark, H. N. Cheng, J. C. W. Chien and G. H. Llinas, Macromolecules 25, 7400 (1992). G. W. Coates and R. M. Waymouth, J. Am. Chem. Sot. 115,91 (1993). P. Pino, B. Rotzinger and E. von Achenbach, Catalytic Polymerization of Olejns (T. Keii and K. Soga Eds.), p. 461, Elsevier, Tokyo (1986). K. Hortmann and H. H. Brintzinger, New J. Chem. 16,51 (1992). P. Burger, K. Hortmann and H. H. Brintzinger, Makromol. Chem., Macromol. Symp. 66, 127 (1993). J. C. W. Chien and R. Sugimoto, Catalytic Olejin Polymerization (T. Keii and K. Soga Eds.), p. 535, Elsevier, Tokyo (1990). D. Fischer, S. Jungling and R. Mulhaupt, Makromol. Chem., Macromol. Symp. 66, 191 (1993). J. M. V. Estrada and A. E. Hamielec, Polymer, 19, 808 (1994). T. Tsutsui and N. Kashiwa, Polym. Commun. 29, 180 (1988). L. Resconi, F. Piemontesi, G. Franciscono, L. Abis and T. Fiorani, J. Am. Chem. Sot. 114, 1025 (1992). T. Tsutsui, A. Mizuno and N. Kashiwa, Polymer 30,428 (1989). L. Resconi, U. Giannini, E. Albizzati and F. Piemontesi, Polym. Prepr. 32(l), 463 (1991). W. Kaminsky, K. Kulper and S. Niedoba, Makromol. Chem., Macromol. Symp. 3, 377 (1986). K. Soga and M. Kaminaka, Makromol. Chem., Rapid Commun. 13,221 (1992). W. Spaleck, M. Antberg, J. Rohrmann, A. Winter, B. Bachmann, P. Kaprof, J. Behm and W. A. Herrmann, Angew. Chem. Int. Ed. Engl. 31(10), 1347 (1992). H. N. Cheng and J. A. Ewen, Makromol. Chem. 190,1931 (1989). H. N. Cheng and M. A. Bennett, Makromol. Chem. 188, 135 (1987). B. Rieger, X. Mu, D. T. Mallin, M. D. Rausch and J. C. W. Chien, Macromolecules 23,3559 (1990). F. A. Bovey and G. V. D. Tiers, J. Polym. Sci. 44, 173 (1960). J. Fukukawa and R. A. Shelden, J. Polym. Sci. B3,23 (1965). Y. Doi and T. Asakura, Makromol. Chem. 176,507 (1975). Y. Doi, Makromol. Chem., Rapid Commun. 3, 635 (1982). H. N. Cheng, Transition Metal Catalyzed Polymerizations (R. P. Quirk Ed.), p. 599, Cambridge University Press, Cambridge (1987). T. Tsutsui, K. Mizuno and N. Kashiwa, Makromol. Chem. 190, 1177 (1989). P. Corradini, Makromol. Chem., Macromol. Symp. 66, 11 (1993). G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica and G. Mazzanti, J. Am. Chem. Sot. 77, 1708 (1955). G. Balbontin, D. Dainelli, M. Galimberti and G. Paganetto, Makromol. Chem. 193, 693 (1992). W. Spaleck, Hoechst High Chem. Mag. 14,44 (1993). G. H. Llinas, S.-H. Dong, D. T. Mallin, M. D. Rausch, Y.-G. Lin, H. H. Winter and J. C. W. Chien, Macromolecules 25, 1242 (1992).
71. W. Kaminsky and M. Schlobohm, Makromol. Chem., Macromol. Symp. 4, 103 (1986). 72. J. Herwig and W. Kaminsky, Polym. Bull. 9, 464 (1983). 73. A. Zambelli and C. Pellecchia, Makromol. Chem., Macromol. Symp. 66, 1 (1993). 74. A. Guyot, Makromol. Chem., Macromol. Symp. 66, 311 (1993). 15. D. Schwank, Modern Plastics Aug., 49 (1993). 76. E. Giannetti, G. M. Nicoletti and R. Mazzocchi, J. Polym. Sci., Polym. Chem. Ed. 23,2117 (1985). 77. C. Pellecchia, A. Grassi and A. Zambelli, J. Mol. Catal. 82, 57 (1993). 78. T. Miyatake, K. Mizunuma and M. Kakugo, Makromol. Chem., Macromol. Symp. 66,203 (1993).




















