
Tumoricidal and Anti-Angiogenic Actions of Gamma-Linolenic Acid and Its Derivatives
27
463 ISSN 1758-4299 10.2217/CLP.11.34 © 2011 Future Medicine Ltd Clin. Lipidol. (2011) 6(4), 463–489 Review Essential fatty acids enhance free radical generation and lipid peroxidation to induce apoptosis of tumor cells It is desirable that anticancer agents preferen- tially kill tumor cells without adverse effects on normal cells. However, current available drugs and radiation do not possess such selec- tive action on tumor cells. Radiation and anti- cancer drugs such as doxorubicin and bleo- mycin enhance free radical generation and, thus, produce their tumoricidal action [1–3] . However, these drugs are potent enough to induce generation of free radicals in normal cells that produce visible side-effects receiv- ing in patients with cancer chemotherapy and radiation treatment. Free radicals (especially superoxide anion, hydroxyl radical and hydrogen peroxide) have the ability to influence cell growth and develop- ment, especially in diseases such as cancer [4–6] . Free radicals are generated during the electron- transport steps of ATP production in mito- chondria as a result of the leakage of electrons. Superoxide anion (O 2 - .) and hydroxyl (OH) radicals lead to the production of hydrogen per- oxide (H 2 O 2 ), which forms the basis for further hydroxyl radical generation that is dependent on the presence of Fe 2+ ions [7] . Free radicals are both beneficial and harmful [4] . Some of the beneficial actions include participation in signal-transduction pathways that regulate cell growth [8,9] , reduction–oxidation (redox) reac- tions [7] and, as a first line of defense against infections, by polymorphonuclear leukocytes and macrophages [4] . Generation of exces- sive amounts of free radicals inactivates vital enzymes, proteins and other subcellular ele- ments required for cell survival and, thus, leads to cell death by apoptosis [4,10–12] . Tumor cells have low rates of lipid peroxidation An inverse relationship exists between the con- centrations of lipid peroxides and the rate of cell proliferation; high rate of lipid peroxida- tion decreases rate of cell division and vice versa (reviewed in [13]). This is an important obser- vation in the light of the fact that, in general, tumor cells are more resistant to lipid peroxida- tion compared with normal cells [14,15] , whereas lipid peroxidation decreases with increasing growth rate [16] . Hepatomas that possess a higher growth rate have lower microsomal phospholipid content and a decreased content of unsaturated fatty acids (Table 1) [13,17–19] . Selective elimination of tumor cells with little or no effects on normal cells is desirable for the treatment of cancer. Tumor cells are deficient in polyunsaturated fatty acids ([PUFAs] especially g‑linolenic, arachidonic, eicosapentaneoic and docosahexaenoic acids) caused by decreased activity of enzymes d‑6 and d‑5 desaturases. Exposure of tumor cells to adequate amounts of these fatty acids induces apoptosis in these tumor cells by augmenting free radical generation and lipid peroxidation, while normal cells remain unaffected. Studies have revealed that even tumor cell drug resistance can be overcome by enhancing the incorporation of PUFAs in the membranes of tumor cells. Therefore, it is suggested that methods designed to selectively deliver PUFAs to tumor cells could be a novel method of inducing apoptosis in tumor cells. Such selective delivery of PUFAs to tumor cells can be achieved by intratumoral injection, intra‑arterial administration into tumor‑feeding vessels, conjugation of PUFAs to growth factors and/or monoclonal antibodies to growth factors and cytokines (e.g., TNF‑ a) and/or complexing PUFAs with conventional anticancer drugs and infusing them parenterally. KEYWORDS: g‑linolenic acid n antioxidants n apoptosis n arachidonic acid n cancer n catalase n docosahexaenoic acid n eicosapentaenoic acid n free radicals n lipid peroxides n normal cells n oncogenes n polyunsaturated fatty acids n superoxide dismutase n vitamin E Undurti N Das UND Life Sciences, 13800 Fairhill Road, #321, Shaker Heights, OH 44120, USA and School of Biotechnology, Jawaharlal Nehru Technological University, Kakinada‑533 003, India Tel.: +1 216 231 5548 Fax: +1 928 833 0316 [email protected]
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Tumoricidal and Anti-Angiogenic Actions of Gamma-Linolenic Acid and Its Derivatives

463ISSN 1758-429910.2217/CLP.11.34 © 2011 Future Medicine Ltd Clin. Lipidol. (2011) 6(4), 463–489
Review
Essential fatty acids enhance free radical generation and lipid peroxidation to induce apoptosis of tumor cells
It is desirable that anticancer agents preferen-tially kill tumor cells without adverse effects on normal cells. However, current available drugs and radiation do not possess such selec-tive action on tumor cells. Radiation and anti-cancer drugs such as doxorubicin and bleo-mycin enhance free radical generation and, thus, produce their tumoricidal action [1–3]. However, these drugs are potent enough to induce generation of free radicals in normal cells that produce visible side-effects receiv-ing in patients with cancer chemotherapy and radiation treatment.
Free radicals (especially superoxide anion, hydroxyl radical and hydrogen peroxide) have the ability to influence cell growth and develop-ment, especially in diseases such as cancer [4–6]. Free radicals are generated during the electron-transport steps of ATP production in mito-chondria as a result of the leakage of electrons. Superoxide anion (O
2-.) and hydroxyl (OH)
radicals lead to the production of hydrogen per-oxide (H
2O
2), which forms the basis for further
hydroxyl radical generation that is dependent on the presence of Fe2+ ions [7]. Free radicals are both beneficial and harmful [4]. Some of
the beneficial actions include participation in signal-transduction pathways that regulate cell growth [8,9], reduction–oxidation (redox) reac-tions [7] and, as a first line of defense against infections, by polymorphonuclear leuko cytes and macrophages [4]. Generation of exces-sive amounts of free radicals inactivates vital enzymes, proteins and other subcellular ele-ments required for cell survival and, thus, leads to cell death by apoptosis [4,10–12].
Tumor cells have low rates of lipid peroxidationAn inverse relationship exists between the con-centrations of lipid peroxides and the rate of cell proliferation; high rate of lipid peroxida-tion decreases rate of cell division and vice versa (reviewed in [13]). This is an important obser-vation in the light of the fact that, in general, tumor cells are more resistant to lipid peroxida-tion compared with normal cells [14,15], whereas lipid peroxidation decreases with increasing growth rate [16]. Hepatomas that possess a higher growth rate have lower microsomal phospholipid content and a decreased content of unsaturated fatty acids (Table 1) [13,17–19].
Selective elimination of tumor cells with little or no effects on normal cells is desirable for the treatment of cancer. Tumor cells are deficient in polyunsaturated fatty acids ([PUFAs] especially g‑linolenic, arachidonic, eicosapentaneoic and docosahexaenoic acids) caused by decreased activity of enzymes d‑6 and d‑5 desaturases. Exposure of tumor cells to adequate amounts of these fatty acids induces apoptosis in these tumor cells by augmenting free radical generation and lipid peroxidation, while normal cells remain unaffected. Studies have revealed that even tumor cell drug resistance can be overcome by enhancing the incorporation of PUFAs in the membranes of tumor cells. Therefore, it is suggested that methods designed to selectively deliver PUFAs to tumor cells could be a novel method of inducing apoptosis in tumor cells. Such selective delivery of PUFAs to tumor cells can be achieved by intratumoral injection, intra‑arterial administration into tumor‑feeding vessels, conjugation of PUFAs to growth factors and/or monoclonal antibodies to growth factors and cytokines (e.g., TNF‑a) and/or complexing PUFAs with conventional anticancer drugs and infusing them parenterally.
Keywords: g‑linolenic acid n antioxidants n apoptosis n arachidonic acid n cancer n catalase n docosahexaenoic acid n eicosapentaenoic acid n free radicals n lipid peroxides n normal cells n oncogenes n polyunsaturated fatty acids n superoxide dismutase n vitamin E
Undurti N DasUND Life Sciences, 13800 Fairhill Road, #321, Shaker Heights, OH 44120, USA and School of Biotechnology, Jawaharlal Nehru Technological University, Kakinada‑533 003, India Tel.: +1 216 231 5548 Fax: +1 928 833 0316 [email protected]

Clin. Lipidol. (2011) 6(4)464 future science groupClin. Lipidol. (2011) 6(4)
For example, reduced rates of lipid peroxida-tion observed in Yoshida hepatoma cells and their microsomes when compared with nor-mal liver tissue under the same pro-oxidant conditions was found to be caused by reduced levels of NADPH-cytochrome C reductase and the NADPH-cytochrome P-450 electron transport chain in the Yoshida hepatoma cells (Tables 2–4) [19]. Similar results were obtained with Novikoff hepatoma cells [20], wherein the low rate of lipid peroxidation was found to be caused by their low content of polyunsaturated fatty acids and cytochrome P-450 and elevated levels of lipid-soluble antioxidant a-tocopherol (Tables 2–5). Furthermore, tumor plasma mem-branes demonstrated extremely low rates of malondialdehyde accumulation and lipoprotein lipid hydroperoxide when exposed to xanthine plus xanthine oxidase induced free radicals compared with the normal rat liver membranes [21]. Such a high degree of resistance to peroxi-dation in the tumor cells could be attributed to a marked decrease in their lipid content [21].
Previously, it was noted that polyunsaturated fatty acids (PUFAs) augmented free radical gen-eration (especially superoxide anion and hydro-gen peroxide) and formation of lipid peroxides selectively in the tumor cells compared with nor-mal cells, a phenomenon that occurred despite the fact that the uptake of fatty acids was at least 2–3-times higher in the normal cells compared with tumor cells [22,23]. These results suggest that a close correlation exists between the rate of lipid peroxidation and degree of malignant deviation of the tumor cell, and the susceptibility of the tumor cell to free radical-induced cytotoxicity, implying that the higher the degree of malignant nature of the tumor cell, the lower the rate of lipid peroxida-tion and higher the degree of susceptibility to free radical-induced toxicity. Resistance to lipid perox-idation is observed even at the premalignant stage of the carcinogenesis process, since administration of diethylnitrosamine and 2-acetylaminofluorene leads to inhibition of peroxidation in normal liver and in preneoplastic nodules, as well as in the neoplasms that result from this treatment [24].
Table 1. Content of total lipid, vitamin e and fatty acids in normal liver, yoshida hepatoma cells and in microsomal suspensions from normal liver and yoshida hepatoma cells.
Measurement Normal intact liver Intact yoshida cells Normal liver microsomes yoshida microsomes ref.
Protein mg/106 cells ND 0.133 ± 0.088 ND 0.028 ± 0.002 [4]
Total lipid mg/mg protein
0.41 ± 0.08 0.26 ± 0.9 0.37 ± 0.03 0.21 ± 0.05 [6–8,10]
Cholesterol µg/mg of lipid
96.0 ± 8.1 215.2 ± 6.2 110.0 ± 12.3 266.85 ± 13.5 [6–8,10]
Fatty acid (%)
16:0 18.5 ± 0.2 18.7 ± 2.0 18.9 ± 1.1 18.5 ± 0.5 [6–8,10]
18:0 17.5 ± 0.5 13.3 ± 1.1 22.0 ± 3.0 13.7 ± 0.2 [7,8,10]
18:1 (OA) 12.1 ± 1.0 21.5 ± 0.8 8.6 ± 1.0 18.1 ± 0.3 [6–8,10]
20:4 (AA) 16.7 ± 2.4 8.7 ± 0.7 19.1 ± 2.4 9.6 ± 0.8 [6–8,10]
22:5 – 2.9 ± 0.1 – 2.4 ± 0.1 [6,10]
24:0 – 1.2 ± 0.1 – 2.9 ± 0.3 [6,10]
22:6 (DHA) 6.3 ± 0.2 5.2 ± 0.6 6.1 ± 0.3 5.3 ± 0.4 [6–8,10]
All values are means ± standard deviations. The numbers of estimations are given in the parentheses. AA: Arachidonic acid; DHA: Docosahexaenoic acid; ND: Not determined; OA: Oleic acid. Data taken from [19].
Table 2. Activities of NAdPH-cytochrome C reductase and 7-ethoxycoumarin de-ethylase in microsomal suspensions derived from normal rat liver and yoshida hepatoma cells.
Measurement Normal liver microsomes yoshida hepatoma microsomes ref.
NADPH-cytochrome C reductase (mmol/min per mg) 80.5 ± 4.2 9.13 ± 1.4 [6,40]
7-ethoxycoumarin de-ethylase activity (mmol/min per mg)
0.14 ± 0.04 ND [4]
Cytochrome P-450 (mmol/mg) 0.81 ± 0.06 ND [6]
All values are means ± standard deviations. The numbers of determinations are given in the parenthesis. ND: Not detectable. Data taken from [19].
Review | Das
www.futuremedicine.com 465future science group
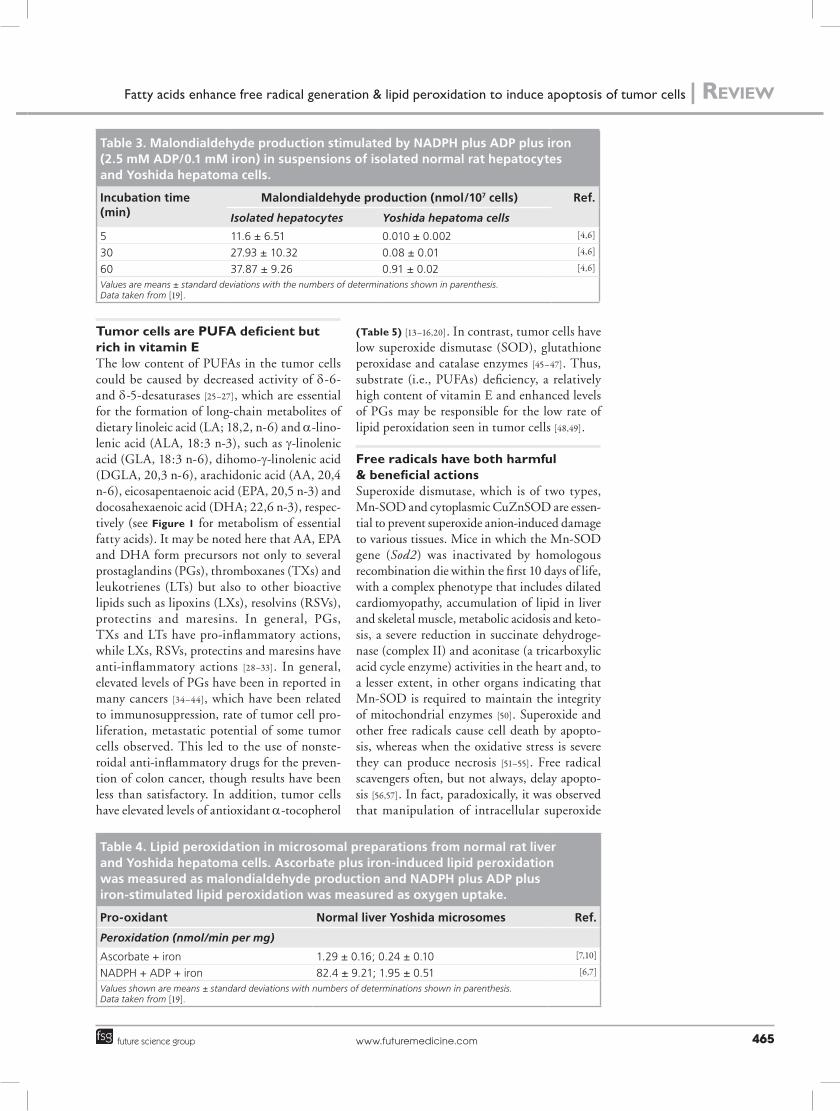
Tumor cells are PUFA deficient but rich in vitamin E The low content of PUFAs in the tumor cells could be caused by decreased activity of d-6- and d-5-desaturases [25–27], which are essential for the formation of long-chain metabolites of dietary linoleic acid (LA; 18,2, n-6) and a-lino-lenic acid (ALA, 18:3 n-3), such as g-linolenic acid (GLA, 18:3 n-6), dihomo-g-linolenic acid (DGLA, 20,3 n-6), arachidonic acid (AA, 20,4 n-6), eicosapentaenoic acid (EPA, 20,5 n-3) and docosahexaenoic acid (DHA; 22,6 n-3), respec-tively (see Figure 1 for metabolism of essential fatty acids). It may be noted here that AA, EPA and DHA form precursors not only to several prostaglandins (PGs), thromboxanes (TXs) and leukotrienes (LTs) but also to other bioactive lipids such as lipoxins (LXs), resolvins (RSVs), protectins and maresins. In general, PGs, TXs and LTs have pro-inflammatory actions, while LXs, RSVs, protectins and maresins have anti-inflammatory actions [28–33]. In general, elevated levels of PGs have been in reported in many cancers [34–44], which have been related to immunosuppression, rate of tumor cell pro-liferation, metastatic potential of some tumor cells observed. This led to the use of nonste-roidal anti-inflammatory drugs for the preven-tion of colon cancer, though results have been less than satisfactory. In addition, tumor cells have elevated levels of antioxidant a-tocopherol
(Table 5) [13–16,20]. In contrast, tumor cells have low superoxide dismutase (SOD), glutathione peroxidase and catalase enzymes [45–47]. Thus, substrate (i.e., PUFAs) deficiency, a relatively high content of vitamin E and enhanced levels of PGs may be responsible for the low rate of lipid peroxidation seen in tumor cells [48,49].
Free radicals have both harmful & beneficial actions Superoxide dismutase, which is of two types, Mn-SOD and cytoplasmic CuZnSOD are essen-tial to prevent superoxide anion-induced damage to various tissues. Mice in which the Mn-SOD gene (Sod2) was inactivated by homologous recombination die within the first 10 days of life, with a complex phenotype that includes dilated cardiomyopathy, accumulation of lipid in liver and skeletal muscle, metabolic acidosis and keto-sis, a severe reduction in succinate dehydroge-nase (complex II) and aconitase (a tricarboxylic acid cycle enzyme) activities in the heart and, to a lesser extent, in other organs indicating that Mn-SOD is required to maintain the integrity of mitochondrial enzymes [50]. Superoxide and other free radicals cause cell death by apopto-sis, whereas when the oxidative stress is severe they can produce necrosis [51–55]. Free radical scavengers often, but not always, delay apopto-sis [56,57]. In fact, paradoxically, it was observed that manipulation of intracellular superoxide
Table 4. Lipid peroxidation in microsomal preparations from normal rat liver and yoshida hepatoma cells. Ascorbate plus iron-induced lipid peroxidation was measured as malondialdehyde production and NAdPH plus AdP plus iron-stimulated lipid peroxidation was measured as oxygen uptake.
Pro-oxidant Normal liver yoshida microsomes ref.
Peroxidation (nmol/min per mg)
Ascorbate + iron 1.29 ± 0.16; 0.24 ± 0.10 [7,10]
NADPH + ADP + iron 82.4 ± 9.21; 1.95 ± 0.51 [6,7]
Values shown are means ± standard deviations with numbers of determinations shown in parenthesis. Data taken from [19].
Table 3. Malondialdehyde production stimulated by NAdPH plus AdP plus iron (2.5 mM AdP/0.1 mM iron) in suspensions of isolated normal rat hepatocytes and yoshida hepatoma cells.
Incubation time (min)
Malondialdehyde production (nmol/107 cells) ref.
Isolated hepatocytes Yoshida hepatoma cells
5 11.6 ± 6.51 0.010 ± 0.002 [4,6]
30 27.93 ± 10.32 0.08 ± 0.01 [4,6]
60 37.87 ± 9.26 0.91 ± 0.02 [4,6]
Values are means ± standard deviations with the numbers of determinations shown in parenthesis. Data taken from [19].
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)466 future science group
in M14 melanoma cells by overexpression or repression of Cu/Zn SOD using a tetracycline-inducible expression system, scavenging intra-cellular superoxide increased tumor cell sensitiv-ity to daunorubicin, etoposide, and pMC540, whereas expression of the antisense SOD mRNA significantly decreased cell sensitivity to drug treatment. Cu/Zn SOD overexpressing cells exhibited higher activation of the executioner caspase 3 upon drug exposure, caspase 3 activa-tion was significantly lower when Cu/Zn SOD was repressed by antisense expression. These data demonstrate that intracellular superoxide anion regulates tumor cell response to drug-induced cell death via a direct or indirect effect on the caspase activation pathway [58]. On the other hand, edelfosine (1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine), a membrane-targeting anticancer ether lipid drug, induced cytotoxicity to human leukemia cells (HL-60 and K562) correlating with the production of free radicals [59]. Thus, it is clear that intracellular oxidation may have a regulatory role in apoptosis. The caspases themselves are cysteine-dependent enzymes and as such, appear to be redox sensi-tive. Indeed, it was observed that prolonged or excessive oxidative stress can actually prevent cas-pase activation. A physiological example of this are NADPH oxidase-derived oxidants generated by stimulated neutrophils that prevent caspase activation in these cells. Stimulated neutrophils appear to use a specialized caspase-independent pathway to initiate phosphatidylserine exposure and subsequent phagocytic clearance. Thus, the dual roles for reactive oxygen species in apop-tosis, that is, induction and inhibition of cas-pases, need to be kept in mind while attributing apoptosis or prevention of apoptosis by various chemotherapeutic drugs on tumor cells [60].
In this context, it is interesting to note that HGF, which suppresses the growth of sar-coma 180 and Meth A cells, enhanced the
generation of reactive oxygen species [61], whereas N-acetylcysteine, a precursor of glutathione and an intracellular free radical scavenger; and reduced form of glutathione, prevented HGF-suppressed growth of the tumor cells, lending support to the involvement of free radicals in the growth-suppressive action of HGF. In contrast to this, donors of nitric oxide (NO) triggered apoptosis of neurons, which could be prevented by growth factors such as IGF-1 and basic FGF (b-FGF). Sodium nitroprusside (SNP)-induced apoptosis of neurons was found to be associated with downregulation of Bcl-2 and upregula-tion of Bax protein levels. Treatment of neurons with a bax antisense oligonucleotide inhibited the caspase-3-like activation and neuronal death induced by SNP. On the other hand, treat-ment of neurons with an inhibitor of caspase-3, together with SNP did not affect the changes in the protein levels, although it inhibited NO-induced cell death. Pretreatment of cultures with either IGF-1 or bFGF prior to NO expo-sure inhibited caspase-3-like activation together with the changes in Bcl-2 and Bax protein levels. These results suggest that the changes in Bcl-2 and Bax protein levels followed by caspase-3-like activation are a component in the cascade of NO-induced neuronal apoptosis and that the neuroprotective actions of IGF-1 and bFGF might be due to inhibition of the changes in the protein levels of the Bcl-2 family [62].
It is interesting to note that HGFs that stimu-late growth, differentiation, and prevent apop-tosis of progenitor cells induce a rapid increase in reactive oxygen species in quiescent cells. HGFs, including granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-3, steel factor and thrombopoietin have their own specific cell surface receptor, which activates both unique and shared signal transduction pathways. When growth factor-deprived cells were exposed to H
2O
2 at concentrations that
Table 5. Content of vitamin e relative to total lipid and bis-allylic methylene units in normal liver, yoshida hepatoma cells and in microsomal suspensions prepared from normal liver and yoshida hepatoma cells.
Measurement Normal intact liver yoshida hepatoma cells
Normal microsomes yoshida microsomes ref.
Vitamin E, lipid (nmol/mg) 2.30 ± 0.53 4.16 ± 0.63 1.67 ± 0.31 3.06 ± 0.42 [6–10]
Vitamin E , bis-allylic methylene groups (×106)
7.6 31.8 5.1 38.6 [19]
Total antioxidant, lipid (nmol/mg)
2.99 ± 0.5 4.21 ± 0.31 ND ND [4,7]
All values are given as means ± standard deviations. The numbers of determinations are shown in parenthesis. ND: Not determined. Data taken from [19].
Review | Das

www.futuremedicine.com 467future science group
increased intracellular ROS, it was noted that H
2O
2 induced a dose-dependent increase in
tyrosine phosphorylation, including increased tyrosine phosphorylation of the GM-CSF recep-tor b-chain (betac), STAT5, and other signaling proteins. H
2O
2 also induced expression of the
early response gene c-FOS, and G1- to S-phase transition, but not S- to G2/M-phase transition of MO7e cells. The cell permeable antioxidant pyrrolidine dithiocarbamate decreased the intracellular levels of ROS and inhibited tyro-sine phosphorylation induced by GM-CSF in MO7e cells, suggesting that ROS generation plays an important role in GM-CSF signaling. Permeable antioxidant pyrrolidine dithiocarba-mate, N-acetyl cysteine and 2-mercaptoethanol,
which are known antioxidants, reduced growth and viability of MO7e cells. It is evident from these results that controlled generation of ROS in response to HGFs contributes to downstream signaling events, especially those involving tyro-sine phosphorylation [63]. Based on these results [51–63], it is reasonable to suggest that free radi-cals have both beneficial and harmful actions, induce apoptosis of normal and tumor cells and participate in the differentiation, growth and prevention of apoptosis of hematopoietic cells in response to growth factors. It is possible that these apparently paradoxical actions (induction and prevention of apoptosis) of free radicals depend on the type of free radical produced, site of their production, amount of the free radicals
Diet
ω-6 series ω-3 series
Cis-linoeic acid(18:2)
α-linolenic acid(18:3)
δ-6 desaturase
γ-linolenic acid(18:3)
Dihomo-GLA(20:3)
1 series ofprostaglandins
δ-5 desaturase
Arachidonic acid(20:4)
Eicosapentaenoic acid(20:5)
Docosahexaenoic acid(22:6)
Resolvins
Lipoxins
Neuroprotectins (NPD1)
Prostaglandins of 3 seriesPGA3, PGE3, PGF3α, PGI3, TXA3, LTB5
Prostaglandins of 2 seriesPGA2, PGE2, PGF2α, PGI2, TXA2, LTB4
Figure 1. Metabolism of essential fatty acids.
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)468 future science group
generated, duration and the rate with which they are produced, stimulus for their produc-tion (whether it is physiological or patho logical stimulus) and the rate with which the free radi-cals are produced in normal and tumor cells. It is likely that on exposure to a toxic stimulus such as anticancer drugs and radiation, excess (in other words high amounts) of free radicals are produced rapidly that disrupt cell and mito-chondrial membrane integrity, activate caspases that lead to apoptosis. In contrast, when the stimulus is a physiological one such as GM-CSF, IL-3, steel factor and thrombopoietin growth factors act on hematopoietic progenitor cells, the production of free radicals will occur in a controlled fashion that does not disrupt cell and mitochondrial cell membranes, induce tyrosine
phosphorylation, STAT5 and other signaling proteins, induce expression of the early response gene c-FOS that lead to G1- to S-phase transi-tion and do not induce the activation of caspases (Figure 2). Thus, a better understanding of the type, amount, site and degree of production of free radicals in response to various stimuli by both normal and tumor cells may help devise better methods to selectively enhance their pro-duction only in the tumor cells, such that selec-tive elimination of tumor cells will become a reality. One such endogenous molecule(s) that could selectively generate free radicals in tumor cells to induce their apoptosis seem to be certain PUFAs such as GLA, AA, EPA and DHA. In this context, the interaction between TNF and PUFAs is interesting.
S�mulus
Physiological (Haematopoie�c growth factors)
Pathological (An�-cancer drugs, Radia�on, TNF-α)
Progenitor cells/ Stem cells
Free Radicals especially, H2O2↑
ROS↑
Tyrosine phosphoryla�on, STAT5, c-FOS↑
Transi�on from G1 to S phase
Differen�a�on and
Growth of cells
Tumor cells/ Virus infected cells
ROS↑↑↑ NO↑↑
Bcl-2↓ Bax↑
Cell membrane and Mitochondrial
membrane disrup�on
Lipid peroxida�on↑↑
Apoptosis and/or
Necrosis of cells
Ac�va�on of Caspases
Stimulus
Physiological(hematopoietic growth factors)
Progenitor cells/stem cells Tumor cells/virus-infected cells
Free radicals,especially H2O2↑
ROS↑↑↑NO↑↑
ROS↑
Tyrosine phosphorylation,STAT5, c-FOS↑
Lipid peroxidation ↑↑
Apoptosis and/or necrosis of cells
Transition from G1 to S phase
Differentiation and growth of cells
Activation of caspases
Bcl-2↓Bax↑
Cell membrane andmitochondrialmembrane disruption
Pathological(anticancer drugs, radiation, TNF-α)
Figure 2. Possible role of free radicals in precursor/stem cell differentiation and growth and tumor cell apoptosis.NO: Nitric oxide; ROS: Reactive oxygen species.
Review | Das

www.futuremedicine.com 469future science group
Interaction between TNF & PUFAs TNF-induced tumor cell death and host toxicity can be related to its ability to induce the produc-tion of free radicals [64], while high intracellular glutathione levels induce tumor cell resistance to recombinant human TNF (rhTNF) and low glu-tathione levels enhanced sensitivity to rhTNF. Conversely, pretreatment of the tumor-bearing hosts with DL-buthionine-(S,R)-sulfoximine, an inhibitor of GSH biosynthesis, resulted in an increased sensitivity of tumor cells to rhTNF [64]. Recombinant IL-1 and TNF have been shown to stimulate human synovial cell phos-pholipase activity and induce the release of AA [65] and stimulate collagenase and PGE2 produc-tion that explains its role in tissue destruction and remodelling seen in inflammatory condi-tions [66]. Monocytes of patients with cancer produce elevated levels of TNF and PGE2 compared with normal subjects [67]. Subsequent studies demonstrated that there is a role for ara-chidonate metabolism in the direct cytolysis of tumor cells by TNF. It was observed that:nCytolysis of human U937 tumor cells by
recombinant TNF was reduced by dexameth-asone and quinacrine, agents that inhibit phospholipase A
2;
nU937 and L929 cells, which are susceptible to TNF cytolysis, released AA and its metabolites within 5 h of TNF challenge, before cell death was apparent, while cells resistant to cytolytic action of TNF such as U937/R and L929/R did not release arachidonate products on TNF challenge.
Surprisingly, rTNF cytolysis of U937 cells was not reduced by inhibitors of the cyclo-oxygenase and lipo-oxygenase pathways of AA metabolism. On the other hand, inhibition of phospholi-pase A
2 activity blocked the cytotoxic action of
TNF, indicating a role for phospholipase A2 but
not for AA or its metabolites in the direct cytoly-sis of tumor cells by TNF [68–70].
Suppression of arachidonate metabolism by steroids effectively inhibited TNF cytolysis and a marked, but late, increase in (malondialdehyde [MDA], an indicator of lipid peroxidation) pro-duction was noted in TNF-treated cells, suggest-ing that free radicals are involved in the cyto-lytic process and arachidonate metabolism is an essential link in the cytolytic process [71]. These results are supported by the observation that TNF stimulated a significant time-, dose-, and temperature-dependent increase in superoxide anion production in response to the chemotactic
peptide, f-methionyl-leucyl-phenylalanine or the tumor promotor, phorbol myristate acetate, by as much as 278%, indicating that brief expo-sures to rhTNF are able to enhance or prime the neutrophil oxidative burst in response to a sec-ond stimulus [72]. In a similar fashion, rhTNF, as a single agent, has only minimal therapeutic activity for the treatment of metastatic disease, but when combined with recombinant murine g-IFN (rM g-IFN) demonstrated significantly more therapeutic activity than when either agent was administered alone. However, this combina-tion also resulted in increased toxicity similar to the generalized Shwartzman’s reaction observed during endotoxin shock, with multifocal micro-thrombi and ischemic necrosis as sequelae, which were observed in the lungs, liver, gastrointestinal tract, testes or uterus and bone marrow. It was observed that TNF (either directly adminis-tered or induced in situ) and its induction of AA metabolites formed one element of toxicity in this model that were reduced by inhibitors of the cyclooxygenase/lipoxygenase pathway [73]. Furthermore, TNF becomes cytotoxic to human fibroblasts (FS-4 cells) when AA present in the culture medium is converted to PGs [74]. These and other results suggested that the activation of phospholipase A
2 with consequent AA release
and its conversion to PGs and leukotrienes and concomitant release of free radicals seem to be essential in bringing about the tumoricidal action of TNF [64–82]. Since AA seems to be cru-cial to the tumoricidal and cytotoxic actions of TNF, it is reasonable to assume that AA by itself could be exploited as an anticancer molecule.
Arachidonic acid has tumoricidal action [12,13]. TNF-a action on proliferation and cell death of differentiated human colon adenocarcinoma HT-29 cells was potentiated by co-treatment of cells with AA metabolism inhibitors (e.g., indomethacin, a cyclo-oxygenase inhibitor), and these effects were more significant in undiffer-entiated cells [70, 83]. TNF-a and indomethacin co-treatment was associated with accumulation of cells in G0/G1 cell cycle phase, increased reactive oxygen species production, elevated caspase-3 activity, and was accompanied by increased formation of the lipid peroxidation end products, such as malondialdehyde and 4-hydroxynonenal [83]. Similarly, AA administra-tion caused apoptosis in Y79 cells and activation of caspase-3 and cleavage of the endogenous cas-pase substrate poly-(ADP-ribose)-polymerase. AA also caused lamin B cleavage, suggesting subsequent caspase-6 activation. AA treatment
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)470 future science group
was accompanied by increased formation of the lipid peroxidation end products malondi-aldehyde and 4-hydroxy-2-nonenal, lowering in reduced glutathione content and in mitochon-drial membrane potential. Inhibiting glutathi-one synthesis sensitized Y79 cells to apoptosis-inducing stimuli, whilst replenishing reduced glutathione attenuated AA toxicity. Similar find-ings were obtained using hydroperoxyeicosatet-raenoic acids (oxygenated metabolites of AA, which deplete the reduced glutathione pool) and nordihydroguaretic acid, a general inhibi-tor of lipoxygenase pathway that may also trigger rapid depletion of reduced glutathione. Melittin, which is known to activate phospholipase A
2,
also potently induced apoptosis. These results indicate a role for oxidative stress in the cytotox-icity induced by AA [84]. We also noted similar results [85,86]. These results [83–86] and the results obtained with TNF with regard to their action on tumor cells [68–70,81] suggest that free AA is responsible for the apoptosis of tumor cells, pos-sibly, by enhancing the formation of lipid perox-ides and other toxic metabolites of AA.
Tumor cells have altered SOD levels & low amounts of lipid peroxidesIt is evident from the preceding discussion that tumor cells have low amounts of PUFA [25–27], have relatively higher amounts of vitamin E [13–16,20] and have low rates of lipid peroxida-tion [14–20]. Since SODs are a class of enzymes that catalyze the dismutation of superoxide into O
2 and H
2O
2, as such they form an important
antioxidant defense in nearly all cells exposed to O
2 and hence, it is of interest to know its activity
in tumor cells.One of the early studies revealed that the abil-
ity of bleomycin to produce DNA strand scis-sion could be inhibited by low concentration of metal ions, especially copper, and is stimu-lated by ferrous (Fe2+) ions, which was related to increased production of superoxide radicals, OH and H
2O
2. It was reported that diethyldi-
thiocarbamate treatment, which is known to inhibit SOD, rendered the Chinese hamster cell line (V79) cells more susceptible to the actions of bleomycin either as a function of bleomycin dose or as a function of bleomycin treatment time, suggesting that reducing Cu2+ levels and/or increasing superoxide concentrations enhances the cytotoxicity of bleomycin [87]. These results coupled with the observation that the decrease in susceptibility to dimethylbenaz(a)anthracene-induced breast cancer were correlated with
increased levels of SOD activity [88] and that there is a relationship between macrophage-induced cytotoxicity against breast cancer cells and the level of superoxide produced [89] sug-gests that SOD may have a role in cancer cell survival. For example, when SOD was added to the normal macrophage-tumor cell suspensions (MA-160 cell line) macrophage mediated tumor cytotoxicity was suppressed. By contrast, SOD often greatly enhanced the cytotoxic ability of macrophages from breast cancer patients. The macrophages obtained from the normal donors produced 46.0 ± 6.7 nanomoles O
2-. /106 macro-
phages per 10 min, while macrophages from the breast cancer patients who possessed cytotoxic macrophages produced 56.4 ± 5.2 nanomoles O
2-./106 macrophages per 10 min whereas those
from the breast cancer patients who possessed noncytotoxic macrophages produced 18.6 ± 3.2 nanomoles O
2-./106 macrophages per 10 min.
Thus, there is not only a relationship between macrophage cytotoxicity and the level of O
2-.
produced but also the inability to generate suffi-cient amounts of O
2-. might explain the inability
of breast cancer patients’ macrophages to kill tumor cells in some cases [89]. In this context, it is important to note that marked increase in protein levels of Mn-SOD was found in TNF-resistant cell lines after treatment with TNF, whereas no such increase was observed in CuZn-SOD protein in either TNF-resistant or sensitive cells. These results support the idea that the Mn-SOD is a rescue protein that induces resistance to TNF and possibly, anticancer drug cytotoxicity in some cancer cells [90], partly by lowering free radical levels in the cells and thus reducing the lipid peroxidation process. In this context, it is interesting to note that hydroxyl radicals, but not superoxide anion or hydro-gen peroxide likely play a role in the antitumor activity of doxorubicin in sensitive and resistant human ovarian cancer cells [91]. The resistance of adriamycin-resistant breast tumor cells to adria-mycin appears to be associated with a developed tolerance to superoxide, because of a twofold increase in SOD activity and a decreased sus-ceptibility to hydrogen peroxide due to a 12-fold augmented selenium-dependent glutathione per-oxidase activity. Acting in concert, these two enzymes (SOD and glutathione peroxidase) decreased the formation of hydroxyl radicals from reduced molecular oxygen intermediates [92]. These results support a role for oxygen radi-cals in the cytotoxic mechanism of adriamycin and possibly, other anticancer drugs, and that
Review | Das

www.futuremedicine.com 471future science group
the resistance of cancer cells to drugs could be related to alterations in their anti oxidant con-tent. This coupled with the observation that the activities of total SOD, Cu, Zn-SOD and Mn-SOD in hepatocellular carcinoma tissue were lower than those in normal liver tissues respectively and SOD activity in poorly dif-ferentiated HCC tissue was lower than that in well differentiated HCC tissue suggests that SOD activity is impaired in some cancers [93]. Similarly, both total SOD and Mn-SOD specific activities were lower in the tumor cell homog-enates compared with normal liver [19], the low-est activity was associated with the fastest grow-ing tumor. Mn-SOD activity was decreased in the fast- and medium-growth rate hepatomas but was slightly increased in the tumor with the slowest growth rate compared with normal liver. This suggests that decreased Mn-SOD specific activity is not a characteristic of all tumors. In addition, it was observed that antimycin A (an inhibitor of the electron transport chain) stimu-lated the production of O
2-. in normal rat liver
and slow-growth-rate tumor cells but not in the submitochondrial particles of fast-growth-rate tumor cells [94], lending support to the concept that the rate of lipid peroxidation is generally low in tumor cells.
By contrast, certain tumors demonstrated SOD activities that were higher than in nor-mal tissues [95,96], and paradoxically in some human gliomas the malignant phenotype could be suppressed by overexpressing Mn-SOD [97]. In gastric and colorectal adenocarcinomas overexpression of Mn-SOD may correlate with aggressiveness of the tumor [98]. Similarly, in both esophageal and gastric cancers, the levels of Mn-SOD mRNA were significantly elevated compared with normal tissue [99]. In some stud-ies it was observed that the activities of Mn-SOD and CuZn-SOD varied greatly among both human and rat glioma cells [100]. Huang et al. reported that the lower levels of Mn-SOD in SV40-transformed cells was related to increased cytosine methylation of the SOD2 intron region (the SOD2 gene contains a large CpG island spanning >3.5 kb that starts near the 5´ edge of the promoter and extends into intron 2) [101].
The paradoxical results noted about the role of free radicals and SOD and other antioxidants in tumor cell proliferation and apoptosis can be explained in terms of the interaction between the caspases and free radicals. The SH groups of the caspases are essential for their catalytic activity, which are inactivated by free radicals. In
M14 melanoma cells, O2
-. and H2O
2 promoted
tumor cell survival due to the inactivation of caspases [102]. On the other hand, enhanced lev-els of SOD may reduce intracellular ROS that may lead to reduced formation of lipid perox-ides that enhances cell proliferation due to the absence of negative feedback control exerted by lipid peroxides on cell proliferation. Thus, the relationship between ROS, antioxidants and cell proliferation is complex and their final affect on cell proliferation depends on the close interac-tion among ROS, antioxidants, caspases, lyso-phosphatic acid signaling [103], Bcl-2 and Bax protein levels, poly-(ADP-ribose)-polymerase and phospholipase A
2 activity and the cellu-
lar content of various PUFAs and eicosanoids formed, mitochondrial membrane integrity, and mTORC1 signaling in the tumor cells [104]. Similar to superoxide anion and H
2O
2, NO also
may have a role both in cancer suppression and progression [6].
Free radicals & lipid peroxides modulate cell proliferation A negative correlation exists between cell pro-liferation and lipid peroxidation. For example, spermatozoa that proliferate rapidly demonstrate low rates of lipid peroxidation, where as neurons, which seldom divide, have high concentrations of lipid peroxides. In primary cultures of smooth muscle cells, cell proliferation is controlled, at least in part, by general peroxidation reactions rather than the specific peroxidation reactions involved in prostanoid synthesis, when these cells were exposed to various PUFAs [105–107]. In particular, it was noted that the inhibition of proliferation of primary cultures of smooth mus-cle cells was related to the structures of differ-ent PUFAs, the highly unsaturated being more growth inhibitory in nature. Since the inhibi-tion of cell proliferation did not correlate with stimulated or inhibited PG synthesis in these studies, it was concluded that fatty acids them-selves modulate cell proliferation. The growth inhibitory actions of PUFAs could be blocked by antioxidants such as vitamin E, butylated hydroxytoluene, a-naphthol, 6-hydroxy-2,5,7,8-tetramethyl-chrom-2-carboxylic acid and dipyri-damole [105–107] indicating a role for free radicals and lipid peroxides.
The reduced rate of lipid peroxidation could be related, in part, to increased levels of the lipid-soluble antioxidant a-tocopherol at times of maximum DNA synthesis [108,109], suggest-ing that lipid peroxidation is decreased prior to
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)472 future science group
cell division. Thus, tilting the cellular balance more towards antioxidant capacity could lead to decrease in the rates of cell death and increase cell proliferation.
Free radicals & lipid peroxides modulate p53 & caspases & induce apoptosis Many of the gene products that control apopto-sis are also regulators of cell cycle progression, indicating that the cell cycle control and cell death are linked. p53 protein is an example of a gene product that affects both cell cycle progres-sion and apoptosis [110]. The p53 tumor suppres-sor maintains the integrity of the genome. Its nuclear localization is critical to this regulation. Martinez et al. reported that ionizing radiation caused a biphasic p53 translocation response, p53 entered the nucleus 1–2 h postirradiation, subsequently emerged from the nucleus, and then again entered the nucleus 12–24 h after the cells had been irradiated [111]. These changes in the subcellular localization of p53 could be completely blocked by the free radical scavenger WR 1065. It was noted that mitomycin C and doxorubicin, two DNA-damaging agents that do not generate free radicals, caused transloca-tion of p53 only after 12–24 h of exposure to the drugs, and this effect could not be blocked by WR 1065, indicating that although all three DNA-damaging agents induced delocalization of p53 to the nucleus, only trans-localization caused by irradiation was sensitive to free radi-cal scavenging. Thus free radicals signal p53 translocation to the nucleus indicating that free radicals damage DNA and p53. This is sup-ported by the observation that brief exposure of oligodendroglia-type cell line (OLN 93) to H
2O
2 produced a marked translocation of
p53 from the cytosolic to the nuclear compart-ment within 20 min of exposure [112]. By 48 h of exposure to H
2O
2, nearly 60% of the cells
exhibited p53 in the nuclei, at which time a large proportion of the cells underwent apop-tosis. The genotoxic-induced p53 relocaliza-tion appeared to be cell cycle-specific, since cells in the G0/G1 stage had more abundant nuclear-associated p53 and were more suscep-tible to H
2O
2-induced apoptosis than the cells
in G1/S phase. It was observed that genes p21 and mdm2 were upregulated following p53 nuclear translocation, and mdm2 enhancement accelerated the exit of p53 from the nucleus to the cytosol [112]. Thus, H
2O
2-induced p53-
dependent apoptosis that correlated with p53
nuclear translocation and is cell cycle related. In glial cells, H
2O
2-induced apoptosis is medi-
ated by p53 protein [113] in a time- and concen-tration-dependent manner that accompanied nucleosomal DNA fragmentation and chro-matin condensation [113]. In A172 cells, H
2O
2
increased the expression of p53 and enhanced the protein levels of Bak, p21(WAF1/CIP1) and GADD45 with no change in the protein levels of Bcl-2 and Bax. In contrast, primary cultured astrocytes from p53-deficient mouse brain grew faster than wild-type and heterozygous astro-cytes and were also resistant to H
2O
2-induced
apoptosis than wild-type and heterozygous astrocytes. Thus, glial proliferation and repair of damaged DNA is regulated by p53 and glial apoptosis caused by oxidative stress induced by H
2O
2 is also mediated by p53 [112]. In sum-
mary, it can be said that free radicals induce p53 activity and thus, produce apoptosis.
This relationship between free radical-induced p53 activity and consequent apoptosis is supported by the report that transformed mouse fibroblasts lacking p53 are resistant to the cytotoxic effect of pro-oxidants [114]. Mn-SOD activity was increased in liver tissue from p53-deficient mice in comparison with wild type. Transient transfection of HeLa cells with p53 led to a significant reduction in steady-state Mn-SOD mRNA levels and enzymatic activity, suggesting that expression of the antioxidant enzyme Mn-SOD is negatively regulated by p53. Increased expression of Mn-SOD rendered HeLa cells resistant to p53-dependent cytotoxic treatments and in co-transfection experiments, counteracted the growth inhibitory effect of p53 [114]. These results indicate that p53 inhib-its the expression of Mn-SOD, and overexpres-sion of p53 decreases the levels of SOD. It is known that tumor cells that have impaired p53 activity have higher SOD activity and thus, antioxidant capacity to these cells will be high. This suggests that p53 has a pro-oxidant type of activity. Thus, tumor cells exposed to free radicals would have enhanced SOD activ-ity and are likely to demonstrate enhanced expression of p53 due to free radical induced stress [112,113]. The increase in the expression of p53, in turn, would suppress SOD, tilting the balance more towards a pro-oxidant state and thus, leads to apoptosis. Furthermore, free radicals and lipid peroxides inactivate various enzymes, denature proteins and deplete cellu-lar ATP content that ultimately induce apopto-sis [113–115]. In addition, free radicals cause ATP
Review | Das

www.futuremedicine.com 473future science group
depletion in the cells by activating poly-ADP-ribose-polymerase, the substrate of caspase-3 [115], though there is some controversy regarding this [116]. Thus, free radicals, especially H
2O
2,
induces apoptosis by enhancing the expression of p53, decreasing SOD levels, activating poly-ADP-ribose-polymerase and by inactivating several cellular enzymes, denaturing cellular proteins and depleting cellular ATP content (Figure 3A & B). In this context, it is interest-ing to note that oxidative stress can shorten the length of telomeres [117–119].
Oxidant stress & Bcl-2 & their role in apoptosisBcl-2 opposes the pro-oxidant action of p53 by its antioxidant action [120] and ability to sup-press SOD activity, suggesting that a balance exists between the levels of p53, inducing a pro-oxidant status in cells and the expression and antioxidant activity of Bcl-2. It is likely that the increased expression of p53 that occurs during exposure to radiation and free radicals, may lead to inhibition of Bcl-2 expression that, in turn, augments increase in free radical generation and apoptosis. This is supported by the observation that Bcl-2 is downregulated by p53 [121,122].
Bcl-2 blocks lipid peroxidation that induces apoptosis [123]. Following an apoptotic signal, progressive lipid peroxidation occurs in the cells, whereas over-expression of Bcl-2 sup-presses lipid peroxidation [123]. Thus, a close association exists between lipid peroxidation, Bcl-2 and apoptosis [124]. When tumor cells are treated with PUFAs, there is not only an increase in the formation of lipid peroxides, but also enhanced phosphorylation of Bcl-2 that causes apoptosis [125]. For instance, in Burkitt’s lym-phoma cells, increased mitochondrial NAD(P)H was detected in Bcl-2 expressing cells that correlated with increased constitutive mito-chondrial production of H
2O
2 [126]. Although
production of H2O
2 was increased by TNF-a in
Bcl-2-negative lymphoma cells commensurate with the early phases of apoptosis, this increase was not observed in Bcl-2-expressing cells, sug-gesting that Bcl-2 allows cells to adapt to an increased state of oxidative stress, fortifying the cellular antioxidant defenses [126].
Based on these evidences, it is reasonable to propose that enhancing free radical generation specifically in tumor cells may lead to their death by apoptosis. 2-methoxy-oestradiol, PUFAs and thalidomide possess such ability [127–131]. TNF, interleukins and many anticancer drugs,
radiation and protoporphyrin derivatives (used in photodynamic therapy) augment free radical generation and lipid peroxidation process in the
p53 ↑
ROS ↑
DNA fragmentation andchromatin condensation ↑
Poly-ADP-ribose-polymerase ↑
Bak, p21(WAF1/CIP1)and GADD45 ↑
p53 deficiency
Apoptosis
Free radicals
Resistance to cytotoxic treatments
MnSOD ↓
MnSOD activity ↑
ATP ↓
DNA damage
(-)
(-)
Irradiation
Mitomycin CDoxorubicin
Translocation of
Mitomycin CDoxorubicin
No change inBcl-2 and Bax
Figure 3. relationship among free radicals, oncogenes, antioncogenes and cancer. (A) Scheme showing relationship between ROS and p53 and consequent induction of apoptosis in tumor cells. For further details see text. (B) Scheme showing the feedback relationship between p53 deficiency and resistance of tumor cells to apoptosis. (-) Indicates negative control, decrease in activity or synthesis. Cells lacking p53 are resistant to the cytotoxic effect of pro-oxidants. MnSOD activity is increased in hepatic cells that are p53-deficient in comparison with wild-type cells. Transient transfection of cells with p53 induced a significant reduction in MnSOD mRNA levels and enzymatic activity, suggesting that expression of the antioxidant enzyme MnSOD is negatively regulated by p53. Increased expression of MnSOD renders cells resistant to p53-dependent cytotoxic treatments and, in co-transfection experiments, counteracted the growth-inhibitory effect of p53, indicating that p53 inhibits the expression of MnSOD and overexpression of p53 decreases the levels of SOD. Tumor cells that have impaired p53 activity have higher SOD activity and thus the antioxidant capacity of these cells will be high, suggesting that p53 has a pro-oxidant type of activity. Tumor cells exposed to free radicals would have enhanced SOD activity and are likely to demonstrate enhanced expression of p53 due to free radical-induced stress. The increase in the expression of p53, in turn, would suppress SOD, tilting the balance more towards a pro-oxidant state and thus leading to apoptosis. ROS: Reactive oxygen species; SOD: Superoxide dismutase.
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)474 future science group
tumor cells and thus, induce tumor cell death. Some PUFAs have significant cytotoxic action on tumor cells and antiangiogenic properties, and augment free radical generation, specifically in the tumor cells and were found to inhibit the growth of human gliomas with few side effects [132–139].
n PUFAs enhance lipid peroxidation & inhibit cell proliferationWhen used at appropriate concentrations, GLA, AA, EPA and DHA, were toxic to tumor cells with little or no effect on normal cells in vitro [140–147]. This selective tumoricidal action of fatty acids was not blocked by cyclo-oxygenase and lipoxygenase inhibitors at least in some tumor cells, suggesting that PGs and leukotrienes are not major players in inducing apoptosis of PUFA-treated tumor cells [140–144]. Furthermore, GLA-, AA- and EPA-treated tumor cells (irrespective of the form in which these fatty acids are delivered) but not normal cells produced almost a two- to threefold increase in free radicals and lipid per-oxidation products [22,23,148–150]. These results suggest that the low rates of lipid peroxidation observed in tumor cells are, in part, due to a defi-ciency of PUFAs and a relative increase in their antioxidant content [19–21,25–29]. Since there is a direct correlation between the rate of lipid peroxi-dation and the degree of deviation in hepatomas [13–16,20] and as the rate of lipid peroxidation is low in several tumors (as discussed previously), it is suggested that lipid peroxidation might act as a physiological inhibitor of mitosis and regulate cell multiplication. Similarly, TNF-a induced apop-tosis of tumor cells by augmenting free radical generation and enhancing the release of endog-enous AA [151,152] could be prevented by removing of unesterified AA [151]. Thus, the cell ular level of unesterified AA may be a general mechanism by which apoptosis is induced in colon and other tumor cells. Since other PUFAs, such as GLA, DGLA, EPA and DHA, also induce apoptosis of tumor cells, it is likely that methods designed to enhance the cellular content of unesterified PUFAs may trigger apoptosis in tumor cells[153], which may explain the beneficial action of EPA- and DHA-rich fish oils and other unsatu-rated fatty acids in the prevention of colon and other cancers [154–157]. Furthermore, tumor cells exposed to PUFAs demonstrate low levels of vari-ous antioxidants [143,144]. PUFAs, especially n-3 fatty acids, suppress carcinogen-induced ras acti-vation [155,156,] Bcl-2 expression [157]; inhibit the activity of cyclo-oxygenase enzyme and VEGF
expression [158] that contributes to their antican-cer actions. In addition, PUFAs and their prod-ucts such as lipoxins, resolvins and protectins possess antiangiogenic action as well [159–161].
n GLA‑activated macrophages possess tumoricidal action Schlager et al. showed that the fatty acid com-position of specific lipids is important for the resistance of tumor cells to killing by antibody and complement [162–164]. Their studies dem-sonatrated that peritoneal macrophages from mice became cytotoxic after incubation with lymphokine (cytokine) [165–168]. Lymphokine-induced marked changes in lipid composition of peritoneal macrophages, cellular content of cholesterol and PUFA content of cellular lipids (especially 18:3, GLA or ALA) increased two- to threefold after 8 h when the cells demonstrated maximal tumoricidal activity. Cellular lipid and fatty-acid content returned to control levels by 24 h, when the peritoneal macrophages had lost tumoricidal activity. When casein-induced peritoneal macrophages were enriched in choles-terol or linolenic acid (18:3, GLA or ALA); the 18:3-enriched cells were markedly tumoricidal and showed a transient increase in superoxide release, suggesting that endogenous levels of 18:3 GLA/ALA and cholesterol regulate tumor cytotoxic activity of peritoneal macrophages solely by their ability to metabolize or mobilize 18:3 fatty acid but not through regulation of their protein synthesis, oxidative metabolism, or augmented capacity for tumor target binding. Since ALA cannot be synthesized by the mam-mals and so also by macrophages and has to be obtained from the diet, and since cells convert LA to GLA by the activity of the enzyme d-6 desaturase, in all probability, the fatty acid iden-tified in the studies by Schlager et al. could be GLA. This is in line with the observation that it is GLA but not ALA that has potent tumoricidal action [22,23,85,86,140–145,169–171].
n PUFAs are involved in various mitochondrial processesFollowing their intake, PUFAs are distributed to cells and enriched in cellular membranes, where they influence cellular metabolism and survival, in part, by modulating various mitochondrial pro-cesses including mitochondrial calcium homeo-stasis, gene expression, respiratory function, ROS production and mitochondrial apoptosis. The complex mechanisms involved in the effects of PUFAs on mitochondrial actions depend on
Review | Das

www.futuremedicine.com 475future science group
structural properties, cellular uptake, shuttling and metabolism, competition with intracellular stores as well as inherent properties of fatty acid metabolites [172]. For example, INS-1 cells (rat insulinoma INS-1 b cells) when incubated with 0, 50, 250 or 500 µM LA/0.5% (w/v) BSA for 48 h under culture conditions of normal (11.1 mM) or high (25 mM) glucose in serum-free RPMI-1640 medium, LA 500 µM significantly suppressed cell viability and induced apoptosis when adminis-tered in 11.1 and 25 mM glucose culture medium. LA 500 µM significantly increased Bax expres-sion in 25 mM glucose culture medium but not in 11.1 mM glucose culture medium and dose-dependently reduced mitochondrial membrane potential (DYm) and significantly promoted cytochrome c release from mitochondria in both 11.1 and 25 mM glucose culture medium, further reducing glucose-stimulated insulin secretion, which is dependent on normal mitochondrial function. With the increase in glucose levels in culture medium, INS-1 b-cell insulin secretion function deteriorated further. These results indi-cate that chronic exposure to LA-induced b-cell dysfunction and apoptosis, which involved a mitochondrial-mediated signal pathway and increased glucose levels, enhancing LA-induced b-cell dysfunction [173].
In a well-defined rat model of myocardial infarction, it was noted that in comparison to a diet that is high in either saturated or n-6 PUFA but poor in plant and marine n-3 PUFA, a diet that is low in saturated fats and n-6 PUFA but rich in plant and marine n-3 PUFA results in smaller myocardial infarct size (p < 0.01), which in turn was associated with a higher degree of accumulation of n-3 PUFA and a parallel decrease in AA, the main n-6 PUFA, in plasma, cell mem-branes, and cardiac mitochondria (p < 0.00001), suggesting that improved myocardial resistance and protection against ischemia-reperfusion injury showed by dietary n-3 PUFA could be, in part, due to the incorporation of n-3 fatty acids in cardiac mitochondria [174].
These results suggest that n-3 fatty acids are beneficial in protecting against oxidative-induced damage that may, in part be caused by their abil-ity to modulate mitochondrial reactive oxygen species production [173,174]. In fat-1 transgenic mice, that are capable of synthesizing n-3 fatty acids that results in decreases in the n-6:n-3 ratio, it was observed that an increase in n-3 fatty acids and a decrease in the n-6:n-3 ratio in liver mito-chondria occurs in the fat-1 compared with con-trol mice. This change in lipid composition in
mitochondria in fat-1 mice was associated with a decrease (p < 0.05) in the activity of electron transport chain (ETC) complex I and increases (p < 0.05) in the activities of complexes III and IV. Mitochondrial H
2O
2 production with either
succinate or succinate/glutamate/malate sub-strates was decreased (p < 0.05) in the fat-1 mice due to a decrease in ROS production from ETC complex I. These results emphasize the fact that fatty acid changes in liver mitochondria (and pos-sibly in other tissues) has the ability to modulate oxidative stress [175] by altering reactive oxygen species production from ETC complex I. Even the chemo preventive actions of fish oil could be attributed to reduced oxidative stress in mam-mary tissue and liver mitochondrial fraction exposed to mammary carcinogen 7,12-dimethyl benz(a)anthracene [176].
By contrast, tumor cells exposed to PUFAs at micromolar concentrations produce a drastic increase in the generation of ROS, mitochondrial dysfunction along with an increase in the inci-dence of apoptotic cell death, a process that is prevented by antioxidants [177–179]. For example, both DHA and EPA have proapoptotic effects on colorectal cancer cells with different molecu-lar phenotypes but not in noncancerous cells. Apoptosis is caspase dependent, and both intrin-sic and extrinsic pathways are implicated. The dimerization of Bax and Bak, the depolarization of the mitochondrial membrane, and the subse-quent release of cytochrome c and Smac/Diablo to the cytosol was reported in support of the activation of the intrinsic pathway. On the other hand, the involvement of the extrinsic pathway is evident from the fact that activation of caspase-8, along with the down-regulation of FLIP occurs in PUFA-treated cancer cells. It was reported that in addition to the involvement of peroxi-some proliferator-activated receptor (PPAR)-g and cyclooxygenase-2 pathways in the protective action of EPA/DHA in colon cancer, the down-regulation of two key regulatory elements of the extrinsic and intrinsic pathways, FLIP and XIAP, respectively, has also been reported [179] suggesting that these fatty acids could potentially be useful as adjuvant anticancer agents in combination with other chemotherapeutic agents.
Based on these findings [172–179], it can be sug-gested that normal and tumor cells metabolize PUFAs in different ways such that in normal cells oxidative stress is decreased whereas tumor cells show increased oxidative stress on exposure to various PUFAs. Such a differential metabo-lism by normal and tumor cells may also explain
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)476 future science group
why GLA, AA, EPA and DHA are able to kill tumor cells selectively with little or no toxic action on normal cells [28,29,86,135,140,141]. This supposition is further supported by the observa-tion that GLA and to a limited extent EPA and DHA have cytoprotective actions against radia-tion and chemicals, whereas these fatty acids are cytotoxic to tumor cells but not normal cells [28,29,85,86,132,180–185]. It was also reported that alloxan-induced cytotoxic action against pancre-atic b-cells can be prevented by AA, GLA, EPA and DHA [186,187]. The cytoprotective action of PUFAs seems to be mediated to some extent by their products such as PGE
1, lipoxins, resolvins
and protectins [181,185]. Thus, some of the ben-eficial actions of PUFAs and their products such as PGE
1, PGI
2, lipoxins, resolvins and protectins
could be ascribed to their cytoprotective actions.
n PUFAs augment cytotoxic action of anticancer drugs & reverse drug resistance Previously, we and others observed that GLA and AA of the w-6 series and EPA and DHA of the w-3 series potentiated the cytotoxicity of anticancer drugs, vincristine, cis-platinum and doxorubicin on human cervical carcinoma (HeLa) cells and other cancer cells in vitro [188–198]. ALA, GLA, EPA and DHA enhanced the uptake of vincristine by HeLa cells. In addi-tion, DHA, EPA, GLA and DGLA were found to be cytotoxic to both vincristine-sensitive (KB-3–1) and vincristine-resistant (KB-ChR-8–5) human cervical carcinoma cells in vitro. Preincubation of vincristine-resistant cells with suboptimal doses of fatty acids enhanced the cytotoxic action of vincristine. GLA, DGLA, AA, EPA and DHA enhanced the uptake and inhibited the efflux of vincristine and thus, augmented the intra cellular concentration of the anticancer drug(s). These and other results suggest that PUFAs not only enhance the cyto-toxicity of anticancer drugs to tumor cells but also reverse tumor cell drug-resistance [186–198].
n PUFAs are differentially metabolized by normal & tumor cells It appears that both normal and tumor cells metabolize fatty acids differentially. For instance, AA is metabolized to produce the 5-lipoxygenase (5-LO) metabolite, 5-hydroxye-icosatetraenoic acid (5-HETE) by prostate cancer cells that stimulated them to grow, sug-gesting that 5-HETE is a survival factor for these cells. Prostate cancer cells constitutively produce 5-HETE and exogenous arachidonate
markedly increases the production of 5-HETE, while inhibition of 5-LO induced apoptosis in both hormone-responsive (LNCaP) and hormone-nonresponsive (PC3) human pros-tate cancer cells. Apoptosis was specific for 5-LO-programmed cell death, since it was not observed with inhibitors of 12-lipoxygenase, cyclooxygenase, or cytochrome P450 path-ways of AA metabolism. Exogenous 5-HETE protected these cells from apoptosis induced by 5-LO inhibitors, confirming a critical role of 5-LO activity in the survival of these cells [199]. These findings suggest that the way free fatty acids are metabolized by the tumor cells may influence survival and progression of can-cer. It was observed that free AA and GLA are tumoricidal but when AA is converted to form 5-HETE by 5-LO, the tumor cells are stimulated to grow [200,201].
Cyclo-oxygenase (COX)-2 is upregulated in many cancers that may explain as to why nonste-roidal anti-inflammatory (NSAIDs) drugs that inhibit COX-2 prevent colon cancer and cause apoptosis. The mechanism for this response could be due to an accumulation of the sub-strate (e.g., AA) or diversion of the substrate into another pathway. For example, colon adeno-carcinomas overexpress AA-utilizing enzyme, fatty acid-CoA ligase (FACL)4, in addition to COX-2. Thus, it is likely that unesterified AA in cells is a signal for induction of apoptosis. Tumor cells engineered with inducible overexpression of COX-2 and FACL4 act as ‘sinks’ for unesterified AA as evidenced by the observation that activa-tion of the enzymatic sinks blocked apoptosis, and the reduction of cell death was inversely cor-related with the cellular level of AA. Cell death caused by TNF-a is prevented by removal of unesterified AA. These results suggest that the cellular level of unesterified AA and other unsaturated fatty acids is a general mechanism by which apoptosis is regulated and that COX-2 and FACL4 promote carcinogenesis by lower-ing this level [202–205]. Furthermore, NSAIDs upregulated 15-LOX-1 and 15-LOX-1 inhibi-tion blocked NSAID-induced apoptosis, which was restored by 13-S-hydroxyoctadecadienoic acid (13-S-HODE; is the product of 15-LOX-1 protein, the other product of 15-LOX-1 is 15-S-HETE, but in this study 15-S-HETE for-mation was not noted) but not by its parent, LA. Thus, NSAIDs induce apoptosis in colon cancer cells via up-regulation of 15-LOX-1 in the absence of COX-2 [206–208]. Hydroperoxides generated by 5-, 12-, or 15-LOs from linoleate,
Review | Das

www.futuremedicine.com 477future science group
linolenate, or arachidonate and the correspond-ing hydroxides induced apoptosis of erythroleu-kemia and neuro blastoma cells in a concentra-tion- and time-dependent manner, while the terminal products of the arachidonate cascade (i.e., leuko trienes, PGs and TXs) were not cyto-toxic [209]. Thus, free unsaturated fatty acids need to be converted to their respective hydroxides to bring about their tumoricidal action. In addition, many AA metabolites serve as growth signaling molecules. 5-LO pathway metabolite 5-HETE has a growth stimulatory action on breast can-cer cells whereas selective reduction in the levels of 5-HETE but not cyclo-oxygenase inhibitors reduced growth, increased apoptosis, downregu-lated bcl-2, upregulated bax, and increased G1 arrest. 5-LO inhibition upregulated PPAR-a and -g expression, and were growth inhibited when exposed to relevant PPAR agonists. These results suggest that disruption of the 5-LO signaling pathway mediates growth arrest and apoptosis in breast cancer cells, partly, by the induction of PPARs and activation of PPARs with shunted endoperoxides [210,211]. These results imply that delivery of free unsaturated fatty acids to the tumor cells and generation of hydroperoxides by 5- 12- or 15-LOs from linoleate, linolenate, or arachidonate, and simultaneous inhibition of COX-2 enzyme could lead to apoptosis of tumor cells. In addition, PUFAs suppress fatty acid synthase enzyme and thus, induce apoptosis of tumor cells [212–218].
n PUFAs inhibit telomerase activity in tumor cellsHigh telomerase activity is present in most can-cer cells. Unsaturated fatty acids significantly inhibited telomerase activity and the inhibi-tory potency was elevated with an increase in the number of double bonds. PUFAs, EPA and DHA were the most potent telomerase inhibi-tors. DLD-1 cells when cultured in the presence of EPA and DHA demonstrated a remarkable decrease in telomerase activity due to down-regulation of hTERT and c-myc expression via protein kinase C inhibition, suggesting that PUFAs directly inhibit the enzymatic activity of telomerase as well as modulate the telomerase at the transcriptional level [219,220].
PUFAs modulate immune response and inhibit the adhesion of tumor cells to endothe-lium [221,222] and suppress tumor cell growth. Several studies showed that GLA and other PUFAs enhance tumor cell chemosensitivity [188–198,223,224].
n PUFAs modulate G‑protein‑mediated signalsPUFAs modulate G-protein-mediated signal transduction [225], mobilize Ca2+ from intra-cellular stores [226], which may induce apop-tosis [227]; activate PKC and enhance NADPH oxidase in macrophages [228] and thus enhance O
2-. generation. EPA decreased Bcl-2 while
increasing Bax in tumor cells [229]. DHA increased p27, inhibited cyclin-associated kinase, reduced pRb phosphorylation and
Figure 4. CT scan of the brain of a patient with glioma treated with intratumoral g-linolenic acid. (A) CT scan of brain with contrast of a 15-year-old male patient with left temporal glioblastoma multiforme prior to the intratumoral injection of g-linolenic acid. Note marked midline shift suggesting raised intracranial tension and mass effect. (B) CT scan of brain with contrast of the same patient taken 24 h after seven intratumoral injections at the rate of 1 mg/day. CT scan demonstrates significant necrosis of the tumor and marked reduction in the midline shift, suggesting reduction in the intracranial tension and mass effect. There were no side effects due to the therapy.
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)478 future science group
cause some melanoma cells to undergo apop-tosis [230,231]. PUFAs (especially EPA) inhib-ited cell division by inhibiting translation ini-tiation, preferentially reducing the synthesis and expression of G1 cyclins both in vitro and in vivo [232]. In addition, free radicals, especially H
2O
2, directly activated purified heterodimeric
Gi and G
0 (small G proteins) [233], which are
important signaling molecules. Thus, PUFAs target several intracellular second messenger molecules and genes either directly or indirectly to enhance free radical generation to induce tumor cell apoptosis.
n Intratumoral administration of GLA for human gliomasPreviously, we have demonstrated that intratu-moral injection of GLA regressed human brain gliomas without any significant side-effects [132–
136], while GLA injected into the normal brain tissue of healthy dogs did not demonstrate any side-effects (Figure 4) [132–136].
n GLA has antivascular & antiangiogenic actionsModified GLA (in the form of lithium GLA coupled to iodized salt solution) when injected intra-arterially occluded only tumor-feeding vessels (Figures 5 & 6) without any action on normal blood vessels and thus, induced tumor necrosis [234,235]. Though it is common for
cytotoxic infusions to demonstrates vasoactivity that results in vasospasm, this is generally tran-sient (that may not last longer than 24–48 h). In our study, the selective occlusion of tumor feeding vessels with little action on normal blood vessels was seen even after 7 to 10 days after the infusion of modified GLA suggesting that occlusion of tumor feeding vessels is rather specific. Although the exact mechanism of this antiangiogenic antivascular action is not clear, it is possible that GLA-induced free radicals act on endothelial cells of tumor-feeding blood vessels and induce their occlusion. It is known that endothelial cells lining the tumor-feeding vessels are different from the endothelial cells lining the normal blood vessels explaining the selective occlusion of tumor-feeding vessels. It remains to be proven, but it is possible that exposure of endothelial cells of tumor-feeding vessels to PUFAs (such as lithium GLA coupled to iodized salt) enhances free radical generation that, in turn, oxidize the fatty acids to produce hydroperoxy fatty acids, blocking the produc-tion of cytoprotective lipoxins, resolvins and protectins. These hydroperoxy fatty acids and the formation of vasoactive PGs, LTs and TXs may produce intense vasospasm and occlusion of tumor-feeding vessels. These hydroperoxy fatty acids may also be generated by tumor cells exposed to PUFAs.
Possible differential metabolism of PUFA by normal & tumor cells One intriguing possibility is that PUFAs are differentially metabolized by normal and tumor cells. This is because PUFAs form precursors to both cytotoxic and cytoprotective molecules. For instance, as discussed above, generation of hydroperoxides by 5- 12-, or 15-LOs from linoleate, linolenate, or arachidonate have cytotoxic actions and induce apoptosis of tumor cells. In contrast, lipoxins, resolvins, and protectins (including neuroprotectin D
1)
have cytoprotective properties by virtue of their anti-inflammatory actions. Thus, it is possible that when tumor cells are exposed to adequate amounts of PUFAs cytotoxic metabolites are generated that induce apoptosis of tumor cells, whereas normal cells convert PUFAs to cyto-protective molecules such as lipoxins, resolvins and protectins [236–247]. This is further sup-ported by the observation that DHA is toxic to tumor cells but protects normal neural cells from stress-induced apoptosis. DHA induces apoptosis of neuroblastoma cells due to its
Figure 5. effect of lithium-g-linolenic acid injection coupled to iodized salt solution on tumor blood supply in giant cell tumor. (A) Angiogram of a patient with a giant cell tumor of the right femur performed just prior to lithium-g-linolenic acid coupled to iodized salt solution (LGIOC) injection. Single arrow shows the tip of the catheter in the popliteal artery and the site of injection of LGIOC. Double arrows show the origin of the tumor-feeding vessels. (B) Angiogram performed immediately after the injection of LGIOC. Arrow shows the site of complete occlusion of the tumor-feeding vessel. Normal blood vessels, which were distal and in the path of the blood flow and much smaller than the main tumor-feeding vessel, remained patent. (C) Angiogram performed 10 days after the injection of LGIOC. Single arrow shows the site of occlusion of the main tumor-feeding vessel. Double arrows show the accumulation of LGIOC in the tumor.
Review | Das

www.futuremedicine.com 479future science group
conversion to 17-hydroxydocosahexaenoic acid (17-HDHA) via 17-hydroperoxydocosahexae-noic acid (17-HpDHA) through 15-LO and autoxidation [248] and as a result tumor cells do not produce (or form negligible amounts) of the anti-inflammatory lipid mediators such as resolvins and protectins. 17-HpDHA is cyto-toxic to tumor cells [236] and DHA itself could inhibit secretion of PGE
2. Thus, the cytotoxic
action of DHA on neuroblastoma cells is caused by the production of hydroperoxy fatty acids and restricted production of resolvins and pro-tectins that are cytoprotective in nature [30–33]. In a similar fashion, it is possible that when normal cells are exposed to AA and EPA sig-nificant amounts of lipoxins and resolvin are formed, whereas tumor cells would accumu-late respective, prostanoids, leukotrienes, TXs and cyclopentanone PGs and respective hydro-peroxy fatty acids. Based on this evidence, it is proposed that normal cells metabolize PUFAs to produce cytoprotective lipids such as lipox-ins, resolvins and protectins while tumor cells generate toxic hydroperoxy fatty acids. This dif-ferential metabolism of PUFAs by normal and tumor cells may explain why PUFAs are toxic to tumor but not to normal cells (Figure 7).
Lipid-based novel therapeutic approaches to cancer It is evident from the preceding discussion that approaches that specifically enhanced the for-mation of lipid peroxides preferentially in the tumor cells but not normal cells could form a novel therapeutic approach to selectively kill cancer cells by apoptosis. Such a selec-tive delivery of PUFAs to tumor cells could be performed as discussed later.
n Intratumoral injection Selective delivery of PUFAs (especially GLA) to glioma cells can be achieved by their intra-tumoral injection into the tumor bed either by direct injection or delivering the fatty acid by Omayya reservoir as demonstrated previously both in experimental animals [212] and humans [132–136]. In these studies, GLA was injected into the tumor bed and produced no significant side effects while tumor regression was observed.
n Conjugating with iodized salt solution to produce lithium‑GLA‑iodized solution complex for intra‑arterial injection/infusionLithium-GLA-iodized solution complex when injected intra-arterially into the major
tumor-feeding vessels occluded only tumor-feeding vessels without any action on normal blood vessels and thus, induced tumor necrosis and regression of liver cancer, renal cell carci-noma and giant cell tumor of bone as described previously [234,235]. Lithium g-linolenic acid is partially water soluble. It is possible to prepare by suitable chemical modifications lithium-GLA-iodized solution complex-like complex that is more water soluble, stable and could, therefore, be given as an intravenous infusion or injection. If such an intravenous prepara-tion is developed, it could be administered as a continuous intravenous infusion for different types of solid tumors and even for leukemias.
n PUFAs plus growth factors (EGF/VEGF)/antigrowth factor(s) antibodies for cancerPolyunsaturated fatty acids could be conjugated to growth factors and/or monoclonal or spe-cific polyclonal antibodies to growth factors that could ensure their delivery to cancer cells to bring about their tumoricidal action. It is envisaged that such a complex of PUFAs plus growth factors and PUFAs plus antigrowth fac-tor antibody (such as Erbitux® and Avastin®)
will potentiate the antiangiogenic action of antiangiogenic molecules and regresses cancer-ous growth. It is postulated that when Avastin and Erbitux are conjugated with PUFAs (pref-erably GLA, EPA and/or DHA) and stabilized, such a complex could be infused intravenously for colon, breast, prostate and bone cancers. Since tumor cells express high numbers of
Figure 6. selective occlusion of tumor-feeding vessels. (A) Angiogram of the normal right kidney. (B) Angiogram of the same patient that shows the enlarged distorted kidney due to the presence of a renal tumor in the upper pole. The lateral margin of the kidney is not clear and an increase in blood supply to the tumor can be seen. (C) Angiogram of the left kidney performed immediately after injection of lithium-g-linolenic acid coupled to iodized salt solution. Occlusion of the tumor-feeding vessels but not those feeding the normal lower pole of the kidney, which is supplied by the two apparently normal blood vessels, can be observed.
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)480 future science group
Figure 7. Metabolism of polyunsaturated fatty acids in normal and tumor cells. Scheme showing the metabolism of AA in normal and tumor cells. Tumor cells produce more PGs, leukotrienes, thromboxanes, HETEs and HHTs, while normal cells generate lipoxins, resolvins and protectins that are cytoprotective. Similar to the metabolism of AA shown here, eicosapentaenoic acid and docosahexaenoic acid may also be metabolized in a similar fashion. It was reported that higher amounts of COX enzyme-derived 12-HHT and a decrease in 12-LOX, 15-LOX-2, 15-LOX-1 and PGE activities corresponded to higher apoptosis and lower mitosis (12-HHT concentrations were higher, PGE
2, 12-HETE, 15-HETE and 13-HODE was lower and the 13-HODE:12-HETE ratio was higher) [238].
12(S)-HETE augments the invasiveness of prostate carcinoma cells via selective activation of PKC-a [239]; both ERK and cellular protein tyrosine kinase activation are involved in 12(S)-HETE-induced pancreatic cancer cell proliferation [240]. Chen et al. showed that colorectal cancer is associated with decreased 15(S)-HETE and treatment of colon cancer cells with 15S-HETE inhibited cell proliferation and induced apoptosis in a peroxisome proliferator-activated receptor (PPAR)-g-dependent pathway involving augmentation of TGF-b-inducible early gene and reduction of Bcl-2 expression [241]. Both AA and 15-l-(S)-hydroxyeicosatetraenoic acid demonstrated cytotoxic action on MCF-7 (breast cancer) cells [242]. 13(S)-HODE, a primary product of 15-LOX-1 metabolism of LA, is decreased in tumor cells. 13(S)-HODE binds to PPAR-d, decreases PPAR-d activation and downregulates PPAR-d expression in colorectal cancer cells; induction of 15-LOX-1 expression is a critical step in nonsteroidal anti-inflammatory drugs such as aspirin downregulation of PPAR-d and the resultant induction of apoptosis, and PPAR-d is an important signaling receptor for 13(S)-HODE-induced apoptosis [243]. Addition of 13(S)-HODE decreased tumor cell proliferation. HCT-116 colorectal cells that were stably transfected with 15-LOX-1 formed smaller tumors in nude mice. Thus, 13(S)-HODE induces changes in gene expression and has antitumorigenic effects [244]. Tumor cells form lipoxins [245] and deficient lipoxin production was paralleled by an attenuated conversion of AA to 12-HETE in cancer cells. In chronic myeloid leukemia patients, blast crisis was associated with reduced lipoxin synthesis, while this capacity improved after retransformation into a second chronic phase [246]. Lipoxins suppress the growth of tumor cells [247], and resolvins and protectins may have similar action. Tumor cells that form substantial amounts of lipoxins, resolvins and protectins may be resistant to chemotherapy and radiation. Tumor cells that form increased amounts of 17-HDHA and 17-HpDHA (formed from docosahexaenoic acid and formation of similar compounds from AA and eicosapentaenoic acid) undergo apoptosis in response to chemotherapeutic drugs and radiation. AA: Arachidonic acid; COX: Cyclo-oxygenase; HDHA: Hydroxydocosahexaenoic acid; HETE: Hydroxyeicosatetraenoic acid; HODE: Hydroxyoctadecadienoic acid; HpDHA: Hydroperoxydocosahexaenoic acid; HPETE: Hydroperoxyeicosatetraenoic acid; LO: Lipoxygenase; LT: Leukotriene; PG: Prostaglandin.
AA
Phospholipase A2
Cell membrane phospholipids
Tumor cell Normal cell
COX-1 and COX-2; 12-LO and 5-LO ↑
15-LO and acetylated COX-2 ↓
COX-1 and COX-2; 12-LO and 5-LO ↓
15-LO and acetylated COX-2 ↑
PGG2, PGH2, 12-HPETE,5-HPETE, 12-HETE, 5-HETE ↓
15-HPETE, 15(R)-HETE, 15(S)HETE ↑
PGG2, PGH2, 12-HPETE,5-HPETE,12-HETE, 5-HETE ↑
15-HPETE, 15(R)-HETE,15(S)HETE, 13(S)-HODE ↓
PGD2, PGE2, PGF2, PGI2, 12-HHT, LTA4, LTB4, LTC4, LTD4, LTE4 ↑
PGD2, PGE2, PGF2, PGI2, 12-HHT,LTA4, LTB4, LTC4, LTD4, LTE4 ↑
Lipoxin A4, lipoxin B4, 15-epi-lipopoxin A4,15-epi-lipoxin B4, 17-HDHA and 17-HpDHA ↓
Lipoxin A4, lipoxin B4, 15-epi-lipopoxin A4,15-epi-lipoxin B4, 17-HDHA and 17-HpDHA ↑
Enhance proliferation, metastasis, inflammationand susceptibility to chemotherapy
Suppress proliferation, metastasis, inflammationand susceptibility to chemoptherapy
Review | Das

www.futuremedicine.com 481future science group
growth factor receptors compared with normal cells, it is expected that a conjugate of growth factors (such as EGF/VEGF) and PUFAs will bind selectively to their respective growth fac-tors such that PUFAs will be released prefer-entially to the tumor cells to bring about their tumoricidal action. Even if such a growth factor plus PUFA complex binds to normal cells, side effects are unlikely since PUFAs are not toxic to normal cells.
n PUFAs conjugated to antiangiogenic molecules such as angiostatin & endostatinBoth angiostatin and endostatin are well estab-lished endogenous antiangiogenic molecules that proved to be successful in the treatment of animal tumor models but subsequently failed in human clinical trials. It is envisaged that conjugation of PUFAs with angiostatin and endostatin will potentiate the actions of angio-genic molecules and, in turn, angiostatin and endostatin potentiate the anticancer actions of PUFAs. Such endostatin plus PUFAs and angiostatin plus PUFAs complexes could be injected into the glioma tumor bed or given intravenously or intra-arterially for other solid tumors.
n TNF‑a plus PUFA complexesTNF-a is a potent anticancer molecule that elicits its cytotoxic actions on tumor cells by enhancing free radical generation and inducing the release of AA. TNF-a activates phospho-lipase A
2 and thus, induces the release of AA
to bring about its action on tumor cells. Since
tumor cells are deficient in PUFAs (especially AA), many tumor cells are resistant to the cyto-toxic action of TNF-a. Studies demonstrated that tumor cell resistance to TNF-a can be overcome by pretreating them with PUFAs or by simultaneous exposure of tumor cells to both PUFAs and TNF-a. Hence, it is envisaged that a combination of TNF-a and PUFAs will be cytotoxic to cancer cells. PUFAs can be conju-gated to TNF-a and infused to potentiate its anticancer action.
n PUFA plus anticancer drug complexesPolyunsaturated fatty acids can be conju-gated to conventional anticancer drugs such as adriamycin, cyclophosphamide, cis-platinum, and given parentarally to potentiate their anticancer actions.
n PUFAs as nanoparticles alone or in combination with growth factors/monoclonal antibodies to growth factors/TNF‑a/angiostatin/endostatin/anticancer drugsPolyunsaturated fatty acids can be delivered as nanoparticles either by themselves and/or in combination with conventional anticancer drugs/growth factors/antibodies to growth factors/TNF-a/angiostatin/endostatin.
n PUFAs in combination with siRNA siRNA to specific oncogenes could be prepared and complexed with PUFAs for their delivery to cancer cells. It has recently been demonstrated that PUFAs form excellent vehicle to deliver siRNA to various cells in vivo [249–253]. Hence, it
executive summary
n Tumor cells have defective activity of d-6 and d-5 desaturase enzymes.n As a result tumor cells have low concentrations of polyunsaturated fatty acids such as g-linolenic acid, arachidonic acid, eicosapentaenoic
acid and docosahexaenoic acid.n This leads to low rates of lipid peroxidation in tumor cells compared with normal cells.n Tumor cells also have relatively high levels of antioxidants such as vitamin E.n Low rate of lipid peroxidation in tumor cells could be one reason for their high mitotic rate.n Tumor cells are highly susceptible to the toxic actions of lipid peroxides.n Various anticancer drugs, TNF and radiation bring about their tumoricidal actions by enhancing free radical generation and formation of
lipid peroxides in cancer cells.n Drug resistance of tumor cells to various chemotherapeutic drugs could be due to their inability to enhance free radical generation and
lipid peroxidation process in the cells.n Selective delivery of polyunsaturated fatty acids to tumor cells induces apoptosis while these fatty acids are not toxic to normal cells.n A combination of polyunsaturated fatty acids and anticancer drugs or TNF enhances apoptosis of tumor cells and reverse
drug resistance.n Polyunsaturated fatty acids are metabolized differentially by normal and tumor cells.n Polyunsaturated are converted to toxic lipid metabolites in tumor cells while normal cells fail to do so.n Increased formation of cytoprotective molecules such as lipoxins may render tumor cells drug resistant.n Polyunsaturated fatty acids and some of their metabolites also have antiangiogenic action.n Selective delivery of polyunsaturated fatty acids and/or their toxic metabolites may form a new approach to cancer in future.
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)482 future science group
Bibliography1 Casppary WJ, Niziak C, Lanzo DA,
Friedman R, Bachur NR. Bleomycin A2: a ferrous oxidase. Mol. Pharmacol. 16, 256–260 (1979).
2 Young, RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N. Engl J. Med. 305, 139–153 (1981).
3 Gajewski E, Rao G, Nackerdien Z, Dizdaroglu M. Modification of DNA bases in mammalian chromatin by radiation-generated free radicals. Biochemistry 29, 7876–7882 (1990).
4 Das UN. Oxy radicals and their clinical implications. Curr. Sci. 65, 964–968 (1993).
5 Das UN. Free radicals, Biology and relevance to disease. J. Assoc. Phy. Ind. 38, 495–498 (1990).
6 Halliwell BA. Superway to kill cancer cells? Nature Med. 6, 1105–1106 (2000).
7 Cleveland JL, Kastan MB. A radical approach to treatment. Nature 407, 309–311 (2000).
8 Sunderesan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H
2O
2
for platelet-derived growth factor signal transduction. Science 270, 296–299 (1995).
9 Suderesan M, Yu ZX, Ferrans VJ et al. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 318, 379–382 (1996).
10 Jayanthi S, Ordonez S, McCoy MT, Cadet JL. Dual mechanism of Fas-induced cell death in neuroglioma cells, a role for reactive oxygen species. Brain Res. Mol. Brain Res. 72, 158–165 (1999).
11 Wang S, Leonard SS, Ye J, Ding M, Shi X. The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 279, C868–C875 (2000).
12 Das UN. Tuning free radical metabolism to kill tumor cells selectively with emphasis on the interaction(s) between essential fatty acids, free radicals, lymphokines and prostaglandins. Ind. J. Pathol. Microbiol. 33, 94–103 (1990).
13 Das UN. Cis-unsaturated fatty acids as potential anti-mutagenic, tumoricidal and anti-metastatic agents. Asia Paci. J. Pharmacol. 7, 305–327 (1992).
14 Galeotti T, Borrello S, Masoti L. Oxy radical sources, scavenger systems and membrane damage in cancer cells. In: Oxygen Radicals, Systemic Events and Disease Processes. Das DK, Essman R (Eds). S Karger, Basel, Switzerland, 129–148 (1990).
15 Dianzani MU, Rossi MA. Lipid peroxidation in tumors. In: Recent Trends in Chemical Carcinogenesis (Volume 1). Pani P, Feo F, Columbano A, Cagliari ESA (Eds). Cagliari, Italy, 243–257 (1981).
16 Bartoli GM, Galeotti T. Growth-related lipid peroxidation in tumor microsomal membranes and mitochondria. Biochim. Biophys. Acta 574, 537–541 (1979).
17 Hostetler KY, Zenner BD, Morris HP. Phospholipid content of mitochondrial and microsomal membranes from Morris hepatomas of varying growth rates. Cancer Res. 39, 2978–2983 (1979).
is possible to prepare PUFA–siRNA complexes and deliver them to tumor cells as nano par-ticles or by intravenous and oral routes to bring about their anticancer actions.
n PUFAs delivered using biodegradable membranesIn addition, especially for glioma, PUFAs (espe-cially GLA) could be incorporated into biode-gradable membranes and inserted into the tumor bed of glioma at the time of debulking surgery so that GLA is slowly released into the unresected tumor bed to induce apoptosis of the tumor cells.
Conclusion & future perspectiveIt is evident that there is reasonable evidence to believe that lipid peroxidation regulates mitosis and cell proliferation rate. Formation of high levels of lipid peroxides in tumor cells leads to their apoptosis. Several anticancer and radia-tion treatments bring about their tumoricidal action by enhancing free radical generation and formation of excess of lipid peroxides in tumor cells. In view of the selective tumoricidal action of polyunsaturated fatty acids and their relatively nontoxic actions on normal cells suggest that they could be exploited as anticancer molecules. It is likely that a better understanding of the metabolism of PUFAs in normal and tumor cells will lead to synthesis and use of specific tumori-cidal lipid molecules as possible anticancer drugs in future. Development of selective delivery of
polyunsaturated fatty acids and their tumori-cidal metabolites may form a new approach in the prevention and treatment of cancer.
AddendumThe arguments presented in this article that selective enhancement of free radicals will lead to elimination of tumor cells [12,22,86,129,143,144] is supported by the recent study in which it was shown that reactive oxygen species metabolism in primary murine cells following the expres-sion of endogenous oncogenic alleles of Kras, Braf and Myc suppress reactive oxygen species generation by increasing the transcription of Nrf2. Increased expression of Nrf2, in turn, elevated the basal Nrf2 antioxidant program and thereby lowered intracellular reactive oxygen species. Furthermore, genetic targeting of the Nrf2 pathway (and thus, enhancing free radi-cal generation) impaired K-RasG12D-induced proliferation and tumorigenesis in vivo [254].
Financial & competing interests disclosureUN Das was in receipt of the Ramalingaswami Fellowship of the Department of Biotechnology, India during the ten-ure of this study. The author has no other relevant affilia-tions or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Review | Das

www.futuremedicine.com 483future science group
18 Hartz JW, Morton RE, Waite MM, Morris HP. Correlation of fatty acyl composition of mitochondrial and microsomal phospholipid with growth rate of rat hepatomas. Lab. Invest. 46, 73–78 (1982).
19 Cheeseman KH, Emery S, Maddix SP, Slater TF, Burton GW, Ingold K. Studies on lipid peroxidation in normal and tumour tissue. The Yoshida rat liver tumour. Biochem. J. 250, 247–252 (1988).
20 Cheeseman KH, Burton GW, Ingold KU, Slater TF. Lipid peroxidation and lipid antioxidants in normal and tumor cells. Toxicol. Pathol. 12, 235–239 (1984).
21 Borrello S, Minotti G, Galeotti T. Factors influencing O
2 and t-Bu OOH-dependent
lipid peroxidation of tumor microsomes. In: Superoxide and Superoxide Dismutase in Chemistry, Biology and Medicine. Rotilio G (Ed.). Elsevier,Amsterdam, The Netherlands, 323–324 (1988).
22 Das UN, Begin ME, Ells G, Huang YS, Horrobin DF. Polyunsaturated fatty acids augment free radical generation in tumor cells in vitro. Biochem. Biophys. Res. Comm. 145, 15–24 (1987).
23 Das UN, Huang YS, Begin ME, Ells G, Horrobin DF. Uptake and distribution of cis-unsaturated fatty acids and their effect on free radical generation in normal and tumor cells in vitro. Free Rad. Biol. Med. 3, 9–14 (1987).
24 Bendetti A. Loss of lipid peroxidation as a histochemical marker for preneoplastic hepatocellular foci of rats. Cancer Res. 44, 5712–5717 (1984).
25 Dunbar LM, Bailey JM. Enzyme deletions and essential fatty acid metabolism in cultured cells. J. Biol. Chem. 250, 1152–1153 (1975).
26 Morton RE, Hartz JW, Reitz RC, Waite BM, Morris H. The acyl-CoA desaturases of microsomes from rat liver and the Morris 7777 hepatoma. Biochim. Biophys. Acta 573, 321–331 (1979).
27 Nassar BA, Das UN, Huang YS, Ells G, Horrobin DF. The effect of chemical hepatocarcinogenesis on liver phospholipid composition in rats fed n-6 and n-3 fatty acid-supplemented diets. Proc. Soc. Exp. Biol. Med. 199, 365–368 (1992).
28 Das UN. Essential fatty acids – a review. Curr. Pharma. Biotechnol. 7, 467–482 (2006).
29 Das UN. Essential fatty acids, biochemistry, physiology, and pathology. Biotechnol. J. 1, 420–439 (2006).
30 Serhan CN, Arita M, Hong S, Gotlinger K. Resolvins, docosatrienes, and neuroprotectins, novel w-3-derived
mediators, and their endogenous aspirin-triggered epimers. Lipids 39, 1125–1132 (2004).
31 Serhan CN, Hong S, Gronert K et al. Resolvins, a family of bioactive products of w-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 (2002).
32 Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from w-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204 (2000).
33 Serhan CN, Takano T, Chiang N, Gronert K, Clish CB. Formation of endogenous “antiinflammatory” lipid mediators by transcellular biosynthesis. Lipoxins and aspirin-triggered lipoxins inhibit neutrophil recruitment and vascular permeability. Am. J. Respir. Crit. Care Med. 161(2 Pt 2), S95–S101 (2000).
34 Cummings KB, Robertson RP. Prostaglandin, increased production by renal cell carcinoma. J. Urol. 118, 720–723 (1977).
35 Hong SL, Wheless CM, Levine L. Elevated prostaglandins synthetase activity in methylcholanthrene-transformed mouse BALB/3T3. Prostaglandins 13, 271–279 (1977).
36 Ylikorkala O, Kauppila A, Viinikka L. Effect of cytostatics on prostaglandin F2 a prostacyclin, and thromboxane in patients with gynecologic malignancies. Obstet. Gynecol. 58, 483–486 (1981).
37 Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M. Prostaglandin in human breast cancer, evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J. Natl Cancer Inst. 64, 1061–1070 (1980).
38 Trevisani A, Ferretti E, Capuzzo A, Tomasi V. Elevated levels of prostaglandin E2 in Yoshida hepatoma and the inhibition of tumour growth by non-steroidal anti-inflammatory drugs. Br. J. Cancer 41, 341–347 (1980).
39 Young MR, Newby M. Enhancement of Lewis lung carcinoma cell migration by prostaglandin E2 produced by macrophages. Cancer Res. 46, 160–164 (1986).
40 Cyran J, Lea MA, Lysz TW. Prostaglandin biosynthetic capacity of hepatomas with different growth rates. Int. J. Biochem. 21, 445–451 (1989).
41 LeFever A, Funahashi A. Elevated prostaglandin E2 levels in bronchoalveolar lavage fluid of patients with bronchogenic carcinoma. Chest 98, 1397–1402 (1990).
42 Baxevanis CN, Reclos GJ, Gritzapis AD, Dedousis GV, Missitzis I, Papamichail M. Elevated prostaglandin E2 production by monocytes is responsible for the depressed levels of natural killer and lymphokine-activated killer cell function in patients with breast cancer. Cancer 72, 491–501 (1993).
43 Qiao L, Kozoni V, Tsioulias GJ et al. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim. Biophys. Acta 1258, 215–223 (1995).
44 Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 62, 403–408 (2002).
45 Oberley L W, Buettner G. Role of SOD in cancer. A review. Cancer Res. 39, 1141–1149 (1979).
46 Tisdale MJ, Mahmoud MD. Activities of free radical metabolizing enzymes in tumours. Br. J. Cancer 47, 809–812 (1983).
47 Bize IB, Oberley LW, Morris HP. Superoxide dismutase and superoxide radical in Morris hepatomas. Cancer Res. 40, 3686–3693 (1980).
48 Hendrickse CW, Kelly RW, Radley S, Donovan IA, Keighley MR, Neoptolemos JP. Lipid peroxidation and prostaglandins in colorectal cancer. Br. J. Surg. 81, 1219–1223 (1994).
49 Mund RC, Pizato N, Bonatto S et al. Decreased tumor growth in Walker 256 tumor-bearing rats chronically supplemented with fish oil involves COX-2 and PGE2 reduction associated with apoptosis and increased peroxidation. Prostaglandins Leukot Essent. Fatty Acids 76, 113–120 (2007).
50 Jang YC, Remmen VH. The mitochondrial theory of aging, insight from transgenic and knockout mouse models. Exp. Gerontol. 44, 256–260 (2009).
51 Gorman A, McGowan A, Cotter TG. Role of peroxide and superoxide anion during tumour cell apoptosis. FEBS Lett. 404, 27–33 (1997).
52 Li M, Beg AA. Induction of necrotic-like cell death by tumor necrosis factor a and caspase inhibitors, novel mechanism for killing virus-infected cells. J. Virol. 74, 7470–7477 (2000).
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)484 future science group
53 Huang TT, Carlson EJ, Raineri I, Gillespie AM, Kozy H, Epstein CJ. The use of transgenic and mutant mice to study oxygen free radical metabolism. Ann. NY Acad. Sci. 893, 95–112 (1999).
54 Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine (3rd Edition). Oxford University Press, UK (1999).
55 Panaretakis T, Shabalina IG, Grandér D, Shoshan MC, DePierre JW. Reactive oxygen species and mitochondria mediate the induction of apoptosis in human hepatoma HepG2 cells by the rodent peroxisome proliferator and hepatocarcinogen, perfluorooctanoic acid. Toxicol. Appl. Pharmacol.173, 56–64 (2001).
56 Hampton MB, Fadeel B, Orrenius S. Redox regulation of the caspases during apoptosis. Ann. NY Acad. Sci. 854, 328–335 (1998).
57 Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature 374, 814–816 (1995).
58 Pervaiz S, Ramalingam JK, Hirpara JL, Clément MV. Superoxide anion inhibits drug-induced tumor cell death. FEBS Lett. 459, 343–348 (1999).
59 Wagner BA, Buettner GR, Oberley LW, Burns CP. Sensitivity of K562 and HL-60 cells to edelfosine, an ether lipid drug, correlates with production of reactive oxygen species. Cancer Res. 58, 2809–2816 (1998).
60 Hampton MB, Fadeel B, Orrenius S. Redox regulation of the caspases during apoptosis. Ann. NY Acad. Sci. 854,328–335 (1998).
61 Arakaki N, Kajihara T, Arakaki R. Involvement of oxidative stress in tumor cytotoxic activity of hepatocyte growth factor/scatter factor. J. Biol. Chem. 274, 13541–13546 (1999).
62 Tamatani M, Ogawa S, Nuñez G, Tohyama M. Growth factors prevent changes in Bcl-2 and Bax expression and neuronal apoptosis induced by nitric oxide. Cell Death Differ. 5, 911–919 (1998).
63 Sattler M, Winkler T, Verma S. Hematopoietic growth factors signal through the formation of reactive oxygen species. Blood 93, 2928–2935 (1999).
64 Zimmerman RJ, Marafino BJ Jr, Chan A, Landre P, Winkelhake JL. The role of oxidant injury in tumor cell sensitivity to recombinant human tumor necrosis factor in vivo. Implications for mechanisms of action. J. Immunol. 142, 1405–1409 (1989).
65 Godfrey RW, Johnson WJ, Hoffstein ST. Recombinant tumor necrosis factor and interleukin-1 both stimulate human synovial cell arachidonic acid release and phospholipid metabolism. Biochem. Biophys. Res. Commun. 142, 235–241 (1987).
66 Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J. Exp. Med. 162, 2163–2168 (1985).
67 Nara K, Odagiri H, Fujii M. Increased production of tumor necrosis factor and prostaglandin E2 by monocytes in cancer patients and its unique modulation by their plasma. Cancer Immunol. Immunother. 25, 126–132 (1987).
68 Neale ML, Fiera RA, Matthews N. Involvement of phospholipase A
2 activation in
tumour cell killing by tumour necrosis factor. Immunology 64, 81–85 (1988).
69 Hepburn A, Boeynaems JM, Fiers W, Dumont JE. Modulation of tumor necrosis factor- a cytotoxicity in L929 cells by bacterial toxins, hydrocortisone and inhibitors of arachidonic acid metabolism. Biochem. Biophys. Res. Commun. 149, 815–822 (1987).
70 Suffys P, Beyaert R, Van Roy F, Fiers W. Reduced tumour necrosis factor-induced cytotoxicity by inhibitors of the arachidonic acid metabolism. Biochem. Biophys. Res. Commun. 149, 735–743 (1987).
71 Matthews N, Neale ML, Jackson SK, Stark JM. Tumour cell killing by tumour necrosis factor, inhibition by anaerobic conditions, free-radical scavengers and inhibitors of arachidonate metabolism. Immunology 62, 153–155 (1987).
72 Berkow RL, Wang D, Larrick JW, Dodson RW, Howard TH. Enhancement of neutrophil superoxide production by preincubation with recombinant human tumor necrosis factor. J. Immunol. 139, 3783–3791 (1987).
73 Talmadge JE, Bowersox O, Tribble H, Lee SH, Shepard HM, Liggitt D. Toxicity of tumor necrosis factor is synergistic with g-interferon and can be reduced with cyclooxygenase inhibitors. Am. J. Pathol. 128, 410–425 (1987).
74 Hori T, Kashiyama S, Hayakawa M et al. Tumor necrosis factor is cytotoxic to human fibroblasts in the presence of exogenous arachidonic acid. Exp. Cell Res. 185, 41–49 (1989).
75 Hayakawa M, Oku N, Takagi T et al. Involvement of prostaglandin-producing pathway in the cytotoxic action of tumor necrosis factor. Cell Struct. Funct. 16, 333–340 (1991).
76 Das UN, Padma M, Sagar PS, Ramesh G, Koratkar R. Stimulation of free radical generation in human leukocytes by various agents including tumor necrosis factor is a calmodulin dependent process. Biochem. Biophys. Res. Commun. 167, 1030–1036 (1990).
77 Palombella VJ, Vilcek J. Mitogenic and cytotoxic actions of tumor necrosis factor in BALB/c 3T3 cells. Role of phospholipase activation. J. Biol.Chem. 264, 18128–18136 (1989).
78 Reid T, Ramesha CS, Ringold GM. Resistance to killing by tumor necrosis factor in an adipocyte cell line caused by a defect in arachidonic acid biosynthesis. J. Biol. Chem. 266, 16580–16586 (1991).
79 Chang DJ, Ringold GM, Heller RA. Cell killing and induction of manganous superoxide dismutase by tumor necrosis factor- a is mediated by lipoxygenase metabolites of arachidonic acid. Biochem. Biophys. Res. Commun. 188, 538–546 (1992).
80 Fletcher JR, Collins JN, Graves ED 3rd et al. Tumor necrosis factor-induced mortality is reversed with cyclooxygenase inhibition. Ann. Surg. 217, 668–674 (1993).
81 Hayakawa M, Ishida N, Takeuchi K et al. Arachidonic acid-selective cytosolic phospholipase A
2 is crucial in the cytotoxic
action of tumor necrosis factor. J. Biol. Chem. 268, 11290–11295 (1993).
82 West M, Mhatre M, Ceballos A et al. The arachidonic acid 5-lipoxygenase inhibitor nordihydroguaiaretic acid inhibits tumor necrosis factor a activation of microglia and extends survival of G93A-SOD1 transgenic mice. J. Neurochem. 91, 133–143 (2004).
83 Kovaríková M, Hofmanová J, Soucek K, Kozubík A. The effects of TNF- a and inhibitors of arachidonic acid metabolism on human colon HT-29 cells depend on differentiation status. Differentiation 72, 23–31 (2004).
84 Vento R, D’Alessandro N, Giuliano M, Lauricella M, Carabillo M, Tesoriere G. Induction of apoptosis by arachidonic acid in human retinoblastoma Y79 cells, involvement of oxidative stress. Exp. Eye Res. 70, 503–517 (2000).
85 Sagar PS, Das UN. Cytotoxic action of cis-unsaturated fatty acids on human cervical carcinoma (HeLa) cells in vitro. Prostaglandins Leukot Essent. Fatty Acids 53, 287–299 (1995).
86 Das UN. Tumoricidal action of cis-unsaturated fatty acids and their relationship to free radicals and lipid peroxidation. Cancer Lett. 56, 235–243 (1991).
87 Lin PS, Kwock L, Goodchild NT. Copper chelator enhancement of bleomycin cytotoxicity. Cancer 46, 2360–2364 (1980).
88 Werts ED, Gould MN. Relationships between cellular superoxide dismutase and susceptibility to chemically induced cancer in the rat mammary gland. Carcinogenesis 7, 1197–1201 (1986).
Review | Das

www.futuremedicine.com 485future science group
89 Cameron DJ. Suppression or enhancement by superoxide dismutase of tumor cell killing by macrophages of normal donors and breast cancer patients. Jpn. J. Exp. Med. 56, 135–140 (1986).
90 Kawaguchi T, Takeyasu A, Matsunobu K et al. Stimulation of Mn-superoxide dismutase expression by tumor necrosis factor- a, quantitative determination of Mn-SOD protein levels in TNF-resistant and sensitive cells by ELISA. Biochem. Biophys. Res. Commun. 171, 1378–1386 (1990).
91 Cervantes A, Pinedo HM, Lankelma J, Schuurhuis GJ. The role of oxygen-derived free radicals in the cytotoxicity of doxorubicin in multidrug resistant and sensitive human ovarian cancer cells. Cancer Lett. 41, 169–177 (1988).
92 Mimnaugh EG, Dusre L, Atwell J, Myers CE. Differential oxygen radical susceptibility of adriamycin-sensitive and -resistant MCF-7 human breast tumor cells. Cancer Res. 49, 8–15 (1989).
93 Huang C, Wu M. Superoxide dismutase activity in tissues from 19 cases of hepatocellular carcinoma. Zhonghua Yi Xue Za Zhi 70, 138–139 (1990).
94 Han YH, Kim SH, Kim SZ, Park WH. Antimycin A as a mitochondrial electron transport inhibitor prevents the growth of human lung cancer A549 cells. Oncol. Rep. 20, 689–693 (2008).
95 Malafa M, Margenthaler J, Webb B, Neitzel L, Christophersen M. Mn-SOD expression is increased in metastatic gastric cancer. J. Surg. Res. 88, 130–134 (2000).
96 Westman NG, Marklund SL. Copper-and zinc-containing superoxide dismutase and manganese-containing superoxide dismutase in human tissues and human malignant tumors. Cancer Res. 41, 2962–2966 (1981).
97 Zhong W, Oberley LW, Oberley TD, St Clair DK. Suppression of the malignant phenotype of human glioma cells by overexpression of manganese superoxide dismutase. Oncogene 14, 481–490 (1997).
98 Toh Y, Kuninaka S, Oshiro T et al. Overexpression of manganese superoxide dismutase mRNA may correlate with aggressiveness in gastric and colorectal adenocarcinomas. Int. J. Oncol. 17, 107–112 (2000).
99 Izutani R, Asano S, Imano M, Kuroda D, Kato M, Ohyanagi H. Expression of manganese superoxide dismutase in esophageal and gastric cancers. J. Gastroenterol. 33, 816–822 (1998).
100 Zhong W, Yan T, Lim R, Oberley LW. Expression of superoxide dismutases, catalase, and glutathione peroxidase in glioma cells. Free Rad. Biol. Med. 27, 1334–1345 (1999).
101 Huang Y, He T, Domann FE. Decreased expression of manganese superoxide dismutase in transformed cells is associated with increased cytosine methylation of the SOD2 gene. DNA Cell Biol. 18, 643–652 (1999).
102 Clement MV, Pervaiz S. Reactive oxygen intermediates regulate cellular apoptosis response to apoptotic stimuli, an hypothesis. Free Rad. Biol. Med. 30, 247–252 (1999).
103 Saunders JA, Rogers LC, Klomsiri C, Poole LB, Daniel LW. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic. Biol. Med. 49, 2058–2067 (2010).
104 Li M, Zhao L, Liu J et al. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell Signal. 22, 1469–1476 (2010).
105 Morisaki N, Lindsey JA, Stitts JM, Zhang H, Cornwell DG. Fatty acid metabolism and cell proliferation. V. Evaluation of pathways for the generation of lipid peroxides. Lipids 19, 381–394 (1984).
106 Morisaki N, Sprecher H, Milo GE, Cornwell DG. Fatty acid specificity in the inhibition of cell proliferation and its relationship to lipid peroxidation and prostaglandin biosynthesis. Lipids 17, 893–899 (1982).
107 Liepkalns VA, Icard-Liepkalns C, Cornwell DG. Regulation of cell division in a human glioma cell clone by arachidonic acid and a-tocopherol quinone. Cancer Lett. 15, 173–178 (1982).
108 Cheeseman KH, Collins M, Maddix S et al. Lipid peroxidation in regenerating rat liver. FEBS Lett. 209, 191–196 (1986).
109 Slater TF, Cheeseman KH, Benedetto C et al. Studies on the hyperplasia (‘regeneration’) of the rat liver following partial hepatectomy. Changes in lipid peroxidation and general biochemical aspects. Biochem. J. 265, 51–59 (1990).
110 Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis, implications for cancer. Cancer Metastasis Rev. 14, 3–15 (1995).
111 Martinez JD, Pennington ME, Craven MT, Warters RL, Cress AE. Free radicals generated by ionizing radiation signal nuclear translocation of p53. Cell Growth Differ. 8, 941–949 (1997).
112 Uberti D, Yavin E, Gil S, Ayasola KR, Goldfinger N, Rotter V. Hydrogen peroxide induces nuclear translocation of p53 and apoptosis in cells of oligodendroglia origin. Brain Res. Mol. Brain Res. 65, 167–175 (1999).
113 Kitamura Y, Ota T, Matsuoka Y et al. Hydrogen peroxide-induced apoptosis mediated by p53 protein in glial cells. Glia. 25, 154–164 (1999).
114 Pani G, Bedogni B, Anzevino R et al. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 60, 4654–4660 (2000).
115 Hilf R, Murant RS, Narayana U, Gibson SL. Relationship of mitochondrial function and cellular adensine triphosphate levels to hematoporphyrin derivative-induced photosensitization in R 3230 AC mammary tumors. Cancer Res. 46, 211–217 (1986).
116 Lee Y, Shacter E. Hydrogen peroxide inhibits activation, not activity, of cellular caspase-3 in vivo. Free Radic. Biol. Med. 29, 684–692 (2000).
117 Saretzki G, von Zglinicki T. Replicative senescence as a model of aging, the role of oxidative stress and telomere shortening-an overview. Z. Gerontol. Geriatr. 32, 69–75 (1999).
118 Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 453, 365–368 (1999).
119 von Zglinicki T, Pilger R, Sitte N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 28, 64–74 (2000).
120 Tyurina YY, Tyurina VA, Certa G, Quinn PJ, Schor NF, Kagan VE. Direct evidence for antioxidant effect of BCL-2 in PC 12 rat pheochromocytoma cells. Arch. Biochem. Biophys. 344, 413–423 (1997).
121 Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down regulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 54, 2095–2097 (1994).
122 Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc. Natl Acad. Sci. USA 92, 4507–4511 (1995).
123 Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241–251 (1993).
124 Das UN. Essential fatty acids, lipid peroxidation and apoptosis. Prostaglandins Leukotrienes Essen. Fatty Acids 61, 157–163 (1999).
125 Padma M, Das UN. Effect of cis-unsaturated fatty acids on the activity of protein kinases and protein phosphorylation in macrophage tumor (AK-5) cells in vitro. Prostaglandins Leukotrienes Essen. Fatty Acids 60, 55–63 (1999).
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)486 future science group
126 Esposti MD, Hatzinisiriou I, McLennan H, Ralph S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J. Biol. Chem. 274, 29831–29837 (1999).
127 Lin HL, Liu TY, Chau GY, Lui WY, Chi CW. Comparison of 2-methoxyestradiol-induced, docetaxel-induced, and paclitaxel-induced apoptosis in hepatoma cells and its correlation with reactive oxygen species. Cancer 89, 983–994 (2000).
128 Huang P, Feng L, Oldham E A, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407, 390–395 (2000).
129 Das UN. A radical approach to cancer. Med. Sci. Monit. 8, RA79–RA92 (2002).
130 Ge Y, Byun JS, De Luca P et al. Combinatorial antileukemic disruption of oxidative homeostasis and mitochondrial stability by the redox reactive thalidomide 2-(2,4-difluoro-phenyl)-4,5,6,7-tetrafluoro-1H-isoindole-1,3(2H)-dione (CPS49) and flavopiridol. Mol. Pharmacol. 74, 872–883 (2008).
131 Colquhoun A. Mechanisms of action of eicosapentaenoic acid in bladder cancer cells in vitro, alterations in mitochondrial metabolism, reactive oxygen species generation and apoptosis induction. J. Urol. 181, 1885–1893 (2009).
132 Naidu MR, Das UN, Kishan A. Intratumoral g-linoleic acid therapy of human gliomas. Prostaglandins Leukot. Essent. Fatty Acids 45, 181–184 (1992).
133 Das UN, Prasad VV, Reddy DR. Local application of g-linolenic acid in the treatment of human gliomas. Cancer Lett. 94, 147–155 (1995).
134 Bakshi A, Mukherjee D, Bakshi A, Banerji AK, Das UN. g-linolenic acid therapy of human gliomas. Nutrition 19, 305–309 (2003).
135 Das UN. g-linolenic acid therapy of human glioma-a review of in vitro, in vivo, and clinical studies. Med. Sci. Monit. 13, RA119–RA31 (2007).
136 Reddy DR, Prasad VS, Das UN. Intratumoural injection of g linolenic acid in malignant gliomas. J. Clin. Neurosci. 5, 36–39 (1998).
137 Smith DL, Willis AL, Mahmud I. Eicosanoid effects on cell proliferation in vitro, relevance to atherosclerosis. Prostaglandins Leukotrienes Med. 16, 1–10 (1984).
138 Sakai T, Yamaguchi N, Shiroko Y, Sekiguchi M, Fujii G, Nishino H. Prostaglandin D
2 inhibits the proliferation of
human malignant tumor cells. Prostaglandins 27, 17–26 (1984).
139 Booyens J, Englebrecht P, Le Roux S, Louwrens CC, Van der Merwe CF, Katzeff IE. Some effects of the essential fatty acids linoleic acid, a-linolenic acid, and of their metabolites g-linolenic acid, arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid and of prostaglandins A and E on the proliferation of human osteogenic sarcoma cells in culture. Prostaglandins Leukotrienes Med. 15, 15–33 (1984).
140 Begin ME, Das UN, Ells G, Horrobin DF. Selective killing of human cancer cells by polyunsaturated fatty acids. Prostaglandins Leukotrienes Med. 19, 177–186 (1985).
141 Begin ME, Ells G, Das UN, Horrobin DF. Differential killing of human carcinoma cells supplemented with n-3 and n-6 polyunsaturated fatty acids. J. Natl Cancer Inst. 77, 1053–1062 (1986).
142 Das UN. Tumoricidal action of cis-unsaturated fatty acids and their relationship to free radicals and lipid peroxidation. Cancer Lett. 56, 235–243 (1991).
143 Sagar PS, Das UN, Koratkar R, Ramesh G, Padma M, Kumar GS. Cytotoxic action of cis-unsaturated fatty acids on human cervical carcinoma (HeLa) cells, relationship to free radicals and lipid peroxidation and its modulation by calmodulin antagonists. Cancer Lett. 63, 189–198 (1992).
144 Kumar GS, Das UN. Free radical-dependent suppression of growth of mouse myeloma cells by a-linolenic and eicosapentaenoic acids in vitro. Cancer Lett. 92, 27–38 (1995).
145 Padma M, Das UN. Effect of cis-unsaturated fatty acids on cellular oxidant stress in macrophage tumor (AK-5) cells in vitro. Cancer Lett. 109, 63–75 (1996).
146 Seigel I, Liu TL, Yaghoubzadeh E, Kaskey TS, Gleicher N. Cytotoxic effects of free fatty acids on ascites tumor cells. J. Natl Cancer Inst. 78, 271–277 (1987).
147 Tolnai S, Morgan JF. Studies on the in vitro anti-tumor activity of fatty acids. V. Unsaturated fatty acids. Can. J. Biochem. Physiol. 40, 869–875 (1962).
148 Rossi MA, Cecchini G. Lipid peroxidation in hepatomas of different degrees of deviation. Cell Biochem. Function 1, 49–54 (1983).
149 Burlakova EB, Palmina NP. On the possible role of free radical mechanism on the regulation of cell replication. Biofizika 12, 82–88 (1967).
150 Gonzalez M, Schemmel R, Dugan L, Gray J, Welsch C. Dietary fish oil inhibits human breast carcinoma growth, A function of increased lipid peroxidation. Lipids 28, 827–832 (1993).
151 Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl Acad. Sci. USA 97, 11280–11285 (2000).
152 Brekke OL, Sagen E, Bjerve KS. Specificity of endogenous fatty acid release during tumor necrosis factor-induced apoptosis in WEHI 164 fibrosarcoma cells. J. Lipid Res. 40, 2223–2233 (1999).
153 Ramesh G, Das UN. Effect of free fatty acids on two stage skin carcinogenesis in mice. Cancer Lett. 100, 199–209 (1996).
154 Calviello G, Palozza O, Piccioni E et al. Supplementation with eicosapentaenoic and docosahexaenoic acid inhibits growth of Morris hepatocarcinoma 3924A in rats, Effects on proliferation and apoptosis. Int. J. Cancer 75, 699–705 (1998).
155 Chapkin RS, Jiang YH, Davidson LA, Lupton JR. Modulation of intracellular second messengers by dietary fat during colonic tumor development. Adv. Exp. Med. Biol. 422, 85–96 (1997).
156 Collett ED, Davidson LA, Fan Y-Y, Lupton JR, Chapkin RS. n-6 and n-3 polyunsaturated fatty acids differentially modulate oncogenic Ras activation in colonocytes. Am. J. Physiol. Cell Physiol. 280, C1066–C1075 (2001).
157 Roynette CE, Calder PC, Dupertuis YM, Pichar C. n-3 Polyunsaturated fatty acids and colon cancer prevention. Clin. Nutr. 23, 139–151 (2004).
158 Calviello G, Di Nicuolo F, Gragnoli S et al. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2induced ERK-1 and -2 and HIF-1a induction pathway. Carcinogenesis 25, 2303–2310 (2004).
159 Cai J, Jiang WG, Mansel RE. Inhibition of angiogenic factor- and tumor-induced angiogenesis by g -linolenic acid. Prostaglandins Leukotrienes Essen. Fatty Acids 60, 21–29 (1999).
160 Rose DP, Connolly JM. Antiangiogenicity of docosahexaenoic acid and its role in the suppression of breast cancer cell growth in nude mice. Int. J. Oncol. 15, 1011–1015 (1999).
161 Jin Y, Arita M, Zhang Q et al. Anti-angiogenesis Effect of the novel anti-inflammatory and pro-resolving lipid mediators. Invest. Ophthalmol. Vis. Sci.50, 4743–4752 (2009).
162 Schlager SI, Ohanian SH. Correlation between lipid synthesis in tumor cells and their sensitivity to humoral immune attack. Science 197, 773–776 (1977).
Review | Das

www.futuremedicine.com 487future science group
163 Schlager SI, Ohanian SH, Borsos T. Correlation between the ability of tumor cells to incorporate specific fatty acids and their sensitivity to killing by a specific antibody plus guinea pig complement. J. Natl Cancer Inst. 61, 931–934 (1978).
164 Schlager SI, Ohanian SH. Modulation of tumor cell susceptibility to humoral immune killing through chemical and physical manipulation of cellular lipid and fatty acid composition. J. Immunol. 125, 1196–1200 (1980).
165 Schlager SI, Madden LD, Meltzer MS, Bara S, Mamula MJ. Role of macrophage lipids in regulating tumoricidal activity. Cell Immunol. 77, 52–68 (1983).
166 Schlager SI, Meltzer MS, Madden LD. Role of membrane lipids in the immunological killing of tumor cells, II. Effector cell lipids. Lipids 18, 483–488 (1983).
167 Schlager SI, Ohanian SH. Role of membrane lipids in the immunological killing of tumor cells, I. Target cell lipids. Lipids 18, 475–482 (1983).
168 Schlager SI, Meltzer MS. Role of macrophage lipids in regulating tumoricidal activity. II. Internal genetic and external physiologic regulatory factors controlling macrophage tumor cytotoxicity also control characteristic lipid changes associated with tumoricidal cells. Cell Immunol. 80, 10–19 (1983).
169 Bell HS, Wharton SB, Leaver HA, Whittle IR. Effects of N-6 essential fatty acids on glioma invasion and growth, experimental studies with glioma spheroids in collagen gels. J. Neurosurg. 91, 989–996 (1999).
170 Leaver HA, Wharton SB, Bell HS, Leaver-Yap IM, Whittle IR. Highly unsaturated fatty acid induced tumour regression in glioma pharmacodynamics and bioavailability of g linolenic acid in an implantation glioma model, effects on tumour biomass, apoptosis and neuronal tissue histology. Prostaglandins Leukot. Essent. Fatty Acids 67, 283–292 (2002).
171 Benadiba M, Miyake JA, Colquhoun A. g-linolenic acid alters Ku80, E2F1, and bax expression and induces micronucleus formation in C6 glioma cells in vitro. IUBMB Life 61, 244–251 (2009).
172 Rohrbach S. Effects of dietary polyunsaturated fatty acids on mitochondria. Curr. Pharm. Des. 15, 4103–4116 (2009).
173 Tuo Y, Wang D, Li S, Chen C. Long-term exposure of INS-1 rat insulinoma cells to linoleic acid and glucose in vitro affects cell viability and function through mitochondrial-mediated pathways. Endocrine 39, 128–138 (2011).
174 Zeghichi-Hamri S, de Lorgeril M, Salen P et al. Protective effect of dietary n-3 polyunsaturated fatty acids on myocardial resistance to ischemia-reperfusion injury in rats. Nutr. Res. 30, 849–857 (2010).
175 Hagopian K, Weber KL, Hwee DT et al. Complex I-associated hydrogen peroxide production is decreased and electron transport chain enzyme activities are altered in n-3 enriched fat-1 mice. PLoS One 5, e12696 (2010).
176 Kansal S, Negi AK, Kaur R et al. Evaluation of the role of oxidative stress in chemopreventive action of fish oil and celecoxib in the initiation phase of 7,12-dimethyl benz(a)anthracene-induced mammary carcinogenesis. Tumour. Biol. 32, 167–177 (2011).
177 Dymkowska D, Wojtczak L. Arachidonic acid-induced apoptosis in rat hepatoma AS-30D cells is mediated by reactive oxygen species. Acta Biochim. Pol. 56, 711–715 (2009).
178 Ribeiro G, Benadiba M, de Oliveira Silva D, Colquhoun A. The novel ruthenium-g-linolenic complex [Ru(2) (aGLA(4)Cl] inhibits C6 rat glioma cell proliferation and induces changes in mitochondrial membrane potential, increased reactive oxygen species generation and apoptosis in vitro. Cell Biochem. Funct. 8, 15–23 (2010).
179 Giros A, Grzybowski M, Sohn VR et al. Regulation of colorectal cancer cell apoptosis by the n-3 polyunsaturated fatty acids docosahexaenoic and eicosapentaenoic. Cancer Prev. Re. (Phila) 2, 732–742 (2009).
180 Ponnala S, Rao KP, Chaudhury JR et al. Effect of polyunsaturated fatty acids on diphenyl hydantoin-induced genetic damage in vitro and in vivo. Prostaglandins Leukot. Essent. Fatty Acids 80, 43–50 (2009).
181 Das UN, Rao KP. Effect of g-linolenic acid and prostaglandins E1 on g-radiation and chemical-induced genetic damage to the bone marrow cells of mice. Prostaglandins Leukot. Essent. Fatty Acids 74, 165–173 (2006).
182 Das UN, Ramadevi G, Rao KP, Rao MS. Prostaglandins and their precursors can modify genetic damage-induced by g-radiation and benzo(a)pyrene. Prostaglandins 29, 911–920 (1985).
183 Das UN. Tumoricidal and anti-angiogenic actions of g-linolenic acid and its derivatives. Curr. Pharm. Biotechnol. 7, 457–466 (2006).
184 Dhayal S, Morgan NG. Pharmacological characterization of the cytoprotective effects of polyunsaturated fatty acids in insulin-secreting BRIN-BD11 cells. Br. J. Pharmacol. 162, 1340–1350 (2011).
185 Bazan NG. w-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 10, 136–141 (2007).
186 Suresh Y, Das UN. Long-chain polyunsaturated fatty acids and chemically induced diabetes mellitus, effect of w-6 fatty acids. Nutrition 19, 93–114 (2003).
187 Suresh Y, Das UN. Long-chain polyunsaturated fatty acids and chemically induced diabetes mellitus. Effect of w-3 fatty acids. Nutrition 19, 213–228 (2003).
188 Sangeetha PS, Das UN. g-linolenic acid and eicosapentaenoic acid potentiate the cytotoxicity of anti-cancer drugs on human cervical carcinoma (HeLa) cells in vitro. Med. Sci. Res. 21, 457–459 (1993).
189 Madhavi N, Das UN. Reversal of KB-3–1 and KB-Ch-8–5 tumor cell drug-resistance by cis-unsaturated fatty acids in vitro. Med. Sci. Res. 22, 689–692 (1994).
190 Madhavi N, Das UN. Effect of n-6 and n-3 fatty acids on the survival of vincristine sensitive and resistant human cervical carcinoma cells in vitro. Cancer Lett. 84, 31–41 (1994).
191 Das UN, Madhavi N, Sravan Kumar G, Padma M, Sangeetha P. Can tumour cell drug resistance be reversed by essential fatty acids and their metabolites? Prostaglandins Leukot. Essent. Fatty Acids 58, 39–54 (1998).
192 Germain E, Chajès V, Cognault S, Lhuillery C, Bougnoux P. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast tumor cell line MDA-MB-231, relationship to lipid peroxidation. Int. J. Cancer 75, 578–583 (1998).
193 Mahéo K, Vibet S, Steghens JP et al. Differential sensitization of cancer cells to doxorubicin by DHA, a role for lipoperoxidation. Free Radic. Biol. Med. 39, 742–751 (2005).
194 Ilc K, Ferrero JM, Fischel JL et al. Cytotoxic effects of two g-linoleic salts (lithium g-linolenate or meglumine g-linolenate) alone or associated with a nitrosourea, an experimental study on human glioblastoma cell lines. Anticancer Drugs 10, 413–417 (1999).
195 Menendez JA, Ropero S, Lupu R, Colomer R. w-6 polyunsaturated fatty acid g-linolenic acid (18,3n-6) enhances docetaxel (Taxotere) cytotoxicity in human breast carcinoma cells, Relationship to lipid peroxidation and HER-2/neu expression. Oncol. Rep. 11, 1241–1252 (2004).
196 Menéndez JA, Ropero S, del Barbacid MM et al. Synergistic interaction between vinorelbine and g-linolenic acid in breast cancer cells. Breast Cancer Res. Treat. 72, 203–219 (2002).
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review

Clin. Lipidol. (2011) 6(4)488 future science group
197 Menendez JA, Ropero S, Mehmi I, Atlas E, Colomer R, Lupu R. Overexpression and hyperactivity of breast cancer-associated fatty acid synthase (oncogenic antigen-519) is insensitive to normal arachidonic fatty acid-induced suppression in lipogenic tissues but it is selectively inhibited by tumoricidal a -linolenic and g-linolenic fatty acids, a novel mechanism by which dietary fat can alter mammary tumorigenesis. Int. J. Oncol. 24, 1369–1383 (2004).
198 Kong X, Ge H, Chen L et al. g-linolenic acid modulates the response of multidrug-resistant K562 leukemic cells to anticancer drugs. Toxicol. In vitro 23, 634–639 (2009).
199 Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc. Natl Acad. Sci. USA 95, 13182–13187 (1998).
200 Rizzo MT, Regazzi E, Garau D et al. Induction of apoptosis by arachidonic acid in chronic myeloid leukemia cells. Cancer Res. 59, 5047–5053 (1999).
201 Wolf LA, Laster SM. Characterization of arachidonic acid-induced apoptosis. Cell. Biochem. Biophys. 30, 353–368 (1999).
202 Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl Acad. Sci. USA 97, 11280–11285 (2000).
203 Cao Y, Dave KB, Doan TP, Prescott SM. Fatty acid CoA ligase 4 is upregulated in colon adenocarcinoma. Cancer Res. 61, 8429–8434 (2001).
204 Sun Y, Tang XM, Half E, Kuo MT, Sinicrope FA. Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res. 62, 6323–6328 (2002).
205 Tang X, Sun YJ, Half E, Kuo MT, Sinicrope F. Cyclooxygenase-2 overexpression inhibits death receptor 5 expression and confers resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human colon cancer cells. Cancer Res. 62, 4903–4908 (2002).
206 Shureiqi I, Chen D, Lotan R et al. 15-lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 60, 6846–6850 (2000).
207 Shureiqi I, Chen D, Lee JJ et al. 15-LOX-1, a novel molecular target of nonsteroidal anti-inflammatory drug-induced apoptosis in colorectal cancer cells. J. Natl Cancer Inst. 92, 1136–1142 (2000).
208 Shureiqi I, Xu X, Chen D et al. Nonsteroidal anti-inflammatory drugs induce apoptosis in esophageal cancer cells by restoring 15-lipoxygenase-1 expression. Cancer Res. 61, 4879–4884 (2001).
209 Maccarrone M, Ranalli M, Bellincampi L et al. Activation of different lipoxygenase isozymes induces apoptosis in human erythroleukemia and neuroblastoma cells. Biochem. Biophys. Res. Commun. 272, 345–350 (2000).
210 Avis I, Hong SH, Martinez A et al. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 15, 2007–2009 (2001).
211 Hong SH, Avis I, Vos MD, Martínez A, Treston AM, Mulshine JL. Relationship of arachidonic acid metabolizing enzyme expression in epithelial cancer cell lines to the growth effect of selective biochemical inhibitors. Cancer Res. 59, 2223–2228 (1999).
212 Leaver HA, Bell HS, Rizzo MT et al. Antitumour and pro-apoptotic actions of highly unsaturated fatty acids in glioma. Prostaglandins Leukot. Essent. Fatty Acids 66, 19–29 (2002).
213 Menendez JA, Ropero S, Mehmi I, Atlas E, Colomer R, Lupu R. Overexpression and hyperactivity of breast cancer-associated fatty acid synthase (oncogenic antigen-519) is insensitive to normal arachidonic fatty acid-induced suppression in lipogenic tissues but it is selectively inhibited by tumoricidal a-linolenic and g-linolenic fatty acids, a novel mechanism by which dietary fat can alter mammary tumorigenesis. Int. J. Oncol. 24, 1369–1383 (2004).
214 Menendez JA, Mehmi I, Atlas E, Colomer R, Lupu R. Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival, Role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-k B. Int. J. Oncol. 24, 591–608 (2004).
215 Menendez JA, Colomer R, Lupu R. Inhibition of fatty acid synthase-dependent neoplastic lipogenesis as the mechanism of g-linolenic acid-induced toxicity to tumor cells, an extension to Nwankwo’s hypothesis. Med. Hypotheses 64, 337–341 (2005).
216 Menendez JA, Colomer R, Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Med. Hypotheses 64, 342–349 (2005).
217 Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010).
218 Levine L. Proteasome inhibitors, their effects on arachidonic acid release from cells in culture and arachidonic acid metabolism in rat liver cells. BMC Pharmacol. 4, 15. (2004).
219 Eitsuka T, Nakagawa K, Suzuki T, Miyazawa T. Polyunsaturated fatty acids inhibit telomerase activity in DLD-1 human colorectal adenocarcinoma cells, a dual mechanism approach. Biochim. Biophys. Acta 1737, 1–10 (2005).
220 Eitsuka T, Nakagawa K, Miyazawa T. Dual mechanisms for telomerase inhibition in DLD-1 human colorectal adenocarcinoma cells by polyunsaturated fatty acids. Biofactors 21, 19–21 (2004).
221 Jiang WG, Bryce RP, Mansel RE. g linolenic acid regulates gap junction communication in endothelial cells and their interaction with tumour cells. Prostaglandins Leukot. Essen. Fatty Acids 56, 307–316 (1997).
222 Sethi S, Eastman AY, Eaton JW. Inhibition of phagocyte-endothelium interactions by oxidized fatty acids, a natural anti-inflammatory mechanism? J. Lab. Clin. Med. 128, 27–38 (1996).
223 Germain E, Chajes V, Cognault S, Lhuillery C, Bougnoux P. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast tumor cell line MDA-MB-231; relationship to lipid peroxidation. Int. J. Cancer 75, 578–583 (1998).
224 Shao Y, Pardini L, Pardini RS. Dietary menhaden oil enhances mitomycin C antitumor activity toward human mammary carcinoma MX-1. Lipids 30, 1035–1045 (1995).
225 Chow SC, Jondal M. Ca2+ entry in T cells is activated by emptying the inositol 1,4,5-triphosphate sensitive Ca2+ pool. Cell Calcium 11, 641–646 (1990).
226 Nakagawa T, Zhu H, Morishima N et al. Caspase-12 mediates endoplasmic reticulum-specific apoptosis and cytotoxicity by amyloid-b. Nature 403, 98–103 (2000).
227 Huang ZH, Hii CS, Rathjen DA, Poulos A, Murray AW, Ferrante A. N-6 and N-3 polyunsaturated fatty acids stimulate translocation of protein kinase Ca, bI,bII and –e and enhance agonist-induced NADPH oxidase in macrophages. Biochem. J. 325 (pt 2), 553–557 (1997).
228 Peterson DA, Mehta N, Butterfield J et al. Polyunsaturated fatty acids stimulate superoxide formation in tumor cells, a mechanism for specific cytotoxicity and a model for tumor necrosis factor? Biochem. Biophys. Res. Commun. 155, 1033–1037 (1988).
Review | Das

www.futuremedicine.com 489future science group
229 Chiu LC, Wan JM. Induction of apoptosis in HL-60 cells by eicosapentaenoic acid (EPA) is associated with downregulation of bcl-2 expression. Cancer Lett. 145, 17–27 (1999).
230 Albino AP, Juan G, Traganos F et al. Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid, association with decreased pRb phosphorylation. Cancer Res. 60, 4139–4145 (2000).
231 Chen ZY, Istfan NW. Docosahexaenoic acid, a major constituent of fish oil diets, prevents activation of cyclin-dependent kinases and S-phase entry by serum stimulation in HT-29 cells. Prostaglandins Leukot. Essent. Fatty Acids 64, 67–73 (2001).
232 Palakurthi SS, Fluckiger R, Aktas H et al. Inhibition of translation initiation mediates the anticancer effect of the n-3 polyunsaturated fatty acid eicosapentaenoic acid. Cancer Res. 60, 2919–2925 (2000).
233 Nishida M, Maruyama Y, Tanaka R, Kontani K, Nagao T, Kurose H. G a
i and
G ao are target proteins of reactive oxygen
species. Nature 408, 492–495 (2000).
234 Das UN. Abrupt and complete occlusion of tumor-feeding vessels by g-linolenic acid. Nutrition 18, 662–664 (2002).
235 Das UN. Occlusion of infusion vessels on g-linolenic acid infusion. Prostaglandins Leukot. Essent. Fatty Acids 70; 23–32 (2004).
236 Calon F, Lim GP, Yang F et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 43, 633–645 (2004).
237 Lukiw WJ, Cui J-G, Marcheselli VL et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783 (2005).
238 Comba A, Maestri DM, Berra MA et al. Effect of w-3 and w-9 fatty acid rich oils on lipoxygenases and cyclooxygenases enzymes and on the growth of a mammary adenocarcinoma model. Lipids Health Dis. 9, 112 (2010).
239 Liu B, Maher RJ, Hannun YA, Porter AT, Honn KV. 12(S)-HETE enhancement of prostate tumor cell invasion, selective role of PKC a. J. Natl Cancer Inst. 86, 1145–1151 (1994).
240 Ding XZ, Tong WG, Adrian TE. 12-lipoxygenase metabolite 12(S)-HETE stimulates human pancreatic cancer cell proliferation via protein tyrosine phosphorylation and ERK activation. Int. J. Cancer 94, 630–636 (2001).
241 Chen GG, Xu H, Lee JF et al. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor g-dependent pathway. Int. J. Cancer 107, 837–843 (2003).
242 Najid A, Beneytout JL, Tixier M. Cytotoxicity of arachidonic acid and of its lipoxygenase metabolite 15-hydroperoxyeicosatetraenoic acid on human breast cancer MCF-7 cells in culture. Cancer Lett. 46, 137–141 (1989).
243 Shureiqi I, Jiang W, Zuo X et al. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-d to induce apoptosis in colorectal cancer cells. Proc. Natl Acad. Sci. USA 100, 9968–9973 (2003).
244 Nixon JB, Kim KS, Lamb PW, Bottone FG, Eling TE. 15-lipoxygenase-1 has anti-tumorigenic effects in colorectal cancer. Prostaglandins Leukot. Essent. Fatty Acids 70, 7–15 (2004).
245 Kim SJ. Lipoxins formation by rat basophilic leukemia (RBL-1) cells. Res. Commun. Chem. Pathol. Pharmacol. 68, 159–174 (1990).
246 Stenke L, Edenius C, Samuelsson J, Lindgren JA. Deficient lipoxin synthesis, a novel platelet dysfunction in myeloproliferative disorders with special reference to blastic crisis of chronic myelogenous leukemia. Blood 78, 2989–2995 (1991).
247 Chen Y, Hao H, He S et al. Lipoxin A4 and its analogue suppress the tumor growth of transplanted H22 in mice, the role of antiangiogenesis. Mol. Cancer Ther. 9, 2164–2174 (2010).
248 Gleissman H, Yang R, Martinod K et al. Docosahexaenoic acid metabolome in neural tumors, identification of cytotoxic intermediates. FASEB J. 24, 906–915 (2010).
249 Wolrum C, Shi S, Jayaprakash KN et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 25, 1149–1157 (2007).
250 Ichim TE, Popov IA, Riordan NH et al. A novel method of modifying immune responses by vaccination with lipiodol-siRNA mixtures. J. Translat. Med. 4, 2 (2006).
251 Resina S, Prevot P, Thierry AR. Physico-chemical characteristics of lipoplexes influence cell uptake mechanisms and transfection efficacy. PLoS ONE 4(6), e6058 (2009).
252 Love KT, Mahon KP, Levins CG et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 107, 1864–1869 (2010).
253 Davis ME, Zuckerman JE, Choi CHJ et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1071 (2010).
254 DeNicola GM, Karreth FA, Humpton TJ et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011).
Fatty acids enhance free radical generation & lipid peroxidation to induce apoptosis of tumor cells | Review




















