
CD4+CD25+ T-Cells Control Autoimmunity in the Absence of B-Cells
Upload
independentCategory
view
3download
0
of July 8, 2014.This information is current as
Diabetic 8.3 TCR Transgenic MiceIs Required for Diabetes in Nonobese Autoimmunity to Both Proinsulin and IGRP
KayLew, Pere Santamaria, Helen E. Thomas and Thomas W. H.Gellert, Peter G. Colman, Leonard C. Harrison, Andrew M. Balasubramanian Krishnamurthy, Lina Mariana, Shane A.
http://www.jimmunol.org/content/180/7/4458doi: 10.4049/jimmunol.180.7.4458
2008; 180:4458-4464; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/7/4458.full#ref-list-1
, 11 of which you can access for free at: cites 30 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 8, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

Autoimmunity to Both Proinsulin and IGRP Is Required forDiabetes in Nonobese Diabetic 8.3 TCR Transgenic Mice1
Balasubramanian Krishnamurthy,* Lina Mariana,* Shane A. Gellert,† Peter G. Colman,†
Leonard C. Harrison,‡ Andrew M. Lew,‡ Pere Santamaria,§ Helen E. Thomas,*and Thomas W. H. Kay2*¶
T cells specific for proinsulin and islet-specific glucose-6-phosphatase catalytic subunit related protein (IGRP) induce diabetes innonobese diabetic (NOD) mice. TCR transgenic mice with CD8� T cells specific for IGRP206–214 (NOD8.3 mice) develop accel-erated diabetes that requires CD4� T cell help. We previously showed that immune responses against proinsulin are necessary forIGRP206–214-specific CD8� T cells to expand. In this study, we show that diabetes development is dramatically reduced in NOD8.3mice crossed to NOD mice tolerant to proinsulin (NOD-PI mice). This indicates that immunity to proinsulin is even required inthe great majority of NOD8.3 mice that have a pre-existing repertoire of IGRP206–214-specific cells. However, protection fromdiabetes could be overcome by inducing islet inflammation either by a single dose of streptozotocin or anti-CD40 agonist Abtreatment. This suggests that islet inflammation can substitute for proinsulin-specific CD4� T cell help to activate IGRP206–214-specific T cells. The Journal of Immunology, 2008, 180: 4458–4464.
T ype 1 diabetes (T1D)3 is a T cell-mediated organ-specificautoimmune disease that results from selective destruc-tion of the � cells in the islets of the pancreas (1). T cells
specific for a number of islet Ags can be detected in humans andnonobese diabetic (NOD) mice (2). Initiation of autoimmunity inNOD mice may be dependent on an immune response against in-sulin alone rather than an immune response against many � cellAgs. We and others have independently shown that transgenicoverexpression of proinsulin 2 in APCs of NOD mice preventsinsulitis and diabetes (3, 4). Knocking out both insulin genes andintroduction of mutated insulin with alanine rather than tyrosine atposition 16 of the insulin B chain prevents insulitis and diabetes(5). Eliminating immune responses to insulin not only blocks de-
velopment of diabetes and insulitis but also immune responses todownstream autoantigens, such as the islet-specific glucose-6-phosphatase catalytic subunit related protein (IGRP) (6). In bothhumans and animal models, the major determinants of T1D aregenes within the MHC [HLA in humans] (7). The remarkable ho-mogeneity of high-risk genotypes across different populations andthe conservation of aspects of the Ag-binding groove suggest thatthere may be specific peptides that for most individuals representthe initiating targets of autoimmunity (8). In NOD mice (and prob-ably also in humans), the primary autoantigen for diabetes appearsto be insulin, specifically the insulin B chain peptide B9-23 (5).
Identification of target autoantigens for T cells is particularlyimportant because manipulation of immune responses to these Agsoffers the hope of specifically removing the cells that damage �
cells. We and others have proposed that autoreactivity to insulin isnecessary and precedes autoreactivity to IGRP and other autoan-tigens (3, 5, 6). If insulin is the primary Ag, a strategy that inducestolerance to insulin should prevent diabetes. Although this pre-vented diabetes in NOD mice (9), major preventive trials witheither oral or i.v. insulin have thus far failed to prevent diabetes inhumans (10–12). Human subjects enrolled in prevention trialshave multiple autoantibodies, suggesting advanced preclinical di-abetes. By the time the intervention occurs, tissue inflammationhas already developed, the immune response has already expandedbeyond the initial “triggering” Ag, and the frequency of diversearray of Ag-specific T cells may have expanded substantially.Therefore, a clinically important question is whether induction oftolerance to the primary Ag will be effective when there is alreadyan expanded pool of Ag-specific T cells. We wished to knowwhether inducing tolerance to insulin would have any effect inNOD mice that already have an expanded number of T cells spe-cific for IGRP. To address this question, we crossed TCR trans-genic mice that have �90% of their CD8� T cells specific forIGRP206–214 (NOD8.3 mice) (13) with NOD mice tolerant to pro-insulin (NOD-PI mice) that express proinsulin 2 in APCs, makingthem tolerant to proinsulin (3). These mice have dramatically re-duced diabetes, indicating that the requirement for immunity to
*St. Vincent’s Institute, Fitzroy, Victoria, Australia; †Department of Diabetes andEndocrinology and Pathology, The Royal Melbourne Hospital, Parkville, Victoria,Australia; ‡The Walter and Eliza Hall Institute of Medical Research, Parkville, Vic-toria, Australia; §Julia McFarlane Diabetes Research Centre and Department of Mi-crobiology and Infectious Disease, University of Calgary Faculty of Medicine, Cal-gary, Alberta, Canada; and ¶The University of Melbourne Department of Medicine,St. Vincent’s Hospital, Fitzroy, Victoria, Australia
Received for publication December 6, 2007. Accepted for publication January25, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 P.S. is a scientist of the Alberta Heritage Foundation for Medical Research and issupported by the Canadian Institutes of Health Research, the Canadian Diabetes As-sociation, and the Juvenile Diabetes Research Foundation. The Julia McFarlane Di-abetes Research Centre is supported by the Diabetes Association (Foothills). Thework was supported by a Program Grant; Career Development Award (to H.T.) andClinical Centre for Research Excellence from the National Health and Medical Re-search Council of Australia; a Program-Project Grant and a post-doctoral fellowship(to B.K.) from the Juvenile Diabetes Research Foundation; and a Millennium Re-search Grant from Diabetes Australia (to T.K.).2 Address correspondence and reprint requests to Dr. Thomas W. H. Kay, St. Vin-cent’s Institute, 41 Victoria Parade, Fitzroy, VIC 3065, Australia. E-mail address:[email protected] Abbreviations used in this paper: T1D, type 1 diabetes; NOD, nonobese diabetic;IGRP, islet-specific glucose-6-phosphatase catalytic subunit related protein; DC, den-dritic cell; NOD-PI, NOD mice tolerant to proinsulin; STZ, streptozotocin; IAA,insulin autoantibody assay; Treg, regulatory T cell.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
proinsulin is even present in the majority of NOD8.3 mice with apre-existing repertoire of IGRP206–214-specific cells.
Materials and MethodsMice
NOD mice expressing mouse proinsulin 2 under the control of an MHCclass II (I-E��) promoter (NOD-PI) and NOD8.3 mice expressing theTCR�� rearrangements of the H-2Kd-restricted CD8� T cell clone NY8.3have been described (3, 5, 6). Perforin knockout NOD mice were obtainedfrom The Jackson Laboratory T1D repository. All animal studies were
conducted at St. Vincent’s Institute (Melbourne, Australia) and were ap-proved by the institutional animal ethics committee.
Peptides and Abs
The peptides IGRP206–214 (VYLKTNVFL) and TUM (KYQAVTTTL)were synthesized by Auspep. H-2Kd tetramers were made by ImmunoID.Tetramer function was validated by staining NOD8.3 splenocytes.
Tetramer staining
Islet infiltrating T cells were stained with IGRP206–214 or TUM H-2Kd
tetramer as previously described (6).
γ
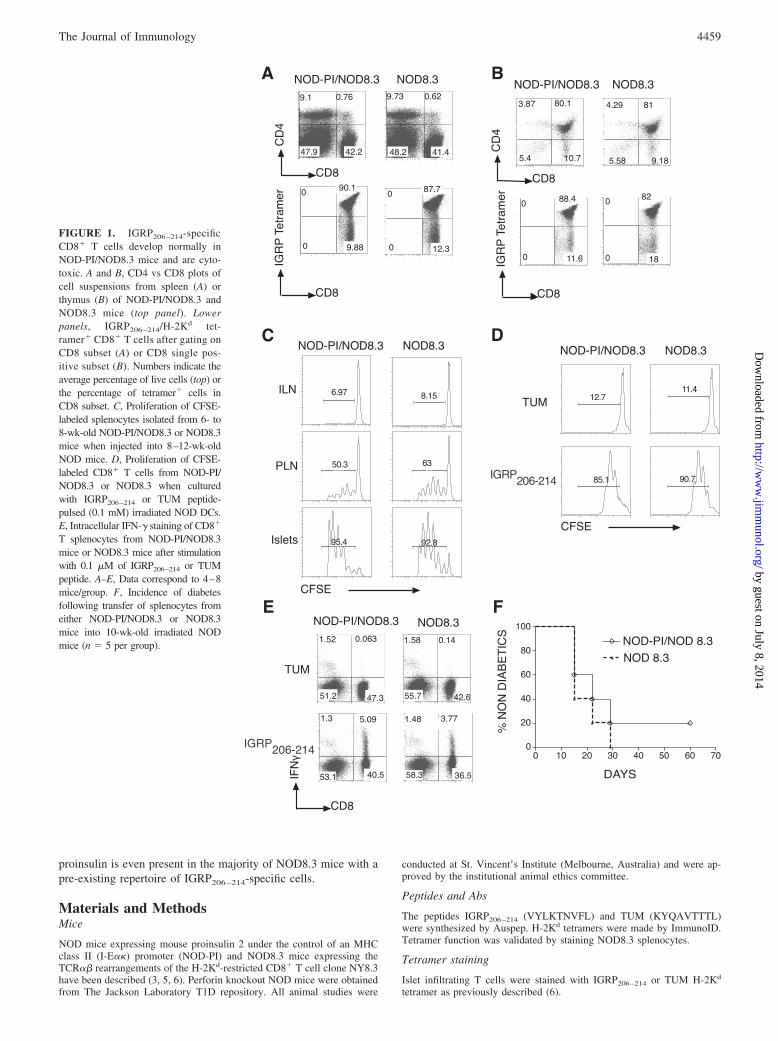
FIGURE 1. IGRP206 –214-specificCD8� T cells develop normally inNOD-PI/NOD8.3 mice and are cyto-toxic. A and B, CD4 vs CD8 plots ofcell suspensions from spleen (A) orthymus (B) of NOD-PI/NOD8.3 andNOD8.3 mice (top panel). Lowerpanels, IGRP206 –214/H-2Kd tet-ramer� CD8� T cells after gating onCD8 subset (A) or CD8 single pos-itive subset (B). Numbers indicate theaverage percentage of live cells (top) orthe percentage of tetramer� cells inCD8 subset. C, Proliferation of CFSE-labeled splenocytes isolated from 6- to8-wk-old NOD-PI/NOD8.3 or NOD8.3mice when injected into 8–12-wk-oldNOD mice. D, Proliferation of CFSE-labeled CD8� T cells from NOD-PI/NOD8.3 or NOD8.3 when culturedwith IGRP206–214 or TUM peptide-pulsed (0.1 mM) irradiated NOD DCs.E, Intracellular IFN-� staining of CD8�
T splenocytes from NOD-PI/NOD8.3mice or NOD8.3 mice after stimulationwith 0.1 �M of IGRP206–214 or TUMpeptide. A–E, Data correspond to 4–8mice/group. F, Incidence of diabetesfollowing transfer of splenocytes fromeither NOD-PI/NOD8.3 or NOD8.3mice into 10-wk-old irradiated NODmice (n � 5 per group).
4459The Journal of Immunology
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
CFSE labeling and adoptive transfer
CD8� T cells from NOD8.3 mouse were labeled with CFSE as previouslydescribed (6). In cotransfer experiments, mice receiving CFSE-labeled 8.3CD8� T cells were injected i.v. (a) on day 0 with 100 �g agonistic anti-CD40 mAb FGK45 or 100 �g isotype control Ab (GL117) in 200 �l PBSand (b) on day �1 with 80 mg/kg streptozotocin (STZ) in 200 ml citratebuffer (pH 4.5) or citrate buffer alone. Hosts were sacrificed on day 3 andtheir pancreatic and inguinal lymph nodes examined for CFSE� cells.
In vitro T cell proliferation assay
Splenic 8.3-CD8� T cells (2 � 104 cells/well) were cultured, in triplicate,either alone or with 2 � 104 cells/well (1:1), 5� 103 cells/well (1:4), or 2 �103 cells/well (1:10) CD4�CD25� or CD4�CD25� T cells from NOD orNOD-PI mice in the presence of IGRP206–214 (0.1 �M)-pulsed NODsplenic irradiated DCs (104 cells/well). The cells were cultured in 96-wellround-bottom plates in 200 �l of RPMI 1640 (Invitrogen Life Technolo-gies) supplemented with antibiotics, 2 mM glutamine, nonessential aminoacids, 50 �M mercaptoethanol, and 10% FCS for 3 days at 37°C in 5%CO2. Wells were pulsed with 1 �Ci [3H]thymidine during the last 18 h ofculture.
MHC class I staining
Islets of Langerhans were isolated from mice according to methods pre-viously described (14). For MHC class I staining, antisera used were anti-mouse H-2Db (28–14-8; BD Pharmingen) followed by allophycocyanin-conjugated streptavidin (Caltag Laboratories). Leukocytes were excludedfrom analysis by staining with anti-CD45 conjugated to PerCp-Cy5.5(3F11; BD Pharmingen), and � cells were identified based on their highautofluorescence (14).
Anti-CD4 Ab treatment
Five 7-wk-old NOD-PI/NOD8.3 mice were injected with anti-CD4 mAb(GK1.5) or isotype control (GL121). The dose was 1 mg followed by 0.5mg after 3 days. Thereafter 0.5 mg was injected every week until the mousebecame diabetic or for 8 wk. Mice were monitored for success of depletionusing noncompeting anti-CD4 (clone RM4-4; BD Pharmingen).
Histological analysis
Immunohistochemical staining and scoring of frozen pancreas sections wasperformed as described (15, 16). Mice were monitored for diabetes asdescribed (6).
Insulin autoantibody assay (IAA)
IAA were measured with a 96-well filtration plate micro IAA assay asdescribed. (6) We have participated in all the Diabetes Autoantibody Stan-
dardization Program workshops. In the murine IAA workshop (2002), thesensitivity and specificity for IAA were 69 and 83%, respectively.
Statistics
Insulin autoantibody levels and insulitis scores were analyzed by Student’st test. Survival curves were analyzed with the log-rank test. Statistical testsused PRISM software (version 3.02; GraphPad). Values of p � 0.05 wasconsidered significant.
ResultsNOD-PI/NOD8.3 mice are protected from diabetes
We have shown that tolerance to proinsulin prevents expansion ofIGRP206–214-specific T cells (6). To study the effect of tolerance toinsulin on an expanded pool of autoreactive T cells, we crossedNOD8.3 TCR transgenic mice to NOD-PI mice.
The proportions of CD4� or CD8� T cells in NOD-PI/NOD8.3mice were similar to NOD8.3 mice, and there was no difference innumbers of IGRP206–214-specific T cells, suggesting normal de-velopment of CD8� T cells in NOD-PI/NOD8.3 mice (Fig. 1, Aand B). IGRP206–214-specific naive T cells from the spleens ofNOD-PI/NOD8.3 mice proliferated in response to the peptide bothin vitro and in vivo similarly to IGRP206–214-specific T cells fromNOD8.3 mice (Fig. 1, C and D). Moreover, IGRP206–214-specificsplenic T cells from NOD-PI/NOD8.3 and NOD8.3 mice secretedIFN-� in response to the peptide to a similar extent, and spleno-cytes from these mice transfer diabetes into irradiated NOD miceto a similar extent, suggesting normal cytotoxic potential (Fig. 1,E and F).
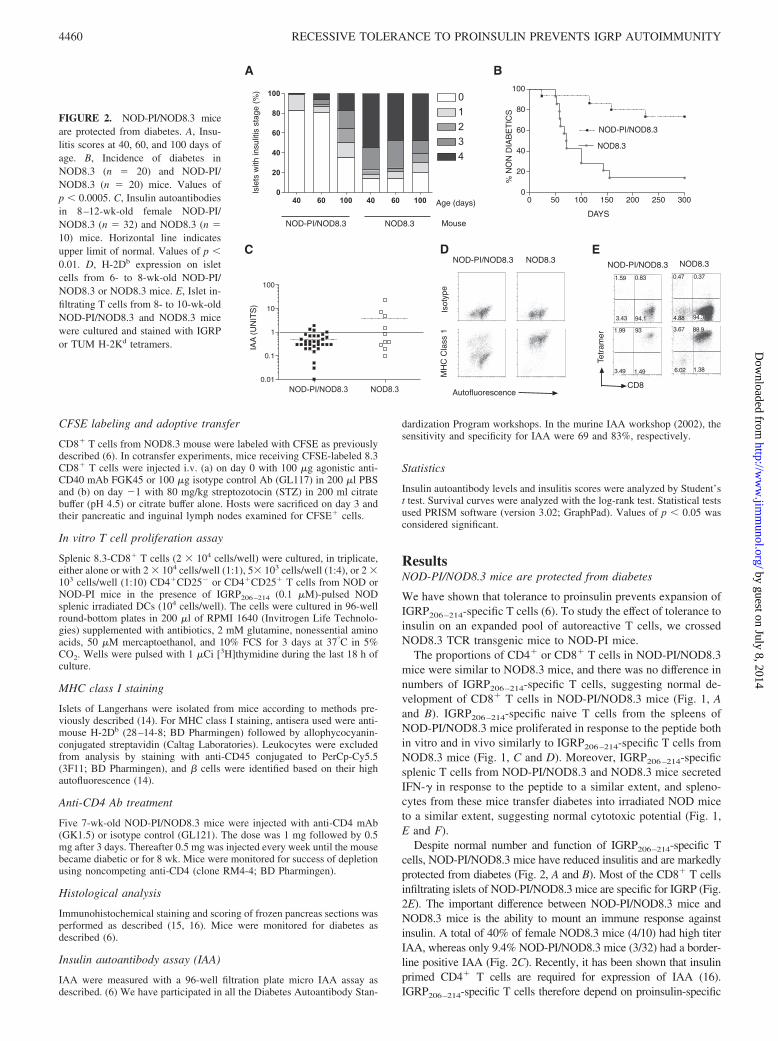
Despite normal number and function of IGRP206–214-specific Tcells, NOD-PI/NOD8.3 mice have reduced insulitis and are markedlyprotected from diabetes (Fig. 2, A and B). Most of the CD8� T cellsinfiltrating islets of NOD-PI/NOD8.3 mice are specific for IGRP (Fig.2E). The important difference between NOD-PI/NOD8.3 mice andNOD8.3 mice is the ability to mount an immune response againstinsulin. A total of 40% of female NOD8.3 mice (4/10) had high titerIAA, whereas only 9.4% NOD-PI/NOD8.3 mice (3/32) had a border-line positive IAA (Fig. 2C). Recently, it has been shown that insulinprimed CD4� T cells are required for expression of IAA (16).IGRP206–214-specific T cells therefore depend on proinsulin-specific
DAYS
FIGURE 2. NOD-PI/NOD8.3 miceare protected from diabetes. A, Insu-litis scores at 40, 60, and 100 days ofage. B, Incidence of diabetes inNOD8.3 (n � 20) and NOD-PI/NOD8.3 (n � 20) mice. Values ofp � 0.0005. C, Insulin autoantibodiesin 8–12-wk-old female NOD-PI/NOD8.3 (n � 32) and NOD8.3 (n �10) mice. Horizontal line indicatesupper limit of normal. Values of p �0.01. D, H-2Db expression on isletcells from 6- to 8-wk-old NOD-PI/NOD8.3 or NOD8.3 mice. E, Islet in-filtrating T cells from 8- to 10-wk-oldNOD-PI/NOD8.3 and NOD8.3 micewere cultured and stained with IGRPor TUM H-2Kd tetramers.
4460 RECESSIVE TOLERANCE TO PROINSULIN PREVENTS IGRP AUTOIMMUNITY
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
immune responses for their full activation and ability to mediate � celldestruction.
Dominant tolerance does not account for protection fromdiabetes in NOD-PI/NOD8.3 mice
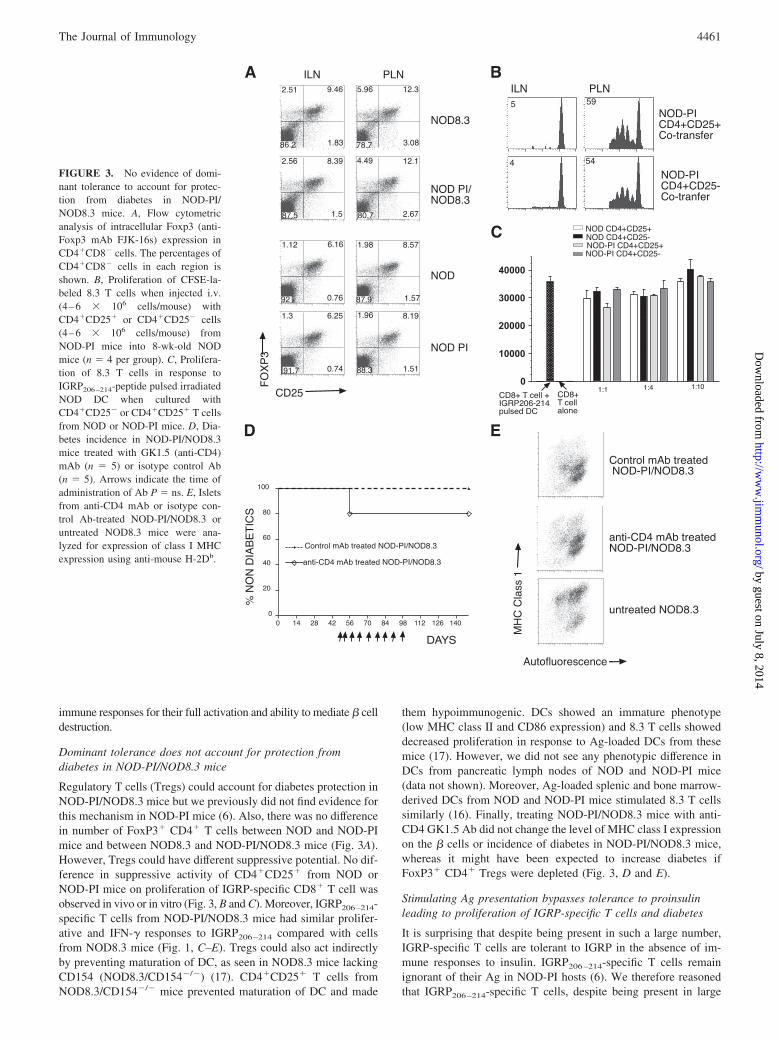
Regulatory T cells (Tregs) could account for diabetes protection inNOD-PI/NOD8.3 mice but we previously did not find evidence forthis mechanism in NOD-PI mice (6). Also, there was no differencein number of FoxP3� CD4� T cells between NOD and NOD-PImice and between NOD8.3 and NOD-PI/NOD8.3 mice (Fig. 3A).However, Tregs could have different suppressive potential. No dif-ference in suppressive activity of CD4�CD25� from NOD orNOD-PI mice on proliferation of IGRP-specific CD8� T cell wasobserved in vivo or in vitro (Fig. 3, B and C). Moreover, IGRP206–214-specific T cells from NOD-PI/NOD8.3 mice had similar prolifer-ative and IFN-� responses to IGRP206–214 compared with cellsfrom NOD8.3 mice (Fig. 1, C–E). Tregs could also act indirectlyby preventing maturation of DC, as seen in NOD8.3 mice lackingCD154 (NOD8.3/CD154�/�) (17). CD4�CD25� T cells fromNOD8.3/CD154�/� mice prevented maturation of DC and made
them hypoimmunogenic. DCs showed an immature phenotype(low MHC class II and CD86 expression) and 8.3 T cells showeddecreased proliferation in response to Ag-loaded DCs from thesemice (17). However, we did not see any phenotypic difference inDCs from pancreatic lymph nodes of NOD and NOD-PI mice(data not shown). Moreover, Ag-loaded splenic and bone marrow-derived DCs from NOD and NOD-PI mice stimulated 8.3 T cellssimilarly (16). Finally, treating NOD-PI/NOD8.3 mice with anti-CD4 GK1.5 Ab did not change the level of MHC class I expressionon the � cells or incidence of diabetes in NOD-PI/NOD8.3 mice,whereas it might have been expected to increase diabetes ifFoxP3� CD4� Tregs were depleted (Fig. 3, D and E).
Stimulating Ag presentation bypasses tolerance to proinsulinleading to proliferation of IGRP-specific T cells and diabetes
It is surprising that despite being present in such a large number,IGRP-specific T cells are tolerant to IGRP in the absence of im-mune responses to insulin. IGRP206–214-specific T cells remainignorant of their Ag in NOD-PI hosts (6). We therefore reasonedthat IGRP206–214-specific T cells, despite being present in large
FIGURE 3. No evidence of domi-nant tolerance to account for protec-tion from diabetes in NOD-PI/NOD8.3 mice. A, Flow cytometricanalysis of intracellular Foxp3 (anti-Foxp3 mAb FJK-16s) expression inCD4�CD8� cells. The percentages ofCD4�CD8� cells in each region isshown. B, Proliferation of CFSE-la-beled 8.3 T cells when injected i.v.(4–6 � 106 cells/mouse) withCD4�CD25� or CD4�CD25� cells(4–6 � 106 cells/mouse) fromNOD-PI mice into 8-wk-old NODmice (n � 4 per group). C, Prolifera-tion of 8.3 T cells in response toIGRP206–214-peptide pulsed irradiatedNOD DC when cultured withCD4�CD25� or CD4�CD25� T cellsfrom NOD or NOD-PI mice. D, Dia-betes incidence in NOD-PI/NOD8.3mice treated with GK1.5 (anti-CD4)mAb (n � 5) or isotype control Ab(n � 5). Arrows indicate the time ofadministration of Ab P � ns. E, Isletsfrom anti-CD4 mAb or isotype con-trol Ab-treated NOD-PI/NOD8.3 oruntreated NOD8.3 mice were ana-lyzed for expression of class I MHCexpression using anti-mouse H-2Db.
4461The Journal of Immunology
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

numbers, might remain ignorant of their Ag, meaning that toler-ance should be broken by increasing T cell activation signals. In-deed, FACS analysis of � cells from NOD-PI/NOD8.3 miceshowed decreased MHC class I expression, which could reducetheir targeting by CD8� T cells (Fig. 2D).
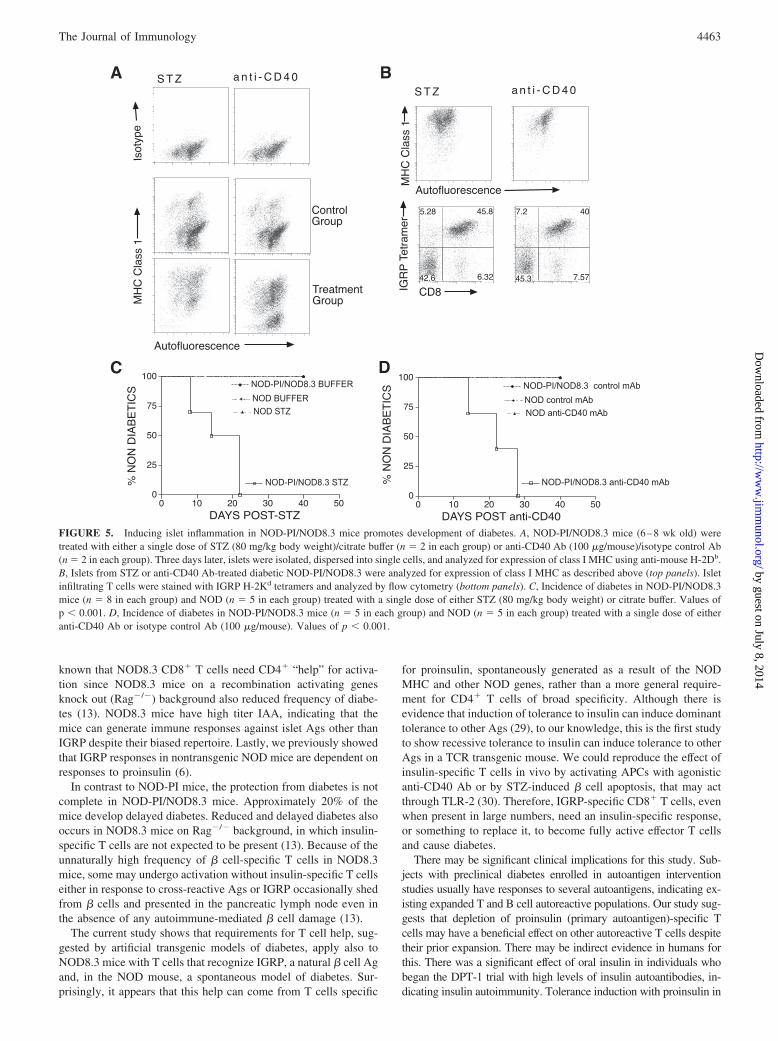
We questioned whether decreased cross-priming of IGRP206–214-specific T cells in NOD-PI hosts could be overcome in response to� cell death or islet inflammation. In the absence of infection, DCmaturation occurs following uptake of Ags from apoptotic cells orby ligation of CD40 by CD154 on CD4� T cells (18–21). Weassessed proliferation of transferred IGRP206–214-specific T cellsfollowing CD40 agonist Ab treatment or induction of � cellapoptosis by low dose STZ. A single dose of 80 mg/kg of STZor 100 �g of CD40 agonist Ab was used based on publishedreports (22–24). Each of these increased proliferation ofNOD8.3 T cells (Fig. 4, A–C).
In addition, we also analyzed IGRP206–214-specific T cells trans-ferred into perforin knockout NOD mice (NOD perforin�/�).These mice have insulitis but very little � cell destruction andreduced and delayed diabetes development (25). TransferredIGRP206–214-specific T cells proliferated to a similar extent as inNOD mice (Fig. 4D), indicating that the reduced � cell destructionin these mice does not decrease Ag presentation. Together, theseexperiments suggested that protection by induction of tolerance toproinsulin could be bypassed.
We next examined whether breaking tolerance by STZ or CD40Ab treatment in NOD-PI/NOD8.3 could induce diabetes. Admin-istration of a single dose of STZ or CD40 Ab treatment increasedMHC class I expression on � cells within 3 days (Fig. 5A) andresulted in diabetes within 1–4 wk in NOD-PI/NOD8.3 mice (Fig.5, C and D). The islet infiltrating cells were predominantlyIGRP206–214 CD8� cells in these mice (Fig. 5B). This suggests
that even with many Ag-specific T cells, immune responses toinsulin are required for full diabetes development. However, thisrequirement can be bypassed by stimuli that induce � cell apopto-sis or promote inflammation. In contrast, this treatment had noeffect on the incidence of diabetes in NOD or NOD-PI mice indi-cating (a) the treatment is stimulating T cells to mediate diabetesand not directly inducing � cell death and (b) mere stimulation of� cell apoptosis or promotion of islet inflammation is not suffi-cient, without a large number of Ag-specific T cells.
DiscussionThis study showed that immune responses against proinsulin, thatcould be eliminated by expression of proinsulin in APCs, are re-quired in the majority of NOD8.3 TCR transgenic mice for theirIGRP-specific T cells to become activated and kill � cells. Diabe-tes was reduced from nearly 100 to 20%, indicating that little T cellactivation and progression occurred in mice with proinsulin im-mune tolerance (confirmed by absent IAA). That an immune re-sponse to one Ag is required for an effective immune response byTCR transgenic T cells specific for a different Ag has not previ-ously been reported. However, aspects of previous studies are con-sistent with the current finding.
Some previous CD8� TCR transgenic mice specific for certainAgs that have been transgenically expressed in the � cell have notshown immune reactivity against that Ag (26). An activation stepis required, either infection with virus, for example LCMV whenLCMV glycoprotein is expressed in � cells (26), or cotransfer ofAg-specific CD4� T cells when a model pathogen or nonpatho-gen-derived Ag, such as influenza hemagglutinin or OVA, is ex-pressed in � cells (27, 28). Therefore exposure to pathogens withactivation of the innate immune system or “help” from CD4� Tcells can promote progression to diabetes. Further, it was already
FIGURE 4. Induction of DC mat-uration or increase in available Ag inNOD-PI mice increases cross-presen-tation of IGRP206–214-specific CD8�
T cells. A, Proliferation of CFSE-la-beled 8.3 T cells when injected intoanti-CD40 mAb or isotype control Abtreated NOD-PI (n � 6 in each group)and NOD (n � 6 in each group) mice.B, Proliferation of CFSE-labeled 8.3T cells when injected into STZ- or ci-trate buffer-treated NOD-PI (n � 8 ineach group) and NOD (n � 8 in eachgroup) mice. C, NOD and NOD-PImice were treated i.v. either with STZ(80 mg/kg) (n � 2 in each group) orcitrate buffer (n � 2 in each group).Islets were harvested 3 h later, incu-bated for 12 h in CMRL medium1066, and dispersed into single cells.Dispersed cells were incubated in aPI-containing hypotonic solution, andnuclei were analyzed by flow cytom-etry. Data show typical profiles andthe percentage of fragmented nuclei.D, Proliferation of CFSE-labeled 8.3T cells when injected into NOD (n �4) and NOD perforin�/� (n � 4)mice. Representative data from 2 to 4independent experiments is shown.
4462 RECESSIVE TOLERANCE TO PROINSULIN PREVENTS IGRP AUTOIMMUNITY
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
known that NOD8.3 CD8� T cells need CD4� “help” for activa-tion since NOD8.3 mice on a recombination activating genesknock out (Rag�/�) background also reduced frequency of diabe-tes (13). NOD8.3 mice have high titer IAA, indicating that themice can generate immune responses against islet Ags other thanIGRP despite their biased repertoire. Lastly, we previously showedthat IGRP responses in nontransgenic NOD mice are dependent onresponses to proinsulin (6).
In contrast to NOD-PI mice, the protection from diabetes is notcomplete in NOD-PI/NOD8.3 mice. Approximately 20% of themice develop delayed diabetes. Reduced and delayed diabetes alsooccurs in NOD8.3 mice on Rag�/� background, in which insulin-specific T cells are not expected to be present (13). Because of theunnaturally high frequency of � cell-specific T cells in NOD8.3mice, some may undergo activation without insulin-specific T cellseither in response to cross-reactive Ags or IGRP occasionally shedfrom � cells and presented in the pancreatic lymph node even inthe absence of any autoimmune-mediated � cell damage (13).
The current study shows that requirements for T cell help, sug-gested by artificial transgenic models of diabetes, apply also toNOD8.3 mice with T cells that recognize IGRP, a natural � cell Agand, in the NOD mouse, a spontaneous model of diabetes. Sur-prisingly, it appears that this help can come from T cells specific
for proinsulin, spontaneously generated as a result of the NODMHC and other NOD genes, rather than a more general require-ment for CD4� T cells of broad specificity. Although there isevidence that induction of tolerance to insulin can induce dominanttolerance to other Ags (29), to our knowledge, this is the first studyto show recessive tolerance to insulin can induce tolerance to otherAgs in a TCR transgenic mouse. We could reproduce the effect ofinsulin-specific T cells in vivo by activating APCs with agonisticanti-CD40 Ab or by STZ-induced � cell apoptosis, that may actthrough TLR-2 (30). Therefore, IGRP-specific CD8� T cells, evenwhen present in large numbers, need an insulin-specific response,or something to replace it, to become fully active effector T cellsand cause diabetes.
There may be significant clinical implications for this study. Sub-jects with preclinical diabetes enrolled in autoantigen interventionstudies usually have responses to several autoantigens, indicating ex-isting expanded T and B cell autoreactive populations. Our study sug-gests that depletion of proinsulin (primary autoantigen)-specific Tcells may have a beneficial effect on other autoreactive T cells despitetheir prior expansion. There may be indirect evidence in humans forthis. There was a significant effect of oral insulin in individuals whobegan the DPT-1 trial with high levels of insulin autoantibodies, in-dicating insulin autoimmunity. Tolerance induction with proinsulin in
FIGURE 5. Inducing islet inflammation in NOD-PI/NOD8.3 mice promotes development of diabetes. A, NOD-PI/NOD8.3 mice (6–8 wk old) weretreated with either a single dose of STZ (80 mg/kg body weight)/citrate buffer (n � 2 in each group) or anti-CD40 Ab (100 �g/mouse)/isotype control Ab(n � 2 in each group). Three days later, islets were isolated, dispersed into single cells, and analyzed for expression of class I MHC using anti-mouse H-2Db.B, Islets from STZ or anti-CD40 Ab-treated diabetic NOD-PI/NOD8.3 were analyzed for expression of class I MHC as described above (top panels). Isletinfiltrating T cells were stained with IGRP H-2Kd tetramers and analyzed by flow cytometry (bottom panels). C, Incidence of diabetes in NOD-PI/NOD8.3mice (n � 8 in each group) and NOD (n � 5 in each group) treated with a single dose of either STZ (80 mg/kg body weight) or citrate buffer. Values ofp � 0.001. D, Incidence of diabetes in NOD-PI/NOD8.3 mice (n � 5 in each group) and NOD (n � 5 in each group) treated with a single dose of eitheranti-CD40 Ab or isotype control Ab (100 �g/mouse). Values of p � 0.001.
4463The Journal of Immunology
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

such subjects might, however, be bypassed if stimuli that enhance Agpresentation occur. Expansion of Ag-specific T cells in NOD8.3 miceis a result of expression of the TCR transgene in T cell precursors andthymic positive selection and is independent of T cell help. This hasallowed us to show that NOD8.3 T cells need T cell help for effectorfunction independent of proliferation. In a nontransgenic T cell rep-ertoire, expansion and activation of Ag-specific T cells are not sepa-rated in this way. The current study suggests there might be beneficialeffects of terminating insulin-specific CD4� T cell help in NOD miceor humans with established preclinical diabetes and autoimmunity tomultiple � cell Ags.
AcknowledgmentsWe thank Jie Lin for help with tetramer production and Rochelle Ayala fortechnical assistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Atkinson, M. A., and N. K. Maclaren. 1994. The pathogenesis of insulin-depen-
dent diabetes mellitus. N. Engl. J. Med. 331: 1428–1436.2. Harrison, L. C. 1992. Islet cell antigens in insulin-dependent diabetes: Pandora’s
box revisited. Immunol. Today 13: 348–352.3. French, M. B., J. Allison, D. S. Cram, H. E. Thomas, M. Dempsey-Collier,
A. Silva, H. M. Georgiou, T. W. Kay, L. C. Harrison, and A. M. Lew. 1997.Transgenic expression of mouse proinsulin II prevents diabetes in nonobese di-abetic mice. Diabetes 46: 34–39.
4. Jaeckel, E., M. A. Lipes, and H. von Boehmer. 2004. Recessive tolerance topreproinsulin 2 reduces but does not abolish type 1 diabetes. Nat. Immunol. 5:1028–1035.
5. Nakayama, M., N. Abiru, H. Moriyama, N. Babaya, E. Liu, D. Miao, L. Yu,D. R. Wegmann, J. C. Hutton, J. F. Elliott, and G. S. Eisenbarth. 2005. Prime rolefor an insulin epitope in the development of type 1 diabetes in NOD mice. Nature435: 220–223.
6. Krishnamurthy, B., N. L. Dudek, M. D. McKenzie, A. W. Purcell, A. G. Brooks,S. Gellert, P. G. Colman, L. C. Harrison, A. M. Lew, H. E. Thomas, andT. W. Kay. 2006. Responses against islet antigens in NOD mice are prevented bytolerance to proinsulin but not IGRP. J. Clin. Invest. 116: 3258–3265.
7. Rewers, M., T. L. Bugawan, J. M. Norris, A. Blair, B. Beaty, M. Hoffman,R. S. McDuffie, Jr., R. F. Hamman, G. Klingensmith, G. S. Eisenbarth, andH. A. Erlich. 1996. Newborn screening for HLA markers associated with IDDM:diabetes autoimmunity study in the young (DAISY). Diabetologia 39: 807–812.
8. Homann, D., and G. S. Eisenbarth. 2006. An immunologic homunculus for type1 diabetes. J. Clin. Invest. 116: 1212–1215.
9. Harrison, L. C., and D. A. Hafler. 2000. Antigen-specific therapy for autoimmunedisease. Curr. Opin. Immunol. 12: 704–711.
10. Atkinson, M. A., N. K. Maclaren, and R. Luchetta. 1990. Insulitis and diabetesin NOD mice reduced by prophylactic insulin therapy. Diabetes 39: 933–937.
11. Skyler, J. S., J. P. Krischer, J. Wolfsdorf, C. Cowie, J. P. Palmer, C. Greenbaum,D. Cuthbertson, L. E. Rafkin-Mervis, H. P. Chase, and E. Leschek. 2005. Effectsof oral insulin in relatives of patients with type 1 diabetes: the diabetes preventiontrial–type 1. Diabetes Care 28: 1068–1076.
12. Diabetes Prevention Trial–Type 1 Diabetes Study Group. 2002. Effects of insulinin relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 346:1685–1691.
13. Verdaguer, J., D. Schmidt, A. Amrani, B. Anderson, N. Averill, andP. Santamaria. 1997. Spontaneous autoimmune diabetes in monoclonal T cellnonobese diabetic mice. J. Exp. Med. 186: 1663–1676.
14. Darwiche, R., M. M. Chong, P. Santamaria, H. E. Thomas, and T. W. Kay. 2003.Fas is detectable on � cells in accelerated, but not spontaneous, diabetes in nono-bese diabetic mice. J. Immunol. 170: 6292–6297.
15. Thomas, H. E., J. L. Parker, R. D. Schreiber, and T. W. Kay. 1998. IFN-� actionon pancreatic � cells causes class I MHC upregulation but not diabetes. J. Clin.Invest. 102: 1249–1257.
16. Nakayama, M., J. N. Beilke, J. M. Jasinski, M. Kobayashi, D. Miao, M. Li,M. G. Coulombe, E. Liu, J. F. Elliott, R. G. Gill, and G. S. Eisenbarth. 2007.Priming and effector dependence on insulin B:9–23 peptide in NOD islet auto-immunity. J. Clin. Invest. 117: 1835–1843.
17. Amrani, A., P. Serra, J. Yamanouchi, B. Han, S. Thiessen, J. Verdaguer, andP. Santamaria. 2002. CD154-dependent priming of diabetogenic CD4� T cellsdissociated from activation of antigen-presenting cells. Immunity 16: 719–732.
18. Albert, M. L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigenfrom apoptotic cells and induce class I-restricted CTLs. Nature 392: 86–89.
19. Rovere, P., C. Vallinoto, A. Bondanza, M. C. Crosti, M. Rescigno,P. Ricciardi-Castagnoli, C. Rugarli, and A. A. Manfredi. 1998. Bystander apo-ptosis triggers dendritic cell maturation and antigen-presenting function. J. Im-munol. 161: 4467–4471.
20. Cella, M., D. Scheidegger, K. Palmer-Lehmann, P. Lane, A. Lanzavecchia, andG. Alber. 1996. Ligation of CD40 on dendritic cells triggers production of highlevels of interleukin-12 and enhances T cell stimulatory capacity: T-T help viaAPC activation. J. Exp. Med. 184: 747–752.
21. Grewal, I. S., and R. A. Flavell. 1998. CD40 and CD154 in cell-mediated im-munity. Annu. Rev. Immunol. 16: 111–135.
22. Hugues, S., E. Mougneau, W. Ferlin, D. Jeske, P. Hofman, D. Homann,L. Beaudoin, C. Schrike, M. Von Herrath, A. Lehuen, and N. Glaichenhaus.2002. Tolerance to islet antigens and prevention from diabetes induced by limitedapoptosis of pancreatic � cells. Immunity 16: 169–181.
23. Serra, P., A. Amrani, J. Yamanouchi, B. Han, S. Thiessen, T. Utsugi,J. Verdaguer, and P. Santamaria. 2003. CD40 ligation releases immature den-dritic cells from the control of regulatory CD4�CD25� T cells. Immunity 19:877–889.
24. Horwitz, M. S., A. Ilic, C. Fine, E. Rodriguez, and N. Sarvetnick. 2002. Presentedantigen from damaged pancreatic � cells activates autoreactive T cells in virus-mediated autoimmune diabetes. J. Clin. Invest. 109: 79–87.
25. Kagi, D., B. Odermatt, P. Seiler, R. M. Zinkernagel, T. W. Mak, andH. Hengartner. 1997. Reduced incidence and delayed onset of diabetes in per-forin-deficient nonobese diabetic mice. J. Exp. Med. 186: 989–997.
26. Ohashi, P. S., S. Oehen, P. Aichele, H. Pircher, B. Odermatt, P. Herrera,Y. Higuchi, K. Buerki, H. Hengartner, and R. M. Zinkernagel. 1993. Induction ofdiabetes is influenced by the infectious virus and local expression of MHC classI and tumor necrosis factor-�. J. Immunol. 150: 5185–5194.
27. Hernandez, J., S. Aung, K. Marquardt, and L. A. Sherman. 2002. Uncoupling ofproliferative potential and gain of effector function by CD8� T cells respondingto self-antigens. J. Exp. Med. 196: 323–333.
28. Kurts, C., F. R. Carbone, M. Barnden, E. Blanas, J. Allison, W. R. Heath, andJ. F. Miller. 1997. CD4� T cell help impairs CD8� T cell deletion induced bycross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 186:2057–2062.
29. Von Herrath, M. G., T. Dyrberg, and M. B. Oldstone. 1996. Oral insulin treat-ment suppresses virus-induced antigen-specific destruction of � cells and pre-vents autoimmune diabetes in transgenic mice. J. Clin. Invest. 98: 1324–1331.
30. Kim, H. S., M. S. Han, K. W. Chung, S. Kim, E. Kim, M. J. Kim, E. Jang,H. A. Lee, J. Youn, S. Akira, and M. S. Lee. 2007. Toll-like receptor 2 senses�-cell death and contributes to the initiation of autoimmune diabetes. Immunity27: 321–333.
4464 RECESSIVE TOLERANCE TO PROINSULIN PREVENTS IGRP AUTOIMMUNITY
by guest on July 8, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















