
Catastrophe and Challenge: Cultural Heritage in Post ... - Opus4
Upload
khangminh22Category
view
3download
0
SCORPIONATE COMPLEXES FOR
COPOLYMERIZATION AND MOLECULAR IMPRINTING
Der Naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. rer. nat.
vorgelegt von
Gazi Türkoglu
aus Nürnberg

Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 28. März 2011
Vorsitzender der Prüfungskommission: Prof. Dr. Rainer Fink
Erstberichterstatter: Prof. Dr. Nicolai Burzlaff
Zweitberichterstatter: Prof. Dr. Dr. h.c. mult. Rudi van Eldik

Die vorliegende Arbeit entstand in der Zeit von November 2007 bis Oktober 2010 im
Department für Chemie und Pharmazie (Lehrstuhl für Anorganische und Analytische
Chemie) der Friedrich-Alexander-Universität Erlangen-Nürnberg unter der Anleitung von
Prof. Dr. Nicolai Burzlaff.
Wesentliche Teile dieser Dissertation wurden bereits veröffentlicht:
"Synthesis and Transition Metal Complexes of Novel N,N,O Scorpionate Ligands Suitable
for Solid Phase Immobilisation", E. Hübner, G. Türkoglu, M. Wolf, U. Zenneck, N. Burzlaff,
Eur. J. Inorg. Chem. 2008, 1226 – 1235.
"Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic Acid: A New Heteroscorpionate Building Block
for Copolymers that Mimic the 2-His-1-carboxylate Facial Triad", G. Türkoglu, C. Pubill
Ulldemolins, R. Müller, E. Hübner, F. W. Heinemann, M. Wolf N. Burzlaff. Eur. J. Inorg.
Chem. 2010, 2962 – 2974.


ANNEM VE BABAM IÇIN


TABLE OF CONTENTS
I. INTRODUCTION ......................................................................................................................... 1
1.1. Bis(pyrazol-1-yl)acetic acids in bioinorganic chemistry ................................................... 2
1.1.1. The 2-His-1-carboxylate facial triad in non-heme iron(II) oxygenases .............. 2
1.1.2. 2-Oxoglutarate dependent iron(II) oxygenases........................................................ 4
1.1.3. Modeling studies for 2-OG dependent iron enzymes............................................. 9
1.2. Immobilization of ligands and complexes ......................................................................... 14
1.2.1. Methods of non-covalent immobilization .............................................................. 14
1.2.2. Covalent immobilization via grafting on supports ............................................... 16
1.2.3. Covalent immobilization via copolymerization .................................................... 19
1.3. Molecular imprinted polymers............................................................................................. 25
1.3.1. General Concept.......................................................................................................... 25
1.3.2. Imprinted polymers as microreactors...................................................................... 26
1.3.3. Imprinting with a transition state analogue .......................................................... 29
1.3.4. Imprinting with catalysts based on transition metals .......................................... 33
II. OBJECTIVE AND AIMS .............................................................................................................39
III. RESULTS AND DISCUSSION .....................................................................................................43
3.1. Substituents at the bridging position of bis(pyrazol-1-yl)acetic acids.......................... 44
3.1.1. Problem statement ...................................................................................................... 44
3.1.2. 2,2-Bis(3,5-dimethlypyrazol-1-yl)propanoic acid as a model ligand ................. 48
3.1.3. Bisligand complexes of type [M(L)2] ....................................................................... 53
3.1.4. Synthesis of [Ru(bdmpzpr)Cl(PPh3)2] and reaction with dinitrogen................. 56
3.1.5. Reaction of [Ru(bdmpzpr)Cl(PPh3)2] with CO and SO2....................................... 64

3.1.6. Conclusion.................................................................................................................... 72
3.2. A new approach: Polymerizable linkers at the pyrazolyl units..................................... 74
3.2.1. Synthesis of 2,2-bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid..................... 74
3.2.2. Transition metal complexes of Hbdmvpza............................................................. 76
3.2.3. Polymerization behavior of Hbdmvpza.................................................................. 81
3.2.4. Incorporation of [M(fac-N,N,O)] fragments .......................................................... 84
3.2.5. Copper and iron containing polymers .................................................................... 89
3.2.6. Conclusion.................................................................................................................... 94
3.3. Towards imprinted polymers............................................................................................... 95
3.3.1. Synthesis of model complexes for 2-OG dependent iron enzymes ................... 95
3.3.2. Polymerization of the template complexes ............................................................ 99
3.3.3. Extraction of the templates and generation of the imprint............................... 101
3.3.4. Conclusion.................................................................................................................. 103
3.4. Further work with bis(pyrazol-1-yl)acetic acids ............................................................ 104
3.4.1. Epoxidation catalysis with carbonyl complexes [Ru(L)Cl(CO)2] .................... 104
3.4.2. Synthesis and characterization of tetragonal [Ni(bdtbpza)Cl]......................... 112
3.4.3. HIF-1α prolyl hydroxylase inhibitor studies........................................................ 117
IV. SUMMARY AND OUTLOOK ...................................................................................................125
V. ZUSAMMENFASSUNG UND AUSBLICK ....................................................................................133
VI. EXPERIMENTAL SECTION .....................................................................................................141
6.1. General Remarks .................................................................................................................. 142
6.1.1. Working techniques ................................................................................................. 142
6.1.2. Chemicals ................................................................................................................... 142
6.1.3. Instrumentation......................................................................................................... 144
6.1.4. Polymer analysis ....................................................................................................... 144
6.2. Synthesis of ligands and organic precursors ................................................................... 146
6.2.1. 2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid (4) .......................................... 146
6.2.2. Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)methane (17) ........................................... 146
6.2.3. 2,2-Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid (Hbdmvpza) (18).......... 147
6.2.4. 2-[4-(Methoxycarbonyl)-oxazol-5-yl]benzoic acid (32)..................................... 148
6.2.5. 4-Hydroxy-1-oxo-1,2-dihydroisoquinoline-3-carboxylate (33) ........................ 149

6.2.6. Methyl 1-chloro-4-hydroxyisoquinoline-3-carboxylate (34)............................. 149
6.2.7. 1-Chloro-4-hydroxyisoquinoline-3-carboxylic acid (35).................................... 150
6.2.8. Ethyl 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido)acetate (36)........... 150
6.2.9. 2-(1-Chloro-4-hydroxyisoquinoline-3-carboxamido)acetic acid (37) .............. 151
6.3. Synthesis of transition metal complexes .......................................................................... 152
6.3.1. [Mn(bdmpzpr)(CO)3] (5) .......................................................................................... 152
6.3.2. [Re(bdmpzpr)(CO)3] (6)............................................................................................ 153
6.3.3. [Cu(bdmpzpr)2] (7).................................................................................................... 153
6.3.4. [Ru(bdmpzpr)Cl(PPh3)2] (9)..................................................................................... 154
6.3.5. [Ru(bdmpzpr)Cl(N2)(PPh3)] (10)............................................................................. 155
6.3.6. [Ru(bdmpzpr)Cl(CO)(PPh3)] (12) ........................................................................... 156
6.3.7. [Ru(bdmpza)Cl(PPh3)(SO2)] (13) ............................................................................ 157
6.3.8. [Ru(bdmpzpr)Cl(PPh3)(SO2)] (14)........................................................................... 158
6.3.9. [Mn(bdmvpza)(CO)3] (19)........................................................................................ 159
6.3.10. [Re(bdmvpza)(CO)3] (20) ......................................................................................... 160
6.3.11. [Ru(bdmvpza)Cl(PPh3)2] (21) .................................................................................. 161
6.3.12. [Ru(bdmvpza)Cl(MeCN)(PPh3)] (22) ..................................................................... 161
6.3.13. [Ru(bdmvpza)Cl(CO)2] (23)..................................................................................... 162
6.3.14. [Ru(bdmpza)Cl(CO)2] (24)....................................................................................... 163
6.3.15. [Cu(bdmvpza)2] (25) ................................................................................................. 164
6.3.16. [Ru(bdmvpza)(BF)(PPh3)] (26) ................................................................................ 165
6.3.17. [Ru(bdmvpza)(NOG)(PPh3)] (27)............................................................................ 166
6.3.18. [Ru(bdmpzpr)Cl(CO)2] (28) ..................................................................................... 167
6.3.19. [Ni(bdtbpza)Cl] (30).................................................................................................. 167
6.4. Synthesis of polymers.......................................................................................................... 169
6.4.1. Copolymerization of ligand 17 with MMA (P17a).............................................. 169
6.4.2. Copolymerization of ligand 18 with MMA (P18a and P18b) ............................ 169
6.4.3. Copolymerization of ligand 18 with EGDMA (P18c) ......................................... 170
6.4.4. Homopolymerization of ligand 18 (P18d) ............................................................. 170
6.4.5. Synthesis of P18a-Mn ............................................................................................... 170
6.4.6. Synthesis of P18a-Re................................................................................................. 170
6.4.7. Synthesis of P18a-Cu................................................................................................ 171
6.4.8. Synthesis of P18a-Fe ................................................................................................. 171

6.4.9. Synthesis of P18c-Mn............................................................................................... 171
6.4.10. Synthesis of P18c-Re................................................................................................. 172
6.4.11. Synthesis of P18c-Fe ................................................................................................. 172
6.4.12. Synthesis of P23......................................................................................................... 172
6.4.13. Synthesis of P26......................................................................................................... 172
6.4.14. Synthesis of P27......................................................................................................... 173
6.4.15. Treatment of P26 or P27 with PMe3 (P26X, P27X).............................................. 173
6.5. Epoxidation catalysis........................................................................................................... 174
VII. APPENDIX ..........................................................................................................................177
7.1. Details of the structure determinations ........................................................................... 178
7.2. List of abbreviations and symbols..................................................................................... 187
7.3. List of compounds ................................................................................................................ 191
VIII. BIBLIOGRAPHY..................................................................................................................195
DANKSAGUNG ...........................................................................................................................213
CURRICULUM VITAE ..................................................................................................................215

I. INTRODUCTION 1
I. INTRODUCTION

2 I. INTRODUCTION
1.1. Bis(pyrazol-1-yl)acetic acids in bioinorganic chemistry
1.1.1. The 2-His-1-carboxylate facial triad in non-heme iron(II) oxygenases
More than two billion years ago, Earth's atmosphere was transformed from an anoxic to an
oxygenic state by means of the photosynthetic action of cyanobacteria.[1-3] Since this
evolutionary change, molecular dioxygen is critical to most life forms on our planet for the
generation of energy and the biosynthesis of important compounds in metabolic pathways.
Although in principle, the uncatalyzed reaction of atmospheric dioxygen with organic
substrates is a thermodynamically feasible process, the kinetic barrier for such a reaction is
very high. This is due to the triplet ground state of molecular 3O2 dioxygen which makes a
direct reaction with singlet ground state molecules (most organic compounds) a spin-
forbidden process. On the other hand, this spin-restriction prevents all life forms from
spontaneous combustion to CO2 and H2O in dioxygen atmosphere.[1, 4-5] Nature has
developed careful strategies to overcome this kinetic barrier in order to control the
oxidizing power of O2 for key metabolic, physiologic and biodegradation processes. The
activation of dioxygen in biological systems is achieved by means of transition metal
cofactors such as iron or copper within metalloenzymes. Besides heme containing iron
enzymes such as cytochrome P45o, recently mononuclear non-heme iron oxygenases have
drawn a lot of attention especially because of the remarkably diversity in oxidative
transformations that these enzymes are able to catalyze.[4-7] During the last 10 – 15 years,
an ever increasing number of protein structures for non-heme iron enzymes has established
the occurrence of a common structural motif which has been named the "2-His-1-
carboxylate facial triad" by L. QUE JR.[6-9] This triad is believed to be one of Nature's
recurring bioinorganic motifs such as the heme cofactor or sulfur clusters.[4, 6]
The structural features of this well-suited platform for the activation of dioxygen are shown
in figure 1 exemplified by the resting state of deacetoxycephalosporin C synthase (DAOCS),
a 2-oxoglutarate (2-OG) dependent iron(II) enzyme.[4, 10] One face of the octahedral ferrous
iron center is occupied by three endogenous protein ligands, i.e. two histidines and one
aspartate or glutamate residue. The remaining three sites opposite of this triad are able to
bind exogenous ligands such as dioxygen, substrates or cofactors at different stages of a
catalytic cycle. In the as-isolated state of the enzymes these remaining sites are usually
occupied by weakly coordinated solvent molecules. Although the 2-His-1-carboxylate triad
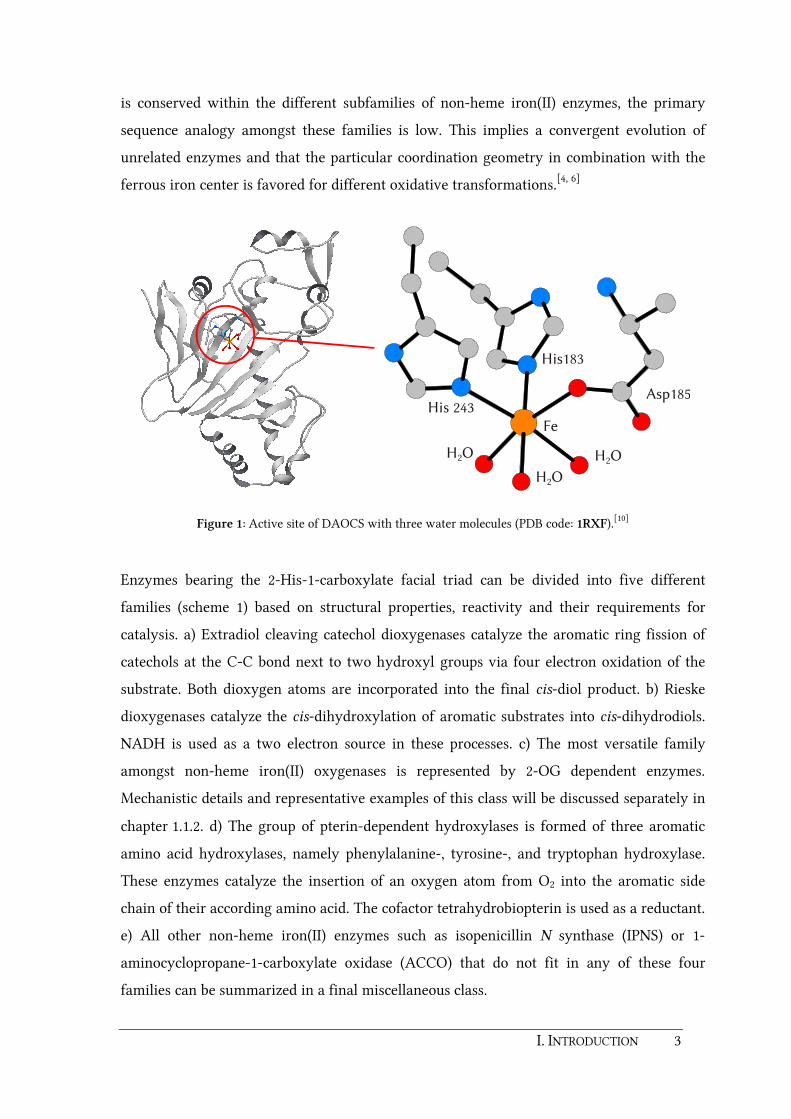
I. INTRODUCTION 3
is conserved within the different subfamilies of non-heme iron(II) enzymes, the primary
sequence analogy amongst these families is low. This implies a convergent evolution of
unrelated enzymes and that the particular coordination geometry in combination with the
ferrous iron center is favored for different oxidative transformations.[4, 6]
His 243
His183
Asp185
Fe
H O2
H O2
H O2
Figure 1: Active site of DAOCS with three water molecules (PDB code: 1RXF).[10]
Enzymes bearing the 2-His-1-carboxylate facial triad can be divided into five different
families (scheme 1) based on structural properties, reactivity and their requirements for
catalysis. a) Extradiol cleaving catechol dioxygenases catalyze the aromatic ring fission of
catechols at the C-C bond next to two hydroxyl groups via four electron oxidation of the
substrate. Both dioxygen atoms are incorporated into the final cis-diol product. b) Rieske
dioxygenases catalyze the cis-dihydroxylation of aromatic substrates into cis-dihydrodiols.
NADH is used as a two electron source in these processes. c) The most versatile family
amongst non-heme iron(II) oxygenases is represented by 2-OG dependent enzymes.
Mechanistic details and representative examples of this class will be discussed separately in
chapter 1.1.2. d) The group of pterin-dependent hydroxylases is formed of three aromatic
amino acid hydroxylases, namely phenylalanine-, tyrosine-, and tryptophan hydroxylase.
These enzymes catalyze the insertion of an oxygen atom from O2 into the aromatic side
chain of their according amino acid. The cofactor tetrahydrobiopterin is used as a reductant.
e) All other non-heme iron(II) enzymes such as isopenicillin N synthase (IPNS) or 1-
aminocyclopropane-1-carboxylate oxidase (ACCO) that do not fit in any of these four
families can be summarized in a final miscellaneous class.

4 I. INTRODUCTION
O2COOH
O2+ 2 H2O
OHC
O2OH
OH
O 2OH + R-O H + CO2
O2
OH
O
R
OH
OH OHR
R+ NADH
+ H+R
+ NAD+
O
OH
O
HO
O
+ R-H
O
HO
O
R + R
a)
b)
c)
d)
e)
NH
NNH
HN
O
NH2
R
+N
NNH
HN
O
NH2
R
H
NH2HO2C
HN
N
H
CO2H
HOO
SH
NH2HO2C
HN
NO
O
S
CO2H
e.g. IPNS
Scheme 1: Catalyzed reactions of iron(II) enzymes of the 2-His-1-carboxylate facial triad.[4, 7, 11] Dioxygen is
marked in red italics to display the disposition of each oxygen atom.
1.1.2. 2-Oxoglutarate dependent iron(II) oxygenases
One of the largest and most versatile families of non-heme iron(II) oxygenases is
represented by the 2-OG dependent enzymes. They are able to catalyze a wide range of
oxidation reactions in animals, plants and microorganisms.[12] Most of these enzymes are
hydroxylases, i.e. the hydroxylation of a substrate is coupled to the oxidative
decarboxylation of 2-OG, yielding CO2 and succinate (scheme 1c). Substrates such as
proteins, methylated nucleotides, lipids or different small molecules are recognized by these
enzymes.[13] Other representatives are capable of catalyzing desaturation, epoxidation, ring

I. INTRODUCTION 5
formation/expansion or halogenation reactions.[13-16] The active site iron in almost all 2-OG
dependent oxygenases is bound facially by the conserved 2-His-1-carboxylate motif as
described above. Sequence analyses predict the presence of more than 60 2-OG oxygenases
in humans, which are involved in important processes including chromatin modification,
fatty acid metabolism, DNA repair, and the hypoxic response system.[14] The first 2-OG
dependent hydroxylase ever identified, prolyl-4-hydroxylase P4H[17], has become one of the
best investigated 2-OG dependent oxygenase enzymes. P4H catalyzes the hydroxylation of
proline residues as depicted in scheme 2. This is an essential reaction in mammals for the
formation of collagens, elastins and other proteins. Non-hydroxylated collagen polypeptide
chains cannot form functional molecules in vivo because only hydroxylated residues are
able to form hydrogen bonds which provide collagen triple helices with thermal stability.
Thus, prolyl hydroxylases are attractive targets for pharmaceutical inhibitors for the
treatment of excessive collagen accumulation in fibrotic diseases.[13, 18]
R'
O
N
R
R'
O
N
R
HO
O
HO
O
OH
P4H
FeII, O 2O
OH
O
HO
O+ CO2
Scheme 2: Hydroxylation of proline residues catalyzed by P4H. 2-OG is stoichiometrically decarboxylated
during this reaction. Dioxygen is marked in red italics to display the disposition of each oxygen atom.
Although prolyl hydroxylases as well as other protein hydroxylases have been investigated
extensively for decades, the interest for these enzymes is still very high. In 2001 it was
found that these enzymes are involved in the hypoxic response system.[19-20] In particular,
prolyl hydroxylases PHD1-3 play a key role in targeting one of the two subunits of the
hypoxia-inducible factor (HIF) for degradation. The HIF pathway and the role of prolyl
hydroxylases will be discussed in detail in the main section (chapter 3.4.3) of this thesis.
This introductory section is more focused on the mechanistic details of 2-OG dependent
enzymes.

6 I. INTRODUCTION
The consensus mechanism[6, 14, 16, 21-22] proposed for the activation of dioxygen by 2-OG
dependent enzymes is shown in scheme 3. a) In the initial state (absence of substrates), the
iron(II) center is bound facially by the 2-His-1-carboxlate triad. Three molecules of water
occupy the remaining positions, completing the slightly distorted octahedral coordination
sphere. b) The cosubstrate 2-OG subsequently displaces two molecules of water by κ2-
coordination. In this stage, the iron center is still six-fold coordinated and therefore
unreactive towards dioxygen. c) The required conversion to an active, square-pyramidal
five-coordinate metal center is achieved by the binding of the substrate (S-H) in proximity
of the metal ion. This change in geometry is accompanied by the displacement of the
remaining water and was crystallographically verified by protein structures for instance of
taurine dioxygenase (TauD), DAOCS and alkylsulfatase in presence of 2-OG and their
native substrates.[4, 23-25] It is also noteworthy, that this increase in reactivity by change of
the geometry only occurs in presence of potential substrates and thus protects the enzyme
from inactivating self-hydroxylation reactions. d) Reaction with dioxygen most likely forms
an adduct with significant iron(III) superoxide radical anion character.[26] e) Nucleophilic
attack of O2 at the keto group of the coordinated 2-OG cosubstrate forms a ferryl peroxo
species. f) This intermediate decomposes under decarboxylation and heterolytic cleavage of
the O-O-bond yielding CO2, succinate and a high valent iron(IV)-oxo species responsible for
substrate oxidation. Once the iron(IV)-oxo intermediate is formed, different reactions are
possible for the various enzymes depending on the specific substrate. g) In case of a
hydroxylase such as TauD for instance, the iron(IV)-oxo intermediate abstracts a hydrogen
atom from the substrate S-H. h) The recombination of the coordinated hydroxyl radical
with the substrate radical gives the hydroxylated product (S-OH) and restores of the iron(II)
form of the enzyme (radical rebound mechanism).

I. INTRODUCTION 7
H2OFeII
His
H2O Asp/Glu
His
H2O
OFeII
His
O Asp/Glu
His
H2OO
R
OFeII
His
O Asp/Glu
His
O
R
S H
OFeIV
His
O Asp/Glu
His
OO
R
OS H
OFeIV
His
OCO Asp/Glu
His
O
O
R
S H
OFeIII
His
OCO Asp/Glu
His
OH
O
R
S
OFeII
His
OCO Asp/Glu
HisO
R
S OH
+ 2-OG
- 2 H2O
+ SH- H2O
+ O 2
a) b)
h) c)
g)
f) e)
OFeIII
His
O Asp/Glu
His
OO
R
O S H
d)
CO2,succinate,SOH
+3H2O
Scheme 3: General mechanism for 2-OG dependent enzymes exemplified for hydroxylases.[6, 14, 16, 21-22]
Dioxygen is marked in red italics to display the disposition of each oxygen atom. R = -CH2CH2CO2H.
It is noteworthy, that this mechanism was already postulated more than two decades ago by
H. M. HANAUSKE-ABEL and V. GÜNZLER based on theoretical considerations.[27] But none of
the proposed oxidized iron intermediates occurring in this cycle had been confirmed prior
to 2002. The first direct evidence for any of these species was given relatively recently by
J. M. BOLLINGER JR., C. KREBS and coworkers.[28-29] On their investigations on TauD form
Escherichia coli, a transient state had been characterized by stopped flow absorption
methods and freeze-quench MÖSSBAUER[29] and EPR spectroscopic measurements.[30-31] This
intermediate has formally an iron(IV) center with an unusual high-spin configuration of
S=2. The position of this species in the catalytic cycle was verified by kinetic measurements

8 I. INTRODUCTION
with a selectively deuterium labeled substrate.[32-33] A large deuterium kinetic isotope effect
(kH/kD ≈ 50) on the decay of this intermediate indicated that it has to be the hydrogen
abstracting species depicted in scheme 3f. The presence of the iron(IV)-oxo group in this
intermediate was confirmed by resonance Raman spectroscopy.[34] A characteristic isotope-
sensitive iron-oxo vibration band was observed in the 800 cm–1 region. Further evidence
came from EXAFS studies revealing a rather short Fe-O interaction of 1.62 Å.[35] More
recently, these experimentally determined spectroscopic parameters have been compared
with those predicted by DFT calculations.[36] Two model structures have found to match the
best with the experimental data: a) a distorted octahedral model in which one of the two
carboxylate ligands is coordinated in an asymmetric bidentate fashion and the other in a
monodentate fashion, b) a trigonal bipyramidal model in which both carboxylates are
coordinated in a monodentate fashion. A second transient state for the TauD mechanism
that was confirmed by J. M. BOLLINGER JR., C. KREBS and coworkers is a high-spin iron(II)
containing product(s) complex.[37] In subsequent investigations of the same group on P4H, a
C-H cleaving iron(IV) complex with nearly identical kinetic and spectroscopic features to
those found in TauD as well as a high-spin iron(II) product was confirmed.[38] This
corroborates the fact that the reaction mechanism of the family of 2-OG dependent
enzymes is conserved.
Very recently, a new subclass of 2-OG depending enzymes was identified, which is able to
catalyze the halogenation of aliphatic carbon centers in the biosynthesis of natural
products.[39-41] A crystal structure of the halogenase SyrB2 (syringomycin biosynthesis
enzyme 2) from Pseudomonas syringae reveals a previously unknown coordination in
which the carboxylate donor of the 2-His-1-carboxylate facial triad is replaced by a chlorido
ligand (figure 2). It is assumed, that the mechanism is similar to the common mechanism for
2-OG dependent hydroxylases with chloride abstraction instead of hydroxyl abstraction.[42]
MÖSSBAUER and absorption spectroscopic investigations on the halogenase CytC3 confirmed
the presence of two high-spin iron(IV) intermediates, which presumably are two rapidly
equilibrating conformers of a chlorido-iron(IV)-oxo complex[28, 43]. The direct coordination
of bromide to the proposed intermediate, forming a bromido-iron(IV)-oxo species was
shown by freeze-quench MÖSSBAUER and EXAFS spectroscopy in the reaction of the
halogenase CytC3.[4, 28-29, 44]

I. INTRODUCTION 9
2-OG
His116
His235
ClFe
H O2
Figure 2: Active site of the syringomycin biosynthesis enzyme 2 (SyrB2) with 2-oxoglutarate.
(PDB-code: 2FCT).[42]
1.1.3. Modeling studies for 2-OG dependent iron enzymes
Since the discovery of the 2-His-1-carboxylate facial triad, many efforts have been devoted
to modeling studies. Different ligands have been used to mimic the facial coordination of
two imidazole groups and the carboxylate donor of the triad. In initial studies, tri- or
tetradentate nitrogen donor ligands such as hydridotris(pyrazol-1-yl)borato (Tp), tris(2-
pyridylmethyl)amine (Tpa) or 1,4,7-triazacyclononane (Tacn) ligands and their derivatives
have been used for structural and functional models.[4, 6, 45] On the quest for ligands more
suitable for modeling studies, bis(pyrazol-1-yl)acetic acids as a new class of monoanionic,
tripodal N,N,O coordinating ligands have been developed. These ligands belong to the
family of heteroscorpionates and are available in a broad variety of sterically more or less
demanding, chiral or achiral derivatives.[45] Structurally they are related to the well-known
Tp ligand which was introduced to coordination chemistry by S. TROFIMENKO over 30 years
ago.[46-49] The first synthesis of a bis(pyrazol-1-yl)acetate ligand was described by A. OTERO
and coworkers. In a multi-step synthesis, bis(3,5-dimethylpyrazol-1-yl)methane was
deprotonated and subsequently treated with CO2 to yield the lithium salt of the bis(pyrazol-
1-yl)acetic acid (scheme 4).[50-53] In addition to these reports, the BURZLAFF group developed
a one-step synthesis (scheme 4). Commercially available dichloroacetic acid is treated with
two equivalents of 3,5-dimethylpyrazole and excess of potassium hydroxide and potassium
carbonate. Benzyltriethylammonium chloride (BTEAC) acts as a phase transfer catalyst
during this reaction.[54-55] In 2005 another versatile class of N,N,O ligands namely 3,3-bis(1-

10 I. INTRODUCTION
alkylimidazol-2-yl)propionates was developed independently by R. J. M. KLEIN GEBBINK and
coworkers and the BURZLAFF group. But since these ligands are out of scope of this thesis,
the reader is referred to comprehensive reviews that have been published very recently.[4, 45]
N
N N
N
R
R
R
R
CO2H
NH
N
R
R
R = H, Me
c)
R = Me, t Bu
a)
N
N N
N
R
R
R
R
b)
N N
N N
R
R
R
R
OO
Fe
NN
NN
R
R
R
R
O O
N
N
N
N
R R
R R
O
OFe
N
N
N
N
RR
RR
O
OFe
Cl
Cl
R = H, Me
R = tBu d)
d)
N N
N N
R
R
R
R
OO
Fe
OPh
OO
e)
Scheme 4: Synthesis of different bis(pyrazol-1-yl)acetic acid derivatives as described by A. OTERO et al. and
N. BURZLAFF et al.[53-55] and formation of ferrous model complexes for non-heme iron(II) oxygenases.[45, 54, 56]
Reaction conditions: a) KOH, K2CO3, CH2Cl2, BTEAC, b) 1. n-BuLi, 2. CO2, 3. H3O+, c) dichloroacetic acid
(0.5 eq.), KOH, K2CO3, BTEAC, THF, d) 1. base, 2. FeII, e) thallous benzoylformate.
During the recent years, structural models for non-heme iron(II) enzymes bearing the facial
2-His-1-carboxylate motif have been synthesized in the BURZLAFF group with bis(pyrazol-1-
yl)acetic acids. The coordination chemistry of these ligands strongly depends on their
sterical demand and the choice of the metal precursors. For instance, deprotonation of 2,2-
bis(3,5-dimethylpyrazol-1-yl)acetic acid (Hbdmpza) and treatment with anhydrous FeCl2
did not yield the desired chlorido complex [Fe(bdmpza)Cl], but a 2:1 bisligand complex
[Fe(bdmpza)2].[54, 56] This is the result of the strong coordination ability of this ligand

I. INTRODUCTION 11
combined with its relatively small sterical demand. It was found that the coordination
sphere around the ferrous iron center is almost octahedral and that the bond lengths and
angles match well with those reported for the active site of non-heme iron(II) enzymes such
as IPNS.[57-58] In contrast, the reaction of the sterically more demanding ligand 2,2-bis(3,5-
di-tert-butylpyrazol-1-yl)acetic acid (Hbdtbpza) with iron(II) yielded a dimeric complex
[Fe(bdtbpza)Cl]2 with a bridging acetato group.[45, 54, 56] Interestingly, the geometry of the
ferrous iron in this complex was found to be trigonal bipyramidal and thus is in good
accordance to one of the two calculated iron(IV)-oxo models for TauD which also revealed a
trigonal bipyramidal geometry (see chapter 1.1.2).
On the quest for 2-OG dependent oxygenase models, the dimeric species [Fe(bdtbpza)Cl]2
was reacted with benzoylformate (BF) to form the complex [Fe(bdtbpza)(BF)].[45] The
observed color change to deep purple (λmax = 544 nm) is characteristic for κ2O
1,O2
coordinated 2-oxocarboxylates and is caused by a metal to ligand charge transfer (MLCT).
One drawback during these structural studies is that coordination of bis(pyrazol-1-
yl)acetates to ferrous iron often result in paramagnetic, high-spin complexes which are
difficult to investigate by NMR spectroscopic methods. Ruthenium as the heavier
homologue of iron is a more versatile transition metal in this context. Ruthenium(II)
complexes are low-spin and thus suitable for NMR characterization. Thus, several
ruthenium(II) complexes bearing the Hbdmpza ligand such as [Ru(bdmpza)Cl(PPh3)2],
carboxylato complexes [Ru(bdmpza)(O2CR)(PPh3)] (R = Me, Ph) and 2-oxocarboxylato
complexes [Ru(bdmpza)(O2CC(O)R)(PPh3)] (R = Me, Et, Ph) have been published by the
BURZLAFF group (scheme 5).[45, 59-61] These complexes have also been used for coordination
studies of inhibitor molecules such as N-oxalylglycine (NOG), as will be described in the
main section of this thesis (chapter 3.3.1).
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
R
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3PO
O
R
Scheme 5: Ruthenium(II) complexes of Hbdmpza with carboxylato or 2-oxocarboxylato co-ligands.[45, 59-61]

12 I. INTRODUCTION
Ferrous 2-oxocarboxylato complexes with Tp and Tpa ligands such as [Fe(BF)(TptBu,iPr)],
[Fe(BF)(TpPh2)], or cationic [Fe(BF)(Tpa)]+ as model complexes for 2-OG dependent
oxygenases are known for more than a decade.[62-70] Almost all of them are able to mimic
half of the typical dioxygenase reaction, i.e. the decarboxylation of the 2-oxoacid by
reaction with O2. However, fully functional examples which are also able to simultaneously
oxidize a substrate are rare. In 1995 J. S. VALENTINE and coworkers reported on the in situ
generation of [Fe(BF)(TpMe2)(MeCN)] which was able to epoxidize cis-stilbene when
exposed to O2. A similar complex [Fe(BF)(TptBu,iPr)] was found to be unreactive probably
due to the increased sterical hindrance caused by the tert-butyl substituents.[67] Furthermore,
in 1999 L. QUE JR et al. could show that the reaction of [Fe(BF)(TpPh2)(MeCN)] leads to a
intramolecular self-hydroxylation of one of the phenyl substituents in the ligand backbone.
Labeling experiments with 18O revealed that one oxygen atom of O2 was incorporated in
the hydroxylated phenyl ring and the other one in benzoic acid.[68]
This particular reaction was recently reinvestigated by E. MÜNCK, L. QUE JR. and coworkers
(scheme 6).[71] It was observed that self-hydroxylation of complex A caused by the putative
FeIV=O intermediate can be intercepted in presence of excess amounts of substrates.
Addition of ten equivalents of thioanisole to compound A for instance completely
suppressed the formation of complex B and favoured the formation of the sulfoxide
complex C. This observation is in close analogy to the "protective mechanism" of 2-OG
dependent enzymes: As described in chapter 1.1.2 (scheme 3), the generation of a reactive
FeIV=O intermediate is favored only in the presence of potential substrates and thus protects
the enzyme of deactivation by self-hydroxylation.[4, 7] Further experiments revealed that
self-hydroxylation of A can also be intercepted by hydrocarbons which in turn become
dehydrogenated. Interestingly, the extent of interception depends not only on the strength
of the C-H bond to be cleaved but also on the shape of the hydrocarbon substrate. Thus,
complex A is a rare example of a simple biomimetic model capable of discriminating
between different substrates. In a very recent work, the group of L. QUE JR. report on the
generation of a high-spin FeIV=O complex bearing a sterically demanding 1,1,1-Tris{2-[N 2-
(1,1,3,3,-tetramethylguanidino)]ethyl}amine (TMG3tren) ligand, which suppresses
intermolecular decay. The complex [Fe(O)TMG3tren]2+ was characterized by resonance
Raman, Mössbauer and EXAFS spectroscopy and accompanied by DFT calculations.[72] This
ligand enforces a trigonal-bipyramidal geometry and an S=2 ground state. As mentioned

I. INTRODUCTION 13
above, comparison of spectroscopical data and DFT calculations on TauD favor also an
octahedral geometry with a κ2-coordinated carboxylate donor.[36]
N
B
N
N N
Ph
Ph
Ph
Ph
Fe
H
N
N
Ph
Ph
OPh
OO
O2
N
B
N
N N
Ph
Ph
Ph
Fe
H
N
N
Ph
Ph
Ph
OO
O
R2S
[Fe(TpPh )(OBz)(OSR2)]
+ [(FeIII)2]
A B
C
2
FeIV=O
Scheme 6: Reactions of [Fe(BF)(TpPh2)] (A) with O2 in benzene as reported by L. QUE JR. et al.[71] Dioxygen is
marked in red italics to display the disposition of each oxygen atom.

14 I. INTRODUCTION
1.2. Immobilization of ligands and complexes
In recent years, immobilization of ligands or complexes has become an important part of
modern catalysis. Homogeneous catalysts show up high selectivity and efficiency. They are
well-defined at the molecular level and thus allow important insights into mechanistic
details of catalytic cycles. Unfortunately, one of their biggest disadvantages is their poor
recycling potential.[73] In many cases, complex procedures are necessary to separate the
usually expensive catalysts from the reaction mixtures. Due to their sensitivity, they have to
be treated carefully and under special conditions like for instance an inert gas atmosphere.
Heterogeneous catalysts however, show up a good stability and are easy to handle in most
cases. Separation from the finished reactions can easily be achieved by simple techniques
such as filtration or centrifugation. But in contrast to homogeneous catalysts, the
preparation of heterogeneous catalysts is complicated and not always reproducible and, as
far as their reactivity and selectivity is concerned, they can not compete with homogeneous
catalysts.[73] Thus, the immobilization of homogeneous catalysts onto polymeric or siliceous
supports is a very elegant way to combine the advantages of homogeneous and
heterogeneous catalysts. In recent years, there have been numerous reviews concerning
aspects and applications of these so called heterogenized homogeneous catalysts.[73-78]
Different methods for immobilization and some selected examples for applications in
catalysis and other research areas will be presented in this chapter. This will also include
the work with grafted N,N,O ligands and complexes so far achieved in the BURZLAFF group.
1.2.1. Methods of non-covalent immobilization
Immobilization of ligands or transition metal complexes via non-covalent bonds is the
easiest method for solid phase fixation. In most cases, this approach does not require a
modification of ligands or complexes in order to make them suitable for fixation. This is a
crucial advantage especially when immobilizing chiral catalysts since manipulations on
chiral ligands can negatively affect the enantioselectivity of the catalyzed reactions.
Depending on the way the catalyst or ligand is connected to the solid support, non-covalent
fixation can be categorized in four general methods namely electrostatic, coordinative,
absorptive, or entrapment methods (figure 3).[79-80] Electrostatic methods are relevant in
case of cationic complexes. The fixation is provided by exchange of the complex counter ion
by a negatively charged solid substrate. Compared to the rather strong substrate-complex
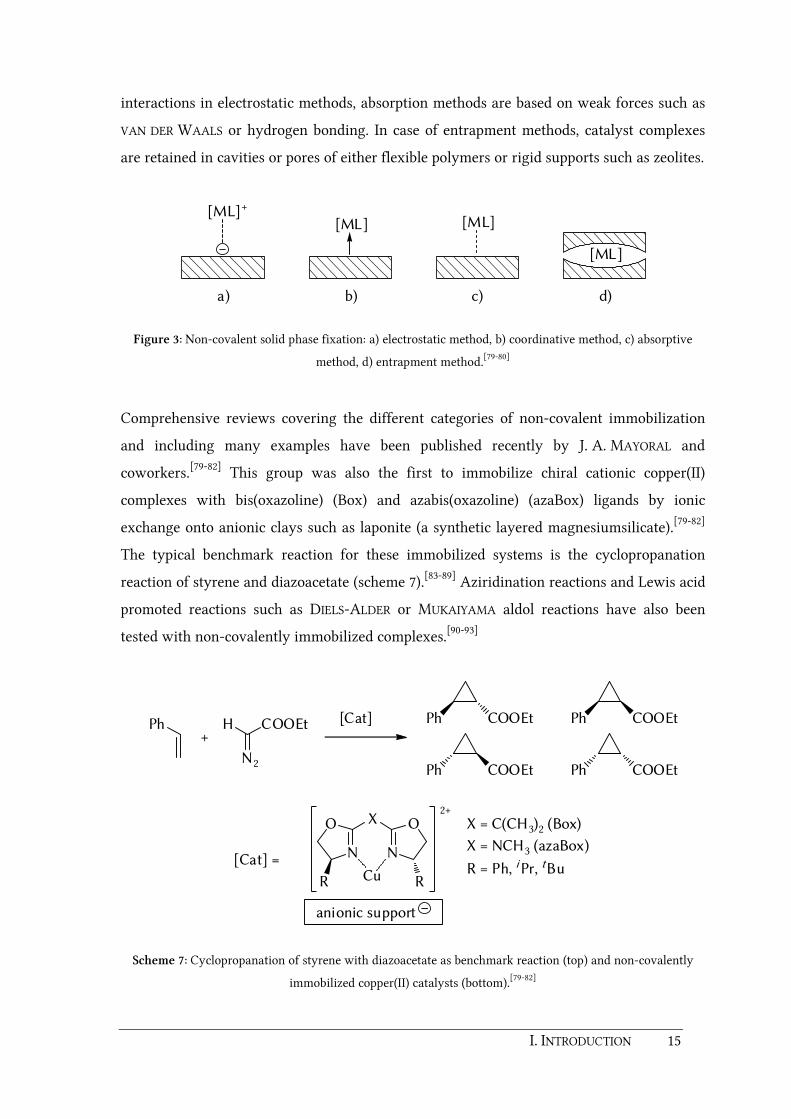
I. INTRODUCTION 15
interactions in electrostatic methods, absorption methods are based on weak forces such as
VAN DER WAALS or hydrogen bonding. In case of entrapment methods, catalyst complexes
are retained in cavities or pores of either flexible polymers or rigid supports such as zeolites.
[ML]+
a)
[ML]
b)
[ML]
c)
[ML]
d)
Figure 3: Non-covalent solid phase fixation: a) electrostatic method, b) coordinative method, c) absorptive
method, d) entrapment method.[79-80]
Comprehensive reviews covering the different categories of non-covalent immobilization
and including many examples have been published recently by J. A. MAYORAL and
coworkers.[79-82] This group was also the first to immobilize chiral cationic copper(II)
complexes with bis(oxazoline) (Box) and azabis(oxazoline) (azaBox) ligands by ionic
exchange onto anionic clays such as laponite (a synthetic layered magnesiumsilicate).[79-82]
The typical benchmark reaction for these immobilized systems is the cyclopropanation
reaction of styrene and diazoacetate (scheme 7).[83-89] Aziridination reactions and Lewis acid
promoted reactions such as DIELS-ALDER or MUKAIYAMA aldol reactions have also been
tested with non-covalently immobilized complexes.[90-93]
Ph H
N2
COOEt+
Ph COOEt Ph COOEt
Ph COOEt Ph COOEt
[Cat]
[Cat] =
anionic support
N
Cu
N
X OO
R R
2+
X = C(CH3)2 (Box)
X = NCH3 (azaBox)
R = Ph, i Pr, tBu
Scheme 7: Cyclopropanation of styrene with diazoacetate as benchmark reaction (top) and non-covalently
immobilized copper(II) catalysts (bottom).[79-82]

16 I. INTRODUCTION
One of the main problems of non-covalently immobilized systems is the leaching of ligands
and/or metal ions from the immobilized catalysts. This leads to the formation of non-chiral
catalytic sites, which in turn are responsible for a decrease in enantioselectivity of the
catalyzed reaction. Thus, the best performances with non-covalently immobilized systems
have so far been obtained with strong coordinating ligands which are able to form stable
complexes. For instance, azaBox ligands have a higher coordinating ability compared to
their Box analogues.[94] The copper complex [Cu(azatBuBox)](OTf)2 immobilized on nafion-
silica shows enantioselectivities of 90 % ee for the trans-products and 84 % ee for the cis-
products during the cyclopropanation benchmark reaction.[95] In contrast to this, an
analogously immobilized system of [Cu(tBuBox)](OTf)2 yields much lower
enantioselectivities of 20 % ee for the trans- and cis-cyclopropanes. It has to be mentioned
that in homogeneous phase, selectivities over 90 % ee have been achieved with the
homogeneous complex [Cu(tBuBox)](OTf)2. The importance of complexation equilibria is
also shown by the fact that addition of free tBuBox ligand to the heterogeneously catalyzed
cyclopropanation reaction with the immobilized complex [Cu(tBuBox)](OTf)2 drastically
improves the enantioselectivity up to a value of 91 % ee for the trans- and 88 % ee for the
cis-cyclopropanes.[82, 85]
1.2.2. Covalent immobilization via grafting on supports
The most common way to immobilize complexes is the formation of a covalent bond
between the solid support and the ligand. The advantage compared to non-covalent
methods is that leaching of ligands is decreased due to irreversible and strong fixation to
the support. In most cases however, covalent immobilization requires an additional
synthetic effort since the desired complexes or ligands have to be functionalized with
suitable groups prior to their heterogenization. As mentioned before, in case of chiral
catalysts or ligands, this required manipulation might negatively influence the
enantioselectivity of catalyzed reactions due to changed steric properties.[82] Covalent
immobilization can either be achieved by grafting onto solid supports or by
copolymerization (as will be discussed in chapter 1.2.3). The general procedure for the
grafting approach is shown in scheme 8. Usually, a linker group (LG) is introduced at the
backbone of a ligand or a complex fragment first. Grafting is then achieved by reaction of
this linker with a suitable anchor group (A) on a preformed organic polymer or a porous

I. INTRODUCTION 17
inorganic support (silica, zeolite, etc.). Solid phases based on silica have excellent
mechanical properties and are usually more stable than flexible polymeric supports.[73, 82]
M
LG
L
LLGA
M
LG
L
L
A
ML
L
Scheme 8: Functionalization of a complex fragment [M(L)2] with a linker group (LG) and grafting a solid
support by means of a matching anchor group (A).
Examples of grafted complexes or catalysts are plenty in literature. For instance,
J. A. MAYORAL and O. REISER et al. have recently immobilized an azatBuBox on a
polystyrene-divinylbenzene (PS-DVB) resin (scheme 9).[81-82, 96] Grafting was achieved by
alkylation of the nitrogen bridge with a bromo functionalized MERRIFIELD resin. The grafted
ligand sites have been treated with Cu(OTf)2 and finally tested in the cyclopropanation
benchmark reaction. Excellent enantioselectivities of up to 99 % ee have been achieved after
optimizing the immobilization conditions.[96] The advantage of azaBox ligands compared to
their Box analogues is on the one hand their strong complexation ability which avoids
leaching of metal ions as mentioned before. On the other hand these ligands are easy to
immobilize by grafting techniques since they posses only one linking point at the bridge
which requires functionalization.
N N
HN OO
tBu tBu
N N
N OO
tBu tBu
N N
N OO
tBu tBuCu
TfO OTf
PS PS
Br
PS
BuLi
Cu(OTf)2
Scheme 9: Grafting of azatBuBox on brominated MERRIFIELD resin.[81, 93]

18 I. INTRODUCTION
The PS-DVB grafted copper azatBuBox catalyst was also tested in the MUKAIYAMA aldol
reaction but showed only low yields due to catalyst poisoning by strong complexation of
products, byproducts or solvents.[81, 92] Interestingly, J. A. MAYORAL and coworkers could
show that the poisoning of the immobilized catalysts in the MUKAIYAMA reaction is not
irreversible and that the recycled catalyst from the MUKAIYAMA reaction could be reused in
other reactions with a completely different mechanism. A recovered grafted catalyst after
three MUKAIYAMA reaction cycles was tested in the cyclopropanation benchmark reaction
and showed the same excellent chemo- and enantioselectivities of a fresh, unused
catalyst.[81, 93]
Examples for covalent immobilization by grafting of bis(pyrazol-1-yl)acetate ligands on
solid supports have been reported previously by the BURZLAFF group. Two new N,N,O
heteroscorpionate ligands, namely 2,2-bis(3,5-dimethylpyrazol-1-yl)-3-hydroxypropanoic
acid (Hbdmpzhp) and 2,2-bis(3,5-dimethylpyrazol-1-yl)pent-4-enoic acid (Hbdmpzpen),
bearing a hydroxymethyl or an allyl linker have been designed starting from Hbdmpza
(scheme 10 and scheme 11).[97]
N
N N
N
Me
Me
Me
Me
CO2HHO
Cl
PS
1. KOtBu, CsBr,
18-crown-6
2.
3. [ReBr(CO)5]
N N
N N
Me
Me
Me
Me
OO
Re
CC CO
OO
O
Hbdmpzhp
PS
Scheme 10: Immobilization of [Re(bdmpzhp)(CO)3] on MERRIFIELD resin.[97]
These functionalized ligands as well as their manganese(I) and rhenium(I) tricarbonyl
complexes [M(L)(CO)3] have been successfully immobilized on MERRIFIELD polymer or
mercaptopropyl functionalized silica, respectively. The accessibility and proper κ3-N,N,O
binding behavior of the incorporated ligand sites was confirmed by comparison of the IR
I. INTRODUCTION 19
spectra of the immobilized [M(L)(CO)3] fragments with those of the corresponding
homogeneous complexes.[97]
N N
N N
Me
Me
Me
Me
OO
M
CC CO
OO
N N
N N
Me
Me
Me
Me
OO
M
CC CO
OO
AIBN
[M(bdmpzpen)(CO)3]M = Mn, Re
O
SiOR
OHS
S Si
O
OR
O
Scheme 11: Immobilization of [M(bdmpzpen)(CO)3] (M = Mn, Re) on mercaptopropyl functionalized silica.[97]
Regardless of whether polymeric or siliceous supports are used, it has to be mentioned that
commercially available solid phases can only be modified to a certain degree. Thus, one
major disadvantage of the grafting approach is that the possibility to tune the degree of
functionalization and the structure of the surrounding solid phase is rather limited.
Furthermore, remaining reactive anchor groups on the support may cause unwanted side
reactions during catalytic cycles.
1.2.3. Covalent immobilization via copolymerization
The copolymerization approach depicted in scheme 12 represents a more elegant way for
covalent immobilization of ligands or complexes. Instead of a linker group, a polymerizable
group (PG) is introduced at the backbone of a ligand. The resulting modified complex
fragment can be copolymerized with different comonomers in a second step. Compared to
grafting methods, the polymerization approach is much cheaper and allows a more detailed
control and fine-tuning. Properties of the resulting solids such as morphology, swelling
behavior or flexibility, strongly depend on polymerization conditions and can be
manipulated for instance by means of additional crosslinking monomers or porogene
solvents. This is especially of importance when it comes to optimize heterogenized systems
for catalytic applications. Several examples for polymerizable transition metal complexes

20 I. INTRODUCTION
and their catalytic application in hydrogenation, oxidation and C-C bond forming and other
reactions have been reviewed a few years ago by C. F. NOBILE et al. as well as P. A. JACOBS
et al.[73, 98]
M
PG
L
LPG
ML
LPG
n
ML
L
Scheme 12: Immobilization via copolymerization with a matching monomer requires the introduction of a
polymerizable group (PG).
For bis(oxazoline) type ligands, the concept of immobilization by copolymerization was first
introduced by S. V. LUIS, J. A. MAYORAL and coworkers.[99-100] Dialkylation of PhBox, tBuBox or IndaBox at their methylene bridge with 4-vinylbenzyl chloride gives rise to
ligands which are suitable for homopolymerization or copolymerization with styrene or
divinylbenzene (DVB) (figure 4). The Cu(OTf)2 complexes of these polymerized ligands
show moderate to good enantioselectivities in the cyclopropanation benchmark reaction.
The best results with regard to enantioselectivity, recycling potential and yield have been
obtained for the homopolymerized systems. The highest enantioselectivity (78 % ee) was
achieved for a homopolymer of the doubly functionalized tBuBox ligand. Nevertheless, a
disadvantage of these polymerized catalysts is that most of the ligand sites remain in the
core of the polymers and thus can not participate in the catalysis.[80-82]
N N
N OO
R R
N N
N OO
PhBox (R = Ph), t BuBox (R = tBu) IndaBox
Figure 4: PhBox, tBuBox and IndaBox ligands functionalized with vinylbenzene groups suitable for
homo- and copolymerization.

I. INTRODUCTION 21
Besides for catalytic applications, immobilization is also of particular interest for model
complexes which can mimic certain metalloenzymes since the polymer support can be used
to obtain structural control over the coordination spheres of the metal binding sites. For
instance, K. SEVERIN and coworkers reported on copper and zinc complexes of the neutral
N,N,N chelating tris[(1-vinylimidazol-2-yl)methyl]amine ligand which have been
copolymerized with ethylene glycol dimethacrylate (EGDMA) (scheme 13).[101] It is known
that such copper(II) and zinc(II) complexes with neutral, chelating N-donor ligands are good
models for hydrolytic enzymes which catalyse DNA scission.[102] However, the problem is
that these model complexes tend to form hydroxy bridged dimers, which are catalytically
inactive.[103-116] K. SEVERIN et al. were able to show that the formation of dimers can be
avoided by copolymerization with EGDMA. The immobilized copper and zinc complexes
are efficient catalysts for the hydrolysis reaction of bis(p-nitrophenyl)phosphate (BNPP)
which is a model for biologically relevant phosphodiesters such as DNA or RNA. The
immobilized copper complex turned out to be 56 times more active than the homogenous
complex. One of the reasons for the efficiency of these immobilized catalysts is a
partitioning effect. In case of the polymer bound complexes, the substrate BNPP is strongly
adsorbed to the EGDMA polymer. This leads to a high local concentration of BNPP in the
polymeric matrix which facilitates the hydrolysis.
N
NN
N
N
NN
1. M2+
2. NH4PF6
3. EGDMA,AIBN
N
N
N
M
N
ClN
N
N
PF6-
Polymer
M = Cu, Zn
Scheme 13: Synthesis of immobilized copper(II) and zinc(II) complexes of tris[(1-vinylimidazol-2-
yl)methyl]amine by copolymerization with EGDMA.[101]
The possibility to control coordination geometries of enzyme relevant model complexes by
immobilization was also successfully proven in the BURZLAFF group. As mentioned in
chapter 1.1.3 sterically less demanding bis(pyrazol-1-yl)acetate ligands tend to form

22 I. INTRODUCTION
complexes of the type [M(L)2] with two ligand molecules coordinated towards one metal
center (see scheme 4). The aim was to control the coordination geometry and avoid the
formation of such 2:1 bisligand complexes by solid phase fixation of the ligand. For this
purpose a new N,N,O ligand bearing a polymerizable metharcryloxy linker, namely 2,2-
bis(3,5-dimethylpyrazol-1-yl)-3-(methacryloxy)propanoic acid (Hbdmpzmp) was
synthesized.[117] Reaction of a soluble MMA (methyl methacrylate) copolymer of
Hbdmpzmp with copper(II) chloride yielded a deep-blue solid phase (scheme 14). The
UV/Vis spectrum of this copper containing polymer showed a absorption maximum at
715 nm which was almost identical to the one found in the homogeneous complex
[Cu(bdmpza)2].[118] Thus, it was assumed that the copper(II) ions act as crosslinking agents
and enable the formation of 2:1 bisligand moieties. In contrast to this, the heterogeneous
reaction of copper(II) with a highly crosslinked EGDMA copolymer of Hbdmpzmp yielded a
lime green polymer (scheme 14). A significant bathochromic shift of the absorption
maximum by 78 nm indicated one-sided bound copper centers and thus the successful
prevention of bisligand formation as desired.
The final example which will be discussed in this introduction for the application of
copolymerized complexes comes from the field of medicinal chemistry. The incorporation
of drugs, prodrugs or proteins into polymeric matrices has become an important method in
the development of so called polymer therapeutics.[119] Immobilization in hydrophilic
polymers for instance is often used to improve the solubility of drugs. It is also known that
polymer-drug conjugates may enhance tumor targeting[119-124] due to a so called enhanced
permeability and retention (EPR) effect, which was first described by H. MAEDA and
coworkers.[125-129] Solid tumor cells exhibit a very high permeability in order to supply their
nutritional demand. Furthermore, the lymphatic function in tumor cells is damaged so that
macromolecular drugs of a molecular weight larger than 45 kDa can accumulate in tumor
tissue and remain there for a long time. Besides therapeutic applications, polymer-drug
conjugates are also used for diagnostic pharmaceuticals. One of the most important
radioisotopes in diagnostic nuclear medicine is 99mTc due to its excellent physical decay
properties (t1/2 = 6 h, Eγ = 140 keV). But also the particle emitting isotope 188Re has
increasingly drawn attention in the last years. Almost a decade ago, R. SCHIBLI and
R. ALBERTO developed methods for the convenient and simple preparation of fac-
[99mTc(H2O)3(CO)3]+ and fac-[188Re(H2O)3(CO)3]
+ as important precursor complexes for
radiopharmaceuticals.[130-131]

I. INTRODUCTION 23
N
N N
N
Me
MeMe
Me CO2H
linear copolymer
NN N
N
Me
MeMe
MeCO2H
O O
O O
OO
MeO O
crosslinked copolymer
1. Base2. CuCl2
N NN N
Me
Me
Me
Me
OO
Cu
NNNN
Me
Me
Me
Me
O O
O
O
O
O
N NN N
Me
Me
Me
Me
OO
Cu
(S)Cl (S)
O
O
O
O
Me
MMA
EDGMA
AIBN
MMAAIBN
1. Base
2. CuCl2
Hbdmpzmp
Scheme 14: Control of the coordination geometry of copolymerized copper(II) complexes of the Hbdmpzmp
ligand.[117]
Since then, the aim of many research groups was to develop ligands which on the one hand
are able to bind the [M(CO)3] fragment and on the other hand posses polymerizable
functional groups for immobilization. For instance P. C. KUNZ and coworkers described the
synthesis of the polymerizable ligand bis(2-pyridylmethyl)-4-vinylbenzylamine (scheme
15).[132] Copolymerization with the biocompatible, water solubler monomer N-(2-
hydroxypropyl)methacrylamide[133] (HPMA) yielded a copolymer with an appropriate size
suitable for the EPR effect (52 kDa). This polymer bound ligand was labeled with
[Re(CO)3Br3]2–. It was successfully proven that the Re(CO)3 fragments remain coordinated
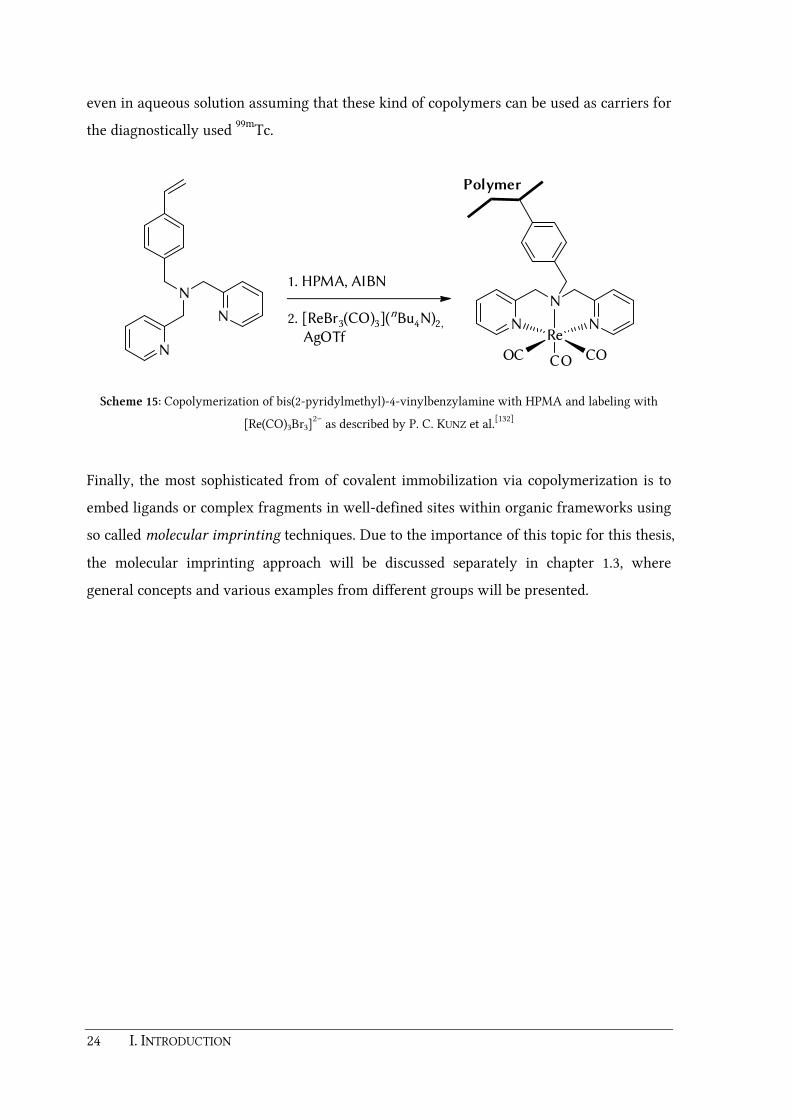
24 I. INTRODUCTION
even in aqueous solution assuming that these kind of copolymers can be used as carriers for
the diagnostically used 99mTc.
N
N
N
1. HPMA, AIBN
2. [ReBr3(CO)3](nBu4N)2,
AgOTf
N
ReNN
CO COOC
Polymer
Scheme 15: Copolymerization of bis(2-pyridylmethyl)-4-vinylbenzylamine with HPMA and labeling with
[Re(CO)3Br3]2– as described by P. C. KUNZ et al.[132]
Finally, the most sophisticated from of covalent immobilization via copolymerization is to
embed ligands or complex fragments in well-defined sites within organic frameworks using
so called molecular imprinting techniques. Due to the importance of this topic for this thesis,
the molecular imprinting approach will be discussed separately in chapter 1.3, where
general concepts and various examples from different groups will be presented.

I. INTRODUCTION 25
1.3. Molecular imprinted polymers
1.3.1. General Concept
Molecular imprinting is an approach, which describes the generation of highly selective
binding sites and specific cavities in synthetic polymers by means of a template molecule.
The concept for molecular imprinting is shown in scheme 16.[134-137] The procedure begins
with an assembly step, where a complex between a template molecule T and different
functional monomers FM is formed. These monomers can be bound either covalently or
non-covalently to the template molecule. The T-FM complex is then copolymerized by
radical initiation with an excess of crosslinking monomers (CM). Typically, DVB or
EGDMA are used for this purpose. The presence of a porogene solvent during the
polymerization step gives rise to macroporous polymers (surface area 100 – 600 m2 g–1) with
a rigid pore structure and thus provides access to the functional sites within the polymer.[73,
136] The subsequent extraction of the template molecule generates a nanosized cavity or
surface recognition site, which is complementary in shape and functionality to the original
template molecule T.
a)
b)
Cavityc)
+
FM
T
T T
CM
Scheme 16: Representation of an imprinting process. a) Assembly of template (T) and functional monomers
(FM), b) copolymerization with crosslinking monomers (CM), c) generation of the cavity by extraction of the
template.[134-136]

26 I. INTRODUCTION
Imprinted polymers are often considered as artificial enzymes because they are able to
recognize template analogue substrates with a high selectivity similar to natures "lock and
key" model. Since the binding sites are embedded in three-dimensional scaffolds, only guest
molecules that fit exactly within the cavities can be accommodated. Compared to natural
enzymes however, they might be tolerant to reaction conditions which usually would
denature most proteins and biopolymers such as heat or certain chemicals and solvents.
Additionally, molecular interactions such as hydrogen bonds which are present in biological
systems can also be utilized in imprinted polymers by introduction of suitable functional
groups.[134]
The first molecular imprinting experiment was described by M. V. POLYAKOV in the 1930s. It
was found that benzene was adsorbed faster than toluene or xylene on silica gel which was
previously dried in a atmosphere of benzene.[135, 138] In 1972 the concept of molecular
imprinting was formalized by G. WULFF and coworkers as a practical methodology and is
now an established research area.[139] Potential applications of molecular imprinted
polymers (MIPs) are widespread. Extensive reviews and books have been published by
different authors, summarizing the general aspects of MIPs as well as their use for
analytical, synthetic, catalytic or biochemical/pharmaceutical purposes.[134-137, 140-151]
Different approaches for molecular imprinting including some selected examples will be
discussed in the following sections.
1.3.2. Imprinted polymers as microreactors
In the beginnings, imprinted polymers have not been used as catalysts but as synthetic aids
for regio- and stereoselective reactions.[136] The representation of a selective synthesis
within a MIP microreactor is given in scheme 17.[152] The reaction of two educts A and B
yields in two regio- or stereoisomers C and D. The idea is to generate a cavity within a
polymer by using the desired isomer C or a structurally related analogue of this isomer as a
template for imprinting. If the reaction between A and B runs within this MIP, the
formation of the desired isomer C should be favoured due to steric restrictions. It is
important to use excess or at least stoichiometric amounts of the MIP in order to avoid non-
selective reactions outside the microreactor. This can also be enforced by binding all of the
reactant A to the imprinted polymer before the reaction with the reactant B occurs.[152]

I. INTRODUCTION 27
D
Imprinting with or an analogue
of CC
+ orA
A
A
B
B
C
C
+A
+B- C
Scheme 17: Usage of a MIP as a microreactor (A, B = reactants; C, D = regio- or stereoisomeric products).[152]
The first examples for an asymmetric synthesis within an imprinted polymer microreactor
have been described by G. WULFF and coworkers.[153-154] Enantioselective C-C bond
formation inside a chiral cavitiy was used to prepare optically active amino acids starting
from glycine. The imprinting was carried out with a polymerizable L-DOPA derivative
bearing boronic acid salicylaldehyde binding sites (scheme 18). Removal of the template by
hydrolysis and subsequent addition of glycine gives a SCHIFF base which can be
deprotonated with a base yielding an ester enolate. If the alkylating agent is placed within
the cavity by means of the boronic acid residue, reaction with the glycine ester enolate will
occur in a stereospecific manner. Amino acids with an enatiomeric excess of up to 36 % ee
have been obtained by this procedure.[134, 136, 152-154]

28 I. INTRODUCTION
BO
O
N
H
O
O
BOH
OH
OB
O
O
N
HO
O
BOH
OH H
N
H
O
O
BOH
OH H
N
O
O
RO
OH
NH3
+ Glycine
Base (B-)
BH
+ RX
X
BH
HBX
OMe
O
N
OB
O
H
OH
Imprinting
Scheme 18: Enantioselective alkylation within an imprinted microreactor.[134, 136, 152-154]
In recent years the concept of reactions within imprinted microreactors was further
expanded. It was found that imprinted polymers that mimic the binding sites of native
enzymes might be used for the synthesis of new bioactive molecules. The discovery of new
drug candidates can be achieved in a so called anti-idiotypic imprinting approach.[134, 145, 155-
156] For this, a lead drug or biologically active compound is imprinted. This forms a cavity
which mimics the receptor or a pocket of a native enzyme. Building blocks are then
introduced and reacted inside these cavities to yield structural and electronic analogues of
the template molecule. This was successfully demonstrated in a recent work of K. MOSBACH

I. INTRODUCTION 29
and coworkers.[155-156] The preparation of new inhibitor molecules for the proteinase
kallikrein with an imprinted microreactor was described. A known inhibitor bearing a
guanidine residue was used as a template for the imprinting attempt. It was shown that the
resulting microreactor can not only be used for the synthesis of the original template but
also for the synthesis of new, structurally related inhibitor molecules. This method
represents an attractive concept for the generation of new pharmaceuticals based on small-
molecule inhibitors, especially when the native enzyme is yet poorly characterized or
difficult to obtain in sufficient quantities for biomedical studies.[134, 152]
1.3.3. Imprinting with a transition state analogue
A very promising approach for the development of new and efficient catalysts is to adopt
principles of the enzymatic catalysis.[136] In native enzymes, the stabilization of a high
energy transition state of a reaction by preferred binding is the key factor for catalysis. This
thesis was already postulated by L. PAULING[157] in 1949, further extended by
W. P. JENCKS[158] and finally verified by R. A. LERNER
[159] and P. G. SCHULTZ[160] on studies
with antibodies against transition state analogues (TSAs) of a certain reaction.[136] The
simplified energy profile for a unimolecular reaction of a substrate (S) to a product (P) is
shown in figure 5a regarding both, the uncatalyzed and the enzyme-catalyzed case.[152] In
the presence of an enzyme, an enzyme-substrate complex (ES) is immediately formed in a
pre-equilibrium step. During the reaction, the bound substrate is converted to an enzyme-
bound product (EP) via a transition state (ES‡). Finally, the product is released from this
enzyme-product complex EP. The difference in energy between ES and ES‡ (=∆Gcat‡) is
smaller than the free activation energy ∆G‡ of the uncatalyzed reaction which means that
the enzyme stabilizes the transition state of the reaction. For sufficient catalytic turnover, it
is also necessary that the enzyme-substrate complex ES is lower in energy (i.e. more stable)
than the product complex EP.

30 I. INTRODUCTION
Reactioncoordinate
E+SES
ES‡
EP
E+P
S‡
∆Gcat
‡
∆G‡
½VMax
KM
VMax
0
Free energy Reaction rate
Substrateconcentration
a) b)
Figure 5: a) Simplified energy profile of a unimolecular reaction S→P (S = substrate, P = product, E = enzyme,
ES = enzyme-substrate complex, EP = enzyme-product complex, S‡ and ∆G‡ = transition state and free
activation energy of the uncatalyzed reaction, ES‡ and ∆Gcat‡ = transition state and free activation energy of
the enzyme catalyzed reaction. b) Schematic diagram of the MICHAELIS-MENTEN saturation kinetics (KM =
MICHAELIS constant).
Any enzyme working this way shows a typical kinetic behavior (figure 5b). With increasing
amounts of substrate, the reaction rate first increases but then levels off. When all active
sites are occupied with substrate molecules, the reaction rate remains constant (Vmax). In
other words, the reaction rate is zero-order with respect to the substrate concentration [S].
This saturation behavior can be described mathematically by the so called MICHAELIS-
MENTEN equation.[136, 152]
E + S ES E + Pd[P]
dt
[E]0 · [S]
KM + [S]= k2 ·
k1
k-1
k2
Equation 1: MICHAELIS-MENTEN equation for a unimolecular reaction.
Keeping this in mind, an effective strategy for the design of enzyme-like catalysts would be
to synthesize a molecule first, which is able to mimic the transition state of a certain
reaction and then determine a matching receptor with a high affinity for this transition
state analogue. This was already successfully applied in the development of new antibodies
which can catalyze transformations such as hydrolysis reactions, DIELS-ALDER reactions,
cyclopropanations or cyclizations.[152, 161-163] A very similar strategy has been used to

I. INTRODUCTION 31
generate imprinted polymers for enzyme-like catalysis. TSA molecules have been used to
generate cavities which are able to selectively bind the transition state of a desired reaction.
The most prominent examples in this context are phosphonic esters which have been used
to simulate the tetrahedral transition state of alkaline hydrolysis reactions, e.g. a
saponification (scheme 19).
R O
O
R'R O
R'
O OH
R OH
O+ R'OH
OH‡
H+
RP
OR'
O OHTSA
Scheme 19: Phosphonic esters as transition state analogues of alkaline mediated hydrolysis reactions.
In pioneering works of G. WULFF and coworkers, the functional monomer N,N'-diethyl-4-
vinylbenzimidamide with a high affinity for the phosphonate and carboylate groups of the
substituted phosphonic monoester was used for the positioning and fixation of the TSA
during the imprinting process (scheme 20).[164] The MIP obtained after polymerization with
EGDMA and removal of the template was used in the saponification reaction of the
depicted ester. Compared to the reaction in homogeneous solution, the rate of the MIP
catalyzed reaction was 100 times faster. Nevertheless, a small turnover number was
observed when using the MIP as catalyst. This is due to inhibition of the system, since the
product of the hydrolysis reaction, homoterephthalic acid, is also tightly bound by the
amindinium functionality.
B. SELLERGREN and K. J. SHEA et al. expanded this concept by utilizing chiral phosphonate
analogues of phenylalanine for the design of MIPs as catalysts for the enantioselective
hydrolysis of D- and L-phenylalanine esters.[165] Additionally, the templates have been
designed in such a way, that the resulting MIPs provided the key elements that are believed
to be responsible for the catalytic action of the proteolytic enzyme chymotrypsin, namely a
stereoselective binding site, a site complementary to a transition state structure as well as a
phenol-, an imidazole- and an acidic group in the binding site. It was shown that the
hydrolysis of the D-phenylalanine derivative was 1.9 times faster than the hydrolysis of the

32 I. INTRODUCTION
L-derivative. Control experiments also indicated that the polymers are able to discriminate
between a planar ground state and a tetrahedral transition state.
TSA
NH
HN O
O
PO O
O
H H
N NEt Et
Et
Et
Me
Me
O
O
Me
CO2H
HO2C HO Me
Me
+
HO2C
Me
H2O
b)
a)
Scheme 20: a) Template for the generation of a catalytic MIP. b) Catalyzed alkaline ester hydrolysis reaction
as reported by G. WULFF et al.[164]
Further research on MIPs for hydrolysis reactions during the last years showed that the
ability for binding of a TSA is not the only key factor for a high reaction rate.[164, 166-168] It is
also important to increase binding of a TSA by steric and electronic effects and to
incorporate and position essential functional groups in a correct way. Recent publications of
G. WULFF and J. LIU describe the construction of very efficient artificial MIP models for the
natural enzyme carboxypeptidase A.[169-171] The catalytic performance of these MIPs was
tested on the hydrolysis reaction of diaryl carbonates (scheme 21a). Phenyl pyridin-2-yl
phosphate was chosen as a TSA. Furthermore, two new polymerizable functional monomers
have been designed.[171] The monomer depicted in scheme 21b has an additional zinc
binding triamine group in close proximity to the amidinium functionality providing a
strong three-fold coordination of the metal center. The monomer shown in scheme 21c has
two amidinium groups as well as an additional amine linker. The advantage of this
monomer is that two TSA molecules per metal ion can be used during the imprinting
process. By imprinting of this template, an extraordinarily efficient copper(II) containing
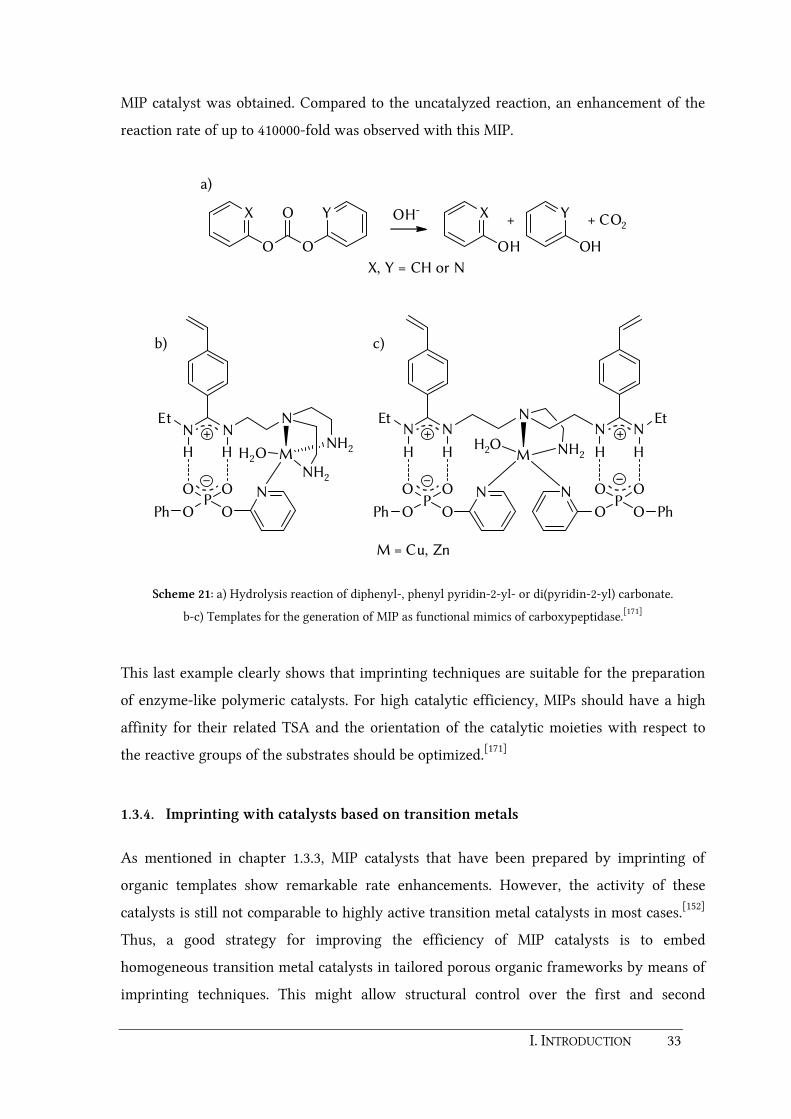
I. INTRODUCTION 33
MIP catalyst was obtained. Compared to the uncatalyzed reaction, an enhancement of the
reaction rate of up to 410000-fold was observed with this MIP.
NEt
NN
H H
O OP
OOPh
N
MNH2
NH2
H2O
NNNN
Et
H H
O O
NEt
HH
O OPP
O O O OPh
N N
Ph
M NH2H2O
b) c)
O
O
O
X Y
OH OH
X Y
X, Y = CH or N
OH-+ CO2+
a)
M = Cu, Zn
Scheme 21: a) Hydrolysis reaction of diphenyl-, phenyl pyridin-2-yl- or di(pyridin-2-yl) carbonate.
b-c) Templates for the generation of MIP as functional mimics of carboxypeptidase.[171]
This last example clearly shows that imprinting techniques are suitable for the preparation
of enzyme-like polymeric catalysts. For high catalytic efficiency, MIPs should have a high
affinity for their related TSA and the orientation of the catalytic moieties with respect to
the reactive groups of the substrates should be optimized.[171]
1.3.4. Imprinting with catalysts based on transition metals
As mentioned in chapter 1.3.3, MIP catalysts that have been prepared by imprinting of
organic templates show remarkable rate enhancements. However, the activity of these
catalysts is still not comparable to highly active transition metal catalysts in most cases.[152]
Thus, a good strategy for improving the efficiency of MIP catalysts is to embed
homogeneous transition metal catalysts in tailored porous organic frameworks by means of
imprinting techniques. This might allow structural control over the first and second

34 I. INTRODUCTION
coordination spheres around reactive metal sites and enable the tuning of functional
properties such as Lewis-acidity, reduction/oxidation behavior and selectivity of the
catalyst.[148] The resulting immobilized catalytic systems should be in close analogy to
metalloenzymes, where reactive metal centers are also embedded in well-defined reaction
sites. The strategy for this approach is depicted in scheme 22.[134, 152] It has to be mentioned
that the functionalization of the ligand periphery with polymerizable side chains is a
prerequisite for this approach. By initial ligand exchange, a pseudo-substrate (PS) is
coordinated to the metal center of the catalyst. Ideally, PS has a structure similar to the
original substrate (S) which will be finally transformed and is also able to mimic the
transition state of the catalyzed reaction. After the polymerization step, the cavity is
generated by removing PS either selectively or together with the metal ions. By the latter
method, it is also possible to exchange inert metal centers for active ones after the
imprinting step.
L M
L
L
X L M
L
L
L M
L
L
L
L
L
L M
L
L
XL M
L
L
PS PS
S
a) b)
c) d)
e)f)
Scheme 22: Procedure towards imprinted metal catalysts: a) Coordination of a pseudo-substrate (PS), b)
polymerization, c) selective removal of PS, d) removal of PS and the (inert) metal center e) addition of an
(active) metal ion, f) original substrate (S) attached to the catalytic site in the final MIP.[134, 152]
K. SEVERIN and coworkers used this strategy to generate MIP catalysts for the transfer
hydrogenation of ketones.[172-173] It is known that half sandwich complexes of ruthenium or
rhodium with amine based auxiliary ligands are highly active catalysts for the reduction of
ketones using formic acid or isopropanol as the reducing agent.[174-178] A six-membered ring

I. INTRODUCTION 35
with a hydrogen bond from the carbonyl O-atom to the amine group as depicted in scheme
23a is proposed as transition state structure for reactions of this kind.[175, 179-180] This
transition state can be imitated with a coordinated diphenylphosphinate as pseudo-substrate
(scheme 23b).
M
NH H O
PO
Ph
RM
NH H O
H
Ph
R
a)
Ph
O
R Ph
OH
R
[Ru]-catalyst
iPrOH
or HCO2H
b) c)
Scheme 23: a) Transfer hydrogenation of aromatic ketones, b) proposed transition state, c) transition state
analogue.[172-173, 181]
A MIP derived from the ruthenium(II) complex shown in figure 6a was seven times more
active in the transfer hydrogenation of benzophenone than a control polymer which was
imprinted without the pseudo-substrate. Furthermore, the obtained MIP was able to
selectively reduce benzophenone out of a mixture of different ketones. In a similar work, a
methylphenylphosphinato Rh(III) complex with a chiral N,N'-chelating co-ligand bearing a
styrene unit (figure 6b) was synthesized and characterized.[181] This complex represents a
TSA for the asymmetric reduction of acetophenone. The cavity of the MIP derived by
imprinting this complex was shown to be highly selective for acetophenone. Excellent
enantioselectivities of up to 95 % ee have been obtained with this MIP catalyst.
N
RuO
NH
SO O
R
O
P
Ph
Ph
iPr
R
N
RhO
NH2
SO O
R
O
P
Ph
Me
i Pr
a) b) R = C6H4CH=CH2
Figure 6: Polymerizable phosphinato complexes of ruthenium(II) and rhodium as TSA for the transfer
hydrogenation of benzophenone or acetophenone.[172-173, 181]
More recent work of the same group describe the synthesis of imprinted porphyrin catalysts
for an oxidation of alcohols and alkanes.[182-183] The ruthenium(II) porphyrin complex
[Ru(L)(CO)], with L = meso-tetra[4-(vinylbenzoxy)benzyl)porphyrin], was synthesized and

36 I. INTRODUCTION
treated with the pseudo substrate aminodiphenylmethane (scheme 24). This template was
polymerized with EGDMA in presence of chloroform as a porogen solvent. A control
polymer without the pseudo-substrate was also polymerized. It was shown that on the one
hand, the MIP catalyst as well as the control polymer displayed a high catalytic activity for
the oxidation of alcohols and alkanes with 2,6-dichloropyridine N-oxide (DCPNO) while the
homogeneous complex [Ru(L)(CO)] was completely inactive under the same reaction
conditions. On the other hand, it was shown that the MIP catalyst derived from the pseudo-
substrate is significantly more active than the control polymer derived without the pseudo-
substrates. Rate enhancements of up to a factor of 16 have been achieved.[183]
R
R
N
N
N
N Ru
CO
R R
R R
R = ORu
CO
R R
R R
=
Ru
CO
R R
R R
Ru
CO
R R
R R
NH
PhPh
Ru
CO
Scheme 24: Ruthenium porphyrin oxidation catalyst imprinted with aminodiphenylmethane as reported by
K. SEVERIN et al.[182-183]



II. OBJECTIVE AND AIMS 39
II. OBJECTIVE AND AIMS

40 II. OBJECTIVE AND AIMS
During the last years, several N,N,O heteroscorpionate ligands for solid phase fixation and
copolymerization as well as their transition metal complexes have been synthesized and
investigated in the BURZLAFF group.[97, 117, 184] So far, the generation of those ligands was
achieved by following one uniform concept. In all cases, the functional group, i.e. a solid
phase linker or a polymerizable group was introduced at the bridging carbon atom of the
well-known 2,2-bis(3,5-dimethylpyrazol-1-yl)acetic acid (Hbdmpza) (1) ligand. In certain
cases however, the substituents at this bridging position caused unexpected problems
during the synthesis of various transition metal complexes. Most conspicuous is the fact,
that so far it was not possible to synthesize bulky ruthenium complexes [Ru(L)Cl(PPh3)2]
with these functionalized N,N,O ligands L, bearing a linker at the bridging carbon atom.
However, ruthenium(II) complexes with such ligands can mimic the active site of 2-OG
dependent iron(II) enzymes (see chapter 1.1.3). And in particular, polymerizable ruthenium
complexes might open the door to molecular imprinting techniques which in turn would be
useful for catalytic applications.
Thus, it was expected to begin this thesis with an in-depth study of the influence of
substituents at the bridging carbon atom of bis(pyrazol-1-yl)acetic acids. A new N,N,O
model ligand had to be synthesized for these investigations by a simple functionalisation of
a well bis(pyrazol-1-yl)acetic acid at the bridging carbon atom. The aim was to synthesize
and spectroscopically characterize transition metal complexes of this model ligand and
compare them to complexes with non-functionalized bis(pyrazol-1-yl)acetic acids in order
to understand and discuss differences in structure and reactivity.
The second aim of this thesis was to develop a new N,N,O heteroscorpionate ligand which is
suitable for immobilization by copolymerization. But in contrast to similar ligands
synthesized before, the task was to introduce functional (polymerizable) groups not at the
bridging carbon atom but somewhere else in the periphery in order to circumvent the
problems of bridge functionalization. There had been attempts earlier in the BURZLAFF
group to follow this concept but unfortunately without satisfying results. Thus, the strategy
for the design of such a ligand was to be reconsidered by a retrosynthetic approach. After a
successful synthesis, polymerization behavior of the new ligand and the incorporation of
transition metal fragments had to be verified using spectroscopical methods. Furthermore,
the possibility to control coordination geometries by immobilization through copoly-
merization had to be investigated.

II. OBJECTIVE AND AIMS 41
To verify that the polymerizable groups do not affect the remaining space of a κ3-N,N,O
bound metal fragment, it was also expected to synthesize and characterize various
transition metal complexes, especially those with bulky co-ligands such as [Ru(L)Cl(PPh3)2].
The third and final aim of this thesis was to use this new polymerizable ligand for the
generation of imprinted polymers. Therefore, it was expected to synthesize and characterize
appropriate template molecules. Ideally, these should be ruthenium(II) model complexes,
which can mimic the active site of 2-OG dependent iron(II) oxygenases, bearing pseudo-
substrate or inhibitor molecules.
Furthermore, two sub-projects had to be fulfilled during this thesis. The first one was to
verify the catalytic activity of the ruthenium(II) dicarbonyl complexes [Ru(L)Cl(CO)2] of
different bis(pyrazol-1-yl)acetic acids L towards the epoxidation of cyclohexene in the
presence of oxidants such as H2O2 or iodosylbenzene. The second sub-project was to
synthesize the isoquinoline based small molecule PHD inhibitor 2-(1-chloro-4-
hydroxyisoquinoline-3-carboxamido)acetic acid (37) according to literature procedures. In
the scope of an interdisciplinary collaboration, this inhibitor was to be provided to the
group of Prof. Dr. K.-U. ECKARDT (Department of Nephrology and Hypertension, Friedrich-
Alexander-University Erlangen-Nürnberg) for studies the on stabilization of HIF in organ
transplants.


III. RESULTS AND DISCUSSION 43
III. RESULTS AND DISCUSSION

44 III. RESULTS AND DISCUSSION
3.1. Substituents at the bridging position of bis(pyrazol-1-yl)acetic acids
3.1.1. Problem statement
During the recent years, several transition metal complexes have been immobilized in the
BURZLAFF group using appropriately functionalized ligands. But there is one major difficulty
one has to consider during these studies. Due to their insolubility, characterization of
immobilized transition metal complexes is limited to a few spectroscopic methods such as
UV/Vis or IR spectroscopy. Thus, for comparison reasons, it is essential to synthesize and
fully characterize corresponding homogeneous complexes of the functionalized ligands
prior to the heterogenization attempt. However the appearance of chirality and the
formation of enantiomers were often observed by synthesizing transition metal complexes
of bridge functionalized ligands. The resulting complexes were difficult to characterize
especially by NMR spectroscopic methods. In particular complexes of ligands like
Hbdmpzmp are affected, where the rotation of a large functional group along the C–C axis
at the bridging carbon atom is restricted. In this case, the linker retains between the
carboxylate group and one pyrazolyl donor. Hence, two enantiomers are conceivable which
can be converted into each other dynamically (figure 7).
Figure 7: Dynamic interconversion of the two enantiomers in case of a coordinated, bridge functionalized
bis(pyrazol-1-yl)acetato ligand (here: Hbdmpzmp).[117, 184]
A typical example for the appearance of chirality was found for the tricarbonyl complex
[Mn(bdmpzmp)(CO)3] (2).[117, 184] This compound crystallized in a chiral conformation and
both possible isomers were found in the cell due to the space group P21/a. For the analogous
rhenium complex [Re(bdmpzmp)(CO)3] the described chirality was also observed in
solution. The 1H NMR spectrum of this complex features an AB resonance system due to
the diastereotopic protons of the -CH2O- group. Furthermore, broad 1H NMR signals as well

III. RESULTS AND DISCUSSION 45
as the appearance of two sets of 13C NMR signals for each pyrazole carbon atom indicate
that there is a dynamic interconversion between the enantiomers in solution. Very similar
results have also been obtained for the bridge functionalized ligand 3-acetoxy-2,2-bis(3,5-
dimethylpyrazol-1-yl)propanoic acid (Hbdmpzap) and its tricarbonyl complexes
[M(bdmpzap)(CO)3] (M=Mn, Re).[117]
A closer view at the molecular structure of tricarbonyl complex 2 reveals a second problem
that is occasionally observed when dealing with bridge functionalized ligands (figure 8).
The carboxylate donor is clearly bent [∡(O2,C1,C2,C3) = 25.6(2)°] in contrast to the
molecular structures reported for analogous complexes with unsubstituted ligands such as
[Mn(bdmpza)(CO)3].[55] This might lead to a weakening of the M–O bond and restrict the
coordination towards certain transition metals.
N22N12
C3
O41
N11 N21
C2
C41
O3
O4
Mn1
C4
C1
O2
O1
C51C31
C6
C5
O51O31
C7
Figure 8: Molecular structure of [Mn(bdmpzmp)(CO)3] (2). Thermal ellipsoids are drawn at the 50 %
probability level. Most hydrogen atoms have been omitted for clarity.[117, 184]

46 III. RESULTS AND DISCUSSION
Table 1: Selected bond lengths and distances for the tricarbonyl complex 2.
Distances in Å Angles in °
d(C1–O1) 1.260(3) ∡(N11,Mn1,C51) 177.12(9)
d(C1–O2) 1.222(3) ∡(N21,Mn1,C31) 173.16(10)
d(Mn1–N11) 2.0806(18) ∡(O1,Mn1,C41) 178.57(9)
d(Mn1–N21) 2.0389(18) ∡(N21,Mn1,O1) 83.66(7)
d(Mn1–C51) 1.809(2) ∡(N11,Mn1,O1) 87.60(7)
d(Mn1–C41) 1.796(2) ∡(N21,Mn1,N11) 83.44(7)
d(Mn1–C31) 1.804(2) ∡(O2,C1,C2,C3) 25.6(2)
d(Mn1–O1) 2.0259(16)
d(C51–O51) 1.146(3)
d(C41–O41) 1.149(3)
d(C31–O31) 1.149(3)
A third disadvantage of bridge functionalized N,N,O ligands is that the introduced linker
groups can compete with the carboxylate donor function for the coordination towards the
metal center. For instance, the molecular structure of the bisligand copper(II) complex
[Cu(bdmpzmp)2] (3) reveals a unusual square planar κ2-N,O coordination of both bdmpzmp
ligands (figure 9 and table 2).[117, 184] Instead of the remaining two nitrogen donors, the
oxygen atoms O3 and O3a of the ester group occupy the free octahedral positions. Moreover,
there is a weak intramolecular hydrogen (IMH) bond between the olefinic hydrogen atom
H6A and oxygen donor O1a of the of the symmetry equivalent ligand.[185] The
hydroxymethyl functionalized ligand Hbdmpzhp synthesized in the BURZLAFF group gives
another example for the concurring effect of linker groups at the bridging position.[97] On
attempts to synthesize manganese(I) and rhenium(I) tricarbonyl complexes of this ligand, it
was observed that several additional bands for the CO vibrations appear in the IR spectrum.
Besides, the NMR spectra show more than one set for the pyrazolyl donors. These
observations indicate that there might be an equilibrium between two species, in which
either the carboxylate group or the hydroxyl linker is coordinated towards the metal center.
Finally, in case of the attempted syntheses of ruthenium(II) complexes with Hbdmpzmp or
Hbdmpzpen it is assumed, that the double bond of the methacryloxy- or the allyl linker
coordinates towards the ruthenium center.[97, 117] This might form an olefin complex instead
of a κ3-N,N,O coordinated complex.

III. RESULTS AND DISCUSSION 47
O2
H6Aa
C6a
O1
N21
N22 N11a
O3a
O4a
Cu1
O4
O3
N12N11
O1a
C6
H6A
O2a
Figure 9: Molecular structure for the bisligand complex [Cu(bdmpzmp)2] (3). Thermal ellipsoids are drawn at
the 50 % probability level. Most hydrogen atoms have been omitted for clarity.[117, 184]
Table 2: Selected bond lengths and distances for the bisligand copper(II) complex 3.
Distances in Å Angles in °
d(O1–Cu) 1.8938(15) ∡(N11,Cu,O1) 87.98(7)
d(N11–Cu) 1.9906(17) ∡(N11,Cu,O1a) 92.02(7)
d(H6A–Cu) 2.94(3) ∡(H6A,Cu,O1) 114.8(6)
d(H6A–O1a) 2.75(3) ∡(H6A,Cu,O1a) 65.2(6)
d(H6A–O1) 4.11(3)
d(C5–C6) 1.339(4)
d(C5–C7) 1.467(3)
d(C5–C4) 1.489(3)
d(O3–H6A) 2.48(3)
d(O3–Cu) 3.3695(15)

48 III. RESULTS AND DISCUSSION
3.1.2. 2,2-Bis(3,5-dimethlypyrazol-1-yl)propanoic acid as a model ligand
In order to gain more information about the influence of linker groups at the bridging
carbon atom of bis(pyrazol-1-yl)acetic acids a very simple bridge functionalized ligand was
to be designed. It has been shown in the past that even a nondescript substituent like a
methyl group which is introduced at the periphery of a ligand can have a major influence
on the stability or reactivity of its metal complexes. An impressive example is represented
by the two FeIV=O complexes [Fe(O)(cyclam-acetate)]+ (cyclam-acetate = 1,4,8,11-
tetraazacyclotetradecane-1-acetate) and [Fe(O)(TMC)(NCCH3)](OTf)2 (TMC = 1,4,8,11-
tetramethyl-1,4,8,11-tetraazacyclotetradecane) reported by K. WIEGHARDT et al. and
L. QUE JR. et al. respectively.[186-187] While [Fe(O)(cyclam-acetate)]+ decays rapidly at –40 °C,
the tetramethyl substituted macrocyclic ligand TMC increases the lifetime of the FeIV=O
moiety drastically, due to a restricted access to the oxo group.
Inspired from these and other examples, 2,2-bis(3,5-dimethylpyrazol-1-yl)propanoic acid
(Hbdmpzpr) (4) bearing a simple methyl group at the bridging carbon atom was considered
an appropriate model ligand. The synthesis of ligand 4 was achieved by deprotonation of
Hbdmpza (1) with lithium diisopropylamide (LDA) at –80 °C followed by methylation by
addition of methyl iodide. After work up with diluted HCl, the new methyl substituted
ligand 4 was extracted from the aqueous phase (scheme 25). The successful formation of 4
was verified by 1H and 13C NMR spectroscopy. The additional 1H resonance at δ = 2.30 ppm
and the 13C resonance at δ = 27.0 ppm can be assigned to introduced methyl group.
Furthermore the mass spectrum of ligand 4 shows a clear peak for the molecular ion [MH]+
at m/z = 263.
The coordination behavior of model ligand 4 was studied by the synthesis of transition
metal complexes of 4 with sterically less demanding co-ligands first. As shown in several
examples before, the reaction of potassium salts of bis(pyrazol-1-yl)acetic acids L with the
manganese(I) and rhenium(I) pentacarbonyl precursors [MnBr(CO)5] and [ReBr(CO)5][188],
leads smoothly to tricarbonyl complexes [M(L)(CO)3] (M = Mn, Re) (scheme 25). Besides
their possible application for pharmaceutical purposes,[189-191] those complexes have turned
out to be useful tools for the characterization of the coordination behavior of tripodal
ligands by IR spectroscopy.[55, 97, 117, 192-194]

III. RESULTS AND DISCUSSION 49
N
N N
N
Me
Me Me
Me
CO2H 1. LDA, THF2. MeI
3. H+
1
1. KOtBu
2. [MBr(CO)5]
N N
N N
Me
Me
Me
Me
OO
M
CC CO
OO
M = Mn (5), Re (6)
Me
N
N N
N
Me
Me Me
Me
CO2H
4
Me
Scheme 25: Synthesis of Hbdmpzpr (4) and manganese(I) and rhenium(I) tricarbonyl complexes 5 and 6.
The formation of the tricarbonyl complexes [Mn(bdmpzpr)(CO)3] (5) and
[Re(bdmpzpr)(CO)3] (6) was monitored by in situ IR spectroscopy. Therefore, samples of the
reaction mixture were taken and investigated on a regular basis. The appearance of a single
A' and two close A'' and A' carbonyl signals indicated the successful formation of the
"piano-stool" type tricarbonyl complexes. The reactions can be considered as finished when
no further changes in the IR spectra of the collected samples are observed. In case of the
manganese complex 5, the reaction was finished after 22 hours. This is in good agreement
with the reaction time required for the formation of the analogous bdmpza manganese
complex [Mn(bdmpza)(CO)3].[55] 1H and 13C NMR spectroscopic analysis of the isolated
compound prove the formation of the tricarbonyl complex 5. A single set of signals for the
pyrazolyl groups was observed in the 1H NMR spectrum and the 13C resonances for the
carbonyl ligands were found at δ = 220.6 and 222.2 ppm. Furthermore, a peak in the FAB-
MS spectrum of tricarbonyl complex 5 at m/z = 401 can be assigned to the molecule ion
[MH+]. The molecular structure of 5 gained by single crystal structure determination finally
exhibits the κ3-N,N,O coordination mode of the bdmpzpr ligand 4 (figure 10 and table 3).
Suitable crystals for this purpose were obtained by layering a solution of 5 in CH2Cl2 with
n-pentane. Complex 5 crystallized in the triclinic space group 1P . A comparison of the

50 III. RESULTS AND DISCUSSION
usual bond lengths of complex 5 with those reported for the analogous complex
[Mn(bdmpza)(CO)3][55] shows no significant differences. The angles ∡(N11,Mn1,N21),
∡(O1,Mn1,N11) and ∡(O1,Mn1,N21) are about 5° smaller than the octahedral angle of 90° as
expected due to the clamp of the tripodal coordinating ligand. Of all three metal-carbonyl
distances, d(Mn1–C32) is the shortest because of the trans influence of both pyrazolyl
donors on the other two M–CO bonds.
Synthesis of the rhenium complex [Re(bdmpzpr)(CO)3] (6) did not proceed as smooth as the
analogous manganese compound 5. After a time of 24 h only a slight progress of the
reaction was visible in the IR spectrum. It took approximately 56 hours until the formation
of complex 6 was completed. This reaction time is almost three-fold longer than the time
required for the formation of [Re(bdmpza)(CO)3] (20 h).[55] An NMR spectroscopic
characterization of rhenium complex 6 was not possible, since the compound is insoluble in
common deuterated solvents. The clearly prolonged reaction time might be explained by a
weaker binding of the bridge functionalized Hbdmpzpr ligand 4 compared to the non-
functionalized ligand Hbdmpza 1 which in turn is the result of two steric effects.
O41
C41
N11
N22N12
N21
C2
Mn1
C3
C31C51
O31O51
C1
O1
O2
Figure 10: Molecular structure of [Mn(bdmpzpr)(CO)3] (5). Thermal ellipsoids are drawn at the 50%
probability level. Most hydrogen atoms have been omitted for clarity.

III. RESULTS AND DISCUSSION 51
Table 3: Selected bond lengths and angles for the Mn(I) tricarbonyl complex 5.
Distances in Å Angles in °
d(Mn1−N11) 2.0537(15) ∡(N11,Mn1,N21) 85.65(6)
d(Mn1–N21) 2.0658(15) ∡(O1,Mn1,N11) 84.86(6)
d(Mn1–O1) 2.0051(13) ∡(O1,Mn1,N21) 84.74(6)
d(Mn1−C31) 1.818(2) ∡(O1,Mn1,C41) 177.92(7)
d(Mn1−C41) 1.7918(19) ∡(N11,Mn1,C51) 177.10(8)
d(Mn1−C51) 1.811(2) ∡(N21,Mn1,C31) 176.04(7)
d(O1−C1) 1.272(2) ∡(C31,Mn1,C41) 90.46(8)
d(O2−C1) 1.228(2) ∡(C31,Mn1,C51) 87.29(8)
d(C1−C2) 1.591(2) ∡(C41,Mn1,C51) 88.24(8)
d(C31−O31) 1.147(2) ∡(O2,C1,C2,C3) –7.7(2)
d(C41−O41) 1.155(2) ∡(Mn1,N21,N22,C2) 13.92(18)
d(C51−O51) 1.146(2)
1) There is a steric hindrance between the new methyl group and the carboxylate
donor functionality. This interaction leads to a slightly twisted coordination mode of the
carboxylate donor (figure 11). The torsion angle ∡(O2,C1,C2,C3) in case of
[Mn(bdmpzpr)(CO)3] (5) is about 5° bigger than the corresponding torsion angle of the
analogous complex [Mn(bdmpza)(CO)3]. This effect seems to depend on the bulkiness of the
introduced linker group. As mentioned earlier, the molecular structure of
[Mn(bdmpzmp)(CO)3] reveals a clearly bent carboxylate donor with even bigger torsion
angle ∡(O2,C1,C2,C3) of 25.6(2)° (figure 8).
NN
NN
Me
Me
Me
Me
O
O
M
Me
Figure 11: Steric hindrance of the new methyl group in ligand 4 when bound facially towards a metal center.
2) The second interaction which might be responsible for a weaker binding of the
model ligand 4 is caused by a steric repulsion between the new methyl group and the
pyrazole methyl substituents in position 5. A closer view at the molecular structures of

52 III. RESULTS AND DISCUSSION
[Mn(bdmpzpr)(CO)3] (5) and [Mn(bdmpza)(CO)3] along their C2–Mn1 axis reveals that
there is a slight twist of one of the pyrazolyl groups in case of complex 5 (figure 12). A
torsion angle ∡(Mn1,N21,N22,C2) = 13.9(2)° was found in the molecular structure of
complex 5 while the same angle in the analogous complex [Mn(bdmpza)(CO)3] is clearly
smaller (2.15°).
Figure 12: Comparison of the molecular structures of [Mn(bdmpza)(CO)3] (left) and [Mn(bdmpzpr)(CO)3] (5)
(right). View along the C2-Mn1 axis.
The differences in the molecular structures are not the only indicators for the changed
binding and donor properties of the Hbdmpzpr ligand 4. The weaker binding and donor
ability of 4 is also evident by comparing absorption bands for tricarbonyl complexes bearing
κ3-N,N,O bound bis(pyrazol-1-yl)acetato ligands, which have been investigated in the past
(table 4).[55, 117, 184] It is known that the wavenumber of the ν(CO) absorption correlates with
both, the strength of the M–C and the C–O bond. By π-backbonding electron density is
withdrawn from the metal center to the antibonding π*(CO) orbital causing a strengthening
of the metal-carbon bond on the one hand, and a weakening of the carbon-oxygen bond on
the other hand. This weakening leads to a shift of the ν(CO) vibration towards lower
wavenumbers. Thus, a change in the electron density at the metal center by other ligands
can have a huge influence on the carbonyl ligand. Compared to the similar complexes
[M(bpza)(CO)3] and [M(bdmpza)(CO)3],[55] tricarbonyl complexes of bridge functionalized
ligands like 4 exhibit ν(CO) vibrations at slightly lower wavenumbers (table 4). This means
that the electron density at the metal center of complexes of bridge functionalized ligands is
higher compared to complexes with non-functionalized ligands such as bis(pyrazol-1-
yl)acetic acid (Hbpza) or Hbdmpza (1). In other words, bridge functionalized ligands are
poorer π acceptors compared to non-functionalized ligands.

III. RESULTS AND DISCUSSION 53
Table 4: Selected IR signals (THF, cm–1) of [Mn(L)(CO)3] and [Re(L)(CO)3] complexes.
Ligand anion (L) ν(CO) [Mn(L)(CO)3] ν(CO) [Re(L)(CO)3]
bdmpzpr (4) 2033, 1938, 1910 2022, 1916, 1892
bdmvpza (18)[a] 2036, 1944, 1917 2026, 1921, 1898
bpza[55] 2041, 1946, 1925 2029, 1924, 1904
bdmpza[55] 2036, 1942, 1915 2025, 1918, 1896
bdmpzmp[117] 2034, 1940, 1913 2024, 1918, 1894
[a] to be discussed in chapter 3.2.
3.1.3. Bisligand complexes of type [M(L)2]
It is well known that sterically less demanding bis(pyrazol-1-yl)acetic acids such as
Hbdmpza (1) tend to form 2:1 bisligand complexes of type [M(L)2]. Examples with different
transition metals (Fe, Zn, Cu, etc.) have been synthesized and investigated in the past.[54, 118]
But once they are formed, such bisligand complexes are chemically inert due to the high
binding affinity of the bis(pyrazol-1-yl)acetates. Any further reactions with additional
ligands or potential substrates have not been observed so far. However, this restriction
might now be eased by means of the changed coordinating behavior of ligand 4. A labile
ligation in bisligand complexes might open a door for their potential use in catalysis and
organometallic chemistry in general. Since the aim of this chapter is to exploit differences
between bridge functionalized and non-functionalized bis(pyrazol-1-yl)acetato ligands, the
2:1 bisligand copper complex [Cu(bdmpzpr)2] 7 was synthesized (scheme 26).
[Cu(OAc)2]
MeCN
N N
N N
Me
Me
Me
Me
OO
Cu
NN
NN
Me
Me
Me
Me
O O
Me
Me
N
N N
N
Me
Me Me
Me
CO2H
4
Me
7
Scheme 26: Synthesis of the 2:1 bisligand complex [Cu(bdmpzpr)2] (7).

54 III. RESULTS AND DISCUSSION
Treatment of Hbdmpzpr (4) with copper(II) acetate in acetonitrile gave a pale blue
precipitate of the bisligand complex 7 after stirring for a few hours at ambient temperature.
The IR spectrum of complex 7 exhibits a strong band at ν ̃ = 1665 cm–1 for the asymmetric
carboxylate vibration which is in good accordance to bisligand complexes of copper(II) and
other transition metals as reported earlier.[54, 117-118, 184] A single crystal X-ray structure
determination of 7 confirms the κ3-N,N,O coordination mode of the bdmpzpr ligand 4
(figure 13 and table 5).
The overall JAHN-TELLER-distorted octahedral geometry of complex 7 is in good accordance
to the analogous copper(II) bisligand complex [Cu(bdmpza)2] described by W. L. DRIESSEN
and J. REEDIJK et al.[118] The copper center is centrosymmetrically surrounded by two
bdmpzpr ligands. The nitrogen atoms N11 and N11a as well as the oxygen atoms O1 and
O1a build up the basal plane of the distorted octahedron. The JAHN-TELLER axis is formed by
the remaining nitrogen atoms N21 and N21a with d(Cu1–N21) = 2.372(3) Å. Nevertheless,
there is a significant difference in the molecular structures of complex 7 and [Cu(bdmpza)2]
as shown in figure 14.
O1a
N11 N21
N22N12
Cu1
N21a
C2
N11a
C3
O1
C1 O2
Figure 13: Molecular structure of the 2:1 bisligand complex [Cu(bdmpzpr)2] (7). Thermal ellipsoids are drawn
at the 30 % probability level. Most hydrogen atoms have been omitted for clarity.

III. RESULTS AND DISCUSSION 55
Table 5: Selected bond lengths and angles for the copper(II) bisligand complex 7.
Distances in Å Angles in °
d(Cu1–N11) 2.071(3) ∡(O1,Cu1,N11) 85.90(11)
d(Cu1–N21) 2.372(3) ∡(N11,Cu1,N21) 84.52(11)
d(Cu1–O1) 1.936(2) ∡(N11,Cu1,N11a) 180.0(2)
d(O1−C1) 1.275(4) ∡(N21,Cu1,N21a) 180.00(12)
d(O2−C1) 1.216(4) ∡(O1,Cu1,O1a) 180.00(19)
d(C1−C2) 1.594(5) ∡(O2,C1,C2,C3) -20.2(4)
∡(Cu1,N11,N12,C2) 22.2(4)
∡(Cu1,N21,N22,C2) 33.9(3)
When looking at both molecules along their C2–C2a axis, a remarkable twist of two
pyrazolyl units can be observed in case of complex 7. The torsion angles
∡(Cu1,N11,N12,C2) = 22.2(7)° and ∡(Cu1,N21,N22,C2) = 33.9(3)° in complex 7 are up to
three times higher than those found in the molecular structure of [Cu(bdmpza)2] (8.6° and
10°). Once again, this observation is the result of a steric repulsion between the introduced
methyl group and the carboxylate donor and/or the pyrazole methyl groups respectively. It
is noteworthy, that the magnitude of this effect seems to be more pronounced in the
bisligand complex 7 than in the tricarbonyl complex [Mn(bdmpzpr)(CO)3] (5). This is
obvious since the steric tension in a bisligand complex is much higher compared to a
complex with sterically less demanding ligands such as the tricarbonyl complex 5.
Figure 14: Comparison of the molecular structures of [Cu(bdmpza)2][118] (left) and [Cu(bdmpzpr)2] (7) (right).
View along the C2–C2a axis.

56 III. RESULTS AND DISCUSSION
In future studies, it has to be tested whether one of the N,N,O ligands in complexes of the
[M(L)2] type can be exchanged by other coordinating moieties such as enzyme relevant
cosubstrates like 2-oxoglutarate or small molecules such as NO, N2, etc. If so, bisligand
complexes might become cheap and easy accessible precursors for organometallic or
catalytic reactions.
3.1.4. Synthesis of [Ru(bdmpzpr)Cl(PPh3)2] and reaction with dinitrogen
As indicated in chapter 3.1.3, the steric influence of a linker function at the bridging
position group strongly seems to depend on the bulkiness of the whole complex. Thus, it
was interesting to investigate, how the new methyl group of model ligand Hbdmpzpr (4)
affects co-ligands in even bulkier complexes than bisligand compounds. A well-known and
often used precursor for sterically demanding ruthenium(II) complexes is [RuCl2(PPh3)3][188,
195]. Reaction of this precursor with Cp (cyclopentadienyl), Tp, Tpm [tris(pyrazolyl)methane]
or bis(pyrazol-1-yl)acetato ligands results in bisphosphine complexes of the type
[Ru(L)Cl(PPh3)2].[59, 196-198] Following the procedures described in the literature, the methyl
substituted ligand 4 was deprotonated with KOtBu and treated with [RuCl2(PPh3)3] (scheme
27). The reaction was monitored by IR-spectroscopy on a regular basis. During a time of 4
hours the band for the asymmetric carboxylate vibration of the potassium salt K[bdmpzpr]
at ν ̃ = 1638 cm–1 disappeared completely. At the same time, the appearance of a band at
ν ̃ =1670 cm–1 indicated the successful coordination of the carboxylate group to the
ruthenium(II) center. It is noteworthy that the synthesis of the analogous bdmpza complex
[Ru(bdmpza)Cl(PPh3)2] (8) is already finished after one hour.[59] The formation of
[Ru(bdmpzpr)Cl(PPh3)2] (9) was doubtlessly confirmed by mass spectrometry by the
appearance of a molecule ion peak at m/z = 922.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
Me
N N
N N
Me
Me
Me
Me
OO
Ru
PPh3Cl PPh3
Me
+1. KOtBu, THF
2. [RuCl2(PPh3)3]
N
N N
N
Me
Me Me
Me
CO2H
4
Me
9a 9b
Scheme 27: Synthesis of the bisphosphine complex [Ru(bdmpzpr)Cl(PPh3)2] (9).

III. RESULTS AND DISCUSSION 57
In order to purify the crude product, the solvent was removed under reduced pressure and
the residue was dissolved in CH2Cl2. On dropwise addition of n-pentane, a yellow solid
started to precipitate, which was collected and further studied by NMR spectroscopy. The 31P NMR spectrum of this compound showed three signals at δ = 35.7, 35.2 and 60.3 ppm
although only two phosphine groups were thought to be present in the isolated compound 9.
Furthermore, three sets of signals for the pyrazolyl groups were found in the 1H and 13C
NMR spectra of 9. This indicated that the isolated solid is a mixture of two isomers, namely
a symmetrical isomer 9a with the chlorido ligand trans towards the carboxylate donor
functionality and an unsymmetrical isomer 9b where the chlorido ligand is bound trans to
one of the pyrazolyl donors (scheme 27). The appearance of two isomers stays in contrast to
the synthesis of the analogous bisphosphine complex [Ru(bdmpza)Cl(PPh3)2] (8) where the
symmetrical isomer was formed exclusively during the reaction.[59] Furthermore, it has to
be mentioned that complex 9 is extremely air sensitive and decomposes rapidly by traces of
oxygen even in a dry solid state. Thus, it was not possible to separate both isomers 9a/9b
and isolate them in a required purity for elemental analysis during this thesis.
This significant lability of compound 9 as well as the formation of two isomers might be
explained by an equilibrium in solution as depicted in (scheme 28). There has to be a
considerable steric repulsion in 9a between the methyl group in position 3 of the pyrazolyl
units and a bulky PPh3 ligands caused by the "twist" of the pyrazole ring as described earlier.
Thus, the dissociation of a PPh3 ligand, i.e. the formation of a coordinatively unsaturated
16 VE intermediate [Ru(bdmpzpr)Cl(PPh3)] 9c seems to be favored to a certain degree. Re-
binding of PPh3 in trans position towards the carboxylate function results in the
unsymmetrical isomer 9b.

58 III. RESULTS AND DISCUSSION
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
Me
N N
N N
Me
Me
Me
Me
OO
Ru
PPh3
Ph3P Cl
Me
+
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P Cl
Me
PPh3
N2, THF
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
Me
9a 9c 9b
10
Scheme 28: Possible equilibrium between the two isomers 9a and 9b and a 16 VE fragment
[Ru(bdmpzpr)Cl(PPh3)] (9c) and formation of the dinitrogen complex [Ru(bdmpzpr)Cl(N2)(PPh3)] (10).
Coordinatively unsaturated complexes such as 9c are important intermediates in
organometallic and inorganic transformations and are of interest as potential catalysts. In
many cases, steric interactions play an important role when it comes to generate and also to
stabilize such coordinatively unsaturated species.[199] It is known that half sandwich type
Cp or Cp* complexes [M(C5R5)X(L)2] are typical precursors for the generation unsaturated
16 VE species.[199] Especially with sterically crowded residues L, ligand dissociation can lead
to either neutral complexes [M(C5R5)X(L)] or positively charged complexes of type
[M(C5R5)(L)2]+ depending whether the halide X– or the ligand L leaves the coordination
sphere.
When steric and electronic conditions are tuned carefully, 16 VE species can even be
isolated as stable compounds such as [Ru(Cp*)Cl(PR3)] with bulky phosphines as co-
ligands.[200-202] Otherwise, species like 9c are not stable and react quickly with any kind of
electron donors to fulfill their 18 VE configuration. This could be any coordinating solvent
or small molecules such as N2. For Tp complexes of type [Ru(Tp)X(L)2] it was not possible

III. RESULTS AND DISCUSSION 59
so far, to isolate stable 16 VE complexes [Ru(Tp)(L)2]+ by halide abstraction. However
M. AKITA and Y. MORO-OKA et al. have shown that the removal of the H2O ligand in the
labile complex [Ru(TpiPr)(H2O)(dppe)]+ [dppe = 1,2-bis(diphenylphosphino)ethane] yields in
the coordinatively unsaturated complex [Ru(TpiPr)(dppe)]+ as depicted in scheme 29.[203]
Nevertheless it has to be mentioned, that this fragment is stabilized by an agostic C-H
interaction which readily can be replaced by a N2 ligand.
N
B
N
N N
R
R
R
R
Ru
PP OH2
H
N
N
R
R
+
N
B
N
N N
R
R
R
Ru
PP
H
N
N
R
R
+
N
B
N
N N
R
R
R
R
Ru
PP N2
H
N
N
R
R
+
CH2H
-H2O N2
PP = Ph2PCH2CH2PPh2 R=iPr
Scheme 29: Water abstraction from the labile complex [Ru(TpiPr)(H2O)(dppe)]+ and reaction of the
coordinatively unsaturated fragment [Ru(TpiPr)(dppe)]+ with dinitrogen.[203]
Thus, this report in mind, it was not surprising that on attempts to crystallize compound 9
under dinitrogen atmosphere, crystals of the N2 complex [Ru(bdmpzpr)Cl(N2)(PPh3)] (10)
were recovered (scheme 28 and figure 15). Similar N2 complexes with Cp, Cp* and Tp
ligands have already been investigated earlier by K. KIRCHNER, K. MEREITER, P. VALERGA,
M. C. PUERTA and others (table 7).[203-214] But as far as the author is aware, the isolated
complex 10 is the first representative dinitrogen complex bearing a κ3-N,N,O bound
bis(pyrazol-1-yl)acetato ligand.
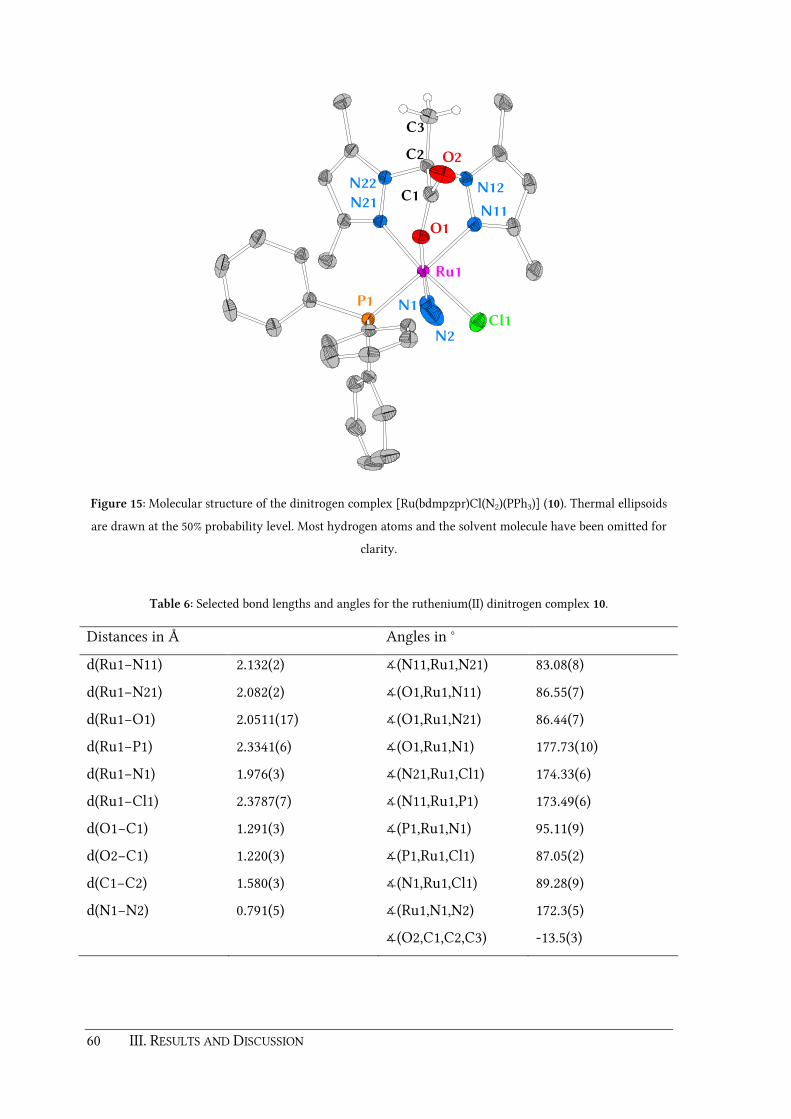
60 III. RESULTS AND DISCUSSION
N2
N1
N21N11
N12N22
Ru1
C2
C3
Cl1
P1
O1
C1
O2
Figure 15: Molecular structure of the dinitrogen complex [Ru(bdmpzpr)Cl(N2)(PPh3)] (10). Thermal ellipsoids
are drawn at the 50% probability level. Most hydrogen atoms and the solvent molecule have been omitted for
clarity.
Table 6: Selected bond lengths and angles for the ruthenium(II) dinitrogen complex 10.
Distances in Å Angles in °
d(Ru1−N11) 2.132(2) ∡(N11,Ru1,N21) 83.08(8)
d(Ru1−N21) 2.082(2) ∡(O1,Ru1,N11) 86.55(7)
d(Ru1−O1) 2.0511(17) ∡(O1,Ru1,N21) 86.44(7)
d(Ru1−P1) 2.3341(6) ∡(O1,Ru1,N1) 177.73(10)
d(Ru1−N1) 1.976(3) ∡(N21,Ru1,Cl1) 174.33(6)
d(Ru1−Cl1) 2.3787(7) ∡(N11,Ru1,P1) 173.49(6)
d(O1−C1) 1.291(3) ∡(P1,Ru1,N1) 95.11(9)
d(O2−C1) 1.220(3) ∡(P1,Ru1,Cl1) 87.05(2)
d(C1−C2) 1.580(3) ∡(N1,Ru1,Cl1) 89.28(9)
d(N1−N2) 0.791(5) ∡(Ru1,N1,N2) 172.3(5)
∡(O2,C1,C2,C3) -13.5(3)

III. RESULTS AND DISCUSSION 61
In order to synthesize the dinitrogen complex 10 methodically, [Ru(bdmpzpr)Cl(PPh3)2] (9)
was dissolved in THF. An instant color change from yellow to red indicated stabilization of
the 16 VE intermediate in solution by the formation of a THF solvent complex
[Ru(bdmpzpr)Cl(PPh3)(THF)]. After a few minutes of stirring under dinitrogen atmosphere
at room temperature, a yellow solid started to precipitate from the solution. The reaction
was monitored by IR spectroscopy. Indeed, the appearance of a strong band in the in situ IR
spectrum at ν ̃ = 2128 cm–1 evidenced the substitution of one phosphine ligand by a N2
molecule (figure 16a).
A control experiment with [Ru(bdmpza)Cl(PPh3)2] (8) instead of bisphosphine complex 9
but with alike conditions showed only traces of a N2 product in the IR spectrum (figure 16b).
This difference in reactivity is surprising since it is known that the sterically hindered
complex 8 is also able to release one of its two PPh3 ligands. Examples of substitution
reactions of 8 leading to carboxylato and 2-oxocarboxylato, carbene, vinylidene, and
allenylidene complexes have been reported earlier by the BURZLAFF group.[60-61, 215-216] It
seems that the labilizing effect of the introduced methyl group of the Hbdmpzpr ligand 4 is
the key factor for the reactivity of [Ru(bdmpzpr)Cl(PPh3)2] (9) towards dinitrogen. This
influence is remarkable especially since the methyl group is 4.654 Å remote from the
ruthenium center.
The dinitrogen complex 10 was isolated as a yellow powder by precipitation with diethyl
ether. NMR spectroscopic analysis revealed the formation of a single, unsymmetrical isomer,
verified by two sets of signals for the pyrazolyl units in the 1H and 13C NMR spectra.
Furthermore, a signal at δ = 38.3 ppm for the remaining PPh3 ligand can be found in the 31P
NMR spectrum of 10. According to the X-ray structure of complex 10, the N2 ligand is
positioned trans towards the carboxylate donor and is bound in an end-on manner (figure
15). In general, this end-on coordination includes a σ-donation from the N2 lone pair to the
metal center as well as a certain though small degree of π-backbonding from the metal to
the unoccupied π* orbitals of the N2 ligand.[217-218] The distance d(Ru–N1) [1.976(3) Å] is in
good agreement to the Ru−N1 distances found in similar N2 complexes (table 7).

62 III. RESULTS AND DISCUSSION
15001700190021002300
Wavenumber [cm-1
]
Ab
sorp
tio
n
b
a
Figure 16: Reaction control via in situ IR spectroscopy: a) [Ru(bdmpzpr)Cl(PPh3)2] (9) when stirred under
dinitrogen atmosphere for 3 h, b) control experiment with [Ru(bdmpza)Cl(PPh3)2] (8). Both measured in THF
solution.
It is known that in most cases the N−N bond length of end-on coordinated N2 is almost the
same as in free N2 (1.0975 Å). Extended Hückel molecular orbital (EHMO) calculations by
K. KIRCHNER et al. suggest that this is due the compensation of the σ-bond strengthening by
a simultaneous π-bond weakening.[214] The σ-bond strengthening is caused by a reduced
anti-bonding character of the σ* MO and the π-bond weakening is due to π-backbonding.
However, the molecular structure of the dinitrogen complex 10 exhibits a rather short
d(N1–N2) distance of 0.791(5) Å compared to similar complexes listed in table 7. This is
caused by a disorder between the N2 and the chlorido ligand Cl1. Due to this disorder, a
more detailed discussion of the molecular structure of 10 was not possible during this thesis.

III. RESULTS AND DISCUSSION 63
Table 7: Selected bond lengths [Å] and angles [°] and ν(N2) vibrations [cm−1] for complexes similar to 10.
Complex ν(N2) d(Ru−N1) d(N1−N2) ∡(Ru,N1,N2)
[Ru(bdmpzpr)(N2)(PPh3)] (10) 2128[a] 1.976(3) 0.791(5) 172.3(5)
[Ru(Cp)(N2)(dippae)][BAr'4][213] 2178[b]
[Ru(Cp)(N2)(dippe)][BAr'4][205] 2158[b] 1.961(3) 1.087(4) 175.8(4)
[Ru(Cp)(N2)(dippe)][BPh4][207] 2145[b]
[Ru(Cp)(N2)(PMeiPr2)(PPh3)][BAr'4][205] 2177[b]
[Ru(Cp)(N2)(PMeiPr2)2][BAr'4][205] 2164[b]
[Ru(Cp)(N2)(R,R-dippach)][BAr'4][213][c] 2186[b] 1.959(5)
1.912(6)
1.096(7)
1.074(9)
175.2(5)
173.6(6)
[Ru(Cp)(N2)(tmeda)][BAr'4][208] not given
[Ru(Cp*)(N2)(dippae)][BAr'4][213] 2201[b]
[Ru(Cp*)(N2)(dippe)][BPh4][207] 2120[b]
[Ru(Cp*)(N2)(dppe)][BAr'4][206] 2159[b]
[Ru(Cp*)(N2)(dppm)][BAr'4][206] 2166[b] 1.975(2) 1.083(4) 175.7(3)
[Ru(Cp*)(N2)(PEt3)2][BPh4][211] 2134[b]
[Ru(Cp*)(N2)(R,R-dippach)][BAr'4][213] 2142[b]
[Ru(Tp)(N2)(dippae)][BAr'4][210] 2157[b] 1.933(4) 0.999(8) 170.2(6)
[Ru(Tp)(N2)(dippe)][BPh4][209] 2165[b]
[Ru(Tp)(N2)(PEt3)2][BPh4][212] 2163[b] 1.91(2) 1.01(2) 166(3)
[Ru(Tp)(N2)(PMeiPr2)2][BPh4][212] 2159[b]
[Ru(Tp)(N2)(pn)][BPh4][214] 2182[b] 1.943(4) 1.097(5) 174.6(4)
[Ru(Tp)(N2)(R,R-dippach)][BAr'4][210] 2160[b]
[Ru(TpiPr)(N2)(dppe)][OTf][203] 2188[d] 1.923(4) 1.105(7) 175.6(4)
[a] THF solution, [b] nujol mull, [c] compound was crystallized with two different cations, [d] KBr pellet.
Abbreviations: dippae = 1,2-bis(diisopropylphosphinoamino)ethane, dippe = 1,2-bis(diisopropylphosphino)-
ethane, dppe = 1,2-bis(diphenylphosphino)ethane, dppm = 1,1-bis(diphenylphosphino)methane, pn = 2-
(diphenylphosphino)-N,N-dimethylethanamine, R,R-dippach = (R,R)-1,2-bis((diisopropyl-phosphino)amino)-
cyclohexane, tmeda = N,N,N',N'-tetramethylethylenediamine.

64 III. RESULTS AND DISCUSSION
3.1.5. Reaction of [Ru(bdmpzpr)Cl(PPh3)2] with CO and SO2
As shown in the last chapter, in the example of the dinitrogen complex 10, the introduced
methyl group in the Hbdmpzpr ligand 4 seems to be the crucial factor for the changed
reactivity of the bisphosphine complex [Ru(bdmpzpr)Cl(PPh3)2] (9) compared to
[Ru(bdmpza)Cl(PPh3)2] (8). To exploit further differences between both ligand systems,
substitution reactions with other small molecules, namely CO and SO2 have been examined.
In contrast to N2, the isoelectronic CO ligand is a much stronger σ-donor and
π-acceptor.[219-221] During the past decades, a large number of monocarbonyl complexes
[Ru(L)(CO)(PR3)] bearing Cp and Tp ligand systems L with different auxiliary phosphine
ligands PR3 was reported (table 9).[202, 222-235] The BURZLAFF group was recently able to
expand this research area for bis(pyrazol-1-yl)acetato ligand systems. It was shown that the
carbonyl ligand is able to displace one PPh3 ligand of the bisphosphine complex 8.[215] NMR
spectroscopic investigations and a successful X-ray structure of the resulting monocarbonyl
complex [Ru(bdmpza)Cl(CO)(PPh3)] (11) proved, that the reaction of 8 with carbon
monoxide leads to a single isomer in which the carbonyl ligand is bound trans to one
pyrazolyl donor (scheme 30).
Following the procedure described in this earlier work, CO gas was passed through a fresh
solution of [Ru(bdmpzpr)Cl(PPh3)2] (9) in THF. After one hour, the IR spectrum of the
reaction mixture showed two new bands at ν̃ = 1956 and 1941 cm–1. This indicated that in
contrast to the experiment with the Hbdmpza ligand 1, two structural isomers 12a and 12b
were formed during the reaction (scheme 30). The appearance of two isomers might be
explained by an enhanced dissociation and re-binding of a PPh3 ligand, similar to the
equilibrium shown in scheme 28. To verify this, the isolated yellow compound 12 was
analyzed by NMR spectroscopy. Indeed, the 1H and 13C NMR spectra of compound 12
exhibited eight signal sets for the pyrazole methyl groups as expected for a mixture of two
unsymmetrical isomers (1H NMR 12a: δ = 1.90, 2.53, 2.54, 2.69 ppm, 12b: δ = 1.72, 2.52, 2.55,
2.73 ppm; 13C NMR 12a: δ = 15.6, 16.2, 17.7, 17.8 ppm, 12b: δ = 14.4, 16.5, 17.6, 18.2 ppm).
Furthermore two signals at δ = 41.3 and 36.1 ppm were observed in the 31P NMR spectrum
for the remaining PPh3 ligands. The characteristic 13C NMR resonances for the CO ligands
were found at δ = 204.4 and 204.7 ppm, respectively. These NMR and IR spectroscopic
values agree well with the ones reported earlier for similar monocarbonyl complexes of
type [Ru(L)(CO)(PR3)] (table 9).[202, 222-235]
III. RESULTS AND DISCUSSION 65
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
R
R = H (8), Me (9)
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (11), SO2 (13)
CO or SO2R =Me
CO or SO2
R = H
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (12a), SO2 (14a)
N N
N N
Me
Me
Me
Me
OO
Ru
LPh3P Cl
Me
L = CO (12b), SO2 (14b)
+
Me
Scheme 30: Reaction of bisphosphine complexes 8 or 9 with CO or SO2.
The integration of the 1H NMR signal sets shows that the structural isomers of complex
[Ru(bdmpzpr)Cl(CO)(PPh3)] (12) were formed in a ratio of 7:3 during the reaction. In order
to clarify the structural properties of the major and minor isomer, the 1H NMR spectrum of
the isolated mixture of isomers was compared with the reference spectrum of the bdmpza
complex 11. Since the signals of the predominantly formed species and 11 match very well,
it can be assumed that in case of the major regioisomer (12a), the CO ligand is bound trans
towards the pyrazolyl donor. Hence the species marked as 12b in scheme 30 has to be the
minor isomer in which the CO ligand is bound trans towards the carboxylate group. A
single crystal of isomer 12a suitable for X-ray structure determination (figure 17 and table 8)
was obtained by slow diffusion of diethyl ether into a CH2Cl2 solution of isomeric mixture
of complex 12 and confirmed these considerations. Complex 12a crystallized in the triclinic
space group 1P . The overall octahedral structure of 12a is very similar to that of the
analogous bdmpza complex 11. The metal-carbonyl distance d(Ru1−C4) [1.836(6) Å] as well
as the distances Ru−N11, Ru−N21 and Ru−O1 are in good agreement with those reported for
similar complexes as shown in table 9.[202, 215, 222-235] On the other hand, the distance d(C4–
O4) [1.139(10) Å] in complex 12a was found to be about 0.02 Å shorter than the

66 III. RESULTS AND DISCUSSION
corresponding distance found for the bdmpza complex [Ru(bdmpza)Cl(CO)(PPh3)] (11).[215]
This effect is most likely caused by the disordered structure.
Cl1
N11 N21
N22N12
Ru1
C2
C4
C3
P1
O4
C1
O1
O2
Figure 17: Molecular structure of carbonyl complex [Ru(bdmpzpr)Cl(CO)(PPh3)] (12a). Thermal ellipsoids are
drawn at the 50% probability level. Most hydrogen atoms and the solvent molecule have been omitted.
Table 8: Selected bond lengths and angles for the ruthenium(II) monocarbonyl complex 12a.
Distances in Å Angles in °
d(Ru1−N11) 2.1503(15) ∡(N11,Ru1,N21) 81.81(6)
d(Ru1−N21) 2.1413(15) ∡(O1,Ru1,N11) 85.06(5)
d(Ru1−O1) 2.0710(12) ∡(O1,Ru1,N21) 85.51(5)
d(Ru1−P1) 2.3388(5) ∡(O1,Ru1,Cl1) 174.81(4)
d(Ru1−C4) 1.836(6) ∡(N11,Ru1,C4) 175.94(13)
d(Ru1−Cl1) 2.4049(12) ∡(N21,Ru1,P1) 173.39(4)
d(O1−C1) 1.285(2) ∡(P1,Ru1,C4) 86.09(12)
d(O2−C1) 1.221(2) ∡(P1,Ru1,Cl1) 96.28(2)
d(C1−C2) 1.588(2) ∡(C4,Ru1,Cl1) 88.22(12)
d(C4−O4) 1.139(10) ∡(Ru1,C4,O4) 176.6(4)
∡(O2,C1,C2,C3) -12.6(2)
III. RESULTS AND DISCUSSION 67
Table 9: Selected bond lengths [Å] and angles [°], ν(CO) vibrations [cm−1] and 13C NMR resonances [ppm] for
ruthenium monocarbonyl complexes similar to [Ru(bdmpzpr)Cl(CO)(PPh3)] (12).
Complex
ν(CO)
13C(CO)
d(Ru1–C4)
d(C4–O4)
∡(Ru1,C4,O4)
[Ru(bdmpzpr)Cl(CO)(PPh3)] (12a)
1966[a]
204.7
1.836(6)
1.139(10)
176.6(4)
[Ru(bdmpza)Cl(CO)(PPh3)][215] (11) 1969[b] 202.6 1.821(5)
1.151(6)
178.0(4)
[Ru(Cp)Cl(CO)(dppm)][222] 1970[d]
[Ru(Cp)Cl(CO)(PBu3)][229] 1900[e] 203.7
[Ru(Cp)Cl(CO)(Ph2PMe)][223] 1956[b] 200.8
[Ru(Cp)Cl(CO)(PPh3)][225, 235] 1958[d]
1959[b]
1.872(6)
1.132(8)
178.3(8)
[Ru(Cp*)Cl(CO)(Cy2PCH2CH2OMe)][228] 1920[f] 207.9
[Ru(Cp*)Cl(CO)(iPr2PPh)][227] 1925[c] 208.9
[Ru(Cp*)Cl(CO)(PCy3)][202] 1910[d]
[Ru(Cp*)Cl(CO)(Ph2PCH2CH2NMe2)][230] 1912 207.1
[Ru(Cp*)Cl(CO)(PiPr3)][202] 1910[d]
[Ru(Cp*)Cl(CO)(PPh3)][226] 1918[d]
[Ru(Tp)Cl(CO)(Ph2PCH2CH2OMe)][231] 203.7 1.868(2)
1.099(2)
176.9(3)
[Ru(Tp)Cl(CO)(Ph2PiPr)][232] 205.7 1.863(4)
1.012(4)
174.5(2)
[Ru(Tp)Cl(CO)(PPh3)][233-234] 1965[d] 203.5 1.872(6)
1.132(8)
178.3(8)
[Ru(Tp)Cl(CO)(PTA)][224][g] 1963[f] 201.8
[a] THF solution, [b] CH2Cl2 solution, [c] toluene-D8 solution, [d] nujol mull, [e] hexane solution, [f] KBr
pellet, [g] PTA = 1,3,5-triaza-7-phosphaadamantane.

68 III. RESULTS AND DISCUSSION
Similar to the CO ligand, sulfur dioxide has also good σ-donating and π-accepting abilities.
It is known that SO2 can bind towards metal centers in many different coordination
modes.[236-239] Typical examples are shown in figure 18. A κ1S-coordination via the sulfur
atom is possible in a coplanar or a pyramidal geometry. In planar κ1S complexes, the sulfur
atom acts as a Lewis base, binding via its lone pair towards the metal center. This σ-bond is
strengthened by additional π-backbonding, where electron density is withdrawn from the
metal center into the empty π* orbital (LUMO) of the SO2 ligand. On the other hand, SO2
can also act as a Lewis acid in pyramidal κ1S coordinated complexes. In this case, the
bonding electron pair is formally provided by a nucleophile metal center. Besides, SO2 can
also coordinate in a κ2S,O mode via a sulfur and a oxygen atom. But it is also possible that
SO2 acts as a bridging ligand or coordinates only with the oxygen atom in a κ1O mode.
Sulfur dioxide complexes of ruthenium bearing Cp ligands are rather rare in literature.
There have been some reports on cationic complexes by W. A. SCHENK et al., J. ELLERMANN
et al. and E. LINDNER et al. (table 10).[240-245] Recently, a molecular structure of the SO2
complex [Ru(bdmpza)(O2CPh)(PPh3)(SO2)] bearing the Hbdmpza ligand 1 was published by
the BURZLAFF group.[61, 216] A rather unusual intramolecular Lewis acid-base interaction was
found between the sulfur atom of the SO2 ligand and the uncoordinated oxygen atom of the
benzoylformato ligand. Furthermore the κ1S bound SO2 ligand was not exactly planar but
slightly distorted in this complex.
Figure 18: Coordination modes of the SO2 ligand towards metal centers: a) κ1S planar, b) κ1
S pyramidal,
c) κ2S,O, d) κ1
O. e) Shape of the HOMO, f) shape of the LUMO orbitals.[236-239]

III. RESULTS AND DISCUSSION 69
Following the procedures of the CO experiments described above, sulfur dioxide was passed
through solutions of complexes [Ru(bdmpza)Cl(PPh3)2] (8) and [Ru(bdmpzpr)Cl(PPh3)2] (9)
respectively (scheme 30). In case of the Hbdmpza ligand 1, this leads to the formation of a
single isomer of the SO2 complex [Ru(bdmpza)Cl(PPh3)(SO2)] (13). Due to the
unsymmetrical nature of this complex, the 1H and 13C NMR spectra exhibit each two sets of
signals for the pyrazolyl residues. The 31P NMR resonance of the remaining PPh3 ligand was
found at δ = 28.8 ppm. The signal for the asymmetric carboxylate vibration shows up at
ν ̃ = 1676 cm−1 in the IR spectrum of complex 13. The two additional bands at ν̃ = 1294 and
1123 cm−1 for the asymmetric and symmetric SO2 vibrations are in good accordance to
similar complexes with a coplanar κ1S bound SO2 ligand (table 10). Usually these values lie
in a range of 1254 – 1296 cm–1 and 1096 – 1128 cm–1 respectively.[61, 216, 240-245]
The result of a single crystal structure analysis of complex 13 is depicted in figure 19. The
bdmpza ligand is facially coordinated in a κ3-N,N,O coordination mode and the SO2 ligand
is bound in trans position to one of the pyrazolyl donors in accordance to the monocarbonyl
complex 11. The Ru1−N11, Ru1−N12 and Ru1−O1 distances are in a similar range of those
found for the monocarbonyl complex 11 (table 11). The bond distance d(Ru1−S1) =
2.1955(15) Å is slightly longer compared to those reported for analogous complexes.
Furthermore, it was found that the S1-O3 distance is about 0.1 Å smaller than reported for
the bdmpza complex [Ru(bdmpza)(O2CPh)(PPh3)(SO2)][61, 216] or the cationic complex
[Ru(Cp)(chir)(SO2)]+[242] [chir = (S,S)-2,3-bis(diphenylphosphino)butane], respectively. It is
also noteworthy that in contrast to the molecular structure of
[Ru(bdmpza)(O2CPh)(PPh3)(SO2)], the SO2 ligand is bound almost perfectly coplanar in
complex 13. This is evidenced by a small dihedral angle ∡(Ru1,O3,O4,S1) of 3.26° and a total
sum of the bond angles around the sulfur atom of 359.8°.

70 III. RESULTS AND DISCUSSION
Table 10: Selected bond lengths [Å], angles [°] and ν(SO2) vibrations [cm−1] for complexes similar to 13 and 14.
Complex ν(SO2) d(Ru–S) d(S–O) ∡(Ru,S,O) ∡(O,S,O)
[Ru(bdmpza)Cl(PPh3)(SO2)] (13) 1294
1123[a]
2.1955(15) 1.370(6)
1.414(6)
123.2(3)
115.9(3)
120.7(4)
[Ru(bdmpzpr)Cl(PPh3)(SO2)] (14) 1291
1125[a]
[Ru(bdmpza)(O2CMe)(PPh3)(SO2)] [61, 216]
1282
1128[a]
[Ru(bdmpza)(O2CPh)(PPh3)(SO2)] [61, 216]
1283
1125[a]
2.182(2) 1.452(5)
1.456(5)
118.1(2)
124.0(2)
114.2(3)
[Ru(Cp)(PPh3)2(SO2)]PF6[244] 1294
1118[b]
[Ru(Cp)(PMe3)2(SO2)]PF6[244] 1268
1118[b]
[Ru(Cp)(dppe)(SO2)]PF6[244] 1296
1117[b]
[Ru(Cp)(dppm)(SO2)]PF6[244] 1287
1121[b]
[Ru(Cp)(chir)(SO2)]PF6[242] 1296
1118[b]
2.128(2) 1.432(6)
1.458(6)
120.9(3)
125.1(3)
113.9(4)
[Ru(Cp)(dppa)(SO2)]Cl[240] 1289
1115[a]
[Ru(Cp*)(PMe3)2(SO2)]PF6[243-244] 1262
1104[b]
[Ru(Cp*)(PPh3)2(SO2)]PF6[244] 1277
1110[b]
[Ru(Cp*)(dppe)(SO2)]PF6[244] 1273
1106[b]
[Ru(Cp*)(dmpm)(SO2)]PF6[245] 1254/1096[b]
[Ru(Cp*)(chir)(SO2)]PF6[245] 1269/1108[b]
[Ru(Cp*)(P~O)(SO)2]BPh4[241] 1277/1109[a]
[a] KBr pellet, [b] nujol mull. Abbreviations: chir = (S,S)-2,3-bis(diphenylphosphino)butane, dmpm =
bis(dimethylphosphino)methane, dppa = N,N-Bis(diphenylphosphino)amine, P~O = κ1P coordinated (1,3-
dioxan-2-ylmethyl)diphenylphosphine.

III. RESULTS AND DISCUSSION 71
Cl1
N21
N12 N22
Ru1
C1
O3
S1
P1
O4
C2
O1
O2
Figure 19: Molecular structure of the SO2 complex [Ru(bdmpza)Cl(PPh3)(SO2)] (13). Thermal ellipsoids are
drawn at the 30% probability level. Most hydrogen atoms as well as the solvent molecule have been omitted
for clarity.
Table 11: Selected bond lengths and angles for the ruthenium(II) SO2 complex 13.
Distances in Å Angles in °
d(Ru1−N11) 2.112(4) ∡(N11,Ru1,N21) 83.15(14)
d(Ru1−N21) 2.154(4) ∡(O1,Ru1,Cl1) 176.79(9)
d(Ru1−O1) 2.065(3) ∡(N11,Ru1,S1) 174.53(10)
d(Ru1−P1) 2.3566(11) ∡(N21,Ru1,P1) 174.11(10)
d(Ru1−S1) 2.1955(15) ∡(P1,Ru1,S1) 88.98(5)
d(Ru1−Cl1) 2.3528(12) ∡(P1,Ru1,Cl1) 96.33(4)
d(O1−C1) 1.280(6) ∡(S1,Ru1,Cl1) 90.65(5)
d(O2−C1) 1.227(5) ∡(Ru1,S1,O3) 123.2(3)
d(C1−C2) 1.549(6) ∡(Ru1,S1,O4) 115.9(3)
d(S1−O3) 1.370(6) ∡(O3,S1,O4) 120.7(4)
d(S1−O4) 1.414(6) ∡(Ru1,O3,O4,S1) 3.26
∡(O2,C1,C2,H2) 12.40

72 III. RESULTS AND DISCUSSION
In agreement to the CO experiments, reaction of [Ru(bdmpzpr)Cl(PPh3)2] (9) with SO2 once
again resulted a mixture of two structural isomers 14a and 14b of the SO2 complex
[Ru(bdmpzpr)Cl(PPh3)(SO2)] (14). This was on the one hand evidenced by the appearance of
four 1H NMR and four 13C NMR signal sets for each of the pyrazolyl groups. On the other
hand the 31P NMR of the isolated mixture of complex 14 revealed two resonances at δ = 26.6
and 27.1 ppm for the PPh3 ligands. This time, both isomers 14a and 14a were formed in the
same quantity as the comparison of the 1H NMR integrals shows. The asymmetric
carboxylate vibration at ν ̃ = 1675 cm−1 in the IR spectrum of complex 14 is almost identical
to the one found for the bdmpza complex 13. Once again, the asymmetrical and
symmetrical SO2 vibrations at ν ̃ = 1291 and 1125 cm−1 are in good accordance to those
reported for similar complexes listed in table 10. Attempts to obtain single crystals suitable
for X-ray structure analysis have not been successful so far.
3.1.6. Conclusion
The investigations in this chapter have shown that substituents at the bridging position of
functionalized bis(pyrazol-1-yl)acetato ligands affect the remaining space at the κ3-N,N,O
bound complex fragment. These substituents can cause chirality and the appearance of
enantiomers in solution which complicates their characterization by NMR spectroscopy.
They might as well compete for a coordination towards the metal center which might be
the major reason for the fact that bulky ruthenium(II) complexes with bridge functionalized
ligands have not been observed so far. By means of the model ligand Hbdmpzpr (4) it was
shown that steric interactions of the linker group at the bridging position and the pyrazole
methyl substituents at position 5 are responsible for a more or less pronounced "twist" of
one pyrazolyl unit depending on the bulkiness of the whole system. This twist is the key
factor for the changed reactivity of complexes with bridge functionalized ligands compared
to those of unmodified bis(pyrazol-1-yl)acetic acids. Prolonged reaction times and changed
geometries are the result. This can also lead to coordinatively unsaturated species by
enforced dissociation of a co-ligand as shown for the bulky bisphosphine complex
[Ru(bdmpzpr)Cl(PPh3)2] (9). On the other hand, this opens the door for species with free
coordination sites which might be able to bind and activate small molecules in order to
fulfill their 18 VE configuration. The synthesized dinitrogen complex
[Ru(bdmpzpr)Cl(N2)(PPh3)] (10) is the first example of a N2 complex bearing a bis(pyrazol-
1-yl)acetato ligand and serves as a proof of concept for these considerations. Other

III. RESULTS AND DISCUSSION 73
experiments with CO and SO2 indicated that bridge functionalized ligands favor the
formation of structural isomers.

74 III. RESULTS AND DISCUSSION
3.2. A new approach: Polymerizable linkers at the pyrazolyl units
One major task of this thesis was to develop a new approach for the functionalization of
pyrazole based ligands considering the observations made so far with bridge functionalized
ligands (chapter 3.1). As mentioned before, it was not possible to synthesize bulky
ruthenium complexes with a κ3-N,N,O coordinated bis(pyrazol-1-yl)acetato ligand bearing a
solid phase linker or polymerization active group at the bridging carbon atom. However,
such model complexes suitable for copolymerization would allow further design of enzyme
mimics and might open the door to molecular imprinting techniques. Thus, the stategy
during this thesis was to generate a bis(pyrazol-1-yl)acetic acid, in which the linker suitable
for copolymerization is no longer located at the bridging position but is attached directly to
the pyrazole rings. A perfect candidate for this approach was considered 2,2-bis(3,5-
dimethyl-4-vinylpyrazol-1-yl)acetic acid (Hbdmvpza) (18) with two vinyl "arms" at in
position 4 of the pyrazolyl groups.
3.2.1. Synthesis of 2,2-bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid
There have been attempts earlier in the BURZLAFF group to synthesize the targeted 4-vinyl
substituted ligand 18 by palladium mediated SUZUKI-MIYAURA coupling of 2,2-bis(4-bromo-
3,5-dimethylpyrazol-1-yl)acetic acid with vinylboronate esters.[246] Unfortunately these
experiments did not lead to satisfying results so far and a retrosynthetic approach of this
problem was necessary. Scissoring of the desired 4-vinyl substituted fragment III in a way
shown in scheme 31 gives 3,5-dimethylpyrazole as a possible synthon (S). Thus, the first
step requires the introduction of a proton-protecting group R in order to generate the
formylated species II by VILSMEIER reaction.
N
N
Me
Me
RN
N
Me
Me
ORN
N
Me
Me
R
HHN
N
Me
Me
H
III SIII
Scheme 31: Retrosynthetic scissoring of the desired 4-vinyl substituted target molecule III.

III. RESULTS AND DISCUSSION 75
During this thesis, an optimized strategy for the synthesis of Hbdmvpza (18) was
successfully developed as shown in scheme 32. In order to bypass the protecting group issue,
two molecules of 3,5-dimethylpyrazole have been connected to form bis(3,5-
dimethylpyrazol-1-yl)methane (15) as an actual starting molecule. This compound was
synthesized by a procedure developed by J. ELGUERO and coworkers using BTEAC as phase
transfer catalyst.[247]
NH
N
Me
Me
2
KOH
K2CO3BTEAC
CH2Cl2
N
N N
N
Me
Me
Me
Me
N
N N
N
Me
Me
Me
Me
O
H
O
H
1. POCl3DMF
2. H2O
15 16
H2C=PPh3
N
N N
N
Me
Me
Me
Me17
N
N N
N
Me
Me
Me
Me
CO2H 1. n-BuLi2. CO2
3. H+
18
Scheme 32: Multi-step synthesis of 4-vinyl substituted Hbdmvpza ligand 18.
In the next step, bis(3,5-dimethyl-4-formylpyrazol-1-yl)methane (16) was generated by a
VILSMEIER reaction according to an procedure recently reported by A. S. POTAPOV, A. I.
KHLEBNIKOV and V. D. OGORODNIKOV.[248] For this purpose, phosphoryl chloride was added
dropwise to a solution of bis(3,5-dimethylpyrazole)methane in dimethylformamide. After
hydrolyzing the mixture with cold water, the precipitated bisaldehyde was collected by
filtration. A singulet 1H NMR resonance at δ = 9.90 ppm can be assigned to the aldehyde
protons and proves the formation of bisaldehyde 16. The dried compound was converted to
the vinyl substituted product bis(3,5-dimethyl-4-vinylpyrazol-1-yl)methane (17) via a
WITTIG reaction. The necessary ylide Ph3P=CH2 was generated in situ by treating a
suspension of methylenetriphenylphosphonium bromide in THF with KOtBu for 1 h at
room temperature. Reaction of the ylide with bisaldehyde 16 yielded the vinyl substituted
product 17 which was purified by column chromatography on silica gel with
n-pentane : ethyl acetate 7 : 3 as eluent. Successful formation of compound 17 was verified

76 III. RESULTS AND DISCUSSION
by NMR spectroscopy. An AMX resonance system[249] was observed for the vinyl group in
the 1H NMR spectrum of compound 17 with three sets of double doublet signals at δ = 5.13
and 5.28 ppm for the terminal protons of the vinyl groups and at δ = 6.46 ppm for the
remaining =CH– proton. The coupling constants of the vinyl groups have been determined
to 1.4 Hz for the geminal 2J coupling, 17.7 Hz for the 3
J (E ) and 11.4 Hz for the 3J (Z )
coupling (figure 20). Furthermore, one set of signals for the pyrazolyl groups was observed
in the 1H as well as in the 13C NMR spectrum of compound 17. This is consistent with CS
symmetry of the molecule in solution.
3J (Z ) = 11.4 Hz
3J (E ) = 17.7 Hz 2J = 1.4 Hz
3J (E ) 3J (Z )
6.6 6.5 6.4 6.3 5.3 5.2 5.1 ppm
Figure 20: AMX resonance system observed in the 1H NMR spectrum of 17.
In the final step of the ligand synthesis, the carboxylate group was introduced at the
bridging carbon atom. Therefore compound 17 was deprotonated with n-BuLi at –80 °C and
subsequently treated with CO2. After aqueous work up, the targeted ligand Hbdmvpza (18)
can be isolated by extraction from the acidified (pH = 2) aqueous phase. NMR spectroscopic
data of ligand 18 are almost identical to those its precursor compound 18 except for an
additional 13C resonance at δ = 165.1 ppm of the introduced carboxylate group. An
assignment of the carbon resonances was possible with a heteronuclear multiple quantum
coherence (HMQC) experiment.
3.2.2. Transition metal complexes of Hbdmvpza
In order to prove the tripodal κ3-N,N,O coordination behaviour of the Hbdmvpza ligand 18,
tricarbonyl complexes of manganese and rhenium were synthesized and investigated first
by deprotonation of ligand 18 and treatment with the precursors [MBr(CO)5] (scheme 33).
Once again, the successful formation of the desired complexes [Mn(bdmvpza)(CO)3] (19)

III. RESULTS AND DISCUSSION 77
and [Re(bdmvpza)(CO)3] (20) was verified by the appearance of a single A' and two close
A'' and A' carbonyl signals (19: ν̃ = 2036, 1944 and 1917 cm−1; 20: 2026, 1921 and 1898 cm−1)
which is typical for unsymmetrical "piano stool" type carbonyl complexes.
N
N N
N
Me
Me
Me
Me
CO2H
18
1. KOtBu, THF2. [MBr(CO)5]
N N
N N
Me
Me
Me
Me
OO
M
CC CO
OO
M = Mn (19), Re (20)1. KOtBu, THF2. [RuCl2(PPh3)3]
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
21
MeCN
- PPh3
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P NCMe
22
Scheme 33: Synthesis of transition metal complexes [Mn(bdmvpza)(CO)3] (19), [Re(bdmvpza)(CO)3] (20),
[Ru(bdmpza)Cl(PPh3)2] (21) and [Ru(bdmpza)Cl(MeCN)(PPh3)] (22).
Complexes 19 and 20 were also investigated by NMR spectroscopy. In both cases, the 13C
NMR spectrum revealed of a clear set of signals for the coordinated bdmvpza ligand 18. The 13C NMR signal for the carboxylate group was found at δ = 165.5 ppm for the manganese
complex 19 and at δ = 163.8 ppm in case of the rhenium complex 20. Furthermore, two 13C
resonances for the carbonyl ligands were observed for each compound (19: δ = 223.1 and
219.9 ppm; 20: δ = 196.0 and 194.9 ppm). The 1H NMR spectrum of the manganese
compound 19 exhibited rather broad signals, due to fast relaxation caused by the
quadrupole moment of 55Mn. Thus, it was not possible to assign any couplings in this case.
On the other hand, the 1H NMR spectrum of the rhenium complex 20 presented sharp
signals with an AMX type resonance system as to expect. Finally, a single crystal X-ray
structure of the manganese complex 19 doubtlessly reveales the κ3-N,N,O coordination
mode of the new 4-vinyl substituted ligand 18 (figure 21 and table 12). Suitable crystals

78 III. RESULTS AND DISCUSSION
have been obtained by layering a dichloromethane solution of 19 with diethyl ether. The
asymmetric unit of the cell contains two molecules of complex 19 of which one was slightly
disordered. The distances and angles of the molecular structure agree well with those of
other manganese tricarbonyl complexes bearing bis(pyrazol-1-yl)acetato ligands (table
12).[55, 97, 117] The distances d(Mn1–C33) and d(Mn1–C35) are slightly longer than d(Mn1–
C34) probably due to the π-acceptor properties of the pyrazolyl donors and the resulting
trans influence.
O34
C34
N41
N42
N51
N52
C31
C35
C33
O35O33
C32
O31
O32
Mn1
Figure 21: Molecular structure of [Mn(bdmvpza)(CO)3] (19). Thermal ellipsoids are drawn at the 50%
probability level. Depicted is only one of the two molecules in the asymmetric unit, which is not disordered.
Most hydrogen atoms have been omitted for clarity.
Table 12: Selected bond lenghts and angles for the molecular structure of the manganese(II) complex 19.
Distances in Å Angles in °
Mn1–N41 2.604(2) ∡(N41,Mn1,N51) 86.12(9)
Mn1–N51 2.066(2) ∡(N41,Mn1,O31) 84.27(8)
Mn1–O31 2.048(2) ∡(N51,Mn1,O31) 85.48(8)
Mn1–C33 1.812(3) ∡(C33,Mn1,C34) 91.55(15)
Mn1–C34 1.800(3) ∡(C33,Mn1,C35) 86.10(15)
Mn1–C35 1.813(3) ∡(C34,Mn1,C35) 91.22(15)

III. RESULTS AND DISCUSSION 79
In the next step the reactivity of ligand 18 for bulky ruthenium(II) model complexes was
tested. In contrast to bridge functionalized bis(pyrazol-1-yl)acetate derivatives, ligand 18
should be able to bind ruthenium facially without any interference of the vinyl groups with
the metal center. Indeed, the bulky bisphosphine ruthenium complex
[Ru(bdmvpza)Cl(PPh3)2] (21) was successfully synthesized by deprotonation of the
Hbdmvpza ligand 18 followed by a reaction with the ruthenium(II) precursor [RuCl2(PPh3)3]
(scheme 33). Formation of bisphosphine complex 21 was verified by NMR spectroscopy. A
single signal in the 31P NMR spectrum at δ = 35.1 ppm as well as one set of signals for the
pyrazolyl groups in the 1H and 13C NMR spectra indicated a symmetric geometry for
complex 21, with the remaining chlorido ligand in trans position to the carboxylate
functionality. Moreover, the successful formation of bisphosphine complex 21 was verified
by elemental analysis and the appearance of a band for the asymmetric carboxylate
vibration in the IR spectrum of complex 21 at ν̃ = 1669 cm−1 which is in good agreement
with the very similar bisphosphine complex [Ru(bdmpza)Cl(PPh3)2] (8) (ν̃ = 1672 cm−1).[59]
On an attempt to crystallize the bisphosphine complex 21 in presence of traces of
acetonitrile a few crystals of the substituted complex [Ru(bdmvpza)Cl(MeCN)(PPh3)] (22)
have been recovered. This is not surprising since similar ruthenium(II) acetonitrile
complexes are already known in literature. Examples such as [Ru(Tp)H(MeCN)(PPh3)] or
[Ru(Tp)Cl(MeCN)(PPh3)] have been reported by C. P. LAU et al. earlier.[250-253] Furthermore,
in a recent work of the BURZLAFF group the analogous complex
[Ru(bdmpza)Cl(MeCN)(PPh3)] was synthesized and investigated.[61] The single crystal
structure analysis (figure 22) of the acetonitrile complex 22 exhibits the κ3-N,N,O
coordination of the Hbdmvpza ligand 18. The distances and angles agree well with those
found for the [Ru(bdmpza)Cl(MeCN)(PPh3)] or [Ru(bdmpza)Cl(PPh3)2] (8).[59, 61]
Furthermore, the structure of 22 reveals that the chlorido ligand is positioned trans to the
carboxylate donor group. This is in contrast to the molecular structure reported for
[Ru(bdmpza)Cl(MeCN)(PPh3)], in which a trans position of the chlorido ligand to one of the
pyrazolyl donors was found.

80 III. RESULTS AND DISCUSSION
Cl1
N11 N21
N12 N22
Ru1
C1
N3
P1C3
C4
O1
C2O2
Figure 22: Molecular structure of [Ru(bdmvpza)Cl(MeCN)(PPh3)] (22). Thermal ellipsoids are drawn at the
50°% probability level. Most hydrogen atoms have been omitted for clarity.
Table 13: Selected bond lenghts and angles for the molecular structure of ruthenium(II) complex 22.
Distances in Å Angles in °
d(Ru1−N11) 2.0903(19) ∡(N11,Ru1,N21) 84.03(7)
d(Ru1−N21) 2.1599(19) ∡(O1,Ru1,N11) 89.53(7)
d(Ru1−O1) 2.0818(14) ∡(O1,Ru1,N21) 85.66(6)
d(Ru1−P1) 2.3018(6) ∡(O1,Ru1,Cl1) 175.80(4)
d(Ru1−Cl1) 2.4050(6) ∡(N11,Ru1,N3) 86.55(7)
d(C1−O1) 1.273(3) ∡(N21,Ru1,P1) 170.26(5)
d(O2−C1) 1.226(3) ∡(P1,Ru1,N3) 93.18(6)
d(C1−C2) 1.547(3) ∡(P1,Ru1,Cl1) 99.19(2)
d(Ru1−N3) 2.012(2) ∡(N3,Ru1,Cl1) 89.38(6)
d(N3−C3) 1.140(3) ∡(Ru1,N3,C3) 169.38(19)
d(C3−C4) 1.459(3) ∡(N3,C3,C4) 177.1(3)

III. RESULTS AND DISCUSSION 81
Complex 22 is also accessible methodically by stirring [Ru(bdmvpza)Cl(PPh3)2] (21) in
acetonitrile (scheme 33). Two sets of signals were observed for the pyrazolyl donors in the 1H and 13C NMR spectrum of complex 22 in agreement to the unsymmetrical geometry of
the molecule. The 13C resonances at δ = 4.0 and 124.5 ppm can be assigned to the
acetonitrile ligand. The 31P resonance for the remaining phosphine ligand was observed at
δ = 49.7 ppm. The IR spectrum of complex 22 exhibited a strong signal for the asymmetric
carboxylate vibration at ν ̃ = 1655 cm−1 as well as a weak signal at ν̃ = 2269 cm−1 which can
be assigned to the C≡N vibration of the coordinated acetonitrile ligand. These NMR and IR
spectroscopic values are in good agreement to the analogous complex
[Ru(bdmpza)Cl(MeCN)(PPh3)].[61]
3.2.3. Polymerization behavior of Hbdmvpza
The heteroscorpionate N,N,O ligand 18 as well as the neutral N,N chelating ligand 17 have
been studied regarding their properties as potential polymer building blocks. It is
noteworthy, that both compounds 17 and 18 as well as their transition metal complexes can
act as crosslinking agents due to the presence of two polymerization active vinyl groups per
molecule. This is another advantage compared to bridge functionalized bis(pyrazol-1-
yl)acetate ligands such as Hbdmpzmp, where the synthesis of highly crosslinked networks
of polymer-ligand hybrid material was only possible by addition of crosslinking monomers
such as ethylene glycol dimethacrylate (EGDMA). Consequently, the Hbdmvpza ligand 18
can also be polymerized with itself forming a unique homopolymer (P18c). The
copolymerization experiments of 17 and 18 with methyl methacrylate (MMA) or EGDMA
as well as the formation of the homopolymer are summerized in scheme 34. In all
polymerization reactions 2,2'-azobis(2-methylproprionitrile) (AIBN) was used as radical
starter.
In case of the copolymerization of the Hbdmvpza ligand 18 with MMA, two different
products can be isolated. A white precipitate P18a is formed during the reaction in xylene
and was separated by filtration. Furthermore, when pouring the remaining filtrate into
methanol, a second polymer P18b starts to precipitate. This observation might be explained
by a smaller reactivity ratio of MMA compared to the crosslinking ligand 18.

82 III. RESULTS AND DISCUSSION
N
N N
N
Me
Me
Me
Me
CO2H
18
N
N N
N
Me
Me
Me
Me
CO2H
P18a, P18b: MMA-copolymerP18c: EGDMA-copolymerP18d: Homopolymer
a, b: AIBN, MMA
c: AIBN, EGDMA ord: AIBN
Xylene, 80 °C
N
N N
N
Me
Me
Me
Me17
N
N N
N
Me
Me
Me
Me
P17: MMA-copolymer
AIBN, MMA
Xylene, 80 °C
Scheme 34: Copolymerization of 17 and 18 with MMA or EGDMA and formation of the homopolymer P18c.
Presumably highly reactive crosslinking ligand molecules 18 will tend to react preferentially
with each other at the beginning of the polymerization, forming a star like polymer[254] with
a highly crosslinked core. As the reaction proceeds, the ratio of MMA and crosslinking
ligand 18 lies in a range where a high molecular weight but soluble polymer P18b is formed.
To corroborate this theory, the incorporation rate of both polymers P18a and P18b was
determined after washing with appropriate solvents and drying. The amount of
incorporation can be determined by the % N value of the elemental analysis of the resulting
polymer according to the following equation:
10mol14g4
%N
Polymer g
Ligand mmol1
⋅⋅⋅
=−
Equation 2: Calculation of the amount of incorporation by th % N value of the elemental analysis.
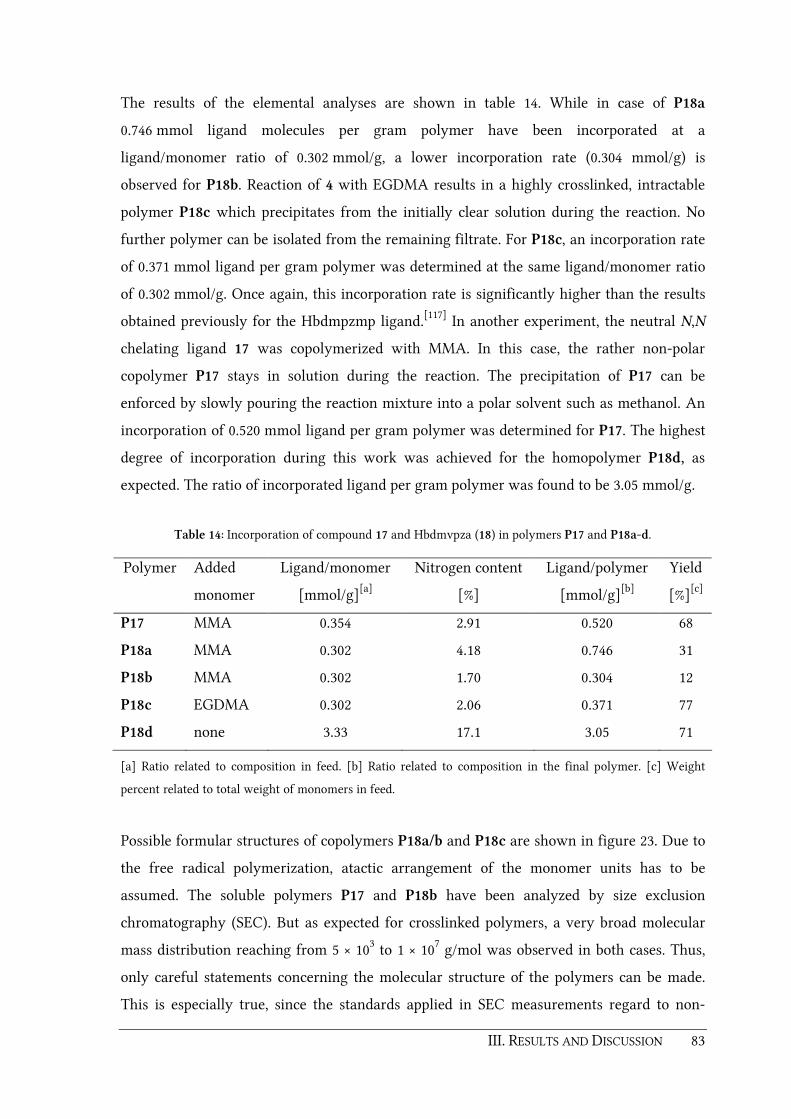
III. RESULTS AND DISCUSSION 83
The results of the elemental analyses are shown in table 14. While in case of P18a
0.746 mmol ligand molecules per gram polymer have been incorporated at a
ligand/monomer ratio of 0.302 mmol/g, a lower incorporation rate (0.304 mmol/g) is
observed for P18b. Reaction of 4 with EGDMA results in a highly crosslinked, intractable
polymer P18c which precipitates from the initially clear solution during the reaction. No
further polymer can be isolated from the remaining filtrate. For P18c, an incorporation rate
of 0.371 mmol ligand per gram polymer was determined at the same ligand/monomer ratio
of 0.302 mmol/g. Once again, this incorporation rate is significantly higher than the results
obtained previously for the Hbdmpzmp ligand.[117] In another experiment, the neutral N,N
chelating ligand 17 was copolymerized with MMA. In this case, the rather non-polar
copolymer P17 stays in solution during the reaction. The precipitation of P17 can be
enforced by slowly pouring the reaction mixture into a polar solvent such as methanol. An
incorporation of 0.520 mmol ligand per gram polymer was determined for P17. The highest
degree of incorporation during this work was achieved for the homopolymer P18d, as
expected. The ratio of incorporated ligand per gram polymer was found to be 3.05 mmol/g.
Table 14: Incorporation of compound 17 and Hbdmvpza (18) in polymers P17 and P18a-d.
Polymer Added
monomer
Ligand/monomer
[mmol/g][a]
Nitrogen content
[%]
Ligand/polymer
[mmol/g][b]
Yield
[%][c]
P17 MMA 0.354 2.91 0.520 68
P18a MMA 0.302 4.18 0.746 31
P18b MMA 0.302 1.70 0.304 12
P18c EGDMA 0.302 2.06 0.371 77
P18d none 3.33 17.1 3.05 71
[a] Ratio related to composition in feed. [b] Ratio related to composition in the final polymer. [c] Weight
percent related to total weight of monomers in feed.
Possible formular structures of copolymers P18a/b and P18c are shown in figure 23. Due to
the free radical polymerization, atactic arrangement of the monomer units has to be
assumed. The soluble polymers P17 and P18b have been analyzed by size exclusion
chromatography (SEC). But as expected for crosslinked polymers, a very broad molecular
mass distribution reaching from 5 × 103 to 1 × 107 g/mol was observed in both cases. Thus,
only careful statements concerning the molecular structure of the polymers can be made.
This is especially true, since the standards applied in SEC measurements regard to non-

84 III. RESULTS AND DISCUSSION
crosslinked polymers. The maximum of the molecular mass distribution of P17 and P18b
was found around 2 × 104 g/mol and 3 × 104 g/mol, respectively. But it remains unclear
whether these values can be assigned to long single chains or to several shorter chains
crosslinked together by ligands 17 or 18. It is very likely that the broad mass distribution is
due to random crosslinking of these units. An SEC analysis of highly crosslinked polymers
P18a and P18c and the homopolymer P18d was not possible, due to their insolubility.
N N
N N
OO
O O
N N
N N
Me Me
Me Me Me Me
Me Me
CO2HCO2H
x y z
Me
Me
N N
N N
O
N N
N N
Me Me
Me Me Me Me
Me Me
CO2HCO2H
x y z
O
Me
Me
Figure 23: Possible formular structure of copolymers of ligand 18 with MMA (top) and/or EGDMA (bottom).
3.2.4. Incorporation of [M(fac-N,N,O)] fragments
After successful copolymerization of the Hbmvpza ligand 18, further experiments intended
to check the coordination behavior of the embedded ligand fragments. Ideally, they should
be able to bind in a κ3-N,N,O fashion just as the free ligand itself. To prove this,
immobilized manganese and rhenium tricarbonyl complexes were synthesized by

III. RESULTS AND DISCUSSION 85
deprotonation of P18a or P18c with KOtBu and subsequent reaction with [MnBr(CO)5] or
[ReBr(CO)5]. The highly crosslinked polymers P18a-Mn/Re and P18c-Mn/Re which are
formed stay insoluble during the reaction (scheme 35).
1. KOtBu, MeOH2. [MBr(CO)5]
N N
N N
Me
Me
Me
Me
OO
M
CC C
OOO
P18a (MMA-copolymer)P18c (EGDMA-copolymer)
M = Mn (P18a-Mn, P18c-Mn)M = Re (P18a-Re, P18c-Re)
N
N N
N
Me
Me
Me
Me
CO2H
Scheme 35: Incorporation of manganese and rhenium tricarbonyl complexes into the copolymers P18a/c.
After washing and drying, IR spectra of the synthesized pale yellow manganese and off-
white rhenium polymers were recorded as nujol mulls. For each metal containing polymer
P18a-Mn, P18a-Re, P18c-Mn, and P18c-Re, IR absorptions characteristic for facial
tricarbonyl complexes (single A' and two close A'' and A') were observed (figure 24 and
table 15). These signals agree well with those of the homogeneous tricarbonyl complexes 19
and 20 and prove the successful formation of the κ3-N,N,O coordinated manganese and
rhenium tricarbonyl moieties. In a control experiment, the reaction product of PMMA
(polymethylmethacrylate) with [ReBr(CO)5] showed no carbonyl signals in the IR spectrum
(figure 24g). In order to investigate the occupancy of the copolymerized ligand sites, the
metal content was determined by AAS (atomic absorption spectrometry) for the manganese
containing polymers P18a-Mn and P18c-Mn and by ICP-AES (inductively coupled plasma
atomic emission spectroscopy) measurements for the rhenium containing polymers P18a-
Re and P18c-Re (table 15).
86 III. RESULTS AND DISCUSSION
1800185019001950200020502100
Wavenumber [cm−1
]
Ab
sorb
ance
a
b
c
d
e
f
g
Figure 24: IR spectra of a) [Mn(bdmvpza)(CO)3] (19) (THF), b) the incorporated manganese complex P18a-Mn
(nujol), c) the incorporated manganese complex P18c-Mn (nujol), d) [Re(bdmvpza)(CO)3] (20) (THF), e) the
incorporated rhenium complex P18a-Re (nujol), f) the incorporated rhenium complex P18c-Re (nujol), g) the
control experiment with PMMA and [ReBr(CO)5] (nujol).
In case of the MMA crosslinked copolymer P18a, the amount of metal was found to be
25.8 mg of Manganese per gram of polymer P18a-Mn and 57.9 mg of Rhenium per gram of
polymer P18a-Re respectively. This equals an occupancy of 63 % (P18a-Mn) and 42 %
(P18a-Re) of the existing N,N,O binding sites of copolymer P18a. For the EGDMA
crosslinked copolymer P18c the occupancies are lower than those of P18a. In P18c-Mn after
all 40 % of the ligand sites are bound to manganese centers, and in P18c-Re 10 % of the
binding sites are occupied by rhenium atoms. The lower incorporation may be explained by
a more restricted access to the ligand sites due to the additional crosslinking by EGDMA.

III. RESULTS AND DISCUSSION 87
Table 15: IR absorptions (nujol) and metal content of the polymers P1a-Mn, P1a-Re, P2-Mn and P2-Re.
Polymer ν(CO)
[cm−1]
Metal/polymer
[mg/g]
Metal/polymer
[mmol/g]
Occupied ligand
sites [%]
P18a-Mn 2038, 1947, 1918 25.8[a] 0.470 63
P18a-Re 2028, 1923, 1898 57.9[b] 0.311 42
P18c-Mn 2041, 1949, 1924 8.10[a] 0.148 40
P18c-Re 2031, 1927, 1908 7.11[b] 0.038 10
[b] Determined by AAS. [b] Determined by ICP-AES.
As shown above, immobilized complexes are accessible by treating the polymer-embedded
ligand fragments with appropriate precursors. Besides this polymer analogous formation,
one could also synthesize polymerizable transition metal complexes first and copolymerize
them with appropriate monomers. Scheme 36 summarizes this second pathway towards
immobilized transition metal complexes as it was accomplished during this thesis.
N
N N
N
Me
Me
Me
Me
R R
CO2H
1. KOt Bu, THF
2. [RuCl2(CO)2]n
R = H N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
241. KOtBu, THF2. [RuCl2(CO)2]n
R =
N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
23
EGDMA, AIBN,MeCN N N
N N
Me
Me
Me
Me
OO
Ru
CCl C
OO
P23
Scheme 36: Synthesis and copolymerization of [Ru(bdmvpza)Cl(CO)2] (23) and synthesis of
[Ru(bdmpza)Cl(CO)2] (24).

88 III. RESULTS AND DISCUSSION
First, the dicarbonyl ruthenium(II) complex [Ru(bdmvpza)Cl(CO)2] (23) was synthesized by
deprotonation of ligand 18 with KOtBu and reaction with the polymeric precursor
[RuCl2(CO)2]n. Parallel to this work, the analogous complex [Ru(bdmpza)Cl(CO)2] (24) was
synthesized by S. TAMPIER from the BURZLAFF group at the same time. Successful formation
of complex 23 was proven by NMR and IR spectroscopy. Two sets of signals for the
pyrazolyl moieties in the 1H and 13C spectra of complex 23 indicated that an unsymmetrical
isomer was formed during the reaction. Hence the chlorido ligand was supposed to be in
trans position towards one pyrazolyl donor. Two clear 13C resonances for the CO ligands
were observed at δ = 192.7 and 196.1 ppm. The IR spectrum of 23 exhibits two ν(CO)
vibrations at ν ̃ = 2068 and 1997 cm−1 as well as an additional weak band for the vinyl C=C
vibration at ν̃ = 1639 cm−1. These values are in good accordance with those found for
analogous complexes [Ru(L)Cl(CO)2] with Cp, Tp and N,N,O ligand systems. This will be
discussed later in chapter 3.4.1. The dicarbonyl complex 23 was immobilized by
copolymerization with EGDMA and AIBN as radical initiator. Acetonitrile was used during
this reaction as porogene. The yellow solid formed during the reaction was washed with
methanol and dried in vacuo. A nujol mull was prepared of a sample of the final polymer
P23 and analyzed by IR spectroscopy (figure 25).
The IR spectrum of the copolymer P23 exhibits two carbonyl vibrations which are nearly
identical to those of the homogeneous complex 23, thus proving the successful
incorporation. The ruthenium content in the copolymer P23 was determined by AAS to
0.103 mmol per gram polymer. The polymerization was achieved using 0.185 mmol of the
complex 23 per gram of monomers. This means that 56 % of the complex is incorporated in
the final polymer. A potential application of this polymer P23 as an oxidation catalyst will
be discussed in chapter 3.4.1.

III. RESULTS AND DISCUSSION 89
190019502000205021002150
Wavenumber [cm−1
]
Ab
sorb
ance
a
b
Figure 25: IR spectra of a) [Ru(bdmvpza)Cl(CO)2] (23) (THF), b) the ruthenium dicarbonyl complex 23
copolymerized with EGDMA (P23) (nujol mull).
3.2.5. Copper and iron containing polymers
One disadvantage of sterically less demanding tripod ligands such as bis(pyrazol-1-yl)acetic
acid or Hbdmpza (1) is their tendency to form undesired 2:1 bisligand complexes. Examples
with different transition metals have been reported recently.[6, 54, 117-118, 184, 255-258] These
bisligand complexes are not suitable for further studies on enzyme models since they are
coordinatively saturated and unreactive towards possible substrate molecules. As shown for
the methacryloxy substituted ligand Hbdmpzmp, a heterogeneous reaction of copper(II)
chloride with a highly crosslinked solid phase allows to control the copper coordination
geometry and favors 1:1 metal to ligand moieties instead of bisligand moieties (see chapter
1.2.3).[117, 184] Following this example, embedded copper(II) complexes seemed to be a useful
target during this work, to verify whether the vinyl substituted ligand Hbdmvpza 18 also
facilitates the control of coordination geometries in crosslinked polymers. In order to

90 III. RESULTS AND DISCUSSION
compare and further investigate these polymers, the homogeneous 2:1 bisligand complex
[Cu(bdmvpza)2] (25) was synthesized first. This was achieved by the reaction of ligand 18
with copper(II) acetate (scheme 37).
[Cu(OAc)2]
MeCN
N N
N N
Me
Me
Me
Me
OO
Cu
NN
NN
Me
Me
Me
Me
O O
N
N N
N
Me
Me Me
Me
CO2H
18
25
Scheme 37: Synthesis of the 2:1 bisligand complex [Cu(bdmvpza)2] (25).
The IR spectrum of the isolated compound exhibits a strong asymmetric carboxylate
vibration at ν ̃ = 1664 cm−1. An additional band at ν ̃ = 1636 cm–1 can be assigned to the vinyl-
C=C vibration. The result of a single-crystal X-ray structure analysis confirms the
successful formation of the bisligand complex 25 and verifies the κ3-N,N,O coordination of
both bdmvpza ligands (figure 26 and table 16). In the molecular structure of complex 25 the
copper(II) center is placed on a special position, with half a molecule of [Cu(bdmvpza)2] in
the asymmetric unit. Complex 25 represents a JAHN-TELLER distorted octahedral geometry.
The basal plane is build up by the nitrogen atoms N11 and N11a [d(N11a–Cu) = 2.141(3) Å]
and the two oxygen atoms O1a and O1b [d(O1a–Cu) = 1.951(2) Å] of the carboxylate
donors. The nitrogen atoms N21a and N21b form the JAHN-TELLER axis with d(N21a–Cu) =
2.278(3) Å. These bond distances are comparable to those of [Cu(bdmpzpr)2] and
[Cu(bdmpza)2].[118] In contrast to the bridge functionalized bisligand complex
[Cu(bdmpzpr)2] (7), the structure of complex 25 shows only a slight twist of the pyrazolyl
moieties [∡(Cu1,N11,N12,C2) = 1.31(1)°, ∡(Cu1,N21,N22,C2) = 7.36(1)°] Furthermore the
carboxylate donor in 25 is considerably less bent compared to complex 7 as evident from
the torsion angle ∡(O2,C1,C2,H2) = 7.05(1)°. Obviously, this is due to a missing steric
hindrance between the pyrazole methyl groups in position 5 and a linker at the bridging
position.

III. RESULTS AND DISCUSSION 91
O2b
O1b
C2b
N21a
N22aN11a
N12a
H1b
C1b
Cu1
C1a
H1a
N12b
N11bN22b
N21b
C2a
O1a
O2a
Figure 26: Molecular structure of [Cu(bdmvpza)2] (25). Thermal ellipsoids are drawn at the 40% probability
level. Most hydrogen atoms and the disordered solvent molecule have been omitted for clarity.
Table 16: Selected bond lenghts and angles for the molecular structure of the copper(II) bisligand complex 25.
Distances in Å Angles in °
Cu−N11a 2.141(3) ∡(N11a,Cu,N21a) 84.82(9)
Cu−N21a 2.278(3) ∡(N11a,Cu,O1a) 85.76(9)
Cu−O1a 1.951(2) ∡(N21a,Cu,O1a) 85.14(9)
∡(O2,C1,C2,H2) 7.05
∡(Cu1,N11,N12,C2) 1.32
∡(Cu1,N21,N22,C2) 7.36
As mentioned earlier, in case of the methacryloxy substituted ligand Hbdmpzmp a
copolymer with 1:1 copper/ligand moieties had been obtained with a solid phase, which was
crosslinked with EGDMA.[117, 184] The reaction of a non-crosslinked polymer with copper(II)
on the other hand led to bisligand moiety formation embedded in the solid phase. In case of
the vinyl substituted ligand 18, copolymers with 1:1 copper/ligand moieties should also be
accessible from the MMA-copolymerized solid phase P18a, since the ligand is a crosslinker
itself. The incorporation of copper was achieved by deprotonation of P18a with KOtBu in

92 III. RESULTS AND DISCUSSION
methanol followed by addition of CuCl2. In doing so, an instant color change of the polymer
to green (scheme 38) was observed.
N N
N N
Me
Me
Me
Me
OO
M
(S)Cl (S)
P18a (MMA-copolymer)P18c (EGDMA-copolymer)
N
N N
N
Me
Me
Me
Me
CO2H
M = Cu (P18a-Cu)M = Fe (P18a-Fe, P18c-Fe)
1. KOtBu, MeOH2. M2+
Scheme 38: Incorporation of copper and iron into copolymers P18a and P18c.
P18a-Cu was isolated as a green powder after filtration and washing with methanol. For
further investigation UV/Vis spectra of copolymer P18a-Cu and the homogeneous 2:1
bisligand complex [Cu(bdmvpza)2] (25) have been recorded and compared with each other
(figure 27). For the copolymer P18a-Cu a bathochromic shift of the absorption maximum by
64 nm was observed. This agrees well with a similar bathochromic shift of 78 nm that was
observed for copper containing copolymers of the methacryloxy substituted ligand
Hbdmpzmp.[117, 184] Due to the bathochromic shift the formation of κ3-N,N,O bound 1:1
copper moieties can be assumed. Nevertheless, other coordination modes such as κ2-N,O
and the coordination of additional solvent molecules can not be excluded entirely.

III. RESULTS AND DISCUSSION 93
500 600 700 800 900 1000
Wavelength [nm]
Ab
sorb
ance
a
b
Figure 27: UV/Vis spectra of a) [Cu(bdmvpza)2] (25) in methanol, b) P18a-Cu (polymer pellet, nujol).
In a further set of experiments, ferrous iron was incorporated into polymers P18a or P18c.
In a similar procedure as described for P18a-Cu, the solid phases P18a or P18c were
deprotonated with KOtBu and treated with a slight excess of an iron(II) salt (FeCl2 or
FeSO4 × 7H2O). The resulting pale greenish, air sensitive polymers were washed with
degassed methanol and dried in vacuo prior to determination of the metal content by AAS
measurement. In order to prove reproducibility with different metal sources, P18a was
treated with FeCl2 or FeSO4 × 7H2O in two separate experiments. The determined amounts
of iron were almost identical for both cases (table 17). AAS measurements show that up to
64 % of the N,N,O binding sites in polymer P18a can be occupied by ferrous centers. It is
noteworthy that a similar high incorporation rate of 0.165 mmol g–1 was achieved in case of
the EGDMA crosslinked polymer P18c-Fe. Surprisingly, nearly 60% of the N,N,O sites in
P18c-Fe were occupied by iron(II) ions, although a restricted access to these sites can be
assumed due to higher crosslinking.

94 III. RESULTS AND DISCUSSION
Table 17: Iron content of the polymers P18a-Fe and P18c-Fe.
Polymer Iron source
Metal/polymer
[mg/g]
Metal/polymer
[mmol/g]
Occupied ligand
sites [%]
P18a-Fe FeCl2 27.4 0.491 64
P18a-Fe FeSO4 × 7H2O 25.0 0.448 65
P18c-Fe FeCl2 9.20 0.165 60
3.2.6. Conclusion
The new polymerizable heteroscorpionate ligand Hbdmvpza (18) is accessible in a four-step
synthesis starting from 3,5-dimethylpyrazole. The vinyl substituents suitable for
copolymerization do not influence the remaining space of κ3-N,N,O bound fragments. This
ligand is able to bind ruthenium(II) in a fac-κ3-N,N,O mode which was proven by the
successful synthesis of bisphosphine complex [Ru(bdmvpza)Cl(PPh3)2] (21). Polymerization
of ligand 18 can be achieved by means of comonomers such as MMA or EGDMA. On the
other hand, self-polymerization of 18 can form a unique homopolymer P18d. The ability of
the MMA or EGDMA derived copolymers of ligand 18 to provide N,N,O binding sites was
verified by a successful incorporation of fac-[Mn(CO)3], fac-[Re(CO)3] and fac-[RuCl(CO)2]
fragments. A heterogeneous reaction of the MMA copolymer of ligand 18 with CuCl2 yields
a solid phase P18a-Cu which reveals a bathochromic shift by 64 nm compared to the
UV/Vis spectrum of the homogeneous complex [Cu(bdmvpza)2] (25). This implies, that
copper(II) centers embedded in this copolymer are mainly coordinated by one ligand moiety.
Successful incorporation of iron(II) was accomplished and proven by AAS measurements.
Up to 65% of the ligand sites in the MMA-copolymer P18a-Fe can be occupied by ferrous
iron centers.

III. RESULTS AND DISCUSSION 95
3.3. Towards imprinted polymers
Imprinted polymers are often considered as artificial enzymes since they can recognize
certain substrates with a high selectivity according to natures "lock and key" principle. Such
highly selective recognition sites surrounded by polymeric matrices are often used for
catalytic applications. As shown in chapter 3.2, the new vinyl substituted Hbdmvpza ligand
18 fulfills two essential preconditions required for the generation of imprinted polymers
which can mimic 2-OG dependent iron(II) enzymes. On the one hand, ligand 18 is suitable
for polymerization due to the vinyl groups. Hence transition metal complexes of 18 can be
immobilized in highly crosslinked polymers. And on the other hand ligand 18 grants access
to κ3-N,N,O bound ruthenium(II) complexes without any interference of the vinyl groups
with the metal center. Such inert ruthenium model complexes could serve as templates for
the generation of cavities. Thus, the final task during this thesis was to prove, whether
molecular imprinting techniques can be established with the Hbdmvpza ligand 18.
3.3.1. Synthesis of model complexes for 2-OG dependent iron enzymes
Usually the strategy for the preparation of molecular imprinted polymers starts with the
design of a template (see chapter 1.3.1). Such "dummy" molecules can be catalytically active
complexes where a substrate or a pseudo-substrate is attached to the metal center. But also
molecules which imitate a transition state of a preferred reaction can be used as templates.
In this project, 2-oxocarboxylato ruthenium complexes have been considered as template
molecules, since the main focus of this work lies on 2-OG dependent iron enzymes.
Examples of 2-oxocarboxylato ruthenium complexes bearing the Hbdmpza ligand 1 as
model complexes for 2-OG dependent iron enzymes have been published by the BURZLAFF
group before.[60-61]
The complex [Ru(bdmvpza)(BF)(PPh3)] (26) was obtained by a reaction of bisphosphine
complex [Ru(bdmvpza)Cl(PPh3)2] (21) with thallous acetate in presence of benzoylformic
acid (scheme 39). During this one pot synthesis, the κ2O
1,O1'-carboxylato complex
[Ru(bdmvpza)(O2CMe)(PPh3)] is formed in situ. As the reaction proceeds, the acetato ligand
is exchanged by the BF ligand and complex 26 starts to precipitate out of the CH2Cl2
solution. Successful formation of 26 was verified by mass spectrometry, elemental analysis
and NMR spectroscopy. The 31P NMR spectrum of complex 26 exhibits a single resonance at

96 III. RESULTS AND DISCUSSION
δ = 57.8 ppm for the remaining PPh3 ligand, thus proving the loss of the other one caused by
κ2O
1,O1' coordination of the BF ligand. Due to the resulting chiral geometry of 26, two sets
of signals assigned to the pyrazolyl groups were detected in the 1H and 13C NMR spectra.
Furthermore six double doublet 1H NMR signals were found for the diastereotopic vinyl
groups of the Hbdmvpza ligand 18. The 1H resonances for the phenyl group of the BF ligand
are observed at δ = 7.23, 7.52 and 8.35 ppm. The 13C signals at δ = 167.9, 169.6 and δ =
202.9 ppm can be assigned to the two carboxylate donors and the keto group of the BF
ligand respectively. A strong signal at ν ̃ = 1652 cm−1 was found in the IR spectrum of
complex 26 for the asymmetric carboxylate vibration of the coordinated Hbdmvpza ligand
18. An additional signal at ν ̃ = 1660 cm−1 is caused by the carboxylate group of the BF
ligand. The UV/Vis spectrum of 26 exhibits an intense maximum at λ = 555 nm
corresponding to the deep purple color of this compound due to a MLCT transition. These
spectroscopic results agree very well with those of the analogous complex
[Ru(bdmpza)(BF)(PPh3)].[60] Unfortunately it was not possible to obtain single crystals of
compound 26 during this thesis in order to clarify its structural properties by X-ray
structure determination.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
21
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
Ph
5a4a
3a3b
4b5b
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
NH
5a4a
3a3b
4b5b
1. TlOAc
2. Benzoylformic acid
1. TlOAc
2. N -oxalylglycine
CO2H
26 27
Scheme 39: Synthesis of [Ru(bdmvpza)(BF)(PPh3)] (26) and [Ru(bdmvpza)(NOG)(PPh3)] (27).

III. RESULTS AND DISCUSSION 97
In a similar one pot reaction, a second template complex [Ru(bdmvpza)(NOG)(PPh3)] (27)
was prepared by using N-oxalylglycine (NOG). It has to be mentioned that NOG is
isostructural to 2-oxoglutaric acid and thus acts as a cosubstrate analogue and inhibitor. In
other words, complex 27 can be considered as a model complex for a 2-OG-dependent
iron(II) enzyme which is inhibited by NOG and therefore is well suited as a "dummy"
complex for an imprinting attempt. The NOG complex 27 was also characterized by NMR,
IR and UV/Vis spectroscopy. Once again due to the chiral geometry caused by the κ2O
1,O1'
coordination of the NOG ligand two sets of signals for the pyrazolyl groups were detected
in the 1H and 13C NMR spectra. The 1H NMR spectrum of 27 also shows an AMX spin
system (δ = 3.49, 3.65 and 9.15 ppm) for the -CH2-NH- unit of the NOG ligand. The 31P
resonance for the remaining PPh3 ligand was observed at δ = 60.7 ppm. The IR signals for
the asymmetric carboxylate vibration of the Hbdmvpza and the NOG ligand were found at
ν ̃ = 1673 and 1623 cm−1. For the orange colored complex 27 an absorption maximum at
λ = 300 nm was observed in the UV/Vis spectrum. An X-ray structure analysis of the NOG
complex 27 reveals the successful κ2O
1,O1' coordination of the NOG inhibitor (figure 28 and
table 18).
O4
C3O3
C4
N1
N11 N21
Ru1
N12
O7
N22
O5
C5
P1
C1
C6
O1
O6
C2O2
Figure 28: Molecular structure of [Ru(bdmvpza)(NOG)(PPh3)] (27). Thermal ellipsoids are drawn at the 50 %
probability level. Only one out of two molecules in the asymmetric unit is depicted. Hydrogen atoms as well
as the disordered solvent molecule have been omitted for clarity.

98 III. RESULTS AND DISCUSSION
Table 18: Selected bond lenghts and angles for the molecular structure of the ruthenium(II) NOG complex 27.
Distances in Å Angles in °
Ru1-N11 2.037(3) ∡(N11,Ru1,N21) 83.37(11)
Ru1-N21 2.180(3) ∡(O1,Ru1,N11) 88.18(9)
Ru1-O1 2.086(2) ∡(O1,Ru1,N21) 86.18(9)
Ru1-O3 2.104(2) ∡(O1,Ru1,P1) 86.39(6)
Ru1-O5 2.134(2) ∡(O1,Ru1,O3) 174.20(8)
Ru1-P1 2.277(9) ∡(O1,Ru1,O5) 96.68(8)
O3-C3 1.272(4) ∡(N21,Ru1,P1) 172.44(7)
O4-C3 1.237(4) ∡(N11,Ru1,O3) 97.02(9)
C3-C4 1.531(4)
C4-N1 1.314(4)
O5-C4 1.258(3)
The molecular structure exhibits a trans geometry regarding both carboxylate donors.
Contrary to this observation, usually the 2-oxo group is trans to the iron-binding aspartate
in the active sites of most 2-oxoglutarate-dependent enzymes. An example showing the
active site of the factor-inhibiting hypoxia-inducible factor (FIH) with the ligated inhibitor
NOG is depicted in figure 29. In a previous project of the BURZLAFF group, R. MÜLLER found
that two structural isomers are formed during the synthesis of 2-oxocarboxylato complexes
in case of prolonged reaction times.[60, 216] Thus, the synthesis and isolation of a second
isomer of 27, which might exhibit a correctly bound NOG inhibitor, will be investigated in
an upcoming project. This might be achieved simply by applying shorter reaction times. A
synthesis of the analogous bdmpza complex [Ru(bdmpza)(NOG)(PPh3)] already resulted in
the correct geometry.[60, 216]
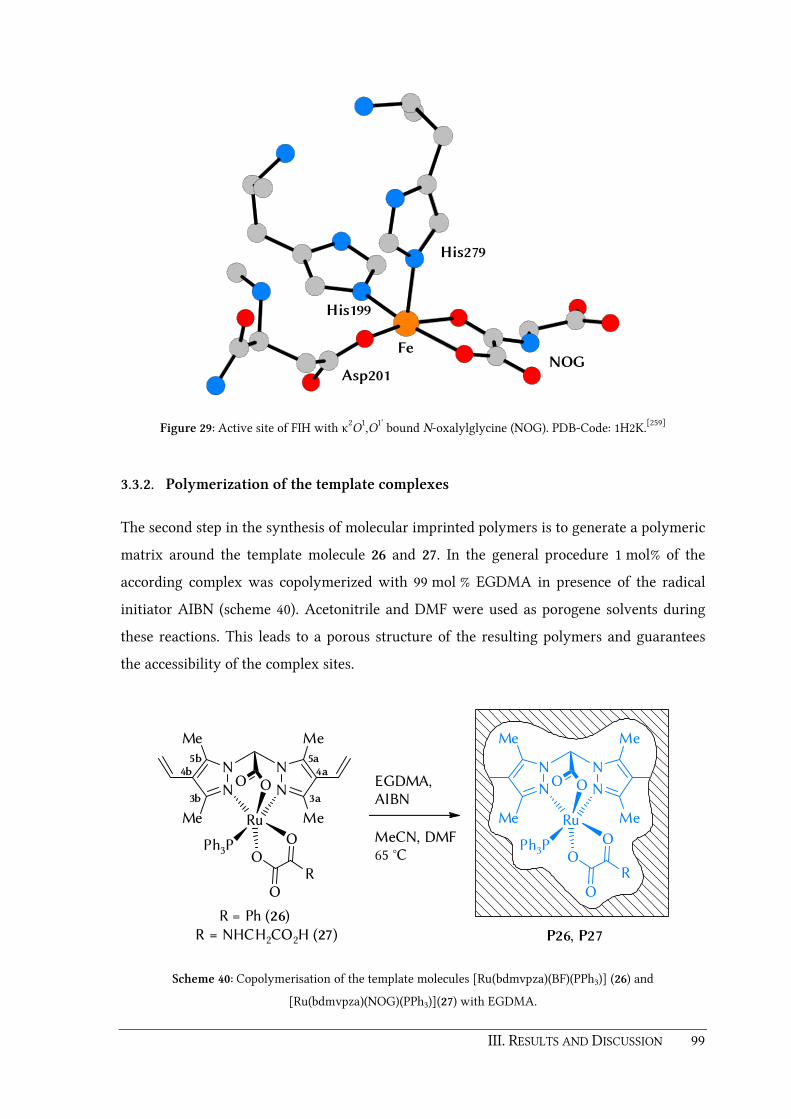
III. RESULTS AND DISCUSSION 99
NOG
His199
Fe
His279
Asp201
Figure 29: Active site of FIH with κ2O
1,O1' bound N-oxalylglycine (NOG). PDB-Code: 1H2K.[259]
3.3.2. Polymerization of the template complexes
The second step in the synthesis of molecular imprinted polymers is to generate a polymeric
matrix around the template molecule 26 and 27. In the general procedure 1 mol% of the
according complex was copolymerized with 99 mol % EGDMA in presence of the radical
initiator AIBN (scheme 40). Acetonitrile and DMF were used as porogene solvents during
these reactions. This leads to a porous structure of the resulting polymers and guarantees
the accessibility of the complex sites.
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
R
EGDMA,AIBN
MeCN, DMF65 °C
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
R
5a4a
3a3b
4b5b
R = Ph (26)R = NHCH2CO2H (27) P26, P27
Scheme 40: Copolymerisation of the template molecules [Ru(bdmvpza)(BF)(PPh3)] (26) and
[Ru(bdmvpza)(NOG)(PPh3)](27) with EGDMA.

100 III. RESULTS AND DISCUSSION
To further investigate the synthesized polymers, UV/Vis spectra of P26 and P27 have been
recorded and compared to the UV/Vis spectra of the free complexes 26 and 27, respectively
(figure 30). In case of P26 and P27, the UV/Vis spectra were recorded using nujol mulls of
the finely ground polymer samples. Prior to these measurements, a baseline was collected
with an analogously prepared mull of PEGDMA [= poly(ethyleneglycol) dimethacrylate]. A
comparison of the recorded UV/Vis spectra shows that the absorption maxima λmax of the
polymers P26 (558.5 nm) and P27 (311.0 nm) are almost identical with the ones recorded for
the corresponding homogeneous complexes 26 (554.5 nm) and 27 (300.4 nm) respectively
which proves the successful incorporation of the complexes into the polymers. Furthermore,
the ruthenium content of both polymers was determined by ICP-AES to analyze the degree
of incorporation. In case of P26, a ruthenium content of 31.1 µmol per gram of polymer was
found. A very similar ruthenium content was found for P27, namely 31.3 µmol ruthenium
per gram of polymer. In both cases, 48.4 µmol of the according complex was copolymerized
per gram of monomers. Thus, the found ICP-AES values equal an incorporation rate of
approximately 65 % for both polymers P26 and P27.
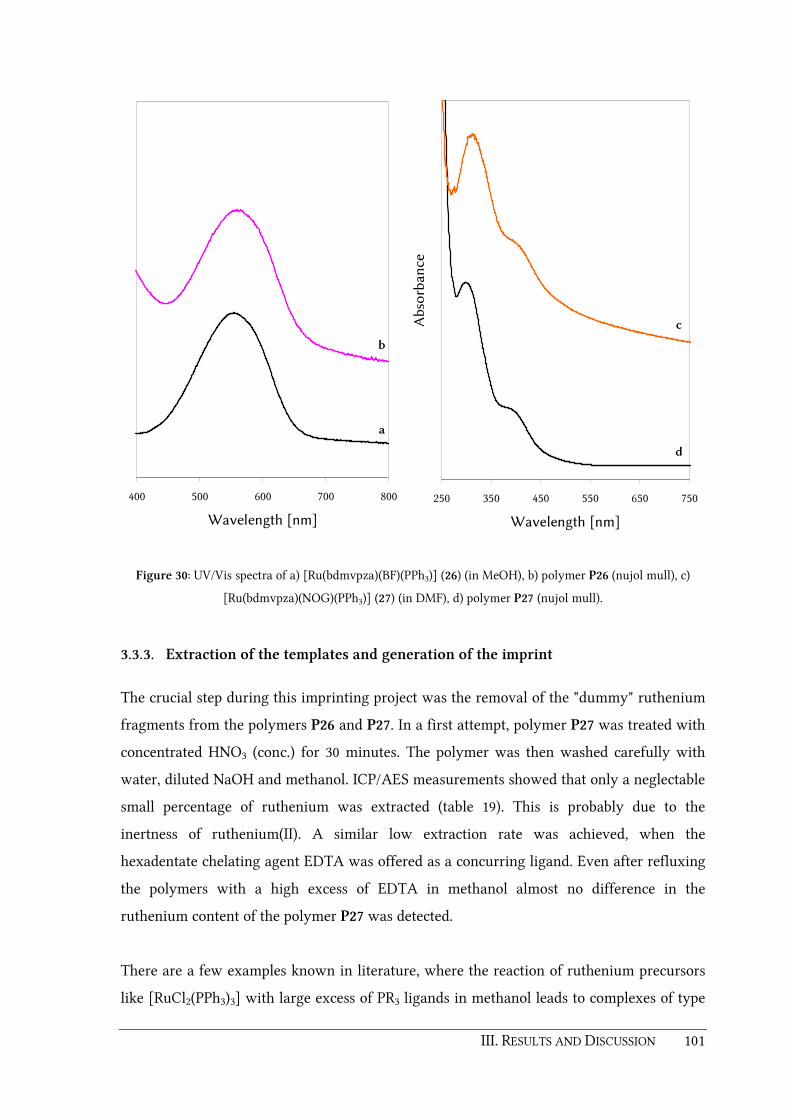
III. RESULTS AND DISCUSSION 101
400 500 600 700 800
Wavelength [nm]
b
a
250 350 450 550 650 750
Wavelength [nm]
Ab
sorb
ance
d
c
Figure 30: UV/Vis spectra of a) [Ru(bdmvpza)(BF)(PPh3)] (26) (in MeOH), b) polymer P26 (nujol mull), c)
[Ru(bdmvpza)(NOG)(PPh3)] (27) (in DMF), d) polymer P27 (nujol mull).
3.3.3. Extraction of the templates and generation of the imprint
The crucial step during this imprinting project was the removal of the "dummy" ruthenium
fragments from the polymers P26 and P27. In a first attempt, polymer P27 was treated with
concentrated HNO3 (conc.) for 30 minutes. The polymer was then washed carefully with
water, diluted NaOH and methanol. ICP/AES measurements showed that only a neglectable
small percentage of ruthenium was extracted (table 19). This is probably due to the
inertness of ruthenium(II). A similar low extraction rate was achieved, when the
hexadentate chelating agent EDTA was offered as a concurring ligand. Even after refluxing
the polymers with a high excess of EDTA in methanol almost no difference in the
ruthenium content of the polymer P27 was detected.
There are a few examples known in literature, where the reaction of ruthenium precursors
like [RuCl2(PPh3)3] with large excess of PR3 ligands in methanol leads to complexes of type

102 III. RESULTS AND DISCUSSION
[Ru(PR3)6]2+.[260] For instance, HIGGINS et al. reported, that [Ru(PMe3)6]
2+ is formed when
[Ru(dmf)6](OTf)3 is treated with a large excess of PMe3 in EtOH and THF.[261] It is
noteworthy that [Ru(PMe3)6]2+ seems to stay in solution during this synthesis. This idea of
generating soluble phosphine complexes of ruthenium(II) was adopted for the extraction
experiments (scheme 41).
N N
N N
Me
Me
Me
Me
OOPMe3
reflux
P26X, P27X
P26 orP27
H FeCl2
N N
N N
Me
Me
Me
Me
OO
Fe
(S)Cl (S)
?
Scheme 41: Extraction of the ruthenium(II) fragments by reaction of P26 and P27 with trimethylphosphine.
For this purpose, the according polymer (P26 or P27) was suspended in a solution of excess
PMe3 in EtOH/THF and heated under reflux conditions for several days. Filtration and
washing with MeOH and acetone gave the polymers P26X and P27X which are slightly
lighter in color compared to the original polymers P26 and P27. Depending on the reaction
conditions and the amount of PMe3 up to 50% of the ruthenium templates can be removed
by this procedure as shown in table 19. This was verified by ICP-AES measurements.
Table 19: Summary of the cleavage attempts with PMe3.
Polymer Reaction conditions Ru/polymer
before [mg/g]
Ru/polymer
after [mg/g]
Cleavage
[%]
P27 HNO3 (xs, conc.), 30 min, RT 3.10 3.00 3
P27 EDTA (xs), 16 h, 70°C 3.10 3.00 3
P27 900 µL PMe3, 72 h, 70°C 3.09 1.77 43
P27 500 µL PMe3, 120 h, 70°C 3.24 2.09 35
P26 250 µL PMe3, 72 h, 70°C 3.30 2.52 24
P26 500 µL PMe3, 120 h, 70°C 3.00 1.48 51

III. RESULTS AND DISCUSSION 103
In upcoming studies, it has to be examined whether generated imprinted polymers are able
to catalytically oxidize substrates such as cyclohexene on addition of iron(II) and 2-OG in
presence of dioxygen or a oxidizing agent such as H2O2. First experiments towards this
were already made in the final phase of this thesis. The extracted polymers P26X and P27X
have been suspended in methanol and treated with a slight excess of iron(II) chloride at
50 °C. After washing with methanol the solid phases have been dried and analyzed by AAS.
Unfortunately the amount of iron was higher than theoretically possible. Thus, in furter
studies it might be necessary to additionally wash the iron containing polymers with
solutions of chelators such as EDTA in order to remove excess of iron.
3.3.4. Conclusion
In this chapter, suitability of the new 4-vinyl substituted ligand 18 for the generation of MIP
was verified. Two template molecules, namely [Ru(bdmvpza)(BF)(PPh3)] (26) and
[Ru(bdmvpza)(NOG)(PPh3)] (27) have been synthesized and characterized by NMR, IR and
UV/Vis spectroscopy. Immobilization of 26 and 27 was achieved using EGDMA as
comonomer and porogene solvents such as MeCN and/or DMF. The UV/Vis spectra of the
obtained polymers P26 and P27 confirmed the successful incorporation of the homogeneous
complexes 26 and 27 into the polymer matrix. The imprint was generated by extraction of
the inactive ruthenium(II) fragments. ICP-AES measurements revealed that up to 50 % of
the inactive sites can be removed by treating P26 and P27 with excess of PMe3 for several
days. Upcomping experiments will investigate the catalytic potential of these MIP in
presence of diverse substrates, atmospheric O2 and the cosubstrate 2-OG.

104 III. RESULTS AND DISCUSSION
3.4. Further work with bis(pyrazol-1-yl)acetic acids
3.4.1. Epoxidation catalysis with carbonyl complexes [Ru(L)Cl(CO)2]
As mentioned in chapter 1.1, the uncatalyzed reaction of organic compounds with
atmospheric dioxygen is thermodynamically feasible but a spin-forbidden process. In nature,
oxygenase enzymes bearing transition metals like iron(II) in a high-spin configuration help
to overcome this spin mismatch by activation of triplet-state dioxygen to a reactive singlet
or doublet (radical) species. Ruthenium(II) complexes with bis(pyrazol-1-yl)acetate are good
structural models for iron(II) enzymes bearing the facial 2-His-1-carboxylate triad. But an
enzyme analogous activation of dioxygen with ruthenium(II) complexes is not favorable
due to their low-spin character. However the necessity of a high-spin center becomes
irrelevant in so called peroxide shunt type reactions. Instead of dioxygen, oxidizing agents
like peroxides or iodosylbenzene are used to generate the FeIV=O species directly. Thus,
dioxygen activation and the overcome of a spin-mismatch are no more essential in these
reactions. Similar peroxide shunt reactions in case of the enzymes have been reported by
J. D. LIPSCOMB et al., for the Rieske-dioxygenases naphthalene dioxygenase and benzoate
1,2-dioxygenase.[262-263] In a more recent publication, P. F. FITZPATRICK et al. were able to
show that in case of tetrahydropterin (PH4) dependent monooxygenases such as
phenylalanine hydroxylase, H2O2 can replace PH4 and O2 which usually are required for the
formation of the hydroxylating intermediate.[264]
It has been well documented that high-valent ruthenium-oxo species, RuVI(=O)2 or RuIV=O
can be used for the catalytic epoxidation of alkenes, oxidation of sulfides and hydroxylation
of alkanes.[265-272] C. M. CHE and coworkers for instance reported on cationic ruthenium(IV)
complexes such as [Ru(Me3tacn)(3,3'-Me2-bpy)(O)]2+ or [Ru(terpy)(tmeda)(O)]2+ that can be
used to epoxidize alkenes stoichiometrically (Me3tacn = 1,4,7-trimethyl-1,4,7-
triazacyclononane, 3,3'-Me2bpy = 3,3'-dimethyl-2,2'-bipyridine, terpy = 2,2':6',2''-terpyridine,
tmeda = N,N,N',N'-tetramethylethylenediamine).[273-276] Furthermore, the complex
[Ru(Me3tacn)(OH2)(O2CCF3)](O2CCF3)2 was shown to be an effective catalyst for
homogeneous oxidation of alkenes by tert-butylhydroperoxide (TBHP) as an oxidant.[277]
More than 6000 product turnovers have been attained in long-term experiments with
reaction times of 300 h. In these studies, it was assumed that the ruthenium(VI) complex
[Ru(Me3tacn)(O)2(O2CCF3)]+ is the actual catalytically active species during the oxidation

III. RESULTS AND DISCUSSION 105
process. In a continuative investigation, [Ru(Me3tacn)(OH2)(O2CCF3)2](O2CCF3) was
immobilized on silica gel by impregnation methods.[278] It was shown that the supported
catalyst effects facile oxidation of alcohols by TBHP and can be reused without any
significant loss of activity. Turnover numbers up to 9000 have been achieved for the
oxidation of 1-phenyl-1-propanol to 1-phenyl-propanone.
The same group also investigated the oxidation chemistry of ruthenium porphyrin based
systems. In particular porphyrins [Ru(por)Cl2], [Ru(por)(CO)] and [Ru(por)(O)2] for
instance with por = tmp (meso-tetramesitylporphyrinato dianion) or 2,6-Cl2tpp [meso-
tetrakis(2,6-dichlorophenyl)porphyrinato dianion] are highly active catalysts for organic
oxidations. Besides homogeneous catalysts, there have been several attempts to improve
catalyst recycling and selectivity by immobilization methods. Heterogenization of
ruthenium porphyrin catalysts was achieved by grafting on surface modified silica gel,
encapsulation into silica gel matrices or attachment on MERRIFIELD's peptide resin. At this
point, the reader is referred to a recently published comprehensive review focused on the
work of the C.-M. CHE group in the area of metalloporphyrin based oxidation catalysis.[279]
In 2007, T. KOJIMA, S. FUKUZUMI and coworkers reported on a series of complexes
[RuCl2(L)]+ with differently substituted tris(2-pyridylmethyl)amine ligands L. Their aim
was to clarify the reaction mechanisms of hydrocarbon oxidation catalyzed by these
complexes with m-chloroperbenzoic acid (mCPBA) as oxidizing agent.[280] It was found that
the reactivity of the catalyst as well as the mechanism of the alkane oxidation can be
altered significantly by electronic effects of substituents on the Tpa ligand backbone. In case
of a catalyst bearing the unsubstituted Tpa ligand, cyclohexanol and cyclohexanon were
formed at comparable rates. This indicates that reaction of this complex with mCPBA gives
free radical species and leads to autooxidation of cyclohexane. A completely different
observation was made for a catalyst bearing a Tpa ligand with electron withdrawing
ethoxycarbonyl groups at the 4-postion of two pyridine moieties. In this case, cyclohexanol
was formed first and cyclohexanon was produced by subsequent oxidation of cyclohexanol.
Labeling experiments and resonance Raman spectroscopy under catalytic conditions
without the substrate confirmed the formation of a RuIV=O intermediate which is finally
responsible for the hydroxylation of cyclohexane. In a more recent work of the same group
a novel ruthenium(II) precursor [Ru(Tpa)(H2O)2]2+ was oxidized with ammonium cerium(IV)
nitrate in water to give a highly efficient catalytically active RuIV=O species evidenced by

106 III. RESULTS AND DISCUSSION
resonance raman and X-ray photoelectron spectroscopy (XPS). Selective oxidation reactions
of organic substrates have been investigated using the bis-aqua precursor, ammonium
cerium(IV) nitrate and water, which can be used as an oxygen source. Cyclohexene for
instance was converted to adipic acid in an 8e– oxidation with a TON up to 2560. The
proposed mechanism involves the epoxidation of cyclohexene, hydrolysis of the epoxide to
yield give cyclohexane-1,2-diol, the 4e– oxidation of the diol to give cyclohexane-1,2-dione
and subsequent Baeyer–Villiger-like oxidation to give an acid anhydride. Final hydrolysis
resulted in adipic acid.
Inspired from these and other results for iron oxygenases, there have been attempts earlier
in the BURZLAFF group to apply ruthenium(II) complexes with bis(pyrazol-1-yl)acetato
ligands as catalysts for oxidation reactions with different oxidizing agents. The complexes
[Ru(bdmpza)Cl(PPh3)2] (8), [Ru(bdmpza)(O2CCH3)(PPh3)], [Ru(bdmpza)(O2CC(O)Ph)(PPh3)]
and [Ru(bdmpza)(O2CC(O)Me)(PPh3)] showed catalytic activity in the epoxidation of
cyclohexene or the oxidation of diphenyl sulfene in presence of hydrogen peroxide or
iodosylbenzene.[216] When using cyclohexene as substrate, it was observed that cyclohexene
oxide is formed selectively during the reaction. Turnover numbers of 2.5 – 5 have been
found for these complexes. To exploit this topic further, one of the tasks during this thesis
was to verify the catalytic activity of the synthesized dicarbonyl complex
[Ru(bdmpza)Cl(CO)2] (24) as well as its immobilized counterpart P23 which have been
described earlier in chapter 3.2.4. This was also motivated by the work of R. SARIEGO et al.
concerning ruthenium(II) complexes of type [Ru(L)Cl2(CO)2] with bipyridine,
phenanthroline, biquinoline and pyridine ligands. It was reported that these compounds
moderately catalyze the epoxidation of cyclohexene in presence of iodosylbenzene.[281]
It was considered reasonable to include the analogous complex [Ru(bdmpzpr)Cl(CO)2] (28)
in the catalytic studies of this thesis, in order to check a possible influence of the methyl
group in bridging position. The synthesis of the dicarbonyl complex 28 (scheme 42) was
achieved by reaction of in situ formed K[bdmpzpr] with the polymeric precursor
[RuCl2(CO)2]n as described chapter 3.2.4. IR monitoring revealed the successful formation of
complex 28 by the appearance of two carbonyl signals at ν̃ = 2063 and 1994 cm−1. Two sets
of 1H and 13C resonances for the pyrazolyl moieties in the NMR spectra of 28 prove the
formation of an unsymmetrical isomer with the chlorido ligand positioned trans to a
pyrazolyl donor.

III. RESULTS AND DISCUSSION 107
N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
N
N N
N
Me
Me
Me
Me
CO2H
4
1. KOtBu, THF2. [RuCl2(CO)2]n
28
Me
Me
Scheme 42: Synthesis of the dicarbonyl complex [Ru(bdmpzpr)Cl(CO)2] (28)
The 13C resonances for the CO ligands were found at δ = 194.3 and 197.4 ppm. Comparable
spectroscopic values have been obtained earlier for analogous complexes of type
[Ru(L)Cl(CO)2] with L = bdmpza, bdmvpza, Cp, Cp* or Tp (see chapter 3.2.4 and table
21).[282-285] The identity of complex 28 was furthermore confirmed by a peak at m/z = 455
for the molecule ion [MH+] and finally by a single crystal X-ray structure determination
(figure 31 and table 20). Suitable crystals therefore can be obtained by layering a CH2Cl2
solution of 28 with diethyl ether. The structure of complex 28 reveals d(Ru–N11), d(Ru–N12)
and d(Ru–O1) distances which do not differ significantly from the corresponding distances
in the analogous complex [Ru(bdmpza)Cl(CO)2] (24). The carboxylate donor is slightly bent
with an angle ∡(C3,C1,C2,O2) of about 10°.

108 III. RESULTS AND DISCUSSION
O5
C5
N12
N11
N22
N21
C3
C2
Ru1
C4
C1
Cl1
O1
O4
O2
Figure 31: Molecular structure of the dicarbonyl complex [Ru(bdmpzpr)Cl(CO)2] (28) Thermal ellipsoids are
drawn at the 50% probability level. Most hydrogen atoms have been omitted for clarity.
Table 20: Selected bond lenghts and angles for the molecular structure of the ruthenium(II) dicarbonyl
complex [Ru(bdmpzpr)Cl(CO)2] (28).
Distances in Å Angles in °
d(Ru1−N11) 2.1265(15) ∡(N11,Ru1,N21) 85.88(6)
d(Ru1−N21) 2.0703(15) ∡(O1,Ru1,N11) 84.28(6)
d(Ru1−O1) 2.0783(13) ∡(O1,Ru1,N21) 84.25(6)
d(Ru1−Cl1) 2.3726(5) ∡(O1,Ru1,C5) 176.08(6)
d(Ru1−C4) 1.922(2) ∡(N11,Ru1,C4) 176.37(7)
d(Ru1−C5) 1.900(2) ∡(N21,Ru1,Cl1) 173.89(4)
d(O1−C1) 1.273(2) ∡(Cl1,Ru1,C4) 86.13(6)
d(O2−C1) 1.222(2) ∡(Cl1,Ru1,C5) 90.63(5)
d(C1−C2) 1.595(2) ∡(C4,Ru1,C5) 91.76(8)
d(C4−O4) 1.098(3) ∡(O2,C1,C2,C3) 10.19
d(C5−O5) 1.091(3)

III. RESULTS AND DISCUSSION 109
Table 21: ν(CO) vibrations [cm−1] and 13C NMR resonances [ppm] of dicarbonyl complexes [Ru(L)Cl(CO)2].
Complex ν(CO) 13C (CO)
[Ru(bdmpzpr)Cl(CO)2] (28) 2063, 1994[a] 194.3, 197.4
[Ru(bdmpza)Cl(CO)2] (24) 2066, 1996[b] 192.6, 196.0
[Ru(bdmvpza)Cl(CO)2] (23) 2068, 1997[a] 192.7, 196.1
[RuCl(Tp)(CO)2][282-283] 2074, 2012[c]
[RuCl(Cp*)(CO)2][284] 2025, 1975[c]
[RuCl(Cp)(CO)2][285] 2056, 2008[d]
[a] THF solution, [b] KBr pellet, [c] CH2Cl2 solution, [d] cyclohexane solution.
Experiments regarding the epoxidation of cyclohexene were carried out in unstablized,
HPLC grade CH2Cl2 under a dinitrogen atmosphere using decane as internal standard.
Besides aqueous hydrogen peroxide, two nucleophilic oxidizing agents, namely
iodosylbenzene (PhIO) and 2,6-dichloropyridine N-oxide (DCPNO) were tested in these
reactions. In order to prove which products are formed during the oxidation reaction,
genuine samples of possible products and of the reactant cyclohexene were analyzed by gas
chromatography (GC). In a preliminary, non-quantitative run, it was found that both
catalysts 24 and 28 are highly selective for the formation of cyclohexene oxide. Other
oxidation products have only been found in traces (scheme 43).
oxidizing agentCH2Cl2
24, 28 or P23 (cat.)
O
OH O
+ + + etc.
traces
Scheme 43: Epoxidation of cyclohexene with [Ru(bdmpza)Cl(CO)2] (24), [Ru(bdmpzpr)Cl(CO)2] (28) or
EGDMA polymer P23.
In a typical catalysis run, the catalyst (0.03 − 0.10 eq.) was dissolved in CH2Cl2 in a schlenk
tube with a Teflon rotaflow stopcock. Cyclohexene (1.0 eq.) and the oxidizing agent (1.0 –
2.5 eq.) were then added consecutively. The schlenk tube was closed and the mixture was
stirred for 24 h at ambient temperature. After this time, the crude mixture was filtrated over
a short plug of silica gel before being analyzed by GC. The quantification was done by
using calibration curves which have been recorded with standard samples of known

110 III. RESULTS AND DISCUSSION
concentration of the reactant and the product cyclohexene oxide. In order to determine the
yield (y), turnover number (TON) and turnover frequency (TOF), following equations were
used.
a) 100%)(t
(t)y
0reactant
product⋅=
n
n
b) catalyst
product (t)TON
n
n=
c) t
TONTOF =
Equation 3: Calculation of yield, turnover number (TON) and turnover frequency (TOF) achieved in the
catalysis experiments.
Table 22 summarizes the results of the epoxidation studies. It is noteworthy that complex 24
with the unsubstiuted bdmpza ligand 1 showed a higher catalytic activity than the
analogous complex 28 with bridge functionalized bdmpzpr ligand 4. A direct comparison
with 10 mol% catalyst reveals that for complex 24 a yield of 65.1 % (6.6 turnovers) can be
obtained while complex 28 only yields in 46.6 % (4.7 turnovers) when PhIO is used as
oxygen donor. This is surprising since actually the opposite was expected due to the results
shown in chapter 3.1. The changed steric properties caused by the introduced methyl group
should have a positive effect on the labilization of co-ligands and thus facilitate the
generation of free coordination sites required for potential substrates in catalytic cycles.
Furthermore, it is conspicuous that both complexes only show activity when
iodosylbenzene is used as oxidant. If H2O2 or DCPNO were utilized as oxidizing agents,
almost no activity was observed. The initially colorless reaction mixtures turned greenish
after 24 hours which is probably due to the decomposition of the catalyst. It is very likely
that 24 and 28 are only pre-catalysts. As mentioned in the beginning of this chapter, usually
RuVI(=O)2 or RuIV=O are considered as the actual catalytic active species in such
reactions.[265-272, 277, 280, 286] R. SARIEGO et al. suppose in their work described earlier that a
RuIV=O species is formed during the catalytic cycle by hydrolysis of the halide ligand and
oxidation of the complex by PhIO.[281] Furthermore, it was shown that complexes without
CO ligands such as [Ru(bpy)2Cl], only reveal very low activity. Thus, the release of a labile

III. RESULTS AND DISCUSSION 111
bound CO ligand seems to be the crucial factor for the catalytic activity. This could also be
true for the dicarbonyl complexes 24 and 28. In order to check whether the presence of
PhIO is essential for the generation of a catalytically active high valent oxo-species, two
experiments using a catalytic amount of PhIO besides a stoichiometric amount of H2O2 or
DCPNO were made. But the yields obtained in these experiments were as low as 3.3 and
4.6 % respectively indicating that PhIO is required in a stoichiometric amount.
Table 22: Results of epoxidation of cyclohexene with dicarbonyl complexes 24 or 28 and polymer P23.
[Ru]
Oxidant t
[h]
neduct (t0)
[mmol]
ncatalyst
[mmol]
neduct (t)
[mmol]
nproduct (t)
[mmol]
y
[%]
TON TOF
[10−5s−1]
24 PhIO 24 0.503 0.050 0.002 0.327 65.1 6.6 7.58
24 PhIO 22.5 0.503 0.025 0.001 0.342 67.9 13.7 16.88
24 PhIO 24 0.750 0.025 0.001 0.515 68.6 20.6 23.83
24 H2O2 23 0.503 0.050 0.488 0.008 1.6 0.2 0.20
24 DCPNO 23 0.503 0.050 0.472 0.000 0.0 0.0 0.00
24 PhIO
/H2O2
24 0.503 0.050 0.440 0.017 3.3 0.3 0.39
24 PhIO
/DCPNO
24 0.503 0.050 0.434 0.023 4.6 0.5 0.53
28 PhIO 22.5 0.503 0.050 0.092 0.234 46.6 4.7 5.79
P23 PhIO 22 0.503 0.018 0.453 0.013 2.6 0.7 0.92
[a] Solution in H2O (35 %) [b] The reaction was initiated with 10 mol% PhIO for 1h before 90 mol% DCPNO
was added to the reaction mixture.
The catalysis experiment with the EGDMA incorporated complex P23 was carried out in
the same way as for the free complexes 24 and 28 with the only difference that P23 stays
insoluble during the reaction. Unfortunately only very low activity was observed for the
reaction with PhIO. This is surely due to the fact, that iodosylbenzene is a polymeric solid
itself and its solubility in dichloromethane is very low.[287-289] Thus, a heterogeneous
reaction of the undissolved catalyst with the solid polymeric oxidant seems quite
unfavorable. Nevertheless, reaction conditions which can help to circumvent this dilemma
are still to be investigated in future studies.

112 III. RESULTS AND DISCUSSION
For instance, T. G. TRAYOR et al. found that the mixture dichloromethane : methanol : water
80 : 18 : 2 provides good solubility and reactivity of PhIO in iron(III) porphyrin catalyzed
epoxidations.[290] More recently Y. KITA et al. were able to show that iodosylbenzene can be
"activated" with catalytic amounts of quaternary ammonium bromide salts or even KBr.[289,
291-292] On the other hand one could also consider the use of highly soluble derivatives of
iodosylbenzene such as such as 1-(tert-butylsulfonyl)-2-iodosylbenzene, which was reported
recently by J. D. PROTASIEWICZ et al.[293-294]
The preliminary studies summarized in this chapter, reveal the promising potential of both
complexes 24 and 28 as well as their immobilized counterpart P23 as epoxidation catalysts.
During this thesis the TON for complex 24 could be improved up to 20.6 by tuning the
reactant/catalyst ratio which is four-fold higher than the best result obtained for
[Ru(bdmpza)(O2CCH3)(PPh3)]. It is not excluded that an even higher TON can be obtained
by optimization of the reaction conditions. Due to time constraints it was not possible
during this thesis to make a statement about mechanism of the epoxidations. Further
experiments will follow in the future which hopefully will reveal the mechanistic details of
the catalytic cycle.
3.4.2. Synthesis and characterization of tetragonal [Ni(bdtbpza)Cl]
As mentioned before, the importance of bis(pyrazol-1-yl)acetic acids lies in their suitability
for model complexes that mimic the active site of zinc and iron enzymes bearing the facial
2-His-1-carboxylate motif. In previous work of the BURZLAFF group, it was shown that the
reaction of the sterically demanding tert-butyl substituted ligand Hbdtbpza (29) with zinc(II)
gives rise to tetrahedrally enforced complex [Zn(bdtbpza)Cl].[45, 54, 56] In order to expand
this research topic, the tetragonal nickel(II) complex [Ni(bdtbpza)Cl] (30) was investigated
during this thesis as potential model for nickel ureases.
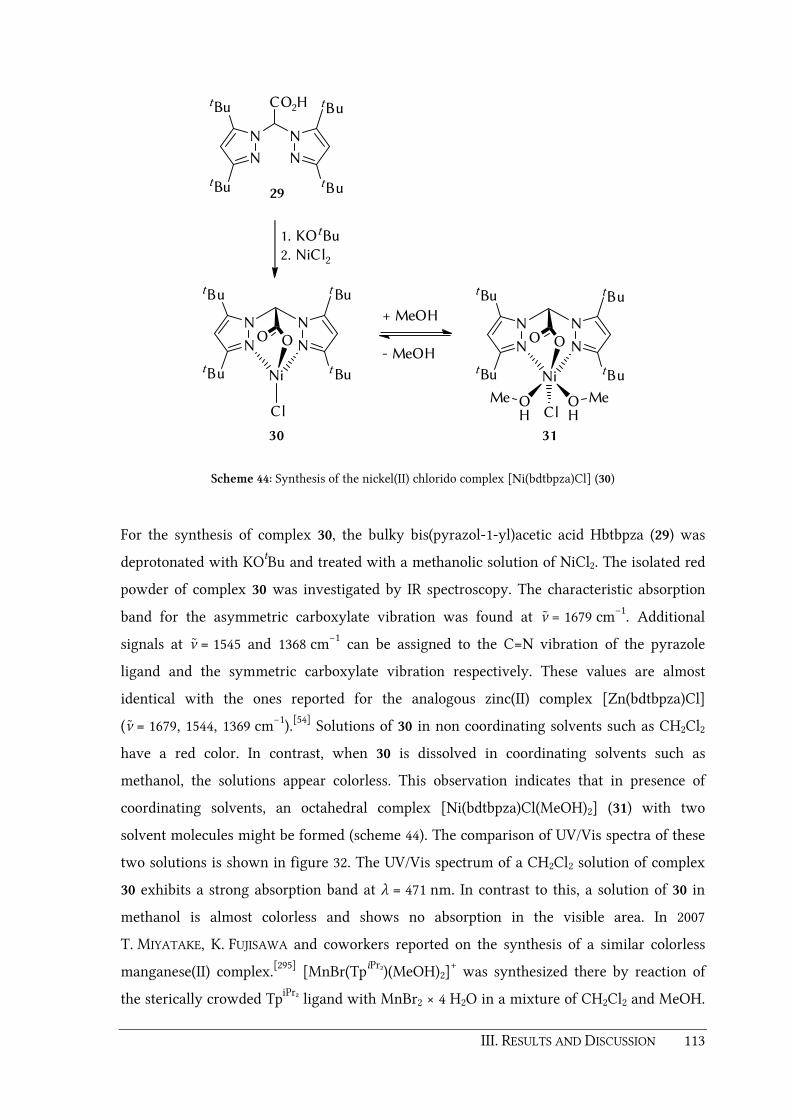
III. RESULTS AND DISCUSSION 113
N
N N
N
tBu
tBu
tBu
tBu
CO2H
29
N N
N N
t Bu
t Bu
tBu
tBu
OO
Ni
Cl
1. KOtBu
2. NiCl2
N N
N N
tBu
tBu
tBu
tBu
OO
Ni
ClO OH
MeH
Me
+ MeOH
- MeOH
3130
Scheme 44: Synthesis of the nickel(II) chlorido complex [Ni(bdtbpza)Cl] (30)
For the synthesis of complex 30, the bulky bis(pyrazol-1-yl)acetic acid Hbtbpza (29) was
deprotonated with KOtBu and treated with a methanolic solution of NiCl2. The isolated red
powder of complex 30 was investigated by IR spectroscopy. The characteristic absorption
band for the asymmetric carboxylate vibration was found at ν ̃ = 1679 cm−1. Additional
signals at ν ̃ = 1545 and 1368 cm−1 can be assigned to the C=N vibration of the pyrazole
ligand and the symmetric carboxylate vibration respectively. These values are almost
identical with the ones reported for the analogous zinc(II) complex [Zn(bdtbpza)Cl]
(ν̃ = 1679, 1544, 1369 cm−1).[54] Solutions of 30 in non coordinating solvents such as CH2Cl2
have a red color. In contrast, when 30 is dissolved in coordinating solvents such as
methanol, the solutions appear colorless. This observation indicates that in presence of
coordinating solvents, an octahedral complex [Ni(bdtbpza)Cl(MeOH)2] (31) with two
solvent molecules might be formed (scheme 44). The comparison of UV/Vis spectra of these
two solutions is shown in figure 32. The UV/Vis spectrum of a CH2Cl2 solution of complex
30 exhibits a strong absorption band at λ = 471 nm. In contrast to this, a solution of 30 in
methanol is almost colorless and shows no absorption in the visible area. In 2007
T. MIYATAKE, K. FUJISAWA and coworkers reported on the synthesis of a similar colorless
manganese(II) complex.[295] [MnBr(TpiPr2)(MeOH)2]+ was synthesized there by reaction of
the sterically crowded TpiPr2 ligand with MnBr2 × 4 H2O in a mixture of CH2Cl2 and MeOH.

114 III. RESULTS AND DISCUSSION
Unfortunately, attempts to confirm the formation of an analogous complex
[Ni(bdtbpza)Cl(MeOH)2] (31) by mass spectroscopy have not been successful so far.
350 400 450 500 550 600 650 700
Wavelength [nm]
Ab
sorb
ance
b
a
Figure 32: UV/Vis spectra of complex [Ni(bdtbpza)Cl] (30) dissolved in CH2Cl2 (a) and MeOH (b).
Furthermore, it has to be mentioned that crystallization of complex 30 with non
coordinating solvents, for instance by layering a CHCl3 solution of 30 with hexane gives red
needles. The result of a X-ray structure analysis of these single crystals confirms both, a κ3-
N,N,O coordination of the Hbdtbpza ligand as well as a tetrahedrally coordinated nickel(II)
center (figure 33 and table 23). The d(N11–Ni1), d(N21–Ni1), and d(Ni1–Cl1) distances are
comparable to those found for the analogous zinc(II) complex [Zn(bdtbpza)Cl][54] and the
similar κ3-N,N,N complex [Ni(TptBu)Cl][296].

III. RESULTS AND DISCUSSION 115
N21
N22
N11
N12 C1
Ni1
Cl1
C2
O1
O2
Figure 33: Molecular structure of the nickel(II) chlorido complex [Ni(bdtbpza)Cl] (30). Thermal ellipsoids are
drawn at the 50% probability level. Most hydrogen atoms and the solvent molecule (CHCl3) have been omitted
for clarity.
Table 23: Selected bond lengths and angles for the molecular structure of the tetrahedral nickel(II) complex 30.
Distances in Å Angles in °
Ni1−N11 1.9881(15) ∡(N11,Ni1,N21) 88.46(6)
Ni1−N21 1.9790(15) ∡(O1,Ni1,N11) 92.36(6)
Ni1−O1 1.9513(14) ∡(O1,Ni1,N21) 91.43(6)
Ni1−Cl1 2.1739(6) ∡(N11,Ni1,Cl1) 131.11(5)
C1–O1 1.220(2) ∡(N21,Ni1,Cl1) 131.23(5)
C1–O2 1.278(2) ∡(O1,Ni1,Cl1) 110.61(4)
Tetrahedral complexes of type [M(Tp)X] with a broad range of sterically demanding Tp
ligands have been extensively studied regarding their catalytic properties. For instance,
O. L. CASAGRANDE JR. et al. described in their work the reaction of [Ni(TpMs)Cl] or
[Ni(TpMs*)Cl] [TpMs = HB(3-mesitylpyrazolyl)3–, TpMs* = HB(3-mesitylpyrazolyl)2(5-
mesitylpyrazolyl)–] with co-catalysts such as methylalumoxane or trimethylaluminium
generating active catalysts for the oligomerization of ethylene with turnover frequencies up

116 III. RESULTS AND DISCUSSION
to 43.1 × 103 h−1 and high selectivity for butane-1.[297]. Similar results have been published
by A. M. ROMANO et al. with the sterically crowded nickel(II) complexes [Ni(Tp*)Cl],
[Ni(TptBu,Me)Cl] and [Ni(TpCum,Me)Cl].[298] To make a contribution to this research area, the
synthesized and characterized κ3-N,N,O complex [Ni(bdtbpza)Cl] (30) is currently
investigated as precatalyst in ethylene polymerization reactions by the group of Prof.
G. LUINSTRA (Institut für Technische und Makromolekulare Chemie, University of
Hamburg).
Some years ago, N. KITAJIMA et al. showed, that a dimeric hydroxido complex
[Ni(TpiPr)2(µ-OH2)2] can be synthesized by reaction of [Ni(TpiPr)Cl] with NaOH (scheme
45).[299] In a recent work, S. HIKICHI et al. reported that condensation of this hydroxo bridged
dimer with TBHP yields in the alkylperoxo complex [Ni(OOtBu)(TpiPr)] which shows
oxidizing ability towards different substrates.[300]
TpiPrNi Cl2 Tpi PrNi
HO
OH
NiTpi PrN
B
N
N N
iPr
iPr
i Pr
i Pr
Ni
OOtBu
H
N
N
iPr
i Pr
2 tBuO2H
-2 H2O2
2 NaOH
-2 NaCl
Scheme 45: Synthesis of the alkylperoxo complex [Ni(OOtBu)(TpiPr)].[299-300]
Inspired from this report, an attempt to synthesize an analogous hydroxido bridged nickel(II)
species with the Hbdtbpza ligand 29 was made (scheme 46). For this purpose, aqueous
NaOH was added dropwise to a solution of complex 30 in CH2Cl2. Unfortunately the IR
monitoring indicated that the formation of the desired dimer [Ni(bdtbpza)2(µ-OH2)2] was
not successful. Instead, the IR spectrum of the isolated green powder revealed the
decomposition of complex 30 to nickel(II) hydroxide.

III. RESULTS AND DISCUSSION 117
N N
N N
tBu
tBu
tBu
tBu
OO
NiNaOH
NN
NN
tBu
tBu
tBu
tBu
O O
Ni
OHHO
N N
N N
tBu
tBu
tBu
tBu
OO
Ni
Cl
30
Scheme 46: Attempted synthesis of [Ni(bdtbpza)2(µ-OH2)2].
3.4.3. HIF-1α prolyl hydroxylase inhibitor studies
The hypoxia-inducible factor (HIF) is an α,β-heterodimeric transcriptional complex which
mediates the response to hypoxia (low oxygen partial pressure) in mammals.[301] Normally,
the HIFβ subunit is present in excess compared to the HIFα subunit. The levels of HIFα are
directly regulated by the availability of oxygen.[301] Under normal oxygen concentrations
(normoxia), levels of the inactive HIFα subunits are downregulated by independent prolyl-
and asparaginyl-hydroxylation pathways (scheme 47).[14, 302-305] Hydroxylation of the
proline residues Pro402 and Pro564 (in human HIF-1α) is catalyzed by HIF prolyl
hydroxylases (PHD1-3). This prolyl hydroxylation generates a binding site for the von
Hippel-Landau tumor suppressor protein (pVHL) which in turn leads to the destruction of
HIFα by the ubiquitin-proteasome degradation pathway.[19-20, 306-307]
Just as with the PHDs, the asparaginyl hydroxylase FIH (factor inhibiting HIF) is a 2-OG
oxygenase. FIH hydroxylates the Asn803 residue of human HIF-1α, a reaction which blocks
the interaction between HIFα and the transcriptional co-activator p300.[308-312] Under
limiting oxygen conditions, prolyl- and asparaginyl-hydroxylation slows and the
heterodimerization of HIFα with HIFβ occurs. This leads to an increased transcription of
hundreds of HIF target genes such as erythropoietin (EPO) or vascular endothelial growth
factor (VEGF) that equilibrate the effects of hypoxia in the hematopoietic, cardiovascular
and respiratory systems.[313-314]

118 III. RESULTS AND DISCUSSION
HYPOXIA
2OG + O 2
succinate
+ CO2
N
O
N
O
HO
2OG + O2
succinate+ CO2
NH2
HN
O
O
NH2
HN
O
O
HO
proteosomal
destruction
no interaction
with p300
HIFαααα
NORMOXIA
HIFβ
p300
HIFα
FIH PHD1-3
transcription
Scheme 47: The HIF pathway under hypoxic and normoxic conditions.[14, 302-304]
Manipulation of the HIF system has great therapeutic potential. Inactivation of HIF could
lead to new therapeutics for pulmonary hypertension or the treatment of tumors. On the
other hand, activation of HIF, e.g. by stabilization of HIFα through inhibition of the PHDs,
could be useful in the treatment of diseases such as myocardial infarction, stroke, peripheral
arterial disease, heart failure, diabetes or respiratory disease.[315] In the past years, several
compounds have been designed and tested regarding their PHD inhibiting properties with
several being in clinical trials.[305, 316-327]
In the scope of an interdisciplinary collaboration, the isoquinoline based small molecule
PHD inhibitor 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido)acetic acid (37) was
synthesized during this thesis and provided to the group of Prof. Dr. K.-U. ECKARDT
(Department of Nephrology and Hypertension, Friedrich-Alexander-University Erlangen-
Nürnberg) for their studies on stabilization of HIF in organ transplants. The overall
synthesis of compound 37 is shown in scheme 48 and was achieved in six steps according to
literature procedures.[328-330]

III. RESULTS AND DISCUSSION 119
O
O
OO
O
NC+
CO2H
O
NMeO2C
32
NH
OH
O
CO2Me
N
OH
Cl
CO2Me
N
OH
Cl
CO2H
b)
c)
35 34 33
N
OH
Cl
NH
O
CO2Et
N
OH
Cl
NH
O
CO2H
e)
d)
f)
a)
3736
Scheme 48: Multi-step synthesis of the PHD inhibitor 37. Reaction conditions: a) DBU, THF, b) HCl, MeOH, c)
POCl3, d) NaOH, EtOH, e) ethyl glycinate hydrochloride, PyBOP, ethyldiisopropylamine, f) NaOH, THF.[328-330]
In the first step, phthalic anhydride was converted with methyl isocyanoacetate as
described by M. SUZUKI et al.[328] The reaction was carried out in the presence of 1,8-
diazabicyclo[5.4.0]undec-7-ene (DBU) using THF as solvent. 2-(4-(Methoxycarbonyl)-
oxazol-5-yl)benzoic acid (32) was obtained in a yield of 89 %. Compound 32 was then
condensed to methyl 4-hydroxy-1-oxo-1,2-dihydroisoquinoline-3-carboxylate (33) by
hydrolysis with a solution of conc. HCl in methanol at 50 °C. The following steps were
synthesized in a slightly modified version of the original procedure described in the patents
EP 1538160 and DE 19746287 by FibroGen Inc. (US) and Hoechst Marion Roussel GmbH (DE)
respectively.[329-330] Selective mono-chlorination of compound 33 was carried out by stirring
in phosphoryl chloride at 70 °C. Aqueous workup gives rise to methyl 1-chloro-4-
hydroxyisoquinoline-3-carboxylate (34) which was deprotected in the next step to 1-chloro-

120 III. RESULTS AND DISCUSSION
4-hydroxyisoquinoline-3-carboxylic acid (35) by alkaline hydrolysis. For the following
peptide coupling reaction, glycine ethyl ester was used together with Benzotriazol-1-
yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) as coupling agent and
ethyl diisopropylamine as auxiliary base. In the original procedure glycine methyl ester was
used but the corresponding ethyl ester is much cheaper. The obtained ethyl 2-(1-chloro-4-
hydroxyisoquinoline-3-carboxamido)acetate 36 was purified by column chromatography on
silica gel with CH2Cl2 as eluent (yield 78.2 %). In the final step the acetate 36 was
deprotected by means of alkaline hydrolysis in THF giving the desired compound inhibitor
37. Successful formation of 37 was verified by 1H NMR spectroscopy. A multiplet 1H
NMRsignal at δ = 4.30 ppm can be assigned to the protons of the methylene group and the
resonances for the aromatic protons show up in the region between δ = 7.96 – 8.33 ppm. The 1H resonances at δ = 8.77 and 13.48 ppm can be assigned to the amide group proton and the
aryl-OH group respectively. The 13C NMR of compound 37 reveals 9 signals between δ =
121.0 and 155.7 ppm for the carbon atoms of the aromatic system. The 13C resonance for the
-CH2- group was found at δ = 41.2 ppm. These spectroscopic values are in good agreement
to the ones reported in literature for compound 37 and its derivatives.[327, 331]
To evaluate the potency of compound 37 to stabilize HIF, several experiments were made in
the ECKARDT group. First, it was applied in vitro and in vivo and compared with the
capacity of HIF activation in response to hypoxia and hypoxia-mimetics. Thus, the renal
tubular cell line HKC-8 was incubated with increasing concentrations of 37 for 6 h.
Exposure to hypoxia (1 % O2) and incubation with dipyridyl (DP; 100 µM) or
dimethyloxalylglycine (DMOG; 1 mM) served as positive controls. Immunoblotting of
protein extracts of cultured cells showed strong accumulation of the HIF-1α isoform after
treatment with compound 37 (figure 34a). As effects of HIF stabilization are mediated by
regulating its target genes, HIF target gene expression was analyzed 6 h after incubation
with compound 37 (500 µM). The glucose transporter 1 (GLUT1) was chosen as a
prototypical HIF target gene. Quantitative real-time PCR of RNA isolates obtained from
HKC-8 cells revealed significant upregulation of GLUT1 mRNA after exposure with
compound 37 (figure 34b). These data show that the provided PHD inhibitor 37 was capable
to stabilize HIF-1α and induce HIF target genes in vitro. Further in vitro analysis showed no
relevant cytotoxicity at least in dosage ranges used in previous experiments.

III. RESULTS AND DISCUSSION 121
HIF-1α
β-Actin
Ctrl H DP
DM
OG
10 50 100
500
Compound 37
Ctrl Hypox DP DMOG 37
0.0
1.0
2.0
3.0
4.0G
LU
T1/
18S
mR
NA
a)
b)
Figure 34: HKC-8 cells were cultivated for 6 h under normoxic, hypoxic conditions or stimulated with
hypoxia-mimetics DP (100 µM), DMOG (1 mM) and increasing concentrations of compound 37 (10 to 500 µM).
a) Western blot analysis using specific antibodies against HIF-1α and β-Actin. (B) Real-time PCR for GLUT-1
mRNA expression in vitro.
In further experiments, compound 37 was injected into C57BL/6 mice at a dose of 40 mg/kg
intraperitoneally based on published data. At normal ambient air conditions in mouse
kidney there is only scattered HIF-1α expression in tubular epithelial cells of the renal
medulla (namely collecting duct cells). Exposure to systemic hypoxia (1%) or functional
anemia (0.1% carbon monoxide) has been shown to markedly increase HIF-1α accumulation
in the kidney.[332] Treatment with compound 37 led to massive accumulation of HIF-1α in
nuclei of tubular epithelial cells in virtually all nephron segments as shown by
immunohistochemistry for HIF-1α (figure 35b). Vehicle-treated animals (figure 35a) showed
HIF-1α staining which was undistinguishable from untreated mice described above. HIF-1α
accumulation was detectable 1 h after application and lasted at least 24 h (not shown).
Compound 37 also stabilized the HIF-2α isoform in interstitial and glomerular endothelial
cells (not shown). As seen under in vitro conditions, compound 37 significantly induced
mRNA transcripts of Glut1 in kidney tissue verified by real-time PCR (figure 35c). These
results show that compound 37 also stabilized HIF-1α and induced HIF target genes in vivo.

122 III. RESULTS AND DISCUSSION
The ECKHARDT group and others previously reported that preconditional HIF activation by
pharmacological inhibition of PHDs can protect the kidney from subsequent (ischemic)
kidney injury.[333]
Ctrl 37
Glu
t1 m
RN
A/1
8S
0.0
1.0
2.0
3.0
4.0
b)a)
c)
Figure 35: Immunhistological staining for HIF-1α in mouse kidney: a) under normoxic conditions, b) 6 h after
intraperitoneal injection of compound 37 (1 mg). c) Real-time PCR for Glut-1 mRNA expression in vivo.
To evaluate the potential protective effect of 37, renal ischemia reperfusion experiments
were performed in mice which were given 40 mg/kg of compound 37 i.p. 6 h before renal
ischemia. Both renal pedicles were occluded for 25 min with vascular clamps, then
reperfusion was allowed by clamp removal and kidney function and morphology was
analyzed 3 days later. Administration of compound 37 before renal ischemia significantly
ameliorated postischemic kidney function and structure. Thus, preconditional HIF
activation using PHD inhibitors might give new opportunities for pharmacological
intervention for ischemic diseases or organ preservation in transplantation.[334]

123


IV. SUMMARY AND OUTLOOK 125
IV. SUMMARY AND OUTLOOK

126 IV. SUMMARY AND OUTLOOK
In previous work of the BURZLAFF group, the design of N,N,O ligands suitable for solid phase
fixation and copolymerization was achieved by manipulation of a bis(pyrazol-1-yl)acetic
acid at the bridging carbon atom. Unfortunately, N,N,O ligands L having a linker at this
position were not useful in the synthesis of aspired model complexes for iron(II) enzymes of
the facial 2-His-1-carboxylate triad such as [Ru(L)Cl(PPh3)2].
Thus, the coordination behavior of bridge functionalized bis(pyrazol-1-yl)acetic acids was
investigated in the first part of this thesis (chapter 3.1). For this purpose, the model ligand
2,2-bis(3,5-dimethylpyrazol-1-yl)propanoic acid (Hbdmpzpr) (4) was synthesized by
methylation of the well known 2,2-bis(3,5-dimethylpyrazol-1-yl)acetic acid (Hbdmpza) (1)
at the bridging carbon atom. Different transition metal complexes of this ligand such as
[M(bdmpzpr)(CO)3] [M = Mn (5), Re (6)] or [Cu(bdmpzpr)2] (7) have been synthesized and
characterized by NMR and/or IR spectroscopy, mass spectrometry and elemental analysis. It
was observed that the introduced methyl group has a crucial influence on the structure and
reactivity of transition metal complexes of ligand 4. Single crystal structure analyses of
synthesized complexes 7 and 5 revealed that steric interactions of the methyl group at the
bridging position and the pyrazolyl substituents at position 5 are responsible for a "twist" of
one pyrazolyl unit. It was found that this effect strongly depends on the bulkiness of the
whole system and is more pronounced in the presence of bulky co-ligands. The steric
tension in the bisphosphine complex [Ru(bdmpzpr)Cl(PPh3)2] (9) for instance is responsible
for the enhanced dissociation of one PPh3 ligand. The resulting coordinatively unsaturated
16 VE fragment [Ru(bdmpzpr)Cl(PPh3)] (9c) readily reacts with N2 molecules of the inert
gas atmosphere to form the dinitrogen complex [Ru(bdmpzpr)Cl(N2)(PPh3)] (10). In contrast,
treatment of [Ru(bdmpza)Cl(PPh3)2] (8) with N2 showed only traces of a N2 product in the
IR spectrum. Although similar dinitrogen complexes with Tp and Cp ligands are known in
literature, complex 10 represents the first example for a N2 complex bearing a bis(pyrazol-1-
yl)acetato ligand and serves as a proof for the changed reactivity of complexes with bridge
functionalized ligands compared to those of unmodified bis(pyrazol-1-yl)acetic acids. This
was further demonstrated by experiments with carbon monoxide and sulfur dioxide. If
complex 8 was treated with CO or SO2, only one isomer [Ru(bdmpza)Cl(CO)(PPh3)] (11) or
[Ru(bdmpza)Cl(PPh3)(SO2)] (13) was formed in which the chlorido ligand was positioned
trans towards the carboxylate donor. In contrast, treatment of complex 9 with CO or SO2
yielded a mixture of two isomers, 12a/b or 14a/b respectively which might be caused by
enhanced dissociation and re-binding of a PPh3 ligand.

IV. SUMMARY AND OUTLOOK 127
In upcoming work coordinatively unsaturated fragments such 9c will be further
investigated, since such compounds are important intermediates in organometallic and
inorganic transformations and are of interest as potential catalysts and for the activation of
small molecules.
N
N N
N
Me
Me Me
Me
CO2H
R = H (1)R = Me (4)
R
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
Me
10
R = Me
N2
R = H
N2
X
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (11), SO2 (13)
R = HCO or SO2
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (12a), SO2 (14a)
N N
N N
Me
Me
Me
Me
OO
Ru
LPh3P Cl
Me
L = CO (12b), SO2 (14b)
+
Me
R = MeCO or SO2
Considering the results obtained above, an alternative strategy for the design of ligands
suitable for copolymerization was investigated in the second part of this work (chapter 3.2).
The new N,N,O ligand 2,2-bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid (Hbdmvpza) (18)
was synthesized in a four-step synthesis starting form commercially available

128 IV. SUMMARY AND OUTLOOK
3,5-dimethylpyrazole. The advantage of this ligand is that the functional units for
immobilization, i.e. the polymerizable vinyl linkers are no longer positioned at the bridging
carbon atom and do not influence the remaining space of κ3-N,N,O bound transition metal
fragments.
In order to investigate its polymerization behavior, ligand 18 was copolymerized with either
MMA or EGDMA in presence of the radical initiator AIBN. The amount of embedded
ligand per gram of polymer (P18a: 0.746 mmol g–1, P18c: 0.341 mmol g–1) was determined
by the nitrogen content of the resulting polymers (% N value of the elemental analysis).
Another advantage of ligand 18 is its crosslinking ability due to the presence of two vinyl
groups per ligand molecule. Thus, a unique homopolymer P18d was synthesized by self-
polymerization of 18. A very high incorporation rate of 3.05 mmol ligand per gram of
polymer was found for P18d. Solid phases P18a/c have been treated with precursor
compounds [MBr(CO)5] (M = Mn, Re) in a heterogeneous reaction to form the incorporated
tricarbonyl manganese and rhenium complexes P18a-Mn/Re and P18c-Mn/Re. Besides this
heterogeneous strategy, one can also embed transition metal fragments into polymeric
matrices by copolymerizing appropriate complexes directly. This was demonstrated by
copolymerization of the dicarbonyl ruthenium complex [Ru(bdmvpza)Cl(CO)2] (23) with
EGDMA. The successful incorporation of the metals was verified by ICP-AES and AAS
measurements of the obtained polymers P18a-Mn/Re, P18c-Mn/Re and P23. On the other
hand, IR spectroscopic investigations on these polymers revealed that the embedded metal
centers are bound in a fac-κ3-N,N,O coordination mode. For comparison purposes, the
homogeneous complexes [M(bdmvpza)(CO)3] [M = Mn (19), Re (20)] and [Cu(bdmvpza)2]
(25) have been synthesized and characterized by NMR and IR spectroscopy, single crystal
structure determination and mass spectrometry.
To investigate whether the fixation of ligand 18 allows the control of coordination
geometries in crosslinked polymers, an embedded copper(II) complex P18a-Cu was
synthesized by heterogeneous reaction of MMA copolymer P18a with CuCl2. UV/Vis
spectroscopic investigation of P18a-Cu revealed a bathochromic shift by 64 nm compared
to the UV/Vis spectrum of the homogeneous complex [Cu(bdmvpza)2] (25). This implies,
that copper(II) centers embedded in this copolymer are mainly coordinated by one ligand
moiety and the unwanted formation of bisligand complexes [M(L)2] can be prevented.
Besides copper, incorporation of iron(II) was also accomplished and proven by AAS

IV. SUMMARY AND OUTLOOK 129
measurements. It was found that up to 65 % of the ligand sites in the MMA-copolymer
P18a-Fe can be occupied by ferrous iron centers.
N
N N
N
Me
Me
Me
Me
CO2H
L (18)
Homopolymer
P18d
EGDMA- bzw. MMA
copolymers
P18a-c
[M(L)(CO)3]-
polymersP18a-Mn/ReP18c-Mn/Re
[M(L)]-
polymersP18a-Fe/CuP18c-Fe
[Ru(L)Cl(PPh3)2](21)
[Ru(L)Cl(CO)2]
(23)
[M(L)(CO)2]-polymer
(P23)
EGDMA[MBr(CO)5] MCl2
[Cu(L)2] (25)[Mn(L)(CO)3] (19)
[Re(L)(CO)3] (20)
In contrast to bridge functionalized ligands for solid phase fixation and copolymerization
which have been investigated in the past, ligand 18 is able to bind ruthenium(II) centers
with a fac-κ3-N,N,O motif. This was verified by synthesis and characterization of the
bisphosphine complex [Ru(bdmvpza)Cl(PPh3)2] (21). This important property of ligand 18
was used for an imprinting approach in the third part of this thesis (chapter 3.3). Two new
2-oxocarboxylato complexes as structural models for the active site of 2-OG dependent
iron(II) oxygenases have been synthesized and characterized, namely
[Ru(bdmvpza)(BF)(PPh3)] (26) (BF = benzoylformate) and [Ru(bdmvpza)(NOG)(PPh3)] (27)
(NOG = N-oxalylglycine). It has to be mentioned that NOG is isostructural to 2-oxoglutaric
acid and thus is a cosubstrate analogue and inhibitor. In other words, complex 27 can be
considered as a model complex for a 2-OG-dependent iron(II) oxygenase inhibited by NOG
and is well suited as a “dummy” complex for the generation attempt which might mimic
the catalytic activity of such an oxygenase enzyme. Both complexes 26 and 27 have been

130 IV. SUMMARY AND OUTLOOK
copolymerized with EGDMA in the presence of AIBN and a porogene solvent and the
resulting solid phases P26 and P27 have been analyzed by UV/Vis spectroscopy. It was
found that the absorption maxima of the polymerized complexes are almost identical to
those recorded for the corresponding homogeneous compexes which proves the successful
incorporation of 26 or 27 into a polymeric matrix. In order to remove the dummy
ruthenium(II) fragments and to generate the imprint, the polymers P26 and P27 have been
treated with excess of PMe3. ICP-AES measurements have confirmed that up to 51% of the
ruthenium templates can be removed by this procedure. In upcoming studies, it has to be
examined whether these imprinted polymers are able to catalytically oxidize substrates
such as cyclohexene on addition of iron(II) and 2-OG in presence of dioxygen or a oxidizing
agent such as H2O2. First experiments of incorporation of the generated cavities with iron(II)
are on their way.
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
R
5a4a
3a3b
4b5b
R = Ph (26)
R = NHCH2CO2H (27)
N N
N N
Me
Me
Me
Me
OO
P26X, P27X
H
Cavity
In the final part of this thesis (chapter 3.4) the catalytic activity of dicarbonyl complexes
[Ru(bdmpza)Cl(CO)2] (24) and [Ru(bdmpzpr)Cl(CO)2] (28) towards the epoxidation of
cyclohexene with different oxidizing agents was investigated. It was found that both
complexes are able to catalyze the selective formation of cyclohexene oxide with iodosyl
benzene. During this thesis the TON for complex 24 could be improved up to 20.6 by tuning
the reactant/catalyst ratio and it is very likely that an even higher TON can be obtained by
further optimization of the reaction conditions. Due to time constraints it was not possible
during this thesis to make a statement about the mechanism of the epoxidations. Further
experiments will follow in the future which hopefully will reveal the mechanistic details of
the catalytic cycle.

IV. SUMMARY AND OUTLOOK 131
PhIO
CH2Cl2
24 or 28
O
OH O
+ + + etc.
traces
Unfortunately the polymer embedded counterpart P23 only revealed very low catalytic
activity under analogous reaction conditions. Obviously, a heterogeneous reaction of the
undissolved catalyst with the also undissolved solid polymeric oxidant seems quite
unfavorable. Reaction conditions which can help to circumvent this problem are still to be
investigated in future studies either by using more suitable solvent mixtures or soluble
iodosyl benzene derivatives such as 1-(tert-butylsulfonyl)-2-iodosylbenzene.[293-294]
In a sub-project of this thesis, the small molecule PHD inhibitor 2-(1-chloro-4-
hydroxyisoquinoline-3-carboxamido)acetic acid (37) was synthesized according to literature
procedures and provided to the group of Prof. Dr. K.-U. ECKARDT (Department of
Nephrology and Hypertension, Friedrich-Alexander-University Erlangen-Nürnberg). It was
found that compound 37 is capable to stabilize HIF 1α and induce HIF target genes in vitro
and in vivo. Preconditional HIF activation using PHD inhibitors such as compound 37 might
give new opportunities for pharmacological intervention for ischemic diseases or organ
preservation in transplantation.


V. ZUSAMMENFASSUNG UND AUSBLICK 133
V. ZUSAMMENFASSUNG UND AUSBLICK

134 V. ZUSAMMENFASSUNG UND AUSBLICK
In früheren Arbeiten in der Arbeitsgruppe BURZLAFF erfolgte die Darstellung von zur
Immobilisierung geeigneten Bis(pyrazol-1-yl)essigsäuren durch eine Funktionalisierung am
verbrückenden Kohlenstoffatom. Es wurde jedoch des Öfteren beobachtet, dass
Substituenten in dieser Position das Koordinationsverhalten des Liganden ungünstig
verändern können. Beispielsweise konnten mit derartigen Liganden L bisher keine
Ruthenium(II)-Komplexe [Ru(L)Cl(PPh3)2] synthetisiert werden, welche jedoch angestrebte
Modellverbindungen für Eisen(II)-Enzyme mit einem facialen 2-His-1-carboxylat Motiv
darstellen.
Um die Ursachen hierfür aufzuklären, wurde im ersten Teil der vorliegenden Arbeit das
Koordinationsverhalten brückenfunktionalisierter Bis(pyrazol-1-yl)essigsäure Liganden
näher untersucht. Hierzu wurde der neue Modell-Ligand 2,2-Bis(3,5-dimethylpyrazol-1-
yl)propansäure (Hbdmpzpr) (4) ausgehend von 2,2-Bis(3,5-dimethylpyrazol-1-yl)essigsäure
(Hbdmpza) (1) synthetisiert und zu verschiedenen Übergangsmetallkomplexen umgesetzt.
Es konnte gezeigt werden, dass sowohl die Struktur als auch die Reaktivität von
Komplexverbindungen des Liganden 4 stark durch die Methylgruppe an der
Brückenposition beeinflusst wird. Röntgenstrukturanalysen der Komplexe [Cu(bdmpzpr)2]
(7) und [Mn(bdmpzpr)(CO)3] (5) zeigen, dass es durch die eingeführte Methylgruppe zu
abstoßenden Wechselwirkungen mit den Methyl-Substituenten der Pyrazoleinheiten
kommt. Dies führt zu einer deutlichen Verdrehung eines der beiden Pyrazoldonoren. Es
stellte sich heraus, dass dieser Effekt umso ausgeprägter ist, je sterisch anspruchsvoller der
gesamte Metall-Komplex ist. Im Bisphosphin-Komplex [Ru(bdmpzpr)Cl(PPh3)2] (9)
beispielsweise führte die Verdrängung eines PPh3-Liganden zur Ausbildung eines
koordinativ ungesättigen 16VE-Fragmentes [Ru(bdmpzpr)Cl(PPh3)] (9c) welches bereitwillig
mit vorhandenen N2-Molekülen des Schutzgases zum Distickstoff-Komplex
[Ru(bdmpzpr)Cl(N2)(PPh3)] (10) reagierte. Analoge Distickstoff-Komplexe mit Tp und Cp
Liganden sind zwar literaturbekannt, jedoch stellt Verbindung 10 das erste bekannte
Beispiel für einen Distickstoff-Komplex eines Bis(pyrazol-1-yl)essigsäure Liganden dar und
ist im Hinblick auf die Aktivierung kleiner Moleküle von besonderem Interesse.
Blindversuche mit dem unmodifizierten Hbdmpza Liganden 1 zeigten, dass dieser nicht zur
Bildung eines analogen Distickstoff-Komplexes [Ru(bdmpza)Cl(N2)(PPh3)] fähig ist. Ein
weiterer Beleg dafür, dass die Reaktivität der Metallkomplexe des funktionalisierten
Liganden 4 gegenüber analogen Komplexen des ursprünglichen Liganden 1 deutlich
verändert ist, zeigten die Umsetzungen der Bisphosphin-Komplexe [Ru(bdmpza)Cl(PPh3)2]

V. ZUSAMMENFASSUNG UND AUSBLICK 135
(8) und [Ru(bdmpzpr)Cl(PPh3)2] (9) mit Kohlenmonoxid bzw. Schwefeldioxid. Die
Umsetzung des Komplexes 8 mit CO bzw. SO2 lieferte jeweils Isomer
[Ru(bdmpza)Cl(CO)(PPh3)] (11) bzw. [Ru(bdmpza)Cl(PPh3)(SO2)] (13) in dem der
Chloridoligand trans zum Carboxylatdonor gebunden ist. Dagegen führte die Umsetzung
des Komplexes 9 mit CO bzw. SO2 jeweils zu einem Gemisch zweier Isomere (12a/b bzw.
14a/b) was vermutlich auf eine einfachere Dissoziation und Wiederanbindung des PPh3-
Liganden zurückzuführen ist.
N
N N
N
Me
Me Me
Me
CO2H
R = H (1)R = Me (4)
R
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
Me
10
R = Me
N2
R = H
N2
X
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (11), SO2 (13)
R = HCO oder SO2
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P L
L = CO (12a), SO2 (14a)
N N
N N
Me
Me
Me
Me
OO
Ru
LPh3P Cl
Me
L = CO (12b), SO2 (14b)
+
Me
R = MeCO oder SO2

136 V. ZUSAMMENFASSUNG UND AUSBLICK
Unter Berücksichtigung dieser Beobachtungen wurde im zweiten Teil der Arbeit (Kapitel
3.2) der zur Copolymerisation geeignete N,N,O-Ligand 2,2-Bis(3,5-dimethyl-4-vinyl-pyrazol-
1-yl)essigsäure (Hbdmvpza) (18) in einer vierstufigen Synthese ausgehend von 3,5-
Dimethylpyrazol dargestellt. Der entscheidende Vorteil dieses neuartigen Liganden ist, dass
sich die polymerisationsaktiven Substituenten nicht wie bisher üblich am
Brückenkohlenstoffatom befinden, sondern als Substituenten in Position 4 der Pyrazolringe
angebracht sind und somit das Koordinationsverhalten des Liganden nicht nachteilig
beeinflussen.
Copolymerisation des Liganden 18 mit MMA oder EGDMA in Anwesenheit des
Radikalstarters AIBN lieferte Festphasen mit einem Ligandanteil von 0.746 mmol bzw.
0.341 mmol pro Gramm Polymer. Zudem war es mit dem neuartigen Liganden erstmals
möglich auch ein Homopolymer P18d mit sehr hohem Ligandgehalt von 3.05 mmol pro
Gramm Polymer zu synthetisieren, da der Ligand zwei Vinylfunktionen pro Molekül besitzt
und somit ein quervernetzendes Monomer darstellt. Die durch Copolymerisation mit MMA
oder EGDMA erzeugten Festphasen P18a/c wurden in heterogenen Reaktionen mit den
Precursorverbindungen [MBr(CO)5] (M = Mn, Re) zu den immobilisierten Mangan- bzw.
Rheniumtricarbonyl-Komplexen P18a-Mn/Re bzw. P18c-Mn/Re umgesetzt. Neben dieser
heterogenen Reaktionsführung gibt es aber auch die Möglichkeit die Immobilisierung direkt
durch Polymerisation entsprechender Komplexe durchzuführen. Dies wurde beispielhaft an
der Copolymerisation des Dicarbonyl-Komplexes [Ru(bdmvpza)Cl(CO)2] (23) mit EGDMA
verdeutlicht. Zum einen konnten mittels AAS und ICP-AES Messungen die Aufnahme der
jeweiligen Übergangsmetalle in die polymeren Festphasen bestätigt werden. Andererseits
konnte durch IR Spektroskopie der Polymere P18a-Mn/Re, P18c-Mn/Re und P23 bewiesen
werden, dass die im Polymer verankerten Ligandfragmente in der Lage sind Metallzentren
wie erwünscht nach einem fac-κ3-N,N,O Motiv zu binden. Für Vergleichszwecke wurden
hierzu auch die homogenen Komplexe [M(bdmvpza)(CO)3] [M = Mn (19), Re (20)] und
[Cu(bdmvpza)2] (25) synthetisiert und mit Hilfe von NMR und IR Spektroskopie,
Röntgenstrukturanalysen und Massenspektrometrie charakterisiert.
Um zu Untersuchen, ob es durch die Immobilisierung des Liganden 18 möglich ist,
Koordiationsgeometrien zu kontrollieren, wurde das MMA Copolymer P18a in einer
heterogenen Reaktion mit CuCl2 umgesetzt. Vergleicht man das UV/Vis-Spektrum des
resultierenden kupferhaltigen Polymers P18a-Cu mit dem des freien Bisligand-Komplexes
V. ZUSAMMENFASSUNG UND AUSBLICK 137
[Cu(bdmvpza)2] (25), so lässt sich für P18a-Cu eine bathochrome Verschiebung des
Absorpionsmaximums um 64 nm feststellen. Dies deutet darauf hin, dass es aufgrund der
Quervernetzung in der polymeren Festphase möglich ist, die Bildung von unerwünschten
Komplexen des Typs [M(L)2] zu umgehen. Die Umsetzung der polymeren Festphasen
P18a/c mit Eisen(II) zu den eisenhaltigen Polymeren P18a-Fe und P18c-Fe zeigte zudem,
dass bis zu 64 % der einpolymerisierten Ligandseiten mit Eisen-Ionen besetzt werden
können.
N
N N
N
Me
Me
Me
Me
CO2H
L (18)
Homopolymer
P18d
EGDMA- bzw. MMA
Copolymere
P18a-c
[M(L)(CO)3]-
Polymere
P18a-Mn/ReP18c-Mn/Re
[M(L)]-
Polymere
P18a-Fe/CuP18c-Fe
[Ru(L)Cl(PPh3)2]
(21)
[Ru(L)Cl(CO)2]
(23)
[M(L)(CO)2]-Polymer
(P23)
EGDMA[MBr(CO)5] MCl2
[Cu(L)2] (25)[Mn(L)(CO)3] (19)
[Re(L)(CO)3] (20)
Im Gegensatz zu brückenfunktionalisierten Liganden zur Festphasenfixierung ist es mit dem
Liganden 18 erstmals möglich, sterisch anspruchsvolle Ruthenium(II)-Zentren mit einem
fac-κ3-N,N,O Motiv zu binden. Dies wurde mit der erfolgreichen Synthese des Bisphosphin-
Komplexes [Ru(bdmvpza)Cl(PPh3)2] (21) bestätigt. Diese wichtige Eigenschaft wurde im
weiteren Verlauf der Promotion (Kaptiel 3.3) zur Erzeugung molekular geprägter Polymere
(molecular imprinted polymers) ausgenutzt. Für dieses Konzept wurden die beiden
Ruthenium(II)-2-Oxocarboxylato-Komplexe [Ru(bdmvpza)(BF)(PPh3)] (26) (BF =
Benzoylformiat) und [Ru(bdmvpza)(NOG)(PPh3)] (27) (NOG = N-Oxalylglycin) als
138 V. ZUSAMMENFASSUNG UND AUSBLICK
strukturelle Modelle für die aktiven Zentren 2-Oxoglutarat-abhängiger Eisen(II)-Enzyme
synthetisiert und spektroskopisch charakterisiert. Besonders hervorzuheben ist, dass
N-Oxalylglycin strukturanalog zum natürlich vorkommenden Cosubstrat 2-Oxoglutarsäure
ist und somit einen Inhibitor für 2-OG-abhängige Eisen(II)-Oxygenasen darstellt. Daher
eignet sich der Komplex 27 besonders gut als Templat für die Erzeugung eines geprägten
Polymers, mit dem die katalytische Funktion solcher Enzyme nachgeahmt werden kann.
Copolymerisation von 26 bzw. 27 erfolgte in Gegenwart von EGDMA und einem
porogenen Solvens. Die UV/Vis-Spektren der so erzeugten Polymere P26 bzw. P27 zeigen
nahezu identische Absorptionsmaxima im Vergleich zu den freien Komplexen 26 bzw. 27
und bestätigen somit die erfolgreiche Einbindung der Komplexe in eine Polymermatrix. Um
die inaktiven Ruthenium(II)-Fragmente zu entfernen und somit die erwünschte Prägung zu
erzeugen, wurden P26 und P27 mit PMe3 behandelt. Durch ICP-AES Messungen konnte ein
deutlicher Rückgang des Rutheniumgehaltes von bis zu 51 % beobachtet werden. Zukünftige
Studien werden zeigen, ob diese geprägten Umgebungen bei Zugabe von Eisen(II) und
2-Oxoglutarat dazu fähig sind, Substrate wie beispielsweise Cyclohexen mit
atmosphärischem Sauerstoff oder einem Oxidationsmittel wie H2O2 katalytisch zu oxidieren.
In ersten Versuchen hierzu wurden geprägten Umgebungen bereits mit Eisen(II) besetzt.
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
R
5a4a
3a3b
4b5b
R = Ph (26)
R = NHCH2CO2H (27)
N N
N N
Me
Me
Me
Me
OO
P26X, P27X
H
Cavity
Im letzten Teil (Kapitel 3.4) der vorliegenden Arbeit wurde unter anderem die Eignung der
Ruthenium(II)-Dicarbonylkomplexe [Ru(bdmpza)Cl(CO)2] (24) und [Ru(bdmpzpr)Cl(CO)2]
(28) als Katalysatoren für die Epoxidierung von Cyclohexen mit Oxidationsmitteln wie
H2O2 und Iodosobenzol untersucht. Gaschromatographische Untersuchungen zeigten, dass
beide Komplexe die selektive Bildung von Cyclohexenoxid katalysieren. Für den Komplex

V. ZUSAMMENFASSUNG UND AUSBLICK 139
[Ru(bdmpza)Cl(CO)2] konnte im Rahmen dieser Promotion eine Wechselzahl (TON,
turnover number) von 20.6 ermittelt werden. Durch weitere Optimierung der
Katalysebedingungen ist eine Steigerung der katalytischen Aktivität derartiger Komplexe
durchaus denkbar.
PhIO
CH2Cl2
24 oder 28
O
OH O
+ + + etc.
Spuren
Der durch Copolymerisation immobilisierte Dicarbonylkomplex P23 zeigte mit
Iodosobenzol fast keine katalytische Aktivität. Dies ist wahrscheinlich darauf
zurückzuführen, dass sowohl die polymere Festphase als auch Iodosobenzol in dem
gewählten Solvens (CH2Cl2) in unlöslicher Form vorliegen und somit eine Reaktion
erschwert ist. In zukünftigen Arbeiten soll daher die Oxidationskatalyse mit dem Polymer
P23 mit löslichen Iodosobenzolderivaten wie z.B. 1-(tert-Butylsulfonyl)-2-iodosobenzol[293-
294] bzw. in besser geeigneten Lösungsmitteln durchgeführt werden.
In einem weiteren Teilprojekt dieser Arbeit, wurde der PHD Inhibitor N-((1-Chlor-4-
hydroxyisochinolin-3-yl)carbonyl)glycin (37) nach Literaturvorschriften synthetisiert und
an die Arbeitsgruppe Prof. Dr. K.-U. ECKARDT (Department of Nephrology and
Hypertension, Friedrich-Alexander-University Erlangen-Nürnberg) für Experimente zur
Stabilisierung von HIF bereitgestellt. Es zeigte sich, dass die Verbindung 37 in der Lage ist,
HIF 1α zu stabilisieren und HIF-Zielgene sowohl in vitro als auch in vivo zu induzieren. Die
Aktivierung von HIF durch Inhibitormoleküle wie z.B. 37 öffnet somit neue Möglichkeiten
für die Behandlung von ischämischen Erkrankungen oder die Konservierung von
Spenderorganen bei Transplantationen.


VI. EXPERIMENTAL SECTION 141
VI. EXPERIMENTAL SECTION

142 VI. EXPERIMENTAL SECTION
6.1. General Remarks
6.1.1. Working techniques
All air sensitive compounds were prepared under dry nitrogen atmosphere (or dry argon
where mentioned) using conventional Schlenk techniques. Purchased solvents (p.a. grade,
<50 ppm H2O) were degassed prior to use and stored under N2 atmosphere. In case of N2
sensitive complexes, all solvents (including deuterated solvents) have been degassed with at
least 5 freeze-pump-thaw cycles and flushed with dry argon immediately before use.
6.1.2. Chemicals
Following chemicals were used as purchased without further purification:
▶ 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU)
▶ 2,2'-Azobis(2-methylproprionitrile) (AIBN)
▶ 2,6-Dichloropyridine N-oxide (DCPNO)
▶ 3,5-Dimethylpyrazole
▶ (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP)
▶ Benzoylformic acid
▶ Benzyltriethylammonium chloride (BTEAC)
▶ Bromine
▶ n-Butyllithium
▶ Copper(II) acetate monohydrate
▶ Cyclohexene
▶ Decane
▶ Dichloroacetic acid
▶ Dimanganese decacarbonyl
▶ Dirhenium decacarbonyl
▶ Ethyl oxalyl chloride
▶ Ethyldiisopropyl amine
▶ Formic acid
▶ Glycine ethyl ester hydrochloride
▶ Hydrogen peroxide (35 wt.% solution in water)
▶ Iron(II) chloride

VI. EXPERIMENTAL SECTION 143
▶ Iron(II) sulfate heptahydrate
▶ Lithium diisopropylamide (LDA)
▶ Methyl iodide
▶ Methyl isocyanoacetate
▶ Methyltriphenylphosphonium bromide
▶ Nickel(II) chloride
▶ Paraformaldehyde
▶ Phosphorous oxychloride (POCl3)
▶ Phthalic anhydride
▶ Potassium tert-butoxide (KOtBu)
▶ Ruthenium(III) chloride hydrate (35 – 40 % Ru)
▶ Thallous acetate
▶ Trimethylphosphine (PMe3)
▶ Triphenylphosphine (PPh3)
The following chemicals were synthesized by literature methods:
▶ 2,2-bis(3,5-dimethylpyrazol-1-yl)acetic acid (Hbdmpza)[54] (1)
▶ Bis(3,5-dimethylpyrazol-1-yl)methane[247] (15)
▶ Bis(3,5-dimethyl-4-formylpyrazol-1-yl)methane[248] (16)
▶ Hbdtbpza[54] (29)
▶ Iodosylbenzene[335]
▶ N-oxalylglycine[318, 336]
▶ [Cu(bdmpzmp)2][117, 184] (3)
▶ [Mn(bdmpzmp)(CO)3][117, 184] (2)
▶ [MnBr(CO)5][337]
▶ [ReBr(CO)5][338]
▶ [Ru(bdmpza)Cl(CO)(PPh3)][215] (11)
▶ [Ru(bdmpza)Cl(PPh3)2][59] (8)
▶ [RuCl2(CO)2]n[339]
▶ [RuCl2(PPh3)3][188]
Prior to their use, methyl methacrylate (MMA) and ethylene glycol dimethacrylate
(EGDMA) were washed with NaOH (1 M, 3×) and brine (3×), dried over anhydrous Na2SO4,
distilled and stored at –30 °C.

144 VI. EXPERIMENTAL SECTION
6.1.3. Instrumentation
Elemental analyses were determined with a Euro EA 3000 (Euro Vector) and EA 1108
(Carlo Erba) instrument (σ = ± 1% of the measured content). IR spectra were recorded with
an Excalibur FTS-3500 FTIR in CaF2 cuvets (0.2 mm), as KBr pellets or as nujol mulls. KBr
pellets were prepared using a Perkin-Elmer hydraulic press (10 t cm2). 1H,
13C,
31P and 2D
NMR spectra were measured with a Bruker AC 250 and a Bruker DPX300 Avance
instrument. The δ values are given relative to tetramethylsilane (1H), the deuterated solvent
(13C) or to PPh3 (δ = –4.72 ppm) as internal standard (31P). Mass spectra were recorded with
a Finnigan MAT 312 and a Jeol JMS-700 instrument by using either FD or FAB technique
with 3-nitrobenyzl alcohol (NBOH) as matrix. UV/Vis spectra were recorded on a Varian
Cary-50 spectrometer in a quartz cuvette (d = 1 cm), as nujol mulls or as polymer films (see
below). X-ray structure determinations were carried out on a modified Siemens P4
diffractometer, a Bruker-Nonius Kappa-CCD and an Enraf Nonius FR590 diffractometer.
Gas chromatography was performed on a Shimadzu GC17A instrument equipped with a
flame ionization detector using a Roticap-5 column (length: 60 m, ID: 0.25 mm, Film:
0.25µm). Melting points were measured with a Electrothermal digital melting point
apparatus (capillary).
6.1.4. Polymer analysis
In case of the polymers P17a and P18a-d, the amount of incorporation was determined by
the nitrogen content (% N) of the elemental analysis of the resulting polymer using the
following equation.
10mol14g4
%N
Polymer g
Ligand mmol1
⋅⋅⋅
=−
Size exclusion chromatography (SEC) was in cooperation with E. HÜBNER (Research
Centre Jülich, Institute of Solid State Research, 52425 Jülich, Germany) The experiments
were carried out using a PL-GPC 220 instrument with three Polypore (Polymer Laboratories)
5 micron colums at 30 °C (Solvent: THF/N,N-dimethylacetamide 85:15 v/v; flow rate: 1
mL/min, calibration: conventional polystyrene calibration).
In case of P26 and P27, the UV/Vis spectra were recorded using nujol mulls of the finely
ground polymer samples. Prior to these measurements, a baseline was collected with an

VI. EXPERIMENTAL SECTION 145
analogously prepared mull of PEGDMA. In case of MMA copolymer P18a-Cu, a thin
polymer pellet was prepared by using a hydraulic press. A drop of mineral oil (spectroscopy
grade) was used to make the pellet transparent. A baseline for this measurement was
collected using a PMMA pellet of the same thickness which was prepared analogously.
Atomic absorption spectrometry (AAS) was carried out using a Perkin–Elmer 5100 F-AAS
with AS-90 sample automation. Method of calibration was standard addition. Flow rates
acetylene/air [L min–1]: Cu 0.9/9.9, Mn 2.0/8.1, Fe 2.0/9.2. Wavelength/spectral band width
[nm]: Cu 324.8/0.7, Mn 279.8/0.7, Fe 248.8/0.2.
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements
were carried out using a Spectro Analytical Instruments Spectroflame instrument in the
radial plasma view mode. RF frequency: 24.12 MHz (1200 W) Argon gas flow rates: 12.8
(coolant), 0.7 (auxiliary), 0.5 (carrier) L min–1. Wavelength for sequential detection mode:
Re 227.525 and 221.426 nm, Ru 267.876 and 240.272 nm. Method of calibration was standard
addition.
Sample preparation for AAS and ICP-AES measurements: A suspension of the according
polymer (50 – 80 mg) in H2SO4 (p.a., conc., 3.00 mL) was heated for 2 h at 140 C in a 25 mL
volumetric flask. The black solution was cooled down to room temperature and treated
carefully with H2O2 (35 wt.% solution in water). The mixture was heated at 140 °C for 12 h
to give a clear colourless solution. If the colour remained brownish, further H2O2 (0.5 mL)
was added and stirring was continued for 2 h. The colourless solution was cooled down to
room temperature, diluted to 25.0 mL with nitric acid (2 wt.% solution in water) and finally
analyzed by AAS or ICP-AES. Every metal analysis was done in duplicates.

146 VI. EXPERIMENTAL SECTION
6.2. Synthesis of ligands and organic precursors
6.2.1. 2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid (4)
Lithium diisopropylamide (LDA) (2 M in heptane/THF/ethylbenzene, 16.0 mL, 32.5 mmol)
was added to a solution of Hbdmpza (1) (2.60 g, 10.5 mmol) in dry THF (50 mL) at –80 °C.
The mixture was warmed up to –40 °C over a period of 2 h and was allowed to stir for 1 h
at this temperature. After addition of methyl iodide (5.90 mL, 94.2 mmol), the reaction
mixture was warmed to room temperature, heated under reflux for 1 h and then stirred
overnight at room temperature. The solvent was removed under reduced pressure, the
residue was dissolved in water and washed with n-pentane (3 × 50 mL) to remove
impurities. The aqueous phase was acidified (pH 2) with diluted HCl and extracted with
diethyl ether (5 × 70 mL). The combined organic layers were dried over anhydrous Na2SO4
and evaporated to dryness. Crystallization from acetone gave compound 4 (2.39 g,
9.11 mmol, 87 %) as colorless needles.
N
N N
N
Me
Me Me
Me
CO2H
4
Me
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.70 (s, 6 H, C5-CH3), 2.28 (s, 6 H, C3-CH3), 2.30 (s, 3 H,
CH3), 5.93 (s, 2 H, C4-H) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 10.5 (C5-CH3), 13.2
(C3-CH3), 27.0 (CH3), 78.6 (Cbridge), 108.7 (C4), 141.4 (C5), 146.8 (C3), 169.7 (CO2H) ppm. IR
(KBr): ν̃ = 1738 (s, CO2H), 1559 (s, C=N) cm–1. FD-MS (CH2Cl2): m/z (%) = 263 (100) [MH+].
Elemental analysis calcd. for C13H18N4O2 (262.31 g mol–1): C 59.53, H 6.92, N 21.36; found
C 59.54, H 6.92, N 21.79 %. M.p.: 123.8 °C.
6.2.2. Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)methane (17)
To a suspension of methyltriphenylphosphonium bromide (8.23 g, 23.1 mmol) in dry THF
was added KOtBu (2.41 g, 21.3 mmol). After stirring at ambient temperature for 1 h, bis(3,5-
dimethyl-4-formylpyrazol-1-yl)methane (16) (2.00 g, 7.68 mmol) was added in small
portions and the mixture was stirred overnight. Solids were filtered off and the solvent was

VI. EXPERIMENTAL SECTION 147
removed under reduced pressure. The crude product was purified by column
chromatography (silica gel, l = 10 cm, d = 4,5 cm, n-pentane : ethyl acetate 7 : 3 v/v).
Evaporation of the solvent gave compound 17 (1.80 g, 7.03 mmol, 91 %) as a white solid.
N
N N
N
Me
Me
Me
Me17
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.26 (s, 6 H, C5-CH3), 2.46 (s, 6 H, C3-CH3), 5.13 (dd,
2JH,H = 1.4 Hz, 3
JH,H = 11.6 Hz, 2 H, (Z )-H2C=), 5.28 (dd, 2JH,H = 1.4 Hz, 3
JH,H = 17.9 Hz, 2 H,
(E )-H2C=), 6.08 (s, 2 H, -CH2-), 6.46 (dd, 3JH,H = 17.9 Hz, 3
JH,H = 11.6 Hz, 2 H, -HC=) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 10.2 (C5-CH3), 13.5 (C3-CH3), 60.7 (-CH2-), 112.8
(=CH2), 116.5 (C4), 127.4 (-HC=), 138.0 (C5), 146.8 (C3) ppm. IR (KBr): ν̃ = 1641 (s, C=Cvinyl),
1547 (m, C=N) cm–1. FD-MS (CH2Cl2): m/z (%) = 257 (100) [MH+]. Elemental analysis calcd.
for C15H20N4 (256.35 g mol–1): C 70.28, H 7.86, N 21.86; found C 70.50, H 7.83, N 21.86. M.p.:
150.8 °C.
6.2.3. 2,2-Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid (Hbdmvpza) (18)
A solution of compound 17 (1.20 g, 4.68 mmol) in dry THF (30 mL) was treated with
n-butyllithium (1.6 M solution in hexanes, 3.0 mL, 4.80 mmol) at −70 °C. The mixture was
allowed to warm to a temperature of −40 °C during a period of 4.5 h. A dry stream of CO2
was passed through the mixture for 1 h. The resulting clear solution was slowly warmed to
room temperature and stirred overnight. After removal of the solvent, the residue was
dissolved in water (250 mL) and the aqueous phase was washed with n-pentane (2 × 50 mL)
to remove impurities. The remaining aqueous phase was acidified (pH 2) with diluted HCl
and extracted with diethylether (2 × 100 mL). The combined organic phases were dried over
anhydrous Na2SO4. Evaporation to dryness gave ligand 18 (1.20 g, 4.00 mmol, 85 %) as a
white solid.
N
N N
N
Me
Me
Me
Me
CO2H
18

148 VI. EXPERIMENTAL SECTION
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.23 (s, 6 H, C5-CH3), 2.31 (s, 6 H, C3-CH3), 5.21 (dd,
2JH,H = 0.8 Hz, 3
JH,H = 11.6 Hz, 2 H, (Z )-H2C=), 5.31 (dd, 2JH,H = 0.8 Hz, 3
JH,H = 17.9 Hz, 2 H,
(E )-H2C=), 6.46 (dd, 3JH,H = 17.9 Hz, 3
JH,H = 11.6 Hz, 2 H, -HC=), 6.91 (s, 1 H, -CH-) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 10.0 (C5-CH3), 13.3 (C3-CH3), 70.9 (-CH2-), 114.5
(=CH2), 117.6 (C4), 126.6 (-HC=), 138.9 (C5), 147.3 (C3), 165.1 (CO2H) ppm. IR (KBr): ν̃ = 1734
(s, CO2H), 1636 (s, C=Cvinyl), 1560 (w, C=N) cm–1. FD-MS (CH2Cl2): m/z (%) = 256 (100)
[MH+ – CO2], 301 (80) [MH+]. Elemental analysis calcd. for C16H20N4O2 (300.36 g mol–1):
C 63.98, H 6.71, N 18.65; found C 64.22, H 6.70, N 18.93. M.p.: 108.6 °C.
6.2.4. 2-[4-(Methoxycarbonyl)-oxazol-5-yl]benzoic acid (32)
This compound was synthesized according to the procedure reported by M. SUZUKI et al.[328]
A mixture of phthalic anhydride (7.40 g, 50.0 mmol) and methyl isocyanoacetate (4.55 mL,
4.95 g, 50.0 mmol) in THF (50 mL) was added dropwise to a sitirred solution of DBU
(7.47 mL, 7.61 g, 50.0 mmol) in THF (40 mL) at 40 °C. After completion of the addition, the
mixture was stirred for 1 h at ambient temperature. The mixture was diluted with H2O
(100 mL) and the organic solvent was removed under reduced pressure. The remaining
aqueous phase was acidified (pH 2) with diluted HCl and extracted with ethyl acetate
(4 × 15 mL). The extract was washed with H2O, dried over Na2SO4 and evaporated to
dryness. Crystallization from ethyl acetate and n-hexane afforded compound 32 (11.0 g,
44.3 mmol, 89 %) as colourless needles.
CO2H
O
NMeO2C
32
1H NMR (DMSO-D6, 300 MHz, 25 °C): δ = 3.66 (s, 3 H, CH3), 7.65 (m, 3 H, Harom), 7.98 (m,
1 H, Harom), 8.56 (s, 1 H, Hoxazole) ppm.

VI. EXPERIMENTAL SECTION 149
6.2.5. 4-Hydroxy-1-oxo-1,2-dihydroisoquinoline-3-carboxylate (33)
This compound was synthesized according to the procedure reported by M. SUZUKI et al.[328]
To a solution of compound 32 (6.70 g, 27.1 mmol) in MeOH was added HCl (conc., 9.00 mL)
and the mixture was stirred for 4 h at 50 °C. After cooling to 0 °C and the white precipitate
was collected by filtration and washed twice with cold MeOH. Drying in vacuo afforded
compound 33 (4.69 g, 21.3 mmol, 79 %) as a white solid.
NH
OH
O
CO2Me
33
1H NMR (CF3OOD, 300 MHz, 25 °C): δ = 4.75 (s, 3 H, CH3), 8.67 (m, 2 H, Harom), 9.14 (m, 2 H,
Harom) ppm.
6.2.6. Methyl 1-chloro-4-hydroxyisoquinoline-3-carboxylate (34)
This compound was synthesized according to a patent procedure.[329-330]
Compound 33 (2.90 g, 13.2 mmol) was stirred in POCl3 (25 mL) for 3 h at 70 °C. After
cooling, the mixture was carefully poured into 500 mL of ice water. On the next day, the
precipitate was collected by filtration, washed with H2O and dried in a desiccator.
Compound 34 (2.99 g, 12.6 mmol, 95 %) was obtained as a white solid.
N
OH
Cl
CO2Me
34
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 4.06 (s, 3 H, CH3), 7.83 (m, 2 H, Harom), 8.34 (m, 2 H,
Harom), 11.8 (s, 1 H, OH) ppm.

150 VI. EXPERIMENTAL SECTION
6.2.7. 1-Chloro-4-hydroxyisoquinoline-3-carboxylic acid (35)
This compound was synthesized according to a patent procedure.[329-330]
Compound 34 (2.97 g, 12.5 mmol) was dissolved in a mixture of ethanol (40 mL) and
aqueous NaOH (2 M, 40 mL) and stirred for 5 h at 90 °C. The mixture was acidified (pH 2)
with diluted HCl, the precipitate was collected by filtration and washed with H2O. After
drying in a desiccator, compound 35 (2.66 g, 11.9 mmol, 95 %) was obtained as a white solid.
N
OH
Cl
CO2H
35
1H NMR (DMSO-D6, 300 MHz, 25 °C): δ = 7.96 (m, 2 H, Harom), 8.24 (m, 2 H, Harom) ppm.
6.2.8. Ethyl 2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido)acetate (36)
This compound was synthesized similar to a patent procedure[329-330]
but using glycine ethyl
ester hydrochloride instead of glycine methyl ester hydrochloride.
To a solution of compound 35 (2.24 g, 10.0 mmol) and glycine ethyl ester hydrochloride
(1.40 g, 10.0 mmol) in CH2Cl2 (60 mL) at 0 °C were added NEt3 (1.40 mL, 1.02 g, 10.1 mmol),
PyBOP (5.20 g, 10.0 mmol) and ethyl diisopropylamine (3.40 mL, 2,66 g, 20.6 mmol). After
stirring for 4 h at ambient temperature, the organic phase was washed with H2O
(3 × 50 mL), dried over anhydrous Na2SO4 and evaporated to dryness. The crude product
was purified by column chromatography (silica gel, l = 18 cm, d = 4,5 cm, CH2Cl2) to yield
ethyl ester 36 (2.41 g, 7.81 mmol, 78 %) as a off-white solid.
N
OH
Cl
NH
O
CO2Et
36
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.30 (t, 3JH,H = 6.9 Hz, 3 H, -CH3), 4.26 (m, 4 H, -CH2-),
7.78 (m, 2 H, Harom), 8.23 (m, 2 H, -NH- and Harom), 8.35 (m, 1 H, Harom), 12.85 (s, 1 H, OH)
ppm.

VI. EXPERIMENTAL SECTION 151
6.2.9. 2-(1-Chloro-4-hydroxyisoquinoline-3-carboxamido)acetic acid (37)
This compound was synthesized similar to a patent procedure.[329-330]
Ethyl ester 36 (1.07 g, 3.47 mmol) was dissolved in a mixture of THF (8 mL) and aqueous
NaOH (1 M, 8 mL). After stirring for 2 h at ambient temperature, the mixture was diluted
with 100 mL H2O and the organic solvent was removed under reduced pressure. The
aqueous phase was washed with CH2Cl2 (2 × 30 mL) and acidified (pH 3) with diluted HCl.
The formed precipitate was collected by filtration, washed with H2O and dried in a
desiccator to yield compound 37 (919 mg, 3,27 mmol, 94 %) as a off-white solid.
N
OH
Cl
NH
O
CO2H
37
1H NMR (Acetone-D6, 300 MHz, 25 °C): δ = 3.75 (br, 1 H, -CO2H), 4.30 (d, 3
JH,H = 6.0 Hz, 2 H,
-CH2-), 7.96 (m, 2 H, Harom), 8.33 (m, 2 H, Harom), 8.77 (br, 1 H, -NH-), 13.48 (s, 1 H, Ar-OH)
ppm. 13C NMR (Acetone-D6, 75.5 MHz, 25 °C): δ = 41.2 (CH2), 121.0, 124.0, 127.1, 130.1, 131.0,
132.2 (2×), 155.4, 155.7 (Carom), 170.3, 170.7 (2× C=O) ppm.

152 VI. EXPERIMENTAL SECTION
6.3. Synthesis of transition metal complexes
6.3.1. [Mn(bdmpzpr)(CO)3] (5)
2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid (Hbdmpzpr) (4) (262 mg, 1.00 mmol) in dry
THF (30 mL) was treated with KOtBu (112 mg, 1.00 mmol) and the mixture was stirred 1 h
at ambient temperature. [MnBr(CO)5] (275 mg, 1.00 mmol) was added and the reaction
mixture was heated under reflux and controlled by IR spectroscopy on a regular basis. After
completion of the reaction (22 h) the solvent was removed under reduced pressure. The
yellow residue was washed with degassed water (5 × 10 mL) and diethyl ether (5 × 10 mL)
and dried in vacuo to yield tricarbonyl complex 5 (337 mg, 0.842 mmol, 84 %) as a yellow
crystal powder. Crystals suitable for X-ray analysis can be obtained by layering a CH2Cl2
solution of complex 5 with n-pentane.
N N
N N
Me
Me
Me
Me
OO
Mn
CC CO
OO
5
Me
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.49 (s, 6 H, Cpz-CH3), 2.58 (s, 9 H, Cpz-CH3 and CH3),
6.00 (s, 2 H, C4-H) ppm. 13C NMR (DMSO-D6, 75.5 MHz, 25 °C): δ = 14.9 (CH3), 16.1 (CH3),
21.9 (Cbridge-CH3), 81.0 (Cbridge), 111.8 (Cpz), 145.1 (Cpz), 152.5 (Cpz), 166.2 (CO2–), 220.6 (CO),
222.2 (CO) ppm. IR (THF): ν̃ = 2033 (s, CO), 1938 (s, CO), 1910 (s, CO), 1682 (m, as-CO2–)
cm–1. IR (KBr): ν ̃ = 2035 (s, CO), 1946 (s, CO), 1924 (s, CO), 1670 (m, as-CO2–), 1561 (w, C=N),
1375 (m, sym-CO2–) cm–1. FAB-MS (NBOH): m/z (%) =272 (100) [M – 3×CO – CO2], 401 (50)
[MH+], 423 (10) [MNa+]. Elemental analysis calcd. for C16H17MnN4O5 (400.27 g mol–1):
C 48.01, H 4.28, N 14.00; found C 48.31, H 4,40, N 14.02 %. M.p.: 231 °C (decomposition).

VI. EXPERIMENTAL SECTION 153
6.3.2. [Re(bdmpzpr)(CO)3] (6)
2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid (Hbdmpzpr) (4) (350 mg, 1.33 mmol) in dry
THF (20 mL) was treated with KOtBu (150 mg, 1.33 mmol) and the mixture was stirred 1 h
at ambient temperature. [ReBr(CO)5] (541 mg, 1.33 mmol) was added and the reaction
mixture was heated under reflux and controlled by IR spectroscopy on a regular basis. After
completion of the reaction (56 h), the solvent was removed under reduced pressure. The
white residue was washed with degassed water (5 × 10 mL) and diethyl ether (5 × 10 mL)
and dried in vacuo to yield dicarbonyl complex 6 (243 mg, 0.457 mmol, 34 %) as a white
crystal powder.
N N
N N
Me
Me
Me
Me
OO
Re
CC CO
OO
6
Me
IR (THF): ν̃ = 2022 (s, CO), 1916 (s, CO), 1892 (s, CO), 1693 (m, as-CO2–), 1560 (w, C=N) cm–1.
IR (KBr): ν̃ = 2025 (s, CO), 1927 (s, CO), 1911 (s, CO), 1677 (m, as-CO2–), 1559 (w, C=N), 1371
(m, sym-CO2–) cm–1. FAB-MS (NBOH): m/z (%) =533 (100) [MH+]. C16H17N4O5Re
(531.54 g mol–1): calcd. C 36.15, H 3.22, N 10.54; found C 35.75, H 3.06, N 10.48 %. M.p.:
255 °C (decomposition).
6.3.3. [Cu(bdmpzpr)2] (7)
To a solution of ligand 4 (300 mg, 1.14 mmol) in acetonitrile (15 mL) was added copper(II)
acetate monohydrate (76.1 mg, 0.381 mmol). The reaction mixture was stirred over night at
ambient temperature. The precipitate was filtered off, washed with water (5 × 5 mL) and
diethylether (5 × 5 mL) and dried in vacuo to yield bisligand complex 7 (154 mg, 0.263 mmol
69 %) as a pale blue powder. Crystals suitable for X-ray structure determination can be
obtained by layering a CH2Cl2 solution of 7 with n-pentane.

154 VI. EXPERIMENTAL SECTION
N N
N N
Me
Me
Me
Me
OO
Cu
NN
NN
Me
Me
Me
Me
O O
Me
Me
7
IR (KBr): ν̃ = 1665 (s, as-CO2–), 1559 (m, C=N), 1367 (m, sym-CO2
–) cm–1. FAB-MS (NBOH):
m/z (%) = 263 (100) [C13H18N4O2H+], 280 (90) [M – C13H17N4O2 – CO2], 587 (5) [MH+].
Elemental analysis calcd. for C26H34CuN8O4 (586.15 g mol–1): C 53.28, H 5.85, N 19.12;
found C 53.16, H 5.82, N 19.36 %. M.p.: 190 °C (decomposition).
6.3.4. [Ru(bdmpzpr)Cl(PPh3)2] (9)
A solution of ligand 4 (200 mg, 0.762 mmol) in dry THF (15 mL) was deprotonated with
KOtBu (84.0 mg, 0.749 mmol) by stirring for 1 h at ambient temperature under an argon
atmosphere. [RuCl2(PPh3)3] (695 mg, 0.725 mmol) was added and the reaction mixture was
stirred for 4 h. The solvent was removed under reduced pressure. The residue was dissolved
in a small amount of dichloromethane, filtered through celite and treated with n-pentane.
The formed yellow precipitate was separated by filtration and washed thoroughly with n-
pentane yielding bisphosphine complex 9 (400 mg 0.434 mmol, 60 %) as a mixture of two
isomers.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
Me
N N
N N
Me
Me
Me
Me
OO
Ru
PPh3Cl PPh3
Me
9a 9b

VI. EXPERIMENTAL SECTION 155
1H NMR (CD2Cl2, 300 MHz, 25 °C) isomer 9a: δ = 1.69 (s, 6 H, CH3), 1.71 (s, 3 H, CH3), 2.15 (s,
6 H, CH3), 5.83 (s, 2 H, Cpz-H), 7.29 (m, 30 H, PPh3) ppm; isomer 9b: δ = 2.60 (m, 9 H, CH3),
2.68 (s, 3 H, CH3), 2.70 (s, 3 H, CH3), 5.97 (s, 1 H, Cpz-H), 5.98 (s, 1 H, Cpz-H), 7.29 (m, 30 H,
PPh3) ppm. 13C NMR (CD2Cl2, 75.5 MHz, 25 °C) isomer 9a: δ = 11.7 (C5-CH3), 13.8 (C3-CH3),
26.1 (CH3), 84.2 (Cbridge), 108.3 (C4), 127.5 (br, m-PPh3), 128.0 (br, p-PPh3), 135.0 (d, 2JC,P = 4.8 Hz, o-PPh3), 135.2 (br, i-PPh3), 142.0 (C5), 155.1 (C3), 168.2 (CO2
–) ppm; isomer 9b:
δ = 13.1 (C5/5'-CH3), 13.4 (C5/5'-CH3), 17.7 (C3/3'-CH3), 17.9 (C3/3'-CH3), 26.1 (CH3), 81.0
(Cbridge), 112.1 (C4/4’), 116.0 (C4/4'), 127.5 (br, m-PPh3), 128.0 (br, p-PPh3), 135.0 (d, 2JC,P = 4.8 Hz, o-PPh3), 135.2 (br, i-PPh3), 143.4 (C5/5'), 143.7 (C5/5'), 154.6 (C3/3'), 155.8 (C3/3'),
169.7 (CO2–) ppm. 31P NMR (CD2Cl2, 121.5 MHz, 25 °C): δ = 35.7, 35.2, 60.3 ppm. IR (CH2Cl2):
ν ̃ = 1659 (s, as-CO2–), 1569 (w, C=N), 1377 (m, sym-CO2
–) cm–1. IR (KBr): ν ̃ = 1655 (s, as-
CO2–), 1569 (w, C=N), 1375 (m, sym-CO2
–) cm–1. FD-MS (CH2Cl2): m/z (%) = 262 (100)
[PPh3], 922 (70) [MH+]. Elemental analysis calcd. for C49H47ClN4O2P2Ru × 0.5 CH2Cl2
(964.86 g mol–1): C 61.62, H 5.01, N 5.81; found C 61.78, H 5.06, N 5.98 %.
6.3.5. [Ru(bdmpzpr)Cl(N2)(PPh3)] (10)
A solution of [Ru(bdmpzpr)Cl(PPh3)2] (9) (175 mg, 0.190 mmol) was dissolved in THF
(10 mL) and stirred vigorously under an atmosphere of nitrogen for 3 h. The reaction was
controlled by IR spectroscopy on a regular basis. The solvent was removed under reduced
pressure and the residue was dissolved in a small amount of dichloromethane. Precipitation
with diethyl ether yields complex 10 (103 mg, 0.150 mmol, 79 %) as a yellow powder.
Crystals suitable for X-ray analysis can be obtained by layering a CH2Cl2 solution of 10
with diethyl ether.
N N
N N
Me
Me
Me
Me
OO
Ru
N2Ph3P Cl
Me
10
1H NMR (CD2Cl2, 300 MHz, 25 °C): δ = 1.73 (s, 3 H, C3'-CH3), 2.56 (s, 3 H, C5-CH3), 2.59 (s,
3 H, C5'-CH3), 2.67 (s, 6 H, C3-CH3), 2,74 (s, 3 H, CH3), 5.83 (s, 1 H, C4-H), 6.05 (s, 1 H, C4'-H),

156 VI. EXPERIMENTAL SECTION
7.33 (m, 9 H, m-PPh3 and p-PPh3), 7.66 (m, 6 H, o-PPh3) ppm. 13C NMR (CD2Cl2, 75.5 MHz,
25 °C): δ = 15.0 (CH3), 15.9 (CH3), 17.5 (CH3), 18.1 (CH3), 24.4 (CH3), 83.9 (Cbridge), 112.9 (C4/4'),
113.0 (C4/4'), 128.3 (d, 3JC,P = 9.0 Hz, m-PPh3), 130.2 (p-PPh3), i-PPh3 not observed, 134.4 (d,
2JC,P = 9.0 Hz, o-PPh3), 145.7 (C5/5'), 145.8 (C5/5'), 155.6 (C3/3'), 155.9 (C3/3'), 168.0 (CO2
–) ppm. 31P NMR (CD2Cl2, 121.5 MHz, 25 °C): δ = 38.3 ppm. IR (THF): ν̃ = 2129 (s, N≡N), 1665 (s, as-
CO2–), 1563 (w, C=N) cm−1. IR (KBr): ν ̃ = 2129 (s, N≡N), 1663 (s, as-CO2
–), 1564 (w, C=N),
1374 (m, sym-CO2–) cm−1. FD-MS (CH2Cl2): m/z (%) = 625 (100) [M –Cl –N2], 661 (30)
[MH+ – N2], 695 (60) [M – N2 + Cl]. Elemental analysis* calcd. for C31H32ClN6O2PRu
(688.12 g mol–1): C 54.11, H 4.69, N 12.21; found C 54.04, H 4.86, N 11.18 %. M.p.: 168 °C
(decomposition).
*The found and calculated percentage values for the nitrogen content do not match, due to
the lability of the N2 ligand in this compound. But carbon and hydrogen microanalysis is
acceptable.
6.3.6. [Ru(bdmpzpr)Cl(CO)(PPh3)] (12)
Ligand 4 (200 mg, 0.762 mmol) in dry THF (15 mL) was treated with KOtBu (84.0 mg,
0.749 mmol) and the mixture was stirred 1 h at ambient temperature under an argon
atmosphere. [RuCl2(PPh3)3] (695 mg, 0.725 mmol) was added and the reaction mixture was
stirred for 4 hours at ambient temperature. Carbon monoxide was then passed through the
solution for 1 hour. The solvent was removed and the yellow residue was washed with
degassed water (3 × 10 mL) and diethyl ether (3 × 10 mL) and dried in vacuo to yield
complex 12 (295 mg, 0.429 mmol, 59 %) as a mixture of two isomers. Crystals of the major
isomer 12a suitable for X-ray analysis can be obtained by layering a CH2Cl2 solution of 12
with diethyl ether.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P C 12a
N N
N N
Me
Me
Me
Me
OO
Ru
CPh3P Cl
Me
12b
Me
OO

VI. EXPERIMENTAL SECTION 157
1H NMR (CD2Cl2, 300 MHz, 25 °C) isomer 12a: δ = 1.90 (s, 3 H, C3'-CH3), 2.53 (s, 3 H,
C5-CH3), 2.54 (s, 3 H, C5'-CH3), 2.69 (s, 6 H, C3-CH3 and CH3), 5.90 (s, 1 H, C4-H), 6.03 (s, 1 H,
C4'-H), 7.47 (m, 15 H, PPh3) ppm; isomer 12b: δ = 1.72 (s, 3 H, C3'-CH3), 2.52 (s, 3 H, C5-CH3),
2.55 (s, 3 H, C5'-CH3), 2.62 (s, 3 H, CH3), 2.73 (s, 3 H, C3-CH3), 5.77 (s, 1 H, Cpz-H), 5.99 (s, 1 H,
Cpz-H), 7.47 (m, 15 H, PPh3) ppm. 13C NMR (CD2Cl2, 75.5 MHz, 25 °C): isomer 12a: δ = 15.6
(CH3), 16.2 (CH3), 17.7 (CH3), 17.8 (CH3), 24.8 (CH3), 83.4 (Cbridge), 113.0 (d, 4JC,P = 3.2 Hz,
C4/4'), 113.3 (C4/4'), 128.5 (d, 3JC,P = 10.3 Hz, m-PPh3), 130.6 (d, 4
JC,P = 2.6 Hz, p-PPh3), 133.4 (d, 1JC,P = 46.4 Hz, i-PPh3), 134.6 (d, 2
JC,P = 9.7 Hz, o-PPh3), 143.9 (d, 4JC,P = 1.9 Hz, C5/5'), 144.5
(C5/5'), 155.8 (C3/3'), 156.1 (C3/3'), 168.2 (CO2–), 204.7 (CO) ppm; isomer 12b: δ = 14.4 (CH3),
16.5 (CH3), 17.6 (CH3), 18.2 (CH3), 24.5 (CH3), 84.1 (Cbridge), 112.6 (d, 4JC,P = 3.2 Hz, C4/4'),
112.8 (C4/4'), 128.4 (d, 3JC,P = 9.7 Hz, m-PPh3), 130.4 (d, 4
JC,P = 2.6 Hz, p-PPh3), 133.7 (d, 1JC,P = 47.0 Hz, i-PPh3), 134.5 (d, 2
JC,P = 9.7 Hz, o-PPh3), 143.8 (d, 4JC,P = 1.9 Hz, C5/5'), 145.8
(C5/5'), 155.3 (C3/3'), 155.8 (C3/3'), 167.5 (CO2–), 204.4 (CO) ppm. 31P NMR (CD2Cl2, 121.5 MHz,
25 °C): δ = 41.3, 36.1 ppm. IR (THF): ν̃ = 1956 (s, CO, isomer 12a), 1941 (s, CO, isomer 12b),
1664 (s, as-CO2–), 1564 (w, C=N) cm–1. IR (KBr): ν ̃ = 1949 (s, CO, isomer 12a), 1936 (s, CO,
isomer 12b), 1668 (s, as-CO2–), 1566 (w, C=N), 1371 (m, sym-CO2
–) cm–1. FD-MS (CH2Cl2):
m/z (%) = 689 (100) [MH+]. Elemental analysis calcd. for C32H32ClN4O3PRu (688.12 g mol–1):
C 55.85, H 4.69, N 8.14; found C 55.66, H 4.66, N 8.01 %. M.p.: 181 °C (decomposition).
6.3.7. [Ru(bdmpza)Cl(PPh3)(SO2)] (13)
Sulfur dioxide gas was passed through a solution of [Ru(bdmpza)Cl(PPh3)2] (8) (400 mg,
0.440 mmol) in THF (10 mL) for 1 h. The solvent was removed under reduced pressure and
the residue was washed with degassed water (3 × 10 mL) and diethyl ether (3 × 10 mL) and
dried in vacuo to yield sulfur dioxide complex 13 (257 mg, 0.361 mmol, 82 %) as an orange
crystal powder. Crystals suitable for X-ray structure determination can be obtained by
layering a CHCl3 solution of 13 with n-pentane.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P SO2
13

158 VI. EXPERIMENTAL SECTION
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.10 (s, 3 H, CH3), 2.49 (m, 9 H, 3 × CH3), 2.74 (s, 3 H,
CH3), 6.01 (s, 1 H, Cpz-H), 6.10 (s, 1 H, Cpz-H), 6.52 (s, 1 H, CH), 7.40 (m, 15 H, PPh3) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 11.0 (CH3), 11.4 (CH3), 15.1 (CH3), 15.7 (CH3), 68.9
(CBridge), 109.5 (C4/4'), 109.9 (C4/4'), 128.3 (d, 3JC,P = 10.3 Hz, m-PPh3), 130.6 (d, 4JC,P = 1.9 Hz, p-
PPh3), 133.9 (d, 2JC,P = 9.0 Hz, o-PPh3), i-PPh3 not observed, 141.0 (C5/5’), 142.2 (C5/5’), 155.4
(C3/3’), 157.9 (C3/3’), 165.2 (CO2–) ppm. 31P NMR (CDCl3, 121.5 MHz, 25 °C): δ = 28.8 ppm. IR
(KBr): ν̃ = 1676 (s, as-CO2–), 1560 (w, C=N), 1395 (m, sym-CO2
–), 1294 (m, S=O), 1123 (s,
S=O) cm–1. FD-MS (CH2Cl2) m/z (%) = 292 (100) [PPh3], 681 (40) [M – SO2 + Cl]. Elemental
analysis calcd. for C30H30ClN4O4PRuS (710.15 g mol–1): C 50.74, H 4.26, N 7.89; found
C 50.45, H 4.17, N 8.20 %.
6.3.8. [Ru(bdmpzpr)Cl(PPh3)(SO2)] (14)
Sulfur dioxide gas was passed through a a solution of [Ru(bdmpzpr)Cl(PPh3)2] (9) (700 mg,
0.759 mmol) in 10 mL THF for 1 h. The solvent was removed under reduced pressure and
the residue was washed with degassed water (3 × 10mL) and diethyl ether (3 × 10mL).
Crystallization from CH2Cl2 and diethyl ether afforded complex 14 (410 mg, 0.566 mmol,
75 %) as a mixture of two isomers.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P SO2
14a
N N
N N
Me
Me
Me
Me
OO
Ru
SO2Ph3P Cl
Me
14b
Me
1H NMR (CD2Cl2, 300 MHz, 25 °C): δ = 1.72 (s, 3 H, CH3), 2.05 (s, 3 H, CH3), 2.57 (s, 3 H,
CH3), 2.58 (s, 6 H, 2 × CH3), 2.62 (s, 3 H, CH3), 2.70 (s, 3 H, CH3), 2.72 (s, 3 H, CH3), 2.74 (s,
3 H, CH3), 2.75 (s, 3 H, CH3), 5.86 (s, 1 H, Cpz-H), 6.00 (s, 1 H, Cpz-H), 6.12 (s, 1 H, Cpz-H), 6.13
(s, 1 H, Cpz-H), 7.41 (m, 30 H, 2 × PPh3) ppm. 13C NMR (DMSO-D6, 75.5 MHz, 25 °C):
δ = 10.7 (CH3), 13.2 (CH3), 15.0 (CH3), 15.4 (CH3), 16.3 (CH3), 16.4 (CH3), 16.5 (CH3), 16.7
(CH3), 16.8 (CH3), 16.9 (CH3), 81.2 (Cbridge), 82.7 (Cbridge), 103.2 (C4/4'), 108.3 (C4/4'), 112.5 (C4/4'),
113.2 (C4/4'), 127.9 (d, 3JC,P = 9.7 Hz, m-PPh3), 128.8 (d, 3
JC,P = 7.1 Hz, m-PPh3), 129.0 (p-PPh3),
130.1 (p-PPh3), 130.3 (d, 1JC,P = 53.4 Hz, i-PPh3), 131.7 (d, 1
JC,P = 46.4 Hz, i-PPh3), 133.2 (d,

VI. EXPERIMENTAL SECTION 159
2JC,P = 19.9 Hz, o-PPh3), 133.8 (d, 2
JC,P = 9.0 Hz, o-PPh3), 140.7 (C5/5'), 144.9 (C5/5'), 145.7 (C5/5'),
146.4 (C5/5'), 153.5 (C3/3'), 153.7 (C3/3'), 156.4 (C3/3'), 156.9 (C3/3'), 166.7 (CO2–), 167.3 (CO2
–)
ppm. 31P NMR (CD2Cl2, 121.5 MHz, 25 °C): δ = 26.6, 27.1 ppm. IR (KBr): ν ̃ = 1675 (s, as-CO2–
), 1560 (w, C=N), 1373 (m, sym-CO2–), 1291 (m, S=O), 1125 (s, S=O) cm–1. FD-MS (CH2Cl2):
m/z (%) = 292 (100) [PPh3], 624 (50) [M – Cl – SO2]. Elemental analysis calcd. for
C31H32ClN4O4PRuS × CH2Cl2 (809.10 g mol–1): C 47.50, H 4.45, N 6.92; found C 47.22, H 4.28,
N 7.18 %.
6.3.9. [Mn(bdmvpza)(CO)3] (19)
Ligand 18 (350 mg, 1.17 mmol) in dry THF (30 mL) was deprotonated with KOtBu (131 mg,
1.17 mmol) for 1 h at ambient temperature. [MnBr(CO)5] (320 mg, 1.17 mmol) was added
and the reaction mixture was heated under reflux and controlled by IR spectroscopy on a
regular basis. After completion of the reaction (approx. 17 h), the solvent was removed
under reduced pressure. The yellow residue was washed with degassed water (5 × 10 mL)
and diethyl ether (5 × 10 mL) and dried in vacuo to yield tricarbonyl complex 19 (270 mg,
0.616 mmol, 53 %) as a yellow crystal powder. Crystals suitable for X-ray analysis can be
obtained by layering a CH2Cl2 solution of complex 19 with diethyl ether.
N N
N N
Me
Me
Me
Me
OO
Mn
CC CO
OO
19
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.44 (s, 6 H, C5-CH3), 2.61 (s, 6 H, C3-CH3), 5.38 (br.,
AMX system, coupling not resolved, 4 H, H2C=), 6.45 (br., AMX system, coupling not
resolved, 3 H, -HC= and -CH-) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 10.1 (C5-CH3),
13.9 (C3-CH3), 66.8 (-CH-), 117.8 (=CH2), 118.6 (C4), 125.5 (-HC=), 138.7 (C5), 152.5 (C3), 165.5
(CO2–), 219.9 (CO), 223.1 (CO) ppm. IR (THF): ν̃ = 2036 (s, CO), 1944 (s, CO), 1917 (s, CO),
1682 (m, CO2–), 1667 (m, C=Cvinyl) cm–1. IR (KBr): ν ̃ = 2039 (s, CO), 1935 (s, CO), 1929 (s, CO),
1670 (m, CO2–), 1640 (m, C=Cvinyl) cm–1. FAB-MS (NBOH): m/z (%) = 439 (50) [MH+], 383
(15) [MH+ – 2×CO], 355 [MH+ – 3×CO], 310 (100) [MH+ – 3×CO – CO2]. Elemental

160 VI. EXPERIMENTAL SECTION
analysis calcd. for C19H19MnN4O5 (438.32 g mol–1): C 52.06, H 4.37, N 12.78; found C 52.17,
H 4.46, N 12.58 %.
6.3.10. [Re(bdmvpza)(CO)3] (20)
Ligand 18 (350 mg, 1.17 mmol) in dry THF (30 mL) was deprotonated with KOtBu (131 mg,
1.17 mmol) for 1 h at ambient temperature. [ReBr(CO)5] (473 mg, 1.17 mmol) was added and
the reaction mixture was heated under reflux and controlled by IR spectroscopy on a
regular basis. After completion of the reaction (approx. 24 h), the solvent was removed
under reduced pressure. The white residue was washed with degassed water (5 × 10 mL)
and diethyl ether (5 × 10 mL) and dried in vacuo to yield tricarbonyl complex 20 (270 mg,
0.474 mmol, 41 %) as a white crystal powder.
N N
N N
Me
Me
Me
Me
OO
Re
CC CO
OO
20
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.45 (s, 6 H, C5-CH3), 2.56 (s, 6 H, C3-CH3), 5.41 (br,
AMX system, coupling not resolved, 4 H, H2C=), 6.44 (dd, 3JH,H = 17.7 Hz, 3
JH,H = 11.5 Hz,
2 H, -HC=), 6.63 (s, 1 H, -CH-) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 10.2 (C5-CH3),
14.7 (C3-CH3), 68.0 (-CH-), 118.5 (C4), 118.7 (=CH2), 125.1 (-HC=), 138.8 (C5), 152.5 (C3), 163.8
(CO2–), 194.9 (CO), 196.0 (CO) ppm. IR (THF): ν̃ = 2026 (s, CO), 1921 (s, CO), 1898 (s, CO),
1697 (m, as-CO2–) cm–1. IR (KBr): ν̃ = 2025 (s, CO), 1913 (s, CO), 1884 (s, CO), 1677 (m, as-
CO2–), 1639 (m, C=Cvinyl) cm–1. FD-MS (CH2Cl2): m/z (%) = 570 (100) [MH+]. Elemental
analysis calcd. for C19H19N4O5Re (569.59 g mol–1): C 40.06, H 3.36, N 9.84; found C 39.92,
H 3.40, N 9.48 %.

VI. EXPERIMENTAL SECTION 161
6.3.11. [Ru(bdmvpza)Cl(PPh3)2] (21)
A solution ligand 18 (450 mg, 1.50 mmol) in THF (25 mL) was deprotonated with KOtBu
(168 mg, 1.50 mmol) at ambient temperature. After one hour [RuCl2(PPh3)3] (1.44 g,
1.50 mmol) was added and the mixture was stirred at ambient temperature for another
45 min. After removal of the solvent under reduced pressure, the residue was washed with
degassed water (3 × 20 mL) and diethyl ether (3 × 20 mL) and dried in vacuo to yield
bisphosphine complex 21 (960 mg, 1.00 mmol, 67 %) as an orange crystal powder.
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P PPh3
21
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.82 (s, 6 H, C3-CH3), 2.46 (s, 6 H, C5-CH3), 5.00 (dd,
2JH,H=1.3 Hz, 3
JH,H=17.9 Hz, 2 H, (E )-H2C=), 5.10 (dd, 2JH,H = 1.3 Hz 3
JH,H = 11.7 Hz, 2 H,
(Z )-H2C=), 6.06 (dd, 3JH,H = 11.5 Hz, 3
JH,H = 17.7 Hz, 2 H, -HC=), 6.64 (s, 1 H, -CH-), 6.95 (m,
12 H, PPh3), 7.10 (m, 6 H, PPh3), 7.41 (m, 12 H, PPh3) ppm. 13C NMR (CDCl3, 75.5 MHz,
25 °C): δ = 10.3 (C5-CH3), 15.2 (C3-CH3), 69.3 (CH), 115.8 (=CH2) , 119.0 (C4), 126.2 (-HC=),
126.8 (vt, JC,P = 4.2 Hz, m-PPh3), 128.3 (s, p-PPh3), 134.8 (vt, JC,P = 4.2 Hz, o-PPh3), 136.4 (t,
JC,P = 39.3 Hz, i-PPh3), 138.1 (C5), 156.0 (C3), 168.2 (CO2−) ppm. 31P NMR (CDCl3, 121.5 MHz,
25 °C): δ = 35.1 ppm. IR (KBr): ν̃ = 1659 (s, CO2–), 1641 (m, C=Cvinyl) cm–1. Elemental
analysis calcd. for C52H49ClN4O2P2Ru (960.44 g mol–1): C 65.03, H 5.14, N 5.83; found.
C 64.89, H 5.09, N 5.71 %.
6.3.12. [Ru(bdmvpza)Cl(MeCN)(PPh3)] (22)
Complex 21 (400 mg, 0.416 mmol) was dissolved in acetonitrile (20 mL) and stirred over
night at ambient temperature. The yellow precipitate was filtered off, washed with diethyl
ether (5 × 10 mL) and dried in vacuo to yield the acetonitrile complex 22 (277 mg,
0.375 mmol, 90 %) as a yellowish brown crystal powder.

162 VI. EXPERIMENTAL SECTION
N N
N N
Me
Me
Me
Me
OO
Ru
ClPh3P NCMe
22
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.84 (s, 3 H, CH3), 1.85 (s, 3 H, CH3), 2.43 (s, 3 H, CH3),
2.47 (s, 3 H, CH3), 2.70 (s, 3 H, CH3), 5.19 (m, 4 H, H2C=), 6.22 (dd, 3JH,H = 11.6 Hz,
3JH,H =17.6 Hz, 1 H, -HC=), 6.40 (dd, 3
JH,H = 11.6 Hz, 3JH,H = 17.8 Hz, 2 H, -HC=), 6.54 (s,
1 H, -CH-), 7.27 (m, 15 H, PPh3) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 4.0 (H3C-CN),
10.2 (CH3), 10.6 (CH3), 14.2 (CH3), 15.0 (CH3), 69.4 (CH), 116.3, 116.5 (2 × =CH2), 118.9, 119.1
(C4 and C4'), 124.5 (H3C-CN), 126.2, 126.5 (2 × -CH=), 127.7 (d, 3JC,P = 8.6 Hz, m-PPh3), 129.3
(p-PPh3), 134.6 (d, 2JC,P = 9.0 Hz, o-PPh3), 134.6 (d, JC,P = 41.2 Hz, i-PPh3), 138.0, 139.1 (C5 and
C5'), 154.0, 157.1 (C3 and C3'), 164.5, 167.7 (2 × CO2−) ppm. 31P NMR (CDCl3, 121.5 MHz,
25 °C): δ = 49.7 ppm. IR (KBr): ν̃ = 2268 (w, C≡N), 1655 (s, as-CO2−), 1639 (m, C=Cvinyl), 1570
(w, C=N). Elemental analysis calcd. for C36H37ClN5O2PRu (739.21 g mol–1): C 58.49, H 5.05,
N 9.47; found C 56.64, H 5.18, N 8.72 %.
6.3.13. [Ru(bdmvpza)Cl(CO)2] (23)
A solution of ligand 18 (450 mg, 1.50 mmol) in dry THF (30 mL) was treated with with
KOtBu (168 mg, 1.50 mmol) for 1 h at ambient temperature. After addition of [RuCl2(CO)2]n
(342 mg, 1.50 mmol), the reaction mixture was heated under reflux and controlled by IR
spectroscopy on a regular basis. After completion of the reaction (approx. 12 h), the solvent
was removed under reduced pressure. The residue was washed with degassed water
(5 × 10 mL) and diethyl ether (5 × 10 mL) and dried in vacuo to yield complex 23 (537 mg,
1.09 mmol, 73 %) as a bright yellow crystal powder.

VI. EXPERIMENTAL SECTION 163
N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
23
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.42 (s, 3 H, CH3), 2.50 (s, 6 H, CH3), 2.70 (s, 3 H, CH3),
5.40 (m, 4 H, H2C=), 6.42 (m, 2 H, -HC=), 6.74 (s, 1 H, -CH-) ppm. 13C NMR (CDCl3, 75.5
MHz, 25 °C): δ = 10.4 (CH3), 10.7 (CH3), 14.9 (CH3), 14.7 (CH3), 68.9 (Cbridge), 118.7, 119.1,
119.2, 119.4 (C4/4' and 2 × H2C=), 125.0, 125.3 (2 × -CH=), 139.3, 140.7 (C5 and C5'), 152.5,
153.6 (C3 and C3'), 164.0 (CO2−), 192.7 (CO), 196.1 (CO) ppm. IR (THF): ν̃ = 2068 (s, CO), 1997
(s, CO), 1690 (m, as-CO2−), 1639 (w, C=Cvinyl) cm−1. IR (KBr): ν̃ = 2064 (s, CO), 1997 (s, CO),
1681 (m, as-CO2−), 1638 (w, C=Cvinyl) cm–1. FD-MS (CH2Cl2): m/z (%) = 493 (40) [MH+], 448
(100) [M − CO2]. Elemental analysis calcd. for C18H19ClN4O4Ru (491.89 g mol–1): C 43.95,
H 3.89, N 11.39; found C 43.60, H 3.94, N 10.99 %. M.p.: 278 °C (decomposition).
6.3.14. [Ru(bdmpza)Cl(CO)2] (24)
Note: This compound was synthesized in collaboration with S. TAMPIER.
A solution of ligand 1 (1.17 g, 4.70 mmol) in THF (50 mL) was treated with KOtBu (527 mg,
4.70 mmol) and stirred for at least 1 h at ambient temperature. After addition of
[RuCl2(CO)2]n (1.07 g, 4.70 mmol), the reaction mixture was heated under reflux and
controlled by IR spectroscopy on a regular basis. After completion of the reaction (approx.
12 h), the solvent was removed under reduced pressure. The residue was extracted with
CH2Cl2 (15 mL) filtered and treated with n-pentane (350 mL), precipitating complex 24
(570 mg, 1.30 mmol, 27 %) as a cream white powder. Crystals suitable for single crystal
structure analysis were obtained by slow evaporation of a CDCl3 solution of complex 24.

164 VI. EXPERIMENTAL SECTION
N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
24
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 2.34 (s, 3 H, CH3), 2.45 (s, 3 H, CH3), 2.46 (s, 3H, CH3),
2.59 (s, 3H, CH3), 6.12 (s, 1 H, Cpz-H), 6.12 (s, 1 H, Cpz-H), 6.60 (s, 1 H, CH) ppm. 13C NMR
(CDCl3, 75.5 MHz, 25 °C): δ = 10.9 (CH3), 11.2 (CH3), 14.5 (CH3), 15.3 (CH3), 68.4 (CH), 109.3,
109.4 (C4 and C4'), 142.6, 144.0 (C5 and C5'), 154.9, 155.8 (C3 and C3'), 165.3 (CO2–), 192.6 (CO),
196.0 (CO) ppm. IR (KBr): ν ̃ = 2066 (s, CO), 1996 (s, CO), 1676 (s, as-CO2–), 1559 (w, C=N)
cm–1. IR (CH2Cl2): ν̃ = 2074 (s, CO), 2005 (s, CO), 1681 (s, as-CO2–), 1563 (w, C=N) cm–1.
FAB MS (NBOH): m/z (%) = 440 (100) [M], 441 (75) [MH+], 880 (21) [M2]. Elemental
analysis calcd. for C14H15ClN4O4Ru (439.82 g mol–1): C 38.23, H 3.44, N 12.74; found C 38.12,
H 3.50, N 12.42 %.
6.3.15. [Cu(bdmvpza)2] (25)
To a solution of ligand Hbdmvpza 18 (300 mg, 1.00 mmol) in acetonitrile (15 mL) was added
copper(II) acetate monohydrate (66.5 mg, 0.333 mmol). The reaction mixture was stirred for
6 h at ambient temperature. The turquoise precipitate was filtered off, washed with water
(5 × 5 mL) and diethylether (5 × 5 mL) and dried in vacuo to yield bisligand complex 25
(135 mg, 0.204 mmol, 61 %) as pale blue powder. Crystals suitable for an X-ray structure
determination were obtained by layering a CH2Cl2 solution of 25 with n-pentane.
N N
N N
Me
Me
Me
Me
OO
Cu
NN
NN
Me
Me
Me
Me
O O
25

VI. EXPERIMENTAL SECTION 165
IR (KBr): ν̃ = 1664 (s, CO2–), 1636 (m, C=Cvinyl), 1560 (w, C=N) cm–1. UV/Vis (MeOH): λmax =
690 nm. FD-MS (CH2Cl2): m/z (%) = 663 (10) [MH+], 617 (10) [MH+ – CO2], 257 (100)
[C16H20N4O2 – CO2]. Elemental analysis calcd. for C32H38CuN8O4 (662.24 g mol–1): C 58.04,
H 5.78, N 16.92; found: C 57.95, H 5.78, N 16.99 %.
6.3.16. [Ru(bdmvpza)(BF)(PPh3)] (26)
To a solution of [Ru(bdmvpza)Cl(PPh3)2] (21) (462 mg, 0.481 mmol) in CH2Cl2 was added
thallous acetate (152 mg, 0.577 mmol) and benzoylformic acid (90.3 mg, 0.601 mmol). The
purple mixture was stirred 2 h at ambient temperature and then filtered through celite. The
solvent was removed in vacuo. Precipitation from a CH2Cl2 solution with n-pentane yields
complex 26 (290 mg, 0.357 mmol, 74 %) as deep purple powder.
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
Ph
5a4a
3a3b
4b5b
26
1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.91 (s, 3 H, C3a-CH3), 1.92 (s, 3 H, C3b-CH3), 2.50 (s,
3 H, C5b-CH3), 2.53 (s, 3 H, C5a-CH3), 5.05 (dd, 2JH,H = 1.0 Hz, 3
JH,H = 17.9 Hz, 1 H, (E )-H2C=),
5.20 (dd, 2JH,H = 1.1 Hz, 3
JH,H = 11.5 Hz, 1 H, (Z )-H2C=), 5,23 (dd, 2JH,H = 1.1 Hz, 3
JH,H =
17.9 Hz, 1 H, (E )-H2C=), 5.29 (dd, 2JH,H = 1.1 Hz, 3
JH,H = 11.5 Hz, 1 H, (Z )-H2C=), 6.23 (dd, 3JH,H = 11.5 Hz, 3
JH,H = 17.7 Hz, 1 H, -HC=), 6.37 (dd, 3JH,H = 11.5 Hz, 3
JH,H = 17.7 Hz, 1 H, -
HC=), 6.60 (s, 1 H, -CH-), 7.20 (t, 6 H, m-PPh3), 7.26 (m, 3 H, p-PPh3), 7.32 (m, 8 H, o-PPh3
and m-Ph), 7.53 (t, 1 H, p-Ph), 8.35 (d, 2 H, o-Ph) ppm. 13C NMR (CDCl3, 75.5 MHz, 25 °C):
δ = 10.3 (2 × C5), 11.7 (C3b), 14.2 (C3a), 68.9 (-CH-), 116.5, 117.4 (2 × =CH2), 119.4 (C4a), 119.0
(C4b), 125.5, 125.8 (2 × -HC=), 128.0 (m-Ph), 128.2 (d, 3JCP = 9.6 Hz, m-PPh3), 129.6 (p-PPh3),
130.3 (o-Ph3), 133.5 (i-Ph), 134.1 (d, 2JCP = 9.0 Hz, o-PPh3), 134.4 (p-Ph), 138.4 (C5b), 140.0 (C5a),
153.1(C3b), 157.2 (C3a), 167.9 (CO2-), 169.6 (CO2
-), 202.9 (C=O) ppm. 31P NMR (CDCl3,
121.5 MHz, 25 °C): δ = 57.8 ppm. IR (KBr): ν̃ = 1653 (s, CO2–) cm–1. IR (CH2Cl2): ν̃ = 1660 (s,
CO2–), 1652 (vs, CO2
–) cm–1. UV/Vis (MeOH): λmax (log ε) = 554.5 (4.77) nm. Elemental

166 VI. EXPERIMENTAL SECTION
analysis calcd. for C42H39N4O5PRu (811.83 g mol–1): C 62.14, H 4.84, N 6.90, found C 61.95,
H 5.16, N 6.75 %.
6.3.17. [Ru(bdmvpza)(NOG)(PPh3)] (27)
To a solution of [Ru(bdmvpza)Cl(PPh3)2] (21) (228 mg, 0.250 mmol) in CH2Cl2 was added
thallous acetate (73.0 mg, 0.275 mmol) and N-oxalylglycine (50.0 mg, 0.305 mmol). The
mixture was stirred 24 h at ambient temperature. The solvent was removed in vacuo, the
residue was washed with degassed water (3 × 10 mL) and diethyl ether (3 × 10 mL) and
dried in vacuo to yield complex 27 (155 mg, 0.192 mmol, 77 %) as an orange crystal powder.
Crystals suitable for an X-ray structure determination were obtained by layering a DMF
solution of 27 with diethyl ether.
N N
N N
Me
Me
Me
Me
OO
Ru
Ph3P
O
OO
NH
5a4a
3a3b
4b5b
CO2H27
1H NMR (DMSO-D6, 300 MHz, 25 °C): δ = 1.70 (s, 3 H, C3b-CH3), 1.98 (s, 3 H, C3a-CH3), 2.43
(s, 3 H, C5-CH3), 2.48 (s, 3 H, C5-CH3), 3.49, 3,65 (ABX, JA,B = 16.9 Hz, 3JH,H = 5.9 Hz,
2 H, -CH2-NH-), 4.97 (d, 3JH,H = 17.7 Hz, 1 H, (E )=CH2), 5.06 (d, 3
JH,H = 11.3 Hz, 1 H,
(Z )=CH2), 5.29 (m, 2 H, =CH2), 6.24 (dd, 3JH,H = 17.6 Hz, 3JH,H = 11.6 Hz, 1 H, -CH=), 6.53 (m,
2 H, CH and -CH=), 7.04 (m, 6 H, o-PPh3), 7.20 (m, 6 H, m-PPh3), 7.31 (m, 6 H, p-PPh3), 9.15
(m, 1 H, NH) ppm. 13C NMR (DMSO-D6, 75.5 MHz, 25 °C): δ = 9.9 (C5a-CH3), 10.0 (C5b-CH3),
10.9 (C3a-CH3), 13.4 (C3b-CH3), 41.5 (-CH2-NH-), 68.4 (CH), 115.4, 116.8 (2 × =CH2), 117.7,
118.0 (2 × C4), 125.9, 126.2 (2 × -CH=), 127.9 (d, 3JC,P = 24 Hz, m-PPh3), 129.3 (p-PPh3), 133.1
(o-PPh3), 139.6 (C5a), 141.4 (C5b), 151.7 (C3a), 155.9 (C3b), 166.3 (CO2–), 167.1 (CO2
–), 167.9
(C=O), 169.3 (CO2H) ppm. 31P NMR (DMSO-D6, 121.5 MHz, 25 °C): δ = 60.7 ppm. IR (KBr):
ν ̃ = 1673 (s, CO2–), 1623 (vs, CO2
–), 1543 w (C=N) cm–1. UV/Vis (DMF): λmax (log ε) =
300.4 (4.88) nm. Elemental analysis calcd. for C38H38N5O7PRu (808.78 g mol–1): C 56.43,
H 4.74, N 8.66; found C 56.69, H 4.91, N 8.26 %.

VI. EXPERIMENTAL SECTION 167
6.3.18. [Ru(bdmpzpr)Cl(CO)2] (28)
A solution of ligand 4 (525 mg, 2.00 mmol) in dry THF (40 mL) is treated with KOtBu
(224 mg, 2.00 mmol) for 1 h at ambient temperature. After addition of [RuCl2(CO)2]n
(456 mg, 2.00 mmol), the reaction mixture was heated under reflux and controlled by IR
spectroscopy on a regular basis. After completion of the reaction (approx. 24 h), the solvent
was removed under reduced pressure. The residue was washed with degassed water (5 × 10
mL) and diethyl ether (5 × 10 mL) and dried in vacuo to yield dicarbonyl complex 28
(633 mg, 1.40 mmol, 70 %)as a yellow crystal powder. Single crystals suitable for an X-ray
structure determination were obtained by layering a CH2Cl2 solution of complex 28 with
diethyl ether.
N N
N N
Me
Me
Me
Me
OO
Ru
CCl CO
O
24
Me
1H NMR (CD2Cl2, 300 MHz, 25 °C): δ = 2.38 (s, 3 H, CH3), 2.55 (s, 6 H, 2 × CH3), 2.64 (s, 3 H,
CH3), 2.67 (s, 3 H, CH3), 6.07 (s, 1 H, Cpz-H), 6.09 (s, 1 H, Cpz-H) ppm. 13C NMR (CD2Cl2,
75.5 MHz, 25 °C): δ = 15.4 (CH3), 16.5 (CH3), 17.5 (CH3), 17.7 (CH3), 23.9 (CH3), 84.1 (Cbridge),
112.7, 112.9 (C4 and C4'), 144.7, 145.9 (C5 and C5'), 154.3, 155.1 (C3 and C3'), 165.9 (CO2–), 194.3
(CO), 197.4 (CO) ppm. IR (THF): ν̃ = 2063 (s, CO), 1994 (s, CO), 1691 (m, as-CO2−) cm–1. IR
(KBr): ν̃ = 2062 (s, CO), 1990 (s, CO), 1676 (s, as-CO2−) cm–1. FD-MS (CH2Cl2): m/z (%) = 455
(100) [MH+]. Elemental analysis calcd. for C15H17ClN4O4Ru (453.84 g mol–1): C 39.70,
H 3.78, N 12.34; found C 39.48, H 3.75, N 12.13 %. M.p.: 266°C (decomposition).
6.3.19. [Ni(bdtbpza)Cl] (30)
2,2-bis(3,5-di-tert-butylpyrazol-1-yl)acetic acid (Hbdtbpza) (29) (350 mg, 0.840 mmol) was
dissolved in dry THF (15 mL) and treated with KOtBu (94.3 mg, 0.840 mmol). After stirring
for 1 h at ambient temperature, a solution of NiCl2 (120 mg, 0.924 mmol) in methanol (5 mL)
was added and the reaction mixture was stirred for another 48 h. The solvent was then
evaporated under reduced pressure. The remaining red solid was dissolved in

168 VI. EXPERIMENTAL SECTION
dichloromethane and centrifuged to remove insoluble salts. The solution was evaporated to
dryness yielding complex 30 (340 mg, 0.667 mmol, 79 %) as a red solid. Crystals suitable for
an X-ray structure determination were obtained by layering a CHCl3 of complex 30 with n-
pentane.
N N
N N
tBu
tBu
t Bu
t Bu
OO
Ni
Cl
30
IR (CH2Cl2): ν ̃ = 1679 (s, CO2–), 1545 (C=N) cm–1. UV/Vis (CH2Cl2): λmax = 471 nm. FD-MS
(CH2Cl2): m/z (%) = 474 (100) [MH+ – Cl], 465 (40) [MH+ – CO2]. Elemental analysis calcd.
for C24H39ClN4NiO2 (509.74 g mol–1): C 56.55, H 7.71, N 10.99; found C 56.83, H 7.72,
N 10.84 %.

VI. EXPERIMENTAL SECTION 169
6.4. Synthesis of polymers
Note: The amount of copolymerized ligand was determined by elemental analysis. The yields
of the polymers are given as weight percent related to the total weight of all monomer units.
6.4.1. Copolymerization of ligand 17 with MMA (P17a)
Ligand 17 (100 mg, 0.332 mmol) and MMA (1.08 mL, 1.00 g, 9.99 mmol) were dissolved in
dry xylene (10 mL) at 80 °C. After addition of AIBN (20.0 mg, 120 µmol), the mixture was
stirred under a dinitrogen atmosphere at 80 °C for 4 h. The resulting polymer solution was
slowly poured into a mixture of MeOH (300 mL) and diluted HCl (3 mL) whereupon a white
precipitate was formed. The precipitate was collected by filtration and washed exhaustively
with MeOH. Drying in vacuo afforded polymer P17 (744 mg, 68 %) as a white powder.
Incorporation: 0.520 mmol g–1.
6.4.2. Copolymerization of ligand 18 with MMA (P18a and P18b)
Ligand 18 (100 mg, 0.332 mmol) and MMA (1.08 mL, 1.00 g, 9.99 mmol) were dissolved in
dry xylene (10 mL) at 80 °C. After addition of AIBN (20.0 mg, 120 µmol), the mixture was
stirred under a dinitrogen atmosphere at 80 °C for 3.5 h. The polymer which precipitated
during the reaction was separated by centrifugation and ground in a mortar; the remaining
xylene solution was put aside. The precipitate was suspended in a mixture of MeOH
(300 mL and diluted HCl (3 mL), filtered off, washed exhaustively with dry MeOH and
finally dried in vacuo to yield polymer P18a (338 mg, 31 %) as a white, powdery mass.
Incorporation: 0.746 mmol g–1.
The remaining clear xylene solution of the experiment was slowly poured into a mixture of
MeOH (100 mL) and diluted HCl (1 mL) whereupon a second polymer started to precipitate.
This precipitate was filtered off, washed exhaustively with MeOH and dried in vacuo to
yield polymer P18b (130 mg, 12 %) as a white powder. Incorporation: 0.304 mmol g–1.

170 VI. EXPERIMENTAL SECTION
6.4.3. Copolymerization of ligand 18 with EGDMA (P18c)
Ligand 18 (100 mg, 0.332 mmol) and EGDMA (950 mL, 1.00 g, 5.04 mmol) were dissolved in
dry xylene (10 mL) at 80 °C. After addition of AIBN (20.0 mg, 120 µmol), the mixture was
stirred under a dinitrogen atmosphere at 80 °C for 2.5 h. The precipitate was collected,
ground in a mortar, and suspended in a mixture of MeOH (300 mL) and diluted HCl
(3 mL). It was then filtered off, washed exhaustively with dry MeOH and finally dried in
vacuo to yield polymer P18c (850 mg, 77 %) as a white, powdery mass. Incorporation:
0.371 mmol g–1
6.4.4. Homopolymerization of ligand 18 (P18d)
Ligand 18 (300 mg, 1.00 mmol) was dissolved in dry xylene (10 mL) at 80°C. After addition
of AIBN (33.0 mg, 200 µmol), the mixture was stirred under a dinitrogen atmosphere for
3.5 h at 80 °C. The precipitiate was collected, ground in a mortar, and suspended in a
mixture of MeOH (300 mL and diluted HCl (3 mL). It was then filtered off, washed
exhaustively with dry MeOH and finally dried in vacuo to yield polymer P18d (213 mg,
71 %) as a white powdery mass. Incorporation: 3.05 mmol g–1.
6.4.5. Synthesis of P18a-Mn
To a suspension of copolymer P18a (100 mg, loading 0.746 mmol g–1, equals 74.6 µmol) in
dry methanol (15 mL) was added KOtBu (8.37 mg, 74.6 µmol) and the suspension was stirred
at 50 °C under a dinitrogen atmosphere. After 1.5 h [MnBr(CO)5] (20.5 mg, 74.6 µmol) was
added and the mixture was stirred for additional 24 h at 50°C. The polymer was collected by
filtration, washed with MeOH and H2O and dried in vacuo to yield P18a-Mn as pale yellow
powder. AAS: 0.470 mmol Mn per g of polymer. IR (nujol): ν̃ = 2038 (CO), 1947 (CO), 1918
(CO) cm–1.
6.4.6. Synthesis of P18a-Re
To a suspension of copolymer P18a (100 mg, loading 0.746 mmol g–1, equals 74.6 µmol) in
dry methanol (15 mL) was added KOtBu (8.37 mg, 74.6 µmol) and the suspension was stirred
at 50 °C under a dinitrogen atmosphere. After 1.5 h [ReBr(CO)5] (30.3 mg, 74.6 µmol) was
added and the mixture was stirred for additional 24 h at 50°C. The polymer was collected by

VI. EXPERIMENTAL SECTION 171
filtration, washed with MeOH and H2O and dried in vacuo to yield P18a-Re as off-white
powder. ICP-AES: = 0.311 mmol Re per g of polymer. IR (nujol): ν̃ = 2028 (CO), 1923 (CO),
1898 (CO) cm–1.
6.4.7. Synthesis of P18a-Cu
To a suspension of P18a (100 mg, loading 0.746 mmol g–1, equals 74.6 µmol) in dry methanol
(10 mL) was added KOtBu (8.37 mg, 74.6 µmol) and the suspension was stirred for 1 h at
50 °C under a dinitrogen atmosphere. A solution of CuCl2 (15 mg, 111 µmol) in methanol
was added whereupon the colour of the suspension immediately turned lime green. The
polymer was collected by filtration, washed exhaustively with dry methanol and dried in
vacuo to yield P18a-Cu as a lime green powder. UV/Vis (polymer film, nujol): λmax =
754 nm.
6.4.8. Synthesis of P18a-Fe
To a suspension of P18a (200 mg, loading 0.746 mmol g–1, equals 0.150 mmol) in dry
methanol (10 mL) was added KOtBu (16.4 mg, 0.150 mmol) is added and the suspension was
stirred for 1 h at 50 °C under a dinitrogen atmosphere. Anhydrous FeCl2 (30.4 mg,
0.240 mmol) was added and the mixture was stirred for 1 h at 50 °C. The polymer was
collected by filtration, washed exhaustively with degassed methanol and dried in vacuo to
yield P18a-Fe as a pale green powder. AAS: 0.491 mmol Fe per g of polymer.
In a parallel experiment FeSO4 × 7H2O was used as instead of FeCl2. In this case, the iron
content was determined to 0.448 mmol Fe per g of polymer.
6.4.9. Synthesis of P18c-Mn
To a suspension of copolymer P18c (150 mg, loading 0.371 mmol g–1, equals 55.7 µmol) in
dry methanol (15 mL) was added KOtBu (6.24 mg, 55.7 µmol) and the suspension was stirred
at 50 °C under a dinitrogen atmosphere. After 1.5 h [MnBr(CO)5] (15.3 mg, 55.7 µmol) was
added and the mixture was stirred for additional 24 h at 50°C. The polymer was collected by
filtration, washed with MeOH and H2O and dried in vacuo to yield P18a-Mn as pale yellow
powder. AAS: 0.148 mmol Mn per g of polymer. IR (nujol): ν̃ = 2041 (CO), 1949 (CO), 1924
(CO) cm–1.

172 VI. EXPERIMENTAL SECTION
6.4.10. Synthesis of P18c-Re
To a suspension of copolymer P18c (150 mg, loading 0.371 mmol g–1, equals 55.7 µmol) in
dry methanol (15 mL) was added KOtBu (6.24 mg, 55.7 µmol) and the suspension was stirred
at 50 °C under a dinitrogen atmosphere. After 1.5 h [ReBr(CO)5] (22.6 mg, 55.7 µmol) was
added and the mixture was stirred for additional 24 h at 50°C. The polymer was collected by
filtration, washed with MeOH and H2O and dried in vacuo to yield P18c-Re as off-white
powder. ICP-AES: 0.038 mmol Re per g of polymer. IR (nujol): ν ̃ = 2031 (CO), 1927 (CO),
1908 (CO) cm–1.
6.4.11. Synthesis of P18c-Fe
To a suspension of P18c (1.00 g, loading 0.371 mmol g–1, equals 0.371 mmol) in dry
methanol (10 mL) was added KOtBu (41.6 mg, 0.371 mmol) and the suspension was stirred
for 1 h at 50 °C under a dinitrogen atmosphere. A solution of FeCl2 (47.0 mg, 0.371 mmol) in
degassed methanol was added and the mixture was stirred for another hour at 50 °C. The
polymer was collected by filtration, washed exhaustively with degassed methanol and dried
in vacuo to yield P18c-Fe as a pale green powder. AAS: 0.165 mmol Fe per g of polymer.
6.4.12. Synthesis of P23
[Ru(bdmvpza)Cl(CO)2] (23) (150 mg, 0.305 mmol) and EGDMA (1.43 mL, 1.50 g, 7.56 mmol)
were dissolved in acetonitrile (12 mL) and heated to 65 °C. AIBN (50.0 mg, 0.304 mmol) was
added and the mixture was stirred for 24 h at 65 °C under a dinitrogen atmosphere. The
polymer was collected by filtration and was washed with acetonitrile (6 × 10 mL) and
MeOH (5 × 10 mL). Drying in vacuo afforded polymer P23 (1.47 g, 89 %) as a pale yellow
powder. ICP-AES: 0.103 mmol Ru per g of polymer. IR (nujol): ν̃ = 2071 (CO), 1999
(CO) cm–1.
6.4.13. Synthesis of P26
To a solution of [Ru(bdmvpza)(BF)(PPh3)] (26) (100 mg, 0.123 mmol) and EGDMA (2.33 mL,
2.44 g, 12.3 mmol) in acetonitrile (2.46 mL) at 65 °C was added AIBN (56.0 mg, 0.341 mmol).
The mixture was allowed to stir for 24 h at this temperature under a dinitrogen atmosphere.
The resulting polymer was collected, ground in a mortar, washed with MeOH and finally

VI. EXPERIMENTAL SECTION 173
dried in vacuo to yield P26 as a purple coloured powder (2.31 g, 91 %). ICP-AES: 31.1 µmol
Ru per g of polymer. UV/Vis (polymer film, nujol): λmax = 558.5 nm.
6.4.14. Synthesis of P27
[Ru(bdmvpza)(NOG)(PPh3)] (27) (100 mg, 0.124 mmol) and EGDMA (2.33 mL, 2.46 g,
12.4 mmol) were dissolved in a mixture of acetonitrile (5.6 mL) and DMF (5.6 mL) at 65 °C
After addition of AIBN (57.0 mg, 0.347 mmol), the mixture was stired for 24 h at 65 °C
under a dinitrogen atmosphere. The resulting polymer was collected, ground in a mortar
and washed thoroughly with DMF and MeOH. Drying in vacuo afforded P27 (2.26 g, 88 %)
as an orange coloured powder. ICP-AES: 31.3 µmol Ru per g of polymer. UV/Vis (polymer
film, nujol): λmax = 311.0 nm.
6.4.15. Treatment of P26 or P27 with PMe3 (P26X, P27X)
General Procedure:
To a suspension of the according polymer (P26 or P27, 500 mg) in a mixture of EtOH (5 mL)
and THF (5 mL) was added PMe3 (500 – 1800 µL/g of polymer). The mixture was allowed to
stir for 72 – 120 h at 70 °C under a dinitrogen atmosphere. After cooling to ambient
temperature, the suspension was poured into a solution of HCl (conc., 1.5 mL) in MeOH
(100 mL). The polymer was collected by filtration and carefully washed with H2O, MeOH
and acetone and finally dried in vacuo. The remaining content of Ru was determined by
ICP-AES. The amount of utilized PMe3, the reaction times and the results of the ICP-AES
measurements are indicated in table 24.
Table 24: Treatment of P26 or P27 with PMe3.
Polymer V(PMe3)
[µL]
Reaction time
[h]
m0(Ru)/m(P)
[mg/g]
mt(Ru)/m(P)
[mg/g]
Cleavage
[%]
P26X 250 72 3.30 2.52 24
P26X 500 120 3.00 1.48 51
P27X 900 72 3.09 1.77 43
P27X 500 120 3.24 2.09 35

174 VI. EXPERIMENTAL SECTION
6.5. Epoxidation catalysis
General procedure:
An evacuated and nitrogen flushed Schlenk tube with a Teflon rotaflow stopcock was
charged with the catalyst (0.03 − 0.10 eq), Cyclohexene (1.0 eq.) and unstabilized CH2Cl2
(5 mL). After addition of the oxidant (1.0 – 2.5 eq.) the tube was closed and stirred for 22 –
24 h at ambient temperature under dinitrogen atmosphere. The crude mixture was filtrated
over a short plug of silica gel and analyzed by GC. The quantification was done by using
calibration curves which have been recorded with standard samples of known
concentration of the reactant and the product cyclohexene oxide. The utilized catalysts, the
type and amount of the oxidant the reaction times as well as the results of the GC analyses
are indicated in table 25.
Table 25: Epoxidation of cyclohexene with dicarbonyl complexes 24 or 28 and polymer P23.
[Ru]
Oxidant t
[h]
neduct (t0)
[mmol]
ncatalyst
[mmol]
neduct (t)
[mmol]
nproduct (t)
[mmol]
y
[%]
TON TOF
[10−5s−1]
24 PhIO 24 0.503 0.050 0.002 0.327 65.1 6.6 7.58
24 PhIO 22.5 0.503 0.025 0.001 0.342 67.9 13.7 16.88
24 PhIO 24 0.750 0.025 0.001 0.515 68.6 20.6 23.83
24 H2O2 23 0.503 0.050 0.488 0.008 1.6 0.2 0.20
24 DCPNO 23 0.503 0.050 0.472 0.000 0.0 0.0 0.00
24 PhIO
/H2O2
24 0.503 0.050 0.440 0.017 3.3 0.3 0.39
24 PhIO
/DCPNO
24 0.503 0.050 0.434 0.023 4.6 0.5 0.53
28 PhIO 22.5 0.503 0.050 0.092 0.234 46.6 4.7 5.79
P23 PhIO 22 0.503 0.018 0.453 0.013 2.6 0.7 0.92
[a] Solution in H2O (35 %) [b] The reaction was initiated with 10 mol% PhIO for 1h before 90 mol% DCPNO
was added to the reaction mixture.

175


VII. APPENDIX 177
VII. APPENDIX

178 VII. APPENDIX
7.1. Details of the structure determinations
Single crystals of the complexes have been obtained by solvent diffuision (layering
technique) as summarized in table 26. Single crystals of [Ru(bdmvpza)Cl(MeCN)(PPh3) (22)
have been obtained by layering a solution of the bisphosphine complex
[Ru(bdmvpza)Cl(PPh3)2] 21 in CH2Cl2 and traces of acetonitrile with diethyl ether.
Table 26: Solvents for the crystallization of the synthesized transition metal complexes.
Complex Dissolved in Layered with
[Mn(bdmpzmp)(CO)3] (2) benzene n-hexane
[Cu(bdmpzmp)2] (3) CH2Cl2 n-hexane
[Mn(bdmpzpr)(CO)3] (5) CH2Cl2 n-pentane
[Cu(bdmpzpr)2] (7) CH2Cl2 n-pentane
[Ru(bdmpzpr)Cl(N2)(PPh3) (10) CH2Cl2 diethyl ether
[Ru(bdmpzpr)Cl(CO)(PPh3)] (12a) CH2Cl2 diethyl ether
[Ru(bdmpza)Cl(PPh3)(SO2)] (13) CHCl3 n-pentane
[Mn(bdmvpza)(CO)3] (19) CH2Cl2 diethyl ether
[Cu(bdmvpza)2] (25) CH2Cl2 n-pentane
[Ru(bdmvpza)(NOG)(PPh3)] (27) dimethylformamide diethyl ether
[Ru(bdmpzpr)Cl(CO)2] (28) CH2Cl2 diethyl ether
[Ni(bdtbpza)Cl] (30) CHCl3 n-pentane
A modified Siemens P4 diffractometer, a Bruker-Nonius Kappa-CCD or an Enraf Nonius
CAD4 Mach 3 diffractometer were used for data collection. Single crystals were mounted
with Paratone-N, glue or perfluorated oil on a glass fibre. The structures were solved by
using direct methods and refined with full-matrix least-squares agains F2 {Siemens
SHELX-97}.[340] A weighting scheme was applied in the last steps of the refinement with
w = 1/[σ2(Fo2) + (aP)2 + bP] and P =[2Fc2 + max(Fo
2,0)]/3. Most hydrogen atoms were
included in their calculated positionsand refined in a riding model. All further details and
parameters of the measurements are summarised in table 27–33. The structure pictures
were prepared with the program Diamond 2.1e.[341-342]

VII. APPENDIX 179
The molecular structure of 27 contains one diethyl ether and two dimethylformamide
solvent molecules per asymmetric unit. Squeeze/Platon was applied to resolve another
severely disordered diethyl ether solvent molecule.[343-344] Within the 265.5 Å3 of void space
accessible to solvent molecules at a special position (x = 0.5, y =1, z = 1), a total of 17.4
electrons were calculated, compared to 21 electrons predicted for the presence of half a
diethyl ether molecule per asymmetric unit.
As parts of this thesis have already been published, the supplementary data for the
complexes 2 (CCDC-661579), 3 (CCDC-661580), 19 (CCDC-729242), 25 (CCDC-729243) and
27 (CCDC-762178) can be obtained free of charge from the Cambridge Crystallographic
Data Centre via www.ccdc.cam.ac.uk/datarequest/cif.

180 VII. APPENDIX
Table 27: Details for the structure determination of 2 and 3.
[Mn(bdmpzmp)(CO)3] (2) [Cu(bdmpzmp)2] (3)
Empirical formula C20H21MnN4O7 C34H42CuN8O8
Formula mass 484.35 754.3
Crystal colour/habit yellow plate blue prism
Crystal system monoclinic monoclinic
Space group P21/a P21/c
a [Å] 12.1154(7) 8.8422(7)
b [Å] 15.3039(8) 12.1958(7)
c [Å] 12.5755(13) 16.7929(7)
α [°] 90.00 90.00
β [°] 116.975(6) 103.152(2)
γ [°] 90.00 90.00
V [Å3] 2078.0(3) 1763.41(19)
θ [°] 3.22 – 27.5 2.9 – 28.49
h –15 to 15 –11 to 11
k –19 to 19 –16 to 16
l –16 to 16 –22 to 22
F(000), Z 1000, 4 790, 2
µ (Mo-Kα) [mm–1] 0.687 0.682
Crystal size [mm] 0.32 × 0.16 × 0.023 0.21 × 0.16 × 0.10
Dcalcd. [g cm–3], T [K] 1.548, 150 1.421, 150
Reflections collected 45570 46808
Independent reflections 4762 4449
Obs. reflections, I > 2σI 3510 3563
Parameter 289 238
Weight parameter a 0.0381 0.0681
Weight parameter b 1.4923 0.8610
R1 (observed) 0.0388 0.0441
R1 (overall) 0.0662 0.0624
wR2 (observed) 0.0825 0.1136
wR2 (overall) 0.0912 0.1222
Diff. peak / hole [e/Å] –0.393 / 0.359 –0.373 / 1.000

VII. APPENDIX 181
Table 28: Details for the structure determination of 5 and 7.
[Mn(bdmpzpr)(CO)3]
(5)
[Cu(bdmpzpr)2]
(7)
Empirical formula C16H17MnN4O5 C26H34CuN8O4
Formula mass 400.28 586.16
Crystal colour/habit yellow prism blue block
Crystal system triclinic monoclinic
Space group 1P P21/c
a [Å] 8.2964(6) 8.8389(10)
b [Å] 9.6530(3) 9.665(2)
c [Å] 11.393 15.970(2)
α [°] 105.882(4) 90
β [°] 99.171(6) 94.446(10)
γ [°] 104.865(3) 90
V [Å3] 821.92(6) 1360.1(4)
θ [°] 3.41 – 28.5 2.31 – 25.98
h –11 to 11 –10 to 10
k –12 to 12 0 to 11
l –15 to 15 –19 to 0
F(000), Z 412, 2 614, 2
µ (Mo-Kα) [mm–1] 0.841 0.851
Crystal size [mm] 0.18 × 0.14 × 0.07 0.23 × 0.15 × 0.13
Dcalcd. [g cm–3], T [K] 1.617, 150 1.431, 293(2)
Reflections collected 20009 2757
Independent reflections 4148 2657
Obs. reflections, I > 2σI 3168 1824
Parameter 235 178
Weight parameter a 0.0210 0.0777
Weight parameter b 0.5353 0.3731
R1 (observed)/R1 (overall) 0.0353/0.0596 0.0481/0.1005
wR2 (observed) 0.0645 0.1188
wR2 (overall) 0.0699 0.1387
Diff. peak / hole [e/Å] 0.351 / -0.357 -0.278 / 0.076

182 VII. APPENDIX
Table 29: Details for the structure determination of 10 and 12a.
[Ru(bdmpzpr)Cl(N2)(PPh3)
(10)
[Ru(bdmpzpr)Cl(CO)(PPh3)]
(12a)
Empirical formula C31H32ClN6O2PRu × CH2Cl2 C32H32Cl1N4O3PRu × CH2Cl2
Formula mass 773.04 773.03
Crystal colour/habit yellow column yellow block
Crystal system triclinic triclinic
Space group 1P 1P
a [Å] 9.8965(8) 10.0431(5)
b [Å] 11.6847(5) 11.6105(5)
c [Å] 15.3070(10) 15.3823(10)
α [°] 82.558(4) 82.767(4)
β [°] 73.358(7) 73.575(5)
γ [°] 76.584(5) 76.413(4)
V [Å3] 1645.9(2) 1668.91(15)
θ [°] 3.05 – 28.5 2.77 – 28.5
h –13 to 13 –13 to 13
k –15 to 15 –15 to 15
l –19 to 20 –20 to 20
F(000), Z 788, 2 788, 2
µ (Mo-Kα) [mm–1] 0.809 0.798
Crystal size [mm] 0.3 × 0.09 × 0.04 0.36 × 0.09 × 0.05
Dcalcd. [g cm–3], T [K] 1.56, 150(2) 1.538, 150(2)
Reflections collected 41656 56113
Independent reflections 8336 8458
Obs. reflections, I > 2σI 6603 7312
Parameter 406 434
Weight parameter a 0.0353 0.0350
Weight parameter b 1.1907 0.7845
R1 (observed)/R1 (overall) 0.0357/0.0557 0.0264/0.0355
wR2 (observed) 0.081 0.0691
wR2 (overall) 0.0751 0.0724
Diff. peak / hole [e/Å] -0.669 / 0.849 -0.634 / 0.54
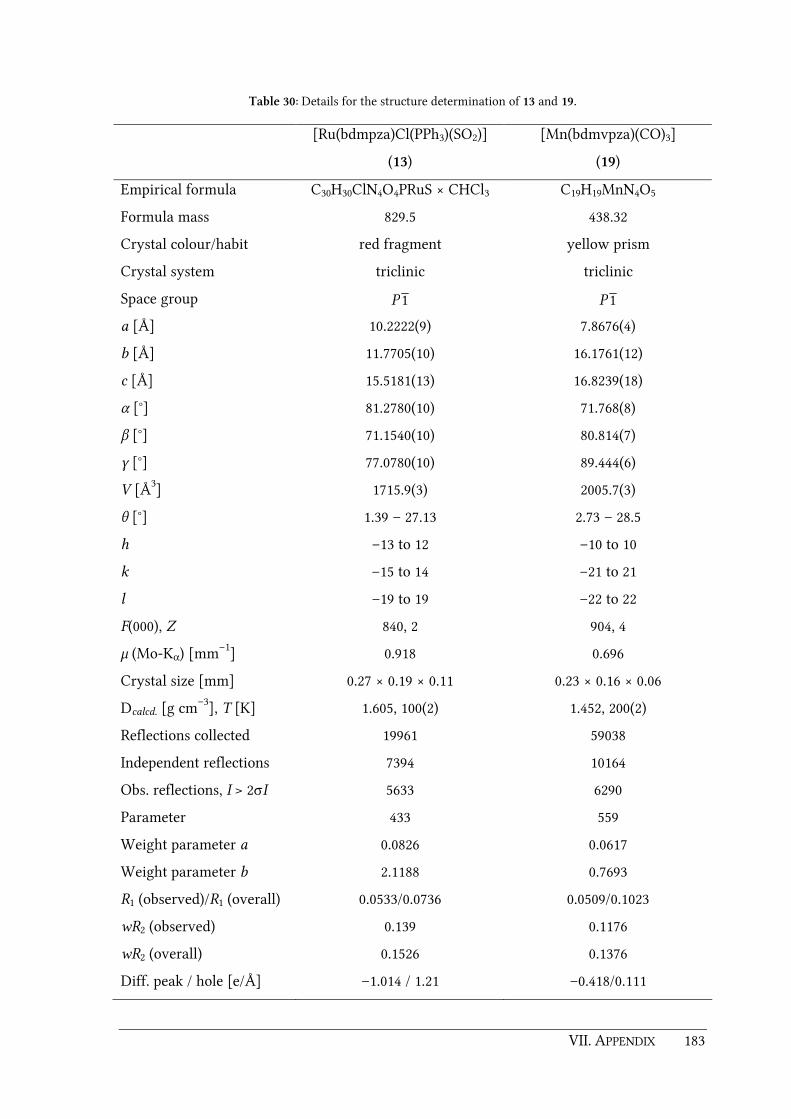
VII. APPENDIX 183
Table 30: Details for the structure determination of 13 and 19.
[Ru(bdmpza)Cl(PPh3)(SO2)]
(13)
[Mn(bdmvpza)(CO)3]
(19)
Empirical formula C30H30ClN4O4PRuS × CHCl3 C19H19MnN4O5
Formula mass 829.5 438.32
Crystal colour/habit red fragment yellow prism
Crystal system triclinic triclinic
Space group 1P 1P
a [Å] 10.2222(9) 7.8676(4)
b [Å] 11.7705(10) 16.1761(12)
c [Å] 15.5181(13) 16.8239(18)
α [°] 81.2780(10) 71.768(8)
β [°] 71.1540(10) 80.814(7)
γ [°] 77.0780(10) 89.444(6)
V [Å3] 1715.9(3) 2005.7(3)
θ [°] 1.39 – 27.13 2.73 – 28.5
h –13 to 12 –10 to 10
k –15 to 14 –21 to 21
l –19 to 19 –22 to 22
F(000), Z 840, 2 904, 4
µ (Mo-Kα) [mm–1] 0.918 0.696
Crystal size [mm] 0.27 × 0.19 × 0.11 0.23 × 0.16 × 0.06
Dcalcd. [g cm–3], T [K] 1.605, 100(2) 1.452, 200(2)
Reflections collected 19961 59038
Independent reflections 7394 10164
Obs. reflections, I > 2σI 5633 6290
Parameter 433 559
Weight parameter a 0.0826 0.0617
Weight parameter b 2.1188 0.7693
R1 (observed)/R1 (overall) 0.0533/0.0736 0.0509/0.1023
wR2 (observed) 0.139 0.1176
wR2 (overall) 0.1526 0.1376
Diff. peak / hole [e/Å] –1.014 / 1.21 –0.418/0.111

184 VII. APPENDIX
Table 31: Details for the structure determination of 22 and 25.
[Ru(bdmvpza)Cl(MeCN)(PPh3)
(22)
[Cu(bdmvpza)2]
(25)
Empirical formula C36H37ClN5O2PRu × CH2Cl2 3(C32H38CuN8O4) × CH2Cl2
Formula mass 824.12 2071.02
Crystal colour/habit yellow prism blue prism
Crystal system triclinic trigonal
Space group 1P R-3
a [Å] 11.2287(4) 27.424(3)
b [Å] 12.5040(6) 27.424(3)
c [Å] 13.1412(7) 10.916(1)
α [°] 85.676(4) 90
β [°] 84.961(4) 90
γ [°] 89.309(3) 120
V [Å3] 1832.68(15) 7110.0(13)
θ [°] 2.74 – 27.5 2.73–28.5
h –14 to 14 –36 to 30
k –16 to 16 –36 to 36
l –17 to 17 –14 to 14
F(000), Z 844, 2 3249, 3
µ (Mo-Kα) [mm–1] 0.731 0.799
Crystal size [mm] 0.23 × 0.12 × 0.05 0.23 × 0.22 × 0.09
Dcalcd. [g cm–3], T [K] 1.493, 150(2) 1.452, 150(2)
Reflections collected 40088 21267
Independent reflections 8429 4003
Obs. reflections, I > 2σI 6927 2828
Parameter 460 217
Weight parameter a 0.0309 0.0607
Weight parameter b 2.0273 27.0636
R1 (observed)/R1 (overall) 0.0303/0.0447 0.0464/0.0802
wR2 (observed) 0.0697 0.1212
wR2 (overall) 0.0756 0.1421
Diff. peak / hole [e/Å] -0.906 / 0.764 –0.481 / 0.974
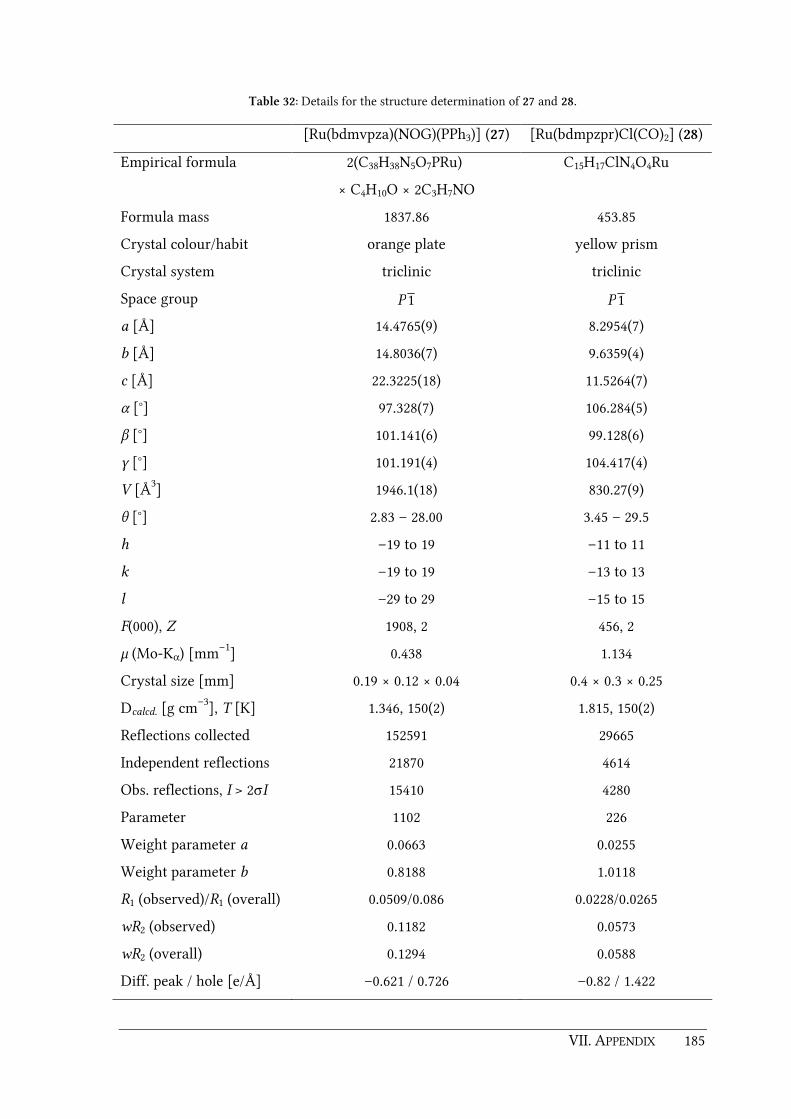
VII. APPENDIX 185
Table 32: Details for the structure determination of 27 and 28.
[Ru(bdmvpza)(NOG)(PPh3)] (27) [Ru(bdmpzpr)Cl(CO)2] (28)
Empirical formula 2(C38H38N5O7PRu)
× C4H10O × 2C3H7NO
C15H17ClN4O4Ru
Formula mass 1837.86 453.85
Crystal colour/habit orange plate yellow prism
Crystal system triclinic triclinic
Space group 1P 1P
a [Å] 14.4765(9) 8.2954(7)
b [Å] 14.8036(7) 9.6359(4)
c [Å] 22.3225(18) 11.5264(7)
α [°] 97.328(7) 106.284(5)
β [°] 101.141(6) 99.128(6)
γ [°] 101.191(4) 104.417(4)
V [Å3] 1946.1(18) 830.27(9)
θ [°] 2.83 – 28.00 3.45 – 29.5
h –19 to 19 –11 to 11
k –19 to 19 –13 to 13
l –29 to 29 –15 to 15
F(000), Z 1908, 2 456, 2
µ (Mo-Kα) [mm–1] 0.438 1.134
Crystal size [mm] 0.19 × 0.12 × 0.04 0.4 × 0.3 × 0.25
Dcalcd. [g cm–3], T [K] 1.346, 150(2) 1.815, 150(2)
Reflections collected 152591 29665
Independent reflections 21870 4614
Obs. reflections, I > 2σI 15410 4280
Parameter 1102 226
Weight parameter a 0.0663 0.0255
Weight parameter b 0.8188 1.0118
R1 (observed)/R1 (overall) 0.0509/0.086 0.0228/0.0265
wR2 (observed) 0.1182 0.0573
wR2 (overall) 0.1294 0.0588
Diff. peak / hole [e/Å] –0.621 / 0.726 –0.82 / 1.422

186 VII. APPENDIX
Table 33: Details for the structure determination of 30.
[Ni(bdtbpza)Cl]
(30)
Empirical formula C24H39ClN4NiO2 × CHCl3
Formula mass 629.12
Crystal colour/habit red plate
Crystal system monoclinic
Space group P21/c
a [Å] 13.594(2)
b [Å] 10.7156(6)
c [Å] 21.572(2)
α [°] 90
β [°] 99.587(10)
γ [°] 90
V [Å3] 3098.5(6)
θ [°] 2.94 – 29.01
h –18 to 18
k –14 to 14
l –29 to 29
F(000), Z 1320, 4
µ (Mo-Kα) [mm–1] 0.999
Crystal size [mm] 0.28 × 0.23 × 0.07
Dcalcd. [g cm–3], T [K] 1.349, 150(2)
Reflections collected 103602
Independent reflections 8241
Obs. reflections, I > 2σI 6458
Parameter 361
Weight parameter a 0.0490
Weight parameter b 2.5874
R1 (observed)/R1 (overall) 0.041/0.0605
wR2 (observed) 0.0951
wR2 (overall) 0.1032
Diff. peak / hole [e/Å] -0.724 / 0.069

VII. APPENDIX 187
7.2. List of abbreviations and symbols
2-OG 2-Oxoglutarate
AAS Atomic absorption spectrometry
AIBN 2,2'-Azobis(2-methylproprionitrile)
arom aromatic
as asymmetric
azaBox Azabis(oxazoline)
BF Benzoylformate
BNPP Bis(p-nitrophenyl)phosphate
Box Bis(oxazoline)
bpy 2,2'-Bipyridine
br broad
BTEAC Benzyltriethylammonium chloride
Bu Butyl
calcd. calculated
chir (S,S)-2,3-Bis(diphenylphosphino)butane
Cp Cyclopentadienyl
d Doublet
DAOCS Deacetoxycephalosporin C synthase
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCPNO 2,6-Dichloropyridine N-oxide
dd Double doublet
DFT Density functional theory
dippae 1,2-Bis(diisopropylphosphinoamino)ethane
dippe 1,2-Bis(diisopropylphosphino)ethane
DMF Dimethylformamide
DMOG Dimethyloxalylglycine
dmpm Bis(dimethylphosphino)methane
dppa N,N-Bis(diphenylphosphino)amine
dppe 1,2-Bis(diphenylphosphino)ethane
dppm 1,1-Bis(diphenylphosphino)methane
DVB Divinylbenzene

188 VII. APPENDIX
e.g. exempli gratia
EDTA Ethylenediaminetetraacetic acid
ee Enantiomeric excess
EGDMA Ethylene glycol dimethacrylate
EPR Enhanced permeability and retention effect
Electron paramagnetic resonance
EXAFS Extended X-ray absorption fine structure
FAB Fast atom bombardment
fac facial
FD Field desorption
FIH Factor-inhibiting hypoxia-inducible factor
GC Gas chromatography
h Hour
Hbdmpza 2,2-Bis(3,5-dimethylpyrazol-1-yl)acetic acid
Hbdmpzap 3-Acetoxy-2,2-bis(3,5-dimethylpyrazol-1-yl)propanoic acid
Hbdmpzhp 2,2-Bis(3,5-dimethylpyrazol-1-yl)-3-hydroxypropanoic acid
Hbdmpzmp 2,2-Bis(3,5-dimethylpyrazol-1-yl)-3-(methacryloxy)propanoic acid
Hbdmpzpen 2,2-Bis(3,5-dimethylpyrazol-1-yl)pent-4-enoic acid
Hbdmpzpr 2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid
Hbdmvpza 2,2-Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid
Hbdtbpza 2,2-Bis(3,5-di-tert-butylpyrazol-1-yl)acetic acid
HIF Hypoxia-inducible Factor
HOMO Highest occupied molecular orbital
Hz Hertz
i iso
i.e. id est
ICP-AES Inductively coupled plasma atomic emission spectroscopy
IPNS Isopenicillin N synthase
IR Infrared spectroscopy
LDA Lithium diisopropylamide
LUMO Lowest unoccupied molecular orbital
m medium (IR spectroscopy)
multiplet (NMR spectroscopy)
m meta

VII. APPENDIX 189
M.p. Melting point
mCPBA m-Chloroperbenzoic acid
Me3Tacn 1,4,7-Trimethyl-1,4,7-triazacyclononane
MIP Molecular imprinted polymer
MLCT Metal to ligand charge transfer
MMA Methyl methacrylate
MS Mass spectrometry
NBOH 3-Nitrobenyzl alcohol
NMR Nuclear magnetic resonance
NOG N-Oxalylglycine
o ortho
p para
P~O κ1P coordinated (1,3-dioxan-2-ylmethyl)diphenylphosphine
P4H Prolyl-4-hydroxylase
PCR Polymerase chain reaction
PEGDMA Poly(ethylene glycol) dimethacrylate
PHD Prolyl hydroxylase domain enzyme
PMMA Poly(methyl methacrylate)
ppm Parts per million
Pr Propyl
PS-DVB Polystyrene-divinylbenzene
PyBOP (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
R,R-dippach (R,R)-1,2-bis((diisopropylphosphino)amino)cyclohexane
s strong (IR spectroscopy)
symmetrical (IR spectroscopy)
Singlet (NMR spectroscopy)
SEC Size exclusion chromatography
t Triplet
Tacn 1,4,7-Triazacyclononane
TauD Taurine dioxygenase
TBHP tert-Butylhydroperoxide
THF Tetrahydrofuran
TMC 1,4,8,11-Tetramethyl-1,4,8,11-tetraazacyclotetradecane
tmeda N,N,N',N'-Tetramethylethylenediamine

190 VII. APPENDIX
TMG3tren 1,1,1-Tris{2-[N 2-(1,1,3,3,-tetramethylguanidino)]ethyl}amine
TOF Turnover frequency
TON Turnover number
Tp Hydridotris(pyrazol-1-yl)borato
Tpa Tris(2-pyridylmethyl)amine
Tpm Tris(pyrazolyl)methane
UV/Vis Ultraviolet-visible spectroscopy
w weak (IR spectroscopy)
y Yield
δ Chemical shift
ε Extinction coefficient
λ Wavelength
ν Stretching vibration
ν ̃ Wavenumber
J Coupling constant

VII. APPENDIX 191
7.3. List of compounds
2,2-Bis(3,5-dimethylpyrazol-1-yl)acetic acid (Hbdmpza) (1)
[Mn(bdmpzmp)(CO)3] (2)
[Cu(bdmpzmp)2] (3)
2,2-Bis(3,5-dimethylpyrazol-1-yl)propanoic acid (Hbdmpzpr) (4)
[Mn(bdmpzpr)(CO)3] (5)
[Re(bdmpzpr)(CO)3] (6)
[Cu(bdmpzpr)2] (7)
[Ru(bdmpza)Cl(PPh3)2] (8)
[Ru(bdmpzpr)Cl(PPh3)2] (9)
[Ru(bdmpzpr)Cl(N2)(PPh3)] (10)
[Ru(bdmpza)Cl(CO)(PPh3)] (11)
[Ru(bdmpzpr)Cl(CO)(PPh3)] (12)
[Ru(bdmpza)Cl(PPh3)(SO2)] (13)
[Ru(bdmpzpr)Cl(PPh3)(SO2)] (14)
Bis(3,5-dimethylpyrazol-1-yl)methane (15)
Bis(3,5-dimethyl-4-formylpyrazol-1-yl)methane (16)
Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)methane (17)
2,2-Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic acid (Hbdmvpza) (18)
[Mn(bdmvpza)(CO)3] (19)
[Re(bdmvpza)(CO)3] (20)
[Ru(bdmvpza)Cl(PPh3)2] (21)
[Ru(bdmvpza)Cl(MeCN)(PPh3)] (22)
[Ru(bdmvpza)Cl(CO)2] (23)
[Ru(bdmpza)Cl(CO)2] (24)
[Cu(bdmvpza)2] (25)
[Ru(bdmvpza)(BF)(PPh3)] (26)
[Ru(bdmvpza)(NOG)(PPh3)] (27)
[Ru(bdmpzpr)Cl(CO)2] (28)
2,2-bis(3,5-di-tert-butylpyrazol-1-yl)acetic acid (Hbdtbpza) (29)
[Ni(bdtbpza)Cl] (30)
[Ni(bdtbpza)Cl(MeOH)2] (31)

192 VII. APPENDIX
MMA copolymer of compound 17 (P17a)
MMA copolymer of ligand 18 (P18a/b)
EGDMA copolymer of ligand 18 (P18c)
Homopolymer of ligand 18 (P18d)
Manganese(I) containing MMA copolymer (P18a-Mn)
Rhenium(I) containing MMA copolymer (P18a-Re)
Copper(II) containing MMA copolymer (P18a-Cu)
Iron(II) containing MMA copolymer (P18a-Fe)
Manganese(I) containing EGDMA copolymer (P18c-Mn)
Rhenium(I) containing EGDMA copolymer (P18c-Re)
Iron(II) containing EGDMA copolymer (P18c-Fe)
EGDMA copolymer of complex 23 (P23)
EDGMA copolymer of complex 26 (P26)
EGDMA copolymer of complex 27 (P27)
PMe3 treated polymer P26 (P26X)
PMe3 treated polymer P27 (P27X)

193


VIII. BIBLIOGRAPHY 195
VIII. BIBLIOGRAPHY

196 VIII. BIBLIOGRAPHY
[1] W. B. Tolman, E. I. Solomon, Inorg. Chem. 2010, 49, 3555-3556.
[2] S. G. E. Lindahl, Anesthesiology 2008, 109, 387-401.
[3] T. W. Lyons, Nature 2007, 448, 1005-1006.
[4] P. C. A. Bruijnincx, G. van Koten, R. J. M. Klein Gebbink, Chem. Soc. Rev. 2008, 37,
2716-2744.
[5] M. M. Abu-Omar, A. Loaiza, N. Hontzedas, Chem. Rev. 2005, 105, 2227-2252.
[6] M. Costas, M. P. Mehn, M. P. Jensen, L. Que Jr., Chem. Rev. 2004, 104, 939-986.
[7] K. D. Koehntop, J. P. Emerson, L. Que Jr., J. Biol. Inorg. Chem. 2005, 10, 87-93.
[8] E. L. Hegg, L. Que Jr., Eur. J. Biochem. 1997, 250, 625-629.
[9] L. Que Jr., Nat. Struct. Biol. 2000, 7, 182-184.
[10] K. Vålegard, A. C. Terwisscha van Scheltinga, M. D. Lloyd, T. Hara, S.
Ramaswamy, A. Perrakis, A. Thompson, H.-J. Lee, J. E. Baldwin, C. J. Schofield, J.
Hajdu, I. Andersson, Nature 1998, 394, 805-809.
[11] L. Que Jr., R. Y. N. Ho, Chem. Rev. 1996, 96, 2607 - 2624.
[12] E. Flashman, C. J. Schofield, Nat. Chem. Biol. 2007, 3, 86-87.
[13] R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21-68.
[14] C. Loenarz, C. J. Schofield, Nat. Chem. Biol. 2008, 4, 152-156.
[15] V. Purpero, G. R. Moran, J. Biol. Inorg. Chem. 2007, 12, 587-601.
[16] J. M. Simmons, T. A. Müller, R. P. Hausinger, Dalton Trans. 2008, 5132-5142.
[17] J. J. Hutton Jr., A. L. Trappel, S. Udenfriend, Biochem. Biophys. Res. Commun. 1966,
24, 179-184.
[18] J. Myllyharju, Ann. Med. 2008, 40, 402-417.
[19] M. Ivan, K. Kondo, H. Yang, W. Kim, J. Valiando, M. Ohh, A. Salic, J. M. Asara, W.
S. Lane, W. G. Kaelin Jr., Science 2001, 292, 464-468.
[20] P. Jaakkola, D. R. Mole, Y.-M. Tian, M. I. Wilson, J. Gielbert, S. J. Gaskell, A. von
Kriegsheim, H. F. Hebestreit, M. Mukherji, C. J. Schofield, P. H. Maxwell, C. W.
Pugh, P. J. Ratcliffe, Science 2001, 292, 468-472.
[21] E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F.
Neese, A. J. Skulan, Y.-S. Yang, J. Zhou, Chem. Rev. 2000, 100, 235-349.
[22] J. M. Bollinger Jr., J. C. Price, L. M. Hoffart, E. W. Barr, C. Krebs, Eur. J. Inorg.
Chem. 2005, 4245-4254.
[23] J. M. Elkins, M. J. Ryle, I. J. Clifton, J. C. Dunning Hotop, J. S. LLoyd, N. I. Burzlaff,
J. E. Baldwin, R. P. Hausinger, P. L. Roach, Biochemistry 2002, 41, 5185-5192.

VIII. BIBLIOGRAPHY 197
[24] K. Vålegard, A. C. Terwisscha van Scheltinga, A. Dubus, G. Ranghino, L. M. Oester,
J. Hajdu, I. Andersson, Nat. Struct. Mol. Biol. 2004, 11, 95-101.
[25] I. Müller, A. Kahnert, T. Pape, G. M. Sheldrick, W. Meyer-Klaucke, T. Dierks, M.
Kertesz, I. Uson, Biochemistry 2004, 43, 3075-3088.
[26] G. Schenk, M. Y. M. Pau, E. I. Solomon, J. Am. Chem. Soc. 2004, 126, 505-515.
[27] H. M. Hanauske-Abel, V. Günzler, J. Theor. Biol. 1982, 94, 421-455.
[28] C. Krebs, D. Galonić Fujimori, C. T. Walsh, J. M. Bollinger Jr., Acc. Chem. Res. 2007,
40, 484-492.
[29] C. Krebs, J. M. Bollinger Jr., Photosynth. Res. 2009, 102, 295-304.
[30] J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger Jr., C. Krebs, Biochemistry 2003,
42, 7497-7508.
[31] C. Krebs, J. C. Price, J. Baldwin, L. Saleh, M. T. Green, J. M. Bollinger Jr., Inorg.
Chem. 2005, 44, 742-757.
[32] J. C. Price, E. W. Barr, T. E. Glass, C. Krebs, J. M. Bollinger Jr., J. Am. Chem. Soc.
2003, 125, 13008-13009.
[33] J. M. Bollinger Jr., C. Krebs, J. Inorg. Biochem. 2006, 100, 586-605.
[34] D. A. Proshlyakov, T. F. Henshaw, G. R. Monterosso, M. J. Ryle, R. P. Hausinger, J.
Am. Chem. Soc. 2004, 126, 1022-1023.
[35] P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger
Jr., C. Krebs, J. Am. Chem. Soc. 2004, 126, 8108-8109.
[36] S. Sinnecker, N. Svensen, E. W. Barr, S. Ye, J. M. Bollinger Jr., F. Neese, C. Krebs, J.
Am. Chem. Soc. 2007, 129, 6168-6179.
[37] J. C. Price, E. W. Barr, L. M. Hoffart, C. Krebs, J. M. Bollinger Jr., Biochemistry 2005,
44, 8138-8147.
[38] L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger Jr., C. Krebs, Proc. Natl. Acad.
Sci. USA 2006, 103, 14738-14743.
[39] F. H. Vaillancourt, J. Yin, C. T. Walsh, Proc. Natl. Acad. Sci. USA 2005, 105, 10111-
10116.
[40] F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova, C. T. Walsh,
Chem. Rev. 2006, 106, 3364-3378.
[41] D. P. Galonić, F. H. Vaillancourt, C. T. Walsh, J. Am. Chem. Soc. 2006, 128, 3900-
3901.
[42] L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh, C. L. Drennan, Nature 2006, 440, 368-
731.

198 VIII. BIBLIOGRAPHY
[43] D. P. Galonić, E. W. Barr, C. T. Walsh, J. M. Bollinger Jr., C. Krebs, Nat. Chem. Biol.
2007, 3, 113-116.
[44] D. Galonić Fujimori, E. W. Barr, M. L. Matthews, G. M. Koch, J. R. Yonce, C. T.
Walsh, J. M. Bollinger Jr., C. Krebs, P. J. Riggs-Gelasco, J. Am. Chem. Soc. 2007, 129,
13408-13409.
[45] N. Burzlaff, Adv. Inorg. Chem. 2008, 60, 101-165.
[46] S. Trofimenko, J. Am. Chem. Soc. 1969, 91, 588-595.
[47] S. Trofimenko, Acc. Chem. Res. 1971, 4, 17-21.
[48] S. Trofimenko, Chem. Rev. 1972, 72, 497-509.
[49] S. Trofimenko, Chem. Rev. 1993, 93, 943-980.
[50] A. Otero, J. Fernández-Baeza, A. Antiñolo, F. Carrillo-Hermosilla, J. Tejeda, E.
Díez-Barra, A. Lara-Sanchez, L. Sanchez-Barba, I. López-Solera, Organometallics
2001, 2428-2430.
[51] A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sanchez, L. Sánchez-
Barba, M. T. Expósito, M. Rodríguez, Dalton Trans. 2003, 1614-1619.
[52] A. Otero, J. Fernández-Baeza, J. Tejeda, A. Antiñolo, F. Carrillo-Hermosilla, E.
Díez-Barra, A. Lara-Sanchez, M. Fernández-López, J. Chem. Soc., Dalton Trans.
2000, 2367-2374.
[53] A. Otero, J. Fernández-Baeza, J. Tejeda, A. Antiñolo, F. Carrillo-Hermosilla, E.
Díez-Barra, A. Lara-Sánchez, M. Fernández-López, M. Lanfranchi, M. A.
Pellinghelli, J. Chem. Soc., Dalton Trans. 1999, 3537-3539.
[54] A. Beck, B. Weibert, N. Burzlaff, Eur. J. Inorg. Chem. 2001, 521-527.
[55] N. Burzlaff, I. Hegelmann, B. Weibert, J. Organomet. Chem. 2001, 626, 16-23.
[56] A. Beck, A. Barth, E. Hübner, N. Burzlaff, Inorg. Chem. 2003, 42, 7182-7188.
[57] P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, A. J. Long, R. W. Strange, S.
S. Hasnain, C. J. Schofield, J. E. Baldwin, J. Hajdu, Eur. J. Biochem. 1996, 242, 736-
740.
[58] P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu, J.
Baldwin, Nature 1997, 387, 827-830.
[59] A. López-Hernández, R. Müller, H. Kopf, N. Burzlaff, Eur. J. Inorg. Chem. 2002,
671-677.
[60] R. Müller, E. Hübner, N. Burzlaff, Eur. J. Inorg. Chem. 2004, 2151-2159.
[61] S. Tampier, R. Müller, A. Thorn, E. Hübner, N. Burzlaff, Inorg. Chem. 2008, 47,
9624-9641.

VIII. BIBLIOGRAPHY 199
[62] N. Burzlaff, Angew. Chem. Int. Ed. 2009, 48, 5580-5582.
[63] Y.-M. Chiou, L. Que Jr., J. Am. Chem. Soc. 1992, 114, 7567-7568.
[64] Y.-M. Chiou, L. Que Jr., J. Am. Chem. Soc. 1995, 117, 3999-4013.
[65] Y.-M. Chiou, L. Que Jr., Inorg. Chem. 1995, 34, 3270-3278.
[66] E. L. Hegg, R. Y. N. Ho, L. Que Jr., J. Am. Chem. Soc. 1999, 121, 1972-1973.
[67] S. Hikichi, T. Ogihara, K. Fujisawa, N. Kitajima, M. Akita, Y. Moro-oka, Inorg.
Chem. 1997, 36, 4539-4547.
[68] M. P. Mehn, K. Fujisawa, E. L. Hegg, L. Que Jr., J. Am. Chem. Soc. 2003, 125, 7828-
7842.
[69] T. Ogihara, S. Hikichi, M. Akita, T. Uchida, T. Kitagawa, Y. Moro-oka, Inorg. Chim.
Acta 2000, 297, 162-170.
[70] E. H. Ha, R. Y. N. Ho, J. F. Kisiel, J. S. Valentine, Inorg. Chem. 1995, 34, 2265-2266.
[71] A. Mukherjee, M. Martinho, E. L. Bominaar, E. Münck, L. Que Jr., Angew. Chem.
Int. Ed. 2009, 48, 1780-1783.
[72] J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck, L.
Que Jr., Angew. Chem. Int. Ed. 2009, 48, 3622-3226.
[73] B. M. L. Dioos, I. F. J. Vankelecom, P. A. Jacobs, Adv. Synth. Catal. 2006, 348, 1413-
1446.
[74] H.-U. Blaser, B. Pugin, M. Studer, in Chiral Catalyst Recycling and Immobilisation
(Eds.: D. De Vos, I. Vankelecom, P. Jacobs), Wiley-VCH Verlag GmbH, Weinheim,
2000.
[75] M. Tada, Y. Iwasawa, Chem. Commun. 2006, 2833-2844.
[76] I. Vankelecom, P. Jacobs, in Chiral Catalyst Recycling and Immobilisation (Eds.: D.
De Vos, I. Vankelecom, P. Jacobs), Wiley-VCH Verlag GmbH, Weinheim, 2000.
[77] L. Alaerts, J. Wahlen, P. A. Jacobs, D. E. De Vos, Chem. Commun. 2008, 1727-1737.
[78] Z. Wang, K. Ding, Y. Uozumi, in Handbook of Asymmetric Heterogeneous
Catalysis (Eds.: K. Ding, Y. Uozumi), Wiley-VCH Verlag GmbH & Co. KGaA,
Weinheim, 2008, pp. 1-24.
[79] J. M. Fraile, J. I. García, J. A. Mayoral, Top. Organomet. Chem. 2005, 15, 149-190.
[80] J. M. Fraile, J. I. García, J. A. Mayoral, Chem. Rev. 2009, 109, 360-417.
[81] J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, E. Pires, L. Salvatella, Catal.
Today 2009, 140, 44-50.
[82] J. M. Fraile, J. I. García, J. A. Mayoral, Coord. Chem. Rev. 2008, 252, 624-646.

200 VIII. BIBLIOGRAPHY
[83] P. J. Alonso, J. M. Fraile, J. García, J. I. García, J. I. Martínez, J. A. Mayoral, M. C.
Sánchez, Langmuir 2000, 16, 5607-5612.
[84] M. J. Fernández, J. M. Fraile, J. I. García, J. A. Mayoral, M. I. Burguete, E. García-
Verdugo, S. V. Luis, M. A. Harmer, Top. Catal. 2000, 13, 303-309.
[85] J. M. Fraile, J. I. García, M. A. Harmer, C. I. Herrerías, J. A. Mayoral, O. Reiser, H.
Werner, J. Mater. Chem. 2002, 12, 3290-3295.
[86] J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, M. A. Harmer, J. Catal. 2004,
221, 532-540.
[87] J. M. Fraile, J. I. García, J. A. Mayoral, T. Tarnai, Tetrahedron: Asymmetry 1997, 8,
2089-2092.
[88] J. M. Fraile, J. I. García, J. A. Mayoral, T. Tarnai, Tetrahedron: Asymmetry 1998, 9,
3997-4008.
[89] J. M. Fraile, J. I. García, J. A. Mayoral, T. Tarnai, M. A. Harmer, J. Catal. 1999, 186,
214-221.
[90] J. M. Fraile, J. García, G. Lafuente, J. A. Mayoral, L. Salvatella, ARKIVOC 2004, 67-
73.
[91] J. M. Fraile, J. I. García, M. A. Harmer, C. I. Herrerías, J. A. Mayoral, J. Mol. Catal.
A: Chem. 2001, 165, 211-218.
[92] J. M. Fraile, I. Pérez, J. A. Mayoral, J. Catal. 2007, 252, 303-311.
[93] J. M. Fraile, I. Pérez, J. A. Mayoral, O. Reiser, Adv. Synth. Catal. 2006, 348, 1680-
1688.
[94] M. Glos, O. Reiser, Org. Lett. 2000, 2, 2045-2048.
[95] J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, O. Reiser, A. Socuellamos, H.
Werner, Chem. Eur. J. 2004, 10, 2997-3005.
[96] H. Werner, C. I. Herrerías, M. Glos, A. Gissibl, J. M. Fraile, I. Pérez, J. A. Mayoral,
O. Reiser, Adv. Synth. Catal. 2006, 348, 125-132.
[97] E. Hübner, T. Haas, N. Burzlaff, Eur. J. Inorg. Chem. 2006, 4989-4897.
[98] P. Mastrorilli, C. F. Nobile, Coord. Chem. Rev. 2004, 248, 377-395.
[99] M. I. Burguete, J. M. Fraile, J. I. García, E. García-Verdugo, C. I. Herrerías, S. V.
Luis, J. A. Mayoral, J. Org. Chem. 2001, 66, 8893-8901.
[100] M. I. Burguete, J. M. Fraile, J. I. García, E. García-Verdugo, S. V. Luis, J. A. Mayoral,
Org. Lett. 2000, 2, 3905-3908.
[101] A. Schiller, R. Scopelliti, K. Severin, Dalton Trans. 2006, 3858-3867.
[102] F. Mancin, P. Scrimin, P. Tecilla, U. Tonellato, Chem. Commun. 2005, 2540-2548.

VIII. BIBLIOGRAPHY 201
[103] M. J. Belousoff, M. B. Duriska, B. Graham, S. R. Batten, B. Moubaraki, K. S. Murray,
L. Spiccia, Inorg. Chem. 2006, 45, 3746-3755.
[104] L. M. Berreau, Eur. J. Inorg. Chem. 2006, 273-283.
[105] K. M. Deck, T. A. Tseng, J. N. Burstyn, Inorg. Chem. 2002, 41, 669-677.
[106] F. H. Fry, A. J. Fischmann, M. J. Belousoff, L. Spiccia, J. Brügger, Inorg. Chem. 2005,
44, 941-950.
[107] F. H. Fry, L. Spiccia, P. Jensen, B. Moubaraki, K. S. Murray, E. R. T. Tiekink, Inorg.
Chem. 2003, 42, 5594-5603.
[108] E. L. Hegg, S. H. Mortimore, C. L. Cheung, J. E. Huyett, D. R. Powell, J. N. Burstyn,
Inorg. Chem. 1999, 38, 2961-2968.
[109] L. A. Jenkins, J. K. Bashkin, J. D. Pennock, J. Florián, A. Warshel, Inorg. Chem. 1999,
38, 3215-3222.
[110] B. Linkletter, J. Chin, Angew. Chem. Int. Ed. Engl. 1995, 34, 472-474.
[111] S. Liu, A. D. Hamilton, Tetrahedron Lett. 1997, 38, 1107-1110.
[112] J. R. Morrow, W. C. Trogler, Inorg. Chem. 1988, 27, 3387-3394.
[113] W. C. Putnam, A. T. Daniher, B. N. Trawick, J. K. Bashkin, Nucl. Acids Res. 2001,
29, 2199-2204.
[114] H. Adams, N. A. Bailey, D. E. Fenton, Q.-Y. He, J. Chem. Soc., Dalton Trans. 1997,
1533-1539.
[115] J. A. Maldonado Calvo, H. Vahrenkamp, Inorg. Chim. Acta 2005, 358, 4019-4026.
[116] N. N. Murthy, K. D. Karlin, J. Chem. Soc., Chem. Commun. 1993, 1236-1238.
[117] E. Hübner, G. Türkoglu, M. Wolf, U. Zenneck, N. Burzlaff, Eur. J. Inorg. Chem.
2008, 1226-1235.
[118] B. Kozlevčar, P. Gamez, R. de Gelder, W. L. Driessen, J. Reedijk, Eur. J. Inorg.
Chem. 2003, 47 - 50.
[119] R. Duncan, Nat. Rev. Drug Discovery 2003, 2, 347-360.
[120] R. Duncan, Nat. Rev. Cancer 2006, 6, 688-701.
[121] R. Duncan, M. J. Vicent, Adv. Drug Delivery Rev. 2010, 62, 272-282.
[122] K. L. Kiick, Science 2007, 317, 1182-1183.
[123] S. Liu, R. Maheshwari, K. L. Kiick, Macromolecules 2009, 42, 3-13.
[124] M. J. Vicent, L. Dieudonné, R. J. Carbajo, A. Pineda-Lucena, Expert Opin. Drug
Discovery 2008, 5, 593-614.
[125] R. Duncan, L. Seymour, J. Drug Targeting 2007, 15, 456.
[126] K. Greish, J. Drug Targeting 2007, 15, 457-464.

202 VIII. BIBLIOGRAPHY
[127] H. Maeda, J. Fang, T. Inutsuka, Y. Kitamoto, Int. Immunopharmacol. 2003, 3, 319-
328.
[128] H. Maeda, J. Wu, P. Sawa, Y. Matsumura, K. Hori, J. Controlled Release 2000, 65,
271-284.
[129] Y. Matsumura, H. Maeda, Cancer Res. 1986, 46, 6387-6392.
[130] R. Alberto, K. Ortner, N. Wheatley, R. Schibli, A. P. Schubiger, J. Am. Chem. Soc.
2001, 123, 3135-3136.
[131] R. Schibli, R. Schwarzbach, R. Alberto, K. Ortner, H. Schmalle, C. Dumas, A. Egli,
A. P. Schubiger, Bioconjugate Chem. 2002, 13, 750-756.
[132] P. C. Kunz, N. E. Brückmann, B. Spingler, Eur. J. Inorg. Chem. 2007, 394-399.
[133] A. Mitra, A. Nan, H. Ghandehari, E. McNeill, J. Mulholland, B. R. Line, Pharm. Res.
2004, 21, 1153-1159.
[134] C. Alexander, L. Davidson, W. Hayes, Tetrahedron 2003, 59, 2025-2057.
[135] M. Tada, Y. Iwasawa, in Model Systems in Catalysis: Single Crystals to Supported
Enzyme Mimics (Ed.: R. M. Rioux), Springer Science + Business Media, LLC., New
York, 2010, pp. 475-493.
[136] G. Wulff, Chem. Rev. 2002, 102, 1-27.
[137] C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A.
Nicholls, J. O'Mahony, M. J. Whitcombe, J. Mol. Recognit. 2006, 19, 106-180.
[138] M. V. Polyakov, Zh. Fiz. Khim. 1931, 2, 799-905.
[139] G. Wulff, A. Sarhan, K. Zabrocki, Tetrahedron Lett. 1973, 4239-4332.
[140] M. Komiyama, T. Takeuchi, T. Mukawa, H. Asanuma, Molecular Imprinting: From
Fundamentals to Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,
2003.
[141] B. Sellergren, in Techniques and Instrumentation in Analytical Chemistry, Vol. 23,
Elsevier, Amsterdam, 2001.
[142] M. Yan, O. Ramstroem, Molecularly Imprinted Materials: Science and Technology,
Marcel Dekker, Inc., New York, N. Y., 2005.
[143] X. Jiang, N. Jiang, H. Zhang, M. Liu, Anal. Bioanal. Chem. 2007, 389, 355-368.
[144] N. M. Maier, W. Lindner, Anal. Bioanal. Chem. 2007, 389, 377-397.
[145] M. Patek, M. Drew, Curr. Opin. Chem. Biol. 2008, 12, 332-339.
[146] O. Ramstroem, K. Mosbach, Curr. Opin. Chem. Biol. 1999, 3, 759-764.
[147] T. Takeuchi, T. Hishiya, Org. Biomol. Chem. 2008, 6, 2459-2467.
[148] L. L. Welbes, A. S. Borovik, Acc. Chem. Res. 2005, 38.

VIII. BIBLIOGRAPHY 203
[149] L. Ye, K. Haupt, Anal. Bioanal. Chem. 2004, 378, 1887-1897.
[150] L. Ye, K. Mosbach, Chem. Mater. 2008, 20, 859-868.
[151] H. Zhang, L. Ye, K. Mosbach, J. Mol. Recognit. 2006, 19, 248-259.
[152] K. Severin, in Molecularly Imprinted Materials: Science and Technology (Eds.: M.
Yan, O. Ramstroem), Marcel Dekker, Inc., New York, N. Y., 2005, pp. 619-640.
[153] G. Wulff, J. Vietmeier, Makromol. Chem. 1989, 190, 1717-1726.
[154] G. Wulff, J. Vietmeier, Makromol. Chem. 1989, 190, 1727-1735.
[155] K. Mosbach, Y. H. Yu, J. Andersch, L. Ye, J. Am. Chem. Soc. 2001, 123, 12420-12421.
[156] Y. Yu, L. Ye, K. Haupt, K. Mosbach, Angew. Chem. Int. Ed. 2002, 41, 4460-4462.
[157] L. Pauling, Chem. Eng. News 1946, 24, 1375-1377.
[158] W. P. Jencks, Catalysis in Chemistry and Enzymology, McGraw-Hill, New York,
1969.
[159] R. A. Lerner, S. J. Benkovic, P. G. Schultz, Science 1991, 252, 659-667.
[160] P. G. Schultz, Angew. Chem. Int. Ed. Engl. 1989, 28, 1283-1444.
[161] D. Hilvert, Annu. Rev. Biochem. 2000, 69, 751-793.
[162] V. Gouverneur, M. Reiter, Adv. Org. Synth. 2005, 1, 519-540.
[163] J.-L. Reymond, Chimia 2001, 55, 330-333.
[164] G. Wulff, T. Gross, R. Schönfeld, Angew. Chem. Int. Ed. Engl. 1997, 36, 1962-1964.
[165] B. Sellergren, R. N. Karmalkar, K. J. Shea, J. Org. Chem. 2000, 65, 4009-4027.
[166] M. Emgenbroich, G. Wulff, Chem. Eur. J. 2003, 9, 4106-4117.
[167] A. G. Strikovsky, D. Kasper, M. Grün, B. S. Green, J. Hradil, G. Wulff, J. Am. Chem.
Soc. 2000, 122, 6295-6296.
[168] G. Wulff, B.-O. Chong, U. Kolb, Angew. Chem. Int. Ed. 2006, 45, 2955-2958.
[169] J.-Q. Liu, G. Wulff, J. Am. Chem. Soc. 2004, 126, 7452-7453.
[170] J.-Q. Liu, G. Wulff, Angew. Chem. Int. Ed. 2004, 43, 1287-1290.
[171] J.-Q. Liu, G. Wulff, J. Am. Chem. Soc. 2008, 130, 8044-8054.
[172] K. Polborn, K. Severin, Chem. Eur. J. 2000, 6, 4604-4611.
[173] K. Polborn, K. Severin, Chem. Commun. 1999, 2481-2482.
[174] M. J. Palmer, M. Willis, Tetrahedron: Asymmetry 1999, 10, 2045-2061.
[175] R. Noyori, S. Hashiguchi, Acc. Chem. Res. 1997, 30, 97-102.
[176] K. Murata, T. Ikariya, R. Noyori, J. Org. Chem. 1999, 64, 2186-2187.
[177] K. Mashima, T. Abe, K. Tani, Chem. Lett. 1998, 1199-1200.
[178] J. Mao, D. C. Baker, Org. Lett. 1999, 1, 841-843.

204 VIII. BIBLIOGRAPHY
[179] D. A. Alonso, P. Brandt, S. J. M. Nordin, P. G. Andersson, J. Am. Chem. Soc. 1999,
121, 9580-9588.
[180] M. Yamakawa, H. Ito, R. Noyori, J. Am. Chem. Soc. 2000, 122, 1466-1478.
[181] K. Polborn, K. Severin, Eur. J. Inorg. Chem. 2000, 1687-1692.
[182] E. Burri, K. Severin, Chimia 2006, 60, 182-184.
[183] E. Burri, M. Öhm, C. Daguenet, K. Severin, Chem. Eur. J. 2005, 11, 5055-5061.
[184] G. Türkoglu, Diploma thesis, Friedrich-Alexander-University (Erlangen-Nürnberg),
2007.
[185] T. S. Thakur, G. R. Desiraju, Chem. Commun. 2006, 552-554.
[186] C. A. Grapperhaus, B. Mienert, E. Bill, T. Weyhermüller, K. Wieghardt, Inorg.
Chem. 2000, 39, 5306-5317.
[187] J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E.
Münck, W. Nam, L. Que Jr., Science 2003, 299, 1037-1039.
[188] P. S. Hallman, T. A. Stephenson, G. Wilkinson, Inorg. Synth. 1970, 12, 237-240.
[189] R. Alberto, Top. Curr. Chem. 2005, 252, 1-44.
[190] R. Alberto, J. Organomet. Chem. 2007, 692, 1179-1186.
[191] B. L. Oliveira, J. D. G. Correia, P. D. Raposinho, I. Santos, A. Ferreira, C. Cordeiro,
A. P. Freire, Dalton Trans. 2009, 125-162.
[192] J. E. Joachim, C. Apostolidis, B. Kanellakopulos, R. Müller, N. Marques, D. Meyer, J.
Müller, A. P. d. Matos, B. Nuber, J. Rebizant, M. L. Ziegler, J. Organomet. Chem.
1993, 448, 119-129.
[193] L. Peters, N. Burzlaff, Polyhedron 2004, 23, 245-251.
[194] L. Peters, E. Hübner, T. Haas, F. W. Heinemann, N. Burzlaff, J. Organomet. Chem.
2009, 694, 2319-2327.
[195] T. A. Stephenson, G. Wilkinson, J. Inorg. Nucl. Chem. 1966, 28, 945-956.
[196] M. I. Bruce, C. Hameister, A. G. Swincer, R. C. Wallis, Inorg. Synth. 1982, 21, 78-84.
[197] N. W. Alcock, I. D. Burns, K. S. Claire, A. F. Hill, Inorg. Chem. 1992, 31, 2906-2908.
[198] L. D. Field, B. A. Messerle, L. Soler, I. E. Buys, T. W. Hambley, J. Chem. Soc.,
Dalton Trans. 2001, 1959-1965.
[199] R. Poli, Chem. Rev. 1996, 96, 2135-2204.
[200] J. T. Poulton, B. E. Hauger, R. L. Kuhlman, K. G. Caulton, Inorg. Chem. 1994, 33,
3325-3330.
[201] B. K. Campion, R. H. Heyn, T. D. Tilley, J. Chem. Soc., Chem. Commun. 1988, 278-
280.

VIII. BIBLIOGRAPHY 205
[202] T. Arliguie, C. Border, B. Chaudret, J. Devillers, R. Poliblanc, Organometallics 1989,
8, 1308-1314.
[203] Y. Takahashi, S. Hikichi, M. Akita, Y. Moro-oka, Organometallics 1999, 18, 2571-
2573.
[204] H. Aneetha, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, V. N. Sapunov, R.
Schmid, K. Kirchner, K. Mereiter, Organometallics 2002, 21, 5334-5346.
[205] H. Aneetha, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, K. Mereiter,
Organometallics 2002, 21, 628-635.
[206] H. Aneetha, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, K. Mereiter,
Organometallics 2003, 22, 1779-1782.
[207] I. De los Ríos, M. Jiménez-Tenorio, J. Padilla, M. C. Puerta, P. Valerga,
Organometallics 1996, 15, 4565-4574.
[208] C. Gemel, J. C. Huffman, K. G. Caulton, K. Mauthner, K. Kirchner, J. Organomet.
Chem. 2000, 593-594, 342-353.
[209] M. Jiménez-Tenorio, M. A. Jiménez-Tenorio, M. C. Puerta, P. Valerga, Inorg. Chim.
Acta 1997, 259, 77-84.
[210] M. Jiménez-Tenorio, M. D. Palacios, M. C. Puerta, P. Valerga, Organometallics 2005,
24, 3088-3098.
[211] M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, J. Organomet. Chem. 2000, 609, 161-
168.
[212] M. A. Jiménez-Tenorio, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, J. Chem. Soc.,
Dalton Trans. 1998, 3601-3607.
[213] M. D. Palacios, M. C. Puerta, P. Valerga, A. Lledós, E. Veilly, Inorg. Chem. 2007, 46,
6958-6967.
[214] G. Trimmel, C. Slugovc, P. Wiede, K. Mereiter, V. N. Sapunov, R. Schmid, K.
Kirchner, Inorg. Chem. 1997, 36, 1076-1083.
[215] H. Kopf, C. Pietraszuk, E. Hübner, N. Burzlaff, Organometallics 2006, 25, 2533 -
2546.
[216] R. Müller, Ph. D. thesis, Universität Konstanz 2006.
[217] M. D. Fryzuk, S. A. Johnson, Coord. Chem. Rev. 2000, 200-202, 379-409.
[218] T. Yamabe, K. Hori, T. Minato, T. Fukui, Inorg. Chem. 1980, 19, 2154-2159.
[219] E. Riedel, Moderne Anorganische Chemie, 2 ed., Walter de Gruyter, Berlin - New
York, 2003.
[220] T. A. Bazhenova, A. E. Shilov, Coord. Chem. Rev. 1995, 144, 69-145.

206 VIII. BIBLIOGRAPHY
[221] B. A. MacKay, M. D. Fryzuk, Chem. Rev. 2004, 104, 385-401.
[222] G. S. Ashby, M. I. Bruce, I. B. Tomkins, R. C. Wallis, Aust. J. Chem. 1979, 32, 1003-
1016.
[223] J. W. Benson, R. J. Angelici, Organometallics 1993, 12, 680-687.
[224] S. Bolaño, M. M. Rodríguez-Rocha, J. Bravo, J. Castro, E. Oñate, M. Peruzzini,
Organometallics 2009, 28, 6020-6030.
[225] M. Cao, L. V. Do, N. W. Hoffman, M.-L. Kwan, J. K. Little, J. M. McGilvray, C. B.
Morris, B. C. Söderberg, A. Wierzbicki, Organometallics 2001, 20, 2270-2279.
[226] F. M. Conroy-Lewis, S. J. Simpson, J. Organomet. Chem. 1987, 322, 221-228.
[227] T. D. Johnson, K. Folting, W. E. Streib, J. D. Martin, J. C. Huffman, S. A. Jackson, O.
Eisenstein, K. G. Caulton, Inorg. Chem. 1995, 34, 488-499.
[228] E. Lindner, M. Haustein, H. A. Mayer, K. Gierling, R. Fawzi, M. Steimann,
Organometallics 1995, 14, 2246-2252.
[229] J. R. Lisko, W. M. Jones, Organometallics 1986, 5, 1890-1896.
[230] K. Mauthner, C. Slugovc, K. Mereiter, R. Schmid, K. Kirchner, Organometallics
1997, 16, 1956-1961.
[231] S. Pavlik, C. Gemel, C. Slugovc, K. Mereiter, R. Schmid, K. Kirchner, J. Organomet.
Chem. 2001, 617-618, 301-310.
[232] S. Pavlik, R. Schmid, K. Kirchner, K. Mereiter, Monatsh. Chem. 2004, 135, 1349-1357.
[233] C. Slugovc, V. N. Sapunov, P. Wiede, K. Mereiter, R. Schmid, K. Kirchner, J. Chem.
Soc., Dalton Trans. 1997, 4209-4216.
[234] N.-Y. Sun, S. J. Simpson, J. Organomet. Chem. 1992, 434, 341-349.
[235] T. Wilczewski, Z. Dauter, J. Organomet. Chem. 1986, 312, 349-356.
[236] G. J. Kubas, Inorg. Chem. 1979, 18, 182-188.
[237] R. R. Ryan, G. J. Kubas, D. C. Moody, P. G. Eller, Struct. Bond. 1981, 46, 47-100.
[238] W. A. Schenk, Angew. Chem. 1987, 99, 101-112.
[239] G. J. Kubas, Acc. Chem. Res. 1994, 27, 183-190.
[240] J. Ellermann, C. Schelle, F. A. Knoch, M. Moll, D. Pohl, Monatsh. Chem. 1996, 127,
783-800.
[241] E. Lindner, M. Haustein, R. Fawzi, M. Steimann, P. Wegner, Organometallics 1994,
13, 5021-5029.
[242] W. A. Schenk, J. Bezler, N. Burzlaff, M. Hagel, B. Steinmetz, Eur. J. Inorg. Chem.
2000, 287-297.
[243] W. A. Schenk, U. Karl, Z. Naturforsch. B 1989, 44, 993-995.

VIII. BIBLIOGRAPHY 207
[244] W. A. Schenk, U. Karl, M. R. Horn, Z. Naturforsch. B 1989, 44, 1513-1518.
[245] B. Steinmetz, W. A. Schenk, Organometallics 1999, 18, 943-946.
[246] C. Pubill Ulldemolins, Experimental Results within the Erasmus Exchange Program
thesis, Friedrich-Alexander-Universität (Erlangen-Nürnberg), 2008.
[247] S. Julià, P. Sala, J. Del Marzo, M. Sancho, J. Heterocycl. Chem. 1982, 19, 1141-1145.
[248] A. S. Potapov, A. I. Khlebnikov, V. D. Ogorodnikov, Russ. J. Org. Chem. 2006, 42,
550-554.
[249] H. Friebolin, Basic one- and two-dimensional NMR spectroscopy, 4 ed., Wiley-VCH,
Weinheim, 2005.
[250] W. C. Chan, C. P. Lau, Y. Z. Chen, Y. Q. Fang, S. M. Ng, Organometallics 1997, 16,
34-44.
[251] C. P. Lau, S. M. Ng, G. Jia, Z. Lin, Coord. Chem. Rev. 2007, 251, 2223-2237.
[252] S. M. Ng, C. Yin, C. H. Yeung, T. C. Chan, C. P. Lau, Eur. J. Inorg. Chem. 2004,
1788-1793.
[253] C. Yin, Z. Xu, S. Y. Yang, S. M. Ng, K. Y. Wong, Z. Lin, C. P. Lau, Organometallics
2001, 20, 1216-1222.
[254] A. K. Ho, I. Iin, P. A. Gurr, M. F. Mills, G. G. Qiao, Polymer 2005, 46, 6727-6735.
[255] A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, Dalton
Trans. 2004, 10, 1499-1510.
[256] A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, J. Tejeda, L. F. Sánchez-Barba, Eur.
J. Inorg. Chem. 2008, 34, 5309-5326.
[257] G. Parkin, Chem. Rev. 2004, 104, 699-767.
[258] C. Pettinari, R. Pettinari, Coord. Chem. Rev. 2005, 249, 663-691.
[259] J. M. Elkins, K. S. Hewitson, L. A. McNeill, J. F. Seibel, I. Schlemminger, C. W.
Pugh, P. J. Ratcliffe, C. J. Schofield, J. Biol. Chem. 2003, 278, 1802-1806.
[260] D. A. Couch, S. D. Robinson, Inorg. Chim. Acta 1974, 9, 39-47.
[261] A. A. La Pensée, J. Bickley, S. J. Higgins, M. Marcaccio, F. Paolucci, S. Roffia, J. M.
Charnock, J. Chem. Soc., Dalton Trans. 2002, 4095-4104.
[262] M. D. Wolfe, J. D. Lipscomb, J. Biol. Chem. 2003, 278, 829-835.
[263] M. B. Neibergall, A. Stubna, Y. Mekmouche, E. Münck, Biochemistry 2007, 46,
8004-8016.
[264] J. A. Pavon, P. F. Fitzpatrick, J. Am. Chem. Soc. 2009, 131, 4582-4583.
[265] G. A. Barf, R. A. Sheldon, J. Mol. Catal. A: Chem. 1995, 102, 23-39.

208 VIII. BIBLIOGRAPHY
[266] M. Chavarot, S. Ménage, O. Hamelin, F. Charnay, J. Pécaut, M. Fontecave, Inorg.
Chem. 2003, 42, 4810-4816.
[267] C.-M. Che, V. W.-W. Yam, Adv. Inorg. Chem. 1992, 39, 233-325.
[268] A. S. Goldstein, R. H. Beer, R. S. Drago, J. Am. Chem. Soc. 1994, 116, 2424-2429.
[269] A. S. Goldstein, R. S. Drago, Chem. Commun. 1991, 21-22.
[270] W. P. Griffith, Chem. Soc. Rev. 1992, 21, 179-185.
[271] X. Hua, A. G. Lappin, Inorg. Chem. 1995, 34, 992-994.
[272] T. Kojima, H. Matsuo, Y. Matsuda, Inorg. Chim. Acta 2000, 300-302, 661-667.
[273] W.-C. Cheng, W.-Y. Yu, K.-K. Cheung, C.-M. Che, J. Chem. Soc., Chem. Commun.
1994, 1063-1064.
[274] W.-C. Cheng, W.-Y. Yu, J. Zhu, K.-K. Cheung, S.-M. Peng, C.-K. Poon, C.-M. Che,
Inorg. Chim. Acta 1996, 242, 105-113.
[275] W.-H. Fung, W.-C. Cheng, W.-Y. Yu, C.-M. Che, T. C. W. Mak, J. Chem. Soc., Chem.
Commun. 1995, 2007-2008.
[276] W.-H. Fung, W.-Y. Yu, C.-M. Che, J. Org. Chem. 1998, 63, 7715-7726.
[277] W.-C. Cheng, W.-H. Fung, C.-M. Che, J. Mol. Catal. A: Chem. 1996, 113, 311-319.
[278] W.-H. Cheung, W.-Y. Yu, W.-P. Yip, N.-Y. Zhu, C.-M. Che, J. Org. Chem. 2002, 62,
7716-7723.
[279] C.-M. Che, J.-S. Huang, Chem. Commun. 2009, 3996-4015.
[280] T. Kojima, K.-I. Hayashi, S.-Y. Iizuka, F. Tani, Y. Naruta, M. Kawano, Y. Ohashi, Y.
Hirai, K. Okhubo, Y. Matsuda, S. Fukuzumi, Chem. Eur. J. 2007, 13, 8212-8222.
[281] P. Aguirre, R. Sariego, S. A. Moya, J. Coord. Chem. 2001, 54, 401-413.
[282] M. I. Bruce, D. N. Sharrocks, F. G. A. Stone, J. Organomet. Chem. 1971, 31, 269-273.
[283] M. M. de V. Steyn, E. Singleton, S. Hietkamp, D. C. Lilies, J. Chem. Soc., Dalton
Trans. 1990, 2991-2997.
[284] H. Nagashima, K. Mukai, Y. Shiota, K. Yamaguchi, K.-I. Ara, T. Fukahori, H. Suzuki,
M. Akita, Y. Moro-oka, K. Itoh, Organometallics 1990, 9, 799-807.
[285] T. Blackmore, J. D. Cotton, M. I. Bruce, F. G. A. Stone, J. Chem. Soc., A 1968, 2931-
2936.
[286] Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa, S. Fukuzumi, Angew.
Chem. Int. Ed. 2008, 47, 5772-5776.
[287] C. J. Carmalt, J. G. Crossley, J. G. Knight, P. Lightfoot, A. Martín, M. P.
Muldowney, N. C. Norman, A. G. Orpen, J. Chem. Soc., Chem. Commun. 1994,
2367-2368.

VIII. BIBLIOGRAPHY 209
[288] V. V. Zhdankin, P. J. Stang, Chem. Rev. 2008, 108, 5299-5358.
[289] V. V. Zhdankin, P. J. Stang, Chem. Rev. 2002, 102, 2523-2584.
[290] T. G. Trayor, J. C. Marsters Jr., T. Nakano, B. E. Dunlap, J. Am. Chem. Soc. 1985,
107, 5537-5539.
[291] H. Tohma, S. Takizawa, T. Maegawa, Y. Kita, Angew. Chem. Int. Ed. 2000, 29, 1306-
1308.
[292] H. Tohma, S. Takizawa, H. Watanabe, Y. Kita, Tetrahedron Lett. 1998, 39, 4547-
4550.
[293] D. Macikenas, E. Skrzypczak-Jankun, J. D. Protasiewicz, J. Am. Chem. Soc. 1999,
121, 7164-7165.
[294] D. Macikenas, E. Skrzypczak-Jankun, J. D. Protasiewicz, Angew. Chem. Int. Ed.
2000, 39, 2007-2010.
[295] M. Nabika, S. Kiuchi, T. Miatake, K.-I. Okamoto, K. Fujisawa, J. Mol. Catal. A:
Chem. 2007, 269, 163-168.
[296] T. R. Belderraín, M. Paneque, E. Carmona, E. Gutiérrez-Puebla, M. Angeles Monge,
C. Ruiz-Valero, Inorg. Chem. 2002, 41, 425-428.
[297] F. A. Kunrath, R. F. de Souza, O. L. Casagrande Jr., N. R. Brooks, V. G. Young Jr.,
Organometallics 2003, 22, 4739-4743.
[298] R. Santi, A. M. Romano, A. Sommazzi, M. Grande, C. Bianchini, G. Mantovani, J.
Mol. Catal. A: Chem. 2005, 229, 191-197.
[299] N. Kitajima, S. Hikichi, M. Tanaka, Y. Moro-oka, J. Am. Chem. Soc. 1993, 115, 5496-
5508.
[300] S. Hikichi, H. Okuda, Y. Ohzu, M. Akita, Angew. Chem. Int. Ed. 2009, 48, 188-191.
[301] G. L. Wang, B.-H. Jiang, E. A. Rue, G. L. Semenza, Proc. Natl. Acad. Sci. USA 1995,
92, 5510-5514.
[302] K. S. Hewitson, N. Granatino, R. W. D. Welford, M. A. McDonough, C. J. Schofield,
Phil. Trans. R. Soc. A 2005, 363, 807-828.
[303] W. G. Kaelin Jr., P. J. Ratcliffe, Mol. Cell 2008, 30, 393-402.
[304] C. J. Schofield, P. J. Ratcliffe, Nat. Rev. Mol. Cell Biol. 2004, 5, 343-354.
[305] S. Nagel, N. P. Talbot, J. Mecinović, T. G. Smith, A. M. Buchan, C. J. Schofield,
Antioxid. Redox Signaling 2010, 12, 481-501.
[306] R. K. Bruick, S. L. McKnight, Science 2001, 294, 1337-1340.
[307] A. C. R. Epstein, J. M. Gleadle, L. A. McNeill, K. S. Hewitson, J. O’Rourke, D. R.
Mole, M. Mukherji, E. Metzen, M. I. Wilson, A. Dhanda, Y.-M. Tian, N. Masson, D.

210 VIII. BIBLIOGRAPHY
L. Hamilton, P. Jaakkola, R. Barstead, J. Hodgkin, P. H. Maxwell, C. W. Pugh, C. J.
Schofield, P. J. Ratcliffe, Cell 2001, 107, 43-54.
[308] K. S. Hewitson, L. A. McNeill, C. J. Schofield, Curr. Pharm. Des. 2004, 10, 821-833.
[309] D. Lando, D. J. Peet, J. J. Gorman, D. A. Whelan, M. L. Whitelaw, R. K. Bruick,
Genes Dev. 2002, 16, 1466-1471.
[310] D. Lando, D. J. Peet, D. A. Whelan, J. J. Gorman, M. L. Whitelaw, Science 2002, 295,
858-861.
[311] L. A. McNeill, K. S. Hewitson, T. D. Claridge, J. F. Seibel, L. E. Horsfall, C. J.
Schofield, Biochem. J. 2002, 367, 571-575.
[312] L. A. McNeill, K. S. Hewitson, J. M. Gleadle, L. E. Horsfall, N. J. Oldham, P. H.
Maxwell, C. W. Pugh, P. J. Ratcliffe, C. J. Schofield, Bioorg. Med. Chem. Lett. 2002,
12, 1547-1550.
[313] G. L. Semenza, Physiology 2008, 24, 97-106.
[314] T. G. Smith, P. A. Robbins, P. J. Ratcliffe, Br. J. Haematol. 2008, 141, 325-334.
[315] K. M. Cook, C. J. Schofield, in Angiogenesis: An integrative approach from science
to medicine (Eds.: W. D. Figg, J. Folkman), Springer Science + Business Media, LLC.,
2008, pp. 359-373.
[316] E. Baader, G. Tschank, K.-H. Baringhaus, H. Burghard, V. Günzler, Biochem. J.
1994, 300, 525-530.
[317] C. J. Cunliffe, T. J. Franklin, Biochem. J. 1986, 239, 311-315.
[318] C. J. Cunliffe, T. J. Franklin, N. J. Hales, G. B. Hill, J. Med. Chem. 1992, 35, 2652-
2658.
[319] N. J. Hales, J. F. Beattie, J. Med. Chem. 1993, 36, 3853-3858.
[320] D. R. Mole, I. Schlemminger, L. A. McNeill, K. S. Hewitson, C. W. Pugh, P. J.
Ratcliffe, C. J. Schofield, Bioorg. Med. Chem. Lett. 2003, 13, 2677-2680.
[321] J. J. Nietfeld, L. De Long, A. Kemp, Biochim. Biophys. Acta 1982, 704, 321-325.
[322] H. Tucker, D. F. Thomas, J. Med. Chem. 1992, 35, 804-807.
[323] J. Wang, J. L. Buss, G. Chen, P. Ponka, K. Pantopoulos, FEBS Lett. 2002, 529, 309-
312.
[324] N. C. Warshakoon, S. Wu, A. Boyer, R. Kawamoto, S. Renock, K. Xu, M. Pokross, A.
G. Evdokimov, S. Zhou, C. Winter, R. Walter, M. Mekel, Bioorg. Med. Chem. Lett.
2006, 16, 5687-5690.

VIII. BIBLIOGRAPHY 211
[325] N. C. Warshakoon, S. Wu, A. Boyer, R. Kawamoto, J. Sheville, S. Renock, K. Xu, M.
Pokross, S. Zhou, C. Winter, R. Walter, M. Mekel, A. G. Evdokimov, Bioorg. Med.
Chem. Lett. 2006, 16, 5517-5522.
[326] R. I. Dowell, E. M. Hadley, J. Med. Chem. 1992, 35, 800-804.
[327] M. Thevis, M. Kohler, N. Schlörer, W. Schänzer, J. Am. Soc. Mass Spectrom. 2008,
19, 151-158.
[328] M. Suzuki, K. Nunami, K. Matsumoto, N. Yoneda, M. Miyoshi, Synthesis 1978, 461-
462.
[329] K. Weidmann, K.-H. Baringhaus, G. Tschank, U. Werner, 1999, DE 19746287.
[330] K. Weidmann, K.-H. Baringhaus, G. Tschank, U. Werner, 2005, EP 1538160.
[331] I. K. H. Leung, E. Flashman, K. K. Yeoh, C. J. Schofield, T. D. W. Claridge, J. Med.
Chem. 2010, 53, 867-875.
[332] C. Rosenberger, S. Mandriota, J. S. Jürgensen, M. S. Wiesener, J. H. Hörstrup, U.
Frei, P. J. Ratcliffe, P. H. Maxwell, S. Bachmann, K.-U. Eckardt, J. Am. Soc. Nephrol.
2002, 13, 1721-1732.
[333] W. M. Bernhardt, V. Câmpean, S. Kany, J. S. Jürgensen, A. Weidemann, C.
Warnecke, M. Arend, S. Klaus, V. Günzler, K. Amann, C. Willam, M. S. Wiesener,
K.-U. Eckardt, J. Am. Soc. Nephrol. 2006, 17, 1970-1978.
[334] W. M. Bernhardt, U. Gottmann, F. Doyon, B. Buchholz, V. Câmpean, J. Schödel, A.
Reisenbuechler, S. Klaus, M. Arend, L. Flippin, M. S. Wiesener, B. Yard, C.
Warnecke, K.-U. Eckardt, Proc. Natl. Acad. Sci. USA 2009, 106, 21276-21281.
[335] H. Saltzman, J. G. Sharefkin, Org. Synth. 1963, 43, 60.
[336] F. J. Fleitz, T. A. Lyle, N. Zheng, J. D. Armstrong III, R. P. Volante, Synth. Commun.
2000, 30, 3171-3180.
[337] E. W. Abel, G. Wilkinson, J. Chem. Soc. 1959, 1501-1505.
[338] S. P. Schmidt, W. C. Trogler, F. Basolo, Inorg. Synth. 1990, 28, 160-165.
[339] M. J. Cleare, W. P. Griffith, J. Chem. Soc., A 1969, 372-380.
[340] G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112-122.
[341] K. Brandenburg, M. Berndt, Diamond – Visual Crystal Structure Information
System, Crystal Impact GbR, Bonn (Germany), 1999.
[342] W. T. Pennington, J. Appl. Crystallogr. 1999, 32, 1028-1029.
[343] P. Van der Sluis, A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr. 1990, 46,
194-201.
[344] A. L. Spek, J. Appl. Crystallogr. 2003, 36, 7-13.


DANKSAGUNG 213
DANKSAGUNG
Mein besonderer Dank gilt meinem Doktorvater Prof. Dr. Nicolai Burzlaff für die
Aufnahme in seine Arbeitsgruppe, den großen akademischen Freiraum zur Bearbeitung des
interessanten Themas und für seine Unterstützung während der Promotionszeit. Danke
auch für das Lösen der zahlreichen Kristallstrukturen und das Korrekturlesen diverser
Manuskripte. Ganz herzlich möchte ich mich auch bei Prof. Dr. Dr. h.c. mult. Rudi van
Eldik bedanken für die Aufnahme in den Lehrstuhl, das positive Arbeitsklima und die
Unterstützung während der Promotion.
Des Weiteren gebührt ein besonderer Dank an alle Mitarbeiter des Departments für Chemie
und Pharmazie, die maßgeblich zum Gelingen dieser Arbeit beigetragen haben: Ich danke
Dr. Achim Zahl (NMR), Dr. Marion Wolf (AAS, ICP-AES), Dr. Frank Heinemann (X-Ray),
Jochen Schmidt (NMR, AAS, ICP-AES), Panagiotis "Panos" Bakatselos (X-Ray), Martin
Bachmüller (MS), Susanne Hoffmann (X-Ray, GC, IR), Christina Wronna (EA), Ronny
Wiefel, Marco Müller (Glasbläserei), Uschi Niegratschka (Sekretariat) sowie allen
Mitarbeitern der Werkstatt und der Chemikalienausgabe. Zudem danke ich Dr. Eike
Hübner (Forschungszentrum Jülich bzw. TU Clausthal) für die SEC Messungen. Den
Mitarbeitern der Medizinischen Klinik 4, insbesondere PD Dr. med. Carsten Willam und Dr.
med. Gunnar Schley danke ich für die HIF Studien und die rege Zusammenarbeit.
Ich danke allen Personen rund um die AC, die mich auf meinem Weg zur Promotion
begleitet haben für die Hilfsbereitschaft und Freundschaft, insbesondere den "Ivanas"
Johanna, Niddi, Felix, Olli und Dominik, den "Dahlenburgs" Harry und Mathias, den
"Eldiks" Raquel, Steffi, Manfred und Thomas. Vielen Dank auch an die
Mitarbeiterpraktikanten und Bachelorstudis Ju, Eva, Sebastian, Atheel und Matthias, die im
A0.2 ihr Unwesen getrieben haben und diverse (teure) Glasgeräte zerdeppert haben ☺.
Den "Burzlaffs" Liv, Fatima, Nina, Eva, Eike, Stefan, Tom, Sascha, und Nico danke ich für
die kollegiale Zusammenarbeit und das positive Arbeitsklima.

214 DANKSAGUNG
Meinem unersetzbaren Laborpartner Andy danke ich für die geniale Zeit im legendären
A0.2 (yes, we can!), die uneingeschränkte Hilfsbereitschaft und die Zusammenarbeit in allen
Belangen. Auf deinem Weg wünsche ich Dir nur das Beste und hoffe dass wir uns nicht aus
den Augen verlieren werden.
Außerdem danke ich Astrid (aka Astridson, aka Wasabison) für....einfach ALLES! Wärst du
nicht gewesen, hätte ich die Zeit hier in Erlangen nicht überlebt ohne durchzudrehen. Für
deine Promotion in Heidelberg wünsche ich dir viel Erfolg, tolle Ergebnisse, viele
spannende Paper, viel Geduld und Kraft. Aber eigentlich weiß ich, dass du das Ding rocken
wirst! (Du schuldest mir noch ein Empfehlungsschreiben).
Den größten Dank schulde ich jedoch meinen Freunden außerhalb der Uni, meiner Familie
und insbesondere meinen Eltern. Ohne euer Verständnis und eure Unterstutzung wäre diese
Arbeit nicht möglich gewesen.

CURRICULUM VITAE 215
CURRICULUM VITAE
Name Gazi Türkoglu
Anschrift Erhardstraße 11, 90482 Nürnberg
Geburtsdatum/-ort 22.06.1981, Nürnberg
Familienstand ledig
Staatsangehörigkeit deutsch
Schul- und Universitätsausbildung
09/1988 – 07/1992 Volksschule Nürnberg Scharrerstraße
09/1992 – 07/2001 Martin-Behaim-Gymnasium, Nürnberg, Abschluss: Abitur
10/2001 Beginn des Studiums der Chemie
Friedrich-Alexander-Universität Erlangen-Nürnberg
01/2007 – 09/2007 Diplomarbeit am Lehrstuhl für Anorganische Chemie
des Departments Chemie & Pharmazie
Friedrich-Alexander-Universität Erlangen-Nürnberg
Betreuer: Prof. Dr. N. Burzlaff
Thema: Synthese neuartiger Bis(pyrazolyl)-acetato-Liganden
für die Koordinationschemie
09/2007 Erlangung des akademischen Grades Diplom-Chemiker Univ.
11/2007 – 10/2010 Promotion am Lehrstuhl für Anorganische Chemie
des Departments Chemie & Pharmazie
Friedrich-Alexander-Universität Erlangen-Nürnberg
Betreuer: Prof. Dr. N. Burzlaff.
Thema: Scorpionate Complexes for Copolymerization and
Molecular Imprinting
Beschäftigung als Wissenschaftlicher Mitarbeiter des SFB 583
(Redoxaktive Metallkomplexe)

216 CURRICULUM VITAE
Publikationen
"Synthesis and Transition Metal Complexes of Novel N,N,O Scorpionate Ligands Suitable
for Solid Phase Immobilisation", E. Hübner, G. Türkoglu, M. Wolf, U. Zenneck, N. Burzlaff,
Eur. J. Inorg. Chem. 2008, 1226 – 1235.
"Scorpionate Complexes Suitable for Enzyme Inhibitor Studies", N. V. Fischer, G. Türkoglu,
N. Burzlaff, Curr. Bioact. Compd. 2009, 5, 277 – 295.
"Bis(3,5-dimethyl-4-vinylpyrazol-1-yl)acetic Acid: A New Heteroscorpionate Building Block
for Copolymers that Mimic the 2-His-1-carboxylate Facial Triad", G. Türkoglu, C. Pubill
Ulldemolins, R. Müller, E. Hübner, F. W. Heinemann, M. Wolf, N. Burzlaff, Eur. J. Inorg.
Chem. 2010, 2962 – 2974.
Posterbeiträge
▶ 4th EuCheMS NLigands Conference, Garmisch-Patenkirchen, 2008.
▶ 5. Koordinationschemie Tagung, Universität Erlangen-Nürnberg, 2009.
▶ 39th Inorganic/Bioinorganic Reaction Mechanism Meeting, Kloster Banz, 2010.
▶ 3rd EuCheMS Chemistry Congress, Nürnberg 2010.
Kurzvorträge
▶ 5. Koordinationschemie Tagung, Universität Erlangen-Nürnberg, 2009.
▶ Tagung des Interdisziplinären Zentrums für Molekulare Materialien (ICMM) der
Universität Erlangen-Nürnberg, Bad Staffelstein, 2009.
▶ Berichtskolloquium des Sonderforschungsbereiches SFB 583, Universität Erlangen-
Nürnberg, 2009.
Copyright © 2022 FDOKUMEN














